-
Die
Bildung von 3-Oxo-delta4-steroiden, ausgehend
von 3β-Hydroxy-delta5-Vorläufern
wird in der Biosynthese aller Klassen der Steroidhormone bei den
Säugern
durch das enzymatische System 3β-Hydroxy-delta5-steroid-Dehydrogenase (EC 1.1.1.145) und
delta5-delta4-Steroid-Isomerase
(EC 5.3.3.1), 3β-HSD genannt,
katalysiert. 3β-HSD
katalysiert z. B. die Umwandlung von Pregnenolon in Progesteron,
von 17α-Hydroxypregnenolon
in 17α-Hydroxyprogesteron,
von Dehydroepiandrosteron in delta4-Androstendion
oder von 5-Androsten-3β-17β-diol in
Testosteron (Simard et al., 1996).
-
Die
3β-HSD stellt
somit eines der Schlüsselenzyme
des Biosynthesewegs von Hydrocortison ausgehend von Cholesterin
in der Nebennierenrinde der Säuger
dar (1).
-
Die
Verwendung von rekombinanten Mikroorganismen, insbesondere von transformierten
Hefen, ermöglicht
die heterologe Expression eines Enzyms oder mehrerer Enzyme der
Säugetiere
für diesen
Biosyntheseweg, um Hydrocortison oder Zwischenprodukte dieser Biosynthese
zu produzieren, und wurde z. B. in der europäischen Patentanmeldung
EP 0 340 878 , dem US-Patent
5,137,822, Dumas et al., 1994 und Cauet et al., 1994, beschrieben.
-
Wenn
die funktionelle 3β-HSD
in der Hefe exprimiert wird, wandeln die transformierten Hefezellen
die 3β-Hydroxysteroide
nicht vollständig
in entsprechende 3-Oxosteroide um, z. B. das Pregnenolon in Progesteron,
sondern akkumulieren eine Verbindung, die ebenfalls im Fall der
Zellen der nicht- transformierten
Hefe beobachtet wird. Die Identifizierung der akkumulierten Verbindung
als 3β-Essigsäureester
des Ausgangssteroids und die Charakterisierung des Enzyms, das Acyltransferaseaktivität hat, die
für diese
Veresterung verantwortlich ist (später als APAT für "Acetylcoenzym A Pregnenolon-Acetyltransferase" bezeichnet) werden
in der vorliegenden Anmeldung beschrieben. Andererseits wurde die
Ansammlung von Pregnenolonacetat durch einen transformierten Hefestamm,
der Pregnenolon produziert, in der europäischen Patentanmeldung
EP 0 727 489 beschrieben.
Diese Beobachtungen erlauben es davon auszugehen, dass die Veresterung
der durch die Hefe produzierten 3β-Hydroxysteroide
unerwünscht
ist, da sie für
Sekundärreaktionen
und Nebenprodukte verantwortlich ist, die zu einer Verringerung
der Ausbeute an akkumulierten 3β-Hydroxysteroiden
führen,
beispielsweise an Pregnenolon oder zu einer Verringerung der Ausbeute
bei der Bioumwandlung von 3β-Hydroxy-delta
5-steroiden in 3-Oxo-delta
4-steroide
führen
und insbesondere bei der Produktion des Progesterons oder des 17α-Hydroxy-progesterons
zu einer Verringerung der letztlichen Produktion des Hydrocortisons
auf dem bereits genannten Biosyntheseweg führen.
-
Als
Folge der erhaltenen Resultate, die oben genannt wurden, beschreibt
die vorliegende Erfindung die Konstruktion von Hefestämmen, die
die unerwünschte
APAT-Aktivität
verloren haben, durch Veränderung des
Gens, das für
diese Aktivität
codiert, und führt
demnach zu einer Stabilisierung der 3β-Hydroxysteroide in Gegenwart dieser.
Diese Stämme
sind demnach als Ausgangsstämme
verwendbar, um rekombinante Stämme
zu konstruieren, die fähig
sind, 3β-Hydroxysteroide
mit verbesserten Ausbeuten in Folgeprodukte umzuwandeln.
-
Die
Erfindung beschreibt auch die Konstruktion von Hefestämmen, die
die APAT-Aktivität
durch Veränderung
des Gens, das für
diese Aktivität
codiert, verloren haben und entweder 3β-HSD oder das Cytochrom P45017α exprimieren
oder 3β-HSD
und das Cytochrom P45017α des Biosynthesewegs des Hydrocortisons
ausgehend von Cholesterin coexprimieren. Die Stämme, die z. B. 3β-HSD exprimieren,
ermöglichen
es, die Ausbeuten der Bioumwandlung der 3β-Hydroxy-delta5-steroide
in 3-Οxo-delta4-steroide zu verbessern und sind somit in
den verbesserten Herstellungsverfahren für Cortison oder seine Zwischenprodukte
in der Hefe einsetzbar.
-
Gegenstand
der vorliegenden Erfindung ist ein modifizierter Hefestamm, bei
dem die Acetyl-CoA-Pregnenolon-Acetyltransferase
(APAT)-Aktivität
durch Veränderung
des Gens, das für
diese Aktivität codiert,
eliminiert ist und bei dem somit eine Stabilisierung der 3β-Hydroxysteroide
resultiert.
-
Die
Veränderung
des Gens, das für
die APAT-Aktivität
codiert, kann z. B. durch Insertion, Deletion oder Substitution
einer DNA-Sequenz in den funktionellen Elementen des Gens, z. B.
der Promotor oder die Sequenz, die für das Protein mit der APAT-Aktivität codiert,
realisiert werden. Der Einbau der so veränderten DNA-Sequenz in einen
Hefewirtsstamm kann dann beispielsweise durch die Methodologie der
homologen Rekombination durchgeführt
werden und führt
zur Erzeugung von Hefechromosomenmutanten, die den modifizierten
Stämmen
der Erfindung entsprechen, in denen das Verschwinden der APAT-Aktivität und die
Stabilisierung der 3β-Hydroxysteroide
sich bemerkbar machen, z. B. durch Kultur der Zellen in Gegenwart
von Pregnenolon und durch Messung des Gehalts an Pregnenolon als
Funktion der Zeit unter den später
im experimentellen Teil beschriebenen Arbeitsbedingungen.
-
Als
Wirtshefestämme,
die für
die Erfindung verwendet werden, kann man insbesondere die Saccharomyces-Stämme, wie
S. cerevisiae, die Candida-Stämme
wie C. maltosa, die Kluyveromyces-Stämme, wie K. lactis, oder die
Pichia-Stämme,
wie z. B. P. pastoris, nennen.
-
Gegenstand
der vorliegenden Erfindung ist insbesondere ein modifizierter Hefestamm,
in dem das veränderte
Gen das Gen ATF2 von S. cerevisiae oder ein Homologes dieses ist.
-
Unter
dem Gen ATF2 versteht man das Gen von S. cerevisiae, das in dem
Genom der Hefe unter dem Locus ATF2 oder YGR177c, "Saccharomyces-Genome
Database" (SGD);
(Cherry et al. http://genome-www.stanford.edu/Saccharomyces/) identifiziert
ist, dessen offenes Leseraster (ORF), YGR177c genannt, in die Aminosäuresequenz
in der Hinterlegungsbank Mips, zugänglich mit der Zugangsnummer
564491 (Hebling U., Hofmann B und Delius H. (Mai 1996)), deren Sequenz
in 4 gezeigt ist, übersetzt wird. Dieses Gen codiert
für ein
Protein, das die APAT-Aktivität
hat, wie es später
im experimentellen Teil gezeigt wird.
-
Unter
einem Gen, das zu dem Gen ATF2 homolog ist, versteht man ein Gen,
das für
ein Protein codiert, das APAT-Aktivität hat und das eine Sequenzidentität von etwa
60% und mehr mit der Sequenz des Proteins YGR177C hat.
-
Gegenstand
der vorliegenden Erfindung ist insbesondere ein modifizierter Hefestamm,
in dem das veränderte
Gen das Gen ATF2 von S. cerevisiae ist und der später mutanter
Stamm atf2 genannt wird.
-
Gegenstand
der Erfindung ist speziell ein Hefestamm, der wie oben modifiziert
ist, in dem das Gen ATF2 durch Insertion einer DNA-Sequenz, die
mindestens ein Nucleotid hat, verändert ist.
-
Die
DNA-Sequenz, die in das Gen ATF2 derart insertiert ist, dass es
die gesamte APAT-Aktivität
verliert, ist z. B. ein auxotrophes Selektionsgen, das eine Nahrungserfordernis
des Wirtsstamms ergänzt,
z. B. das Gen URA3, das Gen LEU2, das Gen TΡP1, das Gen HIS3 oder das Gen
ADE2, beispielsweise ein dominantes Selektionsgen, wie ein Resistenzgen
für ein
Antibiotikum, wie G418, Phleomycin oder Hygromycin oder z. B. ein
Reportergen, wie das Gen βGAL.
-
Die
DNA-Sequenz, die in das Gen ATF2 insertiert wird, kann auch ein
Hefeexpressionsblock sein, der aus einem Promotor und einem Transkriptionsterminator
besteht, z. B. ein Hefepromotor, wie PGK, TDH3, CYC1 oder TEF1,
beispielsweise ein Hefeterminator, wie CYC1, TDH3, TEF1 oder PGK.
Der Expressionsblock kann z. B. eine Kombination der oben angegebenen
Elemente sein, z. B. der Block ΤΕF1prom/PGKterm Gegenstand
der Erfindung ist speziell ein modifizierter Hefestamm, in dem das
Gen ATF2 durch Insertion des Selektionsgens URA3 oder des Expressionsblocks
TEF1prom/PGKterm verändert ist.
-
Gegenstand
der Erfindung ist insbesondere ein modifizierter Hefestamm, in dem
das Gen ATF2 durch Insertion des Selektionsgens URA3 verändert ist.
-
Die
Mutantenstämme
atf2 der Erfindung, die an APAT-Aktivität verarmt sind und in die das
Gen URA3 insertiert ist, die später
als atf2-Δ::URA3
bezeichnet werden, konnten durch Prototrophie für Uracil selektioniert werden.
-
Gegenstand
der vorliegenden Erfindung sind speziell die modifizierten Stämme von
S. cerevisiae, die TGY156 und TGY158 genannt werden, deren detaillierte
Konstruktionen später
im experimentellen Teil angegeben werden.
-
Die
Erfindung hat außerdem
insbesondere einen modifizierten Hefestamm zum Gegenstand, in dem das
Gen ATF2 durch Insertion des Expressionsblocks ΤΕF1prom/PGKterm verändert
ist. Die Mutantenstämme atf2
der Erfindung, die an APAT-Aktivität verarmt sind und in die Expressionsblock ΤΕF1prom/PGKterm eingesetzt wurde,
später
als atf2-Δ::ΤΕF1prom/PGKterm bezeichnet,
konnten wegen des funktionellen Gens URA3, das durch einen Expressionsblock
ersetzt war, durch ihre Resistenz gegenüber 5-Fluor-orotsäure (5-FO)
selektioniert werden.
-
Gegenstand
der vorliegenden Erfindung ist in spezieller Weise der modifizierte
Stamm von S. cerevisiae, TGY186 bezeichnet, dessen detaillierter
Aufbau später
im experimentellen Teil angegeben ist.
-
Gegenstand
der vorliegenden Erfindung ist auch ein transformierter Hefestamm,
in dem die Acetyl-CoA-Pregnenolon-Acetyltransferase (APAT)-Aktivität durch
Veränderung
des Gens, das für
diese Aktivität codiert,
und der mindestens eines der Säugerenzyme
des Biosynthesewegs von Hydrocortison, ausgehend von Cholesterin,
ausgewählt
unter folgenden exprimiert:
- – das Enzym
zur Spaltung der Seitenkette des Cholesterins ( P450SCC),
- – 3β-Hydroxy-delta5-steroid-Dehydrogenase/delta5-delta4-Steroid-Isomerase
(3β-HSD)
und
- – 17α-Steroidhydroxylase
(P45017α).
-
Die
transformierten Hefestämme
der Erfindung können
z. B. durch Transformation der Mutantenstämme atf2 der Erfindung nach
bekannten Methoden, z. B. Transformation eines Expressionsvektors
von P450SCC wie auch von ADX und ADR, durch
einen Expressionsvektor der 3β-HSD
oder durch einen Expressionsvektor der P45017α erhalten
werden. Die Mutantenstämme
atf2 können
gegebenenfalls auch co-transformiert
werden, z. B. durch einen Expressionsvektor der 3β-HSD und
durch einen Expressionsvektor der P45017α, oder können durch
einen Co-Expressionsvektor der 3β-HSD
und der P45017α transformiert werden und werden
z. B. in einem Verfahren der Bioumwandlung von Pregnenolon in 17α-Hydroxyprogesteron
verwendet.
-
Vektoren,
die zur Expression von P
450SCC sowie von
ADX und ADR, 3β-HSD
oder P
45017α vom Rind oder vom Menschen
in Hefezellen konstruiert sind, wurden z. B. von Dumas et al., 1994,
in der europäischen Patentanmeldung
EP 0 340 878 oder im US-Patent
5,137,822 beschrieben.
-
Die
Erfindung betrifft insbesondere einen transformierten Hefestamm,
in dem das veränderte
Gen das Gen ATF2 von S.cerevisiae oder ein Homologes dieses ist.
Gegenstand der vorliegenden Erfindung ist insbesondere ein transformierter
Hefestamm, in dem das veränderte
Gen das Gen ATF2 von S. cerevisiae ist und der einem transformierten
Stamm atf2 entspricht.
-
Gegenstand
der vorliegenden Erfindung ist insbesondere ein transformierter
Hefestamm, in dem das Gen ATF2 durch Insertion einer DNA-Sequenz,
die mindestens ein Nucleotid hat, verändert ist; und ein spezieller
Gegenstand der vorliegenden Erfindung ist ein tranformierter Hefestamm,
in dem das Gen ATF2 durch Insertion des Selektionsgens URA3 verändert ist,
und der einem transformierten Stamm atf2-Δ::URA3 entspricht.
-
Die
Veränderung
des Gens derart, dass es jede APAT verliert, das Gen ATF2 oder ein
Homologes davon, sowie die Wirtsstämme haben die Bedeutungen,
die vorstehend gegeben wurden.
-
Die
vorliegende Erfindung betrifft in ganz spezieller Art den transformierten
Hefestamm atf2-Δ::URA3, der
3β-HSD exprimiert,
und insbesondere den transformierten Stamm von S. cerevisiae, der TGY158/pTG10862
genannt wird, dessen detaillierte Konstruktion später im experimentellen
Teil beschrieben wird.
-
Die
vorliegende Erfindung hat speziell als Gegenstand einen transformierten
Hefestamm, in dem das Gen ATF2 durch Insertion des Expressionsblocks ΤΕF1prom/PGKterm verändert ist,
und der dem transformierten Stamm atf2-Δ::ΤΕF1prom/PGKterm entspricht.
-
Die
Erfindung betrifft in ganz spezieller Weise einen transformierten
Stamm atf2-Δ::ΤΕF1prom/PGKterm der
P45017α exprimiert,
und insbesondere den transformierten Stamm von S. cerevisiae, der TGY186/pTG10435
genannt wird.
-
Die
Erfindung betrifft besonders einen transformierten Hefestamm atf2-Δ::ΤΕF1prom/PGKterm der 3β-HSD und
P45017α gleichzeitig
exprimiert, und in ganz besonderer Weise den transformierten Stamm
von S. cerevisiae, der TGY186/pTG10417 genannt wird.
-
Gegenstand
der vorliegenden Erfindung ist auch ein Verfahren zur in vivo-Oxidation
eines Substrats, das aus einem endogenen Sterol, einem exogenen
Sterol und einem exogenen Steroid ausgewählt ist, wobei man einen transformierten
Hefestamm verwendet, den man entweder allein kultiviert, wenn der
Stamm das endogene Sterol bildet, oder den man mit dem Sterol oder
dem exogenen Steroid inkubiert und gegebenenfalls die erhaltene
oxidierte Verbindung isoliert.
-
Unter
endogenem Sterol versteht man ein Sterol, das im Hefestamm akkumuliert
wird und das ein Substrat für
das Enzym zur Spaltung der Seitenkette (P
450SCC)
ist, wenn die Hefe nach Transformation beispielsweise durch einen
Expressionsvektor von P
450SCC, ADX und ADR
in Abwesenheit von exogenem Sterol kultiviert wird. Die endogenen
Sterole, die zur Durchführung
des erfindungsgemäßen Verfahrens
verwendet werden, können
z. B. Ergosta-5-en-3-ol, Ergosta-5,24(28)-dien-3-ol oder Ergosta-5,22-dien-3-ol
sein. Die europäische
Patentanmeldung
EP 0 727 489 beschreibt
die Akkumulation solcher Sterole in einem Hefestamm und die Spaltung
ihrer Seitenkette in einer Kultur des Stamms nach Transformation
durch einen Expressionsvektor von P
450SCC,
ADX und ADR. Ein derartiger Hefestamm, in dem auch die APAT-Aktivität vorhanden
ist, kann vorher modifiziert werden, um einen Mutantenstamm atf2
gemäß der Erfindung
zu erhalten, kann dann durch einen Expressionsvektor von P
450SCC, ADX und ADR transformiert werden,
um einen transformierten Mutantenstamm atf2 gemäß der Erfindung zu erhalten.
-
Unter
exogenem Sterol versteht man ein Sterol, das ein Substrat für das Spaltungsenzym
P450SCC durch Inkubation mit einem Hefestamm
ist, welcher durch einen Expressionsvektor von P450SCC,
ADX und ADR transformiert wurde, z. B. Cholesterin und Sitosterol.
Ein solcher Stamm kann z. B. ein mutanter Stamm atf2 sein, der durch
einen Expressionsvektor von P450SCC, ADX
und ADR transformiert ist.
-
Das
3β-Hydroxysteroid,
das durch Spaltung der Seitenkette des endogenen oder exogenen Sterols, das
als Substrat verwendet wird, erhalten wird, liegt in den Kulturen
der transformierten Stämme
atf2, die P450SCC, ADX und ADR exprimieren,
in vollständig
freier Form vor, d. h. es ist nicht von dem entsprechenden 3β-Essigsäureester
begleitet.
-
Unter
Steroid versteht man ein Steroid, das ein Substrat des Enzyms 3β-HSD durch
Inkubation mit einem Hefestamm, der z. B. durch einen Expressionsvektor
der 3β-HSD
transformiert ist, ist, z. B. Pregnenolon, 17α-Hydroxypregnenolon oder Dehydroepiandrosteron,
oder ein Steroid, das ein Substrat des Enzyms P45017α durch Inkubation
mit einem Hefestamm, der z. B. durch einen Expressionsvektor von
P45017α transformiert
ist, ist, wie z. B. Progesteron oder Pregnenolon. Ein derartiger
Stamm kann z. B. ein mutanter Stamm atf2 sein, der durch einen Expressionsvektor
der 3β-HSD
oder durch einen Expressionsvektor von P45017α gemäß der Erfindung
transformiert ist.
-
Gegenstand
der vorliegenden Erfindung ist insbesondere das Verfahren zur in
vivo-Oxidation, wobei das Substrat ein β-Hydroxysteroid ist und wobei man den
transformierten Hefestamm atf2-Δ::URA3,
der 3β-HSD
exprimiert, verwendet und gegebenenfalls das erhaltene 3-Oxo-delta4-steroid isoliert; Gegenstand der vorliegenden
Erfindung ist insbesondere ein Verfahren, in dem das 3β-Hydroxysteroid
unter Pregnenolon und 17α-Hydroxypregnenolon
ausgewählt
wird.
-
Das
als Substrat verwendete 3β-Hydroxysteroid
ist stabil, wenn man es mit dem erfindungsgemäßen Stamm atf2-Δ::URA3, der
durch einen Expressionsvektor von 3β-HSD transformiert ist, inkubiert.
Somit liefert die Erfindung ein verbessertes Verfahren zur Herstellung
von 3-Oxo-delta4-steroid in einer Hefe,
da das gesamte 3β-Hydroxysubstrat
in 3-Oxo-delta4-steroid oxidiert werden kann, wie es
später
im experimentellen Teil gezeigt wird.
-
Die
Erfindung hat insbesondere das obige Verfahren zur in vivo-Oxidation
zum Gegenstand, wobei das Substrat ein Steroid ist und wobei man
den transformierten Hefestamm atf2-Δ::ΤΕF1prom/PGKterm verwendet, der P45017α exprimiert,
und gegebenenfalls das erhaltene 17α-hydroxylierte Steroid isoliert;
Gegenstand ist insbesondere ein Verfahren, in dem das Steroidsubstrat
Pregnenolon oder Progesteron ist.
-
Das
als Substrat verwendete Pregnenolon ist stabil, wenn man es mit
dem Stamm atf2-Δ::ΤΕF1prom/PGKterm, der
durch einen Expressionsvektor von P45017α tranformiert
ist, inkubiert.
-
Die
vorliegende Erfindung liefert somit auch ein verbessertes Verfahren
zur Herstellung von 17α-Hydroxysteroiden,
ausgehend von 3β-Hydroxysteroiden,
da die Gesamtheit des 3β-Hydroxysubstrats
17α-hydroxyliert
werden kann.
-
Die
Erfindung hat insbesondere das obige Verfahren zur in vivo-Oxidation
zum Gegenstand, wobei das Substrat ein 3β-Hydroxysteroid ist und wobei man den
transformierten Hefestamm atf2-Δ::ΤΕF1prom/PGKterm verwendet,
der 3β-HSD
und P45017α co-exprimiert, und wobei man
gegebenenfalls das erhaltene 17α-hydroxylierte
3-Oxo-delta4-steroid isoliert. Gegenstand
der vorliegenden Erfindung ist insbesondere das obige Verfahren,
in dem das Steroidsubstrat Pregnenolon ist.
-
Die
transformierten Mutantenhefestämme
atf2 und das erfindungsgemäße Verfahren
erlauben es, ihre vorteilhafte Verwendung in der verbesserten Produktion
von Hydrocortison oder seinen Zwischenprodukten in der Hefe vorauszusagen.
-
Im
später
aufgeführten
experimentellen Teil sind Beispiele des Aufbaus der erfindungsgemäßen Hefen und
der Verwendung des erfindungsgemäßen Verfahrens
beschrieben.
-
Materialien und allgemeine
Verfahren
-
1. Stämme und Milieu
-
Die
zur Durchführung
der Erfindung verwendeten Stämme
von S. cerevisiae sind der Stamm TGY73.4 (MATα, URA3-Δ5, pral-1, prbl-1, prcl-1, cpsl-3,
his)isogenes Leu+-Derivat von c13ABYS86,
beschrieben von Achstetter et al., 1992, und der Stamm Fy1679 (MATa,
URA3-52, trpl-Δ63,
leu2-Δ1,
his3-Δ200, fen1,
GAL), beschrieben von Thierry et al., 1990. Das Wachstum der Stämme wird
in vollständigem
YPD-Milieu (Difco Laboratories), das 2% Glucose enthält, bei
28°C unter
den Bedingungen, die von F. Sherman, 1991, beschrieben wurden, durchgeführt.
-
Für die Transformation
von S. cerevisiae werden die Zellen durch das Lithiumacetatverfahren
kompetent gemacht (Ito et al., 1983). Die Hefen werden in Routinekulturen
auf synthetischem Minimalmedium SD, das 2% Glucose enthält (F. Sherman,
1991), unter Zusatz von Nährstoffbedarf
in einer Konzentration von 100 μg/ml
kultiviert.
-
Der
Stamm E. coli BJ5183 (D. Hanahan, 1983) wurde für die in vivo-Rekombination
verwendet und der Stamm E. coli C600, hsdR (Hubacek et al., 1970)
wurde als Stamm für
die klassischen Ligationsreaktionen verwendet.
-
2. Manipulation der DNA
und in vivo-Rekombination in E. coli
-
Die
allgemeinen Verfahren der Molekularbiologie, die verwendet wurden,
sind bei Sambrook et al., 1989, beschrieben. Das Verfahren zur in
vivo-Rekombination wurde von E. Degryse, 1995 und E. Degryse, 1996,
beschrieben.
-
Test auf enzymatische
APAT-Aktivität
-
Die
Acetyltransferase-Aktivität
der APAT wurde durch Messung des Einbaus von [3H]Acetat
in Pregnenolon, ausgehend von [3H]Acetyl-CoA
(New England Nuclear) bestimmt. Das Reaktionsmilieu (500 μl) enthielt
[3H]Acetyl-CoA (20 μM, 25 Ci/mol) und Pregnenolon
(Sigma) (30 μM).
Das Pregnenolon wird in Lösung
in 2 μl
des Gemisches Tyloxapol (Sigma)/Ethanol (1 : 1) in Kaliumphosphatpuffer
(20 mM) mit pH 7,0 zugesetzt. Nach 15 min Inkubation bei 30°C wird die
Reaktion durch Zusatz von 2 ml Dichlormethan gestoppt.
-
Die
Steroide werden mit Dichlormethan extrahiert, mit Hochdruckflüssigkeitschromatographie
mit Umkehrphase (im Folgenden als RP-HPLC bezeichnet) unter Bedingungen
einer isokratischen Elution mit Acetonitril an einer Ultrasphere
ODS-Säule
(Beckman) mit 45°C
getrennt, wobei ein HP 1090 (Hewlett Packard)-Chromatograph, kombiniert
mit einem Radiodetektor FLO-One 500 (Packard) verwendet wurde, der
die Messung von gebildetem Pregnenolon-[3H]acetat
ermöglicht.
-
Eine
Einheit APAT ist als die Enzymmenge definiert, die 1 nmol Pregnenolonacetat
pro Minute bei 30°C
unter den vorstehend beschriebenen Bedingungen produziert.
-
4. Bestimmung
der Proteinkonzentration
-
Die
Proteinkonzentration wurde unter Verwendung des "Proteinassaykits" (Bio-Rad) mit Rinderserumalbumin als
Standard gemessen.
-
Die
angefügten
Figuren erläutern
bestimmte Aspekte der Erfindung.
-
1 stellt
den Biosyntheseweg von Hydrocortison, ausgehend von Cholesterin,
bei Säugetieren
dar.
-
Die 2A und 2B erläutern die
biologische Umwandlung des Pregnenolons in Pregnenolonacetat bei
S. cerevisiae. Die Analyse wird durch RP-HPLC bei 205 nm durchgeführt:
-
Die 2A zeigt
die Kinetik der Bildung des Pregnenolonacetats und des Verschwindens
des Pregnenolons in Zeitintervallen während der 12-stündigen Inkubation.
-
Die 2B zeigt
das Profil der Steroide bei t = 0 und bei t = 10 h, bezogen auf
ein Profil der Standards Pregnenolon und Pregnenolonacetat.
-
3 veranschaulicht
die Reinigung der APAT durch Chromatographie an MonoP HR 5/20:
- (A) zeigt das Profil der APAT-Aktivität, die in
den Fraktionen 10 bis 20 vorliegt;
- (B) zeigt die Analyse der Fraktionen 14, 15 und 16, die vereinigt
und konzentriert worden waren, durch SDS-PAGE unter Färbung mit
Coomassie-Blau (Bande 2) in Gegenwart von Molekulargewichtsmarkern (Bande
1). Der Pfeil zeigt die MG-Bande,
die bei 62 kDa auftritt.
-
Die 4 stellt
die Aminosäuresequenz
des Proteins YGR177c im Einbuchstabencode dar. Die Peptide, die
ausgehend vom gereinigten und durch Trypsin verdauten Protein APAT
sequenziert wurden, sind unterstrichen.
-
Die 5 stellt
die Strategie der Unterbrechung des Gens ATF2 durch Assoziation
mit dem Gen URA3 durch das Verfahren der Doppelfusions-PCR dar.
Die leeren Balken und die ausgefüllten
Balken stellen die Sequenzen von ATF2 bzw. URA3 dar.
-
Die 6 erläutert die
Wirkung der Unterbrechung des Gens ATF2 in S. cerevisiae auf die
Acetylierung des Pregnenolons. Das Vorliegen von Pregnenolonacetat
wird durch RP-HPLC bei 205 nm, ausgehend von 16 h-Kulturen des Elternstamms
TGY73.4 (A) oder des Mutantenstamms TGY158 (B), deutlich gemacht.
-
Die 7A, 7C und 7C stellen
das Konstruktionsschema der Expressionsplasmide der humanen 3β-HSD bei
der Hefe pTG10832 und pTG10862 dar. Die 7A beschreibt
den Erhalt des Fragments MscI-MluI, das die Sequenz enthält, die
für die
humane 3β-HSD
codiert. Die 7B beschreibt den Erhalt des Fragments
NotI, das den Expressionsblock CYClp/3β-HSD/PGKt enthält.
Die 7C beschreibt den Erhalt des Plasmids pTG10832
und des Plasmids pTG10862.
-
Die 8 stellt
eine Restriktionskarte des Plasmids pTG10832 dar.
-
Die 9 stellt
eine Restriktionskarte des Plasmids pTG10862 dar.
-
Die 10 stellt
das Konstruktionsschema des Expressionsplasmids pTG10435 dar.
-
Die 11 stellt
das Konstruktionsschema des Plasmids pTG10058 dar.
-
Die 12 stellt
eine Restriktionskarte des Plasmids pTG10058 dar.
-
Die 13 stellt
eine Restriktionskarte des Plasmids pTG10293 dar.
-
Die 14 stellt
eine Restriktionskarte des Plasmids pTG10435 dar.
-
Die 15A und 15B stellen
das Konstruktionsschema des Plasmids pTG10274 dar:
-
Die 15A beschreibt den Erhalt des Plasmids pTG10214.
-
Die 15B beschreibt den Erhalt des Plasmids pTG10274.
-
Die 16 stellt
eine Restriktionskarte des Plasmids pTG10274 dar.
-
Die 17 stellt
eine Restriktionskarte des Plasmids pTG10401 dar.
-
Die 18 stellt
das Konstruktionsschema des Expressionsvektors pTG10262 dar.
-
Die 19 stellt
eine Restriktionskarte des Plasmids pTG10262 dar.
-
Die 20 stellt
eine Restriktionskarte des Plasmids pTG10403 dar.
-
Die 21 stellt
eine Restriktionskarte des Plasmids pTG10417 dar.
-
(Abkürzungen
der Restriktionsenzyme: S, SalI; N,NotI; BII,BglII; M,MluI, C,ClaI;
N°, NcoI-Stelle
verloren; XbaI°,
XbaI-Stelle verloren; E,EcoR).
-
Beispiel 1: Identifizierung
der APAT-Aktivität
der Hefe
-
A – in vivo-Acetylierung
von Pregnenolon durch die Hefe
-
Der
Stamm TGY73.4 wurde bei 28°C
in 10 ml YPD-Milieu (Difco), das mit A600 = 0,1 beimpft war, ausgehend
von einer Vorkultur von 24 h und unter Zusatz von 100 μl einer Lösung von
Pregnenolon mit 10 mg/ml in einem Gemisch aus Tergitol (Sigma)/Ethanol
(1 : 1) kultiviert. Die gebildeten Steroide wurden an 250 μl-Aliquots
der Brühe,
die in Zeitintervallen während
10 h entnommen wurde, identifiziert. Nach Extraktion mit 2 ml Dichlormethan
wurden die organischen Phasen unter Stickstoff eingeengt, dann wurden
die erhaltenen Rückstände in Acetonitril
wieder aufgelöst.
Die Steroide wurden durch RP-HPLC an einer Ultrasphere ODS-Säule (Beckman)
analysiert, wobei wie folgt eluiert wurde: 60% Acetonitril in Wasser
während
10 min, dann Acetonitril, variierend von 60 bis 80 in Wasser, während 5
min, dann 80% Acetonitril während
5 min mit einem Durchfluss von 1 ml pro min bei 45°C und bei
Detektion bei 205 nm.
-
Die
erhaltenen Chromatogramme (2B) zeigen,
dass Pregnenolon zu einem Produkt metabolisiert wird, das apolarer
ist und das eine RT hat, die identisch mit der des Pregnenolonacetat-Standards
ist. 2A zeigt, dass Pregnenolon durch die Hefe schnell
in seinen Metaboliten umgewandelt wird.
-
Nach
Behandlung mit Alkali (6% KOH in Methanol) setzt der beobachtete
Metabolit ein Produkt frei, das eine RT hat, die mit der des Pregnenolons
identisch ist. Die Identifizierung des Metaboliten als Pregnenolonacetats
wurde dann durch Massenspektrometrie bestätigt.
-
B – Reinigung des Enzyms, das
APAT-Aktivität
hat
-
Der
Stamm TGY73.4 wurde in einem 10 l-Fermentator in Käppeli-Medium (Fiechter
et al., 1981), das mit 160 g/l Glucose angereichert war, bei 30°C bis A600
= 30 kultiviert. Die Zellen wurden durch Zentrifugation abgetrennt,
mit Wasser gewaschen, dann in 4 l 20 mM Tris-HCl-Puffer, pH 8,0,
mit 4°C
(Puffer A), der 1 mM PMSF enthielt, in Suspension gebracht. Die
Zellen wurden einem Bersten unter einem Druck von 1000 psi in einem
Manton Gaulin-Homogenisator unterworfen. Das erhaltene Zelllysat
wurde mit 12.000 g während
15 min bei 4°C
zentrifugiert, dann wurde Zinkchlorid dem Überstand zu einer Endkonzentration
von 40 mM zugesetzt. Der pH wurde mit 1 N HCl auf 5,5 eingestellt
und die Präzipitation
wurde während
30 min bei 4°C
durchgeführt. Nach
Zentrifugation bei 10.000 g während
10 min bei 4°C
wurde das isolierte Präzipitat
in 3 l Puffer A, der 100 mM EDTA und 1 mM PSFM enthielt, wieder
in Suspension gebracht. Nach Eliminierung des EDTA durch Diafiltration
mit einer Y10S10-Kartusche (Amicon) gegen 30 l Puffer A wurde das
Retentat mit einer Geschwindigkeit von 35 ml/min und bei 4°C auf eine
Säule mit
1,5 l DEAE-Sephacel (Pharmacia), die vorher mit dem Puffer A äquilibriert
worden war, geladen. Nach Waschen der Säule mit Puffer A, dann mit
Puffer A, der 0,15 M NaCl enthielt, wurde die APAT-Aktivität mit dem
Puffer A, der 0,4 M NaCl enthielt, eluiert. Die DEAE-Sephacel-Fraktionen,
die die APAT-Aktivität
enthielten, welche wie in "Materialien
und allgemeine Verfahren" angegeben
gemessen worden war, wurden vereinigt, mit NaCl zu einer Endkonzentration
von 2 M versetzt, dann mit einer Geschwindigkeit von 15 ml pro min
und mit 4°C
auf eine Säule
mit 500 ml Phenyl-Sepharose (Pharmacia) geladen, die vorher mit
dem Puffer A, der 2 M NaCl enthielt, äquilibriert worden war. Nach
Waschen der Säule mit
dem Puffer A, der 0,5 M NaCl enthielt, wurde die APAT-Aktivität mit 1,5
l eines linearen Gradienten von Natriumcholat, der von 0 bis 1%
in Puffer A variierte, eluiert. Die Fraktionen, die die APAT-Aktivität enthielten, wurden
vereinigt, dann durch Ultrafiltration über eine YM10-Membran (Amicon)
konzentriert, danach bis zur Verwendung bei –80°C konserviert.
-
Die
Gesamtheit des Verfahrens wurde einmal wiederholt, so dass Material
in ausreichender Menge hergestellt wurde, um die Reinigung fortzusetzen.
-
Das
gereinigte Material aus den zwei Herstellungen oben wurde aufgetaut,
dann mit einer Geschwindigkeit von 4 ml/min und mit 4°C auf eine
100 ml Säule
Q-Sepharose Fast Flow (Pharmacia) gegeben, die vorher im Puffer
A äquilibriert
worden war. Nach Waschen der Säule
mit dem selben Puffer wurde die APAT-Aktivität mit 500 ml eines linearen
NaCl-Gradienten,
steigend von 0 bis 1 M in dem selben Puffer, eluiert. Die Q-Sepharose-Fraktionen,
die APAT-Aktivität
enthielten, wurden vereinigt, dann mit einer Geschwindigkeit von
2,5 ml/min und mit 4°C
auf eine Säule
mit 7 ml Natriumcholat, das an Sepharose-Perlen immobilisiert ist
(Pharmacia) aufgeladen, wobei diese vorher mit Puffer A, der 0,5
M NaCl enthielt, äquilibriert
worden war. Nach Waschen der Säule
mit dem selben Puffer wurde die APAT-Aktivität mit 100 ml eines linearen
Natriumcholat-Gradienten, der von 0 bis 1% in dem selben Puffer
variierte, eluiert. Die Fraktionen, die APAT-Aktivität enthielten, wurden
vereinigt, durch Ultrafiltration an einer YM10-(Amicon)-Membran
bis zu einer Proteinkonzentration von 1,8 mg/ml konzentriert, dann
bei –80°C konserviert.
-
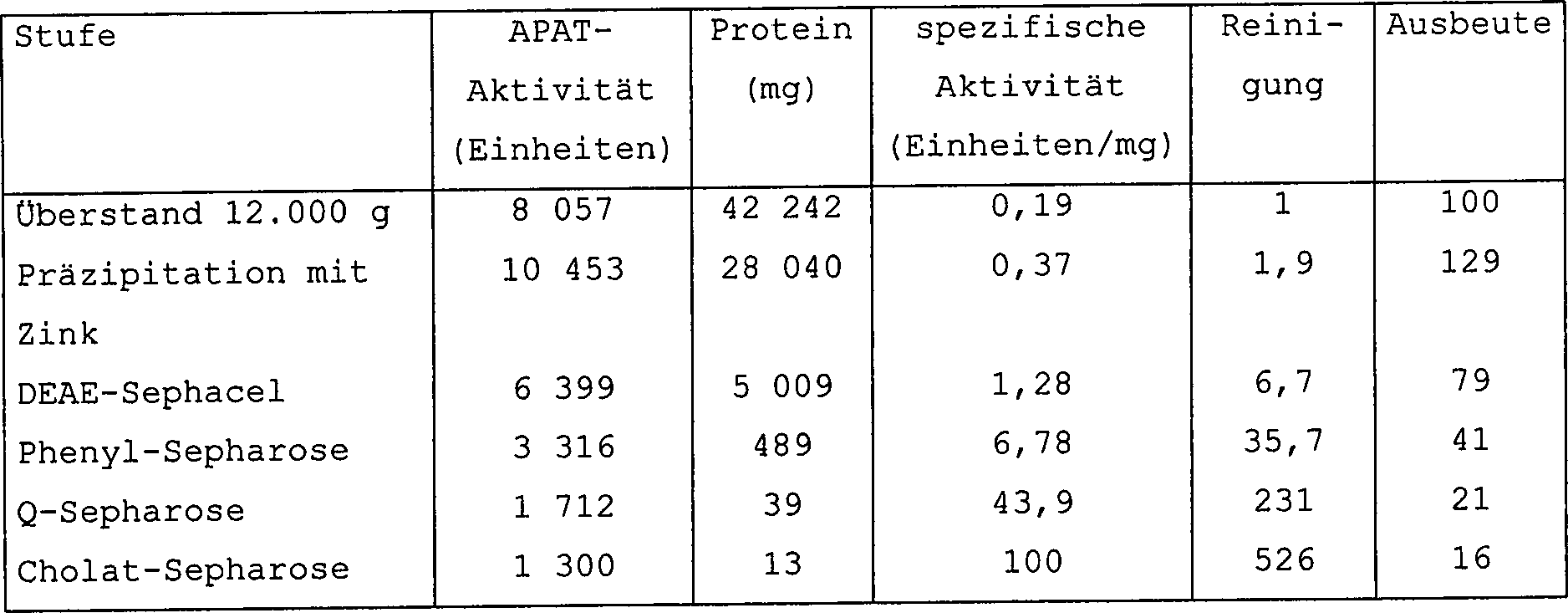
Die
APAT-Aktivität
war so etwa 500-fach, bezogen auf die spezifische Aktivität, gereinigt
worden, und zwar mit einer Ausbeute von etwa 16%, wie es in der
folgenden Tabelle 1 angegeben ist:
-
-
Die
Hälfte
des halb-gereinigten Materials, das oben erhalten worden war (etwa
6 mg Protein), wurde dann aufgetaut, danach mit PEG 4000 (Prolabo)
zu einer Endkonzentration von 20 (G/V) versetzt, um das Cholat und
NaCl zu eliminieren. Nach 30-minütigem
Rühren
bei 4°C
wurde das Präzipitat
durch Zentrifugation mit 12.000 g während 30 min bei 4°C gesammelt,
dann in 4 ml Puffer A erneut aufgelöst. Die so erhaltene Lösung wurde
dann mit einer Geschwindigkeit von 1 ml/min auf eine MonoP HR 5/20-Säule (Pharmacia)
aufgebracht, die vorher mit 25 mM bis-Tris-Puffer, pH 6,3, äquilibriert
worden war. Die APAT-Aktivität
wurde mit dem Puffer Polypuffer 74, pH 4,0, (Pharmacia) eluiert.
1 ml-Fraktionen wurden in Gegenwart von je 50 μl 2 M Tris-HCl-Puffer, pH 8,0,
derart gesammelt, dass die Inaktivierung des Enzyms bei saurem pH
begrenzt wurde. Die Fraktionen 14, 15 und 16 (3(A)),
die eine erhöhte
APAT-Aktivität
enthielten, die wie oben angegeben gemessen worden war, wurden vereinigt,
durch Ultrafiltration mit einer YM10-Membran konzentriert, danach
vor Verwendung bei –80°C konserviert.
Die so erhaltene aktive Fraktion wurde einer SDS-PAGE an 10% Polacrylamid-Gel unterzogen.
Durch Färbung
mit Coomassie-Blau wurden mehrere Banden sichtbar gemacht, und zwar
mit einer Hauptbande mit einem scheinbaren MG von 62 kDa (3(B)), das mit dem MG identisch war, das
durch Superose 6-Gelfiltration (Pharmacia) bestimmt worden war und
der APAT-Aktivität
entsprach.
-
C – Eigenschaften
der APAT
-
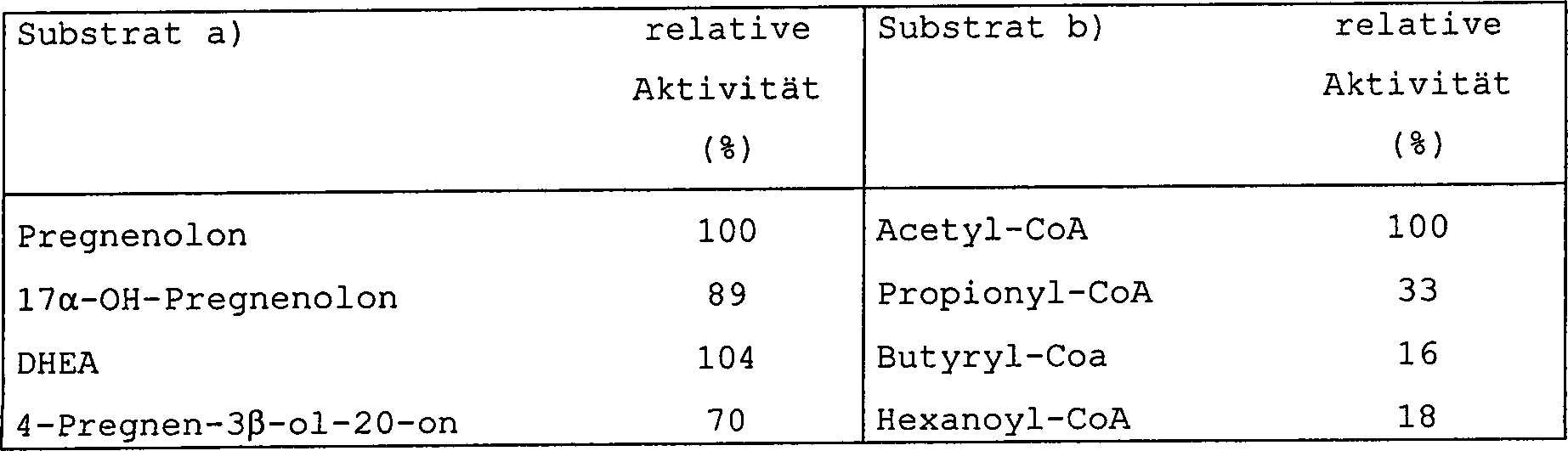
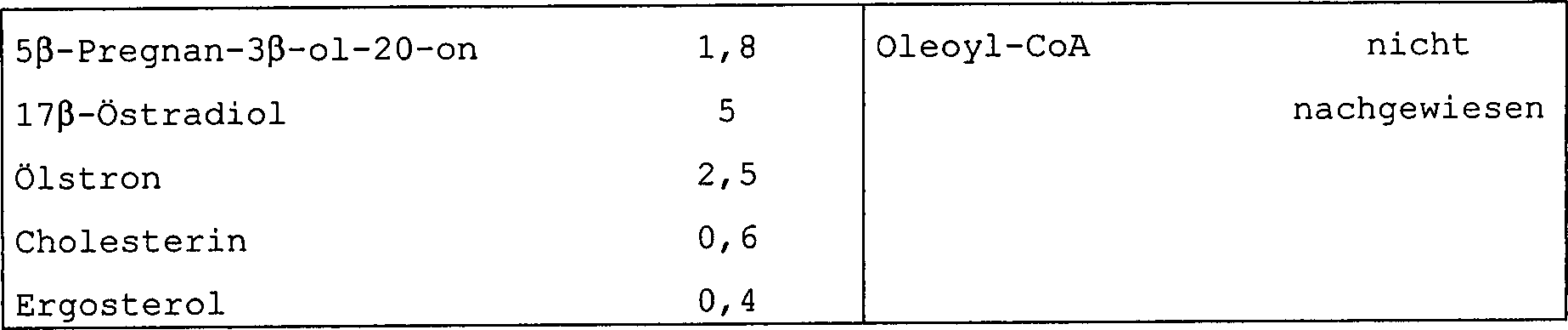
a) Substratspezifität
-
Nach
dem oben für
Acetyl-CoA und Pregnenolon angegebenen Verfahren unter Verwendung
verschiedener Acyl-Donatoren oder unterschiedlicher Steroidsubstrate überträgt halb-gereinigte
APAT Acetat auf 3β-Ol-delta4- oder -delta5-steroide
mit vergleichbarer Wirksamkeit, während der Transfer auf die Östrogene schwach
ist und auf die Sterole nicht nachweisbar ist und wobei eine deutliche
Präferenz
für Acetyl-CoA
als Acyldonator besteht.
-
Die
folgende Tabelle 2 zeigt die erhaltenen Resultate, wobei für a) die
Versuche mit 30 μM
jedes getesteten Steroids und 100 μM [3H]Acetyl-CoA
durchgeführt
wurden und für
b) die Versuche mit 100 μM
jedes Acyldonators und 30 μM
[3H]Pregnenolon durchgeführt wurden:
-
-
-
b) Inhibierung
-
Die
APAT-Aktivität
wird durch die Reagenzien mit Sulfhydryl-Gruppierungen, wie z. B. NEM und DTNB, stark
inhibiert. Die Inhibierung ist in Gegenwart von Zinkchlorid (1 mM)
vollständig.
-
D – Partielle
Aminosäuresequenz
-
Die
partielle Aminosäuresequenz
wurde nach Abbau mit Trypsin an den Gelschnitten nach dem Verfahren,
das von Rosenfeld et al. (1992) beschrieben wurde, bestimmt.
-
Ausgehend
von zwei Drittel des oben erhaltenen Konzentrats, das dann durch
SDS-PAGE getrennt worden war, wurde die 62 kDa-Bande ausgeschnitten, dann mit Trypsin
(Promega) inkubiert. Die erzeugten Peptide wurden dann durch RP-HPLC
an einer Vydac 218TP-Säule
(1,5 × 125
mm) mit einer Geschwindigkeit von 100 μl/min getrennt, wobei ein linearer
Acetonitrilgradient, der von 0 bis 60% in 80 min, dann von 60 bis 100%
in 20 min in einer 0,1% TFA-Lösung
in Wasser, verwendet wurde; dann wurden sie einer Aminosäuresequenzierung
unterworfen.
-
Die
N-terminale Sequenz wurde an einem Proteinsequenzanalysator Modell
477A, der mit einem HPLC-Analysator für PTH-Aminosäuren verbunden
war (Applied Biosystems), bestimmt.
-
Unter
den sequenzierten Proben lieferten zwei Peaks x und y eine eindeutige
Sequenz, die aus den folgenden 10 bzw. 16 Aminosäuren bestehen:
Für den Peak
x: ISEQFKKDDF
für
den Peak y: LIELISPVIIPLGNPK.
-
Die
zwei so erhaltenen Peptidsequenzen wurden verwendet, um die Datenbank
des Genoms von S. cerevisiae durchzumustern. Es konnte ein Protein
mit 62 kDa identifiziert werden, dessen Sequenz genau die Sequenz
der zwei oben genannten enthält
(Mips, Zugangsnummer S64491, Hebling et al., Mai 1996). Dieses Protein,
dessen Sequenz in 4 gezeigt ist und dessen Funktion
noch nicht beschrieben wurde, wird durch ein Gen am Locus YGR177c
codiert. Auf der Grundlage einer Aminosäureidentität von etwa 37% zwischen dem
erfindungsgemäßen Protein,
das Acyltransferase-Aktivität
hat, und dem Produkt des Gens ATF1 bei S. cerevisiae, das von Fujii
et al. (1994) beschrieben wurde und dem Alkohol-Acetyltransferase-Aktivität zugeschrieben
wurde, verwenden wir die Bezeichnung ATF2 für das Gen, das für das Protein
codiert, welches für APAT-Aktivität bei S.
cerevisiae verantwortlich ist.
-
Beispiel 2: Konstruktion
von Hefestämmen,
die das Gen ATFS, unterbrochen durch das Gen URA3 haben, und die
die APAT-Aktivität verloren
haben (atf2-Δ::URA3)
-
A) Targeting des Gens
ATF2
-
Das
Gen URA3 von S. cerevisiae wurde durch Substitution eines ausgewählten Teils
des Gens ATF2 von S. cerevisiae eingeführt, was eine anschließende Selektion
der Mutantenstämme
durch Prototrophie für Uracil
erlaubt.
-
Der
Selektionsmarker URA3 wurde durch Doppelfusions-PCR nach dem Verfahren,
das von Amberg et al., 1995, beschrieben wurde, mit dem Gen ATF2
assoziiert. Die in 5 gezeigte verfolgte Strategie
umfasst insgesamt 4 PCR-Reaktionen. Die beiden ersten Reaktionen
(PCR1 genannt) erlauben es, die Regionen 5' und 3', die die Insertionsstelle des URA3-Markers
in dem unterbrochenen Zielgen ATF2 flankieren, zu amplifizieren
und die dritte Reaktion (PCR2 genannt), erlaubt es, das Markergen
URA3 zu amplifizieren. Die Doppelfusion (PCR3 genannt) erlaubt schließt die Assoziation
der Regionen 5' und
3' des Zielgens
ATF2 an das Markergen URA3 (4'ATF2-URA3-3'ATF2 genannt).
-
Im
ersten Verfahrensschritt wurde eine Probe ganzer Zellen des Stamms
Fy1679, der als DNA-Quelle des Zielgens ATF2 verwendet wurde, im
PCR-Puffer, der 2 mM dNTP (Pharmacia) enthielt, unter den folgenden
Bedingungen amplifiziert: 25 Zyklen; 93°C, 30 s; 54°C, 2 min; 68°C, 3 min, gefolgt von einer
Verlängerung von
5 min bei 72°C;
Polymerase Ampli Taq (Perkin Elmer).
-
Einerseits
wurde die Region 5' des
Gens ATF2 durch PCR amplifiziert, wobei die Oligonucleotide mit den
folgenden Sequenzen als direkte oder indirekte Primer verwendet
wurden:
OTG10841: AAAAGTCGACAAAATGGAAGRTATAGAAGGATACGAACCACRTATCACTC
(SEQ ID No: 1)
und
OTG10844: ATCAATCTCCAATTAGGCCTCTTCGGATTACCC
(SEQ ID NO: 2),
die eine homologe Region zu der Region 5' der Sequenz des
Gens ATF2 (SGD : YGR177c) enthalten, und wobei eine Restriktionsstelle
SalI für
OTG10841 angefügt
wurde.
-
Andererseits
wurde die Region 3' des
Gens ATF2 durch PCR amplifiziert, wobei als direkte und indirekte
Primer die Oligonucleotide mit den folgenden Sequenzen verwendet
wurden:
OTG10846: CATTCGACATTCCCGAAGGTGACAATGACAAG (SEQ ID
NO: 3)
und
OTG10842: AAAAACGCGTAACTATTAAAGCGACGCAAATTCGCCGATGGTTTGG
(SEQ ID NO: 4)
die homologe Regionen der Region 3' der Sequenz des
Gens ATF2 (SGD : YGR177c) enthielten, und wobei eine Restriktionsstelle
MluI für
OTG10842 angefügt
wurde.
-
In
einem zweiten Verfahrensschritt wurde das Gen URA3 von S. cerevisiae
durch PCR amplifiziert, wobei als direkter Beginn das Oligonucleotid
mit der folgenden Sequenz verwendet wurde:
OTG10843:
GGGTAATCCGAAGAGGCCTAATTGGAGATTGATAAGCTTTTCAATTCAATTCATCATTTTTTTTTTATTCTTTTTTTTG
(SEQ ID NO: 5).
-
Diese
enthält
eine Sequenz, die zur Region 5' der
publizierten Sequenz des Gens URA3 homolog ist (Rose et al., 1984;
GenBank: YSCODCD-Zugangsnummer: K02207; SGD : YEL021w), und zwar
assoziiert mit einer Sequenz, die zur Region 5' des Gens ATF2 homolog ist (komplementär zu OTG10844);
als indirekter Primer wurde das Oligonucleotid mit der folgenden
Sequenz verwendet:
OTG10845:
CTTGTCATTGTCACCTTCGGGAATGTCGAATGGGGTAATAACTGATATAATTAAATTGAACTC
(SEQ ID NO: 6)
-
Diese
enthält
eine Sequenz, die zur Region 3' des
Gens URA3 homolog ist, assoziiert mit einer Sequenz, die zur Region
3' des Gens ATF2
homolog ist (komplementär
zu OTG10846). Eine 20 ng-Probe von DNA des Gens URA3, die ausgehend
vom Pendelvektor E. coli-Hefe pTG10021 (Degryse et al., 1995) durch Verdauung
mit dem Restriktionsenzym HindIII isoliert worden war, wurde unter
den oben angegebenen Bedingungen amplifiziert.
-
Die
entsprechend erhaltenen PCR-Produkte wurden gereinigt, indem der "Geneclean Kit" (Bio 101 Inc., La
Jolla, USA) verwendet wurde, dann wurden sie der Doppelfusionsreaktion
unterworfen, wobei als Primer die oben genannten Oligonucleotide
OTG10841 und OTG10842 unter den Amplifikationsbedingungen, welche
für die
früheren
PCR-Reaktionen verwendet
wurden, mit einem Programm aus 20 Zyklen verwendet wurden.
-
Nach
Reinigung des Fusionsendproduktes, das die flankierenden Regionen
des Gens ATF2 fusioniert an das funktionelle Gen URA3 enthielt,
wurde das Vorliegen des Gens URA3 durch Verdauung mit dem Restriktionsenzym
EcoRV bestätigt,
welches das Vorliegen dieser Stelle im amplifizierten Material zeigt.
-
B) Bildung der Hefestämme atf2-Δ::URA3
-
Das
oben erhaltene Fusionsprodukt wurde direkt in die kompetenten Zellen
des Stamms Fy1679 oder des Stamms TGY73.4 transformiert und die
Transformanten wurden durch Wachstum auf dem Milieu SD (F. Sherman,
1991) in Gegenwart des Nährstoffbedarfs
des Stamms und in Abwesenheit von Uracil selektioniert.
-
Ausgehend
von den isolierten Klonen wurde die neue Verbindung zwischen der
Region 5' des Gens ATF2
und dem Gen URA3 (5'ATF2-URA3-3'ATF2) durch Amplifikation
durch PCR an den ganzen Zellen gezeigt, wobei die oben genannten
Primer OTG10841 und OTG10845 verwendet wurden. Die Abwesenheit der APAT-Aktivität wurde
dann nach dem beschriebenen Test, ausgehend vom Zellhomogenat, im
Vergleich zum Elternstamm, der eine deutliche APAT-Aktivität aufweist,
gezeigt.
-
Die
Stämme,
die auf diese Kriterien ansprechen, wurden so als die Mutantenstämme atf2
charakterisiert und als atf2-Δ::URA3
bezeichnet. Ein mutanter Stamm, der so ausgehend vom Elternstamm
Fy1679 erhalten worden war, wurde TGY156 genannt, und ein mutanter
Stamm, der ausgehend vom Elternstamm TGY73.4 erhalten worden war,
wurde TGY158 genannt.
-
Eine
Probe des Stamms TGY156 wurde bei der Collection Nationale de Cultures
de Microorganismes (CNCM) INSTITUT PASTEUR, 25, Rue du Docteur Roux
75724 Paris Cedex 15, Frankreich, am 02. Februar 1998 unter der
Nummer I-1977 hinterlegt.
-
Eine
Probe des Stamms TGY158 wurde bei der Collection Nationale de Cultures
de Microorganismes (CNCM) INSTITUT PASTEUR, 25, Rue du Docteur Roux
75724 Paris Cedex 15, Frankreich, am 02. Februar 1998 unter der
Nummer I-1976 hinterlegt.
-
C) Stabilisierung von
Pregnenolon in vivo in den Kulturen der Hefestämme atf2-Δ::URA3
-
Die
Zellen des Stamms TGY158, die oben erhalten worden waren, wurden
mit A600 = 0,1 in YPD (Difco)-Medium, das 100 μg/ml Pregnenolon enthielt, inokuliert.
Nach 16-stündiger
Inkubation bei 28°C
wurden die Steroide mit Dichlormethan extrahiert und durch RP-HPLC,
wie oben angegeben, analysiert. Die 6(B) zeigt,
dass die Mutante TGY158 die Fähigkeit,
Pregnenolon zu verestern, verloren hat, während der Elternstamm TGY73.4
unter denselben Bedingungen Pregnenolon in Pregnenolonacetat umwandelt
(6(A)).
-
Diese
Resultate lassen den Schluss zu, dass das Produkt des Gens ATF2
für die
Veresterung von Pregnenolon durch die Hefe verantwortlich ist, während die
Unterbrechung des Gens ATF2 zu keinen offensichtlichen Modifikationen
des Zellwachstums unter normalen Bedingungen geführt hat.
-
Beispiel 3: Konstruktion
eines Hefestamms, der das Gen ATF2 durch das Gen URA3 unterbrochen
hat und 3β-HSD
exprimiert
-
Plasmide
2μ, die
eine cDNA, die für
die humane 3β-HSD
codiert, unter der Kontrolle des Promotors CYC1 von S. cerevisae
oder des Promotors TEF1 von S. cerevisae tragen und das Resistenzgen
für G418 tragen,
wurden nach dem Schema der 7A bis 7C aufgebaut,
dann in die Mutantenhefestämme atf2-Δ::URA3 transformiert.
-
Im
ersten Arbeitsgang wurde ein Transfervektor pTG10095, der die cDNA-Sequenz,
die für
die humane 3β-HSD,
Typ II, beschrieben von E. Rheaume et al., 1991, codiert, flankiert
von den SalI- und MluI-Stellen und stromabwärts vom Hefepromotor GAL10/CYC1
gelegen, enthält,
wurde in folgender Weise gebildet:
-
Die
Sequenz, die für
die 3β-HSD
codiert, wurde nach E. Rheaume et al., 1991, als SalI-NotI-Restriktionsfragment
in dieselben Stellen des Vektors Bluescript II (Stratagene) subkloniert.
Der erhaltene Vektor, der von E. Rheaume et al., 1991, beschrieben
wurde, enthält
eine NotI-Stelle am 3'-Ende
der Sequenz, welche für 3β-HSD codiert,
lokalisiert. Dieser Vektor wurde dann durch das Restriktionsenzym
NotI verdaut, danach durch das Klenow-Fragment in Gegenwart von
dNTP behandelt, um die kohäsiven
Enden aufzufüllen,
danach in Gegenwart des Oligonucleotids mit der folgenden Sequenz:
OTG4461:
CACACGCGTGTG (SEQ ID NO: 7),
die vorher phosphoryliert und
mit sich selbst hybridisiert worden war, um eine MluI-Stelle einzuführen, religiert. Der
so erhaltene Vektor pTG10082 (7A) enthält die Sequenz,
die für
3β-HSD codiert,
welche eine BglII-Stelle enthält
und durch die SalI- und MluI-Stellen begrenzt wird, während die
NotI-Stelle verloren wurde. Dieser Vektor enthält noch die nicht-codierende
natürliche
Region 5', welche
durch das Vorliegen einer BglII-Stelle identifiziert worden war.
-
Um
die Stelle SalI des ATG-Initiators stromaufwärts, zu der, an der man den
Promotor GAL10/CYC1 einführen
möchte,
anzunähern,
wurde der Vektor pTG10082 durch das Restriktionsenzym MscI, dessen
Stelle in der nicht-codierenden
Region 5' und genau
stromaufwärts
des ATG-Initiators
gelegen ist, und durch das Restriktionsenzym MluI verdaut. Das Fragment
MscI-MluI mit 1,8 kb, das die codierende Sequenz für 3β-HSD enthält (7A),
wurde isoliert und dann in das Plasmid pTG10033 (E. Degryse et al.,
1995), das den Promotor GAL10/CYC1 enthält, der vorher durch das Restriktionsenzym
SalI verdaut worden war, dann in Gegenwart von dNTP durch das Klenow-Fragment
behandelt, danach durch das Restriktionsenzym MluI verdaut. Auf
diese Weise wird der Vektor pTG10095 (7B) erhalten.
-
In
einem zweiten Verfahrensschritt wird der Rekombinationsvektor pTG10268,
der das Plasmid 2μ von Hefe,
ein Replikon von E. coli, eine Expressionskassette CYClprom-PGKterm und
den Selektionsmarker LEU2 (7B) enthält, konstruiert.
Dieser Vektor ist mit dem beschriebenen Vektor pTG10159 (E. Degryse
et al., 1995) identisch, außer
dass die Stelle XbaI, die in der Region 2μ enthalten war, durch einen
Marker XbaI°,
welcher durch Auffüllen
der natürlichen
Stelle XbaI in Gegenwart des Klenow-Fragments, danach Religation
erhalten worden war, ersetzt war.
-
Das
Expressionsplasmid pTG10268 wurde dann durch homologe Rekombination
durch Einführung des
Expressionsblocks, der den Promotor Gal10/CYClp enthält, welcher
ausgehend vom Plasmid pTG10095, welches oben hergestellt worden
war, dann durch das Restriktionsenzym NotI verdaut worden war, erhalten worden
war, in das Plasmid pTG10260, welches vorher durch die Restriktionsenzyme
SalI und MluI verdaut worden war, erzeugt. Das Plasmid pTG10268
(7B) enthält
die Sequenz, die für
humane 3β-HSD,
Typ II, codiert, unter der Kontrolle des Promotors CYC1.
-
Expressionsplasmid
pTG10862, das die codierende Sequenz für 3β-HSD unter der Kontrolle des
Promotors TEF1 enthält,
wurde danach in folgender Weise konstruiert (7C):
-
Zunächst wurde
das Plasmid pTG10832 (8) durch homologe Rekombination
zwischen dem Fragment NotI, welches ausgehend vom Plasmid pTG10268,
das oben hergestellt worden war, dann durch das Restriktionsenzym
NotI verdaut worden war, erhalten worden war, und dem Rekombinationsplasmid
pTG10164 (E. Degryse et al., 1995), das vorher durch die Restriktionsenzyme
SalI und MluI verdaut worden war, hergestellt.
-
Das
Expressionsplasmid pTG10862 wurde danach durch Einführung des
Promotors TEF1 erhalten, welcher in dem Fragment ClaI-SalI enthalten war,
welches, ausgehend vom Plasmid pTG10085 (E. Degryse et al., 1995),
isoliert worden war, an die Stelle des Promotors CYC1, der durch
Verdau des Plasmids pTG10832, das oben konstruiert worden war, durch
die Restriktionsenzyme ClaI und SalI ausgeschnitten worden war,
erhalten.
-
Das
Plasmid pTG10862, das so erhalten worden war (9)
und die cDNA-Sequenz enthält,
die für humane
3β-HSD,
Typ II, codiert, wurde in den Elternstamm TGY73.4 oder in seine
Mutante atf2-Δ::URA3,
die dem Stamm TGY158, der in Beispiel 2 erhalten worden war, entspricht,
sowie in den Elternstamm FY1679 oder seine Mutante atf2-Δ::URA3, die
dem Stamm TGY156, der in Beispiel 2 erhalten worden war, entspricht,
transformiert. Die Transformanten wurden wie oben angegeben auf
YPD-Medium (Difco), das 250 μg/ml
G418 enthielt, isoliert.
-
Die
so erhaltenen Kolonienkandidaten wurden dann auf SD-Medium, das die für jeden
Stamm notwendigen Nährstoffe
enthielt (Histidin und Uracil für
den Stamm TGY73.4; Histidin für
TGY158; Tryptophan, Histidin, Leucin und Uracil für den Stamm
FY1679; Tryptophan, Histidin und Leucin für den Stamm TGY156; jeweils in
einer Konzentration von 100 μg/ml)
vorkultiviert, dann in ein Medium inokuliert, das 100 μg/ml Pregnenolon enthielt.
Nach 24 h Wachstum und Bioumwandlung bei 28°C wurden die Steroide extrahiert
und wie oben angegeben durch RP-HPLC gemessen. Die erhaltenen Resultate,
die für
jeden Stamm an drei Klonen gemessen wurden, sind in der folgenden
Tabelle 3 angegeben:
-
-
Diese
Resultate zeigen, dass das Substrat Pregnenolon in den Mutantenstämmen TGY156
oder TGY158 fast quantitativ wiedergewonnen wird, wobei in diesen
das Auftreten einer Bioumwandlung von Pregnenolon in Progesteron
beobachtet wird, während
in den Elternstämmen
TGY73.4 oder FY1679 das Verschwinden von Pregnenolon fast vollständig ist,
wobei diese kein oder sehr wenig Progesteron produzieren, das Pregnenolonacetat
allerdings akkumulieren, wie es in Beispiel 2 gezeigt wurde.
-
Eine
Probe des transformierten Stamms TGY158/pTG10862 wurde bei der Collection
Nationale de Cultures de Microorganismes (CNCM) (INSTITUT PASTEUR,
25, Rue du Docteur Roux 75724 Paris Cedex 15, Frankreich, am 02.
Februar 1998 unter der Nummer I-1978
hinterlegt.
-
Beispiel 4: Konstruktion
eines Hefestamms, der das Gen ATF2, das durch TEF1prom/PGKterm unterbrochen ist, hat (atf2-Δ::ΤΕF1/PGK)
-
Der
Stamm TGY186, der ein Stamm ist, der vom Stamm TGY156 abgeleitet
ist (atf2-Δ::URA3)
und der in Beispiel 2 beschrieben ist, wobei das Gen URA3 am Locus
ATF2 durch den Expressionsblock ΤΕF1prom/PGKterm ersetzt
wurde, wurde wie folgt konstruiert:
-
In
einem ersten Verfahrensschritt wurde der Expressionsblock ΤΕF1prom/PGKterm mit
dem Gen ATF2 durch Doppelfusions-PCR unter den in Beispiel 2 beschriebenen
Bedingungen assoziiert, wobei allerdings der Expressionsblock ΤΕF1prom/PGKterm von
S. cerevisiae, der von E. Degryse et al., 1995, beschrieben wurde,
anstelle des Selektionsmarkers URA3 verwendet wurde. Die zwei ersten
PCR-Reaktionen (PCR1), die die Amplifikation der codierenden Regionen
5' und 3' des Gens ATF2, die
die Insertionsstelle des Blocks ΤΕF1prom/PGKterm im unterbrochenen
Zielgen ATF2 flankieren, wurden durchgeführt, indem als direkte oder
indirekte Primer für
die Region 5' die
Oligonucleotide verwendet wurden, die die folgenden Sequenzen haben:
OTG11049:
CTCTCTGTCGACAAAATGGAAGATATAGAAGGATACGAACCACATATCACTC
(SEQ ID NO: 8) und
OTG10844: ATCAATCTCCAATTAGGCCTCTTCGGATTACCC
(SEQ ID NO: 9)
und für
die Region 3', die
Oligonucleotide mit den folgenden Sequenzen verwendet wurden:
OTG10846:
CATTCGACATTCCCGAAGGTGACAATGACAAG. (SEQ ID NO: 10) und
OTG11050:
AACAACACGCGTAACTATTAAAGCGACGCAAATTCGCCGATGCTTTGG (SEQ ID NO: 11).
-
Die
Primer OTG11049 und OTG11050 wurden entwickelt, um jeweils die Restriktionsstellen
SalI und MluI einzuführen.
-
Die
dritte PCR-Reaktion (PCR2), die die Amplifikation des Blocks ΤΕF1prom/PGKterm ermöglicht,
wurde durchgeführte,
indem als direkte oder indirekte Primer die Oligonucleotide verwendet
wurden, die die folgenden Sequenzen haben:
OTG11052:
GGGTAATCCGAAGAGGCCTAATTGGAGATTGATATCGATCACACACCATAGCTTCAAAATGTTTCTAC (SEQ
ID NO: 12) und
OTG11053:
CTTGTCATTGTCACCTTCGGGAATGTCGAAGCTTCGAAACGCAGAATTTTCGAGTTATTAAACTTAR
(SEQ ID NO: 13),
welche die Restriktionsstellen ClaI und HindIII
einführen.
Schließlich
wurde die Verbindung durch Doppelfusion der oben erhaltenen PCR-Produkte
realisiert, wobei als Primer die Oligonucleotide mit den Sequenzen OTG11049
(SEQ ID NO: 8) und OTG11050 (SEQ ID NO: 11) verwendet wurden, welche
die Restriktionsstellen SalI und MluI an ATF2-junction-Stellen einführen.
-
Nach
Reinigung wurde das Fusionsendprodukt mit dem Gen ATF2, welches
in dem Plasmid pTG10885 enthalten war und wie oben angegeben konstruiert
und vorher mit den Restriktionsenzymen BstI und StuI verdaut worden
war, rekombiniert. Auf diese Weise wird das Plasmid pTG10888, das
das Signal ΤΕF1prom/PGKterm an den
Stellen ClaI und HindIII enthält,
welche durch die flankierenden Regionen des Gens ATF2 begrenzt werden.
-
Die
Herstellung des Plasmids pTG10885 umfasst die Amplifikation des
Gens ATF2, ausgehend vom Stamm FY1679, nach den in Beispiel 2 beschriebenen
Bedingungen, aber unter Verwendung als direkte und indirekte Primer,
der Oligonucleotide, die die Sequenzen OTG11049 (SEQ ID NO: 8) und
OTG11050 (SEQ ID NO: 11), die oben angegeben sind, haben, welche
die Restriktionsstellen SalI und MluI einführen. In dem erhaltenen PCR-Produkt
werden diese Stellen dann durch Verdauung mit den Restriktionsenzymen
SalI und MluI, danach Behandlung mit dem Klenow-Fragment der Polymerase
I von E. coli, um die klebrigen Enden aufzufüllen, eliminiert. Das erhaltene
Fragment wurde dann in den Expressionsvektor pTG10031, der von E.
Degryse et al., 1995, beschrieben wurde und der vorher mit den Enzymen
ClaI und HindIII verdaut, danach mit dem Klenow-Fragment behandelt
worden war, ligiert. Durch Transformation in E. coli wird das Plasmid pTG10885
erhalten, das aus der Ligation der SalI-Stelle des PCR-Produktes, das unter
Verwendung des Klenow-Fragments derart aufgefüllt wurde, dass die Sequenz
GTCGA erhalten wurde, mit der HindIII-Stelle des Vektors, die derart
unter Verwendung des Klenow-Fragments aufgefüllt wurde, dass die Sequenz
AGCTT erhalten wurde, resultiert, und zwar derart, dass die Stelle
HindIII (GTCGAAGCTT) (SEQ ID NO: 14) wieder aufgebaut wurde und
die Stelle ClaI verloren ging. Die Stelle ClaI des Vektors, die
unter Verwendung des Klenow-Fragments so aufgefüllt wurde, dass die Sequenz
ATCG erhalten wurde, geht nach Ligation des PCR-Produktes verloren.
-
Das
Signal ΤΕF1prom/PGKterm wurde
dann in Form eines Fragments NotI mit 1,8 kb aus dem Plasmid pTG10888
ausgeschnitten, danach wurde es mit dem Marker URA3 in dem Stamm
TGY156 (atf2-Δ::URA3) ausgetauscht.
-
Der
in Beispiel 2 hergestellte Stamm TGY156, der als Wirtsstamm verwendet
wurde, wurde mit der DNA, die aus dem Plasmid pTG10888 ausgeschnitten
worden war, und mit dem Hefevektor, der einen ARS-Ursprung enthält, Yrp7
genannt, beschrieben von Struhl et al., 1979, co-transformiert,
was es ermöglicht,
den Tryptophanbedarf des Stamms TGY156 zu ergänzen, und eine selektive Detektion
der Kolonien durch ihre Resistenz gegenüber der 5-Fluororotsäure (5-FO)
ermöglicht.
-
2
bis 5 μg
der DNA, die durch Verdau mit dem Restriktionsenzym NotI aus dem
Plasmid pTG10888 und dem Plasmid Yrp7 ausgeschnitten worden war,
wurden durch das Lithiumacetatverfahren (Ito et al., 1983) in den
Stamm TGY156 eingeführt.
Die Selektion auf Komplementation des Bedarfs an Tryptophan des Stamms
wurde dann nach Ausbreitung in Agarschalen in YNGB-Medium (Difco),
das mit Histidin und Leucin angereicht war (jeweils mit 100 μg/ml) durchgeführt. Die
Kandidatenkolonien, die mit Hilfe eines cure-dent entnommen wurden,
wurden dann auf ein Medium gegeben, das 5-FO enthielt und das nach Boeke et al.,
1984, hergestellt worden war; dann wurde die Resistenz gegenüber 5-FO
auf demselben Medium bestätigt,
wobei der eventuelle Verlust des Vektors Yrp7 in den gegenüber 5-FO
resistenten Kolonien durch einen Bedarf an Tryptophan angezeigt
wurde. Unter den so selektionierten Klonen wurde die Assoziation
des Gens ATF2 mit ΤΕF1prom und PGKterm durch
PCR gesteuert. Auf diese Weise wird der Stamm TGY186 erhalten.
-
Beispiel 5: Konstruktion
eines Hefestamms, der das Gen ATF2, unterbrochen durch ΤΕF1prom/PGKterm, hat
und P45017α exprimiert
-
Ein
Plasmid (pTG10435), das einen Hefereplikationsursprung ARSH4/CEN6,
den Selektionsmarker URA3 enthält
und eine cDNA-Sequenz,
die für
das bovine Cytochrom P45017α codiert,
unter der Kontrolle des Promotors TEF1 von S. cerevisiae trägt, wurde
nach dem Schema der 10 aufgebaut, dann in den Mutantenhefestamm
atfs-Δ::ΤΕF1/PGK (TGY186)
transformiert. In einer ersten Stufe wurde das Plasmid pTG10058, das
die cDNA-Sequenz, die für
das bovine Cytochrom P45017α codiert,
das von Zuber et al., 1986, beschrieben wurde, flankiert von den
Stellen SalI und MluI und stromabwärts des Hefepromotors CYC1
gelegen, enthält, nach
dem Schema der 11 gebildet: das Plasmid pGB17α-5, das in
der Patentanmeldung WO 89/10963 beschrieben ist und die Sequenz,
die für
das bovine Cytochrom P45017α codiert,
enthält,
wurde durch Verdau mit dem Restriktionsenzym XhoI geöffnet, dann
mit alkalischer Phosphatase behandelt. Nach Phosphorylierung und
Hybridisierung wurden die Oligonucleotide, die die folgenden Sequenzen
haben:
OTG4511: TCGACGGACGCGTGG (SEQ ID NO: 15) und
OTG4512:
TCGACCACGCGTCCG (SEQ ID NO: 16)
in die Stelle XhoI eingeführt, wodurch
das Plasmid pTG10104 gebildet wurde. Das Plasmid pTG10104 wurde dann
durch die Restriktionsenzyme SalI und MluI behandelt, dann in das
von E. Degryse et al., 1995, beschriebene Plasmid pTG10031 eingeführt, das
den Hefepromotor CYC1 enthielt, der vorher mit den Restriktionsenzymen
SalI und MluI verdaut worden war und mit alkalischer Phosphatase
behandelt worden war. Auf diese Weise wird der Vektor pTG10058,
der die cDNA, die für
das bovine Cytochrom P45017α codiert,
enthält,
erhalten (12). Das Plasmid pTG10058 wurde
dann durch die Restriktionsenzyme SalI und MluI verdaut und mit alkalischer
Phosphatase behandelt. Das Fragment SalI-MluI mit 1,7 kb, das die
für das
bovine Cytochrom P45017α codierende Sequenz enthält, wurde
isoliert, dann in den Expressionsvektor pTG10085, der von E. Degryse
et al., 1995, beschrieben wird und den Hefepromotor TEF1 enthält, vorher
mit den Enzymen SalI und MluI verdaut worden war, ligiert. Auf diese
Weise wird das Plasmid pTG10293 (13), in
dem die für
das Cytochrom P45017α codierende Sequenz unter der
Kontrolle des Promotors TEF1 ist, erhalten. In einer zweiten Stufe
wurde das Expressionsplasmid pTG10435 durch homologe Rekombination
zwischen dem Rekombinationsplasmid TG10434, das von E. Degryse et
al., 1995, beschrieben wurde, und die Sequenz ARSH4/CEN6 enthält, das
vorher durch die Enzyme SalI und MluI verdaut worden war, und dem
Fragment NotI mit 2,8 kb, das ausgehend von dem wie oben beschrieben
hergestellten Plasmid pTG10293 erhalten worden war, gebildet. Das
so erhaltene Plasmid pTG10435 (14), das
die für
das bovine Cytochrom P45017α codierende
Sequenz unter der Kontrolle des Promotors TEF1 enthielt, wurde dann
in den Elternstamm FY1679 oder in den in Beispiel 4 hergestellten
Stam TGY186 (atf2-Δ::TEF1/PGK)
transformiert. Die Transformanten wurden wie oben angegeben auf
YNBG-Medium (Difco), das mit Tryptophan, Histidin und Leucin (jeweils
100 μg/ml)
angereichert worden war, isoliert. Die so erhaltenen Kolonien wurden
dann während
16 h im YNB-Medium (Difco), das 2% Glucose und 0,5% Casaminosäuren enthielt,
vorkultiviert, dann in frischem Medium auf A600 = 0,2 verdünnt. Nach
6 h Wachstum wurden 100 μ/ml
Pregnenolon oder Progesteron zugesetzt. Nach 48 Stunden Wachstum
und Bioumwandlung bei 28°C
wurden die Steroide extrahiert und durch RP-HPLC, wie in Beispiel
1 angegeben, gemessen, wobei jeweils Standards von Pregnenolon und
17α-Hydroxypregenolon
oder Progesteron und 17α-Hydroxyprogesteron
verwendet wurden.
-
Die
erhaltenen Resultate sind in μg/ml
ausgedrückt
und werden in der folgenden Tabelle 4 gezeigt:
-
-
Die
Resultate zeigen, dass das Bioumwandlungsvermögen des Cytochroms P45017α,
das vom Vektor pTG10435 exprimiert wird, etwas das gleiche wie das
im Wildstamm (FY) oder in seiner Mutante atf2 (TGY186) mit Progesteron
als Substrat ist. Andererseits wird mit Pregnenolon als Substrat
dieselbe Bioumwandlung wie bei Progesteron erreicht, allerdings
nur mit der Mutante atf2 (TGY186). Im Wildstamm FY werden das Substrat und
das Produkt gleichzeitig acetyliert, und es wird kein freies Hydroxyprogesteron
nachgewiesen.
-
Eine
Probe des transformierten Stamms TGY186/pTG10435 wurde bei der Collection
Nationale de Cultures de Microorganismes (CNCM) INSTITUT PLASTEUR,
25, Reu du Docteur Roux 75724 Paris Cedex 15, Frankreich, am 20.
Januar 1999 unter der Nummer I-2119 hinterlegt.
-
Beispiel 6: Konstruktion
eines Hefestamms, der das Gen ATF2, das durch TEF1prom/PKGterm unterbrochen ist, hat und der 3β-HSD und
P45017α gleichzeitig
exprimiert
-
Das
Plasmid pTG10417 (21), das das Hefereplikon 2μ und zwei
Expressionsblöcke,
der eine, der für
humane 3β-HSD codiert,
der andere, der für
das bovine Cytochrom P45017α codiert,
wobei beide unter der Kontrolle des Promotors CYC1 von S. cerevisiae
stehen, enthält
und das den Selektionsmarker URA3-d trägt, wurde aufgebaut, indem
sukzessive die Stufen 1 bis 3, die unten beschrieben werden (15A und B), danach
die unten beschriebenen Stufen 4 bis 6 durchgeführt werden, dann wird es in
den Mutantenhefestamm atf2-Δ::TEF1prom/PKGterm (TGY186)
transformiert.
-
Stufe 1: Konstruktion
des Plasmids pTG10210
-
Der
Expressionsvektor pTG10033, der von E. Degryse et al., 1995, beschrieben
wurde und der den Hefehybridpromotor GAL10/CYC1, der vorher durch
das Restriktionsenzym pvuII verdaut worden war, enthielt, wurde
mit alkalischer Phosphatase behandelt und dann in Gegenwart des
Oligonucleotids, das die folgende Sequenz hat:
OTG1050: CCCGAATTCGGG
(SEQ ID NO: 17),
das vorher phosphoryliert und mit sich selbst
hybridisiert worden war, um die Stellen EcoRI am Rand des Expressionsblocks,
der den Promotor GAL10/CYC1 enthält,
einzuführen,
religiert. Auf diese Weise wird der Vektor pTG10210 erhalten.
-
Stufe 2: Konstruktion
des Plasmids pTG10214
-
Der
Expressionsblock, der den Promotor GAL10/CYC1 enthält, der
im Vektor pTG10210 vorliegt, wird dann in den Pendelvektor E. coli-Hefe
pTG10013 eingeführt,
der von E. Degryse et al., 1995, beschrieben wurde und den Selektionsmarker
URA3-d enthält.
-
Der
Vektor pTG10013 wurde nach Behandlung mit dem Restriktionsenzym
EcoRI und Behandlung mit alkalischer Phosphatase in den in Stufe
1 hergestellten Vektor pTG10210, der vorher mit dem Enzym EcoRI verdaut
worden war, ligiert. Der so erhaltene Vektor pTG10214 enthält den Expressionsblock,
der den Promotor GAL10/CYC1 enthält,
welcher zum Replikon 2μ verdaut
worden war.
-
Stufe 3: Konstruktion
des Plasmids pTG10274
-
In
Plasmid pTG10214 wurde dann der Promotor GAL10/CYC1 durch homologe
Rekombination nach Ausschneiden des Plasmids durch Behandlung mit
den Restriktionsenzymen ClaI und SalI durch den Promotor CYC1 ausgetauscht.
Der Expressionsvektor pTG100312, der von E. Degryse et al., 1995,
beschrieben worden war und den Promotor CYC1 enthält, wurde
durch die Restriktionsenzyme HindIII und FspI verdaut, dann mit
Plasmid pTG10214, das in Stufe 2 hergestellt worden war und vorher
mit den Restriktionsenzymen ClaI und SalI verdaut worden war, rekombiniert;
auf diese Weise wurde das Plasmid pTG10274 (16) gebildet.
-
Stufe 4: Konstruktion
des Plasmids pTG10401
-
Ausgehend
vom Plasmid pTG10274, das den Promotor CYC1 enthält, und vom Plasmid pTG10293, das
die Sequenz enthält,
die für
das Cytochrom P45017α unter der Kontrolle des Promotors
TEF1 codiert, wurde dass durch homologe Rekombination neues Plasmid
pTG10401, das die Sequenz enthält,
die unter der Kontrolle des Promotors TEF1 für das Cytochrom P45017α codiert,
gebildet.
-
Der
Promotor CYC1 und ein Teil des Replikons von E. coli wurden aus
dem Plasmid pTG10274, das in sTufe 3 hergestellt worden war, durch
Verdau mit den Restriktionsenzymen MluI und DraI ausgeschnitten. Das
in Beispiel 5 hergestellte Plasmid pTG10293 wurde mit den Restriktionsenzymen
HindIII und PvuII verdaut, dann mit dem Plasmid pTG10274, das mit
den Restriktionsenyzen MluI und DraI verdaut worden war, rekombiniert;
auf diese Weise wurde das Plasmid pTG10401 (17) gebildet.
-
Stufe 5: Konstruktion
des Plasmids pTG10403
-
Die
cDNA, die für
die humane 3β-HSD
des Typs II codiert, wurde dann in das Plasmid pTG10401 unter der
Kontrolle des Promotors CYC1 eingeführt.
-
In
einem ersten Arbeitsschritt wurde der Expressionsvektor pTG10262,
der die cDNA enthielt, die für die
humane 3β-HSD
des Typs II codiert, nach dem Schema der 18 aufgebaut,
und zwar ausgehend vom Transfervektor pTG10095, der in Beispiel
3 hergestellt worden war, und vom Rekombinationsvektor pTG10257,
der das Hefereplikon 2μ,
ein E. coli-Replikon, die Hefe-Expressionskassette CYC1prom-PGKterm und den Selektionsmarker URA3-d enthielt.
Dieser Vektor pTG10257 ist identisch mit dem von E. Degryse et al., 1995,
beschriebenen Rekombinationsvektor pTG10042, mit der Ausnahme der
Stelle XbaI, die in der Region 2μ enthalten
ist, und die durch einen Marker XbaI°, der durch Auffüllen der
natürlichen
Stelle XbaI mit dem Klenow-Fragment und anschließender Religation erhalten
worden war, ersetzt wurde.
-
Der
Expressionsblock der humanen 3β-HSD
des Typs II wurde durch Verdau mit dem Restriktionsenzym NotI aus
dem Transfervektor pTG10095 ausgeschnitten, dann in den Rekombinationsvektor
pTG10257, der vorher mit den Restriktionsenzymen SalI und MluI verdaut
worden war, eingeführt;
auf diese Weise wurde der Expressionsvektor pTG10262 (19)
gebildet. Es ist zu betonen, dass die Rekombination der cDNA des Blocks
GAL10/CYC1, die vom Plasmid pTG10095 stammt, im Rekombinationsvektor
pTG10257, der den Promotor CYC1 enthält, einen Expressionsvektor
produziert, der den Block CYC1-cDNA enthält.
-
In
einem zweiten Verfahren wird der oben hergestellte Expressionsvektor
pTG10262 mit dem Restriktionsenzym XmnI verdaut. Das erhaltene Fragment,
das die cDNA enthält,
welche für
3β-HSD codiert,
wurde mit dem Fragment des in Stufe 4 hergestellten Plasmids pTG10401,
das die cDNA enthält,
welche für
das Cytochrom P45017α codiert, das durch Verdau mit
dem Restriktionsenzym ScalI erhalten worden war. Das so erhaltene
Plasmid pTG10403 (20) enthält zwei Expressionsblöcke, der
eine, der für
3β-HSDH
unter der Kontrolle des Promotors CYC1 codiert, der andere, der
für das
Cytochrom P45017α unter der Kontrolle des Promotors
TEF1 codiert.
-
Stufe 6: Konstruktion
des Plasmids pTG10417
-
Schließlich wurde
in dem obigen Plasmid pTG10403 der Promotor TEF1 durch den Promotor
CYC1 ausgetauscht.
-
Einerseits
wurde das in Beispiel 5 beschriebene Plasmid pTG10058 mit dem Restriktionsenzym
PvuII verdaut, was es ermöglicht,
einen Teil der für
das Cytochrom P45017α codierenden Sequenz, die mit
dem Promotor CYC1 assoziiert ist, sowie den Hauptteil des Replikons
von E. coli freizusetzen. Andererseits wurde ein Teil des Replikons
von E. coli aus dem in Stufe 5 hergestellten Plasmid pTG10403 durch
Verdau mit dem Restriktionsenzym DraI eliminiert. Durch Rekombination
der zwei Plasmide, die vorher so verdaut worden waren, wird schließlich das
Plasmid pTG10417 erhalten. Das Plasmid pTG10417 enthält das Hefereplikon
2μ, den
Selektionsmarker URA3-d und die zwei Expressionsblöcke, von
denen einer für
die humane 3β-HSD
codiert, der andere für
das Cytochrom P45017α vom Rind codiert, wobei die
beiden unter der Kontrolle des Hefepromotors CYC1 sind (21).
-
Das
Plasmid pTG10417 wurde dann in den Elternstamm FY1679 oder in den
in Beispiel 4 hergestellten Stamm TGY186 (atf2-Δ::ΤΕF1prom/PGKterm) transformiert.
-
Die
Transformanten wurden auf einem gelierten YNB-Medium (Difco), das
0,5% Glucose enthielt, mit Tryptophan, Histidin und Leucin angereichert
war (100 μg/ml
jeweils) isoliert.
-
Die
so erhaltenen Kolonien wurden 24 h lang bei 28°C im YNB-Medium (Difco), das 0,5% Glucose und 0,1%
Casaminosäuren
enthält,
vorkultiviert, dann auf A600 = 0,1 verdünnt und mit 100 μg/ml Pregnenolon
ergänzt,
entweder im selben frischen Medium (Medium 1) oder im YNB-Medium
(Difco), das 0,1% Glucose, 2% Glycerin und 0,2% Casaminosäuren enthielt
(Medium 2). Nach 48 Stunden Wachstum und Umwandlung wurden die Steroide
extrahiert und durch RP-HPLC, wie in Beispiel 1 beschrieben, gemessen,
wobei die Standards Pregnenolon, 17α-Hydroxypregnenolon, Progesteron
und 17α-Hydroxyprogesteron
verwendet wurden.
-
Die
erhaltenen Resultate, die in μg/ml
ausgedrückt
sind, werden in Tabelle 5 (Medium 1) und Tabelle 6 (Medium 2), die
folgen, gezeigt:
-
-
-
Diese
Resultate zeigen, dass die Bioumwandlung in den Wildstamm (FY),
der durch das Plasmid pTG10417 transformiert war, zu einer Akkumulierung
von Pregnenolonacetat und 17α-Hydroxypregnenolonacetat
führt,
die letztlich durch die Enzyme 3β-HSDH
oder P45017α nicht transformiert werden,
und dass die Bilanz der Bioumwandlung schlechter ist als die, die
mit der transformierten Mutante atf2 (TGY186) beobachtet wird.
-
Eine
Probe des transformierten Stamms TGY186/pTG10417 wurde bei der Collection
Nationale de Cultures de Microorganismes (CNCM) INSTITUT PASTEUR,
25, Rue du Docteur Roux 75724 Paris Cedex 15, Frankreich, am 20.
Januar 1999 unter der Nummer I-2118 hinterlegt.
-
Literaturstellen
-
- – Achstetter,
T., Nguyen-Julleret M., Findeli A., Merkamm M. und Lemoine Y. (1992),
Gene 110, 25–31.
- – Amberg
D. C., Botstein D. und Beasley E. M., (1995), Yeast 11 : 1275–1280.
- – Boeke
Jef D., Lacroute F. und Fink G. R., (1984) Mol Gen Genet 197 : 345–346.
- – Cauet
G., Dumas B., Degryse E, Spagnoli R. und Achstetter T.. (1994) in
Cytochrome P-450 biochemistry, biophysics and molecular biology
(Lechner M. C., Herausg.), S. 583–586, John Libbey Eurotext.
- – Cherry
J. M., Adler C., Ball C., Dwight S., Chervitz S., Jia Y., Juvik
G., Roe T., Wenig S. und Botstein D., "Saccharomyces Genome Database" http://genome-www.standard.edu/Saccharomyces/
- – Degryse
E., J. Biotech. 39 (1995) 181–187.
- – Degryse
E., Dumas B., Dietrich M., Laruelle L. und Achstetter T. (1995).
Yeast 11 : 629–640.
- – Degryse
E. (1996), Gene 170, 45–50.
- – Dumas
B., Cauet G., Degryse E., Spagnoli R. & Achstetter T. (1994). Cytochrome
P450. 8th International Conference. Herausg. M. C. Lechner. John
Lebbey Eurotext, Paris, S. 527–530.
- – Fiechter
A., Fuhrmann G. F. und Käppeli
O., (1981). Adv. Microb. Physiol. 22, 123–183.
- – Fujii
T., Nagasawa N., Iwamatsu A., Bogaki T., Tamai Y. und Hamachi M.
(1994) Appl. Environ. Microbiol. 60, 2786–2792.
- – Hanahan
D., J. Mol. Biol. 166 (1983) 557–580.
- – Hubacek
J. und Glover S. W., (1970), J. Mol. Biol. 50: 111–127.
- – Ito
H., Fukuda Y., Murata K. und Kimura A., 1983, Journal of Bacteriology,
163–168.
- – Rhéaume E.,
Lachance Y., Zhao H.–F.,
Breton N., Dumont M., de Launoit Y., Trudel C., Luu-The V., Simard J. & Labrie F. (1991).
Mol. Endocrinol. 5, 1147–1157.
- – Rose
M., Grisafi P. und Botstein D., Gene 29, 113–114 (1984).
- – Rosenfeld
J., Capdevielle J., Guillemot J. C. und Ferrara P. (1992) Anal.
Biochem. 203, 173–179.
- – Sambrook
J., Fritsch E. F. und Maniatis T., (1989). Molecular Cloning : A
Laboratory Manual. Cold Spring Harbor University Press, 2. Ausgabe,
Cold Spring Harbor.
- – Sherman
F. (1991) Methods in Enzymology 194, 3–21.
- – Simard
J., Durocher F., Mébarki
F., Turgeon C., Sanchez R., Labrie Y., Couet J., Trudel C., Rheaume
E., Morel Y., Luu-The V. And Labrie F. (1996) J. Endocrinol. 150,
5189–5207.
- – Struhl
K., Stinchomb DT., Scherer S. und Davis RW., 1979, Proc. Natl. Acad.
Sci. USA Bd. 76, Nr. 3, 1035–1039.
- – Thierry
A., Fairhead C. und Dujon B., Yeast; Bd. 6 521–534 (1990).
- – Zuber
MX., Shimpson ER. und Waterman MR., (1986a) Science 234, 1258–1261.
-
-
-
-