-
Die
vorliegende Erfindung betrifft Übergangsmetallverbindungen
und dieselben anwendende Polymerisationskatalysatorsysteme.
-
Die
Verwendung von bestimmten Übergangsmetallverbindungen
zum Polymerisieren von 1-Olefinen, beispielsweise Ethylen, ist auf
dem Fachgebiet gut bekannt. Die Verwendung von Ziegler-Natta-Katalysatoren, beispielsweise
jene Katalysatoren, die durch Aktivieren von Titanhalogeniden mit
Organometallverbindungen, wie Triethylaluminium, hergestellt werden,
ist für
viele industrielle Verfahren zum Herstellen von Polyolefinen von
grundsätzlicher
Bedeutung. Innerhalb der letzten zwanzig oder dreißig Jahre
führten
Fortschritte in der Technologie zu der Entwicklung von Ziegler-Natta-Katalysatoren, die
solche hohen Aktivitäten
aufweisen, dass jene Olefinpolymere und Copolymere, welche sehr
niedrige Konzentrationen von restlichem Katalysator enthalten, direkt
in industriellen Polymerisationsverfahren hergestellt werden können. Die
Mengen von restlichem Katalysator, der in dem hergestellten Polymer
verbleibt, sind so gering, dass ihre Abtrennung und Entfernung für die meisten
industriellen Anwendungen unnötig
wird. Solche Verfahren können
durch Polymerisieren der Monomere in der Gasphase oder in Lösung oder
in Suspension in einem flüssigen
Kohlenwasserstoffverdünnungsmittel
durchgeführt
werden. Die Polymerisation der Monomere kann in der Gasphase (das "Gasphasenverfahren") ausgeführt werden,
beispielsweise durch Fluidisieren eines das Zielpolyolefinpulver
und Teilchen des gewünschten
Katalysators umfassenden Betts unter Polymerisationsbedingungen,
unter Verwendung eines Fluidisierungsgasstroms, der das gasförmige Monomer
umfasst. Bei dem so genannten "Lösungsverfahren" wird die (Co)polymerisation
durch Einführen
des Monomers in eine Lösung
oder Suspension des Katalysators in einem flüssigen Kohlenwasserstoffverdünnungsmittel,
unter solchen Temperatur- und
Druckbedingungen durchgeführt,
dass das hergestellte Polyolefin in dem Kohlenwasserstoffverdünnungsmittel
eine Lösung
bildet. Bei dem "Aufschlämmungsverfahren" sind Temperatur,
Druck und die Auswahl von Verdünnungsmittel
derart, dass das erzeugte Polymer in dem flüssigen Kohlenwasserstoffverdünnungsmittel
eine Suspension bildet. Diese Verfahren werden im Allgemeinen bei
relativ niedrigen Drücken
(beispielsweise 10–50
bar) und niedriger Temperatur (beispielsweise 50 bis 150°C) durchgeführt.
-
Rohstoff-Polyethylene
werden industriell in einer Vielzahl von verschiedenen Arten und
Qualitäten
hergestellt. Homopolymerisation von Ethylen mit auf Übergangsmetall
basierenden Katalysatoren führt
zu der Herstellung von so genannten "hochdichten" Polyethylenqualitäten. Diese Polymere haben relativ
hohe Steifigkeit und sind zum Herstellen von Gegenständen, bei
denen innewohnende Starrheit gefordert ist, verwendbar. Copolymerisation
von Ethylen mit höheren
1-Olefinen (beispielsweise Buten, Hexen oder Octen) wird kommerziell
angewendet, um eine breite Vielzahl von Copolymeren, die sich in
der Dichte und in anderen wichtigen physikalischen Eigenschaften
unterscheiden, bereitzustellen. Besonders wichtige Copolymere, die
durch Copolymerisieren von Ethylen mit höheren 1-Olefinen unter Anwendung
von auf Übergangsmetall
basierenden Katalysatoren hergestellt werden, sind die Copolymere
mit einer Dichte im Bereich von 0,91 bis 0,93. Diese Copolymere,
die auf dem Fachgebiet im Allgemeinen als "linear niederdichtes Polyethylen" bezeichnet werden,
sind in vieler Hinsicht dem so genannten "niederdichten" Polyethylen ähnlich, das durch radikalisch
katalysierte Polymerisation von Ethylen unter hohem Druck hergestellt
wurden. Solche Polymere und Copolymere werden vielfach bei der Herstellung
von biegsamen Blasfolien verwendet.
-
Ein
wichtiges Merkmal der Mikrostruktur der Copolymere von Ethylen und
höheren
1-Olefinen ist die Art, in der polymerisierte Comonomereinheiten
entlang der "Gerüst"kette von polymerisierten
Ethyleneinheiten verteilt sind. Die üblichen Ziegler-Natta-Katalysatoren
erzeugen in der Regel Copolymere, worin die polymerisierten Comonomereinheiten
entlang der Kette miteinander verklumpt sind. Um besonders erwünschte Filmeigenschaften
von solchen Copolymeren zu erreichen, sind die Comonomereinheiten
in jedem Copolymermolekül
vorzugsweise nicht miteinander verklumpt, sondern über die
Länge von
jeder linearen Polyethylenkette deutlich beabstandet. In den letzten
Jahren hat die Verwendung von bestimmten Metallocenkatalysatoren
(beispielsweise Biscyclopentadienylzirkoniumdichlorid, aktiviert
mit Alumoxan) Katalysatoren mit potenziell hoher Aktivität und der
Fähigkeit
zur Bereitstellung einer verbesserten Verteilung von Comonomereinheiten
bereitgestellt. Jedoch haben Metallocenkatalysatoren dieses Typs
eine Vielzahl von Nachteilen, beispielsweise hohe Empfindlichkeit
gegenüber
Verunreinigungen, wenn mit kommerziell verfügbaren Monomeren, Verdünnungsmitteln
und Verfahrensgasströmen
verwendet, Bedarf der Anwendung großer Mengen kostspieliger Alumoxane
zum Erreichen hoher Aktivität
und Schwierigkeiten des Aufbringens des Katalysators auf einen geeigneten Träger.
-
WO98/27124
offenbart, dass Ethylen durch In-Kontakt-Bringen mit bestimmten Eisen- oder Cobaltkomplexen
von ausgewählten
2,6-Pyridincarboxaldehydbis(iminen) und 2,6-Diacylpyridinbis(iminen)
polymerisiert werden kann. Diese Komplexe werden für das Herstellen
von Homopolymeren von Ethylen als geeignet offenbart. Aktivitäten von
6 bis 2985 g/mMol/h/bar werden gezeigt.
-
Wir
haben neue Katalysatoren, unter Anwendung von Komplexen, wie jenen,
offenbart in WO98/27124, entwickelt, welche ausgezeichnete Aktivitäten und
Produkte bereitstellen. Folglich wird gemäß einem ersten Aspekt der Erfindung
ein Katalysator für
die Polymerisation von Olefinen bereitgestellt, umfassend
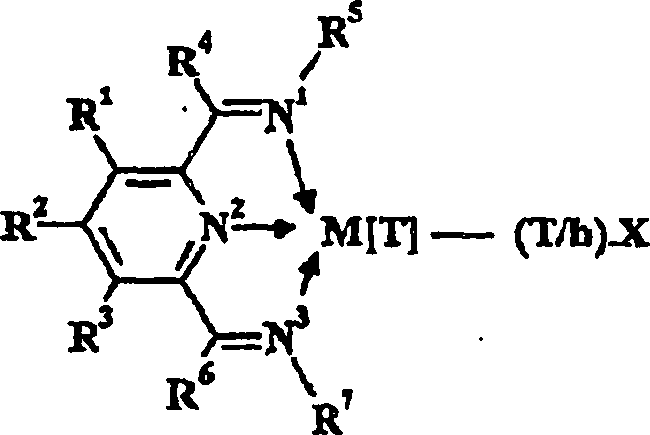
- (1) eine Verbindung der Formel B, Formel
B worin M Fe[II], Fe[III], Co[I], Co[II], Co[III],
Mn[I], Mn[II], Mn[III], Mn[IV], Ru[II], Ru[III] oder Ru[IV] darstellt;
X ein Atom oder eine Gruppe, die kovalent oder ionisch an das Übergangsmetall
M gebunden sind, wiedergibt; T den Oxidationszustand des Übergangsmetalls
M darstellt und b die Wertigkeit des Atoms oder der Gruppe X bedeutet;
R1, R2, R3, R4, R5,
R6 und R7 unabhängig aus
Wasserstoff, Halogen, Kohlenwasserstoff, substituiertem Kohlenwasserstoff,
Heterokohlenwasserstoff oder substituiertem Heterokohlenwasserstoff ausgewählt sind;
und wenn beliebige zwei oder mehr von R1 bis
R7 Kohlenwasserstoff, substituierten Kohlenwasserstoff,
Heterokohlenwasserstoff oder substituierten Heterokohlenwasserstoff
darstellen; die zwei oder mehreren zur Bildung von einem oder mehreren
cyclischen Substituenten verbunden sein können;
- (2) einen Aktivator, der ein Alkylalumoxan darstellt; und
- (3) zusätzlich
zu (2) eine Verbindung der Formel AlR3,
worin jedes R unabhängig
C1-C12-Alkyl oder
Halogen darstellt.
-
Wir
haben gefunden, dass die Einarbeitung von Komponente (3) in den
Katalysator wesentliche Verbesserungen der Aktivität ergeben
kann. Die drei Substituenten R in Verbindung (3), welche gleich
oder verschieden sein können,
sind vorzugsweise Wasserstoff, Methyl, Ethyl, Butyl oder Chlor.
Bevorzugte Verbindungen (3) schließen Trimethylaluminium (TMA),
Triethylaluminium (TEA), Triisobutylaluminium (TIBA), Tri-n-octylaluminium,
Ethylaluminiumdichlorid und Diethylalu miniumchlorid ein. Besonders
bevorzugt sind TMA und TIBA. Jedoch kann die bevorzugte Verbindung
(3) von den Polymerisationsbedingungen abhängen, in denen der Katalysator
angewendet wird: beispielsweise ist TMA beim Verbessern der Katalysatoraktivität in der
Gasphase und auch der Aktivität
von ungetragenen Katalysatoren in der Aufschlämmungsphase besonders wirksam,
während
TIBA im Allgemeinen in der Aufschlämmungsphase der Polymerisation
besonders wirksam ist.
-
Als
Aktivator (2) schließt
der erfindungsgemäße Katalysator
ein Alkylalumoxan ein, das normalerweise ein (C1-C4)Alkylalumoxan
darstellt, wobei die Alkylgruppe im Allgemeinen Methyl, Ethyl, Propyl
oder Isobutyl darstellt. Bevorzugt ist Methylalumoxan (auch bekannt
als Methylaluminoxan oder MAO) oder modifiziertes Methylalumoxan
(MMAO), das zusätzlich
Isobutylalumoxan enthält.
Der wie in dieser Beschreibung verwendete Begriff "Alkylalumoxan" schließt Alkylalumoxane
ein, die kommerziell erhältlich
sind, welche einen Anteil von typischerweise etwa 10 Gewichtsprozent,
jedoch gegebenenfalls bis zu 50 Gewichtsprozent des entsprechenden
Trialkylaluminiums enthalten können;
beispielsweise enthält
kommerzielles MAO gewöhnlich
ungefähr
10 Gewichtsprozent Trimethylaluminium (TMA), während kommerzielles MMAO sowohl
TMA als auch TIBA enthält.
Die Mengen an hierin angeführtem
Alkylalumoxan schließen
solche Trialkylalkylaluminiumverunreinigungen ein, und folglich
wird festgelegt, dass in dieser Erfindung Komponente (3) Verbindungen
der Formel AlR3 zusätzlich zu beliebiger in dem
Alkylalumoxan (2) eingearbeiteter AlR3-Verbindung
enthält
und Mengen von hierin angeführter
Komponente (3) werden auf dieser Basis berechnet.
-
Bei
der Herstellung der erfindungsgemäßen Katalysatorsysteme wird
die Menge an anzuwendender aktivierender Verbindung (2) durch einfaches
Testen, beispielsweise durch die Herstellung von kleinen Testproben,
bestimmt, welche verwendet werden können, um kleine Mengen des/der
Monomers/e zu polymerisieren, um somit die Aktivität des hergestellten
Katalysators zu bestimmen. Es wird im Allgemeinen gefunden, dass die
angewendete Menge ausreichend ist, um 0,1 bis 20000 Atome, vorzugsweise
1 bis 2000 Atome, Aluminium pro Fe-, Co-, Mn- oder Ru-Metallatom
in der Verbindung der Formel B bereitzustellen. Die Menge an Aktivator
(2), die für
optimale Leistung gefordert wird, kann auch von der Menge an vorliegender
Alkylaluminiumverbindung (3) abhängen.
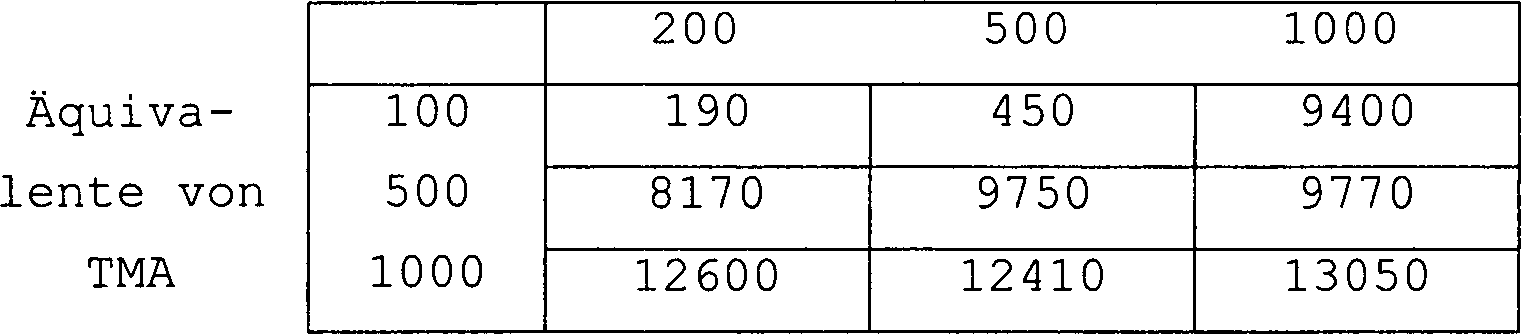
Wenn beispielsweise Verbindung (3) Trimethylaluminium (TMA) darstellt
und die Menge an TMA in dem Katalysator weniger als 500 Moläquivalente,
bezogen auf das Metallatom von Verbindung (1), ist, ist die Menge
an Alkylalumoxan (gewöhnlich
MAO) vorzugsweise mindestens 1000 Moläquivalente. Wenn jedoch 500 Äquivalente
oder mehr TMA vorliegen, ist die optimale Menge von Alkylalumoxan
(gewöhnlich
MAO) 500 bis 1000 Äquivalente.
-
Vorzugsweise
ist in Formel B vorstehend genanntes M Fe[II], Fe[III], Ru[II],
Ru[III] oder Ru[IV]; X gibt ein Atom oder eine Gruppe, die kovalent
oder ionisch an das Übergangsmetall
M gebunden ist, wieder; T ist der Oxidationszustand des Übergangsmetalls
M und b bedeutet die Wertigkeit des Atoms oder der Gruppe X; R1, R2, R3,
R4, R5, R6 und R7 sind unabhängig ausgewählt aus
Wasserstoff, Halogen, Kohlenwasserstoff, substituiertem Kohlenwasserstoff,
Heterokohlenwasserstoff oder substituiertem Heterokohlenwasserstoff;
und wenn beliebige von zwei oder mehreren von R1–R7 Kohlenwasserstoff, substituierter Kohlenwasserstoff,
Heterokohlenwasserstoff oder substituierter Heterokohlenwasserstoff
darstellen; können
zwei oder mehrere zur Bildung eines oder mehrerer cyclischer Substituenten
verbunden sein.
-
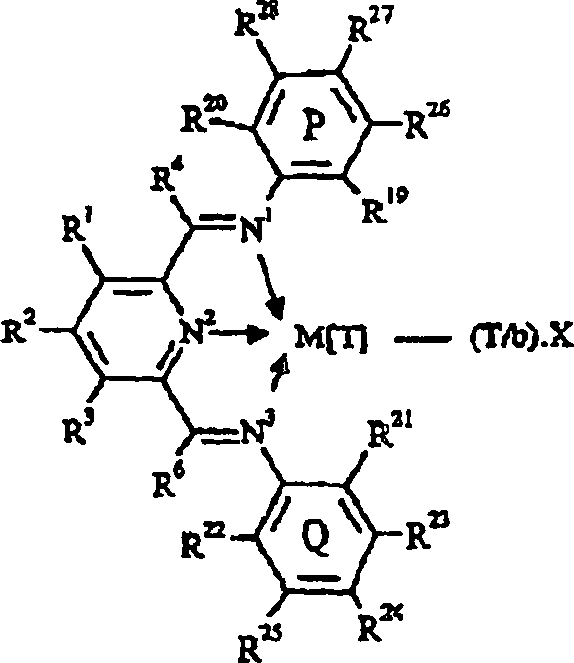
Weitere
Verbindungen zur Verwendung in der vorliegenden Erfindung schließen jene
ein, die die in Formel Z angeführte
Gerüsteinheit
umfassen:
Formel
Z worin M Fe[II], Fe[III], Co[I], Co[II], Co[III],
Mn[I], Mn[II], Mn[III], Mn[IV], Ru[II], Ru[III] oder Ru[IV] darstellt;
X ein Atom oder eine Gruppe, die kovalent oder ionisch an das Übergangsmetall
M gebunden ist, wiedergibt; T den Oxidationszustand des Übergangsmetalls
M bedeutet und b die Wertigkeit des Atoms oder der Gruppe X bedeutet;
R
1 bis R
4, R
6 und R
19 bis R
28 unabhängig
aus Wasserstoff, Halogen, Kohlenwasserstoff, substituiertem Kohlenwasserstoff,
Heterokohlenwasserstoff oder substituiertem Heterokohlenwasserstoff
ausgewählt sind;
wenn beliebige zwei oder mehr von R
1 bis
R
4, R
6 und R
19 bis R
28 Kohlenwasserstoff,
substituierten Kohlenwasserstoff, Heterokohlenwasserstoff oder substituierten
Heterokohlenwasserstoff darstellen; die zwei oder mehreren zur Bildung
von einem oder mehreren cyclischen Substituenten gebunden sein können; mit
der Maßgabe,
dass mindestens einer von R
19, R
20, R
21 und R
22 Kohlenwasserstoff, substituierten Kohlenwasserstoff, Heterokohlenwasserstoff
oder substituierten Heterokohlenwasserstoff darstellt, wenn keines
der Ringsysteme P und Q Teil eines polyaromatisch kondensierten
Ringsystems bildet. In diesem besonderen Aspekt der vorliegenden
Erfindung ist es, wenn keines der Ringsysteme P und Q Teil eines
polyaromatischen Ringsystems bildet, bevorzugt, dass mindestens
einer von R
19 und R
20 und
mindestens einer von R
21 und R
22 aus
Kohlenwasserstoff, substituiertem Kohlenwasserstoff, Heterokohlenwasserstoff
oder substituiertem Heterokohlenwasserstoff ausgewählt ist,
und besonders bevorzugt ist es, dass jener von R
19,
R
20, R
21 und R
22 aus Kohlenwasserstoff, substituiertem
Kohlenwasserstoff, Heterokohlenwasserstoff oder substituiertem Heterokohlenwasserstoff
ausgewählt
ist.
-
Gemäß den vorangehenden
Maßgaben
bezüglich
R19, R20, R21 und R22 in Formel
Z, sind R1 bis R4,
R6 und R19 bis R28 in den in Formeln B und Z der vorliegenden
Erfindung angeführten
Verbindungen, vorzugsweise unabhängig
aus Wasserstoff und C1-C8-Kohlenwasserstoff,
beispielsweise Methyl, Ethyl, n-Propyl, n-Butyl, n-Hexyl und n-Octyl,
ausgewählt.
In Formel B sind R5 und R7 vorzugsweise
unabhängig
aus substituierten oder unsubstituierten alicyclischen, heterocyclischen
oder aromatischen Gruppen, beispielsweise Phenyl, 1-Naphthyl, 2-Naphthyl,
2-Methylphenyl, 2-Ethylphenyl, 2,6-Diisopropylphenyl, 2,3-Diisopropylphenyl,
2,4-Diisopropylphenyl, 2,6-Di-n-butylphenyl, 2,6-Dimethylphenyl,
2,3-Dimethylphenyl, 2,4-Dimethylphenyl, 2-t-Butylphenyl, 2,6-Diphenylphenyl,
2,4,6-Trimethylphenyl, 2,6-Trifluormethylphenyl, 4-Brom-2,6-dimethylphenyl,
3,5-Dichlor-2,6-diethylphenyl und 2,6-Bis(2,6-dimethylphenyl)phenyl,
Cyclohexyl und Pyridinyl ausgewählt.
-
Die
Ringsysteme P und Q in Formel Z sind vorzugsweise unabhängig 2,6-Kohlenwasserstoffphenyl oder
ein polyaromatischer kondensierter Ring, beispielsweise 1-Naphthyl,
2-Naphthyl, 1-Phenanthrenyl und 8-Chinolinyl.
-
In
der Verbindung der Formel B und Z der vorliegenden Erfindung ist
M vorzugsweise Fe[II] oder Co[II].
-
Weitere,
für die
erfindungsgemäßen Katalysatorsysteme
geeignete Verbindungen sind außerdem
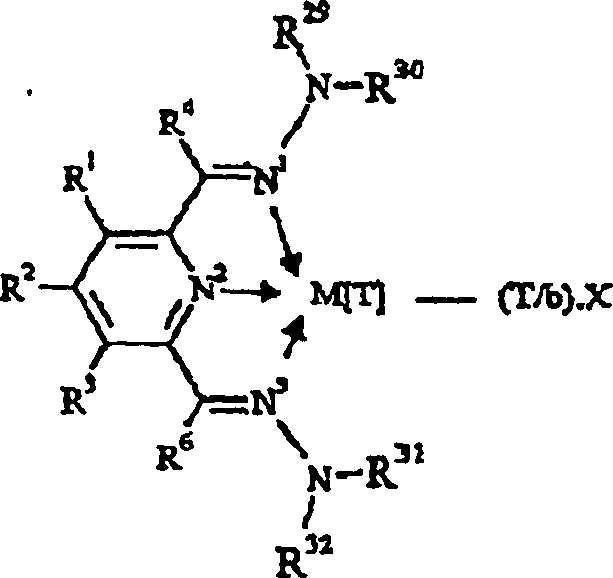
jene, die die in Formel T angeführte
Gerüsteinheit
umfassen:
Formel
T worin M Fe[II], Fe[III], Co[I], Co[II], Co[III],
Mn[I], Mn[II], Mn[III], Mn[IV], Ru[II], Ru[III] oder Ru[IV] darstellt;
X ein Atom oder eine Gruppe, die kovalent oder ionisch an das Übergangsmetall
M gebunden ist, wiedergibt; T den Oxidationszustand des Übergangsmetalls
M bedeutet und b die Wertigkeit des Atoms oder der Gruppe X bedeutet;
R
1 bis R
4, R
6 und R
29 bis R
32 unabhängig
aus Wasserstoff, Halogen, Kohlenwasserstoff, substituiertem Kohlenwasserstoff,
Heterokohlenwasserstoff oder substituiertem Heterokohlenwasserstoff
ausgewählt
sind; wenn beliebige zwei oder mehrere von R
1 bis
R
4, R
6 und R
29 bis R
32 Kohlenwasserstoff,
substituierten Kohlenwasserstoff, Heterokohlenwasserstoff oder substituierten
Heterokohlenwasserstoff darstellen, die zwei oder mehreren zur Bildung
von einem oder mehreren cyclischen Substituenten verbunden sein
können.
-
In
der Verbindung der Formel B der vorliegenden Erfindung ist M vorzugsweise
Fe[II]. In den Verbindungen der Formel Z oder der Formel T der vorliegenden
Erfindung ist M vorzugsweise Fe[II], Mn[II] oder Co[II].
-
Beispiele
für das
Atom oder die Gruppe X in Verbindungen der Formeln B, Z und T sind
Halogenid, beispielsweise Chlorid, Bromid; Hydrid; Kohlenwasserstoffoxid,
beispielsweise Methoxid, Ethoxid, Isopropoxid, Phenoxid; Carboxylat,
beispielsweise Formiat, Acetat, Benzoat; Kohlenwasserstoff, beispielsweise
Methyl, Ethyl, Propyl, Butyl, Octyl, Decyl, Phenyl oder Benzyl;
substituierter Kohlenwasserstoff, Heterokohlenwasserstoff; Tosylat
und Triflat. Vorzugsweise ist X aus Halogenid, Hydrid und Kohlenwasserstoff
ausgewählt.
Chlorid ist besonders bevorzugt.
-
Die
Nachstehenden sind Beispiele von Stickstoffenthaltenden Übergangsmetallkomplexen,
die in dem erfindungsgemäßen Katalysator
angewendet werden können:
2,6-Diacetylpyridinbis(2,6-diisopropylanil)FeCl2
2,6-Diacetylpyridin(2,6-diisopropylanil)MnCl2
2,6-Diacetylpyridin(2,6-diisopropylanil)CoCl2
2,6-Diacetylpyridinbis(2-tert-butylanil)FeCl2
2,6-Diacetylpyridinbis(2,3-dimethylanil)FeCl2
2,6-Diacetylpyridinbis(2-methylanil)FeCl2
2,6-Diacetylpyridinbis(2,4-dimethylanil)FeCl2
2,6-Diacetylpyridinbis(2,6-dimethylanil)FeCl2
2,6-Diacetylpyridinbis(2,4,6-trimethylanil)FeCl2
2,6-Dialdiminpyridinbis(2,6-dimethylanil)FeCl2
2,6-Dialdiminpyridinbis(2,6-diethylanil)FeCl2
2,6-Dialdiminpyridinbis(2,6-diisopropylanil)FeCl2
2,6-Dialdiminpyridinbis(1-naphthil)FeCl2 oder
2,6-Bis(1,1-diphenylhydrazon)pyridin
FeCl2.
-
Ein
bevorzugter erfindungsgemäßer Komplex
ist 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)FeCl2.
-
In
einem weiteren Aspekt der vorliegenden Erfindung kann das Katalysatorsystem
zusätzlich
(4) eine neutrale Lewis-Base umfassen. Neutrale Lewis-Basen sind
auf dem Fachgebiet der Ziegler-Natta-Katalysator-Polymerisationstechnologie
gut bekannt. Beispiele für
Klassen von neutralen Lewis-Basen, die geeigneterweise in der vorliegenden
Erfindung angewendet werden können,
sind natürliche
Kohlenwasserstoffe, beispielsweise Alkene oder Alkine, primäre, sekundäre und tertiäre Amine,
Amide, Phosphoramide, Phosphine, Phosphite, Ether, Thioether, Nitrile,
Carbonylverbindungen, beispielsweise Ester, Ketone, Aldehyde, Kohlenmonoxid
und Kohlendioxid, Sulfoxide, Sulfone und Boroxine. Obwohl 1-Olefine
als neutrale Lewis-Basen wirken können, werden sie für die erfindungsgemäßen Zwecke
als Monomer- oder Comonomer-1-olefine betrachtet und nicht als neutrale
Lewis-Basen an sich. Jedoch werden Alkene, die innere Olefine darstellen,
beispielsweise 2-Buten und Cyclohexen, in der vorliegenden Erfindung
als neutrale Lewis-Basen betrachtet. Bevorzugte Lewis-Basen sind
tertiäre
Amine und aromatische Ester, beispielsweise Dimethylanilin, Diethylanilin, Tributylamin,
Benzoesäureethylester
und Benzoesäurebenzylester.
In diesem besonderen Aspekt der vorliegenden Erfindung können Komponenten
(1), (2) und (4) des Katalysatorsystems gleichzeitig oder in beliebiger gewünschter
Reihenfolge zusammengebracht werden. Wenn jedoch Komponenten (2)
und (4) Verbindungen darstellen, die stark miteinander in Wechselwirkung
treten, beispielsweise eine stabile Verbindung miteinander bilden,
ist es bevorzugt, entweder Komponenten (1) und (2) oder Komponenten
(1) und (4) in einem Anfangsschritt, vor dem Einführen der
definierten Endkomponente, in Kontakt zu bringen. Vorzugsweise werden
Komponenten (1) und (4) miteinander in Kontakt gebracht, bevor Komponente
(2) eingeführt
wird. Die bei der Herstellung dieses Katalysatorsystems angewendeten
Mengen der Komponenten (1) und (2) sind geeigneterweise wie vorstehend
in Bezug auf die erfindungsgemäßen Katalysatoren
beschrieben. Die Menge der neutralen Lewis-Base [Komponente (4)]
ist vorzugsweise derart, dass ein Verhältnis von Komponente (1) :
Komponente (4) im Bereich von 100 : 1 bis 1 : 1000, besonders bevorzugt
im Bereich von 1 : 1 bis 1 : 20, bereitgestellt wird. Komponenten
(1), (2) und (4) des Katalysatorsystems können beispielsweise miteinander
als unverdünnte
Materialien, wie eine Suspension oder Lösung der Materialien in einem
geeigneten Verdünnungsmittel
oder Lösungsmittel
(beispielsweise ein flüssiger
Kohlenwasserstoff) oder, wenn mindestens eine der Komponenten flüchtig ist,
durch Nutzen des Dampfes der Komponente zusammengebracht werden.
Die Komponenten können
bei beliebiger gewünschter
Temperatur zusammengebracht werden. Das Mischen der Komponenten
miteinander bei Raumtemperatur ist im Allgemeinen befriedigend.
Das Erhitzen auf höhere
Temperaturen, beispielsweise bis zu 120°C, kann, falls erwünscht, ausgeführt werden,
beispielsweise, um besseres Mischen der Komponenten zu erreichen.
Es ist bevorzugt, das Zusammenbringen von Komponenten (1), (2) und
(4) in einer Inertatmosphäre
(beispielsweise trockenem Stickstoff) oder im Vakuum auszuführen. Wenn
es erwünscht
ist, den Katalysator auf einem Trägermaterial (siehe nachstehend)
anzuwenden, kann dies beispielsweise durch Vorbilden des Komponenten
(1), (2) und (4) umfassenden Katalysatorsystems und Imprägnieren
des Trägermaterials,
vorzugsweise mit einer Lösung
davon, oder durch Einführen
von einer oder mehreren der Komponenten gleichzeitig oder nacheinander
in das Trägermaterial,
erreicht werden. Falls erwünscht,
kann das Trägermaterial
selbst die Eigenschaften einer neutralen Lewis-Base aufweisen und
kann als oder anstelle von Komponente (4) angewendet werden. Ein
Beispiel eines Trägermaterials
mit neutralen Lewis-Base-Eigenschaften ist Poly(aminostyrol) oder
ein Copolymer von Styrol und Aminostyrol (d. h. Vinylanilin).
-
Die
als Katalysatoren in den erfindungsgemäßen Katalysatorsystemen angewendeten
Verbindungen können,
falls erwünscht,
mehr als eine der vorstehend definierten Übergangsmetallverbindungen
umfassen. Der Katalysator kann beispielsweise ein Gemisch von 2,6-Diacetylpyridinbis(2,6-diisopropylanil)FeCl2-Komplex und 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)FeCl2-Komplex oder ein Gemisch von 2,6-Diacetylpyridin(2,6-diisopropylanil)CoCl2 und 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)FeCl2 umfassen. Zusätzlich zu der/den angeführten einen
oder mehreren definierten Übergangsmetallverbindungen
können
die Katalysatoren auch eine oder mehrere andere Arten von Übergangsmetallverbindungen
oder Katalysatoren, beispielsweise Übergangsmetallverbindungen
des in üblichen
Ziegler-Natta-Katalysatorsystemen verwendeten Typs, auf Metallocen
basierende Katalysatoren oder wärmeaktivierte,
getragene Chromoxidkatalysatoren (z. B. Katalysator vom Phillips-Typ),
einschließen.
-
Die
in der vorliegenden Erfindung angewendeten Katalysatoren können auf
einem Trägermaterial,
beispielsweise Siliziumdioxid, Aluminiumoxid oder Zirkoniumoxid,
oder auf einem Polymer oder Prepolymer, beispielsweise Polyethylen,
Polystyrol oder Poly(aminostyrol), ungetragen oder getragen sein.
Falls erwünscht, können die
Katalysatoren in situ in Gegenwart des Trägermaterials gebildet werden,
oder das Trägermaterial kann
gleichzeitig oder nacheinander mit einer oder mehreren der Katalysatorkomponenten
vorimprägniert
oder vorgemischt werden. Die Katalysatoren können, falls erwünscht, auf
einem heterogenen Katalysator, beispielsweise ein Magnesiumhalogenid-getragener
Ziegler-Natta-Katalysator, ein Phillips-Typ-(Chromoxid)-getragener
Katalysator, oder ein getragener Metallocenkatalysator, getragen
werden. Die Bildung des getragenen Katalysators kann beispielsweise
durch Behandeln der Übergangsmetallverbindungen
mit Alumoxan in einem geeigneten inerten Verdünnungsmittel, beispielsweise
einem flüchtigen
Kohlenwasserstoff, Aufschlämmen
eines teilchenförmigen
Trägermaterials
mit dem Produkt und Verdampfen des flüchtigen Verdünnungsmittels,
erreicht werden. Die Menge des angewendeten Trägermaterials kann stark variieren,
beispielsweise von 100000 bis 1 Gramm pro Gramm des in der Übergangsmetallverbindung
vorliegenden Metalls.
-
Ein
weiterer Aspekt der Erfindung umfasst die Verwendung einer Verbindung
der Formel AlR3, worin jedes R unabhängig C1-C12-Alkyl oder
Halogen darstellt, um die katalytische Aktivität der Polymerisation von 1-Olefinen
einer Verbindung der wie vorstehend definierten Formel B zu verstärken.
-
Die
vorliegende Erfindung stellt weiterhin ein Verfahren für die Polymerisation
und Copolymerisation von 1-Olefinen
bereit, das In-Kontakt-Bringen des monomeren Olefins unter Polymerisationsbedingungen
mit einem Polymerisationskatalysatorsystem, umfassend:
- (1) eine Verbindung der wie vorstehend definierten Formel B,
- (2) einen Alkylalumoxanaktivator,
- (3) zusätzlich
zu (2) eine Verbindung der Formel AlR3,
worin jedes R unabhängig
C1-C12-Alkyl oder
Halogen darstellt, umfasst.
-
In
einem bevorzugten Verfahren wird die Katalysatorverbindung (1) mit
dem Alkylalumoxan (2) vor dem Kontakt mit dem zu polymerisierenden
Monomer aktiviert.
-
Die
Polymerisationsbedingungen können
beispielsweise Lösungsphase,
Aufschlämmungsphase
oder Gasphase sein. Falls erwünscht,
kann das Katalysatorsystem verwendet werden, um Ethylen unter Hochdruck/Hochtemperatur-Verfahrensbedingungen
zu polymerisieren, wobei das Polymermaterial in überkritischem Ethylen eine
Schmelze bildet. Vorzugsweise wird die Po lymerisation unter Gasphasen-Wirbelschicht-Bedingungen
durchgeführt.
Aufschlämmungsphase-Polymerisationsbedingungen
oder Gasphasen-Polymerisationsbedingungen sind für die Herstellung von hochdichten
Polyethylenqualitäten
besonders verwendbar. In diesen Verfahren können die Polymerisationsbedingungen
chargenweise, kontinuierlich oder halbkontinuierlich sein. In dem
Aufschlämmungsphasenverfahren
und dem Gasphasenverfahren wird der Katalysator im Allgemeinen in
Form eines teilchenförmigen
Feststoffs zu der Polymerisationszone gespeist. Dies kann beispielsweise
ein unverdünntes
festes Katalysatorsystem sein, das aus einem Stickstoff-enthaltenden Komplex
und einem Aktivator gebildet wird, oder kann der feste Komplex allein
sein. In der letzteren Situation kann der Aktivator zu der Polymerisationszone,
beispielsweise als eine Lösung,
getrennt von oder zusammen mit dem festen Komplex, zugeführt werden.
Vorzugsweise wird das Katalysatorsystem oder die Übergangsmetallkomplexkomponente
des in der Aufschlämmungspolymerisation
und Gasphasenpolymerisation angewendeten Katalysatorsystems auf
einem Trägermaterial
getragen. Besonders bevorzugt wird das Katalysatorsystem vor seiner
Einführung
in die Polymerisationszone auf einem Trägermaterial getragen. Geeignete
Trägermaterialien
sind zum Beispiel Siliziumdioxid, Aluminiumoxid, Zirkoniumoxid,
Talkum, Kieselgur oder Magnesiumoxid. Die Imprägnierung des Trägermaterials
kann durch übliche
Techniken, beispielsweise durch Bilden einer Lösung oder Suspension der Katalysatorkomponenten
in einem geeigneten Verdünnungs-
oder Lösungsmittel
und Aufschlämmen
des Trägermaterials
damit, ausgeführt
werden. Das so mit Katalysator imprägnierte Trägermaterial kann dann von dem
Verdünnungsmittel,
beispielsweise durch Filtrations- oder Verdampfungstechniken, getrennt
werden.
-
In
dem Aufschlämmungsphasenpolymerisationsverfahren
werden die festen Teilchen des Katalysators oder getragenen Katalysators
zu einer Polymerisationszone entweder als ein trockenes Pulver oder
als eine Aufschlämmung
in dem Polymerisationsverdünnungsmittel
zugeführt.
Vorzugsweise werden die Teilchen zu einer Polymerisationszone als
eine Suspension in dem Polymerisationsverdünnungsmittel zugeführt. Die Polymerisationszone
kann beispielsweise ein Autoklav oder ein ähnliches Reaktionsgefäß oder ein
kontinuierlicher Schleifenreaktor, beispielsweise des bei der Herstellung
von Polyethylen durch das Phillips-Verfahren gut bekannten Typs,
sein. Wenn das erfindungsgemäße Polymerisationsverfahren
unter Aufschlämmungsbedingungen
ausgeführt
wird, wird die Polymerisation vorzugsweise bei einer Temperatur
oberhalb 0°C,
besonders bevorzugt oberhalb 15°C,
ausgeführt.
Die Polymerisationstemperatur wird vorzugsweise unter der Temperatur
gehalten, bei der das Polymer in Gegenwart des Polymerisationsverdünnungsmittels
zu weichen oder sintern beginnt. Wenn die Temperatur über die
letztere Temperatur gelassen wird, kann Ablagerungsbildung im Reaktor
stattfinden. Die Einstellung der Polymerisation innerhalb dieser
definierten Temperaturbereiche kann eine nützliche Maßnahme zum Steuern des mittleren
Molekulargewicht des hergestellten Polymers bereitstellen. Ein weiteres
verwendbares Mittel des Steuerns des Molekulargewichts ist es, die
Polymerisation in Gegenwart von Wasserstoffgas durchzuführen, welches
als ein Kettenübertragungsmittel
wirkt. Im Allgemeinen gilt, je höher
die Konzentration an angewendetem Wasserstoff, um so niedriger ist
das mittlere Molekulargewicht des hergestellten Polymers.
-
Die
Anwendung von Wasserstoffgas als ein Mittel zum Steuern des mittleren
Molekulargewichts des Polymers oder Copolymers gilt allgemein für das erfindungsgemäße Polymerisationsverfahren.
Beispielsweise kann Wasserstoff verwendet werden, um das mittlere
Molekulargewicht der unter Verwendung von Gasphasen-, Aufschlämmungsphasen-
oder Lösungsphasen-Polymerisationsbedingungen
hergestellten Polymere oder Copolymere zu vermindern. Die Menge
an anzuwendendem Wasserstoffgas, um das gewünschte mittlere Molekulargewicht
zu ergeben, kann durch einfache "Versuchs-
und Fehler"-Polymerisationstests
bestimmt werden.
-
Das
erfindungsgemäße Polymerisationsverfahren
stellt Polymere und Copolymere, insbesondere Ethylenpolymere, bei
sehr hoher Produktivität
(bezogen auf die Menge an Polymer oder Copolymer, das pro Gewichtseinheit
von in dem Katalysatorsystem angewendetem, Stickstoff-enthaltendem Übergangsmetallkomplex
hergestellt wurde) bereit. Dies bedeutet, dass relativ kleine Mengen
an Übergangsmetallkomplex
in kommerziellen Verfahren, unter Anwendung des erfindungsgemäßen Verfahrens,
verbraucht werden. Es bedeutet auch, dass, wenn das Polymerisationsverfahren
der vorliegenden Erfindung unter Polymergewinnungsbedingungen durchgeführt wird,
dann kein Katalysatortrennungsschritt angewendet werden muss, wodurch Katalysator
oder Reste davon in dem Polymer zurückbleiben (was beispielsweise
in den meisten kommerziellen Aufschlämmungs- und Gasphasen-Polymerisationsverfahren
stattfindet), wobei die Menge an Übergangsmetallkomplex in dem
hergestellten Polymer sehr klein sein kann. Mit dem erfindungsgemäßen Katalysator ausgeführte Versuche
zeigen, dass beispielsweise die Polymerisation von Ethylen unter
Aufschlämmungs-Polymerisationsbedingungen
ein teilchenförmiges
Polyethylenprodukt bereitstellen kann, das Katalysator enthält, der
durch das gesamte hergestellte Polyethylen so verdünnt ist,
dass die Konzentration an Übergangsmetall auf
beispielsweise 1 ppm oder weniger fällt, wobei "ppm" als
Teile auf das Gewicht von Übergangsmetall
pro Millionen Teile auf das Gewicht Polymer, definiert ist. Somit
kann in einem Polymerisationsreaktor durch das erfindungsgemäße Verfahren
hergestelltes Polyethylen mit dem Polyethylen verdünnten Katalysator
in einem solchen Ausmaß enthalten,
dass der Übergangsmetallgehalt
davon beispielsweise im Bereich von 1–0,0001 ppm, vorzugsweise 1–0,001 ppm,
liegt. Unter Anwendung eines einen erfindungsgemäßen Stickstoff-enthaltenden
Fe-Komplex umfassenden Katalysators, beispielsweise in einer Aufschlämmungspolymerisation,
ist es möglich,
Polyethylenpulver zu erhalten, worin die Fe-Konzen-tration beispielsweise 1,03 bis
0,11 Gewichtsteile Fe pro Million Gewichtsteile Polyethylen ist.
-
Geeignete
Monomere zur Verwendung in dem erfindungsgemäßen Polymerisationsverfahren
sind beispielsweise Ethylen, Propylen, Buten, Hexen, Methacrylsäuremethylester,
Acrylsäuremethylester,
Acrylsäurebutylester,
Acrylnitril, Vinylacetat und Styrol. Bevorzugte Monomere für Homopolymerisationsverfahren sind
Ethylen und Propylen. Der Katalysator kann auch zum Copolymerisieren
von Ethylen mit anderen 1-Olefinen, wie Propylen, 1-Buten, 1-Hexen,
4-Methylpenten-1 und Octen, verwendet werden.
-
Verfahren
zum Arbeiten in Gasphasen-Polymerisationsverfahren sind auf dem
Fachgebiet gut bekannt. Solche Verfahren beinhalten im Allgemeinen
Bewegen (beispielsweise durch Rühren,
Vibrieren oder Wirbelschichtbildung) eines Katalysatorbetts oder
eines Zielpolymerbetts (d. h. Polymer mit den gleichen oder ähnlichen
physikalischen Eigenschaften zu jenem, welches in dem Polymerisationsverfahren
hergestellt werden soll), enthaltend einen Katalysator, und Zuführung dazu
eines Monomerstroms mindestens teilweise in der Gasphase, unter
derartigen Bedingungen, dass mindestens ein Teil des Monomers bei
Kontakt mit dem Katalysator im Bett polymerisiert. Das Bett wird
im Allgemeinen durch die Zugabe von Kühlgas (beispielsweise zurückgeführtes gasförmiges Monomer)
und/oder flüchtige
Flüssigkeit
(beispielsweise ein flüchtiger
inerter Kohlenwasserstoff oder gasförmiges Monomer, das unter Bildung
einer Flüssigkeit
kondensiert werden kann) gekühlt
werden. Das in Gasphasenverfahren hergestellte und daraus isolierte
Polymer bildet direkt einen Feststoff in der Polymerisationszone
und ist frei von oder im Wesentlichen frei von Flüssigkeit.
Dem Fachmann wird gut bekannt sein, dass wenn irgendwelche Flüssigkeit
in die Polymerisationszone eines Gasphasenpolymerisationsverfahrens
gelangen kann, die Flüssigkeitsmenge
in Bezug zu der Menge an in der Polymerisationszone vorliegendem
Polymer klein ist. Dies steht im Gegensatz zu "Lösungsphasen"verfahren, worin
das Polymer in einem Lösungsmittel
gebildet und "Aufschlämmungsphasen"verfahren, worin das
Polymer sich als eine Suspension in einem flüssigen Verdünnungsmittel bildet.
-
Das
Gasphasenverfahren kann unter Chargen-, Halbchargen- oder so genannten "kontinuierlichen" Bedingungen ausgeführt werden.
Es ist bevorzugt, unter derartigen Bedingungen zu arbeiten, dass
Monomer kontinuierlich zu einer bewegten Polymerisationszone, die
Polymerisationskatalysator enthält,
zurückgeführt wird,
wobei Auffüllmonomer
bereitgestellt wird, um polymerisiertes Monomer zu ersetzen, und
kontinuierlich oder unterbrechend hergestelltes Polymer aus der
Polymerisationszone bei einer mit der Bildungsgeschwindigkeit von
Polymer vergleichbaren Geschwindigkeit abgezogen wird, wobei frischer
Katalysator zu der Polymerisationszone gegeben wird, um den aus
der Polymerisationszone mit dem hergestellten Polymer abgezogenen
Katalysator zu ersetzen.
-
In
der bevorzugten Ausführungsform
des erfindungsgemäßen Gasphasenpolymerisationsverfahrens sind
die Gasphasenpolymerisationsbedingungen vorzugsweise Gasphasenwirbelschicht-Polymerisationsbedingungen.
-
Verfahren
zum Arbeiten von Gasphasenwirbelschichtverfahren zum Herstellen
von Polyethylen und Ethylen-Copolymeren sind auf dem Fachgebiet
gut bekannt. Das Verfahren kann beispielsweise in einem vertikalen
zylindrischen Reaktor, der mit einer perforierten Verteilungsplatte
zum Tragen der Schicht und zum Verteilen des hereinkommenden Fluidisierungsgasstroms
durch die Schicht ausgestattet ist, ausgeführt werden. Das durch die Schicht
zirkulierende, fluidisierende Gas dient zum Entfernen der Polymerisationswärme aus der
Schicht und zum Zuführen
von Monomer zur Polymerisation in der Schicht. Somit umfasst das
fluidisierende Gas das/die Monomer(e), normalerweise zusammen mit
etwas Inertgas (beispielsweise Stickstoff), und gegebenenfalls mit
Wasserstoff als Molekulargewichtsmodifizierungsmittel. Das heiße Fluidisierungsgas,
das an der Spitze der Schicht auftaucht, wird gegebenenfalls durch
eine Geschwindigkeitsverminderungszone (dies kann ein zylindrischer
Teil des Reaktors mit einem breiteren Durchmesser sein) und, falls
erwünscht,
einen Zyklon und/oder Filter zum Verhindern von Mitreißen von
feinen festen Teilchen aus dem Gasstrom geführt. Das heiße Gas wird
dann zu einem Wärmeaustauscher
geleitet, um Polymerisationswärme
mindestens teilweise abzuführen.
Der Katalysator wird vorzugsweise kontinuierlich oder in regelmäßigen Abständen zu
der Schicht gespeist. Am Beginn des Verfahrens umfasst die Schicht
fluidisierbares Polymer, das vorzugsweise ähnlich dem Zielpolymer ist.
Polymer wird kontinuierlich innerhalb der Schicht durch die Polymerisation
des/der Monomers(e) erzeugt. Vorzugsweise werden Mittel zum Abziehen
des Polymers aus der Schicht kontinuierlich oder in regelmäßigen Abständen zum
Halten der Wirbelschicht auf der gewünschten Höhe bereitgestellt. Das Verfahren
wird im Allgemeinen bei relativ niedrigem Druck, beispielsweise
bei 10 bis 50 bar, durchgeführt,
obwohl es bei Drücken
von 10 bis 100 bar und bei Temperaturen von beispielsweise zwischen
50 und 120°C
arbeiten kann. Die Temperatur der Schicht wird unterhalb der Sintertemperatur
des fluidisierten Polymers gehalten, um Probleme der Agglomeration
zu vermeiden.
-
Bei
dem Gasphasen-Wirbelschichtverfahren zur Polymerisation von Olefinen
wird die durch die exotherme Polymerisationsreaktion entwickelte
Wärme normalerweise
aus der Polymerisationszone (d. h. der Wirbelschicht) mit Hilfe
des Fluidisierungsgasstroms, wie vorstehend beschrieben, abgeführt. Das
von der Spitze der Schicht auftauchende, heiße Reaktorgas wird durch einen
oder mehrere Wärmeaustauscher,
worin das Gas gekühlt
ist, geleitet. Das gekühlte
Reaktorgas wird dann, zusammen mit beliebigem Auffüllgas, zu dem
Boden der Schicht zurückgeführt. Bei
dem erfindungsgemäßen Gasphasen-Wirbelschicht-Polymerisationsverfahren
ist es erwünscht,
zusätzliches
Kühlen
der Schicht (und dadurch Verbesserung der Raum-Zeit-Ausbeute des Verfahrens) durch
Zuführen
von flüchtiger
Flüssigkeit
zu der Schicht unter derartigen Bedingungen bereitzustellen, dass
die Flüssigkeit
in der Schicht verdampft, wodurch zusätzliche Polymerisationswärme aus
der Schicht durch den "latenten
Verdampfungswärme"effekt abgeführt wird.
Wenn das heiße Rückführgas aus
der Schicht in den Wärmeaustauscher
gelangt, kann die flüchtige
Flüssigkeit
auskondensieren. In einer Ausführungsform
der vorliegenden Erfindung wird die flüchtige Flüssigkeit von dem Rückführgas getrennt
und erneut getrennt in die Schicht eingeführt. Somit kann beispielsweise
die flüchtige
Flüssigkeit
abgetrennt und in die Schicht gesprüht werden. In einer weiteren
Ausführungsform
der vorliegenden Erfindung wird die flüchtige Flüssigkeit zu der Schicht mit
dem Rückführgas zurückgeführt. Somit
kann die flüchtige
Flüssigkeit
aus dem Fluidisierungsgasstrom, der aus dem Reaktor kommt, kondensiert
werden und kann zu der Schicht mit Rückführgas zurückgeführt werden, oder kann von dem
Rückführgas getrennt
und in die Schicht gesprüht
werden.
-
Das
Verfahren zum Kondensieren von Flüssigkeit in dem Rückführgasstrom
und Zurückkehren
des Gemisches von Gas und abgezogener Flüssigkeit zu der Schicht wird
in EP-A-0089691 und EP-A-0241947 beschrieben. Es ist bevorzugt,
die kondensierte Flüssigkeit
in die Schicht getrennt von dem Rückführgas, unter Verwendung des
in unserem US-Patent Nr. 5 541 270, dessen Lehren davon hierin in
diese Beschreibung hierdurch einbezogen sind, wieder einzuführen.
-
Beim
Anwenden der erfindungsgemäßen Katalysatoren
unter Gasphasen-Polymerisationsbedingungen kann der Katalysator
oder eine oder mehrere der angewendeten Komponenten zur Bildung
des Katalysators, beispielsweise in die Polymerisationsreaktionszone
in flüssiger
Form, zum Beispiel als eine Lösung
in einem inerten flüssigen
Verdünnungsmittel,
eingeführt
werden. Somit kann beispielsweise die Übergangsmetallkomponente oder
die Aktivatorkomponente oder beide von diesen Komponenten gelöst oder
in einem flüssigen
Verdünnungsmittel
aufgeschlämmt
und der Polymerisationszone zugeführt werden. Unter diesen Umständen ist
es bevorzugt, die die Komponente(n) enthaltende Flüssigkeit
als feine Tröpfchen
in die Polymerisationszone zu sprühen. Der Tröpfchendurchmesser liegt vorzugs weise
im Bereich von 1 bis 1000 Mikrometern. EP-A-0593083, deren Lehren
hierin hierdurch in diese Beschreibung einbezogen sind, offenbart
ein Verfahren zum Einführen
eines Polymerisationskatalysators in eine Gasphasenpolymerisation.
Die in EP-A-0593083 offenbarten Verfahren können, falls erwünscht, geeigneterweise
in dem erfindungsgemäßen Polymerisationsverfahren
angewendet werden.
-
Die
vorliegende Erfindung wird in den nachstehenden Beispielen erläutert.
-
BEISPIELE
-
Beispiel
1 zeigt die Herstellung einer neuen Eisenverbindung (siehe nachstehende
Formel D) und Beispiel 3 zeigt die Herstellung einer neuen Cobaltverbindung
(siehe Formel K), wobei jede Verbindung gemäß der vorliegenden Erfindung
ist.
-
In
den Beispielen wurden alle Manipulationen von Luft/Feuchtigkeits-empfindlichen
Materialien unter einer üblichen
Vakuum/Inertatmosphären-(Stickstoff)-Leitung
unter Anwendung von Standard-Schlenk-Leitungs-Techniken oder in
einer Inertatmosphären-Glovebox
ausgeführt.
-
Beispiel 1
-
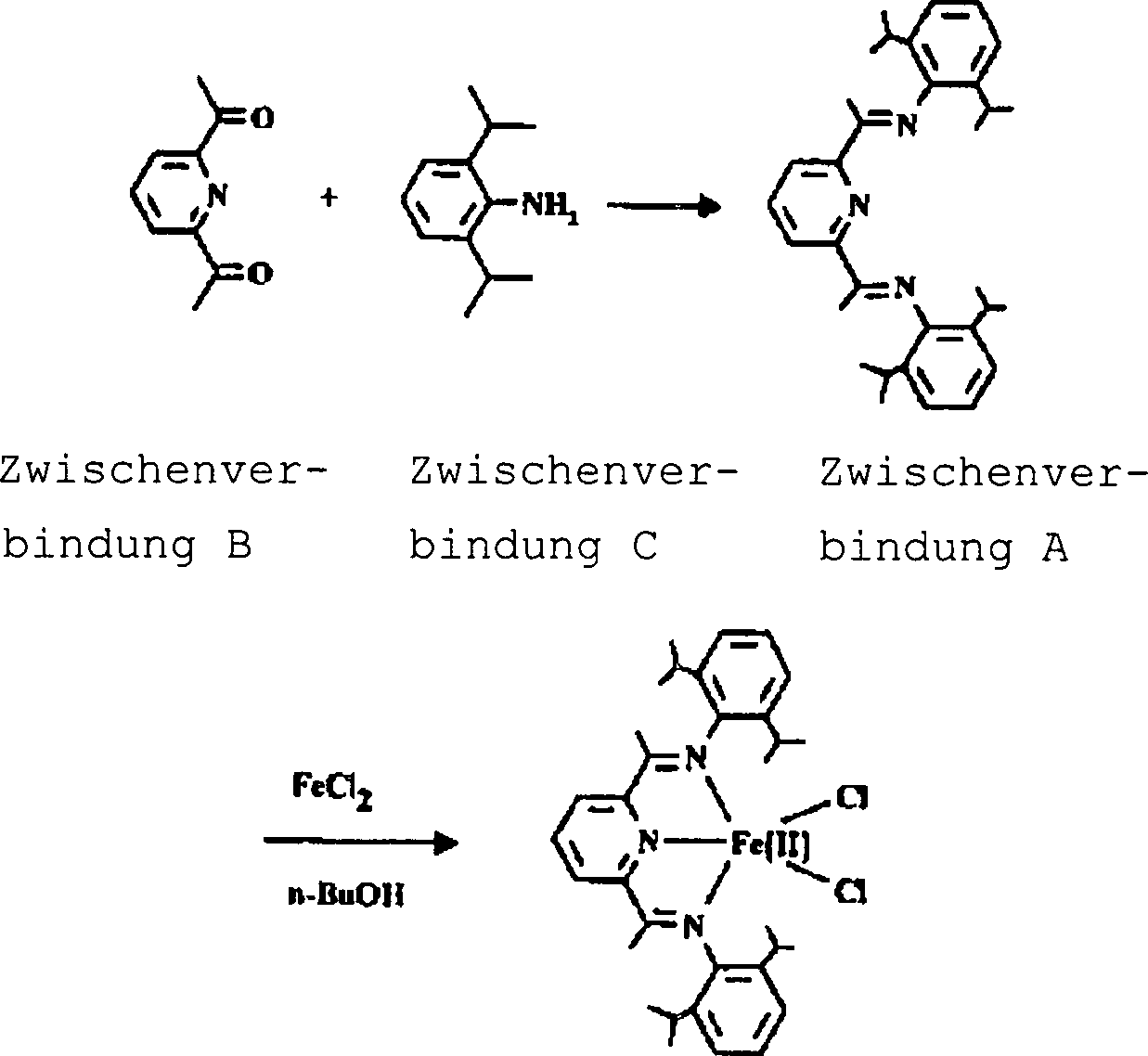
Zwischenprodukt
A [2,6-Diacetylpyridinbis(2,6-diisopropylanil)] wurde durch die
Reaktion von Zwischenprodukt B [2,6-Diacetylpyridin] und Zwischenprodukt
C [2,6-Diisopropylanilin] hergestellt. Zwischenprodukt A wurde dann
mit Eisen(II)chlorid in Butanol umgesetzt, unter Bereitstellung
der Verbindung der Formel D.
-
Herstellung von Zwischenprodukt
A
-
Unter
Verwendung eines Verfahrens, das auf einer verwandten Herstellung
(E. C. Alyea und P. H. Merrell, Synth. React. Inorg. Metal-Org.
Chem., 1974, 4, 535) basiert: wurde 2,6-Diisopropylanilin (3,46
ml, 18,4 mMol) tropfenweise zu einer Lösung von 2,6-Diacetylpyridin
(1,50 g, 9,2 mMol) in absolutem Ethanol (25 ml) [2,6-Diisopropylanilin
und 2,6-Diacetylpyridin
wurden von Aldrich erhalten, wobei das erste davon vor der Anwendung
frisch destilliert wurde] gegeben. Einige Tropfen Eisessig wurden
zugegeben und die Lösung
wurde 48 h unter Rückfluss
erhitzt. Aufkonzentrierung der Lösung
auf ein halbes Volumen und Kühlen
auf –78°C ergab Zwischenprodukt
A als schwach gelbe Kristalle (80%). Berechnet für C33H43N3: C, 82,3; H,
8,9; N, 8,7; gefunden: C, 81,9; H, 8,5; 8,7%. FABMS: M+ (481). 1H NMR (CDCl3): 8,6–7,9 [m,
3H, C5H3N], 7,2–6,9 [m,
6H, C6(CHMe2)H3], 2,73 [Sept., 4H, CHMe2],
2,26 [s, 6H, C5H3N(CMeNAr)2] und 1,16 [m, 24H, CHMe2].
FABMS ist Fast-Atom-Bombardment-Massen-Spektrometrie.
-
-
Herstellung der Verbindung
der Formel D [2,6-Diacetylpyridinbis(2,6-diisopropylanil)FeCl2]
-
FeCl2 (0,24 g; 1,89 mMol) wurde in heißem n-Butanol
(20 ml) bei 80°C
gelöst.
Eine Suspension von 2,6-Diacetylpyridinbis(2,6-diisopropylanil)
(0,92 g; 1,89 mMol) in n-Butanol
wurde tropfenweise bei 80°C
zugegeben. Das Reaktionsgemisch schlug nach Blau um. Nach Rühren bei
80°C für 15 Mi nuten
wurde die Reaktion auf Raumtemperatur herunterkühlen lassen. Das Reaktionsvolumen
wurde auf einige ml vermindert, und Petrolether (40/60) wurde zum
Ausfällen
des Produkts zugesetzt (ein blaues Pulver), welches anschließend dreimal
mit 10 ml Petrolether (40/60) gewaschen wurde. Die Ausbeute war
0,93 g (81%).
Massenspektrum: m/z 607 [M]+, 572 [M – Cl]+,
482 [M – FeCl2]+.
Analyse – berechnet für C33H43N3FeCl2: C, 65,14; H, 7,12; N, 6,91. Gefunden:
C, 64,19; H, 6,90; N, 6,70.
-
Beispiel 3
-
Herstellung von 2,6-Diacetylpyridin(2,6-diisopropylanil)CoCl2 – Formel
K
-
Cobaltchlorid
(CoCl2 – 0,057
g; 0,44 mMol) wurde in heißem
n-Butanol (10 ml) bei 80°C
gelöst.
Eine Suspension von Zwischenprodukt A [2,6-Diacetylpyridinbis(2,6-diisopropylanil)]
(0,21 g; 0,44 mMol) in n-Butanol wurde tropfenweise bei 80°C zugegeben.
Nach Rühren
bei 80°C
für 15
Minuten wurde das hergestellte Reaktionsgemisch auf Raumtemperatur
abkühlen
lassen. Das Reaktionsvolumen wurde auf einige ml vermindert und
Petrolether (40/60) wurde zum Ausfällen des Produkts zugesetzt.
Der olive-grüne,
pulvrige Niederschlag wurde dreimal mit 10 ml aliquoten Mengen von
Petrolether (40/60) gewaschen. Die Ausbeute des Cobaltkomplexes
(Formel K – siehe
nachstehend) war 0,18 g (67% der Theorie). Das Massenspektrum zeigte m/z
575 [M – Cl],
538 [M – 2Cl]+.
-
-
Beispiele 7 bis 9 – Herstellung
von Eisenkomplexen
-
Beispiel 7
-
7.1 – Herstellung von 2,6-Diacetylpyridinbis(2,4-dimethylanil)
-
Das
Verfahren war wie für
Beispiel 4.1, mit der Ausnahme, dass 2,4-Dimethylanilin anstelle
von 2-tertiär-Butylanilin
verwendet wurde. Die Ausbeute war 75% der Theorie.
1H NMR (CDCl3): 8,41,
7,90, 7,05, 6,90, 6,55, (m, 9H, ArH, PyrH), 2,36 (m, 6H, N=CCH3, 6H, CCH3), 2,13
(s, 6H, CCH3).
Massenspektrum: m/z
369 [M]+.
-
7.2 – Herstellung von 2,6-Diacetylpyridinbis(2,4-dimethylanil)FeCl2
-
Das
Verfahren war wie für
Beispiel 4.2, mit der Ausnahme, dass 2,6-Diacetylpyridinbis(2,4-dimethylanil)
anstelle von 2,6-Diacetylpyridinbis(2-tert-butylanil) angewendet
wurde. Die Ausbeute war 75% der Theorie.
Massenspektrum: m/z
496 [M]+, 461 [M – Cl]+,
425 [M – Cl2]+.
-
Beispiel 8
-
8.1 Herstellung von 2,6-Diacetylpyridinbis(2,6-dimethylanil)
-
Das
Verfahren war wie für
Beispiel 4.1, mit der Ausnahme, dass 2,6-Dimethylanilin anstelle
von 2-tertiär-Butylanilin
verwendet wurde. Die Ausbeute war 78% der Theorie.
1H NMR (CDCl3): 8,48,
8,13, 7,98, 7,08, 6,65, (m, 9H, ArH, PyrH), 2,25 (s, 6H, N=CCH3), 2,05 (m, 12H, CCH3).
Massenspektrum:
m/z 369 [M]+.
-
8.2 – Herstellung von 2,6-Diacetylpyridinbis(2,6-dimethylanil)FeCl2
-
Das
Verfahren war wie für
Beispiel 4.2, mit der Ausnahme, dass 2,6-Diacetylpyridinbis(2,6-dimethylanil)
anstelle von 2,6-Diacetylpyridinbis(2-tert-butylanil) verwendet
wurde. Die Ausbeute war 78% der Theorie.
Massenspektrum: m/z
496 [M]+, 461 [M – Cl]+,
425 [M – Cl2]+.
-
Beispiel 9
-
9.1 – Herstellung von 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)
-
Das
Verfahren war wie für
Beispiel 4.1, mit der Ausnahme, dass 2,4,6-Trimethylanilin anstelle
von 2-tertiär-Butylanilin verwendet
wurde. Die Ausbeute war 60% der Theorie.
1H
NMR (CDCl3): 8,50, 7,95, 6,94, (m, 7H, ArH,
PyrH), 2,33 (s, 6H, N=CCH3), 2,28 (s, 6H,
CCH3), 2,05 (s, 12H, CCH3).
Massenspektrum:
m/z 397 [M]+.
-
9.2 – Herstellung von 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)FeCl2
-
Das
Verfahren war wie für
Beispiel 4.2, mit der Ausnahme, dass 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)
anstelle von 2,6-Diacetylpyridinbis(2-tert-butylanil) angewendet
wurde. Die Ausbeute war 64% der Theorie.
Massenspektrum: m/z
523 [M]+, 488 [M – Cl]+,
453 [M – Cl2]+.
-
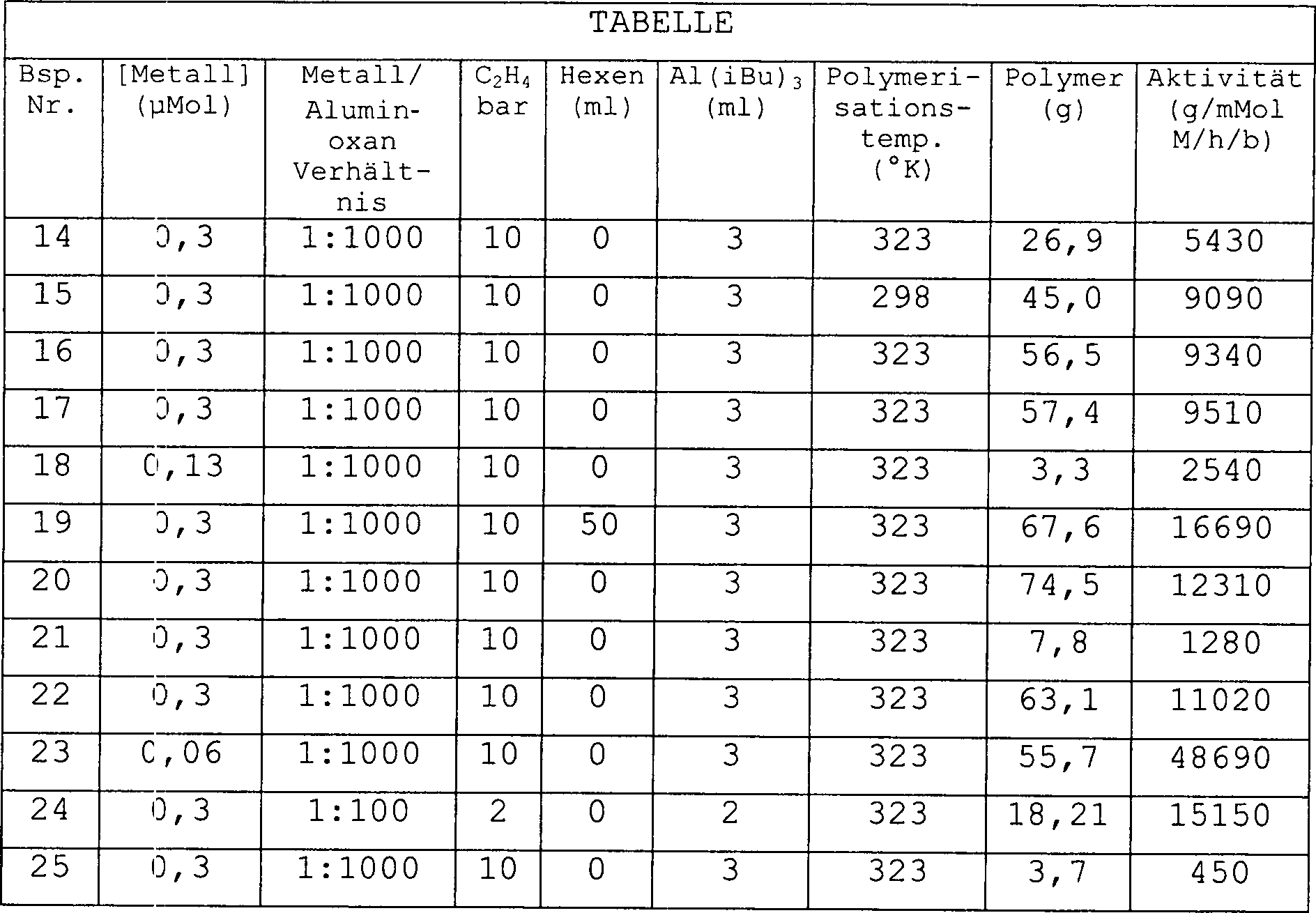
Beispiele 14 bis 25
-
Diese
Beispiele sind eine Reihe von Tests, wo Ethylen oder Ethylen/1-Hexen
unter 10 bar Ethylendruck unter Verwendung der erfindungsgemäßen Katalysatoren
unter "Aufschlämmungs"-Polymerisationsbedingungen
polymerisiert wird.
-
Katalysator-Herstellung
-
Die
als Katalysator in Beispielen 14 bis 25 angewendeten Übergangsmetallkomplexe
waren wie nachstehend:
In Beispielen 14 und 15 war der Komplex
2,6-Diacetylpyridinbis(2,6-diisopropylanil)FeCl2,
hergestellt wie in Beispiel 1 beschrieben (Verbindung Formel D).
In
Beispielen 16 bis 20 war der Komplex 2,6-Diacetylpyridinbis(2,6-dimethylanil)FeCl2, hergestellt wie in Beispiel 8 beschrieben.
In
Beispiel 21 war der Komplex 2,6-Diacetylpyridinbis(2,4-dimethylanil)FeCl2, hergestellt wie in Beispiel 7 beschrieben.
In
Beispielen 22 bis 24 war der Komplex 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)FeCl2, hergestellt wie in Beispiel 9 beschrieben.
In
Beispiel 25 war der Komplex 2,6-Diacetylpyridinbis(2,6-diisopropylanil)CoCl2, hergestellt wie in Beispiel 3 beschrieben
(Formel K).
-
Katalysator-Aktivierung
-
Der Übergangsmetallkomplex
wurde in Toluol (vorher über
Natriummetall getrocknet) unter einer Stickstoffatmosphäre gelöst, und
es wurde eine Lösung
von Aktivator (Co-katalysator)
bei Umgebungstemperatur zugegeben. Das Gemisch wurde bei Raumtemperatur
gerührt,
dann wurde eine aliquote Menge zu der Einspritzeinheit eines Polymerisationsreaktors überführt. Die
Mengen an in der Katalysatoraktivierung angewendeten Reagenzien
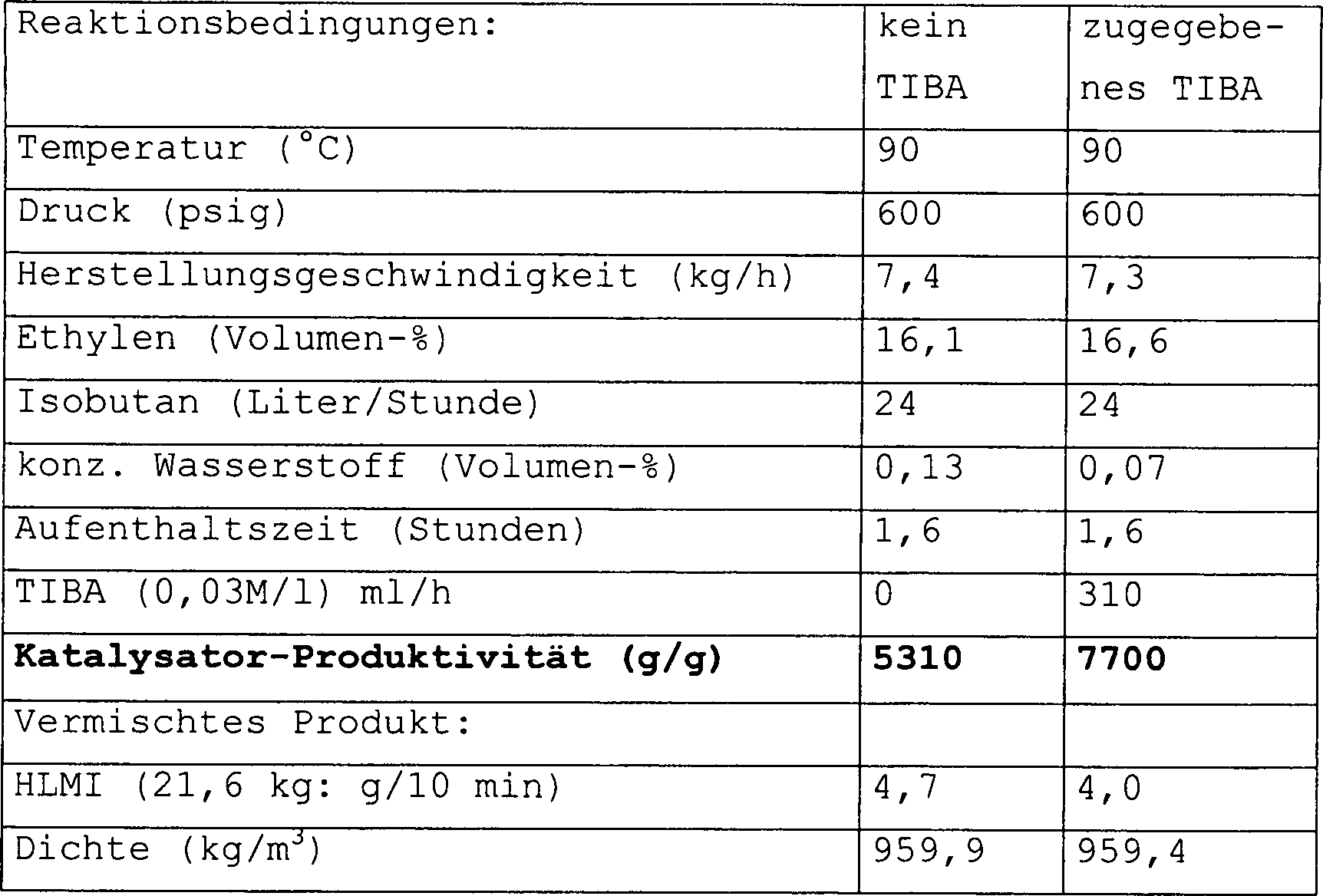
werden in der nachstehenden Tabelle ausgewiesen. Alle Vorgänge wurden
unter einer Stickstoffatmosphäre
durchgeführt,
sofern nicht anders ausgewiesen. "MAO" ist
Methylaluminoxan (1,78 M in Toluol, bezogen von Witco). "MMAO" ist modifiziertes
Methylaluminoxan (10% Gewicht/Gewicht in Heptan – bezogen von Witco). Sie wurden,
wie erhalten, verwendet. Triisobutylaluminium (Al(iBu)3,
als eine 1 M-Lösung in
Toluol), wurde von Aldrich bezogen.
-
-
Polymerisationstests
-
Die
in den Polymerisationstests angewendeten Reagenzien waren Ethylen
Qualität
3,5 (bezogen von Air Products), Hexen (bezogen von Aldrich), destilliert über Natrium/Stickstoff,
und Triisobutylaluminium (1 M in Hexanen, bezogen von Aldrich).
-
Polymerisation von Ethylen
-
Ein
1-Liter-Reaktor wurde unter einem Stickstoffstrom für mindestens
1 Stunde bei > 85°C ausgeheizt. Der
Reaktor wurde dann auf 50°C
gekühlt.
Isobutan (0,5 Liter) und Triisobutylaluminium wurden zugegeben und
der Reaktor wurde in eine Box unter Stickstoff gestellt. Das Alkylaluminium
wurde zum Einfangen von Giften in dem Reaktor für mindestens 1 Stunde belassen.
Ethylen wurde in den Reaktor eingeführt, bis ein vorbestimmter Überdruck
erreicht war, dann wurde die Katalysatorlösung unter Stickstoff eingespritzt.
Der Reaktordruck wurde über
den gesamten Polymerisationsversuch durch Compu ter-gesteuerte Zugabe
von weiterem Ethylen konstant gehalten. Die Polymerisationszeit
war 1 Stunde. Nach Beendigung des Versuchs wurde der Reaktorinhalt
isoliert, mit angesäuertem
Methanol (50 ml HCl/2,5 l Methanol) und Wasser/Ethanol (4 : 1 Volumen/Volumen)
gewaschen und unter Vakuum bei 40°C
für 16
Stunden getrocknet.
-
Co-Polymerisation von
Ethylen/1-Hexen (Beispiel 19)
-
Ein
1-Liter-Reaktor wurde unter einem Stickstoffstrom für mindestens
1 Stunde bei > 85°C ausgeheizt. Der
Reaktor wurde dann auf 50°C
gekühlt.
Isobutan (0,5 Liter), 1-Hexen und Triisobutylaluminium wurden dann
zugegeben und der Reaktor wurde in eine Box unter Stickstoff gestellt.
Das Alkylaluminium wurde zum Einfangen von Giften in dem Reaktor
für mindestens
1 Stunde belassen. Ethylen wurde in den Reaktor eingeführt, bis
ein vorbestimmter Überdruck
erreicht war; dann wurde die Katalysatorlösung unter Stickstoff eingespritzt.
Der Reaktordruck wurde durch den Polymerisationsversuch durch Computer-gesteuerte
Zugabe von Ethylen konstant gehalten. Die Polymerisationszeit war
1 Stunde. Nach Beendigung des Versuchs wurde der Reaktorinhalt isoliert,
mit angesäuertem
Methanol (50 ml HCl/2,5 l Methanol) und Wasser/Ethanol (4 : 1 Volumen/Volumen)
gewaschen und unter Vakuum bei 40°C
für 16
Stunden getrocknet.
-
Die
Daten aus den Polymerisationstests werden nachstehend in der Tabelle
angeführt.
-
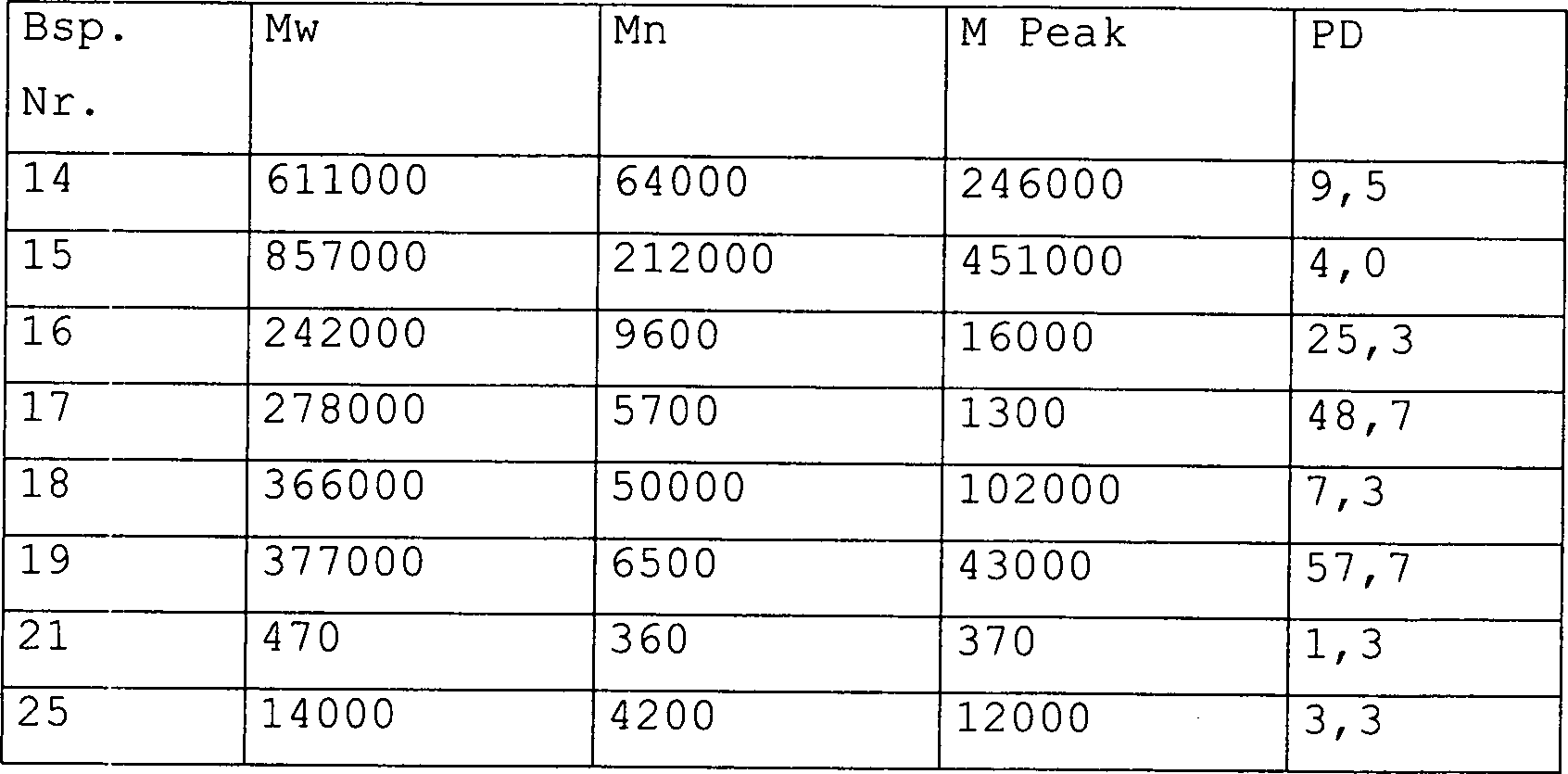
-
Molekulargewichtsdaten
der aus Beispielen 14 bis 25 erhaltenen Polymere werden in der nachstehenden
Tabelle angeführt.
-
-
Beispiele 26 und 27
-
Gas-Phasen-Polymerisationstests
mit getragenen Katalysatoren
-
Beispiele
26 und 27 erläutern
die Verwendung von erfindungsgemäßen Katalysatoren,
die auf Siliziumdioxid-Trägermaterial
getragen werden. Beispiel 26 wendet 2,6-Diacetylpyridinbis(2,6-diisopropylanil)FeCl2 an und Beispiel 27 wendet 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)FeCl2 als die Übergangsmetallkomplexverbindung
an.
-
Beispiel 26
-
Herstellung des getragenen
Katalysators
-
2,6-Diacetylpyridinbis(2,6-diisopropylanil)FeCl2 wurde wie in Beispiel 1 beschrieben hergestellt.
Siliziumdioxid (1,03 g ES70, bezogen von Crosfield), welches unter
Stickstoffstrom auf 700°C
erhitzt wurde, wurde in ein Schlenk-Rohr gegeben und Toluol (10
ml) wurde zugegeben. Das Gemisch wurde auf 50°C erhitzt. Zu einer Lösung von
2,6-Diacetylpyridinbis(2,6-diisopropylanil)FeCl2 (0,036
g) in Toluol (10 ml) wurde Methylaluminoxan (5 ml, 1,78 M in Toluol,
bezogen von Witco) gegeben. Das Gemisch wurde auf 50°C erhitzt
und dann zu einem Siliziumdioxid/Toluolgemisch überführt. Das Siliziumdioxid/MAO/Toluol-Gemisch
wurde bei 50°C
unter regelmäßigem Rühren für 1 Stunde,
bevor das Toluol entfernt wurde, bei 65°C unter Vakuum gehalten, unter
Gewinnung eines rieselfähigen
Pulvers.
-
Beispiel 27
-
Herstellung des getragenen
Katalysators
-
2,6-Diacetylpyridinbis(2,4,6-trimethylanil)FeCl2 wurde wie in Beispiel 9 beschrieben hergestellt.
Siliziumdioxid (1,38 g ES70, bezogen von Crosfield), welches unter
Stickstoffstrom auf 700°C
erhitzt wurde, wurde in ein Schlenk-Rohr gegeben und Toluol (10
ml) wurde zugesetzt. Einer Lösung
von 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)FeCl2 (0,041
g) in Toluol (10 ml) wurde Methylaluminoxan (13,2 ml, 1,78 M in
To luol, bezogen von Witco) zugesetzt. Dieses Gemisch wurde 30 Minuten
zum Auflösen
von soviel wie möglich
Eisenkomplex auf 40°C
erhitzt. Die Lösung
wurde dann zu dem Siliziumdioxid/Toluol überführt. Das Siliziumdioxid/MAO/Toluol-Gemisch
wurde bei 40°C,
unter regelmäßigem Rühren für 30 Minuten
gehalten, bevor das Toluol bei 40°C
unter Vakuum entfernt wurde, unter Gewinnung eines rieselfähigen Pulvers.
-
Analyse
des Feststoffs ergab 16,9% Gewicht/Gewicht Al und 0,144 Gewicht/Gewicht
Eisen.
-
Polymerisationstests – Beispiele
26 und 27
-
Die
bei den Polymerisationstests verwendeten Reagenzien waren: Wasserstoff
Qualität
6.0 (bezogen von Air Products): Ethylen Qualität 3.5 (bezogen von Air Products):
Hexen (bezogen von Aldrich), destilliert über Natrium/Stickstoff: getrocknetes
Pentan (bezogen von Aldrich): Methylaluminium (2 M in Hexanen, bezogen
von Aldrich): und Triisobutylaluminium (1 M in Hexanen, bezogen
von Aldrich).
-
Ein
3-Liter-Reaktor wurde unter Stickstoffstrom für mindestens 1 Stunde bei 77–85°C ausgeheizt,
bevor gepulvertes Natriumchlorid (300 g, vorgetrocknet unter Vakuum,
160°C, > 4 Stunden) zugesetzt
wurde. Das Natriumchlorid wurde als ein fluidisierbares/rührbares
Anfahr-Chargen-Pulver für
die Gasphasenpolymerisation verwendet. Trimethylaluminium (3 ml,
2 M in Hexanen) wurde zu dem Reaktor gegeben und in eine Box unter
Stickstoff gestellt. Das Alkylaluminium wurde zum Einfangen von
Giften in dem Reaktor für
zwischen ½–1 Stunde
belassen, vor dem Entlasten unter Verwendung von 4 × 4 bar
Stickstoffspülungen.
Die für
die Polymerisation zu verwendende Gasphasenzusammensetzung wurde
in den Reaktor eingeführt
und vor der Einspritzung der Katalysatorzusammensetzung auf 77°C vorerhitzt.
Der Katalysator (0,18–0,22
g) wurde unter Stickstoff eingespritzt und die Temperatur wurde
dann auf 80°C
eingestellt. Das Verhältnis
von Hexen und/oder Wasserstoff zu Ethylen während der Polymerisation wurde
durch Verfolgen der Gasphasenzusammensetzung durch ein Massenspektro meter
und, wie erforderlich, Einstellen des Ausgleichs konstant gehalten.
Die Polymerisationstests wurden zum Fortsetzen für zwischen 1 bis 2 Stunden
vor dem Beenden durch Spülen
der Reaktanten aus dem Reaktor unter Stickstoff und Vermindern der
Temperatur auf < 30°C belassen.
Das hergestellte Polymer wurde zum Entfernen von Natriumchlorid
mit Wasser, dann mit angesäuertem
Methanol (50 ml HCl/2,5 l Methanol) und schließlich mit Wasser/Ethanol (4
: 1 Volumen/Volumen) gewaschen. Das Polymer wurde unter Vakuum bei
40°C für 16 Stunden
getrocknet. Einige Versuche unter Verwendung einer Vielzahl von
Arbeitsbedingungen wurden mit jedem der Katalysatoren von Beispielen
26 und 27 ausgeführt.
Alle Polymerisationstests wurden bei einer Polymerisationstemperatur
von 80°C
und einem Ethylendruck von 8 bar ausgeführt. Die Polymerisationsbedingungen
werden in der nachstehenden Tabelle angeführt.
-
-
Molekulargewichtsdaten
der Polymerprodukte werden in der nachstehenden Tabelle angeführt.
-
-
Beispiel 32
-
32.1 – Herstellung einer getragenen
Ziegler-Katalysator-Komponente
-
Kieselgel
(20 kg), Qualität
ES70, bezogen von Crosfield, welches bei 800°C für 5 Stunden in Stickstoffstrom
getrocknet wurde, wurde in Hexan (110 Litern) aufgeschlämmt und
Hexamethyldisilazan (30 Mol), bezogen von Fluka, wurde bei 50°C unter Rühren zugesetzt.
Trockenes Hexan (120 Liter) wurde unter Rühren zugegeben, der Feststoff
absetzen lassen, die Überstandsflüssigkeit
durch Dekantierung entfernt und weiteres trockenes Hexan (130 Liter)
wurde unter Rühren
zugegeben. Die Hexanwaschung wurde weitere 3 Male wiederholt. Dibutylmagnesium
(30 Mol), bezogen von FMC, wurde zugegeben und 1 Stunde bei 50°C gerührt. Tertiär-Butylchlorid
(60 Mol) wurde zugegeben und 1 Stunde bei 50°C gerührt. Zu dieser Aufschlämmung wurde ein äquimolares
Gemisch von Titantetrachlorid (3 Mol) und Titan-tetra-n-propoxid
(3 Mol) unter Rühren
bei 50°C
für 2 Stunden
zugegeben, gefolgt von 5 Waschungen mit trockenem Hexan (130 Liter).
Die Aufschlämmung
wurde unter einem fließenden
Stickstoffstrom getrocknet, unter Gewinnung einer festen, auf Siliziumdioxid
getragenen Ziegler-Katalysator-komponente.
-
32.2 – Herstellung von gemischtem
Katalysator, der eine Ziegler-Komponente und eine Übergangsmetallverbindun
der vorliegenden Erfindung enthält
-
Eine
Lösung
von Methylaluminoxan ("MAO", 10,2 mMol) als
eine 10-gewichtsprozentige Lösung
in Toluol, bezogen von Witco, wurde zu einer Suspension von 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)FeCl2 (0,07 mMol in 5 ml trockenem Toluol), hergestellt
wie in Beispiel 9, gegeben und das Gemisch 5 Minuten geschüttelt. Diese
Lösung
wurde dann zu 2,0 g des vorstehend hergestellten Siliziumdioxid-getragenen
Ziegler-Katalysators, vorstehend hergestelltes Beispiel 32.1, gegeben,
das Gemisch wurde 2 Stunden bei 20°C geschüttelt und dann wurde das Lösungsmittel
unter vermindertem Druck bei 20°C
entfernt, unter Gewinnung des gemischten Katalysators als ein rieselfähiges Pulver.
-
32.3 – Polymerisation von Ethylen/Hexen-Gemisch,
unter Verwendung des gemischten Katalysators
-
Ein
mit einem Helixrührer
ausgestatteter 3-Liter-Reaktor
wurde 1 Stunde unter trockenem durchfließendem Stickstoff auf 95°C erhitzt.
Die Temperatur wurde auf 50°C
vermindert und trockenes Natriumchlorid (300 g) wurde dann mit Trimethylaluminium-(TMA)-Lösung (2
ml 2 molar TMA in Hexan) zugesetzt und der Reaktor 2 Stunden auf
85°C erhitzt.
Der Reaktor wurde mit Stickstoff gespült, auf 50°C gekühlt und TMA-Lösung
(3 ml 2 molar TMA in Hexan) zugegeben. Die Temperatur wurde auf
77°C erhöht und Wasserstoff
(0,5 bar) und Ethylen (8 bar) vor der Zugabe von 1-Hexen (2,6 ml)
zugegeben. Die Reaktion wurde durch Einspritzung des vorher hergestellten
gemischten Katalysators (0,20 g) in den Reaktor begonnen. Die Temperatur
wurde bei 80°C
gehalten und Ethylen zum Halten des konstanten Drucks zugesetzt.
Die Gasphase wurde durch ein Massenspektrometer verfolgt und Wasserstoff
und 1-Hexen, falls notwendig, zum Halten von konstanten Gasphasenkonzentrationen
dieser Komponenten zugesetzt. Die Polymerisation wurde 90 Minuten
ausgeführt. Das
Polymer wurde mit Wasser zum Entfernen von Natriumchlorid, dann
mit angesäuertem
Methanol (50 ml HCl/2,5 Liter Methanol) und schließlich mit
Wasser/Ethanol (4 : 1 Volumen/Volumen) gewaschen. Das Polymer wurde
unter Vakuum bei 40°C
für 16
Stunden getrocknet. 111 g getrocknetes Polymer wurden hergestellt.
Das Polymer hatte eine breite Molekulargewichtsverteilung (wie durch
Gelpermeationschromatographie bestimmt). Die Polydispersität (Mw/Mn)
war 28,5.
-
Beispiel 33
-
33.1 – Vorimprägnierung von Träger mit
Aktivatorverbindung
-
Alle
nachstehend angeführten
Vorgänge
wurden unter einer Stickstoffatmosphäre durchgeführt, sofern nicht anders ausgewiesen.
Siliziumdioxid (Crosfield Qualität
ES70X) wurde unter fließendem
Stickstoff 16 Stunden auf 250° erhitzt.
Eine Probe von diesem Siliziumdioxid (2,5 g) wurde in ein Schlenk-Rohr gegeben und
hatte 12,1 ml 1,78 M Methylaluminoxan, MAO (bezogen von Witco) wurde
zum Bilden einer Aufschlämmung
dazugegeben. Die Aufschlämmung
wurde 4 Stunden auf 50°C
erhitzt, bevor sie 10 Tage bei Raumtemperatur belassen wurde. Die Überstandsflüssigkeit über dem
Siliziumdioxid wurde entfernt und das Siliziumdioxid/MAO dreimal
mit Toluol (3 × 10
ml) bei Raumtemperatur gewaschen, unter Entfernen der Überstandslösung jedes
Mal.
-
33.2 – Tragen des Katalysators
-
(2,6-Diacetylpyridinbis(2,4,6-trimethylanil)eisendichlorid
(0,101 g) (hergestellt wie in Beispiel 9 beschrieben) wurde in Toluol
(20 ml) bei Raumtemperatur aufgeschlämmt und zu dem Siliziumdioxid/MAO
gegeben. Das Gemisch wurde gelegentlich innerhalb eines Zeitraums
von 1 Stunde geschüttelt.
Die Überstandslösung wurde
entfernt und der Siliziumdioxid/MAO/Fe-Komplex wurde mit Toluol
gewaschen, bis das Filtrat farblos war. Der Feststoff wurde unter
Vakuum bei 50°C
getrocknet.
-
33.3 – Gas-Phasen-Polymerisation
von Ethylen
-
Ein
3-Liter-Reaktor wurde unter fließendem Stickstoff für mindestens
1 Stunde bei 77°C
ausgeheizt, bevor Natriumchlorid (300 g, < 1 mm Durchmesser Teilchen, vorgetrocknet
unter Vakuum, 160°C, > 4 Stunden) zugegeben
wurde. Das Natriumchlorid wurde nur als ein Standard-"Chargenpulver" für den Gasphasenpolymerisationsreaktor
angewendet. Trimethylaluminium (3 ml, 2 M in Hexanen, bezogen von
Aldrich) wurde zu dem Reaktor gegeben, der dann verschlossen wurde.
Das Alkylaluminium wurde zum Einfangen von Giften in dem Reaktor
für ½ Stunde
belassen, bevor es durch sukzessives unter Druck setzen und Spülen des
Reaktors mit 4 bar Stickstoff entlastet wird. Ethylen (Qualität 3.5, bezogen
von Air Products) wurde zu dem Reaktor gegeben, unter Gewinnung
eines Drucks von 8 bar bei 77°C,
vor der Katalysatoreinspritzung. Der getragene Katalysator (0,215
g), hergestellt wie in Beispiel 33.2 beschrieben, wurde unter Stickstoff
in den Reaktor eingespritzt und die Temperatur dann auf 80°C eingestellt.
Die Polymerisation wurde 5 Stunden fortsetzen lassen, bevor durch
Spülen
des Ethylens aus dem Reaktor unter Stickstoff und Vermindern der
Temperatur auf unter 30°C
beendet wurde. Das Polymer wurde mit Wasser zum Entfernen von Natriumchlorid
gewaschen, dann mit Methanol (50 ml HCl/2,5 Liter Methanol) angesäuert und
schließlich
mit Wasser/Ethanol (4 : 1 Volumen/Volumen) gewaschen. Das Polymer
wurde unter Vakuum bei 40°C
16 Stunden getrocknet. 161 g getrocknetes Polymer wurden hergestellt.
-
Beispiele 35 bis 38
-
Diese
erläutern
die Herstellung von getragenen Katalysatoren gemäß der vorliegenden Erfindung
und deren Verwendung bei der Polymerisation von Ethylen unter "Aufschlämmungs"-Polymerisationsbedingungen.
-
Beispiel 35
-
35.1 – Herstellung von 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)eisendichlorid,
getragen auf MAO/Siliziumdioxid
-
Siliziumdioxid-getragenes
Material (Qualität
ES70X, bezogen von Crosfield) wurde unter fließendem Stickstoff für 16 Stunden
auf 250°C
erhitzt. Eine Probe von diesem Siliziumdioxid wurde in ein Schlenk-Rohr gegeben
und 12,1 ml 1,78 M Methylaluminoxan ("MAO",
bezogen von Witco) wurden zum Bilden einer Aufschlämmung dazugegeben.
Die Aufschlämmung
wurde 4 Stunden auf 50°C
erhitzt, bevor sie 10 Tage bei Raumtemperatur belassen wurde. Die Überstandsflüssigkeit über dem
Siliziumdioxid wurde dann entfernt und das Siliziumdioxid/MAO 3-mal mit Toluol (10
ml) bei Raumtemperatur gewaschen, unter Entfernen der Überstandslösung jedes
Mal. 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)eisendichloridkomplex
(0,101 g) wurde in Toluol (20 ml) bei Raumtemperatur aufgeschlämmt und
zu dem Siliziumdioxid/MAO gegeben. Das Gemisch wurde gelegentlich
innerhalb eines Zeitraums von 1 Stunde geschüttelt. Die Überstandslösung wurde entfernt und der
hergestellte Siliziumdioxid-getragene MAO/Fe-Komplex mit Toluol
gewaschen, bis die Anfangswaschungen, die in der Farbe hellorange
waren, klar und frei von Farbe wurden. Der hergestellte Siliziumdioxid-getragene
Katalysatorfeststoff wurde unter Vakuum bei 50°C getrocknet.
-
35.3 – Polymerisation von Ethylen
-
Ein
1-Liter-Reaktor wurde unter fließendem Stickstoff für 3 Stunden
auf 80°C
erhitzt. Der Reaktor wurde auf weniger als 30°C gekühlt und 500 ml Isobutan zugegeben.
Trimethylaluminium (3 ml 2 M in Hexanen) wurde zu dem Reaktor gegeben
und dieser wurde dann auf 80°C
erhitzt. Der Druck in dem Reaktor wird auf 13,8 bar erhöht und dann
Ethylen zugesetzt, unter Gewinnung eines Gesamtdrucks von 23,8 bar.
Der in 35.1 vorstehend hergestellte, getragene Katalysator (0,201
g des getragenen Katalysatorfeststoffs in Toluolaufschlämmung) wurde
in den Reaktor unter Stickstoff, unter Verursachen, dass der Reaktordruck
auf 25,4 bar ansteigt, eingespritzt. Die Katalysatoraktivität war etwas
zu hoch für
den Ethyleneinlassstrom, um den Druck konstant zu halten, und dieser
wurde deshalb auf 23,2 bar fallen lassen. Der in dem Reaktor für die Mehrheit der
Polymerisation vorliegende Ethylendruck wurde auf 7,8 bar geschätzt. Der
Test wurde nach 1,75 Stunden beendet und das Polymer mit Methanol/HCl
(2,5 Liter/50 ml), dann Wasser/Ethanol (4 : 1 Volumen/Volumen) gewaschen
und unter Vakuum bei 40°C
getrocknet. 166 g trockenes Polymer wurden gewonnen. Analyse des Polymers
durch GPC wies Mw und Mn mit 182000 bzw. 11000 aus.
-
Beispiel 36
-
36.1 – Herstellung von 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)eisendichlorid,
getragen auf MAO/Siliziumdioxid
-
Eine
Portion (etwa 1–1,5
g) des in Beispiel 35.1 hergestellten getragenen Katalysators wurde
mit 5 × 10
ml aliquoten Mengen Toluol bei 100°C gewaschen. Die Anfangswaschungen
hatten eine tief orange Farbe und diese Färbung wurde mit jedem anschließenden Waschen
geringer, bis die Endwaschung farblos war. Der Feststoff wurde unter
Vakuum bei 100°C
zum Bereitstellen von rieselfähigem,
festem, getragenem Katalysator getrocknet.
-
36.2 – Polymerisation von Ethylen
-
Ein
1-Liter-Reaktor wurde unter fließendem Stickstoff für 1 Stunde
auf 75°C
erhitzt. Trimethylaluminium (3 ml 2 M in Hexanen) wurde zu dem Reaktor
gegeben, der dann auf 50°C
gekühlt
wurde. Isobutan (500 ml) wurde zu dem Reaktor gegeben und die Temperatur
auf 76°C
erhöht.
Der Druck in dem Reaktor wurde auf 13 bar erhöht. Ethylen wurde zu dem Reaktor
gegeben, um 21 bar Gesamtdruck (8 bar Ethylen) zu ergeben. Der in
26.1 vorstehend hergestellte, getragene Katalysator (0,11 g in Toluolaufschlämmung) wurde
in den Reaktor gespritzt und die Druckerhöhung während der Steuerung des Reaktordrucks
während
des Tests in Betracht gezogen. Die Temperatur wurde auf 80°C erhöht. Nach
1 Stunde wurde eine weitere aliquote Menge des gleichen Katalysators
(0,22 g in Hexanaufschlämmung)
eingespritzt und der Test weitere 3,5 Stunden fortgesetzt. 25 g
Polymer wurden gewonnen. Analyse des Polymers durch GPC wiesen Mw
und Mn mit 343000 bzw. 35000 aus.
-
Beispiel 37
-
37.1 – Herstellung von 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)eisendichlorid,
getragen auf MAO/Siliziumdioxid
-
Methylaluminoxan
(24 ml 1,78 M in Toluol, bezogen von Witco) wurde zu Siliziumdioxid
(5 g Qualität ES70X,
bezogen von Crosfield) gegeben, welches unter fließendem Stickstoff
auf 250°C
erhitzt wurde. Das Siliziumdioxid/MAO wurde 1 Stunde auf 80°C erhitzt,
bevor es mit Toluol (5 × 10
ml aliquote Mengen) gewaschen wurde. Die Hälfte der hergestellten Siliziumdioxid/MAO-Aufschlämmung, gekühlt auf
Raumtemperatur, wurde für
die nächste
Stufe der Katalysatorherstellung (die andere Hälfte wurde zur Verwendung in
Beispiel 38 beiseite gestellt) verwendet. 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)eisendichlorid
(73 mg) wurde in Toluol aufgeschlämmt und zu der halben Portion
von Siliziumdioxid/MAO/Toluol überführt und
2 Stunden unter gelegentlichem Mischen reagieren lassen. Der Siliziumdioxid/MAO/Fe-Komplex
wurde mit Toluol (3 × 10
ml aliquote Mengen) bei Raumtemperatur und dann mit Hexan (2 × 10 ml
aliquote Mengen) bei Raumtemperatur, um das Toluol zu entfernen,
vor dem abschließenden
Waschen mit Hexan bei 80°C
(3 × 10
ml aliquote Mengen) gewaschen. Der hergestellte, getragene Katalysatorfeststoff
wurde unter Vakuum bei Raumtemperatur getrocknet.
-
37.2 – Polymerisation von Ethylen
-
Ein
1-Liter-Reaktor wurde unter fließendem Stickstoff für 1 Stunde
auf 80°C
erhitzt. Der Reaktor wurde auf weniger als 30°C gekühlt und 500 ml Isobutan zugegeben.
Der Reaktor wurde auf 77°C
erhitzt und der Druck auf 13,8 bar erhöht. Ethylen wurde zugegeben,
um 21,8 bar Gesamtdruck (8 bar Ethy len) zu ergeben. Triisobutylaluminium
(5 ml 1 M in Hexanen) wurde zu dem Reaktor gegeben und nach 20 Minuten
wurde der vorstehend in 37.1 hergestellte, getragene Katalysator
(0,14 g in Hexanaufschlämmung)
in den Reaktor eingespritzt und die Druckerhöhung bezüglich der Steuerung des Reaktordrucks
während
des Tests genommen. Die Temperatur wurde auf 80°C erhöht. Nach 5 Stunden war die
Polymerisation beendet. 138 g Polymer wurden gewonnen. Analyse des
Polymers durch GPC wiesen Mw und Mn als 567000 bzw. 53000 aus.
-
37.3 – Polymerisation von Ethylen
-
Ein
1-Liter-Reaktor wurde unter fließendem Stickstoff für 1 Stunde
auf 78°C
erhitzt. Der Reaktor wurde auf weniger als 30°C gekühlt und 500 ml Isobutan zugegeben.
Triisobutylaluminium (3 ml 1 M in Hexanen) wurde zu dem Reaktor
gegeben, der dann auf 78°C
erhitzt wurde und der Druck auf 12,1 bar erhöht. Ethylen wurde zugegeben,
um 32,0 bar Gesamtdruck (19,9 bar Ethylen) zu ergeben. Der vorstehend
in 37.1 hergestellte, getragene Katalysator (0,0925 g, aufgeschlämmt in Hexan)
wurde in den Reaktor eingespritzt und der Gesamtdruck wurde bei
31,2 bar gesteuert. Der Ethylendruck während der Polymerisation wurde
auf ungefähr
19,1 bar geschätzt.
Polymerisation wurde 80 Minuten fortsetzen lassen. 181 g Polymer
wurden gewonnen. Analyse des Polymers durch GPC wiesen Mw und Mn
mit 595000 bzw. 44000 aus.
-
37.4 – Polymerisation von Ethylen
-
Ein
1-Liter-Reaktor wurde unter fließendem Stickstoff für 1 Stunde
auf 80°C
erhitzt, bevor er auf weniger als 30°C abgekühlt wurde. Triisobutylaluminium
(3 ml 1 M in Hexanen) wurde zu dem Reaktor gegeben, gefolgt von
500 ml Isobutan. Der Reaktor wurde auf 78°C erhitzt und der Druck auf
13,5 bar erhöht.
Ethylen wurde zugegeben, unter Gewinnung von 17,6 bar Gesamtdruck
(4,1 bar Ethylen). Der vorstehend in 37.1 hergestellte, getragene
Katalysator (0,15 g, aufgeschlämmt
in Hexan) wurde in den Reaktor eingespritzt. Der Ethylendruck wäh rend der
Polymerisation wurde auf ungefähr
4,7 bar geschätzt.
Die Polymerisation wurde 80 Minuten fortsetzen lassen. 21 g Polymer
wurden gewonnen. Analyse des Polymers durch GPC wies Mw und Mn mit
347000 bzw. 26000 aus.
-
Beispiel 38
-
38.1 – Herstellung von 2,6-Diacetylpyridinbis(2,6-diisopropylanil)cobaltdichlorid,
getragen auf MAO/Siliziumdioxid
-
Die
zweite Hälfte
des Siliziumdioxids/MAO, hergestellt in Beispiel 37.1, wurde unter
Vakuum getrocknet. Eine aliquote Menge von dem getrockneten Siliziumdioxid/MAO
(1 g) wurde in ein Schlenk-Rohr gegeben und 2,6-Diacetylpyridinbis(2,6-diisopropylanil)cobaltdichlorid
(40 mg) wurde dazu als trockenes Pulver gegeben. Hexan (10 ml) wurde
dann zu dem Schlenk-Rohr gegeben und der Cobaltkomplex und Siliziumdioxid/MAO
1 Stunde zusammen bei Raumtemperatur aufgeschlämmt. Das Gemisch wurde unter
Vakuum bei Raumtemperatur getrocknet, unter Hinterlassen des hergestellten
getragenen Katalysators als ein trockenes, rieselfähiges Pulver.
-
38.2 – Polymerisation von Ethylen
-
Ein
1-Liter-Reaktor wurde unter fließendem Stickstoff für 1 Stunde
auf 80°C
erhitzt, bevor er auf 30°C gekühlt wurde.
Hexen (250 ml), Triisobutylaluminium (3 ml von 1 M in Hexanen) und
250 ml Isobutan wurden zu dem Reaktor gegeben. Der Reaktor wurde
auf 80°C
erhitzt und der Druck auf 7,1 bar erhöht. Ethylen wurde zugegeben,
unter Gewinnung von 19,2 bar Gesamtdruck (12,1 bar Ethylen). Der
vorstehend hergestellte, getragene Katalysator (0,245 g, aufgeschlämmt in Hexan)
wurde in den Reaktor eingespritzt und die Druckerhöhung in
Bezug auf die Steuerung des Reaktordrucks während des Tests genommen. Polymerisation
wurde für 330
Minuten fortsetzen lassen. 3,3 g Polymer wurden gewonnen. Analyse
des Polymers durch GPC wies Mw = 5300 und Mn = 1500 aus.
-
Beispiel 39
-
Polymerisation von Ethylen
in Aufschlämmungsphase,
unter Verwendung eines getragenen Katalysators
-
Eine
Reihe von Polymerisationstests wurde unter Verwendung eines auf
einem getragenen, auf 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)eisendichlorid
basierenden Katalysators ausgeführt.
-
Beispiel 39.1
-
Ein
1-Liter-Reaktor wurde unter fließendem Stickstoff für 1 Stunde
auf 80°C
erhitzt, bevor er auf 30°C gekühlt wurde.
Isobutan (500 ml), gefolgt von Triisobutylaluminium (3 ml 1 M in
Hexanen) wurde zu dem Reaktor gegeben. Der Reaktor wurde auf 78°C erhitzt
und der Druck auf 13,2 bar erhöht.
Ethylen wurde zugegeben, um 26,2 bar Gesamtdruck zu ergeben. Der
Katalysator von Beispiel 37.1 (0,097 g, aufgeschlämmt in Hexan)
wurde in den Reaktor eingespritzt. Der Reaktordruck wurde bei 26,0
bar während
des Tests (Ethylendruck geschätzt
auf ungefähr
12,8 bar) gesteuert und die Temperatur auf 80°C eingestellt. Die Polymerisation
wurde 60 Minuten fortsetzen lassen. 78 g Polymer wurden gewonnen.
Analyse des Polymers durch GPC wies Mw und Mn als mit 528000 bzw.
40000 aus.
-
Beispiel 39.2
-
Ein
1-Liter-Reaktor wurde unter fließendem Stickstoff für 1 Stunde
auf 80°C
erhitzt, bevor er auf 30°C gekühlt wurde.
Isobutan (500 ml), gefolgt von Triisobutylaluminium (3 ml 1 M in
Hexanen) wurde zu dem Reaktor gegeben. Der Reaktor wurde auf 78°C erhitzt
und der Druck auf 13,4 bar erhöht.
Ethylen wurde zugegeben, um 21,2 bar Gesamtdruck zu ergeben. Der
Katalysator von Beispiel 37.1 (0,124 g, aufgeschlämmt in Hexan)
wurde in den Reaktor eingespritzt. Der Ethylendruck wurde auf ungefähr 8,1 bar
während
der Polymerisation geschätzt
und die Temperatur auf 80°C
eingestellt. Polymerisation wurde 60 Minuten fortsetzen lassen. 47
g Polymer wurden gewonnen. Analyse des Polymers durch GPC wies Mw
und Mn als mit 376000 bzw. 40000 aus.
-
Beispiel 39.3
-
Ein
1-Liter-Reaktor wurde unter fließendem Stickstoff für 1 Stunde
auf 80°C
erhitzt, vor dem Abkühlen auf
30°C. Triisobutylaluminium
(3 ml 1 M in Hexanen) wurde zu dem Reaktor gegeben, gefolgt von
500 ml Isobutan. Der Reaktor wurde auf 78°C erhitzt und der Druck auf
13,0 bar erhöht.
Ethylen wurde zugegeben, um 26,0 bar Gesamtdruck zu ergeben. Der
Katalysator von Beispiel 37.1 (0,0966 g, aufgeschlämmt in Hexan,
und 0,25 ml N,N-Dimethylanilin für
20 Minuten) wurde in den Reaktor gespritzt. Der Druck in dem Reaktor
wurde auf 22,5 bar zum Vermindern der Aktivität des Katalysators fallen lassen.
Der Ethylendruck des Reaktors während
der Mehrheit der Polymerisation wurde auf 9,0 bar geschätzt. Die
Polymerisation wurde 60 Minuten fortsetzen lassen. 88 g Polymer
wurden gewonnen. Analyse des Polymers durch GPC wies Mw und Mn als
mit 430000 bzw. 35000 aus.
-
Beispiel 39.4
-
Ein
1-Liter-Reaktor wurde unter fließendem Stickstoff für 1 Stunde
auf 80°C
erhitzt, bevor er auf 30°C gekühlt wurde.
Triisobutylaluminium (3 ml 1 M in Hexanen) wurde zu dem Reaktor
gegeben, gefolgt von 500 ml Isobutan. Der Reaktor wurde auf 78°C erhitzt
und der Druck auf 12,7 bar erhöht.
Ethylen wurde zugegeben, um 14,7 bar Gesamtdruck zu ergeben. Der
Katalysator von Beispiel 37.1 (0,104 g, aufgeschlämmt in Hexan) wurde
in den Reaktor eingespritzt. Der Ethylendruck während der Polymerisation wurde
als ungefähr
2,2 bar geschätzt.
Die Polymerisation wurde 60 Minuten fortsetzen lassen. 4,8 g Polymer
wurden gewonnen. Analyse des Polymers durch GPC wies Mw und Mn als
mit 340000 bzw. 36000 aus.
-
Beispiel 40 – Polymerisation
von Ethylen in der Aufschlämmungsphase,
unter Verwendung eines getragenen Katalysators
-
Eine
Reihe von Polymerisationstests wurde ausgeführt, unter Verwendung eines
auf einem getragenen 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)eisendichlorid
basierenden Katalysators.
-
Beispiel 40.1
-
Ein
1-Liter-Reaktor wurde unter fließendem Stickstoff für 1 Stunde
auf 80°C
erhitzt, bevor er auf 30°C abgekühlt wurde.
Isobutan (500 ml), gefolgt von Triisobutylaluminium (3 ml 1 M in
Hexanen) wurde zu dem Reaktor gegeben. Der Reaktor wurde auf 78°C erhitzt
und der Druck auf 13,2 bar erhöht.
Ethylen wurde zugegeben, um 26,2 bar Gesamtdruck zu ergeben. Der
Katalysator von Beispiel 37.1 (0,097 g, aufgeschlämmt in Hexan)
wurde in den Reaktor eingespritzt. Der Reaktordruck wurde bei 26,0
bar während
des Tests (Ethylendruck geschätzt
auf ungefähr
12,8 bar) gesteuert und die Temperatur auf 80°C eingestellt. Die Polymerisation wurde
60 Minuten fortsetzen lassen. 78 g Polymer wurden gewonnen. Analyse
des Polymers durch GPC wies Mw und Mn als mit 528000 bzw. 40000
aus.
-
Beispiel 40.2
-
Ein
1-Liter-Reaktor wurde unter fließendem Stickstoff für 1 Stunde
auf 80°C
erhitzt, bevor er auf 30°C abgekühlt wurde.
Isobutan (500 ml), gefolgt von Triisobutylaluminium (3 ml 1 M in
Hexanen) wurde zu dem Reaktor gegeben. Der Reaktor wurde auf 78°C erhitzt
und der Druck auf 13,4 bar erhöht.
Ethylen wurde zugegeben, um 21,2 bar Gesamtdruck zu ergeben. Der
Katalysator von Beispiel 37.1 (0,124 g, aufgeschlämmt in Hexan)
wurde in den Reaktor eingespritzt. Der Ethylendruck wurde auf ungefähr 8,1 bar
während
der Polymerisation geschätzt
und die Temperatur auf 80°C
eingestellt. Die Polymerisation wurde 60 Minuten fortsetzen lassen.
47 g Polymer wurden gewonnen. Analyse des Polymers durch GPC wies
Mw und Mn als mit 376000 bzw. 40000 aus.
-
Beispiel 40.3
-
Ein
1-Liter-Reaktor wurde unter fließendem Stickstoff für 1 Stunde
auf 80°C
erhitzt, bevor er auf 30°C abgekühlt wurde.
Triisobutylaluminium (3 ml 1 M in Hexanen) wurde zu dem Reaktor
gegeben, gefolgt von 500 ml Isobutan. Der Reaktor wurde auf 78°C erhitzt
und der Druck auf 13,0 bar erhöht.
Ethylen wurde zugegeben, um 26,0 bar Gesamtdruck zu ergeben. Der
Katalysator von Beispiel 37.1 (0,0966 g, aufgeschlämmt in Hexan und
0,25 ml N,N-Dimethylanilin für
20 Minuten) wurde in den Reaktor eingespritzt. Der Druck in dem
Reaktor wurde auf 22,5 bar fallen lassen, um die Aktivität des Katalysators
zu vermindern. Der Ethylendruck in dem Reaktor während der Mehrheit der Polymerisation
wurde auf 9,0 bar geschätzt.
Die Polymerisation wurde für 60
Minuten fortsetzen lassen. 88 g des Polymers wurden gewonnen. Analyse
des Polymers durch GPC wies Mw und Mn als mit 430000 bzw. 35000
aus.
-
Beispiel 40.4
-
Ein
1-Liter-Reaktor wurde unter fließendem Stickstoff für 1 Stunde
auf 80°C
erhitzt, bevor er auf 30°C abgekühlt wurde.
Triisobutylaluminium (3 ml 1 M in Hexanen) wurde zu dem Reaktor
gegeben, gefolgt von 500 ml Isobutan. Der Reaktor wurde auf 78°C erhitzt
und der Druck auf 12,7 bar erhöht.
Ethylen wurde zugegeben, um 14,7 bar Gesamtdruck zu ergeben. Der
Katalysator von Beispiel 37.1 (0,104 g, aufgeschlämmt in Hexan) wurde
in den Reaktor eingespritzt. Der Ethylendruck während der Mehrheit der Polymerisation
wurde auf 2,2 bar geschätzt.
Polymerisation wurde 60 Minuten fortsetzen lassen. 4,8 g Polymer
wurden gewonnen. Analyse des Polymers durch GPC wies Mw und Mn als
mit 340000 bzw. 36000 aus.
-
Beispiel 41
-
Dieses
Beispiel zeigt die Verwendung einer Kombination eines Katalysators
vom Metallocen-Typ mit einem Katalysator, der auf einem Eisenkomplex
der vorliegenden Erfindung basiert, zum Polymerisieren von Ethylen
unter Aufschlämmungsbedingungen.
-
41.1 – Herstellung eines getragenen
Metallocen-Katalysators
-
Zu
Siliziumdioxid (Crosfield Qualität
ES70, vorher calciniert bei 200°C,
in fließendem
N2 für
5 h) wurde eine Toluollösung
von Methylaluminoxan (MAO), enthaltend gelöstes Bis(n-butylcyclopentadienyl)ZrCl2, gegeben. Die verwendeten Mengen waren
2,5 mMol MAO pro Gramm Siliziumdioxid und 0,05 mMol Metallocen pro Gramm
Siliziumdioxid. Die erhaltene Aufschlämmung wurde für mindestens
1 Stunde vor dem Trocknen unter vermindertem Druck mild gerührt, unter
Gewinnung eines rieselfähigen
Pulvers.
-
41.2 – Herstellung des kombinierten
Metallocen/Fe-Komplex-Katalysators
-
Der
getragene Metallocen-Katalysator (2,5 g), hergestellt wie vorstehend
in Schritt 41.1 beschrieben, wurde in ein Schlenk-Rohr gegeben und
eine Aufschlämmung
von 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)eisendichlorid
(73 mg) in Hexan (10 ml) wurde bei Umgebungstemperatur dazugegeben.
Das Gemisch wurde auf 80°C
erhitzt und 90 Minuten unter gelegentlichem Schütteln unter Halten einer gut
gemischten Lösung
belassen. Es wurde keine Färbung
in der Überstandslösung oberhalb
des Feststoffs deutlich. Der hergestellte Katalysator wurde bei
80°C unter
Vakuum getrocknet, unter Hinterlassen eines trockenen, rieselfähigen Pulvers.
-
41.3 – Polymerisation von Ethylen
-
Ein
1-Liter-Reaktor wurde unter fließendem Stickstoff für 1 Stunde
auf 80°C
erhitzt, bevor er auf 30°C gekühlt wur de.
Triisobutylaluminium (3 ml 1 M in Hexanen) wurde zu dem Reaktor
gegeben, gefolgt von 500 ml Isobutan. Der Reaktor wurde auf 77°C erhitzt
und der Druck auf 12,9 bar erhöht.
Ethylen wurde zugegeben unter Gewinnung von 20,9 bar Gesamtdruck.
Der Katalysator (0,100 g, aufgeschlämmt in Hexan), hergestellt wie
vorstehend in 41.2 beschrieben, wurde in den Reaktor eingespritzt.
Der Ethylendruck während
der Polymerisation wurde auf ungefähr 8 bar geschätzt. Die
Polymerisation wurde 60 Minuten fortsetzen lassen. 96 g Polymer
wurden gewonnen. Analyse des Polymers durch GPC wies Mw und Mn als
mit 471000 bzw. 30000 aus.
-
41.4 – Vergleich
-
Dies
zeigt die Polymerisation von Ethylen unter Verwendung von nur dem
getragenen Metallocen-Katalysator, der in Schritt 41.1 beschrieben
wird.
-
Ein
1-Liter-Reaktor wurde unter fließendem Stickstoff für 1 Stunde
auf 80°C
erhitzt, bevor er auf 30°C gekühlt wurde.
Triisobutylaluminium (3 ml 1 M in Hexanen) wurde zu dem Reaktor
gegeben, gefolgt von 500 ml Isobutan. Der Reaktor wurde auf 75°C erhitzt
und der Druck auf 12,7 bar erhöht.
Ethylen wurde zugegeben, um 20,7 bar Gesamtdruck zu ergeben. Der
getragene Metallocen-Katalysator (0,094 g, aufgeschlämmt in Hexan),
hergestellt in Schritt 41.1 vorstehend, wurde in den Reaktor eingespritzt.
Der Ethylendruck während
der Polymerisation wurde auf ungefähr 8 bar geschätzt. Polymerisation
wurde 60 Minuten fortsetzen lassen. 49 g Polymer wurden gewonnen.
Analyse des Polymers durch GPC wies Mw und Mn als mit 142000 bzw.
53000 aus.
-
Beispiel 42
-
Gas-Phasen-Polymerisation
-
Dieses
Beispiel zeigt die Verwendung einer Kombination eines Katalysators
vom Metallocen-Typ mit einem auf einem Eisenkomplex der vorliegenden
Erfindung basierenden Katalysa tor zum Polymerisieren von Ethylen
unter Gasphasen-Polymerisationsbedingungen.
-
Ein
3-Liter-Reaktor wurde unter fließendem Stickstoff für mindestens
1 Stunde bei 78°C
ausgeheizt, bevor er auf 30°C
gekühlt
wurde. Pulverförmiges
Natriumchlorid (300 g) Chargenpulver mit einem mittleren Teilchendurchmesser
von weniger als 1 Millimeter (1 mm) und vorgetrocknet unter Vakuum
bei 160°C
für mehr als
4 Stunden, wurde zugegeben, gefolgt von Trimethylaluminium (3 ml,
2 M in Hexanen, bezogen von Aldrich). Der Reaktor wurde dann verschlossen
und auf 78°C
erhitzt. Das Alkylaluminium wurde zum Einfangen von in dem Reaktor
vorliegenden beliebigen Giften für
90 Minuten belassen. Der Reaktor wurde dann viermal durch Unter-Druck-Setzen
auf 4 bar mit Stickstoff gespült
und dann abgelassen. Wasserstoff wurde zu dem Reaktor gegeben, unter
Gewinnung von 0,08 bar Druck, gefolgt von Ethylen (8 bar). Der Katalysator
(0,20 g), wie in vorstehendem Schritt 41.3 hergestellt, wurde unter
Stickstoff eingespritzt und die Temperatur dann auf 80°C eingestellt.
Die Polymerisation wurde 60 Minuten fortsetzen lassen, bevor sie
durch Spülen
des Ethylens aus dem Reaktor unter Verwendung von Stickstoff und
Vermindern der Temperatur auf unter 30°C beendet wurde. Das Polymer
wurde zum Entfernen von Natriumchlorid mit Wasser, dann mit angesäuertem Methanol
(50 ml HCl/2,5 Liter Methanol) und schließlich mit Wasser/Ethanol (4
: 1 Volumen/Volumen) gewaschen. Das Polymer wurde unter Vakuum bei
40°C für 16 Stunden
getrocknet. 64 g getrocknetes Polymer wurden hergestellt. Das GPC
(Gel-Permeations-Chromatogramm) wurde für das Polymerprodukt ausgeführt. Die
erzeugte GPC-Kurve war deutlich bimodal mit Mw = 253000 und einer
Polydispersität
von 64,9.
-
Beispiel 43
-
Aufschlämmungs-Polymerisation,
unter Verwendung von 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)FeCl2 mit und ohne TIBA
-
Dieses
Beispiel zeigt die positive Wirkung auf die Aktivität des durch
Einarbeiten von Triisobutylaluminium (TIBA) als Cokatalysator erhaltenen
Katalysators, im Vergleich mit vollständiger Abwesenheit einer getrennt
zugegebenen Alkylaluminiumverbindung, wie in der vorliegenden Erfindung
als Komponente (3) definiert. Der angewendete Katalysator war jener,
der in vorstehendem Beispiel 37.1 hergestellt wurde.
-
43.1 – Polymerisation mit TIBA
-
Ein
mit einem Rührer
und ummantelt für
die Temperatursteuerung ausgestatteter 2,3-Liter-Reaktor wurde unter
trockenem Stickstoff für
1 Stunde auf 110°C
erhitzt. Er wurde dann auf 85°C
gekühlt
und eine Triisobutylaluminiumlösung
in trockenem Hexan unter Stickstoff eingespritzt. Der Reaktor wurde
dann mit 1 Liter trockenem Isobutan beschickt. Der gerührte Reaktorinhalt
wurde auf 600 psig durch Zugabe von trockenem Ethylen unter Druck
gesetzt, wobei die Temperatur bei 85°C gehalten wurde. Der Katalysator
von Beispiel 37.1 wurde unter Stickstoff in den Reaktor gespritzt
und die Einspritzungsleitung mit ungefähr 50 ml Isobutan gespült. Die
Reaktion wurde dann bei 600 psig durch kontinuierliche Ethylenzugabe
gesteuert und der Umsatz aus dem Ethylenverbrauch verfolgt. Die
Polymerisation wurde für
den in der nachstehenden Tabelle ausgewiesenen Zeitraum durchgeführt, wobei
an dem Punkt die Ethylenzugabe gestoppt wurde und der Reaktor vor
der Polymergewinnung und Stabilisierung auf Atmosphärendruck
entlastet wurde. Das Polymer wurde durch Zugabe einer verdünnten Acetonlösung von
Irganox 1076 stabilisiert, unter Gewinnung von 0,15 zusätzlichem Polymer.
Die Reaktionsbedingungen, Ausbeute und Aktivität werden nachstehend in der
Tabelle angegeben.
-
43.2 – Polymerisation ohne TIBA
(Vergleich)
-
Das
Verfahren von Beispiel 43.1 wurde wiederholt, mit der Ausnahme,
dass kein TIBA zugegeben wurde. Die Einzelheiten werden ebenfalls
nachstehend in der Tabelle gezeigt, woraus ersichtlich werden kann, dass
die Zugabe von TIBA zu dem Katalysator eine wesentliche Verbesserung
der Aktivität
ergibt.
-
-
Ein
Vergleich der Polymerisation mit und ohne TIBA wurde auch in einem
Pilotanlagenmaßstab
ausgeführt.
-
43.3 – Herstellung von 2,6-Diacetylpyridinbis(2,4,6-trimethylanil)eisendichlorid,
getragen auf MAO/Siliziumdioxid
-
Alle
Vorgänge
wurden unter Stickstoff durchgeführt,
sofern nicht anders ausgewiesen. Siliziumdioxid (256,62 g Qualität ES70X,
bezogen von Crosfield), calciniert bei 200°C unter Stickstoffstrom, wurde
in einen 2-Liter-Rundkolben gegeben. Toluol (900 ml) wurde zu dem
Siliziumdioxid gegeben, gefolgt von Methylaluminoxan (441 ml, 1,5
M in Toluol, bezogen von Witco). Das MAO wurde mit dem Siliziumdioxid
bei Raumtemperatur für
10 Minuten umsetzen lassen, wobei zu dem Zeitpunkt die Temperatur
auf 80°C
stieg und die Aufschlämmung
wurde gelegentlich durch manuelles Schütteln des Kolbens vermischt.
Die Temperatur wurde für einen
Zeitraum von 2 Stunden zwischen 80–100°C gehalten.
-
2,6-Diacetylpyridinbis(2,4,6-trimethylanil)eisendichlorid,
hergestellt wie in vorstehendem Beispiel 9 (3,48 g), wurde in Toluol
(50 ml) aufgeschlämmt
und zu der MAO/Siliziumdioxidaufschlämmung bei 80°C gegeben.
Eine weitere aliquote Menge Toluol (20 ml) wurde verwendet, um zu
sichern, dass der gesamte Fe-Komplex zu dem MAO/Siliziumdioxid überführt wurde.
Das Fe/MAO/Siliziumdioxid wurde dann auf 80°C erhitzt, unter gelegentlichem
Schütteln
für 1,5
Stunden und der Feststoff absetzen lassen. Die klare Überstandslösung wurde
von dem Kolben abdekantiert und der Katalysator teilweise unter
Vakuum bei 80°C
für 30 Minuten
getrocknet und dann 16 Stunden bei Raumtemperatur belassen. Trocknen
des Katalysators wurde dann bei 80°C unter Vakuum für weitere
5 Stunden fortgesetzt, bis sich ein trockenes, rieselfähiges Pulver
ergab und kein weiteres Lösungsmittel,
das aus dem Träger
kommt, nachgewiesen werden konnte.
-
43.4 – Pilotanlagenmaßstabs-Polymerisationen
(Aufschlämmung)
-
Ein
kontinuierlicher 93-Liter-Phillips-Polymerisations-Schleifenreaktor
wurde für
die Polymerisationen verwendet. Ethylen, Isobutanverdünnungsmittel,
Wasserstoff und der in vorstehendem Beispiel 1.3 hergestellte Katalysator
wurden in den Reaktor dosiert, um die Reaktionsbedingungen, wie
in nachstehender Tabelle im Einzelnen angegeben, zu halten. Der
Reaktor wurde bei einem Polyethylendurchsatz von ungefähr 7,5 kg/Stunde
arbeiten lassen. Das Polymermolekulargewicht wurde durch Variation
der Wasserstoffzugabegeschwindigkeit gesteuert. Zwei Durchläufe wurden
ausgeführt;
einer ohne getrennt zugegebene Alkylaluminiumverbindung, wie in
Komponente (3) der vorliegenden Erfindung definiert, und einer mit
zugegebenem TIBA. Andere Bedingungen wurden gleichgehalten, mit
der Ausnahme der Wasserstoffzugabegeschwindigkeit, welche mit der
TIBA-Zugabe vermindert wurde, um ein ähnliches HLMI zu halten.
-
-
Die
vorstehende Tabelle zeigt deutlich die signifikante Wirkung auf
die Produktivität
von zugegebenem TIBA.
-
Beispiel 44 – Aufschlämmungspolymerisation,
unter Verwendung verschiedener Mengen MAO und TMA
-
Dieses
Beispiel zeigt die Wirkung auf die Aktivität von variierenden Mengen an
angewendetem Methylalumoxan (MAO) und Trimethylaluminium (TMA).
Das verwendete TMA wurde von Aldrich bezogen. Das MAO wurde von
Aldrich als eine 7-gewichtsprozentige Lösung bezogen und durch Entfernen
aller flüchtigen Stoffe
im Vakuum, und Waschen des zurückbleibenden
Feststoffs dreimal in Pentan, hergestellt.
-
Dies
wurde dann ein weiteres Mal im Vakuum getrocknet und unter Stickstoff
gelagert.
-
44.1 – Polymerisation von Ethylen,
unter Verwendung von 2,6-Diacetylpyridinbis(2,6-diisopropylanil)eisendichlorid
-
Eine
mit einem mechanischen Rührer,
innerer Kühlschleife
und Innendruck- und Temperaturmonitoren ausgestatte te Hochdruck-Glasflasche
wurde unter Vakuum 4 Stunden unter häufigem Spülen mit trockenem Stickstoff
bei 50°C
getrocknet. Ein doppelendiges 10-ml-Edelstahl-Einspritzrohr wurde
unter Stickstoff mit 5 ml Toluol und 1 μMol des vorstehend in Beispiel
1 hergestellten Eisenkatalysators beschickt. Ein zweites, ähnliches
Rohr wurde mit 5 ml Toluol und geeigneten Mengen MAO und TMA (siehe
nachstehende Tabelle für
die Mengen) beschickt.
-
Das
Toluol/MAO/TMA-Gemisch wurde unter einem Strom von 200 ml Toluol
unter 2 bar Stickstoffrückdruck
auf das Glasflaschen-Reaktorgefäß eingespritzt.
Der Reaktor wurde dann entlastet, mit 4 bar Ethylen unter Druck
gesetzt und bei 1000 U/min bei Raumtemperatur gerührt. Nach
15 Minuten wurde das Eisen-Katalysator/Toluol-Gemisch in den Reaktor
unter 4 bar Ethylendruck eingespritzt, der Reaktor entlastet, unter
Bereitstellen des Druckdifferenzials. Die Temperatur wurde 15 Minuten
bei 35–40°C gehalten
und Temperatur und Ethylenfließgeschwindigkeit
jede Minute aufgezeichnet. Nach 15 Minuten wurde die Reaktion durch
die Einspritzung von 10 ml Methanol gestoppt. Der Reaktor wurde
entlastet und weitere 200 ml Methanol zugegeben. Das Polymer wurde
durch Filtration gesammelt und über
Nacht an der Luft getrocknet. Der Versuch wurde einige Male, unter
variierenden Mengen MAO und TMA, bezogen auf die Menge an Eisenkomplex
(1 Äquivalent =
1 μMol),
wiederholt. Die nachstehende Tabelle zeigt die für jeden Versuch gemessenen
Aktivitäten.
-
Für jeden
Versuch gemessene Aktivitäten
(g/mMol·h·bar) Äquivalente
von MAO
- 1000 Äquivalente
MAO
- 58 mg
- 1000 Äquivalente
TMA
- 0,10 ml
-
44.2 – Polymerisation von Ethylen,
unter Verwendung von 2,6-Diacetylpyridinbis(2,6-dimethylanil)eisendichlorid
-
Versuch
44.1 wurde wiederholt, unter Verwendung des vorstehend in Beispiel
8 hergestellten Eisenkomplexes. Die Ergebnisse werden in nachstehender
Tabelle gezeigt.
-
Für jeden
Versuch gemessene Aktivitäten
(g/mMol·h·bar) Äquivalente
von MAO
- 1000 Äquivalente
MAO
- 58 mg
- 1000 Äquivalente
TMA
- 0,10 ml
-
Es
kann aus vorstehenden Tabellen ersichtlich werden, dass, auch wenn
die Gesamtmenge Aluminium (von MAO und TMA vereinigt) in dem System
gleich ist, die Aktivität
noch in Abhängigkeit
von der Al-Quelle variiert. Beispielsweise wird größere Aktivität mit 1000 Äquivalenten
TMA und 500 Äquivalenten
MAO als mit 1000 Äquivalenten
MAO und 500 Äquivalenten
TMA beobachtet.