-
Gebiet der Erfindung:
-
Die
vorliegende Erfindung bezieht sich auf ein Verfahren zur Herstellung
von 2,6-Dialkylnaphthalin (DAN) und insbesondere von 2,6-Dimethylnaphthalin
(2,6-DMN) aus einem Gemisch, das Alkylnapthalin oder Napthalin enthält.
-
Erläuterung des Hintergrunds:
-
Bei
der Herstellung von Hochleistungspolyesterharzen, wie Polyethylennaphthalatpolymer
(PEN) oder Polybutyrennapthalatpolymer (PBN) wird 2,6-DMN als ein
Präkursor
von 2,6-Naphthalindicarbonsäure
verwendet. Dies ist so, da 2,6-DMN leicht zu 2,6-Naphthalindicarbonsäure im Vergleich
zu anderen Präkursoren, wie
2,6-Diisopropylnapthalin oder 2-Methyl-6-isobutyrylnaphthalinen,
oxidiert wird. Es gibt viele Anwendungen für PEN, beispielsweise Filme
und Flaschen, wie Langzeitaufnahmevideofilme, Advanced Photo System, Heißfüllbehälter, Nachfüllflaschen
und Reifenprofile. PEN weist gute physikalische Eigenschaften hinsichtlich der
Festigkeit, Wärmebeständigkeit
und Gasbarriereeigenschaften auf. Typische PBN-Anwendungen umfassen
Elektronik, Isolatoren und Autoteile. PEN und PBN sind bisher jedoch
zu teuer gewesen, um ihren Markt aufgrund der begrenzten kommerziell
realisierbaren Verfahren zur Herstellung von 2,6-DMN effektiv zu
erweitern.
-
Es
hat viele Vorschläge
zur Herstellung von 2,6-DMN gegeben. US-Pat. Nr. 4,795,847 (Weitkamp
et al.) beschreibt ein Verfahren zur Herstellung von 2,6-Dialkylnapthalin
durch Alkylieren von Naphthalin oder 2-Alkylnapthalin mit einem
Alkylierungsmittel in Gegenwart eines Zeoliths (speziell ZSM-5)
als ein Katalysator.
-
US-Pat.
Nr. 5,001,295 (Angevine et al) beschreibt ein Verfahren zur Herstellung
von DMN unter Verwendung von 2-Monomethylnaphthalin (MMN) und Naphthalin
als ein Ausgangsmaterial und einem synthetischen Zeolith (MCM-22)
als ein Katalysator, und es zeigt, daß der MCM-22-Katalysator effektiver
als ZSM-5 bei der Alkylierung von 2-MMN und Naphthalin ist.
-
Jedoch
stellen die obigen Verfahren nur Einheitsoperation (d. h. diskontinuierlich)
zur Alkylierung von 2-MMN bereit, das ein teures Ausgangsmaterial
ist und nicht in großen
Mengen kommerziell erhältlich
ist.
-
US-Pat.
Nr. 4,990,717 (Sikkenga) und 5,073,670 (Sikkenga et al.) beschreibt
ein Mehrstufenverfahren, um 2,6-DMN aus o-Xylen und Butadien herzustellen,
bestehend aus:
- 1) Herstellung von 5-(o-Tolyl)-penten-2(OTP)
durch Alkenylierung von o-Xylen mit Butadien in Gegenwart eines
Katalysators, wie ein Alkalimetallkatalysator,
- 2) Herstellung von 1,5-Dimethyltetralin (1,5-DMT) durch Cyclisierung
von OTP in Gegenwart eines Katalysators, wie Platin und Kupfer,
auf einem ultrastabilen Zeolithkatalysator;
- 3) Herstellung von 1,5-Dimethylnaphthalin (1,5-DMN) durch Dehydrierung
von 1,5-DMT in Gegenwart eines Katalysators, wie Platin und Rhenium
und Gammaaluminiumoxid; und
- 4) Herstellung eines DMN-Gemisches, das reich an dem gewünschtem
2,6-DMN, 1,6-DMN und 1,5-DMN ist, durch Isomerisierung von 1,5-DMN
in Gegenwart eines Katalysators, wie einen beta-Zeolithkatalysator.
-
Wenn
ein Verfahren zum Abtrennen von 2,6-DMN von einem DMN-Gemisch mit
den obigen Schritten kombiniert wurde, konnte ein vollständiges Verfahren,
um gereinigtes 2,6-DMN herzustellen, bereitgestellt werden.
-
Da
mehrere Schritte eine Verfahrensanlage verkomplizieren und die Kosten
erhöhen,
ist es nicht klar, daß die
konventionellen Verfahren ein Verfahren bereitstellen könnten, das
für eine ökonomische
Herstellung von gereinigten 2,6-DMN geeignet ist.
-
Außerdem ist
es sehr schwierig, 2,6-DMN von anderen Isomeren durch konventionelle
Abtrennungsverfahren, wie Destillation und Kühlkristallisation, abzutrennen,
weil:
- 1) Es gibt sehr kleine Unterschiede in
den Siedepunkten von DMN-Isomeren und insbesondere zwischen 2,6-DMN
und 2,7-DMN, wobei der Unterschied in den Siedepunkten nur 0,3°C beträgt, und
es ist nahezu unmöglich,
2,6-DMN durch Destillation abzutrennen.
- 2) Die Kühlung
der DMN-Isomergemischlösung
der 2,6-DMN-Reinigung bildet einen Niederschlag von sehr feinen
2,6-DMN-Kristallen in Suspension, und daher ist die Abtrennung des
2,6-DMNs sehr schwierig.
-
Koide
et al,
US 4,992,619 ,
berichten über
ein Verfahren zur Abtrennung eines Methylderivats von Naphthalin
aus einem Gemisch aus Materialien in hoher Reinheit durch Kristallisation
unter Druck.
-
Moritoki
et al,
US 4,784,766 ,
berichten über
eine Druckkristallisationsvorrichtung.
-
Folglich
werden neue und effizientere Verfahren zur kommerziellen Herstellung
von Dialkylnaphthalinen gesucht.
-
EP 889016 ist eine Erfindung,
die sich auf ein Verfahren zur Herstellung von 2,6 Dialkylnaphthalin (DAN)
aus Naphthalin (NL) als Rohmaterial bezieht. Die Verfügbarkeit
von NL ist ziemlich begrenzt und NL ist teuer. Andererseits bezieht
sich diese Erfindung auf ein Verfahren zur Herstellung von 2,6-DAN
aus Alkylnaphthalinen als ein Gemisch aus NL, MMN (Monomethylnaphthalin),
DMN und PAN (Polyalkylnaphthalinen) usw., die besser erhältlicher
sind und deren Preis niedriger ist. Typischerweise ist dieser Alkylnaphthalinstrom
in geringwertigen Raffinerieströmen
(Typisches Beispiel ist leichtes Kreislauföl, hergestellt aus FFC) oder
Ethylenkrackböden
erhältlich.
Ihr typischer Preis ist ähnlich
dem Heizwert.
-
WO
99/19278, das Stand der Technik gemäß den Artikeln 54(3) und (4)
EPÜ ist,
offenbart ein Verfahren zur Herstellung eines 2,6-Dialkylnaphthalins,
umfassend die Schritte des Alkylierens von Monoalkylnaphthalin mit
einem Alkylierungsmittel in einer Alkylierungszone und Transalkylierens
von Naphthalin mit einem anderen Dialkylnaphthalin als 2,6-Dialkylnaphthalin
in einer Transalkylierungszone.
-
Zusammenfassung der Erfindung
-
Gemäß einem
Gegenstand der Erfindung wird ein Verfahren zur Herstellung von
2,6-Dialkylnaphthalin bereitgestellt.
-
Gemäß einem
anderen Gegenstand der Erfindung wird ein Verfahren zur Herstellung
von 2,6-Dimethylnaphthalin bereitgestellt.
-
Diese
und andere Gegenstände
der vorliegenden Erfindung werden durch ein Verfahren zur Herstellung
von 2,6-Dialkylnaphthalin aus einem Ausgangsmaterial, einschließlich Kohlenwasserstoffe,
das mindestens eine Komponente, ausgewählt aus der Gruppe, bestehend
aus Dialkylnaphthalinisomeren, Monoalkylnaphthalinisomeren, Polyalkylnaphthalinen
und Naphthalin, enthält,
möglich
gemacht, umfassend die folgenden Schritte:
- I.
das Abtrennen eines Produktes, beschickt von Schritt III, und gegebenenfalls
des Ausgangsmaterials in eine Naphthalin umfassende Fraktion, eine
Monoalkylnaphthalin umfassende Fraktion, eine Dialkylnaphthalin
umfassende Fraktion und eine Fraktion, umfassend verbleibende Produkte;
- II. das Abtrennen und Reinigen von 2,6-Dialkylnaphthalin aus
der Dialkylnaphthalinfraktion von Schritt I;
- III. das Dealkylieren des Ausgangsmaterials und der verbleibenden
Produktfraktion von Schritt I und das Beschicken des Dealkylierungsproduktes
zu Schritt I;
- IV. das Alkylieren der Naphthalin und Monoalkylnaphthalin umfassenden
Fraktionen von Schritt I,
wobei das Kohlenwasserstoff-Ausgangsmaterial
zu Schritt III und gegebenenfalls Schritt I beschickt wird.
-
Kurze Beschreibung
der Zeichnungen
-
1 zeigt
das bevorzugte Schema eines Verfahrens nach Anspruch 1.
-
2 zeigt
das bevorzugte Schema eines Vertahrens nach Anspruch 2.
-
3 zeigt
das bevorzugte Schema eines Verfahrens nach Anspruch 3.
-
4 zeigt
das bevorzugte Schema eines Verfahrens nach Anspruch 4.
-
5 zeigt
das bevorzugte Schema eines Verfahrens nach Anspruch 5.
-
6 zeigt
das bevorzugte Schema eines Vertahrens nach Anspruch 6.
-
7 zeigt
das bevorzugte Schema eines Verfahrens nach Anspruch 8.
-
8 zeigt
das bevorzugte Schema eines Verfahrens nach Anspruch 9.
-
9 zeigt
die Ergebnisse von Beispiel 7.
-
10 zeigt
die Ergebnisse von Beispiel 12.
-
11 zeigt
die Ergebnisse von Beispiel 13.
-
Beschreibung
der bevorzugten Ausführungsformen
-
Eine
vollständigere
Einschätzung
der Erfindung und viele der damit verbundenen Vorteile werden ohne
weiteres erhalten, da diese in bezug auf die folgende ausführliche
Beschreibung besser verstanden werden, wenn sie zusammen mit den
beiliegenden Zeichnungen betrachtet werden, wobei keine Einschränkung beabsichtigt
ist, sofern nichts anderes angegeben.
-
Vorzugsweise
kann die vorliegende Erfindung auf jedes Ausgangsmaterial von Kohlenwasserstoffen, das
Alkylnaphthaline, wie Naphthalin, MMN (Monomethylnaphthalin) und
DMN-Isomere, enthält,
angewendet werden. Die vorliegende Erfindung stellt ein effektives
Herstellungsvertahren von 2,6-Dialkylnaphthalin (DAN), insbesondere
2,6-Dimethylnaphthalin (DMN), als hochwertiges zusätzliches
Produkt unter Verwendung eines nicht-wertvollen Ausgangsmaterials
bereit.
-
Insbesondere
ist LCO (leichtes Kreislauföl)
und/oder seine Herzschnittfraktion FCC (Flüssigkatalysatorkracken) oder
HC (Hydrokracker) ein bevorzugtes Beispiel eines Ausgangsmaterials.
Obwohl dieses Ausgangsmaterial Alkylnaphthaline in etwa 20 bis 45
Gew. % des Beschickungsstroms enthält, weist es die folgenden
Probleme und Schwierigkeiten zum weiteren Verarbeiten auf:
-
1) Co-siedende Verbindungen
-
Beispielsweise
enthält
LCO normalerweise viele Komponenten, wie leichte Paraffine und monoaromatische
Verbindungen mit langkettigen Alkylgruppen, die ähnliche Siedepunkte wie Naphthalin,
MMN und DMN aufweisen (Co-siedende Verbindungen). Es ist sehr schwer,
Alkylnaphthaline von ihren co-siedenden Verbindungen durch Destillation
allein zum weiteren Verarbeiten, wie einem Alkylierungsschritt,
abzutrennen.
-
Deshalb
verringern die co-siedenden Verbindungen, wenn unbehandelt, die
Wirksamkeit der Reaktoren. In dem schlechtesten Fall sammeln sich
die co-siedenden Verbindungen in den Rückführungsströmen in dem Verfahren an.
-
2) Polyalkylnaphthaline
(PAN)
-
PANs,
wie Tri-methylnaphthaline, Isopropylnaphthaline und Tetra-methylnaphthaline,
sind normalerweise in dem Beschickungsstrom bei etwa 10 bis 20 Gew.-%
des Beschickungsstroms enthalten; d. h. fast ein Drittel bis eine
Hälfte
der Alkylnaphthalingehalte. PAN wird ebenso bei anderen Verfahrensschritten,
wie Alkylierung und Isomerisierung, als Nebenprodukte hergestellt.
In der konventionellen Technik ist kein wirksames Verfahren, um
PANs zur Herstellung von 2,6-Dialkylnaphthalinen zu verwenden, vorgeschlagen
worden.
-
3) Schwefel- und Stickstoffverbindungen
-
Der
Beschickungsstrom enthält
Schwefel- und Stickstoffverbindungen, die den Katalysator zur Alkylierung
und Isomerisierung vergiften könnten,
und diese Verbindungen sollten aus den Rückführungsströmen und Produkten ausgeschlossen
werden.
-
Die
Erfinder fanden heraus, daß die
Dealkylierung (DA), insbesondere Hydrodealkylierung (HDA), (i) nicht
nur zum Kracken und/oder Reformieren der co-siedenden Verbindungen
zu leichteren Fraktionen wirksam ist, was zur leichteren Abtrennung
von Naphthalinen, Monoalkylnaphthalinen und Dialkylnaphthalinen führt; sondern auch
(ii) zur Herstellung von 2,6-Dialkylnaphthalin, insbesondere 2,6-DMN,
mittels DA, insbesondere HDA, von Polyalkylnaphthalinen (PAN), wie
Tri-methylnaphthaline und schwerere Alkylnaphthaline, die in dem
Ausgangsmaterial enthalten sind und ebenso bei dem Alkylierungsschritt
als unbrauchbares Nebenprodukt hergestellt werden, überraschend
wirksam ist; und (iii) zur Herstellung von Naphthalin und Monoalkylnaphthalin,
das zur Alkylierung des Ausgangsmaterials geeignet ist, effektiv
ist.
-
Die
Entdeckung führt
zur effektiveren Herstellung von 2,6-DMN nicht nur bei dem Alkylierungsschritt, sondern
auch in dem gesamten Verfahren, da die Erfindung eine wirksame Nutzung
von PAN bereitstellt.
-
Obwohl
eine hohe Ausgangsmaterialumwandlung (beispielsweise Umwandlung
von MMN) von rund 50 bis 60 % eine höhere Ausbeute von 2,6-DMN bereitstellt,
erhöht
sich die PAN-Herstellung ebenso, wie in der Tabelle 4 der konventionellen
Verfahren, US-Pat. Nr. 5,744,670 (Motoyuki et al), gezeigt. (Der
DMN- und PAN-Gehalt nach der Alkylierung beträgt 35 % bzw. 23 % bei der MMN-Umwandlung
mit 58,28 %). Dies verursacht Naphthalin-Ringverlust und geringere
Verfahrensausbeuten, wenn PAN nicht erneut in dem rückgeführten Strom
verwendet wird. Um daher den Verlust zu minimieren, wird die MMN-Umwandlung
auf rund 30 % oder so begrenzt, was sich die 2,6-DMN-Ausbeute verringert.
-
Da
die vorliegende Erfindung die effektive Nutzung (eine zusätzliche
Herstellung von 2,6-Dialkylnaphthalin aus DA von PAN) von PAN, das
aus dem Ausgangsmaterial und vorzugsweise aus dem Ausgangsmaterial
und dem Alkylierungsschritt stammt, bereitstellen kann, ermöglicht es
höhere
Ausgangsmaterialumwandlung bei dem Alkylierungsschritt von rund
50 % oder größer, was
ebenso zur höheren
Produktion von 2,6-Dialkylnaphthalin führt. Außerdem fanden die Erfinder
heraus, daß 2,6-armes Dialkylnaphthalin
als die verbleibenden Produkte der Reinigung von 2,6-Dialkylnaphthalin
dealkyliert und auf Gleichgewichtsverteilung von Dialkylnaphthalinisomeren
oder 2,6-reichem Dialkylnaphthalin durch gleichzeitiges Herstellen
von Naphthalin und Monoalkylnaphthalin verändert werden kann.
-
Obwohl
angenommen wird, daß der
Reaktionsmechanismus von DA von 2,6-Dialkylnaphthalin komplett unterschiedlich
ist, sind die Ergebnisse und die Produktzusammensetzung von DA der
Transalkylierung und Isomerisierung ähnlich. Dies bedeutet, daß DA die
Leistung der Transalkylierung, die in den konventionellen Verfahren,
beispielsweise US-Pat. Nr. 5,744,670, gezeigt wird, effektiv übernehmen
kann.
-
Die
vorliegende Erfindung stellt ein effektives Herstellungsverfahren
für 2,6-Dialkylnaphthalin
als ein hochwertiges zusätzliches
Produkt unter Verwendung eines nicht-wertvollen Ausgangsmaterials
bereit.
-
Als
ein bevorzugtes Ausgangsmaterial für das vorliegende Verfahren
kann irgendein Kohlenwasserstoffbeschickungsstrom, enthaltend Alkylnaphthaline,
einschließlich
mindestens eine Komponente, ausgewählt aus der Gruppe, bestehend
aus Dialkylnaphthalinisomeren, Monoalkylnaphthalinisomeren, Polyalkylnaphthalinen
und Naphthalin, wie leichtes Kreislauföl (LCO), das aus katalytisch
gekracktem Erdöl
stammt, verwendet werden. Alternativ wird als Rohmaterial der Kohlenwasserstoff
beschickungsstrom, wie LCO, vorverarbeitet, und danach wird sein
Produkt vorzugsweise als ein Ausgangsmaterial für das vorliegende Verfahren
verwendet. Das Vorverarbeiten umfaßt vorzugsweise Destillation
(beispielsweise Herzschnitt), Konzentration, Hydrotreating (HDT),
um Schwefel- und Stickstoffverbindungen zu reformieren, die normalerweise
in Beschickungsströmen
enthalten sind, und die den Katalysator vergiften könnten, Entschwefelung,
Denitrogenierung und Entwässerung.
-
Bei
der Destillation und Konzentration als Vorverarbeitung wird es bevorzugt,
leichte Komponenten, wie monoaromatische Verbindungen und nicht-aromatische
leichte Paraffine, und schwere Verbindungen, wie triaromatische
und schwerere Verbindungen, von den Alkylnaphthalinkomponenten abzutrennen.
-
Die
bevorzugten Bedingungen von HDT umfassen eine Temperatur von etwa
200 bis 1.000°C,
und stärker
bevorzugt 200 bis 500°C,
und einen Druck von 0 bis 250 atm (25,3 MPa) und vorzugsweise 5
bis 50 atm (0,5066 bis 5,0663 MPa), eine Wasserstoffumwälzgeschwindigkeit
von etwa 500 bis 3000 scf/bbl (89,05 bis 534,3 m3/m3). Die Reaktion wird vorzugsweise unter
Verwendung einer Beschikkungsraumgeschwindigkeit von etwa 0,1 bis
10,0 h-1 erreicht.
-
Ein
bevorzugtes Beispiel eines geeigneten Katalysators für HDT ist
ein aktivierter Aluminiumoxid-getragener Katalysator, der ein Metalloxid
der Gruppe VIII und ein Metalloxid der Gruppe VI-A, vorzugsweise
Nickel bzw. Molybdän,
umfaßt.
Das Oxid kann vorzugsweise bei 600 bis 1200°F (315,6 bis 648,9°C) in Gegenwart
von Schwefelverbindungen behandelt werden.
-
Das
Ausgangsmaterial kann zu der Abtrennung von Schritt I sowie zu der
Dealkylierung von Schritt III beschickt werden. In dem Fall, daß das Ausgangsmaterial
zu Schritt I beschickt wird, kann das Abtrennen leichter Komponenten
und schwerer Verbindungen von Alkylnaphthalinkomponenten in Schritt
I durchgeführt werden.
Andererseits werden bei dem Ausgangsmaterial, das zu Schritt III
beschickt wird, PAN-Komponenten in dem Ausgangsmaterial verringert
und zu DMN- oder MMN-Komponenten
vor der Abtrennung von Schritt I verändert. In beiden Fällen wird
die effektive Herstellung erhalten, da die verbleibenden Produkte
von Schritt I zu Schritt III beschickt werden, und das Produkt von
Schritt III wird zu Schritt I beschickt, wie es aus 1 hervorgeht.
-
Für die Abtrennung
von Schritt 1 können
konventionelle Techniken wie Destillation verwendet werden. In dem
Fall, wo der Beschickungsstrom nicht-aromatische Komponenten mit
Siedepunkten, die dem Naphthalin und/oder MMN sehr ähnlich sind,
enthält,
können
ebenso konventionelle Lösungsmittelextraktionstechniken zusätzlich zu
der obengenannten Destillation in Schritt I angewendet werden.
-
Die
Abtrennung und Reinigung von Schritt II reinigt das 2,6-DAN und
trennt das 2,6-arme-DAN
von der DAN-Fraktion von Schritt I ab, wie aus 1 hervorgeht.
-
Die
Abtrennung und Reinigung von 2,6-Dialkylnaphthalin von Schritt II
kann durch konventionelle Verfahren, die dem Fachmann bekannt sind,
wie Kühlkristallisation
und/oder Adsorption, durchgeführt
werden. Beispielsweise kann die Abtrennung und Reinigung unter Verwendung
eines Verfahrens zur Kristallisation unter hohem Druck ausgeführt werden.
Im allgemeinen wird ein flüssiges
Gemisch, das zwei oder mehrere Substanzen enthält, unter Druck gesetzt, und
eine bestimmte Substanz in dem Gemisch wird verfestigt und aus der
restlichen Flüssigkeit
durch die Wirkung des Drucks abgetrennt. Mit anderen Worten umfaßt dieses
Verfahren eine Abtrennungs- und
Reinigungstechnik, wobei ein flüssiges
Gemisch, das zwei oder mehrere Substanzen enthält, in einen festverschlossenen
Druckbehälter
gegeben wird, ein Teil der gewünschten
Substanz, 2,6-Dialkylnaphthalin, verfestigt wird, um einen Fest-Flüssig-co-existierenden
Zustand zu bilden, die Flüssigkeit
aus dem co-existierenden System während des Aufrechterhaltens
des Drucks des Fest-Flüssig-co-existierenden
Systems bei einem höheren
Niveau als der Gleichgewichtsdruck der Zielsubstanz abgelassen wird, dann
der Feststoff, der in dem Behälter
verbleibt, zum Ablassen der restlichen Flüssigkeit zwischen die Feststoffteilchen
gepreßt
wird und die Feststoffteilchen integriert werden. Diese Technik
wird im allgemeinen in
US 5,220,098 beschrieben.
-
Das
Verfahren umfaßt
das Einspritzen der Aufschlämmung
oder Flüssigkeit
der Temperatur von 70 bis 120°C,
vorzugsweise 80 bis 100°C,
in einen Hochdruckbehälter
zum Durchführen
einer Kristallisation unter hohem Druck; das adiabatische unter
Druck setzen des Behälters
auf einen Druck von 300 bis 4000 kgf/cm2 (29,42
bis 392,3 MPa), vorzugsweise 500 bis 2000 kgf/cm2 (49,03
bis 196,1 MPa), um die Menge zu erhöhen, d. h. die Menge der 2,6-Dialkylnaphthalinkristalle,
wobei die Coexistenz von Flüssig-Fest-Phasen
bei den hohen Druckbedingungen existiert; das Ablassen der Flüssigphasenkomponente
aus dem Hochdruckbehälter, wobei
das Ablassen unter Druck durchgeführt wird, um das Verhältnis der
Festphase in bezug auf die Flüssigphase
innerhalb des Behälters
zu erhöhen;
das Verringern des Drucks der restlichen Flüssigphase, um so das Produkt
teilweise zu lösen
und zu reinigen; Ablassen der restlichen flüssigen Phase durch Ausüben von
Druck auf die Festphase innerhalb des Hochdruckbehälters, wobei
ein 2,6-Dialkylnaphthalinkristallblock mit einer hohen Reinheit
innerhalb des Hochdruckbehälters
erhalten wird. Durch diese Technik kann eine Reinheit von 2,6-Dialkylnaphthalin
(beispielsweise 2,6-Dimethylnaphthalin) von 98 Gew.-%, vorzugsweise
99 Gew.-%, erhalten werden.
-
Bei
der Abtrennung und Reinigung von Schritt II kann vor der Kühlkristallisation
und/oder Kristallisation unter hohem Druck 2,6-Dialkylnaphthalin
aus dem Dialkylnaphthalingemisch durch das System der adsorptiven
Trennung im Festbett vorkonzentriert werden. Wie für die Vorkonzentration
von 2,6-DMN ist es bevorzugt, daß die Adsorption ein Adsorptionsmittel
eines Zeoliths Y, enthaltend ein Alkalimetall, und ein Desorptionsmittel
eines organischen Lösungsmittels,
das hauptsächlich
aus mindestens einer Komponente, ausgewählt aus der Gruppe, bestehend
aus Hexan, Octan, Alkylbenzen und Cyclohexan, besteht, umfaßt. Als
Alkylbenzen werden Mesitylen, o-Xylen und n-Xylen bevorzugt.
-
Bei
der Kühlkristallisation
in dem Abtrennungs- und Reinigungsschritt wird, da 2,6-DMN und 2,7-DMN einen
eutektischen Kristall bei dem Gewichtsverhältnis von 0,7 (=2,6-DMN/2,7-DMN)
bilden, nur eine geringe Ausbeute von 2,6-DMN erreicht. Die theoretische
Abtrennungsausbeute von 2,6-DMN wird durch die folgenden Gleichungen
angegeben:
Ausbeute (%) = (1 – 0,7/k) × 100, wo k = 2,6-DMN/2,7-DMN
bei der Beschickung des Kühlungskristallisators.
-
Daher
ist es insbesondere bevorzugt, das Verhältnis von 2,6-DMN/2,7-DMN für die höhere Ausbeute von
2,6-DMN zu erhöhen.
Die Festbettadsorption kann das Verhältnis von 1,0 bei der Beschickung
zu 2,0 und mehr beim Ablauf erhöhen,
was zu höheren
Abtrennungsausbeuten und geringeren internen Rückführungsmengen des gesamten Verfahrens
führt.
-
Vorzugsweise
kann für
effektivere Produktion die Abtrennung und Reinigung von Schritt
II in den Abtrennungsabschnitt Schritt II-1 und den Reinigungsabschnitt
Schritt II-2 unterteilt werden, wie es aus 5 hervorgeht.
In Schritt II-1 wird die DAN-Fraktion von Schritt I in 2,6-reiches
DAN und 2,6-armes DAN geteilt, und in Schritt II-2 wird 2,6-DAN
aus der 2,6-reichen DAN-Fraktion von Schritt II-1 gereinigt.
-
Beispielsweise
kann die Abtrennung von Schritt II-1 vorzugsweise unter Verwendung
der Destillation durchgeführt
werden, und die Reinigung von Schritt II-2 kann unter Verwendung
der Kühlkristallisation und/oder
Kristallisation unter hohem Druck durchgeführt werden. Durch ein derartiges
System wird das 2,6-arme DAN, das wenig 2,7-DAN enthält, durch
Schritt II-1 abgetrennt, und das 2,6-arme DAN, das viel 2,7-DAN enthält, wird
durch Schritt II-2 als ein verbleibendes Produkt der Reinigung abgetrennt.
-
Die
Bedingungen von HDA von Schritt III umfassen eine Temperatur von
etwa 200 bis 1.000°C
und vorzugsweise 300 bis 700°C,
und einen Druck von 0 bis 250 atm (25,3 MPa) und vorzugsweise 5
bis 150 atm (0,5066 bis 15,1988 MPa), eine Wasserstoffumwälzgeschwindigkeit
von etwa 500 bis 3000 scf/bbl (89,05 bis 534,3 m3/m3). Die Reaktion wird unter Verwendung einer
Beschickungsraumgeschwindigkeit von etwa 0,1 bis 10,0 h-1 geeignet
erreicht.
-
Ein
Beispiel eines geeigneten Katalysators für HDA ist ein aktivierter Aluminiumoxidgetragener
Katalysator, der ein Oxid eines Metalls der Gruppe VIII, vorzugsweise
Chrom, umfaßt.
-
Ein
anderes Beispiel eines geeigneten Katalysators für HDA ist ein aktivierter Aluminiumoxid-getragener
Katalysator, der ein Oxid eines Metalls der Gruppe VIII und eines
Metalls der Gruppe VI, vorzugsweise Cobalt und Molybdän, umfaßt. Das
Oxid kann vorzugsweise bei einer Temperatur von 600 bis 1000°F (315,6
bis 537,8°C)
in Gegenwart eines organischen Sulfids vorbehandelt werden.
-
Andere
bevorzugte Katalysatoren für
HDA umfassen Katalysatoren, einschließlich ein Metall, ausgewählt aus
der Gruppe, bestehend aus Edelmetall, Nickel und Kombinationen davon,
und einen synthetischen Zeolith, gekennzeichnet durch ein Röntgenbeugungsmuster
einschließlich
einem interplanaren d-Abstand und relativer Intensität I/I
o × 100
wie folgt:
| 12,36 ± 0,4 | M-VS |
| 11,03 ± 0,2 | M-S |
| 8,83 ± 0,14 | M-VS |
| 6,18 ± 0,12 | M-VS |
| 6,00 ± 0,10 | W-M |
| 4,06 ± 0,07 | W-S |
| 3,91 ± 0,07 | M-VS |
| 3,42 ± 0,06 | VS. |
-
Das
bevorzugte Edelmetall wird aus der Gruppe, bestehend aus Platin,
Palladium und Kombinationen davon, ausgewählt.
-
Die
Bedingungen der Alkylierung von Schritt IV umfassen vorzugsweise
eine Temperatur von etwa 0 bis 500°C, und vorzugsweise 240 bis
450°C, und
einen Druck zwischen 0 und 250 atm (25,3 MPa) und vorzugsweise 1
atm bis 50 atm (0,101 bis 5,066 MPa). Das Molverhältnis des
Alkylierungsmittels zu der Beschickung von Monoalkylnaphthalin oder
Naphthalin kann etwa 20 : 1 bis 1 : 20, vorzugsweise 10 : 1 bis
1 : 10 betragen. Die Reaktion wird unter Verwendung einer Beschickungsraumgeschwindigkeit
von etwa 0,1 bis 10,0 h-1 erreicht.
-
Bevorzugte
Alkylierungsmittel umfassen Alkohole, Olefine, Aldehyde, Halogenide
und Ether. Beispielsweise werden Methanol, Dimethylether und Polyalkylbenzen
bevorzugt. Methanol und Dimethylether werden besonders bevorzugt.
-
Ein
geeigneter Katalysator zur Alkylierung ist ein synthetischer Zeolith,
gekennzeichnet durch ein Röntgenbeugungsmuster
einschließlich
einem interplanaren d-Abstand und relativer Intensität I/I
o × 100
wie folgt:
| 12,36 ± 0,4 | M-VS |
| 11,03 ± 0,2 | M-S |
| 8,83 ± 0,14 | M-VS |
| 6,18 ± 0,12 | M-VS |
| 6,00 ± 0,10 | W-M |
| 4,
06 ± 0,
07 | W-S |
| 3,91 ± 0,07 | M-VS |
| 3,42 ± 0,06 | VS. |
-
Ein
geeigneter Katalysator ist MCM-22 (Exxon Mobil Chemical Company)
mit dem obigen Röntgenbeugungsmuster.
-
Vorzugsweise
kann die Alkylierung in jedem der bekannten Reaktoren, die normalerweise
für die
Alkylierung eingesetzt werden, durchgeführt werden. Beispielsweise
kann ein Rohrreaktor mit einem Abwärtsstrom von Reaktanten über ein
Festbett des Katalysators eingesetzt werden.
-
Um
die hohe Ausgangsmaterialumwandlung aufrechtzuerhalten, kann die
Einspritzung von Methanol in den Reaktor vorzugsweise in mehreren
Schritten und stärker
bevorzugt in zwei Schritten durchgeführt werden. Beispielsweise
werden ein Reaktor mit Ober- und Mittel-Methanolbeschickung oder
zwei Reaktoren nacheinander mit Ober- und Zwischen-Methanolbeschickung
vorzugsweise verwendet.
-
In
einer bevorzugten Ausführungsform
kann die 2,6-arme DAN-Fraktion aus Schritt II-1 den Isomerisierungsbedingungen
unterzogen werden, um eine Dialkylnaphthalinfraktion bereitzustellen,
die einen größeren Gehalt
an 2,6-Dialkylnaphthalin aufweist, wie aus 6 hervorgeht.
Vorzugsweise kann das Produkt der Isomerisierung zu Schritt I und/oder
Schritt II-2 für
effizientere Rückführung beschickt
werden.
-
Bevorzugte
Isomerisierungsbedingungen werden im allgemeinen in der coanhängigen Anmeldung US-Anmeldung
Serien-Nr. 08/661,114 offenbart und sind für die gleichzeitige Durchführung der
Transalkylierung von Dialkylnaphthalin und Naphthalin und Isomerisierung
von Dialkylnaphthalinen geeignet.
-
Ein
bevorzugter Katalysator zur Isomerisierung ist ein synthetischer
Zeolith, gekennzeichnet durch ein Röntgenbeugungsmuster einschließlich interplanarem
d-Abstand und relativer Intensität
I/I
o × 100
wie folgt:
| 12,36 ± 0,4 | M-VS |
| 11,03 ± 0,2 | M-S |
| 8,83 ± 0,14 | M-VS |
| 6,18 ± 0,12 | M-VS |
| 6,00 ± 0,10 | W-M |
| 4,
06 ± 0,
07 | W-S |
| 3,
91 ± 0,
07 | M-VS |
| 3,42 ± 0,06 | VS. |
-
Ein
geeigneter Katalysator ist MCM-22 (Exxon Mobil Chemical Company)
mit dem obigen Röntgenbeugungsmuster.
-
Vorzugsweise
wird die Isomerisierung bei einer gewichtsstündlichen Raumgeschwindigkeit
(WHSV) von Dialkylnaphthalinen von 0,1 bis 10, vorzugsweise 0,5
bis 5 h-1, stärker bevorzugt 0,75 bis 1,5
h-1, durchgeführt.
-
Vorzugsweise
wird die Isomerisierung bei einer Temperatur von 100 bis 500°C, vorzugsweise
150 bis 350°C,
stärker
bevorzugt 200 bis 300°C,
durchgeführt.
-
Vorzugsweise
wird die Isomerisierung bei einem Druck von atmosphärisch bis
100 kgf/cm2 (9,807 MPa), atmosphärisch bis
vorzugsweise 30 kgf/cm2 (2,942 MPa), durchgeführt.
-
Während der
Isomerisierung ist es gegebenenfalls bevorzugt, Wasserstoff in einer
Menge von 0,1 bis 10 Mol-H2/Mol-Kohlenwasserstoffe
co-einzuspeisen.
-
Gemäß der bevorzugten
Ausführungsform
der 2 oder 3 kann 2,6-Dialkylnaphthalin
aus den Kohlenwasserstoff-Ausgangsmaterialien wie folgt hergestellt
werden:
- I. das Abtrennen eines Produktes, beschickt
von Schritt III, und gegebenenfalls des Kohlenwasserstoff-Ausgangsmaterials
in eine Naphthalin umfassende Fraktion, eine Monoalkylnaphthalin
umfassende Fraktion, eine Dialkylnaphthalin umfassende Fraktion
und eine Fraktion, umfassend verbleibende Produkte;
- II. das Abtrennen und Reinigen von 2,6-Dialkylnaphthalin aus
der Dialkylnaphthalinfraktion von Schritt I;
- IIIa. das Dealkylieren einer Dialkylnaphthalinfraktion, nachdem
2,6-Dialkylnaphthalin davon abgetrennt wurde, in Schritt II und
Rückführen eines
Dealkylierungsproduktes zu Schritt I;
- IIIb. das Dealkylieren des Ausgangsmaterials und der Fraktion,
die verbleibende Produkte enthält,
von Schritt I und das Beschicken des Dealkylierungsproduktes zu
Schritt I;
- IV. das Alkylieren der Naphthalin und Monoalkylnaphthalin umfassenden
Fraktionen von Schritt I.
-
In
diesem Verfahren wird 2,6-armes DAN als verbleibendes Produkt der
Abtrennung/Reinigung von Schritt II dealkyliert und zur Abtrennung
von Schritt I beschickt. So können
2,6-DAN-Isomere in 2,6-armen DAN zu MMN oder NL verändert werden
und können
in Schritt IV alkyliert werden.
-
Was
die bevorzugte Ausführungsform
von 4 angeht, wird das Produkt der Alkylierung von
Schritt IV zur Abtrennung von Schritt I beschickt. Folglich kann
PAN, das in Schritt IV hergestellt wurde, in Schritt I abgetrennt
und dealkyliert in Schritt III beschickt werden. Deshalb ermöglicht es,
die effektive Nutzung von PAN bereitzustellen, und ermöglicht höhere Ausgangsmaterialumwandlung
bei dem Alkylierungsschritt, wie bereits beschrieben.
-
Das
Verfahrensschema von 8 ist eine am stärksten bevorzugte
Ausführungsform
der vorliegenden Erfindung.
-
Beispiele
-
Mit
dieser im allgemeinen beschriebenen Erfindung kann ein weiteres
Verständnis
in bezug auf bestimmte spezielle Beispiele erhalten werden, die
hierin nur zum Zweck der Erläuterung
bereitgestellt werden, und keine Einschränkung beabsichtigen, sofern
nichts anderes angegeben.
-
Beispiel 1: Alkylierung
von MMN und Naphthalin:
-
Eine
Menge von 153 g des MCM-22-Katalysators wird in einen Rohreaktor
(Volumen: 370 cm3) eingespeist. Als ein
Ausgangsmaterial zur Alkylierung werden 1-MMN, 2-MMN und Naphthalin
verwendet und bei einem Molverhältnis
von 2,2 von 2-MMN/1-MMN und einem Gewichtsverhältnis von 3,0 von MMNs (1-MMN
+ 2-MMN) / Naphthalin gemischt.
-
Daraufhin
wird das Ausgangsmaterial dem Reaktor (254°C, 5 kgf/cm2 (0,490
MPa)) bei einer Geschwindigkeit von 153,4 g/h und 1,0 h-1 hinsichtlich
der WHSV mit einer Beschickung von Wasserstoff bei einer Geschwindigkeit
von 1,8 ft3/h (0,051 m3/h)
zugeführt.
Vier Stunden später
wird Methanol als ein Alkylierungsmittel in den Reaktor bei 25,5
g/h eingebracht und die Alkylierung wird für 20 Stunden durchgeführt. Das
erhaltene Produkt wird durch Gaschromatographie analysiert und die
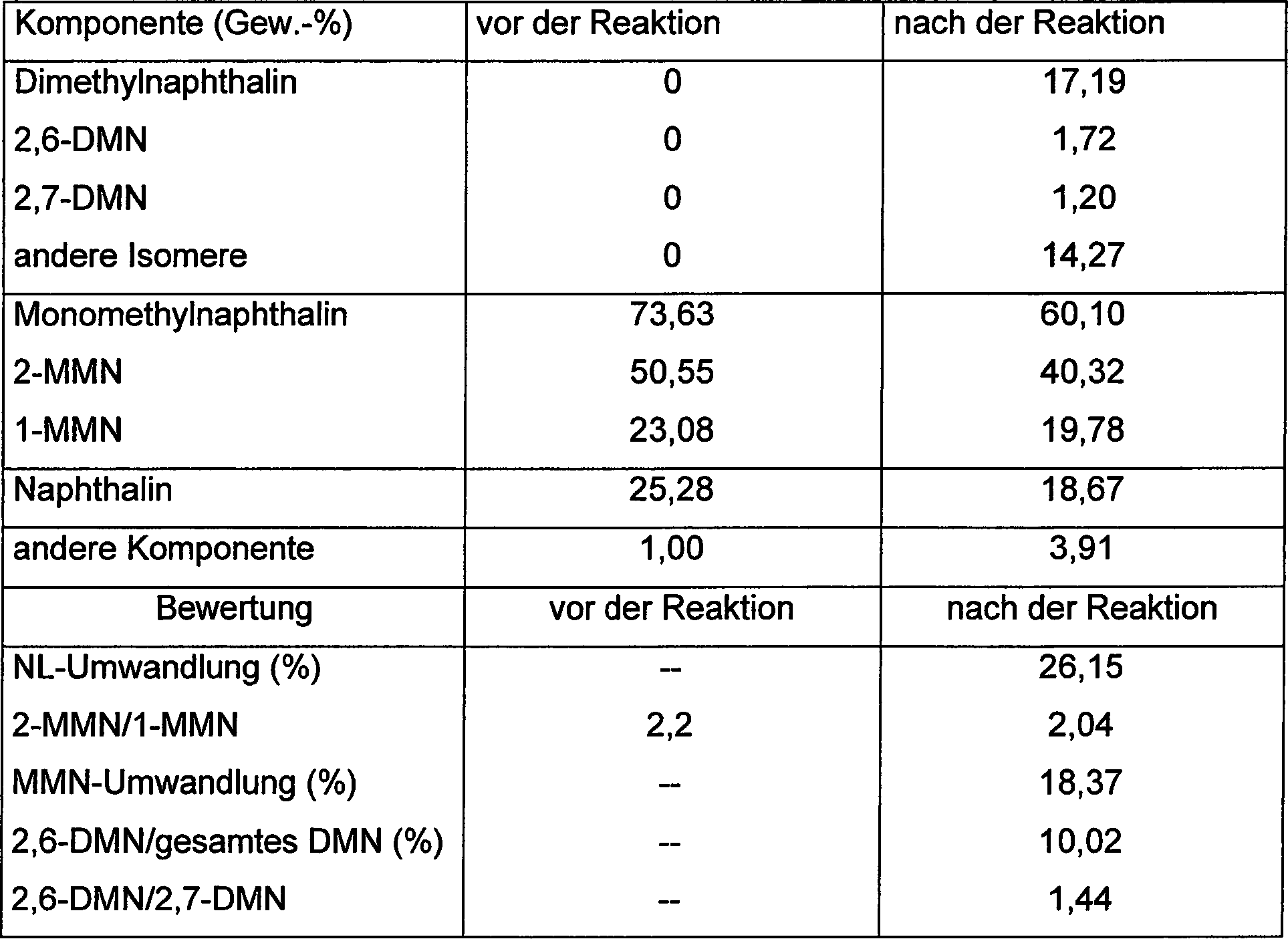
Ergebnisse werden in Tabelle 1 zusammengefaßt.
-
Tabelle
1 (Alkylierung von Monomethylnaphthalin und Naphthalin)
-
Wie
aus Tabelle 1 hervorgeht, liegt das Verhältnis von 2,6-DMN/2,7-DMN über 1,1
und das Verhältnis von
2-MMN/1-MMN liegt über
2,0.
-
Beispiel 2 (Alkylierung)
-
153
g MCM-22 wurden in den Rohrreaktor (Volumen: 370 cm
3)
eingespeist. Als ein Ausgangsmaterial zur Alkylierung wurden 1-MMN
(Reinheit 95,5 %) und 2-MMN (Reinheit 96,6 %) verwendet und bei
dem Molverhältnis
von 2,2 von 2-MMN/1-MMN gemischt. Das Ausgangsmaterial wurde dem
Reaktor (350°C)
bei der Geschwindigkeit von 76,7 g/h und 0,5 h
-1 WHSV
4 Stunden zugeführt.
Danach wurde mit dem Zuführen
von Methanol in den Reaktor bei der Geschwindigkeit von 17,3 g/h
begonnen und die Reaktion wurde für 20 Stunden fortgesetzt. Das
erhaltene Produkt wurde durch Gaschromatographie analysiert und
das Ergebnis wird in Tabelle 2 zusammengefaßt. Tabelle
2 (Alkylierung)
-
Wie
aus Tabelle 2 hervorgeht, liegt das Verhältnis von 2,6-DMN/2,7-DMN über 1,1
und das Verhältnis von
2-MMN/1-MMN liegt über
2,0.
-
Beispiel 3 (Alkylierung
und Destillation)
-
Die
Alkylierung von MMN und Naphthalin ist für mehrere Monate in derselben
Weise, die in Beispiel 1 beschrieben wurde, durchgeführt worden,
und etwa 400 kg des Pro duktes werden gesammelt. Die Destillation des
Produktes wird unter Verwendung eines Batchdestillationsturmes mit
einer gepackten Säule
durchgeführt. Es
wird erwartet, daß die
Anzahl an theoretischen Böden
des Turms mindestens 50 beträgt.
Der Betriebsdruck an der Spitze der Säule wird zwischen 15 und 36
Torr (2 kPa und 4,8 kPa) kontrolliert und die Destillation bei einem
Rückflußverhältnis von
50 bis 75 fortgesetzt.
-
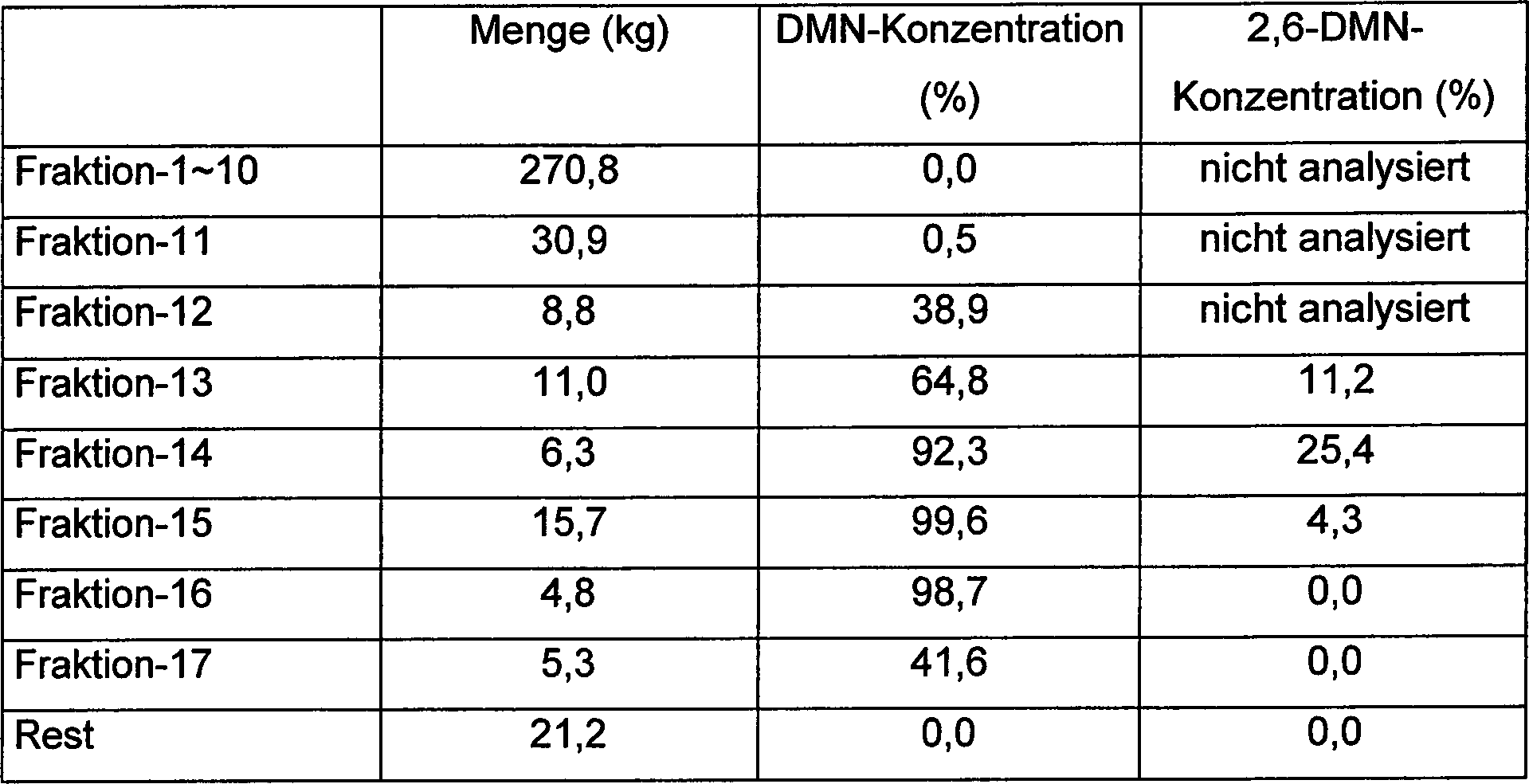
Das
Produkt wird in 17 Fraktionen durch die Unterschiede in den Siedepunkten
geteilt, wie in Tabelle 3 gezeigt. Tabelle
3 (Alkylierung und Destillation)
-
Beispiel 4 (Hydrodealkylierung)
-
Ein
Teil der Fraktion-17 und dem Rest, die in Tabelle 3 gezeigt werden,
werden gemischt, um das Ausgangsmaterial (Blend-A) zur Hydrodealkylierung
herzustellen. Eine Menge von 50 g des Cr2O3/Al2O3-Katalysators,
hergestellt von Süd-Chemie
AG, wird in einen Rohrreaktor eingespeist. Der Reaktor wird allmählich von Umgebungstemperatur
auf 662°F
(350°C)
erwärmt,
um den Katalysator während
des Zuführens
von Wasserstoffgas zu trocknen. Daraufhin wird das Blend-A zu dem
Reaktor bei der Geschwindigkeit von 50 g/h und 1,0 h-1 WHSV
während
des Zuführens
von Wasser stoffgas bei 1,2 scf/h (0,034 m3/h)
beschickt. Die Hydrodealkylierung wird bei 887°F (475°C) und 854 psig (5,99 MPa) durchgeführt. Das
Produkt wird durch GC analysiert und die Ergebnisse der Hydrodealkylierung
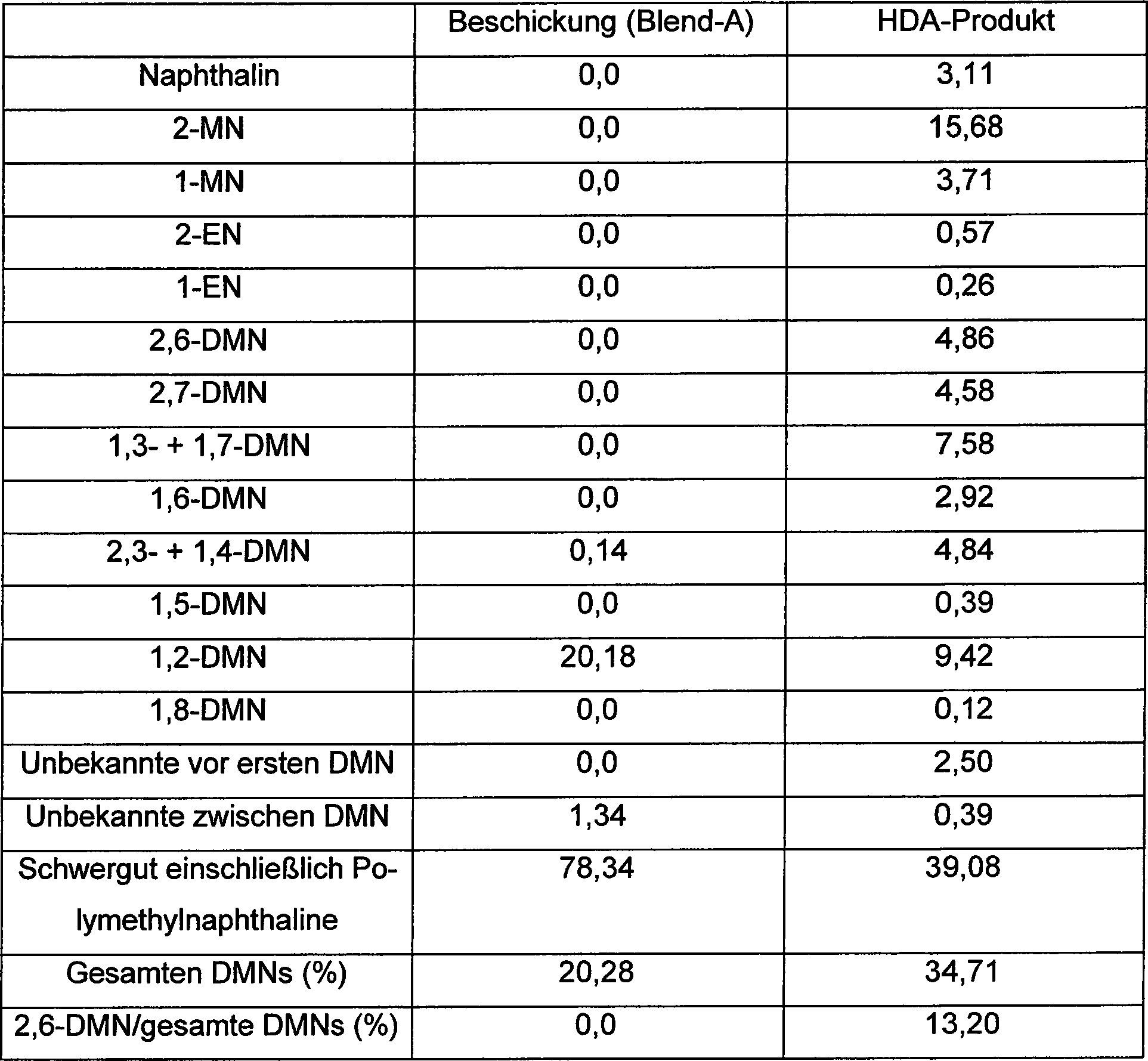
werden in der nachstehenden Tabelle 4 zusammengefaßt.
-
Wie
in Tabelle 4 gezeigt, ist der Cr
2O
3/Al
2O
3-Katalysator
dahingehend effektiv, 2,6-DMN aus der 2,6-DMN-armen Beschickung
anzureichern. Tabelle
4 (Hydrodealkvlierung)
-
Beispiel 5: Isomerisierung
-
Eine
Menge von 25 g des MCM-22-Katalysators wird in den Rohrreaktor (Volumen:
200 cm
3) eingespeist. Der Reaktor wird allmählich von
Umgebungstemperatur auf 400°C
erwärmt,
um den Katalysator während
des Zuführens
von Stickstoffgas zu trocknen, und der Strom an Stickstoffgas wird
eingestellt, wenn die Temperatur bei 400°C stabil wird. Daraufhin wird
2,6-armes DMN dem Reaktor bei der Geschwindigkeit von 25 g/h und
1,0 h
-1 WHSV zugeführt und die Isomerisierung
von DMN wird für
vier Stunden durchgeführt.
Die Gehalte des erhaltenen Produktes werden durch Gaschromatographie
analysiert und die Ergebnisse werden in Tabelle 5 zusammengefaßt. Tabelle
5 (Isomerisierung)
-
Beispiel 6 Abtrennung
und Reinigung:
-
(1) Kristallisation unter
Hochdruckkristallisation
-
Eine
Menge von 1.505 g der DMN-Isomere wird dem Hochdruckkristallisator
(KOBELCO 1,5L-Typ) zugeführt
und 236 g 2,6-DMN-Kristalle (Reinheit 87 %) werden unter der Bedingung
von 2000 kgf/cm2 (196,1 MPa) und 45°C abgetrennt.
-
(2) Kühlkristallisation
-
Unter
Verwendung eines Behälters
zur Kristallisation (3 Liter) werden 2.001 g DMN-Isomere schnell von 50 auf 40°C unter langsamem
Rühren
abgekühlt.
Dann werden 0,5 g Keimkristalle in den Behälter eingespeist, der bei einer
Temperatur bei 40°C
für eine
Stunde gehalten wurde. Daraufhin wird das Ausgangsmaterial auf 10°C bei 2°C/min abgekühlt. Eine
Menge von 360 g 2,6-DMN-Kristallen (Reinheit 68 %) wird durch Filtration
unter Druck abgetrennt.
-
Die
Ergebnisse der Abtrennung durch sowohl Kristallisation unter hohem
Druck als auch Kühlkristallisation
werden in Tabelle 6 zusammengefaßt.
-
Tabelle
6 (Abtrennung und Reinigung)
-
„Rückführung von
2,6-DMN" bedeutet
der Gehalt an 2,6-DMN in den Kristallen gegenüber dem Gehalt an 2,6-DMN in
dem Ausgangsmaterial.
-
„Ausbeute
von 2,6-DMN" bedeutet
der Gehalt an 2,6-DMN in dem Kristall gegenüber dem Gesamtgewicht des Ausgangsmaterials.
-
Wie
in Tabelle 6 gezeigt, ist die Ausbeute von 2,6-DMN durch Kristallisation
unter hohem Druck viel höher
als durch Kühlkristallisation.
Außerdem
beträgt
das Verhältnis
von 2,6-DMN/gesamt-DMN des Filtrats durch Kristallisation unter
hohem Druck weniger als 8 %. Deshalb ist das Filtrat effektiver
als ein Ausgangsmaterial zur Transalkylierung und Isomerisierung
von 2,6-armen DMN.
-
Wenn
außerdem
ein Versuch gemacht wird, die Reinheit von Kristallen durch Kühlkristallisation
zu erhöhen,
verringert sich die Ausbeute von 2,6-DMN drastisch.
-
Beispiel 7 (Reinigung)
-
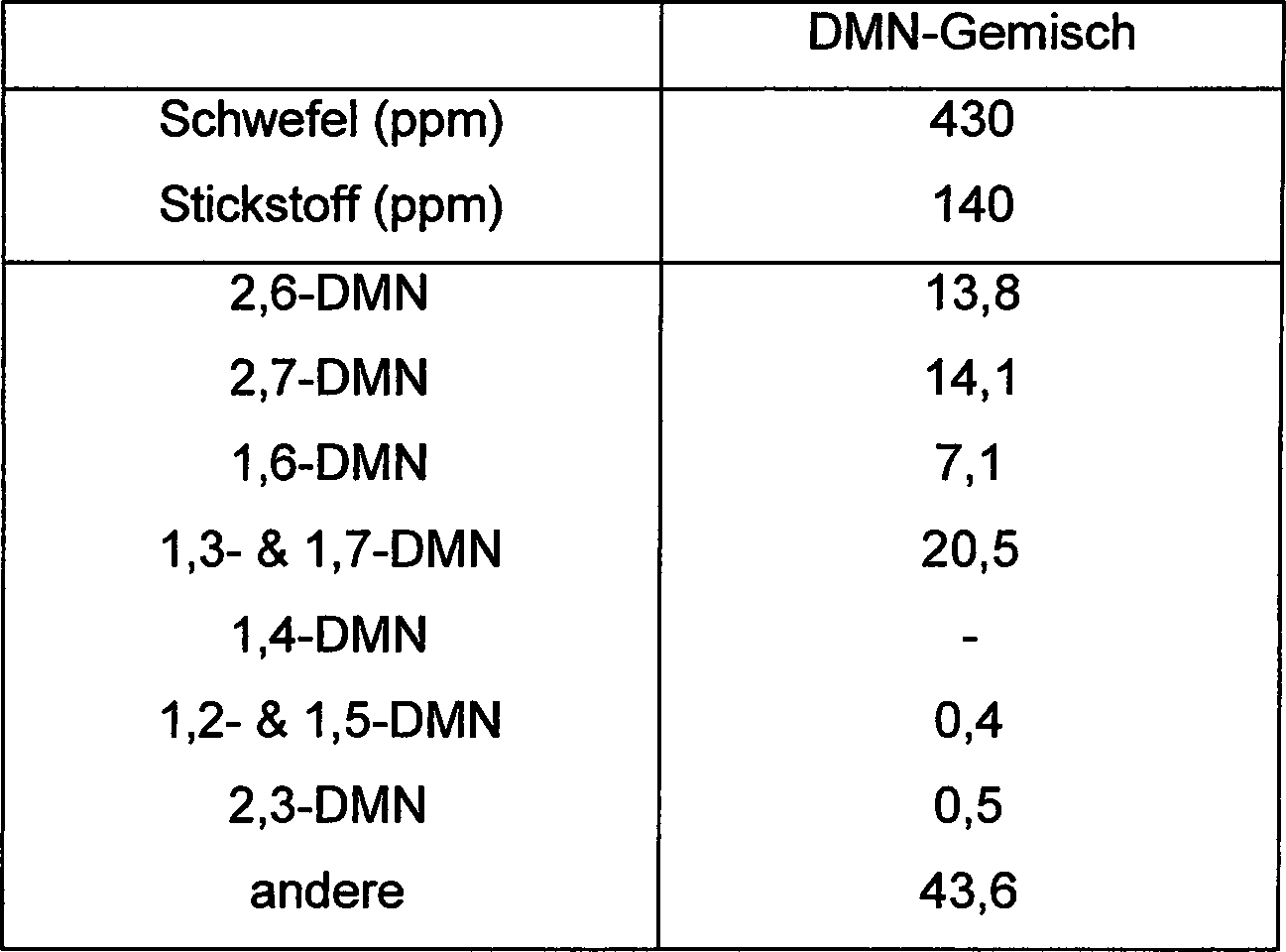
Die
Vorkondensation von 2,6-DMN aus dem DMN-Gemisch (Tabelle 7) wurde
durch Kühlkristallisation versucht,
und ein 2,6-DMN-reicher Kuchen, der als Ausgangsmaterial für die Kristallisation
unter hohem Druck verwendet werden soll, wurde durch Labordruckfiltrationseinheit
abgetrennt.
-
Die
Reinigung von 2,6-DMN aus dem 2,6-DMN-reichen Kuchen wurde durch
das Kristallisationsverfahren unter hohem Druck unter Verwendung
der HPC-Testmaschine von Kobelco durchgeführt.
-
Mehrere
Reihen von Experimenten wurden durchgeführt und die Ergebnisse werden
in 9 zusammengefaßt.
-
Wie
aus 9 hervorgeht, erreicht die Kristallisation unter
hohem Druck effektivere Reinigungsleistung in der Abtrennungsausbeute
und 2,6-DMN-Reinheit durch einstufige Kristallisation, als es die
zweistufige Kühlkristallisation
tut.
-
Tabelle
7 (Zusammensetzung des DMN-Gemisches)
-
Beispiel 8 (Destillation)
-
Zwei
Typen des Batchdestillationsturms werden zur Abtrennung von Alkylnaphthalinen
von LCO verwendet. Einer der Destillationstürme (Fractioneer-A) weist eine
167 l (0,167 m3) Destillieranlage und 32
Fuß (9,75
m) lange Säule
mit PRO-PAK (Scientific Development Company) auf und der andere
Destillationsturm (Fractioneer-B) weist eine 27-Liter-Destillieranlage
mit einer 11 Fuß langen
Säule mit
PRO-PAK auf. 164
kg LCO werden in den Fractioneer-A eingespeist und die Destillation
wird bei einem Rückflußverhältnis von
50 und einem Druck von 60 Torr (8,0 kPa) durchgeführt. 80,5
Liter werden als eine Entnahmerate von 0,7 Litern pro Stunde entnommen.
-
Dann
werden 25 kg des Rests in der Destillieranlage von Fractioneer-A
nach der ersten Destillation entnommen und in den Fractioneer-B
eingespeist. Eine andere Batchdestillation wird bei einem Rückflußverhältnis von
50 und einem Druck von 50 Torr (6,67 kPa) durchgeführt. 14
Liter werden bei einer Entnahmerate von 125 ml pro Stunde entnommen.
Die Komponenten des Produktes (Blend-B), die aus der obengenannten Zweistufendestillation
erhalten wurden, werden in Tabelle 8 gezeigt.
-
Beispiel 9 (Hydrodealkylierung)
-
Eine
Menge von 70 g des Cr2O3/Al2O3-Katalysators,
hergestellt von Süd-Chemie
AG, wird in einen Rohrreaktor eingespeist. Der Reaktor wird allmählich von
Umgebungstemperatur auf 932°F
(500°C)
erwärmt, um
den Katalysator während
des Zuführens
von Wasserstoffgas zu trocknen. Daraufhin wird das Destillationsprodukt
(Blend-B), das aus Beispiel 8 erhalten wurde, dem Reaktor bei der
Geschwindigkeit von 70 g/h und 1,0 h-1 WHSV
während
des Zuführens
von Wasserstoffgas bei 0,98 scf/h (0,028 m3/h)
zugeführt.
Die Hydrodealkylierungsreaktion wird bei 933°F (500,6°C) und 1138 psig (7,95 MPa)
durchgeführt.
Das Produkt wird durch GC analysiert und die Ergebnisse der Hydrodealkylierung
werden in Tabelle 8 zusammengefaßt.
-
Beispiel 10 (Hydrodealkylierung)
-
Eine
Menge von 70 g des CoO/MoO3/Al2O3-Katalysators, hergestellt von Akzo Chemicals
Inc, wird in einen Rohrreaktor eingespeist. Der Reaktor wird allmählich von
Umgebungstemperatur auf 300°F
(148,9°C) mit
einem Stickstoffstrom bei 5 scf/h (0,142 m3/h)
erwärmt.
Dann wird das Fließgas
auf Wasserstoff bei 2 scf/h (0,057 m3/h)
umgeschaltet und der Druck wird auf 500 psig (3,55 MPa) erhöht. Der
Katalysator wird mit einem organischen Sulfid (Kerosin mit 1,0 %
Dimethylsulfid) zum Sulfatisieren während des Zuführens von
Wasserstoffgas in Kontakt gebracht und dann wird die Temperatur
auf 650°F
(343,3°C)
erhöht.
Daraufhin wird das Destillationsprodukt (Blend-B), das aus Beispiel
8 erhalten wurde, zu dem Reaktor bei der Geschwindigkeit von 70
g/h und 1,0 h-1 WHSV während des Zuführens von
Wasserstoffgas bei 5 scf/h (0,028 m3/h)
beschickt. Die Hydrodealkylierung wird bei 932°F (500°C) und 1425 psig (9,93 MPa)
durchgeführt.
Das Produkt wird durch GC analysiert und die Ergebnisse der Hydrodealkylierung
werden in der nachstehenden Tabelle 8 zusammengefaßt.
-
Wie
in Tabelle 8 gezeigt, sind sowohl der Cr2O3/Al2O3-
als auch der CoO/MoO3/Al2O3-Katalysator dahingehend effektiv, DMN-Isomere
aus DMN-armer Beschickung anzureichern.
-
Tabelle
8 (Hydrodealkylierung)
-
Beispiel 11 (Destillation
und Hydrotreating)
-
164
kg LCO werden in den Fractioneer-A eingespeist und die Destillation
wird bei einem Rückflußverhältnis von
50 und einem Druck von 60 Torr (8 kPa) durchgeführt. 120 Liter werden bei einer
Entnahmerate von 0,7 Litern pro Stunde entnommen und zum Hydrotreating
des Ausgangsmaterials als Blend-C hergestellt. NiO/MoO3/Al2O3- Katalysator, hergestellt
von Akzo Chemicals Inc., wird als ein Hydrotreating-Katalysator
ausgewählt
und in den Rohrreaktor eingespeist.
-
Nach
dem Trocknen und Sulfatisieren des Katalysators wird dann Blend-C
zu dem Reaktor bei der Geschwindigkeit von 0,43 h-1 WHSV
beschickt und Hydrotreating wird bei 400 psig (2,86 MPa) und 726°F (385,6°C) während des
Zuführens
von Wasserstoffgas bei 3495 scf/bbl (622,5 m3/m3) durchgeführt. Die Ergebnisse des Hydrotreatings
werden in der nachstehenden Tabelle 9 zusammengefaßt.
-
Wie
in Tabelle 9 gezeigt, ist der NiO/MoO3/Al2O3-Katalysator dahingehend
effektiv, Stickstoff- und/oder Schwefelverbindungen in LCO mit minimalem
Verlust von DMN-Isomeren
zu verringern.
-
Tabelle
9 (Destillation und Hydrotreating)
-
Beispiel 12 (Adsorption)
-
2,6-DMN
und 2,7-DMN werden gemischt bzw. in iso-Octan bei 2,0 Gew.-% Konzentration
gelöst.
Dann wird die DMN-Isooctan-Lösung
zu der Adsorptionssäule
(4,6 mm ID und 500 mm L), gepackt mit K-Y-Zeolith, bei der Geschwindigkeit
von 0,50 ml/min beschickt, während
die Säulentemperatur
bei 158°F
(70°C) kontrolliert
wird. Die Daten des zeitlichen Verlaufs der DMN-Konzentration in
dem Ablauf werden durch GC-Analyse gesammelt und es wird eine Durchbruchskurve
in dem Adsorptionsschritt erhalten.
-
Nach
dem Adsorptionsschritt wird die Flüssigbeschickung zu reinem iso-Octan
umgeschaltet und der Ablauf wird ebenso analysiert, um die Daten
des zeitlichen Verlaufs der DMN-Konzentration in dem Ablauf zu sammeln.
-
Die
Ergebnisse der Durchbruchskurve und Desorptionskurve werden in 10 zusammengefaßt. Wie in
der Figur gezeigt, wird 2,7-DMN im Vergleich zu 2,6-DMN vorher in
Zeolith adsorbiert und es ist offensichtlich möglich, 2,6-/2,7-DMN durch Kontaktieren
der DMN-Isomere mit dem K-Y-Zeolith zu verbessern.
-
Beispiel 13
-
Ein
anderer Adsorptionstest wird in derselben Weise, die in Beispiel
12 beschrieben wird, durchgeführt,
außer
daß Mesitylen
als ein Lösungsmittel
verwendet wird. Die Ergebnisse der Durchbruchskurve und der Desorptionskurve
werden in 11 zusammengefaßt.
-
Offensichtlich
sind zahlreiche Modifikationen und Veränderungen der vorliegenden
Erfindung im Licht der obigen Lehren möglich. Es wird daher verstanden,
daß innerhalb
des Umfangs der beiliegenden Ansprüche die Erfindung anders als
hierin speziell beschrieben praktiziert werden kann.