DE60115051T2 - Immunologische adjuvans verbindungen - Google Patents
Immunologische adjuvans verbindungen Download PDFInfo
- Publication number
- DE60115051T2 DE60115051T2 DE60115051T DE60115051T DE60115051T2 DE 60115051 T2 DE60115051 T2 DE 60115051T2 DE 60115051 T DE60115051 T DE 60115051T DE 60115051 T DE60115051 T DE 60115051T DE 60115051 T2 DE60115051 T2 DE 60115051T2
- Authority
- DE
- Germany
- Prior art keywords
- alkyl
- alkoxy
- alkylene
- hydroxy
- straight
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 0 CCC(C*)**F Chemical compound CCC(C*)**F 0.000 description 15
- FTDVCUHOSZZDIE-VIFPVBQESA-N CC1(C)O[C@@H](CCCC(C)=O)CO1 Chemical compound CC1(C)O[C@@H](CCCC(C)=O)CO1 FTDVCUHOSZZDIE-VIFPVBQESA-N 0.000 description 1
- IKRVQURSPMLGQW-UHFFFAOYSA-N CCCCCCCC(C1OC1Cl)=[IH] Chemical compound CCCCCCCC(C1OC1Cl)=[IH] IKRVQURSPMLGQW-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/08—Esters of oxyacids of phosphorus
- C07F9/09—Esters of phosphoric acids
- C07F9/091—Esters of phosphoric acids with hydroxyalkyl compounds with further substituents on alkyl
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/39—Medicinal preparations containing antigens or antibodies characterised by the immunostimulating additives, e.g. chemical adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/08—Esters of oxyacids of phosphorus
- C07F9/09—Esters of phosphoric acids
- C07F9/113—Esters of phosphoric acids with unsaturated acyclic alcohols
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55572—Lipopolysaccharides; Lipid A; Monophosphoryl lipid A
Description
- ALLGEMEINER STAND DER TECHNIK
- Impfungen haben sich nachweislich als ein erfolgreiches, hochakzeptiertes Verfahren zur Verhinderung von Infektionskrankheiten erwiesen. Sie sind kostengünstig, lösen keine Antibiotikaresistenz gegenüber dem Zielpathogen aus und haben keinen Einfluss auf die normale Flora des Wirtes. In vielen Fällen können, wie bei der Induzierung von Antivirusimmunität, Impfstoffe eine Erkrankung verhindern, für die keine brauchbaren heilenden oder lindernden Behandlungen verfügbar sind.
- Die Funktion von Impfstoffen ist das Anregen des Immunsystems, auf einen Wirkstoff, oder ein Antigen zu reagieren, bei welchem es sich üblicherweise um einen infektiösen Organismus oder einen Teil davon handelt, welcher in einer nichtinfektiösen oder nichtpathogenen Form in den Körper gelangt. Wurde das Immunsystem einmal gegen den Organismus „präpariert" oder sensibilisiert, erfolgt, wenn dass Immunsystem später wieder diesem Organismus als ein infektiöses Pathogen ausgesetzt wird, eine schnelle und robuste Immunreaktion, welche das Pathogen zerstört, bevor es sich vermehren und ausreichend Zellen im Wirtsorganismus infizieren kann, um Krankheitssymptome zu verursachen.
- Der Wirkstoff oder das Antigen, welcher/s zur Vorbereitung des Immunsystems verwendet wird, kann der gesamte Organismus in einem weniger infektiösen Zustand sein, auch bekannt als ein abgeschwächter Organismus, oder in einigen Fällen auch Komponenten des Organismus wie Kohlenhydrate, Proteine oder Peptide, welche verschiedene Strukturkomponenten des Organismus darstellen, oder Nukleinsäuren, welche solche Komponenten codieren.
- In vielen Fällen ist es notwendig, die Immunreaktion gegen die in einem Impfstoff vorliegenden Antigene zu verbessern, um das Immunsystem in einem ausreichenden Umfang zu stimulieren, um den Impfstoff wirksam zu machen, d.h. um Immunität zu verleihen. Viele Protein- und die meisten Peptid- und Kohlenhydratantigene lösen, wenn sie allein verabreicht werden, keine ausreichende Antikörperreaktion aus, um Immunität zu verleihen. Solche Antigene müssen dem Immunsystem so verabreicht werden, dass sie als fremd erkannt werden und eine Immunreaktion auslösen. Zu diesem Zweck wurden Zusatzstoffe (Adjuvanzien) entwickelt, welche die Immunreaktion verbessern.
- Das am besten bekannte Adjuvans, Freund's komplettes Adjuvans, besteht aus einer Mischung aus Mykobakterien in einer Öl/Wasser-Emulsion. Freund's Adjuvans wirkt zweifach: erstens verbessert es die Zell- und humoral-mediierte Immunität und zweitens blockiert es die schnelle Ausbreitung der Antigenbedrohung („Depot-Effekt"). Jedoch kann Freund's Adjuvans aufgrund häufiger toxischer physiologischer und immunologischer Reaktionen auf dieses Material nicht beim Menschen angewandt werden.
- Ein weiteres Molekül, bei welchem immunitätsstimulierende oder Adjuvans-Aktivität nachgewiesen wurde, ist Endotoxin, auch bekannt als Lipopolysaccharid (LPS). LPS stimuliert das Immunsystem durch Auslösung einer „angeborenen" Immunreaktion – eine Reaktion, welche sich entwickelt hat, um es einem Organismus zu ermöglichen, Endotoxin (und die eindringenden Bakterien, von welchen es einen Bestandteil darstellt) zu erkennen, ohne dass der Organismus bereits einmal dem Molekül ausgesetzt war. Während LPS zu toxisch ist, um als brauchbares Adjuvans zu dienen, werden derzeit Moleküle, welche strukturell mit Endotoxin verwandt sind, wie Monophosphoryllipid A („MPL"), in klinischen Versuchen als Adjuvanzien getestet. Jedoch sind derzeit die einzigen von der FDA zugelassenen Adjuvanzien zur Verwendung beim Menschen Aluminiumsalze (Alum), welche zum „Deponieren" von Antigenen durch Präzipitation der Antigene verwendet werden. Alum stimuliert außerdem die Immunreaktion gegen Antigene.
- Weitere Adjuvansverbindungen sind in WO 00/44758 offenbart.
- Somit besteht anerkanntermaßen Bedarf in der Technik für Verbindungen, welche zusammen mit Antigenen verabreicht werden können, um das Immunsystem zu stimulieren, um eine kräftigere Reaktion auf das Antigen zu erzeugen, als wenn das Antigen allein oder mit Alum injiziert werden würde.
- ZUSAMMENFASSUNG DER ERFINDUNG
- In einem Aspekt sieht die Erfindung neuartige Verbindungen vor, welche bei Verabreichung an ein Tier in der Lage sind, eine Immunreaktion bei dem Tier zu verbessern. In einer Ausführungsform funktionieren die Verbindungen der Erfindung als immunologische Adjuvanzien, wenn sie zusammen mit Antigenen verabreicht werden, einschließlich Antigenen, welche als Impfstoffe für eine Erkrankung oder ein Krankheitsbild verwendet werden, die einer Impfung zugänglich sind. Die neuartigen Adjuvansverbindungen der Erfindung weisen Formel I auf:worin

R1 ausgewählt ist aus der Gruppe bestehen aus - (a) -C(O)-;
- (b) -C(O)-C1-14-Alkylen-C(O)- oder -C(O)-C1-14-Alkenylen-C(O)-, wobei das C1-14-Alkylen oder C1-14-Alkenylen wahlweise substituiert ist mit Hydroxy, C1-6-Alkoxy, C1-6-Alkylendioxy, Carboxy, C1-6-Alkoxycarbonyl, C1-6-Carbamoyl, C1-6-Acylamino, C1-6-Alkylamino oder (Aryl)C1-6-Alkyl, wobei der Arylanteil des (Aryl)C1-6-Alkyls wahlweise substituiert ist mit C1-6-Alkyl, C1-6-Alkoxy, C1-6-Alkylamino, (C1-6-Alkoxy)C1-6-Alkylamino, (C1-6-Alkylamino)C1-6-Alkoxy, -O-C1-6-Alkylen-NH-C1-6-Alkylen-O-C1-6-Alkyl, -O-C1-6-Alkylen-NH-C(O)-C1-6-Alkylen-C(O)OH oder -O-C1-6-Alkylen-NH-C(O)-C1-6-Alkylen-C(O)-C1-6-Alkyl;
- (c) C2 bis C15 Alkyl mit gerader oder verzweigter Kette, wahlweise substituiert mit Hydroxy oder Alkoxy; und
- (d) -C(O)-C6-12-Arylen-C(O)-, wobei das Arylen wahlweise substituiert ist mit C1-6-Alkyl, Hydroxy, C1-6-Alkoxy, Halogen, Nitro oder Amino;
- (a) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, welches wahlweise substituiert ist mit Halo, Oxo, Hydroxy oder Alkoxy;
- (b) C2 bis C20 Alkenyl,
Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, welches
wahlweise substituiert ist mit Halo, Oxo, Hydroxy oder Alkoxy; und worin R8 C1-6 Alkyl mit gerader oder verzweigter Kette ist oder C2-6 Alkenyl, Alkenyl oder Dialkenyl mit gerader oder verzweigter Kette; G5 ausgewählt ist aus der Gruppe bestehend aus Sauerstoff, Methylen, Arylen, -NH-, -N(C1-4-Alkyl)-, -N(C(O)-C1-4-Alkyl)-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH-, -O-C(O)-O-, -NH-C(O)-NH- und -S(O)n-, wobei n 0, 1 oder 2 ist; R9 und R10 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus: (i) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, welches wahlweise substituiert ist mit Halo, Oxo, Hydroxy oder Alkoxy; und (ii) C2 bis C20 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, welches wahlweise substituiert ist mit Halo, Oxo, Hydroxy oder Alkoxy; oder eines oder zwei von G1R2, G2R4, G3R5 und G4R7 zusammen ein Wasserstoffatom oder Hydroxyl sein können; oder ein pharmazeutisch akzeptables Salz davon.

- In einem zweiten Aspekt betrifft die vorliegende Erfindung neuartige immunologische Formulierungen, welche zumindest eine der Adjuvansverbindungen der Erfindung aufweisen.
- In einem dritten Aspekt betrifft die Erfindung neuartige immunologische Zusammensetzungen, welche ein Antigen und zumindest eine der Adjuvansverbindungen der Erfindung aufweisen.
- In einem weiteren Aspekt betrifft die vorliegende Erfindung Verfahren zur Verbesserung einer Immunreaktion bei einem Tier, welche das Verabreichen einer Verbindung der Erfindung an ein Tier umfassen.
- DETAILLIERTE BESCHREIBUNG DER BEVORZUGTEN AUSFÜHRUNGSFORMEN
- Die Erfindung sieht neuartige Verbindungen vor, welche bei Verabreichung an ein Tier in der Lage sind, eine Immunreaktion bei dem Tier zu verbessern. Bei bestimmten bevorzugten Ausführungsformen sind die Verbindungen der Erfindung in der Lage, eine immunologische Wirkung zu erzeugen, wenn sie allein verabreicht werden. Bei bestimmten anderen bevorzugten Ausführungsformen funktionieren die Verbindungen der Erfindung als immunologische Adjuvanzien, wenn sie zusammen mit Antigenen verabreicht werden, einschließlich Antigenen, welche als Impfstoffe für eine Erkrankung oder ein Krankheitsbild verwendet werden, welche einer Impfung zugänglich sind. Die Erfindung bietet außerdem immunologische Zusammensetzungen, welche die neuartigen Verbindungen der Erfindung aufweisen und Verfahren zum Immunisieren von Menschen und nichthumane Lebewesen.
- Die Patent- und Wissenschaftsliteratur, auf die hierin verwiesen wird, etabliert Erkenntnisse, welche dem Fachmann zur Verfügung stehen. Die hierin zitierten veröffentlichten Patente, Anmeldungen und Referenzen sind hiermit durch Bezugnahme aufgenommen, und zwar im gleichen Umfang, als wenn spezifisch und individuell bei jeder Erwähnung auf die Aufnahme Einschluss durch Bezugnahme hingewiesen wird. Im Falle von Unstimmigkeiten ist die vorliegende Offenbarung maßgebend.
- Für die Zwecke der vorliegenden Erfindung werden die folgenden Definitionen verwendet:
- Definitionen
- Der Begriff „immunologische Zusammensetzung" soll, wie hierin verwendet, Zusammensetzungen einschließen, welche in der Lage sind, eine Wirkung auf das Immunsystem eines Tieres zu erzeugen, einschließlich, ohne Einschränkung, eine immunprophylaktische, immuntherapeutische, immunpotenzierende oder immunsuppressive Wirkung.
- Der Begriff „Tier" betrifft, wie hierin verwendet, humane Patienten und nichthumane Lebewesen. Zu nichthumanen Lebewesen zählen Tiere, welche in der Lage sind, eine Immunreaktion auf einen Impfstoff zu erzeugen.
- Die Begriffe „Carbonyl" und „Oxo" betreffen, wie hierin verwendet, einen (C=O)-Anteil. Eine Carbonylgruppe kann auch als -C(O)- dargestellt sein.
- Der Begriff „Dicarbonyl" betrifft, wie hierin verwendet, einen Anteil mit der Struktur -C(O)-Alkylen-C(O)- oder -C(O)-Arylen-C(O)-, welcher durch die Kohlenstoffatome beider End-Carbonylanteile an ein Molekül gebunden ist.
- Ein „Alkylester" ist, wie hierin verwendet, ein Anteil mit der Struktur -O-C(O)-Alkyl, welcher durch den einfach gebundenen Sauerstoff der Estergruppe an ein Molekül gebunden ist.
- Der Begriff „Alkoxycarbonyl" betrifft einen Anteil mit der Struktur -C(O)-O-Alkyl, welcher durch das Carbonlykohlenstoffatom an ein Molekül gebunden ist.
- Ein „Alkenylester" ist, wie hierin verwendet, ein Anteil mit der Struktur -O-C(O)-Kohlenstoffkette, wobei die Kohlenstoffkette eine Kohlenstoff-zu-Kohlenstoff-Doppelbindung enthält, wobei der Esteranteil durch sein einfach gebundenes Sauerstoffatom an ein Molekül gebunden ist.
- Der Begriff „Alkylen" steht für eine bivalente Alkylkohlenwasserstoffgruppe mit gerader oder verzweigter Kette.
- Der Begriff „Alkenylen" steht für eine bivalente Kohlenwasserstoffgruppe mit gerader oder verzweigter Kette mit zumindest einer Kohlenstoff-zu-Kohlenstoff-Doppelbindung.
- Der Begriff „Dialkenylen" steht für eine bivalente ungesättigte Kohlenwasserstoffgruppe mit gerader oder verzweigter Kette mit zumindest zwei Kohlenstoff-zu-Kohlenstoff-Doppelbindungen.
- Der Begriff „Arylen" betrifft eine bivalente aromatische Gruppe.
- Wenn die Begriffe „Alkylen", „Alkenylen" oder „Dialkenylen" einen Deskriptor enthalten, welcher die Anzahl der Kohlenstoffatome oder einen Bereich für die Anzahl der Kohlenstoffatome angibt, z.B. C1-14-Alkylen, bezieht sich die Anzahl der Kohlenstoffatome auf die Länge der Kohlenstoffkette, welche die beiden chemischen Gruppen verbindet, zwischen welchen sich die Alkylengruppe befindet. Wenn der Begriff „Arylen" einen Deskriptor enthält, welcher die Anzahl der Kohlenstoffatome oder einen Bereich für die Anzahl der Kohlenstoffatome angibt, bezieht sich die Anzahl der Kohlenstoffatome auf die Anzahl der Kohlenstoffatome in dem aromatischen Ringsystem. Jedes der Kohlenstoffatome einer Alkylen-, Alkenylen-, Dialkenylen- oder Arylengruppe kann wahlweise substituiert sein, wie unten beschrieben, und die Substituenten können weitere Kohlenstoffatome enthalten.
- Der Begriff „Alkyl" betrifft, wie hierin verwendet, aliphatische Gruppen mit gerader oder verzweigter Kette mit 1 bis 20 Kohlenstoffatomen, welche wahlweise mit einem, zwei oder drei Substituenten substituiert sein können.
- Eine „Aryl"-Gruppe ist ein aromatischer C6-14-Anteil, welcher einen bis drei aromatische Ringe aufweist, welche wahlweise substituiert sein können. Bevorzugt ist die Arylgruppe eine C6-10-Arylgruppe. Der Begriff „Aryl" soll außerdem Heteroarylgruppen einschließen, welche 5 bis 14 Ringatome aufweisen, bevorzugt 5, 6, 9 oder 10 Ringatome; mit 6, 10 oder 14 gemeinsamen n Elektronen in einer zyklischen Anordnung; und zusätzlich zu den Kohlenstoffatomen zwischen einem und ungefähr drei Heteroatomen ausgewählt aus der Gruppe bestehend aus N, O und S aufweisen.
- Eine „Aralkyl"- oder „Arylalkyl"-Gruppe umfasst eine Arylgruppe, wie zuvor definiert, welche kovalent an eine Alkylgruppe gebunden ist, wobei jede unabhängig voneinander wahlweise substituiert oder nichtsubstituiert sein kann.
- Der Begriff „Halogen" oder „Halo" betrifft, wie hierin verwendet, Chlor, Brom, Fluor oder Iod.
- Der Begriff „Acylamino" betrifft eine an das Stickstoffatom gebundene Amidgruppe. Der Begriff „Carbamoyl" betrifft eine an das Carbonylkohlenstaffatom gebundene Amidgruppe. Das Stickstoffatom eines Acylamino- oder Carbamoyl-Substituenten kann auch substituiert sein.
- Falls nicht anderweitig ausdrücklich eingeschränkt soll der Begriff „Amino" NH2-, Alkylamino-, Dialkylamino-, Arylamino-, Aralkylamino- und zyklische Aminogruppen einschließen.
- Wie hierin verwendet, betrifft der Begriff „Acyl" einen Alkylcarbonyl- oder Arylcarbonyl-Substituenten.
- Die Abkürzung „Boc" betrifft, wie hierin verwendet, t-Butyloxycarbonyl.
- Der Begriff „Null", wie hierin mit Bezug auf einen bestimmten Substituenten verwendet, steht dafür, dass der Substituent abwesend ist, und die chemischen Gruppen, zwischen welchen der Substituent positioniert ist, sind mittels einer kovalenten chemischen Bindung direkt aneinander gebunden.
- Wie hierin mit Bezug auf Verbindungen und Zusammensetzungen der Erfindung verwendet, betrifft der Begriff „Typ 1" diejenigen Verbindungen der Erfindung, welche Formel I oben entsprechen, wobei die Werte von a und b gleich sind; die Werte von d und e sind gleich; die Werte von d' und e' gleich sind; X1 und Y1 gleich sind; G1 und G3 gleich sind; G2 und C4 gleich sind; R2 und R5 gleich sind; R3 und R6 gleich sind; und R4 und R7 gleich sind.
- Der Begriff „Typ 2" betrifft, wie hierin verwendet, Verbindungen oder Zusammensetzungen der Formel I, wobei eines oder mehrere der Folgenden gilt/gelten: die Werte von a und b sind unterschiedlich; die Werte von d und e sind unterschiedlich; die Werte von d' und e' sind unterschiedlich; X1 und Y1 sind unterschiedlich; G1 und G3 sind unterschiedlich; G2 und G4 sind unterschiedlich; R2 und R5 sind unterschiedlich; R3 und R6 sind unterschiedlich; und R4 und R7 sind unterschiedlich.
- Verbindungen
- In einem Aspekt sieht die vorliegende Erfindung neuartige Verbindungen mit der Formel I vor:worin

R1 ausgewählt ist aus der Gruppe bestehend aus - (a) -C(O)-;
- (b) -C(O)-C1-14-Alkylen-C(O)- oder -C(O)-C1-14-Alkenylen-C(O)-, wobei das C1-14-Alkylen oder C1-14-Alkenylen wahlweise substituiert ist mit Hydroxy, C1-6-Alkoxy, C1-6-Alkylendioxy, Carboxy, C1-6-Alkoxycarbonyl, C1-6-Carbamoyl, C1-6-Acylamino, C1-6-Alkylamino oder (Aryl)C1-6-Alkyl, wobei der Arylanteil des (Aryl)C1-6-Alkyls wahlweise substituiert ist mit C1-6-Alkyl, C1-6-Alkoxy, C1-6-Alkylamino, (C1-6-Alkoxy)C1-6-Alkylamino, (C1-6-Alkylamino)C1-6-Alkoxy, -O-C1-6-Alkylen-NH-C1-6-Alkylen-O-C1-6-Alkyl, -O-C1-6- Alkylen-NH-C(O)-C1-6-Alkylen-C(O)OH oder -O-C1-6-Alkylen-NH-C(O)-C1-6-Alkylen-C(O)-C1-6-Alkyl;
- (c) C2 bis C15 Alkyl mit gerader oder verzweigter Kette, wahlweise substituiert mit Hydroxy oder Alkoxy; und
- (d) -C(O)-C6-12-Arylen-C(O)-, wobei das Arylen wahlweise substituiert ist mit C1-6-Alkyl, Hydroxy, C1-6-Alkoxy, Halogen, Nitro oder Amino;
- (a) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, welches wahlweise substituiert ist mit Halo, Oxo, Hydroxy oder Alkoxy;
- (b) C2 bis C20 Alkenyl,
Alkenyl oder Dialkenyl mit gerader oder verzweigter Kette, welches
wahlweise substituiert ist mit Halo, Oxo, Hydroxy oder Alkoxy; und worin R8 C1-6 Alkyl mit gerader oder verzweigter Kette ist oder C2-6 Alkenyl, Alkenyl oder Dialkenyl mit gerader oder verzweigter Kette; G5 ausgewählt ist aus der Gruppe bestehend aus Sauerstoff, Methylen, Arylen, -NH-, -N(C1-4-Alkyl)-, -N(C(O)-C1-4-Alkyl)-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH-, -O-C(O)-O-, -NH-C(O)-NH- und -S(O)n-, wobei n 0, 1 oder 2 ist; R9 und R10 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus: (i) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, welches wahlweise substituiert ist mit Halo, Oxo, Hydroxy oder Alkoxy; und (ii) C2 bis C20 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, welches wahlweise substituiert ist mit Halo, Oxo, Hydroxy oder Alkoxy; oder eines oder zwei von G1R2, G2R4, G3R5 und G4R7 zusammen ein Wasserstoffatom oder Hydroxyl sein können; oder ein pharmazeutisch akzeptables Salz davon.

- Die Verbindungen der Erfindung sind sauer und sie sind üblicherweise in einer entsprechenden Salzform isoliert. Dementsprechend werden innerhalb des Umfangs der Erfindung Verbindungen mit Formel II spezifisch in Betracht gezogen:worin M ein pharmazeutisch akzeptables Kation ist und alle anderen Variablen wie zuvor für Formel I definiert sind. Bei zweiwertigen Kationen nimmt das Kation die Stelle von zwei M-Variablen in der Formel II oben ein. Pharmazeutisch akzeptable Kationen sind dem Durchschnittsfachmann gut bekannt.

- In einigen bevorzugten Ausführungsformen der Erfindung liegen eine oder mehrere der folgenden Einschränkungen vor: jedes von a und b sind 2; jedes von X1 und Y1 sind NH; jedes von d und e sind 1 oder 2; und jedes von d' und e' sind 0, 1 oder 2. In bestimmten bevorzugten Ausführungsformen sind d und e 1 und d' und e' sind 0. In bestimmten anderen bevorzugten Ausführungsformen sind jedes von d und e 1 und jedes von d' und e' sind 1 oder 2.
- In einigen bevorzugten Ausführungsformen ist R1 -C(O)- oder -C(O)-C1-14-Alkylen-C(O)-, wobei das C1-14-Alkylen wahlweise substituiert ist mit einem oder zwei Substituenten ausgewählt aus der Gruppe bestehend aus Hydroxy, C1-6-Alkoxy, C1-6-Alkylendioxy, C1-6-Alkylamino oder (Aryl)C1-6-Alkyl, wobei der Arylanteil des (Aryl)C1-6-Alkyls wahlweise substituiert ist mit C1-6-Alkyl, C1-6-Alkoxy, C1-6-Alkylamino, (C1-6-Alkoxy)C1-6-Alkylamino, (C1-6-Alkylamino)C1-6-Alkoxy, -O-C1-6-Alkylen-NH-C1-6-Alkylen-O-C1_6-Alkyl, -O-C1-6-Alkylen-NH-C(O)-C1-6-Alkylen-C(O)OH oder -O-C1-6-Alkylen-NH-C(O)-C1-6-Alkylen-C(O)-C1-6-Alkyl.
- In einigen bevorzugten Ausführungsformen sind G1, G2, G3 und G4 unabhängig voneinander ausgewählt aus der Gruppe bestehend -NH-C(O)- und -O-C(O)-.
- In einigen bevorzugten Ausführungsformen sind zumindest zwei von R2-R7, R9 und R10 C6-20 Alkyl, Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, wobei jede der Gruppen wahlweise substituiert sein kann mit einem oder zwei Substituenten ausgewählt aus der Gruppe bestehend aus Halo, Oxo, Hydroxy oder Alkoxy. In bestimmten bevorzugten Ausführungsformen sind zumindest zwei von R2-R7, R9 und R10 C8-15 Alkyl, Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, wobei jede der Gruppen wahlweise substituiert sein kann mit einem oder zwei Substituenten ausgewählt aus der Gruppe bestehend aus Halo, Oxo, Hydroxy oder Alkoxy.
- In einigen bevorzugten Ausführungsformen sind zumindest vier von R2-R7, R9 und R10 C6-20 Alkyl, Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, wobei jede der Gruppen wahlweise substituiert sein kann mit einem oder zwei Substituenten ausgewählt aus der Gruppe bestehend aus Halo, Oxo, Hydroxy oder Alkoxy. In bestimmten bevorzugten Ausführungsformen sind zumindest vier von R2-R7, R9 und R10 C8-15 Alkyl, Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, wobei jede der Gruppen wahlweise substituiert sein kann mit einem oder zwei Substituenten ausgewählt aus der Gruppe bestehend aus Halo, Oxo, Hydroxy oder Alkoxy.
- In einigen bevorzugten Ausführungsformen sind zumindest sechs von R2-R7, R9 und R10 C6-20 Alkyl, Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, wobei jede der Gruppen wahlweise substituiert sein kann mit einem oder zwei Substituenten ausgewählt aus der Gruppe bestehend aus Halo, Oxo, Hydroxy oder Alkoxy. In bestimmten bevorzugten Ausführungsformen sind zumindest sechs von R2-R7, R9 und R10 C8-15 Alkyl, Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, wobei jede der Gruppen wahlweise substituiert sein kann mit einem oder zwei Substituenten ausgewählt aus der Gruppe bestehend aus Halo, Oxo, Hydroxy oder Alkoxy.
- Synthetische Verfahren
- Verbindungen des Typs 1
- Verbindungen des Typs 1 mit der Formel I, bei denen d und e jeweils 1 ist und d' und e' jeweils 0, werden bevorzugt nach dem in Schema 1 dargestellten Syntheseweg synthetisiert. Dabei wird das Aldehyd III, welches mittels aus der Literatur bekannter Verfahren aus L-Serin gewonnen wird, mit einem organometallischen Reagens wie einem Grignard-Reagens behandelt, was den Alkohol IV ergibt. Die Säurebehandlung von IV, z.B. Behandlung mit wasserfreiem HCl-Gas in Methanol, ergibt das entsprechende Aminodiol, welches selektiv am Stickstoff acyliert wird, wodurch V entsteht. Der Schutz des primären Alkohols, gefolgt von Acylierung des sekundären Alkohols, ergibt dann die Verbindung VI. Die Entschützung des primären Alkohols und die Reaktion mit dem phosphorylierenden Reagens, hergestellt wie im Experimentabschnitt beschrieben, ergibt dann VII. Die Behandlung mit Triethylsilan und Trifluoressigsäure bewirkt die Entfernung der Boc-Schutzgruppe, und die Behandlung mit Phosgen ergibt dann das symmetrische Dimer VIII. Abschließend bewirkt die Behandlung mit Phenylsilan und Tetrakis(triphenylphosphin)palladium(0) die Entfernung der Allyl-Schutzgruppe, wodurch IX entsteht.
- Schema 1

- Alternativ kann eine Zwischenstufe IV als ein einzelnes Diastereomer hergestellt werden, und zwar bevorzugt nach dem in Schema 2 dargestellten Syntheseweg.
- Schema 2

- Hier wird das Aldehyd III mit Allylchlorid und (+)-B-Methoxydiisopinocampheylboran behandelt, was X als ein einzelnes Diastereomer ergibt. Die Schließung des Epoxidrings wird durch die Behandlung mit DBU bewirkt, gefolgt von der SN2'-Ringöffnung mit einem Organocuprat-Reagens, wodurch XII entsteht. Die Hydrierung ergibt dann den Alkohol XIII, welcher wie zuvor für IV beschrieben in IX umgewandelt wird.
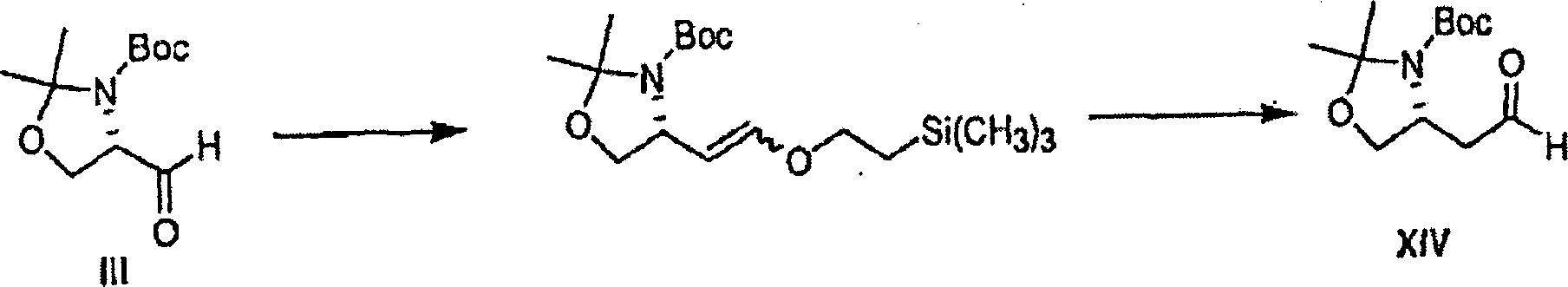
- Verbindungen des Typs 1 mit der Formel I, bei denen d und e jeweils 1 ist und d' und e' jeweils 1, werden bevorzugt durch Verfahren analog zu den in den Schemata 1 und 2 oben beschriebenen hergestellt, jedoch wird anstelle von III mit dem Aldehyd XIV begonnen. Das Aldehyd XIV wird bevorzugt durch Homologierung von III hergestellt, wie in Schema 3 dargestellt. Hier wird das Aldehyd III mit [2-(Trimethylsilyl)ethoxymethyl]triphenylphosphoniumchlorid und n-Butyllithium behandelt, was den Enolether ergibt, welcher mit Essigsäure behandelt wird, um XIV zu erzeugen.
- Schema 3
- Verbindungen des Typs 1 mit der Formel I mit unterschiedlichen Werten für d und e und/oder d' und e' werden bevorzugt durch Verfahren hergestellt, welche im Allgemeinen analog zu den zuvor beschriebenen sind, wie in den Beispielen weiter dargelegt ist. Der Fachmann wird auch leicht erkennen, dass die zuvor geschilderten synthetischen Verfahren für die Synthese von Verbindungen mit der Formel I angepasst werden können, bei denen G3 und/oder G4 nicht -O-C(O)- oder -NH-C(O)- sind, indem anstelle des zuvor beschriebenen Acylierungsschrittes alternative Standardfunktionsgruppenumwandlungen durchgeführt werden.
- Verbindungen des Typs 2
- Verbindungen des Typs 2 mit der Formel I werden bevorzugt durch Verfahren analog zu den für die Synthese der Verbindungen des Typs 1 beschriebenen synthetisiert, bis zu der Stelle kurz nach der Abspaltung der Schutzgruppe von der primären Amingruppe der Phosphatesterverbindung. An dieser Stelle wird die Phosphatesterverbindung mit einer einfach geschützten difunktionalisierten Verbindung wie Dicarbonsäure behandelt. Die entstehende Verbindung ist entschützt und wird mit einer zweiten Phosphatesterverbindung reagieren gelassen. Die abschließende Phosphatentschützung wird dann wie für Verbindungen des Typs 1 beschrieben erreicht.
- Ein alternatives Verfahren für die Synthese der Verbindungen des Typs 2 ist die Bildung der Isocyanat-Zwischenstufe der entschützten primären Amingruppe der Phosphatesterverbindung. Die Isocyanat-Zwischenstufe wird dann mit einem zweiten entschützten, primären Amin einer Phosphatesterverbindung behandelt, gefolgt von der Entschützung wie für die Verbindungen des Typs 1 beschrieben.
- Adjuvans- und Impfstoff-Formulierungen und -Verabreichung
- In einem zweiten Aspekt bietet die Erfindung immunologische Zusammensetzungen, welche eine Verbindung mit der Formel I aufweisen:worin

R1 ausgewählt ist aus der Gruppe bestehend aus - (a) -C(O)-;
- (b) -C(O)-C1-14-Alkylen-C(O)- oder -C(O)-C1-14-Alkenylen-C(O)-, wobei das C1-14-Alkylen oder C1-14-Alkenylen wahlweise substituiert ist mit Hydroxy, C1-6-Alkoxy, C1-6-Alkylendioxy, Carboxy, C1-6-Alkoxycarbonyl, C1-6-Carbamoyl, C1-6-Acylamino, C1-6-Alkylamino oder (Aryl)C1-6-Alkyl, wobei der Arylanteil des (Aryl)C1-6-Alkyls wahlweise substituiert ist mit C1-6-Alkyl, C1-6-Alkoxy, C1-6-Alkylamino, (C1-6-Alkoxy)C1-6-Alkylamino, (C1-6-Alkylamino)C1-6-Alkoxy, -O-C1-6-Alkylen-NH-C1-6-Alkylen-O-C1-6-Alkyl, -O-C1-6-Alkylen-NH-C(O)-C1-6-Alkylen-C(O)OH oder -O-C1-6-Alkylen-NH-C(O)-C1-6-Alkylen-C(O)-C1-6-Alkyl;
- (c) C2 bis C15 Alkyl mit gerader oder verzweigter Kette, wahlweise substituiert mit Hydroxy oder Alkoxy; und
- (d) -C(O)-C6-12-Arylen-C(O)-, wobei das Arylen wahlweise substituiert ist mit C1-6-Alkyl, Hydroxy, C1-6-Alkoxy, Halogen, Nitro oder Amino;
- (a) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, welches wahlweise substituiert ist mit Halo, Oxo, Hydroxy oder Alkoxy;
- (b) C2 bis C20 Alkenyl,
Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, welches
wahlweise substituiert ist mit Halo, Oxo, Hydroxy oder Alkoxy; und worin R8 C1-6 Alkyl mit gerader oder verzweigter Kette ist oder C2-6 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette; G5 ausgewählt ist aus der Gruppe bestehend aus Sauerstoff, Methylen, Arylen, -NH-, -N(C1-4-Alkyl)-, -N(C(O)-C1-4-Alkyl)-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH-, -O-C(O)-O-, -NH-C(O)-NH- und -S(O)n-, wobei n 0, 1 oder 2 ist; R9 und R10 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus: (i) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, welches wahlweise substituiert ist mit Halo, Oxo, Hydroxy oder Alkoxy; und (ii) C2 bis C20 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, welches wahlweise substituiert ist mit Halo, Oxo, Hydroxy oder Alkoxy; oder eines oder zwei von G1R2, G2R4, G3R5 und G4R7 zusammen ein Wasserstoffatom oder Hydroxyl sein können; oder ein pharmazeutisch akzeptables Salz davon; und ein pharmazeutisch akzeptabler/s Träger, Verdünnungsmittel oder Arzneimittelträger enthalten.

- Bevorzugte Ausführungsformen nach diesem Aspekt der Erfindung sind wie die zuvor für den ersten Aspekt beschriebenen.
- Die Erfindung betrifft auch neuartige immunologische Zusammensetzungen, welche eine Verbindung mit der Formel I, wie zuvor beschrieben, ein Antigen und einen pharmazeutisch akzeptablen/s Träger, Verdünnungsmittel oder Arzneimittelträger enthalten.
- Die immunologische Zusammensetzung kann jede geeignete Antigen- oder Impfstoff-Komponente in Kombination mit einer Adjuvansverbindung der Erfindung verwenden. Als ein weiteres Beispiel können solche immunologischen Zusammensetzungen entsprechend einen abgeschwächten Organismus aufweisen, oder eine Komponente eines solchen Organismus, wie ein Kohlenhydrat, Protein oder Peptid, oder eine Nukleinsäure, welche eine solche Komponente codiert.
- Normalerweise wird ein Antigen gemischt mit der Adjuvansverbindung der Erfindung eingesetzt. In bestimmten anderen Ausführungsformen kann es bei einigen Anwendungen von Nutzen sein, ein Antigen kovalent verbunden mit einem Amino-, Carboxyl-, Hydroxyl- und/oder Phosphatanteil der Adjuvansverbindungen der Erfindung einzusetzen. Die spezifische Formulierung therapeutisch effektiver Zusammensetzungen der vorliegenden Erfindung kann so in einer geeigneten Art und Weise ausgeführt werden, wodurch das Adjuvans biologische verfügbar und sicher gemacht wird, und wirksam bei dem Patienten ist, dem die Formulierung verabreicht wird.
- Solche immunologischen Zusammensetzungen können zum Beispiel zumindest ein Antigenagens aufweisen, welches als ein Impfstoff für eine Erkrankung oder ein Krankheitsbild verwendet wird, gegen welchels eine Impfung vorgenommen werden kann, einschließlich, jedoch nicht darauf beschränkt:
- (A) Infektionskrankheiten bei Mensch und Tier, einschließlich solchen, die Bakterien, Viren, Parasiten (z.B. Mycoplasmen, Pilze, Protozoen) und Prione verursacht sind;
- (B) Erkrankungen oder Pathologien, bei denen eine Immunreaktion gegen ein autologes Molekül vorteilhaft sein kann wie, jedoch nicht darauf beschränkt, Alzheimer-Krankheit, bei welchen die Immunisierung gegen Amyloid β42 vorteilhaft sein kann; Magen-Reflux-Krankheit, bei der die Immunisierung gegen Gastrin vorteilhaft sein kann; Krebs, einschließlich, ohne Einschränkung, Melanom, Prostata- und Kolonkrebs, bei welchen die Immunisierung gegen Krebsantigene vorteilhaft sein kann; sowie Autoimmunstörungen, einschließlich, ohne Einschränkung, Diabetes, bei der die Immunisierung gegen Insulin die Entzündungsreaktionen gegen Insulin-produzierende Zellen verringert oder umgekehrt werden kann; und
- (C) nichtpathologische Situationen, in welchen eine Immunreaktion eine gewünschte Veränderung in der Funktion oder Physiologie veranlassen kann, wie z.B., jedoch nicht darauf beschränkt, die Verhütungswirkung induziert durch die Immunisierung gegen hCG.
- Als weitere Beispiele können die immunologischen Zusammensetzungen der Erfindung Antigen- oder Impfstoffkomponenten aufweisen, welche pharmakologisch aktiv für Erkrankungen und Krankheitsbilder sind wie Pocken, Gelbfieber, Staupe, Cholera, Geflügelpocken, Scharlach, Diphtherie, Tetanus, Keuchhusten, Influenza, Tollwut, Mumps, HIV, Windpocken, Röteln, Masern, Maul- und Klauenseuche und Poliomyelitis.
- In der resultierenden Impfstoffformulierung, welche (i) ein Antigen und (ii) zumindest eine Adjuvansverbindung der Erfindung aufweist, sind das Antigen und die Adjuvansverbindung jeweils in einer Menge vorhanden, welche wirksam ist, um eine Immunreaktion auszulösen, wenn die Formulierung einem Wirtstier verabreicht wird oder ein Embryo oder eine Eizelle damit geimpft wird.
- In weiteren Ausführungsformen können die Verbindungen der Erfindung kovalent an Impfstoffantigene gebunden sein, zum Beispiel durch einen Amino-, Thiol-, Carboxyl-, Hydroxyl- oder Phosphatanteil. Die Verfahren der Anbindung der Adjuvanszusammensetzungen der Erfindung an die Impfstoffantigene sind für den Durchschnittsfachmann im Hinblick auf diese Offenbarung verständlich. Die Adjuvanszusammensetzungen können durch jedes der in P. Hoffman et al., Biol. Chem. Hoppe-Sayler, 1989, 370:575-582; K.-H. Wiesmüller et al., Vaccine, 1989, 7:29-33; K.-H. Wiesmüller et al., Int. J. Peptide Protein Res., 1992, 40:255-260; J.-P. Defourt et al., Proc. Natl. Acad. Sci., 1992, 89:3879-3883; T. Tohokuni et al., J. Am. Chem. Soc., 1994, 116:395-396; F. Reichel, Chem. Commun., 1997, 2087-2088; H. Kamitakahara, Angew. Chem. Int. Ed., 1998, 37:1524-1528; W. Dullenkopf et al., Chem. Eur. J., 1999, 5:2432-2438, welche alle durch, Bezugnahme hierin eingefügt sind, beschriebenen Verfahren an Impfstoffe gebunden werden.
- Die entstehenden Impfstoffformulierungen, einschließlich (i) eines Antigens und (ii) einer Adjuvansverbindung werden nutzbringend eingesetzt, um eine Immunreaktion bei einem Tier zu erzeugen, indem einem solchen Tier die Impfstoffformulierung verabreicht wird, und zwar in einer Menge, die ausreichend ist, um eine Antikörperreaktion bei einem solchen Tier zu erzeugen.
- Die Verabreichungsmodi können die Verwendung jedes geeigneten Mittels und/oder Verfahrens einschließen, das geeignet ist zur Abgabe des Adjuvans, des adjuvanshaltigen Impfstoffes oder zur Abgabe des Adjuvans und/oder Antigens an einer oder mehreren Körperstellen des Wirtstieres, an denen das Adjuvans und die zugehörigen Antigene immunstimulatorisch wirksam sind. Zu den Abgabemodi zählen, ohne Einschränkung, parenterale Verabreichungsverfahren wie subkutane (SC) Injektion, transkutane, intranasale (IN), opthalmische, transdermale, intramuskuläre (IM), intradermale (ID), intraperitoneale (IP), intravaginale, pulmonale und rektale Verabreichung sowie nichtparenterale, z.B. orale Verabreichung.
- Die Dosisrate und geeignete Dosierungsformen für die Adjuvans- und Impfstoffzusammensetzungen der vorliegenden Erfindung können durch den Durchschnittsfachmann ohne übermäßige Experimente, leicht bestimmt werden durch Verwendung herkömmlicher Antikörpertiter-Bestimmungsverfahren und herkömmlicher Bioeffizienz-/Biokompatibilitätsprotokolle und in Abhängigkeit von dem bestimmten, mit dem Adjuvans eingesetzten Antigen oder therapeutischen Wirkstoff, der gewünschten therapeutischen Wirkung und der gewünschten Zeitspanne der Bioaktivität.
- Das Adjuvans der vorliegenden Erfindung kann dem Wirtstier mit sämtlichen anderen geeigneten pharmakologisch oder physiologisch aktiven Wirkstoffen, z.B. antigenen und/oder anderen biologisch aktiven Substanzen nutzbringend verabreicht werden.
- Formulierungen der Erfindung können zusätzliche Komponenten enthalten wie Salzlösung, Öl, Squalen, Öl-Wasser-Dispersionen, Liposome und weitere Adjuvanzien wie QS-21, Muramylpeptide, Freund's inkomplettes Adjuvans und ähnliche.
- Die vorliegende Erfindung wird nun durch die folgenden Beispiele erläutert, welche in keiner Weise als Einschränkung zu sehen sind.
- SYNTHETISCHE BEISPIELE
- Alle Reaktionsprodukte lieferten zufriedenstellende NMR-Spektren und Kieselgel-Dünnschicht-Chromatographie (TLC). Alle Chromatographien wurden auf Kieselgel durchgeführt und die Elution durch TLC überwacht. Alle abgeschlossenen Reaktionen wurden durch tlc-Analyse bestimmt. Alle Reaktionen erfolgten unter einer Stickstoffatmosphäre bei Raumtemperatur, falls nicht anders angegeben. Alle Reaktionslösemittel waren wasserfrei, wenn nicht anders vermerkt. Die übliche Aufarbeitung umschließt wässrige Waschungen, Extraktion organischer Lösemittel, Trocknung über wasserfreiem Natriumsulfat und Entfernung des Lösemittels unter verringertem Druck.
- Beispiel 1: Herstellung von ER-805028

- Zu einer gerührten Lösung aus Oxalylchlorid (0,65 ml) in Methylenchlorid (50 ml) wurde bei –78°C tropfenweise DMSO (1,1 ml) zugegeben, gefolgt von Rühren für 30 Minuten. Der chirale Alkohol 1 (908 mg – aus L-Serin über aus der Literatur bekannte Verfahren) in Methylenchlorid (5 ml) wurde tropfenweise zugegeben und dann für eine Stunde zwischen –40°C und –60°C gerührt; nach dieser Zeit wurde tropfenweise Triethylamin (3,48 ml) zugegeben und die Reaktion wurde über einen Zeitraum von einer Stunde auf Raumtemperatur erwärmen gelassen. Das nach der Aufarbeitung erhaltene Rohaldehyd 2 wurde in THF (20 ml) aufgelöst und bei –20°C tropfenweise zu dem zuvor erzeugten Undecanylmagnesiumbromid 3 (erzeugt aus Magnesium-Drehspänen (132 mg) und Undecanylbromid (1.054 g) in THF (4 ml) bei 50°C für 3 Stunden mit einem Kristall aus Iod) zugegeben. Die Grignard-Reaktion wurde auf Raumtemperatur erwärmt und für 2 Stunden gerührt. Die übliche Aufarbeitung und Kieselgelchromatographie-Reinigung ergaben 4 (1,42 g)

- Zu einer gerührten Lösung aus 4 (1,42 g) in Methanol (100 ml) wurde bei 0°C für 10 Minuten wasserfreies HCl-Gas zugegeben; wonach die Reaktionsmischung auf Raumtemperatur erwärmt wurde. Nachdem die Mischung konzentriert wurde bis sie trocken war, wurde das Rohprodukt mit 1 N Natriumhydroxid basisch gemacht und mit Ethylacetat extrahiert, was das Rohmaterial 5 (509 mg) ergab. 5 (509 mg) wurde in THF (4 ml) aufgelöst und gesättigtes Natriumbicarbonat (8 ml) wurde zugegeben, gefolgt von tropfenweiser Zugabe von Tetradecanoylchlorid (0,57 ml) bei 0°C, gefolgt von kräftigem Rühren für 1 Stunde. Normale Aufarbeitung und Kieselgelreinigung ergaben 6 (466 mg).
-

- Zu einer gerührten Lösung aus 6 (466 mg) in Methylenchlorid (10 ml) wurde Imidazol (105 mg) zugegeben, gefolgt von tert-Butyldiphenylsilylchlorid (302 mg). Nach Rühren bei Raumtemperatur für 48 Stunden ergab die normale Aufarbeitung gefolgt von Kieselgelreinigung Isomer 7 (293 mg) getrennt von Isomer 8 (235 mg). Hinweis: Die Stereochemie der beiden Produkte wurde durch direkten Vergleich (1H-NMR und tlc) mit dem aus 17 (Herstellung unten beschrieben) erzeugten chiralen Isomer bestimmt.
-

- Zu einer gerührten Lösung aus 8 (154 mg) in Methylenchlorid (0,5 ml) wurden Laurinsäure (59 mg), 1-[3-(Dimethylamino)propyl]-3-ethylcarbodiimidhydrochlorid (EDC, 70 mg) und N,N-4-Dimethylaminopyridin (DMAP, 3 mg) zugegeben. Nach Rühren für 16 Stunden ergaben wässrige Aufarbeitung und Kieselgelreinigung 9 (197 mg).
-

- Zu einer gerührten Lösung aus 9 (197 mg) in THF (1,0 ml) wurde Essigsäure (20 mg) zugegeben, gefolgt von Tetra-n-butylammoniumfluorid (88 mg) bei Raumtemperatur. Nach Rühren für 16 Stunden wurde die Reaktionsmischung gequencht und mittels Kieselgelchromatographie gereinigt, was 10 (134 mg) ergab.
-

- Zu einer gerührten Lösung aus 10 (134 mg) in Methylenchlorid (0,8 ml) wurde Tetrazol (45 mg) zugegeben, gefolgt von dem phosphorylierenden Reagens (i) (106 mg) bei 0°C. Nach Rühren für 1 Stunde wurde die Reaktionsmischung auf eine gerührte Suspension gegossen, welche Oxon (204 mg) in THF (1 ml) und Wasser (1 ml) enthielt. Nach einer weiteren Stunde Rühren bei 0°C wurde die Mischung aufgearbeitet und über Kieselgel gereinigt, was 11 (140 mg) ergab.
-

- Zum Herstellen des phosphorylierenden Reagens (i) wurde zu einer Lösung aus destilliertem Diisopropylamin (9,0 ml) in Methylenchlorid Tetrazol (4,51 g) bei Raumtemperatur zugegeben, gefolgt von Rühren für 1,5 Stunden. Allylphosphorodiamidit (20,5 ml) wurde tropfenweise mit einer Geschwindigkeit von 6,5 ml/Stunde zugegeben, gefolgt von Rühren für weitere 3 Stunden. N-Boc-2-Aminoethanol (10,36 g) in Methylenchlorid (50 ml) wurde tropfenweise mit einer Geschwindigkeit von 8,4 ml/Stunde zu der obigen Reaktionsmischung zugegeben, gefolgt von Rühren für weitere 18 Stunden. Die weiße Suspension wurde durch Celit 545 gefiltert, mit zwei 20 ml Waschungen mit Methylenchlorid. Das Filtrat wurde konzentriert, gefolgt durch die Suspension und Filterung des Restes mit Hexanen (200 ml). Das entstehende Hexanfiltrat wurde konzentriert bis es trocken war und mit zwei 10 ml Portionen Toluen in eine azeotrope Form gebracht, was das Rohprodukt (i) (21,54 g) als ein Öl ergab.
-

- Zu einer gerührten Lösung aus 11 (58 mg) in Methylenchlorid (0,16 ml) wurde bei Raumtemperatur Triethylsilan (0,16 ml) zugegeben, gefolgt von Trifluoressigsäure (1,0 ml). Nach Rühren für 2 Stunden wurden die Lösemittel konzentriert und zum Trocknen mittels Toluen in eine azeotrope Form gebracht. Das Rohamin 12 wurde in Methylenchlorid (0,6 ml) mit gesättigtem Natriumbicarbonat (0,6 ml) aufgelöst, gefolgt von tropfenweiser Zugabe von Phosgen (0,018 ml einer 1,93 M Lösung in Toluen) bei 0°C. Nach Rühren bei Raumtemperatur für 2 Stunden wurde die Reaktion aufgearbeitet und durch Kieselgel in der üblichen Art und Weise gereinigt, was 13 (55 mg) ergab.
-

- Zu einer gerührten Lösung aus 13 (55 mg) in entgastem Chloroform (3 ml) wurden bei 0°C Phenylsilan (56 mg) und Tetrakis(triphenylphosphin)palladium(0) (54 mg) zugegeben. Nach Rühren für 1 Stunde bei Raumtemperatur wurde die Reaktionsmischung mit einem 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (5 ml) verdünnt und für weitere 30 Minuten gerührt. Die Mischung wurde über DEAE-Cellulose (20 ml) gegossen und mit einer ansteigenden Konzentration von Ammoniumacetat (0,0 – 0,05 M) in einem 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (100 ml) eluiert. Die HPLC-Reinigung (Kieselgel mit Hexan:Isopropanol:Wasser-Gradientenelution) ergab das gewünschte Produkt ER-805028 (2,2 mg).
- Beispiel 2: Herstellung von 8 durch ein chiral-spezifisches Verfahren

- Zu einer gerührten Lösung aus Dicyclohexylamin (634 mg) in THF (5 ml) wurde bei 0°C n-Butyllithium (1,6 M – 2,19 ml) zugegeben. Nach Rühren bei Raumtemperatur für 1 Stunde wurde die Reaktionsmischung tropfenweise zu einer gerührten Lösung aus Allylchlorid (270 mg) und (+)-B-Methoxydiisopinocampheylboran (824 mg) in Ethylether (10 ml) gekühlt auf –95°C zugegeben. Die Mischung wurde für 1 Stunde bei –95°C gerührt; dann wurde Bortrifluoridetherat (827 mg) zugegeben. Nach weiteren 20 Minuten Rühren bei 95°C wurde der Aldehyd 2 (570 mg) in Ethylether (2 ml) zugegeben). Die abschließende Reaktionsmischung wurde langsam auf Raumtemperatur erwärmen gelassen, für 12 Stunden gerührt und mittels Wasserstoffperoxid (9 ml) und Natriumbicarbonat (0,9 g) in einer Lösung aus Methanol (4,5 ml) und gesättigtem Natriumbicarbonat (2,5 ml) oxidativ gequencht. 14 (1,30 g) erhielt man nach der üblichen Aufarbeitung und Kieselgelchromatographie.
-

- Zu einer gerührten Lösung aus 14 (1,3 g) wurde bei 0°C 1,8-Diazabicyclo[5.4.0]undec-7-en (1,77 g) in Methylenchlorid (46 ml) zugegeben. Nach Rühren der Reaktionsmischung bei Raumtemperatur für 72 Stunden ergab eine normale Aufarbeitung mit Kieselgelreinigung 15 (300 mg).
-

- Zu einer gerührten Suspension aus Kupfer-(I)-Cyanid (105 mg) in THF (30 ml) wurde bei –78°C tropfenweise Octylmagnesiumbromid (2,0 M – 7,4 ml) zugegeben, gefolgt von Rühren für 15 Minuten und Raumtemperatur für 2 Stunden. Nachdem die Reaktionsmischung wieder auf –78°C abgekühlt wurde, wurde tropfenweise 15 (198 mg) in THF (2 ml) zugegeben, bei –78°C für 1 Stunde gerührt, gefolgt von Rühren bei 0°C für 1 Stunde. Die normale Aufarbeitung gefolgt von Kieselgelreinigung ergab 16 (168 mg).
-

- Zu einer gerührten Lösung aus 16 (30 mg) in Ethylacetat (50 ml) wurde 10 % Palladium/Kohlenstoff (10 mg) zugegeben; danach wurde die Mischung unter eine Wasserstoffatmosphäre (60 psi) gestellt und bei Raumtemperatur für 16 Stunden geschüttelt. Die Filtrierung gefolgt von Konzentrierung und Kieselgelchromatographie ergab 17 (30 mg).
- Die Stereochemie von 7 und 8 wurde definitiv nach der Umwandlung von 17 in 8 etabliert, und zwar mittels der zuvor ab Verbindung 4 dargelegten Schritte.
- Beispiel 3: Herstellung von ER-804874

- Zu einer gerührten Lösung aus 7 (196 mg) in Methylenchlorid (0,5 ml) wurden Laurinsäure (73,5 mg) und EDC (87 mg), gefolgt von DMAP (3,5 mg) zugegeben. 18 (242 mg) erhielt man nach Rühren bei Raumtemperatur für 16 Stunden, Aufarbeitung und Kieselgelreinigung.
-

- Zu einer gerührten Lösung aus 18 (242 mg) in THF (1,0 ml) wurde Essigsäure (25 mg) zugegeben, gefolgt von Tetra-n-butylammoniumfluorid (109 mg) bei Raumtemperatur. Nach Rühren für 16 Stunden wurde die Reaktionsmischung gequencht und mittels Kieselgelchromatographie gereinigt, was 19 (144 mg) ergab.
-

- Zu einer gerührten Lösung aus 19 (108 mg) in Methylenchlorid (0,8 ml) wurde Tetrazol (36 mg) zugegeben, gefolgt von dem phosphorylierenden Reagens (36 mg) bei 0°C. Nach Rühren für 1 Stunde wurde die Reaktionsmischung auf eine gerührte Suspension gegossen, welche Oxon (204 mg) in THF (1 ml) und Wasser (1 ml) enthielt. Nach einer weiteren Stunde Rühren bei 0°C wurde die Mischung aufgearbeitet und über Kieselgel gereinigt, was 20 (128 mg) ergab.
-

- Zu einer gerührten Lösung aus 20 (40 mg) in Methylenchlorid (0,16 ml) wurde Triethylsilan (0,11 ml) zugegeben, gefolgt von Trifluoressigsäure (1,0 ml) bei Raumtemperatur. Nach Rühren für 2 Stunden wurden die Lösemittel konzentriert und zum Trocknen mittels Toluen in eine azeotrope Form gebracht. Das Rohamin 21 wurde in Methylenchlorid (0,5 ml) mit gesättigtem Natriumbicarbonat (0,5 ml) aufgelöst, gefolgt von tropfenweiser Zugabe von Phosgen (0,012 ml einer 1,93 M Lösung in Toluen) bei 0°C. Nach Rühren bei Raumtemperatur für 2 Stunden wurde die Reaktion aufgearbeitet und durch Kieselgel in der üblichen Art und Weise gereinigt, was 22 (31 mg) ergab.
-

- Zu einer gerührten Lösung aus 22 (31 mg) in entgastem Chloroform (2 ml) wurden bei 0°C Phenylsilan (12 μl) und Tetrakis(triphenylphosphin)palladium(0) (32 mg) zugegeben. Nach Rühren für 1 Stunde bei Raumtemperatur wurde die Reaktionsmischung mit einem 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (5 ml) verdünnt und für weitere 30 Minuten gerührt. Die Mischung wurde über DEAE-Cellulose (20 ml) gegossen und mit einer ansteigenden Konzentration von Ammoniumacetat (0,0 – 0,05 M) in einem 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (100 ml) eluiert. Die HPLC-Reinigung (Kieselgel mit Hexan:Isopropanol:Wasser-Gradientenelution) ergab das gewünschte Produkt ER-804874 (6,0 mg).
- Beispiel 4: Herstellung von ER-804666

- Zu einer gerührten Suspension aus [2-(Trimethylsilyl)ethoxymethyl]triphenylphosphoniumchlorid (1,54 g) in THF (10 ml) wurde bei 0°C n-Butyllithium (1,6 M – 2,25 ml) zugegeben, gefolgt von tropfenweise 2 (649 mg) in THF (5 ml). 23 (371 mg) erhielt man nach Rühren für eine weitere Stunde bei Raumtemperatur, Aufarbeitung und Kieselgelreinigung.
-

- 23 (400 mg) wurde in einer Mischung aus Essigsäure (5 ml) und Wasser (1 ml) aufgelöst und bei Raumtemperatur für 4 Stunden gerührt. 24 (112 mg) erhielt man durch Konzentrierung und Kieselgelreinigung.
-

- Zu einer gerührten Lösung aus 24 (112 mg) in THF (2 ml) wurde bei 0°C tropfenweise n-Decylmagnesiumbromid (1,0 M in THF – 0,6 ml) zugegeben. 25 (148 mg) erhielt man nach Rühren bei Raumtemperatur für 2 Stunden, Aufarbeitung und Kieselgelreinigung.
-

- Zu einer gerührten Lösung aus 25 (148 mg) in Methanol (10 ml) wurde bei 6°C für 15 Minuten wasserfreies Wasserstoffchloridgas zugegeben. Die Mischung wurde für 16 Stunden bei Raumtemperatur gerührt; wonach die Konzentrierung bis zur Trockenheit das rohe 26 (124 mg) ergab. 26 (124 mg) wurde bei 0°C in THF (1 ml) aufgelöst, gefolgt von gesättigtem Natriumbicarbonat (2 ml) und Tetradecanoylchlorid (99 mg). 27 (69 mg) erhielt man nach Rühren bei Raumtemperatur für 1 Stunde, gefolgt von Aufarbeitung und Kieselgelreinigung.
-

- Zu einer gerührten Lösung aus 27 (69 mg) in Methylenchlorid (10 ml) wurde Imidazol (15,4 mg) zugegeben, gefolgt von tert-Butyldiphenylsilylchlorid (39 μl). Nach Rühren bei Raumtemperatur für 48 Stunden ergab die normale Aufarbeitung gefolgt von Kieselgelreinigung 28 (95 mg).
-

- Zu einer gerührten Lösung aus 28 (95 mg) in Methylenchlorid (0,5 ml) wurden Pentadecansäure (41 mg), EDC (40 mg) und DMAP (2 mg) zugegeben. Nach Rühren für 16 Stunden ergab eine wässrige Aufarbeitung und Kieselgelreinigung 29 (121 mg).
-

- Zu einer gerührten Lösung aus 29 (121 mg) in THF (1,0 ml) wurde Essigsäure (18 mg), gefolgt von Tetra-n-butylammoniumfluorid (73 mg) bei Raumtemperatur zugegeben. Nach Rühren für 16 Stunden wurde die Reaktionsmischung gequencht und mittels Kieselgelchromatographie gereinigt, was 30 (81 mg) ergab.
-

- Zu einer gerührten Lösung aus 30 (134 mg) in Methylenchlorid (0,6 ml) wurde Tetrazol (25 ml) zugegeben, gefolgt von dem phosphorylierenden Reagens (60 mg) bei 0°C. Nach Rühren für 1 Stunde wurde die Reaktionsmischung auf eine gerührte Suspension gegossen, welche Oxon (148 mg) in THF (1 ml) und Wasser (1 ml) enthielt. Nach einer weiteren Stunde Rühren bei 0°C wurde die Mischung aufgearbeitet und über Kieselgel gereinigt, was 31 (100 mg) ergab.
-

- Zu einer gerührten Lösung aus 31 (100 mg) in Methylenchlorid (0,10 ml) wurde Triethylsilan (0,10 ml), gefolgt von Trifluoressigsäure (1,0 ml) bei Raumtemperatur zugegeben. Nach Rühren für 2 Stunden wurden die Lösemittel konzentriert und zum Trocknen mittels Toluen in eine azeotrope Form gebracht. Das Rohamin 32 wurde in Methylenchlorid (0,5 ml) mit gesättigtem Natriumbicarbonat (0,5 ml) aufgelöst, gefolgt von tropfenweiser Zugabe von Phosgen (0,013 ml einer 1,93 M Lösung in Toluen) bei 0°C. Nach Rühren bei Raumtemperatur für 2 Stunden wurde die Reaktion aufgearbeitet und durch Kieselgel in der üblichen Art und Weise gereinigt, was 33 (46 mg) ergab.
-

- Zu einer gerührten Lösung aus 33 (46 mg) in entgastem Chloroform (2 ml) wurden bei 0°C Phenylsilan (16 μl) und Tetrakis(triphenylphosphin)palladium(0) (42 mg) zugegeben. Nach Rühren für 1 Stunde bei Raumtemperatur wurde die Reaktionsmischung mit einem 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (5 ml) verdünnt und für weitere 30 Minuten gerührt. Die Mischung wurde über DEAE-Cellulose (20 ml) gegossen und mit einer ansteigenden Konzentration von Ammoniumacetat (0,0 – 0,05 M) im 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (100 ml) eluiert. Die HPLC-Reinigung (Kieselgel mit Hexan:Isopropanol:Wasser-Gradientenelution) ergab das gewünschte Produkt ER-804666 (21,2 mg).
- Beispiel 5: Herstellung von ER-805274

- Zu einer Lösung von handelsüblichem, chiralem 34 (2,69 g) in Dimethylformamid (DMF – 5 ml) wurden 4-Methoxybenzaldehyddimethylacetal (6,19 g) und p-Toluensulfonsäure (0,65 g) zugegeben, gefolgt von Rühren für 16 Stunden bei Raumtemperatur. 35 (3,39 g) erhielt man nach Hochvakuumkonzentration, Aufarbeitung und Kieselgelreinigung.
-

- Zu einer gerührten Lösung aus 35 (1,78) in Methylenchlorid (25 ml) wurde Imidazol (0,81 g) zugegeben, gefolgt von tert-Butyldiphenylsilylchlorid (2,41 g). Nach Rühren bei Raumtemperatur für 16 Stunden ergab die normale Aufarbeitung gefolgt von Kieselgelreinigung 36 (3,55 g).
-

- Zu einer gerührten Lösung aus 36 (3,55 g) in Methylenchlorid (35 ml) wurde bei –78°C tropfenweise Diisobutylaluminiumhydrid (DIBAL – 31 ml einer 1,0 M Lösung in Hexanen) zugegeben. Nach Rühren für weitere 2 Stunden bei –78°C wurde die Reaktionsmischung zuerst mit Methanol:Wasser gequencht, gefolgt von Rochellesalzen. 37 (725 mg) erhielt man nach Aufarbeitung und Kieselgelchromatographie.
-

- Zu einer gerührten Lösung aus Oxalylchlorid (0,11 ml) in Methylenchlorid (8 ml) wurde bei –78°C tropfenweise DMSO (0,17 ml) zugegeben, gefolgt von Rühren für 30 Minuten. 37 (378 mg) in Methylenchlorid (2 ml) wurde tropfenweise zugegeben und dann zwischen –40°C und –60°C für eine Stunde gerührt; wonach wurde tropfenweise Triethylamin (0,57 ml) zugegeben und die Reaktion über einen Zeitraum von einer Stunde auf Raumtemperatur erwärmen gelassen wurde. Das Rohaldehyd 38 (332 mg) erhielt man nach der Aufarbeitung.
-

- Zu einer gerührten Lösung aus 38 (332 mg) in THF (5 ml) wurde bei 0°C tropfenweise Decanylmagnesiumbromid (1,0 M in Ethylether – 1,44 ml) zugegeben. Nach Rühren bei Raumtemperatur für 16 Stunden wurde Decanylmagnesiumbromid (1,0 M in Ethylether – 0,5 ml) zugegeben und die Reaktion wurde für eine weitere Stunde gerührt. 39 (238 mg) erhielt man nach Aufarbeitung und Kieselgelchromatographie.
-

- Zu einer gerührten Lösung aus 39 (238 mg) in Methylenchlorid (4,0 ml) wurden Undecansäure (98 mg), EDC (125 mg) und DMAP (4,4 mg) zugegeben. Nach Rühren für 16 Stunden wurden Undecansäure (75 mg), EDC (94 mg) und DMAP (9 mg) zugegeben und die Reaktion wurde für weitere 24 Stunden gerührt. Eine wässerige Aufarbeitung gefolgt von Kieselgelreinigung ergab 40 (314 mg).
-

- Zu einer gerührten Lösung aus 40 (577 mg) in einer 5:1 Mischung aus Acetonitril:Wasser (7,5 ml) wurde bei 0°C Cerammoniumnitrat (CAN – 1,23 g) zugegeben. Die Reaktionsmischung wurde bei Raumtemperatur für 72 Stunden gerührt, aufgearbeitet und durch Kieselgelchromatographie gereinigt, was 41 (228 mg) und 42 (224 mg) ergab.

- Zu einer gerührten Lösung aus 41 (153 mg) in THF (2,3 ml) wurden Triphenylphosphin (246 mg), Diphenylphosphorylazid (259 mg) und Diethylazodicarboxylat (DEAD – 164 mg) zugegeben. Nach Rühren bei Raumtemperatur für 2 Stunden erhielt man 43 (113 mg) nach Aufarbeitung und Kieselgelchromatographie.
-

- Zu einer gerührten Lösung aus 43 (130 mg) in THF (1,0 ml) wurde Essigsäure (18 mg) zugegeben, gefolgt von Tetra-n-butylammoniumfluorid (79 mg) bei Raumtemperatur. Nach Rühren für 48 Stunden wurde die Reaktionsmischung gequencht und mittels Kieselgelchromatographie gereinigt, was 44 (75 mg) ergab.

- Zu einer gerührten Lösung aus 44 (75 mg) in Methylenchlorid (0,6 ml) wurde Tetrazol (72 mg), gefolgt von dem phosphorylierenden Reagens (170 mg) bei 0°C zugegeben. Nach Rühren für 1 Stunde wurde die Reaktionsmischung auf eine gerührte Suspension gegossen, welche Oxon (627 mg) in THF (1 ml) und Wasser (1 ml) enthielt. Nach einer weiteren Stunde Rühren bei 0°C wurde die Mischung aufgearbeitet und über Kieselgel gereinigt, was 45 (73 mg) ergab.
-

- Zu einer gerührten Lösung aus 45 (37 mg) in Methylenchlorid (0,10 ml) wurde Triethylsilan (0,09 ml), gefolgt von Trifluoressigsäure (1,0 ml) bei Raumtemperatur zugegeben. Nach Rühren für 2 Stunden wurden die Lösemittel konzentriert und zum Trocknen mittels Toluen eine azeotrope Form gebracht. Das Rohamin 46 (33 mg) wurde in Methylenchlorid (0,5 ml) mit gesättigtem Natriumbicarbonat (0,5 ml) aufgelöst, gefolgt von tropfenweiser Zugabe von Phosgen (0,014 ml einer 1,93 M Lösung in Toluen) bei 0°C.
- Nach Rühren bei Raumtemperatur für 2 Stunden wurde die Reaktion aufgearbeitet und durch Kieselgel in der üblichen Art und Weise gereinigt, was 47 (35 mg) ergab.
-
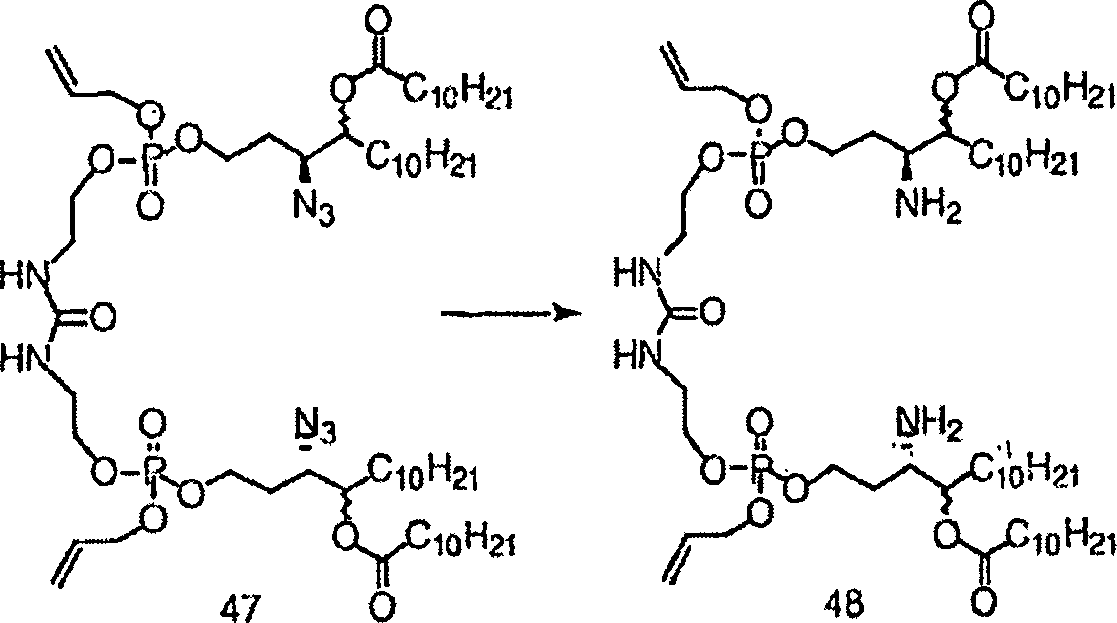
- Zu einer gerührten Lösung aus 47 (35 mg) wurde bei Raumtemperatur ein (PhS)3SnH·Et3N-Komplex (0,5 M – 0,34 ml) in Methylenchlorid zugegeben. Die Reaktionsmischung wurde für 1 Stunde gerührt; wonach wurde sie durch Kieselgelchromatographie gereinigt wurde, was das rohe 48 (29 mg) ergab, welches umgehend im nächsten Schritt verwendet wurde.
-

- Zu einer gerührten Lösung aus 48 (29 mg) in Methylenchlorid (0,5 ml) wurden Tridecansäure (36 mg) und EDC (44 mg) zugegeben. Nach Rühren für 48 Stunden, ergab eine wässrige Aufarbeitung gefolgt von Kieselgelreinigung 49 (21 mg).
-

- Zu einer gerührten Lösung aus 49 (21 mg) in entgastem Chloroform (1,5 ml) wurden bei 0°C Phenylsilan (16 μl) und Tetrakis(triphenylphosphin)palladium(0) (18 mg) zugegeben. Nach Rühren für 10 Minuten bei 0°C wurde die Reaktionsmischung mit einem 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (5 ml) verdünnt und für weitere 30 Minuten gerührt. Die Mischung wurde über DEAE-Cellulose (20 ml) gegossen und mit einer ansteigenden Konzentration von Ammoniumacetat (0,0 – 0,05 M) im 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (100 ml) eluiert. Die HPLC-Reinigung (Kieselgel mit Hexan:Isopropanol:Wasser-Gradientenelution) ergab das gewünschte Produkt ER-805274 (8,9 mg).
- Beispiel 6: Herstellung von ER-805271

- Zu einer gerührten Lösung aus 41 (128 mg) in Methylenchlorid (2,0 ml) wurden Tridecansäure (83 mg), EDC (113 mg) und DMAP (5,0 mg) zugegeben. Nach Rühren für 72 Stunden ergaben eine wässerige Aufarbeitung und Kieselgelreinigung 50 (136 mg).
-

- Zu einer gerührten Lösung aus 50 (136 mg) in THF (0,8 ml) wurde Essigsäure (14 mg) zugegeben, gefolgt von Tetra-n-butylammoniumfluorid (63 mg) bei Raumtemperatur. Nach Rühren für 16 Stunden wurde die Reaktionsmischung gequencht und mittels Kieselgelchromatographie gereinigt, was 51 (82 mg) ergab.
-

- Zu einer gerührten Lösung aus 51 (82 mg) in Methylenchlorid (0,7 ml) wurde Tetrazol (37 mg) zugegeben, gefolgt von dem phosphorylierenden Reagens (89 mg) bei 0°C. Nach Rühren für 2 Stunden wurde die Reaktionsmischung auf eine gerührte Suspension gegossen, welche Oxon (330 mg) in THF (1 ml) und Wasser (1 ml) enthielt. Nach weiteren 2 Stunden Rühren bei 0°C wurde die Mischung aufgearbeitet und über Kieselgel gereinigt, was 52 (70 mg) ergab.
-

- Zu einer gerührten Lösung aus 52 (70 mg) in Methylenchlorid (0,10 ml) wurde Triethylsilan (0,13 ml), gefolgt von Trifluoressigsäure (1,0 ml) bei Raumtemperatur zugegeben. Nach Rühren für 2 Stunden wurden die Lösemittel konzentriert und zum Trocknen mittels Toluen eine azeotrope Form gebildet. Das Rohamin 53 (40 mg) wurde in Methylenchlorid (0,5 ml) mit gesättigtem Natriumbicarbonat (0,5 ml) aufgelöst, gefolgt von tropfenweiser Zugabe von Phosgen (0,014 ml einer 1,93 M Lösung in Toluen) bei 0°C. Nach Rühren bei Raumtemperatur für 2 Stunden wurde die Reaktion aufgearbeitet und durch Kieselgel in der üblichen Art und Weise gereinigt, was 54 (24 mg) ergab.
-

- Zu einer gerührten Lösung aus 54 (24 mg) in entgastem Chloroform (1,5 ml) wurden bei 0°C Phenylsilan (17 mg) und Tetrakis(triphenylphosphin)palladium(0) (18 mg) zugegeben. Nach Rühren für 10 Minuten bei 0°C wurde die Reaktionsmischung mit einem 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (5 ml) verdünnt und für weitere 30 Minuten gerührt. Die Mischung wurde über DEAE-Cellulose (20 ml) gegossen und mit einer ansteigenden Konzentration von Ammoniumacetat (0,0 – 0,05 M) im 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (100 ml) eluiert. Die HPLC-Reinigung (Kieselgel mit Hexan:Isopropanol:Wasser-Gradientenelution) ergab das gewünschte Produkt ER-805271 (3,3 mg).
- Beispiel 7: Herstellung von ER-805270

- Zu einer gerührten Lösung von handelsüblichem D-sphingosinsulphat 55 (120 mg) in Methylenchlorid (5,0 ml) und gesättigtem Natriumbicarbonat (2,5 ml) wurde bei 0°C tropfenweise Tetradecanoylchlorid (99 mg) zugegeben. Nach Rühren für 16 Stunden bei Raumtemperatur wurde die Reaktionsmischung aufgearbeitet und durch Kieselgelchromatographie gereinigt, was 56 (183 mg) ergab.
-

- Zu einer gerührten Lösung aus 56 (183 mg) in Methylenchlorid (3,6 ml) wurde Imidazol (39 mg) zugegeben, gefolgt von tert-Butyldiphenylsilylchlorid (109 mg). Nach Rühren bei Raumtemperatur für 16 Stunden ergab die normale Aufarbeitung gefolgt von Kieselgelreinigung 57 (193 mg).
-

- Zu einer gerührten Lösung aus 57 (193 mg) in Methylenchlorid (0,5 ml) wurden Dodecansäure (74 mg), EDC (70 mg) und DMAP (4 mg) zugegeben. Nach Rühren für 16 Stunden ergaben eine wässrige Aufarbeitung und Kieselgelreinigung 58 (219 mg).
-

- Zu einer gerührten Lösung aus 58 (219 mg) in THF (1,0 ml) wurde Essigsäure (21 mg), gefolgt von Tetra-n-butylammoniumfluorid (93 mg) bei Raumtemperatur zugegeben. Nach Rühren für 48 Stunden wurde die Reaktionsmischung gequencht und mittels Kieselgelchromatographie gereinigt, was 59 (140 mg) ergab.
-

- Zu einer gerührten Lösung aus 59 (140 mg) in Methylenchlorid (1,0 ml) wurde Tetrazol (42 mg), gefolgt von dem phosphorylierenden Reagens (100 mg) bei 0°C zugegeben. Nach Rühren für 2 Stunden wurde die Reaktionsmischung auf eine gerührte Suspension gegossen, welche Oxon (369 mg) in THF (1 ml) und Wasser (1 ml) enthielt. Nach weiteren 16 Stunden Rühren bei Raumtemperatur wurde die Mischung aufgearbeitet und über Kieselgel gereinigt, was 60 (152 mg) ergab.

- Zu einer gerührten Lösung aus 60 (46 mg) in Methylenchlorid (0,2 ml) wurde Triethylsilan (0,12 ml), gefolgt von Trifluoressigsäure (1,0 ml) bei Raumtemperatur zugegeben. Nach Rühren für 1 Stunde wurden die Lösemittel konzentriert und zum Trocknen mittels Toluen eine azeotrope Form gebildet. Das Rohamin 61 wurde in Methylenchlorid (0,6 ml) mit gesättigtem Natriumbicarbonat (0,6 ml) aufgelöst, gefolgt von tropfenweiser Zugabe von Phosgen (0,014 ml einer 1,93 M Lösung in Toluen) bei 0°C.
- Nach Rühren bei Raumtemperatur für 2 Stunden wurde die Reaktion aufgearbeitet und durch Kieselgel in der üblichen Art und Weise gereinigt, was 62 (44 mg) ergab.
-

- Zu einer gerührten Lösung aus 62 (44 mg) in entgastem Chloroform (2 ml) wurden bei 0°C Phenylsilan (40 μl) und Tetrakis(triphenylphosphin)palladium(0) (23 mg) zugegeben. Nach Rühren für 1 Stunde bei Raumtemperatur wurde die Reaktionsmischung mit einem 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (5 ml) verdünnt und für weitere 30 Minuten gerührt. Die Mischung wurde über DEAE-Cellulose (20 ml) gegossen und mit einer ansteigenden Konzentration von Ammoniumacetat (0,0 – 0,05 M) im 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (100 ml) eluiert. Die HPLC-Reinigung (Kieselgel mit Hexan:Isopropanol:Wasser-Gradientenelution) ergab das gewünschte Produkt ER-805270 (7,1 mg).
- Beispiel 8: Herstellung von ER-805328

- Zu einer gerührten Lösung von Oxalylchlorid (0,66 ml) in Methylenchlorid (20 ml) wurde bei –78° tropfenweise DMSO (1,06 ml) zugegeben. Nach Rühren für 15 Minuten wurde tropfenweise handelsübliches geschütztes Glycerol 63 (0,62 g) in Methylenchlorid (5 ml) zugegeben, gefolgt von Rühren für weitere 30 Minuten. Triethylamin (3,5 ml) wurde tropfenweise zugegeben, gefolgt von Erwärmung auf Raumtemperatur und die übliche Aufarbeitung mittels verdünnter HCl. Das Rohaldehyd 64 wurde umgehend im nächsten Schritt verwendet.
-

- Zu einer gerührten Suspension aus gewaschenem Natriumhydrid (0,144 g) in Toluen (30 ml) wurde bei 0°C tropfenweise Phosphonat 65 (1,2 ml) zugegeben, gefolgt von Rühren für 15 Minuten bei Raumtemperatur. Nach Abkühlung auf 0°C wurde tropfenweise das Rohaldehyd 64 (0,62 g) in Toluen (5 ml) zugegeben, gefolgt von Rühren bei Raumtemperatur für 2 Stunden. Die Reaktion wurde in üblicher Art und Weise aufgearbeitet und das Produkt 66 (0,46 g) erhielt man mittels Kieselgelchromatographie.
-

- Zu einer gerührten Lösung des chiralen Esters 66 (3,80 g) in Ethylacetat (100 ml) wurde bei Raumtemperatur 10 % Palladium auf Kohlenstoff (100 mg) zugegeben; danach wurde die Mischung unter eine Wasserstoffatmosphäre (60 psi) gestellt und für 2 Stunden geschüttelt. Filterung gefolgt von Konzentrierung ergab rohes 67 (4,00 g).
-

- Zu einer gerührten Lösung aus 67 (4,00 g) in Hexanen (100 ml) wurde bei 0°C tropfenweise DIBAL (1,0 M in Hexanen – 40 ml) zugegeben. Nach Rühren für weitere 20 Minuten bei 0°C wurde die Reaktionsmischung zuerst mit Methanol:Wasser gequencht, gefolgt von Rochellesalzen. 68 (2,66 g) erhielt man nach Aufarbeitung und Kieselgelchromatographie.
-

- Zu einer gerührten Lösung aus Oxalylchlorid (2,46 ml) in Methylenchlorid (100 ml) wurde bei –78°C tropfenweise DMSO (4 ml) zugegeben, gefolgt von Rühren für 30 Minuten. 68 (2,828 g) in Methylenchlorid (10 ml) wurde tropfenweise zugegeben und dann zwischen –40°C und –60°C für eine Stunde gerührt; danach wurde tropfenweise Triethylamin (13,2 ml) zugegeben und die Reaktion über einen Zeitraum von einer Stunde auf Raumtemperatur erwärmen gelassen. Das Rohaldehyd 69 (3,02 mg), welches man nach der Aufarbeitung erhielt, wurde in THF (20 ml) aufgelöst und bei Raumtemperatur tropfenweise zu Nonanylmagnesiumbromid (4,14 g) in THF (3,82 ml) zugegeben. Nach Rühren für weitere 2 Stunden ergaben die übliche Aufarbeitung und Kieselgelchromatographiereinigung 70 (2,27 g).

- Zu einer gerührten Lösung aus 70 (1,13 mg) in THF (40 ml) wurden bei Raumtemperatur Triphenylphosphin (1,55 g), Diphenylphosphorylazid (1,63 g) und Diethylazodicarboxylat (DEAD – 1,03 g) zugegeben. Nach Rühren für 15 Minuten wurden Triphenylphosphin (1,04 g), Diphenylphosphorylazid (1,09 g) und Diethylazodicarboxylat (0,69 g) zu der Mischung zugegeben und die abschließende Mischung wurde für 20 Minuten gerührt. Rohes 71 (1,83 g) erhielt man nach Aufarbeitung und Kieselgelchromatographie.
-

- Zu dem rohen 71 (1,83 g) wurde bei Raumtemperatur ein (PhS)3SnH·Et3N-Komplex (0,5 M – 20 ml) in Methylenchlorid zugegeben. Die Reaktionsmischung wurde für 20 Minuten gerührt; wonach sie durch Kieselgelchromatographie gereinigt wurde, was rohes 72 (2,01 g) ergab, welches umgehend im nächsten Schritt verwendet wurde.
-

- Zu der gerührten Suspension aus dem rohen 72 (2,01 g) in THF (15 ml) und gesättigtem Natriumbicarbonat (30 ml) wurde bei 0°C tropfenweise Tetradecanoylchlorid (1,74 g) zugegeben. Nach Rühren für 30 Minuten bei Raumtemperatur wurde die Reaktionsmischung aufgearbeitet und durch Kieselgelchromatographie gereinigt, was 73 (1,58 g) ergab.
-

- Zu einer gerührten Lösung aus 73 (785 g) in THF (8 ml) wurde bei Raumtemperatur verdünnte HCl (2,4 N – 10 ml) zugegeben. Nach Rühren der Reaktionsmischung für 2 Sunden erhielt man das Rohprodukt 74 (667 mg) durch eine normale Aufarbeitung.
-

- Zu einer gerührten Lösung aus 74 (667 mg) in Methylenchlorid (16 ml) wurde bei Raumtemperatur Imidazol (161 mg), gefolgt von tert-Butyldiphenylsilylchlorid (435 mg) zugegeben. Nach Rühren für 18 Stunden ergaben normale Aufarbeitung gefolgt von Kieselgelreinigung 75 (850 mg).
-

- Zu einer gerührten Lösung aus 75 (106 mg) in Methylenchlorid (0,5 ml) wurden bei Raumtemperatur Tetradecansäure (42 mg), EDC (44 mg) und DMAP (4 mg) zugegeben. Nach Rühren für 16 Stunden ergaben eine wässrige Aufarbeitung und Kieselgelreinigung 76 (132 mg).
-

- Zu einer gerührten Lösung aus 76 (132 mg) in THF (1,0 ml) wurde bei Raumtemperatur Essigsäure (13 mg) zugegeben, gefolgt von Tetra-n-butylammoniumfluorid (58 mg). Nach Rühren für 16 Stunden wurde die Reaktionsmischung gequencht und mittels Kieselgelchromatographie gereinigt, was 77 (80 mg) ergab.
-

- Zu einer gerührten Lösung aus 77 (80 mg) in Methylenchlorid (0,6 ml) wurde bei 0°C Tetrazol (25 mg) zugegeben, gefolgt von dem phosphorylierenden Reagens (60 mg). Nach Rühren für 1 Stunde wurde die Reaktionsmischung auf eine gerührte Suspension gegossen, welche Oxon (204 mg) in THF (1 ml) und Wasser (1 ml) enthielt. Nach einer weiteren Stunde Rühren bei 0°C wurde die Mischung aufgearbeitet und über Kieselgel gereinigt, was 78 (65 mg) ergab.
-

- Zu einer gerührten Lösung aus 78 (65 mg) in Methylenchlorid (0,1 ml) wurde bei Raumtemperatur Triethylsilan (82 mg) zugegeben, gefolgt von Trifluoressigsäure (1,0 ml). Nach Rühren für 2 Stunden wurden die Lösemittel konzentriert und zum Trocknen mittels Toluen in eine azeotrope Form gebracht. Das Rohamin 79 wurde bei 0°C in Methylenchlorid (0,7 ml) mit gesättigtem Natriumbicarbonat (0,7 ml) aufgelöst, gefolgt von tropfenweiser Zugabe von Phosgen (0,019 ml einer 1,93 M Lösung in Toluen).
- Nach Rühren bei Raumtemperatur für 2 Stunden wurde die Reaktion aufgearbeitet und durch Kieselgel in der üblichen Art und Weise gereinigt, was 80 (50 mg) ergab.
-

- Zu einer gerührten Lösung aus 80 (50 mg) in entgastem Chloroform (3 ml) wurden bei 0°C Phenylsilan (32 mg) und Tetrakis(triphenylphosphin)palladium(0) (26 mg) zugegeben. Nach Rühren für 1 Stunde bei Raumtemperatur wurde die Reaktionsmischung mit einem 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (5 ml) verdünnt und für weitere 30 Minuten gerührt. Die Mischung wurde über DEAE-Cellulose (20 ml) gegossen und mit einer ansteigenden Konzentration von Ammoniumacetat (0,0 – 0,05 M) im 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (100 ml) eluiert. Die HPLC-Reinigung (Kieselgel mit Hexan:Isopropanol:Wasser-Gradientenelution) ergab das gewünschte Produkt ER-805328 (38 mg).
- Beispiel 9: Herstellung von ER-805329

- Zu einer gerührten Lösung aus 75 (0,14 g) in THF (3 ml) wurden bei Raumtemperatur Triphenylphosphin (0,159 g), Diphenylphosphorylazid (0,167 g) und DEAD (0,105 g) zugegeben. Nach Rühren für 1 Stunde erhielt man das Rohazid 81 (0,14 g) nach Aufarbeitung

- Zu dem rohen 81 (0,14 g) wurde bei Raumtemperatur ein (PhS)3SnH·Et3N-Komplex (0,5 M – 5 ml) in Methylenchlorid zugegeben. Die Reaktionsmischung wurde für 20 Minuten gerührt; wonach sie durch Kieselgelchromatographie gereinigt wurde, was das rohe 82 (0,14 g) ergab, welches umgehend im nächsten Schritt verwendet wurde.
-

- Zu einer gerührten Lösung aus 82 (0,14 g) in Methylenchlorid (2 ml) wurden bei Raumtemperatur Tetradecansäure (92 mg) und EDC (116 mg) zugegeben. Nach Rühren für 16 Stunden ergaben eine wässerige Aufarbeitung und Kieselgelreinigung 83 (190 mg).
-

- Zu einer gerührten Lösung aus 83 (190 mg) in THF (2,0 ml) wurde bei Raumtemperatur Essigsäure (20 mg) zugegeben, gefolgt von Tetra-n-butylammoniumfluorid (89 mg). Nach Rühren für 16 Stunden wurde die Reaktionsmischung gequencht und mittels Kieselgelchromatographie gereinigt, was 84 (104 mg) ergab.
-

- Zu einer gerührten Lösung aus 84 (104 mg) in Methylenchlorid (0,8 ml) wurde bei 0°C Tetrazol (42 mg) zugegeben, gefolgt von dem phosphorylierenden Reagens (100 mg). Nach Rühren für 1 Stunde wurde die Reaktionsmischung auf eine gerührte Suspension gegossen, welche Oxon (615 mg) in THF (2 ml) und Wasser (2 ml) enthielt. Nach einer weiteren Stunde Rühren bei 0°C wurde die Mischung aufgearbeitet und über Kieselgel gereinigt, was 85 (82 mg) ergab.
-
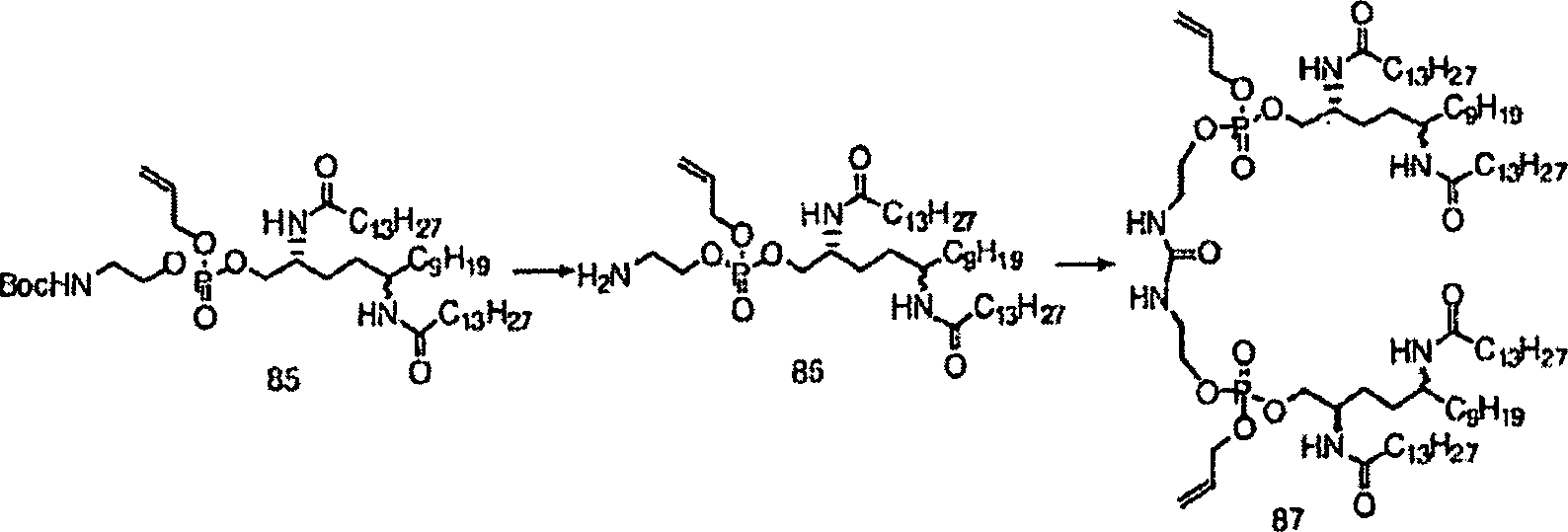
- Zu einer gerührten Lösung aus 85 (82 mg) in Methylenchlorid (0,1 ml) wurde bei Raumtemperatur Triethylsilan (103 mg) zugegeben, gefolgt von Trifluoressigsäure (1,0 ml). Nach Rühren für 2 Stunden wurden die Lösemittel konzentriert und zum Trocknen mittels Toluen in eine azeotrope Form gebracht. Das Rohamin 86 wurde bei 0°C in Methylenchlorid (0,8 ml) mit gesättigtem Natriumbicarbonat (0,8 ml) aufgelöst, gefolgt von tropfenweiser Zugabe von Phosgen (0,023 ml einer 1,93 M Lösung in Toluen).
- Nach Rühren bei Raumtemperatur für 2 Stunden wurde die Reaktion aufgearbeitet und durch Kieselgel in der üblichen Art und Weise gereinigt, was 87 (63 mg) ergab.
-

- Zu einer gerührten Lösung aus 87 (63 mg) in entgastem Chloroform (3 ml) wurden bei 0°C Phenylsilan (40 mg) und Tetrakis(triphenylphosphin)palladium(0) (33 mg) zugegeben. Nach Rühren für 1 Stunde bei Raumtemperatur wurde die Reaktionsmischung mit einem 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (5 ml) verdünnt und für weitere 30 Minuten gerührt. Die Mischung wurde über DEAE-Cellulose (20 ml) gegossen und mit einer ansteigenden Konzentration von Ammoniumacetat (0,0 – 0,05 M) im 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (100 ml) eluiert. Die HPLC-Reinigung (Kieselgel mit Hexan:Isopropanol:Wasser-Gradientenelution) ergab das gewünschte Produkt ER-805329 (5,1 mg).
- Beispiel 10: Herstellung von ER-805517

- Zu einer Lösung von handelsüblichem, chiralem 88 (3,18 g) in Aceton (100 ml) wurden bei Raumtemperatur 2,2-Dimethoxypropan (4,05 ml) und p-Toluensulfonsäure (0,57 g) zugegeben, gefolgt von Rühren für 72 Stunden. 89 (3,67 g) erhielt man nach Aufarbeitung und Kieselgelreinigung.
-

- Zu einer gerührten Lösung aus Methylphosphonat 90 (4,56 g) in THF (100 ml) wurde bei –78°C tropfenweise n-Butyllithium (1,6 M in Hexanen – 18,8 ml) zugegeben. Nach Rühren für 5 Minuten wurde Kupfer-(I)-Jodid (5,7 g) zugegeben, gefolgt von Rühren für weitere 2 Stunden bei –5°C. Das Säurechlorid 91 (5,31 g) wurde tropfenweise zu der klaren gelben Mischung zugegeben, wonach die Reaktion bei Raumtemperatur für 1 Stunde gerührt wurde. 92 (7,28 g) erhielt man nach der üblichen Aufarbeitung und Kieselgelchromatographie.
-

- Zu einer gerührten Lösung aus Oxalylchlorid (2,54 g) in Methylenchlorid (65 ml) wurde bei –78°C tropfenweise DMSO (2,83 ml) zugegeben, gefolgt von Rühren für 30 Minuten. 89 (1,90 g) in Methylenchlorid (5 ml) wurde tropfenweise zugegeben und dann zwischen –40°C und –60°C für eine Stunde gerührt, wonach tropfenweise Triethylamin (9,04 ml) zugegeben und die Reaktion über einen Zeitraum von einer Stunde auf Raumtemperatur erwärmen gelassen wurde. Das Rohaldehyd 93 (3,03 g), welches man nach der Aufarbeitung erhielt, wurde umgehend ohne weitere Reinigung in nächsten Schritt eingesetzt.
-

- Zu einer gerührten Suspension aus gewaschenem Natriumhydrid (79 mg) in Toluen (30 ml) wurde bei 0°C Phosphonat 92 (0,88 g) zugegeben. Nach Rühren bei Raumtemperatur für 30 Minuten wurde die Reaktionsmischung auf –15°C abgekühlt, wonach das Rohaldehyd 93 (0,43 g) zugegeben wurde. Die abschließende Mischung wurde bei Raumtemperatur für 30 Minuten gerührt, gefolgt von der üblichen Aufarbeitung und Kieselgelchromatographiereinigung, was 94 (1,06 g) ergab.
-

- Zu einer gerührten Lösung von 94 (0,726 g) in Ethylacetat (25 ml) wurde bei Raumtemperatur 20 % Palladiumhydroxid (100 mg) zugegeben, wonach die Mischung unter eine Wasserstoffatmosphäre (60 psi) gestellt und für 16-Stunden geschüttelt wurde. Filterung gefolgt von Konzentrierung ergab ohne weitere Reinigung 95 (0,72 g).
-

- Zu einer gerührten Lösung aus 95 (0,72 g) in Methanol (20 ml) bei Raumtemperatur wurde Natriumborhydrid (150 mg) bei 0°C zugegeben, wonach die Mischung für 1,5 Stunden gerührt wurde. Aufarbeitung gefolgt von Konzentrierung ergab ohne weitere Reinigung 96 (0,67 g).
-

- Zu einer gerührten Lösung aus 96 (0,67 g) in Methylenchlorid (10 ml) wurden bei Raumtemperatur Tridecansäure (0,66 g), EDC (0,72 g) und DMAP (29 mg) zugegeben. Nach Rühren für 16 Stunden wurde zusätzliches EDC (0,10 g) zugegeben und die Mischung dann für eine weitere Stunde gerührt. Wässerige Aufarbeitung und Kieselgelreinigung ergaben 97 (1,01 g).
-

- Zu einer gerührten Lösung aus 97 (1,01 g) in THF (21 ml) wurde bei Raumtemperatur 3 N HCl (21 ml) zugegeben. Nach Rühren der Reaktionsmischung für 26 Stunden erhielt man das Rohprodukt 98 (0,92 g) durch eine normale Aufarbeitung.
-

- Zu einer gerührten Lösung aus 98 (0,94 g) in Methylenchlorid (10 ml) wurde bei 0°C Imidazol (214 mg) zugegeben, gefolgt von tert-Butyldiphenylsilylchlorid (608 mg). Nach Rühren bei Raumtemperatur für 72 Stunden ergab die normale Aufarbeitung gefolgt von Kieselgelreinigung 99 (1,44 g).
-

- Zu einer gerührten Lösung aus 99 (552 mg) in THF (8 ml) wurden bei Raumtemperatur Triphenylphosphin (639 mg), Diphenylphosphorylazid (671 mg) und DEAD (425 mg) zugegeben. Nach Rühren für 1 Stunde erhielt man das Rohazid 100 (402 mg) nach Aufarbeitung.
-

- Zu 100 (420 mg) wurde bei Raumtemperatur ein (PhS)3SnH·Et3N-Komplex (0,5 M – 3,43 ml) in Methylenchlorid zugegeben. Die Reaktionsmischung wurde für 20 Stunde gerührt; danach wurde sie durch Kieselgelchromatographie gereinigt, was 101 (434 mg) ergab, welches umgehend im nächsten Schritt verwendet wurde.
-

- Zu einer gerührten Lösung aus 101 (434 mg) in Methylenchlorid (6,0 ml) wurde bei Raumtemperatur und gesättigtem wässerigem Natriumbicarbonat (6,0 ml) Tetradecanoylchlorid (156 mg) zugegeben. Nach Rühren für 15 Minuten ergaben eine Aufarbeitung und Kieselgelreinigung 102 (422 mg).
-

- Zu einer gerührten Lösung aus 102 (422 mg) in THF (2,0 ml) wurde bei Raumtemperatur Essigsäure (43 mg), gefolgt von Tetra-n-butylammoniumfluorid (189 mg) zugegeben. Nach Rühren für 16 Stunden wurde die Reaktionsmischung gequencht und mittels Kieselgelchromatographie gereinigt, was 103 (258 mg) ergab.
-

- Zu einer gerührten Lösung aus 103 (258 mg) in Methylenchlorid (2,0 ml) wurde bei 0°C Tetrazol (112 mg), gefolgt von dem phosphorylierenden Reagens (266 mg) zugegeben. Nach Rühren für 2 Stunden bei Raumtemperatur wurde die Reaktionsmischung auf eine gerührte Suspension gegossen, welche Oxon (1,48 g) in THF (2 ml) und Wasser (2 ml) bei 0°C enthielt. Nach einer weiteren Stunde Rühren bei 0°C wurde die Mischung aufgearbeitet und über Kieselgel gereinigt, was 104 (325 mg) ergab.
-

- Zu einer gerührten Lösung aus 104 (313 mg) in Methylenchlorid (0,1 ml) wurde bei Raumtemperatur Triethylsilan (0,64 μl), gefolgt von Trifluoressigsäure (4,0 ml) zugegeben. Nach Rühren für 2 Stunden wurden die Lösemittel konzentriert und zum Trocknen mittels Toluen in eine azeotrope Form gebracht. Das Rohamin 76 wurde bei 0°C in Methylenchlorid (3,4 ml) mit gesättigtem Natriumbicarbonat (3,4 ml) aufgelöst, gefolgt von tropfenweiser Zugabe von Phosgen (94 μl einer 1,93 M Lösung in Toluen).
- Nach Rühren bei Raumtemperatur für 3 Stunden wurde die Reaktion aufgearbeitet und durch Kieselgel in der üblichen Art und Weise gereinigt, was 105 (143 mg) ergab.
-

- Zu einer gerührten Lösung aus 105 (143 mg) in entgastem Chloroform (8 ml) wurden bei 0°C Phenylsilan (94 mg) und Tetrakis(triphenylphosphin)palladium(0) (75 mg) zugegeben. Nach Rühren für 0,5 Stunden bei Raumtemperatur wurde die Reaktionsmischung mit einem 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (5 ml) verdünnt und für weitere 30 Minuten gerührt. Die Mischung wurde über DEAE-Cellulose (20 ml) gegossen und mit einer ansteigenden Konzentration von Ammoniumacetat (0,0 – 0,05 M) im 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (100 ml) eluiert. Die HPLC-Reinigung (Kieselgel mit Hexan:Isopropanol:Wasser-Gradientenelution) ergab das gewünschte Produkt ER-805517 (84 mg).
- Beispiel 11: Herstellung von ER-805518 und ER-805519

- Zu einer gerührten Lösung von 16 (427 mg) in Methanol (20 ml) wurde bei 0°C für 10 Minuten wasserfreies HCl-Gas zugegeben, wonach die Reaktionsmischung auf Raumtemperatur erwärmt und für weitere 2 Stunden gerührt wurde. Nachdem die Mischung konzentriert wurde, bis sie trocken war, wurde das Rohprodukt lyophilisiert bis es trocken war, was das rohe 106 (307 mg) ergab. 106 (307 mg) wurde in THF (2 ml) aufgelöst und bei 0°C wurde gesättigtes Natriumbicarbonat (4 ml) zugegeben, gefolgt von tropfenweiser Zugabe von Tetradecanoylchlorid (0,31 ml), gefolgt von kräftigem Rühren für 30 Minuten bei Raumtemperatur. Normale Aufarbeitung und Kieselgelreinigung ergaben 107 (266 mg).
-

- Zu einer gerührten Lösung aus 107 (266 mg) in Methylenchlorid (5 ml) wurde bei Raumtemperatur Imidazol (60 mg), gefolgt von tert-Butyldiphenylsilylchlorid (170 mg) zugegeben. Nach Rühren für 20 Stunden ergab die normale Aufarbeitung gefolgt von Kieselgelreinigung Isomer 108 (277 mg).
-

- Zu einer gerührten Lösung aus 108 (154 mg) in Methylenchlorid (2,0 ml) wurden bei Raumtemperatur Laurinsäure (104 mg), EDC (123 mg) und DMAP (5 mg) zugegeben. Nach Rühren für 16 Stunden, wässeriger Aufarbeitung und Kieselgelreinigung erhielt man 109 (361 mg).
-

- Zu einer gerührten Lösung aus 109 (361 mg) in THF (2,0 ml) wurde bei Raumtemperatur Essigsäure (36 mg) zugegeben, gefolgt von Tetra-n-butylammoniumfluorid (157 mg). Nach Rühren für 72 Stunden wurde die Reaktionsmischung gequencht und mittels Kieselgelchromatographie gereinigt, was 110 (134 mg) ergab.
-

- Zu einer gerührten Lösung aus 110 (210 mg) in Methylenchlorid (1,66 ml) wurde bei Raumtemperatur Tetrazol (93 mg), gefolgt von dem phosphorylierenden Reagens (221 mg) zugegeben. Nach Rühren für 2 Stunden wurde die Reaktionsmischung auf eine gerührte Suspension gegossen, welche Oxon (1,48 g) in THF (2 ml) und Wasser (5 ml) bei 0°C enthielt. Nach einer weiteren Stunde Rühren bei 0°C wurde die Mischung aufgearbeitet und über Kieselgel gereinigt, was 111 (174 mg) ergab.
-

- Zu einer gerührten Lösung aus 111 (174 mg) in Methylenchlorid (0,10 ml) wurde bei Raumtemperatur Triethylsilan (0,31 ml) zugegeben, gefolgt von Trifluoressigsäure (2,0 ml). Nach Rühren für 2 Stunden wurden die Lösemittel konzentriert und zum Trocknen mittels Toluen in eine azeotrope Form gebracht. Das Rohamin 112 wurde bei 0°C in Methylenchlorid (2,0 ml) mit gesättigtem Natriumbicarbonat (2,0 ml) aufgelöst, gefolgt von tropfenweiser Zugabe von Phosgen (53 μl einer 1,93 M Lösung in Toluen). Nach Rühren bei Raumtemperatur für 2 Stunden wurde die Reaktion aufgearbeitet und durch Kieselgel in der üblichen Art und Weise gereinigt, was 113 (124 mg) ergab.
-

- Zu einer gerührten Lösung aus 113 (124 mg) in entgastem Chloroform (7 ml) wurden bei 0°C Phenylsilan (825 mg) und Tetrakis(triphenylphosphin)palladium(0) (66 mg) zugegeben. Nach Rühren für 1 Stunde bei Raumtemperatur wurde die Reaktionsmischung mit einem 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (5 ml) verdünnt und für weitere 30 Minuten gerührt. Die Mischung wurde über DEAE-Cellulose (20 ml) gegossen und mit einer ansteigenden Konzentration von Ammoniumacetat (0,0 – 0,05 M) in einem 2:3:1 Verhältnis von Chloroform:Methanol:Wasser (100 ml) eluiert. Die HPLC-Reinigung (Kieselgel mit Hexan:Isopropanol:Wasser-Gradientenelution) ergab zwei isomere Produkte, Isomer A: ER-805518 (68 mg) und Isomer B: ER-805519 (28 mg).
- BIOLOGISCHE BEISPIELE
- Beispiel 12: Induzierung von Cytokinen (in vitro)
- Die Fähigkeit, eine Stimulationsreaktion bei Makrophagen auszulösen, ist sehr wahrscheinlich eine notwendige Eigenschaft von Molekülen mit immunförderlichen Eigenschaften. Daher wurde die Fähigkeit von Verbindungen getestet, die Freisetzung von TNF-α und anderen Zytokinen aus immunen Zellen zu stimulieren, als einen Beleg für ihre Fähigkeit, eine Immunreaktion zu stimulieren, welche in einer förderlichen Aktivität resultieren könnte.
- Das am leichtesten verfügbare humane System zum Testen von Verbindungsaktivität auf Monozyten/Makrophagen ist in Vollblut. Unterschiedliche Konzentrationen von Verbindungen der Erfindung wurden als 10 × Stammlösungen in 50 μl 5 % Dextrose gefolgt von 50 μl 5 % Dextrose in 400 μl heparinisiertem Vollblut erhalten aus normalen Freiwilligen (18-51 Jahre alt, 55-115 kg) in die Kammern von Kunststoffuntersuchungsplatten gegeben, für ein Gesamtvolumen von 500 μl/Kammer (die Endkonzentration des Vollblutes betrug 80 %). Die 10 × Stammlösungen wurden durch Auflösen der Verbindungen auf 1 mM in Wasser und Beschallung für zwei Minuten in einem Eisbad hergestellt. Die Verbindungen wurden dann auf 10 × in 5 % Dextrose gebracht. Nach 3 Stunden Inkubation unter leichtem Schütteln bei 37°C in einer 5 % CO2-Atmosphäre wurden die Untersuchungsplatten bei 1000 × g für 10 Minuten bei 4°C zentrifugiert, und Plasma wurde abgezogen und bei –80°C eingefroren. Die Plasmaproben wurden durch ELISA (R & D Systems, Minneapolis, MN) auf TNF-α analysiert. Jeder Untersuchungspunkt wurde dreifach getestet.
- Wie in Tabelle 1 gezeigt, stimulieren repräsentative Verbindungen der Erfindung blutgetragene Zellen zur Freisetzung von TNF-α, mit Stimulationswerten zwischen 0,008 μM und > 10,0 μM. Der Stimulationswert ist die Verbindungskonzentration, welche erforderlich ist, um eine TNF-α-Freisetzung gleich der Hälfte der Freisetzung zu stimulieren, welche mit 10 ng/ml LPS aus der gleichen Vollblutprobe erreicht wird.
- Tabelle 1. Stimulation der Zytokinfreisetzung durch Verbindungen in vitro

-

-

-

- Beispiel 13: In vivo Induzierung einer Antikörperreaktion
- Der kritischeste Test der Adjuvansaktivität ist die Bestimmung, ob die Zugabe einer Verbindung zu einem Antigenpräparat die Immunreaktion erhöht, indem sie bei Verabreichung an ein lebendes Tier den Pegel der erzeugten Antikörper gegen dieses Antigen ansteigen lässt.
- Zur Untersuchung der Auswirkungen der Verbindungen der Erfindung auf die in vivo Antikörperproduktion wurde Balb/c-Mäusen eine Testverbindung zusammen mit einem Protein wie Tetanustoxoid injiziert. Das Tetanustoxoid wurde als Immunogen mit einer Dosis von 0,25 μg verwendet. Weiblichen Balb/c-Mäusen (Charles River Laboratories; etwa 6-8 Wochen alt (18-25 g)) wurden alle 3 Wochen für insgesamt 3 Injektionen 200 μl einer Mischung aus Antigen und Adjuvans in PBS injiziert. Kontrolltieren wurde Alum oder PBS injiziert. Sämtliche Injektionen erfolgten subkutan im Nacken. Den Mäusen wurde zwei Wochen nach der zweiten und dritten Injektion Blut entnommen. Das Blut wurde durch Einkerben der Schwanzvene genommen, und die Tropfen wurden in Becton Dickinson Markenmikrobehälter-Serumseparatorröhrchen gesammelt. Das Serum wurde durch Mikrozentrifugierung von den roten Blutzellen getrennt und mittels ELISA auf Antigen-spezifische IgG-Pegel getestet.
- Die Immunreaktion auf das Peptid kann durch den heterologen Enzym-Immunoassay (ELISA) getestet werden, welcher die Pegel der Serumantikörper quantitativ bestimmen kann, welche sich an das Tetanustoxoid binden, das auf eine ELISA-Platte beschichtet wurde.
- Die Serumantikörper wurden zwei Wochen nach der zweiten Immunisierung gemessen. Wie in Tabelle 2 gezeigt, zeigten Mäuse, denen die Verbindungen der Erfindung zusammen mit dem Tetanustoxoidantigen injiziert wurden eine größere Immunreaktion (höhere Antikörperpegel) als die, denen nur Tetanustoxoid injiziert wurde.
- Tetanustoxoid von Accurate Chemical (Kat. Nr. sstettox) wurde als ein Immunisierungsantigen verwendet, während das gereinigte Toxoid von List Biologicals (Kat. Nr. 191) als ein Zielantigen für die ELISA-Untersuchung verwendet wurde.
- Für die Herstellung von Antigen/Adjuvans-Mischungen wurden lyophilisierte Testverbindungen auf 2 mg/ml mit phosphatgepufferter Salzlösung (PBS; Kat. Nr. P-3818; Sigma Chemical Co., St. Louis, MO) hergestellt und für zwei Minuten in einem gekühlten Wasserbad beschallt. Imject® Alum, erworben von Pierce Immunochemical, wurde gemäß der Herstelleranweisungen verwendet und umfasste 15 % des Injektionsvolumens. Das Antigen wurde in PBS verdünnt und mit der Testverbindung oder Alum gemischt, so dass die abschließende Konzentration der Verbindung 30 μg in dem 200 μg Injektionsvolumen betrug. Die Mischungen wurden vor der Injektion bei Raumtemperatur für mindestens 40 Minuten inkubiert.
- Die Antigen-spezifischen IgG-Pegel wurden durch direkten ELISA überwacht, bei dem das Antigen passiv auf 96-Kammern Costar EIA/RIA-Platten beschichtet wurde. Die Platten wurden mit 50 μl/Kammer mit Tetanustoxoid mit 3 μg/ml Bicarbonatpuffer beschichtet, über Nacht bei 4°C inkubiert und 3 × mit PBS, welche 0,05 % Tween 20 (PBS-t) enthielt, in einem automatischen Plattenwäscher gewaschen. Die Platten wurden dann mit 200 μl/Kammer 0,5 % Gelatine in PBS für 1 Stunde bei Raumtemperatur (RT) blockiert und 3 × mit PBS-t gewaschen. Das Mäuseserum wurde in PBS-t, welche 1,0 % BSA enthielt, verdünnt und 100 μl unterschiedliche Verdünnungen wurden den Antigen-beschichteten Kammern zugegeben, bei Raumtemperatur für 1 Stunde inkubiert und wieder 3 × mit PBS-t gewaschen. Biotinyliertes Ratten-Anti-Maus IgG (Zymed, South San Francisco, CA) wurde auf 50 ng/ml in PBS-t verdünnt und es wurden 100 μl/Kammer angewandt. Nach Inkubation bei Raumtemperatur für 1 Stunde wurden die Kammern 3 × mit PBS-t gewaschen, gefolgt von der Zugabe von 40 ng/ml Streptavidin-Meerrettichperoxidase-Konjugat (Southern Biotechnology Associates Inc., Birmingham, AL) in PBS-t. Nach Inkubation für 30 Minuten bei RT (Raumtemperatur) wurden die Kammern wieder 3 × mit PBS-t gewaschen. Die Kammern wurden dann für 5 Minuten in 100 μl TMB-Substrat (Kirkegaard and Perry Labs) inkubiert. Eine Farbentwicklung wurde durch die Zugabe eines gleichen Volumens 1M Phosphorsäure gestoppt und die Absorbanz wurde bei 450 nm auf einem Titertek Multiscan Plattenleser mit dem Deltasoft-Softwareanalysepaket abgelesen.
- Für die quantitative Bestimmung der Antigen-spezifischen IgG-Pegel wurden Kurven mit einem bekannten Standard verglichen. Die gesamte IgG-Untersuchung wurde mittels direkt adhärierten bekannten Mengen von gereinigtem IgG (erworben von Southern Biotech) als eine IgG-Standardkurve zusammen mit dem direkten ELISA für das Tetanustoxoid durchgeführt. Dies erlaubte die Ermittlung von gebundenem Antikörper durch die gleichen Reagenzien, welche verwendet wurden, um die Antigen-spezifische Anhaftung von Antikörpern zu messen. Die gleichen Reagenslösungen, welche für den Nachweis von Antikörpern gebunden an Tetanustoxoid verwendet wurden, nämlich biotinylierter Anti-IgG gefolgt von HRP-Streptavidin, wurden simultan auf die gesamte quantitative IgG-Untersuchung und die Antigen-spezifische Untersuchung angewandt. Daher entsprach das Signal von der Bindung der Standardkurve des gereinigten IgG dem, welches für gleiche Mengen an IgG gebunden in der Anti-Tetanustoxoid-Untersuchung erzeugt wurde. Die Menge des Tetanustoxoid-spezifischen Antikörpers im Serum wurde dann mittels einer 4-Parameter-Kurvenpassung (Deltasoft 3 Softwarepaket) von dem gereinigten IgG-Standard interpoliert.
- Tabelle 2. Adjuvansaktivität von Verbindungen in vivo

- Während die vorangehende Erfindung zum Zweck der Klarheit und des Verständnisses recht detailliert beschrieben wurde, wird der Fachmann aus der Lektüre dieser Offenbarung verstehen, dass verschiedene Veränderungen an der Form und in Einzelheiten vorgenommen werden können, ohne sich vom wahren Umfang der Erfindung und den angehängten Ansprüchen zu entfernen.
d und e unabhängig voneinander eine ganze Zahl von 1 bis ungefähr 6 sind;
d' und e' unabhängig voneinander eine ganze Zahl von 0 bis ungefähr 2 sind;
X1 und Y1 unabhängig voneinander ausgewählt aus der Gruppe bestehend aus Null, Sauerstoff, -NH-, -N(C(O)C1-4-Alkyl)- und -N(C1-4-Alkyl)sind;
G1, G2, G3 und G4 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus Sauerstoff, Methylen, -NH-, -N(C1-4-Alkyl)-, -N[C(O)-C1-4-Alkyl]-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH-, -O-C(O)-O-, -NH-C(O)-NH-, -C(O)NH-, C(O)N(C1-4-Alkyl), und -S(O)n-, wobei n 0, 1 oder 2 ist;
R2, R3, R4, R5, R6 und R7 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus:
d und e unabhängig voneinander eine ganze Zahl von 1 bis ungefähr 6 sind;
d' und e' unabhängig voneinander eine ganze Zahl von 0, bis ungefähr 2 sind;
X1 und Y1 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus Null, Sauerstoff, -NH-, -N(C(O)C1-4-Alkyl)- und -N(C1-4-Alkyl)-;
G1, G2, G3 und G4 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus Sauerstoff, Methylen, -NH-, -N(C1-4-Alkyl)-, -N[C(O)-C1-4-Alkyl]-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH-, -O-C(O)-O-, -NH-C(O)-NH-, -C(O)NH-, C(O)N(C1-4-Alkyl), und -S(O)n-, wobei n 0, 1 oder 2 ist;
R2, R3, R4, R5, R6 und R7 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus:
d und e unabhängig voneinander eine ganze Zahl von 1 bis ungefähr 6 sind;
d' und e' unabhängig voneinander eine ganze Zahl von 0 bis ungefähr 2 sind;
X1 und Y1 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus Null, Sauerstoff, -NH-, -N(C(O)C1-4-Alkyl)- und -N(C1-4-Alkyl)-;
G1, G2, G3 und G4 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus Sauerstoff, Methylen, -NH-, -N(C1-4-Alkyl)-, -N[C(O)-C1-4-Alkyl]-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH-, -O-C(O)-O-, -NH-C(O)-NH-, -C(O)NH-, C(O)N(C1-4-Alkyl), und -S(O)n-, wobei n 0, 1 oder 2 ist;
R2, R3, R4, R5, R6 und R7 unabhängig voneinander ausgewählt sind aus der Gruppe bestehend aus:
Claims (19)
- Verbindung der Formel I:wobei R1 ausgewählt ist aus der Gruppe, bestehend aus (a) -C(O); (b) -C(O)-C1-14-Alkylen-C(O)- oder -C(O)-C1-14-Alkenylen-C(O)-, wobei das C1-14-Alkylen oder C1-14-Alkenylen wahlweise mit Hydroxy, C1-6-Alkoxy, C1-6-Alkylendioxy, Carboxy, C1-6-Alkoxycarbonyl, C1-6-Carbamoyl, C1-6-Acylamino, C1-6-Alkylamino oder (Aryl)C1-6-Alkyl substituiert ist, wobei der Arylanteil des (Aryl)C1-6-Alkyls wahlweise durch C1-6-Alkyl, C1-6-Alkoxy, C1-6-Alkylamino, (C1-6-Alkoxy)C1-6-Alkylamino, (C1-4-Alkylamino)C1-6-Alkoxy-, -O-C1-6-Alkylen-NH-C1-6-Alkylen-O-C1-6-Alkyl, -O-C1-6-Alkylen-NH-C(O)-C1-6-Alkylen-C(O)OH oder -O-C1-6-Alkylen-NH-C(O)-C1-6-Alkylen-C(O)-C1-6-Alkyl ersetzt ist; (c) C2 bis C15 Alkyl mit gerader oder verzweigter Kette, wahlweise substituiert durch Hydroxy oder Alkoxy; und (d) -C(O)-C6-12-Arylen-C(O)-, wobei das Arylen wahlweise durch C1-6-Alkyl, Hydroxy, C1-6-Alkoxy, Halogen, Nitro oder Amino ersetzt ist; a und b unabhängig eine ganze Zahl von 0 bis ungefähr 4 sind; d und e unabhängig eine ganze Zahl von 1 bis ungefähr 6 sind; d' und e' unabhängig eine ganze Zahl von 0 bis ungefähr 2 sind; X1 und Y1 unabhängig aus der Gruppe ausgewählt sind, eine aus Null, Sauerstoff, -NH-, -N(C(O)C1-4-Alkyl)- und -N(C1-4-Alkyl)- besteht; G1, G2, G3 und G4 unabhängig aus der Gruppe ausgewählt sind, bestehend aus Sauerstoff, Methylen, -NH-, -N(C1-4-Alkyl)-, -N[C(O)-C1-4-Alkyl]-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH-, -O-C(O)-O-, -NH-C(O)-NH-, -C(O)NH-, C(O)N(C1-4-Alkyl) und -S(O)n-, wobei n 0, 1 oder 2 ist; R2, R3, R4, R5, R6 und R7 unabhängig aus der Gruppe ausgewählt sind, bestehend aus: (a) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist; (b) C2 bis C20 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, das wahlweise durch Halo, Oxo, Hydroxy oder Alkoxy substituiert ist und (c)
 wobei R8 ist C1-6 Alkyl mit gerader oder verzweigter Kette oder C2-6 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette; G5 aus der Gruppe ausgewählt ist, bestehend aus Sauerstoff, Methylen, Arylen, -NH-, -N(C1-4-Alkyl)-, -N(C(O)-C1-4-Alkyl)-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH, -O-C(O)-O-, -NH-C(O)-NH- und -S(O)n-, wobei n 0, 1 oder 2 ist, R9 und R10 unabhängig ausgewählt sind aus der Gruppe, bestehend aus (i) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist und (ii) C2 bis C20 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist, oder eines oder zwei von G1R2, G2R4, G3R5 und G4R7 zusammen ein Wasserstoffatom oder Hydroxyl sein können; oder ein pharmazeutisch akzeptables Salz davon.
wobei R8 ist C1-6 Alkyl mit gerader oder verzweigter Kette oder C2-6 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette; G5 aus der Gruppe ausgewählt ist, bestehend aus Sauerstoff, Methylen, Arylen, -NH-, -N(C1-4-Alkyl)-, -N(C(O)-C1-4-Alkyl)-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH, -O-C(O)-O-, -NH-C(O)-NH- und -S(O)n-, wobei n 0, 1 oder 2 ist, R9 und R10 unabhängig ausgewählt sind aus der Gruppe, bestehend aus (i) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist und (ii) C2 bis C20 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist, oder eines oder zwei von G1R2, G2R4, G3R5 und G4R7 zusammen ein Wasserstoffatom oder Hydroxyl sein können; oder ein pharmazeutisch akzeptables Salz davon.
- Verbindung nach Anspruch 1, wobei a und b jeweils gleich 2 sind.
- Verbindung nach Anspruch 1, wobei X1 und Y1 jeweils NH ist.
- Verbindung nach Anspruch 1, wobei d und e jeweils 1 oder 2 sind.
- Verbindung nach Anspruch 1, wobei d' und e' jeweils 0, 1 oder 2 sind.
- Verbindung nach Anspruch 1, wobei d und e jeweils 1 und d' und e' jeweils 0 sind.
- Verbindung nach Anspruch 1, wobei d und e jeweils 1 und d' und e' jeweils 1 oder 2 sind.
- Verbindung nach Anspruch 1, wobei R1 -C(O)- oder -C(O)-C1-14-Alkylen-C(O)- ist, wobei das C1-14-Alkylen wahlweise durch einen oder zwei Substituenten substituiert ist, die ausgewählt sind aus der Gruppe, bestehend aus Hydroxy, C1-6-Alkoxy, C1-6-Alkylendioxy, C1-6-Alkylamino oder (Aryl)C1-6-Alkyl, wobei der Arylanteil des (Aryl)C1-6-Alkyls wahlweise substituiert ist mit C1-6-Alkyl, C1-6-Alkoxy, C1-6-Alkylamino, (C1-6-Alkoxy)C1-6-Alkylamino, (C1-6-Alkylamino)C1-6-Alkoxy, -O-C1-6-Alkylen-NH-C1-6-Alkylen-O-C1-6-Alkyl, -O-C1-6-Alkylen-NH-C(O)-C1-6-Alkylen-C(O)OH oder -O-C1-6-Alkylen-NH-C(O)-C1-6-Alkylen-C(O)-C1-6Alkyl.
- Verbindung nach Anspruch 1, wobei G1, G2, G3 und G4 jeweils unabhängig aus der Gruppe ausgewählt sind, bestehend aus -NH-C(O)- und -O-C(O)-.
- Verbindung nach Anspruch 1, wobei mindestens zwei von R2-R7, R9 und R10 C6-20 Alkyl, Alkenyl oder Dialkenyl mit gerader oder verzweigter Kette sind, wobei jede dieser Gruppen wahlweise durch einen oder zwei Substituenten substituiert sein kann, die ausgewählt sind aus der Gruppe, bestehend aus Halo, Oxo, Hydroxy und Alkoxy.
- Verbindung nach Anspruch 1, wobei mindestens vier von R2-R7, R9 und R10 C6-20 Alkyl, Alkenyl oder Dialkenyl mit gerader oder verzweigter Kette sind, wobei jede dieser Gruppen wahlweise durch einen oder zwei Substituenten substituiert sein kann, die ausgewählt sind aus der Gruppe, bestehend aus Halo, Oxo; Hydroxy und Alkoxy.
- Verbindung nach Anspruch 1, wobei mindestens sechs von R2-R7, R9 und R10 C6-20 Alkyl, Alkenyl oder Dialkenyl mit gerader oder verzweigter Kette sind, wobei jede dieser Gruppen wahlweise durch einen oder zwei Substituenten substituiert sein kann, die ausgewählt sind aus der Gruppe, bestehend aus Halo, Oxo, Hydroxy und Alkoxy.
- Verbindung nach Anspruch 1, wobei mindestens zwei von R2-R7, R9 und R10 C8-15 Alkyl, Alkenyl oder Dialkenyl mit gerader oder verzweigter Kette sind, wobei jede dieser Gruppen wahlweise durch einen oder zwei Substituenten substituiert sein kann, die ausgewählt sind aus der Gruppe, bestehend aus Halo, Oxo, Hydroxy und Alkoxy.
- Verbindung nach Anspruch 1, wobei mindestens vier von R2-R7, R9 und R10 C8-15 Alkyl, Alkenyl oder Dialkenyl mit gerader oder verzweigter Kette sind, wobei jede dieser Gruppen wahlweise durch einen oder zwei Substituenten substituiert sein kann, die ausgewählt sind aus der Gruppe, bestehend aus Halo, Oxo, Hydroxy und Alkoxy.
- Verbindung nach Anspruch 1, wobei mindestens sechs von R2-R7, R9 und R10 C8-15 Alkyl, Alkenyl oder Dialkenyl mit gerader oder verzweigter Kette sind, wobei jede dieser Gruppen wahlweise durch einen oder zwei Substituenten substituiert sein kann, die ausgewählt sind aus der Gruppe, bestehend aus Halo, Oxo, Hydroxy und Alkoxy.
- Immunologische Adjuvansformulierung, welche eine Verbindung der Formel I aufweist:wobei R1 ausgewählt ist aus der Gruppe, bestehend aus (a) -C(O)-; (b) -C(O)-C1-14-Alkylen-C(O)- oder -C(O)-C1-14-Alkenylen-C(O)-, wobei das C1-14-Alkylen oder C1-14-Alkenylen wahlweise durch Hydroxy, C1-6-Alkoxy, C1-6-Alkylendioxy, Carboxy, C1-6-Alkoxycarbonyl, C1-6-Carbamoyl, C1-6-Acylamino, C1-6-Alkylamino oder (Aryl)C1-6-Alkyl substituiert ist, wobei der Arylanteil des (Aryl)C1-6-Alkyls wahlweise durch C1-6-Alkyl, C1-6-Alkoxy, C1-6-Alkylamino, (C1-6-Alkoxy)C1-6-Alkylamino, (C1-6-Alkylamino)C1-6-Alkoxy-, -O-C1-6-Alkylen-NH-C1-6-Alkylen-O-C1-6-Alkyl, -O-C1-6-Alkylen-NH-C(O)-C1-6-Alkylen-C(O)OH oder -O-C1-6-Alkylen-NH-C(O)-C1-6-Alkylen-C(O)-C1-6-Alkyl ersetzt wird; (c) C2 bis C15 Alkyl mit gerader oder verzweigter Kette, wahlweise substituiert mit Hydroxy oder Alkoxy; und (d) -C(O)-C6-12-Arylen-C(O)-, wobei das Arylen wahlweise durch C1-6-Alkyl, Hydroxy, C1-6-Alkoxy, Halogen, Nitro oder Amino ersetzt wird; a und b unabhängig eine ganze Zahl von 0 bis ungefähr 4 sind; d und e unabhängig eine ganze Zahl von 1 bis ungefähr 6 sind; d' und e' unabhängig eine ganze Zahl von 0 bis ungefähr 2 sind; X1 und Y1 unabhängig aus der Gruppe ausgewählt sind, die aus Null, Sauerstoff, -NH-, -N(C(O)C1-4-Alkyl)- und -N(C1-4-Alkyl)- besteht; G1, G2, G3 und G4 unabhängig aus der Gruppe ausgewählt sind, bestehend aus Sauerstoff, Methylen, -NH-, -N(C1-4-Alkyl)-, -N[C(O)-C1-4-Alkyl]-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH-, -O-C(O)-O-, -NH-C(O)-NH-, und -C(O)NH-, C(O)N(C1-4-Alkyl), -S(O)n-, wobei n 0, 1 oder 2 ist; R2, R3, R4, R5, R6 und R7 unabhängig aus der Gruppe ausgewählt sind, bestehend aus: (a) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist; (b) C2 bis C20 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist und (c)
 wobei R8 C1-6 Alkyl mit gerader oder verzweigter Kette oder C2-6 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette ist; G5 aus der Gruppe ausgewählt ist, bestehend aus Sauerstoff, Methylen, Arylen, -NH-, -N(C1-4-Alkyl)-, -N(C(O)-C1-4-Alkyl)-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH, -O-C(O)-O-, -NH-C(O)-NH- und -S(O)n-, wobei n 0, 1 oder 2 ist, R9 und R10 unabhängig ausgewählt sind aus der Gruppe, bestehend aus (i) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist und (ii) C2 bis C20 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist, oder eines oder zwei von G1R2, G2R4, G3R5 und G4R7 zusammen ein Wasserstoffatom oder Hydroxyl sein können; oder ein pharmazeutisch akzeptables Salz davon und ein pharmazeutisch akzeptabler Träger, Verdünnungsmittel oder Arzneimittelträger.
wobei R8 C1-6 Alkyl mit gerader oder verzweigter Kette oder C2-6 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette ist; G5 aus der Gruppe ausgewählt ist, bestehend aus Sauerstoff, Methylen, Arylen, -NH-, -N(C1-4-Alkyl)-, -N(C(O)-C1-4-Alkyl)-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH, -O-C(O)-O-, -NH-C(O)-NH- und -S(O)n-, wobei n 0, 1 oder 2 ist, R9 und R10 unabhängig ausgewählt sind aus der Gruppe, bestehend aus (i) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist und (ii) C2 bis C20 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist, oder eines oder zwei von G1R2, G2R4, G3R5 und G4R7 zusammen ein Wasserstoffatom oder Hydroxyl sein können; oder ein pharmazeutisch akzeptables Salz davon und ein pharmazeutisch akzeptabler Träger, Verdünnungsmittel oder Arzneimittelträger.
- Impfstoffformulierung, welche ein Antigen und eine Adjuvansverbindung der Formel I enthält:wobei R1 ausgewählt ist aus der Gruppe, bestehend aus (a) -C(O)-; (b) -C(O)-C1-14-Alkylen-C(O)- oder -C(O)-C1-14-Alkenylen-C(O)-, wobei das C1-14-Alkylen oder C1-14-Alkenylen wahlweise mit Hydroxy, C1-6-Alkoxy, C1-6-Alkylendioxy, Carboxy, C1-6-Alkoxycarbonyl, C1-6-Carbamoyl, C1-6-Acylamino, C1-6-Alkylamino oder (Aryl)C1-6-Alkyl substituiert ist, wobei der Arylanteil des (Aryl)C1-6-Alkyls wahlweise durch C1-6-Alkyl, C1-6-Alkoxy, C1-6-Alkylamino, (C1-6-Alkoxy)C1-6Alkylamino, (C1-6-Alkylamino)C1-6-Alkoxy-, -O-C1-6-Alkylen-NH-C1-6-Alkylen-O-C1-6-Alkyl, -O-C1-6-Alkylen-NH-C(O)-C1-6-Alkylen-C(O)OH oder -O-C1-6-Alkylen-NH-C(O)-C1-6-Alkylen-C(O)-C1-6-Alkyl ersetzt wird; (c) C2 bis C15 Alkyl mit gerader oder verzweigter Kette, wahlweise substituiert mit Hydroxy oder Alkoxy; und (d) -C(O)-C6-12-Arylen-C(O)-, wobei das Arylen wahlweise mit C1-6-Alkyl, Hydroxy, C1-6-Alkoxy, Halogen, Nitro oder Amino substituiert ist; a und b unabhängig eine ganze Zahl von 0 bis ungefähr 4 sind; d und e unabhängig eine ganze Zahl von 1 bis ungefähr 6 sind; d' und e' unabhängig eine ganze Zahl von 0 bis ungefähr 2 sind; X1 und Y1 unabhängig aus der Gruppe ausgewählt sind, die aus Null, Sauerstoff, -NH-, -N(C(O)C1-4-Alkyl)- und -N(C1-4-Alkyl)- besteht; G1, G2, G3 und G4 unabhängig aus der Gruppe ausgewählt sind, bestehend aus Sauerstoff, Methylen, -NH-, -N(C1-4-Alkyl)-, -N[C(O)-C1-4-Alkyl]-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH-, -O-C(O)-O-, -NH-C(O)-NH-, -C(O)NH-, C(O)N(C1-4-Alkyl) und -S(O)n-, wobei n 0, 1 oder 2 ist; R2, R3, R4, R5, R6 und R7 unabhängig aus der Gruppe ausgewählt sind, bestehend aus: (a) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist; (b) C2 bis C20 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist und (c)
 wobei R8 C1-6 Alkyl mit gerader oder verzweigter Kette oder C2-6 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette ist; G5 aus der Gruppe ausgewählt ist, bestehend aus Sauerstoff, Methylen, Arylen, -NH-, -N(C1-4-Alkyl)-, -N(C(O)-C1-4-Alkyl)-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH, -O-C(O)-O-, -NH-C(O)-NH- und -S(O)n-, wobei n 0, 1 oder 2 ist, R9 und R10 unabhängig ausgewählt sind aus der Gruppe, bestehend aus (i) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist und (ii) C2 bis C20 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist, oder eines oder zwei von G1R2, G2R4, G3R5 und G4R7 zusammen ein Wasserstoffatom oder Hydroxyl sein können; oder ein pharmazeutisch akzeptables Salz davon.
wobei R8 C1-6 Alkyl mit gerader oder verzweigter Kette oder C2-6 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette ist; G5 aus der Gruppe ausgewählt ist, bestehend aus Sauerstoff, Methylen, Arylen, -NH-, -N(C1-4-Alkyl)-, -N(C(O)-C1-4-Alkyl)-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH, -O-C(O)-O-, -NH-C(O)-NH- und -S(O)n-, wobei n 0, 1 oder 2 ist, R9 und R10 unabhängig ausgewählt sind aus der Gruppe, bestehend aus (i) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist und (ii) C2 bis C20 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist, oder eines oder zwei von G1R2, G2R4, G3R5 und G4R7 zusammen ein Wasserstoffatom oder Hydroxyl sein können; oder ein pharmazeutisch akzeptables Salz davon.
- Wirksame Menge eines Antigens und eine wirksame Menge einer Adjuvansverbindung der Formel I zur Verwendung bei der Stimulierung einer Immunreaktion auf das Antigen:wobei R1 ausgewählt ist aus der Gruppe, bestehend aus: (a) -C(O)-; (b) -C(O)-C1-14-Alkylen-C(O)- oder -C(O)-C1-14-Alkenylen-C(O)-, wobei das C1-14-Alkylen oder C1-14-Alkenylen wahlweise mit Hydroxy, C1-6-Alkoxy, C1-6-Alkylendioxy, Carboxy, C1-6-Alkoxycarbonyl, C1-6-Carbamoyl, C1-6-Acylamino, C1-6-Alkylamino oder (Aryl)C1-6-Alkyl substituiert ist, wobei der Arylanteil des (Aryl)C1-6-Alkyls wahlweise mit C1-6-Alkyl, C1-6-Alkoxy, C1-6-Alkylamino, (C1-6-Alkoxy)C1-6-Alkylamino, (C1-6-Alkylamino)C1-6-Alkoxy-, -O-C1-6-Alkylen-NH-C1-6-Alkylen-O-C1-6-Alkyl, -O-C1-6-Alkylen-NH-C(O)-C1-6-Alkylen-C(O)OH oder -O-C1-6-Alkylen-NH-C(O)-C1-6-Alkylen-C(O)-C1-6-Alkyl substituiert ist; (c) C2 bis C15 Alkyl mit gerader oder verzweigter Kette, wahlweise substituiert mit Hydroxy oder Alkoxy; und (d) -C(O)-C6-12-Arylen-C(O)-, wobei das Arylen wahlweise mit C1-6-Alkyl, Hydroxy, C1-6-Alkoxy, Halogen, Nitro oder Amino substituiert ist; a und b unabhängig eine ganze Zahl von 0 bis ungefähr 4 sind; d und e unabhängig eine ganze Zahl von 1 bis ungefähr 6 sind; d' und e' unabhängig eine ganze Zahl von 0 bis ungefähr 2 sind; X1 und Y1 unabhängig aus der Gruppe ausgewählt sind, die aus Null, Sauerstoff, -NH-, -N(C(O)C1-4-Alkyl)- und -N(C1-4-Alkyl)- besteht; G1, G2, G3 und G4 unabhängig aus der Gruppe ausgewählt sind, bestehend aus Sauerstoff, Methylen, -NH-, -N(C1-4-Alkyl)-, -N[C(O)-C1-4-Alkyl]-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH-, -O-C(O)-O-, -NH-C(O)-NH-, -C(O)NH-, C(O)N(C1-4-Alkyl) und -S(O)n-, wobei n 0, 1 oder 2 ist; R2, R3, R4, R5, R6 und R7 unabhängig aus der Gruppe ausgewählt sind, bestehend aus: (a) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist; (b) C2 bis C20 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist und (c)
 wobei R8 C1-6 Alkyl mit gerader oder verzweigter Kette oder C2-6 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette ist; G5 aus der Gruppe ausgewählt ist, bestehend aus Sauerstoff, Methylen, Arylen, -NH-, -N(C1-4-Alkyl)-, -N(C(O)-C1-4-Alkyl)-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH, -O-C(O)-O-, -NH-C(O)-NH- und -S(O)n-, wobei n 0, 1 oder 2 ist, R9 und R10 unabhängig ausgewählt sind aus der Gruppe, bestehend aus (i) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist und (ii) C2 bis C20 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist, oder eines oder zwei von G1R2, G2R4, G3R5 und G4R7 zusammen ein Wasserstoffatom oder Hydroxyl sein können; oder ein pharmazeutisch akzeptables Salz davon.
wobei R8 C1-6 Alkyl mit gerader oder verzweigter Kette oder C2-6 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette ist; G5 aus der Gruppe ausgewählt ist, bestehend aus Sauerstoff, Methylen, Arylen, -NH-, -N(C1-4-Alkyl)-, -N(C(O)-C1-4-Alkyl)-, -NH-C(O)-, -NH-SO2-, -C(O)-O-, -C(O)-NH-, -O-C(O)-, -O-C(O)-NH, -O-C(O)-O-, -NH-C(O)-NH- und -S(O)n-, wobei n 0, 1 oder 2 ist, R9 und R10 unabhängig ausgewählt sind aus der Gruppe, bestehend aus (i) C1 bis C20 Alkyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist und (ii) C2 bis C20 Alkenyl, Alkynyl oder Dialkenyl mit gerader oder verzweigter Kette, das wahlweise mit Halo, Oxo, Hydroxy oder Alkoxy substituiert ist, oder eines oder zwei von G1R2, G2R4, G3R5 und G4R7 zusammen ein Wasserstoffatom oder Hydroxyl sein können; oder ein pharmazeutisch akzeptables Salz davon.
- Verwendung einer wirksamen Menge eines Antigens und einer wirksamen Menge einer Adjuvansverbindung nach Anspruch 18 bei der Herstellung einer Behandlung zur Verwendung bei der Stimulierung einer Immunreaktion auf das Antigen.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US22175200P | 2000-07-31 | 2000-07-31 | |
| US221752P | 2000-07-31 | ||
| PCT/IB2001/001658 WO2002009752A2 (en) | 2000-07-31 | 2001-07-27 | Immunological adjuvant compounds |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE60115051D1 DE60115051D1 (de) | 2005-12-22 |
| DE60115051T2 true DE60115051T2 (de) | 2006-08-24 |
Family
ID=22829213
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE60115051T Expired - Lifetime DE60115051T2 (de) | 2000-07-31 | 2001-07-27 | Immunologische adjuvans verbindungen |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US6521776B2 (de) |
| EP (1) | EP1307466B1 (de) |
| JP (1) | JP4230765B2 (de) |
| AT (1) | ATE310008T1 (de) |
| AU (1) | AU2001284354A1 (de) |
| DE (1) | DE60115051T2 (de) |
| TW (1) | TWI305775B (de) |
| WO (1) | WO2002009752A2 (de) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040006242A1 (en) | 1999-02-01 | 2004-01-08 | Hawkins Lynn D. | Immunomodulatory compounds and method of use thereof |
| US7915238B2 (en) | 1999-02-01 | 2011-03-29 | Eisai R & D Management Co., Ltd. | Immunomodulatory compounds and methods of use thereof |
| US6835721B2 (en) | 1999-02-01 | 2004-12-28 | Eisai Co., Ltd. | Immunomodulatory compounds and methods of use thereof |
| US6551600B2 (en) * | 1999-02-01 | 2003-04-22 | Eisai Co., Ltd. | Immunological adjuvant compounds compositions and methods of use thereof |
| TW200523235A (en) * | 2003-10-15 | 2005-07-16 | Gtx Inc | Anti-cancer compounds and methods of use thereof |
| JP5185813B2 (ja) | 2005-04-26 | 2013-04-17 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | 癌免疫治療のための組成物および方法 |
| US20070292418A1 (en) * | 2005-04-26 | 2007-12-20 | Eisai Co., Ltd. | Compositions and methods for immunotherapy |
| EP1910387B1 (de) * | 2005-06-30 | 2016-11-16 | Eisai R&D Management Co., Ltd. | Verbindungen zur herstellung von immunologischen adjuvantien |
| KR20080080087A (ko) * | 2005-09-30 | 2008-09-02 | 티티아이 엘뷰 가부시키가이샤 | 신규 약학 담체를 이용한 경피 약물 전달시스템, 장치 및방법 |
| KR20080066712A (ko) * | 2005-09-30 | 2008-07-16 | 티티아이 엘뷰 가부시키가이샤 | 관능화된 미세바늘 경피 약물 전달 시스템, 장치 및 방법 |
| US20070078376A1 (en) * | 2005-09-30 | 2007-04-05 | Smith Gregory A | Functionalized microneedles transdermal drug delivery systems, devices, and methods |
| US20080033398A1 (en) * | 2005-12-29 | 2008-02-07 | Transcutaneous Technologies Inc. | Device and method for enhancing immune response by electrical stimulation |
| WO2009079627A2 (en) * | 2007-12-18 | 2009-06-25 | Eisai R & D Management Co., Ltd. | Reagents and methods for the beta-keto amide synthesis of a synthetic precursor to immunological adjuvant e6020 |
| JPWO2018070543A1 (ja) * | 2016-10-14 | 2019-07-25 | 株式会社ボナック | 新規な配糖体化合物及びその製造方法 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2583892A (en) | 1991-09-11 | 1993-04-05 | Pitman-Moore, Inc. | Method for enhancing the immune system in a host employing liposome-encapsulated polypeptides |
| US5961970A (en) | 1993-10-29 | 1999-10-05 | Pharmos Corporation | Submicron emulsions as vaccine adjuvants |
| AU5543294A (en) | 1993-10-29 | 1995-05-22 | Pharmos Corp. | Submicron emulsions as vaccine adjuvants |
| DE4499552C1 (de) | 1993-12-09 | 1997-07-17 | Heinrich Dr Exner | Adjuvans für Antigene, Verfahren zur Herstellung und Verwendung |
| GB9326253D0 (en) | 1993-12-23 | 1994-02-23 | Smithkline Beecham Biolog | Vaccines |
| DE19511276C2 (de) * | 1995-03-27 | 1999-02-18 | Immuno Ag | Adjuvans auf der Basis kolloidaler Eisenverbindungen |
| US5834015A (en) | 1996-09-11 | 1998-11-10 | Albany Medical College | Protein-lipid vesicles and autogenous vaccine comprising the same |
| US6306404B1 (en) * | 1998-07-14 | 2001-10-23 | American Cyanamid Company | Adjuvant and vaccine compositions containing monophosphoryl lipid A |
| KR100711561B1 (ko) * | 1999-02-01 | 2007-04-27 | 에-자이가부시기가이샤 | 면역학적 보조제 화합물 |
-
2001
- 2001-07-27 EP EP01963334A patent/EP1307466B1/de not_active Expired - Lifetime
- 2001-07-27 AT AT01963334T patent/ATE310008T1/de not_active IP Right Cessation
- 2001-07-27 DE DE60115051T patent/DE60115051T2/de not_active Expired - Lifetime
- 2001-07-27 JP JP2002515305A patent/JP4230765B2/ja not_active Expired - Lifetime
- 2001-07-27 AU AU2001284354A patent/AU2001284354A1/en not_active Abandoned
- 2001-07-27 WO PCT/IB2001/001658 patent/WO2002009752A2/en active IP Right Grant
- 2001-07-30 US US09/919,049 patent/US6521776B2/en not_active Expired - Lifetime
- 2001-07-31 TW TW090118694A patent/TWI305775B/zh not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| ATE310008T1 (de) | 2005-12-15 |
| EP1307466A2 (de) | 2003-05-07 |
| WO2002009752A3 (en) | 2002-06-27 |
| DE60115051D1 (de) | 2005-12-22 |
| JP2004505059A (ja) | 2004-02-19 |
| EP1307466B1 (de) | 2005-11-16 |
| JP4230765B2 (ja) | 2009-02-25 |
| US20020049314A1 (en) | 2002-04-25 |
| TWI305775B (en) | 2009-02-01 |
| US6521776B2 (en) | 2003-02-18 |
| AU2001284354A1 (en) | 2002-02-13 |
| WO2002009752A2 (en) | 2002-02-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE60011571T2 (de) | Verbindungen mit immunologischem adjuvanseffekt | |
| DE60115051T2 (de) | Immunologische adjuvans verbindungen | |
| DE3836006C2 (de) | Monophosphoryllipid-A-Derivate, Verfahren zur Herstellung von Monophosphoryllipid-A-Derivaten und Verwendung von Monophosphoryllipid-A-Derivaten | |
| US4185089A (en) | Immunological adjuvant constituted by the p-amino-phenyl derivative of N-acetyl-muramyl-L-alanyl-D-isoglutamine | |
| DE2655500C2 (de) | ||
| EP0688789B1 (de) | Lipopeptid-Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung | |
| US20020176861A1 (en) | Immunological adjuvant compounds compositions and methods of use thereof | |
| DE2728324A1 (de) | Muramyldipeptidderivate und verfahren zu ihrer herstellung | |
| EP0003833B1 (de) | Antigenderivate, Verfahren zu deren Herstellung, diese enthaltende pharmazeutische Präparate | |
| US6835721B2 (en) | Immunomodulatory compounds and methods of use thereof | |
| EP0431327B2 (de) | Synthetische Vakzine zur spezifischen Induktion zytotoxischer T-Lymphozyten | |
| DE2922533A1 (de) | Oligomere von muramylpeptidartigen verbindungen und sie enthaltende arzneimittel | |
| DE2547104A1 (de) | N-acetyl-muramyl-l-alanyl-d-glutaminsaeure enthaltende oelfreie adjuvanzien | |
| EP0014159B1 (de) | Immunologisch aktive Dipeptidyl-Saccharide und Verfahren zur Herstellung | |
| EP0338437A2 (de) | Synthetische Vakzine gegen die Maul- und Klauenseuche und Verfahren zu deren Herstellung | |
| DE2710455C2 (de) | N-Acetylmuramyl-L-alanyl-D-glutaminsäure-alpha-methylamid, Verfahren zu seiner Herstellung und diese Verbindung enthaltende Arzneimittel | |
| DE2710457A1 (de) | 2-(2-acetamido-2-desoxy-3-0-d-glucopyranosyl)-d-propionyl-l-seryl-isoglutamin, 2-(2-acetamido-2-desoxy-3-0- d-glucopyranosyl)-d-propionyl-l-seryl- d-glutaminsaeure, die amid- und ester- derivate dieser verbindungen, verfahren zu ihrer herstellung und diese verbindungen als wirkstoffe enthaltende arzneimittel | |
| DE2710454C2 (de) | Ester von N-Acetylmuramyl-L-alanyl-D-isoglutamin sowie Mono- und Diester von N-Acetylmuramyl-L-alanyl-D-glutaminsäure, Verfahren zu deren Herstellung und diese Verbindungen enthaltende pharmazeutische Zusammensetzungen | |
| JPS59175495A (ja) | 免疫賦活能を有するジペプチジルd−グルコ−ス誘導体とその製造方法 | |
| AT373266B (de) | Verfahren zur herstellung neuer glucosederivate | |
| AT373267B (de) | Verfahren zur herstellung neuer glucosederivate | |
| DE3710770A1 (de) | Neue alkansaeurealkylester, verfahren zu ihrer herstellung und verwendung von waessrigen emulsionen dieser ester in vaccinen |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition |