-
Technisches
Gebiet der Erfindung
-
Die
Erfindung betrifft Dentalkleberzusammensetzungen zum Binden von
Dental-Restaurierungsmitteln an Dentin und/oder Zahnschmelz. Insbesondere
stellt die Erfindung eine selbstätzende
und selbstgrundierende Dental-Einkomponenten-Kleberzusammensetzung
bereit, die hydrolysestabile polymerisierbare saure Klebermonomere
umfasst.
-
Hintergrund der Erfindung
-
Omura
et al. beschreiben in
US 4 539
382 Zweikomponenten-Kleber. Moszner et al. beschreiben
in
CA 2250333 (
DE 197 46 708 und
EP 0 909 761 ) hydrolysestabile
Monomer. Loehden et al. Beschreibe in in
DE 199 18 974 polymerisierbare Phosphonsäureester.
Haberland beschreibt in
DD 273
846 polymerisierbare Phosphonsäureamide. Selbstätzende selbstgrundierende
Dental-Zweikomponenten-Kleber systeme werden entweder hinter einander
oder in einer Stufe nach Mischen der zwei Komponenten angewendet.
Beide Verfahren haben aufgrund der klinischen Komplikationen, die
zwischen den auf einander folgenden Stufen (Speichel- oder Blutkontamination)
auftreten können,
oder aufgrund von Dosierproblemen, wenn vor der Anwendung des selbstätzenden
Klebers ein Mischen erforderlich ist, inhärente Nachteile. Um diese klinischen
Probleme zu überwinden,
wäre es
von Vorteil, selbstätzende
Kleber als Einkomponenten-System bereitzustellen, um dadurch die
Notwendigkeit einer hinter einander erfolgenden Anwendung oder eines
Vormischens zu eliminieren.
-
US 6 174 935 beschreibt
einen Dentalkleber-Kit, der ein polymerisierbares N-substituiertes
Acrylsäureamidmonomer
mit einer Sulfonsäure-Komponente
enthält.
EP 1 057 468 A1 beschreibt
eine Kleberzusammensetzung, die ein eine Säuregruppe enthaltendes polymerisierbares
Monomer enthält.
WO 02/02057 beschreibt ein Dentalmaterial, das Phosphonsäuren enthält.
-
Beschreibung der Erfindung
-
Die
vorliegende Erfindung betrifft einen hydrolysestabilen selbstätzenden
und selbstgrundierenden Einkomponenten-Dentalkleber, wie er in Anspruch
1 definiert ist.
-
Um
den Effekt und die Adhäsion
zu erhöhen,
kann im hydrolysestabilen selbstätzenden
und selbstgrundierenden Dental-Einkomponenten-Kleber eine organische
und/oder anorganische Säure
zugegeben werden, wie z.B. Methacrylsäure, Acrylsäure, Fumarsäure, Maleinsäure, Citronensäure und
Itaconsäure.
Bevorzugte organische wasserlösliche
Lösungsmittel
können
ausgewählt
werden aus der Gruppe von Alkoholen und Ketonen, wie z.B. Ethanol,
Propanol, Butanol, Aceton und Methylethylketon.
-
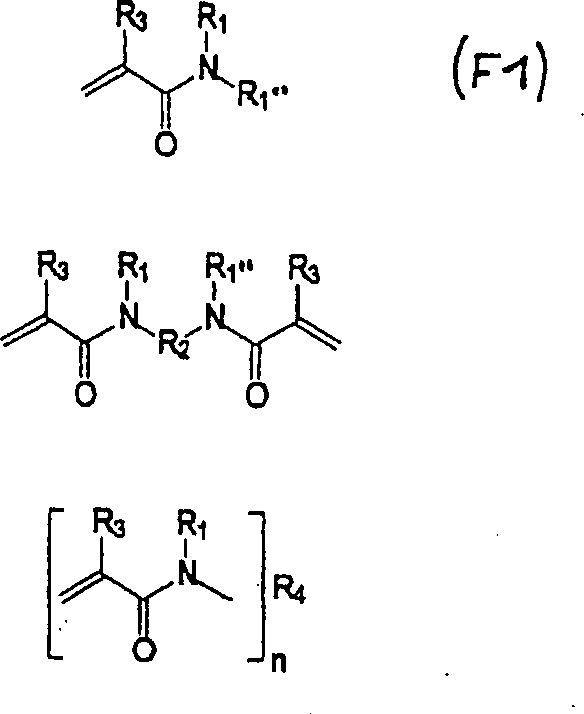
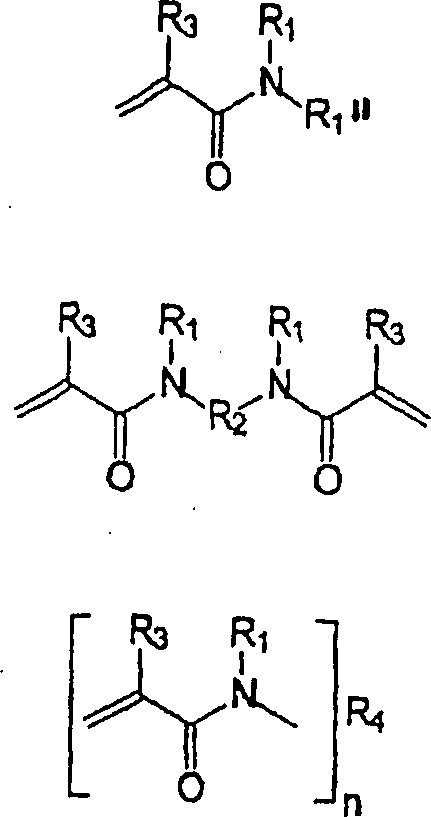
Bevorzugte
polymerisierbare (Meth)acrylamide, die mindestens eine Phosphon-
oder Sulfonsäure-Komponente aufweisen,
zur Verwendung in der hydrolysestabilen selbstätzenden selbstgrundierenden Dental-Einkomponenten-Kleberzusammensetzung
liegen innerhalb des Rahmens der folgenden Formeln.
worin
bedeuten:
R
1 und R
2 unabhängig von
einander Wasserstoff oder ein substituiertes oder unsubstituiertes
C
1-C
18-Alkylen, substituiertes
oder unsubstituiertes Cycloalkylen, substituiertes oder unsubstituiertes
C
5-C
18-Arylen oder -Heteroarylen,
substituiertes oder unsubstituiertes C
5-C
18-Alkylarylen oder -Alkylheteroarylen, substituiertes
oder unsubstituiertes C
7-C
30-Alkylenarylen,
R
3 und R
4 unabhängig von
einander difunktionelles substituiertes oder unsubstituiertes C
1-C
18-Alkylen, difunktionelles
substituiertes oder unsubstituiertes Cycloalkylen, difunktionelles
substituiertes oder unsubstituiertes C
5-C
18-Arylen oder -Heteroarylen, difunktionelles
substituiertes oder unsubstituiertes C
5-C
18-Alkylarylen
oder -Alkylheteroarylen, difunktionelles substituiertes oder unsubstituiertes
C
7-C
30-Alkylenarylen;
n
eine ganze Zahl.
-
Bevorzugte
(Meth)acrylamide zur Verwendung in der hydrolysestabilen selbstätzenden
selbstgrundierenden Dental-Einkomponenten-Kleberzusammensetzung
der Erfindung umfassen Bis- und Mono(meth)acrylamide innerhalb des
Rahmens der folgenden Formeln:
-
Die
erfindungsgemäße hydrolysestabile
selbstätzende
selbstgrundierende Dental-Einkomponenten-Kleberzusammensetzung umfasst vorzugsweise
polymerisierbare hydrolysestabile Monomere innerhalb des Rahmens
der folgenden Formeln:
worin bedeuten:
R
1 und R
3 unabhängig von
einander H oder ein substituiertes oder unsubstituiertes C
1-C
18-Alkylen, substituiertes
oder unsubstituiertes Cycloalkylen, substituieres oder unsubstituiertes
C
5-C
18-Arylen oder
-Heteroarylen, substituiertes oder unsubstituiertes C
5-C
18-Alkylarylen oder -Alkylheteroarylen, substituiertes
oder unsubstituiertes C
7-C
30-Alkylenarylen,
R
2 ein difunktionelles substituiertes oder
unsubstituiertes C
1-C
18-Alkylen,
difunktionelles substituiertes oder unsubstituiertes Cycloalkylen,
difunktionelles substituiertes oder unsubstituiertes C
5-C
18-Arylen oder -Heteroarylen, difunktionelles
substituiertes oder unsubstituiertes C
5-C
18-Alkylarylen oder -Alkylheteroarylen, difunktionelles
substituiertes oder unsubstituiertes C
7-C
30-Alkylenarylen;
R
4 ein
mono- oder polyfunktionelles substituiertes oder unsubstituiertes
C
1-C
18-Alkylen,
mono- oder polyfunktionelles substituiertes oder unsubstituertes
Cycloalkylen, mono- oder polyfunktionelles substituiertes oder unsubstituiertes
C
5-C
18-Arylen oder
-Heteroarylen, mono- oder polyfunktionelles substituiertes oder
unsubstituiertes C
5-C
18-Alkylarylen
oder -Alkylheteroarylen, mono- oder polyfunktionelles substituiertes
oder unsubstituiertes C
7-C
30-Alkylenarylen;
n
eine ganze Zahl.
-
Die
Zusammensetzungen der Erfindung umfassen vorzugsweise mindestens
ein Bis- oder Poly(meth)acrylamid, ein polymerisierbares Monoacrylamid,
einen Initiator, einen Stabilisator, Wasser und/oder ein organisches
Lösungsmittel.
Der Polymerisationsinitiator ist vorzugsweise ein thermischer Initiator,
ein Redox-Initiator oder ein Photoinitiator, und vorzugsweise wird
Campherchinon verwendet. Zur Stabilisierung der Dentalzusammensetzung
kann ein Stabilisator enthalten sein, der Radikale absor biert, wie
z.B. Hydrochinonmonomethylether, 2,6-Di-tert-butyl-p-cresol, Tetramethylpiperidin-N-oxyl-Radikal, Galvanoxyl-Radikal.
-
Vorzugsweise
enthält
der erfindungsgemäße hydrolysestabile
selbstätzende
selbstgrundierende Dental-Einkomponenten-Kleber
5 bis 95 Gew.-% eines hydrolysestabilen polymerisierbaren Monomers
und 0,01 bis 30 Gew.-% einer organischen und/oder anorganischen
Säure.
-
Beste erfindungsgemäße Ausführungsart
-
Die
vorliegende Erfindung stellt eine wässerige selbstätzende und
selbstgrundierende Dental-Einkomponenten-Kleberzusammensetzung
bereit, die einen pH von höchstens
2 aufweist, und wie in Anspruch 1 definiert.
-
Eine
Einkomponenten-Zusammensetzung bedeutet, dass die erfindungsgemäße Zusammensetzung nur
in einem Behälter
enthalten ist, der gelagert werden kann, und ermöglicht die Anwendung der Zusammensetzung
ohne Mischen und ohne spezielle Vorrichtung vor der Anwendung.
-
Selbstätzend bedeutet,
dass die erfindungsgemäße Dentalkleberzusammensetzung
ohne vorhergehendes Ätzen
von Zahnschmelz in einer getrennten Verfahrensstufe auf einem Zahn
appliziert werden kann. Um ein solches selbstätzendes Merkmal zu umfassen,
ist die erfindungsgemäße Zusammensetzung
wässerig und
weist einen pH-Wert von höchstens
2 auf. Vorzugsweise ist der pH-Wert unter 2, insbesondere ist der pH-Wert
unter 1,5, und in erster Linie ist der pH-Wert ca. 1. Das Ätzen von
Zahnschmelz wird somit auf vorteilhafte Weise mit der erfindungsgemäßen Einkomponenten-Zusammensetzung
erreicht. Es ermöglicht
eine Adhäsion
eines aus der Dentalzusammensetzung hergestellten Klebers auf Zahnschmelz
und/oder Dentin mit einer Bindungsfestigkeit von mindestens 8 MPa,
vorzugsweise mindestens 10 MPa. Selbstgrundierend bedeutet, dass
die erfindungsgemäße Dentalkleberzusammensetzung
ohne vorhergehende Anwendung eines Primers an einem Zahn applizier
werden kann.
-
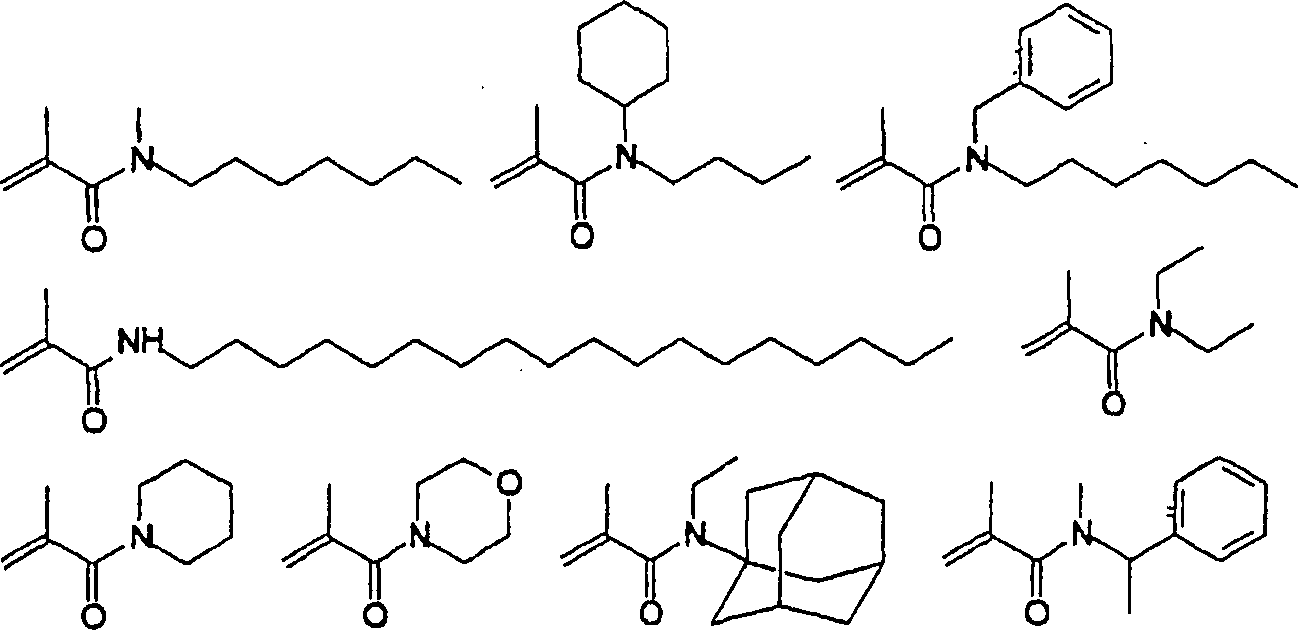
Das
polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidmouomer,
das gegebenenfalls eine anorganische Säurekomponente ausgewählt aus
einer Phosplionsäure-Komponente
oder einer Sulfonsäure-Komponente
enthält,
weist vorzugsweise eine der folgenden Strukturen auf:
worin bedeuten:
R
1, R
1'' und
R
3 unabhängig
von einander Wasserstoff oder eine substituierte oder unsubstituierte
C
1-C
18-Alkylgruppe, eine
substituierte oder unsubstituierte Cycloalkylgruppe, eine substituierte
oder unsubstituierte C
5-C
18-Aryl-
oder -Heteroarylgruppe, eine substituierte oder unsubstituierte
C
5-C
18-Alkylaryl-
oder -Alkylheteroarylgruppe, eine substituierte oder unsubstituierte
C
7-C
30-Aralkylgruppe,
mit
der Maßgabe,
dass, wenn einer der Reste R
1 und R
1'' in Formel
(F1) Wasserstoff ist, der andere nicht Wasserstoff ist;
R
2 ein difunktionelles substituiertes oder
unsubstituiertes C
1-C
18-Alkylen,
difunktionelles substituiertes oder unsubstituiertes Cycloalkylen,
eine difunktionelle substituierte oder unsubstituiere C
5-C
18-Aryl- oder
-Heteroarylgruppe, eine difunktionelle substituierte oder unsubstituierte
C
5-C
18-Alkylaryl-
oder Alkylheteroarylgruppe, eine difunktionelle substituierte oder
unsubstituierte C
7-C
30-Aralkylgruppe,
R
4 eine mono- oder polyfunktionelle substituierte
oder unsubstituierte C
1-C
18-Kohlenstoffkettengruppe,
eine mono- oder polyfunktionelle substituierte oder unsubstituierte
Cycloalkylgruppe, eine mono- oder polyfunktionelle substituierte
oder unsubstituierte C
5-C
18-Aryl-
oder -Heteroarylgruppe, eine mono- oder polyfunktionelle substituierte
oder unsubstituierte C
5-C
18-Alkylaryl-
oder Alkylheteroarylgruppe, eine mono- oder polyfunktionelle substituierte
oder unsubstituierte C
7-C
30-Aralkylgruppe,
und
n eine ganze Zahl, vorzugsweise von 1 bis 10, insbesondere
3 bis 4.
-
Die
optionalen Substituenten an den durch R1,
R1'', R2, R3 und R4 repräsentierten
Gruppen sind vorzugsweise ausgewählt
aus C1-C18-Alkylgruppen.
Besonders bevorzugt sind C1-C6-Alkylgruppen,
und Methylgruppen sind besonders bevorzugt.
-
Bevorzugt
ist es, dass R2 und R4 unabhängig von
einander eine di- oder polyfunktionelle substituierte oder unsubstituierte
C1-C18-Kohlenstoffkettengruppe
oder eine di- oder polyfunktionelle substituierte oder unsubstituierte
Cycloalklengruppe ist, die mindestens eine Ether-, Thioether-, Ester-,
Thiocarbonyl-, Amid-, Carbonyl-, Sulfonyl-, Urethan-Bindung oder
substituierte oder unsubstituierte Amin-Bindungen aufweist.
-
Eine
C
1-C
18-Kohlenstoffkettengruppe
bedeutet, dass ein verzweigter oder geradkettiger Kohlenwasserstoff
mindestens zwei Bindungen oder Valenzen aufweist und 1 bis 18 Kohlenstoffatome.
Im Falle von nur einem Kohlenstoffatom kann die C
1-Kohlenstoffkettengruppe
2 bis 4 Bindungen oder Valenzen aufweisen und 2 bis 0 Wasserstoff-Substituenten,
d.h., die C
1-Kohlenstoffkettengruppe kann
eine der folgenden Strukturen aufweisen:
-
Besonders
bevorzugt ist es, dass R
2 oder R
4-(CH
2)
2-O-(CH
2)
2-O-(CH
2)
2-,
und n
2 ist.
-
In
einer bevorzugten erfindungsgemäßen Ausführungsform
ist das polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidmonomer,
das gegebenenfalls eine anorganische Säure-Komponente, ausgewählt aus
einer Phosphonsäure-Komponente
oder einer Sulfonsäure-Komponente,
enthält,
ausgewählt
aus der Gruppe bestehend aus durch die folgenden Formeln repräsentierten
Verbindungen:
worin
bedeuten:
n eine ganze Zahl von 2 bis 6;
x, y und z unabhängig von
einander eine ganze Zahl von 1 bis 10; und
Z H oder eine C
1-C
18-Alkylgruppe.
Die x, y, 2 aufweisende Formel ist eine Mischung aus Verbindungen.
-
Besonders
bevorzugt ist eine Mischung, worin x+y+z 5,3 ist.
-
In
einer bevorzugten erfindungsgemäßen Ausführungsform
ist das polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidmonomer,
das gegebenenfalls eine anorganische Säure-Komponente ausgewählt aus
einer Phosphonsäure-Komponente
oder einer Sulfonsäure-Komponente
enthält,
ausgewählt aus (Meth)acrylamidmonomeren,
vorzugsweise vom Typ sekundärer
Amine, da diese besonders hydrolysestabil sind.
-
In
einer bevorzugten erfindungsgemäßen Ausführungsform
enthält
das polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidinonomer,
das gegebenenfalls eine anorganische Säure-Komponente ausgewählt aus
einer Phosphonsäure-Komponente
oder einer Sulfonsäure-Komponente
enthält,
mindestens eine solche anorganische Säure-Komponente, vorzugsweise
eine Phosphonsäure-Komponente
oder eine Sulfonsäure-Komponente.
Die durch R1, R1'', R2,
R3 und R4 verstehend
beschriebenen Gruppen sind vorzugsweise durch eine Gruppe substituiert,
die mindestens eine anorganische Säure-Komponente, vorzugsweise eine
Phosphonsäure-Komponente
oder eine Sulfonsäure-Komponente,
enthält.
Besonders bevorzugt ist es, dass R2 und
R4 eine solche anorganische Säure-Komponente
enthält.
Auf diese Weise muss keine getrennte Säure in die erfindungsgemäße Zusammensetzung
eingebaut werden, um einen pH-Wert
von höchstens
2 zu erhalten. Das polymerisierbare N-substituierte Alkylacryl-
oder Acrylsäureamidmonomer,
das gegebenenfalls eine anorganische Säure-Komponente ausgewählt aus
einer Phosphonsäure-Komponente
oder einer Sulfonsäure-Komponente
enthält,
kann dann durch das nachstehend beschriebene polymerisierbare N-substituierte Alkylacryl-
oder Acrylsäureamidinonomer,
das mindestens eine Säure-Komponente
enthält,
repräsentiert
werden.
-
Die
erfindungsgemäße Zusammensetzung
kann jedoch eine organische oder anorganische Säure umfassen, wobei die organische
Säure ausgewählt ist
aus der Gruppe bestehend aus Methacrylsäure, Acrylsäure, Fumarsäure, Maleinsäure, Citronensäure, Itaconsäure und
Ameisensäure,
und wobei die anorganische Säure
ausgewählt
ist aus Phosphorsäure,
Schwefelsäure
und Fluorwasserstoffsäure.
Der Einbau einer Säure ist
in dem Fall, in dem das polymerisierbare N-substituierte Alkylacryl-
oder Acrylsäureamidmonomer,
das gegebenenfalls eine anorganische Säure-Komponente ausgewählt aus
einer Phosphonsäure-Komponente oder einer
Sulfonsäure-Komponente
enthält,
keine Säure-Komponente
enthält,
notwendig. In einer weiteren erfindungsgemäßen Ausführungsform umfasst die erfindungsgemäße Zusammensetzung
außerdem
- (iii) ein polymerisierbares N-substituiertes
Alkylacryl- oder Acrylsäureamidmonomer,
das mindestens eine anorganische Säure-Komponente aufweist, ausgewählt aus
einer Phosphonsäure-Komponente
oder einer Sulfonsäure-Komponente.
-
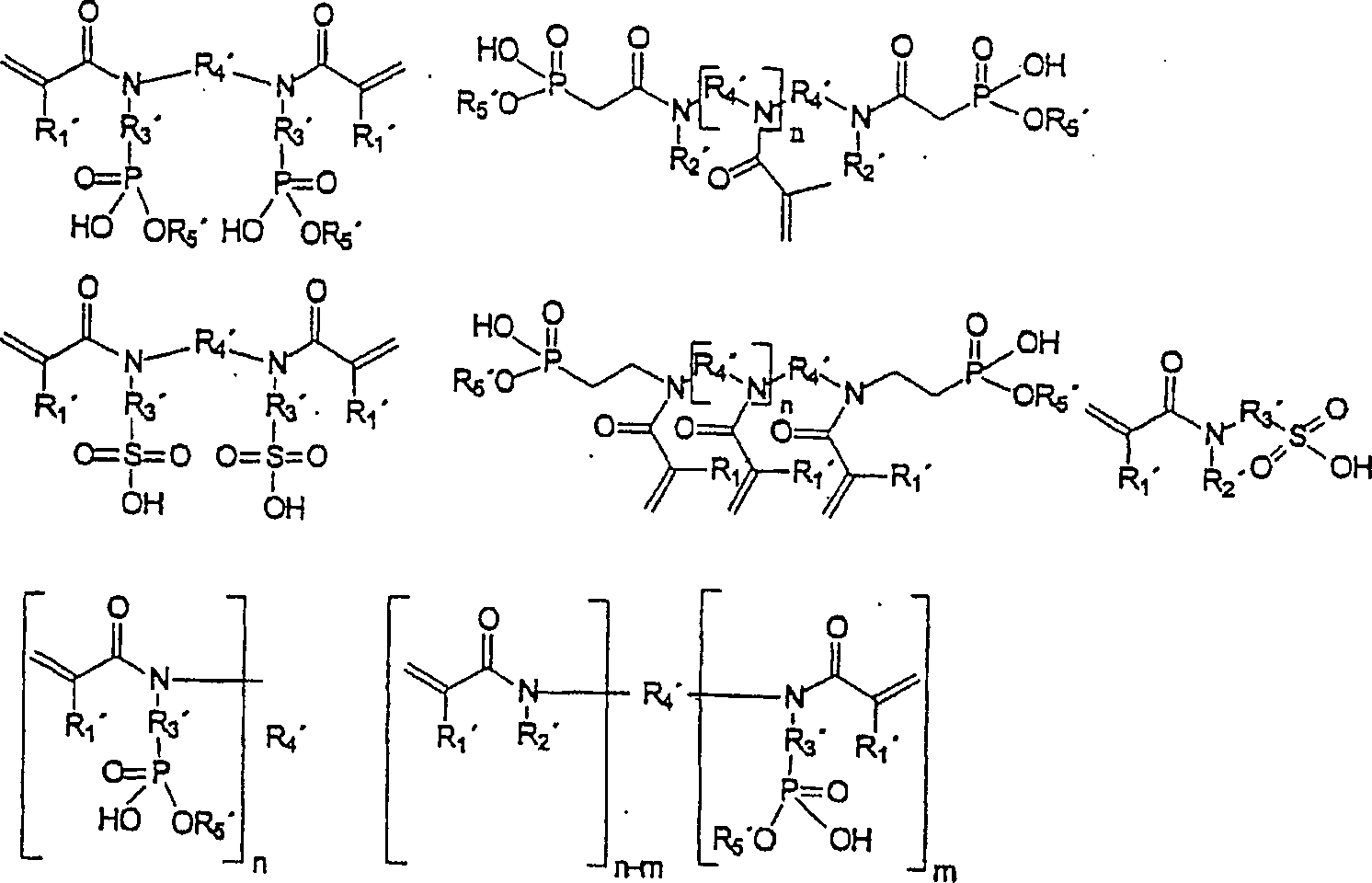
Das
polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidmonomer,
das mindestens eine anorganische Säure-Komponente enthält, ist
vorzugsweise ausgewählt
aus der Gruppe bestehend aus durch die folgenden Formeln repräsentierten
Verbindungen:
worin
bedeuten:
R
1' und R
2' unabhängig von
einander ein Wasserstoffatom oder eine substituierte oder unsubstituierte C
1-C
18-Alkylgruppe, eine substituierte oder unsubstituierte
Cycloalkylgruppe, eine substituierte oder unsubstituierte C
5-C
18-Aryl- oder
-Heteroarylgruppe, eine substituierte oder unsubstituierte C
5-C
18-Alkylaryl- oder Alkylheteroarylgruppe,
eine substituierte oder unsubstituierte C
7-C
30-Aralkylgruppe,
R
3' und R
4' unabhängig von
einander eine difunktionelle substituierte oder unsubstituierte
C
1-C
18-Kohlenstoffkettengruppe,
ein difunktionelles substituiertes oder unsubstituiertes Cycloalkylen,
eine difunktionelle substituierte oder unsubstituierte C
5-C
18-Aryl- oder
-Heteroarylgruppe, eine difunktionelle substituierte oder unsubstituierte
C
5-C
18-Alkylaryl-
oder -Alkylheteroarylgruppe, eine difunktionelle substituierte oder
unsubstituierte C
7-C
30-Aralkylgruppe;
R
5' H
oder eine substituierte oder unsubstituierte C
1-C
18-Alkylgruppe,
n eine ganze Zahl, vorzugsweise
1 bis 18 oder 1 bis 4, und
m eine ganze Zahl, vorzugsweise
von 1 bis 3.
-
Die
optionalen Substituenten an der durch R1', R2',
R3',
R4' und
R5' repräsentierten
Gruppen sind vorzugsweise ausgewählt
aus C1-C18-Alkylgruppen.
Besonders bevorzugt sind C1-C6-Alkylgruppen,
wobei Methylgruppen besonders bevorzugt sind. Die C1-C18-Kohlenstoffkettengruppe weist die vorstehend
angegebene Bedeutung auf.
-
Vorzugsweise
sind R3' und
R4' unabhängig von
einander eine difunktionelle substituierte oder unsubstituierte
C1-C18-Kohlenstoffkettengruppe
oder ein difunktionelles substituiertes oder unsubstituiertes Cycloalkylen,
das mindestens eine Ether-, Thioether-, Ester-, Thiocarbonyl-, Amid-,
Carbonyl-, Sulfonyl-, Urethanbindung oder substituierte oder unsubstituierte
Amin-Bindungen aufweist.
-
Besonders
bevorzugt ist R
3'-(CH
2)
2- oder
und R
4' ist -(CH
2)
2-O-(CH
2)
2-O-(CH
2)
2-.
-
In
einer bevorzugten erfindungsgemäßen Ausführungsform
ist das polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidmonomer,
das mindestens eine anorganische Säure-Komponente enthält, ausgewählt aus
einem der Monomere der folgenden Formeln:
-
In
einer besonders bevorzugten erfindungsgemäßen Ausführungsform ist das mindestens
eine anorganische Säure-Komponente
enthaltende N-substituierte Alkylacryl- oder Acrylsäureamidmonomer
ausgewählt
aus (Meth)acrylamidmonomeren, vorzugsweise vom Typ sekundärer Amide,
insbesondere aus Acrylamidmonomeren vom Typ sekundärer Amide,
da diese besonders hydrolysestabil sind.
-
Das
mindestens eine anorganische Säurekomponente,
ausgewählt
aus einer Phosphonsäure-Komponente
oder einer Sulfonsäure-Komponente,
enthaltende polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidmonomer
(iii) wird in die erfindungsgemäße Zusammensetzung
vorzugsweise in dem Fall eingebaut, in dem keine der vorstehenden
organischen oder anorganischen Säuren
in der erfindungsgemäße Zusammensetzung
eingebaut ist, und/oder in dem Fall, in dem das polymerisierbare
N-substituierte Alkylacryl- oder Acrylsäureamidmonomer, das gegebenenfalls
eine anorganische Säurekomponente,
ausgewählt
aus einer Phosphonsäure-Komponente
oder einer Sulfonsäurekomponente,
enthält
(i), keine Säurekomponente
enthält.
Das polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidinonomer,
das mindestens eine anorganische Säurekomponente, ausgewählt aus
einer Phosphonsäure-Komponente oder einer
Sulfonsäure-Komponente,
enthält
(iii), kann in der erfindungsgemäßen Zusammensetzung
jedoch auch in dem Fall eingebaut sein, in dem die letztere einen
organische oder anorganische Säure
enthält,
oder das polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidmonomer,
das gegebenenfalls eine anorganische Säurekomponente, ausgewählt aus
einer Phosphonsäure-Komponente oder einer
Sulfonsäure-Komponente,
enthält
(i), eine Säurekomponente
enthält.
Das polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidmonomer,
das mindestens eine anorganische Säurekomponente, ausgewählt aus
einer Phosphonsäure-Komponente
oder einer Sulfonsäure-Komponente, enthält (iii),
kann auch in dem Fall in die erfindungsgemäße Zusammensetzung eingebaut
sein, in dem eine anorganische und/oder organische Säure und
das polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidmonomer,
das gegebenenfalls eine anorganische Säurekomponente, ausgewählt aus
einer Phosphonsäure-Komponente
oder einer Sulfonsäure-Komponente,
enthält (i),
eine Säurekomponente
enthält,
in die erfindungsgemäße Zusammensetzung
eingebaut sind.
-
Die
erfindungsgemäße Zusammensetzung
ist vorzugsweise mindestens eine Woche lang bei einer Lagertemperatur
von 50°C
hydrolysestabil, wobei nach einer solchen Lagerung die Bindungsstärke eines
aus einer solchen Kleberzusammensetzung hergestellten Klebers gegenüber Zahnschmelz
und/oder Dentin mindestens 8 MPa, vorzugsweise 10 MPa, ist.
-
Die
erfindungsgemäße wässerige
Zusammensetzung kann neben Wasser ein organisches wasserlösliches
Lösungsmittel
enthalten, vorzugsweise ausgewählt
aus Alkohol und Ketonen.
-
Vorzugsweise
ist das organische wasserlösliche
Lösungsmittel
ausgewählt
aus Ethanol, Propanol, Butanol, Aceton und Methylethylketon.
-
Vorzugsweise
ist das polymerisierbare N-substituierte Alkylacryl- oder Acylsäureamidmonomer,
das gegebenenfalls eine anorganische Säurekomponente ausgewählt aus
aus einer Phosphonsäure-Komponente oder einer
Sulfonsäure-Komponente
enthält
(i), gemäß dem folgenden
Test hydrolysestabil. 1,5 mMol des polymerisierbaren N-substituierten
Alkylacryl- oder Acrylsäureamidmonomer
(i), und für
den Fall, dass das Monomer keine Säurekomponente enthält, 1 Äquivalent
Ethansulfonsäure
pro Alkylacrylamid oder Acrylamidgruppe, werden 5 g eines Lösungsmittelgemisches
bestehend aus 50 Gew.-% Ethanol und 50 Gew.-% Wasser gelöst, unter
Erhalt einer Mischung, die in einem geschlossenen Gefäß in einem
Ofen bei 50°C
gelagert wird; und
nach 2 Wochen einer solchen Lagerung beträgt die Hydrolyse
des getesteten Monomers nach einer HPLC-Analyse weniger als 50%
der absoluten Menge der durch Hydrolyse des getesteten Monomers
gebildeten Alkylacryl- oder Acrylsäure beträgt.
-
In
einer mehr bevorzugten Ausführungsform
ist das polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidmonomer,
das mindestens eine anorganische Säurekomponente ausgewählt aus
einer Phosphonsäure-Komponente
oder einer Sulfonsäure-Komponente
enthält
(iii), gemäß dem folgenden
Test hydrolysestabil:
1,5 mMol des mindestens eine anorganische
Säurekomponente,
ausgewählt
aus einer Phosphonsäure-Komponente oder einer
Sulfonsäure-Komponente
enthaltenden polymerisierbaren N-subsituierten Alkylacryl- oder Acrylsäureamidmonomers
(iii), werden in 5 g eines Lösungsmittelgemisches
bestehend aus 50 Gew.-% Ethanol und 50 Gew.-% Wasser gelöst unter
Erhalt einer Mischung, die in einem geschlossenen Gefäß in einem Ofen
bei 50°C
gelagert wird; und
nach 2 Wochen einer solchen Lagerung beträgt die Hydrolyse
des getesteten polymerisierbaren N-substituierten Alkylacryl- oder Acrylsäureamidmonomers
nach einer HPLC-Analyse weniger als 50% der absoluten Menge der
durch Hydrolyse des getesteten polymerisierbaren N-substituierten
Alkylacryl- oder Acrylsäureamidmonomers
gebildeten Alkylacryl- oder Acrylsäure.
-
In
einer weiteren bevorzugten erfindungsgemäßen Ausführungsform sind das (die) vorstehend
beschriebene(n) Monomer(en) nach dem (den) vorstehenden Test(s)
hydrolysestabil, wenn nach einer Woche der vorstehend beschriebenen
Lagerung die Hydrolyse des getesteten Monomers nach HPLC-Analyse
weniger als 10% der absoluten Menge der durch Hydrolyse des getesteten
Monomers gebildeten Alkylacryl- oder Acrylsäure beträgt.
-
Besonders
bevorzugt ist, dass beide der vorstehend beschriebenen Monomere
(i) und (iii) nach den beschriebenen Tests in dem Fall hydrolysestabil
sind, in dem beide Monomere (i) und (iii) in der erfindungsgemäßen Zusammensetzung
vorhanden sind. Es ist jedoch auch möglich, dass nur eines der Monomere
(i) und (iii) im Falle, dass beide Monomere (i) und (iii) vorhanden
sind, nach dem vorstehenden Test hydrolysestabil ist.
-
Obwohl
das polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidmonomer,
das gegebenenfalls eine anorganische Säurekomponente ausgewählt aus
einer Phosphonsäure-Komponente
oder einer Sulfonsäure-Komponente
enthält,
und/oder das polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidmonomer,
das mindestens eine anorganische Säurekomponente enthält, nach
den vorstehend beschriebenen Tests hydrolysestabil sind, kann eine
Hydrolyse in geringem Ausmaß auftreten.
Das polymerisierbare N-substituierte Alkylacryl- oder Acrylamidsäuremonomer,
das gegebenenfalls eine anorganische Säurekomponente ausgewählt aus
einer Phosphonsäure-Komponente
oder einer Sulfonsäure-Komponente enthält, und/oder
das polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidmonomer,
das mindestens eine anorganische Säurekomponente der vorliegenden
Erfindung enthält,
enthält
deshalb mindestens zwei polymerisierbare Gruppen. Für den Fall,
dass eine Amidbindung hydrolysiert wird, enthält das N-substituierte Alkylacryl-
oder Acrylsäureamidmonomer,
das mindestens zwei polymerisierbare Gruppen enthält, immer
noch mindestens eine polymerisierbare Gruppe, die eine Polymerisation
ermöglicht.
Auf diese Weise wird eine hohe Bindungsfestigkeit gegenüber Zahnschmelz
und/oder Dentin erzielt. Die mindestens zwei polymerisierbaren Gruppen
können
direkt oder indirekt gebunden sein. Vorzugsweise sind sie über eine
Amidbindung gebunden, die die Stabilität der Zusammensetzung und die
Bindungsfestigkeit der Kleberzusammensetzung gegenüber Zahnschmelz
oder Dentin erhöht.
-
Die
erfindungsgemäße Zusammensetzung
kann ferner einen Nanofüllstoff
enthalten.
-
In
einer weiteren bevorzugten erfndungsgemäßen Ausführungsform enthält das polymerisierbare N-substituierte Alkylacryl-
oder Acrylsäureamidmonomer,
das mindestens eine anorganische Säurekomponente enthält, mindestens
zwei anorganische Säurekomponenten.
-
Das
Härtungssystem
in der erfindungsgemäßen Zusammensetzung
umfasst vorzugsweise einen Polymerisationsinitiator, einen Inhibitor
oder Stabilisator, und vorzugsweise ist das Härtungssystem ein Lichthärtungssystem.
-
In
einer weiteren erfindungsgemäßen Ausführungsform
wird eine wässerige
selbstätzende
und selbstgrundierende Dental-Einkomponenten-Kleberzusammensetzung
bereitgestellt, die umfasst:
- (a) ein polymerisierbares
N-substituiertes Alkylacryl- oder Acrylsäureamidmonomer, das mindestens
zwei polymerisierbare Einheiten aufweist,
- (b) ein polymerisierbares N-substituiertes Alkylacryl- oder
Acrylsäureamidmonomer,
das mindestens eine anorganische Säurekomponente enthält, und
- (c) ein Härtungssystem.
-
Vorzugsweise
ist das polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidmonomer,
das mindestens zwei polymerisierbare Einheiten aufweist (a), ausgewählt aus
der Gruppe bestehend aus durch die folgenden Formeln repräsentierten
Verbindungen:
worin
R
1,
R
1'', R
2, R
3 und R
4 und n die vorstehend und in den Ansprüchen 14
bis 16 definierten Bedeutungen besitzen.
-
In
einer besonders bevorzugten Ausführungsform
ist das polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidmonomer,
das mindestens zwei polymerisierbare Einheiten aufweist (a), ausgewählt aus der
Gruppe bestehend aus durch die folgenden Formeln repräsentierten
Verbindungen:
worin
Z H oder eine substituierte oder unsubstituierte C
1-C
18-Alkylgruppe ist, und n, x, y, z die vorstehend
und in Anspruch 17 definierte Bedeutung besitzen. Besonders bevorzugt
sind (Meth)acrylamidmonomere, vorzugsweise vom Typ sekundärer Amide,
da diese besonders hydrolysestabil sind.
-
In
einer weiteren erfindungsgemäßen Ausführungsform
wird eine wässerige
selbstätzende
und selbstgrundierende Dental-Einkomponenten-Kleberzusammensetzung
bereitgestellt, die umfasst:
- (I) ein polymerisierbares
N-substituiertes Alkylacryl- oder Acrylsäureamidinonomer, das gegebenenfalls eine
anorganische Säurekomponente
ausgewählt
aus einer Phosphonsäure-Komponente
oder einer Sulfonsäure-Komponente
enthält,
- (II) ein polymerisierbares N-substituiertes Alkylacryl- oder
Acrylsäureamidmonomer,
das mindestens eine anorganische Säurekomponente enthält, und
- (III) ein Härtungssystem.
-
Besonders
bevorzugt ist es, dass das polymerisierbare N-substituierte Alkylacryl-
oder Acrylsäureamidmonomer,
das mindestens eine anorganische Säurekomponente enthält (II),
mindestens zwei anorganische Säurekomponenten
enthält.
-
Vorzugsweise
ist das polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidmonomer,
das mindestens eine anorganische Säurekomponente enthält (II),
ausgewählt
aus der Gruppe bestehend
aus durch die folgenden
Formeln repräsentierten
Verbindungen mit Phosphonsäure-Komponente(n)
oder Sulfonsäure-Komponente(n).
worin
R
1',
R
2',
R
3',
R
4',
R
5',
n und m die vorstehend angegebene Bedeutung besitzen.
-
Bevorzugt
ist es, dass das polymerisierbare N-substituierte Alkylacryl- oder
Acrylsäureamidmonomer, das
mindestens eine anorganische Säure-Komponente
enthält,
ausgewählt
ist aus (Meth)acrylamidmonomeren, vorzugsweise vom Typ sekundärer Amide,
insbesondere von Acrylamidmonomeren vom Typ sekundärer Amide,
da diese besonders hydrolysestabil sind.
-
Bevorzugt
ist es, dass eine erfindungsgemäße Zusammensetzung
in einem gegenüber
Licht abgeschirmten Behälter
verpackt ist.
-
Erfindungsgemäß sind ein
polymerisierbares N-substituiertes Alkylacryl- oder Acrylsäureamidmonomer,
das gegebenenfalls eine anorganische Säurekomponente ausgewählt aus
einer Phosphonsäure-Komponente oder einer
Sulfonsäure-Komponente
enthält,
und ein polymerisierbares N-substituiertes Alkylacryl- oder Acrylsäureamidmonomer,
das eine anorganische Säurekomponente
enthält,
geeignet zur Herstellung einer wässerigen
selbstätzenden
und selbstgrundierenden Dental-Einkomponenten-Kleberzusammensetzung, die umfasst:
- (i) das polymerisierbare N-substituierte Alkylacryl-
oder Acrylsäureamidmonomer,
das gegebenenfalls eine anorganische Säurekomponente ausgewählt aus
einer Phosphonsäure-Komponente
oder einer Sulfonsäure-Komponente
enthält,
- (ii) das polymerisierbare N-substituierte Alkylacryl- oder Acrylsäureamidmonomer,
das mindestens eine anorganische Säurekomponente enthält, und
ein
- (iii) Härtungssystem.
-
Ein
Verfahren zur Herstellung einer erfindungsgemäßen wässerigen selbstätzenden
und selbstgrundierenden Dental-Einkomponenten-Kleberzusammensetzung
ist gekennzeichnet durch Mischen von
- (A) einem
polymerisierbaren N-substituierten Alkylacryl- oder Acrylsäureamidmonomer,
das gegebenenfalls eine anorganische Säurekomponente ausgewählt aus
einer Phosphonsäure-Komponente
oder einer Sulfonsäure-Komponente
enthält,
- (B) einem Härtungssystem
und
- (C) einem Wasser-enthaltenden Lösungsmittel.
-
Ein
neuer Dentalkleber ist erhältlich
durch Polymerisieren einer der vorstehend beschriebenen erfindungsgemäßen Zusammensetzungen.
-
Erfindungsgemäß wird ein
Verfahren zur Behandlung von menschlichen oder tierischen Zähnen bereitgestellt,
das die Applikation einer der vorstehend beschriebenen Zusammensetzungen
umfasst.
-
Die
vorliegende Erfindung stellt darüber
hinaus einen Kit bereit, der eine der vorstehend beschriebenen Zusammensetzungen
und Verwendungsinstruktionen umfasst.
-
Außerdem wird
erfindungsgemäß ein neues
Monomer bereitgestellt. Es ist ein polymerisierbares N-substituiertes Alkylacryl-
oder Acrylsäureamidinonomer,
das mindestens zwei polymerisierbare Einheiten aufweist, ausgewählt aus
der Gruppe bestehend aus durch die folgenden Formeln repräsentierten
Verbindungen:
worin
R
1,
R
1'', R
2, R
3, R
4 und
n die vorstehend und in einem der Ansprüche 14 bis 16 angegebene Bedeutung
besitzen.
-
Ein
Verfahren zur Herstellung des neuen Monomers umfasst das Umsetzen
einer Di- oder Polyaminverbindung mit einem gegebenenfalls substituierten
Acryloylhalogenid der Formel CH2=CR3-CO-Hal, worin R3 die
vorstehend und in Anspruch 14 angegebene Bedeutung besitzt, und
Hal ein Halogen ist.
-
Ein
Halogen bedeutet Chlor, Brom, Fluor oder Iod, wobei Chlor und Brom
bevorzugt sind.
-
Die
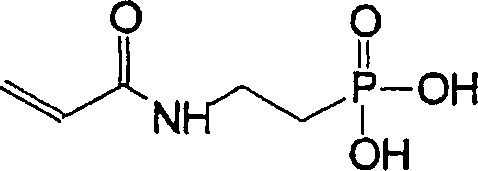
vorliegende Erfindung stellt ferner ein neues Monomer bereit, das
eine saure anorganische Komponente aufweist. Es ist ein polymerisierbares
N-substituiertes Alkylacryl- oder Acrylsäureamidmonomer, das mindestens
eine anorganische Säurekomponente
ausgewählt
aus einer Phosphonsäure-Komponente
oder einer Sulfonsäure-Komponente
enthält,
und ausgewählt
ist aus der Gruppe bestehend aus durch die folgenden Formeln repräsentierten
Verbindungen:
worin
R
1',
R
2',
R
3',
R
4',
R
5' und
n und m die oben angegebene Bedeutung besitzen.
-
Ein
Verfahren zur Herstellung des neuen Monomers, das mindestens eine
anorganische Säurekomponente
aufweist, umfasst:
- (1) Umsetzen einer Di- oder
Polyaminverbindung mit einem Vinylphosphonat oder einem Vinylsulfonsäuresalz,
- (2) Umsetzen des in (1) erhaltenen Produkts mit einem Alkylacryloyl-
oder Aryloylhalogenid der Formel CH2=CR1'-CO-Hal,
worin R1' die
in Anspruch 19 definierte Bedeutung besitzt, und Hal ein Halogenid
ist;
- (3) gegebenenfalls Umsetzen des in (2) erhaltenen Produkts mit
einem Trialkylhalogensilan im Falle des Umsetzens eines Vinylphosphonats
in Stufe (1); und
- (4) Hydrolysieren des in Stufe (2) oder (3) erhaltenen Produkts.
-
Besonders
bevorzugt ist das durch die folgende Formel repräsentierte Monomer:
worin R
1', R
2' und
R
3' die
vorstehend angegebene Bedeutung besitzen. Ein Verfahren zu seiner
Herstellung umfasst:
- (1) Umsetzen eines Alkylacryloyl-
oder Acryloylhalogenids der Formel CH2=CR1-CO-Hal, worin R1 die
vorstehend angegebene Bedeutung besitzt, und Hal ein Halogen ist,
mit N-Hydroxysuccinimid und einem Aminoalkohol der Formel HO-(CH2)n-NH2,
worin n 1 bis 18 ist;
- (2) Umsetzen des in (1) erhaltenen Produkts oder von Methacryloylamid
mit Natriumhydrid und Propansulfon;
- (3) Hydrolysieren des in Stufe (2) erhaltenen Produkts.
-
Die
vorliegende Erfindung wird nun näher
durch die folgenden Beispiele, Tests und Anwendungsbeispiele erläutert. Beispiel
1 N-(3-Sulfopropyl)methacrylamid
(1) und N,N-Bis(3-sulfopropyl)methacrylamid
-
Zu
einer gerührten
Lösung
von 12 g (140,9 mMol) Methacrylamid in 400 ml Methylenchlorid wurden sorgfältig 3,38
g (140,9 mMol) Natriumhydrid stufenweise bei einer Temperatur von
0°C zugegeben.
Die Suspension wurde 3 Stunden lang bei Raumtemperatur gerührt, bevor
18,943 g (155 mMol) 1,3-Propansulfon
zugegeben wurden. Nach 12 Stunden rühren bei Raumtemperatur wurden
sorgfältig
150 ml Wasser in die Reaktioasmischung getropft, während die
Temperatur bei 0°C
gehalten wurde. Die wässerige
Schicht wurde dann abgetrennt und fünfmal mit 100 ml Methylenchlorid
extrahiert. Danach wurde das Wasser in einem Rotationsverdampfer
entfernt und der resultierende weiße Feststoff sorgfältig mit
Aceton gewaschen. Das Sulfonsäurenatriumsalz
wurde wieder in Wasser gelöst
und über
eine Ionen-Austauschersäule (Merck
Ionen-Austauscher I) gegossen. Die resultierende saure wässerige
Lösung
wurde mit 0,025 Mol% Hydrochinon stabilisiert und in einem Rotationsverdampfer
eingedampft. Entfernen des Wassers unter Hochvakuum (8 × 10–3 mbar)
ergab eine Mischung aus N-(3-Sulfopropyl)methacrylamid
1 und N,N'-Bis(3-sulfopropyl)methacrylamid
in einem Verhältnis
von 2,6 : 1 (nach NMR-Spektroskopie) als klares rötliches
hochviskoses Öl
in einer Menge von 16,33 g (35%ige Ausbeute in Bezug auf N-(3-Sulfopropyl)methacrylamid
1).
1H-NMR (250 MHz. D6-DMSO,
ppm) N-(3-Sulfopropyl)methacrylamid: 1,70-1,90 (m, 2H, CH2-CH2-CH2),
1,83 (t, 3H, CH3), 2,57-2,73 (m, 2H, CH2-SO3H), 3,36-3,47
(m, 2H, N-CH2), 5,31 (s, 1H, CH=), 5,65
(s, 1H, CH=), 8,07 (t, 1H, NH)
13C-NMR
(63 MHz, d6-DMSO, ppm) N-(3-Sulfopropyl)methacrylamid:
19,17 (CH3), 25,31 (-CH2),
38,58 (N-CH2), 49,78 (CH2-SO3H), 119,76 (CH2=),
140,35 (=C-CH3), 168,18 (C=O)
-
Beispiel 2
-
N-(6-Hydroxyhexyl)methacrylamid
(2)
-
Eine
Lösung
von 22,56 (0,215 mMol) Methacryloylchlorid in 20 ml Chloroform wurde
langsam in eine gerührte
Lösung
von 24,84 g (0,215 Mol) N-Hydroxysuccinimid in 50 ml Triethylamin
und 500 ml Chloroform bei einer Temperatur von 0°C zugetropft. Nach Rühren der
Lösung
während
3 Stunden bei Raumtemperatur wurden 21,07 g (0,179 Mol) 6-Amino-1-hexanol
in 20 ml Chloroform zugegeben. Die Reaktionsmischung wurde über Nacht
bei Raumtemperatur gerührt,
bevor das Lösungsmittel
unter vermindertem Druck entfernt wurde. Der Rückstand wurde in Methylenchlorid
aufgenommen und der verbleibende Feststoff abfiltriert. Dann wurde
die Lösung
eingedampft und der Niederschlag wieder abfiltriert. Danach wurde
die Lösung
mit 200 ml einer wässerigen
Natruimhydroxidlösung
(20%) gewaschen. Die vereinigten wässerigen Schichten wurden viermal
mit 100 ml Methylenchlorid gewaschen und die vereinigten organischen
Lösungen
wurden dann über Magnesiumsulfat
getrocknet. Filtration und Verdampfen des Lösungsmittels ergaben ein gelbes Öl als Rohprodukt,
das mit 0,025 Mol% BHT stabilisiert wurde. Destillation unter Hochvakuum
(140 bis 143°C,
4 × 10–3 mbar) ergab
die Verbindung 2 als farbloses niedrigviskoses Öl, das durch Zugabe von 0,025
Mol% BHT stabilisiert wurde, in einer Menge von 24,18 g (Ausbeute:
75%).
IR (Film, cm–1), 3118 (s, b), 2930
(s), 1655 (s), 1611 (s), 1534 (s), 1448 (m), 1374 (w), 1318 (w),
1218 (m), 1051 (s), 925 (m)
1H-NMR
(250 MHz, CDCl3, ppm) 1,11-1,14 (m, 4H,
CH2), 1,29-1,1,35 (m, 4H, CH2),
1,72 (s, 3H, =C-CH3), 3,04 (quart., 2H, CH2-N),
3,35 (t, 2H, CH2-O), 4,23 (breit s, 1H,
OH), 5,09 (s, 1H, CH=), 5,49 (s, 1H, CH=), 6,91 (t, 1H, NH)
13C-NMR (63 MHz, CDCl3,
ppm) 18,16 (CH3), 24,91, 26,15, 28,85, 31,94,
39,11 (CH2), 61,50 (CH2-O),
119,08 (CH2=), 139,30 (=C-CH3),
168,54 (C=O)
-
N-[9-(Diethoxyphosphoryl)-7-oxa-nonyl]methacrylamid
(3)
-
Zu
einer Lösung
von 11,63 g (62,8 mMol) N-(6-Hydroxyhexyl)methacrylamid 2 in 250
ml Methylenchlorid wurden 1,5 g (62,8 mMol) Natriumhydrid stufenweise
unter Rühren
bei einer Temperatur von 0°C
zugegeben. Nach 1-stündigem
Rühren
bei Raumtemperatur wurden 10,30 g (62,80 mMol) Diethylvinylphosphonat
zugegeben. Die Reaktionsmischung wurde zusätzliche 4 Tage gerührt, bevor
die Umsetzung durch Zugabe von 200 ml Wasser beendet wurde. Die
Schichten wurden abgetrennt und die organische Schicht wurde mit
100 ml Wasser gewaschen. Die organische Schicht wurde über Magnesiumsulfat
getrocknet und filtriert. Verdampfen des Lösungsmittels unter vermindertem
Druck in einem Rotationsverdampfer und Trocknen unter Hochvakuum
(8 × 10
–3 mbar)
bei 40°C
bis zur Gewichtskonstanz ergab 19,13 g (Ausbeute 87%) eines gelben Öls, das mit
0,025 Mol% BHT stabilisiert wurde.
IR (Film, cm
–1)
3327 (m), 2932 (m), 2863 (m), 1658 (m), 1517 (m), 1525 (m), 1447
(m), 1373 (m), 1310 (w), 1222 (s), 1105 (s), 1025 (s), 957 (s),
792 (s)
1H-NMR (250 MHz, CDCl
3, ppm) 1,06 (t, 6H, CH
3),
1,10-1,17 (m, 4H, CH
2), 1,23-1,37 (m, 4H,
CH
2), 1,68 (s, 3H, =C-CH
3),
1,75-1,88 (m, 2H, CH
2-P), 3,02 (quart.,
2H, CH
2-N), 3,15 (t, 2H, CH
2-O),
3,33-3,44 (m, 2H, CH
2-O), 3,82 (quin., 4H, CH
2-O-P),
5,02 (s, 1H, CH=), 5,44 (s, 1H, CH=), 6,64 (t, 1H, NH)
13C-NMR (63 MHz, CDCl
3,
ppm) 15,57 und 15,67 (d, POCH
2CH
3), 18,10, 25,04, 26,00 und 27,22 (d, CH
2-P), 28,70, 38,77, 60,68 und 60,79 (d, POCH
2CH
3), 63,66 (CH
2-O), 70,00 (CH
2-O),
118,10 (CH
2=), 139,56 (=C-CH
3),
167,82 (C=O) N-[9-(Dihydroxyphosphoryl)-7-oxa-nonyl]methacrylamid
(4)
-
Zu
einer Lösung
von 18,57 g (53,1 mMol) N-[9-(Diethoxyphosphoryl)-7-oxa-nonyl]methacryamid
3 in 100 ml Methylenchlorid wurden 22,06 g (144,1 mMol) Trimethylsilylbromid
tropfenweise bei Raum temperatur zugegeben. Die Mischung wurde für 4 Stunden
am Rückfluss
erhitzt, bevor das Lösungsmittel
in einem Rotationsverdampfer abgedampft wurde und der Rückstand
wieder in 200 ml Methanol gelöst
wurde. Nach Rühren der
Lösung
bei Raumtemperatur während
2 Stunden und Entfernen des Lösungsmittels
wurde ein bräunliches Öl als Rohprodukt
erhalten. Das Material wurde in 200 ml Methylenchlorid gelöst und einmal
mit einer wässerigen
Lösung
von 4,1 g Natriumhydroxid in 120 ml Wasser extrahiert. Nach Abtrennen
der wässerigen
Schicht und viermaligem Waschen mit 100 ml Methylenchlorid wurde
sie über
eine saure Ionenaustauschersäule
(Merck Ionenaustauscher 1) gegossen. Die resultierende saure wässerige
Lösung
wurde in einem Rotationsverdampfer eingeengt und dreimal mit 100
ml Methylenchlorid extrahiert. Bevor die wässerige Lösung im Rotationsverdampfer
aufkonzentriert und dann unter Hochvakuum (3 × 10–3 mbar)
bei 40°C
bis zur Gewichtskonstanz getrocknet wurde, wurden 0,025 Mol% BHT
zugegeben. Dies ergab 11,24 g (Ausbeute: 72%) eines klaren bräunlichen Öls.
IR
(Film, cm–1)
3317(b, m), 2931 (m), 2862 (m), 1648 (m), 1545 (m), 1446 (m), 1373
(m), 1096 (s), 997 (s), 926 (s), 781 (s), 714 (s)
1H-NMR
(250 MHz, d6-DMSO, ppm) 1,14-1,30 (m, 4H,
CH2), 1,33-1,51 (m, 4H, CH2),
1,81 (s, 3H, =C-CH3), 1,75-1,95 (m, 2H, CH2-P),
3,06 (quart., 2H, CH2-N), 3,31 (t, 2H, CH2-O), 3,50 (quart., 2H, CH2-O),
5,26 (s, 1H, CH=), 5,60 (s, 1H, CH=), 7,91 (t, 1H, NH), 10,04 (breit
s, 2H, P-O-H)
13C-NMR (63 MHz, d6-DMSO, ppm) 18,82, 25,51 und 26,39 (d, CH2-P), 27,67, 29,19, 29,79, 38,88, 65,18 (CH2-O), 69,87 (CH2-O),
118,84 (CH2=), 140,14 (=C-CH3),
167,45 (C=O)
-
Beispiel 3
-
N-[2-(Diethoxyphosphoryl)ethyl]acrylamid
(5)
-
Zu
einer Lösung
von 8,09 g (44,7 mMol) 2-Aminoethyl)phosphonsäurediethylester in 150 ml Methylenchlorid
wurden in eine Lösung
von 6,06 g (67 mMol) Acryloylchlorid in 30 ml Methylenchlorid und
eine Lösung von
2,68 g (67 mMol) Natriumhydroxid in 30 ml Wasser gleichzeitig unter
Rühren
so zugegeben, dass die Temperatur bei 0 bis 5°C blieb. Danach wurde die Mischung
bei Raumtemperatur zusätzliche
2 Stunden lang gerührt.
Die Umsetzung wurde durch Zugabe von 100 ml Wasser beendet. Zur
Erzielung einer Trennung der Schichten wurde etwas Natriumchlorid
zugegeben. Die organische Phase wurde abgetrennt und die wässerige Phase
wurde zweimal mit 50 ml Methylenchlorid extrahiert. Die vereinigten
organischen Phasen wurden mit 50 ml 1 N HCl, 50 ml 1 N NaHCO
3 und mit 50 ml Wasser gewaschen. Trocknen über Magnesuimsulfat,
Filtration und Verdampfen des Lösungsmittels
ergab ein gelbes Öl
als Rohprodukt. Zur endgültigen
Reinigung wurde das Material an einer Silikagelsäule mit Ethylacetat als Eluens
chromatographiert (R
1 = 0,27). Dies ergab 6,72
g (Ausbeute: 63%) eines gelblichen Öls, das durch Zugabe von 0,025
Mol% BHT stabilisiert wurde.
IR (Film, cm
–1)
3274 (m), 2937 (w), 1660 (m), 1544 (m), 1444 (m), 1310 (m), 1219
(s), 1021 (s), 954 (s), 828 (m), 788 (m), 698 (m)
1H-NMR
(250 MHz, CDCl
3, ppm) 1,14 (m, 6H, CH
3), 1,81-1,94 (m, 2H, CH
2-P),
3,31-3,45 (m, 2H, CH
2-N), 3,85-3,97 (m, 4H, CH
2-O),
5,40-5,44 (dd, 1H, CH=C-CO), 5,93-6,13 (m, 2H, CH=CH-CO), 7,32 (t,
1H, NH)
13C-NMR (63 MHz, CDCl
3, ppm) 15,93 und 16,04 (d, POCH
2CH
3), 23,96 und 26,17 (d, CH
2-P),
33,26 (CH
2-N), 61,93 und 61,50 (d, POCH
2CH
3), 119,08 (C=),
139,30 (C=), 165,41 (C=O) N-[2-(Dihydroxyphosphoryl)ethyl]acrylamid
(6)
-
8,86
g (56,32 mMol) Trimethylsilylbromid wurden tropfenweise bei Raumtemperatur
unter Rühren
zu einer Lösung
von 5,83 g (24,81 mMol) N-[2-(Diethoxyphosphoryl)ethyl]methacrylamid
5 in 30 ml Methylenchlorid zugegeben. Die Reaktionsmischung wurde
unter Rückfluss
4 Stunden lang erhitzt. Danach wurde das Lösungsmittel in einem Rotationsverdampfer
entfernt und der Rückstand
in Methanol gelöst.
Diese Lösung
wurde 16 Stunden bei Raumtemperatur gerührt. Dann wurde das Lösungsmittel
entfernt und das verbleibende Öl
in 30 ml Wasser gelöst.
Nach zweimaligem Waschen der wässerigen
Lösung
mit 20 ml Methylenchlorid wurde sie durch Zugabe von 0,025 Mol%
BHT stabilisiert und unter vermindertem Druck eingedampft. Nach
Trocknen des Materials unter vermindertem Druck (8 × 10–3 mbar)
bei 40°C
während
19 Stunden wurden 4,385 g (Ausbeute: 98%) eines hochviskosen gelblichen Öls erhalten.
IR
(Film, cm–1)
2812 (b, s), 2359 (m), 1649 (m), 1547 (s), 1444 (m), 1310 (m), 1121
(s), 927 (s), 793 (s), 713 (s)
1H-NMR
(250 MHz, d6-DMSO, ppm) 1,77-1,99 (m, 2H,
CH2-P), 3,25-3,44 (m, 2H, CH2-N),
5,62 (dd, 1H, CH=C-CO), 6,07-6,32 (m, 2H, CH=CH-CO), 8,40 (t, 1H,
NH)
13C-NMR (63 MHz, d6-DMSO,
ppm) 27,11 und 29,23 (d, CH2-P), 34,05 (CH2-N), 125,74 (C=), 131,89 (C=), 165,14 (C=O)
-
Beispiel 4
-
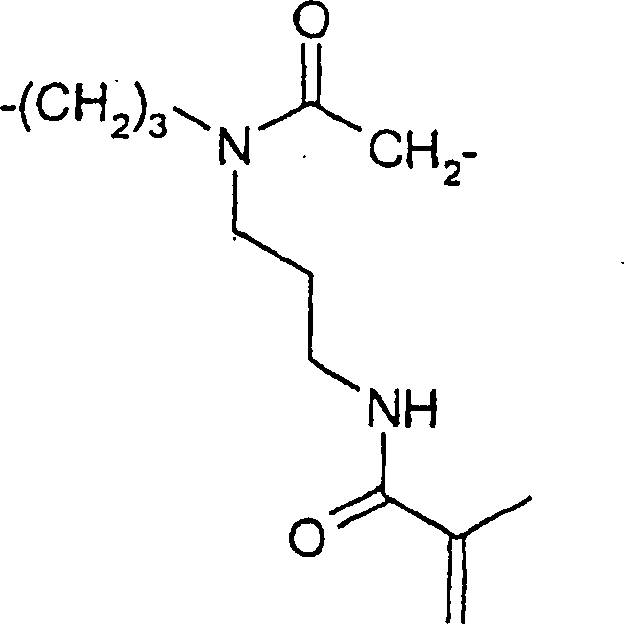
N-[2-(Diethoxyphosphoryl)ethyl]-N-butylamin
(7)
-
30,04
g (0,411 Mol) n-Butylamin und 67,41 g (0,411 Mol) Diethylvinylphosphonat
wurden bei 65°C
24 Stunden lang gerührt.
Dies ergab 97,32 g (Ausbeute: 100%) eines farblosen Öls.
IR
(Film, cm–1)
3411, 3390 (OH), 2973, 2929, 2885 (CH2/CH3), 1390 (CH2/CH3), 1078 cm–1 (OH)
1H-NMR (250 MHz, CDCl3,
ppm) 1,39 (t, 3H, CH3), 1,79-2,20 (m, 10H,
CH2, OCH2CH3), 2,40-2,71 (m, 2H, CH2-P),
3,02-3,29 (m, 2H, CH2-N), 3,39-3,62 (m,
2H, CH2-N), 4,57-4,84 (m, 4H, POCH2)
13C-NMR (63
MHz, CDCl3, ppm) 13,70 (C3H6-CH3), 16,23 (OCH2-CH3), 20,26, 25,18
und 27,36 (d, CH2-P), 31,86, 43,13, 49,08,
61,35 (OCH2-CH3)
-
N-[2-Diethoxyphosphoryl)ethyl]-N-butylacrylamid
(8)
-
Zu
einer Lösung
von 55,27 g (0,233 Mol) N-[2-(Diethoxyphosphoryl)ethyl]-N-butylamin
7 in 100 ml Methylenchlorid wurde eine Lösung von 23,19 g (0,256 Mol)
Acryloylchlorid in 130 ml Methylenchlorid und einer Lösung von
10,25 g (0,256 Mol) Natriumhydroxid in 200 ml Wasser gleichzeitig
unter Rühren
so zugegeben, dass die Temperatur bei 0 bis 5°C blieb. Danach wurde die Mischung
bei Raumtemperatur zusätzliche
2 Stunden lang gerührt.
Die Umsetzung wurde durch Zugabe von 100 ml Wasser beendet. Die organische
Schicht wurde abgetrennt und die wässerige Schicht wurde zweimal
mit 30 ml Methylenchlorid extrahiert. Die vereinigten organischen
Phasen wurden mit 150 ml 1 N HCl, 150 ml 1 N NaHCO
3 und
mit 150 ml Wasser gewaschen. Die Lösung des Produkts wurde mit
0,025 Mol% BHT stabilisiert und über
Natriumsulfat getrocknet. Filtration und Verdampfen des Lösungsmittels
ergab 40,07 g (Ausbeute: 59%) eines farblosen Öls.
IR (Film, cm
–1)
2978/2962/2956 (CH
3/CH
2),
1648 (CO), 1614 (C=C), 1456/1431/1371 (CH
3/CH
2), 794 (C=C)
1H-NMR
(250 MHz, CDCl
3, ppm) 0,79 (t, 3H, CH
3), 1,11-1,31 (m, 8H, CH
2,
OCH
2CH
3), 1,33-1,54
(m, 2H, CH
2), 1,87-2,12 (m, 2H, CH
2-P), 3,12-3,32 (m, 2H, CH
2-N),
3,38-3,57 (m, 2H, CH
2-N), 3,88-4,17 (m,
4H, POCH
2), 5,50-5,67 (m, 1H, CH=C-CO),
6,13-6,30 (m, 1H, CH=C-CO), 6,33-6,52 (m, 1H, C=CH-CO)
13C-NMR (63 MHz, CDCl
3,
ppm) 13,41 (C
3H
6-CH
3), 16,02 und 16,11 (d, POCH
2-CH
3), 19,57, 22,78 und 24,94 (d, CH
2-P), 31,40, 41,35, 48,19, 61,35 und 61,45
(d, POCH
2-CH
3),
127,16 (C=), 127,62 (C=O) N-[2-(Dihydroxyphosphoryl)ethyl]-N-butylacrylamid
(9)
-
33,56
g (0,219 Mol) Trimethylsilylbromid wurden tropfenweise bei Raumtemperatur
unter Rühren
zu einer Lösung
von 31,93 g (0,11 Mol) N-[2-(Diethoxyphosphoryl)ethyl]methacrylamid
8 in 100 ml Methylenchlorid zugegeben. Die Reaktionsmischung wurde
unter Rückfluss
4 Stunden lang erhitzt. Danach wurde das Lösungsmittel in einem Rotationsverdampfer
entfernt und der Rückstand
in 100 ml Methanol gelöst.
Diese Lösung
wurde 4 Stunden lang bei Raumtemperatur gerührt. Die Lösung des Produkts wurde durch
Zugabe von 0,025 Mol% BHT stabilisiert und unter vermindertem Druck
eingedampft. Nach Trocknen des Materials unter vermindertem Druck
(8 × 10
–3 mbar)
bei 40°C
während
24 Stunden wurden 21,65 g (Ausbeute: 84%) eines weißen Feststoffs
erhalten.
IR (ATR, cm
–1) 3411, 3390 (OH),
2973, 2929, 2885 (CH
2/CH
3),
1390 (CH
2/CH
3),
1078cm
–1 (OH)
1H-NMR (250 MHz, d
6-DMSO,
ppm) 0,97 (t, 3H, CH
3), 1,27-1,45 (m, 2H,
CH
2), 1,47-1,66 (m, 2H, CH
2), 1,93-2,17
(m, 2H, CH
2-P), 3,35-3,52 (m, 2H, CH
2-N), 3,55-3,75 (m, 2H, CH
2-N),
5,69-5,83 (m, 1H, CH=C-CO), 6,24 (dd, 1H, CH=C-CO), 6,64-6,81 (m,
1H, C=CH-CO)
13C-NMR (63 MHz, d
6-DMSO, ppm) 14,2 (C
3H
6-CH
3), 21,2, 25,8
und 27,90 (d, CH
2-P), 30,8, 32,6, 43,8,
128,9 (C=), 129,1 (C=), 168,4 (C=O) Beispiel
5 (2,2(4),4)-Trimethylhexamethylenbis(acrylamid)
(10)
-
Zu
einer Lösung
von 60 g (0,379 Mol) (2,2(4),4)-Trimethylhexamethylendiamin in 100
ml Methylenchlorid wurden eine Lösung
von 72,05 g (0,796 Mol) Acryloylchlorid in 130 ml Methylenchlorid
und eine Lösung von
31,84 g (0,796 Mol) Natriumhydroxid in 200 ml Wasser gleichzeitig
unter Rühren
so zugegeben, dass die Temperatur bei 0 bis 5°C blieb. Danach wurde die Mischung
bei Raumtemperatur zusätzliche
2 Stunden lang gerührt.
Die Umsetzung wurde durch Zugabe von 100 ml Wasser beendet. Die
organische Phase wurde abgetrennt und die wässerige Phase wurde zweimal
mit 30 ml Methylenchlorid extrahiert. Die vereinigten organischen
Phasen wurden mit 150 ml 1 N HCl, 150 ml 1 N NaHCO
3 und
mit 150 ml Wasser gewaschen. Die Lösung des Produkts wurde mit
0,025 Mol% BHT stabilisiert und über
Natriumsulfat getrocknet. Filtration und Verdampfen des Lösungsmittels
ergab 87 g (Ausbeute: 86%) eines farblosen hochviskosen Öls.
IR
(Film, cm
–1)
3411, 3278 (NH), 2957 (CH
2/CH
3),
1656 (NHCO), 1546 (C=C), 1241 (CH
2/CH
3), 984/956 (C=C), 703 (C=C)
1H-NMR (250 MHz, CDCl
3,
ppm) 0,7-3,4 (mehrere m, 18H, CH
3, CH
2), 5,51-5,60 (m, 1H, CH=C-CO), 6,26-6,26 (m,
2H, CH=CH-CO), 7,12, 6,84, 6,70, 6,59 (4 × t, 2H, NH)
13C-NMR
(63 MHz, CDCl
3, ppm) 20,80, 22,21, 25,36,
26,10, 26,75, 27,90, 28,02, 29,04, 30,73, 32,80, 35,00, 35,68, 37,21,
38,79, 40,51, 45,16, 46,02, 47,02, 48,70, 125,66 (C=), 131,08 (C=),
165,82 (C=O), 166,09 (C=O) Beispiel
6
-
JEFFAMINE T-403 Polyoxypropylentriamintris(acrylamid)
(11)
-
Zu
einer Lösung
von 73,25 g (0,166 Mol) JEFFAMINE T-403 Polyoxypropylentriamin (x+y+z
= 5,3) in 100 ml Methylenchlorid wurde eine Lösung von 47,46 g (0,524 Mol)I
Acryloylchlorid in 130 ml Methylenchlorid und eine Lösung von
20,98 g (0,524 Mol) Natriumhydroxid in 200 ml Wasser gleichzeitig
unter Rühren
so zugegeben, dass die Temperatur bei 0 bis 5°C verblieb. Danach wurde die
Mischung bei Raumtemperatur zusätzliche
2 Stunden lang gerührt.
Die Umsetzung wurde durch Zugabe von 300 ml Wasser beendet. Die
organische Phase wurde abgetrennt und die wässerige Lösung wurde zweimal mit 100
ml Methylenchlorid extrahiert. Die vereinigten organischen Flüssigkeiten
wurden mit 150 ml 1 N HCl, 150 ml 1 N NaHCO3 und
mit 150 ml Wasser gewaschen. Die Lösung des Produkts wurde mit
0,025 Mol% BHT stabilisiert und über
Natrumsulfat getrocknet. Filtration und Verdampfen des Lösungsmittels
ergaben 86,1 g (Ausbeute: 85%) eines klaren gelblichen hochviskosen Öls.
IR
(Film, cm–1)
3411, 3280 (OH), 2970, 2929, 2872 (CH2/CH3), 1657/1622 (CONH), 1542 (C=C), 1406/1374 (CH2/CH3), 1101 cm–1 (ROR)
1H-NMR (250 MHz, CDCl3,
ppm) 0,74-0,95 (t), 0,99-1,25 (m), 1,27-1,53 (m), 2,90-3,73 (m),
4,04-4,31 (breit s), 5,49-5,73 (m, 1H, CH=C-CO), 5,90-6,43 (m, 2H,
CH=CH-CO)
13C-NMR (63 MHz, CDCl3, ppm) 7,5 (CH2-CH3), 17,0/17,5 (CH-CH3),
22,9, 32,0, 45,1/45,2 (C-N), 71,6/75,1 (CH2-O),
125,7 (C=), 131,1 (C=), 164,9 (C=O)
-
Test 1
-
Untersuchung der hydrolytischen
Stabilität
von N-substituierten Alkylacryl- oder Acrylsäureamidmonomeren, die eine
Säurekomponente
enthalten
-
Als
typisches Beispiel eines N-substituierten Alkylacryl- oder Acrylsäureamidmonomers
wurde N-[2-(Dihydroxyphosphoryl)ethyl]-N-butylacrylamid
9 und als Vergleichsbeispiel Dihydroxyphosphorylmethylmethacrylester
den folgenden Bedingungen unterworfen:
1,5 mMol des Acrylsäureamidmonomers
wurden in 5 g einer Mischung bestehend aus 50 Gew.-% Ethanol und 50
Gew.-% Wasser gelöst.
Die Lösungen
wurden in einem geschlossenen Gefäß bei 50°C gelagert. Die Art und das
Ausmaß der
Hydrolyse wurde mittels HPLC bestimmt.
-
Nach
1 Woche sind 50% und nach 6 Wochen 83% des Esters in Methacrylsäure und
Hydroxymethylphosphonsäure
hydrolysiert, während
das Acrylamid 9 eine Hydrolyse in Acrylsäure und Amin von 0% während 1
Woche und von 7,9% nach 6 Wochen zeigte.
-
Test 2
-
Prüfung der hydrolytischen Stabilität von N-substituierten
Acrylsäureamidmonomeren,
die keine Säurekomponente
enthalten
-
Als
Modellverbindung für
ein N-substituiertes Acrylsäureamidmonomer
wurde n-Butylacrylamid und als Vergleichsbeispiel n-Butylacrylester
den folgenden Bedingungen unterworfen:
1,5 mMal des Acrylamidmonomers
wurden zusammen mit 1,5 mMol Ethansulfonsäure in 5 g einer Mischung bestehend
aus 50 Gew.-% Ethanol und 50 Gew.-% Wasser gelöst. Die Lösungen wurden in einem geschlossenen
Gefäß bei 50°C gelagert.
Die Art und das Ausmaß der
Hydrolyse wurde mittels HPLC bestimmt.
-
Nach
1 Woche sind 62% des Esters zu Acrylsäure und Butanol hydrolysiert,
während
das Acrylamid bis 7 Woschen keine Hydrolyse zeigte.
-
Anwendungsbeispiel 1
-
381,85
mg (2,2(4),4)-Trimethylhexamethylenbis(acrylamid) (10), 416,85 mg
JEFFAMINE T-403 Polyoxypropylentriamintris(acrylamid) (11), 286,00
mg N-[2-Dihydroxyphosphoryl)ethyl]-N-butylacrylamid (9), 17,70 mg
Bis(2,4,6-trimethylbenzol)phenylphosphonoxid, 8,20 mg Dimethylaminoethylbenzoesäu reethylester und
7,10 mg Campherchinon wurden in einer Lösung aus 166,67 mg Ameisensäure, 66,67
mg Ethanol und 266,67 mg Wasser gelöst.
-
Präparation der Zähne
-
Zur
Adhäsion
wird der Zahnschmelz mit 500 Grit-Siliciumcarbid-Papier so abgeschliffen,
dass eine flache Fläche
von Zahnschmelz von ca. 5 min Durchmesser vorhanden ist. Die Zähne werden
dann unter fließendem
Wasser gewaschen und innerhalb von 2 Stunden wie nachstehend angegeben
verwendet.
-
Präparation der Adhäsionsproben
-
Gelatinekapseln
(#5, bezogen von Torpac Inc.) für
die Tests wurden auf ca. zwei Drittel ihrer Länge mit Spectrum TPH gefüllt, und
dieses durch Stellen der Kapseln in einen Lichtofen gehärtet. Für den Test
wurden sechs Zähne
hergestellt.
-
Die
zu beklebende Zahnoberfläche
wird leicht mit einem Papiertuch oder einem 5-sekündigen Luftstrahl
getrocknet, und die Behandlungslösung
wird unter Verwendung einer Applikatorspitze oder einer Bürste appliziert.
Das Material wird mit dem Zahn 20 Sekunden lang in Kontakt gehalten,
mit Luft 5 Sekunden lang getrocknet und 10 Sekunden lang mit einer
Spektrumlampe 800 gehärtet.
Spectrum TPH wird dann in den verbleibenden Raum der vorgefüllten Gelatinekapsel
eingefüllt
und auf die präparierte
Zahnschmelzoberfläche gegeben.
Spectrum TPH wird dann durch dreimaliges Bestrahlen während 20
Sekunden in gleichen Intervallen um die Kapsel gehärtet.
-
Nach
Lagerung bei 37°C
während
2 Stunden beträgt
die Klebefestigkeit am Zahnschmelz 10,5 ± 2,8 MPa.
-
Wenn
die nach dem vorstehend beschriebenen Verfahren hergestellten Proben
1800 Mal zwischen 5 und 55°C
mit einer Verweilzeit in jedem Bad von 20 Sekunden thermozyklisiert
wurden, wurde eine Haftfestigkeit am Zahnschmelz von 10,7 ± 2,2 MPa
gemessen.
-
Weitere Beispiele:
-
Hydrolysestabile polymerisierbare
N-substituierte Alkylacryl- und Acrylsäureamidmonomere
-
Beispiel 7
-
N,N'-Bismethacryloyl-N,N'-dibenryl-5-oxanonandiamin-1,9
-
In
einem 1 l-Vierhalskolben, der mit einem Rührer, einem Thermometer und
zwei 50 ml-Tropftrichtern ausgestattet war, wurden 102,16 g (0,3
Mol) N,N'-Dibenzyl-5-oxanonandiamin-1,9
in 300 ml Methylenchlorid gelöst.
Nach Kühlen
auf 0 bis 5°C
wurden 65,854 g (0,63 Mol) Methacryloylchlorid, gelöst in 30
ml Methylenchlorid, und 25,20 g (0,63 Mol) NaOH, gelöst in 60
ml Wasser, gleichzeitig unter Rühren
während
1,5 Stunden so zugegeben, dass die Temperatur bei 0 bis 5°C verblieb.
Danach wurde die Mischung bei Raumtemperatur zusätzliche 2 Stunden lang gerührt. Dann
wurde die Reaktionsmischung mit 600 ml Eiswasser hydrolysiert. Die
organische Phase wurde abgetrennt und die wässerige Lösung wurde zweimal mit Methylenchlorid
extrahiert. Die vereinigten organischen Flüssigkeiten wurden mit 150 ml
1 N HCl, 150 ml 1 N NaHCO3 und einige Male
mit 150 ml deionisiertem Wasser gewaschen, bis das Was ser einen
pH-Wert von ca. 7 zeigte. Dann wurde die organische Lösung über Natruimsulfat
getrocknet. Danach wurde das NaSO4 abfiltriert
und zur Lösung 0,1346
g 2,6-Di-tert-butyl-p-cresol zugegeben. Das Methylenchlorid wurde
bei 40°C
im Vakuum entfernt und das Bismethacrylamid wurde getrocknet.
Ausbeute:
136,66 g (95,6% Theorie), nD 20 =
1,5383, n = 1,65 Pa·s
C30H40N2O3, 476,65
IR: 2941 (CH2/CH3), 3086/3062/3030 (Ar), 1647 (CONR), 1626
(CH2=CH-), 1119 cm–1 (ROR)
-
Beispiel 8
-
N,N'-Bisacryloyl-N,N'-dibenzylethylendiamin
-
In
einem 1 l-Vierhalskolben, der mit einem Rührer, einem Thermometer und
zwei 50 ml-Tropftrichtern ausgestattet war, wurden 29,198 g (0,12
Mol) N,N'-Dibenzylethylendiamin
in 100 ml Methylenchlorid gelöst. Nach
Kühlen
auf 0 bis 5°C
wurden 21,991 g (0,24 Mol) Acryloylchlorid, gelöst in 30 ml Methylenchlorid,
und 9,718 g (0,24 Mol) NaOH, gelöst
in 40 ml Wasser, gleichzeitig unter Rühren während 1,5 Stunden so zugegeben,
dass die Temperatur bei 0 bis 5°C
verblieb. Danach wurde die Mischung bei Raumtemperatur zusätzliche 2
Stunden lang gerührt.
Dann wurde die Reaktionsmischung mit 600 ml Eiswasser hydrolysiert.
Die organische Phase wurde abgetrennt und die wässerige Lösung wurde zweimal mit Methylenchlorid
extrahiert. Die gesammelten organischen Flüssigkeiten wurden mit 100 ml
1 N HCl, 100 ml 1 N NaHCO3 und einige Male
mit 100 ml deionisiertem Wasser gewaschen, bis das Wasser einen
pH-Wert von ca. 7 zeigte. Dann wurde die organische Lösung über Natriumsulfat
getrocknet. Danach wurde das NaSO4 abfiltriert
und zur Lösung
0,028 g 2,6-Di-tert-butyl-p-cresol zugegeben. Das Methylenchlorid
wurde bei 40°C
im Vakuum entfernt und das Bismethacrylamid wurde getrocknet.
Ausbeute:
27,9 g (65,9% Theorie), Schmp. = 75,5-76,6°C, Tg = –7,2°C, Mn (vpo)
= 350 g/Mol
C22H24N2O2, 348,45 berechnet
C 75,83 H 6,94 N 8,04
gefunden C 76,00 H 7,26 N 8,05
-
Beispiel 9
-
N,N'-Bisacryloyl-N,N'-dibenzyl-4,4'-diaminodicyclohexylamin
-
In
einem 1 l-Vierhalskolben, der mit einem Rührer, einem Thermometer und
zwei 50 ml-Tropftrichtern ausgestattet war, wurden 60,551 g (0,16
Mol) N,N'-Dibenzyl-4,4'-diaminodicyclohexylamin
in 150 ml Methylenchlorid gelöst.
Nach Kühlen
auf 0 bis 5°C
wurden 28,061 g (0,31 Mol) Acryloylchlorid, gelöst in 30 ml Methylenchlorid,
und 12,401 g (0,31 Mol) NaOH, gelöst in 50 ml Wasser, gleichzeitig
unter Rühren
während
1,5 Stunden so zugegeben, dass die Temperatur bei 0 bis 5°C verblieb.
Danach wurde die Mischung bei Raumtemperatur zusätzliche 2 Stunden lang gerührt. Dann
wurde die Reaktionsmischung mit 500 ml Eiswasser hydrolysiert. Die
organische Phase wurde abgetrennt und die wässerige Lösung wurde zweimal mit Methylenchlorid extrahiert.
Die gesammelten organischen Flüssigkeiten
wurden mit 100 ml 1 N HCl, 100 ml 1 N NaHCO3 und einige
Male mit 100 ml deionisiertem Wasser gewaschen, bis das Wasser einen
pH-Wert von ca. 7 zeigte. Dann wurde die organische Lösung über Natriumsulfat
getrocknet. Danach wurde das NaSO4 abfiltriert
und zur Lösung
0,077 g 2,6-Di-tert-butyl- p-cresol
zugegeben. Das Methylenchlorid wurde bei 40°C im Vakuum entfernt und das
Bismethacrylamid wurde getrocknet.
Ausbeute: 54,0 g (69,9%
Theorie), Tg = 47,1 °C
-
Beispiel 10
-
3,(4),8,(9)-Bis(2-propenamidomethyl)tricyclo-5.2.1.02,6-decan
-
In
einem 1 l-Vierhalskolben, der mit einem Rührer, einem Thermometer und
zwei 50 ml-Tropftrichtern ausgestatet war, wurden 96,01 g (0,35
Mol) 3,(4),8,(9)-Bis(aminomethyl)tricyclo-5.2.1.02,6-decan
in 350 ml Methylenchlorid gelöst.
Nach Kühlen
auf 0 bis 5°C
wurden 76,54 g (0,735 Mol) Methacryloylchlorid, gelöst in 35 ml
Methylenchlorid, und 29,40 g (0,735 Mol) NaOH, gelöst in 70
ml Wasser, gleichzeitig unter Rühren
während 1,5
Stunden so zugegeben, dass die Temperatur bei 0 bis 5°C verblieb.
Danach wurde die Mischung bei Raumtemperatur zusätzliche 2 Stunden lang gerührt. Dann
wurde die Reaktionsmischung mit 600 ml Eiswasser hydrolysiert. Die
organische Phase wurde abgetrennt und die wässerige Lösung wurde zweimal mit Methylenchlorid
extrahiert. Die gesammelten organischen Flüssigkeiten wurden mit 150 ml
1 N HCl, 150 ml 1 N NaHCO3 und einige Male
mit 150 ml deionisiertem Wasser gewaschen, bis das Wasser einen
pH-Wert von ca. 7 zeigte. Dann wurde die organische Lösung über Natriumsulfat
getrocknet. Danach wurde das NaSO4 abfiltriert
und zur Lösung
0,1157 g 2,6-Di-tert-butyl-p-cresol
zugegeben. Das Methylenchlorid wurde bei 40°C im Vakuum entfernt und das
Bismethacrylamid wurde getrocknet.
Ausbeute: 106,02 g(91,6%
Theorie)
C20H30N2O2, 330,47
IR:
2941 (CH2/CH3),
3330/1647 (CONHR), 1626 (CH2=CH-)
-
Beispiel 11
-
N,N'-Bismethacryloyl-3,6-dioxaoctandiamin-1,8
-
In
einem 1 l-Vierhalskolben, der mit einem Rührer, einem Thermometer und
zwei 50 ml-Tropftrichtern ausgestattet war, wurden 59,28 g (0,4
Mol) 3,6-Dioxaoctandiamin-1,8 in 300 ml Methylenchlorid gelöst. Nach Kühlen auf
0 bis 5°C
wurden 87,47 g (0,84 Mol) Methacryloylchlorid, gelöst in 40
ml Methylenchlorid, und 33,60 g (0,84 Mol) NaOH, gelöst in 80
ml Wasser, gleichzeitig unter Rühren
während
1,5 Stunden so zugegeben, dass die Temperatur bei 0 bis 5°C verblieb.
Danach wurde die Mischung bei Raumtemperatur zusätzliche 2 Stunden lang gerührt. Dann
wurde die Reaktionsmischung mit 600 ml Eiswasser hydrolysiert. Die
organische Phase wurde abgetrennt und die wässerige Lösung wurde zweimal mit Methylenchlorid
extrahiert. Die gesammelten organischen Flüssigkeiten wurden mit 150 ml
1 N HCl, 150 ml 1 N NaHCO3 und einige Male
mit 150 ml deionisiertem Wasser gewaschen, bis das Wasser einen
pH-Wert von ca. 7 zeigte. Dann wurde die organische Lösung über Natriumsulfat
getrocknet. Danach wurde das NaSO4 abfiltriert
und zur Lösung
0,1137 g 2,6-Di-tert-butyl-p-cresol zugegeben. Das Methylenchlorid
wurde bei 40°C
im Vakuum entfernt und das Bismethacrylamid wurde getrocknet.
Ausbeute:
95,20 g (83,7% Theorie), nD 20 =
1,5042, η =
2,17 Pa·s
C14H24N2O4, 284,35; IR: 2926/2870 (CH2/CH3), 3336/1659 (CONHR), 1620 (CH2=CH-),
1130 cm–1 (ROR)