-
Die
vorliegende Anmeldung bezieht sich auf die US-Voranmeldung mit der
Seriennummer 60/329,236, eingereicht am 12. Oktober 2001.
-
Gebiet der
Erfindung
-
Die
vorliegende Erfindung betrifft neue heterocyclische Verbindungen
und Zusammensetzungen sowie deren Verwendung zur Herstellung eines
Medikaments zur Behandlung oder Vorbeugung von Fettleibigkeit und
mit Fettleibigkeit zusammenhängenden
Krankheiten.
-
Hintergrund
der Erfindung
-
Fettleibigkeit,
die als überschüssiges Körperfett
relativ zu einer mageren Körpermasse
definiert ist, stellt einen allgemein anerkannten Risikofaktor für eine Anzahl
potenziell lebensbedrohender Krankheiten wie für Atherosklerose, Überdruck,
Diabetes, Schlaganfall, Lungenembolie, Schlaf-Atmungsstillstand und für Krebs
dar. Ferner macht sie zahlreiche chronische Bedingungen wie Atmungskrankheiten,
Osteoarthritis, Osteoporose, Gallenblasenkrankheit und Dyslipidämien kompliziert.
Das ungeheure Ausmaß dieses
Problems spiegelt sich am besten in der Tatsache, dass die Todesraten
mit ansteigendem Körpergewicht
eskalieren. Mehr als 50 % der Gesamt-Sterblichkeit kann mit Fettleibigkeit
zusammenhängenden
Bedingungen zugeordnet werden, sobald der Körpermassenindex (body mass
index = BMI) 30 kg/m2 übersteigt, wie dies bei 35
Millionen Amerikanern der Fall ist (Lee, JAMA 268:2045–2049, 1992).
Durch den Beitrag von mehr als 300.000 Todesfällen pro Jahr steht Fettleibigkeit
an zweiter Stelle, lediglich übertroffen
von Tabak-Rauchen als der häufigsten
Ursache eines potenziell vermeidbaren Todes (McGinnis, JAMA 270:2207–2212, 1993).
Mit den ausufernden medizinischen Konsequenzen dieses Problems geht
die ernste finanzielle Belastung einher, die dem Gesundheitsvorsorgesystem
in den Vereinigten Staaten auferlegt ist. Es wird geschätzt, dass
30 bis 50 % der Bevölkerung
mittleren Alters als fettleibig betrachtet werden können (Kuczmarski
et al., JAMA 272:205–211, 1994).
Die wirtschaftlichen Auswirkungen von Fettleibigkeit und ihren zugehörigen Krankheiten
durch medizinisch bedingte Ausgaben und Einkommensverluste werden
mit mehr als 68 Milliarden Dollar/Jahr angegeben (Colditz, Am. J.
Clin. Nutr. 55:503S–507S,
1992). Diese Zahl beinhaltet nicht die mehr als 30 Milliarden Dollar pro
Jahr, die für
Nahrung zur Gewichtsabnahme sowie für entsprechende Produkte und
Programme ausgegeben werden (Wolf, Pharmacoeconomics 5:34–37, 1994).
-
Die
Anhäufung
oder Beibehaltung von Körperfett.
weist eine direkte Beziehung zur Kalorieneinnahme auf. Umfassende
Behandlungsprogramme waren daher auf Verhaltensmodifikationen gerichtet,
um die Kalorieneinnahme zu verringern und körperliche Aktivitäten mit
unzähligen
Systemen zu steigern. Diese Methoden weisen eine nur eingeschränkte Wirksamkeit
auf und stehen in Verbindung mit Rückfallraten von mehr als 95 %
(NIH Technology Assessment Conference Panel, Ann. Intern. Med. 119:764–770, 1993).
-
Fettleibigkeit
ist auch durch Verabreichung spezifischer Mittel, z.B. von anorektischen
Mitteln, an fettleibige Personen behandelt worden. Allerdings stehen
anorektische Mittel wie Dextroamphetamin, die Kombination der Nicht-Amphetamin-Arzneien
von Phentermin mit Fenfluramin (Phen-Fen) sowie von Dexfenfluramin (Redux)
alleine, in Zusammenhang mit ernsthaften Nebenwirkungen. Unverdauliche
Materialien wie Olestra (OLEAN
®, Mineralöl oder Neopentylester
(siehe
US 2,962,419 ))
sind als Ersatzstoffe für
Nahrungsfett vorgeschlagen worden. Garciniasäure und Derivate davon sind
als Behandlungsmittel von Fettleibigkeit durch Eingriff in die Fettsäure-Synthese beschrieben
worden. Quellbare vernetzte Vinylpyridinharze sind als Appetitzügler über den
Mechanismus einer Bereitstellung von Nicht-Nährstoffmasse
beschrieben worden (siehe
US 2,923,662 ).
-
Chirurgische
Eingriffe, wie gastrische Zerteilungsoperationen, Leerdarm-Bypässe und
Vagotomie, sind ebenfalls entwickelt worden, um ernsthafte Fettleibigkeit
zu behandeln (Greenway, Endo. Metab. Clin. N. Amer. 25:1005–1027, 1996).
Obwohl diese chirurgischen Maßnahmen
etwas wirkungsvoller auf lange Sicht sind, hat das Verhältnis von
akutem Risiko zu Nutzen diese invasiven Operationseingriffe für krankhaft
fettleibige Patienten gemäß der National
Health Institutes (NIH)-Konsenskonferenz über Fettleibigkeitschirurgie (BMI > 40 kg/m2)
vorbehalten gelassen (NIH Conference, Ann. Intern. Med. 115:956–961, 1991).
Daher stellt dieser Lösungsansatz
keine Alternative für
die große
Mehrheit von Patienten mit Übergewicht
dar, es sei denn, sie werden tiefgreifend fettleibig und leiden
an den einhergehenden Komplikationen und/oder sie gelangen dahin.
-
Daher
werden neue Vorgehensweisen und Zusammensetzungen dringend benötigt, die
eine Gewichtsabnahme begünstigen
und fördern.
-
Zusammenfassung der Erfindung
-
Die
vorliegende Erfindung betrifft Verbindungen und Zusammensetzungen
sowie deren Verwendung zur Herstellung eines Medikaments zur Behandlung
und Vorbeugung von Fettleibigkeit und damit zusammenhängenden
Krankheiten.
-
Demnach
ist es eine Aufgabe der vorliegenden Erfindung, Verbindungen der
Formeln (Ia) bis (Ie) bereitzustellen:
-
-
Eine
weitere Aufgabe der Erfindung ist es, Zusammensetzungen, die eine
wirkungsvolle Menge mindestens einer Verbindung der Formeln (Ia)
bis (Ie) umfassen, zur Behandlung von Fettleibigkeit eines Säugers bereitzustellen.
-
Diese
und weitere Gegenstände
der Erfindung werden im Lichte der nun folgenden detaillierten Beschreibung
noch klarer.
-
Detaillierte Beschreibung
der Erfindung
-
Die
Erfindung betrifft Verbindungen der Formel (Ia):
worin gilt:
R
1 stellt eine Gruppe der Formel dar:
worin gilt:
Z stellt
O oder S dar,
R
1-1 stellt Wasserstoff
oder (C
1-6)Alkyl dar,
R
1-2 stellt
Wasserstoff oder (C
1-6)Alkyl dar, und
R
1-3 stellt Wasserstoff oder (C
1-6)Alkyl
dar;
R
2 stellt Wasserstoff oder Methyl
dar; oder
R
1 und R
2 stellen
gemein eine Gruppe dar:
die, zusammen mit den Kohlenstoffen,
an die die genannte Gruppe gebunden ist, einen carbocyclischen Ring bildet,
worin
R
1-4 Wasserstoff oder (C
1-6)Alkyl
und
R
1-5 Wasserstoff oder (C
1-6)Alkyl darstellen;
R
3 stellt
Wasserstoff oder Methyl dar;
Y stellt NR
4,
O oder S dar, worin
R
4 Wasserstoff
oder (C
1-6)Alkyl darstellt, das gegebenenfalls
mit 1 oder 2 Substituenten substituiert ist, die unabhängig aus
der Gruppe ausgewählt
sind, bestehend aus (C
1-6)Alkoxy und (C
1-6)Aryloxy;
R
5 stellt
Wasserstoff, (C
1-6)Alkyl oder Phenyl dar,
gegebenenfalls substituiert mit Halogen, (C
1-6)Alkyl
oder mit (C
1-6)Alkoxy;
R
6 stellt
Benzyloxycarbonylamino oder eine Gruppe der Formel dar:
worin gilt:
R
6-1 stellt dar:
– Hydroxy,
– (C
1-6)Alkoxy,
– Benzyloxy,
– eine Gruppe
der Formel:
worin
R
6-1-1 darstellt:
Wasserstoff,
(C
1-6)Alkyl, gegebenenfalls substituiert mit
1 oder 2 Substituenten, unabhängig
ausgewählt
aus der Gruppe, bestehend aus (C
6-10)Aryl
und (C
1-6)Alkoxy, (C
3-8)Cycloalkyl,
gegebenenfalls substituiert mit (C
1-6)Alkyl, (C
6-10)Aryl, gegebenenfalls substituiert mit
1, 2 oder 3 Substituenten, unabhängig
ausgewählt
aus der Gruppe, bestehend aus Halogen, Nitro, Cyano, (C
1-6)Alkyl,
(C
3-8)Cycloalkyl, (C
1-6)Alkoxycarbonyl,
Hydroxy, (C
1-6)Alkoxy, Trifluormethyl, Trifluormethoxy,
Heterocyclyl, (C
6-10)Aryl, (C
6-10)Aryloxy
und aus Benzyl, oder
einen 5- bis 10-gliedrigen heterocyclischen
Rest, umfassend 3 bis 9 Kohlenstoffatome und 1 bis 3 Heteroatome,
ausgewählt
aus O, N oder S, gegebenenfalls substituiert mit Phenyl, Benzyl
oder mit Halogen, und worin
R
6-1-2 Wasserstoff,
(C
1-6)Alkyl oder (C
3-8)Cycloalkyl
darstellt,
– eine
Gruppe der Formel -NH-NH-R
6-2, worin R
6-2 (C
6-10)Aryl darstellt,
oder
– einen
5- bis 10-gliedrigen heterocyclischen Rest, umfassend 3 bis 9 Kohlenstoffatome
und 1 bis 3 Heteroatome, ausgewählt
aus O, N oder S, und gegebenenfalls substituiert mit 1 oder 2 Substituenten,
unabhängig ausgewählt aus
der Gruppe, bestehend aus (C
1-6)Alkyl, Benzyl
oder aus Phenyl, gegebenenfalls substituiert mit (C
1-6)Alkyl;
und
pharmazeutisch geeignete Salze oder Ester davon.
-
Die
Erfindung betrifft auch Verbindungen der Formel (Ib):
worin gilt:
R
7 stellt eine Gruppe der Formel dar:
worin gilt:
Z stellt
S dar,
R
7-1 stellt Wasserstoff oder
(C
1-6)Akyl dar,
R
7-2 stellt
Wasserstoff oder (C
1-6)Alkyl dar,
R
7-3 stellt Wasserstoff oder (C
1-6)Alkyl
dar;
R
8 stellt Wasserstoff oder Methyl
dar;
R
9 stellt stellt Wasserstoff oder
Methyl dar;
R
10 stellt (C
1-6)Alkyl
dar, das gegebenenfalls mit 1 oder 2 Substituenten substituiert
ist, unabhängig
ausgewählt aus
der Gruppe, bestehend aus (C
1-6)Alkoxy und
(C
6-10)Aryloxy;
R
11 stellt
eine Gruppe der Formel dar;
worin gilt:
R
11-1 stellt dar:
– Hydroxy,
– (C
1-6)Alkoxy,
– einen 5- bis 10-gliedrigen
heterocyclischen Rest, umfassend 3 bis 9 Kohlenstoffatome und 1
bis 3 Heteroatome, ausgewählt
aus O, N oder S, gegebenenfalls substituiert mit 1 oder 2 (C
1-6)Alkyl- oder Phenylresten, gegebenenfalls
substituiert mit Halogen, oder
– eine Gruppe der Formel:
worin
R
11-1-1 (C
6-10)Aryl, das gegebenenfalls mit bis zu
3 Substituenten substituiert ist, unabhängig ausgewählt aus Halogen, Nitro, Cyano,
(C
1-6)Alkyl, (C
1-6)Alkoxy,
Trifluormethyl oder aus Phenyl, oder einen 5- bis 10-gliedrigen heterocyclischen
Rest, umfassend 3 bis 9 Kohlenstoffatome und 1 bis 3 Heteroatome,
ausgewählt
aus O, N oder S, und
R
11-1-2 Wasserstoff
darstellen;
R
12 stellt Wasserstoff
oder (C
1-6)Alkyl dar;
und pharmazeutisch
geeignete Salze oder Ester davon.
-
Die
Erfindung betrifft auch Verbindungen der Formel (Ic):
worin gilt:
R
13 stellt eine Gruppe der Formel dar:
worin gilt:
Z stellt
S dar,
R
13-1 stellt Wasserstoff oder
(C
1-6)Alkyl dar,
R
13-2 stellt
Wasserstoff oder (C
1-6)Alkyl dar,
R
13-3 stellt Wasserstoff oder (C
1-6)Alkyl
dar;
R
14 stellt Wasserstoff oder Methyl
dar;
R
15 stellt Wasserstoff oder Methyl
dar;
R
16 stellt eine Gruppe der Formel
dar:
worin gilt:
R
16-1 stelt eine Gruppe der Formel dar:
worin:
R
16-1-1 (C
6-10)Aryl, das gegebenenfalls mit 1, 2 oder
3 Substituenten substituiert ist, unabhängig ausgewählt aus Halogen, Nitro, Cyano,
(C
1-6)Alkyl, (C
1-6)Alkoxy
und aus Trifluormethyl, und
R
16-1-2 Wasserstoff
darstellen;
R
17 stellt (C
1-6)Alkyl
dar, das gegebenenfalls mit 1 oder 2 (C
1-6)-Alkylresten
substituiert ist;
und pharmazeutisch geeignete Salze oder Ester
davon.
-
Die
Erfindung betrifft auch Verbindungen der Formel (Id):
worin gilt:
R
18 stellt eine Gruppe der Formel dar:
worin gilt:
Z stellt
S dar,
R
18-1 stellt Wasserstoff oder
(C
1-6)Alkyl dar;
R
18-2 stellt
Wasserstoff oder (C
1-6)Alkyl dar, und
R
18-3 stellt Wasserstoff oder (C
1-6)Alkyl
dar;
R
19 stellt Wasserstoff oder Methyl
dar;
R
20 stellt Wasserstoff oder Methyl
dar;
R
21 stellt (C
1-6)Alkoxy
dar;
R
22 stellt Wasserstoff, (C
1-6)Alkyl oder Phenyl dar;
R
23 stellt eine Gruppe der Formel dar:
worin
R
23-1 Hydroxy,
(C
1-6)Alkoxy oder Benzyloxy oder eine Gruppe
der Formel darstellt:
worin
R
23-1-1 (C
6-10)Aryl, das gegebenenfalls mit bis zu
3 Substituenten substituiert ist, unabhängig ausgewählt aus Halogen, Nitro, Cyano,
(C
1-6)Alkyl, (C
1-6)Alkoxy
und aus Trifluormethyl, und
R
23-1-2 Wasserstoff
darstellen;
und pharmazeutisch geeignete Salze oder Ester davon.
-
Schließlich betrifft
die Erfindung auch Verbindungen der Formel (Ie):
worin gilt:
R
24 stellt eine Gruppe der Formel dar:
worin gilt:
Z stellt
S dar,
R
24-1 stellt Wasserstoff oder
(C
1-6)Alkyl dar,
R
24-2 stellt
Wasserstoff oder (C
1-6)Alkyl dar,
R
24-3 stellt Natrium, Wasserstoff oder (C
1-6)Alkyl dar;
R
25 stellt
Wasserstoff oder Methyl dar;
R
26 stellt
Wasserstoff oder Methyl dar;
R
27 stellt
Phenyl dar;
R
28 stellt Wasserstoff
dar;
und pharmazeutisch geeignete Salze oder Ester davon.
-
Die
oben identifizierten Begriffe haben durchgängig die folgende Bedeutung:
"*" betrifft den Punkt der Bindung.
"Halogen" bedeutet Fluor,
Chlor, Brom oder Jod.
Der Begriff "(C1-6)Alkyl" bedeutet jeweils
lineare oder verzweigte C1-6-Alkylgruppen.
Beispielsweise schließt
er Gruppen wie Methyl-, Ethyl-, Propyl- oder Isopropylgruppen ein.
Der
Begriff "(C3-8)Cycloalkyl" bedeutet einen Rest eines gesättigten
carbocyclischen Rings mit 3 bis 8 Kohlenstoffatomen, wie Cyclopropyl-,
Cyclopentyl-, Cyclohexyl- oder Cyclooctylgruppen.
Der Begriff "(C1-6)Alkoxy" bedeutet (C1-6)Alkoxyreste wie Methoxy-, Ethoxy-, Isopropoxy-
oder n-Hexyloxygruppen.
Der Begriff "(C1-6)Alkylcarbonyl" bedeutet (C1-6)Alkyl-C(=O)-Reste wie Acetyl, Propanoyl,
Pentanoyl oder Isobutyryl.
Der Begriff "(C6-10)Aryl" bedeutet einen Rest
eines monocyclischen oder kondensierten bicyclischen aromatischen
Rings mit 6 bis 10 Kohlenstoffatomen wie Phenyl oder Naphthyl.
Der
Begriff "(C6-10)Aryloxy" bedeutet (C6-10)Aryloxyreste,
wie Phenoxy oder Naphthyloxy.
Der Begriff "5- bis 10-gliedriger heterocyclischer
Rest" bedeutet ein
monocyclisches oder kondensiertes bicyclisches aromatisches System,
das insgesamt 5 bis 10 Atome enthält, von denen 1 bis 3 Atome
Heteroatome sind, ausgewählt
aus der Gruppe aus Stickstoff, Sauerstoff und Schwefel, wobei die
restlichen Atome Kohlenstoffatome sind.
-
Wird
ein Rest als substituiert beschrieben, kann er 1 oder mehr der angegebenen
Substituenten aufweisen, die an einer verfügbaren Position auf dem Rest
angeordnet sein können.
Liegen 2 oder mehr Substituenten an einem Rest vor, soll jeder unabhängig von
einander in jedem Fall definiert sein. Repräsentative Salze der Verbindungen
der Formeln (Ia) bis (Ie) schließen schließen die herkömmlichen,
nicht-toxischen Salze und die quartären Ammoniumsalze ein, die
z.B. aus anorganischen oder organischen Säuren oder Basen mit im Stand
der Technik gut bekannten Mitteln und Maßnahmen gebildet werden. Beispielsweise
schließen
derartige Säureadditionssalze
Acetat, Adipat, Alginat, Ascorbat, Aspartat, Benzoat, Benzolsulfonat,
Bisulfat, Butyrat, Zitrat, Kamphorat, Kampfersulfonat, Zinnamat,
Cyclopentanpropionat, Digluconat, Dodecylsulfat, Ethansulfonat,
Fumarat, Glucoheptanoat, Glycerophosphat, Hemisulfat, Heptanoat,
Hexanoat, Hydrochlorid, Hydrobromid, Hydrojodid, 2-Hydroxyethansulfonat,
Itaconat, Lactat, Maleat, Mandelat, Methansulfonat, 2-Naphthalinsulfonat,
Nicotinat, Nitrat, Oxalat, Pamoat, Pectinat, Persulfat, 3-Phenylpropionat,
Picrat, Pivalat, Propionat, Succinat, Sulfonat, Tartrat, Thiocyanat,
Tosylat und Undecanoat ein.
-
Basensalze
schließen
Alkalimetallsalze wie Kalium- und Natriumsalze, Erdalkalimetallsalze
wie Calcium- und Magnesiumsalze und Ammoniumsalze mit organischen
Basen wie Dicycloaminsalze und N-Methyl-D-glucamin ein.
-
Außerdem können basische
Stickstoff-haltige Gruppen mit solchen Mitteln wie mit Niederalkylhalogeniden
wie mit Methyl-, Ethyl-, Propyl- und Butylchloriden, -bromiden und
-jodiden, mit Dialkylsulfaten wie mit Dimethyl-, Diethyl-, Dibutyl-
und Diamylsulfaten, mit langkettigen Halogeniden wie mit Decyl-,
Lauryl-, Myristyl- und Stearylchloriden, -bromiden und -jodiden,
mit Aralkylhalogeniden wie mit Benzyl- und Phenethylbromiden und
mit weiteren Mitteln quaterniert werden.
-
Die
Ester in der vorliegenden Erfindung sind nicht-toxische, pharmazeutisch
zulässige
Esterderivate der Alkohole der Formeln (Ia) bis (Ie). Diese schließen Esterderivate
ein, hergestellt aus Essig-, Benzoe-, Mandel-, Stearin-, Milch-,
Salicyl-, Hydroxynaphthoe-, Glucoheptan- und aus Gluconsäure. Die
Alkoholverbindungen der Formeln (Ia) bis (Ie) können mit einer Vielzahl herkömmlicher
Verfahren verestert werden, einschließlich einer Reaktion der entsprechenden
Anhydride, Carbonsäuren
und Säurechloride
mit der Alkoholgruppe der Verbindung der Formeln (Ia) bis (Ie).
Das entsprechende Anhydrid wird mit dem Alkohol in der Gegenwart eines
Acylierungskatalysators wie von 1,8-Bis[dimethylamino]naphthalin
oder von DMAP (N,N-Dimethylaminopyridin) umgesetzt. Eine entsprechende
Carbonsäure
kann mit dem Alkohol in der Gegenwart eines Dehydratisiermittels
wie von Dicyclohexylcarbodiimid, 1-[3-Dimethylaminopropyl]-3-ethylcarbodiimid
oder weiterer wasserlöslicher
Dehydratisierungsmittel, die eingesetzt werden, um die Reaktion
durch Beseitigung des Wassers voranzutreiben, und gegebenenfalls
mit einem Acylierungskatalysator umgesetzt werden. Veresterungen können auch
mit der entsprechenden Carbonsäure
in der Gegenwart von Trifluoressigsäureanhydrid und gegebenenfalls
von Pyridin oder in der Gegenwart von N,N-Carbonyldiimidazol mit
Pyridin durchgeführt
werden. Die Reaktion eines Säurechlorids
mit einem Alkohol kann mit einem Acylierungskatalysator wie mit
DMAP oder mit Pyridin durchgeführt
werden. Der Fachmann erkennt sofort, wie diese sowie weitere Verfahren
zur Veresterung von Alkoholen erfolgreich durchzuführen sind.
Empfindliche oder reaktive Gruppen an der Verbindung der Formeln
(Ia) bis (Ie) können
bei den obigen Verfahren zur Bildung von Estern geschützt werden
müssen, und
die Schutzgruppen können
mit im Stand der Technik gut bekannten herkömmlichen Verfahren gebunden und
wieder abgespalten werden.
-
Anzumerken
ist, dass Diastereomere und Enantiomere der als Beispiele genannten
Strukturen oft möglich
sein werden, und dass reine Isomere bevorzugte Ausgestaltungen darstellen.
Die reinen Stereoisomeren und die Mischungen davon sollen ebenfalls
im Rahmen und Umfang der Erfindung liegen.
-
Die
Verbindungen der vorliegenden Erfindung können, entweder durch die Natur
der asymmetrischen Zentren oder durch eingeschränkte Rotation, in der Form
von Isomeren vorliegen. Jedes Isomer kann in der (R)- oder (S)- oder der (R,S)-Konfiguration
und vorzugsweise in der (R)- oder (S)-Konfiguration vorliegen, welches Isomer
auch immer das aktivste sein mag.
-
Alle
Isomeren, ob getrennt, rein, teilweise rein oder in razemischer
Mischung, der Verbindungen der vorliegenden Erfindung sind vom Umfang
und Rahmen der vorliegenden Erfindung umfasst. Die Reinigung der genannten
Isomeren und die Auftrennung der genannten Isomerenmischungen können mit
im Stand der Technik bekannten Standardverfahren bewerkstelligt
werden.
-
Geometrische
Isomere durch die Natur der Substituenten an einer Doppelbindung
oder einem Ring können
in der cis (= Z)- oder trans (= E)-Form vorliegen, und beide isomere
Formen sind vom Inhalt und Umfang der vorliegenden Erfindung umfasst.
-
Das
besondere Verfahren, das zur Herstellung der Verbindungen der vorliegenden
Erfindung zur Anwendung gelangt, hängt von der spezifischen Verbindung
ab, die angestrebt wird. Solche Faktoren wie die Auswahl der spezifischen
Reste und der spezifischen Substituenten an den verschiedenen Resten
spielen alle eine Rolle im Ablaufweg, der zur Herstellung der spezifischen
Verbindungen der vorliegenden Erfindung einzuschlagen ist. Diese
Faktoren werden vom Durchschnittsfachmann unmittelbar und ohne Weiteres
erkannt.
-
Zur
Synthese einer besonderen Verbindung erkennt der Fachmann, dass
die Verwendung von Schutzgruppen zur Synthese von Verbindungen erforderlich
ist, die bestimmte Substituenten enthalten. Eine Beschreibung geeigneter
Schutzgruppen und entsprechender Verfahren zur Bindung und erneuten
Abspaltung solcher. Gruppen sind zu finden in: Protective Groups
in Organic Synthesis, Zweite Ausgabe, T.W. Greene, John Wiley & Sons, New York,
1991.
-
In
den unten dargestellten Reaktionsschemen erkennt der Fachmann, dass
tatsächlich
eingesetzte Reagenzien und Lösungsmittel
aus mehreren Reagenzien und Lösungsmitteln
ausgewählt
werden können, die
als wirkungsvolle gleichwertige Produkte im Stand der Technik bekannt
sind. Sind spezifische Reagenzien oder Lösungsmittel in einem Reaktionsschema
angegeben, ist daher davon auszugehen, dass diese erläuternde
Beispiele von Bedingungen darstellen, die zur Durchführung des
entsprechenden besonderen Reaktionsschema erwünscht sind. Abkürzungen,
die im Begleittext nicht identifiziert sind, sind weiter unten in
der vorliegenden Beschreibung unter der Überschrift "Abkürzungen" aufgelistet.
-
Eine
weitere Aufgabe der vorliegenden Erfindung ist es, Verfahren zur
Herstellung der Verbindungen der Erfindung anzugeben und zur Verfügung zu
stellen. Die Verbindungen können
aus unmittelbar verfügbaren Materialien
mit den unten in den Reaktionsschemata A bis H dargestellten Verfahren
sowie mit entsprechenden offensichtlichen Modifikationen davon hergestellt
werden.
-
Die
Verbindungen der Formeln (Ia) bis (Ie) der vorliegenden Erfindung
können
mit einfachen und direkten organischen Synthesemaßnahmen
hergestellt werden, die den Fachleuten bekannt sind. Beispiele dieser
Verfahren sind in den unten angegebenen Reaktionsschemata dargestellt,
worin Z und R1 bis R28 wie
hierin oben definiert sind. Außerdem
sind, wie in diesen Schemata angegeben, X Halogen oder eine Austrittsgruppe wie
Mesylat oder Tosylat und R'' Niederalkyl.
-
Mit
in diesen Schemata dargestellten Verfahren sind die Verbindungen
der Formeln (Ia) bis (Ie) ferner in den experimentellen Beispielen
und in den Tabellen 1 bis 11 als Beispiele hergestellt und angegeben.
Die tatsächliche
Struktur der herzustellenden Verbindung bestimmt das anzuwendende
Schema sowie die Ausgangsmaterialien.
-
Beispielsweise
können
Verbindungen der Formel (Ia) mit den in den Reaktionsschemata bis
A bis C dargestellten Verfahren hergestellt werden. Diese Schemata
machen die Alkylierung eines substituierten Phenols oder Thiophenols
und die anschließende Überführung der
Verbindung (III) in einen Borsäureester
(z.B. in die Verbindung IV) sowie die Bereitstellung des 2-Bromheterozyklus,
der gegebenenfalls N-alkyliert ist (z.B. die Verbindung VIIa), erforderlich.
Die Kupplung des Bromheterozyklus mit dem Borsäureester unter Suzuki-Bedingungen
ergibt die Zwischenproduktverbindung (VIII), die in das entsprechende
Säurechlorid
(IX) und dann in eine Vielzahl von Estern, Amiden oder Carbamaten überführt werden
kann, wie dargestellt in den Reaktionsschemata.
-
Reaktionsschema
A Teil
1: Synthese der Verbindung (N)
-
Teil
2: Synthese der Verbindung (VII)
-
Reaktionsschema
A, Fortsetzung Teil
3: Synthese der Verbindungen (Ia-1)
-
-
-
In ähnlicher
Weise können
Strukturen der Formel (Ib) mit dem entsprechenden N-alkylierten
Imidazol als Ausgangsmaterial und unter Anwendung von zu den in
den Reaktionsschemata A, B und C dargestellten analogen Verfahren
hergestellt werden.
-
Reaktionsschema
D Teil
1: Synthese der Verbindung (XV)
-
Teil
2: Synthese der Verbindung (XVIII)
-
Reaktionsschema
D, Fortsetzung Teil
3: Synthese von (Ib-1)
-
Die
Synthesen der Triazolverbindungen der Formel (Ic) sind in den Verfahren
der Reaktionsschemata E und F dargestellt:
-
-
-
Verbindungen
der Formel (Id) werden gemäß dem Reaktionsschema
G hergestellt. Die Reaktion des Carboxylats der Formel (XXV) mit
einem Hydrazinderivat ergibt ein Hydrazin der Formel (XXVI); die
in situ-Abspaltung
der Schutzgruppe und die Kondensation der letzteren Verbindung mit
einem Ketoester ergibt eine Pyrazolonverbindung der Formel (XXVII).
O-Alkylierung und Hydrolyse ergeben die Pyrazolcarbonsäure, die
mit zu den für
(Ia), (Ib) und für
(Ic) beschriebenen Verfahren analogen Verfahren in eine Vielzahl
von Endprodukten (Id) überführt werden
kann. Als spezifischeres Beispiel ist die Herstellung des Amids
der Formel (Id-1) über
ein zweistufiges Verfahren unter Überführung von (XXVII) in ein Säurechlorid
und anschließender
Reaktion mit einem Amin und Base unten angegeben und dargestellt.
Die Hydrolyse des t-Butylesters wird mit TFA durchgeführt.
-
-
Imidazolverbindungen
der Formel (VII), worin Y NR4 ist, zur Herstellung
von Verbindungen der Formel (Ia) können mit den in Reaktionsschema
H dargestellten Verfahren zweckmäßig hergestellt
werden. In diesem Schema wird das Bromimidazol (VIIb), das rasch
durch Bromierung von im Handel verfügbaren Imidazolen erhältlich ist,
N-alkyliert wie dargestellt. Die Verbindung (VIId) kann mit diesem
Verfahren hergestellt werden, z.B. aus (VIIb) und Bromethanol; die
weitere Reaktion von (VIIb) durch Alkylierung ergibt das N-Methoxyethylderivat
der Formel (VIIe).
-
Reaktionsschema
H Herstellung
von Bromimidazol-Ausgangsmaterialien 1. über N-Alkylierung
-
-
Spezifische
Verbindungen, die in die Erfindung eingeschlossen sind, sind die
folgenden:
-
-
-
Experimenteller Abschnitt
-
Elektronimpakt-Massenspektren
(EI-MS) wurden mit einem Hewlett Packard 5989A-Massenspektrometer,
ausgerüstet
mit einem Hewlett Packard 5890 Gas-Chromatograf mit einer J & W DB-5-Säule (0,25 μM Überzug;
30 m × 0,25
mm), erhalten. Die Ionenquelle wurde bei 250°C gehalten, und die Spektren
wurden von 50 bis 800 amu bei 2 s pro Rasterung gerastert.
-
Hochdruck-Flüssigchromatografie-Elektrosprüh-Massenspektren
(LC-MS) wurden erhalten entweder mit einem:
- (A)
Hewlett-Packard 1100 HPLC, ausgerüstet mit einer quaternären Pumpe,
einem variablen Wellenlängendetektor,
gesetzt bei 254 nm, einer YMC-Pro
C-18-säule
(2 × 23
mm, 120A) und mit einem Finnigan-LCQ-Ionenfalle-Massenspektrometer mit Elektrosprüh-Ionisation;
die Spektren wurden von 120 bis 1200 amu mit variabler Ionenzeit
gemäß der Ionenzahl
in der Quelle gerastet. Die Eluierungsmittel waren A: 2 % Acetonitril
in Wasser mit 0,02 % TFA und B: 2 % Wasser in Acetonitril mit 0,018
% TFA; Gradient-Elution
von 10 % bis 95 % über
3,5 min bei einer Fließgeschwindigkeit
von 1,0 mL/min wurde mit einem Anfangshalt von 0,5 min und einem
Endhalt bei 95 % B von 0,5 min angewandt die Gesamtlaufzeit betrug
6,5 min;
oder mit einem:
- (B) Gilson HPLC-System, ausgerüstet mit 2 Gilson 306-Pumpen,
einem Gilson 215-Autosampler, einem Gilson-Diodenarray-Detektor,
einer YMC-Pro C-18-Säule
(2 × 23
mm, 120A) und mit einem Mikromassen-LCZ-Einzel- Quadrupol-Massenspektrometer mit z-Sprüh-Elektrosprühionisation;
die Spektren wurden von 120 bis 800 amu über 1,5 s gerastert. Verdampfungs-Lichtstreu-Detektor-
(ELSD = Evaporative Light Scattering Detector)-Daten wurden als
Analogkanal ebenfalls aufgenommen; die Eluierungsmittel waren A: 2
% Acetonitril in Wasser mit 0,02 % TFA und B: 2 % Wasser in Acetonitril
mit 0,018 % TFA; Gradient-Elution von 10 % B bis 90 % über 3,5
min bei einer Fließgeschwindigkeit
von 1,5 mL/min wurde mit einem Anfangshalt von 0,5 min und einem
Endhalt bei 90 % B von 0,5 min angewandt; die Gesamtlaufzeit betrug
4,8 min. Ein Extra-Schaltventil wurde zum Säulenwechsel und zum Regenerieren
angewandt.
-
Eindimensionale
Routine-NMR-Spektroskopie wurde an 300 MHz Varian Mercury-plus-Spektrometern durchgeführt. Die
Proben wurden in deuterierten Lösungsmitteln,
erhalten von Cambridge Isotope Labs, gelöst und in 5 mm ID Wilmad-NMR-Röhrchen gegeben.
Die Spektren wurden bei 293° K
aufgenommen. Die chemischen Verschiebungen wurden auf der ppm-Skala
aufgenommen und auf entsprechende Lösungsmittelsignale bezogen,
wie auf 2,49 ppm für
DMSO-d6, 1,93 ppm für CD3CN,
3,30 ppm für
CD3OD, 5,32 ppm für CD2Cl2 und auf 7,26 ppm für CDCl3 für die 1H-Spektren, und auf 39,5 ppm für DMSO-d6, 1,3 ppm für CD3CN,
49,0 ppm für
CD3OD, 53,8 ppm für CD2Cl2 und auf 77,0 ppm für CDCl3 für die 13C-Spektren.
-
Zweidimensionale
NMR-Spektroskopie wurde an einem Bruker DMX-600- oder an einem Bruker-DMX-500- oder
an einem Bruker DRX-500-Gerät
ausgerüstet
mit umgekehrten Dreifachresonanzsonden mit Dreifachachsengradienten,
durchgeführt.
Die Messungen wurden in 5 mm ID Wilmad-Röhrchen bei 300° K durchgeführt. COSY1-Versuche wurden mit einer Gradient-verstärkten Pulssequenz
durchgeführt
und aufgenommen (R.E. Hurd, J. Magn. Reson. 87:422, 1990). 2k × 256-Datenpunkte
wurden gesammelt und im Absolutwert-Modus auf eine 512 × 512-Matrix
mit Null-Füllung
in der t1-Dimension
umgerechnet. Zum Erhalt von NOE-Daten wurden entweder die transverse
ROESY-Sequenz von Hwang und Shaka (J. Am. Chem. Soc. 114:3157, 1992)
oder eine reguläre
Gradient-verstärkte
Nuklear-Oberhauser-Effekt-Spektroskopie
(NOESY) (Jenner et al., J. Chem. Phys. 71:4546, 1979) im Phasen-empfindlichen
Modus mit Zeit-proportionalem Phasenzuwachs (time proportional phase
incrementation = TPPI) (Marion et al., J. Magn. Res. 85:393, 1989)
bei Mischzeiten von 300 oder 500 ms angewandt. Endgültige Datensätze von
512 × 512
Punkten wurden nach Sinusglocken-Apodisierung in beiden Dimensionen
erhalten. Kreuz-Peaks wurden qualitativ analysiert und in kleine,
mittlere oder große
Klassen eingeteilt. Phasen-empfindliche HMQC-Daten wurden im States-TPPI
(Marion et al., 1989)-Modus mit einer Pulssequenz, einschließlich bilinearer
Rotationsentkopplung (bilinear rotation decoupling = BIRD) (Garbow
et al., Chem. Phys. Lett. 93:540, 1982) zur Unterdrückung von
an 12C-Kohlenstoffe gekoppelten Protonen
gesammelt. Kohlenstoff-Entkopplung wurde mit global optimierten
Wechselphasen-Rechteckspulsen
(globally optimized alternating-phase rectangular pulses = GARP)
(Shaka et al., J. Magn. Res. 64:547, 1985) durchgeführt. Vor
einer Fourier-Transformation wurde eine quadratische Sinusglocken-Apodisierung
in beiden Dimensionen angewandt. HMBC13-Spektren
wurden mit einer Gradient-verstärkten Pulssequenz
aufgenommen (Wilker et al., Magn. Reson. Chem. 31:287, 1993) und
im Absolutwert-Modus mit quadratischer Sinusglocken-Apodisierung in beiden
Dimensionen für
eine 1k × 1k-Datenmatrix
verarbeitet und umgerechnet. Der Langbereich-Kopplungsentwicklungsverzug
wurde auf 80 ms gesetzt.
-
Abkürzungen:
-
-
- ADDP
- = 1,1'-(Azodicarbonyl)dipiperidin
- CDCl3
- = deuteriertes Chloroform
- DMAP
- = 4-(N,N-Dimethylamino)pyridin
- DMF
- = N,N-Dimethylformamid
- DMSO
- = Dimethylsulfoxid
- DPPA
- = N,N'-Diphenylphosphorylazid
- EI-MS
- = Elektronimpakt-Massenspektroskopie
- h
- = Stunde(n)
- HPLC
- = Hochdruck-Flüssigchromatografie
- LC-MS
- = Flüssigchromatographie-Massenspektroskopie
- min
- = Minuten
- Ms
- = Massenspektroskopie
- NBS
- = N-Bromsuccinimid
- NMR
- = Nuklearmagnetische
Resonanz
- Psi
- = Pfund pro Quadratinch
- rt
- = Raumtemperatur
- RT
- = Retentionszeit
- TEA
- = Triethylamin
- TFA
- = Trifluoressigsäure
- FHF
- = Tetrahydrofuran
- TLC
- = Dünnschichtchromatografie
-
Chemie
-
Abschnitt
A-Imidazole Beispiel
1 Herstellung
von Natrium-2-{[4-(4-{[(2,4-dimethylphenyl)amino]carbonyl}-1-pentyl-1H-imidazol-2-yl)phenyl]sulfanyl}-2-methylpropanoat
-
Stufe
1: Eine Lösung
von Methyl-1H-imidazol-5-carboxylat (10 g, 40 mmol) in Tetrahydrofuran
(25 mL) und in N,N-Dimethylformamid (30 mL) wurde in eine Mischung
aus Natriumhydrid (2,2 g, 44 mmol) in Tetrahydrofuran (30 mL) getropft.
Die Mischung wurde auf rt erwärmt
und 1 h lang gerührt.
Eine Lösung,
enthaltend 1-Jodpentan (11,5 mL, 44 mmol) in Tetrahydrofuran (5
mL), wurde dann zugegeben, und die Reaktionsmischung wurde bei rt
12 h lang gerührt.
Ethylacetat (150 mL × 3)
wurde zugegeben, worauf die organische Schicht mit Wasser (150 mL,
3 ×) und
mit gesättigtem
wässrigen
Natriumbicarbonat gewaschen, mit Natriumsulfat getrocknet und unter
verringertem Druck eingeengt wurde. Der Rückstand wurde durch Blitz-Chromatografie (Kieselgel,
10–50
Ethylacetat in Hexanen) gereinigt, um Methyl-1-pentyl-1H-imidazol-4-carboxylat
(A) (9,8 g, 63 %) und Methyl-1-pentyl-1H-imidazol-5-carboxylat
(B) (2 g, 13 %) zu erhalten.
Methyl-1-pentyl-1H-imidazol-4-carboxylat
(A): Ms = 197,1 (M + H+); 1H-NMR
(CDCl3) δ:
0,89 (t, 3H), 1,31 (m, 4H), 1,81 (m, 2H), 3,89 (s, 3H), 3,96 (t,
2H), 7,62 (s, 1H), 7,71 (s, 1H), TLC: Rf = 0,3 (100 % Ethylacetat); LC-MS:
RT = 1,34 min
Methyl-1-pentyl-1H-imidazol-5-carboxylat (B):
MS = 197,2 (M + H+); 1H-NMR
(CDCl3) δ:
0, 88 (t, 3H) , 1, 30 (m, 4H) , 1, 78 (m, 2H) , 3, 85 (s, 3H) ,
4,30 (t, 2H), 7,74 (s, 1H), 7,78 (s, 1H)
-
-
-
Zwischenproduktverbindung
A-1
Methyl-2-brom-1-pentyl-1H-imidazol-4-carboxylat
-
Stufe
2: Zu einer Lösung
von Methyl-1-pentyl-1H-imidazol-4-carboxylat (7,24 g, 37 mmol) in
Kohlenstofftetrachlorid (700 mL) wurden N-Bromsuccinimid (13,19
g, 74 mmol) und 2,2'-Azobisisobutyronitril
(0,30 g, 1,8 mmol) gegeben. Die Mischung wurde bei 60°C 16 h lang
gerührt.
Nach Beendigung der Reaktion wurde die Mischung filtriert, und das
Filtrat wurde unter verringertem Druck eingeengt. Der Rückstand
wurde durch Blitz-Chromatografie
(Kieselgel, 20–70
% Ethylacetat/Hexane) gereinigt, um Methyl-2-brom-1-pentyl-1H-imidazol-4-carboxylat
(die Zwischenproduktverbindung A-1) zu erhalten (5,1 g, 50 %). Ms
= 275,1 (M + H+); 1H-NMR
(CDCl3) δ:
0,90 (t, 3H), 1,32 (m, 4H), 1,78 (m, 2H), 3,86 (s, 3H), 3,94 (t,
2H), 7,64 (s, 1H)
-
-
-
t-Butyl-2-((4-bromphenyl)sulfanyl]-2-methylpropanoat
-
Stufe
3: Zu einer Lösung
von 4-Brombenzolthiol (92 g, 0,50 mol) in Ethanol wurde Kaliumhydroxid (27,3
g, 0,49 mol) langsam gegeben. Die Mischung wurde auf 0°C abgekühlt, nachdem
das 4-Brombenzolthiol vollständig
aufgelöst
war. t-Butyl-2-brom-2-methylpropanoat (91 mL, 0,49 mol) wurde zur
Lösung
getropft. Die Mischung wurde 1 h lang am Rückfluss erwärmt, auf rt _ abgekühlt und
filtriert. Das Filtrat wurde unter verringertem Druck eingeengt,
um einen Feststoff zu ergeben. Der Feststoff wurde in Dichlormethan
(800 mL) aufgelöst,
worauf die Lösung
mit Wasser gewaschen wurde. Die Schichten wurden getrennt, und die
organische Schicht wurde getrocknet (Natriumsulfat) und eingeengt,
um einen Feststoff zu ergeben. Umkristallisation (wasserfreie Hexane)
ergab t-Butyl-2-[(4- bromphenyl)sulfanyl]-2-methylpropanoat
als farblosen Feststoff (115 g, 71,4 %). 1H-NMR
(CDCl3) δ:
1,41 (s, 15H), 7,35 (d, 2H), 7,44 (d, 2H)
-
Zwischenproduktverbindung
A-2
t-Butyl-2-methyl-2-{[4-(4,4,5,5--tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfanyl}propanoat
-
Stufe
4: Zu einer Mischung aus 4,4,4',4',5,5,5',5'-Octamethyl-2,2'-bi-1,3,2-dioxaborolan
(16,9 g, 66,4 mmol), [1,1'-Bis(diphenylphosphino)ferrocen]dichlorpaladium(II)
(1:1-Komplex mit Dichlormethan) (1,48 g, 1,81 mmol) und aus Kaliumacetat
wurde t-Butyl-2-[(4-bromphenyl)sulfanyl]-2-methylpropanoat in 200 mL Dimethylsulfoxid
gegeben, worauf die Mischung bei 80°C 16 h lang erhitzt wurde. Die
Mischung wurde durch einen Pfropf aus Kieselgel mit Hexanen (1 L)
und mit 5 % Ethylacetat in Hexanen als Eluierungsmittel filtriert, um
t-Butyl-2-methyl-2-{[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfanyl}propanoat
(die Zwischenproduktverbindung A-2) als farblosen Feststoff zu ergeben
(23,63 g, quantitativ). 1H-NMR (CDCl3) δ:
1,32 (s, 12H), 1,41 (s, 9H), 1,44 (s, 6H), 7,44 (d, 2H), 7,74 (d,
2H)
-
Methyl-2-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfonyl]phenyl}-1-pentyl-1H-imidazol-4-carboxylat
-
Stufe
5: Zu einer Mischung aus t-Butyl-2-methyl-2-{[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfanyl}propanoat
(7,45 g, 20 mmol), Methyl-2-brom-1-pentyl-1H-imidazol-4-carboxylat
(4,50 g, 16 mmol) und aus [1,1'-Bis(diphenylphosphino)ferrocen]dichlorpalladium(II)
(1:1-Komplex mit Dichlormethan) (0,560 g, 0,69 mmol) wurden Toluol
(200 mL) und Dioxan (50 mL) gegeben. Die entstandene Lösung wurde
mit Argon 30 min lang gespült.
Natriumbicarbonat-Lösung
(2 M, 50 mL) wurde zugegeben, und die Mischung wurde bei 85°C 48 h lang
erhitzt. Die Reaktionsmischung wurde auf rt abgekühlt und
mit 200 mL Ethylacetat verdünnt.
Die Schichten wurden getrennt, worauf die wässrige Schicht 2 Mal mit Ethylacetat
(50 mL) extrahiert wurde. Die gereinigten organischen Schichten
wurden dann über
Natriumsulfat getrocknet, filtriert und unter verringertem Druck
eingeengt, um ein dunkelbraunes Öl
zu ergeben. Der Rückstand
wurde durch Blitz-Chromatografie
(Kieselgel, 10/90 Ethylacetat/Hexane (1 L) und dann 30/70 Ethylacetat/Hexane)
gereinigt, um Methyl-2-(4-[(2-t-Butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl)phenyl}-1-pentyl-1H-imidazol-4-carboxylat zu
ergeben (7,29 g, 99 %). Ms = 447,1 (M + H+); 1H-NMR (CDCl3) δ: 0,84 (t,
3H), 1,22 (m, 4H), 1,41–1,47
(m, 15H), 1,72 (m, 2H), 3,88 (s, 3H), 3,98 (t, 2H), 7,55 (m, 4H),
7,72 (s, 1H)
-
2-{4-[(2-t-Butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-1-pentyl-1H-imidazol-4-carbonsäure
-
Stufe
6: Zu einer Lösung
von Methyl-2-(4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-1-pentyl-1H-imidazol-4-carboxylat
(7,29 g, 16,3 mmol) in Ethanol wurde eine wässrige Kaliumhydroxid-Lösung (2,5 %ig,
366 mL) gegeben. Die Mischung wurde bei 70°C 1,5 h lang erwärmt. Die
Reaktionsmischung wurde dann auf rt abgekühlt, und der pH-Wert der Lösung wurde
auf ca. 5 mit 0,5 N Salzsäure-Lösung eingestellt.
Die Mischung wurde mit Ethylacetat extrahiert (150 mL × 3). Die
vereinigten organischen Schichten wurden getrocknet (Natriumsulfat)
und unter verringertem Druck eingeengt, um 2-(4-[(2-t-Butoxy-1,1-dinmethyl-2-oxoethyl)sulfanyl]phenyl}-1-pentyl-1H-imidazol-4-carbonsäure als Öl zu erhalten
(6,99 g, 99 %). Ms = 433,5 (M + H+); 1H-NMR (CDCl3) δ: 0,85 (t,
3H), 1,24 (m, 4H), 1,41–1,48
(m, 15H), 1,77 (m, 2H), 4,12 (t, 2H), 7,62 (m, 4H), 7,84 (s, 1H)
-
t-Butyl-2-{[4-(4-{[(2,4-dimethylphenyl)amino]carbonyl}-1-pentyl-1H-imidazol-2-yl)phenyl]sulfanyl}-2-methylpropanoat
-
Stufe
7: Zu einer Lösung
von 2-{4-[(2-Butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-1-pentyl-1H-imidazol-4-carbonsäure (5,15
g, 11,9 mmol) in Dichlormethan (150 mL) wurden Oxalylchlorid (5,2
mL, 60 mmol) und N,N-Dimethylformamid (1 mL) gegeben. Die entstandene
Lösung
wurde bei rt 1 h lang gerührt, bevor
sie unter verringertem Druck eingeengt wurde. Der hellgelbe Rückstand
wurde dann in Dichlorethan (50 mL) gelöst und zu einer Lösung, enthaltend
2,4-Dimethylanilin (4,4 mL, 36 mmol), Dichlorethan (50 mL), 4-Dimethylaminopyridin
(50 mg) und Triethylamin (3 mL), gegeben. Die Reaktionsmischung
wurde bei rt 30 min lang gerührt,
bei 55°C
1 h lang erwärmt,
abgekühlt
und bei rt 16 h lang gerührt.
Die Mischung wurde unter verringertem Druck eingeengt, worauf der
Rückstand
in Ethylacetat gelöst
wurde. Die entstandene Lösung
wurde mit Wasser gewaschen, über
Natriumsulfat getrocknet und unter verringertem. Druck eingeengt.
Der Rückstand
wurde durch Blitz-Chromatografie (Kieselgel, 10/90 bis 30/70 Ethylacetat/Hexane)
gereinigt, um t-Butyl-2-{[4-(4-{[(2,4-dimethylphenyl)amino]carbonyl}-1-pentyl-1H-imidazol-2-yl)phenyl]-sulfanyl}-2-methylpropanoat
als weißen
Feststoff zu ergeben (5,7 g, 89 %). Ms = 536,6 (M + H+); 1H-NMR (CDCl3) δ: 0,86 (t,
3H), 1,27 (m, 4H), 1,45–1,47
(m, 15H), 1,75 (m, 2H), 2,30 (s, 3H), 2,33 (s, 3H), 4,07 (t, 2H),
7,03 (m, 2H), 7,55–7,64
(m, 4H), 7,75 (s; 1H), 7,90 (m, 1H), 8,94 (s, 1H)
-
2-{[4-(4-{[(2,4-Dimethylphenyl)amino]carbonyl}-1-pentyl-1H-imidazol-2-yl)phenyl]sulfanyl}-2-methylpropansäure
-
Stufe
8: Zu einer Lösung
von t-Butyl-2-{[4-(4-{[(2,4-dimethylphenyl)amino]carbonyl}-1-pentyl-1H-imidazol-2-yl)phenyl]sulfanyl}-2-methylpropanoat
(3,00 g, 5,59 mmol) in Dichlormethan (10 mL) wurde Trifluoressigsäure (10
mL) gegeben. Die Mischung wurde bei rt 16 h lang gerührt. Die
Mischung wurde unter verringertem Druck eingeengt, und das Rohmaterial
wurde durch Blitz-Chromatografie (Kieselgel, 100 % Hexane bis 10 %
Ethylacetat in Hexanen) gereinigt, um 2-{[4-(4-{[(2-Dimethylphenyl)amino]carbonyl}-1-pentyl-1H-imidazol-2-yl)phenyl]sulfanyl}2-methylpropansäure als
weißen
Feststoff zu ergeben (1,8 g, 67 %). Ms = 480,4 (M + H+); 1H-NMR (CDCl3) δ: 0,84 (t,
3H), 1,26 (m, 4H), 1,52 (s, 6H), 1,74 (m, 2H), 2,30 (s, 3H), 2,33
(s; 3H), 3,98 (t, 2H), 7,03 (m, 2H), 7,49–7,60 (m, 4H), 7,70 (m, 1H),
7,85 (s, 1H), 9,28 (bs, 1H)
-
Natrium-2-{[4-(4-{[(2,4-dimethylphenyl)amino]carbonyl}-1-pentyl-1H-imidazol-2-yl)phenyl]sulfanyl}-2-methylpropanoat
-
Stufe
9: Zu einer Lösung
von 2-{[4-(4-{[(2,4-Dimethylphenyl)amino]carbonyl}-1-pentyl-1H-imidazol-2-yl)phenyl]sulfanyl}-2-methylpropansäure (0,710
g, 1,48 mmol) in Acetonitril (1 mL) und Wasser (0,5 mL) wurde wässriges
0,1 N Natriumhydroxid (1,48 mL, 1,48 mmol) gegeben. Die Mischung
wurde bei rt 30 min lang gerührt.
Die Lösung
wurde gefriergetrocknet, um Natrium-2-{[4-(4-{((2,4-dimethylphenyl)amino)carbonyl}-1-pentyl-1H- imidazol-2-yl)phenyl]sulfanyl}-2-methylpropanoat
als weißen
Feststoff zu erhalten (0,656 g, 88 %). Ms = 480,4 (M – Na + H)+; 1H-NMR (CDCl3) δ:
0,75 (t, 3H), 1,13 (m, 4H), 1,37 (s, 6H), 1,63 (m, 2H), 2,20 (s,
3H), 2,24 (s, 3H), 3,86 (m, 2H), 6,95 (m, 2H), 7,39–7,55 (m,
4H), 7,62 (m, 1H), 7,79 (s, 1H), 8,68 (s, 1H)
-
Beispiel
2 Herstellung
von 2-Methyl-2-({4-[5-methyl-1-pentyl-4-({[4-(trifluormethyl)phenyl]amino}carbonyl)-1H-imidazol-2-yl]phenyl}sulfanyl)propansäure
-
Unter
Anwendung eines Reaktionsweges ähnlich
dem oben für
Beispiel 1, Abschnitt A, beschriebenen und durch Einsatz der entsprechenden
Ausgangsmaterialien oder Zwischenproduktverbindungen (siehe unten)
wurde die obige Verbindung hergestellt. Ms = 534,2 (M + H+); 1H-NMR (300 MHz,
CDCl3) δ:
0,80 (t, 3H), 1,18 (m, 4H), 1,46 (s, 6H), 1,88 (q, 2H), 2,68 (s;
3H), 3,84 (t, 2H), 7,42 (d, 2H), 7,56 (m, 4H), 7,88 (d, 2H), 9,60 (s,
1H)
-
Beispiel
3 Herstellung
von Natrium-2-methyl-2-({{4-[5-methyl-1-pentyl-4-({[4-(trifluormethyl)phenyl]amino}carbonyl)-1-H-imidazol-2-yl]phenyl}sulfanyl)propanoat
-
Unter
Anwendung eines Syntheseweges ähnlich
dem oben für
Beispiel 1, Abschnitt A, beschriebenen und durch Einsatz der entsprechenden
Ausgangsmaterialien oder Zwischenproduktverbindungen (siehe unten)
wurde die obige Verbindung hergestellt. Ms = 534,1 (M + H+); 1H-NMR (300 MHz,
DMSO) δ:
0,82 (t, 3H), 1,18 (m, 4H), 1,26 (s, 6H), 1,52 (q, 2H), 2,61 (s,
3H), 3,98 (t, 2H), 7,52 (m, 4H), 7,62 (d, 2H), 8,12 (d, 2H), 10,14 (s,
1H)
-
Beispiel
4 Herstellung
von 2-({4-[4-{[(4-Ethylphenyl)amino]carbonyl}-1-(3-methoxypropyl)-5-methyl-1H-imidazol-2-yl]phenyl}sulfanyl)-2-methylpropansäure
-
Unter
Anwendung eines Syntheseweges ähnlich
dem oben für
Beispiel 1, Abschnitt A, beschriebenen und durch Einsatz der entsprechenden
Ausgangsmaterialien oder Zwischenproduktverbindungen (siehe unten)
wurde die obige Verbindung hergestellt. Ms = 496,2 (M + H)+; 1H-NMR (CDCl3) 6: 1,21 (t, 3H), 1,50 (s, 6H), 1,80 (m,
2H), 2,61 (q, 2H), 2,69 (s, 3H), 3,18 (s, 3H), 3,21 (t, 2H), 4,05
(t, 2H), 7,15 (m, 2H), 7,46 (m, 4H), 7,63 (m, 2H), 9,35 (s, 1H)
-
Beispiel
5 Herstellung
von Natrium-2-({4-[4-{[(4-ethylphenyl)amino]carbonyl}-1-(3-methoxypropyl)-5-methyl-1H-imidazol-2-yl]phenyl}sulfanyl)-2-methylpropanoat
-
Unter
Anwendung eines Syntheseweges ähnlich
dem oben für
Beispiel 1, Abschnitt A, beschriebenen und durch Einsatz der entsprechenden
Ausgangsmaterialien oder Zwischenproduktverbindungen (siehe unten)
wurde die obige Verbindung hergestellt. Ms = 496,2 (M – Na + H)+; 1H-NMR (CDCl3) δ:
1,16 (t, 3H), 1,43 (s, 6H), 1,69 (m, 2H), 2,56 (m, 5H), 3,08 (m,
5H), 3,91 (m, 2H), 7,06 (d, 2H), 7,42 (d, 2H), 7,52 (m, 4H), 8,88 (s,
1H)
-
Beispiel
6 Herstellung
von 2-{[4-(4-{[(Benzyloxy)carbonyl]amino}-1-pentyl-1H-imidazol-2-yl)phenyl]sulfanyl}-2-methylpropansäure
t-Butyl-2-{[4-(4-([(benzyloxy)carbonyl]amino}-1-pentyl-1H-imidazol-2-yl)phenyl]sulfanyl}-2-methylpropanoat
-
Stufe
1: eine Lösung
von 0,197 g 2-{4-[(2-t-Butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-1-pentyl-1H-imidazol-4-ylcarbaminsäure (siehe
Beispiel 1, Abschnitt A, zur Herstellung) (0,455 mmol), 0,125 g
Diphenylphosphorylazid (0,455 mmol) und von 0,046 g Triethylamin
(0,455 mmol) in 3,0 mL Toluol wurde bei rt 0,5 h lang und dann bei
85°C 45
min lang gerührt.
Benzylalkohol (0,049 g, 0,455 mmol) wurde zugegeben, und die entstandene
Mischung wurde bei 85°C
14 h lang gerührt.
Die Mischung wurde auf rt abgekühlt,
worauf gesättigtes
wässriges
Natriumcarbonat (1 mL) zugegeben wurde. Die Schichten wurden getrennt,
und die wässrige
Schicht wurde 2 Mal mit 0,5 mL Ethylacetat extrahiert. Die vereinigten
organischen Schichten wurden mit Magnesiumsulfat getrocknet und
unter verringertem Druck eingeengt, um 0,260 g Rohprodukt zu ergeben. Dieses
Material wurde durch Blitz-Chromatografie (Biotage-Säule, 15:85
Ethylacetat:Hexan) gereinigt, um t-Butyl-2-{[(benzyloxy)carbonyl]amino}-1-pentyl-1H-imidazol-2-yl)phenyl]sulfanyl}-2-methylpropanoat
zu ergeben (0,103 g, 42 %). LC-MS = 538,3 (M + H)+,
RT = 3,61 min
-
2-{[4-(4-{[(Benzyloxy)carbonyl]amino}-1-pentyl-1H-imidazol-2-yl)phenyl]sulfanyl}-2-methylpropansäure
-
Stufe
2: Eine Lösung
von 0,046 g t-Butyl-2-{[4-(4-{[(benzyloxy)carbonyl]amino}-1-pentyl-1H-imidazol-2-yl)phenyl]sulfanyl}-2-methylpropanoat
(0,089 mmol) in 0,5 mL 30%iger Bromwasserstoffsäure-Essigsäure wurde 45 min lang bei rt
gerührt.
Wasser (0,3 mL) wurde zugegeben, und die Mischung wurde unter verringertem
Druck eingeengt. Das Rohprodukt wurde mit HPLC gereinigt, um 2-{[4-(4-{[(Benzyloxy)carbonyl]amino}-1-pently-1H-imidazol-2-yl)phenyl]sulfanyl}-2-methylpropansäure als
klares, farbloses Öl
zu ergeben (0,0142 g, 33 % Ausbeute). 1H-NMR
(300 MHz, CDCl3) δ: 0,84 (t, 3H), 1,18–1,33 (m,
4H), 1,57 (s, 6H), 1,76–1,89
(m, 2H), 4,02 (t, 2H), 5,21 (s, 2H), 7,29–7,52 (m, 8H), 7,73 (d, 2H),
11,20 (s, 1H); LC-MS = 482,3 (M + H)+, RT
= 3,63 min
-
Beispiel
7 Herstellung
von 2-[5-(4-{[4-Ethylphenyl)amino]carbonyl}-1-pentyl-1H-imidazol-2-yl)-2,3-dihydro-1H-finden-1-yl]butansäure
Methyl-2-(6-methoxy-1H-inden-3-yl)butanoat
-
Stufe
1: In einem im Ofen getrockneten 5-L-Vierhals-Rundkolben mit Thermoeter,
Kühler,
Tropftrichter und mechanischem Rührer
wurde unter Argon-Schutz eine Suspension von 5-Methoxy-1-indanon
(80,0 g, 499 mmol), Zinkpulver (Lancaster, 56,2 g, 865 mmol) in
2 L wasserfreiem Tetrahydrofuran bei 60°C (Innentemperatur) gerührt, während eine
Lösung
von Methylbrombutyrat (134,1 g, 741 mmol) in 400 mL wasserfreiem
Tetrahydrofuran langsam durch den Tropftrichter zugetropft wurde.
Nach Beendigung der Zugabe wurde die Reaktionsmischung bei 60°C (Innentemperatur)
1 h lang gerührt.
Die Reaktion wurde durch TLC-Analyse von Anteilsmengen nach Aufarbeitung
mit 1 N wässriger
Salzsäure
verfolgt. Nach Beendigung der Reaktion wurde sie in einem Eiswasser-Bad
abgekühlt,
worauf langsam 3 L 1 N Salzsäure-Lösung zugegeben
wurde. Die Topftemperatur wurde unterhalb 20°C gehalten. Die Mischung wurde
dann mit 1 L Ethylacetat extrahiert. Die organische Schicht wurde
mit Wasser bis zum pH-Wert von 6,0 bis 7,0 und dann mit gesättigter
Natriumchlorid-Lösung
gewaschen und über
Natriumsulfat getrocknet. Methyl-2-(6-methoxy-1H-inden-3-yl)butanoat
(127 g, > 99 %) wurde
als gelbes Öl
nach Entfernung des Lösungsmittels
und Trocknung unter Vakuum erhalten. 1H-NMR (DMSO-d6) δ: 7,28 (d,
1H), 7,05 (d, 1H), 6,82 (dd, 1H), 6,22 (s, 1H), 3,72 (s, 3H), 3,60
(m, 1H), 3,58 (s, 3H), 3,28 (s, 2H), 1,95 (m, 1H), 1,80 (m, 1H),
0,88 (t, 3H)
-
Methyl-5-methoxy-2,3-dihydro-1H-inden-1-ylbutanoat
-
Stufe
2: Eine Lösung
aus Methyl-2-(6-methoxy-1H-inden-3-yl)butanoat (105 g, 453 mmol)
und Palladium auf Kohlenstoff (10,0 g, 10 % Äq.) in Ethanol (945 mL) und
Tetrahydrofuran (105 mL) wurde in einer 2-L-Druckflasche unter 60
psi Wasserstoff 16 h lang geschüttelt.
Das Lösungsmittel
wurde unter verringertem Druck entfernt. Methyl-5-methoxy-2,3-dihydro-1H-inden-1-yl-butanoat (101,0 g,
95 % Ausbeute) wurde als hellgelbes Öl erhalten. 1H-NMR (DMSO-d6) δ:
12,20 (s, 1H), 7,04 (d, 1H), 6,78 (d, 1H), 6,66 (dd, 1H), 3,70 (s, 3H),
3,28 (m, 1H), 2,72 (m, 2H), 2,32 (m, 1H), 2,06 (m, 1H), 1, 80 (m,
1H), 1,50 (m, 1H), 1,36 (m, 1H), 0,82 (t, 3H)
-
Methyl-5-hydroxy-2,3-dihydro-1H-inden-1-ylbutanoat
-
Stufe
3: Zu einer kalten Lösung
(Eiswasser-Bad) von Methyl-5-methoxy-2,3-dihydro-1H-inden-1-yl-butanoat (233
g, 0,94 mol) in 2,5 L CH2Cl2 wurde
Aluminiumtrichlorid (630 g, 4,7 mol) langsam unter Argon gegeben.
Die Topftemperatur wurde unterhalb 20°C gehalten, und die Reaktion
färbte
sich purpurfarben. Ethylthiol (345 mL, 4,7 mol) wurde über einen
Tropftrichter zur Reaktionsmischung langsam getropft, wobei die
Innentemperatur unterhalb 15°C
gehalten wurde. Nach 2 h Rühren
bei unterhalb 20°C
war die Reaktion gemäß NMR-Analyse
beendet. Die Topfmischung wurde langsam in 2,5 L Eiswasser unter
starkem Rühren
gegossen. Die organische Schicht wurde abgetrennt, und die wässrige Schicht
wurde mit 1 L Dichlormethan extrahiert. Die vereinigten Dichlormethan-Schichten
wurden mit Wasser (4 × 1
L) gewaschen, bis der pH-Wert 6,0 bis 7,0 betrug, worauf über Natriumsulfat
getrocknet wurde. Methyl-5-hydroxy-2,3-dihydro-1H-inden-1-yl-butanoat (216 g,
98 %) wurde als weißer
Feststoff nach Entfernung des Lösungsmittels
und Vakuumtrocknung erhalten. 1H-NMR (DMSO-d6) δ:
9,10 (s, 1H), 6,78 (d, 1H), 6,58 (d, 1H), 6,50 (dd, 1H), 3,60 (s,
3H), 3,20 (q, 1H), 2,70 (m, 2H), 2,40 (m, 1H), 2,08 (m, 1H9, 1,80
(m, 1H), 1,50 (m, 2H), 0,80 (t, 3H)
-
Methyl-2-(5-{[(trifluormethyl)sulfonyl]oxy}-2,3-dihydro-1H-inden-1-yl)butanoat
-
Stufe
4: Zu einer Mischung aus Methyl-2-(5-hydroxy-2,3-dihydro-1H-inden-1-yl) butanoat (2,0
g, 8,5 mmol) und aus Triethylamin (1,0 g, 9,9 mmol) in Tetrahydrofuran
(20 mL) wurde Trifluormethansulfonylchlorid (1,6 g, 9,5 mmol) gegeben.
Die Reaktionsmischung wurde bei rt 2 h lang gerührt und dann zur Beseitigung eines
Niederschlags filtriert, das Filtrat wurde unter verringertem Druck
eingeengt, um ein klares Öl
zu ergeben. Reinigung durch Blitz-Chromatografie (Kieselgel, Ethylacetat/Hexane)
ergab Methyl-2-(5- {[(trifluormethyl)sulfonyl]oxy}-2,3-dihydro-1H-inden-1-yl)butanoat
als klares Öl
(1,5 g, 50 %). 1H-NMR (CD2Cl2) δ:
7,35 (d, 1H), 7,15 (d, 1H), 7,05 (dd, 1H), 3,60 (s, 3H), 3,40 (m,
1H), 2,80–3,00
(m, 2H), 2,60 (m, 1H), 2,30 (m, 1H), 2,10 (m, 1H), 1,50–1,80 (m,
2H), 0,91 (t, 3H); EI-MS = 366,3 (M+), RT
= 8,40 min
-
Methyl-2-[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,3-dihydro-1H-inden-1-yl]butanoat
-
Stufe
5: Zu einer Lösung
von Methyl-2-(5-{[(trifluormethyl)sulfonyl]oxy}-2,3-dihydro-1H-inden-1-yl)butanoat (1,5
g, 4 mmol) in Dimethylsulfoxid (10 mL) wurden Dichlor[1,1'-bis(diphenylphosphino)ferrocen]palladium(II)
in CH2Cl2 (100 mg),
Bis(pinacolat)dibor (1,2 g, 4,4 mmol) und KOAc (1,2 g, 12 mmol)
gegeben. Die Mischung wurde entgast und über Nacht bei 80°C gerührt. Die
Reaktionsmischung wurde dann einer Kieselgel-Chromatografie (Hexan/Ethylacetat)
unterzogen, um Methyl-2-[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,3-dihydro-1H-inden-1-yl]butanoat
als klares Öl
zu ergeben (1,2 g, 85 %). 1H-NMR (CD2Cl2) δ: 7,60 (d,
1H), 7,50 (dd, 1H), 7,30 (d, 1H), 3,60 (s, 3H), 3,40 (m, 1H), 2,80–3,00 (m,
2H), 2,60 (m, 1H), 2,30 (m, 1H), 2, 10 (m, 1H), 1,50–1,80 (m,
2H), 1,30 (m, 12H), 0,91 (t, 3H); EI-MS (M+):
344, RT = 10,00 min
-
Methyl-2-{1-[1-(methoxycarbonyl)propyl]-2,3-dihydro-1H-inden-5-yl}-1-pentyl-1H-imidazol-4-carboxylat
-
Stufe
6: Zu einer Lösung
von Methyl-2-[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,3-dihydro-1H-inden-1-yl]butanoat
(0,8 g, 2,3 mmol) in Toluol (20 mL) und in Dioxan (5 mL) wurden
Dichlor[1,1'-bis(diphenylphosphino)ferrocen]palladium(II)-Dichlormethan-Addukt
(50 mg), Methyl-N-pentyl-2-bromimidazol-4-carboxylat (0,6 g, 2,3
mmol) (siehe Beispiel 1, Abschnitt A, zur Herstellung) und Natriumcarbonat
(2 M, 5 mL) gegeben. Die Mischung wurde entgast und 48 h lang bei
90°C gerührt. Die
entstandene Mischung wurde mit Salzlösung gewaschen, worauf die
organische Schicht über
Natriumsulfat getrocknet und unter verringertem Druck eingeengt
wurde. Der entstandene Rückstand
wurde durch Kieselgel-Chromatografie
(Hexan/Ethylacetat) gereinigt, um Methyl-2-{1-[1-(methoxycarbonyl)propyl]-2,3-dihydro-1H-inden-5-yl}-1-pentyl-1H-imidazol-4-carboxylat zu ergeben
(0,51 g, 54 % Ausbeute). 1H-NMR (CD2Cl2) δ: 7,60 (s,
1H), 7,30 (d, 1H), 7,20 (m, 2H), 3,90 (t, 2H), 3,70 (s, 3H), 3,60
(s, 3H), 3,40 (m, 1H), 2,80–3,00
(m, 2H), 2,60 (m, 1H), 2,30 (m, 1H), 2,10 (m, 1H), 1,50–1,80 (m,
4H), 1,20 (m, 4H), 0,91 (t, 3H), 0,70 (t, 3H); LC-MS = 413,1 (M
+ H)+, RT = 3,12 min
-
2-{1-[1-(Methoxycarbonyl)propyl]-2,3-dihydro-1H-inden-5-yl}-1-pentyl-1H-imidazol-4-carbonsäure
-
Stufe
7: Zu einer Lösung
von Methyl-2-{1-[1-(methoxycarbonyl)propyl]-2,3-dihydro-1H-inden-5-yl}-1-pentyl-1H-imidazol-4-carboxylat
(0,5 g, 1,2 mmol) in Methanol wurde wässriges Kaliumhydroxid (0,6
g in 1 mL Wasser) gegeben. Die Mischung wurde 6 h lang bei rt gerührt und
dann unter verringertem Druck eingeengt. Salzsäure (1 M) wurde zugegeben,
um den pH-Wert auf 4 einzustellen. Die Mischung wurde mit Ethylacetat
extrahiert, worauf die vereinigten Extrakte getrocknet und eingeengt
wurden, um 2-{1-[1-(Methoxycarbonyl)propyl-2,3-dihydro-1H-inden-5-yl}-1-pentyl-1H-imidazol-4-carbonsäure zu ergeben
(0,45 g, 90 % Ausbeute). 1H-NMR (CD2Cl2) δ: 7,80 (s,
1H), 7,30 (m, 3H), 3,90 (t, 2H), 3,60 (s, 3H), 3,40 (m, 1H), 2,80–3,00 (m,
2H), 2,60 (m, 1H), 2,30 (m, 1H), 2,10 (m, 1H), 1,50–1,80 (m,
4H), 1,20 (m, 4H), 0,91 (t, 3H), 0,70 (t, 3H); LC-MS = 399,2 (M
+ H+), RT = 2,76 min
-
Methyl-2-[5-(4-{[(4-ethylphertyl)amino]carbonyl}-1-pentyl-1H-imidazol-2-yl)-2,3-dihydro-1H-inden-1-yl]butanoat
-
Stufe
8: Methyl-2-[5-(4-{[(4-ethylphenyl)amino]carbonyl}-1-pentyl-1H-imidazol-2-yl)-2,3-dihydro-1H-inden-1-yl]butanoat
wurde mit einem Verfahren ähnlich
dem der Stufe 7, Beispiel 1, Abschnitt A, hergestellt. Ausbeute
= 60 %; 1H-NMR (CD2Cl2) δ:
9,00 (s, 1H), 7,70 (s, 1H), 7,60 (d, 2H), 7,40 (m, 3H), 7,20 (d, 2H),
4,00 (t, 2H), 3,60 (s, 3H), 3,40 (m, 1H9, 2,80–3,00 (m, 2H), 2, 60 (m, 3H),
2,30 (m, 1H), 2,10 (m, 1H), 1,50–1,80 (m, 4H), 1,20 (m, 7H),
0,90 (t, 3H), 0,80 (t, 3H); LC-MS = 502,4 (M + H+),
RT = 3,83 min
-
2-[5-(4-{[4-Ethylophenyl)amino)carbonyl}-1-pentyl-1H-imidazol-2-yl)-2,3-dihydro-1H-inden-1-yl)butansäure
-
Stufe
9: Kaliumhydroxid (10 mg, gelöst
in einer Minimalmenge Wasser) wurde zu einer Lösung von Methyl-2-[5-(4-{[4-ethylphenyl)amino)carbonyl}-1-pentyl-1H-imidazol-2-yl)-2,3-dihydro-1H-inden-1-yl]butanoat
(20 mg) in Methanol (2 mL) gegeben. Die Mischung wurde über Nacht
bei 60°C
gerührt.
Die entstandene Mischung wurde eingeengt, worauf Salzsäure (1 M)
zugegeben wurde, um den pH-Wert auf 5 einzustellen. Die Mischung
wurde mit HPLC (ODS, Wasser/Acetonitril/Trifluoressigsäure) gereinigt,
um 2-[5-(4-{[(4-Ethylphenyl)amino]carbonyl}-1-pentyl-1H-imidazol-2-yl)-2,3-dihydro-1H-incien-1-yl]butansäure zu ergeben
(10 mg, 50 % Ausbeute). 1H-NMR (CD2Cl2) δ: 7,80 (s,
1H), 7,70 (d, 2H), 7,40 (m, 3H), 7,20 (d, 2H), 4,00 (t, 2H), 3,50
(m, 1H), 2,80–3,10
(m, 2H), 2,60 (m, 3H), 2,30 (m, 1H), 2,10 (m, 1H), 1,50–1,80 (m,
4H), 1,20 (m, 7H), 0,90 (t, 3H), 0,80 (t, 3H); LC-MS = 488,4, (M
+ H+), RT = 3,39 min
-
Die
folgenden Verbindungen, deren physikalische Eigenschaften unten
zusammengefasst sind, wurden in ähnlicher
Weise wie die Verbindung des Beispiels 7 hergestellt:
-
Beispiel
8 N-(4-t-Butylphenyl)-2-[(15)-1-(2-hydroxy-2-propenyl)-2,3-dihydro-1H-inden-5-yl]-5-methyl-1-pentyl-1H-imidazol-4-carboxamid-Hydrat
-
- 1H-NMR (CDCl3) δ: 9,98 (s,
1H), 7,62 (d, 2H), 7,40 (s, 1H), 7,28–7,40 (m, 4H), 3,90 (t, 2H),
3,56–3,70
(m, 1H), 2,71–3,05
(m, 3H), 2,68 (s, 3H), 2,35–2,60
(m, 2H), 1,64–1,97
(m, 3H), 1,30 (s, 9H), 1,10–1,28
(m, 4H), 0,89 (t, 3H); LC-MS = 502,3 (M + H+),
RT = 4,30 min
-
Beispiel
9 N-(3,4-Dimethylphenyl)-2-[(1S)-1-(2-hydroxy-2-propenyl)-2,3-dihydro-1H-inden-5-yl]-5-methyl-1-pentyl-1H-imidazol-4-carboxamid-Hydrat
-
- 1H-NMR (CDCl3) δ: 9,92 (s,
1H), 7,32–7,50
(m, 5H), 7,10 (d, 1H), 3,90 (t, 2H), 3,56–3,70 (m, 1H), 2,80–3,05 (m,
2H), 2,71 (s, 3H), 2,45–2,
60 (m, 2H), 2,21 (s, 3H), 2,15 (s, 3H), 1,60–1,95 (m, 3H), 1,10–1,34 (m,
4H), 0,84 (t, 3H); LC-MS = 474,1 (M + H+),
RT = 3,44 min
-
Beispiel
10 2-[(1S)-1-(2-Hydroxy-2-propenyl)-2,3-dihydro-1H-inden-5-yl]-5-methyl-N-[2-methyl-4-(trifluormethoxy)phenyl]-1-pentyl-1H-imidazol-4-carboxamid-Hydrat
-
- 1H-NMR (CDCl3) δ: 9,89 (s,
1H), 7,64 (d, 1H), 7,45 (s, 1H), 7,38 (d, 2H), 7,06 (d, 2H), 3,90
(t, 2H), 3,56–3,70 (m,
1H); 2,79–3,05
(m, 3H), 2,62 (s, 3H), 2,45–2,60
(m, 2H), 2,33 (s, 3H), 1,60–1,95
(m, 3H), 1,10–1,35
(m, 4H), 0,86 (t, 3H); LC-MS = 544,3 (M + H+),
RT = 4,36 min
-
Herstellung von Zwischenproduktverbindungen
-
Herstellung
der Zwischenproduktverbindung A-3 Ethyl-2-brom-1-(3-methoxypropyl)-5-methyl-1H-imidazol-4-carboxylat
Ethyl-2-brom-5-methyl-1H-imidazol-4-carboxylat
-
Stufe
1: Eine Mischung aus Ethyl-5-methyl-1H-imidazol-4-carboxylat (50,0
g, 324 mmol), N-Bromsuccinimid (1,1 Äq., 63,5 g, 357 mmol) und aus
trockenem Acetonitril (400 mL) wurde 16 h lang unter einer Argon-Atomosphäre gerührt. Einengen
der Mischung unter verringertem Druck ergab ein Öl, das in Dichlormethan gelöst wurde.
Die Feststoffe wurden abfiltriert, und das Filtrat wurde unter verringertem
Druck eingeengt, um ein Öl
zu ergeben. Die Reinigung des Öls
durch Kieselgel-Chromatografie (30 % Ethylacetat/Hexane (1 L) und
dann 50 $ Ethylacetat/Hexane) ergab 33 g (44 %) Ethyl-2-brom-5-methyl-1H-imidazol-4-carboxylat
als weißen
Feststoff: 1H-NMR (CDCl3) δ: 1,38 (t,
3H), 2,78 (s, 3H), 4,31 (q, 2H)
-
Ethyl-2-brom-1-(3-hydroxypropyl)-5-methyl-1H-imidazol-4-carboxylat
-
Stufe
2: Zu einer Lösung
von 5,04 g Ethyl-2-brom-5-methyl-1H-imidazol-4-carboxylat (0,0213 mmol) in 50 mL Tetrahydrofuran
wurden unter Argon 0,66 g 95%iges Natriumhydrid gegeben. Nach Rühren der
entstandenen Mischung über
30 min bei rt wurden 7,81 g Brompropan-3-ol (0,0562 mmol) zugegeben,
worauf die Mischung 18 h lang am Rückfluss erwärmt wurde. Die Mischung wurde
dann zur Entfernung von Feststoffen filtriert, und das Filtrat wurde
eingeengt. Das Rohmaterial wurde durch Blitz-Chromatografie (Biotage-Säule, 3:2
Ethylacetat:Hexan) gereinigt, um Ethyl-2-brom-1-(3-hydroxypropyl)-5-methyl-1H-imidazol-4-carboxylat
als klares, farbloses Öl
zu ergeben (4,63 g, 74 %). 1H-NMR (CDCl3) δ:
1,37 (t, 3H), 1,89–1,98
(m, 2H), 2,22 (br t, 1H), 2,58 (s, 3H), 3,65–3,71 (m, 2H), 4,06 (t, 2H),
4,33 (q, 2H); LC-MS = 293,0 (M + H+)
-
Ethyl-2-brom-1-(3-methoxypropyl)-5-methyl-1H-imidazol-4-carboxylat
-
Stufe
3: Bei 0°C
wurden zu einer Lösung
von 4,76 g Ethyl-2-brom-1-(3-hydroxypropyl)-5-methyl-1H-imidazol-4-carboxylat
(0,0291 mmol) in 30 mL Tetrahydrofuran unter Argon 0,47 g 95%iges
Natriumhydrid gegeben. Die entstandene Mischung wurde 30 min lang
gerührt.
Jodmethan (18,51 g, 0,1304 mmol) wurde zugegeben, und die Mischung
wurde 70 min lang bei 0 bis 5°C
gerührt.
Eiswasser (20 mL) wurde zugegeben, und die Mischung wurde mit Ethylacetat
(2 × 20
mL) extrahiert. Die vereinigten Extrakte wurden mit Magnesiumsulfat
getrocknet und eingeengt, um 4,6 g dunkelgelbes Öl zu ergeben. Dieses Material
wurde durch Blitz-Chromatographie (Biotage-Blitz-Säule,
1:1 Ethylacetat:Hexan) gereinigt, um Ethyl-2-brom-1-(3-methoxypropyl)-5-methyl-1H-imidazol-4-carboxylat
als klares, farbloses Öl
zu ergeben (1,71 g, 34 % Ausbeute).
1H-NMR
(CDCl
3) δ:
1,37 (t, 3H), 1,89–1,99
(m, 2H), 2,56 (s, 3H), 3,34 (t, 2H), 3,33 (s, 3H), 4,01 (t, 2H),
4,34 (q, 2H); LC-MS = 307,0 (M + H
+) Herstellung
der Zwischenproduktverbindung A-4 t-Butyl-2-methyl-2-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy]propanoat
t-Butyl-2-(4-bromphenoxy)-2-methylpropanoat
-
Stufe
1: Zu einer Lösung
von 4-Bromphenol (5,0 g, 28,9 mmol) in Ethanol (60 mL) wurde Kaliumhydroxid
(1,62 g, 28,9 mmol) langsam gegeben, und die entstandene Suspension
wurde bei 60°C
erwärmt,
bis alles Kaliumhydroxid aufgelöst
war. Die entstandene Lösung
wurde auf 0°C
abgekühlt,
worauf t-Butyl-2-bromisobutyrat (5,4 mL, 28,9 mmol) zugetropft wurde.
Die Mischung wurde dann 16 h lang am Rückfluss gehalten, bevor sie
auf rt abgekühlt
wurde. Kaliumbromid (weißer
Feststoff) wurde abfiltriert, worauf die Mischung unter verringertem
Druck eingeengt wurde. Der Rückstand
wurde in Dichlormethan gelöst,
und die entstandene Lösung
wurde mit Wasser und Salzlösung
gewaschen, über
wasserfreiem Natriumsulfat getrocknet und unter verringertem Druck
eingeengt. Der Rückstand
wurde durch Blitz-Chromatografie
(Biotage-Blitz-40 M-Säule, 6:1
Hexan:Ethylacetat) gereinigt, um t-Butyl-2-(4-bromphenoxy)-2-methylpropanoat
zu ergeben (3,15 g, 35 %). EI-MS = 314 (M+); 1H-NMR ((300 MHz, CDCl3) δ: 1,44 (s,
9H), 1,55 (s, 6H), 6,71 (m, 2H), 7,31 (d, 2H)
-
t-Butyl-2-methyl-2-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy]propanoat
-
Stufe
2: [1,1'-Bis(diphenylphosphino)ferrocen]dichlorpalladium(II)
(1:1-Komplex mit
Dichlormethan) (245 mg, 0,3 mmol), Kaliumacetat (2,9 g, 29,5 mmol)
und Bis(pinacolato)dibor (2,74 g, 10,81 mmol) wurden in einen trockenen
Kolben unter Argon gegeben. Eine Lösung von t-Butyl-2-(4-bromphenoxy)-2-methylpropanoat (3,1
g, 9,83 mmol) in 30 mL Dimethylsulfoxid wurde zugegeben, und die
entstandene Lösung
wurde bei 80°C 48
h lang erhitzt. Die Mischung wurde dann durch einen Pfropf von Kieselgel
(100 % Hexan zuerst, um Bis(pinacolato)dibor zu eluieren, und dann
5 % Ethylacetat) filtriert, um t-Butyl-2-methyl-2-[4-(4,4,5,5-tetrmethyl-1,3,2-dioxaborolan-2-yl)phenoxy]propanoat
(2,4 g, 67 %) als hellgelbes Öl
zu erhalten. EI-MS = 362 (M+); 1H-NMR
(300 MHz, CDCl3) δ: 1,35 (s, 9H), 1,43 (s, 12H),
1,54 (s, 6H), 6,79 (d, 2H), 7,67 (d, 2H)
-
Herstellung
der Zwischenproduktverbindung A-5 t-Butyl-2-methyl-2-{[3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfanyl}propanoat
-
Stufe
1: Zu einer Lösung
von m-Thiocresol (5,00 g, 40,25 mmol) in Ethanol (81 mL) wurde Kaliumhydroxid
(2,26 g, 40,25 mmol) langsam gegeben, und die entstandene Suspension
wurde bei 60°C
erwärmt,
bis alles Kaliumhydroxid gelöst
war. Die entstandene Lösung
wurde auf 0°C
abgekühlt,
worauf t-Butyl-2-bromisobutyrat
(7,51 mL, 40,25 mmol) zugetropft wurde. Die Mischung wurde dann
am Rückfluss
1 h lang gehalten, bevor sie auf rt abgekühlt wurde. Kaliumbromid (weißer Feststoff)
wurde abfiltriert, worauf die Mischung unter verringertem Druck
eingeengt wurde. Der Rückstand
wurde in Dichlormethan gelöst,
und die entstandene Lösung
wurde mit Wasser und Salzlösung
gewaschen, über
wasserfreiem Natriumsulfat getrocknet und unter verringertem Druck
eingeengt. Der Rückstand
wurde durch Blitz-Chromatografie
(Biotage-Blitz-75-Säule,
4:1 Hexan:Ethylacetat) gereinigt, um t-Butyl-2-methyl-2-[(3-methylphenyl)sulfanyl]propanoat
zu ergeben (8,6 g, 80 %). EI-MS = 266; 1H-NMR
(300 MHz, CDCl3) δ: 1,41 (s, 9H), 1,44 (s, 6H),
2,32 (s, 3H), 7,20 (m, 4H)
-
t-Butyl-2-[(4-brom-3-methylphenyl)sulfanyl]-2-methylpropanoat
-
Stufe
2: Zu einer Lösung
von t-Butyl-2-methyl-2-[(3-methylphenyl)sulfanyl]propanoat (2,0
g, 7,52 mmol) in Acetonitril (75 mL) wurde N-Bromsuccinimid (1,47
g, 8,27 mmol) gegeben. Die entstandene Lösung wurde rt 16 h lang gerührt. Die
Mischung wurde unter verringertem Druck eingeengt, worauf der Rückstand
in Ethylacetat gelöst
wurde. Die entstandene Lösung
wurde mit Salzlösung,
gesättigtem
wässrigen
Natriumthiosulfat und mit Wasser gewaschen, über wasserfreiem Natriumsulfat
getrocknet und unter verringertem Druck eingeengt. Der Rückstand
wurde durch Blitz-Chromatografie
(Biotage-Blitz-40M) mit 95:5 Hexan:Ethylacetat gereinigt, um t-Butyl-2-[(4-brom-3-methylphenyl)sulfanyl]-2-methylpropanoat
zu ergeben (1,79 g, 69 %). EI-MS = 346; 1H-NMR
(300 MHz, CDCl3) δ: 1,41 (s, 15H), 2,35 (s, 3H),
7,14 (dd; 1H), 7,34 (s, 1H), 7,44 (d, 1H)
-
t-Butyl-2-methyl-2-{[3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfanyl}propanoat
-
Stufe
3: [1,1'-Bis(diphenylphosphino)ferrocen]dichlorpalladium(II)
(1:1-Komplex mit Dichlormethan) (213 mg, 0,26 mmol), Kaliumacetat
(1,52 g, 15,45 mmol) und Bis(pinacolato)dibor (1,44 g, 5,67 mmol)
wurden in einen trockenen Kolben unter Argon gegeben. Eine Lösung von
t-Butyl-2-[(4-brom-3-methylphenyl)sulfanyl]-2-methylpropanoat
(1,78 g, 5,15 mmol) in 15 mL Dimethylsulfoxid wurde zugegeben, und
die entstandene Lösung
wurde bei 80°C
18 h lang erhitzt. Die Mischung wurde dann durch einen Pfropf aus
Kieselgel (100 % Hexan zuerst, um überschüssiges Pinacoldibor zu beseitigen,
und dann 95:5 Hexan:Ethylacetat) filtriert, um t-Butyl-2-methyl-2-{[3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfanyl}propanoat
zu erhalten (1,2 g, 59 %). EI-MS = 392; 1H-NMR
(300 MHz, CDCl3 δ: 1,35 (s, 6H), 1,39 (s, 15H),
1,42 (s, 6H), 2,00 (s, 3H), 7,00 (d, 1H), 7,37 (d, 1H), 7,41 (s,
1H)
-
Herstellung
der Zwischenproduktverbindung A-6 t-Butyl-2-methyl-2-{[2-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfanyl}propanoat
t-Butyl-2-methyl-2-[(2-methylphenyl)sulfanyl]propanoat
-
Stufe
1: Zu einer Lösung
von 2-Methylbenzolthiol (5,0 g, 40,25 mmol) in Ethanol (81 mL) wurde
Kaliumhydroxid (2,26 g, 40,25 mmol) langsam gegeben, und die entstandene
Suspension wurde bei 60°C
erwärmt,
bis alles Kaliumhydroxid gelöst
war. Die entstandene Lösung
wurde auf 0°C
abgekühlt,
worauf t-Butyl-2-bromisobutyrat (7,51 mL, 40,25 mmol) zugetropft
wurde. Die Mischung wurde dann 1 h lang am Rückfluss gehalten, bevor sie
auf rt abgekühlt
wurde. Kaliumbromid (weißer
Feststoff) wurde abfiltriert, und das Filtrat wurde unter vermindertem
Druck eingeengt. Der Rückstand
wurde in Dichlormethan gelöst,
worauf die entstandene Lösung
mit Wasser und Salzlösung
gewaschen, über
wasserfreiem Natriumsulfat getrocknet und unter verringertem Druck
eingeengt wurde. Der Rückstand
wurde durch Blitz-Chromatografie
(Biotage-Blitz-75-Säule;
4:1 Hexan:Ethylacetat) gereinigt, um t-Butyl-2-methyl-2-[(2-methylphenyl)sulfanyl]propanoat
zu ergeben (7,9 g, 74 %). EI-MS = 266 1H-NMR
(300 MHz, CDCl3) δ: 1,41 (s, 9H), 1,43 (s, 6H),
2,47 (s, 3H), 7,23 (d, 2H), 7,11 (m, 1H), 7,46 (d, 1H)
-
t-Butyl-2-[(4-brom-2-methylphenyl)sulfanyl]-2-methyl]propanoat
-
Stufe
2: Zu einer Lösung
von t-Butyl-2-methyl-2-[(2-methylphenyl)sulfanyl]propanoat
(3,0 g, 11,28 mmol) in Acetonitril (113 mL) wurde N-Bromsuccinimid
(2,21 g, 12,41 mmol) gegeben. Die entstandene Lösung wurde bei rt 16 h lang
gerührt.
Die Mischung wurde unter verringertem Druck eingeengt, und der Rückstand wurde
in Ethylacetat gelöst.
Die entstandene Lösung
wurde mit Salzlösung,
gesättigtem
wässrigen
Natriumthiosulfat und mit Wasser gewaschen, über wasserfreiem Natriumsulfat
getrocknet und unter verringertem Druck eingeengt. Der Rückstand
wurde durch Blitz-Chromatographie
(Biotage-Blitz-40M-Säule,
95:5 Hexan:Ethylacetat) gereinigt, um t-Butyl-2-[(4-brom-2-methylphenyl)sulfanyl]-2-methylpropanoat
zu ergeben (2,5 g, 65 %). EI-MS = 346; 1H-NMR
(300 MHZ, CDCl3) δ: 1,41 (s, 15H), 2,44 (s, 3H),
7,24 (s, 1H), 7,31 (s, 1H), 7,39 (s, 1H)
-
t-Butyl-2-methyl-2-{[2-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfanyl}propanoat
-
Stufe
3: [1,1'-Bis(diphenylphosphin)ferrocen]dichlorpalladium(II)
(1:1-Komplex mit Dichlormethan) (294 mg, 0,36 mmol), Kaliumacetat
(2,13 g, 21,72 mmol) und Bis(pinacolato)dibor (2,02 g, 7,96 mmol)
wurden in einen trockenen Kolben unter Argon gegeben. Eine Lösung von
MP-03-2 (2,5 g, 7,24 mmol) in 20 mL Dimethylsulfoxid wurde zugegeben,
und die entstandene Lösung
wurde bei 80°C
18 h lang erhitzt. Die Mischung wurde dann durch einen Pfropf aus
Kieselgel (100 % Hexan zuerst, um überschüssiges Bis(pinacolato)dibor zu
beseitigen, und dann 95:5 Hexan:Ethylacetat) filtriert, um t-Butyl-2-methyl-2-{[2-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfanyl}propanoat
zu erhalten (1,1 g, 40 %). EI-MS = 392; 1H-NMR
(300 MHz, CDCl3) δ: 1,33 (s, 12H), 1,38 (s, 9H),
1,40 (s, 6H), 2,45 (s, 3H), 7,42 (d, 1H), 7,53 (d, 1H), 7,66 (s,
1H)
-
Herstellung
der Zwischenproduktverbindung A-7 t-Butyl-{[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfanyl}acetat
-
Mit
einem Syntheseweg ähnlich
dem oben für
die Zwischenproduktverbindung A-2 beschriebenen und unter Einsatz
der entsprechenden Materialien wurde t-Butyl-{[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfanyl}acetat
hergestellt. 1H-NMR (300 MHz, CDCl3) δ:
1,31 (s, 12H), 1,42 (s, 6H), 3,60 (s, 2H), 7,32 (d, 2H), 7,72 (d,
2H)
-
Herstellung
der Zwischenproduktverbindung A-8 t-Butyl-2-{[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfanyl}propanoat
-
Mit
einem Syntheseweg ähnlich
dem oben für
die Zwischenproduktverbindung A-2 beschriebenen und unter Einsatz
der entsprechenden Materialien wurde t-Butyl-2-{[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfanyl}propanat
hergestellt. 1H-NMR (300 MHz, CDCl3) δ:
1,29 (s, 12H), 1,38 (s, 9H), 1,48 (d, 3H), 3,81 (q, 1H), 7,41 (d,
2H), 7,71 (d, 2H)
-
Herstellung
der Zwischenproduktverbindung A-9 Ethyl-2-brom-5-methyl-1-pentyl-1H-imidazol-4-carboxylat
-
Zu
einer Lösung
von Natriumhydrid (0,80 g, 33,1 mmol) in 100 mL Tetrahydrofuran
wurde bei 0°C Ethyl-2-brom-5-methyl-1H-imidazol-4-carboxylat (7,0 g,
30,1 mmol) (siehe oben zur Herstellung), gelöst in 50 mL Tetrahydrofuran,
gegeben. Die Mischung wurde 30 min lang gerührt und dann auf rt erwärmt. Nach
1 h bei rt wurde eine Lösung
von 1-Jodpentan (4,36 g, 33,1 mmol) in 5 mL Tetrahydrofuran zugegeben,
und die Mischung wurde 16 h lang am Rückfluss gehalten. Die Reaktionsmischung
wurde abgekühlt
und filtriert. Einengen des Filtrats unter verringertem Druck ergab
ein Öl,
das in 150 mL Ethylacetat gelöst
wurde. Die entstandene Lösung
wurde mit Wasser und Salzlösung
gewaschen, über
wasserfreiem Natriumsulfat getrocknet und unter verringertem Druck
eingeengt, um ein Öl
zu ergeben. Reinigung durch Blitz-Chromatografie (Kieselgel, 50
% Ethylacetat in Hexanen bis 100 % Ethylacetat) ergab Ethyl-2-brom-5-methyl-1-pentyl-1H-imidazol-4-carboxylat
als farbloses Öl
(8,43 g, 92 %): Ms = 303,1 (M + H+); 1H-NMR (CDCl3) δ: 0,85 (t,
3H), 1,34 (t, 3H), 1,20–1,42
(m, 4H), 1,68–1,80
(m, 2H), 2,58 (s, 3H), 3,85 (t, 2H), 4,32 (q, 2H)
-
-
Herstellung der Zwischenproduktverbindungen
A-10 und A-11
-
Zwischenproduktverbindung
A-10 Ethyl-2-brom-1-(2-methoxyethyl)-5-methyl-1H-imidazol-4-carboxylat
-
Zwischenproduktverbindung
A-11 Ethyl-2-brom-1-(2-methoxyethyl)-4-methyl-1H-imidazol-5-carboxylat
-
Mit
Verfahren ähnlich
denen zur Herstellung der Zwischenproduktverbindung 9 und unter
Einsatz des entsprechenden Elektrophils wurden Ethyl-2-brom-1-(2-methoxyethyl)-5-methyl-1H-imidazol-4-carboxylat (A-10,
LC-MS = 293,0, RT = 1,97 min) und Ethyl-2-brom-1-(2-methoxyethyl)-4-methyl-1H-imidazol-5-carboxylat
(A-11, LC-MS = 293,1, RT = 2,30 min) hergestellt.
-
Abschnitt B-Thiazole und
Oxazole
-
Beispiel
1 Herstellung
von Natrium-2-{[4-(4-{[(4-ethylphenyl)amino]carbonyl}-5-phenyl-1,3-thiazol-2-yl)phenyl]sulfanyl}-2-methylpropanoat
Methyl-2-amino-5-phenyl-1,3-thiazol-4-carboxylat
-
Stufe
1: Eine Lösung
von Ethyldichloracetat (45 mL, 57,60 g, 366,88 mmol) und von Benzaldehyd
(40 mL, 41,76 g, 393,52 mmol) in trockenem Tetrahydrofuran (160
mL) wurde bei –5°C unter Argon
gekühlt,
und dann durch Zutropfen von Natriummethoxid (19,82 g, 366,90 mmol)
in trockenem Methanol (200 mL) behandelt. Die entstandene milchige
Suspension wurde 90 min lang bei –5°C gerührt und dann in Salzlösung (400 mL)
und in Tetrahydrofuran (400 mL) gegossen. Die Schichten wurden getrennt,
und die wässrige
Schicht wurde mit Tetrahydrofuran (200 mL) extrahiert. Die vereinigten
organischen Extrakte wurden über
Natriumsulfat getrocknet und zu einem halb-festen Rückstand
eingeengt, der anschließend
in Methanol (435 mL) gelöst
wurde. Die Lösung
wurde mit Thioharnstoff (23,67 g, 310,96 mmol) behandelt, worauf
das ganze am Rückfluss unter
Argon milde erwärmt
wurde. Nach 18 h wurde die gelbe, opake Lösung auf 5°C abgekühlt, worauf der pH-Wert auf
7 bis 8 mit konzentriertem wässrigen
Ammoniumhydroxid (ca. 15 mL) eingestellt wurde. Das Ganze wurde
dann mit Wasser (200 mL) verdünnt
und filtriert. Der entstandene Kuchen wurde mit Wasser (2 × 300 mL)
gewaschen und dann unter verringertem Druck bei 40°C getrocknet,
um Methyl-2-amino-5-phenyl-1,3-thiazol-4-carboxylat
(64,24 g, 274,21 mmol, 88 %) als gelben Feststoff zu ergeben. 1H-NMR (DMSO-d6, 300 MHz): δ: 3,60 (s,
3H, -CO2CH3); 7,27
(br s, 2H, -NH2); 7,36 (m, 5H, aromatisch);
Ms (HPLC/ES): 235 (M + 1); RT = 1,91 min
-
Zwischenproduktverbindung
B-1 Methyl-2-brom-5-phenyl-1,3-thiazol-4-carboxylat
-
Stufe
2: Eine Lösung
von Kupfer(II)bromid (143,0 g, 223,36 mmol) und t-Butylnitril (38
mL, 33,01 g, 320,13 mmol) in wasserfreiem Acetonitril (500 mL) wurde
bei 60°C
unter Argon erwärmt,
und dann mit anteiliger Zugabe von Methyl-2-aminbo-5-phenyl-1,3-thiazol-4-carboxylat
(50,0 g, 234,28 mmol) behandelt. Eine exotherme und schnelle Entwicklung
von Stickstoffgas wurde bei der Zugabe beobachtet. Das Ganze wurde bei
60°C 60
min lang gerührt,
auf 20°C
abgekühlt
und dann in 2 N wässrige
Salzsäure
(500 mL) und in Ethylacetat (500 mL) gegossen. Die Schichten wurden
getrennt, und die organischen Schichten wurden mit 2 N wässriger
Salzsäure
(500 mL) und mit Salzlösung
(4 × 500
mL) gewaschen, über
Natriumsulfat getrocknet und zu einem orangefarbenen Feststoff eingeengt.
Der Feststoff wurde aus Methanol umkristallisiert und unter stark
verringertem Druck bei 40°C
getrocknet, um Methyl-2-brom-5-phenyl-1,3-thiazol-4-carboxylat (Zwischenproduktverbindung
B-1) (55,36 g, 185,68 mmol, 87 %) als blassgelbe Kristalle zu ergeben. 1H-NMR (DMSO-d6, 300
MHz) δ:
3,69 (s, 3H, -OCH3); 7,49 (m, 5H); Ms (HPLC/ES):
298 (M + H); RT = 2,93 min
-
Methyl-2-[4-(t-butylsulfanyl)phenyl]-5-phenyl-1,3-thiazol-4-carboxylat
-
Stufe
3: Zu einer Lösung
von Methyl-2-brom-5-phenyl-1,3-thiazol-4-carboxylat (0,7 g, 2,3 mmol) wurden
Dichlor[1,1'-bis(diphenylphosphino)ferrocen]palladium(II)-Dichlormethan-Komplex
(50 mg) in Toluol:Dioxan (4:1, 25 mL), t-Butyl-2-methyl-2-{[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfanyl}propanoat
(0,6 g, 2,3 mmol, 1,0 Äq.)
(siehe Abschnitt 1 zur Herstellung) und Natriumcarbonat (2 M, 5
mL) gegeben. Die Mischung wurde entgast und 48 h lang bei 90°C gerührt. Die
entstandene Mischung wurde mit Salzlösung gewaschen, worauf die
organische Schicht über
Natriumsulfat getrocknet und unter verringertem Druck eingeengt
wurde. Der. entstandene Rückstand
wurde durch Blitz-Chromatografie (Kieselgel, Hexan/Ethylacetat)
gereinigt, um Methyl-2-[4-(t-butylsulfanyl)phenyl]-5-phenyl-1,3-thiazol-4-carboxylat
zu ergeben (0,7 g, 50 % Ausbeute). 1H-NMR
(CD2Cl2) δ: 8,00 (d,
2H), 7,50 (m, 5H), 7,40 (d, 2H), 3,80 (s, 3H >), 1,40 (m, 15H); LC-MS = 407,2 (M + H+), RT = 4,31 min
-
2-{[4-{[(4-Ethyl)amino]carbonyl}-5-phenyl-1,3-thiazol-2-yl)phenyl]sulfanyl}-2-methylpropansäure
-
Stufe
4: Kaliumhydroxid (0,2 g in 2 mL Wasser) wurde zu einer Lösung von
Methyl-2-[4-(t-butylsulfanyl)phenyl]-5-phenyl-1,3-thiazol-4-carboxylat
(0,7 g) in Methanol (5 mL) gegeben. Die Mischung wurde 6 h lang
bei rt gerührt.
Der pH-Wert wurde dann auf ca. 4 mit 1 M HCl eingestellt, worauf
die wässrige
Schicht mit Ethylacetat (10 mL) extrahiert wurde. Die Schichten
wurden getrennt, und die organische Schicht wurde getrocknet und
eingeengt. Der entstandene Rückstand
wurde in Methylenchlorid (7 mL) gelöst, und Oxalylchlorid (1 mL)
und DMF (0,1 mL) wurden zugegeben. Die Mischung wurde 12 h lang
gerührt,
bevor sie unter verringertem Druck eingeengt wurde, um die Säurechlorid-Zwischenproduktverbindung
zu ergeben. Das Säurechlorid
(50 mg aus insgesamt 500 mg) wurde in Tetrahydrofuran (2 mL) gelöst, und
4-Ethylanilin (09,1 mL) und Triethylamin (0,2 mL) wurden zugegeben.
Die Mischung wurde 12 h lang gerührt.
Trifluoressigsäure
(2 mL) wurde zugegeben, das Ganze wurde weitere 12 h lang gerührt. Einengen
der Reaktionsmischung unter verringertem Druck ergab ein Rohmaterial,
das durch HPLC (ODS-Säule,
Wasser/Acetonitril/Trifluoressigsäure, aufgebracht als Methanol-Lösung) gereinigt
wurde, um 2-{[4-(4-{[(4-Ethyl)amino]carbonyl}-5-phenyl-1,3-thiazol-2-yl)phenyl]sulfanyl}-2-methylpropansäure zu ergeben. Ausbeute
50 %. 1H-NMR (CD2Cl2) δ:
9,60 (s, 1H), 8,10 (d, 2H), 7,60 (m, 6H), 7,40 (m, 3H), 7,20 (d,
2H), 2,60 (q, 2H), 1,60 (s, 6H), 1,20 (t, 3H); LC-MS = 503,1 (MSäure +
H+), RT = 4,32 min
-
Natrium-2-{[4-(4-{[(4-ethylphenyl)amino]carbonyl}5-phenyl-1,3-thiazol-2-yl)phenyl]sulfanyl}-2-methylpropanoat
-
Stufe
5: Eine Mischung aus 12,98 g Säure
(25,80 mmol) und aus 24,5 mL 1 M NaOH-Lösung in Wasser (24,5 mol) wurde
erwärmt
und bei 60°C
2,5 h lang gerührt.
Das zurückgebliebene,
ungelöste
Feststoffmaterial wurde durch Zugabe von 285 mL Acetonitril und
90 mL Wasser gelöst
und bei 60°C
2 h lang gerührt. Nach
Abkühlung
auf rt wurde die Reaktionsmischung über Ethylacetat (mit 180 mL
und dann mit 100 mL) extrahiert. Die wässrige Schicht wurde gefriergetrocknet,
um einen weißen
sehr hellen Feststoff zu ergeben, der anschließend unter stark verringertem
Druc kbei 45°C
28 h lang getrocknet wurde, um Natrium-2-{[4-(4-{[(4-ethylphenyl)amino]carbonyl}-5-phenyl-1,3-thiazol-2-yl)phenyl]sulfanyl}-2-methylpropanoat
zu ergeben (8,25 g, 64 % Ausbeute). 1H-NMR
(500 MHz, DMSO) δ:
1,17 (t, 3H), 1,36 (s, 6H), 2,57 (q, 2H), 7,15 (d, 2H), 7,41–7,45 (m,
3H), 7,60–7,65
(m, 6H), 7,94 (d, 2H), 10,16 (s, 1H); LC-MS = 503,1 (M + H+), RT = 416 min
-
Beispiel 2
-
Herstellung
von 2-Methyl-2-({4-[4-({[4-(4-morpholinyl)phenyl)amino}carbonyl)-5-phenyl-1,3-thiazol-2-yl]phenyl}sulfanyl)propansäure
-
Zu
einer Lösung
von t-Butyl-2-methyl-2-({4-[4-({[4-(4-morpholinyl)phenyl]amino}carbonyl)-5-phenyl-1,3-thiazol-2-yl]phenyl}sulfanyl)propanoat
(300 mg, 0,49 mmol) (erhalten mit Verfahren ähnlich denen in Beispiel 1,
Abschnitt B, unter Einsatz des entsprechenden Anilins [4-(4-Morphilinyl)anilin]
in Stufe 4) in Dichlormethan (8 mL) wurde Trifluoressigsäure (8 mL)
gegeben, und die entstandene Lösung
wurde 16 h lang bei rt gerührt.
Die Mischung wurde unter verringertem Druck eingeengt, und der Rückstand
wurde in gesättiger
Natriumbicarbonat-Lösung
(8 mL) gelöst
und 1 h lang kräftig
gerührt.
Die Mischung wurde dann unter verringertem Druck eingeengt, worauf
der Rückstand
in Wasser (10 mL) erneut gelöst
wurde. Der pH-Wert der Mischung wurde dann auf 4 bis 5 mit 0,5 M
Phosphorsäure
eingestellt, worauf die saure Mischung mit Ethylacetat extrahiert
wurde. Die vereinigten Extrakte wurden über wasserfreiem Natriumsulfat
getrocknet und unter verringertem Druck eingeengt, um die Säure 2-Methyl-2-({4-(4-[{[4-(4-morpholinyl)phenyl]amino}carbonyl)-5-phenyl-1,3-thiazol-2-yl]phenyl}sulfanyl)propansäure zu ergeben
(260 mg, 94 %). HPLC, RT = 3,50 min; Ms = 560,7 (M + H+); 1H-NMR (300 MHz, CDCl3) δ: 1, 58 (s,
6H), 3,18 (m, 4H), 3,84 (m, 4H), 6,86 (d, 2H), 7,41 (d, 3H), 7,62
(m, 6H), 7,86 (d, 2H), 9,28 (s, 1H)
-
Beisiel 3
-
Herstellung
von Natrium-2-methyl-2-[{4-[4-({[4;(4-morpholinyl)phenyl]amino}carbonyl)-5-phenyl-1,3-thiazol-2-yl]phenyl}sulfanyl)propanoat
-
2-Methyl-2-({4-(4-({[4-(4-morpholinyl)phenyl]amino}carbonyl)-5-phenyl-1,3-thiazol-2-yl]phenyl}sulfanyl)propansäure (240
mg, 0,43 mmol) wurde in Acetonitril (1 mL) gelöst und mit 1 N Natriumhydroxid
(0,41 mL, 0,41 mmol) und Wasser (0,5 mL) behandelt. Die Mischung
wurde bei rt 20 min lang gerührt.
Die Mischung wurde mit Wasser verdünnt, und die wässrige Schicht
wurde mit Ethylacetat zur Beseitigung der restlichen Säure gewaschen.
Die wässrige
Schicht wurde dann unter verringertem Druck eingeengt (Rotationsverdampfer, Hochvakuumpumpe),
um das Natriumsalz zu ergeben, das unter verringertem Druck bei
40°C weiter
getrocknet wurde. Das Salz wurde dann in einer Minimalmenge Wasser
aufgelöst,
worauf Isopropanol zugetropft wurde, bis die Mischung trüb wurde.
Der Kolben wurde bei 0°C
15 min lang gekühlt.
Das Salz wurde durch Filtration gesammelt und mit kaltem Isopropanol
gewaschen und unter Hochvakuum bei 40°C weiter getrocknet, um Natrium-2-methyl-2-({4-[4-({[4-(4-morpholinyl)phenyl]amino}carbonyl)-5-phenyl-1,3-thiazol-2-yl]phenyl}sulfanyl)propanoat
(61 mg, 25 %) als gelben Feststoff zu ergeben. HPLC: RT = 3,52 min;
Ms = 560,2 (M + H+); 1H-NMR
(300 MHz, DMSO) δ:
1;32 (s, 6H), 3,12 (m, 4H), 3,72 (m, 4H), 6,92 (d, 2H), 7,42 (m,
3H), 7,60 (m, 6H), 7,98 (d, 2H); 10,22 (s, 1H)
-
Herstellung von Zwischenproduktverbindungen
-
Herstellung
der Zwischenproduktverbindung B-2 Ethyl-2-jod-5-phenyl-1,3-oxazol-4-carboxylat
Ethyl-5-phenyl-1,3-oxazol-4-carboxylat
-
Stufe
1: Zu einer Mischung von Ethylisocyanoacetat (8,74 mmol) und 1,8-Diazabicyclo(5.4.0)undec-7-en
(8,84 mmol) in Tetrahydrofuran (12 mL) wurde eine Lösung von
Benzoesäureanhydrid
(8,84 mmol) in Tetrahydrofuran (2 mL) bei 10°C unter Rühren gegeben. Die entstandene
Mischung wurde bei rt 18 h lang kräftig gerührt. Die Mischung wurde eingeengt,
um einen Rückstand
zu ergeben, der zwischen Ethylacetat und Wasser verteilt wurde.
Die organische Schicht wurde über
wasserfreiem Natriumsulfat getrocknet und eingeengt, um ein bernsteinfarbenes Öl zu ergeben,
das durch Mitteldruck-Säulenchromatografie
gereinigt wurde (Biotage-40S-Normalphase-Kieselgel-Säule, Hexane:Ethylacetat = 6:1
bis 4:1 bis 2:1), um Ethyl-5-phenyl-1,3-oxazol-4-carboxylat als klares Öl in 42%iger
Ausbeute zu ergeben.
LC-MS = 218,1 (M + H+),
RT = 2,52 min
-
-
Stufe
2: Eine 1 M Lösung
von Lithium(trimethylsilyl)amid in Tetrahydrofuran(1,11 mmol) wurde
mit einer Spritze bei –78°C in eine
Lösung
von Ethyl-5-phenyl-1,3-oxazol-4-carboxylat
(0,921 mmol, 1 Äq.)
in Tetrahydrofuran (7 mL) getropft. Die entstandene Lösung wurde
bei –78°C 1 h lang
gerührt,
wobei eine Lösung
von Jod (1,38 mmol) in 2 mL Tetrahydrofuran mit einer Spritze zugetropft
wurde. Die Reaktionsmischung wurde auf rt erwärmt und bei dieser Temperatur
1,5 h lang gerührt.
Die entstandene Lösung
wurde in 10%iges wässriges Natriumthiosuflat
(15 mL) getropft und mit Ethylacetat extrahiert. Die organischen
Extrakte wurden mit Salzlösung
gewaschen, über
wasserfreiem Natriumsulfat getrocknet und unter verringertem Druck
eingeengt. Der Rückstand
wurde mit Mitteldruck-Säulenchromatografie
(Biotage-40S-Normalphase-Kieselgel-Säule, Hexane:Ethylacetet = 9:1)
gereinigt, um Ethyl-2-jod-5-phenyl-1,3-oxazol-4-carboxylat als blassgelben
Feststoff in 82%iger Ausbeute zu ergeben.
LC-MS = 344,0 (M
+ H+), RT = 3,01 min; Rf = 0,31 (Hexane:Ethylacetat
= 6:1)
-
Mit
Syntheseverfahren ähnlich
den für
die Zwischenproduktverbindung B-1 beschriebenen und unter Einsatz
entsprechender Ausgangsmaterialien wurden die Zwischenproduktverbindungen
B-3, B-4 und B-5 hergestellt:
-
Herstellung
der Zwischenproduktverbindung B-3
-
- RT (LC-MS) = 3,29 min; 1H-NMR (300
MHz, DMSO) δ:
7,52 (dd, 4H), 3,69 (s, 3H)
-
Herstellung
der Zwischenproduktverbindung B-4
-
- RT (LC-MS) = 3,00 min; 1H-NMR (300
MHz, DMSO) δ:
7,46 (d, 2H), 7,00 (d, 2H), 3,80 (s, 3H) und 3,71 (s, 3H)
-
Herstellung
der Zwischenproduktverbindung B-5
-
- Rt (LC-MS) = 3,31 min; 1H-NMR (300 MHz, DMSO) δ: 7,40 (d,
2H), 7,37 (d, 2H), 3,68 (s, 3H), 2,33 (s, 3H)
-
Herstellung
der Zwischenproduktverbindung B-6
-
N,N-Dimethylformamid
(55 mL) und Hexamethylphosphoramid (3,3 mL) wurden zu einer Mischung aus
Methyl-2-{4-[(2-t-butoxy-2-oxoethyl)sulfanyl]phenyl}-5-phenyl-1,3-thiazol-4-carboxylat
(1,41 q, 3,2 mmol, erhalten mit Verfahren ähnlich den in Beispiel 1, Abschnitt
B, beschriebenen unter Einsatz des entsprechenden Elektrophils)
und aus Kaliumjodid (3,18 g, 19,1 mmol) in einem trockenen Kolben
gegeben. Die entstandene Mischung wurde bei 120°C 6 Tage lang erhitzt. Die Mischung
wurde mit Ethylacetat verdünnt,
mit Wasser (2 × 10
mL) und mit Salzlösung
gewaschen und über
Magnesiumsulfat getrocknet. Einengen unter verringertem Druck ergab
die rohe Säure
(1,38 g), die in der nächsten
Stufe ohne weitere Reinigung eingesetzt wurde.
-
Die
Zwischenproduktverbindung B-6 wurde mit einer Verfahrensweise ähnlich der
des Beispiels 1, Abschnitt B, weiter derivatisiert, um die in Tabelle
5 angegebenen Endprodukte zu ergeben.
-
Herstellung
der Zwischenproduktverbindungen B-7 und B-8
-
2-{4-[(2-t-Butoxy-1-methyl-2-oxoethyl)sulfanyl]phenyl}-5-phenyl-1,3-thiazol-4-carbonsäure (B-7)
wurde mit Syntheseverfahren ähnlich
den oben für
die Zwischenproduktverbindung B-6 beschriebenen hergestellt. Die
Säure B-7
wurde mit Verfahren ähnlich
denen des Beispiels 1, Abschnitt B, weiter derivatisiert. Der Ester B-8
wurde als Nebenrprodukt erhalten und mit dem üblichen Verfahren (siehe Beispiel
1, Abschnitt B) zur entsprechenden Säure hydrolysiert.
-
Abschnitt C-Triazole
-
Beispiel
1 Herstellung
von 2-{[4-(5-{[(2,4-Dimethylphenyl)aminojcarbonyl}-1-pentyl-H-1,2,4-triazol-3-yl)phenyl]sulfanyl}-2-methylpropansäure
4-[(2-t-Butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]benzoesäure
-
Stufe
1: Zu einer Lösung
von 4-Mercaptobenzoesäure
(8,0 g, 52 mmol) in Ethanol (80 mL) und in destilliertem Wasser
(20 mL) wurden t-Butyl-2-bromisobutyrat
(12,7 g, 57,1 mmol) und Kaliumhydroxid (6,4 g, 114 mmol) unter einer
Argon-Atmosphäre
gegeben. Die Reaktionsmischung wurde bei 100°C 16 h lang unter Argon erhitzt
und dann unter verringertem Druck eingeengt, um einen gelben Feststoff
zu ergeben. Der Rückstand
wurde mit destilliertem Wasser (100 mL) verdünnt und mit Ethylacetat (3 × 80 mL)
extrahiert. Die vereinigten Extrakte wurden durch Kieselgel filtriert,
worauf das Filtrat unter verringertem Druck eingeengt wurde, um
4-[(2-t-Butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]benzoesäure als
weißen
Feststoff zu ergeben (12,8 g, 83 %).
-
Ethyl-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-1H-1,2,4-triazol-5-carboxylat
-
Stufe
2: Zu einer Lösung
von 4-[(2-t-Butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]benzoesäure (7,30
g, 24,8 mmol) in trockenem Tetrahydrofuran (10 mL) wurden Ethylchlorformiat
(3,0 mL, 32 mmol) und Triethylamin (4,4 mL, 31 mmol) bei 0°C gegeben.
Die Reaktionsmischung wurde bei rt 90 min lang gerührt und
dann filtriert. Das Filtrat wurde mit einer Lösung von Ethyloxamidrazonat
(siehe z.B. J. Org. Chem. 23:1931, 1958, zur Herstellung dieses
Reagens) (2,95 g, 22,5 mmol) in Tetrahydrofuran (2 mL) befiandelt,
und die entstandene Mischung wurde bei rt 3 h lang gerührt und
unten verringertem Druck eingeengt, um einen orangefarbenen Feststoff
zu ergeben. Eine Lösung
des orangefarbenen Rohmaterials in Kohlenstofftetrachlorid (50 mL)
und Acetonitril (30 mL) wurde mit Triphenylphosphin (10 g, 38 mmol)
2 h lang am Rückfluss
gehalten. Die Mischung wurde dann auf rt abgekühlt und unter verringertem
Druck eingeengt. Reinigung durch Blitz-Chromatografie (Kieselgel-Säule, 20
bis 40 % Ethylacetat/Hexane) ergab Ethyl-3-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl)phenyl}-1H-1,2,4-triazol-5-carboxylat
als weißen
Feststoff (1,8 g, 20 %).
1H-NMR (CDCl3) δ:
8, 02 (d, 2H), 7,55 (d, 2H), 4,48 (q, 2H), 1,46 (s, 6H), 1,40 (s,
9H), 1,25 (t, 3H); LC-MS = 392,4 (M + H+),
RT = 3,36 min
-
Ethyl-3-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-1-pentyl-1H-1,2,4-triazol-5-carboxylat
-
Stufe
3: Zu einer von Ethyl-3-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-1H-1,2,4-triazol-5-carboxylat
(0,9 g, 2,55 mmol) in N,N-Dimethylformamid (10 mL) wurden Natriumhydrid
(0,11 g, 2,81 mmol) und 1-Jodpentan (0,5 mL, 3,8 mmol) bei 0°C gegeben.
Die Reaktionsmischung wurde bei 0°C
30 min lang und dann nach Erwärmen
auf rt 4 h lang gerührt.
Die Mischung wurde mit destilliertem Wasser (10 mL) abgeschreckt
und mit Ethylacetat (3 × 10
mL) extrahiert. Die vereinigten Extrakte wurden mit Salzlösung (20 mL)
gewaschen, über
Magnesiumsulfat getrocknet, filtriert und unter verringertem Druck
eingeengt, um ein blassgelbes Öl
zu ergeben. Reinigung durch Blitz-Chromatografie (Kieselgel, 10
bis 20 % Ethylacetat/Hexane) ergab Ethyl-3-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-1-pentyl-1H-1,2,4-triazol-5-carboxylat als
farbloses Öl
(0,56 g, 59 %). 1H-NMR (CDCl3) δ: 8,09 (d,
2H), 7,56 (d, 2H), 4, 63 (t, 2H), 4,51 (q, 2H), 1,95–1,90 (m,
2H), 1,46 (t, 9H), 1,42 (s, 9H), 1,38–1,34 (m, 4H), 0,91 (t, 3H);
LC-MS = 462,3 (M + H+), RT = 4,60 min
-
1-Pentyl-3-{4-[(1,1,4,4-tetramethyl-2-oxopentyl)sulfanyl]phenyl}-1H-1,2,4-triazol-5-carbonsäure
-
Stufe
4: Zu einer Lösung
von Ethyl-3-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-1-pentyl-1H-1,2,4-triazol-5-carboxylat
(0,55 g, 1,19 mmol) in Ethanol (8 mL) wurde 1 N Natriumhydroxid
(2,0 mL) gegeben, worauf die Mischung bei rt 4 h lang gerührt wurde.
Die Reaktionsmischung wurde dann mit 2 N Salzsäure auf einen pH-Wert von 5
bis 6 angesäuert.
Die entstandene Mischung wurde unter verringertem Druck teilweise
eingeengt und dann mit Dichlormethan (3 × 8 mL) extrahiert. Die vereinigten
Extrakte wurden mit destilliertem Wasser (10 mL) und mit Salzlösung (10
mL) gewaschen, über
Magnesiumsulfat getrocknet, filtriert und unter verringertem Druck
eingeengt, um 1-Pentyl-3-{4-[(1,1,4,4-tetramethyl-2-oxopentyl)sulfanyl]phenyl}-1H-1,2,4-triazol-5-carbonsäure als
farbloses Öl
zu ergeben (0,48 g, 93 %). 1H-NMR (CDCl3) δ:
8,04 (d, 2H), 7,56 (d, 2H), 4,18 (t, 2H), 1,96–1,91 (m, 2H), 1,46 (s, 6H),
1,42 (s, 9H), 1,41–1,32
(m, 4H), 0,91 (t, 3H); LC-MS = 390,3 (M + H+ – CO2), RT = 3,99 min. Dieses Material wurde
in der nächsten
Stufe unmittelbar eingesetzt.
-
2-{[4-(5-{[(2,4-dimethylphenyl)amino]carbonyl}-1-pentyl-1H-1,2,4-triazol-3-yl)phenyl]sulfanyl}-2-methylpropansäure
-
Stufe
5: Zu einer Lösung
von 1-Pentyl-3-{4-[(1,1,4,4-tetramethyl-2-oxopentyl)sulfanyl)phenyl}-1H-1,2,4-triazol-5-carbonsäure (0,025
g, 0,058 mmol) in trockenem Dichlormethan (1 mL) wurden N,N-Dimethylformamid
(3 Tropfen) und Oxalylchlorid (0,14 mL, 0,29 mmol) gegeben. Die
Reaktionsmischung wurde bei rt 90 min lang gerührt und dann unter verringertem
Druck eingeengt. Der Rückstand
wurde mit einer Mischung aus 2,4-Dimethylanilin (0,02 mL, 0,12 mmol),
Triethylamin (0,02 mL, 0,12 mmol) und aus Dimethylaminopyridin (0,002
g, 0,01 mmol) in Dichlormethan (1 mL) behandelt, worauf die Reaktionsmischung
bei rt 16 h lang gerührt
wurde. Eine Lösung
von Trifluoressigsäure
(0,8 mL) in Dichlormethan (0,5 mL) wurde zugegeben, und diese Reaktionsmischung
wurde bei rt 4 h lang gerührt.
Die Mischung wurde unter verringertem Druck eingeengt, um ein gelbes Öl zu ergeben.
Reinigung durch Umkehrphase-HPLC (0 bis 70 % Acetonitril) ergab 2-{[4-(5-{[(2,4-Dimethylphenyl)amino]carbonyl}-1-pentyl-1H-1,2,4-triazol-3-yl)phenyl]sulfanyl}-2-methylpropansäure als
farbloses Öl
(0,003 g, 11 %). LC-MS = 481,3 (M + H+),
RT = 4,24 min
-
Beispiel 2
-
Herstellung
von 2-({4-[5-(Anilinocarbonyl)-1-(2-ethoxyethyl)-1H-1,2,4-triazol-3-yl]phenyl}sulfanyl)-2-methylpropansäure
Ethyl-3-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-1-(2-ethoxyethyl)-1H-1,2,4-triazol-5-carboxylat
-
Stufe
1: Auf einem Syntheseweg ähnlich
dem oben in Beispiel 1, Abschnitt C, beschriebenen und unter Einsatz
des entsprechenden Elektrophils wurde Ethyl-3-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-1-(2-ethoxyethyl)-1H-1,2,4-triazol-5-carboxylat
hergesellt. LC-MS 464,3 (M + H+), RT = 3,97
min
-
t-Butyl-2-({4-[1-(2-ethoxyethyl)-1H-1,2,4-triazol-3-yl]phenyl}sulfanyl)-2-methylpropanoat
-
Stufe
2: Zu einer Lösung
von Ethyl-3-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-1-(2-ethoxyethyl)-1H-1,2,4-triazol-5-carboxylat
(0,67 g, 1,45 mmol) in Ethanol (8 mL) wurde 1 N Natriumhydroxid
(2,0 mL) gegeben, und die Mischung wurde bei rt 12 h lang gerührt. Die Reaktionsmischung
wurde dann mit 2 N Salzsäure
auf einen pH-Wert von 5 bis 6 angesäuert. Die entstandene Mischung
wurde unter verringertem Druck teilweise eingeengt und mit Dichlormethan
(3 × 8
mL) extrahiert. Die vereinigten Extrakte wurden mit destilliertem
Wasser (10 mL) und Salzlösung
(10 mL) gewaschen, über
Magnesiumsulfat getrocknet, filtriert und unter verringertem Druck
eingeengt, um das decarboxylierte Produkt t-Butyl-2-({4-[1-(2-ethoxyethyl)-1H-1,2,4-triazol-3-yl]phenyl}sulfanyl)-2-methylpropanoat
als farbloses Öl
zu ergeben (0,54 g, 86 %). 1H-NMR (CDCl3) δ:
8,19 (s, 1H), 8,06 (d, 2H), 7,58 (d, 2H), 4,36 (t, 2H), 3,80 (t,
2H), 3,66 (q, 2H), 1,50 (s, 6H), 1,42 (s, 9H), 1,18 (t, 3H); LC-MS
= 392,2 (M + H+), RT = 3,45 min
-
2-({4-[5-Anilinocarbonyl)-1-(2-ethoxyethyl)-1H-1,2,4-triazol-3-yl]phenyl}sulfanyl)-2-methylpropansäure
-
Stufe
3: Zu einer Lösung
von t-Butyl-2-({4-[1-(2-ethoxyethyl)-1H-1,2,4-triazol-3-yl]phenyl}sulfanyl)-2-methylpropanoat
(0,05 g, 0,13 mmol) in Tetrahydrofuran (2 mL) wurde n-Butyllithium
(0,06 mL, 0,15 mmol) bei –78°C unter Argon
gegeben, und die Reaktionsmischung wurde bei –78°C 1 h lang gerührt. Phenylisocyanat
(0,02 mL, 0,15 mmol) wurde bei –78°C zugegeben,
worauf die Mischung auf rt über
3 h erwärmt wurde.
Destilliertes Wasser (4 mL) wurde zugegeben, und die Mischung wurde
mit Ethylacetat (3 × 4
mL) extrahiert. Die vereinigten Extrakte wurden mit Salzlösung gewaschen, über Magnesiumsulfat
getrocknet, durch Celite filtriert und unter verringertem Druck
eingeengt. Der entstandene Rückstand
wurde dann mit einer Lösung
von Trifluoressigsäure
(1,0 mL) in Dichlormethan (1,0 mL) behandelt und bei rt 2 h lang
gerührt.
Die Mischung wurde unter verringertem Druck eingeengt, um ein gelbes Öl zu ergeben.
Reinigung durch Umkehrphase-HPLC (0 bis 70 % Acetonitril) ergab
(2-({4-[5-(Anilinocarbonyl)-1-(2-ethoxyethyl)-1H-1,2,4-triazol-3-yl]phenyl}sulfanyl)-2-methylpropansäure als
farbloses Öl
(0,004 g, 7 %). LC-MS = 455,1 (M + H+),
RT = 3,56 min
-
Abschnitt D-Pyrazole
-
Beispiel
1 Herstellung
von 2-{[4-(5-Butoxy-3-{[(2,4-dimethylphenyl)amino]carbonyl}-4-methyl-1H-pyrazol-1-yl)phenyl]sulfanyl}-2-methylpropansäure
t-Butyl-2-{[4-[N-t-butoxy)carbonylamino]-N-[(t-butoxy)carbonyl]amino]phenyl]sulfanyl}-2-methylpropanoat
-
Stufe
1: Zu einer gekühlten
(bei –78°C) Lösung von
t-Butyl-2-[(4-bromphenyl)sulfanyl]-2-methylpropanoat
(siehe Abschnitt 1 zur Herstellung) (10,0 g, 31,4 mmol) in wasserfreiem
Tetrahydrofuran (70 mL) wurde eine 1,6 M Lösung von n-Butyllithium in
Tetrahydrofuran (22 mL, 13,8 mmol) getropft (ca. 6 min lang). Es
wurde weitere 10 min lang weiter gerührt, worauf eine Lösung von
Di-t-butylazodicarboxylat (7,94 g, 34,5 mmol) in Tetrahydrofuran
(30 mL) in mehreren Anteilen bei –78°C zugegeben wurde. Die entstandene
Lösung
wurde 15 min lang gerührt,
worauf Essigsäure
(1,4 mL, 34,5 mmol) zugegeben wurde. Die Mischung wurde auf rt erwärmt, worauf
Wasser (30 mL) und Ether (100 mL) zugegeben wurden. Die Schichten
wurden getrennt, und die organische Schicht wurde mit Salzlösung gewaschen,
getrocknet (MgSO4), filtriert und eingeengt.
Das Rohmaterial wurde durch Blitz-Chromatografie (Kieselgel, 20
% Ethylacetat/Hexan) gereinigt, um t-Butyl-2-{(4-[N-(t-butoxy)carbonylamino]-N-[(t-butoxy)carbonyl]amino]phenyl]sulfanyl}-2-methylpropanoat
(4,2 g, 8,7 mmol) als hellgelbes Öl zu ergeben. 1H-NMR
(CDCl3, 400 MHz), δ: 1,39 (m, 15H), 1,48 (s, 18H),
7,33–7,45 (m,
4H)
-
Ethyl-1-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-5-hydroxy-4-methyl-1H-pyrazol-3-carboxylat
-
Stufe
2: Diethyloxalpropionat (2,64 mL, 14,0 mmol) wurde zu einer Lösung von
t-Butyl-2-{[4-[N-(t-butoxy)carbonylamino]-N-[{t-butoxy)carbonyl]amino]phenyl]sulfanyl}-2-methylpropanoat
(4,7 g, 9,75 mmol) und von Salzsäure
(4,0 M in Dioxan, 7,0 mL, 28,0 mmol) in Acetonitril (150 mL) bei
rt gegeben. Die Reaktionsmischung wurde bei 40°C 3 h lang gerührt. Wasser
(30 mL) und Ethylacetat (70 mL) wurden zugegeben, und die Schichten
wurden getrennt. Die wässrige
Schicht wurde mit Ethylacetat extrahiert (3×), und die vereinigten organischen
Schichten wurden getrocknet (Magnesiumsulfat) und eingeengt. Das
Rohmaterial wurde durch Blitz-Chromatografie (Kieselgel, 30 bis
50 % Ethylacetat/Hexan) gereinigt, um Ethyl-1-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-5-hydroxy-4-methyl-1H-pyrazol-3-carboxylat (350 mg,
0,83 mmol) als hellgelben Feststoff zu ergeben.
1H-NMR
(CDCl3, 400 MHz) δ: 1,34 (t, 3H), 1,38 (s, 6H),
1,39 (s, 9H), 2,09 (s, 3H), 4,33–4,45 (m, 2H), 7,46 (d, 2H),
7,64 (m, 2H);
LC-MS = 365,2 (M + H+)
-
Ethyl-5-butoxy-1-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-4-methyl-1H-pyrazol-3-carboxylat
-
Stufe
3: Eine Lösung
von Ethyl-1-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-5-hydroxy-4-methyl-1H-pyrazol-3-carboxylat
(350 mg, 0,83 mmol), n-Butanol (381 μL, 4,15 mmol), Tributylphosphin (410 μL, 1,66 mmol)
und von 1,1'-(Azodicarbonyl)dipiperidin
(419 mg, 1,66 mmol) in Toluol (20 mL) wurde bei 80°C 15 h lang
erhitzt. Die Mischung wurde eingeengt, und der Rückstand wurde durch Blitz-Chromatografie (Kieselgel,
20 % Ethylacetat/Hexan) gereinigt, um Ethyl-5-butoxy-1-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl(sulfanyl]phenyl}-4-methyl-1H-pyrazol-3-carboxylat
(300 mg, 0,63 mmol) als weißen
Feststoff zu erhalten. 1H-NMR (CDCl3), 400 MHz) δ: 0,80 (t, 3H), 1,30–1,45 (m,
20H), 1,56–1,65
(m, 2H), 2,19 (s, 3H), 3,84 (t, 2H), 4,35 (q, 2H), 7,50 (d, 2H),
7,63 (d, 2H);
LC-MS = 477,3 (M + H+)
-
5-Butoxy-1-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-4-methyl-1H-pyrazol-3-carbonsäure
-
Stufe
4: Zu einer Lösung
von Ethyl-5-butoxy-1-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-4-methyl-1H-pyrazol-3-carboxylat
in Tetrahydrofuran (5 mL) und in Methanol (3 mL) wurden 1,89 mL
einer 1 M wässrigen
Lösung
von Natriumhydroxid gegeben. Die entstandene Mischung wurde bei
25°C 15
h lang gerührt.
Die Mischung wurde unter verringertem Druck teilweise eingeengt,
und der pH-Wert des wässrigen Rückstands
wurde mit 2 N Salzsäure
auf 7 eingestellt. Das Ganze wurde dann mit Ethylacetat (3 × 10 mL) extrahiert.
Die vereinigten organischen Schichten wurden getrocknet und eingeengt,
um 5-Butoxy-1-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl]sulfanyl]phenyl}-4-methyl-1H-pyrazol-3-carbonsäure (288
mg, 0,64 mmol) als weißen
Feststoff zu ergeben. Dieser wurde in der nächsten Stufe ohne weitere Reinigung
eingesetzt.
-
2-{[4-(5-Butoxy-3-{[(2,4-dimethylphenyl)amino]carbonyl}-4-methyl-1H-pyrazol-1-yl)phenyl]sulfanyl}-2-methylpropansäure
-
Stufe
5: Zu einer Lösung
von 5-Butoxy-1-{4-[(2-t-butoxy-1,1-dimethyl-2-oxoethyl)sulfanyl]phenyl}-4-methyl-1H-pyrazol-3-carbonsäure (290
mg, 0,65 mmol) in Dichlormethan (4 mL) wurde 2 M Oxalylchlorid (972 μL, 1,95 mmol)
unter Argon getropft. Dimethylformamid (1 Tropfen) wurde zugegeben,
und die Reaktionsmischung wurde bei rt 30 min lang gerührt. Die
Mischung wurde unter verringertem Druck eingeengt, und der Rückstand
wurde in Dichlormethan (5 mL) gelöst. 2,4-Dimethylphenylamin
(121 mL, 0,97 mmol) und Triethylamin (135 mL, 0,97 mmol) wurden
zugegeben, und die Reaktionsmischung wurde 15 h lang bei rt gerührt. Die
Reaktionsmischung wurde eingeengt, und der Rückstand würde durch Blitz-Chromatografie
(Kieselgel, 15 % Ethylacetat/Hexan) gereinigt, um t-Butyl-2-{[4-(5-butoxy-3-{[(2,4-dimethylphenyl)amino]carbonyl}-4-methyl-1H-pyrazol-1-yl)phenyl]sulfanyl}-2-methylpropanoat
(300 mg, 0,54 mmol) als gelbes Öl
zu ergeben. Dieses Öl
wurde dann in 50:50-Trifluoressigäsure/Dichlormethan (20 mL)
gelöst,
worauf diese Mischung bei rt 3 h lang gerührt wurde. Die Reaktionsmischung
wurde eingeengt, und der Rückstand
wurde durch Blitz-Chromatografie
(Kieselgel, 20 % Ethylacetat/Hexan) gereinigt, um 2-{(4-(5-Butoxy-3-{[(2,4-dimethylphenyl)amino]carbonyl}-4-methyl-1H-pyrazol-1-yl)phenyl]sulfanyl}-2-methylpropansäure (260,6
mg, 0,53 mmol) als weißen
Feststoff zu ergeben. 1H-NMR (CDCl3, 400 MHz) δ: 0,84 (t, 3H), 1,30–1,38 (m,
2H), 1,50 (s, 6H), 1,56–1,64 (m,
2H), 2,25 (s, 3H), 2,27 (s, 3H), 2,28 (s, 3H), 3,94 (t, 2H), 7,00
(d, 1H), 7,01 (s, 1H), 7,53 (d, 1H), 7,58 (d, 2H), 7,70 (d, 2H),
8,75 (s, 1H); LC-MS = 496,22 (M + H+)
-
Verbindungen
der Erfindung der Formeln (Ia) bis (Ie) sind ferner in den Tabellen
1 bis 11 dargestellt, worin Z, R1-1, R1-2, R1-3, R3, R4, R5,
R5-1, R6-1, R6-2, R7-3, R10, R11-1, R12, R16-1, R17, R23-1-1, R27 und R28 wie für die obigen
Formeln (Ia) bis (Ie) definiert sind. Die Nomenklatur der in den
Tabellen 1 bis 11 dargestellten und angegebenen Verbindungen ist
in Tabelle 12 beschrieben.
-
Tabellen
-
Die
in den Tabellen 1 bis 11 dargestellten Verbindungen der Formeln
(Ia) bis (Ie) wurden mit Synthesewegen ähnlich den in den Beispielen
(Abschnitte A, B, C und D) beschriebenen und unter Einsatz der entsprechenden
rasch verfügbaren
Ausgangsmaterialien oder der hierin beschriebenen Zwischenproduktverbindungen
hergestellt:
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
Die
vorliegende Erfindung betrifft auch die Verwendung der Verbindungen
der vorliegenden Erfindung zur Behandlung von Bulimie und von Fettleibigkeit,
einschließlich
damit zusammenhängender
Dyslipidämie und
weiterer auf Fettleibigkeit und Übergewicht
bezogener Komplikationen wie z.B. Cholesterin-Gallensteine, Krebs
(z.B. Darm, Rektum, Prostata, Brust, Eierstöcke, Gebärmutter (Endometrium), Nacken/Hals
(Cervix), Gallenblase und Gallengang), menstruale Abnormitäten, Unfruchtbarkeit,
polyzystische Eierstöcke,
Osteoarthritis und Schlaf-Atmungsstillstand, sowie weiterer pharmazeutischer
damit zusammenhängender
Anwendungen, wie zur Regulierung des Appetit, der Nahrungseinnahme,
von Dyslipidämie,
Hypertriglyceridämie,
Syndrom X, Typ-II-Diabetes (nicht-Insulin-abhängiger Diabetis), atherosklerotischer
Krankheiten, wie von Herzversagen, von Hyperlipidämie, Hypercholesterinämie, niedrigen
HDL-Spiegeln, Hochdruck, Herzgefäßkrankheiten
(einschließlich
Atherosklerose, Koronar-Herzkrankheiten,
Koronar-Arteriekrankheiten und Überdruck),
von Gehirngefäß- und peripheralen
Gefäßkrankheiten.
Die Verbindungen der vorliegenden Erfindung können auch zur Behandlung physiologischer
Störungen
angewandt werden, die sich z.B. auf die Regulierung einer Insulin-Empfindlichkeit,
von Entzündungsreaktionen,
Plasma-Triglyceriden, HDL, LDL und von Cholesterin-Spiegeln und
dgl. beziehen.
-
Die
Verbindungen der Formeln (Ia) bis (Ie) der vorliegenden Erfindung
sollten auch wertvoll als therapeutische Mittel sein. Demzufolge
schließt
eine Ausgestaltung der vorliegenden Erfindung ein Verfahren zur Behandlung
der verschiedenen oben identifizierten Bedingungen und Krankheiten
bei einem Patient (einschließlich
Säugern)
ein, wobei man dem genannten Patient eine Zusammensetzung verabreicht,
die eine Menge der Verbindung der Formeln. (Ia) bis (Ie) enthält, welche
zur Behandlung des Zielzustands wirkungsvoll ist.
-
Die
Verbindungen der Formeln (Ia) bis (Ie) können allein oder in Kombination
mit einem oder mehreren zusätzlichen
therapeutischen Mitteln verabreicht werden. Eine Kombinationstherapie
schließt
die Verabreichung einer einzelnen pharmazeutischen Dosierungsformulierung,
die eine Verbindung der Formeln (Ia) bis (Ie) und ein oder mehrere
zusätzliche
therapeutische Mittel enthält,
sowie die Verabreichung der Verbindungen der Formeln (Ia) bis (Ie)
und jeweils zusätzlicher
therapeutischer Mittel in deren eigenen getrennten pharmazeutischen
Dosierungsformulierungen ein. Beispielsweise können eine Verbindung der Formeln
(Ia) bis (Ie) und ein therapeutisches Mittel einem Patient zusammen
in einer einzelnen oralen Dosierungszusammensetzung wie einer Tablette
oder Kapsel oder jedes Mittel in getrennten oralen Dosierungsformulierungen
verabreicht werden.
-
Bei
Anwendung getrennter Dosierungsformulierungen können die Verbindungen der Formeln
(Ia) bis (Ie) und ein oder mehrere zusätzliche therapeutische Mittel
im Wesentlichen gleichzeitig (z.B. mit einander) oder zu getrennten
Zeiten (z.B. nach einander) verabreicht werden.
-
Beispielsweise
können
die Verbindungen der Formeln (Ia) bis (Ie) in Kombination mit weiteren
Therapien und Arzneien zur Behandlung von Fettleibigkeit, z.B. in
Kombination mit β3-Adrenorezeptoragonisten, wie mit CL-316.243,
oder in Kombination mit einer Arzneiverbindung, die die Verdauung
oder den Metabolismus moduliert, wie mit Arzneien angewandt werden,
die die Thermogenese, Lipolyse, Darmbeweglichkeit, Fettabsorption
und das Sättigungsgefühl modulieren.
-
Außerdem können die
Verbindungen der Formeln (Ia) bis (Ie) in Kombination mit einem
oder mehreren der folgenden hypoglycämischen Mitteln zur Behandlung
von Diabetes oder mit Diabetes zusammenhängenden Störungen verabreicht werden:
mit Insulin; mit Biguanidinen wie Metformin oder Buformin; mit Sulfonylharnstoffen
wie Acetohexamid, Chlorpropamid, Tolazamid, Tolbutamid, Glyburid,
Glipizid, Glyclazid; oder mit weiteren Insulin-Sekretagogen wie
z.B. mit Repaglinid und Nateglinid; oder mit α-Glycosidase-Inhibitoren wie mit Acarbose,
Voglibose oder Miglitol. Auch können
die Verbindungen der Formeln (Ia) bis (Ie) in Kombination mit HMG-Co-A-Reduktase-Inhibitoren
(Statinen), Gallensäure-bindendem
Harz oder mit Fibrinsäure-Derivaten zur
Verbesserung des Lipidprofils von Personen, die an Dyslipidämie leiden,
angewandt werden. Verbindungen der Formeln (Ia) bis (Ie) können auch
in Kombination mit Mitteln, die den Hoch- bzw. Überdruck regulieren, z.B. mit
Inhibitoren des Gefäßdruck-Umwandlungsenzyms
(angiotension converting enzyme = ACE), β-Blockern und mit Calciumkanal-Blockern, angewandt
werden.
-
Ferner
wurde herausgefunden, dass Verbindungen der vorliegenden Erfindung
als Folge einer oralen Dosierung bei Nagern in signifikanten Konzentrationen
im Gehirn vorliegen. Daher können
die Verbindungen der vorliegenden Erfindung von Nutzen zur Behandlung
verschiedener CNS-(Zentralnervensystem)-
oder psychologischer Störungen,
wie zur Behandlung von Substanz- oder Verhaltenssucht, und zur Behandlung
von Störungen
sein, die mit der Anwendung psychotroper Substanzen zusammenhängen. Desgleichen
können
die Verbindungen der vorliegenden Erfindung von Nutzen zur Handhabung
und Behandlung von Wahrnehmungs- und Gedächtnisstörungen sein.
-
Die
Verbindungen der Formeln (Ia) bis (Ie) können auch in freier Basenform
oder in Zusammensetzungen sowie in Forschung und Diagnostik oder
als analytische Bezugsstandards und dgl. genutzt und angewandt werden,
welche im Stand der Technik gut bekannt sind. Daher schließt die vorliegende
Erfindung auch Zusammensetzungen ein, die aus einem inerten Träger und
einer wirkungsvollen Menge einer Verbindung der Formeln (Ia) bis
(Ie) oder eines Salzes oder Esters davon zusammengesetzt sind. Ein
inerter Träger
ist ein Material, das mit der Verbindung, die getragen wird, nicht
in Wechselwirkung tritt und auf die enthaltene Verbindung Stütz-, Transport-,
Masse- und Aufspürmaterialeigenschaften
und dgl. überträgt. Eine
wirkungsvolle Menge der Verbindung ist diejenige Menge, die ein
Ergebnis erzeugt oder einen Einfluss auf die besondere Verfahrensweise
ausübt,
die durchgeführt
wird.
-
Es
wird davon ausgegangen, dass sich Vorarznei-Formen der Verbindungen
der vorliegenden Erfindung als nützlich
unter bestimmten Umständen
erweisen, und derartige Verbindungen sollen ebenfalls unter den
Umfang und Rahmen der Erfindung fallen. Vorarznei-Formen können Vorteile
gegenüber
den hier als Beispiele angegebenen Stammverbindungen insofern aufweisen,
als sie besser absorbiert, besser verteilt, rascher in das Zentralnervensystem
eingebracht, langsamer metabolisiert oder geklärt usw. werden. Vorarznei-Formen
können
auch Formulierungsvorteile bezüglich
Kristallinität
und Wasserlöslichkeit
aufweisen. Beispielsweise können
Verbindungen der Erfindung mit einer oder mehreren Hydroxylgruppen
in Ester oder Carbonate mit einer oder mehreren Carboxyl-, Hydroxyl-
oder Aminogruppen überführt werden,
die bei physiologischen pH-Werten hydrolysiert oder durch endogene
Esterasen oder Lipasen in vivo gespalten werden. Siehe z.B.
US 4,942,184, 4,960,790,
5,817,840 und 5,824,701 (welche alle in ihrer Gesamtheit durch Bezugnahme hierin
aufgenommen werden) sowie die darin genannten Bezugszitate.
-
Aufgabe
der vorliegenden Erfindung ist es auch, Medikamente zur Induzierung
eines Gewichtsverlusts in einem Individuum durch Verabreichung einer
Verbindung der Erfindung bereitzustellen. Die Erfindung umfasst
auch Medikamente zur Verhinderung einer Gewichtszunahme in einem
Individuum durch Verabreichung einer Menge von mindestens einer
Verbindung der Erfindung oder einer Vorarznei davon, welche hinreicht,
die Gewichtszunahme zu verhindern und einer solchen vorzubeugen.
-
Bewertung
der biologischen Aktivität
-
Die
Darlegung der biologischen Aktivitäten der Verbindungen der vorliegenden
Erfindung kann durch in vitro-, ex vivo- und in vivo-Assayverfahren erfolgen,
welche im Stand der Technik gut bekannt sind. Beispielsweise können zur
Darlegung des Wirkvermögens
eines pharmazeutischen Mittels zur Behandlung von Fettleibigkeit
und mit Fettleibigkeit zusammenhängenden
Störungen
wie von Diabetes, Syndrom X oder einer atherosklerotischen Krankheit
und damit zusammenhängender
Störungen
wie von Hypertriglyceridämie
und Hypercholesterinämie
die folgenden Assayverfahren angewandt werden.
-
Bewertung des Wirkvermögens einer
Verbindung zur Verringerung des Körpergewichts in Diät-induzierten
fettleibigen Mäusen
-
Der
Zweck dieses Protokolls ist es, den Effekt der chronischen Verabreichung
einer Verbindung auf das Körpergewicht
von Mäusen
zu ermitteln und zu bestimmen, die fettleibig gemacht wurden, indem
sie einer 45 % kcal/g hohen Fett-Diät während mehr als 10 Wochen ausgesetzt
wurden. Das Körpergewicht
der für
die Studien ausgewählten
Mäuse ist
höher als
3 Standardabweichungen vom Gewicht einer Vergleichsgruppe von Mäusen, die
mit einem Standard-Mäusefutter
mit niedrigem Fettgehalt (5 bis 6 % Fett) gefüttert wurden. Diät-induzierte
fettleibige (diet-induced obese = DIO)-Tiere werden häufig zur Bestimmung des Wirkvermögens einer
Verbindung zur Verringerung des Körpergewichts herangezogen.
Dieses Tiermodell ist erfolgreich zur Identifizierung und Charakterisierung
des Wirksamkeitsprofils von Verbindungen angewandt worden, die zum Management
des Körpergewichts
in fettleibigen Menschen herangezogen werden oder worden sind (siehe Brown
et al., British J. Pharmacol. 132:1898–1904, 2001; Guerre-Millo et
al., J. Biol. Chem. 275:16638–16642, 2000;
Han et al., Intl. J. Obesity Rel. Metab, Dis. 23:174–179, 1999;
Surwit et al., Endocrinol. 141:3630–3637, 2000).
-
Eine
typische Studie schließt
60 bis 80 männliche
C57b1/J6-Mäuse
(n = 10/Behandlungsgruppe) mit einem Durchschnittskörpergewicht
von ca. 45 g ein. Die Mäuse
werden in Standard-Tierräumen
unter gesteuerter Temperatur und Feuchte und einem 12 h/12 h-Hell-Dunkel-Zyklus
gehalten. Wasser und Futter sind kontinuierlich verfügbar. Die
Mäuse werden
einzeln in Schuhschachteln untergebracht. Die Tiere werden mit Studien-Vehikel
mindestens 4 Tage vor Erstellung der 2-Tage-Basislinie-Messungen
des Körpergewichts
und einem Futter- und Wasserverbrauch von 24 h Scham dosiert. Die
Mäuse werden
einer von 6 bis 8 Behandlungsgruppen zugeteilt, bezogen auf deren
Körpergewicht
auf Basislinie. Die Gruppen werden so festgelegt, dass der Mittelwert
und der Standardfehler des Mittelwerts des Körpergewichts ähnlich sind.
-
Den
Tieren wird oral (5 mL/kg) täglich
vor der Dunkelphase des Hell-Dunkel-Zyrklus über eine
vorbestimmte Anzahl von Tagen (in typischer Weise 8 bis 14 Tage
lang) ihre zugeteilte Dosis/Verbindung gegeben. Das Körpergewicht
und der Futter- und Wasserverbrauch werden gemessen. Die Daten werden
mit entsprechend geeigneten Statistiken gemäß dem Untersuchungsplan analysiert.
Am letzten Tag werden die Tiere durch CO2-Inhalation euthanisiert.
-
Bewertung des Wirkvermögens der
Verbindungen auf die Verringerung der Nahrungsaufnahme in mageren, über Nacht
am Fasten gehaltenen Ratten Fütterungs-Assay
unter Fasten und erneuter Fütterung
-
Der
Zweck dieses Protokolls ist es, den Effekt einer Einzeldosis einer
unbekannten Verbindung auf den Nahrungsverbrauch von magernen, über Nacht
am Fasten gehaltenen Ratten zu ermitteln. Das Modell mit am Fasten
gehaltenen und erneut gefütterten
Ratten wird häufig
auf dem Gebiet der Fettleibigkeit angewandt, um Verbindungen mit
einem Potenzial für
anorektische Effekte zu identifizieren. Dieses Tiermodell ist erfolgreich
zur Identifikation und Charakterisierung des Wirksamkeitsprofils
von Verbindungen angewandt worden, die im Management des Körpergewichts
bei fettleibigen Menschen angewandt werden oder angewandt worden
sind (siehe z.B. Balvet et al., Gen. Pharmacol. 13:293–297, 1982;
Grignaschi et al., Br. J. Pharmacol. 127:1190–1194, 1999; McTavish and Heel,
Drug 43:713–733,
1992; Rowland et al., Life Sci. 36:2295–2300, 1985).
-
Eine
typische Studie schloss 60 bis 80 männliche Ratten (n = 10/Behandlungsgruppe)
mit einem Durchschnittskörpergewicht
von ca. 280 g ein. Die Ratten wurden in Standard-Tierräumen unter
geregelter Temperatur und Feuchte und einem 12/12-Hell/Dunkel-Zyklus
gehalten. Die Ratten wurden einzeln in aufgehängten Käfigen mit einem Mesh-Boden
untergebracht. Wasser und Nahrung sind kontinuierlich verfügbar, es sei
denn, die Tiere werden für
die Studie am Fasten gehalten.
-
Der
Vehikel-Test: Die Ratten werden auf Basis ihres Leistungsverhaltens
in einem Vehikel-Test in Gruppen eingeteilt. Der Vehikel-Test wird
2 bis 7 Tage lang vor dem Wirksamkeitstest durchgeführt. Die
Ratten werden über
Nacht während
der Dunkelphase (insgesamt 16 bis 18 h lang) am Fasten gehalten.
Das Tier wird mit 0,5 mL entionisiertem Wasser dosiert. 1 h nach
der Dosierung werden vorab gewogene Futternäpfe in den Heimkäfig des
Tieres zurückgestellt.
Den Ratten wird 1 h Fütterungszeit
gewährt.
Nach 1 h wird die verschüttete
Menge in den Futternapf zurückgegeben,
und es wird die Menge an verbrauchter Nahrung bestimmt. Die Ratten
werden so in Gruppen eingeteilt, dass der Mittelwert und der Standardfehler
des Mittelwertes des 1 h-Nahrungsverbrauchs ähnlich unter den Gruppen sind.
-
Der
Wirksamkeitstest: Die Ratten werden über Nacht während der Dunkelphase (insgesamt
16 bis 18 h lang) am Fasten gehalten. Das Tier wird jeweils mit
einer zugeordneten Behandlung (2 mg/mL) dosiert. 1 h nach Dosierung
werden vorab gewogene Futternäpfe
in den Käfig
zurückgestellt.
Die Nahrungsaufnahme wird 30, 60, 90, 180 und 240 min nach der Rückkehr zur
Nahrungsaufnahme ermittelt und festgehalten. Zu jedem Zeitpunkt
wird die verschüttete
Menge in den Futternapf zurückgegeben,
worauf die Futternäpfe
gewogen werden. Die Menge an verbrauchter Nahrung wird für jeden-Zeitpunkt
bestimmt. Die Differenz unter den Behandlungsgruppen wird mit entsprechender
statistischer Analyse ermittelt.
-
Bewertung der Wirksamkeit
der Verbindungen auf eine Verringerung des Körpergewichts und des Nahrungs- und
Wasserverbrauchs in fettleibigen Zucker-fa/fa-Ratten
-
Chronischer Fütterungsassay
-
Der
Zweck dieses Protokolls ist es, den Effekt der chronischen Verabreichung
einer unbekannten Verbindung auf das Körpergewicht und den Nahrungs-
und Wasserverbrauch in fettleibigen Zucker-fa/fa-Ratten zu ermitteln.
Fettleibige Zucker-fa/fa-Ratten werden häufig zur Bestimmung der Wirksamkeit
einer Verbindung zur Herabsetzung des Körpergewichts herangezogen.
Dieses Tiermodell ist erfolgreich zur Identifizierung und Charakterisierung
des Wirksamkeitsprofils der Verbindungen angewandt worden, die im
Management des Körpergewichts
bei fettleibigen Menschen angewandt werden oder angewandt worden
sind (siehe z.B. Al-Barazanji et al., Obes Res. 8:317–323, 2000;
Assimacopoulos-Jeannet et al., Am. J. Physiol. 260 (2 Pt 2):R278–283; 1991;
Dryden et al., Horm. Metab. Res. 31:363–366, 1999; Edwards und Stevens,
Pharmacol. Biochem. Behav. 47:865–872, 1994; Grinker et al.,
Pharmacol. Biochem. Behav. 12:265–275, 1980).
-
Eine
typische Studie schließt
60 bis 80 männliche
Zucker-fa/fa-Ratten (n = 10/Behandlungsgruppe) mit einem Durchschnittskörpergewicht
von ca. 550 g ein. Die Ratten werden in Standard-Tierräumen unter
geregelter Temperatur und Feuchte und einem 12/12-Hell-Dunkel-Zyklus
gehalten. Wasser und Nahrung sind kontinuierlich verfügbar. Die
Ratten werden einzeln in großen
Ratten-Schuhschachteln mit Bodenrost untergebracht. Die Tiere werden
an die Bodenroste gewöhnt
und mit Studie-Vehikel mindestens 4 Tage lang vor Aufnahme der 2-Tage-Basislinie-Messung
des Körpergewichts
und des 24-h-Nahrungs-
und Wasserverbrauchs Scham-dosiert. Die Ratten werden in eine von
6 bis 8 Behandlungsgruppen auf Basis ihres auf die Basislinie bezogenen
Körpergewichts
eingeteilt. Die Gruppen werden so festgelegt, dass der Mittelwert
und der Standardfehler des Mittelwerts des Körpergewichts ähnlich sind.
-
Den
Tieren wird oral (2 mL/kg) täglich
vor der Dunkelphase des HD-Zyklus über eine vorbestimmte Anzahl
von Tagen (typisch 6 bis 14 Tage lang) die ihnen zugeteilte Dosis/Verbindung
gegeben. Zu diesem Zeitpunkt werden das Körpergewicht sowie der Nahrungs-
und Wasserverbrauch gemessen. Am letzten Tag werden die Tiere durch
CO2-Inhalation euthanisiert, und das Körpergewicht
wird gemessen.
-
Biologische Assayverfahren
für mit
Fettleibigkeit zusammenhängenden
Störungen
-
Verfahren zur Messung
des Blutglucosegehalts
-
db/db-Mäusen (erhalten
von Jackson Lboratories, Bar Harbor, ME) wird Blut (entweder aus
dem Auge oder aus der Schwanzvene) entnommen, und sie werden gemäß den äuqivalenten
mittleren Blutglucosegehaltsmengen in Gruppen eingeteilt. Sie werden
oral (durch Gabe in einem pharmazeutisch zulässigen Vehikel) mit der Testverbindung
1 Mal täglich
14 Tage lang dosiert. Zu diesem Zeitpunkt wird den Tieren erneut
Blut aus dem Auge oder aus der Schwanzvene entnommen, und die Blutglucosegehaltsmengen
werden bestimmt. In jedem Fall werden die Glucosegehaltsmengen mit
einem Glucometer Elite XL (Bayer Corporation, Elkhart, IN) gemessen.
-
Verfahren zur Messung
des Trigyceridgehalts
-
hApoAl-Mäusen (erhalten
von Jackson Laboratories, Bar, Harbor, ME) wird Blut (entweder aus
dem Auge oder aus der Schwanzvene) entnommen, und sie werden gemäß gleichwertigen
mittleren Serum-Triglyceridgehaltsmengen in Gruppen eingeteilt.
Sie werden oral (durch Gabe in einem pharmazeutisch zulässigen Vehikel)
mit der Testverbindung 1 Mal täglich
8 Tage lang dosiert. Den Tieren wird dann erneut aus dem Auge oder
der Schwanzvene Blut entnommen, und die Serum-Triglyceridgehaltsmengen
werden bestimmt. In jedem Fall werden die Triglyceridgehaltsmengen
mit einem Technicon Axon Autoanalysegerät (Bayer Corporation, Tarrytown,
NY) gemessen.
-
Verfahren
zur Messung des HDL-Cholesteringehalts Zur Bestimmung des Plasma-HDL-Cholesteringehalts
wird hApoAl-Mäusen
Blut entnommen, und sie werden gemäß gleichwertigen mittleren
Plasma-HDL-Cholesteringehaltsmengen
in Gruppen eingeteilt. Die Mäuse
werden oral 1 Mal täglich
mit Vehikel oder Testverbindung 7 Tage lang dosiert, worauf ihnen
erneut Blut am 8. Tag entnommen wird. Das Plasma wird bezüglich HDL-Cholesterin mit dem
Synchron Clinical System (CX4) (Beckman Coulter, Fullerton, CA) analysiert.
-
Verfahren zur Messung
der Gehaltsmengen von Gesamt-Cholesterin, HDL-Cholesterin, Triglyceriden und von Glucose
-
In
einem weiteren in vivo-Assay wird fettleibigen Affen Blut entnommen,
dann werden sie oral 1 Mal täglich
mit Vehikel oder Testverbindung 4 Wochen lang dosiert, worauf ihnen
erneut Blut entnommen wird. Das Serum wird bezüglich Gesamt-Cholesterin, HDL-Cholesterin,
Triglyceriden und Glucose mit dem Synchron Clinical System (CX4)
(Beckman Coulter, Fullerton, CA) analysiert. Die Lipoprotein-Unterklasse
wird mit NMR-Spektroskopie analysiert, wie beschrieben von Oliver
et al. (Proc. Natl. Acad. Sci. USA 98:5306–5311, 2001).
-
Verfahren zur Messung
des Effekts auf Herzgefäß-Parameter
-
Herzgefäß-Parameter
(z.B. Herztätigkeit
und Blutdruck) werden ebenfalls bewertet. SHR-Ratten werden oral
1 Mal täglich
mit Vehikel oder Testverbindung 2 Wochen lang dosiert. Der Blutdruck
und die Herztätigkeit
werden mit einem Schwanz-Stegreif-Verfahren bestimmt, wie beschrieben
von Grinsell et al. (Am. J. Hypertens. 13:370–375, 2000). Bei Affen werden
der Blutdruck und die Herztätigkeit
aufgenommen und messend verfolgt, wie beschrieben von Shen et al.
(J. Pharmacol. Exp. Therap. 278:1435–1443, 1996).
-
Pharmazeutische Zusammensetzungen
-
Auf
der Grundlage der obigen Tests oder weiterer gut bekannter Assayverfahren
zur Bestimmung der Wirksamkeit für
eine Behandlung identifizierter Bedingungen in Säugern und durch Vergleich dieser
Ergebnisse mit den Ergebnissen bekannter Medikamente, die zur Behandlung
dieser Bedingungen bereits angewandt werden, kann die effektive
Dosierung der Verbindungen der vorliegenden Erfindung unmittelbar
und ohne Weiteres für
die Behandlung einer jeden gewünschten
Indikation ermittelt und bestimmt werden. Die Menge des Wirkbestandteils,
die zur Behandlung einer dieser Bedingungen verabreicht wird, kann
in breitem Umfang gemäß solcher
Gesichtspunkte wie der besonderen Verbindung und angewandten Dosierungseinheit,
dem Verabreichungsmodus, der Behandlungsdauer, dem Alter und Geschlecht
des behandelten Patienten sowie der Natur und dem Ausmaß der behandelten
Bedingung schwanken.
-
Die
zu verabreichende Gesamtmenge des Wirkbestandteils kann ganz allgemein
ca. 0,001 bis 200 und vorzugsweise ca. 0,01 bis ca. 200 mg/kg Körpergewicht
pro Tag ausmachen. Eine Einheitsdosierung kann ca. 0,05 bis ca.
1.500 mg Wirkbestandteil enthalten und 1 oder mehrere Male pro Tag
verabreicht werden. Die tägliche
Dosierung zur Verabreichung durch Injektion, einschließlich intravenöser, intramuskulärer, subkutaner und
parenteraler Injektionen, und zur Anwendung von Infusionstechniken
kann ca. 0,01 bis ca. 200 mg/kg betragen. Das tägliche rektale Dosierungsregimen
kann 0,01 bis 200 mg/kg Gesamtkörpergewicht
betragen. Die transdermale Konzentration kann eine sein, die benötigt wird,
um eine tägliche
Dosierung von 0,01 bis 200 mg/kg aufrecht zu halten.
-
Natürlich schwankt
das spezifische anfängliche
und kontinuierliche Dosierungsregimen für jeden Patient abhängig von
der Natur und Strenge der Bedingung gemäß Bestimmung durch den behandelnden
Diagnostiker, gemäß der Aktivität der spezifischen
angewandten Verbindung, dem Alter des Patienten, der Ernährung des
Patienten, der Verabreichungszeit, dem Verabreichungsweg, der Ausscheidungsrate
der Arznei, der Arzneikombinationen und dgl.. Der gewünschte Behandlungsmodus
und die Anzahl der Dosismengen einer Verbindung der vorliegenden
Erfindung oder eines pharmazeutisch zulässigen Salzes davon können mit
Bestimmtheit von den einschlägigen
Fachleuten unter Anwendung herkömmlicher
Behandlungstests festgelegt werden.
-
Die
Verbindungen der vorliegenden Erfindung können angewandt und genutzt
werden, um den gewünschten
pharmakologischen Effekt durch Verabreichung an einen Patient, der
dessen bedarf, in einer in geeigneter Weise zubereiteten pharmazeutischen
Zusammensetzung zu erzielen. Ein Patient für den Anwendungszweck der vorliegenden
Erfindung ist ein Säuger,
einschließlich
des Menschen, bei denen eine besondere Bedingung oder Krankheit
behandelt werden müssen.
Daher schließt
die vorliegende Erfindung pharmazeutische Zusammensetzungen ein,
die aus einem pharmazeutisch zulässigen
Träger
und einer pharmazeutisch wirksamen Menge einer mit den oben beschriebenen
Verfahren identifizierten Verbindung oder eines pharmazeutisch zulässigen Salzes
oder Esters davon zusammengesetzt sind. Ein pharmazeutisch zulässiger bzw.
geeigneter Träger
ist jeder Träger,
der für
einen Patienten bei Konzentrationen in Übereinstimmung mit der wirkungsvollen
Aktivität
des Wirkbestandteils relativ nicht-toxisch und unschädlich ist,
so dass jegliche Nebeneffekte, die dem Träger zugeschrieben werden könnten, die
vorteilhaften Effekte des Wirkbestandteils nicht beeinträchtigen.
Die pharmazeutisch wirkungsvolle Menge einer Verbindung ist diejenige
Menge, die ein Ergebnis erzeugt oder einen Einfluss auf die besondere
Bedingung ausübt,
die behandelt wird. Die mit den hierin beschriebenen Verfahren identifizierten
Verbindungen können
mit einem pharmazeutisch zulässigen
Träger unter
Anwendung jeder wirkungsvollen herkömmlichen Dosierungseinheitsform
verabreicht werden, einschließlich
z.B. unmittelbarer und zeitlich verzögerter Freisetzzubereitungen,
und zwar oral, parenteral, topisch oder dgl..
-
Zur
oralen Verabreichung können
die Verwendung zu festen oder flüssigen
Zubereitungen wie z.B. Kapseln, Pillen, Tabletten, Pastillen, Rauten,
Schmelzen, Pulvern, Lösungen,
Suspensionen oder zu Emulsionen formuliert und gemäß Verfahren
hergestellt werden, die im Stand der Technik zur Herstellung pharmazeutischer
Zusammensetzungen bekannt sind. Die festen Einheitsdosierungsformen
können
eine Kapsel sein, die den Typ einer gewöhnlichen Hart- oder Weichschalengelatine
aufweist, die z.B. oberflächenaktive
Mittel, Gleitmittel und inerte Füllstoffe
wie Lactose, Saccharose, Calciumphosphat und Maisstärke enthält.
-
In
einer weiteren Ausgestaltung können
die Verbindungen der vorliegenden Erfindung mit herkömmlichen
Tablettengrundlagen wie Lactose, Saccharose und Maisstärke in Kombination
mit Bindern wie Akazie, Maisstärke
oder Gelatine, Zerfallmitteln zur Unterstützung des Aufbrechens und Auflösens der
Tablette nach der Verabreichung wie Kartoffelstärke, Aginsäure, Maisstärke und Guar-Gummi, Gleitmitteln
zur Fließverbesserung
der Tablettengranulierung und zur Verhinderung des Anhaftens von
Tablettenmaterial an den Oberflächen
der Tabletten-Matrizen und -Stanzen, z.B. mit Talkum, Stearinsäure oder
mit Magnesium-, Calcium- oder Zinkstearat, mit Farbstoffen, Färbemitteln
und mit Geschmacksmitteln zur Verbesserung der ästhetischen Qualitäten der
Tabletten und zur besseren Verträglichkeit
beim Patienten tablettiert werden. Geeignete Exzipienten zur Anwendung
in oralen flüssigen
Dosierungsformen schließen
Verdünnungsmittel
wie Wasser und Alkohole, z.B. Ethanol, Benzylalkohol und Polyethylenalkohole,
entweder mit oder ohne die Zugabe pharmazeutisch geeigneter oberflächenaktiver
Mittel, Suspendiermittel oder Emulgatoren, ein. Verschiedene weitere
Materialien können
als Überzüge vorliegen
oder auf sonstige Weise die physikalische Form der Dosierungseinheit modifizieren.
Beispielsweise können
Tabletten, Pillen oder Kapseln mit Schellack, Zucker oder mit beiden überzogen
sein.
-
Dispergierbare
Pulver und Körner
eignen sich zur Zubereitung einer wässrigen Suspension. Sie stellen
den Wirkbestandteil in Abmischung mit einem Dispergier- oder Benetzungsmittel,
einem Suspendiermittel und einem oder mehreren Konservierungsstoffen
bereit. Geeignete Dispergier- und/oder Benetzungs- und Suspendiermittel
sind diejenigen, die bereits oben als Beispiele genannt sind. Zusätzliche
Exzipienten, z.B. die bereits oben beschriebenen Süßungs-,
Geschmacks- und Färbemittel,
können
ebenfalls vorhanden sein.
-
Die
pharmazeutischen Zusammensetzungen der vorliegenden Erfindung können auch
in der Form von Öl-in-Wasser-Emulsionen
vorliegen. Die Öl-Phase
kann ein pflanzliches Öl
wie flüssiges
Paraffin oder eine Mischung. pflanzlicher Öle sein. Geeignete Emulgatoren
können
(1) natürlich
vorkommende Gummiprodukte wie Akazie- und Tragacanth-Gummi, (2)
natürlich
vorkommende Phosphatide wie Sojabohne und Lecithin, (3) Ester oder
Partialester aus Fettsäuren
und Hexitanhydriden; z.B. Sorbitanmonooleat, und (4) Kondensationsprodukte
der genannten Teilester mit Ethylenoxid, z.B. Polyoxyethylensorbitanmonooleat,
sein. Die Emulsionen können
auch Süßungs- und Geschmacksmittel
enthalten.
-
Ölige Suspensionen
können
durch Suspendieren des Wirkbestandteils in einem pflanzlichen Öl, wie z.B.
in Arachisöl
(Erdnussöl),
Oliven-, Sesam- oder
Kokosnussöl,
oder in einem Mineralöl
wie flüssigem
Paraffin formuliert werden. Die öligen
Suspensionen können
Verdickungsmittel wie z.B. Bienenwachs, Hartparaffin oder Cetylalkohol
enthalten. Die Suspensionen können
auch einen oder mehrere Konservierungsstoffe, z.B. Ethyl- oder n-Propyl-p-hydroxybenzoat,
ein oder mehrere Färbemittel,
ein oder mehrere Geschmacksmittel und ein oder mehrere Süßungsmittel
wie Saccharose oder Saccharin enthalten.
-
Sirup-Produkte
und Elixiere können
mit Süßungsmitteln,
wie mit Glycerin, Propylenglykol, Sorbit oder mit Saccharose, formuliert
werden. Solche Formulierungen können
auch Linderungsmittel und Konservierungsstoffe, Geschmacks- und
Färbungsmittel
enthalten.
-
Die
Verbindungen der vorliegenden Erfindung können auch parenteral, d.h.
subkutan, intravenös,
intramuskulär
oder intraperitoneal, als injizierbare Dosierungen der Verbindung
in einem physiologisch zulässigen
Verdünnungsmittel
mit einem pharmazeutischen Träger
verabreicht werden, der eine sterile Flüssigkeit oder Mischung aus
Flüssigkeiten
wie Wasser, Kochsalzlösung,
wässriger
Dextrose und verwandter Zuckerlösungen,
ein Alkohol wie etwa Ethanol, Isopropanol oder Hexadecylalkohol,
ein Glykol wie Propylenglykol oder Polyethylenglykol, ein Glycerylketal
wie 2,2-Dimethyl-1,3-dioxolan-4-methanol,
ein Ether wie Poly(ethylen)glykol 400, ein Öl, eine Fettsäure, ein
Fettsäureester
oder -glycerid oder ein acetyliertes Fettsäureglycerid mit oder ohne die
Zugabe pharmazeutisch geeigneter obenflächenaktiver Mittel wie einer
Seife oder eines Detergens, eines Suspendiermittels wie von Pectin,
Carbomeren, Methylcellulose, Hydroxypropylmethylcellulose oder von
Carboxymethylcellulose oder eines Emulgators und weiterer pharmazeutischer
Hilfsstoffe sein kann.
-
Beispielhafte Öle, die
in den parenteralen Formulierungen der vorliegenden Erfindung verwendet
werden können,
sind diejenigen aus Petroleum, Produkte tierischen, pflanzlichen
oder synthetischen Ursprungs, z.B. aus Erdnuss-, Sojabohnen-, Sesam-,
Baumwollsamen-, Mais-, Olivenöl,
Petrolatum und aus Mineralöl. Geeignete
Fettsäuren
schließen Öl-, Stearin- und Isostearinsäure ein.
Geeignete Fettsäureester
sind z.B. Ethyloleat und Isopropylmyristat. Geeignete Seifen schließen Fett-Alkalimetall-,
-Ammonium- und -Triethanolaminsalze und geeignete Detergenzien schließen kationische
Detergenzien, z.B. Dimethyldialkylammoniumhalogenide, Alkylpyridiniumhalogenide
und Alkylaminacetate, anionische Detergenzien, z.B. Alkyl-, Aryl-
und Olefinsulfonate, Alkylolefinether und Monoglyceridsulfate und
-sulfosuccinate, nicht-ionische Detergenzien, z.B. Fettaminoxide,
Fettsäurealkanolamide
und Polyoxyethylen-Polyoxypropylen-Copolymere, und amphotere Detergenzien,
z.B. Alkyl-β-aminopropionate
und 2-Alkylimidazolinammoniumsalze sowie Mischungen davon ein.
-
Die
parenteralen Zusammensetzungen der vorliegenden Erfindung können in
typischer Weise ca. 0,5 bis ca. 25 Gew.-% des Wirkbestandteils in
Lösung
enthalten. Konservierungsstoffe und Puffer können dabei ebenfalls in vorteilhafter
Weise zur Anwendung gelangen. Zur Minimierung oder Eliminierung
einer Reizung an der Einstichstelle können die entsprechenden Zusammensetzungen
ein nicht-ionisches oberflächenaktives Mittel
mit einer Hydrophil-Lipophil-Bilanz (HLB) von ca. 12 bis ca. 17
enthalten. Die Menge des oberflächenaktiven
Mittels in einer solchen Formulierung macht ca. 5 bis ca. 15 Gew.-%
aus. Das obenflächenaktive
Mittel kann eine Einzelkomponente mit dem obigen HLB-Wert oder eine
Mischung aus 2 oder mehr Komponenten mit dem gewünschten HLB-Wert sein.
-
Beispielhafte
oberflächenaktive
Mittel für
parenterale Formulierungen sind die Klasse der Polyethylensorbitanfettsäureester,
z.B. Sorbitanmonooleat, und der hochmolekularen Addukte von Ethylenoxid
mit einer hydrophoben Base aus der Kondensation von Propylenoxid
mit Propylenglykol.
-
Die
pharmazeutischen Zusammensetzungen können auch in der Form steriler
injizierbarer wässriger Suspensionen
vorliegen. Solche Suspensionen können
gemäß bekannten
Verfahren mit geeigneten Dispergier- oder Benetzungs- und Suspendiermitteln,
wie z.B. mit Natriumcarboxymethylcellulose, Methylcellulose, Hydroxypropylmethylcellulose,
Natriumalginat, Polyvinylpyrrolidon, Tragacanth- und Akazie-Gummi,
mit Dispergier- oder Benetzungsmitteln, die ein natürlich vorkommendes
Phosphatid wie Lecithin, ein Kondensationsprodukt eines Alkylenoxids
mit Fettsäure,
z.B. Polyoxyethylenstearat, ein Kondensationsprodukt von Ethylenoxid mit
einem langkettigen aliphatischen Alkohol, z.B. Heptadecaethylenoxycetanol,
ein Kondensationsprodukt von Ethylenoxid mit einem Partialester
aus einer Fettsäure
und einem Hexit wie Polyoxyethylensorbitmonooleat oder ein Kondensationsprodukt
eines Ethylenoxids mit einem Partialester aus einer Fettsäure und
einem Hexitanhydrid, z.B. Polyoxyethylensorbitanmonooleat, sein
können,
formuliert werden.
-
Die
sterile injizierbare Zubereitung kann auch eine sterile injizierbare
Lösung
oder Suspension in einem nicht-toxischen parenteral geeigneten Verdünnungs-
oder Lösungsmittel
sein. Die Verdünnungs-
und Lösungsmittel,
die eingesetzt werden können,
sind z.B. Wasser, Ringer's
Lösung
und isotonische Natriumchlorid-Lösung.
Außerdem
werden sterile fixierte Öle
in herkömmlicher
Weise als Lösungsmittel
oder Suspendiermedien angewandt. Zu diesem Zweck kann jedes milde,
fixierte Öl,
einschließlich
synthetischer Mono- oder Diglyceride, herangezogen werden. Außerdem können Fettsäuren wie Ölsäure zur
Herstellung injizierbarer Formulierungen verwendet werden.
-
Zusammensetzungen
der Erfindung können
auch in der Form von Suppositorien zur rektalen Verabreichung der
Arznei zubereitet werden. Diese Zusammensetzungen können durch
Vermischen der Arznei mit einem geeigneten nicht-reizenden Exzipient
hergestellt werden, der bei gewöhnlichen
Temperaturen fest, aber bei der Rektaltemperatur flüssig ist,
weil er im Rektum geschmolzen wurde, um die Arznei freizusetzen.
Derartige Materialien sind z.B. Kakaobutter und Polyethylenglykol.
-
Als
weitere Formulierung, die in den Verfahren der vorliegenden Erfindung
zur Anwendung gelangt, werden transdermale Abgabevorrichtungen ("Pflaster") angewandt. Solche
transdermalen Pflaster können verwendet werden,
um eine kontinuierliche oder diskontinuierliche Infusion der Verbindungen
der vorliegenden Erfindung in gesteuerten Mengen zu ergeben. Die
Konstruktion und Verwendung transdermaler Pflaster zur Abgabe pharmazeutischer
Mittel ist im Stand der Technik gut bekannt (siehe z.B. 5,023,252,
die durch Bezugnahme hierin aufgenommen wird). Solche Pflaster können zur
kontinuierlichen pulsatilen oder bedarfsweisen Abgabe der pharmazeutischen
Mittel aufgebaut sein.
-
Es
kann wünschbar
oder notwendig sein, die pharmazeutische Zusammensetzung beim Patient über eine
mechanische Abgabevorrichtung einzuführen. Die Konstruktion und
Verwendung mechanischer Abgabevorrichtungen zur Abgabe pharmazeutischer
Mittel ist im Stand der Technik gut bekannt. Beispielsweise beinhalten
direkte Verfahrenstechniken zur Verabreichung einer Arznei direkt
in das Gehirn gewöhnlich
die Anbringung eines Arznei-Abgabekatheters im Ventrikularsystem
des Patienten, um die Blut-Hirn-Schranke zu umgehen. Ein derartiges
implantierbares Abgabesystem, das zum Transport von Mitteln zu spezifischen
anatomischen Körperregionen
angewandt wird, ist in
US 5,011,472 beschrieben,
die durch Bezugnahme hierin aufgenommen wird.
-
Die
Zusammensetzungen der Erfindung können auch weitere herkömmliche
pharmazeutisch zulässige
Kompoundierbestandteile, die allgemein als Träger oder Verdünnungsmittel
bezeichnet werden, gemäß Notwendigkeit
oder Wunsch enthalten. Jede Zusammensetzung der vorliegenden Erfindung
kann durch Zugabe eines Antioxidans wie von Ascorbinsäure oder
durch weitere geeignete Konservierungsstoffe konserviert werden.
Herkömmliche
Verfahrensweisen zur Herstellung solcher Zusammensetzungen in geeigneten
Dosierungsformen können
angewandt werden.
-
Gewöhnlich verwendete
pharmazeutische Bestandteile, die in entsprechend geeigneter Weise
verwendet werden können,
um die Zusammensetzung im Hinblick auf ihren Verabreichungsweg zu
formulieren und zuzubereiten, schließen ein: Ansäuerungsmittel,
z.B., ohne darauf eingeschränkt
zu sein, Essig-, Zitronen-, Fumar-, Salz- und Salpetersäure, sowie
alkalisch stellende Mittel, wie, ohne darauf eingeschränkt zu sein,
Ammoniak-Lösung,
Ammoniumcarbonat, Diethanolamin, Monoethanolamin, Kaliumhydroxid;
Natriumborat, Natriumcarbonat, Natriumhydroxid, Triethanolamin,
Trolamin.
-
Weitere
pharmazeutische Bestandteile schließen z.B., ohne darauf eingeschränkt zu sein,
Adsorbenzien (z.B. gepulverte Cellulose und Aktivkohle), Aerosol-Treibmittel
(z.B. Kohlendioxid, CCl2F2,
F2ClC-CClF2 und
CClF3), Luft-Verdrängungsmittel (z.B. Stickstoff
und Argon), antifungale Konservierungsstoffe (z.B. Benzoesäure, Butylparaben,
Ethylparaben, Methylparaben, Propylparaben, Natriumbenzoat), antimikrobielle
Konservierungsstoffe (z.B. Benzalkoniumchlorid, Benzethoniumchlorid,
Benzylalkohol, Cetylpyridiniumchlorid, Chlorbutanol, Phenol, Phenylethylalkohol,
Phenylquecksilbernitrat und Thimerosal), Antioxidanzien (z.B. Ascorbinsäure, Ascorbylpalmitat,
butyliertes Hydroxyanisol, butyliertes Hydroxytoluol, Hypophosphorsäure, Monothioglycerin,
Propylgallat, Natriumascorbat, Natriumbisulfit, Natrium-Formaldehydsulfoxylat,
Natriummetabisulfit), Bindermaterialien (d.h. Blockpolymere, natürlichen
und synthetischen Gummi, Polyacrylate, Polyurethane, Silicone und
Styrol-Butadien-Copolymere), Puffermittel (z.B. Kaliummetaphosphat,
monombasisches Kaliumphosphat, Natriumacetat, wasserfreies Natriumzitrat
und Natriumzitrat-Dihydrat), Trägermittel
(z.B. Akazie-Sirup, aromatischen Sirup, aromatisches Elixier, Kirsch-,
Kakao-, Orangen-Sirup, Sirup, Mais-, Mineral-, Erdnuss-, Sesamöl, bakteriostatische
Natriumchlorid-Injektion und bakteriostatisches Wasser zur Injektion), Chelat-Bildner
(z.B. Edetat-Dinatrium und Editinsäure), Färbemittel (z.B. FD&C Rot Nr. 3, RD&C Rot Nr. 20, FD&C Gelb Nr. 6,
FD&C Blau Nr.
2, D&C Grün Nr. 5,
D&C Orange Nr.
5, D&C Rot Nr.
8, Karamel und Eisenoxid-Rot), Klärungsmittel (z.B. Bentonit),
Emulgatoren (ohne aber darauf eingeschränkt zu sein, wie Akazie, Ketomakrogol,
Cetylalkohol, Glycerylmonostearat; Lecithin, Sorbitanmonoleat, Polyethylen-50-stearat),
Verkapselungsmittel (z.B. Gelatine und Celluloseacetatphthalat),
Geschmacksmittel (z.B. Anisöl,
Zimtöl,
Kakao, Menthol, Orangenöl,
Pfefferminzöl
und Vanillin), Feuchtigkeitsmittel (z.B. Glycerin, Propylenglykol
und Sorbit), Glättungsmittel
(z.B. Mineralöl
und Glycerin), Öle
(z.B. Arachis-, Mineralöl,
Oliven-, Erdnuss-, Sesam- und Pflanzenöl), Salbengrundlagen (z.B.
Lanolin, hydrophile Salbe, Polyethylenglykol-Salbe, Petrolatum,
hydrophiles Petrolatum, weiße
Salbe, gelbe Salbe und Rosenwasser-Salbe), Eindringsteigerungsmittel
(transdermale Abgabe) (z.B. Monohydroxy- oder Polyhydroxyalkohole,
gesättigte
oder ungesättigte
Fettalkohole, gesättigte
oder ungesättigte
Fettester, gesättigte
oder ungesättigte
Dicarbonsäuren,
essentielle Öle,
Phosphatidylderivate, Cephalin, Terpene, Amide, Ether, Ketone und
Harnstoffe), Weichmacher (z.B. Diethylphthalat und Glycerin), Lösungsmittel
(z.B. Alkohol, Maisöl,
Baumwollsamenöl,
Glycerin, Isopropylalkohol, Mineralöl, Ölsäure, Erdnussöl, gereinigtes
Wasser, Wasser zur Injektion, steriles Wasser zur Injektion und
steriles Wasser zur Bewässerung),
Versteifungsmittel (z.B. Cetylalkohol, Cetylesterwachs, mikrokristalles
Wachs, Paraffin, Stearinalkohol, weißes und gelbes Wachs), Suppositorien-Grundlagen
(z.B. Kakaobutter und Polyethylenglykole (Mischungen)), oberflächenaktive
Mittel (z.B. Benzalkoniumchlorid, Nonoxynol-10, Oxtoxynol-9, Polysorbat-80,
Natriumlaurylsulfat und Sorbitanmonopalmitat), Suspendiermittel
(z.B. Agar, Bentonit, Carbomere, Carboxymethylcellulose-Natrium,
Hydroxyethylcellulose, Hydroxypropylcellulose, Hydroxypropylmethylcellulose,
Kaolin, Methylcellulose, Tragacanth und Veegum (= MgAl-Silikat)),
Süßungsmittel
(z.B. Aspartam, Dextrose, Glycerin, Mannit, Propylenglykol, Saecharin-Natrium, Sorbit und
Saccharose), Tabletten-Antianhaftmittel (z.B. Magnesiumstearat und
Talkum), Tabletten-Bindemittel (z.B. Akazie, Alginsäure, Carboxymethylcellulose-Natrium,
komprimierbaren Zucker, Ethylcellulose, Gelatine, flüssige Glucose,
Methylcellulose, Povidon und vorgelatinierte Stärke), Tabletten- und Kapsel-Verdünnungsmittel
(z.B. dibasisches Calciumphosphat, Kaolin, Lactose, Mannit, mikrokristalline
Cellulose, pulverisierte Cellulose, gefälltes Calciumcarbonat, Natriumcarbonat,
Natriumphosphat, Sorbit und Stärke),
Tabletten-Überzugsmittel
(z.B. flüssige
Glucose, Hydroxyethylcellulose, Hydroxypropylcellulose, Hydroxypropylmethylcellulose,
Methylcellulose, Ethylcellulose, Celluloseacetatphthalat und Schellack),
Tabletten-Direktkompressionexzipienten
(z.B. dibasisches Calciumphosphat), Tabletten-Zerfallmittel (z.B. Alginsäure, Carboxymethylcellulose-Calcium,
mikrokristalline Cellulose, Polacrillin-Kalium, Natriumalginat,
Natrium-Stärkeglycolat
und Stärke),
Tabletten-Gleitmittel (z.B. kolloidales Silica, Maisstärke und
Talkum), Tabletten-Schmiermittel (z.B. Calciumstearat, Magnesiumstearat,
Mineralöl,
Stearinsäure
und Zinkstearat), opak-machende Mittel für Tabletten/Kapseln (z.B. Titandioxid),
Tabletten-Poliermittel (z.B. Karnuba-Wachs und weißes Wachs),
Verdickungsmittel (z.B. Bienenwachs, Cetylalkohol und Paraffin),
Tonizitätsmittel
(z.B. Dextrose und Natriumchlorid), Viskositätssteigerungsmittel (z.B. Alginsäure, Betonit,
Carbomere, Carboxymethylcellulose-Natrium, Methylcellulose, Povidon,
Natriumalginat und Tragacanth) sowie Netzmittel (z.B. Heptadecaethylenoxycetanol,
Lecithine, Polyethylensorbitmonooleat, Polyoxyethylensorbitmonooleat und
Polyoxyethylenstearat) ein.
-
Die
durch die hierin beschriebenen Verfahren identifizierten Verbindungen
können
als das einzige pharmazeutische Mittel oder in Kombination mit einem
oder mehreren weiteren pharmazeutischen Mitteln verabreicht werden,
wobei die Kombination keine inakzeptablen Nebeneffekte verursacht.
Beispielsweise können die
Verbindungen der vorliegenden Erfindung mit bekannten Anti-Fettleibigkeitsmitteln
oder mit bekannten antidiabetischen oder weiteren Indikationsmitteln
und dgl. sowie mit Abmischungen und Kombinationen davon kombiniert
werden.
-
Die
durch die hierin beschriebenen Verfahren identifizierten Verbindungen
können
auch in freier Basenform oder in Zusammensetzungen in Forschung
und Diagnostik oder als analytische Bezugsstandards und dgl. genutzt
und angewandt werden. Daher schließt die vorliegende Erfindung
auch Zusammensetzungen ein, die aus einem inerten Träger und
einer wirksamen Menge einer durch die hierin beschriebenen Verfahren
identifizierten Verbindung oder eines Salzes oder Esters davon zusammengesetzt
sind. Ein inerter Träger
ist jedes Material, das mit der enthaltenen Verbindung nicht in
Wechselwirkung tritt und ihr Stützung,
Beförderung,
Masse, Aufspürmaterialeigenschaften
und dgl. verleiht. Die wirkungsvolle Menge der Verbindung ist diejenige
Menge, die ein Ergebnis erzeugt oder einen Einfluss auf die besondere
durchzuführende
Verfahrensweise ausübt.
-
Formulierungen,
die sich zur subkutanen, intravenösen, intramuskulären Anwendung
und dgl. eignen, geeignete pharmazeutische Träger sowie die Verfahrenstechniken
zur Formulierung und Verabreichung können mit jedem der im Stand
der Technik gut bekannten Verfahren hergestellt, zubereitet und
angewandt werden (siehe z.B. Remington's Pharmaceutical Sciences, Mack Publishing
Co., Easton, Pa., 20. Ausgabe, 2000).
-
Die
folgenden Beispiele sind zur weiteren Erläuterung der hierin beschriebenen
Erfindung angegeben, sollen aber in keiner Weise den Umfang der
Erfindung einschränken.
-
Kapsel-Formulierung
-
Eine
Kapsel-Formel wird hergestellt aus:
| Verbindung
der Erfindung | 40
mg |
| Stärke | 109
mg |
| Magnesiumstearat | 1
mg |
-
Die
Komponenten werden vermischt, durch ein geeignetes Mesh-Sieb gesiebt
und in Hartgelatine-Kapseln abgefüllt.
-
Tabletten-Formulierung
-
Eine
Tablette wird hergestellt aus:
| Verbindung
der Erfindung | 25
mg |
| Cellulose,
mikrokristallin | 200
mg |
| kolloidales
Siliziumdioxid | 10
mg |
-
Die
Bestandteile werden vermischt und zur Bildung von Tabletten komprimiert.
Geeignete wässrige und
nicht-wässrige Überzüge werden
zur Steigerung der Genießbarkeit,
zur Verbesserung der Eleganz und der Stabilität oder der Absorptionsverzögerung aufgebracht.
-
Steril-IV-Lösung
-
Eine
5 mg/mL-Lösung
der gewünschten
Verbindung der vorliegenden Erfindung wird mit sterilem, injizierbaren
Wasser hergestellt, und der pH-Wert
wird, falls notwendig, eingestellt. Die Lösung wird zur Verabreichung
auf 1 bis 2 mg/mL mit steriler 5%iger Dextrose verdünnt und
als eine IV-Infusion über 60 min
verabreicht.
-
Intramuskuläre Suspension
-
Die
folgende intramuskuläre
Suspension wird hergestellt aus:
| Verbindung
der Erfindung | 50
mg/mL |
| Natriumcarboxymethylcellulose | 5
mg/mL |
| TWEEN
80 | 9
mg/mL |
| Natriumchlorid | 90
mg/mL |
| Benzylalkohol | 9
mg/mL |
-
Die
Suspension wird intramuskulär
verabreicht.
-
Hartschale-Kapseln
-
Eine
große
Zahl von Einheitskapseln wird durch Befüllen von Standard-2-Stück-Hartgelatine-Kapseln mit
jeweils 100 mg pulverisiertem Wirkbestandteil, 150 mg Lactose, 50
mg Cellulose und mit 6 mg Magnesiumstearat hergestellt.
-
Weichgelatine-Kapseln
-
Eine
Mischung des Wirkbestandteils in einem verdaubaren Öl wie Sojabohnen-,
Baumwollsamen- oder Olivenöl
wird zubereitet und mittels einer positiven Verdrängungspumpe
in geschmolzene Gelatine zur Bildung von Weichgelatine-Kapseln gespritzt,
die 100 mg Wirkbestandteil enthalten. Die Kapseln werden gewaschen
und getrocknet. Der Wirkbestandteil kann in einer Mischung aus Polyethylenglykol,
Glycerin und aus Sorbit gelöst
sein, um eine mit Wasser mischbare Mischung der medizinisch anwendbaren
Substanz herzustellen.
-
Tabletten/Kapseln zur
unmittelbaren Freisetzung
-
Diese
stellen feste orale Dosierungsformen dar, die mit herkömmlichen
und neuen Verfahren hergestellt werden. Diese Einheiten werden oral
ohne Wasser zur unmittelbaren Auflösung und Abgabe der Medikation
eingenommen. Der Wirkbestandteil wird in einer Flüssigkeit
vermischt, die einen Bestandteil wie Zucker, Gelatine, Pectin und
Süßstoffe
enthält.
Diese Flüssigkeiten
werden zu festen Tabletten oder Kapletten durch Gefriertrocknung
und Festzustand-Extraktionstechniken verfestigt. Die Arzneiverbindungen
können
mit viskoelastischen und thermoelastischen Zuckern und Polymeren
oder mit Brausekomponenten verpresst werden, um poröse Matrizes
zur unmittelbaren Freisetzung zu erzeugen, ohne Wasser zu benötigen.
-
Die
hierin beschriebenen Strukturen, Materialien, Zusammensetzungen
und Verfahren sollen lediglich repräsentative Beispiele der Erfindung
darstellen, und es ist zu verstehen, dass der Umfang der Erfindung
nicht durch den Umfang der Beispiele eingeschränkt ist. Die Fachleute werden
erkennen, dass die Erfindung mit Variationen bei den offenbarten
Strukturen, Materialien, Zusammensetzungen und Verfahren ausgeführt werden
kann, wobei solche Variationen so anzusehen und zu bewerten sind,
dass sie unter den Rahmen und Umfang der vorliegenden Erfindung
fallen.




















































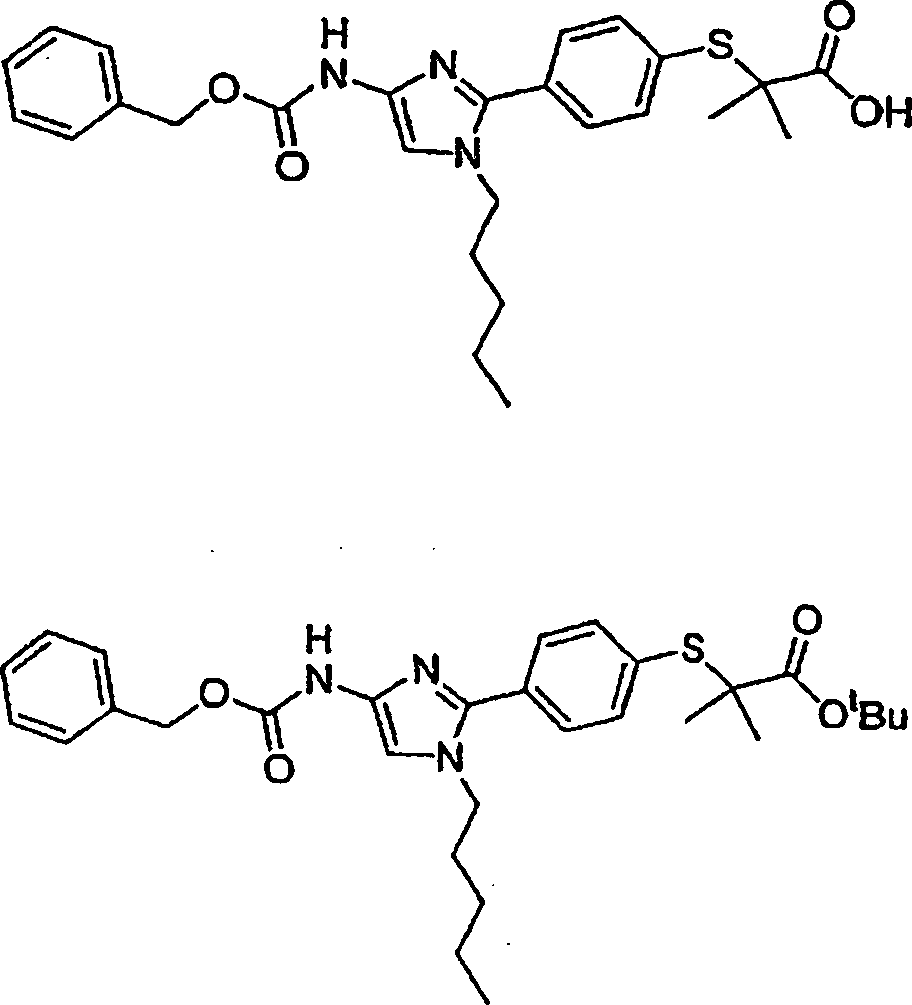















































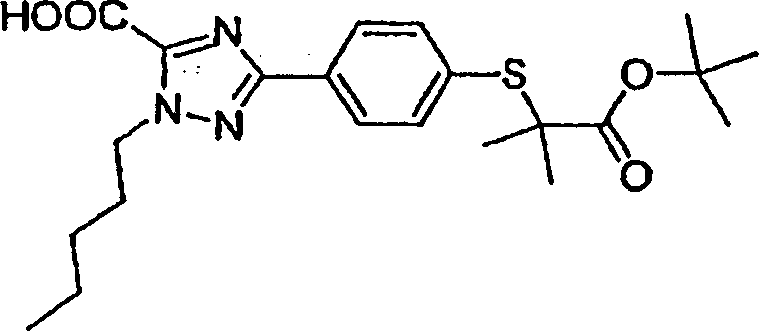







































































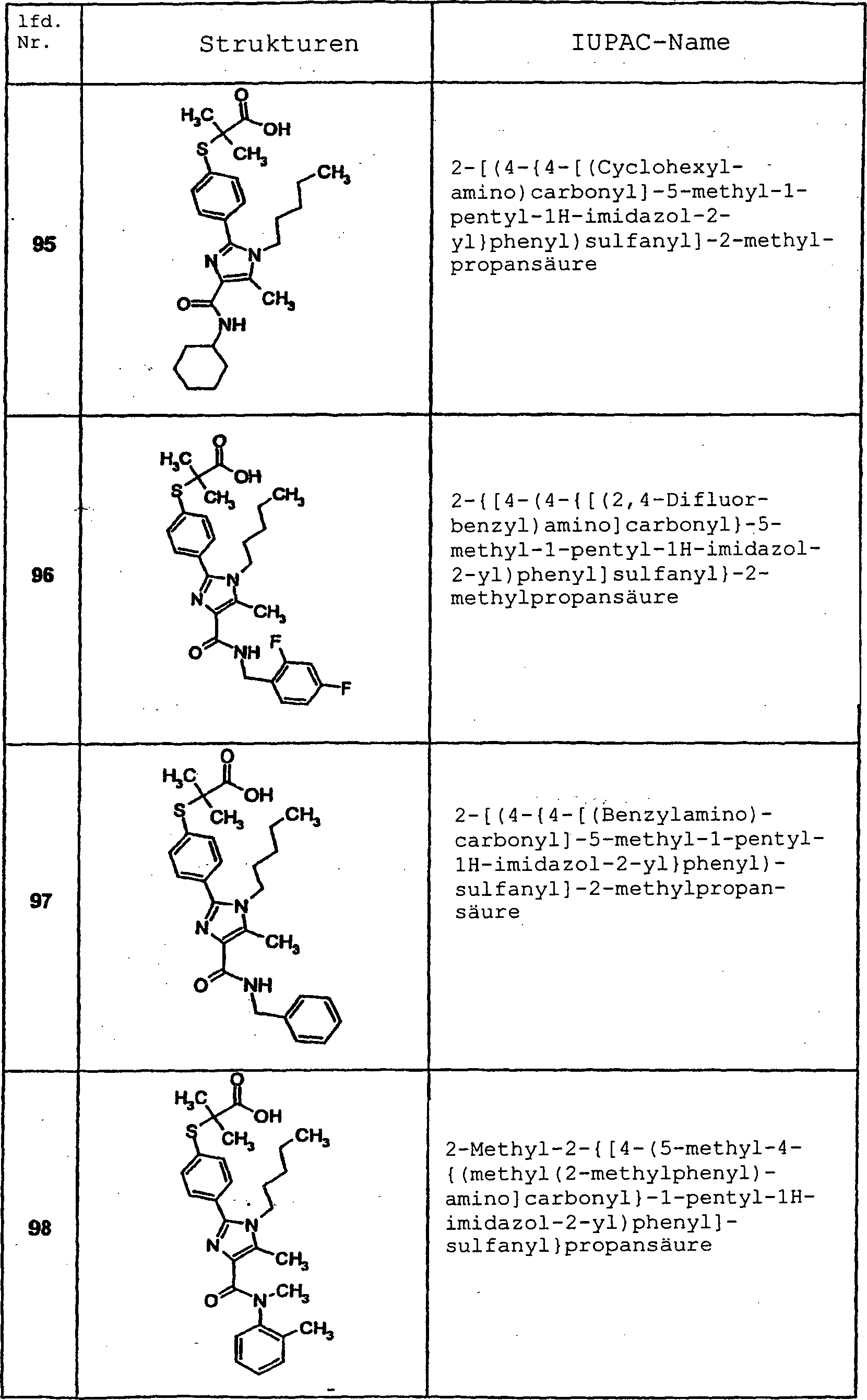



























































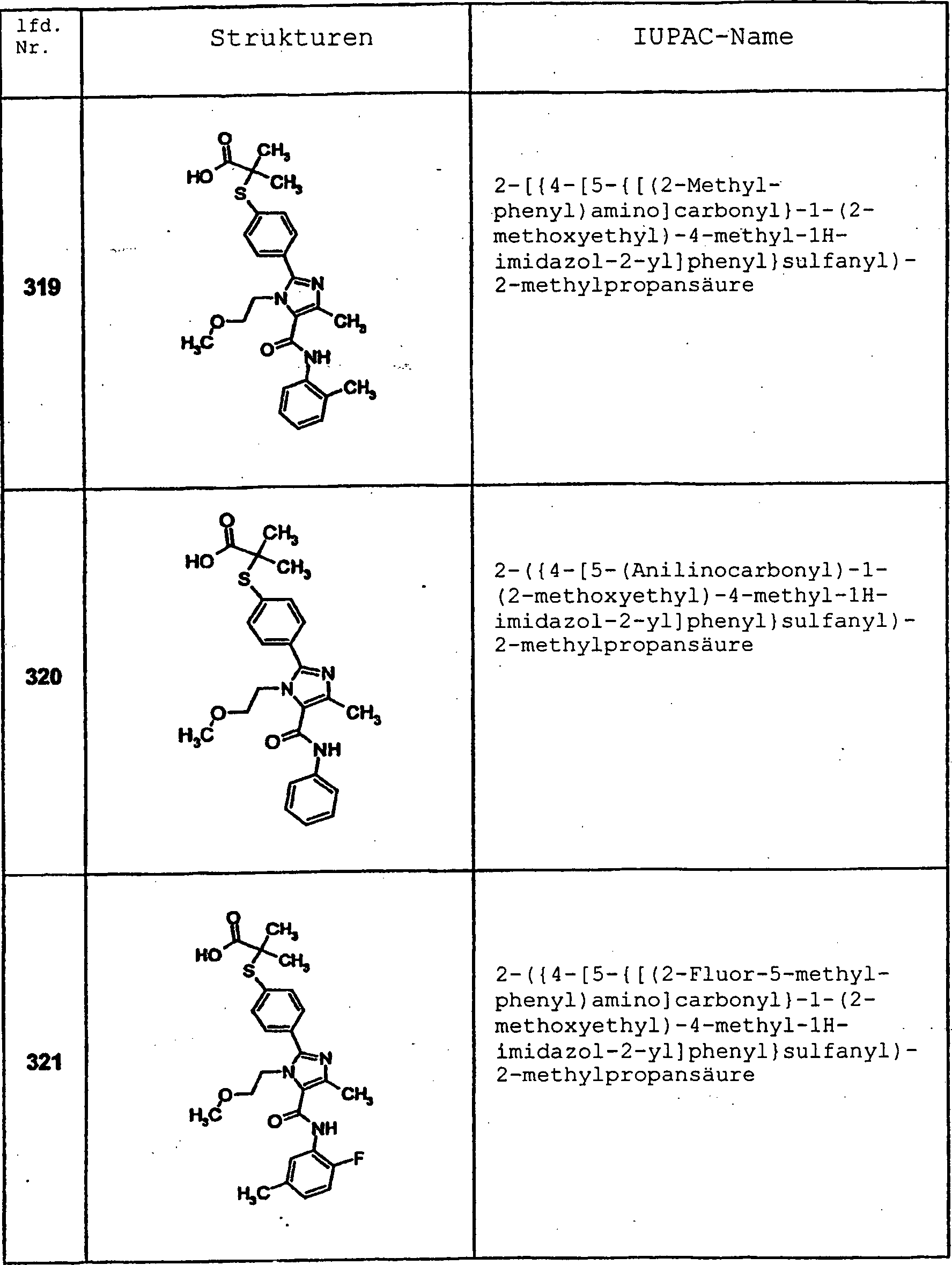








 worin gilt: Z stellt O oder S dar, R1-1 stellt Wasserstoff oder (C1-6)Alkyl dar, R1-2 stellt Wasserstoff oder (C1-6)Alkyl dar, und R1-3 stellt Wasserstoff oder (C1-6)Alkyl dar; R2 stellt Wasserstoff oder Methyl dar; oder R1 und R2 stellen gemeinsam eine Gruppe dar:
worin gilt: Z stellt O oder S dar, R1-1 stellt Wasserstoff oder (C1-6)Alkyl dar, R1-2 stellt Wasserstoff oder (C1-6)Alkyl dar, und R1-3 stellt Wasserstoff oder (C1-6)Alkyl dar; R2 stellt Wasserstoff oder Methyl dar; oder R1 und R2 stellen gemeinsam eine Gruppe dar: die, zusammen mit den Kohlenstoffen, an die die genannte Gruppe gebunden ist, einen carbocyclischen Ring bildet, worin R1-4 Wasserstoff oder (C1-6)Alkyl und R1-5 Wasserstoff oder (C1-6)Alkyl darstellen; R3 stellt Wasserstoff oder Methyl dar; Y stellt NR4, O oder S dar, worin R4 Wasserstoff oder (C1-6)Alkyl darstellt, das gegebenenfalls mit 1 oder 2 Substituenten substituiert ist, die unabhängig aus der Gruppe ausgewählt sind, bestehend aus (C1-6)Alkoxy und (C1-6)Aryloxy; R5 stellt Wasserstoff, (C1-6)Alkyl oder Phenyl dar, gegebenenfalls substituiert mit Halogen, (C1-6)Alkyl oder mit (C1-6)Alkoxy; R6 stellt Benzyloxycarbonylamino oder eine Gruppe der Formel dar:
die, zusammen mit den Kohlenstoffen, an die die genannte Gruppe gebunden ist, einen carbocyclischen Ring bildet, worin R1-4 Wasserstoff oder (C1-6)Alkyl und R1-5 Wasserstoff oder (C1-6)Alkyl darstellen; R3 stellt Wasserstoff oder Methyl dar; Y stellt NR4, O oder S dar, worin R4 Wasserstoff oder (C1-6)Alkyl darstellt, das gegebenenfalls mit 1 oder 2 Substituenten substituiert ist, die unabhängig aus der Gruppe ausgewählt sind, bestehend aus (C1-6)Alkoxy und (C1-6)Aryloxy; R5 stellt Wasserstoff, (C1-6)Alkyl oder Phenyl dar, gegebenenfalls substituiert mit Halogen, (C1-6)Alkyl oder mit (C1-6)Alkoxy; R6 stellt Benzyloxycarbonylamino oder eine Gruppe der Formel dar: worin gilt: R6-1 stellt dar: – Hydroxy, – (C1-6)Alkoxy, – Benzyloxy, – eine Gruppe der Formel:
worin gilt: R6-1 stellt dar: – Hydroxy, – (C1-6)Alkoxy, – Benzyloxy, – eine Gruppe der Formel: worin R6-1-1 darstellt: Wasserstoff, (C1-6)Alkyl, gegebenenfalls substituiert mit 1 oder 2 Substituenten, unabhängig ausgewählt aus der Gruppe, bestehend aus (C6-10)Aryl und (C1-6)Alkoxy, (C3-8)Cycloalkyl, gegebenenfalls substituiert mit (C1-6)Alkyl, (C6-10)Aryl, gegebenenfalls substituiert mit 1, 2 oder 3 Substituenten, unabhängig ausgewählt aus der Gruppe, bestehend aus Halogen, Nitro, Cyano, (C1-6)Alkyl, (C3-8)Cycloalkyl, (C1-6)Alkoxycarbonyl, Hydroxy, (C1-6)Alkoxy, Trifluormethyl, Trifluormethoxy, Heterocyclyl, (C6-10)Aryl, (C6-10)Aryloxy und aus Benzyl, oder einen 5- bis 10-gliedrigen. heterocyclischen Rest, umfassend 3 bis 9 Kohlenstoffatome und 1 bis 3 Heteroatome, ausgewählt aus O, N oder S, gegebenenfalls substituiert mit Phenyl, Benzyl oder mit Halogen, und worin R6-1-2 Wasserstoff, (C1-6)Alkyl oder (C3-8)Cycloalkyl darstellt, – eine Gruppe der Formel -NH-NH-R6-2, worin R6-2 (C6-10)Aryl darstellt, oder – einen 5- bis 10-gliedrigen heterocyclischen Rest, umfassend 3 bis 9 Kohlenstoffatome und 1 bis 3 Heteroatome, ausgewählt aus O, N oder S, und gegebenenfalls substituiert mit 1 oder 2 Substituenten, unabhängig ausgewählt aus der Gruppe, bestehend aus (C1-6)Alkyl, Benzyl oder aus Phenyl, gegebenenfalls substituiert mit (C1-6)Alkyl; und pharmazeutische Salze oder Ester davon.
worin R6-1-1 darstellt: Wasserstoff, (C1-6)Alkyl, gegebenenfalls substituiert mit 1 oder 2 Substituenten, unabhängig ausgewählt aus der Gruppe, bestehend aus (C6-10)Aryl und (C1-6)Alkoxy, (C3-8)Cycloalkyl, gegebenenfalls substituiert mit (C1-6)Alkyl, (C6-10)Aryl, gegebenenfalls substituiert mit 1, 2 oder 3 Substituenten, unabhängig ausgewählt aus der Gruppe, bestehend aus Halogen, Nitro, Cyano, (C1-6)Alkyl, (C3-8)Cycloalkyl, (C1-6)Alkoxycarbonyl, Hydroxy, (C1-6)Alkoxy, Trifluormethyl, Trifluormethoxy, Heterocyclyl, (C6-10)Aryl, (C6-10)Aryloxy und aus Benzyl, oder einen 5- bis 10-gliedrigen. heterocyclischen Rest, umfassend 3 bis 9 Kohlenstoffatome und 1 bis 3 Heteroatome, ausgewählt aus O, N oder S, gegebenenfalls substituiert mit Phenyl, Benzyl oder mit Halogen, und worin R6-1-2 Wasserstoff, (C1-6)Alkyl oder (C3-8)Cycloalkyl darstellt, – eine Gruppe der Formel -NH-NH-R6-2, worin R6-2 (C6-10)Aryl darstellt, oder – einen 5- bis 10-gliedrigen heterocyclischen Rest, umfassend 3 bis 9 Kohlenstoffatome und 1 bis 3 Heteroatome, ausgewählt aus O, N oder S, und gegebenenfalls substituiert mit 1 oder 2 Substituenten, unabhängig ausgewählt aus der Gruppe, bestehend aus (C1-6)Alkyl, Benzyl oder aus Phenyl, gegebenenfalls substituiert mit (C1-6)Alkyl; und pharmazeutische Salze oder Ester davon.
 worin: R6-1 darstellt: – eine Gruppe der Formel:
worin: R6-1 darstellt: – eine Gruppe der Formel: worin gilt: R6-1-1 stellt dar: (C1-6)Alkyl, gegebenenfalls substituiert mit 1 oder 2 Substituenten, unabhängig ausgewählt aus der Gruppe, bestehend aus (C6-10)Aryl und (C1-6)Alkoxy, (C3-8)Cycloalkyl, gegebenenfalls substituiert mit (C1-6)Alkyl, (C6-10)Aryl, gegebenenfalls substituiert mit 1, 2 oder 3 Substituenten, unabhängig ausgewählt aus der Gruppe, bestehend aus Halogen, Nitro, Cyano, (C1-6)Alkyl, (C3-8)Cycloalkyl, (C1-6)Alkoxycarbonyl, Hydroxy, (C1-6)Alkoxy, Trifluormethyl, Trifluormethoxy, Heterocyclyl, (C6-10)Aryl, (C6-10)Aryloxy und aus Benzyl, oder einen 5- bis 10-gliedrigen heterocyclischen Rest, umfassend 3 bis 9 Kohlenstoffatome und 1 bis 3 Heteroatome, ausgewählt aus O, N oder S, gegebenenfalls substituiert mit Phenyl, Benzyl oder mit Halogen, und worin R6-1-2 Wasserstoff oder (C1-6)Alkyl darstellt; und pharmazeutische Salze oder Ester davon.
worin gilt: R6-1-1 stellt dar: (C1-6)Alkyl, gegebenenfalls substituiert mit 1 oder 2 Substituenten, unabhängig ausgewählt aus der Gruppe, bestehend aus (C6-10)Aryl und (C1-6)Alkoxy, (C3-8)Cycloalkyl, gegebenenfalls substituiert mit (C1-6)Alkyl, (C6-10)Aryl, gegebenenfalls substituiert mit 1, 2 oder 3 Substituenten, unabhängig ausgewählt aus der Gruppe, bestehend aus Halogen, Nitro, Cyano, (C1-6)Alkyl, (C3-8)Cycloalkyl, (C1-6)Alkoxycarbonyl, Hydroxy, (C1-6)Alkoxy, Trifluormethyl, Trifluormethoxy, Heterocyclyl, (C6-10)Aryl, (C6-10)Aryloxy und aus Benzyl, oder einen 5- bis 10-gliedrigen heterocyclischen Rest, umfassend 3 bis 9 Kohlenstoffatome und 1 bis 3 Heteroatome, ausgewählt aus O, N oder S, gegebenenfalls substituiert mit Phenyl, Benzyl oder mit Halogen, und worin R6-1-2 Wasserstoff oder (C1-6)Alkyl darstellt; und pharmazeutische Salze oder Ester davon.
 worin R6-1 einen 5- bis 10-gliedrigen heterocyclischen Rest darstellt, umfassend 3 bis 9 Kohlenstoffatome und 1 bis 3 Heteroatome, ausgewählt aus O, N oder S, und gegebenenfalls substituiert mit 1 oder 2 Substituenten, unabhängig ausgewählt aus der Gruppe, bestehend aus (C1-6)Alkyl, Benzyl oder aus Phenyl, gegebenenfalls substituiert mit (C1-6)Alkyl; und pharmazeutische Salze oder Ester davon.
worin R6-1 einen 5- bis 10-gliedrigen heterocyclischen Rest darstellt, umfassend 3 bis 9 Kohlenstoffatome und 1 bis 3 Heteroatome, ausgewählt aus O, N oder S, und gegebenenfalls substituiert mit 1 oder 2 Substituenten, unabhängig ausgewählt aus der Gruppe, bestehend aus (C1-6)Alkyl, Benzyl oder aus Phenyl, gegebenenfalls substituiert mit (C1-6)Alkyl; und pharmazeutische Salze oder Ester davon.
 worin R6-1 darstellt: – eine Gruppe der Formel:
worin R6-1 darstellt: – eine Gruppe der Formel: worin R6-1-1 Hydroxy oder (C1-6)Alkoxy darstellt; und pharmazeutische Salze oder Ester davon.
worin R6-1-1 Hydroxy oder (C1-6)Alkoxy darstellt; und pharmazeutische Salze oder Ester davon.
 worin R6-1 darstellt: – eine Gruppe der Formel:
worin R6-1 darstellt: – eine Gruppe der Formel: worin R6-1-1 (C6-10)Aryl, gegebenenfalls substituiert mit 1, 2 oder 3 Substituenten, unabhängig ausgewählt aus der Gruppe, bestehend aus Halogen, Nitro, Cyano, (C1-6)Alkyl, (C3-8)-Cylcoalkyl, (C1-6)Alkylcarbonyl, Hydroxy, (C1-6)Alkoxy, Trifluormethyl, Trifluormethoxy, Heterocyclyl, (C6-10)Aryl, (C6-10)Aryloxy und aus Benzyl, und R6-1-2 Wasserstoff darstellen; und pharmazeutische Salze oder Ester davon.
worin R6-1-1 (C6-10)Aryl, gegebenenfalls substituiert mit 1, 2 oder 3 Substituenten, unabhängig ausgewählt aus der Gruppe, bestehend aus Halogen, Nitro, Cyano, (C1-6)Alkyl, (C3-8)-Cylcoalkyl, (C1-6)Alkylcarbonyl, Hydroxy, (C1-6)Alkoxy, Trifluormethyl, Trifluormethoxy, Heterocyclyl, (C6-10)Aryl, (C6-10)Aryloxy und aus Benzyl, und R6-1-2 Wasserstoff darstellen; und pharmazeutische Salze oder Ester davon.
 worin R6-1 eine Gruppe der Formal darstellt:
worin R6-1 eine Gruppe der Formal darstellt: worin R6-1-1 (C6-10)Aryl, das gegebenenfalls mit 1, 2 oder 3 Substituenten substituiert ist, unabhängig ausgewählt aus der Gruppe, bestehend aus Halogen, Nitro, Cyano, (C1-6)Alkyl, (C3-8)-Cylcoalkyl, (C1-6)Alkylcarbonyl, Hydroxy, (C1-6)Alkoxy, Trifluormethyl, Trifluormethoxy, Heterocyclyl, (C6-10)Aryl, (C6-10)Aryloxy und aus Benzyl, und R6-1-2 Wasserstoff, (C1-6)Alkyl oder (C3-8)Cycloalkyl darstellen; und pharmazeutische Salze oder Ester davon.
worin R6-1-1 (C6-10)Aryl, das gegebenenfalls mit 1, 2 oder 3 Substituenten substituiert ist, unabhängig ausgewählt aus der Gruppe, bestehend aus Halogen, Nitro, Cyano, (C1-6)Alkyl, (C3-8)-Cylcoalkyl, (C1-6)Alkylcarbonyl, Hydroxy, (C1-6)Alkoxy, Trifluormethyl, Trifluormethoxy, Heterocyclyl, (C6-10)Aryl, (C6-10)Aryloxy und aus Benzyl, und R6-1-2 Wasserstoff, (C1-6)Alkyl oder (C3-8)Cycloalkyl darstellen; und pharmazeutische Salze oder Ester davon.
 worin gilt: R6-1 stellt dar: – Hydroxy, – (C1-6)Alkoxy, – eine Gruppe der Formel:
worin gilt: R6-1 stellt dar: – Hydroxy, – (C1-6)Alkoxy, – eine Gruppe der Formel: worin R6-1-1 (C6-10)Aryl, das gegebenenfalls mit 1, 2 oder 3 Substituenten substituiert ist, unabhängig ausgewählt aus der Gruppe, bestehend aus Halogen, Nitro, Cyano, (C1-6)Alkyl, (C3-8)-Cylcoalkyl, (C1-6)Alkylcarbonyl, Hydroxy, (C1-6)Alkoxy, Trifluormethyl, Trifluormethoxy, Heterocyclyl, (C6-10)Aryl, (C6-10)Aryloxy und aus Benzyl, und R6-1-2 Wasserstoff darstellen; – einen 5- bis 10-gliedrigen heterocyclischen Rest, umfassend 3 bis 9 Kohlenstoffatome und 1 bis 3 Heteroatome, ausgewählt aus O, N oder S, und gegebenenfalls substituiert mit 1 oder 2 Substituenten, unabhängig ausgewählt aus der Gruppe, bestehend aus (C1-6)Alkyl, Benzyl oder aus Phenyl, gegebenenfalls substituiert mit (C1-6)Alkyl; und pharmazeutische Salze oder Ester davon.
worin R6-1-1 (C6-10)Aryl, das gegebenenfalls mit 1, 2 oder 3 Substituenten substituiert ist, unabhängig ausgewählt aus der Gruppe, bestehend aus Halogen, Nitro, Cyano, (C1-6)Alkyl, (C3-8)-Cylcoalkyl, (C1-6)Alkylcarbonyl, Hydroxy, (C1-6)Alkoxy, Trifluormethyl, Trifluormethoxy, Heterocyclyl, (C6-10)Aryl, (C6-10)Aryloxy und aus Benzyl, und R6-1-2 Wasserstoff darstellen; – einen 5- bis 10-gliedrigen heterocyclischen Rest, umfassend 3 bis 9 Kohlenstoffatome und 1 bis 3 Heteroatome, ausgewählt aus O, N oder S, und gegebenenfalls substituiert mit 1 oder 2 Substituenten, unabhängig ausgewählt aus der Gruppe, bestehend aus (C1-6)Alkyl, Benzyl oder aus Phenyl, gegebenenfalls substituiert mit (C1-6)Alkyl; und pharmazeutische Salze oder Ester davon.
 worin gilt: Z stellt S dar, R7-1 stellt Wasserstoff oder (C1-6)Alkyl dar, R7-2 stellt Wasserstoff oder (C1-6)Alkyl dar, R7-3 stellt Wasserstoff oder (C1-6)Alkyl dar; R8 stellt Wasserstoff oder Methyl dar; R9 stellt Wasserstoff oder Methyl dar; R10 stellt (C1-6)Alkyl dar, das gegebenenfalls mit 1 oder 2 Substituenten substituiert ist, unabhängig ausgewählt aus der Gruppe, bestehend aus (C1-6)Alkoxy und (C6-10)Aryloxy; R11 stellt eine Gruppe der Formel dar:
worin gilt: Z stellt S dar, R7-1 stellt Wasserstoff oder (C1-6)Alkyl dar, R7-2 stellt Wasserstoff oder (C1-6)Alkyl dar, R7-3 stellt Wasserstoff oder (C1-6)Alkyl dar; R8 stellt Wasserstoff oder Methyl dar; R9 stellt Wasserstoff oder Methyl dar; R10 stellt (C1-6)Alkyl dar, das gegebenenfalls mit 1 oder 2 Substituenten substituiert ist, unabhängig ausgewählt aus der Gruppe, bestehend aus (C1-6)Alkoxy und (C6-10)Aryloxy; R11 stellt eine Gruppe der Formel dar: worin gilt: R11-1 stellt dar: – Hydroxy, – (C1-6)Alkoxy, – einen 5- bis 10-gliedrigen heterocyclischen Rest, umfassend 3 bis 9 Kohlenstoffatome und 1 bis 3 Heteroatome, ausgewählt aus O, N oder S, gegebenenfalls substituiert mit 1 oder 2 (C1-6)Alkyl- oder Phenylresten, gegebenenfalls substituiert mit Halogen, oder – eine Gruppe der Formel:
worin gilt: R11-1 stellt dar: – Hydroxy, – (C1-6)Alkoxy, – einen 5- bis 10-gliedrigen heterocyclischen Rest, umfassend 3 bis 9 Kohlenstoffatome und 1 bis 3 Heteroatome, ausgewählt aus O, N oder S, gegebenenfalls substituiert mit 1 oder 2 (C1-6)Alkyl- oder Phenylresten, gegebenenfalls substituiert mit Halogen, oder – eine Gruppe der Formel: worin R11-1-1 (C6-10)Aryl, das gegebenenfalls mit bis zu 3 Substituenten substituiert ist, unabhängig ausgewählt aus Halogen, Nitro, Cyano, (C1-6)Alkyl, (C1-6)Alkoxy, Trifluormethyl oder aus Phenyl, oder einen 5- bis 10-gliedrigen heterocyclischen Rest, umfassend 3 bis 9 Kohlenstoffatome und 1 bis 3 Heteroatome, ausgewählt aus O, N oder S, und R11-1-2 Wasserstoff darstellen; R12 stellt Wasserstoff oder (C1-6)Alkyl dar; und pharmazeutische Salze oder Ester davon.
worin R11-1-1 (C6-10)Aryl, das gegebenenfalls mit bis zu 3 Substituenten substituiert ist, unabhängig ausgewählt aus Halogen, Nitro, Cyano, (C1-6)Alkyl, (C1-6)Alkoxy, Trifluormethyl oder aus Phenyl, oder einen 5- bis 10-gliedrigen heterocyclischen Rest, umfassend 3 bis 9 Kohlenstoffatome und 1 bis 3 Heteroatome, ausgewählt aus O, N oder S, und R11-1-2 Wasserstoff darstellen; R12 stellt Wasserstoff oder (C1-6)Alkyl dar; und pharmazeutische Salze oder Ester davon.
 worin: R11-1 eine Gruppe der Formel darstellt:
worin: R11-1 eine Gruppe der Formel darstellt: worin R11-1-1 (C6-10)Aryl, das gegebenenfalls mit bis zu 3 Substituenten substituiert ist, unabhängig ausgewählt aus Halogen, Nitro, Cyano, (C1- 6)Alkyl, (C1-6)Alkoxy, Trifluormethyl oder aus Phenyl, und R11-1-2 Wasserstoff darstellen; R12 stellt Wasserstoff oder (C1-6)Alkyl dar; und pharmazeutische Salze oder Ester davon.
worin R11-1-1 (C6-10)Aryl, das gegebenenfalls mit bis zu 3 Substituenten substituiert ist, unabhängig ausgewählt aus Halogen, Nitro, Cyano, (C1- 6)Alkyl, (C1-6)Alkoxy, Trifluormethyl oder aus Phenyl, und R11-1-2 Wasserstoff darstellen; R12 stellt Wasserstoff oder (C1-6)Alkyl dar; und pharmazeutische Salze oder Ester davon.
 worin gilt: Z stellt S dar, R13-1 stellt Wasserstoff oder (C1-6)Alkyl dar, R13-2 stellt Wasserstoff oder (C1-6)Alkyl dar, R13-3 stellt Wasserstoff oder (C1-6)Alkyl dar; R14 stellt Wasserstoff oder Methyl dar; R15 stellt Wasserstoff oder Methyl dar; R16 stellt eine Gruppe der Formel dar:
worin gilt: Z stellt S dar, R13-1 stellt Wasserstoff oder (C1-6)Alkyl dar, R13-2 stellt Wasserstoff oder (C1-6)Alkyl dar, R13-3 stellt Wasserstoff oder (C1-6)Alkyl dar; R14 stellt Wasserstoff oder Methyl dar; R15 stellt Wasserstoff oder Methyl dar; R16 stellt eine Gruppe der Formel dar: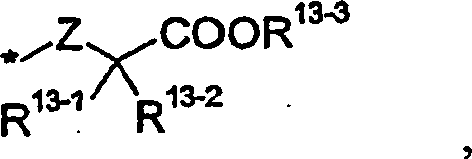 worin R16-1 eine Gruppe der Formel darstellt:
worin R16-1 eine Gruppe der Formel darstellt: worin: R16-1-1 (C6-10)Aryl, das gegebenenfalls mit 1, 2 oder 3 Substituenten substituiert ist, unabhängig ausgewählt aus Halogen, Nitro, Cyano, (C1-6)Alkyl, (C1-6)Alkoxy und aus Trifluormethyl, und R16-1-2 Wasserstoff darstellen; R17 stellt (C1-6)Alkyl dar, das gegebenenfalls mit 1 oder 2 (C1-6)Alkylresten substituiert ist; und pharmazeutische Salze oder Ester davon.
worin: R16-1-1 (C6-10)Aryl, das gegebenenfalls mit 1, 2 oder 3 Substituenten substituiert ist, unabhängig ausgewählt aus Halogen, Nitro, Cyano, (C1-6)Alkyl, (C1-6)Alkoxy und aus Trifluormethyl, und R16-1-2 Wasserstoff darstellen; R17 stellt (C1-6)Alkyl dar, das gegebenenfalls mit 1 oder 2 (C1-6)Alkylresten substituiert ist; und pharmazeutische Salze oder Ester davon.
 worin gilt: Z stellt S dar, R18-1 stellt Wasserstoff oder (C1-6)Alkyl dar, R18-2 stellt Wasserstoff oder (C1-6)Alkyl dar, und R18-3 stellt Wasserstoff oder (C1-6)Alkyl dar; R19 stellt Wasserstoff oder Methyl dar; R20 stellt Wasserstoff oder Methyl dar; R21 stellt (C1-6)Alkoxy dar; R22 stellt Wasserstoff, (C1-6)Alkyl oder Phenyl dar; R23 stellt eine Gruppe der Formel dar:
worin gilt: Z stellt S dar, R18-1 stellt Wasserstoff oder (C1-6)Alkyl dar, R18-2 stellt Wasserstoff oder (C1-6)Alkyl dar, und R18-3 stellt Wasserstoff oder (C1-6)Alkyl dar; R19 stellt Wasserstoff oder Methyl dar; R20 stellt Wasserstoff oder Methyl dar; R21 stellt (C1-6)Alkoxy dar; R22 stellt Wasserstoff, (C1-6)Alkyl oder Phenyl dar; R23 stellt eine Gruppe der Formel dar: worin R23-1 Hydroxy, (C1-6)Alkoxy oder Benzyloxy oder eine Gruppe der Formel darstellt:
worin R23-1 Hydroxy, (C1-6)Alkoxy oder Benzyloxy oder eine Gruppe der Formel darstellt: worin R23-1-1 (C6-10)Aryl, das gegebenenfalls mit bis zu 3 Substituenten substituiert ist, unabhängig ausgewählt aus Halogen, Nitro, Cyano, (C1-6)Alkyl, (C1-6)Alkoxy und aus Trifluormethyl, und R23-1-2 Wasserstoff darstellen; und pharmazeutische Salze oder Ester davon.
worin R23-1-1 (C6-10)Aryl, das gegebenenfalls mit bis zu 3 Substituenten substituiert ist, unabhängig ausgewählt aus Halogen, Nitro, Cyano, (C1-6)Alkyl, (C1-6)Alkoxy und aus Trifluormethyl, und R23-1-2 Wasserstoff darstellen; und pharmazeutische Salze oder Ester davon.
 worin gilt: Z stellt S dar, R24-1 stellt Wasserstoff oder (C1-6)Alkyl dar, R24-2 stellt Wasserstoff oder (C1-6)Alkyl dar, R24-3 stellt Natrium, Wasserstoff oder (C1-6)Alkyl dar; R25 stellt Wasserstoff oder Methyl dar; R26 stellt Wasserstoff oder Methyl dar; R27 stellt Phenyl dar; R28 stellt Wasserstoff dar; und pharmazeutische Salze oder Ester davon.
worin gilt: Z stellt S dar, R24-1 stellt Wasserstoff oder (C1-6)Alkyl dar, R24-2 stellt Wasserstoff oder (C1-6)Alkyl dar, R24-3 stellt Natrium, Wasserstoff oder (C1-6)Alkyl dar; R25 stellt Wasserstoff oder Methyl dar; R26 stellt Wasserstoff oder Methyl dar; R27 stellt Phenyl dar; R28 stellt Wasserstoff dar; und pharmazeutische Salze oder Ester davon.

