ES2818652T3 - Sal de derivado de piridinilaminopirimidina, método de preparación de la misma y aplicación de la misma - Google Patents
Sal de derivado de piridinilaminopirimidina, método de preparación de la misma y aplicación de la misma Download PDFInfo
- Publication number
- ES2818652T3 ES2818652T3 ES17762392T ES17762392T ES2818652T3 ES 2818652 T3 ES2818652 T3 ES 2818652T3 ES 17762392 T ES17762392 T ES 17762392T ES 17762392 T ES17762392 T ES 17762392T ES 2818652 T3 ES2818652 T3 ES 2818652T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- formula
- mesylate salt
- present
- salt
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Una sal mesilato del compuesto de fórmula (I), **(Ver fórmula)**
Description
DESCRIPCIÓN
Sal de derivado de piridinilaminopirimidina, método de preparación de la misma y aplicación de la misma Campo técnico
La presente invención se refiere a una sal de derivado de piridinilaminopirimidina. En particular, la presente invención se refiere a la sal mesilato de N-{2-{[2-(dimetilamino)etil](metil)amino}-6-(2,2,2-trifluoroetoxi)-5-{[4-(1 -metil-1 H-indol-3-il)pirimidin-2-il]amino}piridin-3-il}acrilamida, el método de preparación de la misma, una composición farmacéutica que contiene dicha sal, y el uso de dicha sal en el tratamiento de enfermedades mediadas por mutaciones de activación y resistencia de EGFR, en particular cánceres, en mamíferos, en particular en seres humanos.
Antecedentes
Los receptores de factores de crecimiento de células epidérmicas (EGFR) se identifican como un factor impulsor significativo en el proceso de crecimiento y proliferación celular. La familia de receptores de factores de crecimiento de células epidérmicas está compuesta por EGFR (Erb-B1), Erb-B2 (HER-2/neu), Erb-B3 y Erb-B4. Los receptores de factores de crecimiento de células epidérmicas están asociados con el proceso de la mayoría de los cánceres, tales como el cáncer de pulmón, el cáncer de colon, el cáncer de mama y similares. Se ha demostrado que la sobreexpresión y mutación de EGFR es el principal factor de riesgo de un cáncer de mama con mal pronóstico.
La investigación actual de vanguardia está centrada en un inhibidor de EGFR irreversible de tercera generación. La solicitud de patente CN201410365911.4 describe el siguiente compuesto de fórmula (I), compuesto que tiene una actividad de inhibición sustancialmente más alta para la mutación activadora de EGFR (tal como la mutación activadora de deleción del exón 19, o la mutación activadora de L858R) y la mutación de resistencia T790M que la actividad de inhibición del EGFR de tipo natural (WT EGFR), con una buena selectividad, un efecto secundario de toxicidad relativamente baja y una buena seguridad.

Compendio de la invención
El problema técnico a resolver por la presente invención es proporcionar la sal mesilato del compuesto de fórmula (I), el método de preparación de la misma, una composición farmacéutica que contenga dicha sal, y el uso de dicha sal en el tratamiento de enfermedades mediadas por mutaciones activantes y de resistencia de EGFR, en particular cánceres, en mamíferos, en particular en seres humanos.
La presente invención proporciona la sal mesilato del compuesto de fórmula (I).

La presente invención proporciona el método de preparación de la sal mesilato del compuesto de fórmula (I).
La presente invención proporciona además un método para preparar la sal mesilato del compuesto de fórmula (I), que comprende hacer reaccionar directamente el compuesto de fórmula (I) con ácido metanosulfónico en un disolvente. La presente invención proporciona una composición farmacéutica que comprende la sal mesilato del compuesto de fórmula (I) y un portador farmacéuticamente aceptable.
La presente invención proporciona además una composición farmacéutica, que comprende la sal mesilato del compuesto de fórmula (I) y un portador, excipiente o diluyente farmacéuticamente aceptables.
La presente invención proporciona la sal mesilato del compuesto de fórmula (I) para su uso como medicamento antitumoral.
La presente invención también proporciona el uso de la sal mesilato del compuesto de fórmula (I) en la fabricación de un medicamento para tratar enfermedades mediadas por mutaciones activadoras y de resistencia de EGFR, en particular cánceres en mamíferos, en particular en seres humanos.
La presente invención también proporciona el uso de la sal mesilato del compuesto de fórmula (I) en la fabricación de un medicamento para tratar cánceres.
La presente invención también proporciona el uso de la sal mesilato del compuesto de fórmula (I) en el tratamiento de enfermedades mediadas por mutaciones activadoras y de resistencia de EGFR, en particular cánceres en mamíferos, en particular en seres humanos.
La presente invención también proporciona el uso de la composición farmacéutica que contiene la sal mesilato del compuesto de fórmula (I) en la fabricación de un medicamento para tratar enfermedades mediadas por mutaciones activadoras y de resistencia de EGFR, en particular cánceres en mamíferos, en particular en seres humanos.
La presente invención también proporciona el uso de la composición farmacéutica que contiene la sal mesilato del compuesto de fórmula (I) en la fabricación de un medicamento para tratar el cáncer.
La presente invención también proporciona la sal mesilato del compuesto de fórmula (I), o la composición farmacéutica que comprende una cantidad terapéuticamente eficaz de la sal mesilato del compuesto de fórmula (I) y un portador, excipiente o diluyente farmacéuticamente aceptables para su uso en un método para tratar enfermedades mediadas por mutaciones activadoras y de resistencia de EGFR, en particular cánceres en mamíferos, en particular seres humanos, que incluye administrar a un sujeto la sal mesilato del compuesto de fórmula (I), o la composición farmacéutica que comprende una cantidad terapéuticamente eficaz de la sal mesilato del compuesto de fórmula (I) y un portador, excipiente o diluyente farmacéuticamente aceptables.
La presente invención también proporciona la sal mesilato del compuesto de fórmula (I), o la composición farmacéutica que comprende una cantidad terapéuticamente eficaz de la sal mesilato del compuesto de fórmula (I) y un portador, excipiente o diluyente farmacéuticamente aceptables para su uso en un método para tratar el cáncer, que incluye administrar a un sujeto la sal mesilato del compuesto de fórmula (I), o la composición farmacéutica que comprende una cantidad terapéuticamente eficaz de la sal mesilato del compuesto de fórmula (I) y un portador, excipiente o diluyente farmacéuticamente aceptables.
El cáncer tal como se menciona en la presente invención incluye, pero no se limita a, por ejemplo, cáncer de pulmón, cáncer de ovario, cáncer de cuello uterino, cáncer de mama, cáncer de estómago, cáncer colorrectal, cáncer de páncreas, glioma, glioblastoma, melanoma, cáncer de próstata, leucemia, linfoma, linfoma no Hodgkin, cáncer hepatocelular, tumor del estroma gastrointestinal (GIST), cáncer de tiroides, colangiocarcinoma, cáncer de endometrio, cáncer renal, linfoma anaplásico de células grandes, leucemia mieloide aguda (AML), mieloma múltiple, y mesotelioma. En particular, la presente invención tiene un buen efecto sobre los cánceres que tienen una mutación del receptor del factor de crecimiento epidérmico, mutación que sustituye una treonina por una metionina en la posición 790 (EGFR T790M). Por ejemplo, la sal mesilato del compuesto de fórmula (I) de la presente invención puede usarse como fármaco para tratar el cáncer de pulmón de células no pequeñas (EGFR T790M).
La presente invención proporciona un método para preparar el compuesto de fórmula (I), método que se refiere al método tal como se describe en el ejemplo 1 de la solicitud de patente N.° CN201410365911.4, en donde la preparación del intermedio 1c y del intermedio 2a se refiere directamente a los ejemplos de la solicitud de patente N.° CN201410365911.4.

El intermedio 1c y el intermedio 2a se someten a una reacción de sustitución o acoplamiento para producir el compuesto (II). El grupo nitro del compuesto (II) se reduce para producir el compuesto (III). El compuesto (III) y el
cloruro de acriloílo se someten a acilación para producir el compuesto (I). La reacción de sustitución o acoplamiento del intermedio 1c y el intermedio 2a comprende aquellas reacciones que pueden llevarse a cabo bajo la catálisis por catalizadores de metales de transición, en donde dichos catalizadores de metales de transición incluyen pero no se limitan a tris(dibencilidenacetona)dipaladio/4,5-bis(difenilfosfino)-9,9-dimetilxanteno. La reducción del grupo nitro se lleva a cabo con los catalizadores de reducción convencionales bien conocidos en la técnica, que incluyen pero no se limitan a polvo de hierro, polvo de zinc, sulfuro de sodio, H2/dióxido de platino.
El método de preparación de la sal mesilato del compuesto de fórmula (I) según la presente invención es el siguiente:

En un disolvente, el compuesto de fórmula (I) y el ácido metanosulfónico se hacen reaccionar directamente de manera que se forma una sal, para proporcionar la sal mesilato del compuesto de fórmula (I). Dicho disolvente incluye, pero no se limita a, un disolvente mixto de acetona y agua.
La sal mesilato del compuesto de fórmula (I) según la presente invención puede administrarse a un mamífero, que incluye ser humano, y puede administrarse por vía oral, vía rectal, vía parenteral (intravenosa, intramuscular o subcutánea), vía tópica (tal como en forma de polvos, pomadas o gotas), o por vía intratumoral.
La sal mesilato del compuesto de fórmula (I) según la presente invención puede administrarse en una dosificación de aproximadamente 0,05-50 mg/kg de peso corporal/día, por ejemplo 0,1-45 mg/kg de peso corporal/día, en un ejemplo adicional 0,5-35 mg/kg de peso corporal/día.
Las sales mesilato del compuesto de fórmula (I) de la presente invención pueden formularse en formas farmacéuticas sólidas para su administración oral, que incluyen, pero no se limitan a, cápsulas, comprimidos, píldoras, polvos, gránulos o similares. En estas formas farmacéuticas sólidas, las sales mesilato del compuesto de fórmula (I) de la presente invención como principios activos se mezclan con al menos un excipiente (o portador) inerte convencional, tal como citrato de sodio o fosfato dicálcico, o se mezclan con los siguientes componentes: (1) cargas o extensores, tales como almidón, lactosa, sacarosa, glucosa, manitol y ácido silícico o similares; (2) aglutinantes, tales como hidroximetilcelulosa, aginato, gelatina, polivinilpirrolidona, sacarosa y goma arábiga o similares; (3) humectantes, tales como glicerol o similares; (4) agentes disgregantes, tales como agar, carbonato de calcio, almidón de patata o almidón de tapioca, ácido algínico, determinado silicato compuesto, y carbonato de sodio o similares; (5) agentes retardadores, tales como cera de parafina o similares; (6) potenciadores de la absorción, tales como compuestos de amonio cuaternario o similares; (7) agentes humectantes, tales como cetanol y monoestearato de glicerilo o similares; (8) absorbentes, tales como caolín o similares; y (9) lubricantes, tales como talco, estearato de calcio, estearato de magnesio, polietilenglicol sólido, dodecilsulfato de sodio o similares, o mezclas de los mismos. Las cápsulas, comprimidos y píldoras también pueden comprender tampones.
Dichas formas farmacéuticas sólidas tales como comprimidos, píldoras de azúcar, cápsulas, píldoras y gránulos también pueden recubrirse o microencapsularse mediante recubrimientos y materiales de cubierta tales como recubrimientos entéricos y otros materiales bien conocidos en la técnica. Pueden comprender opacificantes, y la liberación de principios activos en estas composiciones puede llevarse a cabo en una determinada parte del tubo digestivo de manera retardada. Los ejemplos de componentes de incrustación que pueden adoptarse son sustancias a base de polímeros y a base de cera. Si es necesario, también pueden formularse principios activos en forma de microcápsulas con uno o más de los excipientes anteriores.
Las sales mesilato del compuesto de fórmula (I) de la presente invención pueden formularse en formas farmacéuticas líquidas para su administración oral, que incluyen pero no se limitan a, emulsiones, disoluciones, suspensiones, jarabes y tinturas farmacéuticamente aceptables o similares. Además de la sal mesilato del compuesto de fórmula (I) como principios activos, las formas farmacéuticas líquidas pueden comprender diluyentes inertes usados habitualmente en la técnica, tales como agua y otros disolventes, solubilizantes y emulsionantes, tales como etanol, isopropanol, carbonato de etilo, acetato de etilo, propilenglicol, 1,3-butanodiol, dimetilformamida, y aceites, especialmente aceite de semilla de algodón, aceite de cacahuete, aceite de germen de maíz, aceite de oliva, aceite de ricino y aceite de sésamo y similares, o mezclas de los mismos, y similares . Además de estos diluyentes inertes, las formas farmacéuticas líquidas de la presente invención también pueden comprender adyuvantes convencionales, tales como agentes humectantes, emulsionantes y agentes de suspensión, agentes edulcorantes, agentes aromatizantes y fragancias y similares.
Dichos agentes de suspensión incluyen, tales como alcohol isoestearílico etoxilado, polioxietilensorbitol y éster de sorbitán, celulosa microcristalina, metóxido de aluminio y agar y similares, o mezclas de los mismos.
Las sales mesilato del compuesto de fórmula (I) de la presente invención pueden formularse en formas farmacéuticas para su inyección parenteral, que incluyen pero no se limitan a, disoluciones, dispersiones, suspensiones o emulsiones acuosas o no acuosas estériles fisiológicamente aceptables, y polvo estéril para volver a disolver en disoluciones o dispersiones inyectables estériles. Los portadores, diluyentes, disolventes o excipientes adecuados incluyen agua, etanol, alcohol polihidroxilado y mezclas adecuadas de los mismos.
El compuesto de la presente invención o una sal farmacéuticamente aceptable del mismo también puede formularse en formas farmacéuticas para su administración tópica, que incluyen pero no se limitan a, pomadas, polvos, supositorios, gotas, propulsores e inhalantes y similares. Las sales mesilato del compuesto de fórmula (I) de la presente invención como principios activos se mezclan junto con portadores fisiológicamente aceptables y de manera opcional conservantes, tampones o, si es necesario, propulsores, en condiciones estériles.
La presente invención también proporciona una composición farmacéutica que contiene la sal mesilato del compuesto de fórmula (I) de la presente invención, y un portador, excipiente o diluyente farmacéuticamente aceptables. Cuando se prepara la composición farmacéutica, la sal mesilato del compuesto de fórmula (I) de la presente invención se mezcla generalmente con el portador, excipiente o diluyente farmacéuticamente aceptables.
Mediante métodos de preparación convencionales, la composición de la presente invención puede formularse en preparaciones farmacéuticas convencionales, tales como comprimidos, píldoras, cápsulas, polvos, gránulos, emulsiones, suspensiones, dispersiones, disoluciones, jarabes, elixires, pomadas, gotas, supositorios, inhalantes, propulsores y similares.
La sal mesilato del compuesto de fórmula (I) según la presente invención puede administrarse sola o en combinación con otros agentes terapéuticos farmacéuticamente aceptables, especialmente con otros fármacos antitumorales. Los agentes terapéuticos incluyen, pero no se limitan a, fármacos antitumorales que ejercen un efecto sobre la estructura química del ADN, tal como cisplatino, fármacos antitumorales que afectan a la síntesis de ácido nucleico, tal como metotrexato (MTX), 5-fluorouracilo (5FU) y similares, fármacos antitumorales que afectan a la traducción de ácidos nucleicos, tales como adriamicina, epirrubicina, aclacinomicina, mitramicina y similares, fármacos antitumorales que ejercen un efecto sobre la síntesis de tubulina, tales como paclitaxel, vinorrelbina y similares, inhibidores de aromatasa tales como aminoglutetimida, lentaron, letrozol, anastrozol y similares, inhibidores de la ruta de señalización celular tales como inhibidores del receptor del factor de crecimiento epidérmico imatinib, gefitinib, erlotinib, y similares. Cada agente terapéutico a combinar se puede administrar de manera simultánea o secuencial, y puede administrarse en una formulación unitaria o en formulaciones por separado. Tal combinación incluye no solo la combinación del compuesto de la presente invención con otro principio activo, sino también la combinación del compuesto de la presente invención con dos o más de otros principios activos.
El método de determinación de la biodisponibilidad absoluta de la administración intragástrica de las sales mesilato del compuesto de fórmula (I) de la presente invención es el siguiente:
Para administración intravenosa: Se agrupan aleatoriamente ratas SD sanas. El compuesto sometido a prueba se administra en una determinada dosis D a través de la administración intravenosa. Las muestras de sangre se recogen a través del plexo venoso retrobulbar antes de la administración y 5 min, 15 min, 0,5 h, 1,0 h, 2,0 h, 4,0 h, 8,0 h, 12 h y 24 h después de la administración, y se separan para dar plasmas. La concentración del fármaco en plasma se determina con el método de cromatografía líquida-espectrometría de masas en tándem para obtener una curva de concentración de fármaco-tiempo.
Para administración intragástrica: Se agrupan aleatoriamente ratas SD sanas. La sustancia sometida a prueba se administra en una determinada dosis D a través de la administración intragástrica. Las muestras de sangre intravenosas se recogen a través del plexo venoso retrobulbar de la rata antes de la administración y 0,5, 1,0, 2,0, 4,0, 6,0, 8,0, 10, 12 y 24 h después de la administración, y se separan para dar plasmas. La concentración del fármaco en plasma se determina con el método de cromatografía líquida-espectrometría de masas en tándem para obtener una curva de concentración de fármaco-tiempo.
Después de la calibración de la dosificación, la biodisponibilidad absoluta F se calcula mediante el área bajo la curva de concentración de fármaco-tiempo (AUC0-t). La ecuación de cálculo es la siguiente:
F — (A U C ¡ntragástricaXD ¡ntravenosa ) /(A U C ¡n tra v e n o s a X D ¡ ntragástr¡ca)X 100 % .
La farmacodinámica de las sales mesilato del compuesto de fórmula (I) de la presente invención en términos de inhibición del crecimiento de tumores trasplantados en animales puede someterse a ensayo mediante métodos convencionales. Un método de evaluación preferible tiene como objetivo el efecto inhibidor sobre el crecimiento de tumores trasplantados subcutáneamente de ratones desnudos portadores de H1975 con cáncer de pulmón humano. El método experimental es el siguiente: se inocula cepa celular H1975 de cáncer de pulmón humano (5 x 106/cada ratón) a ratones desnudos por vía subcutánea en el lado derecho de el lomo de los mismos. Después de que los tumores crezcan hasta 100-150 mm3 en promedio, los animales se dividen en grupos aleatoriamente según el tamaño del tumor y el peso del animal. Los compuestos sometidos a prueba se administran por administración intragástrica en determinadas dosificaciones, y a los grupos de control de disolvente se les administra la misma cantidad de
disolvente por administración intragástrica, en donde la administración se realiza una vez al día durante un período continuo de 21 días. Durante todo el procedimiento experimental, se mide el peso del animal y el tamaño del tumor dos veces a la semana, para observar si se produce o no la reacción tóxica.
El volumen tumoral se calcula de la siguiente manera: Volumen tumoral (mm3) = 0,5 x diámetro mayor del tumor x diámetro menor del tumor x diámetro menor del tumor
Los efectos técnicos beneficiosos producidos por la presente invención comprenden:
La sal mesilato del compuesto de fórmula (I) según la presente invención tiene una excelente biodisponibilidad en animales.
La sal mesilato del compuesto de fórmula (I) según la presente invención puede inhibir notablemente el crecimiento de tumores trasplantados en animales y mostrar una buena seguridad.
Breve descripción de los dibujos
La figura 1 es la curva de volumen tumoral para tumores trasplantados por vía subcutánea de ratones desnudos portadores de H1975 con cáncer de pulmón humano a la dosificación de administración de 25 mg/kg de la sal mesilato del compuesto de fórmula (I) preparada según el ejemplo 2 y la sustancia del ejemplo comparativo 2 (es decir, el ejemplo 16 de la solicitud de patente CN201410365911.4).
La figura 2 es la curva de peso corporal para ratones desnudos portadores de H1975 con cáncer de pulmón humano a la dosificación de administración de 25 mg/kg de la sal mesilato del compuesto de fórmula (I) preparada según el ejemplo 2 y la sustancia del ejemplo comparativo 2 (es decir, el ejemplo 16 de solicitud de patente CN201410365911.4).
La presente invención se ilustrará adicionalmente a continuación en el presente documento con relación a ejemplos específicos. Debe entenderse que estos ejemplos solo se utilizan para ilustrar la presente invención a modo de ejemplos sin limitar el alcance de la misma. En los siguientes ejemplos, los métodos experimentales sin especificar condiciones se realizan generalmente según condiciones convencionales o basándose en las condiciones recomendadas por el fabricante. Las partes y los porcentajes son las partes y los porcentajes en peso respectivamente, a menos que se especifique de otro modo.
Descripción detallada de la invención
I. Ejemplo de preparación
Ejemplo 1:
N-{2-{[2-(dimetilamino)etil](metil)amino}-6-(2,2,2-trifluoroetoxi)-5-{[4-(1-metil-1 H-indol-3-il)pirimidin-2-il]amino}piridin-3-il}acrilamida
Intermedio 1c: N2-metil-N2-[2-(dimetilamino)etil]-6-(2,2,2-trifluoroetoxi)-3-nitropiridin-2,5-diamina, refiriéndose el método de preparación del mismo al ejemplo descrito en la solicitud de patente N.° CN201410365911.4

(Intermedio 1c)
Intermedio 2a: 3-(2-cloropirimidin-4-il)-1 -metil-1 H-indol, refiriéndose el método de preparación del mismo al ejemplo descrito en la solicitud de patente N.° CN201410365911.4
Compuesto (II): Síntesis de N12 *-metil-N2-[2-(dimetilamino)etil]-6-(2,2,2-trifluoroetoxi)-N5-[4-(1-metil-1H-indol-3-il)pirimidin-2-il]-3-nitropiridin-2,5-diamina

A un matraz de fondo redondo se le añadió 3-(2-cloropirimidin-4-il)-1 -metil-1 H-indol (73 mg, 0,3 mmol), N2-metil-N2-[2-(dimetilamino)etil]-6-(2,2,2-trifluoroetoxi)-3-nitropiridin-2,5-diamina (100 mg, 0,3 mmol), tris(dibencilidenacetona) dipaladio (14 mg, 0,015 mmol) ), 4,5-bis(difenilfosfino)-9,9-dimetilxanteno (14 mg, 0,03 mmol), fosfato de potasio (127 mg, 0,6 mmol) y 8 ml de dioxano. La mezcla se hizo reaccionar bajo la protección de gas argón a 95°C durante 5 horas, y se filtró. El filtrado se evaporó a vacío hasta sequedad, y se purificó mediante cromatografía en columna de gel de sílice (diclorometano:metanol = 20:1) para dar 140 mg del producto con un rendimiento del 86%. EM m/z: 545 [M+1].
Compuesto (III): Síntesis de N2-metil-N2-[2-(dimetilamino)etil]-6-(2,2,2-trifluoroetoxi)-N5-[4-(1-metil-1H-indol-3-il)pirimidin-2-il]piridin-2,3,5-triamina

A un matraz de fondo redondo se le añadió N2-metil-N2-[2-(dimetilamino)etil]-6-(2,2,2-trifluoroetoxi)-N5-[4-(1-metil-1H-indol-3-il)pirimidin-2-il]-3-nitropiridin-2,5-diamina (150 mg, 0,27 mmol), dióxido de platino (60 mg) y 10 ml de metanol. Luego se introdujo hidrógeno. La mezcla se hizo reaccionar a temperatura ambiente durante 1 h, se filtró, y se separó con una placa preparativa (diclorometano:metanol = 10:1) para dar 80 mg del compuesto objetivo con un rendimiento del 56%. EM m/z: 515 [M+1].
Compuesto (I): Síntesis de N-{2-{[2-(dimetilamino)etil](metil)amino}-6-(2,2,2-trifluoroetoxi)-5-{[4-(1-metil-1H-indol-3-il)pirimidin-2-il]amino}piridin-3-il}acrilamida

A un matraz de fondo redondo se le añadió N2-metil-N2-[2-(dimetilamino)etil]-6-(2,2,2-trifluoroetoxi)-N5-[4-(1-metil-1H-indol-3-il)pirimidin-2-il]piridin-2,3,5-triamina (80 mg, 0,16 mmol) y 5 ml de diclorometano, y la mezcla se enfrió en un baño de agua-hielo. Se le añadió 0,5 N de una disolución de cloruro de acriloílo en diclorometano (0,5 ml, 0,25 mmol). La mezcla resultante se hizo reaccionar en un baño de agua-hielo durante 1,5 horas, se diluyó con 50 ml de acetato de etilo, y se lavó con una disolución saturada de bicarbonato de sodio. La fase orgánica se secó con sulfato de sodio anhidro, y se filtró. El filtrado se concentró a vacío, y se purificó separando con una placa preparativa (diclorometano: metanol = 10:1) para dar 20 mg del producto objetivo con un rendimiento del 23%. EM m/z: 569 [M+1].
1H RMN (400 MHz, DMSO-d6) 510,41 (s, 1H), 10,27 (s, 1H), 8,68 (s, 1H), 8,44 (s, 1H), 8,28 (t, J = 8,5 Hz, 2H) , 8,18 (s, 1H), 7,52 (d, J = 8,0 Hz, 1H), 7,29-7,14 (m, 3H), 6,98 (s, 1H), 6,28 (d, J = 17,1Hz, 1H), 5,76 (d, J = 10,4 Hz, 1H), 5,00 (q, J = 9,0 Hz, 2H), 3,89 (s, 3H), 3,61 (s, 2H), 3,28 (s, 2H), 2,80 (s, 3H), 2,73 (s, 6H).
Ejemplo 2: Síntesis de la sal mesilato de N-{2-{[2-(dimetilamino)etil](metil)amino}-6-(2,2,2-trifluoroetoxi)-5-{[4-(1-metil-1 H-indol-3-il)pirimidin-2-il]amino}piridin-3-il}acrilamida

A un matraz de tres bocas se le añadió N-{2-{[2-(dimetilamino)etil](metil)amino}-6-(2,2,2-trifluoroetoxi)-5-{[4-(1-metil-1H-indol-3-il)pirimidin-2-il]amino}piridin-3-il}acrilamida (1 g, 1,76 mmol), acetona (35 ml), agua (7 ml) y ácido metanosulfónico (169 mg). La mezcla se calentó a 50°C hasta que se disolvió, y se evaporó a vacío hasta sequedad. Se le añadió acetonitrilo, y la mezcla resultante se evaporó a vacío hasta sequedad. Al residuo se le añadió acetona. La mezcla resultante se trató ultrasónicamente, y se filtró. La torta de filtración se secó para dar 685 mg del producto objetivo con un rendimiento del 59%.
1H RMN (400 MHz, DMSO-d6) 59,80 (s, 1H), 9,23 (s, 1 H), 8,53 (s, 1H), 8,42 (s, 1H), 8,30 (d, J = 5,4 Hz, 2H) , 8,23 (s, 1 H), 7,52 (d, J = 8,2Hz, 1H), 7,25 (t, J = 7,2Hz, 1 H), 7,22 (d, J = 8,0Hz, 1H), 7,15 (t, J = 7,4 Hz, 1 H), 6,70 (dd, J = 17,0, 10,2Hz, 1 H), 6,34 (dd, J = 17,0, 1,7 Hz, 1 H), 5,83 (dd, J = 10,3, 1,6 Hz, 1H), 5,02 (q, J = 9,1 Hz, 2H), 3,88 (s, 3H), 3,65 (t, J = 6,0 Hz, 2H), 3,33 (t, J = 6,0 Hz, 2H), 2,86 (s, 6H), 2,81 (s, 3H), 2,44 (s, 3H).
II. Ejemplos de prueba de actividad
Ejemplo de prueba 1: experimentos de absorción de fármaco en ratas SD (ratas Sprague Dawley)
Para administración intravenosa: Se asignaron aleatoriamente a 4 grupos 16 ratas SD sanas (mitad machos y mitad hembras) con 200-280 g de peso corporal, proporcionadas por Shanghai Sippr-BK laboratory animal Co. Ltd.. Las sustancias de los anteriores ejemplo 1, el ejemplo 2, ejemplo comparativo 1 y ejemplo comparativo 2 se administraron por vía intravenosa en la dosificación que se muestra en la siguiente tabla. Se recogieron 0,2 ml de las muestras de sangre intravenosas a través del plexo venoso retrobulbar de la rata antes de la administración y 5 min, 15 min, 0,5 h, 1,0 h, 2,0 h, 4,0 h, 8,0 h, 12 h y 24 h después de la administración, y se separaron para dar plasmas. La concentración del fármaco en plasma se determinó con el método de cromatografía líquida-espectrometría de masas en tándem para obtener una curva de concentración de fármaco-tiempo.
Los principales parámetros farmacocinéticos se muestran en la tabla 1 a continuación:
Tabla 1

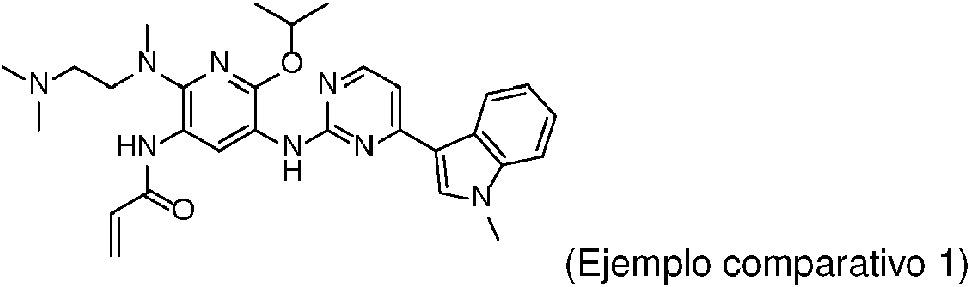
En la tabla 1, la estructura de la sustancia del ejemplo Comparativo 1 se muestra a continuación y se preparó según el ejemplo 2 de la solicitud de patente CN201410365911.4.
La estructura de la sustancia del ejemplo comparativo 2 se muestra a continuación y se preparó según el ejemplo 16 de la solicitud de patente CN201410365911.4.

T1/2 : semivida de eliminación; Cmáx: concentración máxima de fármaco en plasma; AUCo-t: área bajo la curva de concentración de fármaco-tiempo
Para administración intragástrica: Se asignaron aleatoriamente a 4 grupos 16 ratas SD sanas (mitad machos y mitad hembras) con 200-280 g de peso corporal, proporcionadas por Shanghai Sippr-BK laboratory animal Co. Ltd.. Las sustancias de los anteriores ejemplo 1, ejemplo 2, ejemplo comparativo 1 y ejemplo comparativo 2 se administraron por vía intragástrica en la dosificación que se muestra en la siguiente tabla. Se recogieron 0,2 ml de las muestras de sangre intravenosas a través del plexo venoso retrobulbar de la rata antes de la administración y 0,5, 1,0, 2,0, 4,0, 6,0, 8,0, 10, 12 y 24 h después de la administración, y se separaron para dar plasmas. La concentración del fármaco en plasma se determinó con el método de cromatografía líquida-espectrometría de masas en tándem para obtener una curva de concentración de fármaco-tiempo.
Sus principales parámetros farmacocinéticos se muestran en la tabla 2:

En la tabla 2, las estructuras y los métodos de preparación de los ejemplos comparativos 1 y 2 son idénticos a los de la tabla 1.
Tras la calibración de la dosificación, se obtuvo la biodisponibilidad absoluta F mediante el cálculo de la AUC0-t. La ecuación de cálculo fue la siguiente:
F = (AUCintragástrica x Dintravenosa)/(AUCintravenosa x Dintragástrica) x 100%. Los datos obtenidos de biodisponibilidad absoluta F se muestran en la tabla 2 anterior.
Conclusión: la biodisponibilidad absoluta para la administración intragástrica de la sal mesilato del compuesto de fórmula (I) del ejemplo 2 era de hasta el 28,28%, y notablemente mejor que la biodisponibilidad absoluta para la administración intragástrica del compuesto de fórmula (I) según el ejemplo 1, el ejemplo comparativo 1 y el ejemplo comparativo 2.
Ejemplo de prueba 2: Efecto de inhibición sobre el crecimiento de tumores trasplantados por vía subcutánea de ratones desnudos portadores de H1975 con cáncer de pulmón humano
Se observaron los efectos de inhibición de la sal mesilato del compuesto de fórmula (I) preparado según el ejemplo 2 de la presente invención y la sustancia del ejemplo comparativo 2 (la estructura del ejemplo comparativo 2 se muestra a continuación y se preparó con referencia al ejemplo 16 de solicitud de patente CN201410365911.4) sobre tumores trasplantados por vía subcutánea de ratones desnudos portadores de H1975 con cáncer de pulmón humano.

(Ejemplo comparativo 2)
Cultivo celular: Se colocó H1975 en un medio RPMI-1640 que contenía FBS al 10%, y se cultivó en una incubadora de temperatura constante que contenía CO2 al 5% a 37°C. Las células en fase de crecimiento exponencial se recogieron y se contaron para su inoculación.
Animales de prueba: 15 ratones desnudos macho BALB/c, 6 semanas de edad, 18-20 g, 5 ratones en cada grupo, disponibles comercialmente en el Shanghai Experiment Animal Research Center.
Se establecieron tres grupos de prueba: el grupo de control de disolvente de carboximetilcelucosa de sodio al 0,5%, el grupo del ejemplo 2 (25 mg/kg) y el grupo del ejemplo comparativo 2 (25 mg/kg).
Método experimental: se inoculó cepa de células H1975 con cáncer de pulmón humano (5 x 106/cada ratón) a ratones desnudos por vía subcutánea en el lado derecho del lomo de los mismos. El crecimiento tumoral se observó con regularidad. El diámetro del tumor trasplantado se midió con un pie de rey. Después de que los tumores crecieran hasta 100-150 mm3 en promedio, los ratones se dividieron en grupos aleatoriamente según el tamaño del tumor y el peso del ratón. Las sustancias del ejemplo 2 y del ejemplo Comparativo 2 se administraron mediante administración intragástrica en la dosificación de 25 mg/kg, y al grupo de control de disolvente se le administró igual cantidad de disolvente mediante administración intragástrica, en donde la administración se realizó una vez al día durante un período continuo de 21 días. Durante todo el procedimiento experimental, se midieron el peso del ratón y el tamaño del tumor dos veces por semana, para observar si se producía o no la reacción tóxica.
El volumen tumoral se calculó de la siguiente manera: Volumen tumoral (mm3) = 0,5 x diámetro mayor del tumor x diámetro menor del tumor x diámetro menor del tumor.
La curva de crecimiento tumoral de tres grupos experimentales se mostró en la figura 1, y la curva de peso corporal se mostró en la figura 2.
Conclusión: la sal mesilato del compuesto de fórmula (I) según el ejemplo 2 de la presente invención tuvo un buen efecto de inhibición sobre el crecimiento de tumores trasplantados por vía subcutánea de ratones desnudos portadores de H1975 con cáncer de pulmón humano, que fue notablemente mejor que el ejemplo comparativo 2, y mostró una buena seguridad.
Claims (6)
1. Una sal mesilato del compuesto de fórmula (I),

2. Un método para preparar la sal mesilato del compuesto de fórmula (I) según la reivindicación 1, caracterizado por que, en un disolvente, el compuesto de fórmula (I) y ácido metanosulfónico se hacen reaccionar directamente para dar la sal mesilato.
3. El método según la reivindicación 2, caracterizado por que dicho disolvente es un disolvente mezclado de acetona y agua.
4. Una composición farmacéutica, caracterizada por que contiene la sal mesilato del compuesto de fórmula (I) según la reivindicación 1 y un portador farmacéuticamente aceptable.
5. Uso de la sal mesilato del compuesto de fórmula (I) según la reivindicación 1 en la fabricación de un medicamento para tratar cánceres.
6. Uso de la composición farmacéutica según la reivindicación 4 en la fabricación de un medicamento para tratar el cáncer.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201610126987.0A CN107163026B (zh) | 2016-03-07 | 2016-03-07 | 吡啶胺基嘧啶衍生物的盐及其制备方法和应用 |
| PCT/CN2017/000202 WO2017152706A1 (zh) | 2016-03-07 | 2017-03-01 | 吡啶胺基嘧啶衍生物的盐及其制备方法和应用 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2818652T3 true ES2818652T3 (es) | 2021-04-13 |
Family
ID=59790088
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17762392T Active ES2818652T3 (es) | 2016-03-07 | 2017-03-01 | Sal de derivado de piridinilaminopirimidina, método de preparación de la misma y aplicación de la misma |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US10472349B2 (es) |
| EP (1) | EP3428158B1 (es) |
| JP (1) | JP6696670B2 (es) |
| KR (1) | KR102142796B1 (es) |
| CN (1) | CN107163026B (es) |
| CA (1) | CA3016826C (es) |
| ES (1) | ES2818652T3 (es) |
| WO (1) | WO2017152706A1 (es) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105315259B (zh) | 2014-07-29 | 2018-03-09 | 上海艾力斯医药科技有限公司 | 吡啶胺基嘧啶衍生物、其制备方法及应用 |
| CN110590749B (zh) * | 2016-03-07 | 2020-11-06 | 上海艾力斯医药科技股份有限公司 | 吡啶胺基嘧啶衍生物甲磺酸盐的结晶形式及其制备和应用 |
| CN110606842B (zh) * | 2018-06-15 | 2021-06-01 | 上海艾力斯医药科技股份有限公司 | 吡啶胺基嘧啶衍生物的制备方法及其中间体 |
| CN110606841A (zh) * | 2018-06-15 | 2019-12-24 | 上海艾力斯医药科技有限公司 | 吡啶胺基嘧啶衍生物的晶型及其制备方法 |
| CN111747931A (zh) * | 2019-03-29 | 2020-10-09 | 深圳福沃药业有限公司 | 用于治疗癌症的氮杂芳环酰胺衍生物 |
| WO2023035223A1 (zh) * | 2021-09-10 | 2023-03-16 | 上海艾力斯医药科技股份有限公司 | 药物组合物及其用途 |
| CN113896716A (zh) * | 2021-10-27 | 2022-01-07 | 浙江爱索拓科技有限公司 | 一种放射性同位素碳-14双标记甲磺酸伏美替尼合成方法 |
| WO2024059962A1 (en) * | 2022-09-19 | 2024-03-28 | Shanghai Allist Pharmaceuticals Co., Ltd. | Pharmaceutical composition and use thereof |
| CN116478138A (zh) * | 2023-04-21 | 2023-07-25 | 江苏艾力斯生物医药有限公司 | 一种甲磺酸伏美替尼原料药的结晶方法 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105348266B (zh) * | 2011-07-27 | 2018-04-10 | 阿斯利康(瑞典)有限公司 | 取代的3‑氯‑n‑[3‑(嘧啶‑2‑基氨基)苯基]丙酰胺或其盐 |
| CN104761544B (zh) * | 2014-01-03 | 2019-03-15 | 北京轩义医药科技有限公司 | Egfr酪氨酸激酶的临床重要突变体的选择性抑制剂 |
| CN105315259B (zh) * | 2014-07-29 | 2018-03-09 | 上海艾力斯医药科技有限公司 | 吡啶胺基嘧啶衍生物、其制备方法及应用 |
| CN110590749B (zh) * | 2016-03-07 | 2020-11-06 | 上海艾力斯医药科技股份有限公司 | 吡啶胺基嘧啶衍生物甲磺酸盐的结晶形式及其制备和应用 |
-
2016
- 2016-03-07 CN CN201610126987.0A patent/CN107163026B/zh active Active
-
2017
- 2017-03-01 KR KR1020187028797A patent/KR102142796B1/ko active IP Right Grant
- 2017-03-01 WO PCT/CN2017/000202 patent/WO2017152706A1/zh active Application Filing
- 2017-03-01 JP JP2018547437A patent/JP6696670B2/ja active Active
- 2017-03-01 EP EP17762392.3A patent/EP3428158B1/en active Active
- 2017-03-01 ES ES17762392T patent/ES2818652T3/es active Active
- 2017-03-01 US US16/083,209 patent/US10472349B2/en active Active
- 2017-03-01 CA CA3016826A patent/CA3016826C/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| KR20180117697A (ko) | 2018-10-29 |
| WO2017152706A1 (zh) | 2017-09-14 |
| EP3428158A1 (en) | 2019-01-16 |
| EP3428158A4 (en) | 2019-08-07 |
| JP6696670B2 (ja) | 2020-05-20 |
| CA3016826A1 (en) | 2017-09-14 |
| CN107163026A (zh) | 2017-09-15 |
| JP2019507787A (ja) | 2019-03-22 |
| CA3016826C (en) | 2022-06-14 |
| EP3428158B1 (en) | 2020-08-05 |
| US20190100509A1 (en) | 2019-04-04 |
| US10472349B2 (en) | 2019-11-12 |
| KR102142796B1 (ko) | 2020-08-10 |
| CN107163026B (zh) | 2019-07-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2818652T3 (es) | Sal de derivado de piridinilaminopirimidina, método de preparación de la misma y aplicación de la misma | |
| ES2855050T3 (es) | Derivado de piridinamidopirimidina, método de preparación y utilización del mismo | |
| ES2863923T3 (es) | Formas cristalinas de la sal mesilato de derivado de piridinilaminopirimidina, métodos de preparación para las mismas, y aplicaciones de las mismas | |
| ES2707534T3 (es) | Compuestos útiles como inmunomoduladores | |
| ES2868748T3 (es) | Compuesto de piridina | |
| ES2731216T3 (es) | Nuevos compuestos | |
| KR20170049604A (ko) | Bub1 억제제로서의 벤질 치환된 인다졸 | |
| ES2911040T3 (es) | Nuevos derivados de heteroaril amida como inhibidores selectivos de histona deacetilasa 1 y 2 (HDAC1/2) | |
| TWI826535B (zh) | 環狀二核苷酸類似物、其藥物組合物及應用 | |
| EP3967696A1 (en) | Compound used as kinase inhibitor and application thereof | |
| JP6606806B2 (ja) | 重水素化キナゾリノン化合物及び該化合物を含む薬物組成物 | |
| CA2967551A1 (en) | Imidazopyridazine derivatives as pi3k.beta. inhibitors | |
| CN110066272B (zh) | 取代的苯并[d]咪唑类化合物及其药物组合物 | |
| ES2760507T3 (es) | Derivados de imidazopiridazina enlazados a heterociclilo como inhibidores de PI3Kß | |
| CA2891900A1 (en) | Derivatives of indole for the treatment of cancer, viral infections and lung diseases | |
| CN115677666A (zh) | 一种吲哚联嘧啶类化合物、其中间体、制备方法及其应用 |