-
Das
Verfahren besteht aus der Umsetzung geeigneter Nitril-Vorstufen
der Strukturen I oder III mit Stickstoffverbindungen zu den gewünschten
substituierten Pyridinen II.
-
-
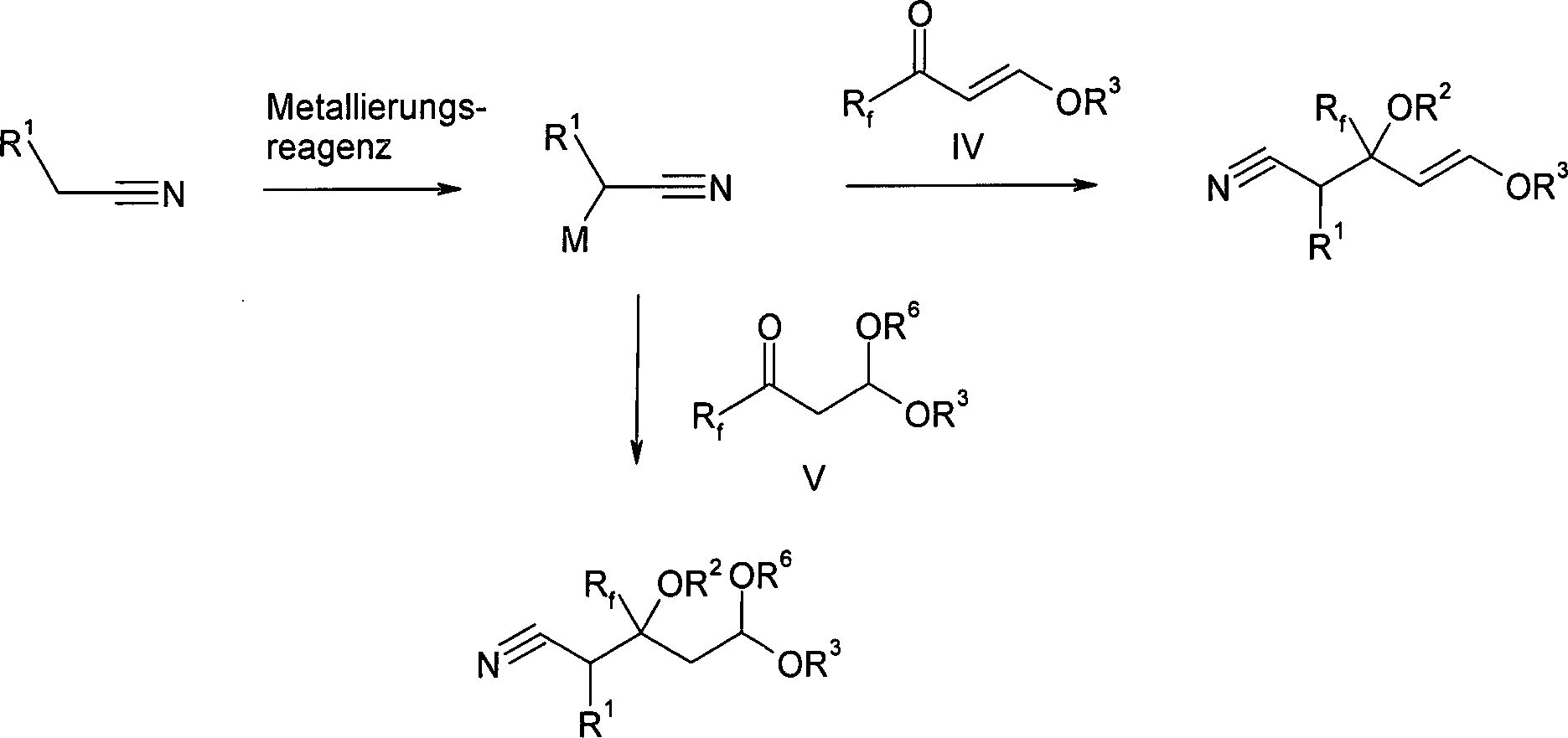
Die
Herstellung der Ausgangprodukte I oder III erfolgt dabei im allgemeinen
durch Addition eines alpha-metallierten Nitrils an eine Carbonylverbindung
IV oder V gemäß einer der beiden folgenden Reaktionsgleichungen:
-
R2 ist dann im allgemeinen gleich Wasserstoff.
Es können aber auch andere geeignete Reste eingeführt
werden (z. B. durch Umsetzung des nach der Additionsreaktion und
vor der wäßrigen Aufarbeitung intermediär
entstehenden Alkoholates mit einem anderen Elektrophil), die die
nachfolgende Umsetzung nicht stören, z. B. um die Stabilität
der Verbindungen I und III zu erhöhen.
-
[Hintergrund]
-
Pyridine
sind wichtige Strukturelemente in einer Vielzahl von Produkten der
chemischen und pharmazeutischen Industrie und es sind in der Literatur
sehr viele verschiedene Verfahren zur Herstellung beschrieben. Diese
können grob unterteilt werden in Verfahren, bei denen der
Pyridinring aufgebaut wird und solche, bei denen Substituenten eingeführt
(z. B. durch elektrophile oder nucleophile Substituion am Aromaten)
oder modifiziert werden. Werden 2-Aminopyridinderivate benötigt,
so erfolgt die Herstellung meistens durch Einführung des
Amin-Substituenten in einen bereits vorhandenen Pyridinring. Beispiele
für solche Reaktionen sind die Tschitschibabin-Reaktion
(siehe z. B.
DE 374291 ),
also die Umsetzung von Pyridinen mit Natriumamid unter Eliminierung
von Natriumhydrid oder die Umsetzung von 2-Halogenpyridinen mit
Stickstoffverbindungen (siehe z. B.
Chem. Ber. 1936, 69,
2593 für die Umsetzung von 3-Amino-2-chlorpyridin
zu 2,3-Diaminopyridin).
-
-
-
Diese
Reaktionen haben jedoch den Nachteil, daß entweder recht
drastische Bedingungen nötig sind (Tschitschibabin-Reaktion,
Reaktion von 2-Halogenpyridinen mit wäßrigem Ammoniak)
oder daß bei den neueren katalytischen Varianten zur Umsetzung
teure Edelmetalle und Liganden benötigt werden (z. B. Org.
Lett. 2001, 3, 3417).
-
Ein
weiterer Nachteil der beschriebenen Reaktionen ist die Tatsache,
daß zur Synthese von schwierig substituierten Pyridinen
die entsprechenden Vorstufen verfügbar sein müssen,
was oft nicht der Fall. Außerdem muß für
das Einführen des gewünschten Substituenten in
die gewünschte Position ein technisch durchführbares
Verfahren zur Verfügung stehen, das die Umwandlung der
Vorstufe auch jenseits des Labormaßstabs erlaubt.
-
Dies
ist insbesondere oft dann nicht der Fall, wenn Perfluoralkylgruppen
(meist Trifluormethylgruppen) in den Pyridin-Ring eingeführt
werden sollen. Zwar sind hier einige Reaktionen in der Literatur
beschrieben, wie die Umsetzung von Iodpyridinen mit (Trifluormethyl)trimethylsilan
oder die Umwandlung von Methylgruppen
in Trifluormethylgruppen durch Einwirkung von Chlor und Flußsäure.
-
-
Alle
bisher bekannten Verfahren haben aber Nachteile, die die Verwendung
für die Herstellung der gesuchten Aminopyridine unattraktiv
macht bzw. ausschließt. So sind die Aminopyridine unter
den drastischen Bedingungen der Umwandlung von Methyl- in Trifluormethylgruppen
nicht stabil. Es müßten also zunächst
andere Pyridinderivate, z. B. Halogenpyridine herstellt werden und
in einem gesonderten Schritt die Umwandlung von Halogen in Amin
durchgeführt werden, was in aufwendigen und teueren Verfahren
resultiert. Die Einführung einer Trifluormethylgruppe durch
Umwandlung eines Iodpyridins mit Hilfe von (Trifluormethyl)trimethylsilan
ist für den technischen Maßstab wegen der hohen
Preise der Startmaterialien ebenfall kaum attraktiv.
-
Gesucht
war daher ein Verfahren, mit dessen Hilfe die gesuchten 2-Aminopyridin-Derivate
mit hoher Flexibilität bezüglich des Substitutionsmusters
hergestellt werden können und mit dem insbesondere Perfluoralkylsubstituenten,
bevorzugt Trifluormethylsubstituenten, in den Pyridinring eingebaut
werden können.
-
Ein ähnliches
Verfahren wurde bereits für 2-Halogenpyridine ausgearbeitet.
Bei diesem Verfahren wird ein Nitril zunächst metalliert
und dann mit einer geeigneten Carbonylverbindung zum Hydroxynitril
umgesetzt. Der finale Ringschluß erfolgt dann unter stark
sauren Bedingungen mit HX (HCl, HBr, HI) oder anorganischen Ester
dieser Substanzen (z. B. SOCl2, POCl3, PCl5, PBr3 etc.) unter sehr stark sauren Bedingungen.
Beispielhaft ist diese Reaktion für die Synthese von 4-Trifluormethyl-2-chlorpyridin
erläutert.
-
-
Um
hiervon ausgehend das entsprechende Aminopyridin herstellen zu können,
ist jedoch eine weitere Umsetzung mit Ammoniak nötig, die
unter drastischen Bedingungen abläuft (siehe
EP 228846 B1 oder
Dunn et
al. in J. Fluor. Chem 1999, Seite 153) und hohe Temperaturen
und hohen Druck erfordert.
-
-
Es
bestand daher die Aufgabe, ein ökonomisches und technisch
einfach durchführbares Verfahren für die Herstellung
von 2-Amino-4-(fluoralkyl)pyridin-Derivaten zu entwickeln.
-
Es
wurde nun völlig überraschend gefunden, daß sich
diese Aufgabe durch direkte Umsetzung der beschriebenen Nitrilvorstufen
mit Ammonak oder anderen geeigneten Stickstoffverbindungen lösen
läßt.
-
-
Dies
ist vor allem auch deshalb sehr überraschend, weil das
Arbeiten unter stark sauren Bedingungen in der weiter oben beschriebenen
Umsetzung zum Halogenpyridin als entscheidend für den Erfolg
angesehen wurde.
-
[Beschreibung]
-
Es
resultiert so ein allgemeines und flexibles Verfahren, mit dem 2-Aminopyridine
mit Perfluoralkylsubstituenten durch Aufbau des Pyridinringes hergestellt
werden können:
-
Bei
den verschiedenen Resten R im obigen Schema kann es sich um folgende
Substituenten handeln:
Rf: CnH(2n+1-m)Xm
X: F, Cl, Br
n: positive ganze
Zahl
m: positive ganze Zahl kleiner oder gleich 2n + 1
R1: Wasserstoff, Alkyl, Aryl, Heteroaryl,
COOR, CN, SO2R, SOR, PO(OR)2
R:
Alkyl, Aryl, Heteroaryl
R2, Wasserstoff
oder typische Schutzgruppen für Alkohole, z. B. Acyl, Alkyl,
2-tetrahydropyranyl, R3Silyl
R3, R6:Alkyl, Acyl,
Aryl, R3Silyl R4,
R5:Wasserstoff, Alkyl, Aryl, Heteroaryl,
Acyl, -CONR2, beide Substituenten können
dabei auch Teil eines Ringsystems sein
-
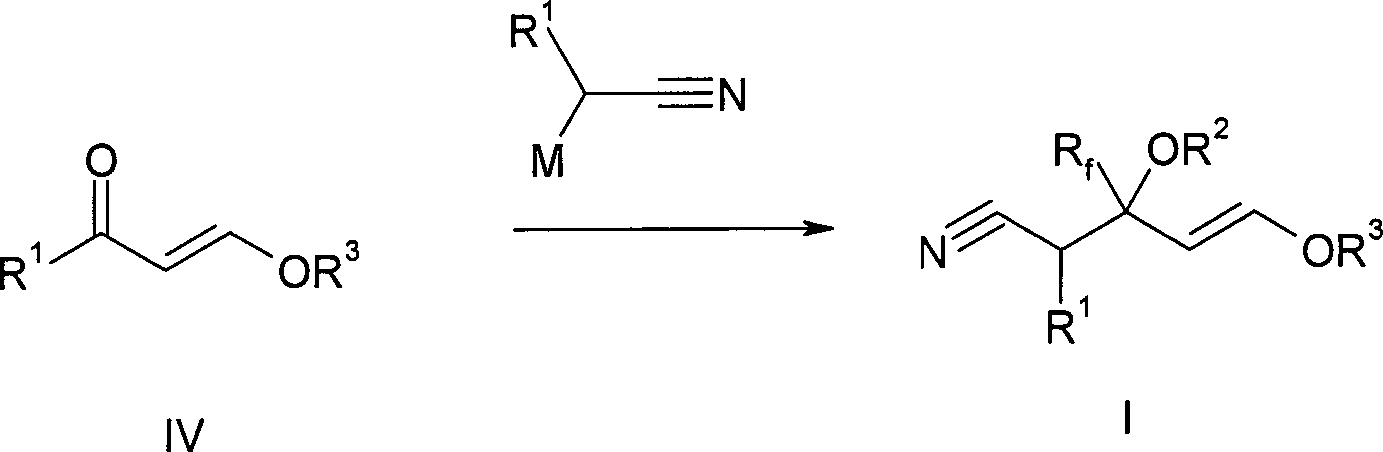
Die
Herstellung der benötigten Nitrilvorstufen kann dabei nach
jedem beliebigen Verfahren erfolgen. Meist dürfte aber
die Herstellung nach einem der beiden folgenden Schemata ausgehend
vom ungesättigten Keton IV oder vom Acetal V besonders ökonomisch
sein:
-
Die
Bedeutung der Reste R ist dabei die gleiche, wie oben erläutert.
Zusätzlich steht M für die folgenden Metalle:
M:
Li, Na, K, MgY, Mg0,5, CaY, Ca0,5,
ZnY, Zn0,5, CdY, Cd0,5,
Cu, TiY3
Y: X (wie oben), I, OR, O-CO-R
-
[Synthese der zur Cyclisierung benötigten
Nitrile]
-
Zur
Synthese der zur Cyclisierung benötigten Nitrile ist, wie
oben in den Formelschemata beschrieben, im allgemeinen der Zugang
aus den Ketonen IV oder V und einem Salz eines Acetonitril-Derivates
der günstigste Weg. Dazu wird zunächst Acetonitril
oder ein substituiertes Derivat in einem geeigneten Lösungsmittel metalliert
und das entstandene Salz dann mit einem Keton der allgemeinen Formel
IV oder V umgesetzt.
-
-
Für
diese Reaktion sind alle Lösungsmittel geeignet, die für
Metallierungsreaktionen eingesetzt werden können. Dies
sind insbesondere Ether wie Tetrahydrofuran, 2-Methyltetrahydrofuran,
Diethylether, Diisopropylether, Di-n-butylether, Dioxan, 1,2-Dimethoxyethan,
Diethylengylcoldimethylether, Diethylenglcoldi-n-butylether, Tetraethylenglycoldimethylether
oder Mischungen dieser Lösungsmittel untereinander oder
mit einem inerten anderen Lösungsmittel wie Benzol, Toluol,
Xylol, Cyclohexan oder Petrolethern (Kohlenwasserstoffgemische).
In besonderen Fällen können aber auch reine Kohlenwasserstoffe
wie Benzol, Toluol, Xylol, Cyclohexan oder Petrolether geeignet
sein oder im Falle von stark aciden Acetonitril-Derivaten (R1 starker Akzeptor-Substituent) sogar Alkohole
wie Methanol, Ethanol, Isopropanol oder Butanole.
-
Als
Metallierungsreagenzien kommen alle Basen in Frage, die ausreichend
basisch sind, um ein Wasserstoffatom von dem gegebenenfalls substituierten
Acetonitrilen zu abstrahieren. Bei Acetonitril selbst oder Alkyl-substituierten
Acetonitrilen kommen dafür hauptsächlich sehr
starke Basen wie n-Butyllithium, sec-Butyllithium, t-Butyllithium,
n-Hexyllithium, Lithium-N,N-diisopropylamid (LDA), Lithium-2,2,6,6-tetramethylpiperidid (Li-TMP), Lithiumhexamethyldisilazan
(LiHMDS), Natriumhexamethyldisilazan (NaHMDS) oder Kaliumhexamethyldisilazan
(KHMDS) in Frage. Bei etwas acideren Acetonitril-Derviaten wie beispielsweise
Aryl-substituierten (R5 = Aryl) sind Basen
wie Natriumamid, Lithiumhydrid, Natriumhydrid oder Kaliumhydrid
zusätzlich zu den oben genannten geeignet. Bei den am stärksten
aciden Acetonitril-Derivaten (R5 = COOR, CN, SO2R, SOR,
PO(OR)2) sind zusätzlich zu den
bereits genannten starken Basen auch Alkoxide wie die Lithium- Natrium-,
oder Kaliumsalze von Methanol, Ethanol oder t-Butanol als Basen
geeignet.
-
Die
Reaktionsbedingungen, die bei der Metallierung einzuhalten sind,
hängen wiederum von den verwendeten Acetonitril-Derivaten
ab. So wird bei den am wenigsten aciden Acetonitril-Derivaten (R1 = Alkyl oder Wasserstoff) bevorzugt bei
Temperaturen unter –25°C gearbeitet und besonders
bevorzugt unter –45°C, um die Zersetzung der gebildeten
Salze zu vermeiden. Die acideren Acetonitril-Derivate können
wegen der größeren Stabilität der gebildeten
Salze auch bei höheren Temperaturen metalliert werden (R1 = Aryl bis zu ca. 0°C; R1 = CN, COOR, SO2R,
SOR auch bei Raumtemperatur oder sogar darüber).
-
Die
sich anschließende Umsetzung mit geeigneten Ketonen mit
den allgemeinen Formeln IV oder V wird am besten bei der gleichen
Temperatur durchgeführt wie die Metallierung und erfolgt
im allgemeinen durch Zugabe der Ketone zum metallierten Acetonitril
oder Acetonitril-Derivat. Die Zugabe-Reihenfolge kann jedoch auch
vertauscht sein. Die Aufarbeitung des Reaktionsgemisches schließlich
erfolgt meist durch Neutralisieren der enthaltenen Base mit einer
geeigneten Säure (z. B. Schwefelsäure, Essigsäure,
Zitronensäure, Salzsäure) und Entfernen des gebildeten
Salzes mit Wasser. Das so entstandene Produkt wird mit üblichen
Techniken wie Destillation oder Kristallisation gereinigt oder kann
oft auch roh in die Folgestufe eingesetzt werden. In manchen Fällen
kann es auch vorteilhaft sein, nicht mit einer Protonenquelle zu
quenchen, sondern mit anderen Elektrophilen. Es entstehen dann nicht
die Alkohole (R2 = H), sondern entsprechende
Derivate (R2 = Alkyl, Acyl, 2-Tetrahydropyranyl,
R3Silyl) als Ausgangsmaterialien für
die Cyclisierung.
-
[Cyclisierung]
-
Die
Cyclisierung der Vorstufen I oder III zu den gewünschten
Aminopyridinderivaten II kann mit allen geeigneten Stickstoffverbindungen
erfolgen, d. h. mit solchen, bei denen R4 und
R5 wie oben angegeben unabhängig
voneinander Wasserstoff, Alkyl, Aryl, Heteroaryl, Acyl, oder -CONR2 sind. Beide Substituenten können
dabei auch Teil eines Ringsystems sein. Die Cyclisierungsreaktion
wird im allgemeinen in einem geeigneten Lösungsmittel durchgeführt.
Dies ist im einfachsten Fall die Stickstoffverbindung selbst oder
eine Mischung der Stickstoffverbindung mit anderen Lösemitteln
oder Lösemittelgemischen. Geeignete Lösemittel
sind alle, die die Reaktion nicht behindern also z. B. Ether (Dioxan,
THF, MTBE, Diisopropylether, Di-n-butylether), Aromaten (Toluol,
Xylol, Benzol, Chlorbenzol, Anisol), Alkohole (Methanol, Ethanol,
Propanol, Isopropanol, Butanol) oder Wasser. Die Reaktion wird durchgeführt
durch simples Erwärmen der Vorstufen I oder III mit den Stickstoff-Verbindungen
in einem optional zu verwendenden Lösemittel. Typische
Temperaturen sind dabei 40°C bis 250°C, bevorzugt
60°C bis 200°C und besonders bevorzugt 80°C
bis 150°C. Der Zusatz eines Katalysators ist zur Erzielung
der Cyclisierung normalerweise nicht nötig, es können
aber bei Bedarf saure oder alkalische Additive zugesetzt werden,
um die Cyclisierungsreaktion zu beschleunigen. Als saure Additive
werden bevorzugt Salze der verwendeten Stickstoffverbindungen eingesetzt,
besonders bevorzugt Salze von Salzsäure, Schwefelsäure,
Phosphorsäure, p-Toluolsulfonsäure, Essigsäure
oder Zitronensäure. Als basische Additive eignen sich Hydroxide,
Carbonate, Oxide, Alkoholate und andere stark basische Verbindungen,
bevorzugt solche, die stärker basisch sind, als die verwendeten
Stickstoffverbindungen.
-
Die
Aufarbeitung erfolgt in Abhängigkeit von den Eigenschaften
des Produktes durch Destillation oder Kristallisation. Beispiel
1: Herstellung von 5-Ethoxy-3-hydroxy-3-(trifluormethyl)-pent-4-ennitril
als Cyclisierungsvorstufe
-
500
ml 1,2-Dimethoxyethan wurden auf –72°C gekühlt
und bei dieser Temperatur zunächst mit 126 ml n-BuLi (2,5
molar in Hexan) und dann innerhalb von 2 h ebenfalls bei –72°C
mit 12,8 g Acetonitril versetzt. Das Gemisch wurde nun 90 min nachrühren
gelassen, um die Bildung des Anions zu vervollständigen.
Anschließend wurde bei –72°C innerhalb
von 2 h mit einer Lösung von 50 g 1,1,1-Trifluoro-but-3-en-2-one
(Herstellung gemäß
Chem. Ber. 1989, 122,
1179–1186) in 100 ml 1,2-Dimethoxyethan versetzt
und dann 1 h bei dieser Temperatur nachrühren gelassen.
Anschließend wurde das Gemisch auf 0°C erwärmt
und zum Neutralisieren mit einer Lösung von 16,1 g Schwefelsäure
(96%ig) in 50 ml Wasser versetzt. Anschließend wurden 500
ml Toluol zugegeben, die Phasen getrennt und die wäßrige
Phase zweimal mit weiteren 100 ml Toluol gegenextrahiert. Die vereinigten
organischen Phasen wurden mit Natriumsulfat getrocknet und dann
am Rotationsverdampfer konzentriert. Schließlich wurde
das Produkt im vollen Ölpumpenvakuum (ca. 0,2 mbar) destilliert.
Es konnten so 48,5 g Produkt (78%) vom Siedepunkt 95 bis 110°C
gewonnen werden. Dieses wurde anhand seines Massenspektrums identifiziert
(M+ = 209, weitere Fragmente bei m/e = 169, 141 und 71). Beispiel
2: Herstellung von 2-Amino-4-(trifluormethyl)pyridin aus 5-Ethoxy-3-hydroxy-3-(trifluormethyl)-pent-4-ennitril
mit wäßrigem Ammoniak
-
Es
wurden 50 g 5-Ethoxy-3-hydroxy-3-(trifluormethyl)-pent-4-en-nitril
mit 600 g wäßrigem Ammoniak (25%ig) gemischt und
die resultierende Mischung wurde 24 h in einem Autoklaven auf 125°C
erhitzt, wobei sich ein Druck von ca. 14 bar aufbaute. Anschließend
wurde das Reaktionsgemisch abgekühlt und das resultierende
zweiphasige Gemisch mehrmals mit Dichlormethan extrahiert. Die vereinigten
organischen Phasen wurden vorsichtig einrotiert und das Produkt
anschließend aus Cyclohexan umkristallisiert. Es konnten
so 26,3 g 2-Amino-4-(trifluormethyl)pyridin (68%) als gelblichbrauner
Feststoff gewonnen werden. Die spektroskopischen Daten stimmten
mit den in der Literatur angegebenen überein (
A.
D. Dunn et al. in J. Fluorine Chem. 1999, 93, 153–157). Beispiel
3: Herstellung von Dimethyl-(4-trifluormethyl-pyridin-2-yl)-amin
aus 5-Ethoxy-3-hydroxy-3-(trifluormethyl)-pent-4-ennitril mit wäßriger
Dimethylamin-Lösung
-
Es
wurde analog zu Beispiel 2 gearbeitet, aber statt 600 g wäßrigem
Ammoniak wurden 600 g wäßrige Dimethylamin-Lösung
(40%ig) eingesetzt. Es konnten so 28,1 g (62%) Produkt isoliert
werden. Beispiel
4: Herstellung von 5-Ethoxy-3-hydroxy-2-phenyl-3-trifluormethyl-pent-4-enenitril
als Cyclisierungsvorstufe
-
500
ml THF wurden auf –72°C gekühlt und bei
dieser Temperatur zunächst mit 31,9 g Diisopropylamin und
dann bei derselben Temperatur mit 126 ml n-BuLi (2,5 molar in Hexan)
versetzt. Anschließend wurden 35,1 g Benzylcyanid gelöst
in weiteren 250 ml THF innerhalb von 1 h zugetropft. Es wurde weitere
2 h nachrühren gelassen, um die Bildung des Anions zu vervollständigen.
Anschließend wurde bei –72°C innerhalb
von 2 h mit einer Lösung von 50 g 1,1,1-Trifluoro-but-3-en-2-one
versetzt und dann 1 h bei dieser Temperatur nachrühren
gelassen. Anschließend wurde das Gemisch auf 0°C
erwärmt und zum Neutralisieren mit einer Lösung von
16,1 g Schwefelsäure (96%ig) in 50 ml Wasser versetzt.
Anschließend wurden 500 ml Toluol zugegeben, die Phasen
getrennt und die wäßrige Phase zweimal mit weiteren
100 ml Toluol gegenextrahiert. Die vereinigten organischen Phasen
wurden mit Natriumsulfat getrocknet und dann am Rotationsverdampfer
konzentriert. Es wurden so ca. 77 g eines Rohproduktes gewonnen,
das so in der Folgestufe eingesetzt wurde. Beispiel
5: Herstellung von 3-Phenyl-4-trifluormethyl-pyridin-2-ylamin durch
Cyclisierung von 5-Ethoxy-3-hydroxy-2-phenyl-3-trifluormethyl-pent-4-enenitril
mit Ammoniakwasser

-
Es
wurden 50 g 5-Ethoxy-3-hydroxy-2-phenyl-3-trifluoromethyl-pent-4-enenitrile
mit 600 g wäßrigem Ammoniak (25%ig) gemischt und
die resultierende Mischung wurde 24 h in einem Autoklaven auf 125°C
erhitzt. Anschließend wurde das Reaktionsgemisch abgekühlt
und das resultierende zweiphasige Gemisch mehrmals mit Dichlormethan
extrahiert. Die vereinigten organischen Phasen wurden einrotiert
und das Produkt anschließend aus Heptan umkristallisiert.
Es konnten so 24,1 g 3-Phenyl-4-trifluoromethyl-pyridin-2-ylamin
(52% über beide Stufen) isoliert werden.
-
ZITATE ENTHALTEN IN DER BESCHREIBUNG
-
Diese Liste
der vom Anmelder aufgeführten Dokumente wurde automatisiert
erzeugt und ist ausschließlich zur besseren Information
des Lesers aufgenommen. Die Liste ist nicht Bestandteil der deutschen
Patent- bzw. Gebrauchsmusteranmeldung. Das DPMA übernimmt
keinerlei Haftung für etwaige Fehler oder Auslassungen.
-
Zitierte Patentliteratur
-
- - DE 374291 [0004]
- - EP 2288461 B [0011]
-
Zitierte Nicht-Patentliteratur
-
- - Chem. Ber.
1936, 69, 2593 [0004]
- - Org. Lett. 2001, 3, 3417 [0005]
- - Dunn et al. in J. Fluor. Chem 1999, Seite 153 [0011]
- - Chem. Ber. 1989, 122, 1179–1186 [0026]
- - A. D. Dunn et al. in J. Fluorine Chem. 1999, 93, 153–157 [0027]