EP0014963A2 - Verfahren zur Herstellung von bicyclischen Enoläthern sowie Äther von 2-(3-Hydroxyprop-1-yl)-cycloalkanonen - Google Patents
Verfahren zur Herstellung von bicyclischen Enoläthern sowie Äther von 2-(3-Hydroxyprop-1-yl)-cycloalkanonen Download PDFInfo
- Publication number
- EP0014963A2 EP0014963A2 EP80100747A EP80100747A EP0014963A2 EP 0014963 A2 EP0014963 A2 EP 0014963A2 EP 80100747 A EP80100747 A EP 80100747A EP 80100747 A EP80100747 A EP 80100747A EP 0014963 A2 EP0014963 A2 EP 0014963A2
- Authority
- EP
- European Patent Office
- Prior art keywords
- mol
- tert
- ethers
- allyl
- preparation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/94—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems condensed with rings other than six-membered or with ring systems containing such rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/68—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
- C07C45/69—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by addition to carbon-to-carbon double or triple bonds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/385—Saturated compounds containing a keto group being part of a ring
- C07C49/517—Saturated compounds containing a keto group being part of a ring containing ether groups, groups, groups, or groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/04—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D307/18—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D307/20—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/08—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D309/10—Oxygen atoms
- C07D309/12—Oxygen atoms only hydrogen atoms and one oxygen atom directly attached to ring carbon atoms, e.g. tetrahydropyranyl ethers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/74—Benzo[b]pyrans, hydrogenated in the carbocyclic ring
Definitions
- the present invention relates to an improved process for the preparation of bicyclic enol ethers of the general formula I. in which n has a value from 3 to 12 and in which the radicals R 1 - R 3 are hydrogen or a C 1 - C 4 alkyl radical.
- the invention further relates to new ethers of 2- (3-hydroxy-prop-1-yl) cycloalkanes of the general formula II in which R 4 denotes the tert-butyl group, the tetrahydrofuran-2-yl group or the tetrahydropyran-2-yl group.
- the invention was therefore based on the object of the basic method known from DE-PS 21 36 496 and To improve the avoidance of the disadvantages mentioned.
- bicyclic enol ethers of the general formula I in which n has a value of 3 to 12 and in which the radicals R 1 - R 3 represent hydrogen or a C 1 - C 4 alkyl radical, by radical addition of an allyl compound to a cyclic ketone of the general formula IV and subsequent acid-catalyzed cyclization to (I) is advantageously obtained if an allyl ether of the general formula III is used as the allyl compound used, in which R 4 is the tert-butyl group, the tetrahydrofuran-2-yl group or the tetrahydropyran-2-yl group.
- the starting compounds (III) are derived in accordance with the mainly desired process products I from the allyl alcohol.
- but-2-en-1-ol, but-1-en-3-ol and methallyl alcohol are of particular importance.
- the ethers of these alcohols according to the definition can be obtained in a simple manner by reacting the alcohols with isobutene, 2,3-dihydrofuran or 2,3-dihydropyran in the presence of acidic catalysts, it being particularly expedient to use acidic ion exchangers as catalysts.
- This etherification is generally carried out at normal pressure or at elevated pressure - approximately up to 50 bar - and at temperatures of 0-100 ° C., preferably 40-80 ° C.
- the radical catalysts for the reaction of (III) with (IV) can in principle be of any type, but preference is given to using those which only become active at those temperatures, i.e. decay into radicals in which the ⁇ addition of (III) to (IV) proceeds with sufficient speed. These temperatures are usually between 80 and 180 ° C, especially between 120 and 150 ° C. In general, this reaction can be carried out at 50-200 ° C, although it slows down noticeably at temperatures lower than 80 ° C and even if the formation is generally higher than 150 ° C. of by-products.
- suitable catalysts for the particularly preferred temperature range of 120-150 ° C. are, for example, dibenzoyl peroxide, tert-butyl peroctoate and azodiisobutyronitrile, and especially di-tert-butyl peroxide.
- (III) and (IV) react with one another in equimolar proportions, but in order to suppress side reactions, it is generally advantageous to use (IV) in a molar excess of up to 20 times.
- the amount of the radical catalyst is preferably in the range of 0.05-0.7 mol, particularly 0.1-0.3 mol, per mole (III).
- reaction of (III) with (IV) gives the compounds.
- (II) which, if desired, can be isolated in the customary manner, but is preferably subjected directly to the acid-catalyzed cyclization reaction which proceeds with the elimination of water and isobutene, 2,3-dihydrofuran or 2,3-dihydropyran.
- any acids i.e. protonic acids and Lewis acids
- a homogeneous or heterogeneous phase e.g. acidic ion exchangers
- acids e.g. acidic ion exchangers
- semi-volatile organic strong acids in a homogeneous phase have proven particularly useful, with cheap p-toluenesulfonic acid being the first to be mentioned.
- the amount of the acid is advantageously 0.001-0.3, particularly 0.05-0.2 mol equivalents. per mole (II).
- the cyclization is preferably carried out at 60-150 ° C., particularly 80-130 ° C. and under a pressure of 0.05 mbar-50 bar, particularly 0.1-1 mbar.
- the removal of the water can be done by ongoing azeotropic distillation, e.g. with toluene. After the water has been separated off, the olefinic cleavage products can be returned to the stage of the preparation of (III).
- the process can be carried out discontinuously as well as continuously according to the usual techniques, so that further details are not necessary. This also applies to the processing for the process products (I).
- the main advantage of the process is that the allyl alcohols (III ') are converted into the ethers (III) at relatively low temperatures, at the reaction temperatures the radical addition is much easier to handle than the free alcohols. Despite the additional etherification stage, the process is more economical than when the alcohols (III ') are used directly.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Furan Compounds (AREA)
- Pyrane Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract

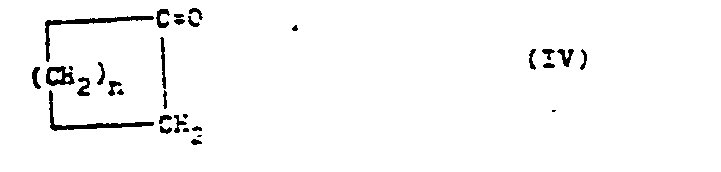
Description
- Die vorliegende Erfindung betrifft ein verbessertes Verfahren zur Herstellung bicyclischer Enoläther der allgemeinen Formel I


- Aus der DE-PS 21 36 496 ist es bekannt, die Verbindungen I, die als Zwischenprodukte für die Synthese von moschusartigen Duftstoffen Bedeutung haben, durch radikalische Addition eines Allylalkohols oder eines Allylesters (III')



- Dieses Verfahren ist wegen der großen Empfindlichkeit des Allylalkohols und seiner Homologen nachteilig. Geht man hingegen von den Estern dieser Alkohole aus, so erfordert dies nach der Additionsreaktion den zusätzlichen Verfahrensschritt der hydrolytischen Abspaltung der Säure.
- Ferner ist aus Isvest. Akad. Nauk, SSSR, Otdel. Khim. Nauk 1961, Seite 2065 f die radikalische α-Addition von Allyl- äthern von n-Alkan-1-olen an Cyclopentanon und Cyclohexanon zu Verbindungen des Typs (II) bekannt, jedoch lassen sich diese Verbindungen nicht zu bicyclischen Enoläthern cyclisieren.
- Der Erfindung lag daher die Aufgabe zugrunde, das aus der DE-PS 21 36 496 in seinen Grundzügen bekannte Verfahren un- r ter Vermeidung der genannten Nachteile zu verbessern.
- Es wurde nun gefunden, daß man bicyclische Enoläther der allgemeinen Formel I



- Die Ausgangsverbindungen (III) leiten sich, entsprechend den hauptsächlich gewünschten Verfahrensprodukten I, vor allem vom Allylalkohol ab. Daneben haben besonders das But-2-en-1--ol, das But-1-en-3-ol und der Methallylalkohol Bedeutung.
- Die definitionsgemäßen Äther dieser Alkohole sind in einfacher Weise durch Umsetzung der Alkohole mit Isobuten, 2,3-Dihydrofuran bzw. 2,3-Dihydropyran in Gegenwart saurer Katalysatoren erhältlich, wobei es besonders zweckmäßig ist, als Katalysatoren saure Ionenaustauscher zu verwenden. Man nimmt diese Verätherung im allgemeinen bei Normaldruck oder bei erhöhtem Druck - etwa bis zu 50 bar - und bei Temperaturen von 0 - 100°C, vorzugsweise 40 - 80°C, vor.
- Unter den Ausgangsverbindungen (IV) sind das Cyclopentanon, Cyclohexanon, Cyclooctanon und vor allem das Cyclododecanon hervorzuheben.
- Die radikalischen Katalysatoren für die Umsetzung von (III) mit (IV) können prinzipiell beliebig sein, jedoch verwendet man vorzugsweise solche, die erst bei denjenigen Temperaturen aktiv werden, d.h. in Radikale zerfallen, bei denen die α-Addition von (III) an (IV) mit genügender Geschwindigkeit verläuft. Diese Temperaturen liegen in der Regel zwischen 80 und 180°C, besonders zwischen 120 und 150°C. Allgemein läßt sich diese Reaktion bei 50 - 200°C ausführen, wenngleich sie sich bei tieferen Temperaturen als 80°C merklich verlangsamt und wenngleich man über 150°C in der Regel mit der vermehrten Bildung. von Nebenprodukten zu rechnen hat. Für die Wahl der Temperatur ist es weiterhin maßgebend, daß man die Umsetzung nach Möglichkeit bei Normaldruck ausführt, weil sowohl geringerer Druck - etwa bis zu 200 mbar absolut - als auch höherer Druck - etwa bis zu 50 bar - den wirtschaftlichen Nachteil der Verwendung von Druckapparaturen bedingt.
- Als Katalysatoren für den besonders bevorzugten Temperaturbereich von 120 - 150°C eignen sich aus den genannten Gründen beispielsweise Dibenzoylperoxid, tert.-Butylperoctoat und Azodiisobutyronitril sowie besonders Di-tert.-butylperoxid.
- (III) und (IV) reagieren in äquimolaren Mengenverhältnissen miteinander, jedoch ist es im allgemeinen zur Unterdrückung von Nebenreaktionen vorteilhaft, (IV) im etwa bis zu 20-fachen molaren Überschuß einzusetzen. Die Menge des radikalischen Katalysators liegt vorzugsweise im Bereich von 0,05 - 0,7 mol, besonders 0,1 - 0,3 mol pro Mol (III).
- Soweit (III) und (IV) bei den Reaktionstemperaturen flüssig sind und der Katalysator im Reaktionsgemisch ausreichend löslich ist, erübrigt sich in aller Regel die Mitverwendung eines inerten Lösungsmittels. Möchte man trotzdem in Gegenwart eines Lösungsmittels arbeiten, etwa weil die Ausgangsverbindungen bereits in Lösung vorliegen oder weil sich die Bildung von Nebenprodukten mit Hilfe von Lösungsmitteln zuweilen vermindern läßt, so eignen sich hierfür z.B. Petrol- äther, Cyclohexan, Benzol und Chlorbenzol.
- Bei der Umsetzung von (III) mit (IV) erhält man die Verbindungen.(II)

- Als Säuren lassen sich grundsätzlich beliebige Säuren, also Protonensäuren und Lewissäuren, in homogener oder heterogener Phase (z.B. saure Ionenaustauscher) verwenden. Aus verfahrenstechnischen Gründen haben sich jedoch schwerflüchtige organisch starke Säuren in homogener Phase besonders bewährt, wobei die billige p-Toluolsulfonsäure an erster Stelle zu nennen ist.
- Die Menge der Säure beträgt zweckmäßigerweise 0,001 - 0,3, besonders 0,05 - 0,2 moläqu. pro Mol (II).
- Vorzugsweise nimmt man die Cyclisierung bei 60 - 150°C, besonders bei 80 - 130°C und unter einem Druck von 0,05 mbar - 50 bar, besonders von 0,1 - 1 mbar vor. Die Entfernung des Wassers kann durch laufende Azeotropdestillation, z.B. mit Toluol, begünstigt werden. Die olefinischen Spaltprodukte lassen sich nach Abtrennung des Wassers wieder in die Stufe der Herstellung von (III) zurückführen.
- Sieht man von der erfindungsgemäßen Verbesserung ab, so läßt sich das Verfahren sowohl diskontinuierlich als auch kontinuierlich nach den üblichen Techniken ausführen, so daß sich nähere Angaben hierzu erübrigen. Dies gilt auch für die Aufarbeitung auf die Verfahrensprodukte (I).
- Das Verfahren bietet vor allem den Vorteil, daß man die Allylalkohole (III') bei relativ niedrigen Temperaturen in die Äther (III) überführt, die bei den Reaktionstemperaturen der radikalischen Addition wesentlich leichter zu handhaben sind als die freien Alkohole. Trotz der zusätzlichen Ver- ätherungsstufe gestaltet sich das Verfahren wirtschaftlicher als bei unmittelbarer Verwendung der Alkohole (III').
- Herstellung von 13-Oxabicyclo-[10.4.0]-hexadec-Δ-1,12-en 1820 g (10 mol) Cyclododecanon wurden unter Stickstoffatmo- sphäre bei 140°C im Laufe von 4 Stunden mit einer Lösung aus 114 g (1 mol) Allyl-tert.-butyläther und 29,2 g (0,2 mol) Di-tert.-butylperoxid versetzt und noch eine weitere Stunde bei dieser Temperatur gehalten. Das überschüssige Cyclododecanon wurde sodann abdestilliert, wonach der Rückstand zusammen mit 10 g (58 mmol) p-Toluolsulfonsäure eine Stunde lang bei 300 mbar auf 130 C erhitzt wurde. Hierbei wurden 0,7 mol Wasser und 0,7 mol Isobuten abgespalten. Die übliche Aufarbeitung lieferte die oben genannte Verbindung in 56%iger Ausbeute.
- Sdp. 112 - 114°C / 0,1 mbar; n

- Herstellung von 13-Oxabicyclo-[10.4.0]-hexadec-Δ-1,12-en Cyclododecanon und Allyl-tert.-butyläther wurden analog Beispiel 1 im Laufe von 6 Stunden miteinander umgesetzt. Nach Entfernung des überschüssigen Cyclododecanons wurde der Rückstand zusammen mit 10 g p-Toluolsulfonsäure und 1 1 Toluol bei Normaldruck 3 Stunden lang unter Rückflußkühlung erhitzt, wobei das Wasser laufend azeotrop abdestilliert wurde. Die Lösung wurde mit verdünnter wäßriger NaHCO3-Lösung neutral gewaschen und danach wie üblich aufgearbeitet. Die Ausbeute an dem oben genannten Verfahrensprodukt betrug 65 %.
- 638 g (7,6 mol) Cyclopentanon wurden unter Stickstoffatmosphäre bei 125 - 130°C im Laufe von 4 Stunden mit einer Lösung aus 87 g (0,76 mol) Allyl-tert.-butyläther und 33 g (0,23 mol) Di-tert.-butylperoxid versetzt und anschließend noch 4 Stunden auf 120°C gehalten.
- Die destillative Aufarbeitung des Reaktionsgemisches lieferte das 2-(3-tert.-Butaxy-prop-1-yl)-cyclopentanon in 50%iger Ausbeute.
- Sdp. 95 - 90°C / 0,4 mbar; n

- Herstellung von 6-Oxabicyclo-[3,4,0]-non-Δ-1,5-en
- 37 g (0,19 mol) des Verfahrensproduktes von Beispiel 3 wurden zusammen mit 5 g (29 mmol) p-Toluolsulfonsäure 1 Stunde lang bei 18 mbar auf 100°C erhitzt. Danach wurde das Reaktionsgemisch wie üblich aufgearbeitet; die Ausbeute an dem oben genannten bicyclischen Enoläther betrug 30 %, bezogen auf den eingesetzten Allyläther.
- Sdp. 68 - 70°C / 0,15 mbar
- 980 g (10 mol) Cyclohexanon wurden unter Rühren bei 140°C im Laufe von 2 Stunden mit einer Lösung aus 114 g (1 mol) Allyl-tert.-butyläther und 29,2 g (0,2 mol) Di-tert.--butylperoxid versetzt und noch eine Stunde bei dieser Tem- peratur gehalten. Die destillative Aufarbeitung des Reaktionsgemisches lieferte das 2-(3-tert.-Butoxy-prop-1-yl)--cyclohexanon in 60%iger Ausbeute.
- Sdp. 85 - 90°C / 0,05 mbar; n

- Herstellung von 7-Ox2bicyclo-[4.4.0]-dec-Δ-1,5-en
- 180 g (0,85 mol) des Verfahrensproduktes von Beispiel 5 wurden zusammen mit 10 g (58 mmol) p-Toluolsulfonsäure und 1 1 Toluol analog Beispiel 2 der Cyclisierungsreaktion unterworfen. Die Ausbeute an dem oben genannten Verfahrensprodukt betrug 42 %, bezogen auf den Allyläther.
- Sdp. 66 - 70°C / 0,05 mbar; n

- 504 g (4 mol) Cyclooctanon, 46 g (0,4 mol) Allyl-tert.--butyläther und 23 g (0,16 mol) Di-tert.-butylperoxid wurden analog Beispiel 1 umgesetzt. Die Ausbeute an 2-(3-tert.--Butoxy-prop-1-yl)-cyclooctanon betrug 84 %.
- Sdp. 105 - 110°C / 0,15 mbar; n

- Herstellung von 9-Oxabicyclo-[6.4.0]-dodec-Δ-1,8-en
- 49 g (0,2 mol) des Verfahrensprodukts von Beispiel 7 wurden zusammen mit 5 g (29 mmol) p-Toluolsulfonsäure und 0,5 Toluol auf die in Beispiel 2 beschriebene Weise der Cyclisierungsreaktion unterworfen. Die Aufarbeitung lieferte die oben genannte Verbindung in 75%iger Ausbeute, bezogen auf den Allyläther.
- Sdp. 55°C / 0,15 mbar; n

- Herstellung von 13-Oxabicyclo-[10.4.0]-hexadec-Δ-1,12-en
- 237 g (1,3 mol) Cyclcdodecanon wurden bei 140°C im Laufe von 6 Stunden mit 142 g (1 mol) 2-Allyloxytetrahydropyran und 56 g (0,38 mol) Di-tert.-butylperoxid versetzt und anschließend noch 5 Stunden bei dieser Temperatur gehalten.
- Nach Entfernung des überschüssigen Cyclododecanons wurde das Additionsprodukt zusammen mit 1 g (6 mmol) p-Toluolsulfonsäure bei 0,1 mbar auf 125°C erhitzt, wobei die oben genannte Verbindung überdestillierte und in 65%iger Ausbeute anfiel.
- Unter gleichen Bedingungen, jedoch mit 10 mol Cyclodedecanon konnte die Ausbeute auf 75 % erhöht werden.
- Herstellung von 7-Oxabicyclo-[4.4.0]-dec-Δ-1,6-en
- 980 g (10 mol) Cyclohexanon, 142 g (1 mol) 2-(Allyloxy)--tetrahydropyran und 56 g (0,38 mol) Di-tert.-butylperoxid wurden analog Beispiel 6 zu der oben genannten Verbindung umgesetzt; die Ausbeute betrug 55 %.
- Herstellung von 14-Methyl-13-oxabicyclo-[10.4.0]-hexadec-Δ--1,12-en
- 910 g (5 mol) Cyclododecanon, 78 g (0,5 mol)-2-(But-1-en-3--yloxy)-tetrahydropyran und 28 g (0,18 mol) Di-Tert.-butylperoxid wurden analog Beispiel 6 zu der oben genannten Verbindung umgesetzt. Die Ausbeute betrug 44 %.
- Sdp. 110 - 1120C / 0,1 mbar.
- Herstellung von 13-Oxabicyclo-[10.4.0]-hexadec-Δ-1,12-en
- 455 g (2,5 mol) Cyclododecanon wurden unter Stickstoffatmosphäre bei 100°C im Laufe von 4 Stunden mit einer Lösung aus 57 g (0,5 mol) Allyl-tert.-butyläther und 23 g (0,1 mol) tert.-Butylperoctoat versetzt und noch eine weitere Stunde bei dieser Temperatur gehalten. Das überschüssige Cyclododecanon wurde sodann abdestilliert, wonach der Rückstand zusammen mit 3 g (17,4 mmol) p-Toluolsulfonsäure eine Stunde lang bei 20 mbar auf 100°C erhitzt wurde. Danach wurde das Reaktionsgemisch wie üblich aufgearbeitet; die Ausbeute an dem oben genannten bicyclischen Enoläther betrug 30 %, bezogen auf den Allyläther.
- Kontinuierliche Herstellung von 13-Oxabicyclo-[10.4.0]-hexa- dec-Δ-1,12-en
- Eine Lösung aus 1 400 g Cyclohexan, 310 g (5 mol) Cyclododecanon, 114 g (1 mol) Allyl-tert.-butyläther und 58,4 g (0,4 mol) Di-tert.-butylperoxid wurden im Verlauf von 25 Stunden bei 150°C und 18 bar durch einen 0,3-1-Rührautoklaven gepumpt. Aus dem Reaktionsaustrag wurde das Lösungsmittel und das überschüssige Cyclododecanon abdestilliert, wonach der Rückstand zusammen mit 15 g (87 mmol) p-Toluolsulfonsäure eine Stunde lang bei 300 mbar auf 120°C erhitzt wurde. Die übliche Aufarbeitung lieferte die oben genannte Verbindung in 40%iger Ausbeute.
Claims (2)




Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19792906296 DE2906296A1 (de) | 1979-02-19 | 1979-02-19 | Verfahren zur herstellung von bicyclischen enolaethern sowie aether von 2-(3-hydroxyprop-1-yl)-cycloalkanonen |
| DE2906296 | 1979-02-19 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| EP0014963A2 true EP0014963A2 (de) | 1980-09-03 |
| EP0014963A3 EP0014963A3 (en) | 1980-11-26 |
| EP0014963B1 EP0014963B1 (de) | 1983-07-13 |
Family
ID=6063293
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP80100747A Expired EP0014963B1 (de) | 1979-02-19 | 1980-02-14 | Verfahren zur Herstellung von bicyclischen Enoläthern sowie Äther von 2-(3-Hydroxyprop-1-yl)-cycloalkanonen |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US4268445A (de) |
| EP (1) | EP0014963B1 (de) |
| JP (1) | JPS55111437A (de) |
| DE (2) | DE2906296A1 (de) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3137939A1 (de) * | 1981-09-24 | 1983-05-19 | Consortium für elektrochemische Industrie GmbH, 8000 München | "substituierte macrobicyclische ether, deren herstellung und verwendung" |
| JPS6212737A (ja) * | 1985-07-10 | 1987-01-21 | Ube Ind Ltd | 新規な10員環化合物及びその製造方法 |
| US4999439A (en) * | 1990-03-22 | 1991-03-12 | International Flavors & Fragrances Inc. | Alkyl-substituted tetra- or hexahydrobenzopyran derivatives, organoleptic uses thereof and process for preparing same |
| EP2197865B1 (de) * | 2007-09-07 | 2012-08-22 | Furanix Technologies B.V | Hydroxymethylfurfuralether aus hmf und olefinen |
| EP2540713A1 (de) | 2011-06-30 | 2013-01-02 | Basf Se | Makrocyclische Lactone |
| US8648031B2 (en) | 2011-06-30 | 2014-02-11 | Basf Se | Macrocyclic lactones |
| US8410293B2 (en) | 2011-06-30 | 2013-04-02 | Basf Se | Process for the preparation of cyclic enol ethers |
| EP2540712A1 (de) | 2011-06-30 | 2013-01-02 | Basf Se | Verfahren zu Herstellung cyclischer Enolether |
| EP3050869B1 (de) * | 2015-01-30 | 2019-10-02 | Symrise AG | Verfahren zur Herstellung von substituierten Alkylcycloalkanonen |
| EP3339298B1 (de) * | 2016-12-20 | 2021-07-07 | International Flavors & Fragrances Inc. | Verfahren zur herstellung oxa-bicycloalken |
| US20230257679A1 (en) * | 2020-07-31 | 2023-08-17 | S H Kelkar And Company Limited | Odorants and compositions comprising odorants |
| CN118908931B (zh) * | 2024-06-28 | 2025-09-09 | 安徽华业香料合肥有限公司 | 一种环十五内酯中间体双环烯醚的制备方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3907831A (en) * | 1969-05-29 | 1975-09-23 | Firmenich & Cie | Process for preparing lactones |
| BE786518A (fr) * | 1971-07-21 | 1973-01-22 | Haarmann & Reimer Gmbh | Procede de preparation d'oxa-bicyclo-alcenes |
-
1979
- 1979-02-19 DE DE19792906296 patent/DE2906296A1/de not_active Withdrawn
-
1980
- 1980-01-31 US US06/117,128 patent/US4268445A/en not_active Expired - Lifetime
- 1980-02-13 JP JP1558080A patent/JPS55111437A/ja active Granted
- 1980-02-14 DE DE8080100747T patent/DE3064073D1/de not_active Expired
- 1980-02-14 EP EP80100747A patent/EP0014963B1/de not_active Expired
Also Published As
| Publication number | Publication date |
|---|---|
| JPS6326758B2 (de) | 1988-05-31 |
| US4268445A (en) | 1981-05-19 |
| JPS55111437A (en) | 1980-08-28 |
| DE2906296A1 (de) | 1980-08-28 |
| EP0014963B1 (de) | 1983-07-13 |
| DE3064073D1 (en) | 1983-08-18 |
| EP0014963A3 (en) | 1980-11-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0014963B1 (de) | Verfahren zur Herstellung von bicyclischen Enoläthern sowie Äther von 2-(3-Hydroxyprop-1-yl)-cycloalkanonen | |
| DE2216974C3 (de) | Verfahren zur Herstellung höhermolekularer ungesättigter Ketone | |
| EP0191935A1 (de) | Verfahren zur Herstellung von 2-Alkyl-1,4-butandial | |
| EP0032576B1 (de) | Verfahren zur Herstellung von p-substituierten Phenylpropanolen und deren Estern sowie von p-substituierten 3-Phenyl-2-methyl-propanalen | |
| DE2608932C2 (de) | Verfahren zur Herstellung von 3-Oxy-4H-pyran-4-on-Derivaten | |
| EP0038480B1 (de) | Verfahren zur Herstellung von 1-(4-Hydroxy-phenyl)-butan-3-on sowie neue Zwischenprodukte dieses Verfahrens | |
| DE2715208B2 (de) | Verfahren zur Herstellung von 3-Methyl-2-buten-l-al | |
| DE2225612C2 (de) | Cyclische Methyl-fumardialdehyd-monoacetale und Verfahren zu ihrer Herstellung | |
| DE2759994C2 (de) | Ketale von 4-(1-Hydroxy-1-methylethyl)-3-cyclohexen-1-on und Verfahren zu ihrer Herstellung | |
| DE2804597C2 (de) | Verfahren zur Herstellung von β-Damascon und β-Damascenon | |
| DE2536503B2 (de) | M-DichloM-methyl-S-hydroxypenten-(l) | |
| EP0512348B1 (de) | Derivate cyclischer Lactone, Verfahren zu ihrer Herstellung und Verfahren zur Herstellung von 15-Pentadecanolid und seinen Homologen | |
| EP0596246B1 (de) | Neue 2-tert.-Amylverbindungen | |
| DE2852587C3 (de) | Verfahren zur Herstellung von Methylheptenon | |
| DE2831385C2 (de) | ||
| EP0436860A1 (de) | Verfahren zur Herstellung von 2-(4-Chlorphenyl)-3-methyl-buttersäure | |
| EP0021013A1 (de) | Neue 2,4-disubstituierte Pyranderivate, deren Herstellung und deren Verwendung als Riechstoffe | |
| EP0075866B1 (de) | Substituierte macrobicyclische Ether, deren Herstellung und Verwendung | |
| DE2719976C2 (de) | Bicyclische Aldehyde und Verfahren zu deren Herstellung | |
| DE2157035A1 (de) | Verfahren zur herstellung hoehermolekularer alpha-beta-ungesaettigter aldehyde | |
| DE2925176A1 (de) | Verfahren zur herstellung von beta -damascon und beta -damascenon | |
| EP0579113A1 (de) | Verbessertes Verfahren zur Herstellung von cyclischen Acetalen von 3-formyl-2-butenyl-triphenylphosphoniumchlorid | |
| DE10055653A1 (de) | Verfahren zur Oxidation von Ketonen und Aldehyden mit Wasserstoffperoxid über eine Baeyer-Villiger-Oxidation | |
| DE69701054T2 (de) | Verfahren zum Herstellen von 4-substituierten-2-Butenalen | |
| EP0252389B1 (de) | 1,1-Dialkoxy-2-methyl-4,4- diacyloxy-2-butene |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Designated state(s): AT BE CH DE FR GB IT LU NL SE |
|
| PUAL | Search report despatched |
Free format text: ORIGINAL CODE: 0009013 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR GB IT NL |
|
| 17P | Request for examination filed |
Effective date: 19801209 |
|
| ITF | It: translation for a ep patent filed | ||
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR GB IT NL |
|
| REF | Corresponds to: |
Ref document number: 3064073 Country of ref document: DE Date of ref document: 19830818 |
|
| ET | Fr: translation filed | ||
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed | ||
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 19890117 Year of fee payment: 10 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 19890120 Year of fee payment: 10 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 19890123 Year of fee payment: 10 |
|
| ITTA | It: last paid annual fee | ||
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 19890228 Year of fee payment: 10 Ref country code: GB Payment date: 19890228 Year of fee payment: 10 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 19890303 Year of fee payment: 10 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Effective date: 19900214 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19900221 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CH Effective date: 19900228 Ref country code: BE Effective date: 19900228 |
|
| BERE | Be: lapsed |
Owner name: BASF A.G. Effective date: 19900228 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Effective date: 19900901 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee | ||
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee | ||
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Effective date: 19901031 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |