ES2771151T3 - Derivados de piperidina como inhibidor de señalización wnt - Google Patents
Derivados de piperidina como inhibidor de señalización wnt Download PDFInfo
- Publication number
- ES2771151T3 ES2771151T3 ES14832281T ES14832281T ES2771151T3 ES 2771151 T3 ES2771151 T3 ES 2771151T3 ES 14832281 T ES14832281 T ES 14832281T ES 14832281 T ES14832281 T ES 14832281T ES 2771151 T3 ES2771151 T3 ES 2771151T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- methyl
- oxo
- piperidin
- dihydroquinazolin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- XAZKFISIRYLAEE-UHFFFAOYSA-N CC1CC(C)CC1 Chemical compound CC1CC(C)CC1 XAZKFISIRYLAEE-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D475/00—Heterocyclic compounds containing pteridine ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/052—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being six-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/056—Ortho-condensed systems with two or more oxygen atoms as ring hetero atoms in the oxygen-containing ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Abstract
Un compuesto heterocíclico de anillo condensado representado por la fórmula general (I) o una sal farmacéuticamente aceptable del mismo: **(Ver fórmula)** [en donde n1 representa 0 o 1; cada uno de n2 y n3 es 2; R1 representa opcionalmente arilo sustituido, un grupo heterocíclico aromático opcionalmente sustituido o un grupo heterocíclico alifático opcionalmente sustituido; R2 representa un átomo de hidrógeno o hidroxi; R3 representa un grupo heterocíclico aromático opcionalmente sustituido; X1, X2, X3, y X4 pueden ser iguales o diferentes y cada uno representa N o CR4 (en donde R4 representa un átomo de hidrógeno, alquilo inferior, ciano, halógeno, hidroxi, alcoxi inferior, alcanoílo inferior o alquilsulfonilo inferior); Y1 representa CH2 o C(=O); Y2 representa CH o N y L representa CH2 o NH].
Description
DESCRIPCIÓN
Derivados de piperidina como inhibidor de señalización wnt
Campo técnico
La presente invención se refiere un derivado heterocíclico de anillo condensado o una sal farmacéuticamente aceptable del mismo, que tiene una actividad inhibidora de la señalización Wnt y es útil como agente terapéutico y/o preventivo para, por ejemplo, cáncer, fibrosis pulmonar, fibromatosis, osteoartritis y similares, y similares.
Técnica antecedente
En quimioterapia para el cáncer, se usan diversos agentes antitumorales tales como los agonistas de microtúbulos tales como los taxanos, alcaloides Vinca y similares; inhibidores de topoisomerasa, agentes alquilantes y similares. Estos agentes antitumorales tienen diversos problemas, por ejemplo, los tipos de cáncer para los cuales pueden usarse estos agentes antitumorales son limitados, se observan efectos adversos tales como mielotoxicidad, neuropatía y similares, surgen tumores resistentes a fármacos y similares (Nature Reviews Cancer 2003, 3, 502). Recientemente, se ha informado de un agente antitumoral dirigido molecularmente que muestra eficacia contra un tipo específico de cáncer. Imatinib o gefitinib, que es un inhibidor de la tirosina quinasa, muestra eficacia también en la leucemia mieloide crónica o el cáncer de pulmón de células no microcíticas contra los cuales los agentes antitumorales existentes son ineficaces. Sin embargo, los tipos de cáncer contra los cuales el agente muestra eficacia son limitados, y también, se ha informado un caso en el que se observa la adquisición de resistencia (Nature Reviews Drug Discovery 2004, 3, 1001). Por lo tanto, se ha demandado un agente antitumoral novedoso en el cual se mejoren tales problemas.
La señalización de Wnt/p-catenina es una vía importante asociada al desarrollo, la diferenciación y el mantenimiento de organismos vivos (Nature Reviews Drug Discovery 2006, 5, 997). Por otro lado, se sabe que la señalización anormal de Wnt/p-catenina también está asociada a diversas enfermedades tales como cáncer y similares. En ausencia de señalización de Wnt, la p-catenina citoplasmática se mantiene a un nivel bajo. La Axina y Poliposis Adenomatosa Coli (APC) forman un andamio para acelerar la fosforilación de la p-catenina intracelular por la caseína quinasa 1a (CK1a) y la glucógeno sintasa quinasa 3p (GSK3p). La p-catenina fosforilada se ubiquitina y se degrada por el proteasoma. Debido a esto, la p-catenina se mantiene en un nivel bajo y, por lo tanto, no puede desempeñar un papel como activador transcripcional. En presencia de un ligando Wnt, cuando el ligando Wnt se une a un receptor Frizzled (Fzd) y a un receptor de proteína relacionada con el receptor de lipoproteína de baja densidad (LRP), un complejo Axina-APC-CK1 a-GSK3p se inactiva a través de Deshevelled (Dv1). La p-catenina desfosforilada es estable y se acumula en las células y se transfiere al núcleo y después se une a un factor de transcripción de la familia del factor de células T (Tcf)/factor de potenciador linfoide (Lef). Este complejo del factor de transcripción induce la activación transcripcional de diversos genes diana asociados con la proliferación, la supervivencia y la diferenciación de células.
Se ha informado de la activación anormal de la señalización de Wnt/p-catenina en diversos tejidos tumorales. La activación de la señalización de Wnt/p-catenina en un tumor está asociada a una mutación genética de una molécula que constituye esta señalización o un aumento o disminución en el nivel de expresión de un producto génico del mismo (Nature Reviews Drug Discovery 2006, 5, 997, Nature Reviews Cancer 2008, 8, 387). Por ejemplo, en cáncer de intestino grueso y poliposis adenomatosa familiar coli, se ha informado una mutación de pérdida de función del gen APC. En el cáncer de intestino grueso, carcinoma hepatocelular, hepatoblastoma y meduloblastoma, se ha informado una mutación de pérdida de función del gen Axina. En el cáncer de intestino grueso, cáncer de estómago, carcinoma hepatocelular, hepatoblastoma, tumor de Wilms, cáncer de ovario y cáncer de páncreas, se ha informado una mutación de ganancia de función del gen de la p-catenina. En el cáncer de intestino grueso, cáncer de mama, melanoma, cáncer de cabeza y cuello, cáncer de pulmón no microcítico, cáncer de estómago, mesotelioma y cáncer de páncreas, se ha informado un aumento en la expresión de un ligando de Wnt. En el cáncer de intestino grueso, cáncer de mama, cáncer de cabeza y cuello, cáncer de estómago, sarcoma sinovial y cáncer de páncreas, se ha informado un aumento en la expresión de un receptor Fzd. En mesotelioma, cáncer de pulmón de células no microcíticas y cáncer de cuello de útero, se ha informado un aumento en la expresión de un miembro de la familia Dvl. En el cáncer de intestino grueso, cáncer de mama, cáncer de estómago, mesotelioma, cáncer de pulmón no microcítico, cáncer de próstata, cáncer de esófago y leucemia, se ha informado una disminución en la expresión de un miembro secreto de la familia de la proteína relacionada con el frizz secretado (SFRP), que es un factor inhibidor del ligando de Wnt. En el cáncer de intestino grueso, cáncer de mama, cáncer de próstata, cáncer de pulmón, cáncer de vejiga y mesotelioma, se ha informado una disminución en la expresión de un miembro de la familia del factor inhibidor de Wnt (WIF). La inhibición de la señalización de Wnt/p-catenina inhibe la proliferación de una línea celular de cáncer en donde la señalización de Wnt/p-catenina se activa de esta manera (Cell 2002, 111,241, Oncogene 2005, 24, 3054, Neoplasia 2004, 6, 7, Clinical Cancer Research 2003, 9, 1291, Cancer Research 2004, 64, 5385, Cancer Cell 2004, 5, 91, Proceedings of the National Academy of Sciences of the United States of America 2004, 101, 12682). Por lo tanto, una molécula que inhibe la vía Wnt/p-catenina se considera prometedora como agente antitumoral. Ha habido un informe de que otras enfermedades además del cáncer incluyendo fibrosis pulmonar, fibromatosis y osteoartritis están asociadas a la señalización de Wnt/p-catenina (The American Journal of Pathology 2003, 162, 1393, Proceedings of the National Academy of Sciences of the United States of America 2002, 99, 6973, Proceedings of the National Academy of Sciences of the United States of America 2004, 101, 9757). Por lo tanto, se espera que una molécula que inhibe la vía
Wnt/p-catenina sea útil como agente terapéutico en estos campos.
Como un compuesto que inhibe la señalización de Wnt/p-catenina, se ha informado un inhibidor de la tanquirasa (Nature 2009, 461, 614). La tanquirasa pertenece a la familia de las poli-(ADP-ribosa) polimerasas (PARP) y también se conoce como "PARP5" (Nature Reviews Molecular Cell Biology 2006, 7, 517). Se ha informado que la tanquirasa se une a Axina, que está asociada a la degradación de la p-catenina citoplasmática para realizar la poli-ADP ribosilación, acelerando de esta manera la degradación de Axina (Nature 2009, 461, 614). Se ha informado que un inhibidor de la tanquirasa acelera la degradación de la p-catenina al estabilizar Axina e inhibe la vía Wnt/p-catenina, inhibiendo así la proliferación de una línea celular de cáncer en donde se activa la señalización de Wnt/p-catenina (Nature 2009, 461, 614). Por lo tanto, se espera que dicho inhibidor de la tanquirasa sea útil como agente terapéutico para una enfermedad en donde la señalización de Wnt/p-catenina se activa como se describió anteriormente.
Por otro lado, se sabe que un compuesto representado por la fórmula (A) siguiente, tiene una actividad de absorción de adenosina (documento de patente 1).

También se sabe que un compuesto representado por la fórmula (B) siguiente tiene una actividad cardiotónica (documento no de patente 1).

Como un compuesto que tiene una actividad inhibidora de la ruta de Wnt, se conoce un compuesto representado por la fórmula (C) siguiente (documento no de patente 2).

Como un compuesto que tiene una actividad inhibidora de la tanquirasa, se conocen un compuesto representado por la fórmula (D) siguiente (documento n o de patente 3), un compuesto representado por la fórmula (E) (documento no de patente 4) siguiente y similares.

Documentos de la técnica anterior
Documento de patente
documento de patente 1: WO96/06841
Documentos de no patente
documento de no patente 1: Chemical and Pharmaceutical Bulletin (Chem. Pharm. Bull.), 1990, vol. 38, pág. 1591 documento de no patente 2: Nature Chemical Biology (Nat. Chem. Biol.), 2009, vol. 5, pág. 100
documento de no patente 3: Journal of Medicinal Chemistry (J. Med. Chem.), 2012, vol. 55, pág. 1127 documento de no patente 4: Nature, 2009, vol. 461, pág. 61
Sumario de la invención
Problemas a solucionar por la invención
Un objetivo de la presente invención es para proporcionar un compuesto heterocíclico de anillo condensado o una sal farmacéuticamente aceptable del mismo, que tiene una actividad inhibidora de la señalización Wnt y es útil como agente terapéutico y/o preventivo para, por ejemplo, cáncer, fibrosis pulmonar, fibromatosis, osteoartritis y similares, y similares.
Medios para resolver los problemas
Un primer aspecto de la invención se refiere a un compuesto heterocíclico de anillo condensado representado por la fórmula general (I) o una sal farmacéuticamente aceptable del mismo:

[en donde n1 representa 0 o 1;
cada uno de n2 y n3 es 2;
R1 representa opcionalmente arilo sustituido, un grupo heterocíclico aromático opcionalmente sustituido o un grupo heterocíclico alifático opcionalmente sustituido;
R2 representa un átomo de hidrógeno o hidroxi;
R3 representa un grupo heterocíclico aromático opcionalmente sustituido;
X1, X2, X3, y X4 pueden ser iguales o diferentes y cada uno representa N o CR4 (en donde R4 representa un átomo de hidrógeno, alquilo inferior, ciano, halógeno, hidroxi, alcoxi inferior, alcanoílo inferior o alquilsulfonilo inferior); Y1 representa CH2 o C(=O);
Y2 representa CH o N y
L representa CH2 o NH].
Un segundo aspecto de la invención se refiere a una composición farmacéutica que comprende, como principio activo, el primer aspecto de la invención.
Un tercer aspecto de la invención se refiere al primer aspecto de la invención, para su uso en el tratamiento y/o prevención de cáncer, fibrosis pulmonar, fibromatosis u osteoartritis.
En el presente documento se divulga un inhibidor de la señalización Wnt, que comprende, como principio activo, un compuesto heterocíclico de anillo condensado representado por la fórmula general (IA) o una sal farmacéuticamente aceptable de mismo:

[en donde n1A representa 0 o 1;
n2A y n3A pueden ser iguales o diferentes y cada uno representa 1 o 2;
R0A representa un átomo de hidrógeno, arilo opcionalmente sustituido, un grupo heterocíclico aromático opcionalmente sustituido o un grupo heterocíclico alifático opcionalmente sustituido;
R2A representa un átomo de hidrógeno o hidroxi;
R3A representa un grupo heterocíclico aromático opcionalmente sustituido o un grupo heterocíclico alifático opcionalmente sustituido;
X1A, x2A, X3A y X4A pueden ser iguales o diferentes y cada uno representa N o CR4A (en donde R4A representa un átomo de hidrógeno, alquilo inferior, ciano, halógeno, hidroxi, alcoxi inferior, alcanoílo inferior o alquilsulfonilo inferior);
Y1A representa CH2 o C(=O);
Y2A representa CH o N y
LA representa CH2 o NH].
En el presente documento se divulga un compuesto heterocíclico de anillo condensado representado por la fórmula general (I) o una sal farmacéuticamente aceptable del mismo:
[Quím. 6]

[en donde n1 representa 0 o 1;
n2 y n3 pueden ser iguales o diferentes y cada uno representa 1 o 2;
R1 representa opcionalmente arilo sustituido, un grupo heterocíclico aromático opcionalmente sustituido o un grupo heterocíclico alifático opcionalmente sustituido;
R2 representa un átomo de hidrógeno o hidroxi;
R3 representa un grupo heterocíclico aromático opcionalmente sustituido o un grupo heterocíclico alifático opcionalmente sustituido;
X1, X2, X3, y X4 pueden ser iguales o diferentes y cada uno representa N o CR4 (en donde R4 representa un átomo de hidrógeno, alquilo inferior, ciano, halógeno, hidroxi, alcoxi inferior, alcanoílo inferior o alquilsulfonilo inferior); Y1 representa CH2 o C(=O);
Y2 representa CH o N y
L representa CH2 o NH].
A continuación se describirán los modos preferidos de los aspectos anteriores.
En el compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con el primer aspecto de la invención, cada uno de n2 y n3 es 2.
Preferentemente, Y2 es N y L es CH2.
Preferentemente, Y1 es CH2.
Preferentemente, n1 es 0.
Preferentemente, R1 es (i) arilo opcionalmente sustituido, en donde el arilo es fenilo o (ii) un grupo heterocíclico aromático opcionalmente sustituido, en donde el grupo heterocíclico aromático es piridilo, piridonilo o pirimidinilo. Como alternativa, R1 es arilo opcionalmente sustituido o un grupo heterocíclico aromático opcionalmente sustituido y el grupo es un grupo representado por la fórmula (al) siguiente:

[en donde R5 representa un átomo de hidrógeno, alquilo C1-10 que puede estar sustituido con hidroxi, alcoxicarbonilo C1-10, alcanoílo C2-11, alquilsulfonilo C1-10, -NR6aR6b (en donde R6a y R6b pueden ser iguales o diferentes y cada uno representa un átomo de hidrógeno, alcanoílo C2-11 o alquilo C1-10), -CONR6cR6d (en donde R6c y R6d pueden ser iguales o diferentes y cada uno representa un átomo de hidrógeno o alquilo C1-10), -SO2NR6eR6f (en donde R6e y R6f pueden ser iguales o diferentes y cada uno representa un átomo de hidrógeno o alquilo C1-10), halógeno, ciano, carboxi o nitro y Z1, Z2, Z3 y Z4 pueden ser iguales o diferentes y cada una representa N o CR7 (en donde R7 representa un átomo de hidrógeno, carboxi o halógeno)] o un grupo representado por la fórmula (a2) siguiente:

(en donde R5, Z1 y Z4 tienen las mismas definiciones que se han descrito anteriormente, respectivamente).
Preferentemente, R5 es ciano, -CONH2 o -SO2NH2.
Más preferentemente, R5 es ciano.
Preferentemente, R7 es un átomo de hidrógeno o un átomo de flúor.
En el compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con el primer aspecto de la invención, R3 es un grupo heterocíclico aromático opcionalmente sustituido.
Preferentemente, el grupo heterocíclico aromático es un grupo heterocíclico aromático bicíclico.
Más preferentemente, el grupo heterocíclico aromático es quinazolinilo.
En el presente documento se divulga un compuesto o la sal farmacéuticamente aceptable del mismo, en donde R3 es un grupo heterocíclico alifático opcionalmente sustituido.
El segundo aspecto de la invención se refiere a una composición farmacéutica, que comprende, como principio activo, el compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con el primer aspecto de la invención. En el presente documento se divulga un inhibidor de la señalización Wnt, que comprende, como principio activo, el compuesto o la sal farmacéuticamente aceptable del mismo descritos anteriormente.
En el presente documento se divulga un inhibidor de la señalización Wnt, en donde la inhibición de la señalización Wnt es inhibición de la señalización Wnt mediante inhibición de la tanquirasa.
En el presente documento se divulga un agente terapéutico y/o preventivo para una enfermedad asociada con señalización la Wnt, que comprende, como principio activo, el compuesto o la sal farmacéuticamente aceptable del mismo descritos anteriormente.
Tal como se divulga en el presente documento, la enfermedad asociada con la señalización Wnt es cáncer, fibrosis pulmonar, fibromatosis u osteoartritis.
En el presente documento se divulga un método para inhibir la señalización Wnt, que comprende administrar una cantidad eficaz de un compuesto heterocíclico de anillo condensado representado por la fórmula general (IA) o una sal farmacéuticamente aceptable del mismo:
iQ u ím . 9 ]

(en donde n1A, n2A, n3A, R0A, R2A, R3A, X1A, X2A, X3A, X4A, Y1A, Y2A y LA tienen las mismas definiciones que se han descrito anteriormente, respectivamente).
En el presente documento se divulga un método para inhibir la señalización Wnt, que comprende administrar una cantidad eficaz del compuesto o de la sal farmacéuticamente aceptable del mismo descritos anteriormente.
Tal como se divulga en el presente documento, el método para inhibir la señalización Wnt es un método para inhibir la señalización Wnt mediante inhibición de la tanquirasa.
En el presente documento se divulga un método para tratar y/o prevenir una enfermedad asociada con la señalización Wnt, que comprende administrar una cantidad eficaz del compuesto o de la sal farmacéuticamente aceptable del mismo descritos anteriormente.
Tal como se divulga en el presente documento, la enfermedad asociada con la señalización Wnt es cáncer, fibrosis pulmonar, fibromatosis u osteoartritis.
En el presente documento se divulga un compuesto heterocíclico de anillo condensado representado por la fórmula general (IA) o una sal farmacéuticamente aceptable del mismo para su uso en la inhibición de la señalización Wnt:
[Q u ím . 10 ]

(en donde n1A, n2A, n3A, R0A, R2A, R3A, X1A, X2A, X3A, X4A, Y1A, Y2A y LA tienen las mismas definiciones que se han descrito anteriormente, respectivamente).
En el presente documento se divulga un compuesto o la sal farmacéuticamente aceptable del mismo descritos anteriormente para su uso en la inhibición de la señalización Wnt.
Tal como se divulga en el presente documento, la inhibición de la señalización Wnt es inhibición de la señalización Wnt mediante inhibición de la tanquirasa.
En el presente documento se divulga el compuesto o la sal farmacéuticamente aceptable del mismo descritos
anteriormente para su uso en el tratamiento y/o prevención de una enfermedad asociada con señalización Wnt.
Tal como se divulga en el presente documento, la enfermedad asociada con la señalización Wnt es cáncer, fibrosis pulmonar, fibromatosis u osteoartritis.
En el presente documento se divulga el uso de un compuesto heterocíclico de anillo condensado representado por la fórmula general (IA) o una sal farmacéuticamente aceptable del mismo para la fabricación de un inhibidor de la señalización de Wnt:
[Quim. 11]

(en donde y LA ti descrito anteriormente, respectivamente).
y LA ti descrito anteriormente, respectivamente).
En el presente documento se divulga el uso del compuesto o la sal farmacéuticamente aceptable del mismo descritos anteriormente, para la fabricación de un inhibidor de la señalización Wnt.
En el presente documento se divulga el uso de un compuesto o de la sal farmacéuticamente aceptable del mismo descritos anteriormente, en donde la inhibición de la señalización Wnt es inhibición de la señalización Wnt mediante inhibición de la tanquirasa.
En el presente documento se divulga el uso del compuesto o de la sal farmacéuticamente aceptable del mismo descritos anteriormente, para la fabricación de un agente terapéutico y/o preventivo para una enfermedad asociada con la señalización Wnt.
Tal como se divulga en el presente documento, la enfermedad asociada con la señalización Wnt es cáncer, fibrosis pulmonar, fibromatosis u osteoartritis.
Efectos de la invención
Un compuesto heterocíclico de anillo condensado o una sal farmacéuticamente aceptable del mismo de acuerdo con la presente invención tiene una actividad inhibidora de la señalización Wnt y es útil como agente terapéutico y/o preventivo para, por ejemplo, cáncer, fibrosis pulmonar, fibromatosis, osteoartritis y similares.
Modo de llevar a cabo la invención o la divulgación
En lo sucesivo en el presente documento, un compuesto representado por la fórmula general (I) se denomina Compuesto (I). Los compuestos que tienen los otros números de fórmula se mencionan de la misma manera.
En las definiciones de los grupos respectivos en la fórmula general (I) y la fórmula general (IA), los ejemplos del alquilo inferior; los restos alquilo inferior del alcoxi inferior, el alcanoílo inferior y el alquilsulfinilo inferior; el alquilo C1-10; y los restos alquilo C1-10 del alcoxicarbonilo C1-10, el alcanoílo C2-11 y el alquilsulfonilo C1-10 incluyen alquilo lineal o ramificado que tiene cada uno de 1 a 10 átomos de carbono y, más específicamente, incluyen metilo, etilo, propilo, isopropilo, butilo, isobutilo, sec-butilo, tere-butilo, pentilo, isopentilo, neopentilo, hexilo, heptilo, octilo, nonilo, decilo y similares.
Los ejemplos del arilo incluyen arilo que tiene cada uno de 6 a 14 átomos de carbono y, más específicamente, incluyen fenilo, naftilo, azulenilo, antrilo y similares.
Los ejemplos del grupo heterocíclico alifático incluyen un grupo heterocíclico alifático monocíclico de 5 o 6 miembros que tiene al menos un átomo seleccionado entre un átomo de nitrógeno, un átomo de oxígeno y un átomo de azufre, un grupo heterocíclico alifático de anillo condensado, bicíclico o tricíclico en el que los anillos de 3 a 8 miembros están condensados y al menos contiene un átomo seleccionado entre un átomo de nitrógeno, un átomo de oxígeno y un átomo de azufre, y similares y, más específicamente incluye aziridinilo, azetidinilo, pirrolidinilo, piperidino, piperidinilo, azepanilo, 1,2,5,6-tetrahidropiridilo, imidazolidinilo, pirazolidinilo, piperazinilo, homopiperazinilo, pirazolinilo, oxiranilo, tetrahidrofuranilo, tetrahidro-2H-piranilo, 5,6-dihidro-2H-piranilo, oxazolidinilo, morfolino, morfolinilo, tioxazolidinilo,
tiomorfolinilo, 2H-oxazolilo, 2H-tioxazolilo, dihidroindolilo, dihidroisoindolilo, dihidrobenzofuranilo, benzoimidazolidinilo, dihidrobenzoxazolilo, dihidrobenzotioxazolilo, benzodioxolinilo, tetrahidroquinolilo, tetrahidroisoquinolilo, dihidro-2H-cromanilo, dihidro-IH-cromanilo, dihidro-2H-tiocromanilo, dihidro-IH-tiocromanilo, tetrahidroquinoxalinilo, tetrahidroquinazolinilo, dihidrobenzodioxanilo, 7,8-dihidro-5H-pirano[4,3-d]pirimidinilo, 5,6,7,8-tetrahidropirido[4,3-d]pirimidinilo, dioxoloquinazolinilo, 6-oxo-6,7-dihidro-5H-pirimido[4,5-b][1,4]oxazin-4-ilo y similares.
los ejemplos del grupo heterocíclico aromático incluyen un grupo heterocíclico aromático, monocíclico, de 5 o 6 miembros, que tiene al menos un átomo seleccionado entre un átomo de nitrógeno, un átomo de oxígeno y un átomo de azufre, un grupo heterocíclico aromático de anillo condensado, bicíclico o tricíclico, en el que los anillos de 3 a 8 miembros están condensados y contiene al menos un átomo seleccionado entre un átomo de nitrógeno, un átomo de oxígeno y un átomo de azufre, y similares, y más específicamente incluye furilo, tienilo, pirrolilo, imidazolilo, pirazolilo, oxazolilo, isoxazolilo, oxadiazolilo, tiazolilo, isotiazolilo, tiadiazolilo, triazolilo, tetrazolilo, piridilo, piridonilo, piridazinilo, pirimidinilo, pirazinilo, triazinilo, benzofuranilo, benzotiofenilo, benzoxazolilo, benzotiazolilo, isoindolilo, indolilo, indazolilo, benzoimidazolilo, benzotriazolilo, oxazolopirimidinilo, tiazolopirimidinilo, pirrolopiridinilo, pirrolopirimidinilo, imidazopiridinilo, purinilo, quinolinilo, isoquinolinilo, cinnolinilo, ftalazinilo, quinazolinilo, quinoxalinilo, naftiridinilo, piridopiridinilo, 7-oxidopirido[4,3-d]pirimidinilo, benzo[d][1,2,3]triazinilo, [1,2,4]triazolo[4,3-a]piridin-3(2H)-onilo, 8-oxo-8,9-dihidro-7H-purin-6-ilo, 3-oxo-2,3-dihidro-[1,2,4]triazolo[4,3-a]piridin-5-ilo, 4-oxo-3,4-dihidropirido[4,3-d]pirimidin-5-ilo, 4-oxo-3,4-dihidropirido[4,3-d]pirimidin-7-ilo, 4-oxo-3,4-dihidropirido[3,4-d]pirimidin-5- ilo, 4-oxo-3,4-dihidropirido[3,4-d]pirimidin-8-ilo, 3-oxo-2,3-dihidro-[1,2,4]triazolo[4,3-a]piridin-6-ilo, 3-oxo-2,3-dihidro-[1.2.4] triazolo[4,3-a]pirazin-6-ilo, 3-oxo-2,3-dihidro-[1,2,4]triazolo[4,3-a]pirazin-8-ilo, imidazo[1,2-a]pirazinilo y similares.
Los ejemplos de grupo heterocíclico aromático bicíclico incluyen, entre los anillos heterocíclicos aromáticos mencionados anteriormente, benzofuranilo, benzotiofenilo, benzoxazolilo, benzotiazolilo, isoindolilo, indolilo, indazolilo, benzoimidazolilo, benzotriazolilo, oxazolopirimidinilo, tiazolopirimidinilo, pirrolopiridinilo, pirrolopirimidinilo, imidazopiridinilo, purinilo, quinolinilo, isoquinolinilo, cinnolinilo, ftalazinilo, quinazolinilo, quinoxalinilo, naftiridinilo, piridopirimidinilo, 7-oxidopirido[4,3-d]pirimidinilo, benzo[d][1,2,3]triazinilo, [1,2,4]triazolo[4,3-a]piridin-3(2H)-onilo, 8-oxo-8,9-dihidro-7H-purin-6-ilo, 3-oxo-2,3-dihidro-[1,2,4jtriazolo[4,3-a]piridin-5-ilo, 4-oxo-3,4-dihidropirido[4,3-d]pirimidin-5-ilo, 4-oxo-3,4-dihidropirido[4,3-d]pirimidin-7-ilo, 4-oxo-3,4-dihidropirido[3,4-d]pirimidin-5-ilo, 4-oxo-3,4-dihidropirido[3,4-d]pirimidin-8-ilo, 3-oxo-2,3-dihidro-[1,2,4]triazolo[4,3-a]piridin-6-ilo, 3-oxo-2,3-dihidro-[1.2.4] triazolo[4,3-a]pirazin-6-ilo, 3-oxo-2,3-dihidro-[1,2,4]triazolo[4,3-a]pirazin-8-ilo, imidazo[1,2-a]pirazinilo y similares.
El halógeno se refiere a cada átomo de flúor, cloro, bromo o yodo.
Los sustituyentes en el arilo opcionalmente sustituido y el grupo heterocíclico aromático opcionalmente sustituido, que pueden ser iguales o diferentes, incluyen sustituyentes seleccionados entre el grupo que comprende halógeno, hidroxi, nitro, ciano, carboxi, sulfamoílo, alquilo C1-10 que puede estar sustituido con hidroxi, trifluorometilo, cicloalquilo C3-8, arilo C6-14, un grupo heterocíclico alifático, un grupo heterocíclico aromático, alcoxi C1-10, cicloalcoxi C3-8, ariloxi Ca-14, aralquiloxi C7-16, alcanoiloxi C2-11, aroiloxi C7-15, alquilsulfanilo C1-10, -NRXaRYa (en donde RXa y RYa pueden ser iguales o diferentes y cada uno representa un átomo de hidrógeno, alquilo C1-10, cicloalquilo C3-8, arilo C6-14, un grupo heterocíclico aromático, aralquilo C7-16, alcanoílo C2-11, aroílo C7-15, alcoxicarbonilo C1-10 o aralquiloxicarbonilo C7-16 o, RXa y RYa se combinan junto con el átomo de nitrógeno adyacente al mismo para formar un grupo heterocíclico que contiene nitrógeno, el cual puede estar sustituido con alquilo C1-10), alcanoílo C2-11, aroílo C7-15, alcoxicarbonilo C1-10, ariloxicarbonilo C6-14, alquilsulfonilo C1-10, -CONRXbRYb (en donde RXb y RYb pueden ser iguales o diferentes y cada uno representa un átomo de hidrógeno, alquilo C1-10, cicloalquilo C3-8, arilo C6-14, un grupo heterocíclico aromático o aralquilo C7-16 o, RXb y RYb se combinan junto con el átomo de nitrógeno adyacente al mismo para formar un grupo heterocíclico que contiene nitrógeno que puede estar sustituido con alquilo C1-10), -SO2NRXcRYc (en donde RXc y RYc pueden ser iguales o diferentes y cada uno representa un átomo de hidrógeno o alquilo C1-10 o, RXc y RYc se combinan junto con el átomo de nitrógeno adyacente al mismo para formar un grupo heterocíclico que contiene nitrógeno que puede estar sustituido con alquilo C1-10) y similares.
Los sustituyentes del grupo heterocíclico alifático opcionalmente sustituido incluyen sustituyentes seleccionados entre el grupo que comprende oxo, halógeno, hidroxi, nitro, ciano, carboxi, sulfamoílo, alquilo C1-10 que puede estar sustituido con hidroxi, trifluorometilo, cicloalquilo C3-8, arilo C6-14, un grupo heterocíclico alifático, un grupo heterocíclico aromático, alcoxi C1-10, cicloalcoxi C3-8, ariloxi C6-14, aralquiloxi C7-16, alcanoiloxi C2-11, aroiloxi C7-15, alquilsulfanilo C1-10, -NRXdRYd (en donde RXd y RYd pueden ser iguales o diferentes y cada uno representa un átomo de hidrógeno, alquilo C1-10, cicloalquilo C3-8, arilo C6-14, un grupo heterocíclico aromático, aralquilo C7-16, alcanoílo C2-11, aroílo C7-15, alcoxicarbonilo C1-10 o aralquiloxicarbonilo C7-16 o, RXd y RYd se combinan junto con el átomo de nitrógeno adyacente al mismo para formar un grupo heterocíclico que contiene nitrógeno que puede estar sustituido con alquilo C1-10), alcanoílo C2-11, aroílo C7-15, alcoxicarbonilo C1-10, ariloxicarbonilo C6-14, -CONRXeRYe (en donde RXe y RYe pueden ser iguales o diferentes y cada uno representa un átomo de hidrógeno, alquilo C1-10, cicloalquilo C3-8, arilo C6-14, un grupo heterocíclico aromático o aralquilo C7-16 o, RXe y RYe se combinan junto con el átomo de nitrógeno adyacente al mismo para formar un grupo heterocíclico que contiene nitrógeno que puede estar sustituido con alquilo C1-10), alquilsulfonilo C1-10, -SO2NRXfRYf (en donde RXf y RYf pueden ser iguales o diferentes y cada uno representa un átomo de hidrógeno o alquilo C1-10 o, RXf y RYf se combinan junto con el átomo de nitrógeno adyacente al mismo para formar un grupo
heterocíclico que contiene nitrógeno que puede estar sustituido con alquilo C1-10) y similares.
Los ejemplos del alquilo C1-10 y los restos C1-10 del alcoxi C1-10, el alcanoiloxi C2-11, el alquilsulfanilo C1-10, el alcanoílo C2-11, el alquilsulfonilo C1-10 y el alcoxicarbonilo C1-10 aquí mostrados incluyen los grupos ejemplificados como el alquilo inferior descrito anteriormente.
Los ejemplos del cicloalquilo C3-8 y los restos cicloalquilo del cicloalcoxi C3-8 incluyen ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, ciclooctilo y similares.
Los ejemplos del arilo C6-14 arilo y los restos arilo del ariloxi C6-14, el aroílo C7-15, el aroiloxi C7-15 y el ariloxicarbonilo C6-14 incluyen los grupos ejemplificados como el arilo descrito anteriormente.
Los ejemplos de los restos arilo del aralquiloxi C7-16, el aralquilo C7-16 y el aralquiloxicarbonilo C7-16 incluyen los grupos ejemplificados como arilo descritos anteriormente y los ejemplos de los restos alquilo de los mismos incluyen alquileno C1-10 y, más específicamente, incluyen grupos en los que un átomo de hidrógeno se elimina de los grupos ejemplificados como alquilo inferior descritos anteriormente.
El grupo heterocíclico alifático, el grupo heterocíclico aromático y el halógeno tienen las mismas definiciones que se han descrito anteriormente, respectivamente.
Los ejemplos del grupo heterocíclico que contiene nitrógeno formado junto con el átomo de nitrógeno adyacente, incluyen un grupo heterocíclico monocíclico de 5 o 6 miembros que tiene al menos un átomo de nitrógeno (el grupo heterocíclico monocíclico puede contener otro átomo de nitrógeno, un átomo de oxígeno o un átomo de azufre), un grupo heterocíclico de anillo condensado, bicíclico o tricíclico, en el que los anillos de 3 a 8 miembros están condensados y contiene al menos un átomo de nitrógeno (el grupo heterocíclico de anillo condensado puede contener otro átomo de nitrógeno, un átomo de oxígeno o un átomo de azufre) y similares y, más específicamente, incluyen aziridinilo, azetidinilo, pirrolidinilo, piperidino, azepanilo, pirrolilo, imidazolidinilo, imidazolilo, pirazolidinilo, pirazolinilo, pirazolilo, piperazinilo, homopiperazinilo, oxazolidinilo, 2H-oxazolilo, tioxazolidinilo, 2H-tioxazolilo, morfolino, tiomorfolinilo, dihidroindolilo, dihidroisoindolilo, indolilo, isoindolilo, tetrahidroquinolilo, tetrahidroisoquinolilo, dihidrobenzooxazolilo, dihidrobenzotioxazolilo, benzoimidazolidinilo, benzoimidazolilo, dihidroindazolilo, indazolilo, benzotriazolilo, pirrolopiridinilo, pirrolopirimidinilo, imidazopiridinilo, purinilo y similares.
Las sales farmacéuticamente aceptables de los compuestos (IA) y (I) incluyen, por ejemplo, sales de adición de ácidos, sales de metales, sales de amonio, sales de adición de amina orgánica, una sal de adición de aminoácido farmacéuticamente aceptables y similares. Los ejemplos de las sales de adición de ácidos farmacéuticamente aceptables de los compuestos (IA) y (I) incluyen sales de ácidos inorgánicos tales como clorhidratos, bromhidratos, nitratos, sulfatos, fosfatos y similares, sales de ácidos orgánicos tales como acetatos, oxalatos, maleatos, fumaratos, citratos, benzoatos, metanosulfonatos y similares. Los ejemplos de las sales de metal farmacéuticamente aceptables incluyen sales de metales alcalinos tales como sales de sodio, sales de potasio y similares, sales de metales alcalinotérreos, tales como sales de magnesio, sales de calcio, sales de aluminio, sales de cinc y similares. Los ejemplos de sales de amonio farmacéuticamente aceptables incluyen sales de amonio, tetrametilamonio y similares. Los ejemplos de sales de adición de amina orgánica farmacéuticamente aceptables incluyen sales de adición de morfolina, piperidina y similares. Los ejemplos de sales de adición de aminoácidos farmacéuticamente aceptables incluyen sales de adición de lisina, glicina, fenilalanina, ácido aspártico, ácido glutámico y similares.
A continuación, se explicarán los procesos de producción para los compuestos (IA) y (I).
A propósito, en los procesos de producción mostrados a continuación, cuando un grupo definido cambia en las condiciones de los procesos de producción o es inapropiado para llevar a cabo los proceso de producción, se puede producir un compuesto diana usando los métodos para introducir y eliminar un grupo protector usado habitualmente en la química sintética orgánica [por ejemplo, Protective Groups in Organic Synthesis, tercera edición, escrito por T. W. Greene, John Wiley & Sons, Inc. (1999) y similares] y similares. Además, si fuera necesario, es posible cambiar el orden de las etapas de reacción para introducir un sustituyente y similares.
Los compuestos (IA) y (I) se pueden producir de acuerdo con, por ejemplo, las etapas siguientes.
Proceso de producción 1
Entre los compuestos (I), se puede producir el compuesto (I-a) en donde Y1 es CH2, Y2 es N y L es CH2 de acuerdo con, por ejemplo, las etapas siguientes.

(en donde P1 representa un grupo protector para un átomo de nitrógeno normalmente usado en la química sintética orgánica, por ejemplo, metoxicarbonilo, etoxicarbonilo, ferc-butoxicarbonilo, 9-fluorenilmetoxicarbonilo, 2,2,2-tridoroetoxicarbonilo, viniloxicarbonilo, aliloxicarbonilo o similares, X5 representa un átomo de cloro, un átomo de bromo, un átomo de yodo, metanosulfoniloxi, trifluorometanosulfoniloxi, bencenosulfoniloxi, p-toluenosulfoniloxi o similares y X1, X2, X3, X4, R1, R2, R3, n1, n2 y n3 tienen las mismas definiciones que se han descrito anteriormente, respectivamente)
Etapa 1
El compuesto (a-1) se puede producir mediante, por ejemplo, un método modificado del método para eliminar un grupo protector descrito en Protective Groups in Organic Synthesis, escrito por T. W. Greene, John Wiley & Sons, Inc. (1981) y similares.
Por ejemplo, en el caso en el que P1 es ferc-butoxicarbonilo, El compuesto (a-1) se puede producir tratando el compuesto (A-0), por ejemplo, sin disolvente o en un disolvente con 1 equivalente hasta una gran cantidad en exceso de un ácido a una temperatura entre -30 °C y 100 °C durante 5 minutos a 72 horas.
Los ejemplos del ácido incluyen ácido clorhídrico, ácido sulfúrico, ácido trifluoroacético, ácido metanosulfónico y similares. Los ejemplos del disolvente incluyen metanol, etanol, 1-propanol, 2-propanol, tetrahidrofurano (THF), 1,4-dioxano, 1,2-dimetoxietano (DME), tolueno, acetato de etilo, diclorometano, 1,2-dicloroetano, agua y similares y estos se usan en solitario o mezclados.
Los compuestos (A-0) se pueden producir de acuerdo con las etapas mencionadas a continuación.
Etapa 2
El compuesto (I-a) se puede producir haciendo reaccionar el compuesto (a-1) con preferentemente de 1 a 10 equivalentes del compuesto (a-2) sin disolvente o en un disolvente y, si fuera necesario, en presencia de, preferentemente, de 1 a 10 equivalentes de una base a una temperatura entre -20 °C y 150 °C durante 5 minutos a 72 horas.
Los ejemplos de la base incluyen carbonato de potasio, hidróxido de potasio, hidróxido sódico, metóxido sódico, hidruro sódico, ferc-butóxido de potasio, trietilamina, diisopropiletilamina, N-metilmorfolina, piridina, 1,8-di-azabiciclo[5.4.0]-7-undeceno (DBU) y similares.
Los ejemplos del disolvente incluyen metanol, etanol, 2-propanol, diclorometano, cloroformo, 1,2-dicloroetano, tolueno, acetato de etilo, acetonitrilo, éter dietílico, THF, DME, 1,4-dioxano, N,N-dimetilformamida (DMF), N,N-dimetilacetamida (DMA), N-metilpirrolidona (NMP), piridina, agua y similares y estos se usan en solitario o mezclados.
El compuesto (a-2) se obtiene en forma de un producto disponible en el mercado o se puede obtener por un método conocido [por ejemplo, Jikken Kagaku Koza (Encyclopedia of Experimental Chemistry), 5a ed., vol. 13, p. 341, Maruzen Co., Ltd. (2003) o similar] o un método modificado del mismo.
El compuesto (I-a) se puede producir tratando el compuesto (a-1) con preferentemente de 1 a 10 equivalentes del compuesto (a-3) en un disolvente, en presencia de preferentemente de 1 a 10 equivalentes de un agente de condensación y, si fuera necesario, en presencia de, preferentemente, de 1 a 10 equivalentes de una base a una temperatura entre -20 °C y 150 °C durante 5 minutos a 72 horas.
El compuesto (a-3) se obtienen en forma de un producto disponible en el comercio o se puede obtener mediante un método conocido (por ejemplo, Journal of Medicinal Chemistry, 2010, 53, 8089 o similar) o un método modificado del mismo.
Los ejemplos del agente de condensación incluyen hexafluorofosfato de benzotriazol-1-iloxitris(dimetilamino)fosfonio (BOP), hexafluorofosfato de benzotriazol-1-iloxitris(pirrolidino)fosfonio (PyBOP), hexafluorofosfato de
bromotris(pirrolidin)fosfonio (PyBroP) y similares y, preferentemente, incluyen BOP y similares. Los ejemplos de la base incluyen trietilamina, W,A-diisopropiletilamina, DBU, 1,5-diazabicido[4.3.0]non-5-eno (DBN), N-metilpiperidina, N-metilmorfolina y similares y, preferentemente, incluyen DBU y similares. Los ejemplos del disolvente incluyen metanol, etanol, diclorometano, cloroformo, 1,2-dicloroetano, tolueno, acetato de etilo, acetonitrilo, éter dietílico, THF, DME, 1,4-dioxano, DMF, DMA, NMP, agua y similares y estos se usan en solitario o mezclados.
El compuesto (A-0) para usar en el proceso de producción 1 anterior se puede producir de acuerdo con las etapas siguientes.
Entre los compuestos (A-0), el compuesto (A-1), en el que n1 es 1 y el compuesto (A-2), en el que n1 es 0 y R1 es arilo opcionalmente sustituido o un grupo heterocíclico aromático opcionalmente sustituido, se pueden producir de acuerdo con, por ejemplo, las etapas siguientes.

(en donde, R1A representa arilo opcionalmente sustituido o un grupo heterocíclico opcionalmente sustituido en la definición de R1, X5A representa un átomo de cloro, un átomo de bromo, un átomo de yodo, metanosulfoniloxi, trifluorometanosulfoniloxi, bencenosulfoniloxi, p-toluenosulfoniloxi o similares, X6 representa un átomo de cloro, un átomo de bromo, un átomo de yodo, metanosulfoniloxi, trifluorometanosulfoniloxi, bencenosulfoniloxi, ptoluenosulfoniloxi, B(ORB1) (ORB2) (en donde RB1 y RB2 pueden ser iguales o diferentes y cada uno representa un átomo de hidrógeno, alquilo C1-6 o similar o, RB1 y RB2 se combinan para representar alquileno C1-6 o similar) o similar y X1, X2, X3, X4, R2, P1, n2 y n3 tienen las mismas definiciones que se han descrito anteriormente, respectivamente)
Etapa 3
El compuesto (a-6) se puede producir haciendo reaccionar el compuesto (a-4) con preferentemente de 1 a 10 equivalentes del compuesto (a-5) en un disolvente, en presencia de preferentemente de 1 a 10 equivalentes de un agente reductor y preferentemente de 1 a 10 equivalentes de un ácido a una temperatura entre -20 °C y 150 °C durante de 5 minutos a 72 horas.
Los ejemplos del agente de reducción incluyen triacetoxiborohidruro sódico, cianoborohidruro sódico y similares.
Los ejemplos del ácido incluyen ácido clorhídrico, ácido sulfúrico, ácido fórmico, ácido acético, ácido trifluoroacético, ácido p-toluenosulfónico, tetracloruro de titanio y similares.
Los ejemplos del disolvente incluyen metanol, etanol, diclorometano, cloroformo, 1,2-dicloroetano, tolueno, acetato de etilo, acetonitrilo, éter dietílico, t Hf , DME, 1,4-dioxano, DMF, DMA, NMP, agua y similares y estos se usan en solitario o mezclados.
El compuesto (a-5) se obtiene en forma de un producto disponible en el mercado o se puede obtener por un método conocido [por ejemplo, Jikken Kagaku Koza, 5a ed., vol. 14, p. 351, Maruzen Co., Ltd. (2003) o similar] o un método modificado del mismo.
El compuesto (a-4) se puede obtener en forma de un producto disponible en el comercio.
Etapa 4
El compuesto (a-7) se puede producir tratando el compuesto (a-6) en un disolvente en presencia de 1 a 30 equivalentes de un aditivo a una temperatura de entre -20 °C y el punto de ebullición del disolvente para usarse durante de 5 minutos a 72 horas o, mediante tratamiento del compuesto (a-6) en atmósfera de hidrógeno o en presencia de una fuente de hidrógeno en presencia de un catalizador a una temperatura de entre -20 °C y el punto de ebullición del disolvente para usarse a presión normal o con incremento de presión durante de 5 minutos a 72 horas.
Los ejemplos del aditivo incluyen hierro reducido, cloruro de estaño (II) y similares.
Los ejemplos del catalizador incluyen paladio sobre carbono, paladio, hidróxido de paladio, acetato de paladio, negro de paladio y similares y estos se usan en una cantidad de preferentemente el 0,01 al 50 % en peso con respecto al compuesto (a-6).
Los ejemplos de la fuente de hidrógeno incluyen ácido fórmico, formiato de amonio, formiato de sodio, ciclohexadieno, hidrazina y similares y estos se usan en una cantidad de preferentemente 2 equivalentes hasta una gran cantidad en exceso con respecto al compuesto (a-6).
Los ejemplos del disolvente incluyen metanol, etanol, tolueno, acetato de etilo, acetonitrilo, éter dietílico, THF, DME, 1,4-dioxano, DMF, DMA, NMP, ácido acético, agua y similares y estos se usan en solitario o mezclados.
Etapa 5
El compuesto (a-8) se puede producir haciendo reaccionar el compuesto (a-7) en un disolvente en presencia de preferentemente de 1 a 10 equivalentes de fosgeno o 1,1-carbonildiimidazol y, si fuera necesario, en presencia de preferentemente de 1 a 10 equivalentes de una base a una temperatura entre -20 °C y el punto de ebullición del disolvente para usarse de 5 minutos a 72 horas.
Los ejemplos de la base incluyen carbonato de potasio, hidróxido de potasio, hidróxido sódico, metóxido sódico, hidruro sódico, ferc-butóxido de potasio, trietilamina, diisopropiletilamina, DBU y similares.
Los ejemplos del disolvente incluyen diclorometano, cloroformo, 1,2-dicloroetano, tolueno, acetato de etilo, acetonitrilo, éter dietílico, THF, DMF, NMP, piridina y similares y estos se usan en solitario o mezclados.
Etapa 6
El compuesto (A-1) se puede obtener de la misma manera que en la etapa 2 mencionada anteriormente usando el compuesto (a-8) y, preferentemente, de 1 a 10 equivalentes del compuesto (a-9).
El compuesto (a-9) se puede obtener en forma de un producto disponible en el comercio.
Etapa 7
El compuesto (A-2) se puede producir haciendo reaccionar el compuesto (a-8) con de 1 a 10 equivalentes del compuesto (a-10) en un disolvente en presencia de una cantidad catalítica hasta 10 equivalentes de un catalizador de cobre o un catalizador de paladio a una temperatura entre temperatura ambiente y 140 °C durante 5 minutos a 72 horas. La reacción también se puede realizar en presencia de una cantidad catalítica hasta 10 equivalentes de una base y también se puede realizar en presencia de una cantidad catalítica hasta 10 equivalentes de un compuesto organofosforado.
Los ejemplos del catalizador de cobre incluyen cobre (0), yoduro de cobre (I), yoduro de cobre (II), acetato de cobre (II), óxido de cobre (II), cloruro de cobre (I), cloruro de di-|j-hidroxo-bis[(N,N,N',N'-tetrametiletilendiamina)cobre (II)] y similares y, preferentemente, incluyen yoduro de cobre (I), acetato de cobre (II) y similares.
Los ejemplos del catalizador de paladio incluyen acetato de paladio (II), cloruro de bis(trifenilfosfina)paladio (II), tetraquis(trifenilfosfina)paladio (0), cloruro de [1,2-bis(difenilfosfino)etano]paladio (II), cloruro de [1,1'-bis(difenilfosfino)ferroceno]paladio (II), tris(dibencilidenoacetona)dipaladio (0) y similares y, preferentemente, incluyen acetato de paladio (II), cloruro de bis(trifenilfosfina)paladio (II), tetraquis(trifenilfosfina)paladio (0), tris(dibencilidenacetona)dipaladio (0) y similares.
Los ejemplos de la base incluyen carbonato de potasio, carbonato de cesio, cloruro de litio, cloruro de potasio, fercbutóxido de potasio, ferc-butóxido de sodio, trietilamina, acetato de potasio, etóxido de sodio, carbonato sódico, hidróxido sódico, fosfato de potasio, etilendiamina, glicina, N-metilpirrolidina, piridina, 1,2-diaminociclohexano y similares y, preferentemente, incluyen carbonato de potasio, carbonato de cesio, ferc-butóxido de potasio, fosfato de potasio, etilendiamina, 1,2-diaminociclohexano, trietilamina y similares.
Los ejemplos del compuesto organofosforado incluyen trifenilfosfina, tri(2-furil)fosfina, 2-diciclohexilfosfin-2'-(N,N-dimetilamino)bifenilo, difenilfosfinoferroceno, 2-diciclohexilfosfin-2'4'6'-triisopropilbifenilo (Xphos) y similares y, preferentemente, incluyen 2-diciclohexilfosfin-2'-(N,N-dimetilamino)bifenilo, XPhos y similares.
Los ejemplos del disolvente incluyen éter dietílico, THF, 1,4-dioxano, DMF, DMA, dimetilsulfóxido (DMSO), benceno, tolueno, xileno, diclorometano, cloroformo, tetracloruro de carbono, 1,2-dicloroetano, acetonitrilo, acetato de etilo, acetato de metilo, metil etil cetona, metanol, etanol, propanol, 2-propanol, butanol, hexano y similares y, preferentemente, incluyen THF, 1,4-dioxano, DMF y similares.
El compuesto (a-10) se obtiene en forma de un producto disponible en el mercado o se puede obtener por un método conocido [por ejemplo, Jikken Kagaku Koza, 5a ed., vol. 13, p. 341, Maruzen Co., Ltd. (2003) o similar] o un método modificado del mismo.
Entre los compuestos (A-0), el compuesto (A-3) en el que n1 es 0 y R1 es un grupo heterocíclico opcionalmente sustituido se puede producir de acuerdo con, por ejemplo, las etapas siguientes.

(en donde R1B representa un grupo heterocíclico alifático opcionalmente sustituido en la definición de R1 y X1, X2, X3, X4, R2, P1, n2 y n3 tienen las mismas definiciones que se han descrito anteriormente, respectivamente)
Etapa 8
El compuesto (a-12) se puede producir de la misma manera que en la etapa 3 mencionada anteriormente, usando el compuesto (a-7) obtenido en la etapa 4 y el compuesto (a-11).
El compuesto (a-11) se obtiene en forma de un producto disponible en el mercado o se puede obtener por un método conocido [por ejemplo, Jikken Kagaku Koza, 5a ed., vol. 15, p. 154, Maruzen Co., Ltd. (2003) o similar] o un método modificado del mismo.
Etapa 9
El compuesto (A-3) se puede producir de la misma manera que en la etapa 5 mencionada anteriormente, usando el compuesto (a-12).
Proceso de producción 2
Entre los compuestos (I), el compuesto (I-b) y el compuesto (I-c), en el que R2 es un átomo de hidrógeno, Y1 es CH2, Y2 es CH y L es NH y (i) n1 es 1 (compuesto (I-b)) o (ii) n1 es 0 y R1 es arilo opcionalmente sustituido o un grupo heterocíclico aromático opcionalmente sustituido (compuesto (I-c)), se pueden producir de acuerdo con, por ejemplo, las etapas siguientes.
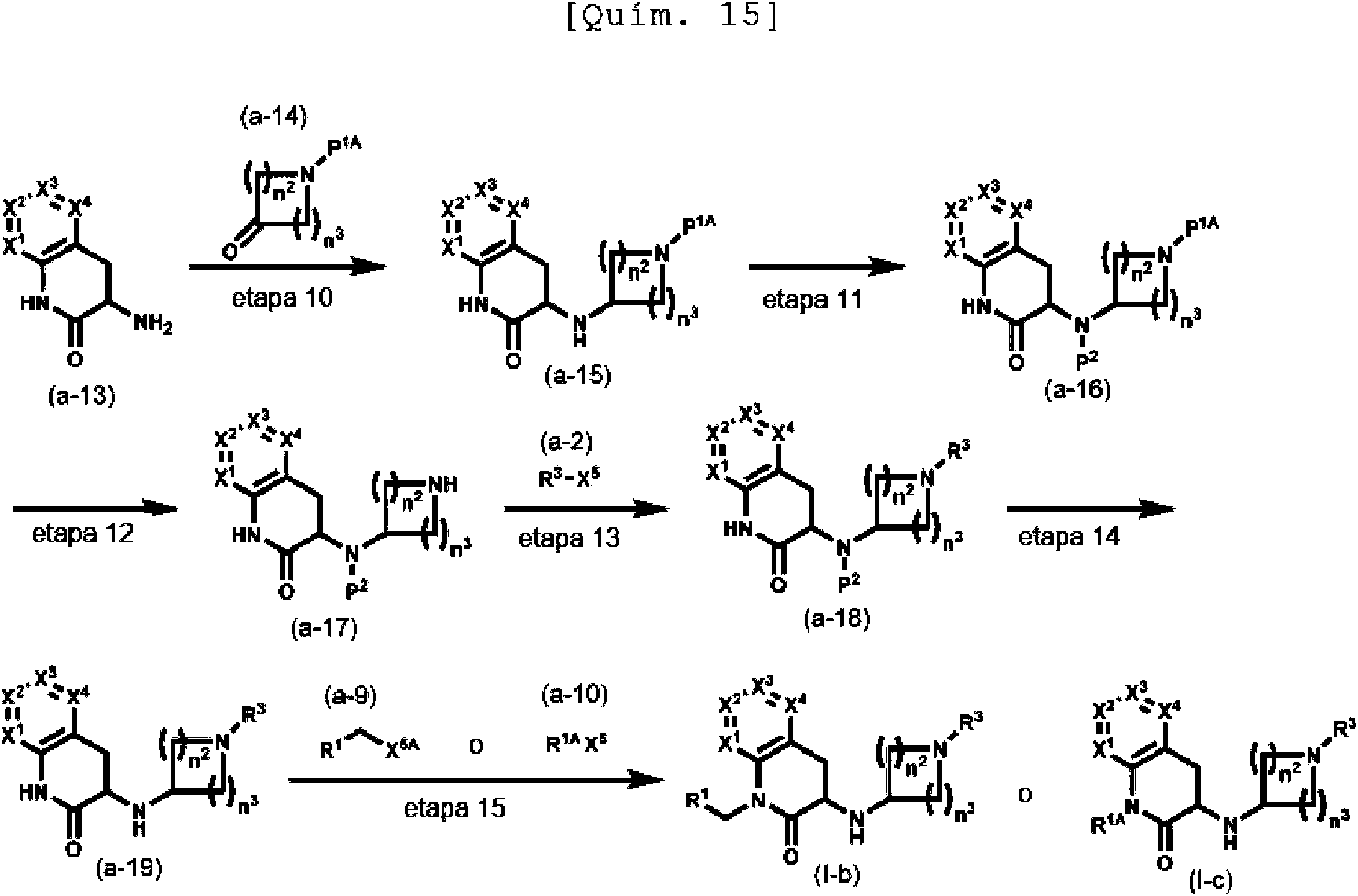
(en donde P1A representa un grupo protector que se puede eliminar con un ácido, entre los grupos representados por P1, por ejemplo, ferc-butoxicarbonilo o similar, P2 representa un grupo protector para un átomo de nitrógeno usado habitualmente en la química sintética orgánica, por ejemplo, acilo tal como formilo, acetilo, monocloroacetilo, trifluoroacetilo, tricloroacetilo, benzoílo o similar y X1, X2, X3, X4, X5, X5A, X6, R1, R1A, R3, n2 y n3 tienen las mismas definiciones que se han descrito anteriormente, respectivamente)
Etapa 10
El compuesto (a-15) se puede producir de la misma manera que en la etapa 3 mencionada anteriormente, usando el compuesto (a-13) y el compuesto (a-14).
El compuesto (a-13) se obtiene en forma de un producto disponible en el comercio o se puede obtener mediante un método conocido (por ejemplo, documento WO2004/98589 o similar) o un método modificado del mismo.
El compuesto (a-14) se obtiene en forma de un producto disponible en el mercado o se puede obtener por un método conocido [por ejemplo, Jikken Kagaku Koza, 5a ed., vol. 15, p. 153, Maruzen Co., Ltd. (2003) o similar] o un método modificado del mismo.
Etapa 11
El compuesto (a-16) se puede producir mediante, por ejemplo, un método modificado del método para introducir un grupo protector descrito en Protective Groups in Organic Synthesis, escrito por T. W. Greene, John Wiley & Sons, Inc. (1981) o similar, usando el compuesto (a-15).
Por ejemplo, en el caso en el que P2 es trifluoroacetilo, el compuesto (a-16) se puede producir haciendo reaccionar el compuesto (a-15) con preferentemente de 1 a 10 equivalentes de anhídrido trifluoroacético sin disolvente o en un disolvente en presencia de preferentemente de 1 a 10 equivalentes de una base a una temperatura entre -78 °C y 150 °C durante 5 minutos a 72 horas.
Los ejemplos de la base incluyen trietilamina, W,W-diisopropiletilamina, piridina, N-metilpiperidina, N-metilmorfolina y similares.
Los ejemplos del disolvente incluyen metanol, etanol, diclorometano, cloroformo, 1,2-dicloroetano, tolueno, acetato de etilo, acetonitrilo, éter dietílico, THF, DME, 1,4-dioxano, DMF, DMA, NMP, agua y similares y estos se usan en solitario o mezclados.
Etapa 12
El compuesto (a-17) se puede obtener de la misma manera que en la etapa 1 mencionada anteriormente, usando el compuesto (a-16).
Etapa 13
El compuesto (a-18) se puede producir de la misma manera que en la etapa 2 mencionada anteriormente, usando el compuesto (a-17) y el compuesto (a-2).
Etapa 14
El compuesto (a-19) se puede producir mediante, por ejemplo, un método modificado del método para eliminar un grupo protector descrito en Protective Groups in Organic Synthesis, escrito por T. W. Greene, John Wiley & Sons, Inc. (1981) o similar.
Por ejemplo, en el caso en el que P2 es trifluoroacetilo, El compuesto (a-19) se puede producir tratando el compuesto (a-18) en un disolvente que contiene agua con preferentemente de 1 equivalente a una gran cantidad en exceso de una base a una temperatura entre -30 °C y el punto de ebullición del disolvente para usarse durante 5 minutos a 72 horas.
Los ejemplos de la base incluyen hidróxido sódico, hidróxido de potasio, hidróxido de litio, hidróxido de bario, carbonato sódico, carbonato potásico y similares. Los ejemplos del disolvente incluyen metanol, etanol, propanol, THF, 1,4-dioxano, DME, tolueno, diclorometano, DMF, agua y similares y estos se usan en solitario o mezclados.
Etapa 15
Los compuestos (I-b) y (I-c) se pueden obtener de la misma manera que en la etapa 6 o la etapa 7 usando el compuesto (a- 19) y el compuesto (a-9) o el compuesto (a-10).
Proceso de producción 3
Entre los compuestos (I), El compuesto (I-d) y el compuesto (I-e), en el que Y1 es C(=O), Y2 es N y L es CH2 y (i) n1 es 1 (compuesto (I-d)) o (¡i) n1 es 0 y R1 es arilo opcionalmente sustituido o un grupo heterocíclico aromático opcionalmente sustituido (compuesto (I-e)), se pueden producir de acuerdo con, por ejemplo, las etapas siguientes.
(en donde X7 representa un átomo de cloro, un átomo de bromo, un átomo de yodo o similar, RP representa alquilo Ci-10, aralquilo C7-16 o similar y X1, X2, X3, X4, X5, X5A, X6, R1, R1A, R2, R3, P1, n2 y n3 tienen las mismas definiciones que se han descrito anteriormente, respectivamente)
Etapa 16
El compuesto (a-21) se puede producir haciendo reaccionar el compuesto (a-20) con preferentemente de 1 a 10 equivalentes del compuesto (a-5) en un disolvente en presencia de preferentemente de 1 a 10 equivalentes de una base a una temperatura entre -20 °C y 150 °C durante 5 minutos a 72 horas.
Los ejemplos de la base incluyen hidróxido sódico, hidróxido de potasio, hidróxido de litio, carbonato sódico, carbonato potásico, trietilamina, diisopropiletilamina, N-metilmorfolina, piridina, DBU y similares.
Los ejemplos del disolvente incluyen metanol, etanol, propanol, THF, 1,4-dioxano, DME, acetato de etilo, tolueno, diclorometano, DMF, agua y similares y estos se usan en solitario o mezclados.
El compuesto (a-20) se puede obtener en forma de un producto disponible en el comercio.
Etapa 17
El compuesto (a-23) se puede producir haciendo reaccionar el compuesto (a-21) con preferentemente de 1 a 10 equivalentes del compuesto (a-22) en un disolvente en presencia de preferentemente de 1 a 10 equivalentes de una base a una temperatura entre -20 °C y 150 °C durante 5 minutos a 72 horas.
Los ejemplos de la base incluyen hidróxido sódico, hidróxido de potasio, hidróxido de litio, carbonato sódico, carbonato potásico, trietilamina, diisopropiletilamina, N-metilmorfolina, piridina, 4-dimetilaminopiridina, DBU y similares.
Los ejemplos del disolvente incluyen metanol, etanol, propanol, THF, 1,4-dioxano, DME, acetato de etilo, tolueno, diclorometano, 1,2-dicloroetano, DMF, agua y similares y estos se usan en solitario o mezclados.
El compuesto (a-22) se puede obtener en forma de un producto disponible en el comercio.
Etapa 18
El compuesto (a-24) se puede producir tratando el compuesto (a-23) en un disolvente en presencia de preferentemente de 1 a 10 equivalentes de una base a una temperatura entre -20 °C y el punto de ebullición del disolvente para usarse durante 5 minutos a 72 horas.
Los ejemplos de la base incluyen hidróxido sódico, hidróxido de potasio, hidróxido de litio, carbonato sódico, carbonato potásico, trietilamina, diisopropiletilamina, N-metilmorfolina, piridina, 4-dimetilaminopiridina, DBU y similares.
Los ejemplos del disolvente incluyen metanol, etanol, propanol, THF, 1,4-dioxano, DME, acetato de etilo, tolueno, diclorometano, 1,2-dicloroetano, DMF, agua y similares y estos se usan en solitario o mezclados.
Etapa 19
El compuesto (a-25) se puede producir de la misma manera que en la etapa 1 mencionada anteriormente, usando el compuesto (a-24).
Etapa 20
El compuesto (a-26) se puede producir de la misma manera que en la etapa 2 mencionada anteriormente, usando el compuesto (a-25) y el compuesto (a-2).
Etapa 21
El compuesto (I-d) y el compuesto (I-e) se pueden producir de la misma manera que en la etapa 6 o la etapa 7 mencionadas anteriormente, usando el compuesto (a-26) y el compuesto (a-9) o el compuesto (a-10).
Proceso de producción 4
Entre los compuestos (I), El compuesto (I-f), en el que R3 es un grupo heterocíclico aromático opcionalmente sustituido o un grupo heterocíclico alifático opcionalmente sustituido y el grupo es un grupo heterocíclico aromático sustituido con -NR8R9 (en donde R8 y R9 pueden ser iguales o diferentes y cada uno representa un átomo de hidrógeno, alquilo C1-10, cicloalquilo C3-8, arilo C6-14, un grupo heterocíclico aromático o aralquilo C7-16 o, R8 y R9 se combinan junto con
el átomo de nitrógeno adyacente al mismo para formar un grupo heterocíclico que contiene nitrógeno que puede estar sustituido con alquilo C1-10 ) (el grupo heterocíclico aromático además puede tener otro sustituyente) o un grupo heterocíclico alifático sustituido con -NR8R9 (en donde R8 y R9 tienen las mismas definiciones que se han descrito anteriormente, respectivamente) (el grupo heterocíclico alifático puede tener además otro sustituyente), también se puede producir de acuerdo con, por ejemplo, el siguiente método.

(en donde, X5B representa un átomo de cloro, un átomo de bromo, un átomo de yodo, metanosulfoniloxi, trifluorometanosulfoniloxi, bencenosulfoniloxi, p-toluenosulfoniloxi o similares, el anillo A representa un resto de grupo heterocíclico aromático de un grupo heterocíclico aromático opcionalmente sustituido (el resto de grupo heterocíclico aromático puede tener además un sustituyente) o un resto de grupo heterocíclico alifático de un grupo heterocíclico alifático opcionalmente sustituido (el resto de grupo heterocíclico alifático puede tener además un sustituyente) en la definición de R3 y, X1, X2, X3, X4, X5, R1, R2, R8, R9, n1, n2 y n3 tienen las mismas definiciones que se han descrito anteriormente, respectivamente)
Etapa 22
El compuesto (a-28) se puede producir de la misma manera que en la etapa 2 mencionada anteriormente, usando el compuesto (a-1) y el compuesto (a-27).
El compuesto (a-27) se obtiene en forma de un producto disponible en el mercado o se puede obtener por un método conocido [por ejemplo, Jikken Kagaku Koza, 5a ed., vol. 13, p. 341, Maruzen Co., Ltd. (2003) o similar] o un método modificado del mismo.
Etapa 23
El compuesto (I-f) se puede producir haciendo reaccionar el compuesto (a-28) con de 1 a 10 equivalentes del compuesto (a-29) en un disolvente en presencia de una cantidad catalítica hasta 10 equivalentes de un catalizador de paladio a una temperatura entre temperatura ambiente y 140 °C durante 5 minutos a 72 horas. La reacción también se puede realizar en presencia de una cantidad catalítica hasta 10 equivalentes de una base y también se puede realizar en presencia de una cantidad catalítica hasta 10 equivalentes de un compuesto organofosforado.
Los ejemplos del catalizador de paladio incluyen acetato de paladio (II), cloruro de bis(trifenilfosfina)paladio (II), tetraquis(trifenilfosfina)paladio (0), cloruro de [1,2-bis(difenilfosfino)etano]paladio (II), cloruro de [1,1'-bis(difenilfosfino)ferroceno]paladio (II), tris(dibencilidenoacetona)dipaladio (0) y similares y, preferentemente, incluyen acetato de paladio (II), cloruro de bis(trifenilfosfina)paladio (II), tetraquis(trifenilfosfina)paladio (0), tris(dibencilidenacetona)dipaladio (0) y similares.
Los ejemplos de la base incluyen carbonato de potasio, carbonato de cesio, cloruro de litio, cloruro de potasio, terc-butóxido de potasio, terc-butóxido de sodio, trietilamina, acetato de potasio, etóxido de sodio, carbonato sódico, hidróxido sódico, fosfato de potasio, etilendiamina, glicina, N-metilpirrolidina, piridina, 1,2-diaminociclohexano y similares y, preferentemente, incluyen carbonato de potasio, carbonato de cesio, terc-butóxido de potasio, fosfato de potasio, trietilamina y similares.
Los ejemplos del compuesto organofosforado incluyen trifenilfosfina, tri(2-furil)fosfina, 2-diciclohexilfosfin-2'-(N,N-dimetilamino)bifenilo, difenilfosfinoferroceno, Xphos y similares y, preferentemente, incluyen 2-diciclohexilfosfin-2'-(N,N-dimetilamino)bifenilo, XPhos y similares.
Los ejemplos del disolvente incluyen éter dietílico, THF, 1,4-dioxano, DMF, DMA, DMSO, benceno, tolueno, xileno, diclorometano, cloroformo, tetracloruro de carbono, 1,2-dicloroetano, acetonitrilo, acetato de etilo, acetato de metilo, metil etil cetona, metanol, etanol, propanol, 2-propanol, butanol, hexano y similares y, preferentemente, incluyen THF, 1,4- dioxano, DMF y similares.
El compuesto (a-29) se obtiene en forma de un producto disponible en el mercado o se puede obtener por un método conocido [por ejemplo, Jikken Kagaku Koza, 5 a ed., vol. 14, p. 351, Maruzen Co., Ltd. (2003) o similar] o un método modificado del mismo.
Proceso de producción 5
Entre los compuestos (I), el compuesto (I-g) y el compuesto (I-h), en donde R3 es un grupo heterocíclico aromático opcionalmente sustituido o un grupo heterocíclico alifático opcionalmente sustituido y el grupo es un grupo heterocíclico aromático sustituido con carboxi o -CONR8'R9' (en donde R8' y R9' pueden ser iguales o diferentes y cada uno representa un átomo de hidrógeno, alquilo C1-10, cicloalquilo C3-8, arilo Ca-14, un grupo heterocíclico aromático o aralquilo C7-1a o, R8' y R9' se combinan junto con el átomo de nitrógeno adyacente al mismo para formar un grupo heterocíclico que contiene nitrógeno que puede estar sustituido con alquilo C1-10) (el grupo heterocíclico aromático puede tener además otro sustituyente) o un grupo heterocíclico alifático sustituido con carboxi o -CONR8'R9' (en donde R8' y R9' tiene las mismas definiciones que se han descrito anteriormente, respectivamente) (el grupo heterocíclico alifático puede tener además otro sustituyente), también se puede producir de acuerdo con, por ejemplo, el siguiente método.
[Q uim . 18]

(en donde RP' representa alquilo C1-10, aralquilo C7-1a o similar y, el anillo A, X1, X2, X3, X4, R1, R2, R8', R9', n1, n2 y n3 tienen las mismas definiciones que se han descrito anteriormente, respectivamente)
Etapa 24
El compuesto (a-30) se puede producir haciendo reaccionar el compuesto (a-28) en un disolvente en atmósfera de monóxido de carbono en presencia de preferentemente 1 equivalente a una gran cantidad en exceso de RP' OH (en donde RP' tiene la misma definición que se ha descrito anteriormente) y, preferentemente, del 1 al 100 % en moles de un catalizador de paladio y, si fuera necesario, en presencia de preferentemente el 1 al 100 % en moles de un compuesto organofosforado y/o preferentemente de 1 a 10 equivalentes de una base a una temperatura entre -20 °C y el punto de ebullición del disolvente para usarse a presión normal o a presión aumentada durante de 5 minutos a 72 horas.
Los ejemplos de la base incluyen carbonato de potasio, fosfato de potasio, hidróxido de potasio, trietilamina, diisopropiletilamina, N-metilmorfolina, piridina, DBU, acetato de potasio, acetato de sodio y similares. Los ejemplos del catalizador de paladio incluyen acetato de paladio, tetraquis(trifenilfosfina)paladio y similares.
Los ejemplos del compuesto organofosforado incluyen trifenilfosfina, 1,1'-bis(difenilfosfino)ferroceno, 1,3-bis(difenilfosfino)propano y similares.
Los ejemplos del disolvente incluyen diclorometano, cloroformo, 1,2-dicloroetano, tolueno, acetato de etilo, acetonitrilo, éter dietílico, THF, DME, 1,4-dioxano, DMF, DMA, NMP, agua y similares y estos se usan en solitario o mezclados.
Etapa 25
El compuesto (I-g) se puede producir mediante, por ejemplo, un método modificado del método para eliminar un grupo protector descrito en Protective Groups in Organic Synthesis, tercera edición, escrito por T. W. Greene, John Wiley & Sons, Inc. (1999) o similar, usando el compuesto (a-30).
Por ejemplo, en el caso en el que RP' es metilo, etilo o n-propilo, el compuesto (I-g) se puede producir tratando el compuesto (a-30) en un disolvente que contiene agua con, preferentemente, de 1 equivalente a una gran cantidad en exceso de una base a una temperatura entre 0 °C y el punto de ebullición del disolvente para usarse durante 5 minutos a 72 horas.
Los ejemplos de la base incluyen hidróxido sódico, hidróxido de potasio, hidróxido de litio y similares.
Los ejemplos del disolvente incluyen metanol, etanol, propanol, THF, 1,4-dioxano, DME, tolueno, diclorometano, DMF, agua y similares y estos se usan en solitario o mezclados.
Además, por ejemplo, en el caso en el que RP' es ferc-butilo, el compuesto (I-g) se puede producir tratando el compuesto (a-30) sin disolvente o en un disolvente con de 1 equivalente a una gran cantidad en exceso de un ácido a una temperatura entre -30 °C y 100 °C durante 5 minutos a 72 horas.
Los ejemplos del ácido incluyen ácido clorhídrico, ácido sulfúrico, ácido trifluoroacético, ácido metanosulfónico y similares.
Los ejemplos del disolvente incluyen metanol, etanol, propanol, THF, 1,4-dioxano, DME, tolueno, acetato de etilo, diclorometano, DMF, agua y similares y estos se usan en solitario o mezclados.
Etapa 26
El compuesto (I-h) se puede producir haciendo reaccionar el compuesto (I-g) con, preferentemente, de 1 a 30 equivalentes del compuesto (a-29) sin disolvente o en un disolvente, en presencia de preferentemente de 1 a 30 equivalentes de un agente de condensación y, si fuera necesario, en presencia de, preferentemente, de 1 a 30 equivalentes de un aditivo a una temperatura entre -30 °C y 150 °C durante 5 minutos a 72 horas.
Los ejemplos de agente de condensación incluyen diciclohexilcarbodiimida (DCC), diisopropilcarbodiimida, N-(3-dimetilaminopropil)-N'-etilcarbodiimida (EDC), clorhidrato de EDC, hexafluorofosfato de O-(7-aza-1H-benzotriazol-1-il)-N,N,N',N'-tetrametiluronio (HATU) y similares.
Los ejemplos del aditivo incluyen 1-hidroxibenzotriazol monohidrato (HOBtH2O), trietilamina, diisopropiletilamina, 4-dimetilaminopiridina (DMAP) y similares y estos se usan en solitario o mezclados.
Los ejemplos del disolvente incluyen acetonitrilo, diclorometano, 1,2-dicloroetano, cloroformo, DME, DMF, DMA, 1,4-dioxano, THF, éter dietílico, diisopropil éter, benceno, tolueno, xileno, piridina, NMP, agua y similares y estos se usan en solitario o mezclados.
Proceso de producción 6
Entre los compuestos (I), el compuesto (I-i) y el compuesto (I-j), en los que R3 es un grupo heterocíclico aromático opcionalmente sustituido y el grupo heterocíclico aromático es un grupo heterocíclico aromático que contiene un átomo de nitrógeno y es un grupo en el que oxo está unido al nitrógeno del grupo heterocíclico aromático (el grupo puede tener además otro sustituyente) y (i) n1 es 1 (compuesto (I-i)) o (ii) n1 es 0 y R1 es arilo opcionalmente sustituido o un grupo heterocíclico aromático opcionalmente sustituido (compuesto (I-j)), también se puede producir de acuerdo con, por ejemplo, el siguiente método.

(en donde el anillo B representa un grupo heterocíclico aromático opcionalmente sustituido y el grupo heterocíclico aromático es un grupo heterocíclico aromático que contiene un átomo de nitrógeno (el resto del grupo heterocíclico aromático también puede tener un sustituyente) en la definición de R3 y, X1, X2, X3, X4, X5, X5A, X6, P1, R1, R1A, R2, n2 y n3 tienen las mismas definiciones que se han descrito anteriormente, respectivamente)
Etapa 27
El compuesto (a-31) se puede producir de la misma manera que en la etapa 1 mencionada anteriormente, usando el compuesto (a-8).
Etapa 28
El compuesto (a-33) se puede producir de la misma manera que en la etapa 2 mencionada anteriormente, usando el compuesto (a-31) y el compuesto (a-32).
El compuesto (a-32) se obtiene en forma de un producto disponible en el mercado o se puede obtener por un método conocido [por ejemplo, Jikken Kagaku Koza, 5a ed., vol. 13, p. 341, Maruzen Co., Ltd. (2003) o similar] o un método modificado del mismo.
Etapa 29
El compuesto (a-34) se puede producir tratando el compuesto (a-33) en un disolvente con 1 equivalente a una gran cantidad en exceso, preferentemente, de 1 a 10 equivalentes de un agente oxidante, a una temperatura entre 0 °C y el punto de ebullición del disolvente para usarse durante 5 minutos a 72 horas.
Los ejemplos del disolvente incluyen diclorometano, cloroformo, 1,2-dicloroetano, THF, 1,4-dioxano, dimetoxietano, éter dietílico, diisopropil éter, metanol, etanol, alcohol isopropílico, benceno, tolueno, xileno, acetonitrilo, acetato de etilo, agua y similares y estos se usan en solitario o mezclados.
Los ejemplos del agente oxidante incluyen ácido meta-cloroperoxibenzoico, peróxido de benzoílo, ácido peracético, peróxido de hidrógeno, peryodato de sodio, oxona y similares.
Etapa 30
El compuesto (I-i) y el compuesto (I-j) se pueden producir de la misma manera que en la etapa 6 o la etapa 7 mencionadas anteriormente, usando el compuesto (a-34) y el compuesto (a-9) o el compuesto (a-10).
Proceso de producción 7
Entre los compuestos (I), el compuesto (I-k), en el que R3 está representado por la fórmula siguiente:
[Quím. 20]

también se puede producir de acuerdo con, por ejemplo, el siguiente método.

(en donde X5C representa un átomo de cloro, un átomo de bromo, un átomo de yodo, metanosulfoniloxi, trifluorometanosulfoniloxi, bencenosulfoniloxi, p-toluenosulfoniloxi o similares y X1, X2, X3, X4, X5, R1, R2, n1, n2 y n3 tienen las mismas definiciones que se han descrito anteriormente, respectivamente)
Etapa 31
El compuesto (a-36) se puede producir de la misma manera que en la etapa 2 mencionada anteriormente, usando el compuesto (a-1) y el compuesto (a-35).
El compuesto (a-35) se obtiene en forma de un producto disponible en el mercado o se puede obtener por un método conocido [por ejemplo, bibliografía de patente (US2009/286816) o similar] o un método modificado del mismo.
Etapa 32
El compuesto (I-k) se puede producir tratando el compuesto (a-36) en un disolvente en atmósfera de hidrógeno o en presencia de una fuente de hidrógeno en presencia de un catalizador y una base, a una temperatura entre -20 °C y el punto de ebullición del disolvente, para usarse a presión normal o a presión aumentada, durante 5 minutos a 72 horas. Los ejemplos del catalizador incluyen paladio sobre carbono, paladio, hidróxido de paladio, acetato de paladio, negro de paladio y similares y estos se usan en una cantidad de preferentemente el 0,01 al 50 % en peso con respecto al compuesto (a-36).
Los ejemplos de la fuente de hidrógeno incluyen ácido fórmico, formiato de amonio, formiato de sodio, ciclohexadieno, hidrazina y similares y estos se usan en una cantidad de preferentemente 2 equivalentes hasta una gran cantidad en exceso con respecto al compuesto (a-36).
Los ejemplos de la base incluyen trietilamina, diisopropiletilamina, piridina, N-metilmorfolina y similares y estos se usan en una cantidad de preferentemente de 1 a 30 equivalentes con respecto al compuesto (a-36).
Los ejemplos del disolvente incluyen metanol, etanol, tolueno, acetato de etilo, acetonitrilo, éter dietílico, THF, DME, 1,4-dioxano, DMF, DMA, NMP, ácido acético, agua y similares y estos se usan en solitario o mezclados.
Proceso de producción 8
Entre los compuestos (I), el compuesto (I-1) y el compuesto (I-m), en los que n1 es 0 y R1 es arilo opcionalmente sustituido o un grupo heterocíclico aromático opcionalmente sustituido y el grupo es arilo sustituido con carboxi o -CONR8” R9” (en donde R8'' y R9'' pueden ser iguales o diferentes y cada uno representa un átomo de hidrógeno, alquilo C1-10, cicloalquilo C3-8, arilo C6-14, un grupo heterocíclico aromático o aralquilo C7-16 o R8'' y R9'' se combinan junto con el átomo de nitrógeno adyacente a los mismos para formar un grupo heterocíclico que contiene nitrógeno) (el arilo puede tener además otro sustituyente) o un grupo heterocíclico aromático sustituido con carboxi o -CONR8'' R9'' (en donde R8'' y R9'' tiene las mismas definiciones que se han descrito anteriormente, respectivamente) (el grupo heterocíclico aromático puede tener además otro sustituyente), también se puede producir de acuerdo con, por ejemplo, el siguiente método.

(en donde X7 representa un átomo de cloro, un átomo de bromo, un átomo de yodo, metanosulfoniloxi, trifluorometanosulfoniloxi, bencenosulfoniloxi, p-toluenosulfoniloxi o similares, el anillo C representa un resto arilo de arilo opcionalmente sustituido (el resto arilo puede tener además un sustituyente) o un resto de grupo heterocíclico aromático de un grupo heterocíclico aromático opcionalmente sustituido (el resto de grupo heterocíclico aromático
puede tener además un sustituyente) en la definición de R1 y, X1, X2, x3, X4, x5, X6, R2,  R8'', R9'', mismas definiciones que se han descrito anteriormente, respectivamente)
R8'', R9'', mismas definiciones que se han descrito anteriormente, respectivamente)
Etapa 33
El compuesto (a-37) se puede producir de la misma manera que en la etapa 2 mencionada anteriormente, usando el compuesto (a-31) y el compuesto (a-2).
Etapa 34
El compuesto (a-39) se puede producir de la misma manera que en la etapa 7 mencionada anteriormente, usando el compuesto (a-37) y el compuesto (a-38).
El compuesto (a-38) se obtiene en forma de un producto disponible en el mercado o se puede obtener por un método conocido [por ejemplo, Jikken Kagaku Koza, 5a ed., vol. 13, p. 341, Maruzen Co., Ltd. (2003) o similar] o un método modificado del mismo.
Etapa 35
El compuesto (a-40) se puede producir de la misma manera que en la etapa 24 mencionada anteriormente, usando el compuesto (a-39).
Etapa 36
El compuesto (I-1) se puede producir de la misma manera que en la etapa 25 mencionada anteriormente, usando el compuesto (a-40).
Etapa 37
El compuesto (I-m) se puede producir de la misma manera que en la etapa 26 mencionada anteriormente, usando el compuesto (I-1) y el compuesto (a-29).
Proceso de producción 9
Entre los compuestos (I), el compuesto (I-n), en el que n1 es 0 y R1 es arilo opcionalmente sustituido o un grupo heterocíclico aromático opcionalmente sustituido y el grupo es arilo sustituido con -CONH2 (el grupo arilo puede tener además otro sustituyente) o un grupo heterocíclico aromático sustituido con -CONH2 (el grupo heterocíclico aromático puede tener además otro sustituyente), también se puede producir de acuerdo con, por ejemplo, el siguiente método.

(en el que el anillo C, X1, X2, X3, X4, X6, R2, R3, n2 y n3 tienen las mismas definiciones que se han descrito anteriormente, respectivamente) Etapa 38
El compuesto (a-42) se puede producir de la misma manera que en la etapa 7 mencionada anteriormente, usando el compuesto (a-37) y el compuesto (a-41).
El compuesto (a-41) se obtiene en forma de un producto disponible en el mercado o se puede obtener por un método conocido [por ejemplo, Jikken Kagaku Koza, 5 a ed., vol. 13, p. 341, Maruzen Co., Ltd. (2003) o similar] o un método modificado del mismo.
Etapa 39
El compuesto (I-n) se puede producir de la misma manera que en la etapa 25 mencionada anteriormente usando el compuesto (a-42).
Proceso de producción 10
El compuesto (IA) se puede producir de acuerdo con el proceso de producción 1 a 11 mencionado anteriormente. Además, entre los compuestos (IA), se puede producir un compuesto en el que n1 es 0 y R1 es un átomo de hidrógeno mediante el método descrito en Chemical & Pharmaceutical Bulletin 1990, 38 (6), 1591 o un método modificado del mismo.
La conversión de un grupo funcional contenido en R1A, R2A o R3A en el compuesto (IA) y en R1, R2 o R3 en el compuesto (I) también se puede realizar mediante un método conocido [por ejemplo, el método descrito en Comprehensive Organic Transformations, 2a edición, escrito por R. C. Larock, Vch Verlagsgesellschaft Mbh (1999) o similares] o un método modificado del mismo.
Los intermedios y los compuestos objetivo en los procesos de producción respectivos mencionados anteriormente, se pueden aislar y purificar sometiéndolos a un método de separación y purificación usado habitualmente en la química sintética orgánica, por ejemplo, filtración, extracción, lavado, secado, concentración, recristalización, diversos tipos de cromatografía o similares. Además, los intermedios se pueden someter a la reacción posterior sin estar particularmente purificados.
Entre los compuestos (IA) y (I), algunos compuestos pueden existir en forma de un estereoisómero tal como un isómero geométrico o un isómero óptico, un tautómero o similar. La presente invención abarca todos los isómeros posibles y mezclas de los mismos incluyendo estos isómeros del compuesto (I).
Parte o todos los átomos respectivos en el compuesto (I) se pueden sustituir por el o los átomos de isótopos correspondientes, respectivamente, y la presente invención también comprende dichos compuestos sustituidos por un átomo o átomos de isótopos. Por ejemplo, parte o todos los átomos de hidrógeno en el compuesto (I) pueden ser un átomo o átomos de hidrógeno que tienen un peso atómico de 2 (átomo o átomos de deuterio).
Los compuestos en los que parte o todos los átomos respectivos en los compuestos (IA) y (I) está o están sustituidos por el o los átomos de isótopo correspondientes, respectivamente, se pueden producir de la misma manera que en los procesos de producción respectivos mencionados anteriormente, usando componentes básicos disponibles en el comercio. Además, los compuestos en los que parte o todos los átomos de hidrógeno en los compuestos (IA) y (I) se sustituyen por un átomo o átomos de deuterio, se pueden sintetizar mediante, por ejemplo, 1) un método para deuterar un ácido carboxílico y similar en condiciones básicas, usando peróxido de deuterio (US3849458), 2) un método para deuterar un alcohol, un ácido carboxílico y similar, usando complejo de iridio como catalizador y usando también agua pesada como fuente de deuterio [Journal of the American Chemical Society 2002, 124(10), 2092], 3) un método para deuterar un ácido graso usando paladio sobre carbono como catalizador y usando también solamente un gas de deuterio como fuente de deuterio [LIPIDS 1974, 9 (11), 913], 4) un método para deuterar un ácido acrílico, acrilato de metilo, ácido metacrílico, metil metacrilato y similar, usando un metal tal como platino, paladio, rodio, rutenio, iridio y similar como catalizador y usando también agua pesada o agua pesada y un gas de deuterio, como fuente de deuterio (JPH5-19536, JPS61-277648 y JPS61-275241), 5) un método para deuterar ácido acrílico, metil metacrilato y similar, usando un catalizador tal como paladio, níquel, cobre, cromita de cobre y similares y usando también agua pesada como fuente de deuterio (JPS63-198638) y similares.
En el caso en el que se desea obtener una sal del compuesto (IA) o (I), cuando se obtiene el compuesto (IA) o (I) en forma de una sal, la sal se puede purificar directamente. O, cuando se obtiene el compuesto (IA) o (I) en una forma libre, el compuesto (IA) o (I) se disuelve o se suspende en un disolvente adecuado y se le añade un ácido o una base para formar una sal y, después, la sal se puede aislar y purificar.
Además, los compuestos (IA) y (I) y sus sales farmacéuticamente aceptables pueden existir en la forma de aductos con agua o cualquiera de diversos disolventes y la presente invención también comprende estos aductos del compuesto (I).
Los ejemplos específicos de los compuestos (IA) y (I) obtenidos de acuerdo con la presente invención se muestran en la tabla 1 a la tabla 5. Los ejemplos marcados con * están fuera del alcance de la invención. Sin embargo, los compuestos de la presente invención no se limitan a los mismos.
[Tabla 1]

[Tabla 2]
Tabla 2

[Tabla 3]


[Tabla 4]
Tabla 4

[Tabla 5]
Tabla 5

A continuación, la actividad farmacológica del compuesto representativo (I) se describirá específicamente mediante los ejemplos de prueba.
Ejemplo de ensayo 1: Actividad inhibitoria contra el factor de células T (TCF)-indicador de luciferasa utilizando la vía Wnt como índice
La actividad inhibitoria de los compuestos de prueba contra la vía Wnt se evaluó mediante el siguiente método.
Se cultivó una línea celular de adenocarcinoma colorrectal humano DLD-1 (Japanese Collection of Research Bioresources) en medio RPMI-1640 (Gibco/life Technologies, Inc.) que contiene un 10 % de suero bovino fetal (Gibco/life Technologies, Inc.), 10 mmol/l de una solución tampón de ácido 4-(2-hidroxietil)-1-piperazinetansulfónico (HEPES) (Gibco/life Technologies, Inc.), 1 mmol/l de una solución de piruvato de sodio (Gibco/life Technologies, Inc.), 4,5 g/l de una solución de D-(+)-glucosa (Sigma-Aldrich, Inc.), 100 unidades/ml de penicilina (Gibco/life Technologies, Inc.) y 100 |jg/ml de estreptomicina (Gibco/life Technologies, Inc.) en condiciones de 37 °C y 5 % de dióxido de carbono gaseoso. Las células DLD-1 se sembraron en un plato de 10 cm, y 20 jg de un plásmido del gen de luciferasa pGL4.27 que tenía una secuencia sensible a TCF insertada en el mismo se transfectaron en las células usando 10 j l de Attractene (Qiagen, Inc.) de acuerdo con el protocolo adjunto al producto. Se seleccionó una línea celular de expresión estable (DLD-1/TCF-Luc) usando 600 jg / j l de higromicina B (Wako Pure Chemical Industries Ltd.). Las células DLD-1/TCF-Luc se separaron con tripsina y se sembraron en una placa de 384 pocillos, y se añadió un compuesto de prueba a diferentes concentraciones. Después de 18 horas, la actividad luciferasa se midió usando el sistema de ensayo de luciferasa Steady-Glo™ (Promega, Inc.).
La relación de inhibición se obtuvo de acuerdo con la siguiente fórmula 1. La relación de inhibición (%) del compuesto de la presente invención o la divulgación a 1 jmol/l contra el indicador TCF-luciferasa usando la vía Wnt como índice se muestra en la Tabla 6.
[Fórm. matem. 1]
Fórmula 1
Relación de inhibición (%) = 100 - {(actividad luciferasa cuando
se añadió el compuesto de prueba) - (actividad luciferasa del
blanco)} / { (actividad luciferasa del control) - (actividad
luciferasa del blanco)} x 100
[Tabla 6]
Tabla 6

continuación

A partir de los resultados anteriores, se demostró que los Compuestos (IA) y (I) y las sales farmacéuticamente aceptables de los mismos inhiben la señalización de Wnt y, por lo tanto, son útiles como agente terapéutico y/o preventivo para una enfermedad asociada a la señalización de Wnt, por ejemplo, cáncer, fibrosis pulmonar, fibromatosis, osteoartritis y similares.
Ejemplo de ensayo 2: Prueba de inhibición de la enzima tanquirasa-2
La actividad enzimática de tanquirasa-2 se evaluó usando el kit de ensayo quimioluminiscente Tankyrase-2 (BPS Bioscience, Inc., N.° de Catálogo 80566). El kit de ensayo de quimioluminiscencia Tankyrase-2 es un kit para evaluar la actividad enzimática de la tanquirasa-2 usando la autoribosilación de la proteína de fusión glutatión-S-transferasa (GST)-tanquirasa-2 como índice. Todos los materiales experimentales, excepto el tampón PBS (PBST) que contiene solución salina tamponada con fosfato (PBS) y Tween 20 al 0,05 % están incluidos en el kit. La enzima GST-tanquirasa-2 diluida con 50 pl de tampón tanquirasa 1x se añadió a los pocillos de una placa de 96 pocillos recubierta con glutatión. Después de dejar la placa en reposo durante la noche a 4 °C, la placa se lavó 3 veces con el tampón PBST. Se añadieron 150 pl de tampón de bloqueo a la misma y la placa se dejó reposar a temperatura ambiente durante 30 minutos para bloquear los pocillos. La placa se lavó 3 veces con el tampón PBST. Antes de la reacción de ribosilación, se mezclaron una mezcla de ensayo que contenía un sustrato biotinilado y un compuesto de prueba diluido con tampón tanquirasa 1x, por lo que se preparó una mezcla de reacción. Para comenzar la reacción de ribosilación, se añadieron 50 pl de la mezcla de reacción a los pocillos. En un pocillo de blanco, se añadió tampón tanquirasa 1x en lugar de la mezcla de reacción que contenía un sustrato biotinilado. La placa se dejó reposar a 30 °C durante 1 hora. Después de la reacción, la placa se lavó 3 veces con el tampón PBST. La estreptavidina-peroxidasa de rábano picante (HRP) se diluyó a 50 veces con el tampón de bloqueo. La estreptavidina-HRP diluida se añadió a los pocillos y la placa se dejó en reposo a temperatura ambiente durante 30 minutos. La placa se lavó 3 veces con el tampón PBSt . Inmediatamente antes de su uso, se mezclaron 50 pl del sustrato quimioluminiscente HRP A y 50 pl del sustrato quimioluminiscente HRP B y se añadieron 100 pl de la mezcla resultante a los pocillos. La quimioluminiscencia se midió usando un aparato de medición de quimioluminiscencia. La relación de inhibición se obtuvo de acuerdo con la siguiente fórmula 2. La actividad inhibitoria de la enzima tanquirasa-2 del compuesto de la presente invención o divulgación se muestra en la Tabla 7.
[Fórm. matem. 2]
Fórmula 2
Relación de inhibición (%) = 100 - {(intensidad de
quimioluminiscencia cuando se añadió el compuesto de prueba)
(intensidad de quimioluminiscencia del blanco)} / {(intensidad de quimioluminiscencia del control) - (intensidad de quimioluminiscencia del blanco)} x 100
[Tabla 7]
Tabla 7

A partir de los resultados anteriores, se demostró que los compuestos (IA) y (I) y las sales farmacéuticamente aceptables de los mismos inhiben la enzima tanquirasa-2. Es decir, se demostró que los compuestos (IA) y (I) y las sales farmacéuticamente aceptables de los mismos inhiben la señalización Wnt mediante la inhibición de la tanquirasa y, por lo tanto, son útiles como agente terapéutico y/o preventivo para una enfermedad asociada con la señalización Wnt, por ejemplo, cáncer, fibrosis pulmonar, fibromatosis, osteoartritis y similares.
Los compuestos (IA) y (I) y las sales farmacéuticamente aceptables de los mismos se pueden administrar solas tal cual están, pero en general es deseable proporcionarlos en forma de diversas preparaciones farmacéuticas. Además, dichas preparaciones farmacéuticas se usan para animales y seres humanos.
La preparación farmacéutica de acuerdo con la presente invención puede contener, como principio activo, el compuesto (I) o una sal farmacéuticamente aceptable del mismo solo o en forma de una mezcla con un ingrediente activo para cualquier otro tratamiento. Además, dicha preparación farmacéutica se prepara mezclando el ingrediente activo con uno o más vehículos farmacéuticamente aceptables (por ejemplo, un diluyente, un disolvente, un excipiente y similares) y sometiendo después la mezcla a cualquier método bien conocido en el campo técnico del estudio de la formulación de fármacos.
En cuanto a la vía de administración, se prefiere el uso de la vía de administración más eficaz en el tratamiento. Los ejemplos de la vía de administración incluyen administración oral y administración parenteral tal como administración intravenosa o similar.
Los ejemplos de la forma de administración incluyen un comprimido, una inyección y similares.
Una forma de administración adecuada para la administración oral, por ejemplo, un comprimido o similar, se puede preparar usando un excipiente tal como lactosa y similar, un disgregante tal como almidón o similar, un lubricante tal como estearato de magnesio y similar, un aglutinante tal como hidroxipropil celulosa y similar, y similares.
Una forma de administración adecuada para administración parenteral, por ejemplo, una inyección o similar, se puede preparar usando un diluyente o un disolvente tal como una solución salina, una solución de glucosa o una solución mezclada de una solución salina y una solución de glucosa y similar, y similares.
La dosis y la frecuencia de administración del compuesto (IA) o (I) o una sal farmacéuticamente aceptable del mismo, puede variar dependiendo de la forma de administración, la edad y el peso corporal de un paciente, la naturaleza o gravedad del síntoma a tratar y similares. Sin embargo, en la administración oral, en general, una dosis de 0,01 a 1000 mg, preferentemente, de 0,05 a 100 mg, se administra a un paciente adulto una o varias veces al día. En la administración parenteral, tal como administración intravenosa, una dosis de 0,001 a 1000 mg, preferentemente, de 0,01 a 100 mg, se administra a un paciente adulto una o varias veces al día. Sin embargo, dicha dosis y frecuencia de administración varía dependiendo de las distintas condiciones anteriormente mencionadas.
En lo sucesivo en el presente documento, la presente invención se describirá más específicamente mediante los ejemplos y ejemplos de referencia, sin embargo, el alcance de la presente invención no se limita a estos ejemplos. Los ejemplos marcados con * están fuera del alcance de la invención.
Ocasionalmente, el espectro de resonancia magnética nuclear de protones (RMN 1H) usado en los ejemplos y ejemplos de referencia se midió a 270 MHz, 300 MHz o 400 MHz y los protones intercambiables pueden no observarse claramente, dependiendo del compuesto y de las condiciones de medición. Ocasionalmente, la multiplicidad de las señales se expresa en términos convencionales y el término "a" indica una señal amplia aparente.
Además, cada compuesto sintetizado se nombró usando ChemBioDraw Ultra versión12.0.
Ejemplo de referencia 1
Clorhidrato de 3-(piperidin-4-ilmetil)-3,4-dihidroquinazolin-2(1H)-ona (compuesto R1)
Etapa 1: Después de agitar 2-nitrobenzaldehído (5,60 g, 37,1 mmol) y 4-(aminometil)piperidin-1-carboxilato de terc-butilo (8,00 g, 37,3 mmol) en metanol a temperatura ambiente durante 1 hora, se le añadió cianoborohidruro sódico (4,70 g, 74,8 mmol) y la mezcla resultante se agitó durante una noche a temperatura ambiente. Después de que la mezcla de reacción se concentrase a presión reducida, se le añadió agua y la mezcla resultante se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturad y después se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo 4-[(2-nitrobencilamino)metil]-piperidin-1-carboxilato de terc-butilo (5,00 g, rendimiento: 40 %).
Etapa 2: El 4-[(2-nitrobencilamino)metil]-piperidin-1-carboxilato de terc-butilo (10,0 g, 28,6 mmol) obtenido en la etapa 1 se disolvió en metanol (100 ml) y se le añadió paladio-carbono (10,0 % en peso, 1,00 g) y la mezcla resultante se agitó en atmósfera de gas hidrógeno (presión atmosférica) a temperatura ambiente durante 12 horas. Después de finalizar la reacción, la mezcla de reacción se trató con tierra de diatomeas y, después, el disolvente se evaporó a presión reducida, con lo que se obtuvo 4-[(2-aminobencilamino)metil]-piperidin-1-carboxilato de terc-butilo (8,00 g, rendimiento: 88 %).
Etapa 3: El 4-[(2-aminobencilamino)metil]-piperidin-1-carboxilato de terc-butilo (10,6 g, 27,7 mmol) obtenido en la etapa 2, N,N'-carbonildiimidazol (11,2 g, 69,3 mmol) y trietilamina (8,11 ml, 58,2 mmol) se calentaron a reflujo en acetonitrilo (110 ml) durante 2 horas. Después de que la mezcla de reacción se enfriase a temperatura ambiente, se le añadió agua y el sólido depositado se recogió por filtración, con lo que se obtuvo 4-(2-oxo-1,4-dihidro-2H-quinazolin-3-ilmetil)piperidin-1-carboxilato de terc-butilo (8,63 g, rendimiento: 93 %).
ESI-MS m/z: 346 (M+H)+, RMN 1H (300 MHz, CDCla, 8): 7,20-7,15 (m, 1H), 7,05-7,03 (m, 1H), 6,97-6,88 (m, 2H), 6,67 (d, J = 7,2 Hz, 1H), 4,46 (s, 2H), 4,09 (s a, 2H), 3,31 (s a, 2H), 2,73-2,64 (m, 2H), 1,97-1,84 (m, 1H), 1,70-1,66 (m, 2H), 1,45 (s, 9H), 1,28-1,12 (m, 2H)
Etapa 4: Al 4-(2-oxo-1,4-dihidro-2H-quinazolin-3-ilmetil)piperidin-1-carboxilato de terc-butilo (13,0 g, 37,6 mmol) obtenido en la etapa 3, se le añadió una solución de ácido clorhídrico-dioxano (4,00 mol/l, 150 ml) en un baño de hielo, y la mezcla resultante se agitó a temperatura ambiente durante 4 horas. La mezcla de reacción se concentró a presión reducida, con lo que se obtuvo el compuesto R1 (10,5 g, rendimiento: 99 %).
ESI-MS m/z: 246 (M+H)+
Ejemplo de referencia 2
Clorhidrato de 2-{[2-oxo-3-(piperidin-4-ilmetil)-3,4-dihidroquinazolin-1(2H)-il]metil}benzonitrilo (Compuesto R2)
Etapa 1: Se obtuvo 4-{[1-(2-cianobencil)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-carboxilato de terc-butilo (2,27 g, 85 %) de la misma manera que en la etapa 1 del ejemplo 1 usando el 4-(2-oxo-1,4-dihidro-2H-quinazolin-3-ilmetil)piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 3 del ejemplo de referencia 1 y bromuro de 2-cianobencilo.
ESI-MS m/z: 461 (M+H)+, RMN 1H (400 MHz, CDCb, 8): 7,68 (d, J = 7,8 Hz, 1H), 7,49 (t, J = 7,3 Hz, 1H), 7,33 (t, J = 7,3 Hz, 1H), 7,22 (d, J = 7,8 Hz, 1H), 7,14-7,08 (m, 2H), 6,97 (t, J = 7,3 Hz, 1H), 6,55 (d, J = 7,8 Hz, 1H), 5,33 (s, 2H), 4,51 (s, 2H), 4,14-4,09 (m a, 2H), 3,43-3,35 (m a, 2H), 2,74-2,67 (m a, 2H), 1,97-1,90 (m, 1H), 1,74-1,67 (m, 2H), 1,45 (s, 9H), 1,26-1,19 (m, 2H)
Etapa 2: El compuesto del título R2 (1,79 g, 92 %) se obtuvo de la misma manera que en la etapa 4 del ejemplo de referencia 1 usando el 4-{[1-(2-cianobencil)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-carboxilato de terc-butilo obtenido en la etapa 1.
ESI-MS m/z: 361 (M+H)+
Ejemplo de referencia 3
5-yodo-2-oxo-1-{[2-(trimetilsilil)etoxi]metil}-1,2-dihidropiridin-3-carbonitrilo (Compuesto R3)
Etapa 1: Se agitaron 2-hidroxinicotinonitrilo (1,00 g, 8,33 mmol), yodo (2,54 g, 9,99 mmol) y carbonato potásico (1,38 g, 9,99 mmol) durante una noche en DMF (10 ml) a temperatura ambiente. A la mezcla de reacción, se le añadió agua y la mezcla resultante se extrajo con a un disolvente mixto de cloroformo/2-propanol. La capa orgánica se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. Al residuo resultante, se le añadió (2-clorometoxietil)trimetilsilano (SEM-C1) (1,67 g, 10,0 mmol) e hidróxido potásico (560 mg, 10,0 mmol) y la mezcla resultante se agitó en THF (25,0 ml) a temperatura ambiente durante 3 horas. A la mezcla de reacción, se le añadió agua y la mezcla resultante se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturad y después se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de acetato de etilo/heptano), con lo que se obtuvo el compuesto del título R3 (372 mg, rendimiento: 12 %).
ESI-MS m/z: 377 (M+H)+, RMN 1H (300 MHz, CDCla, 8): 7,89 (d, J = 2,6 Hz, 1H), 7,86 (d, J = 2,6 Hz, 1H), 5,31 (s, 2H), 3,64 (t, J = 8,3 Hz, 2H), 0,95 (t, J = 8,3 Hz, 2H), -0,01 (s, 9H)
Ejemplo de referencia 4
3- yodofenilsulfonil{[2-(trimetilsilil)etoxi]metil}carbamato de terc-butilo (Compuesto R4)
Etapa 1: 3-yodobencenosulfonamida (100 mg, 0,353 mmol), dicarbonato de di-terc-butilo (Boc2O) (116 mg, 0,530 mmol), DMAP (8,63 mg, 0,071 mmol) y trietilamina (54,0 mg, 0,530 mmol) se agitaron en diclorometano (1,50 ml) a temperatura ambiente durante 3 horas. A la mezcla de reacción, se le añadió agua y la mezcla resultante se extrajo con cloroformo. La capa orgánica se lavó con salmuera saturad y después se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de metanol/cloroformo), con lo que se obtuvo 3-yodofenilsulfonilcarbamato de terc-butilo (100 mg, rendimiento: 74 %).
ESI-MS m/z: 384 (M+H)+
Etapa 2: El compuesto del título R4 (120 mg, rendimiento: 90 %) se obtuvo de la misma manera que en la etapa 1 del ejemplo 14 usando el 3-yodofenilsulfonilcarbamato de terc-butilo obtenido en la etapa 1.
RMN 1H (300 MHz, CDCb, 8): 8,36-8,35 (m, 1H), 8,02-7,98 (m, 1H), 7,95-7,91 (m, 1H), 7,28-7,23 (m, 1H), 5,30 (s, 2H), 3,59-3,54 (m, 2H), 1,37 (s, 9H), 1,03-0,98 (m, 2H), 0,00 (s, 9H) ESI-MS m/z: 514 (M+H)+
Ejemplo de referencia 5
4- oxo-3,4-dihidroquinazolin-7-carboxilato de terc-butilo (Compuesto R5)
El ácido 4-terc-butil-1-metil-1-aminotereftálico (55 mg, 0,22 mmol) obtenido por el método descrito en Journal of Medicinal Chemistry 1999, 42, 545 y acetato de formamidina (46 mg, 0,44 mmol) se calentaron a reflujo durante una noche en etanol (2,0 ml). Después de evaporar el disolvente a presión reducida, el residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo el compuesto del título R5 (28 mg, rendimiento: 51 %).
ESI-MS m/z: 247 (M+H)+, RMN 1H (300 MHz, CDCb, 8): 8,37 (s, 1H), 8,34 (d, J = 8,4 Hz, 1H), 8,11-8,07 (m, 2H), 1,64 (s, 9H)
Ejemplo de referencia 6
4-cloro-5H-pirido[4,5-b][1,4]oxazin-6(7H)-ona (Compuesto R6)
Etapa 1: Se disolvió metil glicolato (488 mg, 5,4 mmol) en DMF (20 ml) y, en un baño de hielo, se le añadió hidruro de sodio (aproximadamente 60 % en peso, 268 mg) y la mezcla resultante se agitó durante 30 minutos. Después de esto, se le añadió una solución en DMF (2,0 ml) de 4,6-dicloro-5-nitropirimidina (1,0 g, 5,1 mmol) gota a gota y la mezcla resultante se agitó durante una noche a temperatura ambiente. A la mezcla de reacción, se le añadió agua helada y, después, la mezcla resultante se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturada y se secó sobre sulfato de magnesio anhidro y, después, el disolvente se evaporó a presión reducida. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de hexano/acetato de etilo), con lo que se obtuvo 2-(6-cloro-5-nitropirimidin-4-iloxi)acetato de metilo (584 mg, 46 %).
RMN 1H (400 MHz, CDCla 8): 8,62 (s, 1H), 5,08 (s, 2H), 3,80 (s, 3H)
Etapa 2: El 2-(6-cloro-5-nitropirimidin-4-iloxi)acetato de metilo (0,58 g, 2,3 mmol) obtenido en la etapa 1 y hierro reducido (654 mg, 12 mmol) se calentaron en ácido acético (15 ml) a 80 °C durante 6 horas. La mezcla de reacción se trató con tierra de diatomeas y un residuo obtenido evaporando el disolvente a presión reducida se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de hexano/acetato), con lo que se obtuvo el compuesto del título R6 (270 mg, rendimiento: 62 %).
RMN 1H (300 MHz, DMSO-d6, 8): 11,0 (s a, 1H), 8,28 (s, 1H), 4,95 (s, 2H)
Ejemplo de referencia 7
8-cloro-[1,2,4]triazolo[4,3-a]pirazin-3(2H)-ona (Compuesto R7)
La 2-cloro-3-hidrazinilpirazina (710 mg, 0,49 mmol) obtenida mediante el método descrito en el documento WO2008/130951 se disolvió en acetonitrilo (12 ml) y después se le añadió 1,1-carbonildiimidazol (1,6 g, 9,8 mmol) y, después, la mezcla resultante se agitó a temperatura ambiente durante 4 horas. A la mezcla de reacción, se le añadió agua y el sólido depositado se recogió por filtración, con lo que se obtuvo el compuesto del título R7 (117 mg, rendimiento: 14 %).
RMN 1H (400 MHz, DMSO-d6, 8): 13,2 (s a, 1H), 7,93 (d, J = 4,0 Hz, 1H), 7,34 (d, J = 4,0 Hz, 1H)
Ejemplo de referencia 8
2,3-difluoroisonicotinamida (Compuesto R8)
Ácido 2,3-difluoroisonicotínico (330 mg, 2,1 mmol), una solución de amoniaco-metanol (aproximadamente 7,0 mol/l, 5,9 ml), hexafluorofosfato O-(7-aza-1H-benzotriazol-1-il)-N,N,N',N'-tetrametiluronio (1,6 g, 4,2 mmol) y diisopropiletilamina (1,4 ml) se agitaron en DMF (3,0 ml) a temperatura ambiente durante 5 horas. A la mezcla de reacción, se le añadió agua y la mezcla resultante se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturada y se secó sobre sulfato de magnesio anhidro. Un residuo obtenido por concentración del disolvente a presión reducida se purificó mediante cromatografía en columna sobre gel de sílice (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo el compuesto del título R8 (239 mg, rendimiento: 73 %).
RMN 1H (400 MHz, CDCla, 8): 8,11 (d, J = 6,8 Hz, 1H), 7,82-7,77 (m, 1H), 6,59 (s a, 1H), 6,24 (s a, 1H)
Ejemplo de referencia 9
8-cloropirido[3,4-d]pirimidin-4(3H)-ona (Compuesto R9)
La 3-amino-2-cloroisonicotinamida (170 mg, 0,99 mmol) obtenida mediante el método descrito en Journal of Heterociclic Chemistry, 2001,38, 99 se agitó en ortoformiato de etil trietilo (3,0 ml) a 150 °C durante 6 horas. La mezcla de reacción se concentró a presión reducida y se le añadió un disolvente mixto de éter dietílico/acetato de etilo (1/1) y el sólido resultante se recogió por filtración, con lo que se obtuvo el compuesto del título R9 (148 mg, rendimiento: 82 %).
RMN 1H (300 MHz, DMSO-d6, 8): 12,8 (s a, 1H), 8,42 (d, J = 5,1 Hz, 1H), 8,30 (s, 1H), 7,95 (d, J = 5,1 Hz, 1H)
Ejemplo de referencia 10
7,8-dihidro-3H-pirano[4,3-d]pirimidin-4(5H)-ona (Compuesto R10)
El 4-oxotetrahidro-2H-piran-3-carboxilato de etilo (500 mg, 2,9 mmol) obtenido mediante el método descrito en el documento US2011/82138, acetato de formamidina (300 mg, 2,9 mmol) y metóxido sódico (500 mg, 9,3 mmol) se calentaron a reflujo en metanol (20 ml) durante 6 horas. A la mezcla de reacción, se le añadió agua y después, la mezcla resultante se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturada y se secó sobre sulfato de magnesio anhidro y, después, el disolvente se evaporó a presión reducida, con lo que se obtuvo el compuesto del título R10 (200 mg, rendimiento: 45 %).
ESI-MS m/z: 153 (M+H)+
Ejemplo de referencia 11
4,7-didoropirido[4,3-d]pirimidina (Compuesto R11)
Etapa 1: El 4-amino-6-cloronicotinato de metilo (15 g, 80 mmol) obtenido mediante el método descrito en US2012/184562 e hidróxido sódico (13 g, 322 mmol) se agitaron en una solución mixta de metanol (100 ml) y agua (50 ml) a temperatura ambiente durante 12 horas. La mezcla de reacción se ajustó a pH 6 con 6,0 mol/l de una solución acuosa de ácido clorhídrico y el sólido resultante se recogió por filtración, con lo que se obtuvo ácido 4-amino-6-cloronicotínico (8,0 g, rendimiento: 58 %).
RMN 1H (300 MHz, DMSO-d6, 8): 8,47 (s, 1H), 7,52 (s a, 2H), 6,75 (s, 1H)
Etapa 2: el ácido 4-amino-6-cloronicotínico (7,0 g, 41 mmol) obtenido en la etapa 1 se agitó en cloruro de tionilo (100 ml) a 80 °C durante 12 horas. La mezcla de reacción se concentró a presión reducida, con lo que se obtuvo cloruro de 4-amino-6-cloronicotinoílo en bruto. Este compuesto se usó en la siguiente reacción sin realizar más purificación en particular.
Etapa 3: El cloruro de 4-amino-6-cloronicotinoílo en bruto obtenido en la etapa 2 se agitó en una solución acuosa de amoniaco (aproximadamente 28 %, 70 ml) a temperatura ambiente durante 4 horas. La mezcla de reacción se extrajo con acetato de etilo y después, la capa orgánica se lavó con salmuera saturada y se secó sobre sulfato de magnesio anhidro. Después de evaporar el disolvente a presión reducida, el residuo resultante se purificó por cromatografía en columna sobre gel de sílice usando un disolvente mixto (diclorometano/metanol), con lo que se obtuvo 4-amino-6-cloronicotinamida (4,5 g, rendimiento: 72 %).
RMN 1H (300 MHz, DMSO-d6, 8): 8,37 (s, 1H), 7,97 (s a, 1H), 7,51 (s a, 2H), 7,24 (s a, 1H), 6,65 (s, 1H)
Etapa 4: la 4-amino-6-cloronicotinamida (4,5 g, 25 mmol) obtenida en la etapa 3 se agitó en ortoformiato de trimetilo (20 ml) a 150 °C durante 5 horas. La mezcla de reacción se enfrió a 0 °C y el sólido resultante se recogió por filtración, con lo que se obtuvo 7-cloropirido[4,3-d]pirimidin-4(3H)-ona (3,2 g, rendimiento: 70 %).
RMN 1H (300 MHz, DMSO-d6, 8): 12,8 (s a, 1H), 9,10 (s, 1H), 8,34 (s, 1H), 7,73 (s, 1H)
Etapa 5: la 7-cloropirido[4,3-d]pirimidin-4(3H)-ona (2,0 g, 11 mmol) obtenida en la etapa 4 y N,N-dimetilanilina (0,1 ml) se calentaron a reflujo en oxicloruro de fósforo (60 ml) durante 15 horas. Después de la mezcla de reacción se diluyó con diclorometano, se le añadió agua helada y la mezcla resultante se extrajo. Después de secar la capa orgánica sobre sulfato de magnesio anhidro, el disolvente se evaporó a presión reducida, con lo que se obtuvo el compuesto del título R11 (1,7 g) en bruto. Este compuesto se usó en la siguiente reacción sin realizar más purificación en particular.
Ejemplo de referencia 12
3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-2(1H)-ona (Compuesto 53)
El compuesto del título 53 se sintetizó de acuerdo con el método descrito en Chemical & Pharmaceutical Bulletin 1990, 38(6), 1591.
Ejemplo 1
2-[(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)metil]benzamida (Compuesto 1)
Etapa 1: la 3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-2(1H)-ona (300 mg, 0,69 mmol) obtenida mediante el método descrito en Chemical & Pharmaceutical Bulletin 1990, 38(6), 1591 se disolvió en DMF (3,0 ml) y se le añadió secuencialmente hidruro sódico (aproximadamente 60 % en peso, 33 mg) y 2-(bromometil)-benzoato de metilo (190 mg, 0,83 mmol) en un baño de hielo. Después de agitar la mezcla resultante a temperatura ambiente durante 2 horas, se le añadió una solución acuosa saturada de bicarbonato sódico a la mezcla de reacción y la mezcla resultante se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturad y después se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo 2-[(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)metil]benzoato de metilo (355 mg, rendimiento: 88 %).
ESI-MS m/z: 582 (M+H)+, RMN 1H (400 MHz, CDCla, 8): 8,66 (s, 1H), 8,04 (d, J = 7,8 Hz, 1H), 7,41-7,36 (m, 1H), 7,32 7,27 (m, 2H), 7,15-7,06 (m, 4H), 6,99-6,94 (m, 1H), 6,57 (d, J = 8,8 Hz, 1H), 5,52 (s, 2H), 4,57 (s, 2H), 4,22-4,15 (m a, 2H), 4,02 (s, 3H), 3,97 (s, 3H), 3,93 (s, 3H), 3,50 (d, J = 7,8 Hz, 2H), 3,12-3,02 (m, 2H), 2,18-2,08 (m, 1H), 1,96-1,87 (m a, 2H), 1,66-1,55 (m a, 2H)
Etapa 2: el 2-[(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)metil]benzoato de metilo (100 mg, 0,17 mmol) obtenido en la etapa 1 e hidróxido de litio monohidrato (12 mg, 0,52 mmol) se agitaron en un disolvente mixto de etanol (0,50 ml)/agua (0,50 ml) a temperatura ambiente durante 2 horas. A la mezcla de reacción, se le añadió 3,0 mol/l de ácido clorhídrico con enfriamiento en hielo y el sólido depositado se recogió por filtración y se secó a presión reducida, con lo que se obtuvo ácido 2-[(3-{[1-(6,7
dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)metil]benzoico (90 mg, rendimiento: 92 %).
RMN 1H (400 MHz, DMSO-d6, 5): 13,16 (s a, 1H), 8,70 (s, 1H), 7,96 (d, J = 6,8 Hz, 1H), 7,47-7,42 (m, 1H), 7,36-7,32 (m, 1H), 7,30-7,18 (m, 3H), 7,13-7,08 (m, 1H), 7,02-6,94 (m, 2H), 6,51 (d, J = 7,8 Hz, 1H), 5,38 (s, 2H), 4,70-4,50 (m, 4H), 3,97 (s, 3H), 3,92 (s, 3H), 3,48-3,36 (m, 4H), 2,26-2,19 (m, 1H), 1,92-1,84 (m, 2H), 1,49-1,39 (m, 2H)
Etapa 3: el ácido 2-[(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)metil]benzoico (40 mg, 0,07 mmol) obtenido en la etapa 2 , clorhidrato de e Dc ( 20 mg, 0 ,11 mmol), HOBtH2O (16 mg, 0,11 mmol) y una solución acuosa de amoniaco (aproximadamente 28 %, 0,04 ml) se agitaron en DMF (1,0 ml) a temperatura ambiente durante 3 horas. A la mezcla de reacción, se le añadió una solución acuosa saturada de bicarbonato sódico y la mezcla resultante se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturad y después se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo el compuesto del título 1 (35 mg, rendimiento: 88 %).
ESI-MS m/z: 567 (M+H)+, RMN 1H (300 MHz, CDCb, 5): 8,65 (s, 1H), 7,58-7,53 (m, 1H), 7,33-7,28 (m, 1H), 7,28-7,24 (m, 2H), 7,23 (s, 1H), 7,20-7,16 (m, 1H), 7,11-7,09 (m, 1H), 7,07 (s, 1H), 7,01-6,97 (m, 1H), 6,84 (d, J = 7,7 Hz, 1H), 6,65 (s a, 1H), 5,72 (s a, 1H), 5,34 (s, 2H), 4,53 (s, 2H), 4,21-4,11 (m a, 2H), 4,02 (s, 3H), 3,97 (s, 3H), 3,45 (d, J = 7,3 Hz, 2H), 3,11-3,00 (m, 2H), 2,13-2,04 (m, 1H), 1,90-1,85 (m, 2H), 1,55-1,50 (m, 2H)
Ejemplo 2
3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-1-[(tetrahidrofuran-2-il)metil]-3,4-dihidroquinazolin-2(1H)-ona (Compuesto 2)
El compuesto del título 2 (95 mg, rendimiento: 40 %) se obtuvo de la misma manera que en el ejemplo 1 usando 2-(bromometil)tetrahidrofurano.
ESI-MS m/z: 518 (M+H)+, RMN 1H (300 MHz, CDCla, 5): 8,64 (s, 1H), 7,26-7,23 (m, 2H), 7,12-6,95 (m, 4H), 4,50 (d, J = 14 Hz, 1H), 4,35 (d, J = 14 Hz, 1H), 4,26-4,11 (m, 5H), 4,02 (s, 3H), 3,98 (s, 3H), 3,95-3,84 (m, 2H), 3,79-3,71 (m, 1H), 3,51-3,34 (m, 1H), 3,09-2,98 (m, 2H), 2,10-1,50 (m, 9H)
Ejemplo 3
2-[(3-{[1-([1,3]dioxo[4,5-g]quinazolin-8-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)metil]benzonitrilo (Compuesto 3)
La [1,3]dioxo[4,5-g]quinazolin-8(7H)-ona (24 mg, 0,13 mmol) obtenida mediante el método descrito en Journal of Medicinal Chemistry, 2010, 53, 8089, BOP (84 mg, 0,19 mmol) y DBU (58 mg, 0,38 mmol) se agitaron en DMF (1,0 ml) a temperatura ambiente durante 1 hora. Después de esto, se le añadió el compuesto R2 (50 mg, 0,18 mmol) obtenido en el ejemplo de referencia 2 y la mezcla resultante se agitó a 80 °C durante 2 horas. A la mezcla de reacción, se le añadió una solución acuosa saturada de bicarbonato sódico y la mezcla resultante se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturad y después se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo el compuesto del título 3 (26 mg, rendimiento: 39 %). ESI-MS m/z: 533 (M+H)+, RMN 1H (400 MHz, CDCb, 5): 8,63 (s, 1H), 7,69 (d, J = 6 ,8 Hz, 1H), 7,52-7,47 (m, 1H), 7,36 7,32 (m, 1H), 7,26-7,23 (m, 1H), 7,20 (s, 1H), 7,16-7,10 (m, 3H), 7,01-6,97 (m, 1H), 6,57 (d, J = 7,8 Hz, 1H), 6,11 (s, 2H), 5,36 (s, 2H), 4,57 (s, 2H), 4,14-4,09 (m a, 2H), 3,50 (d, J = 6 ,8 Hz, 2H), 3,06-2,99 (m, 2H), 2,14-2,07 (m, 1H), 1,94-1,88 (m, 2H), 1,65-1,61 (m, 2H)
Ejemplo 4
2-[(3-1[1-(benzo[d][1,2,3]triazin-4-il)piperidin-4-il]metil-2-oxo-3,4-dihidroquinazolin-1(2H)-il)metil]benzonitrilo (Compuesto 4)
El compuesto del título 4 (18 mg, rendimiento: 29 %) se obtuvo de la misma manera que en el ejemplo 3 usando el compuesto R2 y benzo[d][1,2,3]triazin-4(3H)-ona.
ESI-MS m/z: 490 (M+H)+, RMN 1H (300 MHz, CDCb, 5): 8,26 (d, J = 8,1 Hz, 1H), 7,94-7,87 (m, 2H), 7,77-7,67 (m, 2H), 7 ,54 -7 ,47 (m, 1H), 7,37-7,24 (m, 2H), 7,16-7,10 (m, 2H), 7,02-6,96 (m, 1H), 6,58 (d, J = 8,1 Hz, 1H), 5,35 (s, 2H), 4,57 4,54 (m, 4H), 3,50 (d, J = 7,3 Hz, 2H), 3,34-3,23 (m, 2H), 2,25-2,18 (m, 1H), 2,02-1,94 (m, 2H), 1,72-1,59 (m, 2H)
Ejemplo 5
2-[(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-6-(metilsulfonil)-2-oxo-3,4-dihidroquinazolin-1(2H)-il)metil]benzonitrilo (Compuesto 5)
Etapa 1: A una solución en THF (10,0 ml) de ácido 5-(metilsulfanil)-2-nitrobenzoico (800 mg, 3,75 mmol), se le añadió un complejo de borano-sulfuro de dimetilo (1,14 g, 15,0 mmol) a temperatura ambiente y la mezcla resultante se
calentó a reflujo durante 1,5 horas. La mezcla de reacción se enfrió a 0 °C y se le añadió ácido clorhídrico (1,00 mol/l, 10.0 ml) y la mezcla resultante se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturada y después se secó sobre sulfato de magnesio anhidro. Un residuo obtenido mediante la evaporación del disolvente a presión reducida se disolvió en cloroformo (20,0 ml) y DMF (1,00 ml) y se le añadió dióxido de manganeso (6,00 g, 69.0 mmol) y la mezcla resultante se agitó durante una noche a temperatura ambiente. La mezcla de reacción se trató con tierra de diatomeas y el disolvente se evaporó a presión reducida, obteniendo de este modo 5-(metiltio)-2-nitrobenzaldehído. Usando este compuesto en bruto, se obtuvo 4-{[6-(metiltio)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo (850 mg, rendimiento: 58 %) de la misma manera que en el ejemplo de referencia 1. ESI-MS m/z: 392 (M+H)+
Etapa 2: El 4-{[6-(metiltio)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo (400 mg, 1,02 mmol) obtenido en la etapa 1 se disolvió en diclorometano (10,0 ml) y se le añadió secuencialmente una solución acuosa saturada de bicarbonato sódico (10,0 ml) y ácido meta-cloroperoxibenzoico (aproximadamente 70 % en peso, 630 mg) en un baño de hielo y la mezcla resultante se agitó a temperatura ambiente durante 2 horas. A la mezcla de reacción, se le añadió una solución acuosa saturada de bicarbonato sódico y la capa orgánica se separó y la capa acuosa se extrajo con cloroformo. Las capas orgánicas se combinaron y se lavaron con salmuera saturada después se secaron sobre sulfato de magnesio anhidro. El disolvente se evaporó a presión reducida y el residuo resultante se purificó mediante cromatografía en columna sobre gel de sílice (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo 4-{[6-(metil-sulfonil)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo (433 mg, rendimiento cuantitativo).
ESI-MS m/z: 424 (M+H)+
Etapa 3: El 4-{[6-(metilsulfonil)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo (200 mg, 0,47 mmol) obtenido en la etapa 2 se disolvió en acetato de etilo (3,0 ml) y se le añadió una solución de ácido clorhídrico-acetato de etilo (4,0 mol/l, 3,5 ml) en un baño de hielo. Después de agitar la mezcla resultante a temperatura ambiente durante 2 horas, el disolvente se evaporó a presión reducida. Al residuo resultante, se le añadió 4-cloro-6,7-dimetoxiquinazolina (117 mg, 0,52 mmol) y diisopropiletilamina (183 mg, 1,4 mmol) y la mezcla resultante se calentó a reflujo en 2-propanol (5,0 ml) durante 2 horas. A la mezcla de reacción, se le añadió una solución acuosa saturada de bicarbonato sódico y la mezcla resultante se extrajo con cloroformo. La capa orgánica se lavó con salmuera saturada y se secó sobre sulfato de magnesio anhidro y, después, el disolvente se concentró a presión reducida. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo 3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-6-(metilsulfonil)-3,4-dihidroquinazolin-2(1H)-ona (116 mg, rendimiento: 48 %).
ESI-MS m/z: 512 (M+H)+
Etapa 4: El compuesto del título 5 (23 mg, rendimiento: 24 %) se obtuvo de la misma manera que en el ejemplo 1 usando la 3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-6-(metilsulfonil)-3,4-dihidroquinazolin-2(1H)-ona obtenida en la etapa 3.
ESI-MS m/z: 627 (M+H)+, RMN 1H (300 MHz, CDCla, 8): 8,66 (s, 1H), 7,72-7,69 (m, 3H), 7,55-7,50 (m, 1H), 7,41-7,36 (m, 1H), 7,26-7,22 (m, 2H), 7,09 (s, 1H), 6,73-6,70 (m, 1H), 5,39 (s, 2H), 4,62 (s, 2H), 4,25-4,20 (m, 2H), 4,03 (s, 3H), 3,99 (s, 3H), 3,52 (d, J = 7,2 Hz, 2H), 3,15-3,07 (m, 2H), 3,03 (s, 3H), 2,24-2,11 (m, 1H), 1,94-1,90 (m, 2H), 1,66-1,55 (m, 2H)
Ejemplo 6
3-(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)benzonitrilo (Compuesto 6)
Etapa 1: la 3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-2(1H)-ona (50 mg, 0,12 mmol) obtenida mediante el método descrito en Chemical & Pharmaceutical Bulletin 1990, 38(6), 1591, yoduro de cobre (I) (22 mg, 0,12 mmol), trans-1,2-ciclohexanodiamina (13 mg, 0,12 mmol), 3-yodobenzonitrilo (53 mg, 0,23 mmol) y fosfato tripotásico (49 mg, 0,23 mmol) se agitaron en 1,4-dioxano (1,0 ml) a 100 °C durante 5 horas. A la mezcla de reacción, se le añadió una solución acuosa saturada de bicarbonato sódico y la mezcla resultante se extrajo con acetato de etilo. La capa orgánica se trató con tierra de diatomeas y se concentró a presión reducida. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo el compuesto del título 6 (51 mg, rendimiento: 82 %).
ESI-MS m/z: 535 (M+H)+, RMN 1H (300 MHz, CDCla, 8): 8,66 (s, 1H), 7,73-7,59 (m, 4H), 7,24 (s, 1H), 7,16-7,00 (m, 4H), 6,17 (d, J = 8,4 Hz, 1H), 4,62 (s, 2H), 4,24-4,14 (m a, 2H), 4,02 (s, 3H), 3,98 (s, 3H), 3,47 (d, J = 7,3 Hz, 2H), 3,10 3,06 (m a, 2H), 2,18-2,10 (m, 1H), 1,93-1,90 (m, 2H), 1,62-1,54 (m, 2H)
Ejemplo 7
5-(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)-2-fluorobenzonitrilo (Compuesto 7)
El compuesto del título 7 (40 mg, rendimiento: 63 %) se obtuvo de la misma manera que en el ejemplo 6 usando 2fluoro-5-yodobenzonitrilo.
ESI-MS m/z: 553 (M+H)+, RMN 1H (300 MHz, CDCla, 8): 8,67 (s, 1H), 7,66-7,57 (m, 2H), 7,39-7,31 (m, 1H), 7,24 (s, 1H), 7,16-7,01 (m, 4H), 6,19 (d, J = 8,4 Hz, 1H), 4,61 (s, 2H), 4,21-4,17 (m, 2H), 4,02 (s, 3H), 3,98 (s, 3H), 3,46 (d, J = 7,0 Hz, 2H), 3,12-3,05 (m, 2H), 2,14-2,09 (m, 1H), 1,93-1,89 (m, 2H), 1,65-1,52 (m, 2H)
Ejemplo 8
3- {[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-1-(3-nitrofenil)-3,4-dihidroquinazolin-2(1H)-ona (Compuesto 8)
El compuesto del título 8 (40 mg, rendimiento: 63 %) se obtuvo de la misma manera que en el ejemplo 6 usando 1-yodo-3-nitrobenceno.
ESI-MS m/z: 555 (M+H)+, RMN 1H (300 MHz, CDCla, 8): 8,67 (s, 1H), 8,32-8,27 (m, 1H), 8,26-8,23 (m, 1H), 7,74-7,66 (m, 2H), 7,26-7,24 (m, 1H), 7,17-7,01 (m, 4H), 6,20 (d, J = 8,1 Hz, 1H), 4,63 (s, 2H), 4,24-4,15 (m, 2H), 4,02 (s, 3H), 3.98 (s, 3H), 3,48 (d, J = 7,0 Hz, 2H), 3,15-3,03 (m, 2H), 2,20-2,08 (m, 1H), 1,97-1,88 (m, 2H), 1,66-1,49 (m, 2H)
Ejemplo 9
4- (3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (Compuesto 9)
El compuesto del título 9 (29 mg, rendimiento: 47 %) se obtuvo de la misma manera que en el ejemplo 6 usando 4-yodopicolinonitrilo.
ESI-MS m/z: 536 (M+H)+, RMN 1H (300 MHz, CDCls, 8): 8,80 (d, J = 5,2 Hz, 1H), 8,66 (s, 1H), 7,84 (d, J = 1,9 Hz, 1H), 7,66 (dd, J = 5,2, 1,9 Hz, 1H), 7,24-7,07 (m, 5H), 6,39 (d, J = 8,1 Hz, 1H), 4,59 (s, 2H), 4,23-4,15 (m a, 2H), 4,02 (s, 3H), 3,99 (d, J = 3,3 Hz, 3H), 3,47 (d, J = 7,0 Hz, 2H), 3,13-3,03 (m, 2H), 2,19-2,05 (m, 1H), 1,94-1,85 (m, 2H), 1,66 1,49 (m, 2H)
Ejemplo 10
2- (3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)isonicotinonitrilo (Compuesto 10)
El compuesto del título 10 (580 mg, rendimiento: 94 %) se obtuvo de la misma manera que en el ejemplo 6 usando 2-yodoisonicotinonitrilo.
ESI-MS m/z: 536 (M+H)+, RMN 1H (400 MHz, CDCls, 8): 8,78 (d, J = 4,9 Hz, 1H), 8,66 (s, 1H), 7,86 (s, 1H), 7,55 (d, J = 4,9 Hz, 1H), 7,25 (s, 1H), 7,17-7,03 (m, 4H), 6,27 (d, J = 7,8 Hz, 1H), 4,62 (s, 2H), 4,24-4,14 (m, 2H), 4,02 (s, 3H), 3.98 (s, 3H), 3,48 (d, J = 7,8 Hz, 2H), 3,12-3,03 (m, 2H), 2,20-2,08 (m, 1H), 1,97-1,85 (m, 2H), 1,67-1,52 (m, 2H)
Ejemplo 11
4- (3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)pirimidin-2-carbonitrilo (Compuesto 11)
Etapa 1: El compuesto del título 11 (8,0 mg, rendimiento: 13 %) se obtuvo de la misma manera que en el ejemplo 6 usando la 3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-2(1H)-ona obtenida mediante el método descrito en Chemical & Pharmaceutical Bulletin 1990, 38(6), 1591 y 4-bromopirimidin-2-carbonitrilo.
ESI-MS m/z: 537 (M+H)+, RMN 1H (400 MHz, CDCls, 8): 8,77 (d, J = 5,9 Hz, 1H), 8,66 (s, 1H), 8,19 (d, J = 5,9 Hz, 1H), 7,31-7,21 (m, 4H), 7,14 (d, J = 7,8 Hz, 1H), 7,07 (s, 1H), 4,49 (s, 2H), 4,22-4,14 (m, 2H), 4,02 (s, 3H), 3,98 (s, 3H), 3,50 (d, J = 7,8 Hz, 2H), 3,12-3,01 (m, 2H), 2,13-2,03 (m, 1H), 1,89-1,79 (m, 2H), 1,64-1,52 (m, 2H)
Ejemplo 12
5- (3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)-2-oxo-1,2-dihidropiridin-3- carbonitrilo (Compuesto 12)
Etapa 1: se obtuvo 5-(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)-2-oxo-1-{[2-(trimetilsilil)etoxi]metil}1,2-dihidropiridin-3-carbonitrilo (80 mg, rendimiento: 51 %) de la misma manera que en el ejemplo 6 usando el compuesto R3 obtenido en el ejemplo de referencia 3. ESI-MS m/z: 682 (M+H)+
Etapa 2: el 5-(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)-2-oxo-1-{[2-(trimetilsilil)etoxi]metil}1,2-dihidropiridin-3-carbonitrilo (80 mg, 0,12 mmol) obtenido en la etapa 1 y ácido trifluoroacético (740 mg, 6,5 mmol) se agitaron en diclorometano (1,0 ml) durante 5 horas con enfriamiento en hielo. A la mezcla de reacción, se le añadió una solución acuosa saturada de bicarbonato sódico y la mezcla resultante se extrajo con cloroformo. La capa orgánica se lavó con salmuera saturad y después se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de metanol/cloroformo), con lo que se obtuvo el compuesto del título 12 (20 mg,
rendimiento: 31 %).
ESI-MS m/z: 552 (M+H)+, RMN 1H (300 MHz, CDCla, 8): 8,66 (s, 1H), 7,93 (d, J = 2,7 Hz, 1H), 7,79 (d, J = 2,7 Hz, 1H), 7,25 (s, 1H), 7,21-7,14 (m, 2H), 7,10-7,04 (m, 2H), 6,44 (d, J = 8,1 Hz, 1H), 4,58 (s, 2H), 4,24-4,16 (m, 2H), 4,02 (s, 3H), 3,99 (s, 3H), 3,46 (d, J = 7,3 Hz, 2H), 3,15-3,04 (m, 2H), 2,18-2,09 (m, 1H), 1,94-1,85 (m, 2H), 1,66-1,51 (m, 2H)
Ejemplo 13
3- (3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)bencenosulfonamida (Compuesto 13)
Etapa 1: se obtuvo 3-(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)fenilsulfonil{[2-(trimetilsilil)etoxi]metil}carbamato de ferc-butilo (98 mg, rendimiento: 86 %) de la misma manera que en el ejemplo 6 usando el compuesto R4 obtenido en el ejemplo de referencia 4.
ESI-MS m/z: 820 (M+H)+, RMN 1H (300 MHz, CDCla, 8): 8,66 (s, 1H), 8,08-8,04 (m, 1H), 7,99-7,97 (m, 1H), 7,66-7,62 (m, 2H), 7,24 (s, 1H), 7,14-6,99 (m, 4H), 6,18-6,14 (m, 1H), 5,28 (s, 2H), 4,59 (s, 2H), 4,21-4,11 (m, 2H), 4,02 (s, 3H), 3,98 (s, 3H), 3,61-3,55 (m, 2H), 3,46 (d, J = 7,0 Hz, 2H), 3,13-3,03 (m, 2H), 2,16-2,06 (m, 1H), 1,95-1,87 (m, 2H), 1,65 1,53 (m, 2H), 1,37-1,28 (m, 9H), 0,90-0,85 (m, 2H), -0,03 (s, 9H)
Etapa 2: El compuesto del título 13 (25 mg, rendimiento: 37 %) se obtuvo de la misma manera que en la etapa 2 del ejemplo 12 usando 3-(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)fenilsulfonil[2-(trimetilsilil)etoxi]metil}carbamato de ferc-butilo obtenido en la etapa 1.
ESI-MS m/z: 589 (M+H)+, RMN 1H (300 MHz, CDCb, 8): 8,64 (s, 1H), 7,96-7,90 (m, 2H), 7,65-7,59 (m, 1H), 7,56-7,52 (m, 1H), 7,24 (s, 1H), 7,14-6,97 (m, 4H), 6,20-6,15 (m, 1H), 5,16 (s a, 2H), 4,61 (s, 2H), 4,22-4,13 (m, 2H), 4,01 (s, 3H), 3,97 (s, 3H), 3,45 (d, J = 7,3 Hz, 2H), 3,13-3,03 (m, 2H), 2,18-2,07 (m, 1H), 1,94-1,85 (m, 2H), 1,64-1,49 (m, 2H)
Ejemplo 14
4- (3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinamida (Compuesto 14)
Etapa 1: se obtuvo 4-(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinato de etilo (235 mg, rendimiento: 70 %) de la misma manera que en el ejemplo 6 usando 4-yodopicolinato de etilo. ESI-MS m/z: 555 (M+H)+, RMN 1H (300 MHz, CDCb, 8): 8,89 (d, J = 5,1 Hz, 1H), 8,67 (s, 1H), 8,17 (s, 1H), 7,59 (d, J = 5,1 Hz, 1H), 7,28-7,03 (m, 5H), 6,31 (d, J = 8,1 Hz, 1H), 4,61 (s, 2H), 4,49 (c, J = 7,1 Hz, 2H), 4,25-4,14 (m, 2H), 4,02 (s, 3H), 3,98 (s, 3H), 3,48 (d, J = 7,3 Hz, 2H), 3,14-3,01 (m, 2H), 2,20-2,06 (m, 1H), 1,97-1,85 (m, 2H), 1,63 1,55 (m, 2H), 1,44 (t, J = 7,1 Hz, 3H)
Etapa 2: El compuesto del título 14 (40 mg, rendimiento: 80 %) se obtuvo de la misma manera que en el ejemplo 1 usando 4-(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinato de etilo obtenido en la etapa 1.
ESI-MS m/z: 554 (M+H)+, RMN 1H (300 MHz, CDCb, 8): 8,71 (d, J = 5,1 Hz, 1H), 8,66 (s, 1H), 8,19 (d, J = 2,0 Hz, 1H), 7,86 (s a, 1H), 7,62 (dd, J = 5,1, 2,0 Hz, 1H), 7,23 (s, 1H), 7,17-7,01 (m, 4H), 6,32 (d, J = 8,1 Hz, 1H), 5,71 (s a, 1H), 4,59 (s, 2H), 4,23-4,14 (m, 2H), 4,02 (s, 3H), 3,99 (s, 3H), 3,47 (d, J = 7,3 Hz, 2H), 3,15-3,04 (m, 2H), 2,19-2,06 (m, 1H), 1,96-1,85 (m, 2H), 1,66-1,51 (m, 2H)
Ejemplo 15
Ácido 2-cian-4-(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)benzoico (Compuesto 15)
Etapa 1: se obtuvo 2-bromo-5-(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)benzonitrilo (630 mg, rendimiento: 89 %) de la misma manera que en el ejemplo 6 usando 2-bromo-5-yodobenzonitrilo.
ESI-MS m/z: 613 (M+H)+, RMN 1H (300 MHz, CDCb, 8): 8,66 (s, 1H), 7,81 (d, J = 8,4 Hz, 1H), 7,68 (d, J = 2,6 Hz, 1H), 7,47 (dd, J = 8,4, 2,6 Hz, 1H), 7,23 (s, 1H), 7,16-7,04 (m, 4H), 6,22 (d, J = 8,1 Hz, 1H), 4,60 (s, 2H), 4,21-4,16 (m, 2H), 4,02 (s, 3H), 3,98 (s, 3H), 3,46 (d, J = 7,0 Hz, 2H), 3,13-3,03 (m, 2H), 2,17-2,07 (m, 1H), 1,94-1,87 (m, 2H), 1,61-1,54 (m, 2H)
Etapa 2: el 2-bromo-5-(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)benzonitrilo (200 mg, 0,33 mmol) obtenido en la etapa 1, acetato de paladio (7,3 mg, 0,03 mmol), 1,3-bis(difenilfosfino)propano (DPPP) (130 mg, 0,03 mmol), carbonato potásico (90 mg, 0,65 mmol) y 1-propanol (3,0 ml) se agitaron en DMF (1,0 ml) en atmósfera de monóxido de carbono (presión atmosférica) a 80 °C durante 4 horas. A la mezcla de reacción, se le añadió una solución acuosa saturada de bicarbonato sódico y la mezcla resultante se trató con tierra de diatomeas y después, el filtrado se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturad y después se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de acetato de
etilo/heptano), con lo que se obtuvo 2-cian-4-(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)benzoato de propilo (175 mg, rendimiento: 86 %).
ESI-MS m/z: 621 (M+H)+, RMN 1H (300 MHz, CDCb, 8): 8,65 (s, 1H), 8,28-8,25 (m, 1H), 7,84-7,82 (m, 1H), 7,73-7,69 (m, 1H), 7,23 (s, 1H), 7,18-7,02 (m, 4H), 6,23-6,20 (m, 1H), 4,61 (s, 2H), 4,41 (t, J = 6,6 Hz, 2H), 4,22-4,15 (m, 2H), 4,02 (s, 3H), 3,98 (s, 3H), 3,49-3,44 (m, 2H), 3,13-3,03 (m, 2H), 2,18-2,08 (m, 1H), 1,94-1,83 (m, 2H), 1,64-1,53 (m, 2H), 1,28-1,24 (m, 2H), 1,07 (t, J = 7,5 Hz, 3H)
Etapa 3: El compuesto del título 15 (24 mg, rendimiento: 16 %) se obtuvo de la misma manera que en la etapa 2 del ejemplo 1 usando 2-cian-4-(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)benzoato de propilo obtenido en la etapa 2.
ESI-MS m/z: 579 (M+H)+, RMN 1H (300 MHz, DMSO-d6, 8): 8,59 (s, 1H), 8,30 (s, 1H), 8,20 (d, J = 8,1 Hz, 1H), 8,01 (d, J = 1,8 Hz, 1H), 7,77 (dd, J = 8,4, 2,2 Hz, 1H), 7,28-7,15 (m, 3H), 7,13-6,98 (m, 2H), 6,16 (d, J = 8,1 Hz, 1H), 4,62 (s, 2H), 4,41-4,30 (m, 2H), 3,94 (s, 3H), 3,89 (s, 3H), 3,40-3,19 (m, 4H), 2,16-2,13 (m, 1H), 1,86-1,82 (m, 2H), 1,43 1,39 (m, 2H)
Ejemplo 16
3- {[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-1-(tetrahidro-2H-piran-4-il)-3,4-dihidroquinazolin-2(1H)-ona (Compuesto 16)
Etapa 1: El 4-[(2-aminobencilamino)metil]-piperidin-1-carboxilato de ferc-butilo (1,0 g, 3,1 mmol) obtenido en la etapa 2 del ejemplo de referencia 1 se disolvió en metanol (50 ml) y, en un baño de hielo, se le añadió tetrahidro-4H-piran-4- ona (627 mg, 6,3 mmol) y borohidruro sódico (481 mg, 12 mmol) y la mezcla resultante se agitó durante 2 horas y después se agitó adicionalmente a temperatura ambiente durante 2 horas. La mezcla de reacción se diluyó con agua, y después se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturada y se secó sobre sulfato de magnesio anhidro. Un residuo obtenido mediante la evaporación del disolvente a presión reducida se purificó mediante cromatografía en columna sobre gel de sílice (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo 4-{[2-(tetrahidro-2H-piran-4-ilamino)bencilamida]metil}piperidin-1-carboxilato de ferc-butilo. Este compuesto se disolvió en dioxano (50 ml) y se le añadió 1,1'-carbonildiimidazol (0,4 g, 2,5 mmol) y la mezcla resultante se agitó a 100 °C durante 2 días. A la mezcla de reacción, se le añadió agua y la mezcla resultante se extrajo con acetato de etilo. Después, la capa orgánica se lavó con salmuera saturada y se secó sobre sulfato de magnesio anhidro. Después de evaporar el disolvente a presión reducida, el residuo resultante se purificó por HPLC preparativa de fase inversa (un disolvente mixto de acetonitrilo/agua), con lo que se obtuvo 4-{[2-oxo-1-(tetrahidro-2H-piran-4-il)-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo (50 mg, rendimiento: 4 %).
ESI-MS m/z: 430 (M+H)+
Etapa 2: El compuesto del título 16 (10 mg, rendimiento: 8 %) se obtuvo de la misma manera que en la etapa 3 del Ejemplo 5 usando 4-{[2-oxo-1-(tetrahidro-2H-piran-4-il)-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 1.
ESI-MS m/z: 518 (M+H)+, RMN 1H (300 MHz, CDCb, 8): 8,65 (s, 1H), 7,36-7,26 (m, 2H), 7,19-7,06 (m, 4H), 4,29 (s, 2H), 4,28-4,17 (m, 3H), 4,17-4,07 (m, 2H), 4,03 (s, 3H), 3,90 (s, 3H), 3,60-3,45 (m, 2H), 3,40 (d, J = 7,2 Hz, 2H), 3,16 3,05 (m, 2H), 2,86-2,72 (m, 2H), 2,17-1,96 (m, 1H), 1,93-1,72 (m, 4H), 1,65-1,42 (m, 2H)
* Ejemplo 17
4-(3-{[1-(6,7-dimetoxiquinazolin-4-il)azetidin-3-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (Compuesto 17)
El compuesto del título 17 (12 mg, rendimiento: 13 %) se obtuvo de la misma manera que en la etapa 3 del Ejemplo 5 después de realizar un tratamiento de acuerdo con el ejemplo de referencia 1, usando 2-nitrobenzaldehído y 3-(aminometil)azetidin-1-carboxilato de ferc-butilo.
ESI-MS m/z: 508 (M+H)+, RMN 1H (400 MHz, CDCla, 8): 8,81 (d, J = 4,8 Hz, 1H), 8,51 (s, 1H), 7,81-7,80 (m, 1H), 7,64 7,63 (m, 1H), 7,23-7,09 (m, 5H), 6,37 (d, J = 8,0 Hz, 1H), 4,68-4,61 (s, 4H), 4,36-4,32 (m, 2H), 4,01 (s, 3H), 3,94 (s, 3H), 3,84 (d, J = 8,0 Hz, 2H), 3,32-3,23 (m, 1H)
Ejemplo 18
4-(3-{[1-(6,7-dimetoxiquinazolin-4-il)-4-hidroxipiperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (Compuesto 18)
Etapa 1: el 4-(aminometil)-4-hidroxipiperidin-1-carboxilato de ferc-butilo (3,1 g, 13 mmol) obtenido mediante el método descrito en el documento W02005/000837 se disolvió en metanol (150 ml) y se le añadió 2-nitrobenzaldehído (2,0 g, 13 mmol) y cianoborohidruro sódico (1,3 g, 26 mmol) y la mezcla resultante se agitó a temperatura ambiente durante 12 horas. Después de que la mezcla de reacción se concentrase a presión reducida, se le añadió agua y la mezcla resultante se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturada y después se secó sobre sulfato de magnesio anhidro. Un residuo obtenido por evaporación del disolvente a presión reducida se purificó por
cromatografía en columna sobre gel de sílice (un disolvente mixto de hexano/acetato de etilo), con lo que se obtuvo 4-hidroxi-4-[(2-nitrobencilamino)metil]piperidin-1-carboxilato de ferc-butilo (2,2 g, rendimiento: 40 %).
RMN 1H (400 MHz, CDCls, 8): 7,97 (dd, J = 8,0, 0,8 Hz, 1H), 7,64-7,45 (m, 3H), 5,32 (s, 1H), 4,10 (s, 2H), 3,86 (s a, 2H), 3,18 (t, J = 12 Hz, 2H), 2,59 (s, 2H), 1,55-1,40 (m, 13H)
Etapa 2: El compuesto del título 18 (14 mg, rendimiento: 17 %) se obtuvo realizando los mismos tratamientos que en el ejemplo de referencia 1, etapa 3 del ejemplo 5 y el ejemplo 6 de manera secuencial, usando el 4-hidroxi-4-[(2-nitrobencilamino)metil]piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 1.
ESI-MS m/z: 552 (M+H)+, RMN 1H (400 MHz, DMSO-d6, 8): 8,89 (s, 1H), 8,53 (s, 1H), 8,22 (d, J = 7,6 Hz, 1H), 7,42 7,34 (m, 4H), 7,22 (s, 1H), 7,10 (s, 1H), 7,06 (d, J = 3,2 Hz, 1H), 6,80 (dd, J = 7,6, 3,2 Hz, 1H), 4,39 (s, 2H), 3,92 (s, 3H), 3,90 (s, 3H), 3,77-3,72 (m, 2H), 3,51-3,49 (m, 2H), 3,26 (s, 2H), 1,89 (s a, 4H)
Ejemplo 19
4-{3-[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-ilamino]-2-oxo-3,4-dihidroquinolin-1(2H)-il}picolinonitrilo (Compuesto 19)
Etapa 1: el clorhidrato de 3-amino-3,4-dihidroquinolin-2(1H)-ona (490 mg, 2,5 mmol) obtenido mediante el método descrito en el documento WO2004/98589, 4-oxopiperidin-1-carboxilato de ferc-butilo (590 mg, 3,0 mmol), triacetoxi borohidruro de sodio (1,6 g, 7,4 mmol), trietilamina (300 mg, 3,0 mmol) y ácido acético (0,1 ml) se agitaron durante una noche en 1,2-dicloroetano (20 ml) a temperatura ambiente. A la mezcla de reacción, se le añadió una solución acuosa saturada de bicarbonato sódico y la capa orgánica se trató con tierra de diatomeas. Después de evaporar el disolvente a presión reducida, el residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo 4-(2-oxo-1,2,3,4-tetrahidroquinolin-3-ilamino)piperidin-1-carboxilato de ferc-butilo (626 mg, rendimiento: 74 %). ESI-MS m/z: 346 (M+H)+
Etapa 2: el 4-(2-oxo-1,2,3,4-tetrahidroquinolin-3-ilamino)piperidin-1-carboxilato de ferc-butilo (0,64 g, 1,9 mmol) obtenido en la etapa 1 y diisopropiletilamina (599 mg, 4,6 mmol) se disolvieron en tetrahidrofurano (7,0 ml), y se le añadió anhídrido trifluoroacético (778 mg, 3,7 mmol) a 0 °C. Después de agitar la mezcla resultante a temperatura ambiente durante 30 minutos, se le añadió una solución acuosa saturada de bicarbonato sódico a la mezcla de reacción y la mezcla resultante se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturada y después se secó sobre sulfato de magnesio anhidro y el disolvente se concentró a presión reducida. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo 4-[2,2,2-trifluoro-N-(2-oxo-1,2,3,4-tetrahidroquinolin-3-il)acetamida]piperidin-1-carboxilato de ferc-butilo (644 mg, rendimiento: 79 %).
ESI-MS m/z: 442 (M+H)+
Etapa 3: se obtuvo N-[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]-2,2,2-trifluoro-N-(2-oxo-1,2,3,4-tetrahidroquinolin-3-il)acetamida (226 mg, rendimiento: 98 %) de la misma manera que en la etapa 3 del ejemplo 5 usando el 4-[2,2,2-trifluoro-N-(2-oxo-1,2,3,4-tetrahidroquinolin-3-il)acetamida]piperidin-1-carboxilato de ferc-butilo (192 mg, 0,43 mmol) obtenido en la etapa 2.
ESI-MS m/z: 530 (M+H)+
Etapa 4: la N-[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]-2,2,2-trifluoro-N-(2-oxo-1,2,3,4-tetrahidroquinolin-3-il)acetamida (226 mg, 0,427 mmol) obtenida en la etapa 3 e hidróxido de litio monohidrato (36 mg, 0,85 mmol) se agitaron durante una noche en un disolvente mixto de metanol/agua (1/1, 4,0 ml) a 60 °C. Después de concentrar la mezcla de reacción a presión reducida, se le añadió agua y la mezcla resultante se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturada y después se secó sobre sulfato de magnesio anhidro y el disolvente se evaporó a presión reducida. El residuo resultante se purificó por cromatografía preparativa de capa fina (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo 3-[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-ilamino]-3,4-dihidroquinolin-2(1H)-ona (132 mg, rendimiento: 71 %).
ESI-MS m/z: 434 (M+H)+
Etapa 5: El compuesto del título 19 (12 mg, rendimiento: 19 %) se obtuvo de la misma manera que en el ejemplo 6 usando la 3-[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-ilamino]-3,4-dihidroquinolin-2(1H)-ona obtenida en la etapa 4 y 4-yodopicolinonitrilo.
ESI-MS m/z: 536 (M+H)+, RMN 1H (400 MHz, CDCla, 8): 8,83 (d, J = 6,0 Hz, 1H), 8,67 (s, 1H), 7,70-7,69 (m, 1H), 7,53 7,51 (m, 1H), 7,34-7,32 (m, 1H), 7,27 (s, 1H), 7,21-7,14 (m, 2H), 7,10 (s, 1H), 6,50 (d, J = 11 Hz, 1H), 4,17-4,11 (m, 2H), 4,03 (s, 3H), 3,99 (s, 3H), 3,77 (dd, J = 13, 6,0 Hz, 1H), 3,22-3,01 (m, 5H), 1,73-1,67 (m, 2H), 1,28-1,24 (m, 2H)
Ejemplo 20
3-(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2,4-dioxo-3,4-dihidroquinazolin-1(2H)-il)benzonitrilo (Compuesto 20)
Etapa 1: Se disolvió anhídrido isatoico (5,0 g, 31 mmol) en 1,4-dioxano (40 ml), y se le añadió 4-(aminometil)piperidin1-carboxilato de ferc-butilo (6,6 g, 31 mmol) y diisopropiletilamina (8,3 g, 64 mmol) y la mezcla resultante se agitó a 80 °C durante 2 horas. Después de que la mezcla de reacción se concentrase a presión reducida, se le añadió agua y la mezcla resultante se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturada y después se secó sobre sulfato de magnesio anhidro y el disolvente se evaporó a presión reducida, con lo que se obtuvo 4-[(2-aminobenzamida)metil]piperidin-1-carboxilato de ferc-butilo (10,2 g, rendimiento cuantitativo).
RMN 1H (300 MHz, CDCb, 8): 7,32-7,29 (m, 1H), 7,23-7,17 (m, 1H), 6,69-6,60 (m, 2H), 6,27 (s a, 1H), 5,50 (s a, 2H), 4,15-4,08 (m, 2H), 3,32-3,27 (m, 2H), 2,73-2,64 (m, 2H), 1,76-1,70 (m, 3H), 1,20-1,10 (m, 2H)
Etapa 2: El 4-[(2-aminobenzamida)metil]piperidin-1-carboxilato de ferc-butilo (500 mg, 1,5 mmol) obtenido en la etapa 1 se disolvió en 1,2-dicloroetano (4,0 ml) y se le añadió diisopropiletilamina (388 mg, 3,0 mmol) y DMAP (9,2 mg, 0,08 mmol). Después de enfriar la mezcla resultante a 0 °C, se le añadió cloroformiato de etilo (195 mg, 1,8 mmol) y después, la mezcla resultante se agitó a temperatura ambiente durante 1 hora. A la mezcla de reacción, se le añadió una solución acuosa saturada de bicarbonato sódico y la mezcla resultante se extrajo con cloroformo. La capa orgánica se lavó con salmuera saturada y se secó sobre sulfato de magnesio anhidro y, después, el disolvente se evaporó a presión reducida. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de hexano/acetato de etilo), con lo que se obtuvo 4-{[2-(etoxicarbonilamino)benzamida]metil}piperidin-1-carboxilato de ferc-butilo (577 mg, rendimiento: 95 %).
RMN 1H (300 MHz, CDCla, 8): 10,4 (s a, 1H), 8,36 (dd, J = 8,4, 0,9 Hz, 1H), 7,49-7,40 (m, 2H), 7,04-6,98 (m, 1H), 6,36 6,34 (m, 1H), 4,21 (c, J = 7,2 Hz, 2H), 4,17-4,09 (m, 2H), 3,33 (s a, 2H), 2,74-2,66 (m, 2H), 1,75-1,71 (m, 3H), 1,46 (s, 9H), 1,31 (t, J = 7,2 Hz, 3H), 1,21-1,11 (m, 2H)
Etapa 3: el 4-{[2-(etoxicarbonilamino)benzamida]metil}piperidin-1-carboxilato de ferc-butilo (0,47 g, 1,2 mmol) obtenido en la etapa 2 e hidróxido potásico (390 mg, 7,0 mmol) se calentaron a reflujo en etanol (12 ml) durante 30 minutos. A la mezcla de reacción, se le añadió agua y la mezcla resultante se enfrió a 0 °C y después se neutralizó con ácido clorhídrico diluido. Después, el sólido resultante se recogió por filtración, con lo que se obtuvo 4-[(2,4-dioxo-1,2-dihidroquinazolin-3(4H)-il)metil]piperidin-1-carboxilato de ferc-butilo (343 mg, rendimiento: 82 %).
RMN 1H (400 MHz, CDCla, 8): 9,00 (s a, 1H), 8,13 (d, J = 7,6 Hz, 1H), 7,65-7,60 (m, 1H), 7,25-7,23 (m, 1H), 7,06-7,04 (m, 1H), 4,13-4,10 (m, 2H), 4,00 (d, J = 6,8 Hz, 2H), 2,70-2,64 (m, 2H), 2,04 (s a, 1H), 1,67-1,62 (m, 2H), 1,45 (s, 9H), 1,39-1,27 (m, 2H)
Etapa 4: se obtuvo 3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}quinazolin-2,4(1H,3H)-diona (414 mg, rendimiento: 98 %) de la misma manera que en la etapa 3 del ejemplo 5 usando el 4-[(2,4-dioxo-1,2-dihidroquinazolin-3(4H)-il)metil]piperidin-1 -carboxilato de ferc-butilo obtenido en la etapa 3.
RMN 1H (400 MHz, CDCb, 8): 9,57 (s a, 1H), 8,66 (s, 1H), 8,15 (d, J = 7,6 Hz, 1H), 7,65-7,61 (m, 1H), 7,28-7,24 (m, 2H), 7,10-7,09 (m, 2H), 4,20-4,11 (m, 4H), 4,02 (s, 3H), 3,99 (s, 3H), 3,07-3,01 (m, 2H), 2,24 (s a, 1H), 1,91-1,88 (m, 2H), 1,74-1,65 (m, 2H)
Etapa 5: la 3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}quinazolin-2,4(1H,3H)-diona (30 mg, 0,07 mmol) obtenida en la etapa 4, ácido 3-cianofenilborónico (39 mg, 0,27 mmol), acetato de cobre (II) (49 mg, 0,27 mmol) y trietilamina (27 mg, 0,27 mmol) se mezclaron y se agitaron en diclorometano (1,5 ml) a temperatura ambiente durante 5 horas y posteriormente se agitaron durante una noche a 35 °C. La mezcla de reacción se trató con tierra de diatomeas y el disolvente se evaporó a presión reducida. Después, el residuo resultante se purificó por cromatografía preparativa de capa fina (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo el compuesto del título 20 (18 mg, rendimiento: 48 %).
ESI-MS m/z: 549 (M+H)+, RMN 1H (300 MHz, CDCb, 8): 8,64 (s, 1H), 8,29 (dd, J = 7,5, 1,5 Hz, 1H), 7,88-7,84 (m, 1H), 7,78-7,73 (m, 1H), 7,70-7,69 (m, 1H), 7,65-7,62 (m, 1H), 7,56-7,50 (m, 1H), 7,34-7,27 (m, 2H), 7,18-7,10 (m, 1H), 6,50 (d, J = 8,1 Hz, 1H), 4,23-4,13 (m, 4H), 4,00 (s, 3H), 3,99 (s, 3H), 3,10-3,02 (m, 2H), 2,24 (s a, 1H), 1,92-1,88 (m, 2H), 1,75-1,62 (m, 2H)
Ejemplo 21
4-(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidropirido[3,4-d]pirimidin-1(2H)-il)picolinonitrilo (Compuesto 21)
El compuesto del título 21 (62 mg, rendimiento: 60 %) se obtuvo realizando los mismos tratamientos que en el ejemplo de referencia 1, etapa 3 del ejemplo 5 y el ejemplo 6 secuencialmente, usando 3-aminoisonicotinaldehído.
ESI-MS m/z: 537 (M+H)+, RMN 1H (300 MHz, CDCb, 8): 8,85 (d, J = 5,4 Hz, 1H), 8,66 (s, 1H), 8,36 (d, J = 4,8 Hz, 1H), 7,85-7,84 (m, 1H), 7,76 (s, 1H), 7,65-7,62 (m, 1H), 7,24 (s, 1H), 7,14 (d, J = 4,8 Hz, 1H), 7,08 (s, 1H), 4,63 (s, 2H), 4,22-4,18 (m, 2H), 4,02 (s, 3H), 3,98 (s, 3H), 3,48 (d, J = 7,5 Hz, 2H), 3,13-3,05 (m, 2H), 2,16-2,08 (m, 1H), 1,90-1,86 (m, 2H), 1,65-1,53 (m, 2H)
Ejemplo 22
4-(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidropirido[2,3-d]pirimidin-1(2H)-il)picolinonitrilo (Compuesto 22)
Etapa 1: el (3-formilpiridin-2-il)carbamato de etilo (150 mg, 0,77 mmol) obtenido mediante el método descrito en el documento US2007/259850 y 4-(aminometil)piperidin-1-carboxilato de ferc-butilo (182 mg, 0,85 mmol) se agitaron en metanol (2,0 ml) a 60 °C durante 2 horas. Después de que la mezcla de reacción se enfriase a temperatura ambiente, se le añadió borohidruro de sodio (35 mg, 0,93 mmol) se añadió y la mezcla resultante se agitó durante 40 minutos. Se le añadió tolueno (3,0 ml) y ácido acético (0,5 ml) y la mezcla resultante se agitó a 110 °C durante 3 horas. A la mezcla de reacción, se le añadió agua y la mezcla resultante se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturada y se secó sobre sulfato de magnesio anhidro y, después, el disolvente se evaporó a presión reducida. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de hexano/acetato de etilo), con lo que se obtuvo 4-[(2-oxo-1,2-dihidropirido[2,3-d]pirimidin-3(4H)-il)metil]piper^idin-1-carboxilato de ferc-butilo (246 mg, rendimiento: 92 %). RMN 1H (400 MHz, DMSO-d6, 8): 9,58 (s, 1H), 8,07 (d, J = 4,0 Hz, 1H), 7,49 (d, J = 8,0 Hz, 1H), 6,92-6,89 (m, 1H), 4,44 (s, 2H), 3,93-3,90 (m, 2H), 3,20 (d, J = 7,6 Hz, 2H), 2,67 (s a, 2H), 1,89 (s a, 1H), 1,58-1,56 (m, 2H), 1,39-1,37 (m, 11H)
Etapa 2: El compuesto del título 22 (45 mg, rendimiento: 61 %) se obtuvo realizando los mismos tratamientos que en la etapa 3 del ejemplo 5 y el ejemplo 6 secuencialmente, usando el 4-[(2-oxo-1,2-dihidropirido[2,3-d]pirimidin-3(4H)-il)metil]piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 1.
ESI-MS m/z: 537 (M+H)+, RMN 1H (400 MHz, CDCla, 8): 8,79 (d, J = 6,0 Hz, 1H), 8,66 (s, 1H), 8,18-8,11 (m, 1H), 7,76 (d, J = 2,0 Hz, 1H), 7,57 (dd, J = 6,0, 2,0 Hz, 1H), 7,51-7,49 (m, 1H), 7,24-7,23 (m, 1H), 7,08-7,00 (m, 2H), 4,62 (s, 2H), 4,22- 4,19 (m, 2H), 4,02 (s, 3H), 3,95 (s, 3H), 3,49 (d, J = 7,6 Hz, 2H), 3,12-3,07 (m, 2H), 2,14 (s a, 1H), 1,92-1,89 (m, 2H), 1,64-1,55 (m, 2H)
Ejemplo 23
4-(6-fluoro-2-oxo-3-{[1-(pirido[3,4-d]pirimidin-4-il)piperidin-4-il]metil}-3,4-dihidropirido[2,3-d]pirimidin-1(2H)-il)picolinonitrilo (Compuesto 23)
Etapa 1: se obtuvo 4-[(6-fluoro-2-oxo-1,2-dihidropirido[2,3-d]pirimidin-3(4H)-il)metil]piperidin-1-carboxilato de fercbutilo (876 mg, rendimiento: 63 %) de la misma manera que en el ejemplo de referencia 1 usando 2-amino-5-fluoronicotinaldehído.
ESI-MS m/z: 365 (M+H)+
Etapa 2: se obtuvo 4-{[1-(2-cianopiridin-4-il)-6-fluoro-2-oxo-1,2-dihidropirido[2,3-d]pirimidin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo (588 mg, rendimiento cuantitativo) de la misma manera que en el ejemplo 6 usando el 4-[(6-fluoro-2-oxo-1,2-dihidropirido[2,3-d]pirimidin-3(4H)-il)metil]piperidin-1 -carboxilato de ferc-butilo obtenido en la etapa 1 y 4-yodopicolinonitrilo.
ESI-MS m/z: 467 (M+H)+
Etapa 3: el compuesto del título (34 mg, rendimiento: 17 %) se obtuvo de la misma manera que en la etapa 3 del ejemplo 5 usando 4-{[1-(2-cianopiridin-4-il)-6-fluoro-2-oxo-1,2-dihidropirido[2,3-d]pirimidin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 2 y la 4-cloropirido[3,4-d]pirimidina obtenida mediante el método descrito en el documento US2006/199804.
ESI-MS m/z: 496 (M+H)+, RMN 1H (300 MHz, CDCla, 8): 9,31 (s, 1H), 8,80-8,77 (m, 2H), 8,56 (d, J = 5,9 Hz, 1H), 7,98 (d, J = 2,7 Hz, 1H), 7,73 (t, J = 1,1 Hz, 1H), 7,61 (dd, J = 5,9, 0,9 Hz, 1H), 7,55 (dd, J = 5,4, 2,3 Hz, 1H), 7,29 (dd, J = 7,5, 2,9 Hz, 1H), 4,63 (s, 2H), 4,50 (d, J = 14 Hz, 2H), 3,48 (d, J = 7,2 Hz, 2H), 3,25-3,18 (m, 2H), 2,25-2,17 (m, 1H), 1,94 (t, J = 6,6 Hz, 2H), 1,57 (m, 2H)
Ejemplo 24
4-(3-{[1-(6,7-dimetoxiquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidropteridin-1(2H)-il)picolinonitrilo (Compuesto 24)
El compuesto del título 24 (60 mg, rendimiento: 84 %) se obtuvo realizando los mismos tratamientos que en el ejemplo de referencia 1, etapa 3 del ejemplo 5 y ejemplo 6 secuencialmente, usando 3-aminopirazin-2-carboxaldehído. ESI-MS m/z: 538 (M+H)+, RMN 1H (300 MHz, CDCb, 8): 8,82 (d, J = 5,1 Hz, 1H), 8,66 (s, 1H), 8,24 (d, J = 2,4 Hz, 1H), 8,07 (d, J = 2,4 Hz, 1H), 7,73 (d, J = 2,1 Hz, 1H), 7,55 (dd, J = 5,1,2,1 Hz, 1H), 7,25 (s, 1H), 7,08 (s, 1H), 4,79 (s, 2H), 4,23- 4,18 (m, 2H), 4,02 (s, 3H), 3,99 (s, 3H), 3,54 (d, J = 7,2 Hz, 2H), 3,13-3,05 (m, 2H), 2,16 (s a, 1H), 1,94-1,90 (m, 2H), 1,68-1,56 (m, 2H)
Ejemplo 25
Ácido 4-(4-{[1-(3-cianofenil)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)quinazolin-7-carboxílico (Compuesto 25)
Etapa 1: se obtuvo 3-{[1-(7-bromoquinazolin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-2(1H)-ona (9,0 g, rendimiento: 54 %) de la misma manera que en la etapa 3 del ejemplo 5 usando el compuesto R1 obtenido en el ejemplo de referencia 1 y 7-bromo-4-cloroquinazolina.
ESI-MS m/z: 452 (M+H)+
Etapa 2: se obtuvo 3-(3-{[1-(7-bromoquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)benzonitrilo (0,6 g, rendimiento: 60 %) de la misma manera que en el ejemplo 6 usando 3-{[1-(7-bromoquinazolin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-2(1H)-ona obtenida en la etapa 1 y 3-yodobenzonitrilo.
ESI-MS m/z: 553 (M+H)+
Etapa 3: se obtuvo 4-(4-{[1-(3-cianofenil)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)quinazolin-7-carboxilato de propilo (1,6 g, rendimiento: 50 %) de la misma manera que en la etapa 2 del Ejemplo 15 usando el 3-(3-{[1-(7-bromoquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)benzonitrilo obtenido en la etapa 2. ESI-MS m/z: 561 (M+H)+
Etapa 4: El compuesto del título 25 (1,0 g, rendimiento: 68 %) se obtuvo de la misma manera que en la etapa 2 del ejemplo 1 usando el 4-(4-{[1-(3-cianofenil)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)quinazolin-7-carboxilato de propilo obtenido en la etapa 3.
ESI-MS m/z: 519 (M+H)+, RMN 1H (400 MHz, CDCb, 8): 9,03 (s, 1H), 8,91 (s, 1H), 8,23 (d, J = 6,0 Hz, 1H), 7,87 (d, J = 6,6 Hz, 1H), 7,80-7,62 (m, 4H), 7,21-7,09 (m, 2H), 7,09-6,99 (m, 1H), 6,21 (d, J = 6,0 Hz, 1H), 4,65 (s, 2H), 4,63 (d, J = 10 Hz, 2H), 3,49 (d, J = 1,6 Hz, 2H), 3,37-3,27 (m, 2H), 2,39-2,13 (m, 1H), 2,08-1,91 (m, 2H), 1,81-1,61 (m, 2H)
Ejemplo 26
4-(4-{[1-(3-cianofenil)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)-N-metilquinazolin-7-carboxamido (Compuesto 26)
El compuesto del título 26 (36 mg, rendimiento: 23 %) se obtuvo de la misma manera que en la etapa 3 del ejemplo 1 usando el compuesto 25 obtenido en el ejemplo 25 y metilamina.
ESI-MS m/z: 532 (M+H)+, RMN 1H (300 MHz, CDCla, 8): 8,73 (s, 1H), 8,15 (s, 1H), 7,93 (s, 2H), 7,79-7,62 (m, 4H), 7,17-7,01 (m, 3H), 6,41 (s a, 1H), 6,19 (d, J = 4,5 Hz, 1H), 4,63 (s, 2H), 4,47 (d, J = 13 Hz, 2H), 3,48 (d, J = 7,2 Hz, 2H), 3,26-3,11 (m, 2H), 3,09 (d, J = 4,8 Hz, 3H), 2,25-2,07 (m, 1H), 2,04-1,85 (m, 2H), 1,68-1,41 (m, 2H)
Ejemplo 27
4-[3-({1-[7-(4-metilpiperazin-1-carbonil)quinazolin-4-il]piperidin-4-il}metil)-2-oxo-3,4-dihidroquinazolin-1(2H)-il]picolinonitrilo (Compuesto 27)
Etapa 1: se obtuvo 4-{[1-(2-cianopiridin-4-il)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-carboxilato de terc-butilo (578 mg, rendimiento: 74 %) de la misma manera que en el ejemplo 6 usando el 4-(2-oxo-1,4-dihidro-2H-quinazolin-3-ilmetil)piperidin-1-carboxilato de terc-butilo obtenido en la etapa 3 del ejemplo de referencia 1 y 4-yodopicolinonitrilo. e S|-MS m/z: 448 (M+H)+
Etapa 2: se obtuvo diclorhidrato de 4-[2-oxo-3-(piperidin-4-ilmetil)-3,4-dihidroquinazolin-1(2H)-il]picolinonitrilo (94 mg, rendimiento cuantitativo) de la misma manera que en la etapa 4 del ejemplo de referencia 1 usando el 4-{[1-(2-cianopiridin-4-il)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1 -carboxilato de terc-butilo obtenido en la etapa 1. ESI-MS m/z: 348 (M+H)+
Etapa 3: se obtuvo 4-(4-{[1-(2-cianopiridin-4-il)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)quinazolin-7-carboxilato de terc-butilo (22 mg, rendimiento: 6,4 %) de la misma manera que en el ejemplo 3 usando el clorhidrato de 4-[2-oxo-3-(piperidin-4-ilmetil)-3,4-dihidroquinazolin-1 (2H)-il]picolinonitrilo obtenido en la etapa 2 y el compuesto R5 obtenido en el ejemplo de referencia 5.
ESI-MS m/z: 576 (M+H)+, RMN 1H (300 MHz, CDCb, 8): 8,80 (d, J = 5,1 Hz, 1H), 8,75 (s, 1H), 8,49 (d, J = 1,8 Hz, 1H), 7,98 (dd, J = 8,6, 1,6 Hz, 1H), 7,87 (d, J = 8,8 Hz, 1H), 7,84 (d, J = 1,8 Hz, 1H), 7,66 (dd, J = 5,3, 2,0 Hz, 1H), 7,14 (dc, J = 5,3, 18 Hz, 3H), 6,38 (d, J = 8,4 Hz, 1H), 4,59 (s, 2H), 4,39 (d, J = 13 Hz, 2H), 3,47 (d, J = 7,3 Hz, 2H), 3,15 (t, J = 12 Hz, 2H), 2,16 (dt, J = 4,6, 14 Hz, 1H), 1,91 (d, J = 11 Hz, 2H), 1,68-1,51 (m, 11H)
Etapa 4: Después de agitar el 4-(4-{[1-(2-cianopiridin-4-il)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)quinazolin-7-carboxilato de terc-butilo (22 mg, 0,038 mmol) obtenido en la etapa 3 en un disolvente mixto de ácido trifluoroacético (0,18 ml)/diclorometano (0,20 ml) a temperatura ambiente durante 1,5 horas, el disolvente se evaporó a presión reducida. Usando el residuo resultante y 1-metilpiperazina, se obtuvo el compuesto del título 27 (7,4 mg, rendimiento: 32 %) de la misma manera que en la etapa 3 del ejemplo 1.
ESI-MS m/z: 602 (M+H)+, RMN 1H (300 MHz, CDCb, 8): 8,81 (d, J = 5,0 Hz, 1H), 8,73 (s, 1H), 7,90 (d, J = 8,6 Hz, 1H), 7,84 (d, J = 1,4 Hz, 2H), 7,65 (dd, J = 5,2, 2,0 Hz, 1H), 7,47 (dd, J = 8,6, 1,4 Hz, 1H), 7,20-7,09 (m, 3H), 6,38 (d, J = 8,2 Hz, 1H), 4,60 (s, 2H), 4,39 (d, J = 13 Hz, 2H), 3,86 (s a, 2H), 3,92-3,76 (m, 4H), 3,15 (t, J = 12 Hz, 2H), 2,54 (s a, 2H), 2,43-2,30 (m, 5H), 2,21-2,11 (m, 1H), 1,91 (d, J = 11,3 Hz, 2H), 1,58 (dd, J = 21, 12 Hz, 2H)
Ejemplo 28
4-(4-{[1-(2-cianopiridin-4-il)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)quinazolin-7-carboxamido (Compuesto 28)
El compuesto del título 28 (19 mg, rendimiento: 43 %) se obtuvo de la misma manera que en la etapa 4 del ejemplo 27 usando el 4-(4-{[1-(2-cianopiridin-4-il)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)quinazolin-7-carboxilato de ferc-butilo obtenido en la etapa 3 del ejemplo 27 y cloruro de amonio.
ESI-MS m/z: 519 (M+H)+, RMN 1H (400 MHz, DMSO-d6, 8): 8,88 (d, J = 5,6 Hz, 1H), 8,64 (s, 1H), 8,29 (d, J = 1,6 Hz, 2H), 8,20 (d, J = 1,6 Hz, 1H), 8,02-8,00 (m, 1H), 7,94-7,91 (m, 1H), 7,82 (dd, J = 5,2, 2,0 Hz, 1H), 7,64 (s, 1H), 7,30 (d, J = 7,6 Hz, 1H), 7,15-7,06 (m, 2H), 6,36 (d, J = 7,2 Hz, 1H), 4,64 (s, 2H), 4,35-4,32 (m, 2H), 3,39-3,33 (m, 2H), 3,21 3,15 (m, 2H), 2,16 (s a, 1H), 1,83-1,80 (m, 2H), 1,48-1,43 (m, 2H)
Ejemplo 29
4-(4-{[1-(3-cianofenil)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)pirido[3,4-d]pirimidin-7-óxido (Compuesto 29)
Etapa 1: se obtuvo 3-{[1-(pirido[3,4-d]pirimidin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-2(1H)-ona (83 mg, rendimiento: 48 %) de la misma manera que en el ejemplo 3 usando el compuesto R1 y 4-hidroxipirido[3,4-d]pirimidina. ESI-MS m/z: 375 (M+H)+
Etapa 2: la 3-{-[1-(pirido[3,4-d]pirimidin-4-il)piperidin-4-il]metil}3,4-dihidroquinazolin-2(1H)-ona (598 mg, 1,6 mmol) obtenida en la etapa 1 se disolvió en diclorometano (10 ml) y se le añadió ácido meta-cloroperoxibenzoico (aproximadamente 70 % en peso, 394 mg) y la mezcla resultante se agitó a 0 °C durante 1 hora. A la mezcla de reacción, se le añadió una solución acuosa saturada de bicarbonato sódico y la mezcla resultante se extrajo con cloroformo. La capa orgánica se lavó con salmuera saturad y después se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. El residuo resultante se purificó por HPLC preparativa de fase inversa (un disolvente mixto de acetonitrilo/agua), con lo que se obtuvo 4-{4-[(2-oxo-1,2-dihidroquinazolin-3(4H)-il)metil]piperidin-1-il}pirido[3,4-d]pirimidin-7-óxido (133 mg, rendimiento: 21 %).
ESI-MS m/z: 391 (M+H)+
Etapa 3: El compuesto del título 29 (32 mg, rendimiento: 21 %) se obtuvo de la misma manera que en el ejemplo 6 usando 4-{4-[(2-oxo-1,2-dihidroquinazolin-3(4H)-il)metil]piperidin-1-il}pirido[3,4-d]pirimidin-7-óxido obtenido en la etapa 2. ESI-MS m/z: 492 (M+H)+, RMN 1H (300 MHz, CDCla, 8): 8,70 (d, J = 1,8 Hz, 1H), 8,66 (s, 1H), 8,08-8,05 (m, 1H), 7,66-7,59 (m, 5H), 7,15-7,00 (m, 3H), 6,18 (d, J = 8,1 Hz, 1H), 4,61 (s, 2H), 4,43-4,39 (m, 2H), 3,46 (d, J = 7,5 Hz, 2H), 3,26-3,19 (m, 2H), 2,25-2,18 (m, 1H), 1,97-1,94 (m, 2H), 1,64-1,47 (m, 2H)
Ejemplo 30
4-(3-{[1-(Imidazo[1,2-a]pirazin-8-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (Compuesto 30)
El compuesto del título 30 (12 mg, rendimiento: 19 %) se obtuvo de la misma manera que en el ejemplo 5 usando el clorhidrato de 4-[2-oxo-3-(piperidin-4-ilmetil)-3,4-dihidroquinazolin-1(2H)-il]picolinonitrilo obtenido en la etapa 2 del ejemplo 27 y 8-cloroimidazo[1,2-a]pirazina.
ESI-MS m/z: 465 (M+H)+, RMN 1H (300 MHz, CDCla, 8): 8,80-8,78 (m, 1H), 7,84-7,83 (m, 1H), 7,67-7,65 (m, 1H), 7,53 (d, J = 1,5 Hz, 1H), 7,48-7,47 (m, 2H), 7,33 (d, J = 4,8 Hz, 1H), 7,19-7,07 (m, 3H), 6,40-6,37 (m, 1H), 5,48-5,43 (m, 2H), 4,56 (s, 2H), 3,39 (d, J = 6,9 Hz, 2H), 3,10-3,00 (m, 2H), 2,17-2,10 (m, 1H), 1,88-1,85 (m, 2H), 1,52-1,38 (m, 2H)
Ejemplo 31
4-(3-{[1-(7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (Compuesto 31)
Etapa 1: se obtuvo 4-(2-oxo-3-{[1-(7-{[2-(trimetilsilil)etoxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (63 mg, rendimiento: 67 %) de la misma manera que en el ejemplo 5 usando diclorhidrato de 4-[2-oxo-3-(piperidin-4-ilmetil)-3,4-dihidroquinazolin-1(2H)-il]picolinonitrilo obtenido en la etapa 2 del ejemplo 27 y la 4-cloro-7-{[2-(trimetilsilil)etoxi]metil}-7H-pirrolo[2,3-d]pirimidina obtenida mediante el método descrito en Organic Letters 2009, 11, 1999.
RMN 1H (300 MHz, CDCb, 8): 8,86-8,84 (m, 1H), 8,40 (s, 1H), 7,89 (dd, J= 2,4, 0,9 Hz, 1H), 7,71 (dd, J = 5,4, 2,4 Hz, 1H), 7,25-7,13 (m, 4H), 6,57 (d, J = 3,6 Hz, 1H), 6,45-6,42 (m, 1H), 5,63 (s, 2H), 4,86-4,82 (m, 2H), 4,62 (s, 2H), 3,61 3,55 (m, 2H), 3,45 (d, J = 6,9 Hz, 2H), 3,20-3,11 (m, 2H), 2,30-2,17 (m, 1H), 1,94-1,91 (m, 2H), 1,54-1,41 (m, 2H), 0,99-0,91 (m, 2H), -0,05 (s, 9H)
Etapa 2: El compuesto del título 31 (25 mg, rendimiento: 64 %) se obtuvo de la misma manera que en la etapa 2 del ejemplo 12 usando el 4-(2-oxo-3-{[1-(7-{[2-(trimetilsilil)etoxi]metil}-7H-pirrolo[2,3-d]pirimidin-4-il)piperidin-4-il]metil}-3,4- dihidroquinazolin-1 (2H)-il)picolinonitrilo obtenido en la etapa 1.
ESI-MS m/z: 465 (M+H)+, RMN 1H (300 MHz, CDCla, 5): 10,0 (s a, 1H), 8,80 (d, J = 5,7 Hz, 1H), 8,32 (s, 1H), 7,84 7,83 (m, 1H), 7,67-7,64 (m, 1H), 7,19-7,05 (m, 4H), 6,50 (d, J = 3,3 Hz, 1H), 6,40-6,37 (m, 1H), 4,84-4,79 (m, 2H), 4,57 (s, 2H), 3,40 (d, J = 6,9 Hz, 2H), 3,16-3,08 (m, 2H), 2,25-2,12 (m, 1H), 1,90-1,86 (m, 2H), 1,49-1,36 (m, 2H)
Ejemplo 32
4-(3-{[1-(5-metoxipirimidin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (Compuesto 32)
Etapa 1: se obtuvo 3-{[1-(6-cloro-5-metoxipirimidin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-2(1H)-ona (504 mg, rendimiento: 73 %) de la misma manera que en el ejemplo 5 usando clorhidrato de 4-[2-oxo-3-(piperidin-4-ilmetil)-3,4-dihidro-quinazolin-1(2H)-il]picolinonitrilo obtenido en la etapa 2 del ejemplo 27 y 4,6-didoro-5-metoxipirimidina. ESI-MS m/z: 388 (M+H)+
Etapa 2: la 3-{[1-(6-cloro-5-metoxipirimidin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-2(1H)-ona (100 mg, 0,26 mmol) obtenida en la etapa 1, paladio-carbono (10 % en peso, 27 mg) y trietilamina (52 mg, 0,52 mmol) se mezclaron y se agitaron en acetato de etilo (3,0 ml) en atmósfera de hidrógeno (presión atmosférica) a temperatura ambiente durante 3 horas. La mezcla de reacción se trató con tierra de diatomeas y el disolvente se evaporó a presión reducida. Al residuo resultante, se le añadió éter dietílico y el sólido resultante se recogió por filtración, con lo que se obtuvo 3-{-[1-(5-metoxipirimidin-4-il)piperidin-4-il]metil}-3,4dihidroquinazolin-2(1H)-ona (82 mg, rendimiento: 90 %). ESI-MS m/z: 354 (M+H)+
Etapa 3: El compuesto del título 32 (72 mg, rendimiento: 86 %) se obtuvo de la misma manera que en el ejemplo 6 usando la 3-{-[1-(5-metoxipirimidin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-2(1H)-ona obtenida en la etapa 2 y 4-yodopicolinonitrilo.
ESI-MS m/z: 456 (M+H)+, RMN 1H (400 MHz, CDCla, 5): 8,79 (d, J = 5,6 Hz, 1H), 8,32 (s, 1H), 7,88-7,83 (m, 2H), 7,66 7,64 (m, 1H), 7,19-7,08 (m, 3H), 6,38 (d, J = 7,6 Hz, 1H), 4,59-4,56 (m, 4H), 3,85 (s, 3H), 3,39 (d, J = 6,8 Hz, 2H), 2,91 2,85 (m, 2H), 2,09-2,04 (m, 1H), 1,81-1,78 (m, 2H), 1,45-1,31 (m, 2H)
Ejemplo 33
4-(3-{[1-(6-aminopirimido[3,4-d]pirimidin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (Compuesto 33)
Etapa 1: El compuesto R1 (841 mg, 3,0 mmol), 4,6-dicloropirido[3,4-d]pirimidina (716 mg, 3,6 mmol) obtenida mediante el método descrito en Bioorganic & Medicinal Chemistry Letters, 2001, 11, 1401 y diisopropiletilamina (2,3 g, 18 mmol) se agitaron en 2-propanol (8,0 ml) a 100 °C durante 6 horas. A la mezcla de reacción, se le añadió una solución acuosa saturada de bicarbonato sódico y la mezcla resultante se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturad y después se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo 3-[1-(6-cloropirimido[3,4-d]pirimidin-4-il)piperidin-4-ilmetil]-3,4-dihidroquinazolin-2(1H)-ona (600 mg, rendimiento: 49 %).
ESI-MS m/z: 409 (M+H)+, RMN 1H (300 MHz, CDCls, 5): 9,09 (s, 1H), 8,73 (s, 1H), 7,66 (s, 1H), 7,58 (s a, 1H), 7,21 7,15 (m, 1H), 7,07-7,02 (m, 1H), 6,98-6,93 (m, 1H), 6,74-6,70 (m, 1H), 4,51 (s, 2H), 4,49-4,41 (m, 2H), 3,44 (d, J = 7,7 Hz, 2H), 3,27-3,18 (m, 2H), 2,22-2,11 (m, 1H), 1,98-1,89 (m, 2H), 1,63-1,49 (m, 2H)
Etapa 2: se obtuvo 4-(3-{[1-(6-cloropirimido[3,4-d]pirimidin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (370 mg, rendimiento: 74 %) de la misma manera que en el ejemplo 6 usando la 3-[1-(6-cloropirimido[3,4-d]pirimidin-4-il)piperidin-4-ilmetil]-3,4-dihidroquinazolin-2(1H)-ona obtenido en la etapa 1 y 4-yodopicolinonitrilo.
ESI-MS m/z: 511 (M+H)+, RMN 1H (300 MHz, CDCls, 5): 9,10 (s, 1H), 8,81 (d, J = 5,9 Hz, 1H), 8,74 (s, 1H), 7,84-7,83 (m, 1H), 7,67-7,64 (m, 2H), 7,21-7,09 (m, 3H), 6,41-6,37 (m, 1H), 4,59 (s, 2H), 4,48-4,43 (m, 2H), 3,46 (d, J = 7,3 Hz, 2H), 3,27-3,18 (m, 2H), 2,24-2,16 (m, 1H), 1,98-1,90 (m, 2H), 1,61-1,48 (m, 2H)
Etapa 3: el 4-(3-{[1-(6-cloropirimido[3,4-d]pirimidin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (300 mg, 0,59 mmol) obtenido en la etapa 2, tris(dibencilidenoacetona)dipaladio (0) (Pd2dba3) (27 mg, 0,03 mmol), 4,5-bis(difenilfosfin)-9,9-dimetilxanteno (Xantphos) (68 mg, 0,12 mmol), carbonato de cesio (268 mg, 0,82 mmol) y benzofenona imina (128 mg, 0,71 mmol) se agitaron en un disolvente mixto de 1,4-dioxano (3,0 ml)/tolueno (3,0 ml) a 100 °C durante 6 horas. A la mezcla de reacción, se le añadió una solución acuosa saturada de bicarbonato sódico y la mezcla resultante se extrajo con cloroformo. La capa orgánica se lavó con salmuera saturad y después se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo 4-[3-({1-[6-(difenilmetilenamino)pirimido[3,4-d]pirimidin-4-il]piperidin-4-il}metil)-2-oxo-3,4-dihidroquinazolin-1(2H)-il]picolinonitrilo (44 mg).
Etapa 4: A una solución en THF (0,5 ml) del 4-[3-({1-[6-(difenilmetilenamino)pirimido[3,4-d]pirimidin-4-il]piperidin-4-il}metil)-2-oxo-3,4-dihidroquinazolin-1(2H)-il]picolinonitrilo en bruto (40 mg) obtenido en la etapa 3, se añadió ácido
clorhídrico concentrado (12 mol/l, 0,02 ml) y la mezcla resultante se agitó a temperatura ambiente durante 1 hora. A la mezcla de reacción, se le añadió una solución acuosa saturada de bicarbonato sódico y la mezcla resultante se extrajo con cloroformo. La capa orgánica se lavó con salmuera saturad y después se secó sobre sulfato de magnesio anhidro y se concentró a presión reducida. El residuo resultante se purificó por cromatografía preparativa de capa fina (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo el compuesto del título 33 (8,4 mg, rendimiento: 3 %).
ESI-MS m/z: 492 (M+H)+, RMN 1H (300 MHz, CDCla, 8): 8,92 (s, 1H), 8,81 (d, J = 4,9 Hz, 1H), 8,56 (s, 1H), 7,84 (s, 1H), 7,69-7,62 (m, 1H), 7,23-7,09 (m, 3H), 6,65 (s, 1H), 6,38 (d, J = 7,8 Hz, 1H), 4,67-4,56 (m, 4H), 4,36-4,27 (m, 2H), 3,50-3,45 (m, 2H), 3,11-3,01 (m, 2H), 2,19-2,08 (m, 1H), 1,94-1,85 (m, 2H), 1,63-1,50 (m, 2H)
Ejemplo 34
4-(2-oxo-3-{[1-(4-oxo-3,4-dihidropirido[4,3-d]pirimidin-5-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (Compuesto 34)
El diclorhidrato de 4-[2-oxo-3-(piperidin-4-ilmetil)-3,4-dihidroquinazolin-1(2H)-il]picolinonitrilo (61 mg, 0,15 mmol) obtenido en la etapa 2 del ejemplo 27, la 5-cloropirido[4,3-d]pirimidin-4(3H)-ona (58 mg, 0,32 mmol) obtenida mediante el método descrito en Synthesis, 2010, 42, 30, y diisopropiletilamina (827 mg, 0,64 mmol) se mezclaron y se agitaron en NMP (2,0 ml) a 150 °C durante 1 hora usando un reactor de microondas (fabricado por CEM Corporation) a 300 W. Después de evaporar el disolvente a presión reducida, el residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de cloroformo/metanol), con lo que se obtuvo el compuesto del título 34 (67 mg, rendimiento: 86 %). ESI-MS m/z: 493 (M+H)+, RMN 1H (300 MHz, CDCla, 8): 10,7 (s a, 1H), 8,80 (d, J = 5,4 Hz, 1H), 8,33 (d, J = 5,1 Hz, 1H), 8,08 (s, 1H), 7,86-7,85 (m, 1H), 7,68 (d, J = 2,1, 5,1 Hz, 1H), 7,22-7,07 (m, 3H), 6,96 (d, J = 5,1 Hz, 1H), 6,38 (d, J = 7,8 Hz, 1H), 4,58 (s, 2H), 4,03-3,99 (m, 2H), 3,45 (d, J = 7,2 Hz, 2H), 3,04-2,96 (m, 2H), 2,06 (s a, 1H), 1,84-1,80 (m, 2H), 1,69-1,61 (m, 2H)
Ejemplo 35
4-(2-oxo-3-{[1-(6-oxo-6,7-dihidro-5H-pirimido[4,5-b][1,4]oxazin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (Compuesto 35)
El compuesto del título 35 (21 mg, rendimiento: 19 %) se obtuvo de la misma manera que en el ejemplo 5 usando el compuesto R6 obtenido en el ejemplo de referencia 6.
ESI-MS m/z: 497 (M+H)+, RMN 1H (300 MHz, CDCb, 8): 8,81-8,79 (m, 1H), 8,26 (s, 1H), 7,83 (s, 1H), 7,66-7,65 (m, 1H), 7,34 (s a, 1H), 7,20-7,10 (m, 3H), 6,38 (d, J = 7,5 Hz, 1H), 4,83 (s, 2H), 4,57 (s, 2H), 3,76-3,72 (m, 2H), 3,44-3,42 (m, 2H), 3,00-2,93 (m, 2H), 2,02 (s a, 1H), 1,89-1,85 (m, 2H), 1,58-1,46 (m, 2H)
Ejemplo 36
4-(2-oxo-1-{[1-(8-oxo-8,9-dihidro-7H-purin-6-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (Compuesto 36)
El compuesto del título 36 (67 mg, rendimiento: 63 %) se obtuvo de la misma manera que en el ejemplo 34 usando la 6-cloro-7H-purin-8(9H)-ona (84 mg, 0,49 mmol) obtenida mediante el método descrito en el documento WO2007/125315.
ESI-MS m/z: 482 (M+H)+, RMN 1H (300 MHz, DMSO-d6, 8): 11,4 (s, 1H), 10,7 (s, 1H), 8,86 (d, J = 5,4 Hz, 1H), 8,18 8,17 (m, 1H), 8,06 (s, 1H), 7,19-7,79 (m, 1H), 7,28 (d, J = 6,9 Hz, 1H), 7,17-7,05 (m, 2H), 6,35 (d, J = 7,8 Hz, 1H), 4,60 (s, 2H), 4,23-4,19 (d, J = 13 Hz, 2H), 3,32-3,30 (m, 2H), 2,93-2,85 (m, 2H), 2,01 (s a, 1H), 1,71-1,67 (m, 2H), 1,24-1,16 (m, 2H)
Ejemplo 37
4-(3-{[1-(7-morfolinoquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (Compuesto 37)
Etapa 1: se obtuvo 4-(3-{[1-(7-bromoquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (200 mg, rendimiento: 67 %) de la misma manera que en el ejemplo 5 usando 7-bromo-4-cloroquinazolina. ESI-MS m/z: 554 (M+H)+
Etapa 2: 4-(3-{[1-(7-bromoquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (200 mg, 0,36 mmol) obtenido en la etapa 1, morfolina (63 mg, 0,72 mmol), aducto de tris(dibencilidenacetona)dipaladio (0)-cloroformo (Pd2dba3CHCb) (37 mg, 0,04 mmol), Xantphos (21 mg, 0,04 mmol) y carbonato de cesio (235 mg, 0,72 mmol) se agitaron durante una noche en 1,4-dioxano (10 ml) a 100 °C. La mezcla de reacción se enfrió a temperatura ambiente y después se diluyó con agua y se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturada y después se secó sobre sulfato de magnesio anhidro y el disolvente se evaporó a presión reducida. El residuo resultante se purificó por HPLC preparativa de fase inversa (un disolvente
mezclado de acetonitrilo/agua), con lo que se obtuvo el compuesto del título 37 (50 mg, rendimiento: 24 %).
ESI-MS m/z: 561 (M+H)+, RMN 1H (400 MHz, DMSO-d6, 8): 8,87 (d, J = 5,2 Hz, 1H), 8,45 (s, 1H), 8,20 (d, J = 1,6 Hz, 1H), 7,83-7,75 (m, 2H), 7,30 (d, J = 5,1 Hz, 2H), 7,15-7,08 (m, 2H), 6,99 (d, J = 2,4 Hz, 1H), 6,36 (d, J = 8,0 Hz, 1H), 4,64 (s, 2H), 4,22-4,19 (m, 2H), 3,77 (t, J = 4,8 Hz, 4H), 3,38-3,33 (m, 6H), 3,10-3,04 (m, 2H), 2,08 (s a, 1H), 1,80-1,78 (m, 2H), 1,45-1,37 (m, 2H)
Ejemplo 38
4- (2-oxo-3-{[1-(3-oxo-2,3-dihidro-[1,2,4]triazolo[4,3-a]piridin-5-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (Compuesto 38)
El compuesto del título 38 (50 mg, rendimiento: 47 %) se obtuvo de la misma manera que en el ejemplo 34 usando la 5- bromo-[1,2,4]triazolo[4,3-a]piridin-3(2H)-ona obtenida mediante el método descrito en el documento WO2005/85226. ESI-MS m/z: 481 (M+H)+, RMN 1H (400 MHz, CDCb, 8): 8,78 (d, J = 6,0 Hz, 1H), 7,85-7,83 (m, 2H), 7,64 (dd, J = 6,0, 2,0 Hz, 1H), 7,18-7,07 (m, 4H), 6,44 (d, J = 8,4 Hz, 1H), 6,36 (d, J = 8,0 Hz, 1H), 4,55 (s, 2H), 4,44-4,41 (m, 2H), 3,38 3,36 (m, 2H), 2,96-2,91 (m, 2H), 2,09 (s a, 1H), 1,82-1,79 (m, 2H), 1,34-1,26 (m, 2H)
Ejemplo 39
4-(2-oxo-3-{[1-(3-oxo-2,3-dihidro-[1,2,4]triazolo[4,3-a]pirazin-8-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (Compuesto 39)
El compuesto del título 39 (50 mg, rendimiento: 46 %) se obtuvo de la misma manera que en el ejemplo 34 usando el compuesto R7 obtenido en el ejemplo de referencia 7.
ESI-MS m/z: 482 (M+H)+, RMN 1H (400 MHz, CDCla, 8): 11,0 (s a, 1H), 8,79 (d, J = 5,4 Hz, 1H), 7,83 (d, J = 1,8 Hz, 1H), 7,65 (dd, J = 5,4, 1,8 Hz, 1H), 7,19-7,08 (m, 5H), 6,38 (d, J = 9,8 Hz, 1H), 5,17-5,13 (m, 2H), 4,56 (s, 2H), 3,39 (d, J = 7,2 Hz, 2H), 3,06-2,99 (m, 2H), 2,13 (s a, 1H), 1,87-1,83 (m, 2H), 1,46-1,32 (m, 2H)
Ejemplo 40
3- (4-{[1-(2-cianopiridin-4-il)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)-2-fluoroisonicotinamida (Compuesto 40)
El compuesto del título 40 (50 mg, rendimiento: 46 %) se obtuvo de la misma manera que en el ejemplo 34 usando el compuesto R8 obtenido en el ejemplo de referencia 8.
ESI-MS m/z: 486 (M+H)+, RMN 1H (270 MHz, CDCb, 8): 8,79 (d, J = 5,1 Hz, 1H), 8,07 (d, J = 5,1 Hz, 1H), 7,83 (d, J = 1,9 Hz, 1H), 7,65 (dd, J = 5,1, 1,9 Hz, 1H), 7,31-7,27 (m, 1H), 7,19-7,07 (m, 3H), 6,49 (s a, 1H), 6,38 (d, J = 8,1 Hz, 1H), 5,92 (s a, 1H), 4,57 (s, 2H), 4,05-4,00 (m, 2H), 3,42 (d, J = 8,1 Hz, 2H), 2,95-2,87 (m, 2H), 2,03 (s a, 1H), 1,84 1,80 (m, 2H), 1,55-1,50 (m, 2H)
Ejemplo 41
4- (3-{[1-(7-aminopirido[3,2-d]pirimidin-4-il)piperidin-4-i1]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (Compuesto 41)
Etapa 1: se obtuvo 4-(3-{[1-(7-bromopirido[3,2-d]pirimidin-4-il)piperidin-4-i1]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (89 mg, rendimiento: 90 %) de la misma manera que en la etapa 1 del ejemplo 37 usando 7-bromo-4-cloropirido[3,2-d]pirimidina.
ESI-MS m/z: 555 (M+H)+
Etapa 2: 4-(4-{[1-(2-cianopiridin-4-il)-4-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)pirido[3,2-d]pirimidin-7-ilcarbamato de ferc-butilo (87 mg, rendimiento: 92 %) se obtuvo de la misma manera que en la etapa 3 del ejemplo 33 usando el 4-(3-{[1-(7-bromopirido[3,2-d]pirimidin-4-il)piperidin-4-i1]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo obtenido en la etapa 1 y carbamato de ferc-butilo.
ESI-MS m/z: 555 (M+H)+
Etapa 3: El compuesto del título 41 (20 mg, rendimiento: 28 %) se obtuvo de la misma manera que en la etapa 4 del ejemplo 33 usando el 4-(4-{[1-(2-cianopiridin-4-il)-4-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)pirido[3,2-d]pirimidin-7-ilcarbamato de ferc-butilo obtenido en la etapa 2.
ESI-MS m/z: 492 (M+H)+, RMN 1H (300 MHz, CDCb, 8): 8,79 (d, J = 5,4 Hz, 1H), 8,44 (s, 1H), 8,20 (d, J = 2,4 Hz, 1H), 7,84 (d, J = 1,5 Hz, 1H), 7,65 (dd, J = 5,1, 1,8 Hz, 1H), 7,19-7,07 (m, 4H), 6,38 (d, J = 7,5 Hz, 1H), 5,62-5,58 (m, 2H), 4,57 (s, 2H), 4,23 (s a, 2H), 3,40 (d, J = 6,9 Hz, 2H), 3,17-3,10 (m, 2H), 2,19 (s a, 1H), 1,89-1,84 (m, 2H), 1,52-1,42 (m, 2H) Ejemplo 42
4-(2-oxo-3-{[1-(4-oxo-3,4-dihidropirido[3,4-d]pirimidin-8-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (Compuesto 42)
El compuesto del título 42 (13 mg, rendimiento: 25 %) se obtuvo de la misma manera que en el ejemplo 34.
ESI-MS m/z: 493 (M+H)+, RMN 1H (400 MHz, CDCb, 8): 11,5 (s a, 1H), 8,79 (d, J = 4,8 Hz, 1H), 8,25 (d, J = 5,1 Hz, 1H), 8,05 (s, 1H), 7,85 (d, J = 1,5 Hz, 1H), 7,65 (dd, J = 5,7, 1,8 Hz, 1H), 7,46 (d, J = 5,1 Hz, 1H), 7,19-7,07 (m, 3H), 6,38 (d, J = 7,5 Hz, 1H), 4,63-4,59 (m, 4H), 3,43 (d, J = 7,5 Hz, 2H), 3,04-2,96 (m, 2H), 2,07 (s a, 1H), 1,86-1,82 (m, 2H), 1,70-1,57 (m, 2H)
Ejemplo 43
3-(3-{[1-(7,8-dihidro-5H-pirano[4,3-d]pirimidin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)benzonitrilo (Compuesto 43)
Etapa 1: se obtuvo 3-{[1-(7,8-dihidro-5H-pirano[4,3-d]pirimidin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-2(1H)-ona (40 mg, rendimiento: 11 %) de la misma manera que en el ejemplo 3 usando el compuesto R1 obtenido en el ejemplo de referencia 1 y el compuesto R10 obtenido en el ejemplo de referencia 10.
ESI-MS m/z: 380 (M+H)+, RMN 1H (400 MHz, CDCla, 8): 8,57 (s, 1H), 7,20 (d, J = 8,0 Hz, 1H), 7,07 (d, J = 8,0 Hz, 1H), 7,00-6,90 (m, 1H), 6,85 (s, 1H), 6,69 (d, J = 8,0 Hz, 1H), 4,56 (s, 2H), 4,51 (s, 2H), 4,08 (t, J = 6,0 Hz, 2H), 3,79 (d, J = 13 Hz, 2H), 3,50 (d, J = 7,2 Hz, 2H), 3,00-2,85 (m, 4H), 2,10-1,95 (m, 1H), 1,80-1,70 (m, 2H), 1,50-1,35 (m, 2H)
Etapa 2: El compuesto del título 43 (26 mg, rendimiento: 26 %) se obtuvo de la misma manera que en el ejemplo 6 usando la 3-{[1-(7,8-dihidro-5H-pirano[4,3-d]pirimidin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-2(1H)-ona obtenida en la etapa 1 y 3-yodobenzonitrilo.
ESI-MS m/z: 481 (M+H)+, RMN 1H (400 MHz, DMSO-d6, 8): 8,44 (s, 1H), 7,96-7,86 (m, 2H), 7,73 (t, J = 7,8 Hz, 1H), 7,70-7,63 (m, 1H), 7,25 (d, J = 7,2 Hz, 1H), 7,12-7,06 (m, 1H), 7,05-6,95 (m, 1H), 6,07 (d, J =8,0 Hz, 1H), 4,61 (s, 2H), 4,52 (s, 2H), 3,97 (t, J = 6,2 Hz, 2H), 3,76 (d, J = 13 Hz, 2H), 3,41-3,20 (m, 2H), 2,88 (t, J = 12 Hz, 2H), 2,77 (t, J = 6,0 Hz, 2H), 2,11-1,92 (m, 1H), 1,79-1,45 (m, 2H), 1,36-1,19 (m, 2H)
Ejemplo 44
5-(3-{[1-(6-acetil-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)-2-fluorobenzonitrilo (Compuesto 44)
Etapa 1: 4-(4-{[2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de ferc-butilo (150 mg, rendimiento: 59 %) se obtuvo de la misma manera que en el ejemplo 3 usando el compuesto R1 obtenido en el ejemplo de referencia 1 y el 4-oxo-3,5,7,8-tetrahidro-5H-pirido[4,3-d]pirimidin-6-carboxilato de ferc-butilo obtenido mediante el método descrito en el documento WO2010/066684.
ESI-MS m/z: 479 (M+H)+, RMN 1H (300 MHz, CDCla, 8): 8,57 (s, 1H), 7,20 (t, J = 7,5 Hz, 1H), 7,08-6,93 (m, 2H), 6,80 (s, 1H), 6,69 (d, J = 8,1 Hz, 1H), 4,51 (s, 2H), 4,44 (s, 2H), 3,89-3,85 (m, 2H), 3,74 (s a, 2H), 3,39-3,34 (m, 2H), 2,95 (s a, 4H), 2,07 (s a, 1H), 1,88-1,84 (m, 2H), 1,50 (s a, 11H)
Etapa 2: el 4-(4-{[2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de ferc-butilo (800 mg, 1,7 mmol) obtenido en la etapa 1 se agitó en ácido clorhídrico-dioxano (4,0 mol/l, 20 ml) a temperatura ambiente durante 2 horas. Después, el disolvente se evaporó a presión reducida, con lo que se obtuvo el clorhidrato de 3-{[1-(5,6,7,8-tetrahidropirido[4,3-d]pirimidin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-2(1H)-ona en bruto (600 mg). Este compuesto se usó en la siguiente reacción sin realizar más purificación en particular.
Etapa 3: El clorhidrato de 3-{[1-(5,6,7,8-tetrahidropirido[4,3-d]pirimidin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-2(1H)-ona en bruto (100 mg) obtenido en la etapa 2, cloruro de acetilo (25 mg, 0,25 mmol) y trietilamina (40 mg, 0,4 mmol) se agitaron en diclorometano (5,0 ml) a temperatura ambiente durante 5 horas. A la mezcla de reacción, se le añadió agua y la mezcla resultante se extrajo con diclorometano. La capa orgánica se lavó con salmuera saturada y se secó sobre sulfato de magnesio anhidro. Después de evaporar el disolvente a presión reducida, el residuo resultante se purificó por HPLC preparativa de fase inversa (un disolvente mixto de acetonitrilo/agua), con lo que se obtuvo 3-{[1-(6-acetil-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-2(1H)-ona (20 mg, rendimiento: 19 %).
ESI-MS m/z: 421 (M+H)+, RMN 1H (400 MHz, CDCb, 8): 8,56 (s, 1H), 7,22-7,18 (m, 1H), 7,09-7,07 (m, 1H), 7,02-6,96 (m, 1H), 6,69 (s a, 2H), 4,58 (s, 2H), 4,43 (s, 2H), 3,93-3,86 (m, 2H), 3,80-3,73 (m, 2H), 3,44-3,36 (m, 2H), 2,98-2,93 (m, 4H), 2,19 (s, 3H), 2,08 (s a, 1H), 1,86-1,83 (m, 2H), 1,53-1,44 (m, 2H)
Etapa 4: El compuesto del título 44 (44 mg, rendimiento: 34 %) se obtuvo de la misma manera que en el ejemplo 6 usando la 3-{[1 -(6-acetil-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-4-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-2(1 H)-ona obtenida en la etapa 3 y 2-fluoro-5-yodobenzonitrilo.
ESI-MS m/z: 540 (M+H)+, RMN 1H (400 MHz, DMSO-d6, 8): 8,46 (s, 1H), 8,03-8,02 (m, 1H), 7,81-7,65 (m, 2H), 7,25 (d, J = 7,2 Hz, 1H), 7,10 (t, J = 7,6 Hz, 1H), 7,01 (t, J = 7,2 Hz, 1H), 6,14 (d, J = 8,0 Hz, 1H), 4,70 (s, 2H), 4,62-4,45 (m, 2H), 3,81-3,74 (m, 4H), 3,32 (s a, 2H), 2,93-2,87 (m, 4H), 2,07-2,04 (m, 4H), 1,91-1,84 (m, 2H), 1,38-1,24 (m, 2H)
Ejemplo 45
4-(4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)pirido[3,4-d]pirimidin-6-carboxamido (Compuesto 45)
Etapa 1: 4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo (400 mg, rendimiento: 61 %) se obtuvo de la misma manera que en el ejemplo 6 usando el compuesto R1 obtenido en el ejemplo de referencia 1 y 2-fluoro-5-yodobenzonitrilo.
ESI-MS m/z: 465 (M+H)+
Etapa 2: se obtuvo 5-(3-{[1-(6-cloropirido[3,4-d]pirimidin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)-2-fluorobenzonitrilo (100 mg, rendimiento: 30 %) de la misma manera que en el ejemplo 3 usando el 4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 1 y la 6-cloropirido[3,4-d]pirimidin-4(3H)-ona obtenida mediante el método descrito en el documento WO2005/16926.
ESI-MS m/z: 528 (M+H)+
Etapa 3: se obtuvo 4-(4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)pirido[3,4-d]pirimidin-6-carboxilato de propilo (150 mg, rendimiento: 53 %) de la misma manera que en la etapa 2 del Ejemplo 15 usando el 5-(3-{[1-(6-cloropirido[3,4-d]pirimidin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidroquinazolin-1(2H)-il)-2-fluorobenzonitrilo obtenido en la etapa 2.
Etapa 4: se obtuvo ácido 4-(4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)pirido[3,4-d]pirimidin-6-carboxílico (1,0 mg, rendimiento: 1 %) de la misma manera que en la etapa 3 del ejemplo 15 usando el 4-(4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)pirido[3,4-d]pirimidin-6-carboxilato de propilo obtenido en la etapa 3. ESI-MS m/z: 538 (M+h )+
Etapa 5: el ácido 4-(4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-il)pirido[3,4-d]pirimidin-6-carboxílico (20 mg, 0,04 mmol) obtenido en la etapa 4 se mezcló con cloruro de amonio (6,0 mg, 0,12 mmol), HATU (21 mg, 0,06 mmol) y diisopropiletilamina (14 mg, 0,12 mmol) en THF (4,0 ml) y la mezcla resultante se agitó a temperatura ambiente durante 15 horas. La mezcla de reacción se concentró a presión reducida y el residuo resultante se purificó por HPLC preparativa de fase inversa (un disolvente mixto de acetonitrilo/agua), con lo que se obtuvo el compuesto del título 45 (11 mg, 54 %).
ESI-MS m/z: 537 (M+H)+, RMN 1H (400 MHz, DMSO-d6, 8): 9,15 (s, 1H), 8,73 (s, 1H), 8,46 (s, 1H), 8,26 (s, 1H), 8,04 8,02 (m, 1H), 7,81-7,66 (m, 3H), 7,27 (d, J = 7,6 Hz, 1H), 7,13-7,10 (m, 1H), 7,04-7,01 (m, 1H), 6,15 (d, J = 8,0 Hz, 1H), 4,65 (s, 2H), 4,50 (d, J = 13 Hz, 2H), 3,45-3,20 (m, 4H), 2,20 (s a, 1H), 1,89-1,86 (m, 2H), 1,51-1,29 (m, 2H)
Ejemplo 46
4-(4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidropirido[3,2-d]pirimidin-3(4H)-il]metil}piperidin-1-il)pirido[3,4-d]pirimidin-6-carboxamido (Compuesto 46)
Etapa 1: se obtuvo 4-[(2-oxo-1,2-dihidropirido[3,2-d]pirimidin-3(4H)-il)metil]piperidin-1-carboxilato de ferc-butilo (2,0 g, rendimiento: 62 %) de la misma manera que en el ejemplo de referencia 1 usando 3-aminopicolinaldehído.
ESI-MS m/z: 347 (M+H)+
Etapa 2: se obtuvo 4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidropirido[3,2-d]pirimidin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo (400 mg, rendimiento: 54 %) de la misma manera que en el ejemplo 6 usando el 4-[(2-oxo-1.2- dihidropirido[3,2-d]pirimidin-3(4H)-il)metil]piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 1 y 2-fluoro-5-yodobenzonitrilo.
ESI-MS m/z: 466 (M+H)+
Etapa 3: se obtuvo el compuesto del título (13 mg, rendimiento: 71 %) de la misma manera que en el ejemplo 45 usando el 4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidropirido[3,2-d]pirimidin-3(4H)-il]metil}piperidin-1 -carboxilato de ferc-butilo obtenido en la etapa 2.
ESI-MS m/z: 538 (M+H)+, RMN 1H (400 MHz, DMSO-d6, 8): 9,14 (s, 1H), 8,72 (s, 1H), 8,46 (s, 1H), 8,26 (s, 1H), 8,17 (dd, J = 4,8, 1,2 Hz, 1H), 8,06 (dd, J = 6,0, 2,8 Hz, 1H), 7,89-7,65 (m, 2H), 7,70 (t, J = 9,0 Hz, 1H), 7,15 (dd, J = 8,4, 3,6 Hz, 1H), 6,59 (d, J = 8,4 Hz, 1H), 4,73 (s, 2H), 4,50 (d, J = 13 Hz, 2H), 3,49-3,20 (m, 4H), 2,35-2,13 (m, 1H), 1,89 (d, J = 11 Hz, 2H), 1,53-1,30 (m, 2H)
Ejemplo 47
2-fluoro-5-(2-oxo-3-{[1-(4-oxo-3,4-dihidropirido[4,3-d]pirimidin-5-il)piperidin-4-il]metil}-3,4-dihidropirido[2,3-d]pirimidin-1(2H)-il)benzonitrilo (Compuesto 47)
Etapa 1: se obtuvo 4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidropirido[2,3-d]pirimidin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo (350 mg, rendimiento: 52 %) de la misma manera que en el ejemplo 6 usando el 4-[(2-oxo-1.2- dihidropirido[2,3-d]pirimidin-3(4H)-il)metil]piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 1 del ejemplo
22 y 2-fluoro-5-yodobenzonitrilo.
ESI-MS m/z: 466 (M+H)+
Etapa 2: se obtuvo el compuesto del título 47 (40 mg, rendimiento: 56 %) de la misma manera que en el ejemplo 34 usando el 4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidropirido[2,3-d]pirimidin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 1 y 5-cloropirido[4,3-d]pirimidin-4(3H)-ona.
ESI-MS m/z: 511 (M+H)+, RMN 1H (400 MHz, DMSO-d6, 8): 12,1 (s, 1H), 8,23 (d, J = 5,2 Hz, 1H), 8,11 (s, 1H), 8,03 (d, J = 5,2 Hz, 1H), 7,94 (dd, J = 6,0, 2,4 Hz, 1H), 7,74-7,66 (m, 2H), 7,61 (t, J = 8,8 Hz, 1H) 7,04 (dd, J = 8,0, 4,8 Hz, 1H), 6,82 (d, J = 5,2 Hz, 1H), 4,64 (s, 2H), 3,90-3,87 (m, 2H), 3,35-3,33 (m, 2H), 2,87 (t, J = 8,4 Hz, 2H), 1,99 (s a, 1H), 1,73-1,70 (m, 2H), 1,43-1,38 (m, 2H)
Ejemplo 48
4-(6-metoxi-2-oxo-3-{[1-(4-oxo-3,4-dihidropirido[4,3-d]pirimidin-5-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-1(2H)-il)picolinamida (Compuesto 48)
Etapa 1: se obtuvo 4-[(6-metoxi-2-oxo-1,2-dihidroquinazolin-3(4H)-il)metil]piperidin-1-carboxilato de ferc-butilo (1,5 g, rendimiento: 73 %) de la misma manera que en el ejemplo de referencia 1 usando 5-metoxi-2-nitrobenzaldehído. ESI-MS m/z: 376 (M+H)+, RMN 1H (400 MHz, DMSO-d6, 8): 8,97 (s, 1H), 6,75-6,69 (m, 3H), 4,38 (s, 2H), 3,93-3,90 (m, 2H), 3,90 (s, 3H), 3,18 (d, J = 7,2 Hz, 2H), 2,68 (s a, 2H), 1,84 (s a, 1H), 1,58-1,55 (m, 2H), 1,39 (s, 9H), 1,07-1,00 (m, 2H)
Etapa 2: se obtuvo 4-{[1-(2-cianopiridin-4-il)-6-metoxi-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo (580 mg, rendimiento: 76 %) de la misma manera que en el ejemplo 6 usando el 4-[(6-metoxi-2-oxo-1,2-dihidroquinazolin-3(4H)-il)metil]piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 1 y 4-yodopiridin-2-carbonitrilo.
ESI-MS m/z: 478 (M+H)+
Etapa 3: se obtuvo 4-(6-metoxi-2-oxo-3-{[1-(4-oxo-3,4-dihidropirido[4,3-d]pirimidin-5-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (300 mg, rendimiento: 47 %) de la misma manera que en el ejemplo 36 después de realizar un tratamiento de la misma manera que en la etapa 4 del ejemplo de referencia 1 usando el 4-{[1-(2-cianopiridin-4-il)-6-metoxi-2-oxo-1,2-dihidroquinazolin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 2.
ESI-MS m/z: 523 (M+H)+, RMN 1H (400 MHz, DMSO-d6, 8): 12,1 (s, 1H), 8,82 (d, J = 5,2 Hz, 1H), 8,22 (d, J = 5,2 Hz, 1H), 8,17 (s, 1H), 8,11 (s, 1H), 7,80 (d, J = 5,2 Hz, 1H), 6,91 (s, 1H), 6,82 (d, J = 5,2 Hz, 1H), 6,75-6,73 (m, 1H), 6,39 6,37 (m, 1H), 4,57 (s, 2H), 3,89-3,77 (m, 2H), 3,73 (s, 3H), 3,34-3,32 (m, 2H), 2,89-2,83 (m, 2H), 1,96 (s a, 1H), 1,67 1,66 (m, 2H), 1,43-1,34 (m, 2H)
Etapa 4: el 4-(6-metoxi-2-oxo-3-{[1-(4-oxo-3,4-dihidropirido[4,3-d]pirimidin-5-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (80 mg, 0,15 mmol) obtenido en la etapa 3 e hidróxido de litio monohidrato (14 mg, 0,33 mmol) se agitaron en un disolvente mixto de tetrahidrofurano-agua (1/1, 3,0 ml) a temperatura ambiente durante 15 horas. La mezcla de reacción se concentró a presión reducida y el residuo resultante se purificó por HPLC preparativa de fase inversa, con lo que se obtuvo el compuesto del título 48 (34 mg, rendimiento: 40 %).
ESI-MS m/z: 541 (M+H)+, RMN 1H (400 MHz, DMSO-d6+ D2O, 8): 8,73 (d, J = 5,2 Hz, 1H), 8,32-8,17 (m, 2H), 8,08 (s, 1H), 7,94 (s, 1H), 7,74-7,69 (m, 1H), 7,62 (d, J = 4,4 Hz, 1H), 7,06 (s, 1H), 6,90 (s, 1H), 6,82 (d, J = 5,6 Hz, 1H), 6,76 6,66 (m, 1H), 6,21 (d, J = 8,4 Hz, 1H), 4,56 (s, 2H), 3,83 (d, J = 13 Hz, 2H), 3,67 (s, 3H), 3,31 (d, J = 7,2 Hz, 2H), 2,92 2,80 (m, 2H), 2,05-1,82 (m, 1H), 1,76-1,58 (m, 2H), 1,45-1,27 (m, 2H)
Ejemplo 49
4-(6-fluoro-2-oxo-3-{[1-(4-oxo-3,4-dihidropirido[4,3-d]pirimidin-5-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-1(2H)-il)picolinonitrilo (Compuesto 49)
Etapa 1: se obtuvo 4-[(6-fluoro-2-oxo-1,2-dihidroquinazolin-3(4H)-il)metil]piperidin-1-carboxilato de ferc-butilo (720 mg, rendimiento: 73 %) de la misma manera que en el ejemplo de referencia 1 usando 5-fluoro-2-nitrobenzaldehído. ESI-MS m/z: 364 (M+H)+
Etapa 2: se obtuvo 4-{[1-(2-cianopiridin-4-il)-6-fluoro-2-oxo-1,2,3,4-tetrahidroquinazolin-3-il]metil}piperidin-1-carboxilato de ferc-butilo (500 mg, rendimiento: 78 %) de la misma manera que en el ejemplo 6 usando el 4-[(6-fluoro-2-oxo-1,2-dihidroquinazolin-3(4H)-il)metil]piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 1 y 4-yodopicolinonitrilo. ESI-MS m/z: 466 (M+H)+
Etapa 3: se obtuvo el compuesto del título 49 (80 mg, rendimiento: 54 %) de la misma manera que en el ejemplo 34 después de realizar un tratamiento de la misma manera que en la etapa 4 del ejemplo de referencia 1 usando el 4-{[1-(2-cianopiridin-4-il)-6-fluoro-2-oxo-1,2,3,4-tetrahidroquinazolin-3-il]metil}piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 2.
ESI-MS m/z: 511 (M+H)+, RMN 1H (300 MHz, DMSO-d6, 5): 9,87 (s a, 1H), 8,86 (d, J = 5,1 Hz, 1H), 8,27-8,14 (m, 2H), 8,11 (s, 1H), 7,85-7,77 (m, 1H), 7,25-7,15 (m, 1H), 7,04-6,91 (m, 1H), 6,81 (d, J = 5,4 Hz, 1H), 6,46-6,34 (m, 1H), 4,61 (s, 2H), 3,87 (d, J = 13 Hz, 2H), 3,45-3,21 (m, 2H), 2,86 (t, J = 12 Hz, 2H), 2,08-1,82 (m, 1H), 1,78-1,61 (m, 2H), 1,51 1,29 (m, 2H)
Ejemplo 50
N-[4-(4-{[1-(2-cianopiridin-4-il)-2-oxo-1,2-dihidropirido[2,3-d]pirimidin-3(4H)-il]metil}piperidin-1-il)quinazolin-6-il]acetamida (Compuesto 50)
Etapa 1: el 4-[(2-oxo-1,2-dihidropirido[2,3-d]pirimidin-3(4H)-il)metil]piperidin-1-carboxilato de ferc-butilo (2,0 g, 5,8 mmol) obtenido en la etapa 1 del ejemplo 22, 4-yodopicolinonitrilo (2,0 g, 8,7 mmol), óxido de cobre (I) (3,4 g, 24 mmol) y fosfato tripotásico (2,6 g, 12 mmol) se agitaron en DMA (20 ml) a 120 °C durante 7 horas. La mezcla de reacción se diluyó con diclorometano y se extrajo mediante la adición de agua a la misma. La capa orgánica se lavó con salmuera saturada y se secó sobre sulfato de magnesio anhidro y, después, el disolvente se evaporó a presión reducida. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de diclorometano/metanol), con lo que se obtuvo 4-{[1-(2-cianopiridin-4-il)-2-oxo-1,2-dihidropirido[2,3-d]pirimidin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo (1,6 g, rendimiento: 61 %).
RMN 1H (300 MHz, DMSO-d6, 5): 8,83 (d, J = 5,4 Hz, 1H), 8,12 (d, J = 1,5 Hz, 1H), 8,05 (dd, J = 5,4, 1,5 Hz, 1H), 7,75 (dd, J = 5,4, 2,1 Hz, 1H), 7,69 (d, J = 7,2 Hz, 1H), 7,09 (dd, J = 7,2, 2,1 Hz, 1H), 4,62 (s, 2H), 4,10-3,80 (m, 2H), 3,45 3,15 (m, 2H), 2,90-2,50 (m, 2H), 2,00-1,75 (m, 1H), 1,70-1,50 (m, 2H), 1,38 (s, 9H), 1,15-0,90 (m, 2H)
Etapa 2: se obtuvo 4-(3-{[1-(6-bromoquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidropirido[2,3-d]pirimidin-1(2H)-il)picolinonitrilo (100 mg, rendimiento: 76 %) de la misma manera que en la etapa 3 del ejemplo 5 usando el 4-{[1-(2-cianopiridin-4-il)-2-oxo-1,2-dihidropirido[2,3-d]pirimidin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 1 y 6-bromo-4-cloroquinazolina.
ESI-MS m/z: 555 (M+H)+
Etapa 3: se obtuvo el compuesto del título 50 (25 mg, rendimiento: 26 %) de la misma manera que en la etapa 3 del ejemplo 33 usando el 4-(3-{[1-(6-bromoquinazolin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidropirido[2,3-d]pirimidin-1(2H)-il)picolinonitrilo obtenido en la etapa 2 y acetamida.
ESI-MS m/z: 534 (M+H)+, RMN 1H (400 MHz, DMSO-d6, 5): 10,3 (s, 1H), 8,84 (d, J = 5,2 Hz, 1H), 8,55 (s, 1H), 8,47 (s, 1H), 8,15 (s, 1H), 8,07 (d, J = 4,8 Hz, 1H), 7,85-7,77 (m, 4H), 7,13-7,10 (m, 1H), 4,68 (s, 2H), 4,27-4,23 (m, 2H), 3,42 (d, J = 6,8 Hz, 2H), 3,08 (t, J = 13 Hz, 2H), 2,12 (s a, 4H), 1,87-1,84 (m, 2H), 1,51-1,45 (m, 2H)
Ejemplo 51
4-(6-cian-2-oxo-3-{[1-(4-oxo-3,4-dihidropirido[4,3-d]pirimidin-5-il)piperidin-4-il]metil}-3,4-dihidroquinazolin-1(2H)-il)picolinamida (Compuesto 51)
Etapa 1: se obtuvo 4-{[(5-bromo-2-nitrobencil)amino]metil}piperidin-1-carboxilato de ferc-butilo (7,0 g, rendimiento: 63 %) de la misma manera que en la etapa 1 del ejemplo de referencia 1 usando 5-bromo-2-nitrobenzaldehído.
RMN 1H (300 MHz, CDCla, 5): 7,84 (d, J = 8,4 Hz, 2H), 7,60-7,50 (m, 1H), 5,30 (s, 1H), 4,22-4,05 (m, 2H), 4,03 (s, 2H), 2,79-2,60 (m, 2H), 2,52 (d, J = 6,6 Hz, 2H), 1,80-1,50 (m, 3H), 1,46 (s, 9H), 1,22-1,02 (m, 2H)
Etapa 2: el 4-{[(5-bromo-2-nitrobencil)amino]metil}piperidin-1-carboxilato de ferc-butilo (3,0 g, 7,0 mmol) obtenido en la etapa 1, cinc (1,5 mg), cianuro de cinc (540 mg, 4,6 mmol) y tetraquis(trifenilfosfina)paladio (0) (100 mg, 0,09 mmol) se agitaron en DMF (100 ml) a 100 °C durante 12 horas. A la mezcla de reacción, se le añadió agua y la mezcla resultante se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturada y se secó sobre sulfato de magnesio anhidro. Después, un residuo obtenido por evaporación del disolvente a presión reducida se purificó por cromatografía en columna sobre gel de sílice (un disolvente mixto de hexano/acetato de etilo), con lo que se obtuvo 4-{[(5-cian-2-nitrobencil)amino]metil}piperidin-1-carboxilato de ferc-butilo (1,2 g, rendimiento: 46 %).
RMN 1H (300 MHz, CDCla, 5): 8,09 (s, 1H), 8,01 (d, J = 8,4 Hz, 1H), 7,80-7,68 (m, 1H), 4,25-4,00 (m, 4H), 3,50 (s, 1H), 2,71 (t, J = 12 Hz, 2H), 2,54 (d, J = 3,3 Hz, 2H), 1,80-1,60 (m, 2H), 1,75-1,51 (m, 1H), 1,47 (s, 9H), 1,29-1,01 (m, 2H)
Etapa 3: se obtuvo 4-[(6-cian-2-oxo-1,2-dihidroquinazolin-3(4H)-il)metil]piperidin-1-carboxilato de ferc-butilo (600 mg, rendimiento: 70 %) de la misma manera que en el ejemplo de referencia 1 usando el 4-{[(5-cian-2-nitrobencil)amino]metil}piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 2.
RMN 1H (300 MHz, CDCb, 5): 7,58 (s, 1H), 7,52-7,41 (m, 1H), 7,34 (s, 1H), 6,76 (d, J = 8,4 Hz, 1H), 4,48 (s, 2H), 4,25 4,01 (m, 2H), 3,45-3,25 (m, 2H), 2,80-2,59 (m, 2H), 2,00-1,55 (m, 3H), 1,45 (s, 9H), 1,33-1,10 (m, 2H)
Etapa 4: se obtuvo 4-{[6-cian-1-(2-cianopiridin-4-il)-2-oxo-1,2,3,4-tetrahidroquinazolin-3-il]metil}piperidin-1-carboxilato de ferc-butilo (220 mg, rendimiento: 57 %) de la misma manera que en el ejemplo 6 usando el 4-[(6-ciano-2-oxo-1,2-dihidroquinazolin-3(4H)-il)metil]piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 3 y 4-yodopicolinonitrilo. ESI-MS m/z: 473 (M+H)+
Etapa 5: se obtuvo 1-(2-cianopiridin-4-il)-2-oxo-3-{[1-(4-oxo-3,4-dihidropirido[4,3-d]pirimidin-5-il)piperidin-4-il]metil}-1.2.3.4- tetrahidroquinazolin-6-carbonitrilo (55 mg, rendimiento: 26 %) realizando los mismos tratamientos que en la etapa 4 del ejemplo de referencia 1 y el ejemplo 34 secuencialmente usando el 4-{[6-cian-1-(2-cianopiridin-4-il)-2-oxo-1.2.3.4- tetrahidroquinazolin-3-il]metil}piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 4.
ESI-MS m/z: 518 (M+H)+, RMN 1H (300 MHz, DMSO-d6, 8): 12,1 (s a, 1H), 8,91 (d, J = 5,1 Hz, 1H), 8,25-8,18 (m, 2H), 8,10 (s, 1H), 7,86-7,73 (m, 2H), 7,62-7,52 (m, 1H), 6,81 (d, J = 5,4 Hz, 1H), 6,46 (d, J = 5,4 Hz, 1H), 4,67 (s, 2H), 3,88 (d, J = 13 Hz, 2H), 3,40-3,27 (m, 2H), 2,95-2,78 (m, 2H), 2,05-1,85 (m, 1H), 1,77-1,63 (m, 2H), 1,49-1,30 (m, 2H)
Etapa 6: El compuesto del título 51 (28 mg, rendimiento: 74 %) se obtuvo de la misma manera que en la etapa 4 del Ejemplo 48 usando el 1-(2-cianopiridin-4-il)-2-oxo-3-{[1-(4-oxo-3,4-dihidropirido[4,3-d]pirimidin-5-il)piperidin-4-il]metil}-1.2.3.4- tetrahidroquinazolin-6-carbonitrilo obtenido en la etapa 5.
ESI-MS m/z: 536 (M+H)+, RMN 1H (400 MHz, DMSO-d6, 8): 12,1 (s a, 1H), 8,81 (d, J = 5,2 Hz, 1H), 8,22 (d, J = 5,2 Hz, 2H), 8,10 (s, 1H), 8,00 (s, 1H), 7,78 (s, 2H), 7,28-7,66 (m, 1H), 7,55 (d, J = 8,4 Hz, 1H), 6,82 (d, J = 5,2 Hz, 1H), 6,33 (d, J = 8,4 Hz, 1H), 4,68 (s, 2H), 3,88 (d, J = 13 Hz, 2H), 3,34-3,32 (m, 2H), 2,87 (m, 2H), 2,05-1,93 (m, 1H), 1,82-1,62 (m, 2H), 1,45-1,29 (m, 2H)
Ejemplo 52
4-(4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidropirido[4,3-d]pirimidin-3(4H)-il]metil}piperidin-1-il)pirido[4,3-d]pirimidin-7-carboxamido
Etapa 1: Se obtuvo 4-[(2-oxo-1,2-dihidropirido[4,3-d]pirimidin-3(4H)-il)metil]piperidin-1-carboxilato de ferc-butilo (3,6 g, rendimiento: 61 %) de la misma manera que en el ejemplo de referencia 1 usando 4-aminonicotinaldehído.
ESI-MS m/z: 347 (M+H)+
Etapa 2: Se obtuvo 4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidropirido[4,3-d]pirimidin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo (450 mg, rendimiento: 70 %) de la misma manera que en la etapa 1 del ejemplo 50 usando el 4-[(2-oxo-1,2-dihidropirido[4,3-d]pirimidin-3(4H)-il)metil]piperidin-1 -carboxilato de ferc-butilo obtenido en la etapa 1 y 2-fluoro-5-yodobenzonitrilo.
ESI-MS m/z: 449 (M+H)+
Etapa 3: se obtuvo 5-(3-{[1-(7-cloropirido[4,3-d]pirimidin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidropirido[4,3-d]pirimidin-1(2H)-il)-2-fluorobenzonitrilo (300 mg, 23 %) de la misma manera que en la etapa 3 del ejemplo 5 el usando 4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidropirido[4,3-d]pirimidin-3(4H)-il]metil}piperidin-1-carboxilato de ferc-butilo obtenido en la etapa 2 y el compuesto R11 obtenido en el ejemplo de referencia 11.
ESI-MS m/z: 529 (M+H)+
Etapa 4: se obtuvo 4-(4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidropirido[4,3-d]pirimidin-3(4H)-il]metil}piperidin-1-il)pirido[4,3-d]pirimidin-7-carboxilato de propilo (130 mg, rendimiento: 53 %) de la misma manera que en la etapa 2 del ejemplo 15 usando el 5-(3-{[1-(7-cloropirido[4,3-d]pirimidin-4-il)piperidin-4-il]metil}-2-oxo-3,4-dihidropirido[4,3-d]pirimidin-1(2H)-i1)-2-fluorobenzonitrilo obtenido en la etapa 3.
ESI-MS m/z: 581 (M+H)+
Etapa 5: se obtuvo ácido 4-(4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidropirido[4,3-d]pirimidin-3(4H)-il]metil}piperidin-1-il)pirido[4,3-d]pirimidin-7-carboxílico (35 mg, rendimiento: 30 %) de la misma manera que en la etapa 3 del ejemplo 15 usando el 4-(4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidropirido[4,3-d]pirimidin-3(4H)-il]metil}piperidin-1-il)pirido[4,3-d]pirimidin-7-carboxilato de propilo obtenido en la etapa 4.
ESI-MS m/z: 539 (M+H)+, RMN 1H (400 MHz, DMSO-d6, 8): 9,32 (s, 1H), 8,67 (s, 1H), 8,37 (s, 1H), 8,20 (d, J = 5,6 Hz, 1H), 8,15-8,05 (m, 2H), 7,89-7,72 (m, 1H), 7,72 (t, J = 9,0 Hz, 1H), 6,17 (d, J = 5,6 Hz, 1H), 4,71 (s, 2H), 4,57 (d, J = 13 Hz, 2H), 3,49-3,20 (m, 4H), 2,31-2,12 (m, 1H), 1,88 (d, J = 10 Hz, 2H), 1,51-1,33 (m, 2H)
Etapa 6: El compuesto del título 52 (12 mg, rendimiento: 34 %) se obtuvo de la misma manera que en la etapa 5 del ejemplo 45 usando el ácido 4-(4-{[1-(3-cian-4-fluorofenil)-2-oxo-1,2-dihidropirido[4,3-d]pirimidin-3(4H)-il]metil}piperidin-1-il)pirido[4,3-d]pirimidin-7-carboxílico obtenido en la etapa 5.
ESI-MS m/z: 538 (M+H)+, RMN 1H (300 MHz, DMSO-d6, 8): 9,23 (s, 1H), 8,65 (s, 1H), 8,33 (s, 1H), 8,25-8,13 (m, 2H), 8,08 (s, 1H), 8,11-8,00 (m, 1H), 7,85 (s, 1H), 7,85-7,62 (m, 2H), 6,13 (d, J = 5,4 Hz, 1H), 4,67 (s, 2H), 4,54 (d, J = 12 Hz, 2H), 3,58-3,00 (m, 4H), 2,25-2,12 (m, 1H), 1,85 (d, J = 12 Hz, 2H), 1,51-1,30 (m, 2H)
* Ejemplo 53
Comprimidos (Compuesto 53)
Se preparan comprimidos que tienen los siguientes ingredientes, de acuerdo con la manera habitual. Se mezclan el compuesto 53 (40 g), lactosa (286,8 g) y almidón de patata (60 g) y se le añade una solución de hidroxipropil celulosa acuosa al 10 % (120 g). La mezcla resultante se amasa, se granula, se seca y se dimensiona de acuerdo con la manera convencional, con lo cual se preparan los gránulos para la formación de los comprimidos. Se le añade
estearato de magnesio (1,2 g) y se mezcla y la mezcla resultante se comprime usando una prensa de comprimidos con una mano de mortero que tiene un diámetro de 8 mm (modelo RT-15, fabricado por Kikusui Seisakusho Ltd.), con lo cual se obtienen comprimidos (que contienen 20 mg del ingrediente activo por comprimido).
[Tabla 8]
Formulación
Compuesto 53 20 mg
Lactosa 143,4 mg
Almidón de patata 30 mg
Hidroxipropilcelulosa 6mg
Estearato de magnesio______ 0,6 mg
200 mg
Aplicabilidad industrial
De acuerdo con la presente invención, se puede proporcionar un compuesto heterocíclico de anillo condensado o una sal farmacéuticamente aceptable del mismo, que tiene una actividad inhibidora de la señalización Wnt y es útil como agente terapéutico y/o preventivo para, por ejemplo, cáncer, fibrosis pulmonar, fibromatosis, artrosis.
Claims (13)
1. Un compuesto heterocíclico de anillo condensado representado por la fórmula general (I) o una sal farmacéuticamente aceptable del mismo:

[en donde n1 representa 0 o 1;
cada uno de n2 y n3 es 2;
R1 representa opcionalmente arilo sustituido, un grupo heterocíclico aromático opcionalmente sustituido o un grupo heterocíclico alifático opcionalmente sustituido;
R2 representa un átomo de hidrógeno o hidroxi;
R3 representa un grupo heterocíclico aromático opcionalmente sustituido;
X1, X2, X3, y X4 pueden ser iguales o diferentes y cada uno representa N o CR4 (en donde R4 representa un átomo de hidrógeno, alquilo inferior, ciano, halógeno, hidroxi, alcoxi inferior, alcanoílo inferior o alquilsulfonilo inferior); Y1 representa CH2 o C(=O);
Y2 representa CH o N y
L representa CH2 o NH].
2. El compuesto o la sal farmacéuticamente aceptable del mismo, de acuerdo con la reivindicación 1, en donde Y2 es N y L es CH2.
3. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de la reivindicación 1 o la reivindicación 2, en donde Y1 es CH2.
4. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las reivindicaciones 1 a 3, en donde n1 es 0.
5. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las reivindicaciones 1 a 4, en donde R1 es (i) arilo opcionalmente sustituido, en donde el arilo es fenilo o (ii) un grupo heterocíclico aromático opcionalmente sustituido, en donde el grupo heterocíclico aromático es piridilo, piridonilo o pirimidinilo.
6. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las reivindicaciones 1 a 4, en donde R1 es arilo opcionalmente sustituido o un grupo heterocíclico aromático opcionalmente sustituido y el grupo es un grupo representado por la fórmula (a1) siguiente:

[en donde R5 representa un átomo de hidrógeno, alquilo C1-10 que puede estar sustituido con hidroxi, alcoxicarbonilo C1-10, alcanoílo C2-11, alquilsulfonilo C1-10, -NR6aR6b (en donde R6a y R6b pueden ser iguales o diferentes y cada uno representa un átomo de hidrógeno, alcanoílo C2-11 o alquilo C1-10), -CONR6cR6d (en donde R6c y R6d pueden ser iguales o diferentes y cada uno representa un átomo de hidrógeno o alquilo C1-10), -SO2NR6eR6f (en donde R6e y R6f pueden ser iguales o diferentes y cada uno representa un átomo de hidrógeno o alquilo C1-10), halógeno, ciano, carboxi o nitro y Z1, Z2, Z3 y Z4 pueden ser iguales o diferentes y cada una representa N o CR7 (en donde R7 representa un átomo de hidrógeno, carboxi o halógeno)] o un grupo representado por la fórmula (a2) siguiente:

(en donde R5, Z1 y Z4 tienen las mismas definiciones que se han descrito anteriormente, respectivamente).
7. El compuesto o la sal farmacéuticamente aceptable del mismo, de acuerdo con la reivindicación 6, en donde R5 es ciano, -CONH2 o -SO2NH2.
8. El compuesto o la sal farmacéuticamente aceptable del mismo, de acuerdo con la reivindicación 6, en donde R5 es ciano.
9. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las reivindicaciones 6 a 8, en donde R7 es un átomo de hidrógeno o un átomo de flúor.
10. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las reivindicaciones 1 a 9, en donde R3 es un grupo heterocíclico aromático bicíclico.
11. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las reivindicaciones 1 a 9, en donde R3 es quinazolinilo.
12. Una composición farmacéutica, que comprende, como principio activo, el compuesto o la sal farmacéuticamente aceptable del mismo descrito en una cualquiera de las reivindicaciones 1 a 11.
13. El compuesto o la sal farmacéuticamente aceptable del mismo descrito en una cualquiera de las reivindicaciones 1 a 11 para su uso en el tratamiento y/o prevención de cáncer, fibrosis pulmonar, fibromatosis u osteoartritis.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013156458 | 2013-07-29 | ||
| PCT/JP2014/069872 WO2015016195A1 (ja) | 2013-07-29 | 2014-07-29 | Wntシグナル阻害剤 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2771151T3 true ES2771151T3 (es) | 2020-07-06 |
Family
ID=52431728
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14832281T Active ES2771151T3 (es) | 2013-07-29 | 2014-07-29 | Derivados de piperidina como inhibidor de señalización wnt |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US9771351B2 (es) |
| EP (1) | EP3028703B1 (es) |
| JP (1) | JP6522502B2 (es) |
| ES (1) | ES2771151T3 (es) |
| WO (1) | WO2015016195A1 (es) |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3849458A (en) | 1966-01-21 | 1974-11-19 | Incentive Res & Dev Ab | Method for deuterating organic compounds |
| JPS61275241A (ja) | 1985-05-29 | 1986-12-05 | Mitsubishi Rayon Co Ltd | 重水素化アクリル酸又は重水素化メタクリル酸の製造方法 |
| JPS61277648A (ja) | 1985-06-03 | 1986-12-08 | Mitsubishi Rayon Co Ltd | 重水素化アクリル酸又は重水素化メタクリル酸の製造方法 |
| DE3701302A1 (de) | 1987-01-17 | 1988-07-28 | Hoechst Ag | Verfahren zur herstellung von deuterierten organischen verbindungen |
| JP3107100B2 (ja) | 1991-07-11 | 2000-11-06 | 大日本インキ化学工業株式会社 | 静電荷像現像用カラートナーの組合せ |
| JPH09165385A (ja) | 1994-08-26 | 1997-06-24 | Kyowa Hakko Kogyo Co Ltd | キナゾリン誘導体 |
| DE69722630T2 (de) | 1996-02-19 | 2004-04-29 | Kyowa Hakko Kogyo Co., Ltd. | Therapeutisches mittel zur behandlung von nierenerkrankungen |
| CA2524269A1 (en) | 2003-05-02 | 2004-11-18 | Elan Pharmaceuticals, Inc. | 4- bromo - 5 - (2- chloro - benzoylamino) - 1h - pyrazole - 3 - carboxylic acid amide derivatives and related compounds as bradykinin b1 receptor antagonists for the treatment of inflammatory diseases |
| JP4916878B2 (ja) | 2003-06-19 | 2012-04-18 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | 5ht4−拮抗薬としてのアミノスルホニル置換4−(アミノメチル)−ピペリジンベンズアミド |
| WO2005016926A1 (en) | 2003-08-19 | 2005-02-24 | Warner-Lambert Company Llc | Pyrido [3,4-d] pyrimidine derivatives as matrix metalloproteinase-13 inhibitors |
| WO2005085226A1 (en) | 2004-03-10 | 2005-09-15 | Janssen Pharmaceutica N.V. | Mtp inhibiting aryl piperidines or piperazines substituted with 5-membered heterocycles |
| GB0503962D0 (en) | 2005-02-25 | 2005-04-06 | Kudos Pharm Ltd | Compounds |
| WO2007125315A2 (en) | 2006-04-25 | 2007-11-08 | Astex Therapeutics Limited | Pharmaceutical compounds |
| ES2368456T3 (es) | 2006-05-02 | 2011-11-17 | Bristol-Myers Squibb Company | Compuestos restringidos como antagonistas del receptor de cgrp. |
| TW200811147A (en) | 2006-07-06 | 2008-03-01 | Arena Pharm Inc | Modulators of metabolism and the treatment of disorders related thereto |
| WO2008130951A1 (en) | 2007-04-17 | 2008-10-30 | Bristol-Myers Squibb Company | Fused heterocyclic 11-beta-hydroxysteroid dehydrogenase type i inhibitors |
| EP2185556A1 (en) * | 2007-08-27 | 2010-05-19 | Wyeth a Corporation of the State of Delaware | Imidazopyridine analogs and their use as agonists of the wnt-beta-catenin cellular messaging system |
| CA2726164A1 (en) | 2008-05-27 | 2009-12-23 | The Board Of Regents Of The University Of Texas System | Wnt protein signalling inhibitors |
| AU2009278442B2 (en) | 2008-08-05 | 2013-09-26 | Daiichi Sankyo Company, Limited | Imidazopyridin-2-one Derivatives |
| JO3265B1 (ar) | 2008-12-09 | 2018-09-16 | Novartis Ag | مثبطات بيريديلوكسى اندولات vegf-r2 واستخدامها لعلاج المرض |
| EP2485728B1 (en) | 2009-10-07 | 2013-07-10 | Siena Biotech S.p.a. | Wnt pathway antagonists |
| US20120184562A1 (en) | 2011-01-19 | 2012-07-19 | Kin-Chun Luk | 1,6- and 1,8-naphthyridines |
-
2014
- 2014-07-29 EP EP14832281.1A patent/EP3028703B1/en active Active
- 2014-07-29 WO PCT/JP2014/069872 patent/WO2015016195A1/ja active Application Filing
- 2014-07-29 ES ES14832281T patent/ES2771151T3/es active Active
- 2014-07-29 JP JP2015529571A patent/JP6522502B2/ja not_active Expired - Fee Related
- 2014-07-29 US US14/908,945 patent/US9771351B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2015016195A1 (ja) | 2017-03-02 |
| EP3028703B1 (en) | 2019-12-04 |
| WO2015016195A1 (ja) | 2015-02-05 |
| US9771351B2 (en) | 2017-09-26 |
| US20160168125A1 (en) | 2016-06-16 |
| EP3028703A1 (en) | 2016-06-08 |
| JP6522502B2 (ja) | 2019-05-29 |
| EP3028703A4 (en) | 2017-01-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2935746T3 (es) | Inhibidores de desmetilasa-1 específica de lisina | |
| AU2016228660B2 (en) | Substituted 2-hydrogen-pyrazole derivative serving as anticancer drug | |
| RU2553681C2 (ru) | N-содержащие гетероарильные производные в качестве ингибиторов jak3 киназы | |
| ES2666353T3 (es) | Inhibidores de JAK3 de imidazopiridazina y su uso para el tratamiento de enfermedades inflamatorias y autoinmunitarias | |
| ES2908412T3 (es) | Derivados de piperidin-4-IL azetidina como inhibidores de JAK1 | |
| EP3746435A1 (en) | Substituted quinazoline and pyridopyrimidine derivatives useful as anticancer agents | |
| JP2021518395A (ja) | Shp2阻害剤およびその使用 | |
| AU2018237123B2 (en) | Bruton's tyrosine kinase inhibitors | |
| EP3414234A1 (en) | Bruton's tyrosine kinase inhibitors | |
| BR112019012927A2 (pt) | Composto, uso de um composto, combinação de um composto, composição farmacêutica e dispositivo | |
| KR20150059647A (ko) | 키나제 억제제로서 유용한 이미다조트리아진카르보니트릴 | |
| WO2019074809A1 (en) | INDAZOLYL-SPIRO [2.2] PENTANE-CARBONITRILE DERIVATIVES AS LRRK2 INHIBITORS, PHARMACEUTICAL COMPOSITIONS AND USES THEREOF | |
| JP2022502440A (ja) | モノアシルグリセロールリパーゼ調節因子 | |
| TW202208379A (zh) | 新穎的巨環lrrk2激酶抑制劑 | |
| ES2963339T3 (es) | Compuestos de pirazolopiridinona | |
| WO2022140769A1 (en) | Lactam (hetero)arylfusedpyrimidine derivatives as inhibitors of erbb2 | |
| KR20200092987A (ko) | 피라졸로피리디논 화합물 | |
| KR20230051500A (ko) | Alk5 억제제로서 피리다진일 아미노 유도체 | |
| KR20160086930A (ko) | 피롤로피롤론 유도체 및 bet 억제제로서의 그의 용도 | |
| ES2771151T3 (es) | Derivados de piperidina como inhibidor de señalización wnt | |
| KR20210108433A (ko) | 티에노피리디논 화합물 | |
| TWI793877B (zh) | 作為甘油二酯激酶抑制劑的雜環化合物及其用途 | |
| WO2023154124A1 (en) | Acylated heterocyclic quinazoline derivatives as inhibitors of erbb2 | |
| TW202246261A (zh) | 作為抗癌劑的化合物 | |
| CN116262750A (zh) | 一种芳杂环类化合物及其制备方法和用途 |