ES2825035T3 - Nucleósidos, nucleótidos y análogos de estos sustituidos con 4-azidoalquilo - Google Patents
Nucleósidos, nucleótidos y análogos de estos sustituidos con 4-azidoalquilo Download PDFInfo
- Publication number
- ES2825035T3 ES2825035T3 ES18174398T ES18174398T ES2825035T3 ES 2825035 T3 ES2825035 T3 ES 2825035T3 ES 18174398 T ES18174398 T ES 18174398T ES 18174398 T ES18174398 T ES 18174398T ES 2825035 T3 ES2825035 T3 ES 2825035T3
- Authority
- ES
- Spain
- Prior art keywords
- optionally substituted
- compound
- alkyl
- mmol
- hydrogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CCC(CC(C)CC(CC1)CC1C(C1)C11C2=CC12)C(C)CCC1*C1 Chemical compound CCC(CC(C)CC(CC1)CC1C(C1)C11C2=CC12)C(C)CCC1*C1 0.000 description 9
- QDQVXVRZVCTVHE-YFKPBYRVSA-N CC(C)OC([C@H](C)N)=O Chemical compound CC(C)OC([C@H](C)N)=O QDQVXVRZVCTVHE-YFKPBYRVSA-N 0.000 description 1
- RDOXTESZEPMUJZ-UHFFFAOYSA-N COc1ccccc1 Chemical compound COc1ccccc1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 1
- FDFQGGVYSMLKSP-VETPBDLOSA-N C[C@@](COP(C)(Oc1ccccc1)=O)(CC1)O[C@H]1N(C=CC(N1)=O)C1=O Chemical compound C[C@@](COP(C)(Oc1ccccc1)=O)(CC1)O[C@H]1N(C=CC(N1)=O)C1=O FDFQGGVYSMLKSP-VETPBDLOSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
- A61K31/7072—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid having two oxo groups directly attached to the pyrimidine ring, e.g. uridine, uridylic acid, thymidine, zidovudine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
- A61K31/708—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid having oxo groups directly attached to the purine ring system, e.g. guanosine, guanylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/10—Pyrimidine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/20—Purine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- Virology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, **(Ver fórmula)** donde: **(Ver fórmula)** B1A es RA es hidrógeno; R1A se selecciona del grupo constituido por hidrógeno, un acilo opcionalmente sustituido, un aminoácido conectado a **(Ver fórmula)** través de O opcionalmente sustituido, y Ra1 y Ra2 son independientemente hidrógeno o deuterio; R2A es un azidoalquilo C1-6; R3A se selecciona del grupo constituido por OH, -OC(=O)R"A y un aminoácido conectado a través de O opcionalmente sustituido; R4A es halógeno; R5A es hidrógeno; R6A y R7A se seleccionan independientemente del grupo constituido por ausente, hidrógeno, **(Ver fórmula)** R6A es **(Ver fórmula)** m y R7A está ausente o es hidrógeno; R8A es un arilo opcionalmente sustituido; R9A es un aminoácido conectado a través de N opcionalmente sustituido o un derivado de tipo éster de un aminoácido conectado a través de N opcionalmente sustituido; R10A y R11A son independientemente un aminoácido conectado a través de N opcionalmente sustituido o un derivado de tipo éster de un aminoácido conectado a través de N opcionalmente sustituido; R12A, R13A y R14A pueden independientemente estar ausentes o ser hidrógeno; R22A y R23A se seleccionan independientemente del grupo constituido por hidrógeno, un alquilo C1-24 opcionalmente sustituido y un arilo opcionalmente sustituido; R24A se selecciona del grupo constituido por hidrógeno, un alquilo C1-24 opcionalmente sustituido, un arilo opcionalmente sustituido, un -O-alquilo C1-24 opcionalmente sustituido, un -O-arilo opcionalmente sustituido, un -Oheteroarilo opcionalmente sustituido y un -O-heterociclilo monocíclico opcionalmente sustituido; R25A se selecciona independientemente del grupo constituido por hidrógeno, un alquilo C1-24 opcionalmente sustituido y un arilo opcionalmente sustituido; R"A es un alquilo C1-24 opcionalmente sustituido; m es 0 o 1; s es 0; t es 0; y Z1A, Z2A, Z3A y Z4A son independientemente O o S.
Description
DESCRIPCIÓN
Nucleósidos, nucleótidos y análogos de estos sustituidos con 4’-azidoalquilo
REFERENCIA A LA LISTA DE SECUENCIAS
La presente solicitud se presenta con una Lista de secuencias en formato electrónico. La Lista de secuencias se proporciona como un archivo titulado ALIOS076.txt, creado el 23 de junio de 2014, que tiene un tamaño de aproximadamente 4 kb.
ANTECEDENTES
Campo
La presente solicitud se refiere a los campos de la química, bioquímica y medicina. Más concretamente, en la presente se describen nucleósidos, nucleótidos y análogos de estos, composiciones farmacéuticas que incluyen uno o más nucleósidos, nucleótidos y análogos de estos, y métodos para sintetizarlos. También se describen en la presente métodos para mejorar y/o tratar una infección vírica provocada por paramixovirus con uno o más nucleósidos, nucleótidos y análogos de estos.
Descripción
Las infecciones víricas respiratorias, incluidas las infecciones víricas del aparato respiratorio superior e inferior, se contagian y representan la causa principal de fallecimientos de millones de personas cada año. Las infecciones víricas del aparato respiratorio superior implican la nariz, los senos nasales, la faringe y/o la laringe. Las infecciones víricas del aparato respiratorio inferior implican el sistema respiratorio situado por debajo de las cuerdas vocales, que incluye la tráquea, los bronquios primarios y los pulmones.
Los análogos de nucleósidos son una clase de compuestos que se ha demostrado que ejercen actividad antivírica tanto in vivo como in vitro y, por lo tanto, han sido objeto de una investigación generalizada para el tratamiento de infecciones víricas. Normalmente, los análogos de nucleósidos son compuestos terapéuticamente inactivos que se convierten por acción de enzimas víricas o del huésped en sus anti-metabolitos activos respectivos, los cuales, a su vez, pueden inhibir polimerasas que participan en la proliferación celular o vírica. La activación se produce mediante varios mecanismos tales como la adición de uno o más grupos fosfato y, o de forma combinada con, otros procesos metabólicos.
GORE, K.R. ET AL.: «Influence of 2'-fluoro versus 2'O-methyl substituent on the sugar puckering of 4'-C-aminomethyluridine», JOURNAL OFORGANIC CHEMISTRY, vol. 78, 9 de septiembre de 2013 (2013-09-09), páginas 9956-9962 se refiere a la síntesis de 4'-C-aminometil-2'desoxi-2'-fluorouridina.
PFUNDHELLER, H.M. ET AL.: «Oligonucleotides containing novel 4'-C- or 3'-C-(aminoalkyl)-branches thymidines», HELVETICA CHIMICA ACTA., vol. 83, 2000, páginas 128-151 se refiere a cuatro nucleósidos con ramificaciones 3'-C y 4’ y a su transformación en los correspondientes bloques estructurales de tipo 3'-O-fosforamidito para la síntesis automatizada de oligonucleótidos.
El documento WO 2012/040124 se refiere a nucleósidos, nucleótidos y análogos de estos, composiciones farmacéuticas que incluyen uno o más de los nucleósidos, nucleótidos y análogos de estos, y métodos para sintetizarlos.
S.-Y. PARK ET AL.: «Efficacy of Oral Ribavirin in Hematologic Disease Patients with Paramyxovirus Infection: Analytic Strategy Using Propensity Scores», ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, vol. 57, n ° 2, 1 de febrero de 2013 (2013-02-01), páginas 983-989 se refiere al efecto de la ribavirina oral sobre los resultados clínicos de infecciones provocadas por paramixovirus en pacientes con enfermedades hematológicas.
COMPENDIO
La protección requerida para esta invención es como se define en las reivindicaciones adjuntas. Por consiguiente, algunas realizaciones divulgadas en la presente se refieren a un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este. Algunas realizaciones divulgadas en la presente se refieren a una composición farmacéutica que comprende una cantidad eficaz de un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, y un portador, diluyente o excipiente farmacéuticamente aceptable o una combinación de estos. En la presente también se divulgan métodos para mejorar y/o tratar una infección vírica provocada por paramixovirus que pueden incluir administrar a un sujeto que padece la infección vírica provocada por paramixovirus una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, o una composición farmacéutica que incluye uno o más compuestos de Fórmula (I) o una sal farmacéuticamente aceptable de estos. En
la presente también se divulga el uso de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, en la elaboración de un medicamento para mejorar y/o tratar una infección vírica provocada por paramixovirus. En la presente también se divulgan compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, que se pueden utilizar para mejorar y/o tratar una infección vírica provocada por paramixovirus. En la presente también se divulgan métodos para mejorar y/o tratar una infección vírica provocada por paramixovirus que pueden incluir poner en contacto una célula infectada con el paramixovirus con una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, o una composición farmacéutica que incluye uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos. En la presente también se divulgan métodos para inhibir la replicación de un paramixovirus que pueden incluir poner en contacto una célula infectada con el paramixovirus con una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, o una composición farmacéutica que incluye uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos.
En la presente también se divulgan métodos para mejorar y/o tratar una infección vírica provocada por paramixovirus que pueden incluir administrar a un sujeto que padece la infección vírica una cantidad eficaz de un compuesto descrito en la presente o una sal farmacéuticamente aceptable de este (por ejemplo, uno o más compuestos de Fórmula (I) o una sal farmacéuticamente aceptable de estos), o una composición farmacéutica que incluye uno o más compuestos descritos en la presente, combinados con uno o más agentes descritos en la presente. En la presente también se divulgan métodos para mejorar y/o tratar una infección vírica provocada por paramixovirus que pueden incluir poner en contacto una célula infectada con el virus con una cantidad eficaz de un compuesto descrito en la presente o una sal farmacéuticamente aceptable de este (por ejemplo, uno o más compuestos de Fórmula (I) o una sal farmacéuticamente aceptable de estos), o una composición farmacéutica que incluye uno o más compuestos descritos en la presente, combinados con uno o más agentes descritos en la presente.
BREVE DESCRIPCIÓN DE LOS DIBUJOS
La Figura 1 muestra ejemplos de agentes anti-VRS.
DESCRIPCIÓN DETALLADA
La familia paramyxoviridae es una familia de virus de ARN monocatenario. Varios géneros de la familia paramyxoviridae incluyen respirovirus, rubulavirus, pneumovirus y metapneumovirus. Estos virus se pueden transmitir de persona a persona mediante el contacto directo o cercano con microgotas respiratorias o fómites contaminados.
El virus respiratorio sincitial humano (VRS) es una especie de pneumovirus y un virus de ARN monocatenario negativo. El VRS puede provocar infecciones respiratorias y se puede asociar con la bronquiolitis y la neumonía. Los síntomas de una infección provocada por el VRS incluyen tos, estornudos, secreción nasal, fiebre, reducción del apetito, dolor de garganta, cefalea y sibilancia. El VRS es la causa más común de bronquiolitis y neumonía en los niños menores de un año a nivel mundial y puede ser la causa de traqueobronquitis en niños mayores y adultos. En los Estados Unidos, entre 75000 y 125000 niños ingresan en los hospitales cada año con el VRS. Entre los adultos mayores de 65 años, se estima que se pueden atribuir 14000 fallecimientos y 177000 ingresos en los hospitales al VRS.
En la actualidad, las opciones de tratamiento para las personas infectadas con el VRS son limitadas. Los antibióticos, que se prescriben normalmente para tratar infecciones bacterianas, y la medicación sin receta médica no son eficaces para tratar el VRS y pueden ayudar tan solo a aliviar algunos de los síntomas. En los casos graves, se puede prescribir un bronquiodilatador nebulizado, tal como albuterol, para aliviar algunos de los síntomas tales como la sibilancia. RespiGam® (RSV-IGIV, MedImmune, aprobado para niños de alto riesgo menores de 24 meses) y Synagis® (palivizumab, MedImmune, aprobado para niños de alto riesgo menores de 24 meses) han sido aprobados para el uso profiláctico contra el VRS, y Virzole® (ribavirina en aerosol, ICN pharmaceuticals) ha sido aprobado para el tratamiento del VRS.
Los virus de parainfluenza son habitualmente virus de ARN de sentido negativo. Las especies de respirovirus incluyen los virus de parainfluenza humana 1 y 3; y las especies de rubulavirus incluyen los virus de parainfluenza humana 2 y 4. Los virus de parainfluenza humana incluyen cuatro tipos de serotipo (VPIH-1, VPIH-2, VPIH-3 y VPIH-4) y el virus de parainfluenza humana 4 (VPIH-4) incluye dos subgrupos antigénicos, A y B. Los virus de parainfluenza humana pueden provocar infecciones del aparato respiratorio superior e inferior. El virus de parainfluenza humana 1 (VPIH-1) y el virus de parainfluenza humana 2 (VPIH-2) se pueden asociar con la laringotraqueobronquitis; el virus de parainfluenza humana 3 (VPIH-3) se puede asociar con la bronquiolitis y la neumonía. De acuerdo con los Centros para el Control y la Prevención de Enfermedades (CDC, por sus siglas en inglés), no existen vacunas contra los virus de parainfluenza humana.
Una especie de metapneumovirus es el metapneumovirus humano. El metapneumovirus humano es un virus de ARN monocatenario negativo. Los metapneumovirus humanos pueden provocar infecciones del aparato respiratorio tales como infecciones del aparato respiratorio superior e inferior, en seres humanos, por ejemplo, niños pequeños.
Las infecciones respiratorias incluyen resfriados, laringotraqueobronquitis, neumonía, bronquitis y bronquiolitis. Los síntomas pueden incluir tos, secreción nasal, congestión nasal, dolor de garganta, fiebre, dificultad para respirar, respiración anómalamente rápida, sibilancia, vómitos, diarrea e infecciones de oído.
Definiciones
A menos que se defina de otro modo, todos los términos técnicos y científicos utilizados en la presente tienen el mismo significado que interpreta habitualmente un experto en la técnica. Todas las patentes, solicitudes, solicitudes publicadas y otras publicaciones a las que se hace referencia en la presente se incorporan por referencia en su totalidad a menos que se indique lo contrario. En el caso de que exista una pluralidad de definiciones para un término de la presente, prevalecerán las de esta sección a menos que se indique de otro modo.
Tal como se utiliza(n) en la presente, cual(es)quiera grupo(s) «R» tal(es) como, sin carácter limitante, R1A, R2A, R3A, R4A R5A R6A R7A R8A R9A R10A R11A R12A R13A R14A R15A R16A R17A R18A R19A R20A R21A R22A R23A R24A R25A R26A, r27a, r28a, R29a, R30a, r31a, r32a, R33a, R34a, R35a, R36a, R37a y r38a representan sustituyentes que se pueden unir al átomo indicado. Un grupo R puede estar sustituido o no sustituido. Si se describe que dos grupos «R» se consideran «conjuntamente», los grupos R y los átomos a los cuales están unidos pueden formar un cicloalquilo, cicloalquenilo, arilo, heteroarilo o heterociclo. Por ejemplo, sin carácter limitante, si se indica que Ra y Rb de un grupo NRa Rb se consideran «conjuntamente», esto significa que están enlazados covalentemente entre sí para formar un anillo:

Además, si se describe que dos grupos «R» se consideran «conjuntamente» con el o los átomos a los cuales están unidos para formar un anillo como alternativa, los grupos R no se limitan a las variables o sustituyentes que se han definido previamente.
Siempre que se describa que un grupo está «opcionalmente sustituido», ese grupo puede estar no sustituido o sustituido con uno o más de los sustituyentes indicados. Del mismo modo, cuando se describe que un grupo está «no sustituido o sustituido», si está sustituido, el o los sustituyentes se pueden seleccionar entre uno o más de los sustituyentes indicados. Si no se indican sustituyentes, esto significa que el grupo «opcionalmente sustituido» o «sustituido» indicado puede estar sustituido con uno o más grupos seleccionados individual e independientemente entre alquilo, alquenilo, alquinilo, cicloalquilo, cicloalquenilo, arilo, heteroarilo, heterociclilo, aril(alquilo), heteroaril(alquilo), heterociclil(alquilo), hidroxi, alcoxi, acilo, ciano, halógeno, tiocarbonilo, O-carbamilo, N-carbamilo, O-tiocarbamilo, N-tiocarbamilo, C-amido, N-amido, S-sulfonamido, N-sulfonamido, C-carboxi, O-carboxi, isocianato, tiocianato, isotiocianato, azido, nitro, sililo, sulfenilo, sulfinilo, sulfonilo, haloalquilo, haloalcoxi, trihalometanosulfonilo, trihalometanosulfonamido, un amino, un grupo amino monosustituido y un grupo amino disustituido.
La expresión «Ca a Cb», tal como se utiliza en la presente, donde «a» y «b» son números enteros, se refiere al número de átomos de carbono en un grupo alquilo, alquenilo o alquinilo, o el número de átomos de carbono en el anillo de un grupo cicloalquilo, cicloalquenilo, arilo, heteroarilo o heteroaliciclilo. Es decir, el alquilo, alquenilo, alquinilo, anillo(s) del cicloalquilo, anillo(s) del cicloalquenilo, anillo(s) del arilo, anillo(s) del heteroarilo o anillo(s) del heterociclilo pueden contener de «a» a «b», inclusive, átomos de carbono. Así, por ejemplo, un grupo «alquilo C1 a C4» se refiere a todos los grupos alquilo que tienen de 1 a 4 carbonos, es decir, CH3-, CH3CH2-, CH3CH2CH2-, (CH3)2CH-, CH3CH2CH2CH2-, CH3CH2CH(CH3)- y (CH3)3C-. Si no se designan «a» ni «b» con respecto a un grupo alquilo, alquenilo, alquinilo, cicloalquilo, cicloalquenilo, arilo, heteroarilo o heterociclilo, se debe asumir el intervalo más amplio descrito en estas definiciones.
El término «alquilo», tal como se utiliza en la presente, se refiere a una cadena hidrocarbonada lineal o ramificada que comprende un grupo hidrocarbonado totalmente saturado (sin dobles ni triples enlaces). El grupo alquilo puede tener de 1 a 20 átomos de carbono (siempre que aparezca en la presente, un intervalo numérico tal como «de 1 a 20» se refiere a cada número entero en el intervalo dado, por ejemplo, «de 1 a 20 átomos de carbono» significa que el grupo alquilo puede estar constituido por 1 átomo de carbono, 2 átomos de carbono, 3 átomos de carbono, etc. hasta 20 átomos de carbono inclusive, aunque la presente definición también cubre el caso del término «alquilo» donde no se designa ningún intervalo numérico). El grupo alquilo también puede ser un alquilo de tamaño medio que tenga de 1 a 10 átomos de carbono. El grupo alquilo también podría ser un alquilo inferior que tenga de 1 a 6 átomos de carbono. El grupo alquilo de los compuestos se puede designar como «alquilo C1-C4» o con designaciones similares. A modo de ejemplo únicamente, «alquilo C1-C4» indica que hay de uno a cuatro átomos de carbono en la cadena de alquilo, es decir, la cadena de alquilo se selecciona entre metilo, etilo, propilo, isopropilo, n-butilo, isobutilo, sec-butilo y f-butilo. Los grupos alquilo habituales incluyen, sin carácter limitante, metilo, etilo, propilo, isopropilo, butilo, isobutilo, butilo terciario, pentilo y hexilo. El grupo alquilo puede estar sustituido o no sustituido.
El término «alquenilo», tal como se utiliza en la presente, se refiere a un grupo alquilo que contiene en la cadena hidrocarbonada lineal o ramificada uno o más dobles enlaces. Los ejemplos de grupos alquenilo incluyen alenilo, vinilmetilo y etenilo. Un grupo alquenilo puede estar sustituido o no sustituido.
El término «alquinilo», tal como se utiliza en la presente, se refiere a un grupo alquilo que contiene en la cadena hidrocarbonada lineal o ramificada uno o más triples enlaces. Los ejemplos de alquinilos incluyen etinilo y propinilo. Un grupo alquinilo puede estar sustituido o no sustituido.
El término «cicloalquilo», tal como se utiliza en la presente, se refiere a un sistema anular hidrocarbonado mono- o multicíclico completamente saturado (sin dobles ni triples enlaces). Cuando se compone de dos o más anillos, los anillos pueden estar unidos entre sí de manera fusionada. Los grupos cicloalquilo pueden contener de 3 a 10 átomos en el o los anillos o de 3 a 8 átomos en el o los anillos. Un grupo cicloalquilo puede estar sustituido o no sustituido. Los grupos cicloalquilo habituales incluyen, sin carácter limitante, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo y ciclooctilo.
El término «cicloalquenilo», tal como se utiliza en la presente, se refiere a un sistema anular hidrocarbonado mono- o multicíclico que contiene uno o más dobles enlaces en al menos un anillo; aunque, si hay más de uno, los dobles enlaces no pueden formar un sistema de electrones pi completamente deslocalizado en todos los anillos (de lo contrario, el grupo sería un «arilo», tal como se define en la presente). Cuando se compone de dos o más anillos, los anillos pueden estar conectados entre sí de manera fusionada. Un cicloalquenilo puede contener de 3 a 10 átomos en el o los anillos o de 3 a 8 átomos en el o los anillos. Un grupo cicloalquenilo puede estar sustituido o no sustituido.
El término «arilo», tal como se utiliza en la presente, se refiere a un sistema anular aromático monocíclico o multicíclico carbocíclico (todo carbono) (que incluye sistemas anulares fusionados donde dos anillos carbocíclicos comparten un enlace químico) que tiene un sistema de electrones pi completamente deslocalizado en todos los anillos. El número de átomos de carbono en un grupo arilo puede variar. Por ejemplo, el grupo arilo puede ser un grupo arilo C6-C14, un grupo arilo C6-C10 o un grupo arilo C6. Los ejemplos de grupos arilo incluyen, sin carácter limitante, benceno, naftaleno y azuleno. Un grupo alilo puede estar sustituido o no sustituido.
El término «heteroarilo», tal como se utiliza en la presente, se refiere a un sistema anular aromático monocíclico o multicíclico (un sistema anular con sistema de electrones pi completamente deslocalizado) que contiene uno o más heteroátomos (por ejemplo, de 1 a 5 heteroátomos), es decir, un elemento distinto de carbono, que incluye, sin carácter limitante, nitrógeno, oxígeno y azufre. El número de átomos en el o los anillos de un grupo heteroarilo puede variar. Por ejemplo, el grupo heteroarilo puede contener de 4 a 14 átomos en el o los anillos, de 5 a 10 átomos en el o los anillos o de 5 a 6 átomos en el o los anillos. Además, el término «heteroarilo» incluye sistemas anulares fusionados en los que dos anillos, tales como al menos un anillo de arilo y al menos un anillo de heteroarilo, o al menos dos anillos de heteroarilo, comparten al menos un enlace químico. Los ejemplos de anillos de heteroarilo incluyen, sin carácter limitante, furano, furazano, tiofeno, benzotiofeno, ftalazina, pirrol, oxazol, benzoxazol, 1,2,3-oxadiazol, 1,2,4-oxadiazol, tiazol, 1,2,3-tiadiazol, 1,2,4-tiadiazol, benzotiazol, imidazol, bencimidazol, indol, indazol, pirazol, benzopirazol, isoxazol, benzoisoxazol, isotiazol, triazol, benzotriazol, tiadiazol, tetrazol, piridina, piridazina, pirimidina, pirazina, purina, pteridina, quinolina, isoquinolina, quinazolina, quinoxalina, cinolina y triazina. Un grupo heteroarilo puede estar sustituido o no sustituido.
El término «heterociclilo» o «heteroaliciclilo», tal como se utiliza en la presente, se refiere a un sistema anular monocíclico, bicíclico y tricíclico de tres, cuatro, cinco, seis, siete, ocho, nueve, diez y hasta 18 miembros donde los átomos de carbono junto con de 1 a 5 heteroátomos constituyen dicho sistema anular. Un heterociclo puede contener opcionalmente uno o más enlaces insaturados situados de tal manera, sin embargo, que no se produzca un sistema de electrones pi completamente deslocalizado en todos los anillos. El o los heteroátomos son un elemento distinto de carbono que incluye, sin carácter limitante, oxígeno, azufre y nitrógeno. Un heterociclo puede contener además una o más funcionalidades de carbonilo o tiocarbonilo, para hacer que la definición incluya sistemas oxo y tio tales como lactamas, lactonas, imidas cíclicas, tioimidas cíclicas y carbamatos cíclicos. Cuando se compone de dos o más anillos, los anillos pueden estar unidos entre sí de manera fusionada. Además, cualesquiera nitrógenos en un heterociclilo o un heteroaliciclilo se pueden cuaternizar. Los grupos heterociclilo o heteroalicíclicos pueden estar sustituidos o no sustituidos. Los ejemplos de tales grupos «heterociclilo» o «heteroaliciclilo» incluyen, sin carácter limitante, 1,3-dioxina, 1,3-dioxano, 1,4-dioxano, 1,2-dioxolano, 1,3-dioxolano, 1 ,4-dioxolano, 1,3-oxatiano, 1,4-oxatiino, 1,3-oxatiolano, 1,3-ditiol, 1,3-ditiolano, 1,4-oxatiano, tetrahidro-1,4-tiazina, 2H-1,2-oxazina, maleimida, succinimida, ácido barbitúrico, ácido tiobarbitúrico, dioxopiperazina, hidantoína, dihidrouracilo, trioxano, hexahidro-1,3,5-triazina, imidazolina, imidazolidina, isoxazolina, isoxazolidina, oxazolina, oxazolidina, oxazolidinona, tiazolidina, morfolina, oxirano, W-óxido de piperidina, piperidina, piperazina, pirrolidina, pirrolidona, pirrolidiona, 4-piperidona, pirazolina, pirazolidina, 2-oxopirrolidina, tetrahidropirano, 4H-pirano, tetrahidrotiopirano, tiamorfolina, sulfóxido de tiamorfolina, tiamorfolinosulfona y sus análogos benzofusionados (por ejemplo, bencimidazolidinona, tetrahidroquinolina y 3,4-metilenodioxifenilo).
Los términos «aralquilo» y «aril(alquilo)», tal como se utilizan en la presente, se refieren a un grupo arilo conectado, como sustituyente, a través de un grupo alquileno inferior. El grupo arilo y alquileno inferior de un aralquilo puede estar sustituido o no sustituido. Los ejemplos incluyen, sin carácter limitante, bencilo, 2-fenil(alquilo), 3-fenil(alquilo) y naftil(alquilo).
Los términos «heteroaralquilo» y «heteroaril(alquilo)», tal como se utilizan en la presente, se refieren a un grupo heteroarilo conectado, como sustituyente, a través de un grupo alquileno inferior. El grupo heteroarilo y alquileno inferior de un heteroaralquilo puede estar sustituido o no sustituido. Los ejemplos incluyen, sin carácter limitante, 2-tienil(alquilo), 3-tienil(alquilo), furil(alquilo), tienil(alquilo), pirrolil(alquilo), piridil(alquilo), isoxazolil(alquilo), imidazolil(alquilo) y sus análogos benzofusionados.
Un «heteroaliciclil(alquilo)» y «heterociclil(alquilo)» se refieren a un grupo heterocíclico o heteroaliciclílico conectado, como sustituyente, a través de un grupo alquileno inferior. El grupo heterociclilo y alquileno inferior de un heteroaliciclil(alquilo) puede estar sustituido o no sustituido. Los ejemplos incluyen, sin carácter limitante, tetrahidro-2H-piran-4-il(metilo), piperidin-4-il(etilo), piperidin-4-il(propilo), tetrahidro-2H-tiopiran-4-il(metilo) y 1,3-tiazinan-4-il(metilo).
Los «grupos alquileno inferior» son grupos conectores -CH2- de cadena lineal, que forman enlaces para conectar fragmentos moleculares a través de sus átomos de carbono terminales. Los ejemplos incluyen, sin carácter limitante, metileno (-CH2-), etileno (-CH2CH2-), propileno (-CH2CH2CH2-) y butileno (-CH2CH2CH2CH2-). Se puede sustituir un grupo alquileno inferior reemplazando uno o más hidrógenos del grupo alquileno inferior por uno o más sustituyentes enumerados en la definición de «sustituido».
El término «alcoxi», tal como se utiliza en la presente, se refiere a la fórmula -OR donde R es un alquilo, un alquenilo, un alquinilo, un cicloalquilo, un cicloalquenilo, arilo, heteroarilo, heterociclilo, aril(alquilo), heteroaril(alquilo) o heterociclil(alquilo) como se define en la presente. Una lista no limitante de alcoxis son metoxi, etoxi, n-propoxi, 1-metiletoxi (isopropoxi), n-butoxi, isobutoxi, sec-butoxi, ferf-butoxi, fenoxi y benzoxi. Un alcoxi puede estar sustituido o no sustituido.
El término «acilo», tal como se utiliza en la presente, se refiere a un hidrógeno, un alquilo, un alquenilo, un alquinilo, un cicloalquilo, un cicloalquenilo, arilo, heteroarilo, heterociclilo, aril(alquilo), heteroaril(alquilo) o heterociclil(alquilo) conectados, como sustituyentes, a través de un grupo carbonilo. Los ejemplos incluyen formilo, acetilo, propanoílo, benzoílo y acrilo. Un acilo puede estar sustituido o no sustituido.
El término «hidroxialquilo», tal como se utiliza en la presente, se refiere a un grupo alquilo en el que uno o más de los átomos de hidrógeno se han reemplazado por un grupo hidroxi. Los ejemplos de grupos hidroxialquilo incluyen, sin carácter limitante, 2-hidroxietilo, 3-hidroxipropilo, 2-hidroxipropilo y 2,2-dihidroxietilo. Un hidroxialquilo puede estar sustituido o no sustituido.
El término «haloalquilo», tal como se utiliza en la presente, se refiere a un grupo alquilo en el que uno o más de los átomos de hidrógeno se ha reemplazado por un halógeno (por ejemplo, mono-haloalquilo, di-haloalquilo y trihaloalquilo). Tales grupos incluyen, sin carácter limitante, clorometilo, fluorometilo, difluorometilo, trifluorometilo, 1-cloro-2-fluorometilo y 2-fluoroisobutilo. Un haloalquilo puede estar sustituido o no sustituido.
El término «haloalcoxi», tal como se utiliza en la presente, se refiere a un grupo alcoxi en el que uno o más de los átomos de hidrógeno se han reemplazado por un halógeno (por ejemplo, mono-haloalcoxi, di-haloalcoxi y trihaloalcoxi). Tales grupos incluyen, sin carácter limitante, clorometoxi, fluorometoxi, difluorometoxi, trifluorometoxi, 1-cloro-2-fluorometoxi y 2-fluoroisobutoxi. Un haloalcoxi puede estar sustituido o no sustituido.
Un grupo «sulfenilo» se refiere a un grupo «-SR» en el que R puede ser hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, cicloalquenilo, arilo, heteroarilo, heterociclilo, aril(alquilo), heteroaril(alquilo) o heterociclil(alquilo). Un sulfenilo puede estar sustituido o no sustituido.
Un grupo «sulfinilo» se refiere a un grupo «-S(=O)-R» en el que R puede ser el mismo que se ha definido respecto a sulfenilo. Un sulfinilo puede estar sustituido o no sustituido.
Un grupo «sulfonilo» se refiere a un grupo «SO2R» en el que R puede ser el mismo que se ha definido respecto a sulfenilo. Un sulfonilo puede estar sustituido o no sustituido.
Un grupo «O-carboxi» se refiere a un grupo «RC(=O)O-» en el que R puede ser hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, cicloalquenilo, arilo, heteroarilo, heterociclilo, aril(alquilo), heteroaril(alquilo) o heterociclil(alquilo), tal como se define en la presente. Un O-carboxi puede estar sustituido o no sustituido.
Los términos «éster» y «C-carboxi» se refieren a un grupo «-C(=O)OR» en el que R puede ser el mismo que se ha definido respecto a O-carboxi. Un éster y un C-carboxi pueden estar sustituidos o no sustituidos.
Un grupo «tiocarbonilo» se refiere a un grupo «-C(=S)R» en el que R puede ser el mismo que se ha definido respecto a O-carboxi. Un tiocarbonilo puede estar sustituido o no sustituido.
Un grupo «trihalometanosulfonilo» se refiere a un grupo «X3CSO2-» en el que cada X es un halógeno.
Un grupo «trihalometanosulfonamido» se refiere a un grupo «X3CS(O)2N(Ra)-» en el que cada X es un halógeno, y R a es hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, cicloalquenilo, arilo, heteroarilo, heterociclilo, aril(alquilo), heteroaril(alquilo) o heterociclil(alquilo).
El término «amino», tal como se utiliza en la presente, se refiere a un grupo -NH2.
El término «hidroxi», tal como se utiliza en la presente, se refiere a un grupo -OH.
Un grupo «ciano» se refiere a un grupo «-CN».
El término «azido», tal como se utiliza en la presente, se refiere a un grupo -N3.
Un grupo «isocianato» se refiere a un grupo «-NCO».
Un grupo «tiocianato» se refiere a un grupo «-CNS».
Un grupo «isotiocianato» se refiere a un grupo «-NCS».
Un grupo «carbonilo» se refiere a un grupo C=O.
Un grupo «S-sulfonamido» se refiere a un grupo «-S02N(RaRb)» en el que Ra y Rb pueden ser independientemente hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, cicloalquenilo, arilo, heteroarilo, heterociclilo, aril(alquilo), heteroaril(alquilo) o heterociclil(alquilo). Un S-sulfonamido puede estar sustituido o no sustituido.
Un grupo «N-sulfonamido» se refiere a un grupo «RS02N(Ra)-» en el que R y Ra pueden ser independientemente hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, cicloalquenilo, arilo, heteroarilo, heterociclilo, aril(alquilo), heteroaril(alquilo) o heterociclil(alquilo). Un N-sulfonamido puede estar sustituido o no sustituido.
Un grupo «O-carbamilo» se refiere a un grupo «-0C(=0)N(RaRb)» en el que Ra y Rb pueden ser independientemente hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, cicloalquenilo, arilo, heteroarilo, heterociclilo, aril(alquilo), heteroaril(alquilo) o heterociclil(alquilo). Un O-carbamilo puede estar sustituido o no sustituido.
Un grupo «N-carbamilo» se refiere a un grupo «R0C(=0)N(Ra)-» en el que R y Ra pueden ser independientemente hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, cicloalquenilo, arilo, heteroarilo, heterociclilo, aril(alquilo), heteroaril(alquilo) o heterociclil(alquilo). Un N-carbamilo puede estar sustituido o no sustituido.
Un grupo «O-tiocarbamilo» se refiere a un grupo «-0C(=S)N(RaRb)» en el que Ra y Rb pueden ser independientemente hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, cicloalquenilo, arilo, heteroarilo, heterociclilo, aril(alquilo), heteroaril(alquilo) o heterociclil(alquilo). Un O-tiocarbamilo puede estar sustituido o no sustituido.
Un grupo «N-tiocarbamilo» se refiere a un grupo «R0C(=S)N(Ra)-» en el que R y Ra pueden ser independientemente hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, cicloalquenilo, arilo, heteroarilo, heterociclilo, aril(alquilo), heteroaril(alquilo) o heterociclil(alquilo). Un N-tiocarbamilo puede estar sustituido o no sustituido.
Un grupo «C-amido» se refiere a un grupo «-C(=0)N(RaRb)» en el que Ra y Rb pueden ser independientemente hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, cicloalquenilo, arilo, heteroarilo, heterociclilo, aril(alquilo), heteroaril(alquilo) o heterociclil(alquilo). Un C-amido puede estar sustituido o no sustituido.
Un grupo «N-amido» se refiere a un grupo «RC(=0)N(Ra)-» en el que R y Ra pueden ser independientemente hidrógeno, alquilo, alquenilo, alquinilo, cicloalquilo, cicloalquenilo, arilo, heteroarilo, heterociclilo, aril(alquilo), heteroaril(alquilo) o heterociclil(alquilo). Un N-amido puede estar sustituido o no sustituido.
La expresión «átomo de halógeno» o «halógeno», tal como se utiliza en la presente, significa cualquiera de los átomos radioestables de la columna 7 de la Tabla periódica de los elementos tal como flúor, cloro, bromo y yodo.
Cuando no se especifica el número de sustituyentes (por ejemplo, haloalquilo), puede haber uno o más sustituyentes presentes. Por ejemplo, «haloalquilo» puede incluir uno o más halógenos iguales o diferentes. A modo de otro ejemplo, «(alcoxi C1-C3)fenilo» puede incluir uno o más grupos alcoxi iguales o diferentes que contienen uno, dos o tres átomos. Tal como se utilizan en la presente, las abreviaturas para cualesquiera grupos protectores, aminoácidos y otros compuestos, a menos que se indique lo contrario, están de acuerdo con su uso común, abreviaturas reconocidas o la Comisión de Nomenclatura Bioquímica de la IUPAC-IUB (remítase a Biochem. 11: 942 -944 (1972)).
El término «nucleósido» se utiliza en la presente en su sentido ordinario tal como lo interpretan los expertos en la técnica y se refiere a un compuesto constituido por un resto de pentosa opcionalmente sustituido o un resto de pentosa modificado unido a una base heterocíclica o tautómero de esta a través de un enlace N-glicosídico, tal como unido
mediante la posición 9 de una base de purina o la posición 1 de una base de pirimidina. Los ejemplos incluyen, sin carácter limitante, un ribonucleósido que comprende un resto de ribosa y un desoxirribonucleósido que comprende un resto de desoxirribosa. Un resto de pentosa modificado es un resto de pentosa en el que un átomo de oxígeno ha sido reemplazado por un carbono y/o un carbono ha sido reemplazado por un átomo de azufre o de oxígeno. Un «nucleósido» es un monómero que puede tener una base sustituida y/o un resto de azúcar. Además, un nucleósido se puede incorporar en polímeros y oligómeros de ADN y/o ARN más grandes. En algunos casos, el nucleósido puede ser un fármaco análogo a un nucleósido.
El término «nucleótido» se utiliza en la presente en su sentido ordinario tal como lo interpretan los expertos en la técnica y se refiere a un nucleósido que tiene un éster de tipo fosfato unido al resto de pentosa, por ejemplo, en la posición 5'.
La expresión «base heterocíclica», tal como se utiliza en la presente, se refiere a un heterociclilo que contiene nitrógeno opcionalmente sustituido que se puede unir a un resto de pentosa opcionalmente sustituido o resto de pentosa modificado. En algunas realizaciones, la base heterocíclica se puede seleccionar entre una base de purina opcionalmente sustituida, una base de pirimidina opcionalmente sustituida y una base de triazol opcionalmente sustituida (por ejemplo, un 1,2,4-triazol). La expresión «base de purina» se utiliza en la presente en su sentido ordinario tal como lo interpretan los expertos en la técnica e incluye sus tautómeros. De manera similar, la expresión «base de pirimidina» se utiliza en la presente en su sentido ordinario tal como lo interpretan los expertos en la técnica e incluye sus tautómeros. Una lista no limitante de bases de purina opcionalmente sustituidas incluye purina, adenina, guanina, hipoxantina, xantina, aloxantina, 7-alquilguanina (por ejemplo, 7-metilguanina), teobromina, cafeína, ácido úrico e isoguanina. Los ejemplos de bases de pirimidina incluyen, sin carácter limitante, citosina, timina, uracilo, 5,6-dihidrouracilo y 5-alquilcitosina (por ejemplo, 5-metilcitosina). Un ejemplo de una base de triazol opcionalmente sustituida es 1,2,4-triazol-3-carboxamida. Otros ejemplos no limitantes de bases heterocíclicas incluyen diaminopurina, 8-oxo-N6-alquiladenina (por ejemplo, 8-oxo-N6-metiladenina), 7-desazaxantina, 7-desazaguanina, 7-desazaadenina, N4,N4-etanocitosina, N6,N6-etano-2,6-diaminopurina, 5-halouracilo (por ejemplo, 5-fluorouracilo y 5-bromouracilo), pseudoisocitosina, isocitosina, isoguanina y otras bases heterocíclicas descritas en las Patentes de EE. UU. N.os 5 432 272 y 7 125 855, que se incorporan a la presente por referencia con el propósito limitado de divulgar bases heterocíclicas adicionales. En algunas realizaciones, una base heterocíclica puede estar opcionalmente sustituida con una amina o un grupo o grupos protectores de enol.
La expresión «aminoácido conectado a través de N» se refiere a un aminoácido que está unido al resto indicado a través de un grupo amino de la cadena principal o un grupo amino monosustituido. Cuando el aminoácido está unido en un aminoácido conectado a través de N, uno de los hidrógenos que forma parte del grupo amino de la cadena principal o el grupo amino monosustituido no está presente y el aminoácido está unido a través del nitrógeno. Los aminoácidos conectados a través de N pueden estar sustituidos o no sustituidos.
La expresión «derivado de tipo éster de un aminoácido conectado a través de N» se refiere a un aminoácido en el que un grupo ácido carboxílico de la cadena principal se ha convertido en un grupo éster. En algunas realizaciones, el grupo éster tiene una fórmula seleccionada entre alquil-O-C(=O)-, cicloalquil-O-C(=O)-, aril-O-C(=O)- y aril(alquil)-O-C(=O)-. Una lista no limitante de grupos éster incluye versiones sustituidas o no sustituidas de los siguientes: metil-O-c (=o )-, etil-O-C(=O)-, n-propil-O-C(=O)-, isopropil-O-C(=O)-, n-butil-O-C(=O)-, isobutil-O-C(=O)-, ferí-butil-O-C(=O)-, neopentil-O-C(=O)-, ciclopropil-O-C(=O)-, ciclobutil-O-C(=O)-, ciclopentil-O-C(=O)-, ciclohexil-O-C(=O)-, fenil-O-C(=O)-, bencil-O-C(=O)- y naftil-O-C(=O)-. Los derivados de tipo éster de aminoácidos conectados a través de N pueden estar sustituidos o no sustituidos.
La expresión «aminoácido conectado a través de O» se refiere a un aminoácido que está unido al resto indicado mediante el hidroxi de su grupo ácido carboxílico de la cadena principal. Cuando el aminoácido está unido en un aminoácido conectado a través de O, el hidrógeno que forma parte del hidroxi de su grupo ácido carboxílico de la cadena principal no está presente y el aminoácido está unido a través del oxígeno. Los aminoácidos conectados a través de O pueden estar sustituidos o no sustituidos.
El término «aminoácido», tal como se utiliza en la presente, se refiere a cualquier aminoácido (aminoácidos tanto estándar como no estándar), que incluye, sin carácter limitante, a-aminoácidos, p-aminoácidos, Y-aminoácidos y 5-aminoácidos. Los ejemplos de aminoácidos adecuados incluyen, sin carácter limitante, alanina, asparagina, aspartato, cisteína, glutamato, glutamina, glicina, prolina, serina, tirosina, arginina, histidina, isoleucina, leucina, lisina, metionina, fenilalanina, treonina, triptófano y valina. Ejemplos adicionales de aminoácidos adecuados incluyen, sin carácter limitante, ornitina, hipusina, ácido 2-aminoisobutírico, deshidroalanina, ácido gamma-aminobutírico, citrulina, betaalanina, alfa-etilglicina, alfa-propilglicina y norleucina.
Los términos «fosforotioato» y «fosfotioato» se refieren a un compuesto de fórmula general



El término «fosfato», tal como se utiliza en la presente, se utiliza en su sentido ordinario tal como lo interpretan los
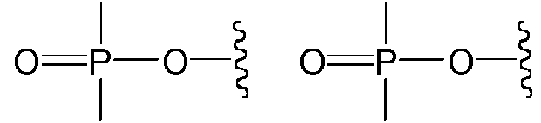
expertos en la técnica e incluye sus formas protonadas (por ejemplo, u y UM ). Los términos «monofosfato», «difosfato» y «trifosfato», tal como se utilizan en la presente, se utilizan en su sentido ordinario tal como lo interpretan los expertos en la técnica e incluyen las formas protonadas.
Las expresiones «grupo protector» y «grupos protectores», tal como se utilizan en la presente, se refieren a cualquier átomo o grupo de átomos que se añade a una molécula para evitar que los grupos existentes en la molécula experimenten reacciones químicas no deseadas. Se describen ejemplos de restos de grupos protectores en T. W. Greene y P. G. M. Wuts, Protective Groups in Organic Synthesis, 3.a Ed. John Wiley & Sons, 1999, y en J.F.W. McOmie, Protective Groups in Organic Chemistry Plenum Press, 1973, los cuales se incorporan a la presente por referencia con el propósito limitado de divulgar grupos protectores adecuados. El resto del grupo protector se puede seleccionar de tal manera que sea estable en ciertas condiciones de reacción y se elimine fácilmente en una etapa conveniente utilizando una metodología conocida en la técnica. Una lista no limitante de grupos protectores incluye bencilo; bencilo sustituido; alquilcarbonilos y alcoxicarbonilos (por ejemplo, t-butoxicarbonilo (BOC), acetilo o isobutirilo); arilalquilcarbonilos y arilalcoxicarbonilos (por ejemplo, benciloxicarbonilo); éter metílico sustituido (por ejemplo, éter metoximetílico); éter etílico sustituido; un éter bencílico sustituido; éter tetrahidropiranílico; sililos (por ejemplo, trimetilsililo, trietilsililo, triisopropilsililo, t-butildimetilsililo, triisopropilsililoximetilo, [2-(trimetilsilil)etoxi]metilo o tbutildifenilsililo); ésteres (por ejemplo, éster de tipo benzoato); carbonatos (por ejemplo, metoximetilcarbonato); sulfonatos (por ejemplo, tosilato o mesilato); cetal acíclico (por ejemplo, acetal dimetílico); cetales cíclicos (por ejemplo, 1,3-dioxano, 1,3-dioxolanos y los descritos en la presente); acetal acíclico; acetal cíclico (por ejemplo, los descritos en la presente); hemiacetal acíclico; hemiacetal cíclico; ditiocetales cíclicos (por ejemplo, 1,3-ditiano o 1,3-ditiolano); ortoésteres (por ejemplo, los descritos en la presente) y grupos triarilmetilo (por ejemplo, tritilo; monometoxitritilo (MMTr); 4,4'-dimetoxitritilo (DMTr); 4,4',4"-trimetoxitritilo (TMTr); y los descritos en la presente).
La expresión «sal farmacéuticamente aceptable» se refiere a una sal de un compuesto que no provoca irritación significativa a un organismo al que se administra y no anula la actividad biológica ni las propiedades del compuesto. En algunas realizaciones, la sal es una sal de adición de ácido del compuesto. Las sales farmacéuticas se pueden obtener haciendo reaccionar un compuesto con ácidos inorgánicos tales como un ácido halhídrico (por ejemplo, ácido clorhídrico o ácido bromhídrico), ácido sulfúrico, ácido nítrico y ácido fosfórico. Las sales farmacéuticas también se pueden obtener haciendo reaccionar un compuesto con un ácido orgánico tal como ácidos carboxílicos o sulfónicos alifáticos o aromáticos, por ejemplo, ácido fórmico, acético, succínico, láctico, málico, tartárico, cítrico, ascórbico, nicotínico, metanosulfónico, etanosulfónico, p-toluensulfónico, salicílico o naftalenosulfónico. Las sales farmacéuticas también se pueden obtener haciendo reaccionar un compuesto con una base para formar una sal tal como una sal de amonio, una sal de un metal alcalino, tal como una sal de sodio o potasio, una sal de un metal alcalinotérreo, tal como una sal de calcio o magnesio, una sal de bases orgánicas tales como diciclohexilamina, W-metil-D-glucamina, tris(hidroximetil)metilamina, alquilamina C1-C7, ciclohexilamina, trietanolamina, etilendiamina, y sales con aminoácidos tales como arginina y lisina.
Los términos y expresiones que se utilizan en esta solicitud, y variaciones de estos, especialmente en las reivindicaciones adjuntas, a menos que se indique expresamente lo contrario, se deben interpretar como abiertos y no como limitantes. A modo de ejemplo de lo anterior, se debe interpretar que la expresión «que incluye» significa «que incluye, sin limitación», «que incluye, pero no se limita a» o similares; la expresión «que comprende», tal como se utiliza en la presente, es sinónima de «que incluye», «que contiene» o «que se caracteriza por» y es inclusiva o abierta y no excluye elementos o pasos del método adicionales no mencionados; la expresión «que tiene» se debe interpretar como «que tiene al menos»; la expresión «incluye» se debe interpretar como «incluye pero no se limita a»; el término «ejemplo» se utiliza para proporcionar casos ilustrativos del artículo en discusión, y no una lista exhaustiva ni limitante de este; y el uso de términos tales como «preferentemente», «preferido», «deseado» o «deseable» y términos de significado similar no se debe interpretar como que implique que ciertas características sean críticas, esenciales o incluso importantes para la estructura o función sino que en su lugar se debe interpretar como que pretende únicamente resaltar características alternativas o adicionales que pueden utilizarse o no en una realización particular. Además, la expresión «que comprende» se debe interpretar de forma sinónima a las expresiones «que tiene al menos»
o «que incluye al menos». Cuando se utiliza en el contexto de un proceso, la expresión «que comprende» significa que el proceso incluye al menos los pasos mencionados, pero puede incluir pasos adicionales. Cuando se utiliza en el contexto de un compuesto, composición o dispositivo, la expresión «que comprende» significa que el compuesto, composición o dispositivo incluye al menos las características o componentes mencionados, pero puede incluir también características o componentes adicionales. Del mismo modo, un grupo de artículos enlazados con la conjunción «y» no se debe interpretar como que requiera que cada uno de esos artículos esté presente en la agrupación, sino que en su lugar se debe interpretar como «y/o» a menos que se indique expresamente lo contrario. De forma similar, un grupo de artículos enlazados con la conjunción «o» no se debe interpretar como que requiera exclusividad mutua entre ese grupo, sino que en su lugar se debe interpretar como «y/o» a menos que se indique expresamente lo contrario.
Con respecto al uso de sustancialmente cualesquiera términos en singular y/o plural en la presente, los expertos en la técnica pueden traducir del plural al singular y/o del singular al plural según sea apropiado para el contexto y/o la aplicación. Las diferentes permutaciones de singular/plural se pueden exponer expresamente en la presente con fines de claridad. El artículo indefinido «un» o «una» no excluye una pluralidad. Un único procesador u otra unidad puede cumplir las funciones de varios artículos mencionados en las reivindicaciones. El mero hecho de que ciertas medidas se mencionen en reivindicaciones dependientes mutuamente diferentes no indica que una combinación de estas medidas no se pueda utilizar para obtener ventajas. Cualesquiera signos de referencia en las reivindicaciones no se deben interpretar como limitantes del alcance.
Se debe sobreentender que, en cualquier compuesto descrito en la presente que tenga uno o más centros quirales, si no se indica expresamente una estereoquímica absoluta, entonces cada centro puede tener independientemente una configuración R o una configuración S o una mezcla de estas. De este modo, los compuestos proporcionados en la presente pueden ser enantioméricamente puros, enantioméricamente enriquecidos, una mezcla racémica, diastereoméricamente puros, diastereoméricamente enriquecidos o una mezcla estereoisomérica. Además, se sobreentiende que, en cualquier compuesto descrito en la presente que tenga uno o más dobles enlaces que generen isómeros geométricos que se pueden definir como E o Z, cada doble enlace puede ser independientemente E o Z o una mezcla de estos.
Del mismo modo, se sobreentiende que, en cualquier compuesto descrito, también se pretende que todas las formas tautoméricas estén incluidas. Por ejemplo, se pretende que todos los tautómeros de un grupo fosfato y un grupo fosforotioato estén incluidos. Los ejemplos de tautómeros de un fosforotioato incluyen los siguientes:

Además, se pretende que queden incluidos todos los tautómeros de bases heterocíclicas que se conocen en la técnica, incluidos los tautómeros de bases de purina y bases de pirimidina naturales y no naturales.
Se debe sobreentender que, cuando los compuestos divulgados en la presente tienen valencias sin rellenar, entonces las valencias deben rellenarse con hidrógenos o sus isótopos, por ejemplo, hidrógeno-1 (protio) e hidrógeno-2 (deuterio).
Se debe sobreentender que los compuestos descritos en la presente se pueden marcar isotópicamente. La sustitución con isótopos tales como deuterio puede proporcionar ciertas ventajas terapéuticas que den como resultado una mayor estabilidad metabólica, como, por ejemplo, una semivida in vivo mayor o unos requisitos de dosis reducidos. Cada elemento químico tal como se representa en la estructura de un compuesto puede incluir cualquier isótopo de dicho elemento. Por ejemplo, en la estructura de un compuesto, un átomo de hidrógeno puede estar descrito explícitamente o se puede sobreentender que está presente en el compuesto. En cualquier posición del compuesto en la que esté presente un átomo de hidrógeno, el átomo de hidrógeno puede ser cualquier isótopo de hidrógeno, que incluye, sin carácter limitante, hidrógeno-1 (protio) e hidrógeno-2 (deuterio). De este modo, la referencia en la presente a un compuesto engloba todas las formas isotópicas potenciales a menos que el contexto dicte claramente lo contrario.
Se sobreentiende que los métodos y las combinaciones que se describen en la presente incluyen formas cristalinas (también conocidas como polimorfos, que incluyen las diferentes disposiciones de empaquetamiento cristalino de la misma composición elemental de un compuesto), fases amorfas, sales, solvatos e hidratos. En algunas realizaciones, los compuestos descritos en la presente existen en formas solvatadas con disolventes farmacéuticamente aceptables tales como agua, etanol o similares. En otras realizaciones, los compuestos descritos en la presente existen en una forma no solvatada. Los solvatos contienen cantidades estequiométricas o no estequiométricas de un disolvente y se pueden formar durante el proceso de cristalización con disolventes farmacéuticamente aceptables tales como agua, etanol o similares. Se forman hidratos cuando el disolvente es agua o se forman alcoholatos cuando el disolvente es un alcohol. Además, los compuestos proporcionados en la presente pueden existir en formas no solvatadas así como
solvatadas. En general, se considera que las formas solvatadas son equivalentes a las formas no solvatadas a los efectos de los compuestos y métodos proporcionados en la presente.
Cuando se proporciona un intervalo de valores, se sobreentiende que el límite superior e inferior, y cada valor intermedio entre el límite superior e inferior del intervalo está englobado dentro de las realizaciones.
Compuestos
Algunas realizaciones divulgadas en la presente se refieren a un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable del anterior:

donde:  hidrógeno; R1A se selecciona entre hidrógeno, un acilo opcionalmente sustituido, un aminoácido conectado a través de O opcionalmente sustituido,
hidrógeno; R1A se selecciona entre hidrógeno, un acilo opcionalmente sustituido, un aminoácido conectado a través de O opcionalmente sustituido,

un azidoalquilo C1-6 ; R3A se selecciona entre OH, -OC(=O)R"A y un aminoácido conectado a través de O opcionalmente sustituido; R4A es halógeno; R5A es hidrógeno; R6A y R7A se seleccionan independientemente del grupo constituido por

R9A es un aminoácido conectado a través de N opcionalmente sustituido o un derivado de tipo éster de un aminoácido conectado a través de N opcionalmente sustituido; R10A y R11A son independientemente un aminoácido conectado a través de N opcionalmente sustituido o un derivado de tipo éster de un aminoácido conectado a través de N opcionalmente sustituido; R12A, R13A y R14A independientemente están ausentes o son hidrógeno; R22A y R23A se seleccionan independientemente entre hidrógeno, un alquilo C1-24 opcionalmente sustituido y un arilo opcionalmente sustituido; R24A se selecciona entre hidrógeno, un alquilo C1-24 opcionalmente sustituido, un arilo opcionalmente sustituido, un -O-alquilo C1-24 opcionalmente sustituido, un -O-arilo opcionalmente sustituido, un -O-heteroarilo opcionalmente sustituido y un -O-heterociclilo monocíclico opcionalmente sustituido; R25A se selecciona entre hidrógeno, un alquilo C1-24 opcionalmente sustituido y un arilo opcionalmente sustituido; R"A es un alquilo C1-24 opcionalmente sustituido; m es 0 o 1; t es 0; s es 0; y Z1A, Z2A, Z3A y Z4A son independientemente O o S.
En algunas realizaciones, R1A puede ser algunas realizaciones, R6A y R7A pueden ser ambos hidrógeno. En otras realizaciones, R6A y R7A pueden estar ambos ausentes. En otras realizaciones más, al menos uno de entre R6A y R7A puede estar ausente. En todavía otras realizaciones más, al menos uno de entre R6A y R7A puede ser hidrógeno. Los expertos en la técnica sobreentienden que, cuando R6A y/o R7A están ausentes, el o los oxígenos asociados tendrán una carga negativa. Por ejemplo, cuando R6A está ausente, el oxígeno asociado a R6A tendrá una carga negativa. En algunas realizaciones, Z1A puede ser O (oxígeno). En otras realizaciones, Z1A puede ser S (azufre). En algunas realizaciones, R1A puede ser un monofosfato. En otras realizaciones, R1A puede ser un monotiofosfato.
algunas realizaciones, R6A y R7A pueden ser ambos hidrógeno. En otras realizaciones, R6A y R7A pueden estar ambos ausentes. En otras realizaciones más, al menos uno de entre R6A y R7A puede estar ausente. En todavía otras realizaciones más, al menos uno de entre R6A y R7A puede ser hidrógeno. Los expertos en la técnica sobreentienden que, cuando R6A y/o R7A están ausentes, el o los oxígenos asociados tendrán una carga negativa. Por ejemplo, cuando R6A está ausente, el oxígeno asociado a R6A tendrá una carga negativa. En algunas realizaciones, Z1A puede ser O (oxígeno). En otras realizaciones, Z1A puede ser S (azufre). En algunas realizaciones, R1A puede ser un monofosfato. En otras realizaciones, R1A puede ser un monotiofosfato.

En algunas realizaciones, tanto R6A como R7A pueden ser
Cuando uno de entre R6A y R7A o ambos son pueden seleccionar independientemente entre hidrógeno, un alquilo C1-24 opcionalmente sustituido y un arilo opcionalmente sustituido; R24A se puede seleccionar independientemente entre hidrógeno, un alquilo C1-24 opcionalmente sustituido, un arilo opcionalmente sustituido, un -O-alquilo C1-24 opcionalmente sustituido, un -O-arilo opcionalmente sustituido, un -O-heteroarilo opcionalmente sustituido y un -O-heterociclilo monocíclico opcionalmente sustituido; y Z4A puede ser independientemente O (oxígeno) o S (azufre). En algunas realizaciones, R22A y R23A pueden ser hidrógeno. En otras realizaciones, al menos uno de entre R22A y R23A puede ser un alquilo C1-24 opcionalmente sustituido o un arilo opcionalmente sustituido. En algunas realizaciones, R24A puede ser un alquilo C1-24 opcionalmente sustituido. En otras realizaciones, R24A puede ser un arilo opcionalmente sustituido. En otras realizaciones más, R24A puede ser un -O alquilo C1-24 opcionalmente sustituido o un -O-arilo opcionalmente sustituido. En algunas realizaciones, Z4A puede ser O (oxígeno). En otras realizaciones, Z4A puede ser o S (azufre). En algunas realizaciones, uno de entre R6A y R7A o ambos pueden ser isopropiloxicarboniloximetilo (POC). En algunas realizaciones, uno de entre R6A y R7A o ambos pueden ser pivaloiloximetilo (POM). En algunas realizaciones, R6A y R7A pueden ser ambos un grupo isopropiloxicarboniloximetilo y formar un profármaco de tipo bis(isopropiloxicarboniloximetilo) (bis(POC)). En algunas realizaciones, R6A y R7A pueden ser ambos un grupo pivaloiloximetilo y formar un profármaco de tipo bis(pivaloiloximetilo) (bis(POM)).
pueden seleccionar independientemente entre hidrógeno, un alquilo C1-24 opcionalmente sustituido y un arilo opcionalmente sustituido; R24A se puede seleccionar independientemente entre hidrógeno, un alquilo C1-24 opcionalmente sustituido, un arilo opcionalmente sustituido, un -O-alquilo C1-24 opcionalmente sustituido, un -O-arilo opcionalmente sustituido, un -O-heteroarilo opcionalmente sustituido y un -O-heterociclilo monocíclico opcionalmente sustituido; y Z4A puede ser independientemente O (oxígeno) o S (azufre). En algunas realizaciones, R22A y R23A pueden ser hidrógeno. En otras realizaciones, al menos uno de entre R22A y R23A puede ser un alquilo C1-24 opcionalmente sustituido o un arilo opcionalmente sustituido. En algunas realizaciones, R24A puede ser un alquilo C1-24 opcionalmente sustituido. En otras realizaciones, R24A puede ser un arilo opcionalmente sustituido. En otras realizaciones más, R24A puede ser un -O alquilo C1-24 opcionalmente sustituido o un -O-arilo opcionalmente sustituido. En algunas realizaciones, Z4A puede ser O (oxígeno). En otras realizaciones, Z4A puede ser o S (azufre). En algunas realizaciones, uno de entre R6A y R7A o ambos pueden ser isopropiloxicarboniloximetilo (POC). En algunas realizaciones, uno de entre R6A y R7A o ambos pueden ser pivaloiloximetilo (POM). En algunas realizaciones, R6A y R7A pueden ser ambos un grupo isopropiloxicarboniloximetilo y formar un profármaco de tipo bis(isopropiloxicarboniloximetilo) (bis(POC)). En algunas realizaciones, R6A y R7A pueden ser ambos un grupo pivaloiloximetilo y formar un profármaco de tipo bis(pivaloiloximetilo) (bis(POM)).

,
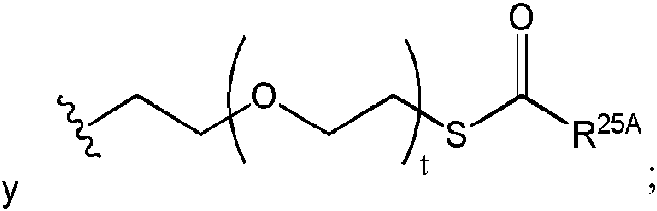
ser hidrógeno. En otras realizaciones, R25A puede ser un alquilo C1-24 opcionalmente sustituido. En otras realizaciones más, R25A puede ser un arilo opcionalmente sustituido. En algunas realizaciones, R25A puede ser un alquilo C1-6, por ejemplo, metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, te/t-butilo, pentilo (de cadena lineal y ramificada) y hexilo (de cadena lineal y ramificada). En algunas realizaciones, uno de entre R6A y R7A o ambos pueden ser S-aciltioetilo (SATE).
En algunas realizaciones, R1A puede ser puede ser
puede ser puede estar ausente o ser hidrógeno; R12A, R13A y R14A pueden independientemente estar ausentes o ser hidrógeno; y m puede ser 0 o 1. En algunas realizaciones, m puede ser 0, y R7A, R12A y R13A pueden independientemente estar ausentes o ser hidrógeno. En otras realizaciones, m puede ser 1, y R7A, R12A, R13A y R14A pueden independientemente estar ausentes o ser hidrógeno. Los expertos en la técnica sobreentienden que, cuando m es 0, R6A puede ser difosfato,
cuando Z1A es oxígeno, o un alfa-tiodifosfato, cuando Z1A es azufre. Del mismo modo, los expertos en la técnica sobreentienden que, cuando m es 1, R6A puede ser trifosfato, cuando Z1A es oxígeno, o un alfa-tiotrifosfato, cuando Z1A es azufre.
puede estar ausente o ser hidrógeno; R12A, R13A y R14A pueden independientemente estar ausentes o ser hidrógeno; y m puede ser 0 o 1. En algunas realizaciones, m puede ser 0, y R7A, R12A y R13A pueden independientemente estar ausentes o ser hidrógeno. En otras realizaciones, m puede ser 1, y R7A, R12A, R13A y R14A pueden independientemente estar ausentes o ser hidrógeno. Los expertos en la técnica sobreentienden que, cuando m es 0, R6A puede ser difosfato,
cuando Z1A es oxígeno, o un alfa-tiodifosfato, cuando Z1A es azufre. Del mismo modo, los expertos en la técnica sobreentienden que, cuando m es 1, R6A puede ser trifosfato, cuando Z1A es oxígeno, o un alfa-tiotrifosfato, cuando Z1A es azufre.
En algunas realizaciones, R6A y R7A pueden ser iguales. En algunas realizaciones, R6A y R7A pueden ser diferentes.
En algunas realizaciones, Z1A puede ser oxígeno. En otras realizaciones, Z1A puede ser azufre.
En algunas realizaciones, R1A puede ser
En algunas realizaciones, R9A se puede seleccionar entre alanina, asparagina, aspartato, cisteína, glutamato, glutamina, glicina, prolina, serina, tirosina, arginina, histidina, isoleucina, leucina, lisina, metionina, fenilalanina, treonina, triptófano, valina y derivados de tipo éster de estos. Los ejemplos de derivados de tipo éster de un aminoácido conectado a través de N opcionalmente sustituido incluyen versiones opcionalmente sustituidas de los siguientes: éster isopropílico de alanina, éster ciclohexílico de alanina, éster neopentílico de alanina, éster isopropílico de valina

y éster isopropílico de leucina. En algunas realizaciones, R9A puede tener la estructura ^ donde R33A se puede seleccionar entre hidrógeno, un alquilo C1-6 opcionalmente sustituido, un cicloalquilo C3-6 opcionalmente sustituido, un arilo opcionalmente sustituido, un aril(alquilo C1-6) opcionalmente sustituido y un haloalquilo opcionalmente sustituido; R34A se puede seleccionar entre hidrógeno, un alquilo C1-6 opcionalmente sustituido, un haloalquilo C1-6 opcionalmente sustituido, un cicloalquilo C3-6 opcionalmente sustituido, un arilo C6 opcionalmente sustituido, un arilo C10 opcionalmente sustituido y un aril(alquilo C1-6) opcionalmente sustituido; y R35A puede ser hidrógeno o un alquilo C1-4 opcionalmente sustituido; o R34A y R35A se pueden considerar conjuntamente para formar un cicloalquilo C3-6 opcionalmente sustituido.
Cuando R34A está sustituido, R34A puede estar sustituido con uno o más sustituyentes seleccionados entre N-amido, mercapto, alquiltio, un arilo opcionalmente sustituido, hidroxi, un heteroarilo opcionalmente sustituido, O-carboxi y amino. En algunas realizaciones, R34A puede ser un alquilo C1-6 no sustituido tal como los que se describen en la presente. En algunas realizaciones, R34A puede ser hidrógeno. En otras realizaciones, R34A puede ser metilo. En algunas realizaciones, R33A puede ser un alquilo C1-6 opcionalmente sustituido. Los ejemplos de alquilos C1-6 opcionalmente sustituidos incluyen variantes opcionalmente sustituidas de los siguientes: metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, ferf-butilo, pentilo (de cadena lineal y ramificada) y hexilo (de cadena lineal y ramificada). En algunas realizaciones, R33A puede ser metilo o isopropilo. En algunas realizaciones, R33A puede ser etilo o neopentilo. En otras realizaciones, R33A puede ser un cicloalquilo C3-6 opcionalmente sustituido. Los ejemplos de cicloalquilo C3-6 opcionalmente sustituido incluyen variantes opcionalmente sustituidas de los siguientes: ciclopropilo, ciclobutilo, ciclopentilo y ciclohexilo. En una realización, R33A puede ser un ciclohexilo opcionalmente sustituido. En otras realizaciones más, R33A puede ser un arilo opcionalmente sustituido tal como fenilo y naftilo. En todavía otras realizaciones más, R33A puede ser un aril(alquilo C1-6) opcionalmente sustituido. En algunas realizaciones, R33A puede ser un bencilo opcionalmente sustituido. En algunas realizaciones, R33A puede ser un haloalquilo C1-6 opcionalmente sustituido, por ejemplo, CF3. En algunas realizaciones, R35A puede ser hidrógeno. En otras realizaciones, R35A puede ser un alquilo C1-4 opcionalmente sustituido tal como metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo y ferf-butilo. En una realización, R35A puede ser metilo. En algunas realizaciones, R34A y R35A se pueden considerar conjuntamente para formar un cicloalquilo C3-6 opcionalmente sustituido. Los ejemplos de cicloalquilo C3-6 opcionalmente sustituido incluyen variantes opcionalmente sustituidas de los siguientes: ciclopropilo, ciclobutilo, ciclopentilo y ciclohexilo. Dependiendo de los grupos que se seleccionan para R34A y R35A, el carbono al cual están unidos R34A y R35A puede ser un centro quiral. En algunas realizaciones, el carbono al cual están unidos R34A y R35A puede ser un centro quiral (R). En otras realizaciones, el carbono al cual están unidos R34A y R35A puede ser un centro quiral (S).
En algunas realizaciones, cuando puede ser O (oxígeno). En otras realizaciones, cuando
puede ser O (oxígeno). En otras realizaciones, cuando


R1A es * Z2A puede ser S (azufre). En algunas realizaciones, cuando R1A es ■' > un compuesto de Fórmula (I) puede ser un profármaco de tipo fosforamidato tal como un profármaco de tipo arilfosforamidato.
En algunas realizaciones, R1A puede ser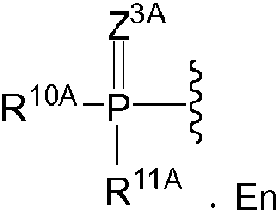 algunas realizaciones, R10A y R11A pueden ser ambos un aminoácido conectado a través de N opcionalmente sustituido o un derivado de tipo éster de un aminoácido conectado a través de N opcionalmente sustituido. En algunas realizaciones, R10A y R11A se pueden seleccionar independientemente entre alanina, asparagina, aspartato, cisteína, glutamato, glutamina, glicina, prolina, serina, tirosina, arginina, histidina, isoleucina, leucina, lisina, metionina, fenilalanina, treonina, triptófano, valina y derivados de tipo éster de estos. En algunas realizaciones, R10A y R11A pueden ser una versión opcionalmente sustituida de los siguientes: éster isopropílico de alanina, éster ciclohexílico de alanina, éster neopentílico de alanina, éster isopropílico de valina y éster isopropílico de leucina. En algunas realizaciones, R10A y R11A pueden tener independientemente la
algunas realizaciones, R10A y R11A pueden ser ambos un aminoácido conectado a través de N opcionalmente sustituido o un derivado de tipo éster de un aminoácido conectado a través de N opcionalmente sustituido. En algunas realizaciones, R10A y R11A se pueden seleccionar independientemente entre alanina, asparagina, aspartato, cisteína, glutamato, glutamina, glicina, prolina, serina, tirosina, arginina, histidina, isoleucina, leucina, lisina, metionina, fenilalanina, treonina, triptófano, valina y derivados de tipo éster de estos. En algunas realizaciones, R10A y R11A pueden ser una versión opcionalmente sustituida de los siguientes: éster isopropílico de alanina, éster ciclohexílico de alanina, éster neopentílico de alanina, éster isopropílico de valina y éster isopropílico de leucina. En algunas realizaciones, R10A y R11A pueden tener independientemente la

estructura ^ donde R36A se puede seleccionar entre hidrógeno, un alquilo C1-6 opcionalmente sustituido, un cicloalquilo C3-6 opcionalmente sustituido, un arilo opcionalmente sustituido, un aril(alquilo C1-6) opcionalmente sustituido y un haloalquilo opcionalmente sustituido; R37A se puede seleccionar entre hidrógeno, un alquilo C1-6 opcionalmente sustituido, un haloalquilo C1-6 opcionalmente sustituido, un cicloalquilo C3-6 opcionalmente sustituido, un arilo C6 opcionalmente sustituido, un arilo C10 opcionalmente sustituido y un aril(alquilo C1-6) opcionalmente sustituido; y R38A puede ser hidrógeno o un alquilo C1-4 opcionalmente sustituido; o R37A y R38A se pueden considerar conjuntamente para formar un cicloalquilo C3-6 opcionalmente sustituido.
Cuando R37A está sustituido, R37A puede estar sustituido con uno o más sustituyentes seleccionados entre N-amido, mercapto, alquiltio, un arilo opcionalmente sustituido, hidroxi, un heteroarilo opcionalmente sustituido, O-carboxi y amino. En algunas realizaciones, R37A puede ser un alquilo C1-6 no sustituido tal como los que se describen en la presente. En algunas realizaciones, R37A puede ser hidrógeno. En otras realizaciones, R37A puede ser metilo. En algunas realizaciones, R36A puede ser un alquilo C1-6 opcionalmente sustituido. Los ejemplos de alquilos C1-6 opcionalmente sustituidos incluyen variantes opcionalmente sustituidas de los siguientes: metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, fe/f-butilo, pentilo (de cadena lineal y ramificada) y hexilo (de cadena lineal y ramificada). En algunas realizaciones, R36A puede ser metilo o isopropilo. En algunas realizaciones, R36A puede ser etilo o neopentilo. En otras realizaciones, R36A puede ser un cicloalquilo C3-6 opcionalmente sustituido. Los ejemplos de cicloalquilo C3-6 opcionalmente sustituido incluyen variantes opcionalmente sustituidas de los siguientes: ciclopropilo, ciclobutilo, ciclopentilo y ciclohexilo. En una realización, R36A puede ser un ciclohexilo opcionalmente sustituido. En otras realizaciones más, R36A puede ser un arilo opcionalmente sustituido tal como fenilo y naftilo. En todavía otras realizaciones más, R36A puede ser un aril(alquilo C1-6) opcionalmente sustituido. En algunas realizaciones, R36A puede ser un bencilo opcionalmente sustituido. En algunas realizaciones, R36A puede ser un haloalquilo C1-6 opcionalmente sustituido, por ejemplo, CF3. En algunas realizaciones, R38A puede ser hidrógeno. En otras realizaciones, R38A puede ser un alquilo C1-4 opcionalmente sustituido tal como metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo y fe/f-butilo. En una realización, R38A puede ser metilo. En algunas realizaciones, R37A y R38A se pueden considerar conjuntamente para formar un cicloalquilo C3-6 opcionalmente sustituido. Los ejemplos de cicloalquilo C3-6 opcionalmente sustituido incluyen variantes opcionalmente sustituidas de los siguientes: ciclopropilo, ciclobutilo, ciclopentilo y ciclohexilo. Dependiendo de los grupos que se seleccionan para R37A y R38A, el carbono al cual están unidos R37A y R38A puede ser un centro quiral. En algunas realizaciones, el carbono al cual están unidos R37A y R38A puede ser un centro quiral (R). En otras realizaciones, el carbono al cual están unidos R37A y R38A puede ser un centro quiral (S).
Los ejemplos de grupos adecuados incluyen los siguientes:
adecuados incluyen los siguientes:

En algunas realizaciones, R10A y R11A pueden ser iguales. En algunas realizaciones, R10Ay R11A pueden ser diferentes.
En algunas realizaciones, Z3A puede ser O (oxígeno). En otras realizaciones, Z3A puede ser S (azufre). En algunas
realizaciones, cuando un compuesto de Fórmula (I) puede ser un profármaco de tipo diamida fosfónica.
un compuesto de Fórmula (I) puede ser un profármaco de tipo diamida fosfónica.
En algunas realizaciones, R1A puede ser hidrógeno. En algunas realizaciones, R1A puede ser un acilo opcionalmente sustituido. En otras realizaciones, R1A puede ser -C(=O)R39A, donde R39A se puede seleccionar entre un alquilo C1-12 opcionalmente sustituido, un alquenilo C2-12 opcionalmente sustituido, un alquinilo C2-12 opcionalmente sustituido, un cicloalquilo C3-8 opcionalmente sustituido, un cicloalquenilo C5-8 opcionalmente sustituido, un arilo C6-10 opcionalmente sustituido, un heteroarilo opcionalmente sustituido, un heterociclilo opcionalmente sustituido, un aril(alquilo C1-6) opcionalmente sustituido, un heteroaril(alquilo C1-6) opcionalmente sustituido y un heterociclil(alquilo C1-6) opcionalmente sustituido. En algunas realizaciones, R39A puede ser un alquilo C1-12 sustituido. En otras realizaciones, R39A puede ser un alquilo C1-12 no sustituido.
En otras realizaciones más, R1A puede ser un aminoácido conectado a través de O opcionalmente sustituido. Los ejemplos de aminoácidos conectados a través de O adecuados incluyen alanina, asparagina, aspartato, cisteína, glutamato, glutamina, glicina, prolina, serina, tirosina, arginina, histidina, isoleucina, leucina, lisina, metionina, fenilalanina, treonina, triptófano y valina. Ejemplos adicionales de aminoácidos adecuados incluyen, sin carácter limitante, ornitina, hipusina, ácido 2-aminoisobutírico, deshidroalanina, ácido gamma-aminobutírico, citrulina, betaalanina, alfa-etilglicina, alfa-propilglicina y norleucina. En algunas realizaciones, el aminoácido conectado a través de

O puede tener la estructura ~ ^ donde r4oa se puede seleccionar entre hidrógeno, un alquilo C1-6 opcionalmente sustituido, un haloalquilo C1-6 opcionalmente sustituido, un cicloalquilo C3-6 opcionalmente sustituido, un arilo C6 opcionalmente sustituido, un arilo C10 opcionalmente sustituido y un aril(alquilo C1-6) opcionalmente sustituido; y R41A puede ser hidrógeno o un alquilo C1-4 opcionalmente sustituido; o R40A y R41A se pueden considerar conjuntamente para formar un cicloalquilo C3-6 opcionalmente sustituido. Los expertos en la técnica sobreentienden que, cuando R1A es un aminoácido conectado a través de O opcionalmente sustituido, el oxígeno de R1AO- de la Fórmula (I) forma parte del aminoácido conectado a través de O opcionalmente sustituido. Por ejemplo, cuando R1A

es el oxígeno indicado con un «*» es el oxígeno de R1A0- de la Fórmula (I).
Cuando R40A está sustituido, R40A puede estar sustituido con uno o más sustituyentes seleccionados entre N-amido, mercapto, alquiltio, un arilo opcionalmente sustituido, hidroxi, un heteroarilo opcionalmente sustituido, O-carboxi y amino. En algunas realizaciones, R40A puede ser un alquilo C1-6 no sustituido tal como los que se describen en la presente. En algunas realizaciones, R40A puede ser hidrógeno. En otras realizaciones, R40A puede ser metilo. En algunas realizaciones, R41A puede ser hidrógeno. En otras realizaciones, R41A puede ser un alquilo C1-4 opcionalmente sustituido tal como metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo y íert-butilo. En una realización, R41A puede ser metilo. Dependiendo de los grupos que se seleccionan para R40A y R41A, el carbono al cual están unidos R40A y R41A puede ser un centro quiral. En algunas realizaciones, el carbono al cual están unidos R40A y R41A puede ser un centro quiral (R). En otras realizaciones, el carbono al cual están unidos R40A y R41A puede ser un centro quiral (S).


Los ejemplos de grupos 0 N H 2 adecuados incluyen los siguientes:

Tal como se describe en la presente, R2A es un azidoalquilo Ci-6. Por ejemplo, R2A puede ser un azidometilo, azidoetilo, azidopropilo, azidobutilo, azidopentilo o azidohexilo.
Los grupos unidos a la posición 3’ del anillo de pentosa pueden variar. En algunas realizaciones, R3A puede ser OH. En otras realizaciones, R3A puede ser un aminoácido conectado a través de O opcionalmente sustituido. Los ejemplos de aminoácidos conectados a través de O adecuados incluyen alanina, asparagina, aspartato, cisteína, glutamato, glutamina, glicina, prolina, serina, tirosina, arginina, histidina, isoleucina, leucina, lisina, metionina, fenilalanina, treonina, triptófano y valina. Ejemplos adicionales de aminoácidos adecuados incluyen, sin carácter limitante, ornitina, hipusina, ácido 2-aminoisobutírico, deshidroalanina, ácido gamma-aminobutírico, citrulina, beta-alanina, alfa-etilglicina, alfa-propilglicina norleucina. En algunas realizaciones, el aminoácido conectado a través de O puede tener la

estructura ’ donde R42A se puede seleccionar entre hidrógeno, un alquilo C1-6 opcionalmente sustituido, un haloalquilo Ci-6 opcionalmente sustituido, un cicloalquilo C3-6 opcionalmente sustituido, un arilo C6 opcionalmente sustituido, un arilo C10 opcionalmente sustituido y un aril(alquilo C1-6) opcionalmente sustituido; y R43A puede ser hidrógeno o un alquilo C1-4 opcionalmente sustituido; o R42A y R43A se pueden considerar conjuntamente para formar un cicloalquilo C3-6 opcionalmente sustituido.
Cuando R42A está sustituido, R42A puede estar sustituido con uno o más sustituyentes seleccionados entre N-amido, mercapto, alquiltio, un arilo opcionalmente sustituido, hidroxi, un heteroarilo opcionalmente sustituido, O-carboxi y amino. En algunas realizaciones, R42A puede ser un alquilo C1-6 no sustituido tal como los que se describen en la presente. En algunas realizaciones, R42A puede ser hidrógeno. En otras realizaciones, R42A puede ser metilo. En algunas realizaciones, R43A puede ser hidrógeno. En otras realizaciones, R43A puede ser un alquilo C1-4 opcionalmente sustituido tal como metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo y íert-butilo. En una realización, R43A puede ser metilo. Dependiendo de los grupos que se seleccionan para R42A y R43A, el carbono al cual están unidos R42A y R43A puede ser un centro quiral. En algunas realizaciones, el carbono al cual están unidos R42A y R43A puede ser un centro quiral (R). En otras realizaciones, el carbono al cual están unidos R42A y R43A puede ser un centro quiral (S).

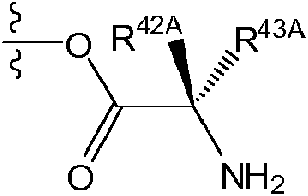
Los ejemplos de grupos adecuados incluyen los siguientes:

En otras realizaciones más, R3A puede ser -OC(=O)R"A, donde R"A puede ser un alquilo C1-24 opcionalmente sustituido. En algunas realizaciones, R"A puede ser un alquilo C1-8 sustituido. En otras realizaciones, R"A puede ser un alquilo C1-8 no sustituido. En otras realizaciones más, R3A puede ser un -O-acilo opcionalmente sustituido. En todavía otras realizaciones más, R3A puede ser -OC(=O)R44A, donde R44A se puede seleccionar entre un alquilo C1-12 opcionalmente sustituido, un alquenilo C2-12 opcionalmente sustituido, un alquinilo C2-12 opcionalmente sustituido, un cicloalquilo C3-8 opcionalmente sustituido, un cicloalquenilo C5-8 opcionalmente sustituido, un arilo C6-10 opcionalmente sustituido, un heteroarilo opcionalmente sustituido, un heterociclilo opcionalmente sustituido, un aril(alquilo C1-6) opcionalmente sustituido, un heteroaril(alquilo C1-6) opcionalmente sustituido y un heterociclil(alquilo C1-6) opcionalmente sustituido.
En algunas realizaciones, R44A puede ser un alquilo C1-12 sustituido. En otras realizaciones, R44A puede ser un alquilo C1-12 no sustituido.
En la posición 2’ del anillo de pentosa puede haber varios sustituyentes presentes. En algunas realizaciones, R4A puede ser fluoro o cloro. En algunas realizaciones, R4A puede ser fluoro. En otras realizaciones, R4A puede ser cloro.
En la posición 5’ del anillo de pentosa también puede haber varios sustituyentes presentes. En algunas realizaciones, tanto Ra1 como R32 pueden ser hidrógeno. En otras realizaciones, Ra1 puede ser hidrógeno y Ra2 puede ser deuterio. En otras realizaciones más, tanto Ra1 como Ra2 pueden ser deuterio. Para la posición 1’, en algunas realizaciones, RA puede ser hidrógeno. En otras realizaciones, RA puede ser deuterio.

. , . realizaciones, R1A no puede ser hidrógeno cuando R2A es azidometilo, R3A es hidroxi, R4A es halógeno (por ejemplo, fluoro), R5A es hidrógeno, RA es hidrógeno y B1A es uracilo. En algunas realizaciones, cuando R2A es azidometilo, R3A es hidroxi, R4A es halógeno (por ejemplo, fluoro), R5A es hidrógeno y RA es hidrógeno, entonces B1A no puede ser uracilo. En algunas realizaciones, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, no
puede ser
En algunas realizaciones, B1A puede ser otras realizaciones, B1A puede ser
otras realizaciones, B1A puede ser
En algunas realizaciones, un compuesto de Fórmula (I) puede tener la estructura:

realizaciones de este párrafo, B1A puede ser citosina. En otras realizaciones de este párrafo, B1A puede ser uracilo. En algunas realizaciones de este párrafo, R1A puede ser hidrógeno. En otras realizaciones de este párrafo, R1A puede ser un acilo opcionalmente sustituido. En otras realizaciones más de este párrafo, R1A puede ser mono-, di- o trifosfato. En otras realizaciones más de este párrafo, R1A puede ser un profármaco de tipo fosforamidato tal como un profármaco de tipo arilfosforamidato. En algunas realizaciones de este párrafo, R1A puede ser un profármaco de tipo fosfato de un éster aciloxialquílico. En otras realizaciones de este párrafo, R1A puede ser un profármaco de tipo S-aciltioetilo (SATE). En otras realizaciones más, R1A puede ser un profármaco de tipo diamida fosfónica.
Los ejemplos de compuestos de Fórmula (I) adecuados incluyen, sin carácter limitante, los siguientes:


los anteriores.
Ejemplos adicionales de un compuesto de Fórmula (I) incluyen los siguientes:

farmacéuticamente aceptable de cualquiera de los anteriores.
Otros ejemplos de un compuesto de Fórmula (I) incluyen, sin carácter limitante, los siguientes:
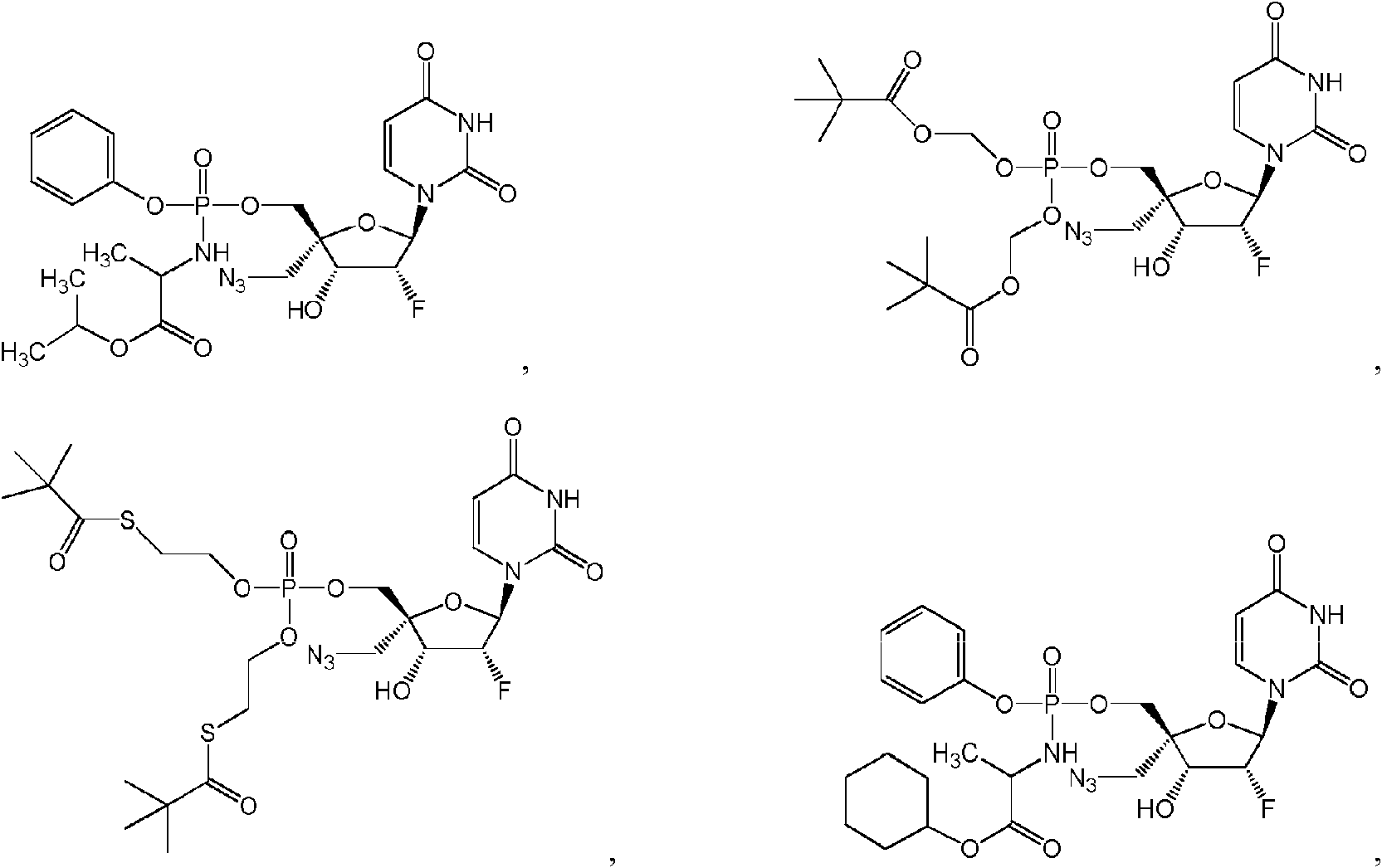


cualquiera de los anteriores.
Síntesis
Los compuestos de Fórmula (I) y los que se describen en la presente se pueden preparar de varias maneras. Algunos compuestos de Fórmula (I) se pueden obtener de proveedores comerciales y/o se pueden preparar utilizando procedimientos sintéticos conocidos. En la presente se muestran y describen rutas sintéticas generales para obtener los compuestos de Fórmula (I) y algunos ejemplos de materiales de partida utilizados para sintetizar los compuestos de Fórmula (I). Las rutas mostradas y descritas en la presente son únicamente ilustrativas y no se pretende, ni se debe interpretar, que limiten el alcance de las reivindicaciones de ningún modo en absoluto. Los expertos en la técnica serán capaces de reconocer modificaciones de las síntesis divulgadas y diseñar rutas alternativas basándose en las divulgaciones de la presente; todas estas modificaciones y rutas alternativas se encuentran dentro del alcance de las reivindicaciones.

Los compuestos de Fórmula (I), donde R2A es un azidoalquilo C1-6, se pueden preparar a partir de un nucleósido, por ejemplo, un nucleósido de Fórmula (A). En el Esquema 1, Ra, R3a, R4a, R5a y B1a pueden ser los mismos que RA, R3A,
R4A, R5A y B1A tal como se han descrito en la presente para la Fórmula (I), PG1 puede ser un grupo protector adecuado y LG1 puede ser un grupo saliente adecuado. La posición 5’ del nucleósido se puede oxidar para obtener un aldehído utilizando métodos conocidos por los expertos en la técnica. Las condiciones de oxidación adecuadas incluyen, sin carácter limitante, una oxidación de Moffatt, oxidación de Swern y oxidación de Corey-Kim; y los agentes oxidantes adecuados incluyen, sin carácter limitante, el peryodinano de Dess-Martin, IBX (ácido 2-yodoxibenzoico), TPAP/NMO (perrutenato de tetrapropilamonio/N-óxido de N-metilmorfolina), reactivo de la oxidación de Swern, PCC (clorocromato de piridinio), PDC (dicromato de piridinio), peryodato de sodio, reactivo de Collin, nitrato de amonio cérico CAN, Na2Cr2O7 en agua, Ag2CO3 en celite, HNO3 caliente en dimetoxietano acuoso, O2-piridina CuCl, Pb(OAc)4-piridina y peróxido de benzoílo-NiBr2. Se puede añadir un grupo hidroximetilo a la posición 4’ del anillo de pentosa junto con la reducción del aldehído hasta obtener un alcohol. El grupo hidroximetilo se puede añadir mediante una reacción de condensación utilizando formaldehído y una base tal como hidróxido de sodio. Después de la adición del grupo hidroximetilo, se puede llevar a cabo una reducción del compuesto intermedio con un grupo 4’-hidroximetilo utilizando un agente reductor. Los ejemplos de agentes reductores adecuados incluyen, sin carácter limitante, NaBH4 y LiAlH4. Se puede formar un grupo saliente adecuado, tal como un triflato, reemplazando el hidrógeno del grupo hidroximetilo unido a la posición 4’, y se puede proteger el oxígeno unido a la posición 5’ con un grupo protector adecuado (por ejemplo, mediante una ciclación con la base, B1a, o con un grupo protector diferente). El grupo saliente se puede reemplazar por un grupo azido utilizando un reactivo que sea una ácida de un metal, por ejemplo, azida sódica.

Los compuestos de Fórmula (I) que tienen un grupo que contiene fósforo unido a la posición 5’ del anillo de pentosa se pueden preparar utilizando varios métodos conocidos por los expertos en la técnica. En los Esquemas 2 y 3 se muestran ejemplos de los métodos. En los Esquemas 2 y 3, Ra, R2a, R3a, R4a, R5a y B1a pueden ser los mismos que RA, R2A, R3A, R4A, R5A y B1A tal como se han descrito en la presente para la Fórmula (I). Se puede acoplar un precursor que contenga fósforo al nucleósido, por ejemplo, un compuesto de Fórmula (B). Según se muestra en el Esquema 2, tras el acoplamiento del precursor que contiene fósforo, cualesquiera grupos salientes se pueden escindir en condiciones adecuadas tales como una hidrólisis. Se pueden añadir otros grupos que contengan fósforo utilizando métodos conocidos por los expertos en la técnica, por ejemplo, utilizando un pirofosfato.
En algunas realizaciones, se puede generar un alcóxido a partir de un compuesto de Fórmula (C) utilizando un reactivo organometálico tal como un reactivo de Grignard. El alcóxido se puede acoplar al precursor que contiene fósforo. Los reactivos de Grignard adecuados son conocidos por los expertos en la técnica e incluyen, sin carácter limitante, cloruros de alquilmagnesio y bromuros de alquilmagnesio. En algunas realizaciones, se puede utilizar una base adecuada. Los ejemplos de bases adecuadas incluyen, sin carácter limitante, una base de tipo amina tal como una alquilamina (incluidas las mono-, di- y trialquilaminas (por ejemplo, trietilamina)), opcionalmente piridinas sustituidas (por ejemplo, colidina) e imidazoles opcionalmente sustituidos (por ejemplo, W-metilimidazol)). Como alternativa, se puede añadir un precursor que contenga fósforo al nucleósido y formar un fosfito. El fosfito se puede oxidar para obtener un fosfato utilizando condiciones conocidas por los expertos en la técnica. Las condiciones adecuadas incluyen, sin carácter limitante, ácido meta-cloroperoxibenzoico (MCPBA) y yodo como agente oxidante y agua como dador de oxígeno.
Un azidoalquilo C1-6 en la posición 4’ se puede reducir para obtener un aminoalquilo C1-6. Se pueden utilizar varios agentes /condiciones de reducción que son conocidos por los expertos en la técnica. Por ejemplo, el grupo azido se puede reducir para obtener un grupo amino mediante una hidrogenación (por ejemplo, H2-Pd/C o HCÜ2NH4-Pd/C), reacción de Staudinger, NaBH4/CoCl2^6 H2O, Fe/NH4Cl o Zn/NH4Cl.
Cuando en los compuestos de Fórmula (I) Z1A, Z2A o Z3A es azufre, el azufre se puede añadir de varias maneras conocidas por los expertos en la técnica. En algunas realizaciones, el azufre puede formar parte del precursor que
R6A0 — S P— Cl or OH R8A0 f P ci
contiene fósforo, por ejemplo, R7AQ o r9A • Como alternativa, el azufre se puede añadir utilizando un reactivo de sulfurización. Los agentes de sulfurización adecuados son conocidos por los expertos en la técnica e incluyen, sin carácter limitante, azufre elemental, reactivo de Lawesson, ciclooctaazufre, 1,1 -dióxido de 3H-1,2-benzoditiol-3-ona (reactivo de Beaucage), 3-((W,W-dimetilaminometiliden)amino)-3H-1,2,4-ditiazol-5-tiona (DDTT) y tetrasulfuro de bis(3-trietoxisilil)propilo (TEST).
Los precursores que contienen fósforo adecuados se pueden obtener de proveedores comerciales o se pueden preparar mediante métodos sintéticos conocidos por los expertos en la técnica. En los Esquemas 2 y 3 se muestran ejemplos de estructuras generales de precursores que contienen fósforo.
Durante la síntesis de cualquiera de los compuestos descritos en la presente, si se desea, cualesquiera grupos hidroxi unidos al anillo de pentosa, y cualesquiera grupos -NH y/o NH2 presentes en el B1a, se pueden proteger con uno o más grupos protectores adecuados. En la presente se describen grupos protectores adecuados. Por ejemplo, cuando R3a es un grupo hidroxi, R3a se puede proteger con un grupo triarilmetilo o un grupo sililo. Del mismo modo, cualesquiera grupos -NH y/o grupos NH2 presentes en B1a se pueden proteger, por ejemplo, con un grupo o grupos triarilmetilo y sililo. Los ejemplos de grupos triarilmetilo incluyen, sin carácter limitante, tritilo, monometoxitritilo (MMTr), 4,4'-dimetoxitritilo (DMTr), 4,4',4"-trimetoxitritilo (TMTr), 4,4',4"-tris(benzoiloxi)tritilo (TBTr), 4,4',4"-tris(4,5-dicloroftalimido)tritilo (CPTr), 4,4',4"-tris(levuliniloxi)tritilo (TLTr), p-anisil-1 -naftilfenilmetilo, di-o-anisil-1 -naftilmetilo, ptolildifenilmetilo, 3-(imidazolilmetil)-4,4'-dimetoxitritilo, 9-fenilxanten-9-ilo (Pixilo), 9-(p-metoxifenil)xanten-9-ilo (Mox), 4-deciloxitritilo, 4-hexadeciloxitritilo, 4,4'-dioctadeciltritilo, 9-(4-octadeciloxifenil)xanten-9-ilo, 1,1'-bis-(4-metoxifenil)-1 '-pirenilmetilo, 4,4',4''-tris(tert-butilfenil)metilo (TTTr) y 4,4'-di-3,5-hexadienoxitritilo. Los ejemplos de grupos sililo incluyen, sin carácter limitante, trimetilsililo (TMS), tert-butildimetilsililo (TBDMS), triisopropilsililo (TIPS), tert-butildifenilsililo (TBDPS), triisopropilsililoximetilo y [2-(trimetilsilil)etoxi]metilo. Los expertos en la técnica apreciarán que los grupos unidos al anillo de pentosa y cualesquiera grupos -NH y/o NH2 presentes en B1a se pueden proteger con varios grupos protectores y cualesquiera grupos protectores presentes se pueden intercambiar por otros grupos protectores. La selección y el intercambio de los grupos protectores se encuentran dentro de las competencias de los expertos en la técnica. Cual(es)quiera grupo(s) protector(es) se puede(n) eliminar mediante métodos conocidos en la técnica, por ejemplo, con un ácido (por ejemplo, un ácido orgánico o mineral), una base o una fuente de fluoruro.
Composiciones farmacéuticas
En la presente también se describen composiciones farmacéuticas, que pueden incluir una cantidad eficaz de uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) y un portador, diluyente, excipiente farmacéuticamente aceptable o una combinación de estos.
La expresión «composición farmacéutica» se refiere a una mezcla de uno o más compuestos divulgados en la presente con otros componentes químicos tales como diluyentes o portadores. La composición farmacéutica facilita la administración del compuesto a un organismo. Las composiciones farmacéuticas también se pueden obtener haciendo reaccionar los compuestos con ácidos orgánicos o inorgánicos tales como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico, ácido metanosulfónico, ácido etanosulfónico, ácido p-toluenosulfónico y ácido salicílico. Por lo general, las composiciones farmacéuticas se adaptarán a la vía de administración deseada específica.
La expresión «fisiológicamente aceptable» define un portador, diluyente o excipiente que no cancela la actividad biológica ni las propiedades del compuesto.
Un «portador», tal como se utiliza en la presente, se refiere a un compuesto que facilita la incorporación de un compuesto en células o tejidos. Por ejemplo, sin limitación, el sulfóxido de dimetilo (DMSO) es un portador utilizado habitualmente que facilita la captación de muchos compuestos orgánicos en las células o tejidos de un sujeto.
Un «diluyente», tal como se utiliza en la presente, se refiere a un ingrediente en una composición farmacéutica que carece de actividad farmacológica pero que puede ser necesario o deseable desde un punto de vista farmacéutico. Por ejemplo, se puede utilizar un diluyente para incrementar el volumen de un fármaco potente cuya masa sea demasiado pequeña para su elaboración y/o administración. También podría ser un líquido para la disolución de un fármaco que se ha de administrar mediante inyección, ingesta o inhalación. Una forma habitual de diluyente en la técnica es una solución acuosa tamponada tal como, sin limitación, solución salina tamponada con fosfato que imita la composición de la sangre humana.
Un «excipiente», tal como se utiliza en la presente, se refiere a una sustancia inerte que se añade a una composición farmacéutica para proporcionar, sin limitación, volumen, consistencia, estabilidad, capacidad de unión, lubricación, capacidad de desintegración, etc. a la composición. Un «diluyente» es un tipo de excipiente.
Las composiciones farmacéuticas descritas en la presente se pueden administrar a un paciente humano per se o en composiciones farmacéuticas en las que se mezclan con otros principios activos, como en una terapia combinada, o portadores, diluyentes, excipientes o combinaciones de estos. Una formulación adecuada depende de la vía de administración seleccionada. Las técnicas para la formulación y administración de los compuestos descritos en la presente son conocidas por los expertos en la técnica.
Las composiciones farmacéuticas divulgadas en la presente se pueden elaborar de un modo que es conocido de por sí, por ejemplo, mediante procesos convencionales de mezcla, disolución, granulación, elaboración de grageas, levitación, emulsificación, encapsulación, atrapamiento o formación de comprimidos. Además, los principios activos están contenidos en una cantidad eficaz para conseguir su fin deseado. Muchos de los compuestos utilizados en las
combinaciones farmacéuticas divulgadas en la presente se pueden proporcionar como sales con contraiones farmacéuticamente compatibles.
En la técnica existen múltiples técnicas para administrar un compuesto que incluyen, sin carácter limitante, la vía oral, rectal, tópica, aerosol, inyección y suministro parenteral, que incluye inyecciones intramusculares, subcutáneas, intravenosas, intramedulares, intratecales, intraventriculares directas, intraperitoneales, intranasales e intraoculares.
También se puede administrar el compuesto de forma local en lugar de sistémica, por ejemplo, mediante la inyección del compuesto directamente en el área afectada, a menudo en una formulación de liberación sostenida o prolongada (depot). Además, se puede administrar el compuesto en un sistema de suministro farmacológico dirigido, por ejemplo, en un liposoma recubierto con un anticuerpo específico para un tejido. Los liposomas se dirigirán selectivamente al órgano y serán captados selectivamente por este.
Si se desea, las composiciones se pueden presentar en un envase o dispositivo dispensador que puede contener una o más formas farmacológicas unitarias que contienen el principio activo. El envase puede comprender, por ejemplo, una lámina metálica o de plástico tal como un envase de tipo blíster. El envase o dispositivo dispensador puede venir acompañado de instrucciones de administración. El envase o dispensador también puede venir acompañado de una nota asociada con el paquete en una forma prescrita por una agencia gubernamental que regula la elaboración, el uso o la venta de productos farmacéuticos, donde dicha nota refleja la autorización por parte de la agencia de la forma del fármaco para administración humana o veterinaria. Tal nota puede ser, por ejemplo, la etiqueta aprobada por la Administración de Fármacos y Alimentos de los Estados Unidos para fármacos recetados o el prospecto del producto aprobado. También se pueden preparar composiciones que pueden incluir un compuesto descrito en la presente formulado en un portador farmacéutico compatible, se pueden colocar en un envase adecuado y se pueden etiquetar para el tratamiento de una afección indicada.
Métodos de uso:
En la presente también se describen métodos para mejorar, tratar y/o prevenir una infección vírica provocada por paramixovirus, que pueden incluir administrar a un sujeto una cantidad eficaz de uno o más compuestos descritos en la presente, o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este). El sujeto se puede identificar como que padece una infección vírica provocada por paramixovirus.
En la presente también se describen métodos para inhibir la replicación vírica de un paramixovirus, que pueden incluir poner en contacto una célula infectada con el virus con una cantidad eficaz de un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este). Por ejemplo, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, puede actuar como un terminador de la cadena e inhibir la replicación del virus.
En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una infección provocada por paramixovirus. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para prevenir una infección provocada por paramixovirus. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para inhibir la replicación de un paramixovirus. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para inhibir el complejo de polimerasas de un paramixovirus.
En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una infección vírica del aparato respiratorio superior provocada por una infección por paramixovirus. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una infección vírica del aparato respiratorio inferior provocada por una infección por paramixovirus. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para
tratar y/o mejorar uno o más síntomas de una infección provocada por una infección por paramixovirus (tal como las que se describen en la presente).
En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una bronquiolitis y/o traqueobronquitis debida a una infección provocada por paramixovirus. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una neumonía debida a una infección provocada por paramixovirus. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una laringotraqueobronquitis debida a una infección provocada por paramixovirus.
En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una infección vírica respiratoria sincitial (VRS). En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para prevenir una infección vírica respiratoria sincitial. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para inhibir la replicación de un virus respiratorio sincitial. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para inhibir el complejo de polimerasas de un VRS.
En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una infección vírica del aparato respiratorio superior provocada por una infección por VRS. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una infección vírica del aparato respiratorio inferior provocada por una infección por VRS. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar uno o más síntomas de una infección provocada por una infección por VRS (tal como las que se describen en la presente).
En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una bronquiolitis y/o traqueobronquitis debida a una infección provocada por VRS. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una neumonía debida a una infección provocada por VRS. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una laringotraqueobronquitis debida a una infección provocada por VRS.
En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una infección provocada por VPIH-1 y/o una infección provocada por VPIH-3. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para prevenir una infección provocada por VPIH-1 y/o una infección provocada por VPIH-3. En algunas realizaciones, una cantidad eficaz
de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para inhibir la replicación de VPIH-1 y/o VPIH-3. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para inhibir el complejo de polimerasas de VPIH-1 y/o el complejo de polimerasas de VPIH-3.
En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una infección provocada por VPIH-2 y/o una infección provocada por VPIH-4. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para prevenir una infección provocada por VPIH-2 y/o una infección provocada por VPIH-4. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para inhibir la replicación de VPIH-2 y/o VPIH-4. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para inhibir el complejo de polimerasas de VPIH-2 y/o el complejo de polimerasas de VPIH-4.
En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una infección metapneumovírica. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para prevenir una infección metapneumovírica. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para inhibir la replicación de un metapneumovirus. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para inhibir el complejo de polimerasas de un metapneumovirus. En algunas realizaciones, incluidas las de este párrafo, el metapneumovirus puede ser un metapneumovirus humano.
En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una infección vírica del aparato respiratorio superior provocada por un virus seleccionado entre un virus de tipo VRS, un virus de parainfluenza y un metapneumovirus. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una infección vírica del aparato respiratorio inferior provocada por un virus seleccionado entre un virus de tipo VRS, un virus de parainfluenza y un metapneumovirus. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar uno o más síntomas de una infección provocada por un virus seleccionado entre un virus de tipo VRS, un virus de parainfluenza y un metapneumovirus (tal como los que se describen en la presente).
En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una bronquiolitis y/o traqueobronquitis debida a una infección provocada por un virus de tipo VRS, una infección provocada por el virus de parainfluenza y una infección provocada por un metapneumovirus. En algunas realizaciones, una cantidad eficaz de uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una neumonía debida a una infección provocada por un virus de tipo VRS, una infección provocada por el virus de parainfluenza y una infección provocada por un metapneumovirus. En algunas realizaciones, una cantidad eficaz de
uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, y/o una composición farmacéutica que incluye uno o más compuestos descritos en la presente (por ejemplo, un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este) se puede utilizar para tratar y/o mejorar una laringotraqueobronquitis debida a una infección provocada por un virus de tipo VRS, una infección provocada por el virus de parainfluenza y una infección provocada por un metapneumovirus.
Dichos uno o más compuestos de Fórmula (I), o una sal farmacéuticamente aceptable de estos, que se pueden utilizar para tratar, mejorar y/o prevenir una infección vírica provocada por paramixovirus pueden ser un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, proporcionado en cualquiera de las realizaciones descritas en el párrafo que sigue inmediatamente al subencabezamiento «Compuestos» hasta el párrafo que empieza por «Otros ejemplos de un compuesto de Fórmula (I) incluyen, sin carácter limitante, los siguientes:».
Los términos «prevenir» y «que previene», tal como se utilizan en la presente, se refieren a que se reduce la eficacia de la replicación vírica y/o se inhibe la replicación vírica en mayor grado en un sujeto que recibe el compuesto en comparación con un sujeto que no recibe el compuesto. Los ejemplos de formas de prevención incluyen la administración profiláctica a un sujeto que ha sido o puede ser expuesto a un agente infeccioso tal como un paramixovirus (por ejemplo, VRS).
Los términos y expresiones «tratar», «que trata», «tratamiento», «terapéutico» y «terapia», tal como se utilizan en la presente, no se refieren necesariamente a la anulación o cura total de la enfermedad o afección. Cualquier alivio de cualesquiera signos o síntomas no deseados de una enfermedad o afección, en cualquier medida se puede considerar como tratamiento y/o terapia. Además, el tratamiento puede incluir actos que pueden empeorar la sensación de bienestar o apariencia general del sujeto.
Las expresiones «cantidad terapéuticamente eficaz» y «cantidad eficaz» se utilizan para indicar una cantidad de un compuesto, o agente farmacéutico, activo, que suscita la respuesta biológica o médica indicada. Por ejemplo, una cantidad eficaz de compuesto puede ser la cantidad necesaria para prevenir, aliviar o mejorar los síntomas de una enfermedad o prolongar la supervivencia del sujeto que está siendo tratado. Esta respuesta puede ocurrir en un tejido, sistema, animal o ser humano e incluye el alivio de los signos o síntomas de la enfermedad que se esté tratando. La determinación de una cantidad eficaz se encuentra totalmente dentro de las competencias de los expertos en la técnica, teniendo en cuenta la divulgación proporcionada en la presente. La cantidad terapéuticamente eficaz de los compuestos divulgados en la presente requerida como dosis dependerá de la vía de administración, el tipo de animal, incluido el ser humano, que se esté tratando, y las características físicas del animal específico en consideración. La dosis se puede adaptar para conseguir el efecto deseado, pero dependerá de factores tales como el peso, dieta, medicación concurrente y otros factores que los expertos en la técnica médica reconocerán.
Los expertos en la técnica conocen varios indicadores para determinar la eficacia de un método para tratar una infección vírica provocada por paramixovirus. Los ejemplos de indicadores adecuados incluyen, sin carácter limitante, una reducción de la carga vírica, una reducción de la replicación vírica, una reducción del tiempo de seroconversión (virus indetectable en el suero del paciente), una reducción de la morbilidad o mortalidad en los resultados clínicos y/u otro indicador de la respuesta de la enfermedad.
En algunas realizaciones, una cantidad eficaz de un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, es una cantidad que es eficaz para reducir las titulaciones víricas hasta niveles no detectables, por ejemplo, hasta de aproximadamente 1000 a aproximadamente 5000, hasta de aproximadamente 500 a aproximadamente 1000, o hasta de aproximadamente 100 a aproximadamente 500 copias del genoma/mL de suero. En algunas realizaciones, una cantidad eficaz de un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, es una cantidad que es eficaz para reducir la carga vírica en comparación con la carga vírica antes de la administración del compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este. Por ejemplo, donde la carga vírica se mide antes de la administración del compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, y de nuevo tras la finalización del régimen de tratamiento con el compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este (por ejemplo, 1 semana tras la finalización). En algunas realizaciones, una cantidad eficaz de un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, puede ser una cantidad que es eficaz para reducir la carga vírica hasta menos de aproximadamente 100 copias de genoma/mL de suero. En algunas realizaciones, una cantidad eficaz de un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, es una cantidad que es eficaz para conseguir una reducción en la titulación vírica en el suero del sujeto comprendida en el intervalo de una reducción de aproximadamente 1.5-log a aproximadamente 2.5-log, una reducción de aproximadamente 3-log a aproximadamente 4-log o una reducción superior a aproximadamente 5-log en comparación con la carga vírica antes de la administración del compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este. Por ejemplo, donde la carga vírica se mide antes de la administración del compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, y de nuevo tras la finalización del régimen de tratamiento con el compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este (por ejemplo, 1 semana tras la finalización).
En algunas realizaciones, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, puede dar como resultado una reducción de al menos 1, 2, 3, 4, 5, 10, 15, 20, 25, 50, 75, 100 veces o más en la replicación de un paramixovirus respecto a los niveles previos al tratamiento en un sujeto, tal como se determina tras la finalización
del régimen de tratamiento (por ejemplo, 1 semana después de la finalización). En algunas realizaciones, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, puede dar como resultado una reducción de la replicación de un paramixovirus respecto a los niveles previos al tratamiento comprendida en el intervalo de aproximadamente 2 a aproximadamente 5 veces, de aproximadamente 10 a aproximadamente 20 veces, de aproximadamente 15 a aproximadamente 40 veces o de aproximadamente 50 a aproximadamente 100 veces. En algunas realizaciones, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, puede dar como resultado una reducción de la replicación de un paramixovirus comprendida en el intervalo de una reducción de 1 a 1.5 log, de 1.5 log a 2 log, de 2 log a 2.5 log, de 2.5 a 3 log, de 3 log a 3.5 log o de 3.5 a 4 log más de la replicación del paramixovirus en comparación con la reducción del paramixovirus conseguida con ribavirina (Virazole®), o puede conseguir la misma reducción que la terapia con ribavirina (Virazole®) en un periodo de tiempo más corto, por ejemplo, en una semana, dos semanas, un mes, dos meses o tres meses, en comparación con la reducción conseguida después de seis meses de una terapia con ribavirina (Virazole®).
En algunas realizaciones, una cantidad eficaz de un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, es una cantidad que es eficaz para conseguir una respuesta vírica sostenida, por ejemplo, que se obtenga un ARN del paramixovirus no detectable o sustancialmente no detectable (por ejemplo, menos de aproximadamente 500, menos de aproximadamente 400, menos de aproximadamente 200 o menos de aproximadamente 100 copias de genoma por mililitro de suero) en el suero del sujeto durante un periodo de al menos aproximadamente una semana, dos semanas, un mes, al menos aproximadamente dos meses, al menos aproximadamente tres meses, al menos aproximadamente cuatro meses, al menos aproximadamente cinco meses o al menos aproximadamente seis meses tras el cese de la terapia.
Después de un periodo de tiempo, los agentes infecciosos pueden desarrollar resistencia a uno o más agentes terapéuticos. El término «resistencia», tal como se utiliza en la presente, se refiere a una cepa vírica que muestra una respuesta retardada, reducida y/o nula a uno o más agentes terapéuticos. Por ejemplo, después del tratamiento con un agente antivírico, la carga vírica de un sujeto infectado con un virus resistente se puede reducir en un grado menor en comparación con la magnitud de la reducción de la carga vírica mostrada por un sujeto infectado con una cepa no resistente. Un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, se puede administrar a un sujeto infectado con un VRS que es resistente a uno o más agentes anti-VRS diferentes (por ejemplo, ribavirina) para mejorar y/o tratar una infección provocada por VRS. En algunas realizaciones, el desarrollo de una o más cepas de VRS resistentes se puede retardar cuando los sujetos se tratan con un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, en comparación con el desarrollo de una o más cepas de VRS resistentes a otros agentes anti-VRS.
En algunas realizaciones, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, puede reducir el porcentaje de sujetos que experimentan complicaciones debidas a una infección vírica provocada por VRS en comparación con el porcentaje de sujetos que experimentan complicaciones al ser tratados con ribavirina. Por ejemplo, el porcentaje de sujetos que están siendo tratados con un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, que experimentan complicaciones puede ser un 10%, 25%, 40%, 50%, 60%, 70%, 80% y 90% inferior en comparación con sujetos que están siendo tratados con ribavirina.
Un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, o una composición farmacéutica que incluye un compuesto descrito en la presente, se puede utilizar combinado con uno o más agentes adicionales. Un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, se puede utilizar combinado con uno o más agentes utilizados actualmente para tratar un VRS. Por ejemplo, el agente adicional puede ser ribavirina, palivizumab y RSV-IGIV. Para el tratamiento de un VRS, los agentes adicionales incluyen, sin carácter limitante, ALN-RSV01 (un agente de tipo ARNip con la secuencia de la hebra sentido (de 5' a 3') GGCUCUUAGCAAAGUCAAGdTdT (SEQ ID NO. 1) y la secuencia de la hebra antisentido (de 5' a 3') CUUGACUUUGCUAAGAGCCdTdT (SEQ ID NO.
2), Alnylam Pharmaceuticals, Publicación de EE. UU. N ° 2009/0238772, presentada el 15 de diciembre de 2008), b Ms -433771 (1 -ciclopropil-3-[[1 -(4-hidroxibutil)bencimidazol-2-il]metil]imidazo[4,5-c]piridin-2-ona), RFI-641 (ácido 4,4''-bis-{4,6-bis-[3-(bis-carbamoilmetil-sulfamoil)fenilamino]-(1,3,5)triazin-2-ilamino}bifenil-2,2"-disulfónico), RSV604 ((S)-1-(2-fluorofenil)-3-(2-oxo-5-fenil-2,3-dihidro-1 H-benzo[e][1,4]diazepin-3-il)urea), MDT-637 5,5'-bis[1-(((5-amino-1 H-tetrazolil)imino)metil)]2,2',4''-metilidinotrisfenol), BTA9881 ((R)-9¿>-(4-clorofenil)-1 -(4-fluorobenzoil)-2,3-dihidro-1 H-imidazo[1 ',2':1,2]pirrolo[3,4-c]piridin-5(9¿>H)-ona), Tm C-353121 (2-[[6-[[[2-(3-hidroxipropil)-5-metilfenil]amino]metil]-2-[[3-(morfolin-4-il)propil]amino]bencimidazol-1 -il]metil]-6-metilpiridin-3-ol) (Tibotec), MBX-300 ([2,2-bis(docosiloxioximetil)propil-5-acetamido-3,5-didesoxi-4,7,8,9-tetra-0-(sodio-oxisulfonil)-D-glicero-D-galacto-2-nonulopiranosid]onato), YM-53403 (6- {4-[(bifenil-2-ilcarbonil)amino]benzoil}-W-ciclopropil-5,6-dihidro-4H-tieno[3,2-ad[1]benzazepino-2-carboxamida), motavizumab (Medi-524, MedImmune), Medi-559 (VRS recombinante A2 cp248/404/1030/ASH), Medi-534 (candidato de vacuna que es un vector del virus de parainfluenza humano/bovino recombinante de tipo 3 (VPI3)/VRS F2), Medi-557, RV568 y una vacuna de partículas de VRS-F (Novavax).
Un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, se puede administrar con uno o más agentes adicionales conjuntamente en una única composición farmacéutica. Un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, se puede administrar con uno o más agentes adicionales como dos o más composiciones farmacéuticas separadas. Por ejemplo, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, se puede administrar en una composición farmacéutica y al menos uno de los agentes adicionales
se puede administrar en una segunda composición farmacéutica. Si hay al menos dos agentes adicionales, uno o más de los agentes adicionales pueden estar en una primera composición farmacéutica que incluye un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, y al menos uno de los demás agentes adicionales puede estar en una segunda composición farmacéutica.
El orden de administración de un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, con uno o más agentes adicionales puede variar. Un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, se puede administrar antes de todos los agentes adicionales. Un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, se puede administrar antes de al menos un agente adicional. Un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, se puede administrar de forma concomitante con uno o más agentes adicionales. Un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, se puede administrar de forma posterior a la administración de al menos un agente adicional. Un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, se puede administrar de forma posterior a la administración de todos los agentes adicionales.
Una posible ventaja del hecho de utilizar un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, combinado con uno o más agentes adicionales que se describen en el párrafo que empieza por «En algunas realizaciones, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, o una composición farmacéutica que incluye un compuesto descrito en la presente, se puede utilizar combinado con uno o más agentes adicionales...», incluidas las sales y profármacos farmacéuticamente aceptables de estos, puede ser una reducción en la(s) cantidad(es) requerida(s) de uno o más compuestos del párrafo que empieza por «En algunas realizaciones, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, o una composición farmacéutica que incluye un compuesto descrito en la presente, se puede utilizar combinado con uno o más agentes adicionales...» (incluidas las sales y profármacos farmacéuticamente aceptables de estos) que es (son) eficaz (eficaces) en el tratamiento de una afección patológica divulgada en la presente (por ejemplo, VRS), en comparación con la cantidad requerida para conseguir el mismo resultado terapéutico cuando uno o más compuestos descritos en el párrafo que empieza por «En algunas realizaciones, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, o una composición farmacéutica que incluye un compuesto descrito en la presente, se puede utilizar combinado con uno o más agentes adicionales...», incluidas las sales y profármacos farmacéuticamente aceptables de estos, se administran sin un compuesto de Fórmula (I) o una sal farmacéuticamente aceptable de este. Por ejemplo, la cantidad de un compuesto descrito en el párrafo que empieza por «En algunas realizaciones, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, o una composición farmacéutica que incluye un compuesto descrito en la presente, se puede utilizar combinado con uno o más agentes adicionales...», incluida una sal y un profármaco farmacéuticamente aceptable de estos, puede ser inferior en comparación con la cantidad del compuesto descrito en el párrafo que empieza por «En algunas realizaciones, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, o una composición farmacéutica que incluye un compuesto descrito en la presente, se puede utilizar combinado con uno o más agentes adicionales...», incluida una sal y un profármaco farmacéuticamente aceptable de estos, necesaria para conseguir la misma reducción de la carga vírica cuando se administra como una monoterapia. Otra posible ventaja del hecho de utilizar un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, combinado con uno o más agentes adicionales descritos en el párrafo que empieza por «En algunas realizaciones, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, o una composición farmacéutica que incluye un compuesto descrito en la presente, se puede utilizar combinado con uno o más agentes adicionales...», incluidas las sales y profármacos farmacéuticamente aceptables de estos, consiste en que el uso de dos o más compuestos que tienen mecanismos de acción diferentes puede crear una barrera más alta para el desarrollo de cepas víricas resistentes en comparación con la barrera obtenida cuando el compuesto se administra como monoterapia.
Otras ventajas del hecho de utilizar un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, combinado con uno o más agentes adicionales que se describen en el párrafo que empieza por «En algunas realizaciones, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, o una composición farmacéutica que incluye un compuesto descrito en la presente, se puede utilizar combinado con uno o más agentes adicionales...», incluidas las sales y profármacos farmacéuticamente aceptables de estos, pueden incluir una resistencia cruzada escasa o nula entre un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, y uno o más agentes adicionales descritos en el párrafo que empieza por «En algunas realizaciones, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, o una composición farmacéutica que incluye un compuesto descrito en la presente, se puede utilizar combinado con uno o más agentes adicionales...» (incluidas las sales y profármacos farmacéuticamente aceptables de estos); diferentes rutas para eliminar un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, y uno o más agentes adicionales descritos en el párrafo que empieza por «En algunas realizaciones, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, o una composición farmacéutica que incluye un compuesto descrito en la presente, se puede utilizar combinado con uno o más agentes adicionales...» (incluidas las sales y profármacos farmacéuticamente aceptables de estos); unas toxicidades solapadas de escasas a nulas entre un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, y uno o más agentes adicionales descritos en el párrafo que empieza por «En algunas realizaciones, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, o una composición farmacéutica que incluye un compuesto descrito en la presente, se puede utilizar combinado con uno o más agentes adicionales...» (incluidas las sales y profármacos farmacéuticamente aceptables de estos); unos efectos de escasos a no significativos sobre el citocromo P450; y/o unas interacciones farmacocinéticas de escasas a nulas entre un compuesto
de Fórmula (I), o una sal farmacéuticamente aceptable de este, y uno o más agentes adicionales descritos en el párrafo que empieza por «En algunas realizaciones, un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este, o una composición farmacéutica que incluye un compuesto descrito en la presente, se puede utilizar combinado con uno o más agentes adicionales...» (incluidas las sales y profármacos farmacéuticamente aceptables de estos).
Como será muy evidente para un experto en la técnica, la dosis in vivo útil que se ha de administrar y el modo particular de administración variarán dependiendo de la edad, el peso, la gravedad de la aflicción y la especie de mamífero que se esté tratando, los compuestos particulares empleados y el uso específico para el que se empleen estos compuestos. Un experto en la técnica puede lograr la determinación de los niveles de dosis eficaces, es decir, los niveles de dosis necesarios para conseguir el resultado deseado, utilizando métodos rutinarios, por ejemplo, ensayos clínicos con seres humanos y estudios in vitro.
La dosis puede variar ampliamente, dependiendo de los efectos deseados y la indicación terapéutica. Como alternativa, las dosis se pueden basar en el área superficial del paciente y se pueden calcular en función de esta, según interpretan los expertos en la técnica. Aunque la dosis exacta se determinará en función de cada fármaco, en la mayoría de los casos se pueden realizar ciertas generalizaciones respecto a la dosis. La pauta posológica diaria para un paciente humano adulto puede ser, por ejemplo, una dosis oral comprendida entre 0.01 mg y 3000 mg de cada principio activo, preferentemente entre 1 mg y 700 mg, por ejemplo, entre 5 y 200 mg. La dosis puede ser solo una o una serie de dos o más administradas en el transcurso de uno o más días, según necesite el sujeto. Los compuestos se pueden administrar durante un periodo de terapia continuada, por ejemplo, durante una semana o más, o durante meses o años.
En los casos en los que se han establecido dosis para seres humanos de los compuestos para al menos alguna afección, se pueden utilizar estas mismas dosis, o dosis que estén comprendidas entre aproximadamente un 0.1% y un 500%, más preferentemente entre aproximadamente un 25% y un 250% de la dosis establecida para seres humanos. Cuando no se han establecido dosis para seres humanos, como será el caso para composiciones farmacéuticas recientemente descubiertas, se puede inferir una dosis adecuada para seres humanos a partir de los valores de DE50 o DI50, u otros valores adecuados derivados de estudios in vivo o in vitro, que se cualifican mediante estudios de toxicidad y estudios de eficacia en animales.
En casos de administración de una sal farmacéuticamente aceptable, las dosis se pueden calcular como la base libre. Como sobreentenderán los expertos en la técnica, en ciertas situaciones puede ser necesario administrar los compuestos divulgados en la presente en cantidades que excedan, o incluso que excedan en gran medida, el intervalo de dosis preferido mencionado anteriormente con el fin de tratar de forma eficaz y agresiva infecciones o enfermedades particularmente agresivas.
La cantidad y el intervalo de la dosis se pueden ajustar de forma individual para proporcionar unos niveles en plasma del resto activo que sean suficientes para mantener los efectos moduladores o una concentración mínima eficaz (CME). La CME variará para cada compuesto pero se puede estimar a partir de datos in vitro. Las dosis necesarias para conseguir la CME dependerán de las características del individuo y la vía de administración. A pesar de ello, se pueden utilizar ensayos de HPLC o bioensayos para determinar las concentraciones en plasma. Los intervalos de la dosis también se pueden determinar utilizando el valor de CME. Las composiciones se deben administrar utilizando un régimen que mantenga los niveles en plasma por encima de la CME durante un 10-90% del tiempo, preferentemente entre un 30-90% y de la forma más preferida entre un 50-90%. En casos de administración local o captación selectiva, puede ser que la concentración local eficaz del fármaco no esté relacionada con la concentración en plasma.
Cabe destacar que el médico responsable sabrá cómo y cuándo terminar, interrumpir o ajustar la administración debido a toxicidad o disfunciones de los órganos. A la inversa, el médico responsable también sabrá ajustar el tratamiento a niveles más elevados si la respuesta clínica no fuera adecuada (excluyendo la toxicidad). La magnitud de una dosis administrada en la gestión del trastorno de interés variará según la gravedad de la afección que se ha de tratar y la vía de administración. La gravedad de la afección se puede evaluar, por ejemplo, en parte, mediante métodos de evaluación de pronóstico estándar. Además, la dosis y quizás la frecuencia de la dosis también variarán de acuerdo con la edad, el peso corporal y la respuesta del paciente individual. En medicina veterinaria, se puede utilizar un programa comparable al expuesto anteriormente.
Los compuestos divulgados en la presente se pueden evaluar para determinar su eficacia y toxicidad utilizando métodos conocidos. Por ejemplo, la toxicología de un compuesto particular, o de un subconjunto de los compuestos, que comparten ciertos restos químicos, se puede establecer determinando la toxicidad in vitro respecto a una línea celular, tal como una línea celular de mamífero y preferentemente de ser humano. Los resultados de tales estudios son a menudo predictivos de la toxicidad en animales tales como mamíferos o, más específicamente, seres humanos. Como alternativa, la toxicidad de compuestos particulares en un modelo animal tal como con ratones, ratas, conejos o monos, se puede determinar utilizando métodos conocidos. La eficacia de un compuesto particular se puede establecer utilizando varios métodos reconocidos tales como métodos in vitro, modelos animales o ensayos clínicos con seres humanos. A la hora de seleccionar un modelo para determinar la eficacia, el experto se puede guiar por el estado de la técnica para elegir un modelo, dosis, vía de administración y/o régimen que sean adecuados.
EJEMPLOS
En los siguientes ejemplos, se describen realizaciones adicionales con más detalle, las cuales no pretenden limitar de ningún modo el alcance de las reivindicaciones.
EJEMPLO 1
Preparación del Compuesto 1A

Preparación de (1-2): A una solución de 1-1 (50 g, 203 mmol) en piridina anhidra (200 mL), se añadió TBDPS-Cl (83.7 g, 304 mmol). Se dejó que la reacción procediera durante toda la noche a T.A. La solución se concentró a baja presión para obtener un residuo, el cual se repartió entre acetato de etilo y agua. La fase orgánica se separó, se lavó con salmuera, se secó con sulfato de magnesio y se concentró a presión reducida para obtener un éter de tipo 5'-OTBDPS como una espuma blanca (94 g).
A una solución del éter de tipo 5'-OTBDPS (94.0 g, 194.2 mmol) en DCM anhidro (300 mL), se añadió nitrato de plata (66.03 g, 388.4 mmol) y colidina (235 mL, 1.94 mol). La mezcla se agitó a T.A. Después de 15 min, la mezcla se enfrió hasta 0 °C y se añadió cloruro de monometoxitritilo (239.3 g, 776.8 mmol) en una única porción. Después de agitar durante toda la noche a T.A., la mezcla se filtró a través de Celite y el filtrado se diluyó con TBME. La solución se lavó sucesivamente con ácido cítrico 1 M, salmuera diluida y bicarbonato de sodio al 5%. La solución orgánica se secó con sulfato de sodio y se concentró al vacío para obtener el intermedio totalmente protegido como una espuma amarilla.
Este intermedio totalmente protegido se disolvió en tolueno (100 mL) y la solución se concentró a presión reducida. El residuo se disolvió en THF anhidro (250 mL) y se trató con TBAF (60 g, 233 mmol). La mezcla se agitó durante 2 h a T.A. y se eliminó el disolvente a presión reducida. El residuo se arrastró con acetato de etilo y la solución se lavó en primer lugar con bicarbonato de sodio saturado y a continuación con salmuera. Después de secar con sulfato de magnesio, se eliminó el disolvente al vacío y el residuo se purificó mediante cromatografía en columna (50% de EA en PE) para obtener 1-2 (91 g, 86.4%) como una espuma blanca.
Preparación de (1-3): A una solución de 1-2 (13.5 g, 26 mmol) en DCM (100 mL), se añadió piridina (6.17 mL, 78 mmol). La solución se enfrió hasta 0 °C y se añadió peryodinano de Dess-Martin (33.8 g, 78 mmol) en una única porción. La mezcla de reacción se agitó durante 4 h a T.A. y se desactivó mediante la adición de una solución de Na2S2O3 (al 4%) y una solución acuosa de bicarbonato de sodio (al 4%) (se ajustó el pH de la solución hasta un pH de 6, ~150 mL). La mezcla se agitó durante 15 min. La fase orgánica se separó, se lavó con salmuera diluida y se concentró a presión reducida. El residuo se disolvió en dioxano (100 mL) y la solución se trató con formaldehído acuoso al 37% (21.2 g, 10 eq.) e hidróxido sodio acuoso 2 N (10 eq.). La mezcla de reacción se agitó a T.A. durante toda la noche. Después de agitar durante 0.5 h a T.A., el exceso de hidróxido de sodio acuoso se eliminó con NH4Cl saturado (~150 mL). La mezcla se concentró a presión reducida y el residuo se repartió entre acetato de etilo y bicarbonato de sodio al 5%. La fase orgánica se separó, se lavó con salmuera, se secó con sulfato de magnesio y se concentró. El residuo se purificó mediante cromatografía en columna (2% de MeOH en DCM) para obtener el diol 1-3 (9.2 g, 83.6%) como una espuma blanca.
Preparación de (1-4): El compuesto 1-3 (23 g, 42.0 mmol) se coevaporó con tolueno dos veces. El residuo se disolvió en DCM anhidro (250 mL) y piridina (20 mL). La solución se enfrió hasta 0 °C y se añadió anhídrido tríflico (24.9 g, 88.1 mmol) gota a gota durante 10 min. A esta temperatura, la reacción se agitó durante 40 min. La reacción se monitorizó mediante TLC (PE: EA = 2:1 y DCM: MeOH = 15:1). Una vez finalizada, la mezcla de reacción se desactivó
con agua (50 mL) a 0 °C. La mezcla se agitó durante 30 min y se extrajo con EA. La fase orgánica se secó con Na2SÜ4 y se filtró a través de un lecho de gel de sílice. El filtrado se concentró a presión reducida y el residuo se purificó mediante cromatografía en columna (50% de EA en PE) para obtener 1-4 (30.0 g, 88.3%) como una espuma marrón.
Preparación de (1-5): A una solución agitada de 1-4 (4.4 g, 5.42 mmol) en DMF anhidro (50 mL), se añadió NaH (260 mg, 6.5 mmol) a 0 °C en atmósfera de nitrógeno. La solución se agitó a T.A. durante 1.5 h. La solución se utilizó para el siguiente paso sin ningún tratamiento adicional.
Preparación de (1-6): Se añadió NaN3 (1.5 g, 21.68 mmol) a la solución agitada a 0 °C en atmósfera de nitrógeno y la solución resultante se agitó a T.A. durante 1.5 h. La reacción se desactivó con agua, se extrajo con EA, se lavó con salmuera y se secó con MgSÜ4. La fase orgánica concentrada se utilizó para el siguiente paso sin purificación adicional.
Preparación de (1-7): A una solución de 1-6 (3.0 g, 5.4 mmol) en 1,4-dioxano anhidro (18 mL), se añadió NaÜH (5.4 mL, 2 M en agua) a T.A. La mezcla de reacción se agitó a T.A. durante 3 h. La reacción se diluyó con EA, se lavó con salmuera y se secó con MgSÜ4. La fase orgánica concentrada se purificó en una columna de gel de sílice (30% de EA en PE) para obtener 1-7 (2.9 g, 93%) como una espuma blanca.
Preparación de (1A): Se disolvió el compuesto 1-7 (520 mg, 0.90 mmol) en HCÜÜH al 80% (20 mL) a T.A. La mezcla se agitó durante 3 h y se monitorizó mediante TLC. Se eliminó el disolvente y el residuo se trató con MeÜH y tolueno 3 veces. Se añadió NH3/MeÜH y la mezcla de reacción se agitó a T.A. durante 5 min. Se concentró el disolvente a sequedad y el residuo se purificó mediante cromatografía en columna para obtener 1A (120 mg, 44.4%) como un sólido blanco. ESI-LCMS: m/z 302.0 [M+H]+, 324.0 [M+Na]+.
EJEMPLO 2
Preparación del Compuesto 2A

Preparación de (2-1): A una solución agitada de 1-7 (1.1 g, 2.88 mmol) en DCM anhidro (10 mL), se añadió MMTrCl (1.77 g, 5.76 mmol), AgNÜ3 (1.47 g, 8.64 mmol) y colidina (1.05 g, 8.64 mmol) a 25 °C en atmósfera de N2. La reacción se calentó a reflujo durante 12 h. Se añadió MeÜH (20 mL) y se eliminó el disolvente a sequedad. El residuo se purificó en una columna de gel de sílice (20% de EA en PE) para obtener 2-1 (1.6 g, 85.1%) como una espuma blanca.
Preparación de (2-2): A una solución agitada de 2-1 (800 mg, 0.947 mmol) en MeCN anhidro (10 mL), se añadió TPSCl (570 mg, 1.89 mmol), DMAP (230 mg, 1.89 mmol) y TEA (190 mg, 1.89 mmol) a T.A. La mezcla se agitó durante 12 h. Se añadió NH4ÜH (25 mL) y la mezcla se agitó durante 2 h. Se eliminó el disolvente y el residuo se purificó en una columna de gel de sílice como una espuma amarilla. Una purificación adicional mediante prep-TLC proporcionó 2-2 (700 mg, 87.1%) como un sólido blanco.
Preparación de (2A): Se disolvió el compuesto 2-2 (300 mg, 0.355 mmol) en HCÜÜH al 80% (5 mL) a T.A. La mezcla se agitó durante 3 h y se monitorizó mediante TLC. A continuación, se eliminó el disolvente y el residuo se trató con MeÜH y tolueno (3 veces). Se añadió NH3/MeÜH y la mezcla se agitó a T.A. durante 5 min. Se eliminó el disolvente y el residuo se purificó mediante cromatografía en columna para obtener 2A (124 mg, 82.6%) como un sólido blanco. ESI-LCMS: m/z 301.0 [M+H]+, 601.0 [2M+H]+.
EJEMPLO 3
Preparación del Compuesto 14A

Preparación de (AA-2): Se disolvió AA-1 (2.20 g, 3.84 mmol) en HCOOH al 80% (40 mL) a T.A. (18 °C). La mezcla se agitó a T.A. durante 12 h. Se eliminó el disolvente a presión reducida. El residuo se purificó mediante cromatografía en columna utilizando un 50% de EA en hexano para obtener AA-2 (1.05 g, 91.3%) como un sólido blanco.
Preparación de (AA-3): A una solución agitada de AA-2 (1 g, 3.32 mmol) en piridina anhidra (20 mL), se añadió TBSCl (747 mg, 4.98 mmol) e imidazol (451 mg, 6.64 mmol) a T.A. (16 °C) en atmósfera de N2. La mezcla se agitó a T.A. durante 4 h. La solución resultante se concentró hasta sequedad a presión reducida y el residuo se disolvió en EA (100 mL). La solución se lavó con una solución sat. de NaHCO3 y salmuera, y se secó con MgSO4 anhidro. La solución se concentró a sequedad y el residuo se purificó en una columna de gel de sílice utilizando un 20% de EA en hexano para obtener AA-3 (1.4 g, 79.5%) como un sólido blanco.
Preparación de (AA-4): A una solución agitada de AA-3 (1.50 g, 2.83 mmol, 1.00 eq.) en CH3CN anhidro (28 mL), se añadió TPSCl (1.71 g, 5.80 mmol, 2.05 eq.), DMAP (691.70 mg, 5.66 mmol, 2.00 eq.) y TEA (573.00 mg, 5.66 mmol, 2.00 eq.) a T.A. (15 °C). La mezcla se agitó durante 2 h. Se añadió NH3.H2O (20 mL) y la mezcla se agitó durante 3 h. La mezcla se extrajo con EA (3 x 60 mL). La fase orgánica se lavó con salmuera, se secó con Na2SO4 anhidro y se concentró a baja presión. El residuo se purificó en una columna de gel de sílice (30% de EA en PE) para obtener AA-4 (2.3 g, crudo) como una espuma amarilla.
Preparación de (AA-5): A una solución agitada de AA-4 (1.90 g, 2.34 mmol) en DCM anhidro (20 mL), se añadió DMTrCl (1.82 g, 3.49 mmol) y 2,4,6-trimetilpiridina (1.00 g, 8.25 mmol) a T.A. (15 °C) en atmósfera de N2. La mezcla se agitó a T.A. durante 12 h. Se añadió MeOH (20 mL). La mezcla se filtró y el filtrado se concentró a sequedad. El residuo se disolvió en EA (80 mL). La solución se lavó con salmuera, se secó con Na2SO4 anhidro y se concentró a baja presión. El residuo se purificó en una columna de gel de sílice (5% de MeOH en DCM) para obtener AA-5 (1.4 g, crudo) como un sólido blanco.
Preparación de (AA): Se disolvió AA-5 (2.40 g, 2.60 mmol) en TBAF (10 mL, 1 M en THF). La mezcla se agitó a T.A. (15 °C) durante 30 min. La mezcla se concentró a sequedad y el residuo se disolvió en EA (60 mL). La solución se lavó con salmuera, se secó con MgSO4 y se concentró a presión reducida. El residuo se purificó en una columna de gel de sílice (5% de MeOH en DCM) para obtener AA (1.50 g, 95.8%) como un sólido blanco. ESI-MS: m/z 625.3 [M+Na]+.
Preparación de (14-1): A una solución de AA (60.0 mg, 99.57 gmol, 1.00 eq.) en piridina (1 mL), se añadió anhídrido isobutírico (31.50 mg, 199.13 gmol, 2.00 eq.) en 1 porción a T.A. (15 °C) en atmósfera de N2. La mezcla se agitó a T.A. durante 12 h. La mezcla se concentró y el residuo se repartió entre EA y agua. Las fases orgánicas combinadas se lavaron con agua y salmuera, y se secaron con Na2SO4 anhidro. La mezcla se filtró y el filtrado se concentró a sequedad. El residuo se purificó mediante cromatografía en gel de sílice (30% de EA en PE) para proporcionar 14-1 (59.00 mg, 79.77%) como un sólido blanco.
Preparación de (14A): Se disolvió 14-1 (57.00 mg, 76.74 gmol, 1.00 eq.) en CH3COOH al 80% (8 mL). La solución se agitó a T.A. (15 °C) durante12 h. La mezcla se concentró a sequedad. El residuo se purificó en una columna de gel de sílice (2.5% de MeOH en DCM) para obtener 14A (23.00 mg, 68.05%) como una espuma blanca. ESI-MS: m/z 441.2 [M+H]+, 463.2 [M+Na]+.
EJEMPLO 4
Preparación del Compuesto 15A

Preparación de (15-1): 15-1 se preparó de un modo similar a 14-1 utilizando AA (60.00 mg, 99.57 gmol, 1.00 eq.) en piridina (1 mL) y anhídrido propiónico (25.92 mg, 199.13 gmol, 2.00 eq.). 15-1 (sólido blanco, 56.00 mg, 78.69%).
Preparación de (15A): El compuesto 15A se preparó de un modo similar a 14A utilizando 15-1 (54.00 mg, 75.55 gmol, 1.00 eq.). 15A (espuma blanca, 18.00 mg, 57.78%). ESI-MS: m/z 413.1 [M+H]+.
EJEMPLO 5
Preparación del Compuesto 16A

Preparación de (16-1): 16-1 se preparó de un modo similar a 14-1 utilizando AA (62.00 mg, 102.89 gmol, 1.00 eq.) en piridina (1 mL) y anhídrido pentanoico (38.32 mg, 205.77 gmol, 2.00 eq.). 16-1 (sólido blanco, 60.00 mg, 75.65%).
Preparación de (16A): El compuesto 16A se preparó de un modo similar a 14A utilizando 16-1 (75.00 mg, 97.30 gmol, 1.00 eq.). 16A (espuma blanca, 28.00 mg, 61.43%). ESI-MS: m/z 469.2 [M+H]+.
EJEMPLO 6
Preparación del Compuesto 24A

Preparación de (24-1): A una solución agitada de AA-1 (300.0 mg, 497.83 gmol) en piridina anhidra (0.5 mL), se añadió DMTrCl (337.36 mg, 995.66 gmol) a T.A. (17 °C) en atmósfera de N2. La solución se agitó a 50 °C ~60 °C durante 12
h. La mezcla se concentró a sequedad a presión reducida y el residuo se disolvió en EA (40 mL). La solución se lavó con salmuera, se secó con MgSO4 anhidro y se concentró a sequedad a baja presión. El residuo se purificó en una columna de gel de sílice utilizando un 20% de EA en PE para obtener 24-1 (300 mg, 66.59%) como un sólido blanco.
Preparación de (24-2): A una solución agitada de 24-1 (100.00 mg, 110.50 pmol) en piridina anhidra (0.5 mL), se añadió DMAP (6.75 mg, 55.25 pmol), DCC (22.80 mg, 110.50 pmol) y ácido n-actanoico (31.87 mg, 221.00 pmol) a T.A. (18 °C) en atmósfera de N2. La solución se agitó a T.A. durante 12 h. La solución se concentró a sequedad a presión reducida. El residuo se purificó en una columna de gel de sílice utilizando un 15% de EA en PE para obtener 24-2 (98.00 mg, 86.0%) como una espuma blanca.
Preparación de (24A): Se disolvió 24-2 (90.00 mg, 87.28 pmol) en CH3COOH al 80% (20 mL) a T.A. (16 °C). La mezcla se agitó a T.A. durante 12 h. La reacción se desactivó con MeOH y la mezcla se concentró a sequedad. El residuo se purificó en una columna de gel de sílice (5% de MeOH en DCM) para obtener 24A (33.00 mg, 88.7%) como un sólido blanco. ESI-MS: m/z 427.2 [M+H]+.
EJEMPLO 7
Preparación del Compuesto 25A

Preparación de (BB-2): A una solución agitada de BB-1 (500.00 mg, 0.87 mmol) en piridina anhidra (1 mL), se añadió TBSCl (236.5 mg, 1.57 mmol) a 20 °C en atmósfera de N2. La solución se agitó a 50 °C ~60 °C durante 12 h. La solución se concentró a sequedad a presión reducida. El residuo se disolvió en EA (50 mL). La solución se lavó con una solución sat. de NaHCO3 y salmuera, y se secó con MgSO4 anhidro. La solución se filtró y el filtrado se concentró a sequedad. El residuo se purificó en una columna de gel de sílice para obtener BB-2 (510.00 mg, 85.06%) como un sólido blanco.
Preparación de (BB-3): A una solución agitada de BB-2 (430.00 mg, 625.15 mmol) en MeCN anhidro (6 mL), se añadió TPSCl (368.65 mg, 1.25 mmol), DMAP (152.75 mg, 1.25 mmol) y TEA (126.52 mg, 1.25 mmol) a T.A. La mezcla se agitó durante 2 h. Se añadió NH4OH (8 mL) y la mezcla se agitó durante 3 h. La mezcla se extrajo con EA (3 x 40 mL). La fase orgánica se lavó con salmuera, se secó con Na2SO4 anhidro y se concentró a baja presión. El residuo se purificó en una columna de gel de sílice (25% de EA en PE) para obtener BB-3 (500 mg de crudo) como una espuma amarilla.
Preparación de (BB-4): A una solución agitada de BB-3 (500 mg de crudo, 0.72 mmol) en DCM anhidro (7 mL), se añadió DMTrCl (365 mg, 1.0 mmol) y colidina (305 mg, 2.5 mmol) y AgNO3 (184 mg, 1.08 mmol) a T.A. (15 °C) en atmósfera de N2. La mezcla se agitó a T.A. durante 12 h. Se añadió MeOH (5 mL). La mezcla se filtró y el filtrado se concentró a sequedad. El residuo se disolvió en EA (50 mL). La solución se lavó con salmuera, se secó con Na2SO4 anhidro y se concentró a baja presión. El residuo se purificó en una columna de gel de sílice (5% de MeOH en DCM) para obtener BB-4 (500 mg, 70.3%) como un sólido blanco.
Preparación de (BB): Se disolvió BB-4 (1.00 g, 1.01 mmol) en TBAF (5 mL, 1 M en THF) y se agitó a T.A. durante 30 min. La mezcla se diluyó con EA (100 mL). La mezcla se lavó con agua y salmuera, y se secó con MgSO4 anhidro. La fase orgánica se concentró a sequedad. El residuo se purificó en una columna de gel de sílice (30% de EA en PE) para obtener BB (0.80 g, 91.5%) como un sólido blanco. ESI-MS: m/z 873.7 [M+1]+.
Preparación de (25-1): A una solución de BB (100.00 mg, 114.29 pmol) en piridina anhidra (1.5 mL), se añadió DMAP (2.79 mg, 22.86 pmol), DCC (70.75 mg, 342.88 pmol) y ácido n-octanoico (49.45 mg, 342.88 pmol) a T.A. (18 °C) en atmósfera de N2. La solución se agitó a T.A. durante 12 h. La solución se concentró a sequedad a presión reducida. El residuo se purificó en una columna de gel de sílice utilizando un 15% de EA en PE para obtener 25-1 (95.00 mg, 83.03%) como una espuma blanca.
Preparación de (25A): Se disolvió 25-1 (110.00 mg, 109.87 pmol) en CH3COOH al 80% (25 mL) a T.A. (15 °C). La mezcla se agitó durante 12 h. La reacción se desactivó con MeOH y la solución se concentró a sequedad. El residuo se purificó en una columna de gel de sílice (5% de MeOH en DCM) para obtener 25A (30.00 mg, 64.03%) como un sólido blanco. ESI-MS: m/z 427.2 [M+H]+.
EJEMPLO 8
Preparación del Compuesto 26A

Preparación de (26-1): A una solución de N-Boc-L-Valina (620.78 mg, 2.86 mmol) y TEA (144.57 mg, 1.43 mmol) en THF anhidro (2.5 mL), se añadió BB (250.00 mg, 285.73 pmol). La mezcla se coevaporó con piridina y tolueno para eliminar el agua. El residuo se disolvió en THF (2.5 mL). Se añadió DIPEA (369.28 mg, 2.86 mmol) seguida de la adición de BOP-Cl (363.68 mg, 1.43 mmol) y 3-nitro-1 H-1,2,4-triazol (162.95 mg, 1.43 mmol) a T.A. (18 °C). La mezcla se agitó a T.A. durante 12 h y a continuación se diluyó con EA (40 mL). La solución se lavó con salmuera, se secó con Na2SO4 anhidro y se concentró a sequedad a baja presión. El residuo se purificó en una columna de gel de sílice (30% de EA en PE) para obtener 26-1 (220 mg, crudo) como una espuma blanca.
Preparación de (26-2): Se disolvió 26-1 (250.0 mg, 232.73 pmol) en CH3COOH al 80% (30 mL). La solución se calentó hasta 50 °C y se agitó durante 12 h. La reacción se desactivó con MeOH y la solución se concentró a sequedad. El residuo se purificó en una columna de gel de sílice (5% de MeOH en DCM) para obtener 26-2 (80.00 mg, 68.82%) como una espuma blanca.
Preparación de (26A): Se disolvió 26-2 (78.00 mg, 156.16 pmol) en HCl/dioxano (1.5 mL) y EA (1.5 mL) a T.A. (19 °C). La mezcla se agitó a T.A. durante 30 min. La solución se concentró a sequedad a baja presión. El residuo se purificó mediante HPLC prep. para obtener 26A (23 mg, 31.25%) como un sólido blanco. ESI-MS: m/z 400.20 [M+H]+,799.36 [2M+H]+.
EJEMPLO 9
Preparación del Compuesto 27A

Preparación de (27-1): 27-1 se preparó de un modo similar a 26-1 utilizando BB (250.0 mg, 276.25 pmol), ácido (2S)-2-(te/t-butoxicarbonilamino)-3-metilbutanoico (360.11 mg, 1.66 mmol) y TEA (83.86 mg, 828.75 pmol). 27-1 (espuma blanca, 220.0 mg, 72.12%).
Preparación de (27-2): 27-2 se preparó de un modo similar a 26-2 utilizando 27-1 (230.00 mg, 208.29 pmol, 1.00 eq.).
27-2 (espuma blanca, 80.00 mg, 77.66%).
Preparación de 27A: 27A se preparó de un modo similar a 26 utilizando 27-2 (100.00 mg, 200.20 pmol, 1.00 eq.). 27A (sólido blanco, 56 mg, 59.57%). ESI-MS: m/z 400.0 [M+H]+, 422.1 [M+Na]+; 799.1 [2M+H]+, 821.2 [2M+Na]+.
EJEMPLO 10
Preparación del Compuesto 13A

Preparación de (13-1): A una solución de 2A (200 mg, 0.67 mmol) en piridina anhidra (5 mL), se añadió TBSCl (120 mg, 0.8 mmol) a T.A. La mezcla se agitó durante toda la noche y la mezcla de reacción se diluyó con EA. La mezcla se lavó con una solución ac. de NaHCÜ3 y salmuera. La fase orgánica se secó, se filtró y se concentró para obtener un residuo, el cual se purificó mediante cromatografía en columna de gel de sílice (de un 5% de MeÜH en DCM a un 25% de MeÜH en DCM) para obtener 13-1 (153 mg, 55%) como un sólido blanco.
Preparación de (13-2): A una solución de 13-1 (54 mg, 0.13 mmol) en DCM anhidro (2 mL), se añadió colidina (95 pL, 0.78 mmol), DMTrCl (262 mg, 0.78 mmol) y AgNÜ3 (66 mg, 0.39 mmol) a T.A. La mezcla se agitó durante toda la noche y a continuación se diluyó con DCM (5 mL). La mezcla se filtró a través de un embudo empaquetado previamente con celite y el filtrado se lavó con una solución ac. de NaHCÜ3, una solución de ácido cítrico 1.0 M y a
continuación salmuera. La fase orgánica se secó con Na2SÜ4 y se concentró a baja presión para obtener un residuo. El residuo se purificó mediante cromatografía en columna de gel de sílice (de un 25% de EA en PE a un 100% de EA) para obtener 13-2 (83.5 mg, 63.6%).
Preparación de (13-3): A una solución de 13-2 (83 mg, 0.081 mmol) en THF (1 mL), se añadió una solución 1 M de TBAF en THF (0.122 mL, 0.122 mmol) a la temperatura de un baño de hielo. La mezcla se agitó durante 1.5 h. La mezcla se diluyó con EA y se lavó con agua y salmuera. La fase orgánica se secó y se concentró para obtener el producto crudo, el cual se purificó mediante cromatografía en columna de gel de sílice (desde DCM hasta un 5% de MeÜH en DCM) para obtener 13-3 (66.6 mg, 91%) como una espuma blanca.
Preparación de (13-4): 13-3 (66.6 mg, 0.074 mmol) se coevaporó con tolueno y THF (3x). Se añadió Bis(POC)fosfato (33 mg, 0.96 mmol) y a continuación se coevaporó con tolueno (3x). La mezcla se disolvió en THF anhidro (1.5 mL) y se enfrió en un baño de hielo (de 0 a 5 0C). Se añadió sucesivamente 3-nitro-1,2,4-triazol (13 mg, 0.11 mmol), diisopropiletilamina (54 pL, 0.3 mmol) y BOP-Cl (28 mg, 0.11 mmol). La mezcla se agitó durante 2 h a una temperatura comprendida entre 0 y 5 °C, se diluyó con EtOAc, se lavó con ácido cítrico 1.0 M, NaHCÜ3 ac. sat. y salmuera, y se secó con Na2SÜ4. El residuo se purificó en sílice (columna de 10 g) con CH2Cl2 :i-PrOH (gradiente de 4-10%) para obtener 13-4 (68 mg, 76%) como un sólido blanco.
Preparación de (13A): Se disolvió 13-4 (68 mg, 0.07 mmol) en HCOOH al 80%. La mezcla se agitó a T.A. durante 2 h. Se evaporaron los disolventes a T.A. y se coevaporó con tolueno (3x). El residuo se disolvió en un 50% de CH3CN/H2O, se purificó en un HPLC de fase inversa (C18) utilizando CH3CN y H2O. El producto se sometió a liofilización para obtener 13A (4.8 mg, 14%) como una espuma blanca. ESI-LCMS: m/z = 613.1 [M+H]+, 1225.2 [2M+H]+.
EJEMPLO 11
Preparación del Compuesto 17A

Preparación de (17-1): 17-1 (40.7 mg, 53%) se preparó del mismo modo a partir de 1-7 (50 mg, 0.087 mmol) y fosfato de bis(isopropiloxicarboniloximetilo) (58 mg,0.175 mmol) con DIPEA (75 pL, 0.52 mmol), BOP-Cl (66.2 mg, 0.26 mmol) y 3-nitro-1,2,4-triazol (30 mg, 0.26 mmol) en THF (0.4 mL) de un modo similar a 13-4.
Preparación de (17A): Se disolvió 17-1 (40 mg, 0.045 mmol) en CH3CN anhidro (0.5 mL) y se añadió HCl 4 N en dioxano (34 pL, 0.135 mmol) a una temperatura comprendida entre 0 y 5 °C. La mezcla se agitó a T.A. durante 3 h. Se añadió EtOH anhidro (200 pL). Se evaporaron los disolventes a T.A. y se coevaporó con tolueno (3x). El residuo se purificó en sílice (columna de 10 g) con MeOH/CH2Cl2 (gradiente de 5-7%) y se liofilizó para obtener 17A (15.4 mg, 76%) como una espuma blanca. ESI-LCMS: m/z = 614.15 [M+H]+, 1227.2 [2M+H]+.
EJEMPLO 12
Preparación del Compuesto 18A

Preparación de (18-1): A una solución agitada de 1-7 (80 mg, 0.14 mmol) en CH3CN anhidro (2.0 mL), se añadió N-metilimidazol (0.092 mL, 1.12 mmol) a 0 °C (baño de hielo/agua). A continuación, se añadió una solución de fosforocloridato de fenilo e (isopropoxi-L-alaninilo) (128 mg, 0.42 mmol, disuelto en CH3CN (0.5 mL)) (que se preparó de acuerdo con un procedimiento general tal como se describe en McGuigan et al., J. Med. Chem. (2008) 51:5807-5812). La solución se agitó a una temperatura comprendida entre 0 y 5 °C durante h y a continuación se agitó a T.A. durante 16 h. La mezcla se enfrió hasta una temperatura comprendida entre 0 y 5 °C, se diluyó con EA y a continuación se añadió agua (5 mL). La solución se lavó con ácido cítrico 1.0 M, NaHCO3 ac. sat. y salmuera, y se secó con MgSO4.
El residuo se purificó en sílice (columna de 10 g) con EA/hexanos (gradiente de 25-100%) para obtener 18-1 (57.3 mg, 49%) como una espuma.
Preparación de (18A): Se disolvió 18-1 (57.3 mg, 0.07 mmol) en CH3CN anhidro (0.5 mL) y se añadió HCl 4 N en dioxano (68 pL, 0.27 mmol) a una temperatura comprendida entre 0 y 5 °C. La mezcla se agitó a T.A. durante 2 h y se añadió EtOH anhidro (100 pL). Se evaporaron los disolventes a T.A. y se coevaporó con tolueno (3x). El residuo se purificó en sílice (columna de 10 g) con MeOH/CH2Cl2 (gradiente de 1-7%) y se liofilizó para obtener 18A (27.8 mg, 72%) como una espuma blanca. ESl-LCMS: m/z = 571.1 [M+H]+, 1141.2 [2M+H]+.
EJEMPLO 13
Preparación del Compuesto 28A

Preparación de (28-1): 28-1 (68.4 mg, 44.7%) se preparó a partir de 1-7 (100 mg, 0.174 mmol) y fosfato de bis(tertbutoxicarboniloximetilo) (126 mg, 0.35 mmol) con DIPEA (192 pL, 1.04 mmol), BOP-Cl (133 mg, 0.52 mmol) y 3-nitro-1,2,4-triazol (59 mg, 0.52 mmol) en THF (1.5 mL) del mismo modo que 13-4.
Preparación de (28A): 28A (31.4 mg, 67%) se preparó a partir de 28-1 (68 mg, 0.077 mmol) del mismo modo que 17A.
ESl-LCMS: m/z = 627.15 [M+Na]+, 1219.25 [2M+H]+.
EJEMPLO 14
Preparación del Compuesto 19A

Preparación de (19-1): A una solución de 1-7 (100 mg, 0.175 mmol) en CH3CN anhidro (2 mL), se añadió 5-etiltio-1 H-tetrazol en CH3CN (0.25 M; 0.84 mL, 0.21 mmol). Se añadió gota a gota Bis-SATE-fosforamidato (95 mg, 0.21 mmol) en CH3CN (1 mL) a una temperatura comprendida entre 0 y 5 °C. La mezcla se agitó durante 2 h a una temperatura comprendida entre 0 y 5 °C en atmósfera de Ar. Se añadió una solución de m-CPBA (78 mg, 0.35 mmol) al 77% en DCM (1 mL) y la mezcla se agitó durante 2 h a una temperatura comprendida entre 0 y 5 °C en atmósfera de Ar. La mezcla se diluyó con EtOAc (50 mL), se lavó con ácido cítrico 1.0 M, NaHCO3 sat. y salmuera, y se secó con MgSO4. La mezcla se filtró y los disolventes se evaporaron al vacío. El residuo se purificó en sílice (columna de 10 g) con EA/hexanos (gradiente de 20-100%) para obtener 19-1 (105 mg, 63.6%) como una espuma blanca.
Preparación de (19A): Se disolvió 19-1 (105 mg, 0.112 mmol) en CH3CN anhidro (0.8 mL) y se añadió HCl 4 N en dioxano (84 pL, 0.334 mmol) a una temperatura comprendida entre 0 y 5 °C. La mezcla se agitó a T.A. durante 2 h. Se añadió EtOH anhidro (100 pL). Se evaporaron los disolventes a T.A. y se coevaporó con tolueno (3x). El residuo se purificó en sílice (columna de 10 g) con MeOH/CH2Cl2 (gradiente de 1-7%) y se liofilizó para obtener 19A (42.7 mg, 57%) como una espuma blanca. ESl-LCMS: m/z = 692.15 [M+Na]+, 1339.30 [2M+H]+.
EJEMPLO 15
Preparación del Compuesto 20A

Preparación de (20-2): Se coevaporó 1-7 (100 mg, 0.174 mmol) con piridina anhidra (3x), tolueno (3x) y CH3CN (3x), y se secó con un vació elevado durante toda la noche. Se disolvió 1-7 en CH3CN (2 mL). Se añadió Proton-sponge 012 mg, 0.52 mmol) y POCl3 (49 uL, 0.52 mmol) a una temperatura comprendida entre 0 y 5 °C. La mezcla se agitó durante 3 h a una temperatura comprendida entre 0 y 5 °C para obtener el intermedio 20-1. Se añadió a esta solución el clorhidrato del éter isopropílico de la L-alanina (146 mg, 0.87 mmol) y TEA (114 uL, 1.74 mmol). La mezcla se agitó durante 4 h a una temperatura comprendida entre 0 y 5 °C. La mezcla se agitó durante 2 h a una temperatura comprendida entre 0 y 5 °C, a continuación se diluyó con EtOAc. La mezcla se lavó con ácido cítrico 1.0 M, NaHCO3 ac. sat. y salmuera, y se secó con Na2SO4. El residuo se purificó en sílice (columna de 10 g) con CH2Cl2/MeOH (gradiente de 0-7%) para obtener 20-2 (67 mg, 43.7%) como un sólido blanco.
Preparación de (20A): Se disolvió 20-2 (65 mg, 0.074 mmol) en CH3CN anhidro (0.5 mL) y se añadió HCl 4 N en dioxano (55 pL, 0.22 mmol) a una temperatura comprendida entre 0 y 5 °C. La mezcla se agitó a T.A. durante 1.5 h. Se añadió una segunda porción de HCl 4 N en dioxano (15 pL) y la mezcla se agitó a T.A. durante 2 h. Se añadió EtOH anhidro (300 pL). Se evaporaron los disolventes a T.A. y se coevaporó con tolueno (3x). El residuo se disolvió en un 50% de CH3CN/H2O, se purificó en un HPLC de fase inversa (C18) con CH3CN y agua, y se liofilizó para obtener 20A (9 mg, 20%) como una espuma blanca. ESI-LCMS: m/z = 608.15 [M+H]+, 1215.3 [2M+H]+.
EJEMPLO DE REFERENCIA 16
Preparación del Compuesto 23A

Preparación de (23-2): A una solución agitada de 1-7 (100 mg, 0.175 mmol) en CH3CN anhidro (2.0 mL), se añadió N-metilimidazol (0.14 mL, 1.4 mmol) a 0 °C (baño de hielo/agua). Se añadió una solución de 23-1 (220 mg, 0.53 mmol, disuelto en 0.5 mL de CH3CN) (que se preparó de acuerdo con un procedimiento general descrito en Bondada, L. et al., ACS Medicinal Chemistry Letters, (2013) 4(8):747-751). La solución se agitó a una temperatura comprendida entre 0 y 5 °C durante 1 h y a continuación se agitó a T.A. durante 16 h. La mezcla se enfrió hasta una temperatura comprendida entre 0 y 5 °C, se diluyó con EA y a continuación se añadió agua (5 mL). La solución se lavó con ácido cítrico 1.0 M, NaHCO3 ac. sat. y salmuera, y se secó con MgSO4. El residuo se purificó en sílice (columna de 10 g) con EA/hexanos (gradiente de 25-100%) para obtener 23-2 (56,4 mg, 33.7%) como una espuma blanca.
Preparación de (23A): Se disolvió 23-2 (56 mg, 0.0585 mmol) en CH3CN anhidro (0.7 mL) y se añadió HCl 4 N en dioxano (44 gL, 0.176 mmol) a una temperatura comprendida entre 0 y 5 °C. La mezcla se agitó a T.A. durante 2 h. Se añadió HCl 4 N en dioxano (20 gL). La mezcla se agitó a T.A. durante 2 h. Se añadió EtOH anhidro (100 gL). Se evaporaron los disolventes a T.A. y se coevaporó con tolueno (3x). El residuo se purificó en sílice (columna de 10 g) con MeOH/CH2Cl2 (gradiente de 1 -7%) y se liofilizó para obtener 23A (27.6 mg, 69%) como una espuma blanca. ESI-LCMS: m/z = 685.2 [M+H]+.
EJEMPLO 17
Preparación del Compuesto 29A

Preparación de (29-1): A una solución de BB (100 mg, 0.114 mmol) en CH3CN anhidro (2 mL), se añadió una solución de bis-SATE-fosforamidato (62.2 mg, 0.14 mmol) en CH3CN (1 mL) y a continuación 5-etiltio-1 H-tetrazol en CH3CN (0.25 M; 0.56 mL, 0.14 mmol) a una temperatura comprendida entre 0 y 5 °C gota a gota. La mezcla se agitó durante 2 h a una temperatura comprendida entre 0 y 5 °C en atmósfera de Ar. Se añadió una solución de m-CPBA (49 mg, 0.22 mmol) al 77% en DCM (1 mL) y la mezcla se agitó durante 2 h a una temperatura comprendida entre 0 y 5 °C en atmósfera de Ar. La mezcla se diluyó con EtOAc (50 mL), se lavó con ácido cítrico 1.0 M, NaHCO3 sat. y salmuera, y se secó con MgSO4. La mezcla se filtró y los disolventes se evaporaron al vacío. El residuo se purificó en sílice (columna de 10 g) con EA/hexanos (gradiente de 10-100%) para obtener 29-1 (72 mg, 50.8%) como un sólido blanco.
Preparación de (29A): Se disolvió 29-1 (72 mg, 0.056 mmol) en CH3CN anhidro (1.0 mL) y se añadió HCl 4 N en dioxano (87 gL, 0.35 mmol) a una temperatura comprendida entre 0 y 5 °C. La mezcla se agitó a T.A. durante 2 h. Se observó el intermedio 29-2 mediante LCMS. Se evaporaron los disolventes a T.A. y se coevaporó con tolueno (3x). El residuo obtenido se disolvió de nuevo en HCOOH al 80% (2 mL). La mezcla se agitó a T.A. durante 4.5 h. Se evaporaron los disolventes a T.A. y se coevaporó con tolueno (3x). Se añadió EtOH anhidro (3 x 5 mL). El residuo se disolvió en un 50% de CH3CN/H2O, se purificó en un HPLC de fase inversa (C18) utilizando CH3CN y H2O, y se liofilizó para obtener 29A (19.2 mg) como una espuma blanca. ESI-LCMS: m/z = 669.2 [M+H]+, 1337.25 [2M+H]+.
EJEMPLO 18
Preparación del Compuesto 30A
Preparación de (30-1): 30-1 (98 mg, 72.6%) se preparó del mismo modo a partir de BB (100 mg, 0.114 mmol) y fosfato de bis(tert-butoxicarboniloximetilo) (83 mg, 0.35 mmol) con DIPEA (126 pL, 0.69 mmol), BOP-Cl (87 mg, 0.34 mmol) y 3-nitro-1,2,4-triazol (39 mg, 0.34 mmol) en THF (1.5 mL) del mismo modo que 13-4.
Preparación de (30A): 30A (30.2 mg, 60%) se preparó a partir de 30-1 (98 mg, 0.083 mmol) del mismo modo que 17A.
ESI-LCMS: m/z = 609.15 [M+H]+, 1217.3 [2M+H]+.
EJEMPLO 19
Preparación del Compuesto 21A

Preparación de (21-3): Una solución de 21-1 (4.7 g, 11.2 mmol; que se preparó de acuerdo con el procedimiento de Villard etal., Bioorg. Med. Chem. (2008) 16:7321-7329) y Et3N (3.4 mL, 24.2 mmol) en THF (25 mL) se añadió gota a gota durante 1 h a una solución agitada de W,W-fosforodicloridito de diisopropilo (1.0 mL, 5.5 mmol) en THF (35 mL) a -75 °C. La mezcla se agitó a T.A. durante 4 h. La mezcla se filtró y se concentró el filtrado. El residuo oleoso se purificó en una columna de gel de sílice con EtOAc/hexanos (gradiente de 2-20%) para obtener 21-3 (1.4 g, 26%).
Preparación de (21-4): A una solución de 21-2 (50 mg, 0.08 mmol) y 21-3 (110 mg, 0.11 mmol) en CH3CN (1.0 mL), se añadió 5-(etiltio)tetrazol (0.75 mL, 0.16 mmol; 0.25 M en CH3CN). La mezcla se agitó a T.A. durante 1 h. La mezcla se enfrió hasta -40 °C y se añadió una solución de ácido 3-cloroperoxibenzoico (37 mg, 0.16 mmol) en CH2G 2 (0.3 mL). La mezcla se calentó hasta T.A. durante 1 h. La reacción se desactivó con una solución de Na2S2O3 al 7% en NaHCO3 ac. sat. La mezcla se diluyó con EtOAc y se separaron las fases. La fase orgánica se lavó con salmuera y se secó con Na2SO4. Se evaporó el disolvente y el residuo se purificó en una columna de gel de sílice con EtOAc/hexanos (gradiente de 30-100%) para obtener 21-4 (52 mg, 45%).
Preparación de (21A): Una solución de 21-4 (52 mg, 0.036 mmol) en MeCN (0.5 mL) y HCl (45 pL; 4 N en dioxano) se agitó durante 20 h a T.A. La reacción se desactivó con MeOH y se evaporaron los disolventes. El residuo se coevaporó con tolueno y se purificó en una columna de gel de sílice con MeOH/CH2Cl2 (gradiente de 4-10%) para obtener 21A (14 mg, 51%). ESI-LCMS: m/z = 702 [M+H]+.
EJEMPLO DE REFERENCIA 20
Preparación del Compuesto 22A

Preparación de (22-2): Una mezcla de 22-1 (0.14 g, 0.24 mmol; que se preparó de acuerdo con el procedimiento descrito en el documento WO 2008/082601, presentado el 28 de diciembre de 2007) y 21-2 (120 mg, 0.2 mmol) se anhidrizó evaporándola con piridina y a continuación se disolvió en piridina (3 mL). Se añadió cloruro de pivaloílo (48 pL) gota a gota a -15 °C. La mezcla se agitó a -15 °C durante 2 h. La reacción se desactivó con una solución ac. sat. de NH4Cl y se diluyó con CH2G 2. La fase orgánica se lavó con salmuera y se secó con Na2SO4. Se evaporaron los disolventes y el residuo se purificó en una columna de gel de sílice con EtOAc/hexanos (gradiente de 30-100%) para obtener 22-2 (50 mg, 24%).
Preparación de (22-3): Una mezcla de 22-2 (43 mg; 0.04 mmol) en CCL (0.8 mL), clorhidrato del éster isopropílico de la L-valina (20 mg, 0.12 mmol) y Et3N (33 pL, 0.24 mmol) se agitó a T.A. durante 2 h. La mezcla se diluyó con EtOAc. La mezcla se lavó con NaHCO3 ac. sat. y salmuera, y se secó con Na2SO4. Se evaporaron los disolventes y el residuo se purificó en una columna de gel de sílice con i-PrOH/CH2Cl2 (gradiente de 2-10%) para obtener 22-3 (35 mg, 75%). Preparación de (22A): Una solución de 22-3 (35 mg, 0.03 mmol) en MeCN (0.4 mL) y HCl (40 pL; 4 N en dioxano) se agitó durante 4 h a T.A. La reacción se desactivó con la adición de MeOH y se evaporaron los disolventes. El residuo se coevaporó con tolueno y se purificó en una columna de gel de sílice con MeOH/CH2Cl2 (gradiente de 4-10%) para obtener 23A (11 mg, 56%). ESI-LCMS: m/z = 655 [M+H]+.
EJEMPLO DE REFERENCIA 21
Preparación del Compuesto 7A

Preparación de (7-2): A una solución de 7-1 (20.0 g, 70.1 mmol) en piridina anhidra (230 mL), se añadió imidazol (19.1 g, 280.7 mmol) y TBSCl (42.1 g, 280.7 mmol) a 25 °C. La solución se agitó a 25 °C durante 15 h. La mezcla se concentró a sequedad a presión reducida y el residuo se disolvió en EA. Se obtuvo un sólido blanco y se filtró. La masa húmeda retenida sobre el filtro se concentró a sequedad para obtener 7-2 (30.1 g, 83%) como un sólido blanco. Preparación de (7-3): Se disolvió 7-2 (30.1 g, 58.7 mmol) en THF (120 mL) y H2O (80 mL). Se añadió HOAc (260 mL) y a continuación se agitó a 80 °C durante 13 h. La mezcla se enfrió hasta T.A. y se concentró a sequedad a presión reducida. El residuo se disolvió en EA y se filtró. La masa húmeda retenida sobre el filtro se concentró a sequedad para obtener 7-3 (20.1 g, 86%) como un sólido blanco.
Preparación de (7-4): Se disolvió 7-3 (20.1 g, 50.4 mmol) en piridina anhidra (200 mL). Se añadió Ac2O (7.7 g, 75.5 mmol) y a continuación se agitó a 25 °C durante 18 h. Se añadió MMTrCl (46.5 g, 151.1 mmol) y AgNO3 (25.5 g, 151.1 mmol). La solución se agitó a 25 °C durante 15 h. La reacción se desactivó con agua. La mezcla se concentró a sequedad a presión reducida y el residuo se disolvió en EA. La solución se lavó con salmuera. La fase orgánica se secó con Na2SO4 y se filtró. El filtrado se concentró al vacío a sequedad. El residuo se purificó en una columna de gel de sílice (2% de MeOH en DCM) para obtener 7-4 (21.5 g, 60%) como una espuma blanca.
Preparación de (7-5): Se disolvió 7-4 (4.3 g, 6.0 mmol) en NH3/MeOH (40 mL). La mezcla se agitó a 25 °C durante 20 h. La solución se evaporó a sequedad. El residuo se purificó en una columna de gel de sílice (2% de MeOH en DCM) para obtener 7-5 (3.1 g, 76,5%) como un sólido amarillo.
Preparación de (7-6): A una solución de 7-5 (3.1 g, 4.6 mmol) en DCM anhidro (50 mL), se añadió el reactivo de Dess-Martin (3.5 g, 8.2 mmol) a 0 °C. La mezcla se agitó a 0 °C durante 2 h y a continuación se agitó a T.A. durante 2 h. La reacción se desactivó con una solución de Na2S2O3 y NaHCO3 saturado. La fase orgánica se lavó con salmuera (2x) y se secó con Na2SO4 anhidro. Se evaporó el disolvente para obtener 7-6 crudo (2.8 g) como una espuma amarilla. Preparación de (7-7): A una solución de 7-6 (2.8 g, 4.2 mmol) en 1,4-dioxano (40 mL), se añadió HCHO al 37% (2.7 g, 33.5 mmol) y una solución acuosa de NaOH 2.0 N (3.0 mL, 6.0 mmol). La mezcla se agitó durante 12 h a 25 °C. La mezcla se trató con EtOH (20 mL) y NaBH4 (2.5 g, 66.9 mmol) y se agitó durante 30 min. La reacción se desactivó con NH4Cl ac. sat. y se extrajo con EA (50 mL). La fase orgánica se secó con Na2SO4. La fase orgánica concentrada se
purificó en una columna de gel de sílice (2% de MeOH en DCM) para obtener 7-7 (2.1 g, 72.4%) como un sólido amarillo.
Preparación de (7-8): A una solución de 7-7 (2.1 g, 3.0 mmol) en DCM (20 mL), se añadió piridina (5 mL) y DMTrCl (1.0 g, 3.0 mmol) a 0 °C. La solución se agitó a 25 °C durante 1 h. La mezcla se trató con MeOH (8 mL) y se concentró a presión reducida. El residuo se purificó en una columna de gel de sílice (2% de MeOH en DCM) para obtener 7-8 (1.1 g, 36.7%) como un sólido amarillo.
Preparación de (7-9): A una solución de 7-8 (1.1 g, 1.1 mmol) en piridina anhidra (10 mL), se añadió TBDPSCl (0.9 g, 3.3 mmol) y AgNO3 (0.6 g, 3.3 mmol). La mezcla se agitó a 25 °C durante 15 h. El sólido se retiró mediante filtración y el filtrado se concentró a baja presión. El residuo se disolvió en EA. La solución resultante se lavó con salmuera. La fase orgánica se secó con Na2SO4 y se concentró a baja presión. El residuo se purificó mediante cromatografía en columna (2% de MeOH en DCM) para obtener 7-9 (1.2 g, 88.2%) como una espuma blanca.
Preparación de (7-10): A una solución de 7-9 (1.2 g, 1.0 mmol) en DCM anhidro (15 mL), se añadió CbCHCOOH (0.6 mL) a -78 °C. La mezcla se agitó a -20 °C durante 1 h. La reacción se desactivó con NaHCO3 ac. sat. y se extrajo con DCM. La fase orgánica se secó con Na2SO4 y se concentró a baja presión. El residuo se purificó en una columna de gel de sílice (2% de MeOH en DCM) para obtener 7-10 (693 mg, 76.3%) como una espuma blanca.
Preparación de (7-11): A una solución de 7-10 (693 mg, 0.74 mmol) en DCM anhidro (25 mL) y piridina (291 mg, 3.70 mmol), se añadió Tf2O (312 mg, 1.1 mmol) en DCM (1 mL) gota a gota a 0°C. La mezcla se agitó a 0 °C durante 15 min. La reacción se desactivó con agua-hielo. La fase orgánica se separó y se lavó con salmuera. La fase orgánica se secó con Na2SO4 anhidro y se evaporó para obtener 7-11 (442 mg, crudo) como una espuma amarilla.
Preparación de (7-12): A una solución de 7-11 (442 mg, 0.41 mmol) en DMF anhidro (5 mL), se añadió NaN3 (134 mg, 2.1 mmol). La mezcla se agitó a T.A. Durante 12 h. La reacción se desactivó con agua y se extrajo con EA (20 mL, 2x). La fase orgánica se lavó con salmuera, se secó con Na2SO4 anhidro y se concentró a baja presión. El residuo se purificó en una columna de gel de sílice (1% de MeOH en DCM) para obtener 7-12 puro (313 mg, 78.6%) como una espuma blanca.
Preparación de (7-13): Una mezcla de 7-12 (313 mg, 0.32 mmol) y NH4F (240 mg, 6.5 mmol) en MeOH (10 mL) se agitó a 80 °C durante 12 h. La mezcla se enfrió hasta T.A. El sólido se eliminó por filtración. Se eliminó el disolvente a presión reducida y el residuo se purificó en una columna de gel de sílice (5% de MeOH en DCM) para obtener 7-13 (102 mg, 52%) como una espuma blanca.
Preparación de (7A): Se disolvió 7-13 (102 mg, 0.17 mmol) en CH3COOH (80%). La mezcla se agitó a 60 °C durante 2 h y a continuación se enfrió hasta T.A. La mezcla se concentró a sequedad a presión reducida. El residuo se purificó en una columna de gel de sílice (de un 5% a un 10% de MeOH en DCM) para obtener el producto crudo (67 mg). El producto crudo se purificó mediante HPLC prep. (0.1% de NH4HCO3 en agua y CH3CN) para obtener 7A (37.5 mg, 66%) como un sólido blanco. MS: m/z 341 [M+H]+.
EJEMPLO DE REFERENCIA 22
Preparación del Compuesto 31A

Preparación de (31-2): A una solución agitada de 7-7 (1.92 g, 27.3 mmol), PPh3 (1.43 g, 54.7 mmol), EtOH (0.25 g, 54.7 mmol) en dioxano anhidro (20 mL), se añadió DIAD (1.11 g, 54.7 mmol) gota a gota a 0 °C. La solución se agitó a 25 °C durante 15 h. La reacción se desactivó con agua y se extrajo con EA. La mezcla se lavó con agua y salmuera. La fase orgánica se secó con Na2SO4 y se filtró. El filtrado se concentró al vacío a sequedad y el residuo se purificó en una columna de gel de sílice (de un 2% a un 5% de MeOH en DCM) para obtener 31-1 (1.43 g, 71%) como una espuma blanca.
Preparación de (31-2): A una solución agitada de 31-1 (1.43 g, 19.6 mmol) en DMF (15 mL), se añadió TEA (0.59 g, 58.8 mmol) y DMTrCl (0.99 g, 29.4 mmol) a 0 °C. La solución se agitó a 25 °C durante 12 h. La mezcla se trató con MeOH (1 mL) y se diluyó con EA. La solución se lavó con agua y salmuera. La fase orgánica se secó con NaSO4 anhidro y se concentró a sequedad. El residuo se purificó en una columna de gel de sílice (2% de MeOH en DCM) para obtener 31-2 (1.13 g, 56%) como un sólido amarillo.
Preparación de (31-3): A una solución agitada de 31-2 (1.13 g, 1.1 mmol) en piridina anhidra (10 mL), se añadió TBDPSCl (0.91 g, 3.3 mmol) y AgNO3 (0.61 g, 3.3 mmol). La mezcla se agitó a 25 °C durante 15 h. El sólido se eliminó por filtración y el filtrado se diluyó con EA (50 mL). La solución se lavó con salmuera. La fase orgánica se secó con Na2SO4 anhidro y se concentró a baja presión. El residuo se purificó en una columna de gel de sílice (2% de MeOH en DCM) para obtener 31-3 (1.22 g, 88%) como una espuma blanca.
Preparación de (31-4): A una solución agitada de 31-3 (1.22 g, 1.0 mmol) en DCM anhidro (15 mL), se añadió Cl2CHCOOH (0.6 mL) a -78 °C. La mezcla se agitó a 20 °C durante 1 h. La reacción se desactivó con NaHCO3 ac. sat. y se extrajo con DCM. La fase orgánica se secó con Na2SO4 anhidro y se concentró a baja presión. El residuo se purificó mediante cromatografía en columna (2% de MeOH en DCM) para obtener 31-4 (0.52 g, 56%) como una espuma blanca.
Preparación de (31-5): A una solución agitada de 31-4 (0.52 g, 0.5 mmol) en DCM anhidro (15 mL) y piridina (0.21 g, 2.5 mmol), se añadió Tf2O (0.30 g, 1.0 mmol) en DCM (1 mL) gota a gota a 0°C. La mezcla se agitó a 0 °C durante 15 min. La reacción se desactivó con agua-hielo. La fase orgánica se separó y se lavó con agua. La fase orgánica se secó con Na2SO4 anhidro y se concentró a baja presión para obtener 31-5 (442 mg, crudo) como una espuma amarilla.
Preparación de (31-6): A una solución agitada de 31-5 (442 mg, 0.4 mmol) en DMF anhidro (5 mL), se añadió NaN3 (131 mg, 2.0 mmol). La mezcla se agitó a T.A. durante 12 h. La reacción se desactivó con agua y se extrajo con EA (20 mL, 2x). La fase orgánica se lavó con agua y se secó con Na2SO4. La fase orgánica se evaporó a sequedad a presión reducida. El residuo se purificó en una columna de gel de sílice (1% de MeOH en DCM) para obtener 31-6 (352 mg, 88%) como una espuma blanca.
Preparación de (31-7): Una mezcla de 31-6 (352 mg, 0.35 mmol) y NH4F (392 mg, 10.6 mmol) en MeOH (10 mL) se agitó a 80 °C durante 12 h. La mezcla se enfrió hasta T.A. El sólido se eliminó por filtración. Se concentró el disolvente a presión reducida. El residuo se purificó en una columna de gel de sílice (de un 2% a un 5% de MeOH en DCM) para obtener 31-7 crudo (151 mg). El producto crudo se purificó mediante HPLC prep. (0.1% de NH4HCO3 en agua y CH3CN) para obtener 31-7 (71.5 mg, 32%) como un sólido blanco. MS: m/z 641 [M+H]+.
Preparación de (31-8): Una mezcla de 31-7 (64 mg, 0.1 mmol) y bis(pivaloiloximetil)fosfato, después de anhidrizarla evaporándola con tolueno, se disolvió en CH3CN (1 mL) y se enfrió hasta 0 °C. Se añadió BopCl (40 mg, 0.15 mmol) y NMI (40 gL, 0.5 mmol). La mezcla se agitó a 0 °C durante 2 h. Se añadió EtOAc y la mezcla se lavó con ácido cítrico ac. 0.5 N, NaHCO3 ac. sat. y salmuera, y a continuación se secó con Na2SO4. Se eliminaron los disolventes y el residuo se purificó en una columna de gel de sílice con un 3% de /-PrOH en CH2 G 2 para obtener 31-8 (38 mg, 40%).
Preparación de (31A): Una solución de 31-8 (30 mg, 0.03 mmol) en CH3CN (0.3 mL) y HCl (30 gL; 4 N en dioxano) se agitó a T.A. durante 100 min. La reacción se desactivó con EtOH y se evaporó la mezcla. El residuo crudo se purificó en una columna de gel de sílice con /-PrOH/CH2Cl2 (gradiente de 3-10%) para proporcionar 31A (10 mg, 50%). ESI-LCMS: m/z = 681 [M+H]+.
EJEMPLO DE REFERENCIA 23
Preparación del Compuesto 32A

Se hidrógeno 2A (30 mg, 0.1 mmol) en MeOH con Pd/C al 10% a presión normal. El catalizador se separó por filtración y el filtrado se purificó mediante RP HPLC en una columna Synergy Hydro-RP de 4 micras (Phenominex). Se utilizó un gradiente lineal de MeOH de un 0% a un 20% en un tampón de acetato de trietilamonio 50 mM (pH 7.5) para la elución. Las fracciones correspondientes se combinaron, se concentraron y se liofilizaron (3x) con el fin de eliminar el exceso de tampón para proporcionar 32A (17 mg, 63%). ESI-LCMS: m/z = 275.2 [M+H]+, 297.1 [M+Na]+.
EJEMPLO DE REFERENCIA 24
Preparación del Compuesto 8A

Preparación de (8-2): A una solución de 8-1 (3.0 g, 11.15 mmol) en piridina anhidra (90 mL), se añadió imidazol (3.03 g, 44.59 mmol) y TBSCl (6.69 g, 44.59 mmol) a 25 °C en atmósfera de N2. La solución se agitó a 25 °C durante 15 h. La solución se concentró a sequedad a presión reducida. El residuo se disolvió en EA. La solución se lavó con NaHCO3 sat. y salmuera, y se secó con MgSO4 anhidro. El disolvente se eliminó a baja presión para obtener 8-2 crudo (4.49 g, 90%) como un sólido blanco.
Preparación de (8-3): A una solución agitada de 8-2 (3.5 g, 7.04 mmol) en una mezcla de EA y EtOH (1:1,55 mL), se añadió TsOH (10.7 g, 56.34 mmol) a 0 °C. La mezcla se agitó a 30 °C durante 8 h. Se añadió agua (30 mL) y se eliminó la solución a sequedad. El residuo se purificó en una columna de gel de sílice (10% de MeOH en DCM) para obtener 8-3 (1.75 g, 65%) como una espuma blanca.
Preparación de (8-4): A una solución de 8-3 (3.4 g, 8.88 mmol) en piridina anhidra (17 mL), se añadió colidina (4.3 g, 35.51 mmol), AgNO3 (5.50 g, 35.51 mmol) y MMTrCl (8.02 g, 26.63 mmol) a 25 °C en atmósfera de N2. La mezcla se agitó a 25 °C durante 12 h. Se añadió MeOH (20 mL) y se eliminó el disolvente a sequedad a baja presión. El residuo se purificó en una columna de gel de sílice (10% de EA en PE) para obtener 8-4 (5.76 g, 70%) como una espuma blanca.
Preparación de (8-5): A una solución de 8-4 (2.0 g, 2.16 mmol) en DCM anhidro (10 mL), se añadió G 2CHCOOH (2.8 g, 21.57 mmol) gota a gota a -78 °C. La mezcla se calentó hasta -10 °C y se agitó a esta temperatura durante 20 min. La reacción se desactivó con NaHCO3 sat. a -10 °C. La mezcla se extrajo con DCM, se lavó con salmuera y se secó con MgSO4 anhidro. La solución se concentró a baja presión. El residuo se purificó en una columna de gel de sílice (10% de EA en PE) para obtener 8-5 (0.99 g, 70%) como una espuma blanca.
Preparación de (8-6): A una solución agitada de 8-5 (3.5 g, 5.34 mmol) en DMSO anhidro (35 mL), se añadió DCC (3.30 g, 16.03 mmol) y Py-TFA (1.03 g, 5.34 mmol). La mezcla se agitó a 30 °C durante 1 h. La reacción se desactivó con agua fría a 0 °C y se extrajo con EA (3 x 60 mL). El precipitado se filtró. Las fases orgánicas se lavaron con salmuera (3x) y se secaron con MgSO4 anhidro. La fase orgánica se concentró a baja presión para obtener 8-6 crudo (3.5 g) como un aceite amarillo.
Preparación de (8-7): A una solución agitada de 8-6 (3.5 g, 5.34 mmol) en MeCN (35 mL), se añadió HCHO al 37% (11.1 mL) y TEA (4.33 g, 42.7 mmol). La mezcla se agitó a 25 °C durante 12 h. La mezcla se trató con EtOH (26 mL) y NaBH4 (3.25 g, 85.5 mmol) y a continuación se agitó durante 30 min. La reacción se desactivó con NH4G ac. sat. y
se extrajo con EA (3 x 60 mL). La fase orgánica se secó con MgSO4 anhidro y se concentró a baja presión. El residuo se purificó mediante cromatografía en columna (de un 10% de EA en PE a un 50% de DCM en PE) para obtener 8-7 (1.46 g, 40%) como un sólido blanco.
Preparación de (8-8): A una solución agitada de 8-7 (1.85 g, 2.7 mmol) en piridina (24 mL) y DCM (9.6 mL), se añadió DMTrCl (1.3 g, 3.9 mmol) a -35 °C en atmósfera de N2. La solución se agitó a 25 °C durante 16 h. La mezcla se trató con MeOH (15 mL) y se concentró a baja presión. El residuo se purificó mediante cromatografía en columna (EA en PE de un 10% a un 30%) para obtener 8-8 (1.60 g, 60 %) como un sólido blanco.
Preparación de (8-9): A una solución de 8-8 (1.07 g, 1.08 mmol) en piridina anhidra (5 mL), se añadió AgNO3 (0.65 g, 3.79 mmol) y TBDPSCl (1.04 g, 3.79 mmol). La mezcla se agitó a 25 °C durante 16 h. El disolvente se eliminó a presión reducida. El residuo se disolvió en EA (50 mL). La solución resultante se lavó con salmuera. La fase orgánica se secó con MgSO4 anhidro y se concentró a baja presión. El residuo se purificó en una columna de gel de sílice (10% de EA en PE) para obtener 8-9 (0.93 g, 70%) como una espuma blanca.
Preparación de (8-10): A una solución agitada de 8-9 (1 g, 0.82 mmol) en DCM anhidro (13.43 mL), se añadió CbCHCOOH (2.69 mL) a -78 °C. La mezcla se agitó a -10 °C durante 20 min. La reacción se desactivó con. NaHCO3 ac. sat. y se extrajo con DCM. La fase orgánica se secó con Na2SO4 anhidro y se concentró a baja presión. La fase orgánica se purificó mediante cromatografía en columna (MeOH en DCM de un 0.5% a un 2%) para obtener 8-10 (0.48 g, 65%) como un sólido.
Preparación de (8-11): A una solución enfriada con hielo de 8-10 (0.4 g, 0.433 mmol) en DCM anhidro (2.7 mL), se añadió piridina (171 mg, 2.17 mmol) y Tf2O (183 mg , 0.65 mmol) gota a gota a -35 °C. La mezcla se agitó a -10 °C durante 20 min. La reacción se desactivó con agua-hielo y se agitó durante 30 min. La mezcla se extrajo con DCM (3 x 20 mL). La fase orgánica se lavó con salmuera (100 mL), se secó con Na2SO4 anhidro y se concentró a baja presión para obtener 8-11 crudo (0.46 g), que se utilizó para el siguiente paso sin purificación adicional.
Preparación de (8-12): A una solución de 8-11 (0.46 g, 0.43 mmol) en DMF anhidro (2.5 mL), se añadió NaN3 (42 mg, 0.65 mmol). La mezcla se agitó a 30 °C durante 16 h. La solución se diluyó con agua y se extrajo con EA (3 x 30 mL). Las fases orgánicas combinadas se secaron con Na2SO4 anhidro y se concentraron a baja presión. El residuo se purificó en una columna de gel de sílice (EA en PE de un 5% a un 15%) para obtener 8-12 (0.31 g, 70%) como un sólido.
Preparación de (8-13): A una solución de 8-12 (0.31 g, 0.33 mmol) en MeOH (5 mL), se añadió NH4F (0.36 g, 9.81 mmol) a 70 °C. La mezcla se agitó a esta temperatura durante 24 h. La mezcla se evaporó a sequedad. El residuo se purificó en una columna de gel de sílice (MeOH en DCM de un 0.5% a un 2.5%) para obtener 8-13 (117 mg, 60%) como un sólido blanco.
Preparación de (8A): Se disolvió 8-13 (300 mg, 0.50 mmol) en HOAc al 80% (20 mL). La mezcla se agitó a 55 °C durante 1 h. La reacción se desactivó con MeOH y se concentró a baja presión. El residuo se purificó mediante HPLC prep para obtener 8A (100 mg, 61.3 %) como un sólido blanco. ESI-LCMS: m/z 325.1 [M H]+.
EJEMPLO 25
Preparación del Compuesto 33A

El compuesto 33-3 se preparó de acuerdo con el esquema proporcionado anteriormente. El compuesto 33A se puede obtener utilizando métodos conocidos por los expertos en la técnica, incluidos los descritos en la Publicación de EE. UU. N ° 2012/0071434, presentada el 19 de septiembre de 2011.
EJEMPLO 26
Preparación de compuestos de tipo trifosfato
Compuestos 3A, 4A, 9A y 11A: Se disolvió el nucleósido anhidro (0.05 mmol) en una mezcla de PO(OMe)3 (0.7 mL) y piridina (0.3 mL). La mezcla se evaporó al vacío durante 15 min con una temperatura del baño de 42 °C y a continuación se enfrió hasta T.A. Se añadió W-metilimidazol (0.009 mL, 0.11 mmol) seguido de POCl3 (9 gL, 0.11 mmol) y la mezcla se mantuvo a T.A. durante 40 min. La reacción se controló mediante LCMS y se monitorizó mediante la aparición del correspondiente 5’-monofosfato del nucleósido. Después de que se consiguiera más de un 50% de transformación, se añadió la sal de tetrabutilamonio del pirofosfato (150 mg) y a continuación DMF (0.5 mL) para obtener una solución homogénea. Después de 1.5 horas a temperatura ambiente, la reacción se diluyó con agua (10 mL) y se introdujo en una columna HiLoad 16/10 con Q Sepharose de alto rendimiento. La separación se llevó a cabo con un gradiente lineal de NaCl desde 0 hasta 1 N en tampón de TRIS 50 mM (pH 7.5). El trifosfato se eluyó a un 75-80% de B. Se concentraron las fracciones correspondientes. La eliminación de las sales se consiguió mediante RP HPLC en una columna Synergy Hydro-RP de 4 micras (Phenominex). Se utilizó un gradiente lineal de metanol de un 0% a un 30% en un tampón de acetato de trietilamonio 50 mM (pH 7.5) para la elución. Las fracciones correspondientes se combinaron, se concentraron y se liofilizaron 3 veces para eliminar el exceso de tampón.
Compuestos 5A, 6A, 10A y 12A: Los 5’-trifosfatos de los nucleósidos con un grupo 4’-azidoalquilo se disolvieron en agua (0.1 mL), y se añadió metanol (3 mL) seguido de Pd/C al 10% (3 mg). Se burbujeó hidrógeno a través de la solución durante 2 h. El catalizador se separó por filtración y el filtrado se purificó mediante RP HPLC en una columna Synergy Hydro-RP de 4 micras (Phenominex). Se utilizó un gradiente lineal de metanol de un 0% a un 20% en un tampón de acetato de trietilamonio 50 mM (pH 7.5) para la elución. Las fracciones correspondientes se combinaron, se concentraron y se liofilizaron 3 veces para eliminar el exceso de tampón.
Tabla 1 - Trifosfatos obtenidos del Ejemplo 25


EJEMPLO 27
Compuestos adicionales
Las síntesis anteriores son ilustrativas y se pueden utilizar como punto de partida para preparar un gran número de compuestos adicionales. A continuación se proporcionan ejemplos de compuestos de Fórmula (I) que se pueden preparar de varias maneras, incluidos los esquemas sintéticos que se muestran y describen en la presente. Los expertos en la técnica serán capaces de reconocer modificaciones de las síntesis divulgadas y diseñar rutas
basándose en las divulgaciones de la presente; siempre que tales modificaciones y rutas alternativas se encuentren dentro del alcance de las reivindicaciones.
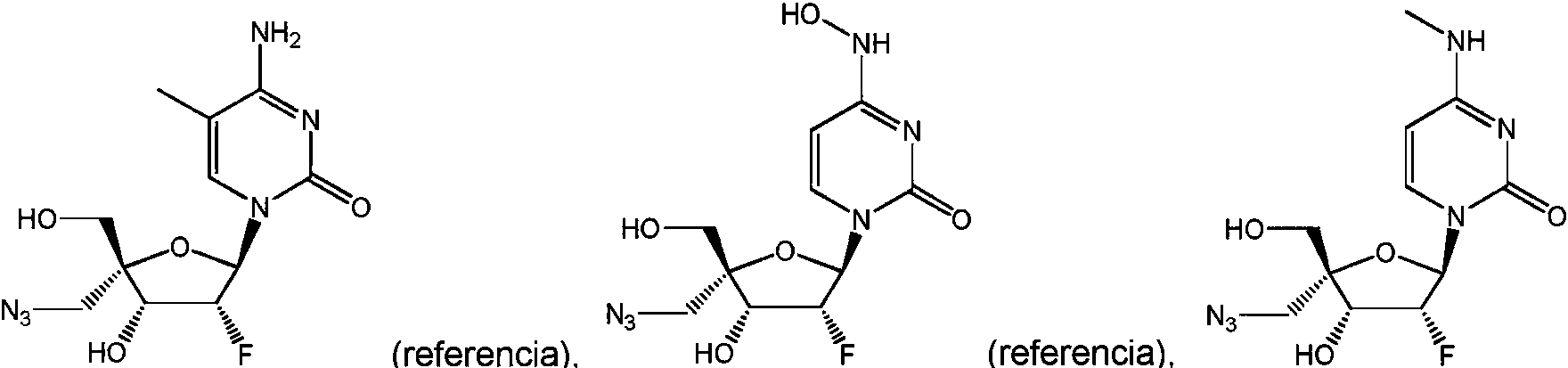
(referencia)  (referencia)
(referencia)
(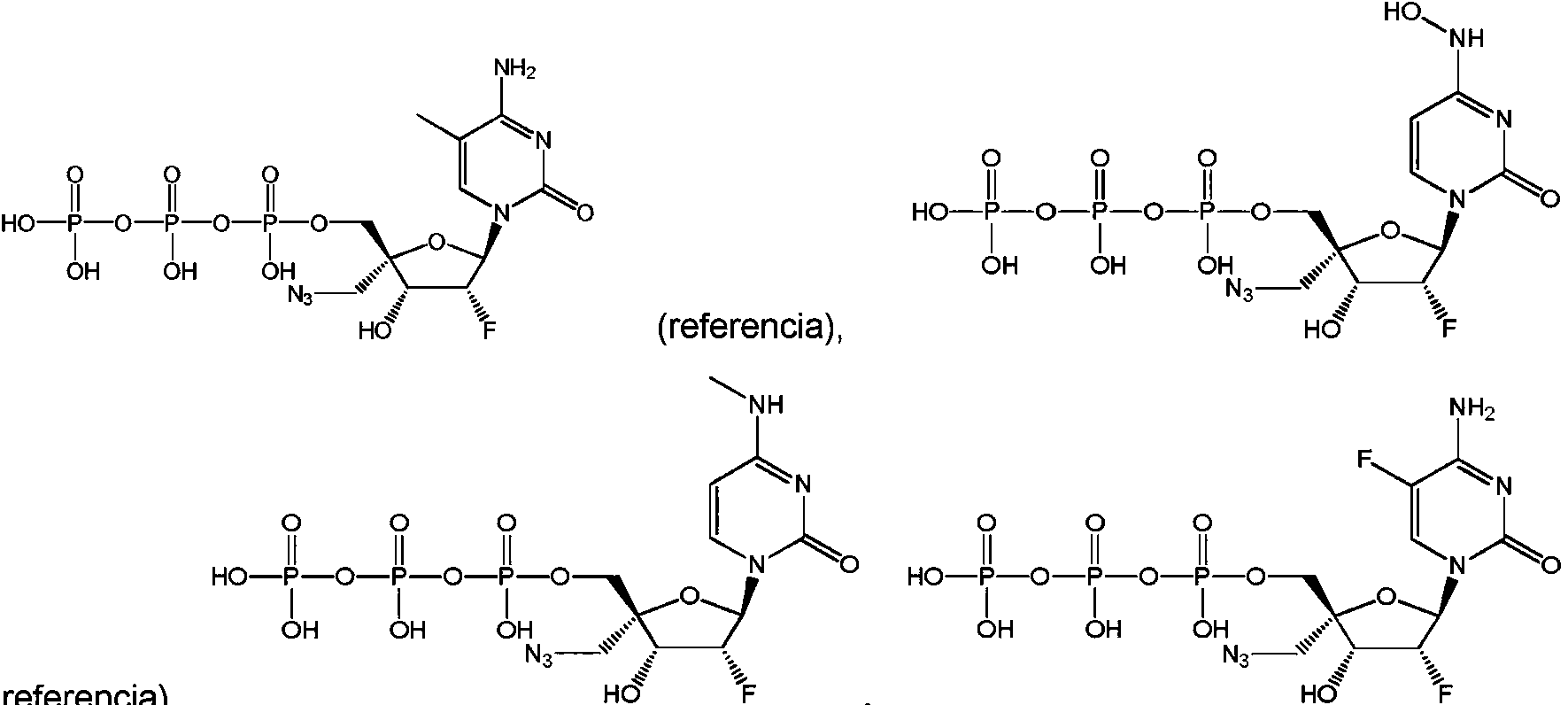 ,
,
(referencia) 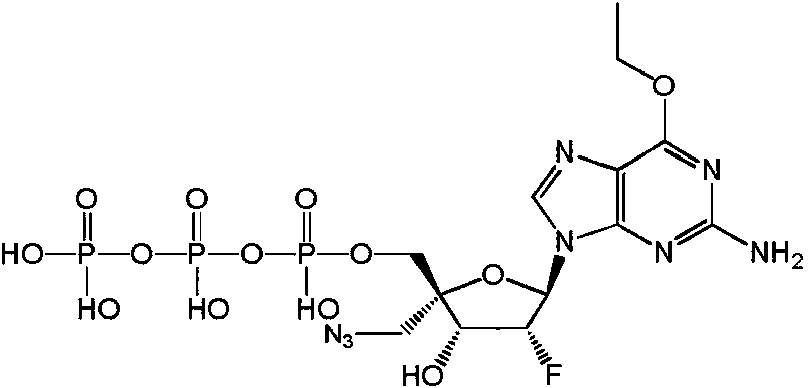 (referencia)
(referencia) 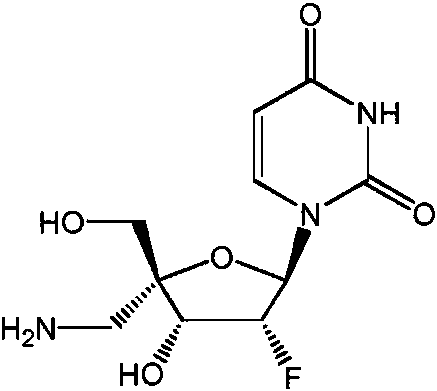

Ensayo con el VRS
Se obtuvo una licencia para el replicón subgenómico del VRS 395 HeLa de Apath (Brooklyn, NY) y este fue desarrollado originariamente por el Dr. Mark Meeples del Centro de Vacunas e Inmunidad, del Instituto de Investigación del Hospital Nacional de Niños en Columbus, Ohio. Para generar el replicón del VRS subgenómico, se eliminaron tres genes de glicoproteínas, los de SH, G y F, de un ADNc antigenómico del VRS recombinante que expresaba GFP (rg) de longitud completa. En su lugar, se insertó un gen de blasticidina S desaminasa (bsd). A través de múltiples pasos, el replicón del VRS se estableció en las células HeLa. Las células 395 HeLa se cultivaron en medio Eagle modificado de Dulbecco (DMEM) que contenía 4500 mg/L de D-glucosa, L-glutamina y 110 mg/L de piruvato de sodio (Invitrogen, n ° de cat. 11995-040). El medio se suplementó adicionalmente con un 10% (v/v) de suero fetal bovino (FBS) (Mediatech, n ° de cat. 35-010-CV), un 1% (v/v) de penicilina/estreptomicina (Mediatech, n.° de cat. 30-002-CI) y 10 pg/mL de blasticidina (BSD) (Invivogen, código de cat. ant-bl-1). Las células se mantuvieron a 37 °C en una atmósfera humidificada de CO 2 al 5%.
La determinación del 50% de la concentración inhibitoria (CE50), el 90% de la concentración inhibitoria (CE90) y el 50% de la concentración citotóxica (CC50) en células con replicón del VRS se llevó a cabo mediante el siguiente procedimiento. El primer día, se colocaron 5000 células con replicón del VRS por pocillo en una placa de 96 pocillos. Al día siguiente, los compuestos que se iban a evaluar se solubilizaron en DMSO al 100% hasta obtener 100 veces la concentración de ensayo final deseada. Cada compuesto se diluyó en serie (1:3) hasta obtener 9 concentraciones distintas. Los compuestos en DMSO al 100% se redujeron a DMSO al 10% (v/v) diluyendo con un factor de 1:10 en medio de cultivo celular. Se utilizó una muestra de 10 pL de los compuestos diluidos hasta DMSO al 10% (v/v) con medio de cultivo celular para tratar las células con replicón del VRS en formato de 96 pocillos. La concentración final de DMSO fue de un 1% (v/v). Las células se incubaron con los compuestos durante 7 días a 37 °C en una atmósfera de CO2 al 5%. En cada ensayo, se incluyó un control positivo que se había caracterizado previamente en el ensayo con el replicón del VRS.
Se utilizó el sistema de ensayo de la luciferasa de Renilla (Promega, n.° de cat. E2820) para medir la actividad del replicón anti-VRS. Las placas de ensayo se configuraron como se ha mencionado anteriormente. Se registró la luminiscencia utilizando un contador multimarca Victor 3V de Perkin Elmer. La CE50, que es la concentración del fármaco requerida para reducir el ARN del replicón del VRS en un 50% respecto al valor de control de células no tratadas, se calculó a partir del gráfico de reducciones porcentuales del valor de densidad óptica (DO) frente a las concentraciones del fármaco utilizando la función de pronóstico de Microsoft Excel.
Se utilizó el ensayo de proliferación de células 395 HeLa (Promega; ensayo de viabilidad celular luminiscente CellTiter-Glo, n ° de cat. G7572) para medir la viabilidad celular. El ensayo de viabilidad celular luminiscente CellTiter-Glo® es un método homogéneo para determinar el número de células viables en un cultivo en función de la cuantificación del ATP presente, que señala la presencia de células metabólicamente activas. Las placas de ensayo se configuraron en el mismo formato que se ha indicado anteriormente para el ensayo del replicón. Se añadió el reactivo de CellTiter-Glo (100 pL) a cada pocillo y se incubó a temperatura ambiente durante 8 minutos. Se registró la luminiscencia utilizando un contador multimarca Victor 3V de Perkin Elmer. La CC50, que es la concentración del fármaco requerida para reducir las células viables en un 50% respecto al valor de control de células no tratadas, se calculó a partir del gráfico de reducciones porcentuales del valor de luminiscencia frente a las concentraciones del fármaco utilizando la función de pronóstico de Microsoft Excel.
La Tabla A1 incluye compuestos con un valor de CE50 que es inferior a 1 pM. La Tabla A2 incluye compuestos con un valor de CE50 que es igual o superior a 1 pM e inferior a 50 pM. Otros compuestos evaluados divulgados en la presente presentaron un valor de CE50 de 50 pM o superior
Tabla A1



Tabla A2



(donde los compuestos 7A, 22A y 23A son compuestos de referencia).
Se llevaron a cabo ensayos de polimerasa del VRS estándar en presencia de 3 pL de extracto de células infectadas con VRS en un tampón de reacción que contenía tris-acetato 50 mM de pH 8, K-acetato 120 mM, MgCl24.5 mM, glicerol al 5%, EDTA 2 mM, 50 pg/mL de BSA y DTT 3 mM. Se utilizaron diferentes concentraciones de los compuestos de prueba para iniciar la síntesis de ARN durante 120 min a 30 °C, y se utilizó 33P GTP radiactivo (15 uCi) como marcador. La reacción se detuvo mediante la adición de EDTA 50 mM, y las muestras de ARN se purificaron a través de columnas de centrifugación de exclusión por tamaño G-50 y extracción con fenolcloroformo. Los productos de ARN radiomarcados se resolvieron mediante electroforesis en un gel de poliacrilamida TBE al 6%, y se visualizaron y cuantificaron después de exponerlos en la pantalla de un phosphorImager. Se llevaron a cabo experimentos de inhibición de la polimerasa (CI50) del mismo modo en presencia de una concentración cada vez mayor de los compuestos de prueba.
La Tabla A3 incluye compuestos con un valor de CI50 que es inferior a 1 pM frente a la polimerasa. La Tabla A4 incluye compuestos con un valor de CI50 que es igual o superior a 1 pM e inferior a 50 pM frente a la polimerasa. Otros compuestos evaluados divulgados en la presente presentaron un valor de CI50 de 50 pM o superior frente a la polimerasa.
Tabla A3


Tabla A4


(donde los compuestos 5A, 6A, 11A y 12A son compuestos de referencia).
EJEMPLO 29
Ensayo en placa con el virus de parainfluenza 3 (VPI-3)
Se cultivaron células MA-104 en placas de 24 pocilios hasta obtener una confluencia de un 90% en presencia de medio esencial mínimo (MEM) suplementado con un 10% de suero bovino fetal y antibióticos (C-EMEM). A continuación, las células se lavaron dos veces con medio esencial mínimo no completo (NC-EMEM). Los artículos de prueba se disolvieron en DMSO hasta obtener una concentración patrón de 10 mM.
A continuación, se inoculó una alícuota de 0.5 mL del artículo de prueba con diferentes concentraciones en pocillos por triplicado y se incubó durante 60 min a 37 °C con un 5% de CO2 para la difusión del artículo de prueba en las células MA-104. Después del periodo de incubación, se descongeló un patrón de VPI humano de tipo 3 y se diluyó con NC-EMEM para conseguir una concentración vírica de 104 pfu/mL. A continuación, se inoculó una alícuota de 0.1 mL en todos los pocillos, con la excepción de los pocillos de control de toxicidad del artículo de prueba y negativo. Tras la infección, las placas se incubaron durante 72 h a 37 °C con un 5% de CO2. Después de la incubación, las placas se examinaron por microscopía para registrar la citotoxicidad. Los sobrenadantes se recolectaron para llevar a cabo una cuantificación vírica utilizando un ensayo de placa estándar que empleó células MA-104 como células indicadoras.
Para realizar el ensayo de placas, se cultivaron células MA-104 hasta obtener confluencia en placas de 24 pocillos. Las células se lavaron con medio exento de suero antes de la inoculación de pocillos por duplicado con diluciones en serie de 10 veces de la muestra de sobrenadante. Después de 1 h de incubación a 37 °C, las muestras se aspiraron y se añadió 1.0 mL de medio de recubrimiento de metilcelulosa a cada pocillo. Después de 6 días de cultivo, las células se fijaron y se tiñeron con un 0.06% de violeta cristalino en glutaraldehído al 1% y se enumeraron las placas víricas. Los datos se analizaron con el software Prism, definiendo CE50 como la concentración de fármaco que redujo la carga vírica en un 50% respecto al control vírico (CV). La Tabla B1 proporciona una lista de compuestos de Fórmula (I) que son activos frente al VPI-3 con un valor de CE50 < 20 pM.
Tabla B1


Se cultivaron células LLC-MK2 en placas de 24 pocillos hasta obtener una confluencia de un 90% en presencia de medio esencial mínimo (MEM) suplementado con un 10% de suero bovino fetal y antibióticos (C-EMEM). A continuación, las células se lavaron dos veces con medio esencial mínimo no completo (NC-EMEM). Los artículos de prueba se disolvieron en DMSO hasta obtener una concentración patrón de 10 mM.
A continuación, se inoculó una alícuota de 0.5 mL del artículo de prueba con diferentes concentraciones en pocillos por triplicado y se incubó durante 60 min a 37 °C con un 5% de CO2 para la difusión del artículo de prueba en las células LLC-MK2. Después del periodo de incubación, se descongeló un patrón de metapneumovirus humano y se diluyó con NC-EMEM para conseguir una concentración vírica de 104 pfu/mL. A continuación, se inoculó una alícuota de 0.1 mL en todos los pocillos, con la excepción de los pocillos de control de toxicidad del artículo de prueba y negativo. Tras la infección, las placas se incubaron durante 7 días a 37 °C con un 5% de CO2. Después de la incubación, las placas se examinaron por microscopía para registrar la citotoxicidad. Los sobrenadantes se recolectaron para llevar a cabo una cuantificación vírica utilizando un ensayo de DICT50 estándar que empleó células LLC-MK2 como células indicadoras. Los datos se analizaron con el software Prism, definiendo CE50 como la concentración de fármaco que
redujo la carga vírica en un 50% respecto al control vírico (CV). La Tabla C1 proporciona una lista de compuestos de Fórmula (I) que son activos frente al metapneumovirus humano, con un valor de CE50 < 20 pM
Tabla C1




(donde los compuestos 7A y 8A son compuestos de referencia).
Aunque lo anterior se ha descrito con cierto detalle a modo de ilustraciones y ejemplos con fines de claridad y comprensión, los expertos en la técnica entenderán que se pueden realizar modificaciones numerosas y diversas sin alejarse de la presente divulgación. Por lo tanto, debe entenderse claramente que las formas divulgadas en la presente son únicamente ilustrativas y no se pretende que limiten el alcance de la presente divulgación.
A continuación, se exponen características de la presente divulgación en las siguientes cláusulas numeradas, las cuales contienen la materia en cuestión de las reivindicaciones de la solicitud original tal como se presentó inicialmente.
1. Un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este,

B1A es una base heterocíclica opcionalmente sustituida o una base heterocíclica opcionalmente sustituida con un grupo amino protegido;
RA es hidrógeno o deuterio;
R1A se selecciona del grupo constituido por hidrógeno, un acilo opcionalmente sustituido, un aminoácido conectado a
través de O opcionalmente sustituido,
Ra1 y Ra2 son independientemente hidrógeno o deuterio;
R2A es un azidoalquilo C1-6 o un aminoalquilo C1-6;
R3A se selecciona del grupo constituido por OH, -OC(=O)R"A y un aminoácido conectado a través de O opcionalmente sustituido;
R4A es halógeno;
R5A es hidrógeno o halógeno;
R6A, R7A y R8A se seleccionan independientemente del grupo constituido por ausente, hidrógeno, un alquilo C1-24 opcionalmente sustituido, un alquenilo C3-24 opcionalmente sustituido, un alquinilo C3-24 opcionalmente sustituido, un cicloalquilo C3-6 opcionalmente sustituido, un cicloalquenilo C3-6 opcionalmente sustituido, un arilo opcionalmente sustituido, un heteroarilo opcionalmente sustituido, un aril(alquilo C1-6) opcionalmente sustituido, un *-(CR15AR16A)p-O(alquilo C i-24 ) opcionalmente sustituido, un *-(CR17AR18A)q-0-(alquenilo C-1-24) opcionalmente sustituido,


R6A y R7A se consideran conjuntamente para formar un resto seleccionado del grupo constituido por un
opcionalmente sustituido y un > opcionalmente sustituido, donde los oxígenos conectados a R6A y R7A, el fósforo y el resto forman un sistema anular de seis miembros a diez miembros;
> opcionalmente sustituido, donde los oxígenos conectados a R6A y R7A, el fósforo y el resto forman un sistema anular de seis miembros a diez miembros;
R9A se selecciona independientemente del grupo constituido por un alquilo C1-24 opcionalmente sustituido, un alquenilo C2-24 opcionalmente sustituido, un alquinilo C2-24 opcionalmente sustituido, un cicloalquilo C3-6 opcionalmente sustituido, un cicloalquenilo C3-6 opcionalmente sustituido, NR30AR31A, un aminoácido conectado a través de N opcionalmente sustituido y un derivado de tipo éster de un aminoácido conectado a través de N opcionalmente sustituido;
R10A y R11A son independientemente un aminoácido conectado a través de N opcionalmente sustituido o un derivado de tipo éster de un aminoácido conectado a través de N opcionalmente sustituido;
R12A, R13A y R14A pueden independientemente estar ausentes o ser hidrógeno;
cada R15A, cada R16A, cada R17A y cada R18A son independientemente hidrógeno, un alcoxi o alquilo C1-24 opcionalmente sustituido;
R19A, R20A, R22A y R23A se seleccionan independientemente del grupo constituido por hidrógeno, un alquilo C1-24 opcionalmente sustituido y un arilo opcionalmente sustituido;
R21A y R24A se seleccionan independientemente del grupo constituido por hidrógeno, un alquilo C1-24 opcionalmente sustituido, un arilo opcionalmente sustituido, un -O-alquilo C1-24 opcionalmente sustituido, un -O-arilo opcionalmente sustituido, un -O-heteroarilo opcionalmente sustituido, un -O-heterociclilo monocíclico opcionalmente sustituido y

R25A y R29A se seleccionan independientemente del grupo constituido por hidrógeno, un alquilo C1-24 opcionalmente sustituido y un arilo opcionalmente sustituido;
R26A y R27A son independientemente -C=N o un sustituyente opcionalmente sustituido seleccionado del grupo constituido por organilcarbonilo C2-8, alcoxicarbonilo C2-8 y organilaminocarbonilo C2-8;
R28A se selecciona del grupo constituido por hidrógeno, un alquilo C1-24 opcionalmente sustituido, un alquenilo C2-24 opcionalmente sustituido, un alquinilo C2-24 opcionalmente sustituido, un cicloalquilo C3-6 opcionalmente sustituido y un cicloalquenilo C3-6 opcionalmente sustituido;
R30A y R31A se seleccionan independientemente del grupo constituido por hidrógeno, un alquilo C1-24 opcionalmente sustituido, un alquenilo C2-24 opcionalmente sustituido, un alquinilo C2-24 opcionalmente sustituido, un cicloalquilo C3-6 opcionalmente sustituido y un cicloalquenilo C3-6 opcionalmente sustituido;
R"A es un alquilo C1-24 opcionalmente sustituido;
m y t son independientemente 0 o 1;
p y q se seleccionan independientemente del grupo constituido por 1,2 y 3;
r es 1 o 2;
s es 0, 1, 2 o 3;
u es 1 o 2; y
Z1A, z 2A, Z3A y Z4A son independientemente O o S.
2. El compuesto de la Cláusula 1, donde RZA es azidometilo.
3. El compuesto de la Cláusula 1, donde R2A es aminometilo.
4. El compuesto de cualquiera de las Cláusulas 1-3, donde
5. El compuesto de la Cláusula 4, donde R6A y R7A son ambos hidrógeno.
6. El compuesto de la Cláusula 4, donde R6A y R7A están ambos ausentes.
7. El compuesto de la Cláusula 4, donde R6A y R7A se seleccionan ambos independientemente del grupo constituido por un alquilo C1-24 opcionalmente sustituido, un alquenilo C3-24 opcionalmente sustituido, un alquinilo C3-24 opcionalmente sustituido, un cicloalquilo C3-6 opcionalmente sustituido, un cicloalquenilo C3-6 opcionalmente sustituido, un arilo opcionalmente sustituido, un heteroarilo opcionalmente sustituido y un aril(alquilo C1-6) opcionalmente sustituido.
8. El compuesto de la Cláusula 4, donde R6A y R7A son ambos un alquilo C1-24 opcionalmente sustituido.
9. El compuesto de la Cláusula 4, donde R6A y R7A son ambos un alquenilo C3-24 opcionalmente sustituido.
10. El compuesto de la Cláusula 4, donde R6A y R7A son ambos *-(CR15AR16A)p-O-(alquilo C1-24).
11. El compuesto de la Cláusula 4, donde R6A y R7A son ambos *-(CR17AR18A)p-O-(alquenilo C2-24).
12. El compuesto de la Cláusula 4, donde R6A y R7A son ambos un arilo opcionalmente sustituido.
13. El compuesto de la Cláusula 4, donde R6A y R7A son ambos un aril(alquilo C1-6) opcionalmente sustituido.
14. El compuesto de la Cláusula 4, donde R6A y R7A son ambos
15. El compuesto de la Cláusula 4, donde R6A y R7A son ambos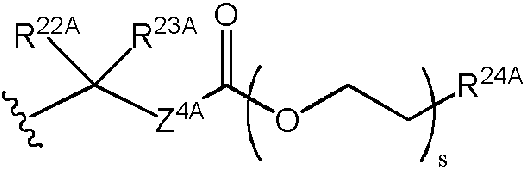

16. El compuesto de la Cláusula 4, donde R6A y R7A son ambos
17. El compuesto de la Cláusula 4, donde R6A y R7A son ambos
18. El compuesto de la Cláusula 4, donde R6A y R7A son ambos
19. El compuesto de la Cláusula 4, donde R6A y R7A se pueden considerar conjuntamente para formar un resto
seleccionado del grupo constituido por un opcionalmente sustituido y un
opcionalmente sustituido y un > opcionalmente sustituido, donde los oxígenos conectados a R6A y R7A, el fósforo y el resto forman un sistema anular de seis miembros a diez miembros;
> opcionalmente sustituido, donde los oxígenos conectados a R6A y R7A, el fósforo y el resto forman un sistema anular de seis miembros a diez miembros;
20. El compuesto de cualquiera de las Cláusulas 1 a 19, donde Z1A es O.
21. El compuesto de cualquiera de las Cláusulas 1 a 19, donde Z1A es S.
22. El compuesto de la Cláusula 1, donde
23. El compuesto de la Cláusula 22, donde R8A es hidrógeno y R9A es NR30AR31A, donde R30 y R31 se seleccionan independientemente del grupo constituido por hidrógeno, un alquilo C1-24 opcionalmente sustituido, un alquenilo C2-24 opcionalmente sustituido, un alquinilo C2-24 opcionalmente sustituido, un cicloalquilo C3-6 opcionalmente sustituido y un cicloalquenilo C3-6 opcionalmente sustituido.
24. El compuesto de la Cláusula 22, donde R8A es un arilo opcionalmente sustituido; y R9A es un aminoácido conectado a través de N opcionalmente sustituido o un derivado de tipo éster de un aminoácido conectado a través de N opcionalmente sustituido.
25. El compuesto de la Cláusula 22, donde R8A es un arilo opcionalmente sustituido y R9A tiene la estructura R 33A0 R34A R 35A

donde R33A se selecciona del grupo constituido por hidrógeno, un alquilo C1-6 opcionalmente sustituido, un cicloalquilo C3-6 opcionalmente sustituido, un arilo opcionalmente sustituido, un aril(alquilo C1-6) opcionalmente sustituido y un haloalquilo opcionalmente sustituido; R34A se selecciona del grupo constituido por hidrógeno, un alquilo C1-6 opcionalmente sustituido, un haloalquilo C1-6 opcionalmente sustituido, un cicloalquilo C3-6 opcionalmente sustituido, un arilo C6 opcionalmente sustituido, un arilo C10 opcionalmente sustituido y un aril(alquilo C1-6) opcionalmente sustituido; y R35A es hidrógeno o un alquilo C1-4 opcionalmente sustituido; o R34A y R35A se consideran conjuntamente para formar un cicloalquilo C3-6 opcionalmente sustituido.
26. El compuesto de cualquiera de las Cláusulas 22 a 25, donde Z2A es O.
27. El compuesto de cualquiera de las Cláusulas 22 a 25, donde Z2A es S.
28. El compuesto de la Cláusula 1, donde
29. El compuesto de la Cláusula 28, donde R10A y R11A son ambos un aminoácido conectado a través de N opcionalmente sustituido o un derivado de tipo éster de un aminoácido conectado a través de N opcionalmente sustituido.
30. El compuesto de la Cláusula 28, donde R10A y R11A tienen independientemente la estructura donde R36A se selecciona del grupo constituido por hidrógeno, un alquilo C1-6 opcionalmente susti
C3-6 opcionalmente sustituido, un arilo opcionalmente sustituido, un aril(alquilo C1-6) opcionalmente sustituido y un haloalquilo opcionalmente sustituido; R37A se selecciona del grupo constituido por hidrógeno, un alquilo C1-6 opcionalmente sustituido, un haloalquilo C1-6 opcionalmente sustituido, un cicloalquilo C3-6 opcionalmente sustituido, un arilo C6 opcionalmente sustituido, un arilo C10 opcionalmente sustituido y un aril(alquilo C1-6) opcionalmente sustituido; y R38A es hidrógeno o un alquilo C1-4 opcionalmente sustituido; o R37A y R38A se consideran conjuntamente para formar un cicloalquilo C3-6 opcionalmente sustituido.
31. El compuesto de cualquiera de las Cláusulas 28 a 30, donde Z3A es O.
32. El compuesto de cualquiera de las Cláusulas 28 a 30, donde Z3A es S.
33. El compuesto de la Cláusula 1, donde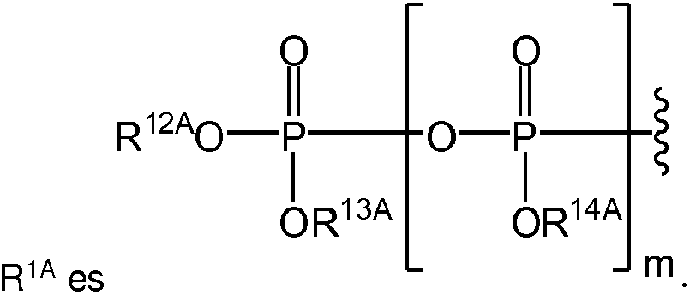
34. El compuesto de la Cláusula 33, donde m es 0, y R12A y R13A independientemente están ausentes o son hidrógeno.
35. El compuesto de la Cláusula 33, donde m es 1, y R12A, R13A y R14A independientemente están ausentes o son hidrógeno.
36. El compuesto de la Cláusula 1, donde R1A es H.
37. El compuesto de la Cláusula 1, donde R1A es un acilo opcionalmente sustituido.
38. El compuesto de la Cláusula 1, donde R1A es un aminoácido conectado a través de O opcionalmente sustituido.
39. El compuesto de la Cláusula 1, donde 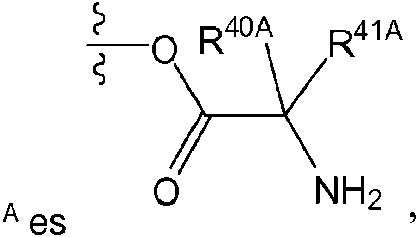 donde R40A se selecciona del grupo constituido por hidrógeno, un alquilo C1-6 opcionalmente sustituido, un haloalquilo C1-6 opcionalmente sustituido, un cicloalquilo C3-6 opcionalmente sustituido, un arilo C6 opcionalmente sustituido, un arilo C10 opcionalmente sustituido y un aril(alquilo C1-6) opcionalmente sustituido; y R41A es hidrógeno o un alquilo C1-4 opcionalmente sustituido; o R40A y R41A se consideran conjuntamente para formar un cicloalquilo C3-6 opcionalmente sustituido.
donde R40A se selecciona del grupo constituido por hidrógeno, un alquilo C1-6 opcionalmente sustituido, un haloalquilo C1-6 opcionalmente sustituido, un cicloalquilo C3-6 opcionalmente sustituido, un arilo C6 opcionalmente sustituido, un arilo C10 opcionalmente sustituido y un aril(alquilo C1-6) opcionalmente sustituido; y R41A es hidrógeno o un alquilo C1-4 opcionalmente sustituido; o R40A y R41A se consideran conjuntamente para formar un cicloalquilo C3-6 opcionalmente sustituido.
40. El co m p u e s to de c u a lq u ie ra de las C láusu la s 1 a 39, dond e B 1A se s e le c c io n a de l g ru po co n s titu id o por:

donde:
RA2 se selecciona del grupo constituido por hidrógeno, halógeno y NHRJ2, donde RJ2 se selecciona del grupo constituido por hidrógeno, -C(=O)RK2 y -C(=O)ORL2,
RB2 es halógeno o NHRW2, donde RW2 se selecciona del grupo constituido por hidrógeno, un alquilo C1-6 opcionalmente sustituido, un alquenilo C2-6 opcionalmente sustituido, un cicloalquilo C3-8 opcionalmente sustituido, -C(=O)RM2 y -C(=O)ORN2;
RC2 es hidrógeno o NHRO2, donde RO2 se selecciona del grupo constituido por hidrógeno, -C(=O)RP2 y -C(=O)ORQ2; RD2 se selecciona del grupo constituido por hidrógeno, deuterio, halógeno, un alquilo C1-6 opcionalmente sustituido, un alquenilo C2-6 opcionalmente sustituido y un alquinilo C2-6 opcionalmente sustituido;
RE2 se selecciona del grupo constituido por hidrógeno, hidroxi, un alquilo C1-6 opcionalmente sustituido, un cicloalquilo C3-8 opcionalmente sustituido, -C(=O)RR2 y -C(=O)ORS2;
RF2 se selecciona del grupo constituido por hidrógeno, halógeno, un alquilo C1-6 opcionalmente sustituido, un alquenilo C2-6 opcionalmente sustituido y un alquinilo C2-6 opcionalmente sustituido;
Y2 e Y3 son independientemente N o CR12, donde R12 se selecciona del grupo constituido por hidrógeno, halógeno, un alquilo C1-6 opcionalmente sustituido, un alquenilo C2-6 opcionalmente sustituido y un alquinilo C2-6 opcionalmente sustituido;
RG2 es un alquilo C1-6 opcionalmente sustituido;
RH2 es hidrógeno o NHRT2, donde RT2 se selecciona independientemente del grupo constituido por hidrógeno, -C(=O)RU2 y -C(=O)ORV2; y
RK2’ RL2, RM2, RN2, RP2, RQ2 RR2, RS2’ RU2 y RV2 se seleccionan independientemente del grupo constituido por alquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6, cicloalquenilo C3-6, arilo C6-10, heteroarilo, heterociclilo, aril(alquilo C1-6), heteroaril(alquilo C1-6) y heterociclil(alquilo C1-6).
41. El c o m p u e s to de la C lá u su la 40,
42. El compuesto de la Cláusula 40, donde
43. El compuesto de la Cláusula 40, donde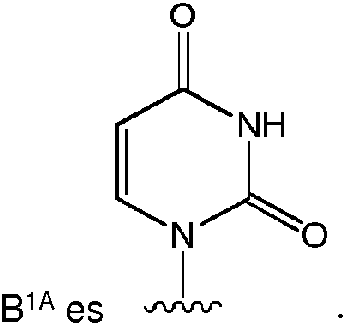
44. El compuesto de la Cláusula 40, donde
45. El compuesto de la Cláusula 40, donde
46. El compuesto de la Cláusula 40, donde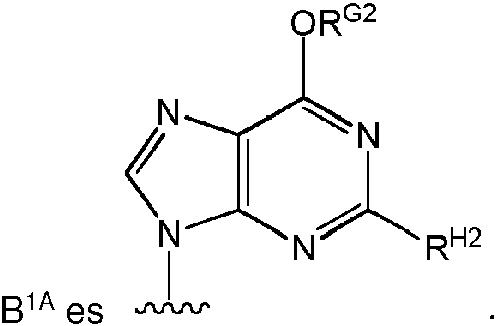
47. El compuesto de cualquiera de las Cláusulas 1 a 46, donde R3A es OH.
48. El compuesto de cualquiera de las Cláusulas 1 a 46, donde R3A es -OC(=O)R"A.
49. El compuesto de cualquiera de las Cláusulas 1 a 46, donde R3A es un aminoácido conectado a través de O.
50. El compuesto de cualquiera de las Cláusulas 1 a 46, donde R3A e del grupo constituido por hidrógeno, un alquilo C1-6 opcionalmente
sustituido, un cicloalquilo C3-6 opcionalmente sustituido, un arilo C6 opcionalmente sustituido, un arilo C10 opcionalmente sustituido y un aril(alquilo C1-6) opcionalmente sustituido; y R43A es hidrógeno o un alquilo C1-4 opcionalmente sustituido; o R42A y R43A se consideran conjuntamente para formar un cicloalquilo C3-6 opcionalmente sustituido.
51. El compuesto de cualquiera de las Cláusulas 1 a 50, donde R5A es hidrógeno.
52. El compuesto de cualquiera de las Cláusulas 1 a 50, donde R5A es halógeno.
53. El compuesto de la Cláusula 52, donde R5A es fluoro o cloro.
uiera de las Cláusulas 1 a 53, donde R4A es fluoro.
uiera de las Cláusulas 1 a 53, donde R4A es cloro.
lquiera de las Cláusulas 1 a 55, donde Ra1 y Ra2 son ambos hidrógeno.
lquiera de las Cláusulas 1 a 55, donde Ra1 y Ra2 son ambos deuterio.
lquiera de las Cláusulas 1 a 57, donde RA es hidrógeno.
lquiera de las Cláusulas 1 a 57, donde RA es deuterio.
usula 1, donde el compuesto de Fórmula (I) es:

láusula 1, donde el compuesto de Fórmula (I) es:

usula 1, donde el compuesto de Fórmula (I) se selecciona del grupo constituido por:
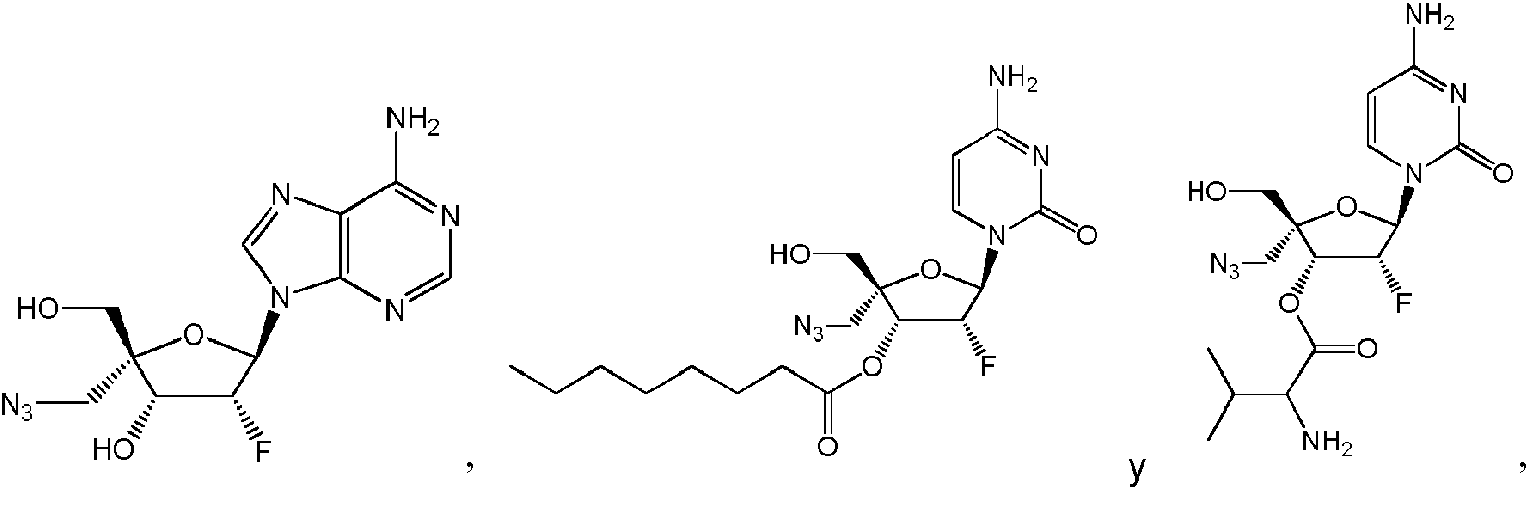
o una sal farmacéuticamente aceptable de lo anterior.
63. El compuesto de la Cláusula 1, donde el compuesto de Fórmula (I) se selecciona del grupo constituido por:

64. El compuesto de la Cláusula 1, donde el compuesto de Fórmula (I) se selecciona del grupo constituido por:

65. El compuesto de la Cláusula 1, donde el compuesto de Fórmula (I) se selecciona del grupo constituido por:

66. El c o m p u e s to de la C lá u su la 1, d o n d e el co m p u e s to de F ó rm u la (I) se se le c c io n a de l g ru p o co n s titu id o por:



67. Una composición farmacéutica que comprende una cantidad eficaz de un compuesto de cualquiera de las Cláusulas 1 a 66, o una sal farmacéuticamente aceptable de este, y un portador, diluyente o excipiente farmacéuticamente aceptable o una combinación de estos.
68. Un método para mejorar o tratar una infección vírica provocada por paramixovirus que comprende administrar a un sujeto que se identifica que padece la infección vírica provocada por paramixovirus una cantidad eficaz de un compuesto de cualquiera de las Cláusulas 1 a 66 o una sal farmacéuticamente aceptable de este, o una composición farmacéutica de la Cláusula 67.
69. Un método para inhibir la replicación de un paramixovirus que comprende poner en contacto una célula infectada con el paramixovirus con una cantidad eficaz de un compuesto de cualquiera de las Cláusulas 1 a 66 o una sal farmacéuticamente aceptable de este, o una composición farmacéutica de la Cláusula 67.
70. Un método para mejorar o tratar una infección vírica provocada por paramixovirus que comprende poner en contacto una célula infectada con el paramixovirus en un sujeto que se identifica que padece la infección vírica con una cantidad eficaz de un compuesto de cualquiera de las Cláusulas 1 a 66 o una sal farmacéuticamente aceptable de este, o una composición farmacéutica de la Cláusula 67.
71. Un método para mejorar o tratar una infección vírica provocada por paramixovirus en combinación con uno o más agentes que comprende administrar a o poner en contacto una célula en un sujeto que se identifica que padece la infección vírica provocada por paramixovirus con una cantidad eficaz de un compuesto de cualquiera de las Cláusulas 1 a 66 o una sal farmacéuticamente aceptable de este, o una composición farmacéutica de la Cláusula 67.
72. El método de cualquiera de las Cláusulas 68 a 71, donde la infección vírica provocada por paramixovirus es una infección provocada por el virus sincitial respiratorio humano.
73. El método de cualquiera de las Cláusulas 68 a 71, donde la infección vírica provocada por paramixovirus es una infección provocada por el virus sincitial respiratorio humano; y donde dicho uno o más agentes se selecciona del grupo constituido por ribavirina, palivizumab, RSV-IGIV, ALN-RSV01, BMS-433771, RFI-641, RSV604, MDT-637, BTA9881, TMC-353121, MBX-300, YM-53403, RV568 y una vacuna de partículas de VRS-F.
74. El método de cualquiera de las Cláusulas 68 a 71, donde la infección vírica provocada por paramixovirus es una infección provocada por el virus de parainfluenza humana.
75. El método de la cláusula 74, donde la infección provocada por el virus de parainfluenza humana es una infección provocada por el virus de parainfluenza humana 3.
76. El método de cualquiera de las Cláusulas 68 a 71, donde la infección vírica provocada por paramixovirus es una infección provocada por un metapneumovirus humano.
77. El uso de una cantidad eficaz de un compuesto de cualquiera de las Cláusulas 1 a 66 o una sal farmacéuticamente aceptable de este, o una composición farmacéutica de la Cláusula 67 en la preparación de un medicamento para mejorar o tratar una infección vírica provocada por paramixovirus en un sujeto que se identifica que padece la infección vírica provocada por paramixovirus.
78. El uso de una cantidad eficaz de un compuesto de cualquiera de las Cláusulas 1 a 66 o una sal farmacéuticamente aceptable de este, o una composición farmacéutica de la Cláusula 67 en la preparación de un medicamento para inhibir la replicación de un paramixovirus.
79. El uso de una cantidad eficaz de un compuesto de cualquiera de las Cláusulas 1 a 66 o una sal farmacéuticamente aceptable de este, o una composición farmacéutica de la Cláusula 67 en la preparación de un medicamento para ponerlo en contacto con una célula infectada con un paramixovirus.
Claims (1)
- REIVINDICACIONES1. Un compuesto de Fórmula (I), o una sal farmacéuticamente aceptable de este,
 RA es hidrógeno;R1A se selecciona del grupo constituido por hidrógeno, un acilo opcionalmente sustituido, un aminoácido conectado através de O opcionalmente sustituido,
RA es hidrógeno;R1A se selecciona del grupo constituido por hidrógeno, un acilo opcionalmente sustituido, un aminoácido conectado através de O opcionalmente sustituido,
 Ra1 y Ra2 son independientemente hidrógeno o deuterio;R2A es un azidoalquilo C1-6 ;R3A se selecciona del grupo constituido por OH, -OC(=O)R"A y un aminoácido conectado a través de O opcionalmente sustituido;R4A es halógeno;R5A es hidrógeno;R6A R7A se seleccionan independientemente del rupo constituido por ausente, hidrógeno,o;
Ra1 y Ra2 son independientemente hidrógeno o deuterio;R2A es un azidoalquilo C1-6 ;R3A se selecciona del grupo constituido por OH, -OC(=O)R"A y un aminoácido conectado a través de O opcionalmente sustituido;R4A es halógeno;R5A es hidrógeno;R6A R7A se seleccionan independientemente del rupo constituido por ausente, hidrógeno,o; R8A es un arilo opcionalmente sustituido;R9A es un aminoácido conectado a través de N opcionalmente sustituido o un derivado de tipo éster de un aminoácido conectado a través de N opcionalmente sustituido;r 10A y R11A son independientemente un aminoácido conectado a través de N opcionalmente sustituido o un derivado de tipo éster de un aminoácido conectado a través de N opcionalmente sustituido;R12A, R13A y R14A pueden independientemente estar ausentes o ser hidrógeno;R22A y R23A se seleccionan independientemente del grupo constituido por hidrógeno, un alquilo C1-24 opcionalmente sustituido y un arilo opcionalmente sustituido;R24A se selecciona del grupo constituido por hidrógeno, un alquilo C1-24 opcionalmente sustituido, un arilo opcionalmente sustituido, un -O-alquilo C1-24 opcionalmente sustituido, un -O-arilo opcionalmente sustituido, un -O-heteroarilo opcionalmente sustituido y un -O-heterociclilo monocíclico opcionalmente sustituido;R25A se selecciona independientemente del grupo constituido por hidrógeno, un alquilo C1-24 opcionalmente sustituido y un arilo opcionalmente sustituido;R"A es un alquilo C1-24 opcionalmente sustituido;m es 0 o 1;s es 0;t es 0; yZ1A, Z2A, Z3A y Z4A son independientemente O o S.2. El compuesto de la Reivindicación 1, donde Z1A, Z2A, Z3A y Z4A son cada uno O.3. El compuesto de la Reivindicación 1 o 2, donde R2A es azidometilo.4. El compuesto de cualquiera de las Reivindicaciones 1-3, donde R1A es un acilo no sustituido.5. El compuesto de la Reivindicación 4, donde el acilo es -C(=O)R39A, donde R39A es un alquilo C1-12 no sustituido.
R8A es un arilo opcionalmente sustituido;R9A es un aminoácido conectado a través de N opcionalmente sustituido o un derivado de tipo éster de un aminoácido conectado a través de N opcionalmente sustituido;r 10A y R11A son independientemente un aminoácido conectado a través de N opcionalmente sustituido o un derivado de tipo éster de un aminoácido conectado a través de N opcionalmente sustituido;R12A, R13A y R14A pueden independientemente estar ausentes o ser hidrógeno;R22A y R23A se seleccionan independientemente del grupo constituido por hidrógeno, un alquilo C1-24 opcionalmente sustituido y un arilo opcionalmente sustituido;R24A se selecciona del grupo constituido por hidrógeno, un alquilo C1-24 opcionalmente sustituido, un arilo opcionalmente sustituido, un -O-alquilo C1-24 opcionalmente sustituido, un -O-arilo opcionalmente sustituido, un -O-heteroarilo opcionalmente sustituido y un -O-heterociclilo monocíclico opcionalmente sustituido;R25A se selecciona independientemente del grupo constituido por hidrógeno, un alquilo C1-24 opcionalmente sustituido y un arilo opcionalmente sustituido;R"A es un alquilo C1-24 opcionalmente sustituido;m es 0 o 1;s es 0;t es 0; yZ1A, Z2A, Z3A y Z4A son independientemente O o S.2. El compuesto de la Reivindicación 1, donde Z1A, Z2A, Z3A y Z4A son cada uno O.3. El compuesto de la Reivindicación 1 o 2, donde R2A es azidometilo.4. El compuesto de cualquiera de las Reivindicaciones 1-3, donde R1A es un acilo no sustituido.5. El compuesto de la Reivindicación 4, donde el acilo es -C(=O)R39A, donde R39A es un alquilo C1-12 no sustituido.
 m es 1; y R7A, R12A, R13A y R14A independientemente están ausentes o son hidrógeno.
m es 1; y R7A, R12A, R13A y R14A independientemente están ausentes o son hidrógeno.
 R6A y R7A son cada uno donde R24A es un -O-alquilo C1-24 no sustituido; o donde R1A es
R6A y R7A son cada uno donde R24A es un -O-alquilo C1-24 no sustituido; o donde R1A es
 independientemente éster isopropílico de N-alanina, éster ciclohexílico de N-alanina, éster neopentílico de N-alanina, éster isopropílico de N-valina o éster isopropílico de N-leucina.8. El compuesto de cualquiera de las Reivindicaciones 1-3, donde R1A es H.9. El compuesto de cualquiera de las Reivindicaciones 1-8, donde
independientemente éster isopropílico de N-alanina, éster ciclohexílico de N-alanina, éster neopentílico de N-alanina, éster isopropílico de N-valina o éster isopropílico de N-leucina.8. El compuesto de cualquiera de las Reivindicaciones 1-3, donde R1A es H.9. El compuesto de cualquiera de las Reivindicaciones 1-8, donde 10. El compuesto de cualquiera de las Reivindicaciones 1-9, donde R3A es OH.11. El compuesto de cualquiera de las Reivindicaciones 1-9, donde R3A es -OC(=O)R"A; donde R"A es un alquilo C1-8 no sustituido.12. El compuesto de cualquiera de las Reivindicaciones 1-11, donde R4A es fluoro.13. El compuesto de cualquiera de las Reivindicaciones 1-12, donde Ra1 y Ra2 son cada uno hidrógeno.14. El compuesto de la Reivindicación 1, donde el compuestouna sal farmacéuticamente aceptable de este.
10. El compuesto de cualquiera de las Reivindicaciones 1-9, donde R3A es OH.11. El compuesto de cualquiera de las Reivindicaciones 1-9, donde R3A es -OC(=O)R"A; donde R"A es un alquilo C1-8 no sustituido.12. El compuesto de cualquiera de las Reivindicaciones 1-11, donde R4A es fluoro.13. El compuesto de cualquiera de las Reivindicaciones 1-12, donde Ra1 y Ra2 son cada uno hidrógeno.14. El compuesto de la Reivindicación 1, donde el compuestouna sal farmacéuticamente aceptable de este. 15. El compuesto de la Reivindicación 14, donde
15. El compuesto de la Reivindicación 14, donde 16. El compuesto de la Reivindicación 14, donde R1A es hidrógeno.17. El compuesto de la Reivindicación 14, donde R1A es -C(=O)R39A, donde R39A es un alquilo C1-12 no sustituido. 18. El compuesto de la Reivindicación 14, donde R3A es OH.19. El compuesto de la Reivindicación 14, donde R3A es -OC(=O)R"A, donde R"A es un alquilo C1-8 no sustituido.20. El compuesto de la Reivindicación 1, donde el compuestouna sal farmacéuticamente aceptable de este.
16. El compuesto de la Reivindicación 14, donde R1A es hidrógeno.17. El compuesto de la Reivindicación 14, donde R1A es -C(=O)R39A, donde R39A es un alquilo C1-12 no sustituido. 18. El compuesto de la Reivindicación 14, donde R3A es OH.19. El compuesto de la Reivindicación 14, donde R3A es -OC(=O)R"A, donde R"A es un alquilo C1-8 no sustituido.20. El compuesto de la Reivindicación 1, donde el compuestouna sal farmacéuticamente aceptable de este. 21. Una composición farmacéutica que comprende una cantidad eficaz de un compuesto de cualquiera de las Reivindicaciones 1-20, o una sal farmacéuticamente aceptable de este, y un portador, diluyente o excipiente farmacéuticamente aceptable o una combinación de estos.
21. Una composición farmacéutica que comprende una cantidad eficaz de un compuesto de cualquiera de las Reivindicaciones 1-20, o una sal farmacéuticamente aceptable de este, y un portador, diluyente o excipiente farmacéuticamente aceptable o una combinación de estos.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361839756P | 2013-06-26 | 2013-06-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2825035T3 true ES2825035T3 (es) | 2021-05-14 |
Family
ID=52116188
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14817999.7T Active ES2685591T3 (es) | 2013-06-26 | 2014-06-24 | Nucleósidos, nucleótidos sustituidos y análogos de los mismos |
| ES18174398T Active ES2825035T3 (es) | 2013-06-26 | 2014-06-24 | Nucleósidos, nucleótidos y análogos de estos sustituidos con 4-azidoalquilo |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14817999.7T Active ES2685591T3 (es) | 2013-06-26 | 2014-06-24 | Nucleósidos, nucleótidos sustituidos y análogos de los mismos |
Country Status (22)
| Country | Link |
|---|---|
| US (3) | US9422322B2 (es) |
| EP (2) | EP3424938B1 (es) |
| JP (2) | JP6453322B2 (es) |
| KR (1) | KR20160023711A (es) |
| CN (1) | CN105408341B (es) |
| AP (1) | AP2015008954A0 (es) |
| AU (2) | AU2014302715B2 (es) |
| BR (1) | BR112015031763A2 (es) |
| CA (1) | CA2913210C (es) |
| CL (1) | CL2015003615A1 (es) |
| DK (2) | DK3424938T3 (es) |
| EA (1) | EA201592075A1 (es) |
| ES (2) | ES2685591T3 (es) |
| GE (1) | GEP201706793B (es) |
| MX (1) | MX2015017556A (es) |
| NZ (1) | NZ714415A (es) |
| PE (1) | PE20160205A1 (es) |
| PH (1) | PH12015502799A1 (es) |
| SG (1) | SG11201509585YA (es) |
| TW (1) | TWI655199B (es) |
| UA (1) | UA118760C2 (es) |
| WO (1) | WO2014209983A1 (es) |
Families Citing this family (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2619215B1 (en) | 2010-09-22 | 2018-09-05 | Alios Biopharma, Inc. | Azido nucleosides and nucleotide analogs |
| CA2819041A1 (en) | 2010-12-22 | 2012-06-28 | Alios Biopharma, Inc. | Cyclic nucleotide analogs |
| CA2860234A1 (en) | 2011-12-22 | 2013-06-27 | Alios Biopharma, Inc. | Substituted phosphorothioate nucleotide analogs |
| SG10201913554YA (en) | 2011-12-22 | 2020-03-30 | Alios Biopharma Inc | Substituted nucleosides, nucleotides and analogs thereof |
| USRE48171E1 (en) | 2012-03-21 | 2020-08-25 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| HK1206362A1 (zh) | 2012-03-21 | 2016-01-08 | Alios Biopharma, Inc. | 硫代氨基磷酸酯核苷酸前藥的固體形式 |
| US9441007B2 (en) | 2012-03-21 | 2016-09-13 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| MX2014014323A (es) | 2012-05-25 | 2015-02-12 | Janssen R & D Ireland | Nucleosidos de espirooxetano de uracilo. |
| US9598457B2 (en) | 2012-12-21 | 2017-03-21 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| BR112015014457A2 (pt) | 2012-12-21 | 2017-11-21 | Alios Biopharma Inc | composto ou sal farmaceuticamente aceitável do mesmo e composição farmacêutica e respectivos usos e processos para melhorar ou tratar infecção de hcv, para inibir a atividade da ns5b polimerase do vírus da hepatite c e a replicação de vírus da hepatite c |
| CA2908313A1 (en) | 2013-04-05 | 2014-10-09 | Alios Biopharma, Inc. | Hepatitis c viral infection treatment using a combination of compounds |
| AU2014302711A1 (en) | 2013-06-26 | 2015-12-10 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| MX2015017556A (es) | 2013-06-26 | 2016-05-09 | Alios Biopharma Inc | Nucleosidos sustituidos, nucleotidos y analogos de los mismos. |
| WO2015054465A1 (en) | 2013-10-11 | 2015-04-16 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| MA40031A (fr) * | 2014-06-24 | 2015-12-30 | Alios Biopharma Inc | Nucléosides substitués, nucléotides et analogues de ceux-ci |
| HUE051986T2 (hu) | 2014-06-24 | 2021-04-28 | Janssen Biopharma Inc | Helyettesített nukleozidok, nukleotidek és analógjaik virális fertõzés kezelésére való alkalmazásra |
| PE20170673A1 (es) * | 2014-08-05 | 2017-05-22 | Alios Biopharma Inc | Terapia de combinacion para tratar una paramixovirus |
| WO2016069489A1 (en) | 2014-10-28 | 2016-05-06 | Alios Biopharma, Inc. | Methods of preparing substituted nucleoside analogs |
| CN105254695B (zh) * | 2014-11-10 | 2019-01-18 | 南京曼杰生物科技有限公司 | 核苷氨基磷酸酯衍生物及其应用 |
| MA41213A (fr) | 2014-12-19 | 2017-10-24 | Alios Biopharma Inc | Nucléosides substitués, nucléotides et analogues de ceux-ci |
| MA41441A (fr) | 2014-12-19 | 2017-12-12 | Alios Biopharma Inc | Nucléosides substitués, nucléotides et analogues de ceux-ci |
| US10208045B2 (en) | 2015-03-11 | 2019-02-19 | Alios Biopharma, Inc. | Aza-pyridone compounds and uses thereof |
| CA2984421C (en) * | 2015-05-01 | 2024-04-09 | Cocrystal Pharma, Inc. | Nucleoside analogs for treatment of the flaviviridae family of viruses and cancer |
| DK3324977T3 (da) | 2015-07-22 | 2022-10-17 | Enanta Pharm Inc | Benzodiazepinderivater som rsv-inhibitorer |
| US10202412B2 (en) | 2016-07-08 | 2019-02-12 | Atea Pharmaceuticals, Inc. | β-D-2′-deoxy-2′-substituted-4′-substituted-2-substituted-N6-substituted-6-aminopurinenucleotides for the treatment of paramyxovirus and orthomyxovirus infections |
| BR112020006334A2 (pt) * | 2017-09-29 | 2020-09-24 | Enanta Pharmaceuticals, Inc. | combinação de agentes farmacêuticos como inibidores de rsv |
| CN110938104B (zh) * | 2018-09-21 | 2024-07-12 | 上海迪诺医药科技有限公司 | 环状二核苷酸类似物、其药物组合物及应用 |
| BR112021018335A2 (pt) | 2019-03-18 | 2021-11-23 | Enanta Pharm Inc | Derivados de benzodiazepina como inibidores de rsv |
| US11505558B1 (en) | 2019-10-04 | 2022-11-22 | Enanta Pharmaceuticals, Inc. | Antiviral heterocyclic compounds |
| WO2021216722A1 (en) * | 2020-04-23 | 2021-10-28 | Atea Pharmaceuticals, Inc. | Stereoselective process of manufacture of purine phosphoramidates |
| US11945824B2 (en) | 2020-10-19 | 2024-04-02 | Enanta Pharmaceuticals, Inc. | Heterocyclic compounds as anti-viral agents |
| US12274700B1 (en) | 2020-10-30 | 2025-04-15 | Accencio LLC | Methods of treating symptoms of coronavirus infection with RNA polymerase inhibitors |
| US12612412B2 (en) | 2021-02-26 | 2026-04-28 | Enanta Pharmaceuticals, Inc. | Antiviral heterocyclic compounds |
| CN114773417B (zh) * | 2022-04-06 | 2023-08-22 | 郑州大学 | 一种虫草素磷酸酯及其制备方法与应用 |
| AR129003A1 (es) | 2022-04-07 | 2024-07-03 | Enanta Pharm Inc | Compuestos heterocíclicos antivirales |
| WO2023211997A1 (en) | 2022-04-27 | 2023-11-02 | Enanta Pharmaceuticals, Inc. | Antiviral compounds |
Family Cites Families (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5674998A (en) | 1989-09-15 | 1997-10-07 | Gensia Inc. | C-4' modified adenosine kinase inhibitors |
| US5432272A (en) | 1990-10-09 | 1995-07-11 | Benner; Steven A. | Method for incorporating into a DNA or RNA oligonucleotide using nucleotides bearing heterocyclic bases |
| SK286630B6 (sk) | 2001-01-22 | 2009-02-05 | Merck & Co., Inc. | Nukleozidové deriváty, farmaceutický prostriedok s ich obsahom a ich použitie |
| GB0114286D0 (en) | 2001-06-12 | 2001-08-01 | Hoffmann La Roche | Nucleoside Derivatives |
| US20080261913A1 (en) | 2006-12-28 | 2008-10-23 | Idenix Pharmaceuticals, Inc. | Compounds and pharmaceutical compositions for the treatment of liver disorders |
| US7964580B2 (en) | 2007-03-30 | 2011-06-21 | Pharmasset, Inc. | Nucleoside phosphoramidate prodrugs |
| NZ585784A (en) | 2007-12-13 | 2012-09-28 | Alnylam Pharmaceuticals Inc | siRNAs for the treatment and prevention of respiratory syncytial virus (RSV) infection |
| AU2009329872B2 (en) | 2008-12-23 | 2016-07-07 | Gilead Pharmasset Llc | Synthesis of purine nucleosides |
| JP2012521359A (ja) | 2009-03-20 | 2012-09-13 | アリオス バイオファーマ インク. | 置換されたヌクレオシドアナログおよびヌクレオチドアナログ |
| NZ607996A (en) | 2010-09-22 | 2014-07-25 | Alios Biopharma Inc | Substituted nucleotide analogs |
| EP2619215B1 (en) | 2010-09-22 | 2018-09-05 | Alios Biopharma, Inc. | Azido nucleosides and nucleotide analogs |
| WO2012040126A1 (en) | 2010-09-22 | 2012-03-29 | Alios Biopharma, Inc. | Substituted nucleotide analogs |
| CA2819041A1 (en) | 2010-12-22 | 2012-06-28 | Alios Biopharma, Inc. | Cyclic nucleotide analogs |
| CA2860234A1 (en) | 2011-12-22 | 2013-06-27 | Alios Biopharma, Inc. | Substituted phosphorothioate nucleotide analogs |
| SG10201913554YA (en) | 2011-12-22 | 2020-03-30 | Alios Biopharma Inc | Substituted nucleosides, nucleotides and analogs thereof |
| WO2013142159A1 (en) | 2012-03-21 | 2013-09-26 | Alios Biopharma, Inc. | Pharmaceutical combinations comprising a thionucleotide analog |
| GEP201706721B (en) | 2012-03-21 | 2017-08-25 | Alios Biopharma Inc | Substituted nucleosides, nucleotides and analogs thereof |
| US8846896B2 (en) | 2012-03-21 | 2014-09-30 | Alios Biopharma, Inc. | Methods of preparing substituted nucleotide analogs |
| HK1206362A1 (zh) | 2012-03-21 | 2016-01-08 | Alios Biopharma, Inc. | 硫代氨基磷酸酯核苷酸前藥的固體形式 |
| US9441007B2 (en) | 2012-03-21 | 2016-09-13 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| WO2013142157A1 (en) | 2012-03-22 | 2013-09-26 | Alios Biopharma, Inc. | Pharmaceutical combinations comprising a thionucleotide analog |
| BR112015014457A2 (pt) | 2012-12-21 | 2017-11-21 | Alios Biopharma Inc | composto ou sal farmaceuticamente aceitável do mesmo e composição farmacêutica e respectivos usos e processos para melhorar ou tratar infecção de hcv, para inibir a atividade da ns5b polimerase do vírus da hepatite c e a replicação de vírus da hepatite c |
| US9598457B2 (en) | 2012-12-21 | 2017-03-21 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| WO2014134251A1 (en) | 2013-02-28 | 2014-09-04 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions |
| WO2014164533A1 (en) | 2013-03-11 | 2014-10-09 | Vertex Pharmaceuticals Incorporated | Methods of stereoselective synthesis of substituted nucleoside analogs |
| CA2908313A1 (en) | 2013-04-05 | 2014-10-09 | Alios Biopharma, Inc. | Hepatitis c viral infection treatment using a combination of compounds |
| MX2015017556A (es) | 2013-06-26 | 2016-05-09 | Alios Biopharma Inc | Nucleosidos sustituidos, nucleotidos y analogos de los mismos. |
| AU2014302711A1 (en) | 2013-06-26 | 2015-12-10 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| WO2015054465A1 (en) | 2013-10-11 | 2015-04-16 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| AU2015280245A1 (en) | 2014-06-24 | 2017-01-05 | Alios Biopharma, Inc. | Methods of preparing substituted nucleotide analogs |
| MA40031A (fr) | 2014-06-24 | 2015-12-30 | Alios Biopharma Inc | Nucléosides substitués, nucléotides et analogues de ceux-ci |
| HUE051986T2 (hu) | 2014-06-24 | 2021-04-28 | Janssen Biopharma Inc | Helyettesített nukleozidok, nukleotidek és analógjaik virális fertõzés kezelésére való alkalmazásra |
| CA2955604A1 (en) | 2014-07-22 | 2016-01-28 | Alios Biopharma, Inc. | Methods for treating paramyxoviruses |
| PE20170673A1 (es) | 2014-08-05 | 2017-05-22 | Alios Biopharma Inc | Terapia de combinacion para tratar una paramixovirus |
| WO2016069489A1 (en) | 2014-10-28 | 2016-05-06 | Alios Biopharma, Inc. | Methods of preparing substituted nucleoside analogs |
| MA41441A (fr) | 2014-12-19 | 2017-12-12 | Alios Biopharma Inc | Nucléosides substitués, nucléotides et analogues de ceux-ci |
| MA41213A (fr) | 2014-12-19 | 2017-10-24 | Alios Biopharma Inc | Nucléosides substitués, nucléotides et analogues de ceux-ci |
| TW201811339A (zh) | 2016-08-12 | 2018-04-01 | 美商艾洛斯生物製藥公司 | 經取代之核苷、核苷酸及其類似物 |
| US20180117042A1 (en) | 2016-10-27 | 2018-05-03 | Alios Biopharma, Inc. | Methods for treating respiratory syncytial virus infection |
| CA3075950A1 (en) | 2017-09-18 | 2019-03-21 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
-
2014
- 2014-06-24 MX MX2015017556A patent/MX2015017556A/es unknown
- 2014-06-24 TW TW103121826A patent/TWI655199B/zh not_active IP Right Cessation
- 2014-06-24 DK DK18174398.0T patent/DK3424938T3/da active
- 2014-06-24 EP EP18174398.0A patent/EP3424938B1/en active Active
- 2014-06-24 AP AP2015008954A patent/AP2015008954A0/xx unknown
- 2014-06-24 WO PCT/US2014/043841 patent/WO2014209983A1/en not_active Ceased
- 2014-06-24 SG SG11201509585YA patent/SG11201509585YA/en unknown
- 2014-06-24 UA UAA201511862A patent/UA118760C2/uk unknown
- 2014-06-24 ES ES14817999.7T patent/ES2685591T3/es active Active
- 2014-06-24 JP JP2016523850A patent/JP6453322B2/ja not_active Expired - Fee Related
- 2014-06-24 DK DK14817999.7T patent/DK3013843T3/en active
- 2014-06-24 CA CA2913210A patent/CA2913210C/en active Active
- 2014-06-24 KR KR1020157036462A patent/KR20160023711A/ko not_active Withdrawn
- 2014-06-24 NZ NZ714415A patent/NZ714415A/en not_active IP Right Cessation
- 2014-06-24 EA EA201592075A patent/EA201592075A1/ru unknown
- 2014-06-24 GE GEAP201414043A patent/GEP201706793B/en unknown
- 2014-06-24 PE PE2015002648A patent/PE20160205A1/es not_active Application Discontinuation
- 2014-06-24 AU AU2014302715A patent/AU2014302715B2/en not_active Ceased
- 2014-06-24 US US14/312,990 patent/US9422322B2/en not_active Expired - Fee Related
- 2014-06-24 EP EP14817999.7A patent/EP3013843B1/en active Active
- 2014-06-24 CN CN201480038065.3A patent/CN105408341B/zh not_active Expired - Fee Related
- 2014-06-24 BR BR112015031763A patent/BR112015031763A2/pt not_active Application Discontinuation
- 2014-06-24 ES ES18174398T patent/ES2825035T3/es active Active
-
2015
- 2015-12-14 CL CL2015003615A patent/CL2015003615A1/es unknown
- 2015-12-16 PH PH12015502799A patent/PH12015502799A1/en unknown
-
2016
- 2016-07-20 US US15/215,434 patent/US9932363B2/en active Active
-
2018
- 2018-02-13 US US15/895,872 patent/US10696708B2/en not_active Expired - Fee Related
- 2018-12-12 JP JP2018232331A patent/JP2019059766A/ja not_active Withdrawn
-
2019
- 2019-03-06 AU AU2019201536A patent/AU2019201536A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2825035T3 (es) | Nucleósidos, nucleótidos y análogos de estos sustituidos con 4-azidoalquilo | |
| ES2701020T3 (es) | Nucleósidos azido y análogos nucleotídicos | |
| DK2794627T3 (en) | SUBSTITUTED NUCLEOSIDES, NUCLEOTIDES AND ANALOGUES THEREOF | |
| ES2710506T3 (es) | Nucleósidos sustituidos, nucleótidos y sus análogos | |
| EP3177299A1 (en) | Combination therapy for treating a paramyxovirus | |
| JP2017538722A (ja) | 置換ヌクレオシド、ヌクレオチド、及びそのアナログ | |
| CA2819041A1 (en) | Cyclic nucleotide analogs | |
| HK40000768A (en) | 4'-azidoalkyl-substituted nucleosides, nucleotides and analogs thereof | |
| HK1222178B (en) | Substituted nucleosides, nucleotides and analogs thereof | |
| OA17648A (en) | Substituted nucleosides, nucleotides and analogs thereof. |