ES2775673T3 - Composición para el tratamiento de enfermedades inflamatorias del tracto urogenital que contiene sulfato de condonita (4,5 mg/ml), ácido hialurónico (16 mg/ml) y tampón de fosfato (pH 6,1 a 7,9) con mayor estabilidad de almacenamiento para tratar la cistitis - Google Patents
Composición para el tratamiento de enfermedades inflamatorias del tracto urogenital que contiene sulfato de condonita (4,5 mg/ml), ácido hialurónico (16 mg/ml) y tampón de fosfato (pH 6,1 a 7,9) con mayor estabilidad de almacenamiento para tratar la cistitis Download PDFInfo
- Publication number
- ES2775673T3 ES2775673T3 ES17173823T ES17173823T ES2775673T3 ES 2775673 T3 ES2775673 T3 ES 2775673T3 ES 17173823 T ES17173823 T ES 17173823T ES 17173823 T ES17173823 T ES 17173823T ES 2775673 T3 ES2775673 T3 ES 2775673T3
- Authority
- ES
- Spain
- Prior art keywords
- composition
- kda
- range
- component
- concentration
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000000203 mixture Substances 0.000 title claims abstract description 346
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 title claims abstract description 108
- 229920002674 hyaluronan Polymers 0.000 title claims abstract description 67
- 229960003160 hyaluronic acid Drugs 0.000 title claims abstract description 67
- 238000003860 storage Methods 0.000 title claims description 84
- 201000003146 cystitis Diseases 0.000 title claims description 70
- 238000011282 treatment Methods 0.000 title claims description 46
- 239000008363 phosphate buffer Substances 0.000 title claims description 45
- 208000027866 inflammatory disease Diseases 0.000 title claims description 29
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 title description 2
- SQDAZGGFXASXDW-UHFFFAOYSA-N 5-bromo-2-(trifluoromethoxy)pyridine Chemical compound FC(F)(F)OC1=CC=C(Br)C=N1 SQDAZGGFXASXDW-UHFFFAOYSA-N 0.000 claims abstract description 106
- 229920001287 Chondroitin sulfate Polymers 0.000 claims abstract description 79
- 229940059329 chondroitin sulfate Drugs 0.000 claims abstract description 75
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 claims abstract description 48
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 claims abstract description 48
- 239000007853 buffer solution Substances 0.000 claims abstract description 34
- 150000003839 salts Chemical class 0.000 claims abstract description 22
- WCDDVEOXEIYWFB-VXORFPGASA-N (2s,3s,4r,5r,6r)-3-[(2s,3r,5s,6r)-3-acetamido-5-hydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-4,5,6-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@@H]1C[C@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](C(O)=O)O[C@@H](O)[C@H](O)[C@H]1O WCDDVEOXEIYWFB-VXORFPGASA-N 0.000 claims abstract description 17
- 229940014041 hyaluronate Drugs 0.000 claims abstract description 17
- 210000003932 urinary bladder Anatomy 0.000 claims description 96
- 208000005615 Interstitial Cystitis Diseases 0.000 claims description 67
- 239000004480 active ingredient Substances 0.000 claims description 38
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 37
- 229910001868 water Inorganic materials 0.000 claims description 26
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 claims description 22
- 230000001684 chronic effect Effects 0.000 claims description 18
- 239000003792 electrolyte Substances 0.000 claims description 18
- 239000003513 alkali Substances 0.000 claims description 16
- 230000001580 bacterial effect Effects 0.000 claims description 13
- 239000007857 degradation product Substances 0.000 claims description 13
- 230000001225 therapeutic effect Effects 0.000 claims description 12
- 230000000699 topical effect Effects 0.000 claims description 12
- 239000011780 sodium chloride Substances 0.000 claims description 11
- 238000004806 packaging method and process Methods 0.000 claims description 9
- 230000000069 prophylactic effect Effects 0.000 claims description 8
- 230000000306 recurrent effect Effects 0.000 claims description 8
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 claims description 7
- 239000007864 aqueous solution Substances 0.000 claims description 7
- 239000008213 purified water Substances 0.000 claims description 7
- 229910052708 sodium Inorganic materials 0.000 claims description 7
- 239000011734 sodium Substances 0.000 claims description 7
- 206010063057 Cystitis noninfective Diseases 0.000 claims description 5
- 201000005661 acute cystitis Diseases 0.000 claims description 5
- 230000001154 acute effect Effects 0.000 claims description 5
- 230000032683 aging Effects 0.000 claims description 5
- 201000003139 chronic cystitis Diseases 0.000 claims description 5
- 159000000011 group IA salts Chemical class 0.000 claims description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 claims description 4
- 206010011796 Cystitis interstitial Diseases 0.000 claims description 4
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 claims description 4
- 229910000403 monosodium phosphate Inorganic materials 0.000 claims description 4
- 235000019799 monosodium phosphate Nutrition 0.000 claims description 4
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 claims description 4
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 claims description 3
- 229910052783 alkali metal Inorganic materials 0.000 claims description 3
- 150000001340 alkali metals Chemical class 0.000 claims description 3
- 239000007900 aqueous suspension Substances 0.000 claims description 3
- 238000011109 contamination Methods 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 3
- 229910052700 potassium Inorganic materials 0.000 claims description 3
- 239000011591 potassium Substances 0.000 claims description 3
- 229920002683 Glycosaminoglycan Polymers 0.000 description 21
- 201000010099 disease Diseases 0.000 description 21
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 21
- 239000010410 layer Substances 0.000 description 21
- 238000000034 method Methods 0.000 description 21
- 210000003741 urothelium Anatomy 0.000 description 21
- 239000013543 active substance Substances 0.000 description 19
- 210000002700 urine Anatomy 0.000 description 19
- 208000002193 Pain Diseases 0.000 description 18
- 230000036407 pain Effects 0.000 description 18
- 239000000126 substance Substances 0.000 description 18
- 239000000243 solution Substances 0.000 description 15
- 208000024891 symptom Diseases 0.000 description 15
- 230000001965 increasing effect Effects 0.000 description 14
- 239000003814 drug Substances 0.000 description 13
- 239000000872 buffer Substances 0.000 description 12
- 230000006641 stabilisation Effects 0.000 description 12
- 238000011105 stabilization Methods 0.000 description 12
- 229920002385 Sodium hyaluronate Polymers 0.000 description 11
- 229940010747 sodium hyaluronate Drugs 0.000 description 11
- YWIVKILSMZOHHF-QJZPQSOGSA-N sodium;(2s,3s,4s,5r,6r)-6-[(2s,3r,4r,5s,6r)-3-acetamido-2-[(2s,3s,4r,5r,6r)-6-[(2r,3r,4r,5s,6r)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2- Chemical compound [Na+].CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 YWIVKILSMZOHHF-QJZPQSOGSA-N 0.000 description 11
- KXKPYJOVDUMHGS-OSRGNVMNSA-N chondroitin sulfate Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](OS(O)(=O)=O)[C@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](C(O)=O)O1 KXKPYJOVDUMHGS-OSRGNVMNSA-N 0.000 description 10
- 230000009471 action Effects 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- 230000015556 catabolic process Effects 0.000 description 8
- 238000006731 degradation reaction Methods 0.000 description 8
- 229940079593 drug Drugs 0.000 description 8
- 238000005227 gel permeation chromatography Methods 0.000 description 8
- 230000003993 interaction Effects 0.000 description 8
- 239000008194 pharmaceutical composition Substances 0.000 description 8
- 238000002560 therapeutic procedure Methods 0.000 description 8
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 230000008859 change Effects 0.000 description 7
- 230000006870 function Effects 0.000 description 7
- 230000027939 micturition Effects 0.000 description 7
- 159000000000 sodium salts Chemical class 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 239000000017 hydrogel Substances 0.000 description 6
- 239000004615 ingredient Substances 0.000 description 6
- 239000011159 matrix material Substances 0.000 description 6
- 210000004877 mucosa Anatomy 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 230000009467 reduction Effects 0.000 description 6
- 230000002829 reductive effect Effects 0.000 description 6
- 238000001356 surgical procedure Methods 0.000 description 6
- 230000003139 buffering effect Effects 0.000 description 5
- 238000011049 filling Methods 0.000 description 5
- 230000006872 improvement Effects 0.000 description 5
- 150000002500 ions Chemical class 0.000 description 5
- 230000035699 permeability Effects 0.000 description 5
- 241000894006 Bacteria Species 0.000 description 4
- 229920002567 Chondroitin Polymers 0.000 description 4
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 4
- 206010061218 Inflammation Diseases 0.000 description 4
- 230000002921 anti-spasmodic effect Effects 0.000 description 4
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 4
- DLGJWSVWTWEWBJ-HGGSSLSASA-N chondroitin Chemical compound CC(O)=N[C@@H]1[C@H](O)O[C@H](CO)[C@H](O)[C@@H]1OC1[C@H](O)[C@H](O)C=C(C(O)=O)O1 DLGJWSVWTWEWBJ-HGGSSLSASA-N 0.000 description 4
- 238000002574 cystoscopy Methods 0.000 description 4
- 229920000669 heparin Polymers 0.000 description 4
- 229960002897 heparin Drugs 0.000 description 4
- 208000015181 infectious disease Diseases 0.000 description 4
- 230000004054 inflammatory process Effects 0.000 description 4
- 239000000825 pharmaceutical preparation Substances 0.000 description 4
- 239000011241 protective layer Substances 0.000 description 4
- 208000019206 urinary tract infection Diseases 0.000 description 4
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 241000194017 Streptococcus Species 0.000 description 3
- 150000001447 alkali salts Chemical class 0.000 description 3
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 3
- 150000001342 alkaline earth metals Chemical class 0.000 description 3
- 239000000739 antihistaminic agent Substances 0.000 description 3
- 230000009286 beneficial effect Effects 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 230000006378 damage Effects 0.000 description 3
- 230000036541 health Effects 0.000 description 3
- 230000002757 inflammatory effect Effects 0.000 description 3
- 230000007774 longterm Effects 0.000 description 3
- 159000000003 magnesium salts Chemical class 0.000 description 3
- 230000014759 maintenance of location Effects 0.000 description 3
- 210000004400 mucous membrane Anatomy 0.000 description 3
- 229940127557 pharmaceutical product Drugs 0.000 description 3
- 210000002307 prostate Anatomy 0.000 description 3
- 230000005855 radiation Effects 0.000 description 3
- 230000008929 regeneration Effects 0.000 description 3
- 238000011069 regeneration method Methods 0.000 description 3
- 230000008439 repair process Effects 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 230000002485 urinary effect Effects 0.000 description 3
- 206010002091 Anaesthesia Diseases 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 208000035143 Bacterial infection Diseases 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- BHPQYMZQTOCNFJ-UHFFFAOYSA-N Calcium cation Chemical compound [Ca+2] BHPQYMZQTOCNFJ-UHFFFAOYSA-N 0.000 description 2
- 241000251730 Chondrichthyes Species 0.000 description 2
- 229920002307 Dextran Polymers 0.000 description 2
- 206010020751 Hypersensitivity Diseases 0.000 description 2
- JLVVSXFLKOJNIY-UHFFFAOYSA-N Magnesium ion Chemical compound [Mg+2] JLVVSXFLKOJNIY-UHFFFAOYSA-N 0.000 description 2
- BPQQTUXANYXVAA-UHFFFAOYSA-N Orthosilicate Chemical compound [O-][Si]([O-])([O-])[O-] BPQQTUXANYXVAA-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 108010072866 Prostate-Specific Antigen Proteins 0.000 description 2
- 102100038358 Prostate-specific antigen Human genes 0.000 description 2
- 229920001218 Pullulan Polymers 0.000 description 2
- 239000004373 Pullulan Substances 0.000 description 2
- 208000025865 Ulcer Diseases 0.000 description 2
- 210000001015 abdomen Anatomy 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 230000006978 adaptation Effects 0.000 description 2
- 230000037005 anaesthesia Effects 0.000 description 2
- 230000003110 anti-inflammatory effect Effects 0.000 description 2
- 239000000935 antidepressant agent Substances 0.000 description 2
- 229940005513 antidepressants Drugs 0.000 description 2
- 229940125715 antihistaminic agent Drugs 0.000 description 2
- 229940124575 antispasmodic agent Drugs 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 208000022362 bacterial infectious disease Diseases 0.000 description 2
- 230000004888 barrier function Effects 0.000 description 2
- 230000006399 behavior Effects 0.000 description 2
- 230000000740 bleeding effect Effects 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910001424 calcium ion Inorganic materials 0.000 description 2
- 159000000007 calcium salts Chemical class 0.000 description 2
- 210000000845 cartilage Anatomy 0.000 description 2
- 150000001768 cations Chemical class 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000000812 cholinergic antagonist Substances 0.000 description 2
- 229940006423 chondroitin sulfate sodium Drugs 0.000 description 2
- 229940107200 chondroitin sulfates Drugs 0.000 description 2
- 208000037976 chronic inflammation Diseases 0.000 description 2
- 201000003147 chronic interstitial cystitis Diseases 0.000 description 2
- 239000007979 citrate buffer Substances 0.000 description 2
- 239000002537 cosmetic Substances 0.000 description 2
- 238000009799 cystectomy Methods 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 239000008367 deionised water Substances 0.000 description 2
- 229910021641 deionized water Inorganic materials 0.000 description 2
- 230000001627 detrimental effect Effects 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- 238000002405 diagnostic procedure Methods 0.000 description 2
- 235000015872 dietary supplement Nutrition 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- 206010013990 dysuria Diseases 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 210000000981 epithelium Anatomy 0.000 description 2
- 238000002695 general anesthesia Methods 0.000 description 2
- 230000001632 homeopathic effect Effects 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 238000009434 installation Methods 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 229910001425 magnesium ion Inorganic materials 0.000 description 2
- 239000013081 microcrystal Substances 0.000 description 2
- 210000004126 nerve fiber Anatomy 0.000 description 2
- 210000004197 pelvis Anatomy 0.000 description 2
- 230000008092 positive effect Effects 0.000 description 2
- 235000019423 pullulan Nutrition 0.000 description 2
- SQGYOTSLMSWVJD-UHFFFAOYSA-N silver(1+) nitrate Chemical compound [Ag+].[O-]N(=O)=O SQGYOTSLMSWVJD-UHFFFAOYSA-N 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 238000012549 training Methods 0.000 description 2
- 231100000397 ulcer Toxicity 0.000 description 2
- 230000000007 visual effect Effects 0.000 description 2
- 239000008215 water for injection Substances 0.000 description 2
- 230000003442 weekly effect Effects 0.000 description 2
- WHTVZRBIWZFKQO-AWEZNQCLSA-N (S)-chloroquine Chemical compound ClC1=CC=C2C(N[C@@H](C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-AWEZNQCLSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 206010006784 Burning sensation Diseases 0.000 description 1
- 229940127291 Calcium channel antagonist Drugs 0.000 description 1
- 208000009458 Carcinoma in Situ Diseases 0.000 description 1
- 206010010904 Convulsion Diseases 0.000 description 1
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 1
- 108010036949 Cyclosporine Proteins 0.000 description 1
- 206010011793 Cystitis haemorrhagic Diseases 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- 238000013494 PH determination Methods 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229920000388 Polyphosphate Polymers 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 208000025747 Rheumatic disease Diseases 0.000 description 1
- 208000019802 Sexually transmitted disease Diseases 0.000 description 1
- 206010072005 Spinal pain Diseases 0.000 description 1
- GUGOEEXESWIERI-UHFFFAOYSA-N Terfenadine Chemical compound C1=CC(C(C)(C)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 GUGOEEXESWIERI-UHFFFAOYSA-N 0.000 description 1
- 241001122767 Theaceae Species 0.000 description 1
- 229940123445 Tricyclic antidepressant Drugs 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- 206010046543 Urinary incontinence Diseases 0.000 description 1
- 206010046555 Urinary retention Diseases 0.000 description 1
- RVCSYOQWLPPAOA-CVPHZBIISA-M [(5s)-spiro[8-azoniabicyclo[3.2.1]octane-8,1'-azolidin-1-ium]-3-yl] 2-hydroxy-2,2-diphenylacetate;chloride Chemical compound [Cl-].[N+]12([C@H]3CCC2CC(C3)OC(=O)C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CCCC1 RVCSYOQWLPPAOA-CVPHZBIISA-M 0.000 description 1
- 230000003187 abdominal effect Effects 0.000 description 1
- 239000008351 acetate buffer Substances 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 238000001467 acupuncture Methods 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000005456 alcohol based solvent Substances 0.000 description 1
- 235000013334 alcoholic beverage Nutrition 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 229960000836 amitriptyline Drugs 0.000 description 1
- KRMDCWKBEZIMAB-UHFFFAOYSA-N amitriptyline Chemical compound C1CC2=CC=CC=C2C(=CCCN(C)C)C2=CC=CC=C21 KRMDCWKBEZIMAB-UHFFFAOYSA-N 0.000 description 1
- 230000000202 analgesic effect Effects 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 210000003484 anatomy Anatomy 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 230000001387 anti-histamine Effects 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 230000001174 ascending effect Effects 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- 230000000721 bacterilogical effect Effects 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 230000003542 behavioural effect Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 208000029162 bladder disease Diseases 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 239000000480 calcium channel blocker Substances 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 231100000357 carcinogen Toxicity 0.000 description 1
- 239000003183 carcinogenic agent Substances 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 229960003677 chloroquine Drugs 0.000 description 1
- WHTVZRBIWZFKQO-UHFFFAOYSA-N chloroquine Natural products ClC1=CC=C2C(NC(C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-UHFFFAOYSA-N 0.000 description 1
- 229940094517 chondroitin 4-sulfate Drugs 0.000 description 1
- 230000006020 chronic inflammation Effects 0.000 description 1
- 208000037893 chronic inflammatory disorder Diseases 0.000 description 1
- 229960001265 ciclosporin Drugs 0.000 description 1
- 235000016213 coffee Nutrition 0.000 description 1
- 235000013353 coffee beverage Nutrition 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- 230000001120 cytoprotective effect Effects 0.000 description 1
- HXGBXQDTNZMWGS-RUZDIDTESA-N darifenacin Chemical compound C=1C=CC=CC=1C([C@H]1CN(CCC=2C=C3CCOC3=CC=2)CC1)(C(=O)N)C1=CC=CC=C1 HXGBXQDTNZMWGS-RUZDIDTESA-N 0.000 description 1
- 229960002677 darifenacin Drugs 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 239000000645 desinfectant Substances 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000000378 dietary effect Effects 0.000 description 1
- 230000010339 dilation Effects 0.000 description 1
- IPZJQDSFZGZEOY-UHFFFAOYSA-N dimethylmethylene Chemical group C[C]C IPZJQDSFZGZEOY-UHFFFAOYSA-N 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 230000035622 drinking Effects 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 238000011010 flushing procedure Methods 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 230000003862 health status Effects 0.000 description 1
- 201000002802 hemorrhagic cystitis Diseases 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 230000009610 hypersensitivity Effects 0.000 description 1
- 230000037189 immune system physiology Effects 0.000 description 1
- 230000002055 immunohistochemical effect Effects 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 201000004933 in situ carcinoma Diseases 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 230000036512 infertility Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 230000030214 innervation Effects 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 239000002085 irritant Substances 0.000 description 1
- 231100000021 irritant Toxicity 0.000 description 1
- 230000000622 irritating effect Effects 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 210000001931 lesser pelvis Anatomy 0.000 description 1
- 238000012153 long-term therapy Methods 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 238000009115 maintenance therapy Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 238000000691 measurement method Methods 0.000 description 1
- 229940127554 medical product Drugs 0.000 description 1
- 238000010197 meta-analysis Methods 0.000 description 1
- 230000002906 microbiologic effect Effects 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 210000003097 mucus Anatomy 0.000 description 1
- 239000003158 myorelaxant agent Substances 0.000 description 1
- 230000001272 neurogenic effect Effects 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 230000004007 neuromodulation Effects 0.000 description 1
- 229960001597 nifedipine Drugs 0.000 description 1
- HYIMSNHJOBLJNT-UHFFFAOYSA-N nifedipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=O HYIMSNHJOBLJNT-UHFFFAOYSA-N 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 230000035764 nutrition Effects 0.000 description 1
- 229940126701 oral medication Drugs 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 238000006213 oxygenation reaction Methods 0.000 description 1
- 230000008058 pain sensation Effects 0.000 description 1
- 230000008533 pain sensitivity Effects 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 210000002640 perineum Anatomy 0.000 description 1
- 230000004526 pharmaceutical effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 230000000079 pharmacotherapeutic effect Effects 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 230000037081 physical activity Effects 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 230000010399 physical interaction Effects 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 239000001205 polyphosphate Substances 0.000 description 1
- 235000011176 polyphosphates Nutrition 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000009993 protective function Effects 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 208000020016 psychiatric disease Diseases 0.000 description 1
- 230000003236 psychic effect Effects 0.000 description 1
- 201000007608 radiation cystitis Diseases 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 230000011514 reflex Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 230000037390 scarring Effects 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 229910001961 silver nitrate Inorganic materials 0.000 description 1
- 230000000391 smoking effect Effects 0.000 description 1
- 229910001415 sodium ion Inorganic materials 0.000 description 1
- SNXUWJAFSLKIMF-UHFFFAOYSA-M sodium;hypochlorous acid;4-tetradecylbenzenesulfonate Chemical compound [Na+].ClO.CCCCCCCCCCCCCCC1=CC=C(S([O-])(=O)=O)C=C1 SNXUWJAFSLKIMF-UHFFFAOYSA-M 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 235000021259 spicy food Nutrition 0.000 description 1
- 238000013112 stability test Methods 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 238000011146 sterile filtration Methods 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 230000035882 stress Effects 0.000 description 1
- 230000009469 supplementation Effects 0.000 description 1
- 238000011477 surgical intervention Methods 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 235000013616 tea Nutrition 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 239000003440 toxic substance Substances 0.000 description 1
- 239000003029 tricyclic antidepressant agent Substances 0.000 description 1
- 229960001530 trospium chloride Drugs 0.000 description 1
- 239000000439 tumor marker Substances 0.000 description 1
- 238000002604 ultrasonography Methods 0.000 description 1
- 210000003708 urethra Anatomy 0.000 description 1
- 210000001635 urinary tract Anatomy 0.000 description 1
- 210000001215 vagina Anatomy 0.000 description 1
- 238000011121 vaginal smear Methods 0.000 description 1
- 238000012800 visualization Methods 0.000 description 1
- 230000003313 weakening effect Effects 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 230000036642 wellbeing Effects 0.000 description 1
- 230000029663 wound healing Effects 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/08—Solutions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/715—Polysaccharides, i.e. having more than five saccharide radicals attached to each other by glycosidic linkages; Derivatives thereof, e.g. ethers, esters
- A61K31/726—Glycosaminoglycans, i.e. mucopolysaccharides
- A61K31/728—Hyaluronic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/715—Polysaccharides, i.e. having more than five saccharide radicals attached to each other by glycosidic linkages; Derivatives thereof, e.g. ethers, esters
- A61K31/737—Sulfated polysaccharides, e.g. chondroitin sulfate, dermatan sulfate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K33/00—Medicinal preparations containing inorganic active ingredients
- A61K33/14—Alkali metal chlorides; Alkaline earth metal chlorides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/02—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0034—Urogenital system, e.g. vagina, uterus, cervix, penis, scrotum, urethra, bladder; Personal lubricants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61M—DEVICES FOR INTRODUCING MEDIA INTO, OR ONTO, THE BODY; DEVICES FOR TRANSDUCING BODY MEDIA OR FOR TAKING MEDIA FROM THE BODY; DEVICES FOR PRODUCING OR ENDING SLEEP OR STUPOR
- A61M3/00—Medical syringes, e.g. enemata; Irrigators
- A61M3/02—Enemata; Irrigators
- A61M3/0233—Enemata; Irrigators characterised by liquid supply means, e.g. from pressurised reservoirs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Dermatology (AREA)
- Molecular Biology (AREA)
- Urology & Nephrology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Inorganic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Reproductive Health (AREA)
- Gynecology & Obstetrics (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Heart & Thoracic Surgery (AREA)
- Hematology (AREA)
- Biomedical Technology (AREA)
- Anesthesiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Composición, en la que la composición contiene en combinación y, en cada caso, en cantidades eficaces (a) sulfato de condroitina y/o una sal de sulfato de condroitina fisiológicamente aceptable en una concentración de (4,5±0,5) mg/ml (componente (a)); y (b) ácido hialurónico y/o una sal de ácido hialurónico fisiológicamente aceptable (hialuronato) en una concentración de (16±1,6) mg/ml (componente (b)); donde la composición presenta un valor pH en el intervalo de 6,1 a 7,9 y/o donde la composición se ajusta a un valor pH en el intervalo de 6,1 a 7,9, caracterizada por que la composición (c) contiene un sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)).
Description
DESCRIPCIÓN
Composición para el tratamiento de enfermedades inflamatorias del tracto urogenital que contiene sulfato de condonita (4,5 mg/ml), ácido hialurónico (16 mg/ml) y tampón de fosfato (pH 6,1 a 7,9) con mayor estabilidad de almacenamiento para tratar la cistitis
La presente invención se refiere al campo médico-terapéutico del tratamiento de enfermedades preferiblemente inflamatorias del tracto urogenital, en particular cistitis, tal como cistitis intersticial.
En particular, la presente invención se refiere a una composición, en particular a una composición farmacéutica, que es particularmente adecuada para el tratamiento profiláctico o terapéutico de enfermedades preferiblemente inflamatorias del tracto urogenital, en particular de enfermedades inflamatorias de la vejiga urinaria, preferiblemente cistitis. Del mismo modo, la presente invención también se refiere a un dispositivo de almacenamiento o aplicación que contiene la composición según la invención. La presente invención también se refiere a una unidad de embalaje que contiene el dispositivo de almacenamiento o aplicación según la invención, así como a un kit que contiene el dispositivo de almacenamiento o aplicación según la invención, a la composición según la invención, así como a un dispositivo de instilación que puede conectarse al dispositivo de almacenamiento o aplicación.
El cuadro clínico de la cistitis generalmente se puede dividir en dos grupos. Además de dicha cistitis, que es causada en particular por infecciones bacterianas y que generalmente se trata con antibióticos o mediante intervención quirúrgica para eliminar las causas de la infección, por ejemplo, en caso de obstrucciones o reflujo, hay una serie de inflamaciones de la vejiga que no son causadas por infecciones. Se cuentan entre ellas la cistitis radiogénica (= cistitis por radiación) y la cistitis intersticial.
La cistitis radiogénica ocurre en aproximadamente el 5% de los pacientes irradiados debido a tumores malignos en la pelvis menor. Esta cistitis hemorrágica generalmente ocurre de seis meses a diez años después de la radiación y puede atribuirse a cambios tisulares probablemente irreversibles.
Las opciones de tratamiento para la cistitis radiogénica incluyen terapia con antiespasmódicos, por ejemplo, cloruro de trospio, darifenacina, etc., o la llamada oxigenación hiperbárica.
Un área mucho más grande es la cistitis intersticial, especialmente porque afecta a un gran grupo de personas. La cistitis intersticial a veces también se define como “cistitis idiopática crónica de origen desconocido”. La cistitis intersticial es difícil de diagnosticar y sus opciones de tratamiento se consideran difíciles. La cistitis intersticial también está comprendida incluye bajo la expresión “síndrome de vejiga dolorosa” ("painful bladder syndrome").
Con respecto a la etiología, a veces no hay conocimientos completamente confirmados. Al respecto, se discuten varias hipótesis, como una liberación de sustancias inflamatorias por la activación de mastocitos debido a diferentes estímulos; infecciones ocultas; aumento de la permeabilidad de la pared de la vejiga para sustancias tóxicas; procesos inmunológicos, así como hipersensibilidad de las fibras nerviosas con el aumento de la densidad de las fibras nerviosas.
El espectro diagnóstico de la cistitis intersticial incluye el protocolo de micción y el diario de dolor con VAS (escala analógica visual), así como el examen bacteriológico para descartar una infección del tracto urinario y cistología urinaria para descartar carcinoma in situ y la cistoscopia que se realizará bajo anestesia con una biopsia acompañante.
La cistitis intersticial es una enfermedad inflamatoria crónica de la vejiga sin bacterias detectables en la orina. Como resultado, se trata en este caso de una cistitis de origen no bacteriano. Se trata de una enfermedad que aún no se ha aclarado por completo, con la cual los pacientes a menudo sufren más que con una afección tumoral. El National Health Institute de los Estados Unidos también ha clasificado a la cistitis intersticial como una enfermedad de mayor prioridad. La calidad de vida puede verse extremadamente afectada por una fuerte necesidad de orinar, micción frecuente durante el día y por la noche y un aumento del dolor.
Según encuestas recientes, la frecuencia de la enfermedad parece estar aumentando. Los pacientes que experimentan la enfermedad son en su mayoría de mediana edad. La enfermedad es más común en mujeres que en hombres. Una razón para esto se puede ver en la anatomía de la mujer, cuya uretra es más corta que la del hombre. Esto conduce a una mayor susceptibilidad a las infecciones ascendentes del tracto urinario. En mujeres con infecciones repetidas (recurrentes) del tracto urinario, la mucosa de la vejiga está más gravemente dañada. Esta irritación constante puede conducir a una cistitis intersticial no bacteriana, especialmente crónica. La cistitis intersticial se diagnostica predominantemente en mujeres, aunque los hombres también pueden verse afectados por la enfermedad.
Los síntomas más comunes de la cistitis intersticial son (en orden descendente):
- aumento de la necesidad de orinar;
- micción frecuente;
- dolor pélvico, abdominal bajo e intestinal;
- presión pélvica;
- micción dolorosa asociada con las cantidades más pequeñas de orina;
- dolor intenso durante y después del coito;
- sensación dolorosa de ardor;
- problemas masivos para dormir debido al dolor;
- sangre en la orina.
Los principales síntomas de la cistitis intersticial son la pérdida de capacidad de la vejiga con dolor dependiente del llenado y la necesidad frecuente y masiva de orinar. Se habla de la tríada: “frequency, urgency, pain" (frecuencia, urgencia, dolor).
Los síntomas de cistitis intersticial mencionados con anterioridad pueden aumentar, por ejemplo, por una mayor sensibilidad al dolor o por factores psíquicos. Se mostró una alteración en la composición de la capa de glicosaminoglicanos, y los estudios inmunohistoquímicos muestran, entre otros, una disminución de la tinción con sulfato de condroitina.
Sin desear limitarse o comprometerse con una teoría, la aparición y el desarrollo de una cistitis intersticial se pueden explicar de la siguiente manera: a través de huecos en la capa protectora de la mucosa de la vejiga, la llamada capa de glicosaminoglicanos, las bacterias, microcristales, proteínas y/o componentes nocivos de la orina disueltos, como la urea, ingresan directamente en las capas más profundas de la mucosa de la vejiga, causando allí más daño.
El daño a la mucosa de la vejiga y la inflamación crónica resultante conducen a procesos de reparación que, a menudo, van acompañados de cicatrices. Esto puede conducir a una reducción de la elasticidad de la pared de la vejiga y una pérdida creciente de la capacidad de la vejiga. En la etapa tardía de la cistitis intersticial, se puede desarrollar una vejiga retráctil y la extirpación quirúrgica de la vejiga puede ser necesaria en ciertas circunstancias. Por lo tanto, la detección temprana y la terapia para evitar la etapa tardía de la cistitis intersticial son de gran importancia.
La causa principal de la cistitis intersticial es el daño a la mucosa de la vejiga. La mucosa de la vejiga está protegida por una capa protectora, que contiene ácido hialurónico, entre otras cosas, contra microorganismos, sustancias cancerígenas y otras sustancias nocivas que se producen en la orina. Esta capa protectora, que también se conoce como la capa de glicosaminoglicanos (capa GAG) y que, además del ácido hialurónico, también contiene sulfato de condroitina, heparina y polifosfato de pentosano como componentes importantes, es extremadamente resistente al agua y forma una “película de agua”, por así decirlo, y, por lo tanto, una barrera física adicional contra sustancias dañinas en la orina.
En pacientes con cistitis y especialmente con cistitis intersticial, hay defectos en esta capa protectora de la mucosa vesical. En particular, se ha detectado una pérdida de ácido hialurónico.
Otras causas de cistitis intersticial pueden incluir, por ejemplo, reacciones autoinmunes, que se dirigen contra las propias células del cuerpo en la vejiga, o infecciones bacterianas crónicas previas.
Los síntomas típicos de la cistitis intersticial son micción frecuente, aumento de la necesidad de orinar y, en algunos casos, micción incontrolada (incontinencia urinaria) y sangre en la orina. El dolor intenso se desarrolla especialmente cuando la vejiga está llena; una disminución en la sensación de dolor después de orinar es típica. Otros signos son dolor en la pelvis, la parte inferior del abdomen y el perineo, presión pélvica y dolor al orinar, combinado con el hecho de que la orina solo se puede liberar en gotas. El dolor intenso a menudo ocurre durante y después del coito. En muchos casos, los dolores del paciente causan un sufrimiento tan enorme que incluso son necesarios los procedimientos quirúrgicos, incluida la cistectomía.
Para diagnosticar la cistitis intersticial, es importante descartar otros trastornos de la vejiga con síntomas similares. El primer paso es aclarar si el paciente siente dolor debido a una cirugía previa (por ejemplo, en la parte inferior del abdomen), si la infección de la vejiga fue causada por radiación o quimioterapia, o si había o hay infecciones recurrentes o repetidas. Posteriormente, debe examinarse si pueden excluirse las enfermedades ginecológicas, neurológicas, psiquiátricas y/o reumáticas. Además, deben excluirse los dolores de columna y alergias.
Como parte del examen o diagnóstico de cistitis intersticial, se puede realizar en un laboratorio, por ejemplo, un cultivo de orina y un examen de los componentes celulares en la orina (citología de orina). En pacientes mujeres, se debe realizar un frotis vaginal para descartar enfermedades de transmisión sexual.
La sensibilidad al dolor se evalúa mediante un examen táctil de la vagina. En pacientes hombres, se crea un cultivo bacteriano a partir de la eyaculación para descartar inflamación bacteriana de la próstata. Para descartar el cáncer de próstata, se determina el valor del marcador tumoral específico de próstata (PSA = antígeno prostático específico). La orina residual se determina mediante un examen de ultrasonido, y se descarta un crecimiento de la próstata en la vejiga.
También se puede realizar un examen adicional utilizando un examen de la vejiga (cistoscopia). El examen de la vejiga se puede realizar bajo anestesia. Los signos típicos de cistitis intersticial, que pueden manifestarse mediante cistoscopia, son aumento del crecimiento de vasos sanguíneos en la mucosa de la vejiga, acumulación de líquido en la membrana mucosa, rotura de la membrana mucosa (glomérulos), sangrado puntiforme después de la dilatación de la vejiga bajo presión por lavado de agua (hidrodistensión) y signos de úlceras de vejiga (úlcera de Hunner) en alrededor del 10 al 20% de los pacientes.
Con la cistitis intersticial, los pacientes sienten una fuerte necesidad de orinar incluso con pequeñas cantidades de orina; su capacidad de vejiga se reduce. Para el diagnóstico de cistitis intersticial, puede realizarse la determinación del volumen máximo de llenado y luego una medición comparativa de la capacidad de la vejiga (cistometría).
Los estudios sobre cistitis intersticial muestran que el epitelio de la vejiga o el urotelio de la vejiga es deficiente en presencia de cistitis. Este debilitamiento contribuye significativamente a los síntomas clínicos de la cistitis intersticial.
En cuanto a la terapia de la cistitis intersticial, actualmente no existe una cura o método de tratamiento que sea eficaz para todos los pacientes.
Por lo tanto, una composición a base de polisulfato de pentosano sódico se conoce en la técnica anterior. Se cree que el modo de acción es reparar una pared de vejiga delgada o dañada. Sin embargo, los éxitos no siempre son satisfactorios.
Los antidepresivos, como los antidepresivos tricíclicos, también han demostrado ser algo eficaces para aliviar el dolor y la micción en la cistitis intersticial. Sin embargo, en la cistitis intersticial, estos medicamentos se usan solo debido a sus propiedades analgésicas.
Otros medicamentos orales incluyen agentes antiinflamatorios, antiespasmódicos, antihistamínicos y miorrelajantes. Sin embargo, tales medicamentos solo pueden aliviar la enfermedad en cierta medida. Generalmente, no es posible un éxito terapéutico completo con estos medicamentos.
La instilación de la vejiga también se puede realizar con ciertas sustancias. De esta manera, se puede lograr el estiramiento de la vejiga, llenando la vejiga con agua bajo anestesia general para el estiramiento. Si bien esto es principalmente parte del procedimiento de diagnóstico para la cistitis intersticial, también se puede usar terapéuticamente.
Además, el DMSO (dimetilsulfóxido) se puede llenar directamente en la vejiga como medicamento. Se dice que tiene un efecto antiinflamatorio y, por lo tanto, reduce el dolor. El DMSO se puede mezclar con esteroides, heparina y otros ingredientes para hacer un “cóctel para la vejiga”. Sin embargo, los efectos secundarios a menudo son altos.
Otras instilaciones en la vejiga, por ejemplo, que usan oxicloroseno de sodio, son en su mayoría muy dolorosas y requieren anestesia general. El nitrato de plata rara vez se usa y se considera una terapia desactualizada.
Otros tratamientos, como la nutrición dirigida, en los que se evitan ciertos alimentos, especialmente los alimentos ácidos y picantes, solo pueden aliviar ligeramente la gravedad de los síntomas. La cistitis intersticial también puede exacerbarse al fumar, tomar café o té y bebidas alcohólicas.
Las técnicas de autoayuda pueden mejorar la calidad de vida en pequeña medida y reducir la incidencia y la gravedad de las convulsiones. Incluyen, por ejemplo, un cambio en el estilo de vida, reducción del estrés, visualización, biorretroalimentación, entrenamiento de la vejiga y actividad física. Sin embargo, el éxito de la terapia a largo plazo a menudo no es posible con estos métodos.
La cirugía de vejiga se puede tener en cuenta para un pequeño número de pacientes con síntomas graves que no responden a otros tratamientos. En algunos casos, sin embargo, esto no mejora los síntomas. Se han utilizado varios tipos de operaciones para tratar la cistitis intersticial, incluida la cistectomía y la derivación urinaria. Sin embargo, debido a la gravedad del procedimiento quirúrgico, la cirugía siempre debe ser el último recurso.
Las opciones terapéuticas son tan diversas como insatisfactorias en general. En resumen y además de las realizaciones anteriores, hasta ahora se han considerado los siguientes métodos de tratamiento para la cistitis intersticial. Medicamentos que influyen en la inervación (antiespasmódicos, antihistamínicos); una terapia citodestructiva con regeneración posterior (por ejemplo, DMSO, hidrodistensión); una terapia citoprotectora para restaurar la capa de glicosaminoglicanos (heparina, polisulfato de pentosulfano). En el contexto de la terapia conservadora, se trata de la reducción de los síntomas por medio de sustancias administradas por vía oral, como los antiespasmódicos (bajo éxito); antihistamínicos; antidepresivos, especialmente tricíclicos (amitriptilina); citoprotectores, como el polisulfato de pentosulfano (latencia muy larga de hasta dos años hasta que se pueda medir el éxito); inmunosupresores tales como azatioprina, ciclosporina, cloroquina; bloqueadores de los canales de calcio, por ejemplo, nifedipina. La hidrodistensión es el estiramiento de la vejiga usando un globo insertado intravesicalmente. Como regla general, el estiramiento se lleva a cabo durante un período de tres horas, pero el efecto terapéutico es leve y solo de corta duración. También hay varias medidas farmacoterapéuticas intravesicales, en particular para restaurar la capa de glicosaminoglicanos. Como se discutió con anterioridad, para ello se usan polisulfato de
pentosano, heparina o DMSO.
También se llevan a cabo procedimientos de tratamiento alternativos. Estos incluyen ejercicios de relajación, entrenamiento conductual, acupuntura, neuromodulación y medidas dietéticas.
En la técnica anterior, se proponen otras composiciones o procedimientos para el tratamiento de la cistitis, tales como la cistitis intersticial; sin embargo, no siempre tienen la eficacia deseada o requerida. Además, tales composiciones de la técnica anterior a veces no son suficientemente estables en almacenamiento.
El documento WO 2004/073584 A2 se refiere a una composición farmacéutica para usar en la vejiga de un paciente, donde la composición comprende sulfato de condroitina. Este documento se centra en el uso de un solo ingrediente activo.
Además, el documento US 6.083.933 A se refiere a composiciones que contienen sulfato de condroitina que pueden usarse en el tratamiento de la cistitis intersticial. Sin embargo, al usar una monocomposición que no contiene otros ingredientes activos aparte del sulfato de condroitina, la acción con respecto a las cistitis por tratar no siempre es suficiente.
El documento US 5880 108 A1 se refiere a un procedimiento para el tratamiento de la cistitis, donde la vejiga urinaria y las estructuras asociadas a esta se deben poner en contacto con una solución, donde la solución comprende ácido hialurónico. La composición también puede contener ciertas sustancias que han de ser adecuadas para el tratamiento de enfermedades subyacentes relacionadas con la cistitis.
El documento CN 105982912 A se refiere a una composición farmacéutica que contiene hialuronato de sodio con un peso molecular medio y sulfato de condroitina con un peso molecular relativamente bajo. El peso molecular del hialuronato de sodio está en el intervalo de 200 a 1.000 kDa y la concentración está en el intervalo del 1 al 2%. El peso molecular del sulfato de condroitina está en el intervalo de 5 a 10 kDa y la concentración está en el intervalo del 1,5 al 3%. La composición puede usarse para el tratamiento de cistitis no bacteriana, cistitis bacteriana crónica y recurrente, cistitis debida a radiación o irradiación, cistitis intersticial e infecciones neurogénicas del tracto urinario.
Además, la publicación científica según Pyo y Cho describe: “Systematic Review and Meta-Analysis of Intravesical Hyaluronic Acid and Hyaluronic Acid/Chondroitin Sulfate Instillation for Interstitial Cystitis/Painful Biadder Syndrome", publicado en “Cellular Physiology and Biochemistry”, septiembre de 2016, páginas 1618-1625, composiciones para el tratamiento de la cistitis intersticial que contienen ácido hialurónico y sulfato de condroitina. El tratamiento se lleva a cabo por instilación vesical.
Además, en función del producto de mercado “iAIuRil®”, se vende una jeringa de inyección para instilación vesical, que contiene una composición a base de hialuronato de sodio con una concentración de 800 mg/50 ml y sulfato de condroitina de sodio con una concentración de 1 g/50 ml. La composición se usa para tratar la cistitis intersticial.
El documento WO 2007/138014 A1 también describe composiciones a base de glicosaminoglicanos, en particular ácido hialurónico, en combinación con sulfato de condroitina para el tratamiento de la cistitis intersticial.
Finalmente, el documento WO 2013/144867 A1 describe composiciones farmacéuticas con sulfato de condroitina y derivados del ácido hialurónico, que se usan para tratar la cistitis intersticial y estabilizar un tampón fosfato y que contienen un tampón de fosfato para la estabilización, de modo que la composición presenta un valor de pH estable de aproximadamente 7 en el intervalo fisiológico.
Con respecto a los requisitos médico-terapéuticos para las composiciones de instilación correspondientes y su manipulabilidad, la técnica anterior, además de proporcionar un alto nivel de eficacia en relación con la enfermedad subyacente, también tiene una gran necesidad de composiciones estables o composiciones con alta durabilidad, es decir, en tales composiciones para instilación, que disponen de propiedades (de producto) definidas o constantes durante un período de tiempo correspondientemente largo, por ejemplo, con respecto a la estabilidad de las sustancias activas, con pequeñas cantidades asociadas de productos de degradación a veces tóxicos, estabilidad del pH, viscosidad constante, o similares. En particular, existe una necesidad correspondiente de composiciones con alta estabilidad de almacenamiento, es decir, aquellas composiciones para instilación que son estables o constantes durante un largo período de tiempo con respecto a sus sustancias o ingredientes activos o sus propiedades fisicoquímicas.
Esto se debe a que las composiciones conocidas de la técnica anterior, que se administran en el curso del tratamiento de, en particular, enfermedades inflamatorias del tracto urogenital, como la cistitis, a veces tienen un comportamiento de estabilidad no óptimo, donde, además, no siempre hay una eficacia óptima. En este contexto, existe un riesgo particular de que, en el curso del tratamiento subyacente, se puedan usar composiciones degradadas en forma prematura con una cantidad posiblemente reducida de sustancia activa o una proporción correspondientemente aumentada de productos de degradación potencialmente dañinos, pero esto puede ser perjudicial para el éxito de un tratamiento completo.
En este contexto, es un objeto de la presente invención proporcionar una composición correspondiente, en particular
una composición farmacéutica, preferiblemente para el tratamiento profiláctico o terapéutico de tratamientos preferiblemente inflamatorios del tracto urogenital, por ejemplo, cistitis, en particular cistitis intersticial, que al menos evita ampliamente o al menos debilita las desventajas de la técnica anterior descrita con anterioridad.
En particular, dicha composición debería tener una estabilidad mejorada, en particular estabilidad al almacenamiento, en comparación con las composiciones farmacéuticas o preparaciones convencionales que son seguras para el tratamiento, en particular, de enfermedades inflamatorias del tracto urogenital, en particular cistitis, como la cistitis intersticial, especialmente con respecto a cantidades constantes de ingrediente activo y propiedades fisicoquímicas de las composiciones subyacentes, incluso durante períodos de tiempo más largos.
Además, otro objeto de la presente invención consiste en proporcionar una composición correspondiente que también tenga una alta eficacia de acción en el tratamiento de enfermedades preferiblemente inflamatorias del tracto urogenital, en particular cistitis, tal como cistitis intersticial, al tiempo que debería mejorar aún más su manejo y tolerabilidad.
Para lograr el objeto descrito con anterioridad, la presente invención propone, según un primer aspecto de la presente invención, una composición, en particular una composición farmacéutica, según la reivindicación 1; además, en particular, las realizaciones ventajosas de la composición según la invención son objeto de las subreivindicaciones relacionadas.
Además, según un segundo aspecto de la presente invención, la presente invención se refiere a un dispositivo de almacenamiento o aplicación que presenta o que contiene la composición según la invención, como se define en la reivindicación independiente relevante relacionada con el dispositivo de almacenamiento o aplicación; además, en particular, las realizaciones ventajosas del dispositivo de almacenamiento o aplicación según la invención son objeto de la subreivindicación relacionada.
Además, según un tercer aspecto de la presente invención, la presente invención también se refiere a una unidad de embalaje que contiene el dispositivo de almacenamiento o aplicación según la invención, según la reivindicación independiente relevante relacionada con la unidad de embalaje.
Finalmente, según un cuarto aspecto de la presente invención, la presente invención también se refiere a un kit, en particular a un sistema de instalación, como se define en la reivindicación independiente relacionada con el kit.
En relación con las siguientes realizaciones, no hace falta decir que las configuraciones, realizaciones, ventajas y similares, que se enumeran a continuación con el fin de evitar repeticiones para un solo aspecto de la invención, por supuesto, también se aplican en consecuencia con respecto a los otros aspectos de la invención, sin que esto requiera una mención por separado.
Con toda la información relativa o porcentual relacionada con el peso mencionada a continuación, en particular los datos de cantidad, también debe tenerse en cuenta que, dentro del alcance de la presente invención, el experto en la técnica debe seleccionarlos de tal manera que lleguen al 100% o sumen el 100% en peso, incluidos todos los componentes o ingredientes, en particular como se define a continuación; esto es evidente para el experto en la técnica.
Además, el experto en la técnica, dependiendo de la aplicación o caso por caso, puede desviarse de la información de concentración, peso, cantidad e intervalo que se proporciona a continuación, sin apartarse del alcance de la presente invención.
Además, todos los valores o parámetros o similares mencionados a continuación se pueden calcular o determinar en principio utilizando métodos de determinación normalizados o estandarizados o explícitamente especificados o utilizando métodos de determinación o medición que son per se familiares para el experto en la técnica en este campo.
El término “fármaco” o “medicamento” (sinónimo también de “producto farmacéutico”), como se usa en el contexto de la presente invención, debe entenderse de una manera muy extensa e incluye no solo medicamentos o productos farmacéuticos como tales (es decir, en términos de la ley farmacéutica), sino también especialmente los llamados dispositivos médicos y también suplementos homeopáticos y nutricionales, así como cosméticos y artículos de uso diario. En otras palabras, la composición según la invención puede estar, por lo tanto, en forma de un medicamento (producto farmacéutico), dispositivo médico, producto homeopático, suplemento nutricional, cosmético o en forma de un objeto de uso diario.
Con eso en mente, la presente invención se explicará ahora en detalle a continuación.
Es objeto de la presente invención, según un primer aspecto de la presente invención, por lo tanto, una composición, donde la composición contiene en combinación y, en cada caso, en cantidades eficaces
(a) sulfato de condroitina y/o una sal de sulfato de condroitina fisiológicamente aceptable en una concentración de (4,5±0,5) mg/ml (componente (a)); y
(b) ácido hialurónico y/o una sal de ácido hialurónico fisiológicamente aceptable (hialuronato) en una concentración de (16±1,6) mg/ml (componente (b));
donde la composición tiene un pH en el intervalo de 6,1 a 7,9 y/o donde la composición se ajusta a un pH en el intervalo de 6,1 a 7,9,
caracterizada por que la composición (c) contiene un sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)).
Sorprendentemente, se encontró dentro del alcance de la presente invención que, basándose en el concepto según la invención, se puede proporcionar una composición especial, es decir, que contiene como componente (a) sulfato de condroitina o una sal de sulfato de condroitina fisiológicamente aceptable con una concentración definida, como componente (b) ácido hialurónico o una sal de ácido hialurónico fisiológicamente aceptable (también conocido como “hialuronato”) con una concentración definida, como componente (c) un sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (sinónimo de “(sistema de) tampón H2 PO4YHPO42-” o “(sistema de) tampón fosfato” o “sistema tamponante químico) y, opcionalmente, como el componente (d) al menos un electrolito fisiológicamente aceptable, donde la composición también presenta un valor de pH definido que se establece o se mantiene constante por medio del sistema tamponante químico, para una aplicación intencional, en particular la instilación, dentro del marco del tratamiento de enfermedades preferiblemente inflamatorias del tracto urogenital, como la cistitis, en particular la cistitis intersticial, que, respecto a la técnica anterior, al mismo tiempo con alta eficacia o acción y muy buena tolerabilidad, presenta una estabilidad mejorada, en particular estabilidad al almacenamiento.
Sobre la base de la composición según la invención, se proporciona un fármaco o medicamento o producto (médico) altamente eficaz que, debido a la combinación y coordinación específicas de los componentes subyacentes y al ajuste de un pH definido, tiene una excelente estabilidad, en particular estabilidad al almacenamiento, por lo que incluso después de períodos de almacenamiento correspondientemente largos, no hay degradación significativa de los ingredientes o principios activos que tienen un efecto negativo tanto en la eficacia como en la tolerancia, donde, más allá de ello, las propiedades fisicoquímicas y la eficacia de la composición también permanecen al menos sustancialmente sin cambios.
En cuanto a la mejora en la estabilidad, en particular la estabilidad al almacenamiento, encontrada en el contexto de la presente invención de una manera completamente sorprendente, y al mismo tiempo alta eficacia de la composición según la invención, las concentraciones de los componentes (a) y (b) presentes en la composición según la invención también tienen gran importancia. En este contexto, la solicitante ha encontrado que, con respecto a las concentraciones previstas según la invención y las cantidades asociadas de sustancias, en particular sobre la base de los componentes (a) y (b), existe una estabilidad y eficacia óptimas a este respecto.
Las medidas según la invención también se encadenan, por así decirlo, con respecto al uso de un sistema tamponante especial según el componente (c) y el ajuste de un pH especial, y se refuerzan entre sí y el efecto de las medidas individuales con respecto a garantizar una alta estabilidad, en particular la estabilidad de almacenamiento, y eficacia, de modo que hay un efecto sinérgico en este sentido, como también se muestra usando los ejemplos de realización enumerados más abajo. Sin desear limitarse o comprometerse con la siguiente teoría, con respecto a la composición según la invención como resultado de las medidas relevantes, como se mencionó con anterioridad, existe, por así decirlo, una formación óptima de una matriz o un hidrogel sobre la base de los componentes subyacentes, en particular sobre la base de los componentes (a) y (b), que contrarresta la degradación de las sustancias o ingredientes activos subyacentes o una modificación en las propiedades fisicoquímicas relacionada con el almacenamiento o el tiempo, como la viscosidad o similares. Como resultado, hay un aumento correspondiente en la estabilidad, junto con períodos de almacenamiento más largos.
El sistema tamponante químico según el componente (c) se usa en el contexto de la presente invención, en particular para estabilizar la composición según la invención. En particular, el ajuste de un valor de pH constante por medio del sistema tamponante conduce sorprendentemente a una degradación no deseada de las sustancias activas, en particular los componentes (a) y (b), que se contrarresta de manera eficaz durante el almacenamiento, incluso durante un período de tiempo relativamente largo. De esta manera, la estabilidad o la vida útil de la composición según la invención se mejora aún más.
Con respecto al comportamiento de estabilidad, también se ha encontrado en el contexto de la presente invención de una manera completamente sorprendente que el uso de un (sistema tamponante) tampón de fosfato especial según el componente (c) es superior a otros tampones o sistemas tamponantes, de modo que, debido al uso del tampón (sistema tamponante) de fosfato especial, hay una mejora adicional en las propiedades de estabilidad subyacentes. Igualmente, sin desear restringirse o definir esta teoría, el uso de un tampón especial también conduce a una mayor estabilización de la matriz resultante de los componentes (a) y (b) o del hidrogel relacionado, también porque, sobre la base del sistema tamponante especial, existe una estabilización óptima del valor de pH, que es beneficioso para la estabilidad general de la composición.
Además, la composición según la invención también es notable por su excelente eficacia o eficacia con una simultánea buena tolerabilidad con respecto al tratamiento de enfermedades preferiblemente inflamatorias del tracto urogenital, en particular cistitis, tal como cistitis intersticial. Debido a las propiedades fisicoquímicas constantes definidas de la composición según la invención y que también están presentes después de largos períodos de almacenamiento, se mejora la manipulabilidad o la aplicación, en particular con respecto a la instilación en la vejiga urinaria, lo que también
conduce a una mayor aceptación por parte del paciente y una reducción en el mal uso. Sobre la base del concepto según la invención, se contrarresta en general una degradación temporal o una degradación relacionada con el tiempo de los ingredientes activos o principios activos correspondientes, en particular también con respecto a los componentes (a) y (b), de modo que incluso después de períodos de tiempo correspondientemente largos, están presentes concentraciones de principios activos elevadas o constantes a un pH temporalmente constante. Esto también asegura que a veces no estén presentes productos de degradación tóxicos o dañinos o solo lo estén en pequeñas cantidades, lo que es igualmente positivo para la eficacia y la tolerancia de la composición según la invención.
En general, sobre la base del concepto según la invención, se proporciona una composición que es extraordinariamente adecuada para el tratamiento de enfermedades preferiblemente inflamatorias del tracto urogenital, como la cistitis, y que tiene propiedades significativamente mejoradas en comparación con la técnica anterior.
Con respecto al componente (a) utilizado según la invención en forma de sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (también conocida como sinónimo de sal de polisulfato de condroitina o sal de polisulfato de condroitina), ha resultado ventajoso según la invención cuando la composición presenta o contiene el sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) en una concentración de (4,5±0,4) mg/ml, en particular en una concentración de (4,5±0,3) mg/ml, preferiblemente en una concentración de (4,5±0,15) mg/ml, más preferiblemente, en una concentración de aproximadamente 4,5 mg/ml. En particular, la solicitante ha encontrado que, como se mencionó con anterioridad, con respecto a los intervalos de concentración en cuestión, en particular también en interacción con el componente (b), existe un máximo correspondiente de estabilidad y eficacia con respecto a la composición según la invención.
Además, el peso molecular (también conocido como sinónimo de “masa molar”) del componente (a) es de gran importancia:
Según la invención, se puede prever que el sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) tenga un peso molecular medio en número (masa molar) Mn en el intervalo de 2 kDa a 200 kDa, en particular en el intervalo de 5 kDa a 150 kDa, preferiblemente en el intervalo de 10 kDa a 100 kDa, preferiblemente en el intervalo de 12 kDa a 75 kDa.
En particular, el sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) puede tener un peso molecular medio en peso (masa molar) Mw en el intervalo de 10 kDa a 200 kDa, en particular en el intervalo de 15 kDa a 175 kDa, preferiblemente en el intervalo de 20 kDa a 150 kDa, preferiblemente en el intervalo de 30 kDa a 120 kDa.
Además, el sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) puede tener un peso molecular medio centrifugal (masa molar) Mz en el intervalo de 30 kDa a 1000 kDa, en particular en el intervalo de 40 kDa a 800 kDa, preferiblemente en el intervalo de 50 kDa a 600 kDa, preferiblemente en el intervalo de 100 kDa a 450 kDa.
En este contexto, el índice de polidispersidad (PDI) tiene una correspondiente importancia: a este respecto, según la invención, puede preverse que el sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) tenga un índice de polidispersidad (PDI), calculado como el cociente a partir de peso molecular medio en peso Mw y el peso molecular medio en número Mn (Mw:Mn), de al menos 1, en particular de al menos 1,2, preferiblemente de al menos 1,5, preferiblemente de al menos 2.
Además, el sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) puede tener un índice de polidispersidad (PDI), calculado como el cociente del peso molecular medio en peso Mw y el peso molecular medio en número Mn, de como máximo 30, en particular máximo 20, preferiblemente máximo 10, preferiblemente máximo 8.
Además, el sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) puede tener un índice de polidispersidad (PDI), calculado como el cociente del peso molecular medio en peso Mw y el peso molecular medio en número Mn, en el intervalo de 1 a 30, en particular en el intervalo de 1,2 a 20, preferiblemente en el intervalo de 1,5 a 10, preferiblemente en el intervalo de 2 a 8.
Las propiedades mencionadas con anterioridad del componente (a) con respecto al peso molecular o a la masa molar o al índice de polidispersidad relacionado conducen -sin desear limitar o establecer esta teoría- a una conformación mejorada adicional de la matriz en la que se basa la composición según la invención, en particular también con respecto a la formación de un hidrogel, con la correspondiente estabilización de los ingredientes activos o principios activos respectivos. Igualmente, los pesos moleculares o las masas molares en cuestión en relación con el componente (a) también conducen a una buena eficacia de la composición según la invención con respecto a la enfermedad subyacente, en particular -igualmente sin desear restringir o establecer esta teoría- como resultado de una buena u óptima interacción o adherencia al urotelio de la vejiga.
Sin desear limitar o establecer ninguna teoría a este respecto, este sulfato de condroitina especial es particularmente
adecuado debido a su estructura molecular para regenerar o, en cierta medida, reponer la capa de glicosaminoglicanos del urotelio, en particular en relación con el componente (b), por lo que la permeabilidad de esta capa se reduce. Esto aumenta la función de protección de la capa de glicosaminoglicanos o la capa de mucosa del urotelio (capa de mucina), de modo que los síntomas de la enfermedad pueden aliviarse significativamente o incluso curarse.
Con respecto a la determinación del peso molecular del componente (a) utilizado según la invención, el peso molecular medio en número Mn o el peso molecular medio en peso Mw o el peso molecular medio centrifugal Mz y/o el índice de polidispersidad (PDI) del sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) determinado en particular por medio de cromatografía de permeación en gel (GPC) o según la norma DIN 55672-3:2016-03.
En particular, el peso molecular medio en número Mn o el peso molecular medio en peso Mw o el peso molecular medio centrifugal Mz o el índice de polidispersidad (PDI) del sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) se puede determinar mediante cromatografía de permeación en gel (GPC), en particular a una temperatura en el intervalo de 20 °C a 40 °C o con 0,1 mol/l de solución de NaCl en agua desionizada como eluyente y/o en una solución de sulfato de condroitina y/o la sal de sulfato de condroitina fisiológicamente aceptable con una concentración de 3 g/l y/o utilizando un estándar de dextrano/pululano como reactivo de calibración y/o según la norma DIN 55672-3:2016-03.
De manera preferida según la invención, el sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) pueden ser de origen marino. Al respecto, el sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) puede obtenerse a partir de peces de cartílago, en particular tiburones, preferiblemente cartílago de tiburón. Al respecto, la solicitante ha encontrado, de manera bastante sorprendente, que un sulfato de condroitina de origen marino, particularmente en el tratamiento de enfermedades preferiblemente inflamatorias del tracto urogenital, como la cistitis, conduce a resultados particularmente buenos, en particular si se usa en una combinación apropiada con el ácido hialurónico según el componente (b). El aislamiento de las sustancias activas a este respecto es familiar para el experto en la técnica como tal, de modo que no se requieren explicaciones adicionales a este respecto.
Según la invención, se prevé particularmente que el sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) esté en forma de una sal alcalina, preferiblemente en forma de una sal de sodio, lo que conduce a resultados particularmente buenos en el contexto de la presente invención.
Según la invención, el sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) está preferiblemente en forma de sulfato sódico de condroitina.
Según la invención, el sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) también se puede seleccionar del grupo de condroitina 4-sulfato, condroitina 6-sulfato, condroitina 2,6-sulfato, condroitina 4,6-sulfato y sus combinaciones o mezclas, preferiblemente condroitina 2,6-sulfato (sulfato de condroitina D). Los sulfatos de condroitina especiales antes mencionados también se usan según la invención de una manera preferida en forma de sus sales alcalinas, preferiblemente sales de sodio, que se acompaña de resultados particularmente buenos con respecto al tratamiento de enfermedades preferiblemente inflamatorias del tracto urogenital, tales como la cistitis.
En el contexto de la presente invención, el componente (a) de la composición según la invención puede basarse preferiblemente en una solución o suspensión estéril y/o altamente purificada, en particular de la sal de sodio de sulfato de condroitina.
El sulfato de condroitina o la sal de sulfato de condroitina que se pueden usar en el contexto de la presente invención está generalmente disponible en comercios, por ejemplo, de Nexira, Rouen (Francia), Artesan Pharma GmbH & Co. KG o de Pharma Greven GmbH, Greven (Alemania).
Para más detalles sobre la expresión sulfatos de condroitina, se puede hacer referencia a ROMPP Chemielexikon, 10a edición, volumen 1, 1996, Georg Thieme Verlag Stuttgart/Nueva York, página 736, palabra clave: “Chondroitinsulfate”, y a la literatura a la que se hace referencia allí, donde la totalidad del contenido de divulgación de la literatura mencionada con anterioridad se incorpora por referencia en el presente documento.
Con respecto al componente adicional en forma del ácido hialurónico o la sal de ácido hialurónico fisiológicamente aceptable según el componente (b), que se usa para la composición según la invención, se debe mencionar a este respecto lo siguiente:
Según la invención, generalmente se puede prever que la composición contenga el ácido hialurónico o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) en una concentración de (16±1,2) mg/ml, en particular en una concentración de (16±0,8) mg/ml, preferiblemente en una concentración de (16±0,4) mg/ml, más preferiblemente, en una concentración de aproximadamente 16 mg/ml. Se obtienen propiedades particularmente buenas con respecto a la estabilidad y la eficacia en los intervalos de concentración mencionados con anterioridad, en particular sobre la base de una interacción, en particular con los componentes (a) y/o (c) y el valor del pH especial de la composición según la invención.
En particular con respecto a la estabilidad, preferiblemente estabilidad al almacenamiento, de la composición según la invención y su eficacia en el tratamiento de enfermedades preferiblemente inflamatorias del tracto urogenital, en particular cistitis, preferiblemente cistitis intersticial, también se obtienen resultados particularmente buenos si el ácido hialurónico presenta un peso molecular especial o una masa molar especial.
En este contexto, se prefiere según la invención si el ácido hialurónico o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) presenta un peso molecular medio en número (masa molar) Mn en el intervalo de 10 kDa a 300 kDa, en particular en el intervalo de 20 kDa a 275 kDa, preferiblemente en el intervalo de 30 kDa a 260 kDa, preferiblemente en el intervalo de 50 kDa a 250 kDa, más preferiblemente, en el intervalo de 75 kDa a 200 kDa.
En particular, el ácido hialurónico o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) puede tener un peso molecular medio en peso (masa molar) Mw en el intervalo de 10 kDa a 500 kDa, en particular en el intervalo de 20 kDa a 450 kDa, preferiblemente en el intervalo de 50 kDa hasta 425 kDa, preferiblemente en el intervalo de 100 kDa a 400 kDa, más preferiblemente, en el intervalo de 150 kDa a 395 kDa.
Además, el ácido hialurónico o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) puede tener un peso molecular medio centrifugal (masa molar) Mz en el intervalo de 80 kDa a 1500 kDa, en particular en el intervalo de 100 kDa a 1250 kDa, preferiblemente en el intervalo de 200 kDa a 1000 kDa, preferiblemente en el intervalo de 300 kDa a 750 kDa.
Sobre la base de los pesos moleculares mencionados con anterioridad, una estabilización y eficacia particularmente buenas de la composición según la invención da como resultado en particular una coordinación y combinación intencionadas con los pesos moleculares correspondientes del componente (a), en particular porque, sin desear restringir o establecer esta teoría, sobre la base de los pesos moleculares mencionados con anterioridad, se garantiza una formación óptima de matriz o hidrogel con una estabilización adecuada y una interacción optimizada adicional, en particular con el urotelio de la vejiga.
Al respecto, el índice de polidispersidad del componente (b) utilizado según la invención también es importante:
Según la invención, se puede prever que el ácido hialurónico o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) tenga un índice de polidispersidad (PDI), calculado como el cociente del peso molecular medio en peso Mw y el peso molecular medio en número Mn, de al menos 1, en particular de al menos 1,1, preferiblemente de al menos 1,2, preferiblemente de al menos 1,3, más preferiblemente, de al menos 1,4.
En particular, el ácido hialurónico o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) puede tener un índice de polidispersidad (PDI), calculado como el cociente del peso molecular medio en peso Mw y el peso molecular medio en número Mn, de como máximo 50, en particular máximo 25, preferiblemente máximo 10, preferiblemente máximo 5, más preferiblemente máximo 3.
Además, según la invención, puede preverse que el ácido hialurónico o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) tenga un índice de polidispersidad (PDI), calculado como el cociente del peso molecular medio en peso Mw y el peso molecular medio en número Mn, en el intervalo de 1 a 50, en particular en el intervalo de 1,1 a 25, preferiblemente en el intervalo de 1,2 a 10, preferiblemente en el intervalo de 1,3 a 5, más preferiblemente, en el intervalo de 1,4 a 3.
Los pesos moleculares mencionados con anterioridad se pueden determinar usando métodos conocidos per se por los expertos en la técnica. Según la invención, el peso molecular medio en número Mn o el peso molecular medio en peso Mw y/o el peso molecular medio centrifugal Mz y/o el índice de polidispersidad (PDI) del ácido hialurónico y/o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) se puede determinar en particular por medio de cromatografía de permeación en gel (GPC) y/o según la norma DIN 55672-3:2016-03.
Además, el peso molecular medio en número Mn o el peso molecular medio en peso Mw o el peso molecular medio centrifugal Mz o el índice de polidispersidad (PDI) del ácido hialurónico y/o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) se pueden determinar mediante cromatografía de permeación en gel (GPC), en particular a una temperatura en el intervalo de 20 °C a 40 °C y/o con 0,1 mol/l de solución de NaCl en agua desionizada como eluyente y/o en una solución de ácido hialurónico y/o la sal de ácido hialurónico fisiológicamente aceptable con una concentración de 3 g/l y/o utilizando un estándar de dextrano/pululano como reactivo de calibración y/o según la norma DIN 55672-3:2016-03.
Según la invención, en particular se puede prever que la relación del peso molecular respectivo Mn, Mw o Mz de sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) al peso molecular correspondiente Mn, Mw o Mz del ácido hialurónico y/o el sal de ácido hialurónico fisiológicamente aceptable (componente (b)) esté en un intervalo de 1:3 a 1:4, en particular de 1:1 a 1:100, preferiblemente de 1:1,5 a 1:50, preferiblemente de 1:2 a 1:25, más preferiblemente, de 1:3 a 1:10. Como resultado de la coordinación dirigida de los respectivos pesos moleculares o masas molares del componente (a) y el componente (b), se obtienen resultados particularmente buenos con respecto a la estabilización y la eficacia de la composición según la invención, en particular porque, sin querer restringir o establecer esta teoría, a este respecto, existe una suplementación de la acción o alta compatibilidad de los componentes (a) y (b) en cuestión entre sí.
Según la invención, también se puede prever en particular que el ácido hialurónico o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) sea de origen no animal.
En este contexto, el ácido hialurónico o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) puede ser de origen bacteriano o fermentativo. Al respecto, el ácido hialurónico o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) se puede obtener preferiblemente en forma fermentativa de bacterias del género Streptococcus, en particular Streptococcus lancefields, preferiblemente Streptococcus lancefields cepa A. La extracción o el aislamiento de las sustancias activas a este respecto es familiar para el experto en la técnica como tal, de modo que no se requieren más explicaciones en este sentido.
En este sentido, la solicitante ha encontrado sorprendentemente que el uso de un ácido hialurónico no animal del tipo antes mencionado también mejora de modo significativo la eficacia farmacéutica de la composición según la invención en relación con el tratamiento de enfermedades inflamatorias de la vejiga urinaria, tales como cistitis, preferiblemente cistitis intersticial. Sin querer restringir o establecer una teoría específica, esto puede justificarse por el hecho de que el ácido hialurónico no animal, en particular obtenido bacterianamente, es un producto particularmente puro con propiedades químicas o físicas definidas, que es altamente eficaz. La formación definida del ácido hialurónico de alta pureza es igualmente beneficiosa para la estabilidad de la composición, ya que no hay impurezas que sean perjudiciales para la estabilidad. Además, el ácido hialurónico no animal es bien tolerado, ya que no existe contaminación ni polución con otras sustancias, como suele ser el caso con los productos derivados de animales. El ácido hialurónico de origen no animal tiene, por lo tanto, una pureza y homogeneidad particularmente altas, lo que también es beneficioso para la estabilidad, en particular la estabilidad al almacenamiento, de la composición según la invención.
Según una realización preferida según la invención, el ácido hialurónico o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) puede estar en forma de una sal alcalina, preferiblemente en forma de una sal de sodio, y/o en forma de un hialuronato alcalino. Asimismo, se obtienen resultados particularmente buenos si el ácido hialurónico o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) está en forma de hialuronato de sodio.
El componente (b) de la composición según la invención se puede usar en particular sobre la base de una solución o suspensión estéril o altamente purificada, en particular de la sal de sodio del ácido hialurónico.
El ácido hialurónico o la sal de ácido hialurónico que se pueden usar en el contexto de la presente invención generalmente está disponible en comercios, por ejemplo, de Vivatis Pharma GmbH, Hamburgo (Alemania), Contipro SA, Dolni Dobrouc (República Checa) o de GFN Herstellung von Naturextrakten GmbH, Wald-Michelbach (Alemania).
Para más detalles sobre la expresión ácido hialurónico y sus sales fisiológicamente aceptables, ver ROMPP Chemistry Lexicon, 10a edición, volumen 3, 1997, Georg Thieme Verlag Stuttgart/Nueva York, página 1820, palabra clave: “Hyaluronsaure”, así como la literatura a la que se hace referencia allí, donde el contenido total de la divulgación de la literatura mencionada con anterioridad se incorpora por referencia en el presente documento.
Según la invención, también se puede prever que el sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)), por un lado, y el ácido hialurónico y/o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)), por otro lado, en la composición estén en una relación en peso (relación de concentración) del componente (a), por un lado, al componente (b), por el otro, [(a):(b)] en un intervalo de 1:3 a 1:4, en particular 1:3,2 a 1:3,8, preferiblemente 1:3,4 a 1:3,6, más preferiblemente, aproximadamente 1:3,55.
En general, la solicitante ha logrado lograr una eficacia particularmente buena con respecto al tratamiento de las enfermedades subyacentes mediante la adaptación y coordinación específicas de las sustancias activas respectivas en función de los componentes (a) y (b) en relación con la capa de glicosaminoglicanos del urotelio de la vejiga, ya que, sin desear restringir o establecer ninguna teoría en particular, especialmente con las proporciones cuantitativas mencionadas con anterioridad, existe una interacción o integración particularmente buena de las sustancias activas en la capa de glicosaminoglicanos del urotelio de la vejiga o una buena regeneración de la capa de glicosaminoglicanos del urotelio de la vejiga, en particular en relación con los pesos moleculares o masas molares especiales de los componentes (a) o (b) mencionados con anterioridad. Como resultado, la permeabilidad del urotelio se reduce significativamente, lo que conduce a una reducción significativa de los síntomas asociados con la enfermedad subyacente, en particular la cistitis, especialmente porque las sustancias irritantes ya no pueden penetrar tan profundamente en el urotelio o en las capas inferiores. Las relaciones en peso previamente mencionadas también tienen un efecto positivo sobre la estabilidad de la composición según el sistema tamponante químico.
En lo que respecta al componente (c) utilizado según la invención, la composición puede contener el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) en una concentración total de sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) de (1,75±1,65) mg/ml, en particular en una concentración total de (1,75±1,5) mg/ml, preferiblemente en una concentración total de (1,75±1,25) mg/ml, preferiblemente en una concentración total de (1,75±1) mg/ml, más preferiblemente, en una concentración total de (1,75±0,9) mg/ml, siendo lo más preferible, en una concentración total de (1,75±0,8) mg/ml, incluso más preferiblemente, en una concentración total de aproximadamente 1,75 mg/ml.
En el contexto de la presente invención, el uso dirigido de un sistema tamponante especial, en particular un sistema tamponante químico, según el componente (c) en función del sistema tamponante de dihidrogenofosfato/monohidrogenofosfato ha demostrado ser particularmente ventajoso. Si se utiliza este sistema tamponante especial, particularmente si las concentraciones correspondientes del tampón o los componentes del tampón se cumplen al mismo tiempo, se contrarresta un cambio no deseado en el valor del pH o una degradación no deseada de los ingredientes activos durante el almacenamiento, particularmente durante un período de tiempo más largo, en un grado particular y así se mejora la vida útil de la composición según la invención en particular medida, como se indicó con anterioridad.
En particular, la composición puede contener el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) en una cantidad total de sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) de (87,5±85) mg, en particular en una cantidad total de (87,5±75) mg, preferiblemente en una cantidad total de (87,5±62,5) mg, preferiblemente en una cantidad total de (87,5±50) mg, más preferiblemente, en una cantidad total de (87,5±45) mg, siendo lo más preferible, en una cantidad total de (87,5±40) mg, aún más preferiblemente, en una cantidad total de aproximadamente 87,5 mg.
Además, la composición puede contener el dihidrogenofosfato del sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)), en particular en función de la cantidad pesada cuando se fabricó y/o proporcionó la composición, en una concentración de (0,2±0,19) mg/ml, en particular en una concentración de (0,2±0,15) mg/ml, preferiblemente en una concentración de (0,2±0,125) mg/ml, preferiblemente en una concentración de (0,2±0,1) mg/ml, más preferiblemente, en una concentración de aproximadamente 0,2 mg/ml.
Asimismo, se ha demostrado que es ventajoso si la composición contiene el monohidrogenofosfato del sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)), en particular en función del peso de la muestra cuando la composición se fabrica y/o proporciona, en una concentración de (1,5±1,4) mg/ml, en particular en una concentración de (1,5±1,25) mg/ml, preferiblemente en una concentración de (1,5±1,1) mg/ml, preferiblemente en una concentración de (1,5±1) mg/ml, más preferiblemente, en una concentración de (1,5±0,75) mg/ml, siendo lo más preferible, en una concentración de aproximadamente 1,5 mg/ml.
Con respecto a la estabilización de la composición según la invención, también se ha demostrado que es particularmente ventajoso si la composición contiene el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) en una relación en peso (relación de concentración) de dihidrogenofosfato a monohidrogenofosfato [dihidrogenofosfato:monohidrogenofosfato], en particular en función del pesaje en la preparación y/o provisión de la composición, en el intervalo de 2:1 a 1:100, preferiblemente en el intervalo de 1:1 a 1:75, preferiblemente en el intervalo de 1:2 a 1:50, más preferiblemente, en el intervalo de 1:5 a 1:25.
Según una realización de la invención, el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) puede estar presente o formado como un sistema tamponante de dihidrogenofosfato alcalino/monohidrogenofosfato alcalino. El metal alcalino puede seleccionarse preferiblemente de sodio y/o potasio, en particular sodio.
Además, el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) puede estar presente o formado como un sistema tamponante de dihidrogenofosfato de sodio/monohidrogenofosfato de sodio.
Además, el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) se estar presente o formado como un sistema tamponante de NaH2(PO4)/Na2H(PO4), en particular como un sistema tamponante de NaH2(PO4) • 2 H2O/Na2H(PO4) • 2 H2O.
En general, el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) puede usarse o usarse para ajustar o mantener constante el pH de la composición. Esto da como resultado una estabilización adicional de la composición según la invención, acompañada de una tolerancia particularmente buena de la composición según la invención cuando se usa o aplica.
Los tampones de fosfato que se pueden usar en el contexto de la presente invención están generalmente disponibles en comercios, por ejemplo, de Merck KGaA, Darmstadt (Alemania).
El término tampón o sistema tamponante químico puede referirse en particular a ROMPP Lexikon Chemie, 10a edición, Georg-Thieme-Verlag, Stuttgart/Nueva York, Volumen 5, 1998, páginas 3618/3619, palabra clave: “Puffer”, y a la literatura a la que se hace referencia allí, donde todo el contenido de divulgación de la literatura antes mencionada se incorpora por referencia en el presente documento.
Además de las sustancias o ingredientes activos mencionados con anterioridad, la composición según la invención según el componente (d) puede tener al menos un electrolito fisiológicamente aceptable: en este contexto, la composición puede contener el electrolito (componente (d)) en una concentración de (8±6) mg/ml, en particular en una concentración de (8±4) mg/ml, preferiblemente en una concentración de (8±2) mg/ml, preferiblemente en una concentración de (8±1) mg/ml, más preferiblemente, en una concentración de aproximadamente 8 mg/ml.
En particular, el electrolito (componente (d)) puede estar en forma de una sal alcalina, en particular en forma de cloruro
alcalino, preferiblemente en forma de cloruro de sodio. Según la invención, se prefiere que el electrolito (componente (d)) sea cloruro de sodio.
Los electrolitos que pueden usarse en el contexto de la presente invención están generalmente disponibles en comercios, por ejemplo, de Merck KGaA, Darmstadt (Alemania).
Como resultado del uso de un electrolito, los posibles efectos osmóticos en el curso de la aplicación o el tratamiento con la composición según la invención pueden mantenerse lo más bajos posible, lo que aumenta aún más la compatibilidad de la composición según la invención.
Según una realización preferida según la invención, la presente invención también se refiere, según el presente aspecto, a una composición, en particular una composición farmacéutica, preferiblemente para el tratamiento profiláctico o terapéutico de enfermedades preferiblemente inflamatorias del tracto urogenital, en particular enfermedades inflamatorias de la vejiga urinaria, preferiblemente cistitis, en particular una tal como una composición descrita con anterioridad, donde la composición contiene en combinación y en cada caso en cantidades eficaces, en particular farmacéuticamente eficaces
(a) sulfato sódico de condroitina (condroitinsulfato sódico) a una concentración de (4,5±0,5) mg/ml (componente (a));
(b) hialuronato de sodio a una concentración de (16±1,6) mg/ml (componente (b));
(c) un sistema tamponante de dihidrogenofosfato alcalino/monohidrogenofosfato alcalino (componente (c)), en particular en una concentración total de sistema tamponante de dihidrogenofosfato alcalino/monohidrogenofosfato alcalino de (1,75±1,65) mg/ml;
(d) al menos un electrolito fisiológicamente aceptable (componente (d)), preferiblemente cloruro de sodio, en particular en una concentración de (8±6) mg/ml;
donde la composición tiene un pH en el intervalo de 6,1 a 7,9 y/o donde la composición se ajusta a un pH en el intervalo de 6,1 a 7,9.
Con respecto al pH de la composición según la invención en general, la composición también puede tener un pH en el intervalo de 6,6 a 7,7, en particular en el intervalo de 6,9 a 7,6, preferiblemente en el intervalo de 7,1 a 7,4. En particular, el pH de la composición puede mantenerse constante o ajustado en el intervalo de 6,6 a 7,7, en particular en el intervalo de 6,9 a 7,6, preferiblemente en el intervalo de 7,1 a 7,4. El pH se puede establecer o predefinir preferiblemente por medio del sistema tamponante de dihidrogenofosfato alcalino/monohidrogenofosfato alcalino (componente (c)).
Si se cumplen los intervalos de valores de pH mencionados con anterioridad, se contrarresta una degradación no deseada de los ingredientes activos durante el almacenamiento en un grado particular incluso durante un período de tiempo más largo y la vida útil de la composición según la invención también se mejora en un grado particular.
Debido a las propiedades previamente mencionadas con respecto al pH, la composición según la invención también es particularmente bien tolerada.
En el contexto de la presente invención, el valor del pH puede determinarse usando métodos conocidos per se por los expertos en la técnica. En particular, el valor del pH se puede determinar sobre la base de un análisis potenciométrico. El pH puede determinarse preferiblemente según el método según la Ph. Eur. [Pharmacopoea Europaea], novena edición (9.0), 2017, novena edición en inglés, sección 2.2.3. “Potentiometric determination of pH”.
Para más detalles sobre el concepto del valor del pH, se puede hacer referencia en particular a ROMPP Lexikon Chemie, 10a edición, Georg-Thieme-Verlag, Stuttgart/Nueva York, Volumen 4, 1998, páginas 3230 a 3232, palabra clave: “pH”, y a la literatura a la que se hace referencia allí, donde todo el contenido de divulgación de la literatura mencionada con anterioridad se incorpora por referencia en el presente documento.
En lo que respecta a la composición según la invención, puede tener una viscosidad dinámica de al menos 2000 mPas, en particular al menos 4000 mPas, preferiblemente al menos 5000 mPas, preferiblemente al menos 5250 mPas, a una temperatura de 20 °C. Además, la composición a una temperatura de 20 °C puede tener una viscosidad dinámica de como máximo 7900 mPas, en particular máximo 6900 mPas, preferiblemente máximo 6000 mPas, preferiblemente máximo 5750 mPas.
Según la invención, en particular se puede prever que la composición a una temperatura de 20 °C presente una viscosidad dinámica en el intervalo de 2000 mPas a 7900 mPas, en particular en el intervalo de 4000 mPas a 6900 mPas, preferiblemente en el intervalo de 5000 mPas a 6000 mPas, preferiblemente en el intervalo de 5250 mPas a 5750 mPas.
Debido a la viscosidad prevista o establecida según la invención, se asegura una instilación particularmente simple y menos dolorosa de la composición en la vejiga. Además, debido a la viscosidad prevista según la invención, existe una interacción particularmente buena del componente activo en el que se basa la composición según la invención con el urotelio, que tiene un efecto positivo sobre la eficacia. Además, el ajuste dirigido de la viscosidad también
conduce a una mayor estabilización de la composición, en particular con respecto a la formación de una matriz definida o un hidrogel definido, sin restringir o querer establecer esta teoría, lo que conlleva una estabilidad de almacenamiento de la composición según la invención.
La viscosidad dinámica se puede determinar utilizando métodos conocidos por el experto en la técnica. En particular, la viscosidad dinámica según el método según la Ph. Eur. [Pharmacopoea Europaea], novena edición (9.0), 2017, novena edición en inglés, sección 2.2.8. “Viscosity” y sección 2.2.9. “Capillary viscometer method”.
Además, la composición según la invención puede tener una osmolaridad en el intervalo de 150 mosmol/kg a 600 mosmol/kg, en particular en el intervalo de 200 mosmol/kg a 550 mosmol/kg, preferiblemente en el intervalo de 250 mosmol/kg a 500 mosmol/kg, preferiblemente en el intervalo de 275 mosmol/kg a 450 mosmol/kg, más preferiblemente, en el intervalo de 300 mosmol/kg a 400 mosmol/kg. La configuración específica de la osmolaridad también sirve para estabilizar aún más y, en particular, para mejorar la tolerancia de la composición según la invención.
La osmolaridad se puede determinar según métodos conocidos per se por el experto en la técnica. En particular, la osmolaridad se puede determinar según el método según la Ph. Eur. [Pharmacopoea Europaea], novena edición (9.0), 2017, novena edición en inglés, sección 2.2.35. “Osmolality”. La osmolaridad también se puede establecer, por ejemplo, en función del electrolito mencionado con anterioridad (componente (d)), por ejemplo, a base de iones alcalinos, tales como iones sodio e iones cloruro, en particular por medio de cloruro de sodio.
Además, la composición a una temperatura de 20 °C y a una presión de 1013,25 mbar (presión atmosférica) puede presentar una densidad en el intervalo de 1,001 g/cm3 a 1,5 g/cm3, en particular en el intervalo de 1,005 g/cm3 a 1,25 g/cm3, preferiblemente en el intervalo de 1,0075 g/cm3 a 1,1 g/cm3, preferiblemente en el intervalo de 1,0075 g/cm3 a 1,075 g/cm3, más preferiblemente, en el intervalo de 1,0075 g/cm3 a 1,05 g/cm3.
En particular, la composición puede presentar una densidad relativa, en función del agua pura, en el intervalo de 1,001 a 1,5, en particular en el intervalo de 1,005 a 1,25, a una temperatura de 20 °C y a una presión de 1.013,25 mbar (presión atmosférica), preferiblemente en el intervalo de 1,0075 a 1,1, preferiblemente en el intervalo de 1,0075 a 1,075, más preferiblemente, en el intervalo de 1,0075 a 1,05.
Dentro del alcance de la presente invención, la densidad se puede determinar usando métodos conocidos por los expertos en la técnica. En particular, la densidad relativa se puede determinar según el método según la Ph. Eur.
[Pharmacopoea Europaea], novena edición (9.0), 2017, novena edición en inglés, sección 2.2.5. “Relative density”. El manejo y la tolerancia de la composición según la invención pueden mejorarse adicionalmente mediante el ajuste apropiado de la densidad.
Además, se prefiere en el contexto de la presente invención si la composición está en forma de una composición acuosa. En este contexto, se puede prever en particular que la composición tenga base acuosa o se formule en forma acuosa.
En particular, la composición puede estar en forma de una solución acuosa o una suspensión acuosa, preferiblemente en forma de una solución acuosa.
Al respecto, la composición puede contener agua, en particular agua purificada. En particular, la composición puede contener agua como vehículo (excipiente) farmacéuticamente aceptable. En general, la composición puede por consiguiente estar formada a base de agua, en particular usando agua para fines de inyección.
En cuanto a la cantidad de agua utilizada, ella puede variar ampliamente. Según la invención, se prefiere que la composición según la invención presente un contenido de agua, en particular agua purificada, de al menos el 50% en peso, en particular al menos el 75% en peso, preferiblemente al menos el 80% en peso, preferiblemente al menos el 90% en peso, más preferiblemente, al menos el 95% en peso, en función de la composición.
El agua que se puede usar en el contexto de la presente invención, en particular agua para fines de inyección, generalmente está disponible en comercios, por ejemplo, de Fresenius Kabi Deutschland GmbH, Bad Homburg (Alemania).
Según una realización de la invención, se puede prever que la composición según la invención consista en los componentes mencionados con anterioridad (a), (b), (c) y, en caso necesario, (d) y, en caso necesario, agua, en particular agua purificada.
Además, se puede prever según la invención que la composición según la invención esté al menos sustancialmente libre de disolventes orgánicos y/o dispersantes orgánicos, en particular disolventes a base de alcohol y/o dispersantes a base de alcohol. En particular, se puede prever según la invención que la composición esté al menos sustancialmente libre de alcoholes. Esto aumenta aún más la tolerancia de la composición según la invención.
No menos importante debido a la alta estabilidad, en particular la estabilidad de almacenamiento, de la composición según la invención, también se puede prever en el contexto de la presente invención que la composición esté al menos esencialmente libre de conservantes o al menos esencialmente libre de desinfectantes. Esto también mejora la
tolerancia de la composición según la invención, ya que según la invención se puede prescindir del uso de aditivos que pueden irritar el urotelio o la membrana mucosa de la vejiga urinaria.
Como se mencionó con anterioridad, ha sido posible dentro del alcance de la presente invención proporcionar una composición que se distingue por una excelente estabilidad, de modo que la composición pueda almacenarse durante largos períodos de almacenamiento. Sobre esta base, la seguridad de la aplicación se incrementa aún más.
En particular, la composición puede ser estable, en particular estable al almacenamiento, a temperaturas en el intervalo de 20 °C a 45 °C, a una presión de 1.013,25 mbar (presión atmosférica) y a una humedad atmosférica relativa en el intervalo del 50% al 90% durante al menos 6 meses, en particular al menos 12 meses, preferiblemente al menos 24 meses, preferiblemente al menos 36 meses. En este contexto, la composición al momento de almacenamiento respectivo puede tener un contenido total de productos de degradación de los componentes (a) y (b) de como máximo el 5%, en particular máximo el 4%, preferiblemente máximo el 3%, preferiblemente máximo el 2%, más preferiblemente, máximo el 1%, en función de la concentración total de los componentes (a) y (b) o de los eductos.
En particular, la composición puede tener una estabilidad, en particular estabilidad de almacenamiento, en condiciones de envejecimiento acelerado según la norma ASTM F 1980 [ASTM F 1980: Standard Guide for Accelerated Aging of Sterile Barrier Systems for Medical Devices; 2007-04] a una temperatura de envejecimiento de 55 °C de al menos 6 meses, en particular al menos 12 meses, preferiblemente al menos 24 meses, preferiblemente al menos 36 meses. En particular, la composición en este contexto, al momento de almacenamiento respectivo, puede presentar un contenido total de productos de degradación de los componentes (a) y (b) de como máximo el 5%, en particular máximo el 4%, preferiblemente máximo el 3%, preferiblemente máximo el 2%, más preferiblemente, máximo el 1%, en función de la concentración total de los componentes (a) y (b) o los eductos.
Además, la composición según la invención, si está presente, se puede distinguir por un bajo contenido de constituyentes alcalinotérreos:
Según una realización de la invención, la composición puede tener una concentración de sal alcalinotérrea, en particular una concentración de sal de calcio o concentración de sal de magnesio, como máximo 1 mg/ml, en particular máximo 0,5 mg/ml, preferiblemente máximo 0,1 mg/ml, preferiblemente máximo 0,05 mg/ml, más preferiblemente máximo 0,01 mg/ml.
En el contexto de la presente invención, la composición puede estar además al menos sustancialmente libre de sales alcalinotérreas, en particular al menos esencialmente libre de sales de calcio y/o sales de magnesio.
En general, la composición según la invención puede tener una concentración de metal alcalinotérreo, en particular concentración de calcio o concentración de magnesio, de como máximo 1 mg/ml, en particular máximo 0,5 mg/ml, preferiblemente máximo 0,1 mg/ml, preferiblemente máximo 0,05 mg/ml, más preferiblemente, máximo 0,01 mg/ml.
En particular, la composición según la invención puede estar al menos esencialmente libre de metales alcalinotérreos, en particular al menos esencialmente libre de sales de calcio o de magnesio.
Además, la composición puede tener una concentración de iones bivalentes, en particular cationes bivalentes, preferiblemente iones alcalinotérreos, preferiblemente iones calcio o iones magnesio, de como máximo 1 mg/ml, en particular máximo 0,5 mg/ml, preferiblemente máximo 0,1 mg/ml, preferiblemente máximo 0,05 mg/ml, más preferiblemente, máximo 0,01 mg/ml.
En particular, la composición puede estar al menos sustancialmente libre de iones bivalentes, en particular cationes bivalentes, preferiblemente iones alcalinotérreos, preferiblemente iones calcio y/o iones magnesio.
El concepto según la invención, según el cual la composición de la invención puede estar al menos sustancialmente libre de sales alcalinotérreas o metales alcalinotérreos, en particular en forma iónica, según las realizaciones anteriores, conduce en particular al hecho de que se reduce o previene una interacción a este respecto con los componentes (a) o (b) o (c), en particular con el fin de evitar la formación de complejos no deseados o similares. En consecuencia, la precipitación de componentes activos o componentes de tampón se puede contrarrestar sobre esta base. En particular, un cambio indeseable en la viscosidad, en particular como resultado de la formación no controlada de complejos o similares, también se puede evitar sobre esta base.
Según una realización de la invención, la composición, en particular lista para usar, dosificar y/o aplicar, puede estar presente con un volumen de (50±10) ml, en particular (50±5) ml, preferiblemente (50±2) ml, preferiblemente (50±1) ml, más preferiblemente (50±0,5) ml, siendo lo más preferible, aproximadamente 50 ml.
En este contexto, la composición, en particular lista para usar, dosificar y/o aplicar, puede estar presente y/o preparada para la administración, en particular la instilación en la vejiga urinaria, de un volumen de la composición de (50±10) ml, en particular (50±5) ml, preferiblemente (50±2) ml, preferiblemente (50±1) ml, más preferiblemente, (50±0,5) ml, siendo lo más preferible, aproximadamente 50 ml. En particular, la composición, en particular lista para usar, dosificar y/o aplicar, en particular por instilación en la vejiga urinaria, se puede administrar con un volumen de la composición de (50±10) ml, en particular (50±5) ml, preferiblemente (50±2) ml, preferiblemente (50±1) ml, más preferiblemente,
(50±0,5) ml, siendo lo más preferible, aproximadamente 50 ml.
Igualmente, la composición, en particular lista para usar, dosificar o aplicar, puede estar presente con una cantidad de sustancia activa de sulfato de condroitina y/o sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) de (225±25) mg, en particular (225±15) mg, preferiblemente (225±10) mg, preferiblemente de aproximadamente 225 mg.
La composición según la invención también puede estar presente o preparada, en particular, lista para su uso, dosificación y/o aplicación, para la administración de una cantidad de ingrediente activo de sulfato de condroitina o sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) de (225±25) mg, en particular (225±15) mg, preferiblemente (225±10) mg, preferiblemente, de aproximadamente 225 mg.
En este contexto, la composición, en particular lista para usar, dosificar y/o aplicar, se puede administrar con una cantidad de ingrediente activo de sulfato de condroitina y/o sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) de (225±25) mg, en particular (225±15) mg, preferiblemente (225±10) mg, preferiblemente de aproximadamente 225 mg.
En este contexto, la composición según la invención también puede estar presente, en particular, lista para usar, dosificar o aplicar, con una cantidad de ingrediente activo de ácido hialurónico o sal de ácido hialurónico fisiológicamente aceptable (hialuronato) (componente (b)) de (800±80) mg, en particular (800±60) mg, preferiblemente (800±40) mg, preferiblemente de aproximadamente 800 mg.
Según la invención, la composición en particular puede estar presente y/o preparada, lista para usar, dosificar o aplicar, para la administración de una cantidad de ingrediente activo de ácido hialurónico y/o sal de ácido hialurónico fisiológicamente aceptable (hialuronato) (componente (b)) de (800±80) mg, en particular (800±60) mg, preferiblemente (800±40) mg, preferiblemente de aproximadamente 800 mg. En particular, la composición, en particular lista para usar, dosificar y/o aplicar, se puede administrar con una cantidad de ingrediente activo de ácido hialurónico o sal de ácido hialurónico fisiológicamente aceptable (hialuronato) (componente (b)) de (800±80) mg, en particular (800±60) mg, preferiblemente (800±40) mg, preferiblemente de aproximadamente 800 mg.
Según la invención, por lo tanto, de manera específica, se presta particular atención a una composición especialmente medida con respecto a los componentes activos (a) y (b), según qué componente (a) está presente de manera intencional en cantidades o dosis relativamente pequeñas, mientras que para el componente (b) se prevén cantidades o dosis relativamente grandes, donde la composición también está preferiblemente lista para usar con un volumen definido, como se definió previamente. En este sentido, la solicitante ha descubierto, de una manera completamente sorprendente, que las cantidades correspondientes y coordinadas de ingrediente activo, combinadas con una alta estabilidad de la composición según la invención, conducen a una eficacia particularmente buena con respecto a las enfermedades subyacentes del tracto urogenital, en particular cistitis, preferiblemente cistitis intersticial, de modo que, en este sentido, existe un óptimo de estabilidad o eficacia. Al respecto, el volumen especial, como se definió con anterioridad, es de gran importancia en la medida en que, sobre esta base, existe una adaptación óptima de las cantidades de administración de la composición por administrar o instilar a la situación patológica, en particular en el caso de cistitis, preferiblemente cistitis intersticial, después de lo cual a menudo puede haber una capacidad reducida de la vejiga o una llamada vejiga retráctil. Mediante la administración o instilación del volumen especial subyacente, se puede aumentar el tiempo de retención o la duración y, por lo tanto, el resto de la composición en la vejiga, lo que mejora aún más la eficacia de la composición según la invención.
En el contexto de la presente invención, se puede prever que la composición se introduzca o esté presente en un dispositivo de almacenamiento o aplicación, en particular en un recipiente de almacenamiento o aplicación, en particular listo para usar, dosificar o aplicar.
En este contexto, la composición puede así estar introducida, en particular lista para usar, dosificar o aplicar, en un dispositivo de almacenamiento o aplicación, en particular en un recipiente de almacenamiento y/o aplicación, preferiblemente en tamaños de volumen y/o con un volumen de la composición de (50±10) ml, en particular (50±5) ml, preferiblemente (50±2) ml, preferiblemente (50±1) ml, más preferiblemente, (50±0,5) ml, siendo lo más preferible, aproximadamente 50 ml, por recipiente o unidad de aplicación (unidad de dosificación).
En este contexto, en particular, el dispositivo de almacenamiento o aplicación según la invención puede ser una jeringa preferiblemente estéril, en particular una jeringa de aplicación, preferiblemente una jeringa de aplicación desechable, en particular con un volumen de retención o llenado de (50±10) ml, en particular (50±5) ml, preferiblemente (50±2) ml, preferiblemente (50±1) ml, más preferiblemente, (50±0,5) ml, siendo lo más preferible, aproximadamente 50 ml. Por el contrario, el dispositivo de almacenamiento o aplicación también puede estar en forma de un vial, preferiblemente con cierre estéril, o similar.
Según el presente aspecto, la presente invención también se refiere a la composición según la invención para instilación o preferiblemente aplicación tópica en el área urogenital, en particular en la vejiga urinaria.
En particular, la composición según la invención puede prepararse para administración o para instilación o preferiblemente aplicación tópica en el tracto urogenital, en particular en la vejiga urinaria. Asimismo, la composición
según la invención puede administrarse en este contexto por instilación o, preferiblemente, para aplicación tópica en el tracto urogenital, en particular en la vejiga urinaria.
Asimismo, la presente invención también se refiere a la composición según la invención para uso en el tratamiento profiláctico o terapéutico de enfermedades preferiblemente inflamatorias del tracto urogenital, en particular enfermedades inflamatorias de la vejiga urinaria, preferiblemente cistitis, en particular cistitis aguda o crónica, preferiblemente cistitis intersticial, cistitis radiogénica, crónica cistitis recurrente, quimiocistitis, cistitis abacteriana crónica y cistitis bacteriana crónica, más preferiblemente, cistitis intersticial.
Asimismo, la presente invención también se refiere a la composición según la invención para el tratamiento profiláctico o terapéutico de enfermedades preferiblemente inflamatorias del tracto urogenital, en particular enfermedades inflamatorias de la vejiga urinaria, preferiblemente de cistitis, en particular cistitis aguda o crónica, preferiblemente cistitis intersticial, cistitis radiogénica, cistitis recurrente crónica, quimiocistitis, cistitis abacteriana crónica y cistitis bacteriana crónica, más preferiblemente, cistitis intersticial.
Por lo tanto, la composición según la invención es adecuada para su uso en el tratamiento profiláctico o terapéutico de enfermedades preferiblemente inflamatorias del tracto urogenital, en particular enfermedades inflamatorias de la vejiga urinaria, preferiblemente cistitis, en particular cistitis aguda o crónica, preferiblemente cistitis intersticial, cistitis radiogénica, cistitis recurrente crónica, quimiocistitis, cistitis abacteriana crónica y cistitis bacteriana crónica, más preferiblemente, cistitis intersticial.
La composición según la invención, como se definió con anterioridad, también es adecuada para producir un fármaco o medicamento para el tratamiento profiláctico o terapéutico de enfermedades preferiblemente inflamatorias del tracto urogenital, en particular enfermedades inflamatorias de la vejiga urinaria, preferiblemente de cistitis, en particular cistitis aguda o crónica, preferiblemente cistitis intersticial, cistitis radiogénica, cistitis recurrente crónica, quimiocistitis, cistitis abacteriana crónica y cistitis bacteriana crónica, más preferiblemente, cistitis intersticial.
La composición o combinación según la invención, que contiene sulfato de condroitina o una sal de sulfato de condroitina fisiológicamente aceptable (componente (a)), por un lado, y ácido hialurónico o una sal de ácido hialurónico fisiológicamente aceptable (componente (b)), por otro lado, en una concentración específica y también un sistema tamponante de fosfato especial (componente (c)), además de la sorprendente alta estabilidad encontrada, en particular la estabilidad al almacenamiento, también conduce a una eficacia particularmente buena con respecto a las enfermedades mencionadas con anterioridad. Se puede ver una posible explicación de la excelente acción de la composición según la invención, sin restringirse ni desear establecer esta teoría, en el hecho de que las sustancias activas de la composición según la invención interactúan de una manera particularmente eficaz, en particular con el urotelio de la vejiga urinaria, o el almacenamiento de las sustancias activas en esta capa, lo que conduce a una reparación de la capa de glicosaminoglicanos dañados por la enfermedad o a una regeneración relacionada. La reducción en la permeabilidad del urotelio asociada con este modo de acción, que en cierto sentido equivale a un efecto de “sellado” contra la orina presente en la vejiga, conduce a un alivio significativo de los síntomas causados por la enfermedad, en particular también con respecto a los síntomas de dolor subyacentes. Por lo tanto, el uso de la composición según la invención conduce a un estado de salud significativamente mejorado después de solo unos pocos tratamientos en pacientes afectados por las enfermedades mencionadas con anterioridad. La composición según la invención, por lo tanto, en cierta medida, también puede servir al menos para reemplazar temporalmente una capa defectuosa de glicosaminoglicanos del urotelio.
En este contexto, el modo de acción de la composición según la invención, sin desear estar sujeto a ella, se puede ver en particular en una interacción física significativa en la que los ingredientes activos según el componente (a) o (b) se incorporan o depositan en el urotelio o se adhieren a él, para que una pérdida de sulfato de condroitina o ácido hialurónico en la pared de la vejiga o en el urotelio causada por reacciones inflamatorias en particular sea equilibrada o compensada. Hasta cierto punto, existe una regulación de la permeabilidad de la pared de la vejiga, lo que conduce a una contención de la reacción inflamatoria y, por lo tanto, a apoyar la curación de la herida y, por lo tanto, a una mejora general del estado de salud. La composición según la invención forma casi una protección del epitelio de la vejiga contra sustancias irritantes, tales como bacterias, microcristales o similares, actuando la composición según la invención como un reemplazo y protección para la capa de glicosaminoglicanos en la vejiga y las vías urinarias eferentes.
Como parte de su uso, la composición según la invención puede instilarse o aplicarse tópicamente en el área urogenital, en particular en la vejiga urinaria. La instilación o aplicación debe realizarse preferiblemente en la vejiga urinaria, que preferiblemente se vacía previamente. Al respecto, la composición según la invención puede permanecer en la vejiga urinaria, por ejemplo, durante un período de varios minutos a unas pocas horas para permitir que las sustancias activas tengan un efecto óptimo sobre el urotelio. La composición especialmente concentrada según la invención permite una interacción particularmente eficaz de las sustancias activas presentes en las cantidades o dosis especiales respectivas en forma de componentes (a) o (b) con el urotelio, con una acumulación o incorporación correspondientemente alta de las sustancias activas en cuestión sobre o en la capa subyacente de la vejiga.
En este contexto, puede llevarse a cabo en particular de tal manera que el volumen subyacente de la composición, que se encuentra en particular en el dispositivo de almacenamiento o aplicación que aún se describe a continuación,
se instile en la vejiga después de que la vejiga urinaria se haya vaciado por completo, donde el volumen subyacente debería ser en particular de aproximadamente 50 ml. Para lograr resultados óptimos, la composición según la invención debe permanecer en la vejiga el mayor tiempo posible, como se describió con anterioridad, durante un período de varios minutos a varias horas.
Por ejemplo, en el curso del tratamiento de la cistitis, la instilación de la composición según la invención se puede llevar a cabo una vez por semana durante un período de cuatro semanas, siendo la dosis única respectiva de sulfato de condroitina o componente (a) de aproximadamente 225 mg y la dosis única de ácido hialurónico o el componente (b) debe tener aproximadamente 800 mg en un volumen de composición de aproximadamente 50 ml. En el contexto de la terapia de mantenimiento, la composición según la invención se puede usar, por ejemplo, una vez al mes, en particular hasta que los síntomas hayan desaparecido por completo. Sin embargo, también puede haber desviaciones del esquema de terapia de ejemplo mencionado con anterioridad, si esto es necesario caso por caso.
Otra ventaja de los usos de la composición según la invención es que es segura de usar y que no se producen efectos secundarios significativos durante el tratamiento, en particular dado que las sustancias utilizadas son sustancias biocompatibles. La tolerancia particularmente buena está relacionada en especial con el uso de sulfato de condroitina de origen marino o ácido hialurónico de origen no animal.
Mediante el uso apropiado de las dosis o cantidades especiales de sustancias activas mencionadas con anterioridad a base de los componentes activos (a) y (b), la composición de la invención también es altamente eficaz, que tampoco se ve afectada negativamente por el sistema tamponante especial de fosfato. Más bien, el uso dirigido del sistema tamponante en cuestión según el componente (c) en combinación con las otras medidas según la invención conduce a una estabilización efectiva de la composición, lo que conduce a un alto nivel de seguridad de la aplicación y el mantenimiento de la eficacia o la acción.
En resumen, en el caso de la composición según la invención, se trata preferiblemente de un producto médico que, según una realización preferida según la invención, contiene una solución o dispersión especialmente dosificada de los principios activos a base de los componentes (a) y (b).
En general, la composición se puede administrar al menos una vez a la semana, preferiblemente durante un período de preferiblemente al menos un mes, por instilación o para aplicación tópica en la vejiga.
En lo que respecta a la composición según la invención como tal o sus usos, la composición puede administrarse o instilarse en particular con un volumen de la composición de (50±10) ml, en particular (50±5) ml, preferiblemente (50±2) ml, preferiblemente (50±1) ml, más preferiblemente, (50±0,5) ml, siendo lo más preferible, aproximadamente 50 ml, y/o una cantidad de sustancia activa en sulfato de condroitina y/o sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) de (225±25) mg, en particular (225±15) mg, preferiblemente (225±10) mg, preferiblemente de aproximadamente 225 mg, o una cantidad de ingrediente activo de ácido hialurónico o sal de ácido hialurónico fisiológicamente aceptable (hialuronato) (componente (b)) de (800±80) mg, en particular (800±60) mg, preferiblemente (800±40) mg, preferiblemente de aproximadamente 800 mg, en particular al menos una vez a la semana, preferiblemente durante un período de preferiblemente al menos un mes, o puede aplicarse preferiblemente en la vejiga urinaria en forma tópica o puede estar preparada para al menos una administración semanal (al menos una vez a la semana), preferiblemente durante un período de preferiblemente al menos un mes, por instilación o aplicación tópica en la vejiga.
En este contexto, la composición según la invención también se distingue por una excelente estabilidad de almacenamiento y por una alta eficacia con cantidades optimizadas de sustancia activa, como se definió previamente.
Según aún otro aspecto de la presente invención, la presente invención se refiere igualmente a un dispositivo de almacenamiento o aplicación, en particular un recipiente de almacenamiento o aplicación, en particular en forma de una jeringa preferiblemente estéril, en particular una jeringa de aplicación, preferiblemente una jeringa de aplicación desechable, preferiblemente para instilación o preferiblemente para aplicación tópica en el área urogenital, en particular en la vejiga urinaria, que contiene una composición según la invención, como se definió previamente.
En este contexto, la presente invención en este aspecto también se refiere a un dispositivo de almacenamiento o aplicación, en particular un recipiente de almacenamiento o aplicación, en particular en forma de una jeringa preferiblemente estéril, en particular una jeringa de aplicación, preferiblemente una jeringa de aplicación desechable, preferiblemente para instilación o preferiblemente para aplicación tópica en el área urogenital, en particular en la vejiga urinaria, que contiene una composición como se definió previamente,
donde la composición contiene en combinación y, en cada caso, en cantidades eficaces, en particular farmacéuticamente eficaces
(a) sulfato de condroitina y/o una sal de sulfato de condroitina fisiológicamente aceptable en una cantidad de ingrediente activo de (225±25) mg (componente (a));
(b) ácido hialurónico y/o una sal de ácido hialurónico fisiológicamente aceptable (hialuronato) en una cantidad de ingrediente activo de (800±80) mg (componente (b));
(c) un sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c));
(d) opcionalmente al menos un electrolito fisiológicamente aceptable (componente (d));
donde la composición presenta un valor de pH en el intervalo de 6,1 a 7,9 y/o donde la composición se ajusta a un pH en el intervalo de 6,1 a 7,9.
Según la invención, se prefiere si el dispositivo de almacenamiento o aplicación tiene un volumen de retención o llenado de (50±10) ml, en particular (50±5) ml, preferiblemente (50±2) ml, preferiblemente (50±1) ml, más preferiblemente, (50±0,5) ml, siendo lo más preferible, aproximadamente 50 ml.
En particular, la composición puede tener una cantidad de ingrediente activo de (a) sulfato de condroitina o sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) de (225±15) mg, preferiblemente (225±10) mg, preferiblemente de aproximadamente 225 mg.
Además, la composición puede tener una cantidad de ingrediente activo (b) ácido hialurónico o sal de ácido hialurónico fisiológicamente aceptable (hialuronato) (componente (b)) de (800±60) mg, preferiblemente (800±40) mg, preferiblemente de aproximadamente 800 mg.
Según la invención, también se puede prever que la composición presente el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) en una cantidad total de sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) de (87,5±85) mg, en particular en una cantidad total de (87,5±75) mg, preferiblemente en una cantidad total de (87,5±62,5) mg, preferiblemente en una cantidad total de (87,5±50) mg, más preferiblemente, en una cantidad total de (87,5±45) mg, siendo lo más preferible, en una cantidad total de (87,5±40) mg, incluso más preferiblemente, en una cantidad total de aproximadamente 87,5 mg.
En particular, la composición puede contener el electrolito (componente (d)) en una cantidad de (400±300) mg, en particular en una cantidad de (400±200) mg, preferiblemente en una cantidad de (400±100) mg, preferiblemente en una cantidad de (400±50) mg, más preferiblemente, en una cantidad de aproximadamente 400 mg.
Según la invención, también se puede prever que el sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) esté en forma de una sal alcalina, preferiblemente en forma de una sal de sodio.
Del mismo modo, se puede prever según la invención que el sulfato de condroitina o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) esté presente en forma de sulfato de condroitina de sodio (sulfato de condroitina de sodio).
Además, el ácido hialurónico o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) puede estar presente en forma de una sal alcalina, preferiblemente en forma de una sal de sodio, y/o en forma de un hialuronato alcalino.
En particular, el ácido hialurónico o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) puede estar presente en forma de hialuronato de sodio.
Además, el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) puede estar presente o formado como un sistema tamponante de dihidrogenofosfato alcalino/monohidrogenofosfato alcalino. Al respecto, el metal alcalino puede seleccionarse de entre sodio y/o potasio, en particular sodio.
En particular, el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) puede estar presente o formado como un sistema tamponante de dihidrogenofosfato de sodio/monohidrogenofosfato de sodio.
Además, el electrolito (componente (d)) puede estar presente en forma de una sal alcalina, en particular en forma de cloruro alcalino, preferiblemente en forma de cloruro de sodio. En particular, el electrolito (componente (d)) puede ser cloruro de sodio.
En lo que respecta a la composición, puede tener un pH en el intervalo de 6,6 a 7,7, en particular en el intervalo de 6,9 a 7,6, preferiblemente en el intervalo de 7,1 a 7,4.
En particular, el valor del pH de la composición puede mantenerse constante o ajustarse en el intervalo de 6,6 a 7,7, en particular en el intervalo de 6,9 a 7,6, preferiblemente en el intervalo de 7,1 a 7,4. El valor del pH se puede establecer o predefinir preferiblemente por medio del sistema tamponante de dihidrogenofosfato alcalino/monohidrogenofosfato alcalino (componente (c)).
En particular, la composición puede presentar una viscosidad dinámica de al menos 2.000 mPas, en particular al menos 4.000 mPas, preferiblemente al menos 5.000 mPas, preferiblemente al menos 5.250 mPas, a una temperatura de 202C.
Asimismo, la composición a una temperatura de 20 °C puede presentar una viscosidad dinámica de como máximo
7.900 mPas, en particular máximo 6.900 mPas, preferiblemente máximo 6.000 mPas, preferiblemente máximo 5.750 mPas.
Además, la composición puede presentar una viscosidad dinámica a una temperatura de 20 °C en el intervalo de 2.000 mPas a 7.900 mPas, en particular en el intervalo de 4.000 mPas a 6.900 mPas, preferiblemente en el intervalo de 5.000 mPas a 6.000 mPas, preferiblemente en el intervalo de 5.250 mPas a 5.750 mPas.
Además, la composición puede presentar una osmolaridad en el intervalo de 150 mosmol/kg a 600 mosmol/kg, en particular en el intervalo de 200 mosmol/kg a 550 mosmol/kg, preferiblemente en el intervalo de 250 mosmol/kg a 500 mosmol/kg, preferiblemente en el intervalo de 275 mosmol/kg a 450 mosmol/kg, más preferiblemente, en el intervalo de 300 mosmol/kg a 400 mosmol/kg.
Además, a una temperatura de 20 °C y a una presión de 1.013,25 mbar (presión atmosférica), la composición puede presentar una densidad en el intervalo de 1,001 g/cm3 a 1,5 g/cm3, en particular en el intervalo de 1,005 g/cm3 a 1,25 g/cm3, preferiblemente en el intervalo de 1,0075 g/cm3 a 1,1 g/cm3, preferiblemente en el intervalo de 1,0075 g/cm3 a 1,075 g/cm3, más preferiblemente, en el intervalo de 1,0075 g/cm3 a 1,05 g/cm3.
En particular, la composición puede presentar una densidad relativa, en función del agua pura, en el intervalo de 1,001 a 1,5, en particular en el intervalo de 1,005 a 1,25, a una temperatura de 20 °C y a una presión de 1.013,25 mbar (presión atmosférica), preferiblemente en el intervalo de 1,0075 a 1,1, preferiblemente en el intervalo de 1,0075 a 1,075, más preferiblemente, en el intervalo de 1,0075 a 1,05.
Según la invención, se prefiere si el dispositivo de almacenamiento o aplicación contiene la composición, en particular lista para usar, dosificar y/o aplicar, con un volumen de (50±10) ml, en particular (50±5) ml, preferiblemente (50±2) ml, preferiblemente (50±1) ml, más preferiblemente, (50±0,5) ml, siendo lo más preferible, aproximadamente 50 ml.
En particular, la composición puede estar presente con un volumen de (50±10) ml, en particular (50±5) ml, preferiblemente (50±2) ml, preferiblemente (50±1) ml, más preferiblemente, (50±0,5) ml, más preferiblemente de aproximadamente 50 ml.
Según la invención, también se prefiere si la composición está presente en forma de una composición acuosa. En particular, la composición puede tener una base particularmente acuosa o puede formularse en forma acuosa, en particular en forma de una solución acuosa o suspensión acuosa, preferiblemente en forma de una solución acuosa.
En particular, la composición puede contener agua, en particular agua purificada. En particular, la composición puede contener agua como vehículo (excipiente) farmacéuticamente aceptable. Según la invención, la composición puede estar formada con base acuosa. En este contexto, la composición puede presentar un contenido de agua de al menos 50% en peso, en particular al menos 75% en peso, preferiblemente al menos 80% en peso, preferiblemente al menos 90% en peso, más preferiblemente, al menos 95% en peso, en función de la composición.
Según la invención, la composición según la invención prevista para el dispositivo de almacenamiento o aplicación puede consistir en los componentes mencionados con anterioridad (a), (b), (c) y, en caso necesario, (d) y agua.
Para más detalles sobre el dispositivo de almacenamiento o aplicación según la invención, que contiene la composición según la invención, también se puede hacer referencia a las realizaciones de los otros aspectos de la invención, que se aplican correspondientemente en relación con el dispositivo de almacenamiento o aplicación según la invención.
Además, según otro aspecto más de la presente invención, la presente invención también se refiere a la unidad de embalaje según la invención, que contiene al menos un dispositivo de almacenamiento o aplicación, como se definió previamente. Al respecto, el dispositivo de almacenamiento o aplicación puede estar presente en un embalaje externo que protege contra la contaminación. Para más detalles sobre la unidad de embalaje según la invención, puede hacerse referencia a las realizaciones con respecto a los otros aspectos de la invención, que se aplican en consecuencia a la unidad de embalaje según la invención.
Finalmente, según un aspecto adicional de la presente invención, la presente invención también se refiere al kit según la invención, en particular un sistema de instilación, que comprende (i) al menos un dispositivo de almacenamiento o aplicación, como se definió previamente, y (ii) al menos una composición, como se definió previamente, en particular cuando la composición está presente preferiblemente lista para usar, dosificar o aplicar en el dispositivo de almacenamiento y/o aplicación, y (iii) al menos un dispositivo de instilación que puede conectarse al dispositivo de almacenamiento o aplicación, en particular en forma de una manguera de instilación o similar.
Sobre la base del kit según la invención, en particular se proporciona un sistema de instalación correspondiente con simple manipulabilidad, preparación operativa rápida y alta seguridad de aplicación.
Para más detalles sobre el kit según la invención, puede hacerse referencia a las realizaciones anteriores con respecto a los otros aspectos de la invención, que se aplican correspondientemente al kit según la invención.
Dentro del alcance de la presente invención, se logra así un poderoso concepto general sobre la base de la
composición según la invención con los componentes de ingredientes activos especialmente dosificados, a saber, que el componente (a) está presente en cantidades o dosis relativamente pequeñas y el componente (b) está presente en cantidades o dosis relativamente grandes, para el tratamiento de enfermedades preferiblemente inflamatorias del tracto urogenital, tales como cistitis, preferiblemente cistitis intersticial, siempre que la composición según la invención, además de ser altamente eficaz en relación con las enfermedades subyacentes, con bajos efectos secundarios y excelente manipulabilidad, tenga una estabilidad significativamente mayor, en particular estabilidad al almacenamiento,
Las configuraciones, modificaciones y variaciones adicionales, así como las ventajas de la presente invención, son fácilmente reconocibles y realizables por el experto en la técnica al leer la descripción, sin por ello abandonar el alcance de la presente invención.
Los siguientes ejemplos de realización solo pretenden ilustrar la presente invención, pero sin restringir la presente invención a ellos.
Ejemplos de realización
1. Ejemplos de preparación
Para preparar 50 ml de una solución acuosa de composiciones según la invención transparente, opcionalmente ligeramente amarillenta, se procede según el procedimiento conocido en sí por el experto en la técnica.
Primero, a temperatura ambiente y presión ambiente, se dispone una porción definida (en particular, aproximadamente el 75% del volumen final deseado) de agua purificada en un recipiente de vidrio con un dispositivo de agitación. Mientras se agita, se añaden un electrolito en forma de cloruro de sodio y luego los componentes del sistema tamponante en forma de dihidrogenofosfato de sodio e hidrogenofosfato disódico. Después de que los componentes se hayan disuelto completamente, se determina el valor del pH y se ajusta al valor deseado (por ejemplo, valor del pH de aproximadamente 7), si es apropiado, usando ácido fosfórico o lejía de sosa.
Las siguientes cantidades y tipos de los ingredientes y principios activos adicionales especificados en los ejemplos de recetas se agregan luego con agitación, es decir, el componente (a) en forma de sal de sulfato de condroitina (sulfato de condroitina de sodio) y el componente (b) en forma de hialuronato de sodio. La solución se completa hasta el volumen final deseado de 50 ml con más agua.
El valor del pH de la composición obtenida se determina nuevamente y se reajusta si es necesario. Las otras especificaciones correspondientes de las soluciones también se verifican para los intervalos de valores establecidos o preseleccionados (por ejemplo, osmolaridad; densidad relativa; pureza microbiológica y esterilidad; exclusión de impurezas, en particular productos de degradación de los ingredientes y principios activos; viscosidad; aspecto).
La solución obtenida de esta manera se puede someter a filtración estéril, por ejemplo, con la ayuda de nitrógeno a través de un cartucho de filtro con un sistema de filtro especial, y posteriormente se llena en un receptáculo o recipiente de aplicación particularmente estéril, como una jeringa de plástico o similar. La composición obtenida puede usarse para estudios de estabilidad adicionales o para estudios de aplicación o eficacia correspondientes.
Según esta especificación de preparación general, las recetas o composiciones especificadas a continuación se preparan disolviendo en agua los siguientes componentes con las cantidades relevantes (peso en la preparación de la composición):
Composición A1 (información por composición de 50 ml)

El valor del pH de la composición A1 también es de aproximadamente 7,2. Además, la presente composición presenta una viscosidad dinámica de aproximadamente 5.600 mPas y la densidad es de aproximadamente 1,022 g/cm3 (20 °C y presión atmosférica). La osmolaridad de la presente composición también es de aproximadamente 370 mosmol/kg.
Composiciones B1 a B4
Se producen composiciones adicionales correspondientes a la composición A1, pero con la condición de que se usen diferentes cantidades de sulfato de condroitina sódico:

Composiciones B5 a B8
Se producen nuevamente composiciones adicionales correspondientes a la composición A1, pero con la condición de que se usen diferentes cantidades de hialuronato de sodio:

Composiciones C1 y C2
Se producen nuevamente composiciones adicionales correspondientes a la composición A1, pero con la condición de que el sulfato de condroitina sódico (CS-Na) se use con una cantidad respectiva de 225 mg cada una con diferentes masas molares:

Composiciones D1 y D2
A su vez, otras composiciones corresponden a la composición A1, pero con la condición de que se use hialuronato de sodio (HA-Na) en una cantidad empleada al respecto de 800 mg con distintas masas molares:

Composiciones E1 a E4
En el caso de composiciones adicionales a base de la composición A1, el valor del pH se varía como se representa a continuación:

Composiciones F1 a F4
Con aún más composiciones, se varía el sistema tamponante; con este fin, se usan cantidades correlativas de sistemas tamponantes a base de ácido carbónico/bicarbonato, ácido acético/acetato, ácido carbónico/silicato y ácido cítrico/citrato para el sistema tamponante fosfato usado en la composición A1; las composiciones F1 a F4 corresponden así a la composición A1 con la condición de que se use un sistema tamponante diferente a este respecto (comparación):

2. Estudios de estabilidad
Las pruebas de estabilidad se llevan a cabo en los respectivos lotes de la composición según las recetas descritas con anterioridad.
Para este propósito, el contenido total de productos de degradación de los componentes utilizados en las composiciones m
encionadas se determina en los tiempos o momentos de almacenamiento apropiados, al comienzo de los estudios y con los tiempos de almacenamiento correspondientes de 3 meses, 6 meses, 12 meses, 18 meses, 24 meses, 36 meses y 39 meses. Por otro lado, el valor de pH y la viscosidad también se determinan durante el mismo período de almacenamiento en los tiempos de almacenamiento mencionados con anterioridad.
El almacenamiento se realiza en este caso para un primer lote de las composiciones a una temperatura de (25±2) °C y a una humedad atmosférica relativa de (60±5)% r.H. (siguientes tablas 1A a 1F).
Además, el almacenamiento para un lote adicional de las composiciones se lleva a cabo a una temperatura de (40±2) °C y a una humedad atmosférica relativa de (75±5)% r.H. (siguientes tablas 2A a 2F).
Las siguientes tablas 1A a 1F y 2A a 2F muestran los resultados obtenidos al respecto. En las siguientes tablas, “++” significa un contenido total de productos de degradación (“degradación”) en el tiempo de almacenamiento respectivo de como máximo el 3%, en función de la concentración total de los componentes, o un cambio en el valor del pH (“pH”) o la viscosidad (“Visco”) en el momento de almacenamiento respectivo de como máximo el 3%, en función del valor inicial respectivo. Además, “+” significa un contenido total de productos de degradación en el momento de almacenamiento respectivo de como máximo el 5%, en función de la concentración total de los componentes, o un cambio en el pH o la viscosidad en el momento de almacenamiento respectivo de como máximo el 5%, en función del valor inicial respectivo. Finalmente, un contenido total de productos de degradación en el momento de almacenamiento respectivo significa más del 5%, en función de la concentración total de los componentes, o un cambio en el pH o la viscosidad en el momento de almacenamiento respectivo de más del 5%, en función del valor inicial respectivo.
Tabla 1A [(25±2) °C y (60±5)% r.H.]:

Tabla 1B [(25±2) °C y (60±5)% r.H.]:

Tabla 1C [(25±2) °C y (60±5)% r.H.]:

Tabla 1D [(25±2) °C y (60±5)% r.H.]:

Tabla 1E [(25±2) °C y (60±5)% r.H.]:

Tabla 1F [(25±2) °C y (60±5)% r.H.]:

Tabla 2A [(40±2) °C y (75±5)% r.H.]:

Tabla 2B [(40±2) °C y (75±5)% r.H.]:


Tabla 2C [(40±2) °C y (75±5)% r.H.]:

Tabla 2D [(40±2) °C y (75±5)% r.H.]:

Tabla 2E [(40±2) °C y (75±5)% r.H.]:


Tabla 2F [(40±2) °C y (75±5)% r.H.]:
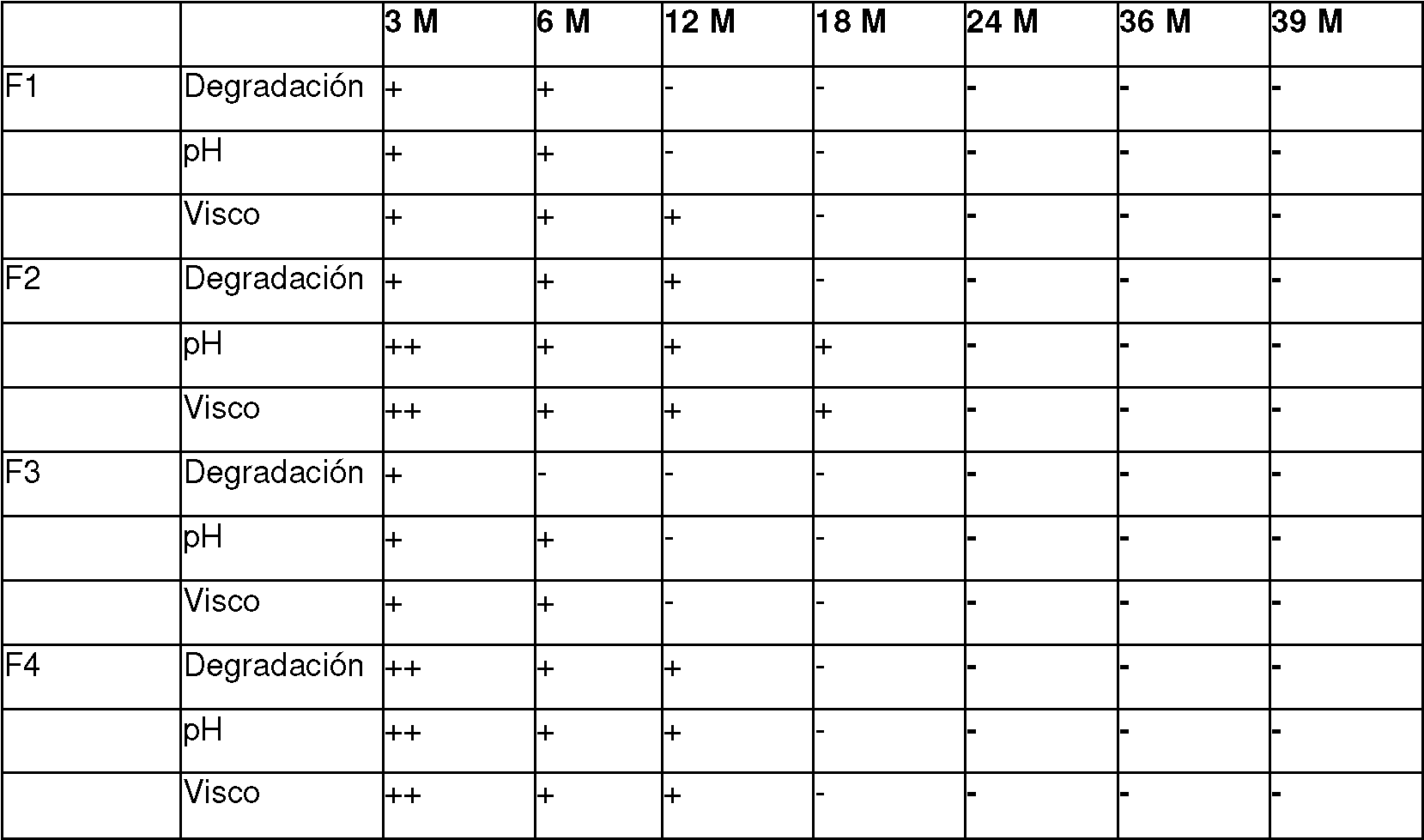
Una medida característica de la estabilidad de una composición es, por un lado, la constancia del valor del pH o la viscosidad y la constancia del contenido de principios activos (es decir, el componente (a) y el componente (b)). Además, el contenido de productos de degradación en la composición puede determinarse para evaluar la estabilidad de la composición.
En este contexto, los estudios de estabilidad realizados muestran que tanto la cantidad de los componentes del ingrediente activo en la composición como su peso molecular específico y, además, el valor del pH y, en gran medida, también el sistema tamponante utilizado tienen una influencia significativa sobre la estabilidad, en particular la estabilidad al almacenamiento, de las composiciones subyacentes, donde la composición A1 con la coordinación especial de los componentes presenta las mejores propiedades en este sentido.
Además, las investigaciones que utilizan diferentes sistemas tamponantes muestran que una estabilidad confiable a largo plazo solo puede lograrse con el sistema tamponante de fosfato utilizado según la invención. Porque, como los estudios de estabilidad de la solicitante muestran de manera sorprendente, solo un sistema tamponante de fosfato de este tipo, en comparación con otros posibles sistemas tamponantes que se pueden usar, produce la estabilización deseada y confiable a largo plazo de la composición según la invención, incluso durante largos períodos de tiempo. Con otros sistemas tamponantes, por otro lado, que pueden usarse en el intervalo de pH comparable, por ejemplo, un sistema tamponante de ácido carbónico/bicarbonato, un sistema tamponante de ácido carbónico/silicato, un sistema tamponante de ácido acético/acetato, un sistema tamponante de ácido cítrico/citrato o similares, tales buenos resultados de estabilidad no pueden obtenerse o no siempre de manera confiable. Sin desear limitarse a ninguna teoría en particular, este efecto del sistema tamponante fosfato utilizado según la invención también puede remitirse a efectos secundarios, por ejemplo, con respecto a la estabilización adicional de la matriz o del hidrogel sobre la base de los componentes (a) o (b) en la composición.
Como lo demuestran los resultados de los estudios de estabilidad de la solicitante que se muestran más arriba, para encontrar una composición estable para el tratamiento de enfermedades preferiblemente inflamatorias del tracto urogenital, en particular enfermedades inflamatorias de la vejiga urinaria, preferiblemente cistitis, de la manera de la presente invención, se requirió un esfuerzo de investigación intensivo por parte de la solicitante.
Los estudios de estabilidad anteriores también muestran la complejidad del enfoque de solución encontrado por la solicitante. Esto incluye la presencia conjunta de los ingredientes activos o componentes (a) y (b) en una cantidad definida mientras se conserva o mantiene contante el valor del pH de la composición en el intervalo previamente definido, así como otros factores como la selección del sistema tamponante apropiado.
El enfoque de solución según la invención sorprendentemente se maneja con solo unos pocos ingredientes adicionales que, sin embargo, no perjudican la eficacia farmacológica de los ingredientes activos subyacentes; esto es de crucial importancia en vista del efecto farmacéutico previsto.
Por último, pero no menos importante, los resultados en su conjunto demuestran la excelente estabilidad a largo plazo de la composición según la invención.
3. Observaciones de aplicación y estudios de eficacia
A los grupos respectivos de sujetos con cistitis intersticial diagnosticada (es decir, no bacteriana) se les administran las composiciones A1 y B1, B4 (cantidad de sulfato de condroitina sódico) durante un período de dos meses; B5, B8 (cantidad de hialuronato de sodio); C1, C2 (masa molar de sulfato de condroitina sódico); D1, D2 (masa molar de hialuronato de sodio) y E1 y E4 (valor de pH). La administración se realiza por instilación en la vejiga. La administración se repite semanalmente durante un período de dos meses. El volumen de la composición instilada es de 50 ml cada uno. Se examinan 10 sujetos por lote o composición. Los intervalos de examen son 1 semana, 2 semanas, 1 mes y, finalmente, 2 meses, cada uno en función del día de la instilación inicial. En este caso, se llevan a cabo los siguientes complejos de investigación:
a) Como parte del complejo del primer examen, los sujetos de prueba expresan su sentimiento subjetivo en una escala de grado escolar de 1 a 6 (1 = muy bueno a 6 = insatisfactorio), siendo posibles también los valores intermedios. Este examen se centra en particular en el dolor en la vejiga urinaria y la pelvis, la necesidad excesiva de orinar y en una pequeña capacidad de la vejiga urinaria. Se determinan los valores medios respectivos y las desviaciones estándar asociadas.
b) En un segundo examen, un mes después de la primera aplicación de las composiciones mencionadas con anterioridad, se realiza una citoscopia (reflejo de la vejiga) en los respectivos grupos de prueba, donde se realiza un examen visual del urotelio de la vejiga usando un endoscopio.
c) Además, se toman muestras de orina al comienzo de la prueba y después de un mes y el contenido de glicosaminoglicanos en la orina se basa en el procedimiento según Whitley et al. (ver C. B., Ridnour, M. D., Draper, K. A., Dutton, C. M. and Negila, J. P.: Diagnostic test for mucoplysaccharidosis. I. Direct method for quantifying excessive urinary glycosaminoglycan excretion. Clin. Chem., 35: 374, 1989), donde los glicosaminoglicanos presentes en la orina se tiñen con azul de dimetilmetileno y sus concentraciones se determinan luego espectroscópicamente. En este contexto, se sabe que en pacientes diagnosticados con cistitis intersticial existe una concentración de glicosaminoglicanos presentes en la orina que se aparta de la concentración en pacientes sanos, es decir, en pacientes sin evidencia de cistitis intersticial. Al respecto, también se sabe que el contenido de glicosaminoglicano en la orina se reduce en pacientes con cistitis intersticial avanzada o crónica en comparación con un grupo de control sin hallazgos, mientras que el contenido de glicosaminoglicano en la orina en sujetos con cistitis intersticial en la etapa inicial o en una etapa no muy avanzada se incrementa.
Las investigaciones y los estudios de eficacia muestran que, para la composición A1, en comparación con las otras composiciones, como se indicó con anterioridad, está presente la mejor eficacia en relación con el tratamiento de la enfermedad subyacente en forma de cistitis intersticial. En particular, se puede observar una mejora significativa en el bienestar de la salud, donde en el caso de los sujetos del grupo tratado con la composición A1 también existe un moco vesical casi intacto y, además, no hay lesiones ni sangrado en el urotelio. También hay una mejora significativa en el contenido de glicosaminoglicanos. En general, los estudios también muestran la excelente eficacia terapéutica del concepto según la invención.
Claims (15)
1. Composición, en la que la composición contiene en combinación y, en cada caso, en cantidades eficaces
(a) sulfato de condroitina y/o una sal de sulfato de condroitina fisiológicamente aceptable en una concentración de (4,5±0,5) mg/ml (componente (a)); y
(b) ácido hialurónico y/o una sal de ácido hialurónico fisiológicamente aceptable (hialuronato) en una concentración de (16±1,6) mg/ml (componente (b));
donde la composición presenta un valor pH en el intervalo de 6,1 a 7,9 y/o donde la composición se ajusta a un valor pH en el intervalo de 6,1 a 7,9,
caracterizada por que la composición
(c) contiene un sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)).
2. Composición según la reivindicación 1,
en la que la composición presenta el sulfato de condroitina y/o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) en una concentración de (4,5±0,4) mg/ml, en particular en una concentración de (4,5±0,3) mg/ml, preferiblemente en una concentración de (4,5±0,15) mg/ml, más preferiblemente, en una concentración de aproximadamente 4,5 mg/ml; y/o
en la que el sulfato de condroitina y/o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) presenta un peso molecular medio en número (masa molar) Mn en el intervalo de 2 kDa a 200 kDa, en particular en el intervalo de 5 kDa a 150 kDa, preferiblemente en el intervalo de 10 kDa a 100 kDa, preferiblemente en el intervalo de 12 kDa a 75 kDa; y/o
en la que el sulfato de condroitina y/o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) presenta un peso molecular medio en peso (masa molar) Mw en el intervalo de 10 kDa a 200 kDa, en particular en el intervalo de 15 kDa a 175 kDa, preferiblemente en el intervalo de 20 kDa a 150 kDa, preferiblemente en el intervalo de 30 kDa a 120 kDa; y/o
en la que el sulfato de condroitina y/o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) presenta un peso molecular medio centrifugal (masa molar) Mz en el intervalo de 30 kDa a 1000 kDa, en particular en el intervalo de 40 kDa a 800 kDa, preferiblemente en el intervalo de 50 kDa a 600 kDa, preferiblemente en el intervalo de 100 kDa a 450 kDa; y/o
en la que el sulfato de condroitina y/o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) presenta un índice de polidispersidad (PDI), calculado como el cociente del peso molecular medio en peso Mw y el peso molecular medio en número Mn, de al menos 1, en particular al menos 1,2, preferiblemente al menos 1,5, preferiblemente al menos 2; y/o
en la que el sulfato de condroitina y/o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) presenta un índice de polidispersidad (PDI), calculado como el cociente del peso molecular medio en peso Mw y el peso molecular medio en número Mn, de como máximo 30, en particular máximo 20, preferiblemente máximo 10, preferiblemente máximo 8; y/o
en la que el sulfato de condroitina y/o la sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) presenta un índice de polidispersidad (PDI), calculado como el cociente del peso molecular medio en peso Mw y el peso molecular medio en número Mn, en el intervalo de 1 a 30, en particular en el intervalo de 1,2 a 20, preferiblemente en el intervalo de 1,5 a 10, preferiblemente en el intervalo de 2 a 8.
3. Composición según la reivindicación 1 o 2,
en la que la composición contiene el ácido hialurónico y/o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) en una concentración de (16±1,2) mg/ml, en particular en una concentración de (16±0,8) mg/ml, preferiblemente en una concentración de (16±0,4) mg/ml, más preferiblemente en una concentración de aproximadamente 16 mg/ml; y/o
en la que el ácido hialurónico y/o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) presenta un peso molecular medio en número (masa molar) Mn en el intervalo de 10 kDa a 300 kDa, en particular en el intervalo de 20 kDa a 275 kDa, preferiblemente en el intervalo de 30 kDa a 260 kDa, preferiblemente en el intervalo de 50 kDa a 250 kDa, más preferiblemente, en el intervalo de 75 kDa a 200 kDa; y/o
en la que el ácido hialurónico y/o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) presenta un peso molecular medio en peso (masa molar) Mw en el intervalo de 10 kDa a 500 kDa, en particular en el intervalo de 20 kDa a 450 kDa, preferiblemente en el intervalo de 50 kDa a 425 kDa, preferiblemente en el intervalo de 100 kDa a
400 kDa, más preferiblemente, en el intervalo de 150 kDa a 395 kDa; y/o
en la que el ácido hialurónico y/o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) presenta un peso molecular medio centrifugal (masa molar) Mz en el intervalo de 80 kDa a 1500 kDa, en particular en el intervalo de 100 kDa a 1250 kDa, preferiblemente en el intervalo de 200 kDa a 1000 kDa, preferiblemente en el intervalo de 300 kDa a 750 kDa; y/o
en la que el ácido hialurónico y/o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) presenta un índice de polidispersidad (PDI), calculado como el cociente del peso molecular medio en peso Mw y el peso molecular medio en número Mn, de al menos 1, en particular de al menos 1,1, preferiblemente de al menos 1,2, preferiblemente de al menos 1,3, más preferiblemente, de al menos 1,4; y/o
en la que el ácido hialurónico y/o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) presenta un índice de polidispersidad (PDI), calculado como el cociente del peso molecular medio en peso Mw y el peso molecular medio en número Mn, de como máximo 50, en particular máximo 25, preferiblemente máximo 10, preferiblemente máximo 5, más preferiblemente, máximo 3; y/o
en la que el ácido hialurónico y/o la sal de ácido hialurónico fisiológicamente aceptable (componente (b)) presenta un índice de polidispersidad (PDI), calculado como el cociente del peso molecular medio en peso Mw y el peso molecular medio en número Mn, en el intervalo de 1 a 50, en particular en el intervalo de 1,1 a 25, preferiblemente en el intervalo de 1,2 a 10, preferiblemente en el intervalo de 1,3 a 5, más preferiblemente, en el intervalo de 1,4 a 3.
4. Composición según cualquiera de las reivindicaciones anteriores,
en la que la composición contiene el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) en una concentración total de sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) de (1,75±1,65) mg/ml, en particular en una concentración total de (1,75±1,5) mg/ml, preferiblemente en una concentración total de (1,75±1,25) mg/ml, preferiblemente en una concentración total de (1,75±1) mg/ml, más preferiblemente, en una concentración total de (1,75±0,9) mg/ml, siendo lo más preferible, en una concentración total de (1,75±0,8) mg/ml, incluso más preferiblemente, en una concentración total de aproximadamente 1,75 mg/ml; y/o
en la que la composición contiene el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) en una cantidad total de sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) de (87,5±85) mg, en particular en una cantidad total de (87,5±75) mg, preferiblemente en una cantidad total de (87,5±62,5) mg, preferiblemente en una cantidad total de (87,5±50) mg, más preferiblemente, en una cantidad total de (87,5±45) mg, siendo lo más preferible, en una cantidad total de (87,5±40) mg, incluso más preferiblemente, en una cantidad total de aproximadamente 87,5 mg; y/o
en la que la composición contiene el dihidrogenofosfato del sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)), en particular en función del peso de la muestra al producir y/o proporcionar la composición, en una concentración de (0,2±0,19) mg/ml, en particular en una concentración de (0,2±0,15) mg/ml, preferiblemente en una concentración de (0,2±0,125) mg/ml, preferiblemente en una concentración de (0,2±0,1) mg/ml, más preferiblemente, en una concentración de aproximadamente 0,2 mg/ml; y/o
en la que la composición contiene el monohidrogenofosfato del sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)), en particular en función del peso de la muestra al producir y/o proporcionar la composición, en una concentración de (1,5±1,4) mg/ml, en particular en una concentración de (1,5±1,25) mg/ml, preferiblemente en una concentración de (1,5±1,1) mg/ml, preferiblemente en una concentración de (1,5±1) mg/ml, más preferiblemente, en una concentración de (1,5±0,75) mg/ml, siendo lo más preferible, en una concentración de aproximadamente 1,5 mg/ml; y/o
en la que la composición contiene el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) en una relación en peso (relación de concentración) de dihidrogenofosfato a monohidrogenofosfato [dihidrogenofosfato:monohidrogenofosfato], en particular en función del peso de la muestra al producir y/o proporcionar la composición, en el intervalo de 2:1 a 1:100, preferiblemente en el intervalo de 1:1 a 1:75, preferiblemente en el intervalo de 1:2 a 1:50, más preferiblemente, en el intervalo de 1:5 a 1:25.
5. Composición según cualquiera de las reivindicaciones anteriores,
en la que el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) está presente y/o se forma como un sistema tamponante de dihidrogenofosfato alcalino/monohidrogenofosfato alcalino; en particular, en la que el metal alcalino se selecciona de sodio y/o potasio, en particular de sodio; y/o
en la que el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) está presente y/o se forma como sistema tamponante de dihidrogenofosfato de sodio/monohidrogenofosfato de sodio, y/o
en la que el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) está presente y/o se forma como sistema tamponante de NaH2(PO4)/Na2H(PO4), en particular como sistema tamponante de NaH2(PO4) ■ 2
H2O/Na2H(PO4) ■ 2 H2O, y/o
en la que el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) sirve y/o se usa para ajustar y/o mantener constante el valor pH de la composición.
6. Composición según cualquiera de las reivindicaciones anteriores,
en la que la composición contiene el electrolito (componente (d)) en una concentración de (8±6) mg/ml, en particular en una concentración de (8±4) mg/ml, preferiblemente en una concentración de (8±2) mg/ml, preferiblemente en una concentración de (8±1) mg/ml, más preferiblemente, en una concentración de aproximadamente 8 mg/ml; y/o
en la que el electrolito (componente (d)) está en forma de una sal alcalina, en particular en forma de un cloruro alcalino, preferiblemente en forma de cloruro de sodio, y/o en la que el electrolito (componente (d)) es cloruro de sodio.
7. Composición según cualquiera de las reivindicaciones anteriores,
en la que la composición presenta un valor pH en el intervalo de 6,6 a 7,7, en particular en el intervalo de 6,9 a 7,6, preferiblemente en el intervalo de 7,1 a 7,4; y/o
en la que el valor pH de la composición se mantiene constante y/o se ajusta en el intervalo de 6,6 a 7,7, en particular en el intervalo de 6,9 a 7,6, preferiblemente en el intervalo de 7,1 a 7,4; y/o
en la que el pH se ajusta y/o se predetermina por medio del sistema tamponante de dihidrogenofosfato alcalino/monohidrogenofosfato alcalino (componente (c)); y/o
en la que la composición está presente como composición acuosa y/o en la que la composición es de base acuosa y/o está formulada en forma acuosa, en particular en forma de una solución acuosa y/o suspensión acuosa, preferiblemente en forma de una solución acuosa; y/o
en la que la composición contiene agua, en particular agua purificada, y/o en la que la composición contiene agua como un vehículo (excipiente) farmacéuticamente aceptable y/o en la que la composición está formada a base de agua;
y/o en la que la composición presenta un contenido de agua de al menos el 50% en peso, en particular de al menos el 75% en peso, preferiblemente de al menos el 80% en peso, preferiblemente de al menos el 90% en peso, más preferiblemente de al menos el 95% en peso, en función de la composición; y/o
en la que la composición consiste en los componentes (a), (b), (c) antes mencionados y, en caso necesario, (d), así como, en caso necesario, agua, en particular agua purificada.
8. Composición según cualquiera de las reivindicaciones anteriores,
en la que la composición es estable, en particular estable al almacenamiento, a temperaturas comprendidas en el intervalo de 20 °C a 45 °C, a una presión de 1013,25 mbar (presión atmosférica) y a una humedad atmosférica relativa comprendida en el intervalo del 50% al 90% durante al menos 6 meses, en particular al menos 12 meses, preferiblemente al menos 24 meses, preferiblemente al menos 36 meses; en particular en la que la composición presenta al momento del almacenamiento respectivo un contenido total de productos de degradación de los componentes (a) y (b) de como máximo el 5%, en particular máximo el 4%, preferiblemente máximo el 3%, preferiblemente máximo el 2%, más preferiblemente máximo el 1%, en función de la concentración total de los componentes (a) y (b); y/o
en la que la composición presenta una estabilidad, en particular estabilidad de almacenamiento, en condiciones de envejecimiento acelerado según la norma ASTM F 1980 a una temperatura de envejecimiento de 55 °C de al menos 6 meses, en particular al menos 12 meses, preferiblemente al menos 24 meses, preferiblemente al menos 36 meses; en particular en la que la composición presenta al momento del almacenamiento respectivo un contenido total de productos de degradación de los componentes (a) y (b) de como máximo el 5%, en particular de máximo el 4%, preferiblemente máximo el 3%, preferiblemente máximo el 2%, más preferiblemente máximo el 1%, en función de la concentración total de los componentes (a) y (b).
9. Composición según cualquiera de las reivindicaciones anteriores,
en la que la composición, particularmente lista para usar, dosificar y/o aplicar, está presente con un volumen de (50±10) ml, en particular de (50±5) ml, preferiblemente de (50±2) ml, preferiblemente de (50±1) ml, más preferiblemente de (50±0,5) ml, siendo lo más preferible de aproximadamente 50 ml; y/o
en la que la composición, particularmente lista para usar, dosificar y/o aplicar, está presente y/o preparada para la administración, en particular la instilación en la vejiga, de un volumen de la composición de (50±10) ml, en particular de (50±5) ml, preferiblemente de (50±2) ml, preferiblemente de (50±1) ml, más preferiblemente de (50±0,5) ml, siendo lo más preferible de aproximadamente 50 ml, o en la que la composición, en particular lista para usar, dosificar y/o
aplicar, en particular por instilación en la vejiga urinaria, se administra con un volumen de la composición de (50±10) ml, en particular de (50±5) ml, preferiblemente de (50±2) ml, preferiblemente de (50±1) ml, más preferiblemente de (50±0,5) ml, siendo lo más preferible de aproximadamente 50 ml; y/o
en la que la composición, en particular lista para usar, dosificar y/o aplicar, está presente con una cantidad de ingrediente activo en sulfato de condroitina y/o sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) de (225±25) mg, en particular de (225±15) mg, preferiblemente de (225±10) mg; y/o
en la que la composición, en particular lista para usar, dosificar y/o aplicar, está presente y/o preparada para la administración de una cantidad de ingrediente activo en sulfato de condroitina y/o sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) de (225±25) mg, en particular de (225±15) mg, preferiblemente de (225±10) mg, o en la que la composición, en particular lista para usar, dosificar y/o aplicar, se administra con una cantidad de ingrediente activo en sulfato de condroitina y/o sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) de (225±25) mg, en particular de (225±15) mg, preferiblemente de (225±10) mg; y/o
en la que la composición, en particular lista para usar, dosificar y/o aplicar, está presente con una cantidad de ingrediente activo de ácido hialurónico y/o sal de ácido hialurónico fisiológicamente aceptable (hialuronato) (componente (b)) de (800±80) mg, en particular de (800±60) mg, preferiblemente de (800±40) mg; y/o
en la que la composición, en particular lista para usar, dosificar y/o aplicar, está presente y/o preparada para administrar una cantidad de ingrediente activo de ácido hialurónico y/o sal de ácido hialurónico fisiológicamente aceptable (hialuronato) (componente (b)) de (800±80) mg, en particular de (800±60) mg, preferiblemente de (800±40) mg, o en la que la composición, en particular lista para usar, dosificar y/o aplicar, se administra con una cantidad de ingrediente activo de ácido hialurónico y/o sal de ácido hialurónico fisiológicamente aceptable (hialuronato) (componente (b)) de (800±80) mg, en particular de (800±60) mg, preferiblemente de (800±40) mg.
10. Composición según cualquiera de las reivindicaciones anteriores
para la instilación y/o preferiblemente la aplicación tópica en el área urogenital, en particular en la vejiga urinaria; y/o
en la que la composición está preparada para la administración y/o para la instilación y/o para la aplicación tópica preferiblemente en el tracto urogenital, en particular en la vejiga urinaria, o en la que la composición se administra por instilación y/o para la aplicación tópica preferiblemente en el tracto urogenital, en particular en la vejiga urinaria.
11. Composición según cualquiera de las reivindicaciones anteriores para su uso en el tratamiento profiláctico y/o terapéutico de enfermedades preferiblemente inflamatorias del tracto urogenital, en particular enfermedades inflamatorias de la vejiga urinaria, preferiblemente de cistitis, en particular cistitis aguda o crónica, preferiblemente cistitis intersticial, cistitis radiogénica, cistitis recurrente crónica, quimiocistitis, cistitis abacteriana crónica y cistitis bacteriana crónica, más preferiblemente, cistitis intersticial.
12. Dispositivo de almacenamiento y/o aplicación, en particular recipiente de almacenamiento y/o aplicación, en particular en forma de una jeringa preferiblemente estéril, en particular jeringa de aplicación, preferiblemente jeringa de aplicación desechable, preferiblemente para la instilación y/o la aplicación tópica preferiblemente en el área urogenital, en particular en la vejiga urinaria, que contiene una composición según cualquiera de las reivindicaciones anteriores.
13. Dispositivo de almacenamiento y/o aplicación según la reivindicación 12,
en el que la composición presenta una cantidad de ingrediente activo de (a) sulfato de condroitina y/o sal de sulfato de condroitina fisiológicamente aceptable (componente (a)) de (225±15) mg, preferiblemente de (225±10) mg; y/o
en el que la composición presenta una cantidad de ingrediente activo de (b) ácido hialurónico y/o sal de ácido hialurónico fisiológicamente aceptable (hialuronato) (componente (b)) de (800±60) mg, preferiblemente de (800±40) mg; y/o
en el que la composición presenta el sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) en una cantidad total de sistema tamponante de dihidrogenofosfato/monohidrogenofosfato (componente (c)) de (87,5±85) mg, en particular en una cantidad total de (87,5±75) mg, preferiblemente en una cantidad total de (87,5±62,5) mg, preferiblemente en una cantidad total de (87,5±50) mg, más preferiblemente en una cantidad total de (87,5±45) mg, siendo lo más preferible en una cantidad total de (87,5±40) mg, aún más preferiblemente en una cantidad total de aproximadamente 87,5 mg; y/o
en el que la composición contiene el electrolito (componente (d)) en una cantidad de (400±300) mg, en particular en una cantidad de (400±200) mg, preferiblemente en una cantidad de (400±100) mg, preferiblemente en una cantidad contiene de (400±50) mg, más preferiblemente en una cantidad de aproximadamente 400 mg; y/o
en el que el dispositivo de almacenamiento y/o aplicación contiene la composición, en particular lista para usar, dosificar y/o aplicar, con un volumen de (50±10) ml, en particular de (50±5) ml, preferiblemente de (50±2) ml, preferiblemente de (50±1) ml, más preferiblemente de (50±0,5) ml, siendo lo más preferible de aproximadamente 50 ml; y/o
en el que la composición está presente con un volumen de (50±10) ml, en particular de (50±5) ml, preferiblemente de (50±2) ml, preferiblemente de (50±1) ml, más preferiblemente, de (50±0,5) ml, siendo lo más preferible de aproximadamente 50 ml.
14. Unidad de embalaje que contiene al menos un dispositivo de almacenamiento y/o aplicación según la reivindicación 12 o 13, en particular en la que el dispositivo de almacenamiento y/o aplicación está presente en un embalaje externo que protege contra la contaminación.
15. Kit, en particular sistema de instilación, que comprende (i) al menos un dispositivo de almacenamiento y/o aplicación según la reivindicación 12 o 13, (ii) al menos una composición según cualquiera de las reivindicaciones anteriores, en particular en el que la composición, preferiblemente lista para usar, dosificar y/o aplicar, está presente en el dispositivo de almacenamiento y/o aplicación, y (iii) al menos un dispositivo de instilación conectable al dispositivo de almacenamiento y/o aplicación, en particular en forma de una manguera de instilación, o similar.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17000821 | 2017-05-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2775673T3 true ES2775673T3 (es) | 2020-07-27 |
Family
ID=58709189
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17173823T Active ES2775673T3 (es) | 2017-05-12 | 2017-05-31 | Composición para el tratamiento de enfermedades inflamatorias del tracto urogenital que contiene sulfato de condonita (4,5 mg/ml), ácido hialurónico (16 mg/ml) y tampón de fosfato (pH 6,1 a 7,9) con mayor estabilidad de almacenamiento para tratar la cistitis |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US11351113B2 (es) |
| EP (1) | EP3400951B1 (es) |
| KR (1) | KR102312625B1 (es) |
| CA (1) | CA3061271A1 (es) |
| ES (1) | ES2775673T3 (es) |
| RU (1) | RU2742277C1 (es) |
| WO (1) | WO2018206358A1 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7124041B2 (ja) * | 2020-11-25 | 2022-08-23 | 株式会社朋 | ハンナ病変の指摘のためのプログラム |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IN181358B (es) * | 1995-02-14 | 1998-05-30 | Bioniche Inc | |
| US5880108A (en) | 1995-02-14 | 1999-03-09 | Bioniche, Inc. | Method for treating the internal urinary bladder and associated structures using hyaluronic acid |
| US6083933A (en) | 1999-04-19 | 2000-07-04 | Stellar International Inc. | Treatment of cystitis-like symptoms with chondroitin sulfate following administration of a challenge solution |
| US20040161476A1 (en) | 2003-02-19 | 2004-08-19 | Hahn Sungtack Samuel | Cystitis treatment with high dose chondroitin sulfate |
| JP2008520392A (ja) * | 2004-11-23 | 2008-06-19 | アルコン,インコーポレイティド | 天然ポリマーの粘弾性組成物 |
| ITMI20061030A1 (it) * | 2006-05-26 | 2007-11-27 | Altergon Sa | Nuova composizione comprendente glicosamminoglicani a viscosita' controllata e uso di tale composizione nella terapuia della cistite cronica |
| DE102006060953A1 (de) * | 2006-12-12 | 2008-08-07 | Farco-Pharma Gmbh | Pharmazeutische Zubereitung für die Behandlung entzündlicher Erkrankungen des Urogenitaltraktes |
| US20080275015A1 (en) * | 2007-05-01 | 2008-11-06 | Potter Jeffrey A | Formulation and Method for Treating Interstitial Cystitis and Related Bladder Conditions |
| ITPD20120098A1 (it) | 2012-03-30 | 2013-10-01 | Fidia Farmaceutici | "nuove formulazioni faramaceutiche contenenti condroitin solfato e derivati dell'acido ialuronico" |
| EP2948154A4 (en) * | 2013-01-28 | 2016-09-14 | Urigen Pharmaceuticals Inc | STABLE COMPOSITIONS WITH HEPARINOID, A DIRECT ANESTHETIC, AND A BUFFER |
| KR20150001070A (ko) * | 2013-06-26 | 2015-01-06 | (주)아모레퍼시픽 | 피부재생 촉진용 조성물 |
-
2017
- 2017-05-31 ES ES17173823T patent/ES2775673T3/es active Active
- 2017-05-31 EP EP17173823.0A patent/EP3400951B1/de active Active
-
2018
- 2018-05-02 US US16/611,827 patent/US11351113B2/en active Active
- 2018-05-02 CA CA3061271A patent/CA3061271A1/en not_active Abandoned
- 2018-05-02 KR KR1020197034864A patent/KR102312625B1/ko not_active Expired - Fee Related
- 2018-05-02 RU RU2019140863A patent/RU2742277C1/ru active
- 2018-05-02 WO PCT/EP2018/061148 patent/WO2018206358A1/de not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| KR20200003015A (ko) | 2020-01-08 |
| RU2742277C1 (ru) | 2021-02-04 |
| US20210145733A1 (en) | 2021-05-20 |
| KR102312625B1 (ko) | 2021-10-15 |
| US11351113B2 (en) | 2022-06-07 |
| CA3061271A1 (en) | 2018-11-15 |
| EP3400951A1 (de) | 2018-11-14 |
| EP3400951B1 (de) | 2019-12-11 |
| WO2018206358A1 (de) | 2018-11-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102079794B (zh) | 用于医药装置和药物制剂中的含有木葡聚糖的粘膜粘附性制剂 | |
| ES2664951T3 (es) | Composiciones que comprenden glicosaminoglicanos de baja viscosidad y uso de dicha composición en terapia de cistitis crónica | |
| ES2769059T3 (es) | Composición de instilación de la vejiga que contiene sulfato de condonitina (20 mg/ml), ácido hialurónico (16 mg/ml) y buffer de fosfato (pH 6,1 a 7,9) con mayor estabilidad de almacenamiento para el tratamiento de la cistitis | |
| ES2775673T3 (es) | Composición para el tratamiento de enfermedades inflamatorias del tracto urogenital que contiene sulfato de condonita (4,5 mg/ml), ácido hialurónico (16 mg/ml) y tampón de fosfato (pH 6,1 a 7,9) con mayor estabilidad de almacenamiento para tratar la cistitis | |
| US6286513B1 (en) | Methods for treating superficial bladder carcinoma | |
| JP2018538315A (ja) | 骨関節炎治療のための親水化されたスルファサラジンおよびヒアルロン酸を含む組成物およびその製造方法 | |
| CN116077512A (zh) | 曼那斯汀减轻化疗药物顺铂导致的多器官损伤的应用 | |
| HUP0900717A2 (en) | Pharmaceutical composition for urological use containing zinc hyaluronate | |
| Cabral et al. | Toxic epidermal necrolysis-Lyell's syndrome | |
| ES2367454T3 (es) | Preparación farmacéutica para el tratamiento de enfermedades inflamatorias del tracto genitourinario. | |
| EP3415163B1 (de) | Zusammensetzung mit lokalanästhetischer wirkung und deren verwendung | |
| ES2983732T3 (es) | Composición para el tratamiento de enfermedades de la piel y de las membranas mucosas | |
| RU2810419C1 (ru) | Способ лечения хронического пародонтита | |
| ES2577885B1 (es) | Composición de doxiciclina en liposomas para la prevención, mejora y/o tratamiento de patologías oculares. | |
| RU2776878C1 (ru) | Способ комбинированного лечения клещевого риккетсиоза, обусловленного Хейлунцзянской риккетсией, органическим селеном | |
| DE202017103288U1 (de) | Zusammensetzung für die Behandlung entzündlicher Erkrankungen des Urogenitaltraktes in Form einer hochdosierten Wirkstoffkombination | |
| DE202017104675U1 (de) | Zusammensetzung mit lokalanästhetischer Wirkung und deren Verwendung | |
| KR20040066316A (ko) | 안질환 치료용 약물전달필름 및 그 제조방법 | |
| RU2455008C1 (ru) | Пролонгированная жидкая лекарственная форма на основе бишофита | |
| ES2245611A1 (es) | Composicion farmaceutica para uso ototopico. | |
| WO2024063715A1 (en) | Effervescent tablet comprising l-arginine used for healing wounds | |
| US10350232B1 (en) | Medicinal drops | |
| Telmasre | A Comparative Clinical Study with Kushtha Taila and Chlorhexidine (Savlon) in Umbilical Cord Care | |
| WO2020210608A1 (en) | Liquid concentrates of calcium and magnesium | |
| DE202017103289U1 (de) | Zusammensetzung für die Behandlung entzündlicher Erkrankungen des Urogenitaltraktes in Form einer Wirkstoffkombination |