CN111635359B - 一种通过氟烷基亚磺酰基制备芳香族烯基化合物的方法 - Google Patents
一种通过氟烷基亚磺酰基制备芳香族烯基化合物的方法 Download PDFInfo
- Publication number
- CN111635359B CN111635359B CN202010411494.8A CN202010411494A CN111635359B CN 111635359 B CN111635359 B CN 111635359B CN 202010411494 A CN202010411494 A CN 202010411494A CN 111635359 B CN111635359 B CN 111635359B
- Authority
- CN
- China
- Prior art keywords
- structural formula
- compound
- reaction
- compound represented
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/22—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the nitrogen-containing ring
- C07D217/24—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D221/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00
- C07D221/02—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00 condensed with carbocyclic rings or ring systems
- C07D221/20—Spiro-condensed ring systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
本发明公开了一种通过氟烷基亚磺酰基制备芳香族烯基化合物的方法,属于有机合成领域。本发明以氟烷基亚磺酰基为无痕导向基团,烯烃为偶联试剂,在过渡金属催化剂催化下进行邻位碳氢键烯基化反应,制得单烯基化芳香族化合物,反应过程中,氟烷基亚磺酰基导向基团直接从酰胺底物中脱除,故反应后无需再进行脱除导向基团的处理,大大节省了反应步骤,提高了步骤经济性,且底物兼容性好,能以较高产率得到相应的单烯基化产物。
Description
技术领域
本发明属于有机合成领域,具体涉及一种通过氟烷基亚磺酰基制备芳香族烯基化合物的方法。
背景技术
传统的过渡金属催化的交叉偶联反应提供了一种高效的合成碳碳键以及碳杂键的方法。很多经典的偶联反应都被越来越广泛应用,但仍具有难以忽视的缺点:该类反应都需要事先将底物进行预官能团化,制成亲电试剂(如卤化物或类卤化物)以及亲核试剂等。增加了反应的步骤,降低了原子经济性。因此,是否能直接实现碳氢键的官能团,使底物直接参与反应,这将大大提高的效率。
由于具有步骤和原子经济性,近年来在碳氢键活化领域最常用的策略就是导向基导向的碳氢键活化。传统导向基导向的碳氢键官能团化反应有苯和苯乙烯的氧化偶联反应(Tetrahedron Lett.1967,8,1119;Tetrahedron Lett.1968,5,633)。1993年,Murai小组报道了过渡金属Ru催化的芳基酮邻位烷基化反应(Nature,1993,366,529),该反应的效率和产率都大为提高。2002年,Leeuwen小组报道了乙酰苯胺作为导向基的C-H键烯基化反应(J.Am.Chem.Soc.2002,124,8,1586)。2010年,Daugulis小组报道了以N,S双原子双配位的导向基碳氢键活化反应(J.Am.Chem.Soc.2010,132,3965),此后还有C-H键的芳基化(J.Org.Chem.,2013,78,3030),烷基化(Org.Lett.,2011,13,4850),炔基化[Org.Lett.,2012,14,354],酰基化(Org.Lett.,2012,14,1238-1241),氨基化(J.Am.Chem.Soc.,2012,134,7),卤代(J.Am.Chem.Soc.,2013,135,9342)等双配位导向基官能团化反应。此外,还有以酰胺类(J.Am.Chem.Soc.,2010,132(11),3680)、吡啶类(J.Am.Chem.Soc.2011,133,6541)和羧酸类(J.Org.Chem.2011,76,3024)作为导向基进行直接的烯基化反应。除了氮和氧原子导向基外,还有一些杂原子导向基团也能实现C-H键烯基化反应,如硫醚(Org.Lett.,2012,14,2164)导向和苯基磷酸单酯(Org.Lett.,2013,15,1910)导向的C-H键活化的烯基化反应。传统的导向基导向的烯基化反应总体上有着反应温和,选择性高和反应产率高等优点,但是有着需要预先安装导向基,且难以脱除的特点。
如能在反应过程中,直接将底物上的导向基脱除或者导向基不在反应底物上,可以减少反应步骤,提高反应的步骤经济性,无痕导向基导向的碳氢键官能团化反应应运而生。1997年,Jun课题组报道了用Ru催化亚胺作为瞬时导向基的烯烃氢芳基化反应(J.Org.Chem.1997,62,1200)。2016年,林国强院士课题组报道了以溴作为无痕导向基团,钯催化的芳基溴和连硼酸频哪醇酯的偶联反应(J.Am.Chem.Soc.2016,138,2897)。此后,又有全氟酯基导向(J.Am.Chem.Soc.2018,140,1502)的脂肪烃氧化为醇的反应。
碳氢键活化烯基化反应在最近十年得到了快速的发展,导向基导向的sp2烯基化反应已经发展的相对较成熟。然而,如何更高效地对导向基进行安装或脱除仍是一个亟需解决的课题。
发明内容
为解决上述现有技术中存在的缺点和不足,本发明的目的在于提供一种通过以氟烷基亚磺酰基为无痕导向基团的邻位碳氢键烯基化反应来制备芳香族烯基化合物的方法,导向基氟烷基亚磺酰基在反应后即进行脱除,可以大大节省反应步骤,提高步骤经济性,且底物兼容性好,能以较高产率得到相应的单烯基化产物。
本发明提供了一种通过氟烷基亚磺酰基制备芳香族烯基化合物的方法,所述芳香族烯基化合物的结构式如式(5)所示,所述方法包括以下步骤:将结构式(1)所示的化合物与结构式(2)所示的化合物在过渡金属催化剂、氧化剂以及碱的存在下于有机溶剂A中进行反应而得所述芳香族烯基化合物;

其中,R1为氢原子、卤原子、烷基、芳基、烷氧基中的一种,n=1或2;R2以及R3各自独立地为烷基或者R2与R3共同形成环烷基;R4为烷基、环烷基、芳基、苄基或任选具有取代基的烷基;所述氧化剂为醋酸盐、氯化盐中的至少一种;所述碱为钠盐、钾盐中的至少一种;所述有机溶剂A为含氟有机试剂。当R4表示烷基时,可为直链烷基或支链烷基。
上述方法利用无痕导向基导向的策略,以氟烷基亚磺酰基为无痕导向基团,烯烃为偶联试剂,在过渡金属催化剂催化下,制得单烯基化芳香族化合物;反应过程中,氟烷基亚磺酰基导向基团直接从酰胺底物中脱除,故反应后无需再进行脱除导向基团的处理。
优选地,所述具有取代基的烷基为羟基烷基、烷氧基烷基、杂环基烷基中的一种。
优选地,所述R1为氢原子、氯原子、溴原子、甲基、乙基、叔丁基、芳基、甲氧基中的一种;所述R2与R3各自独立地为甲基、乙基中的一种,或者所述R2与R3共同形成环丙基、环丁基、环戊基、环己基中的一种;所述R4为甲基、乙基、正丁基、异丁基、环己基、芳基、苄基、4-羟基丁基、2-甲氧基乙基、四氢-2-呋喃基甲基中的一种。
优选地,所述氧化剂为氯化铜、醋酸铜、醋酸银中的至少一种。
优选地,所述碱为碳酸钠、碳酸钾、磷酸钾、磷酸氢钾、醋酸钠、三氟醋酸钠中的至少一种。发明人采用单因素试验(即只改变碱的种类)来研究碱的种类对结构如式(5)所示的化合物产率的影响,发现当碱为碳酸锂时,得不到结构如式(5)所示的化合物;当碱为碳酸钠时产率最高,碱为碳酸钾时产率次之,碱为磷酸钾、磷酸氢钾、醋酸钠或三氟醋酸钠时产率再次之。
优选地,所述过渡金属催化剂为醋酸钯。
优选地,所述有机溶剂A为六氟异丙醇、三氟乙醇中的至少一种。
优选地,结构式(1)所示的化合物、结构式(2)所示的化合物、过渡金属催化剂、氧化剂、碱以及有机溶剂A的比例为结构式(1)所示的化合物:结构式(2)所示的化合物:过渡金属催化剂:氧化剂:碱:有机溶剂A=1mol:2-5mol:0.05-0.2mol:2-5mol:1-3mol:(0.8-1.5)*104mL;所述反应温度为60-120℃,反应时间为0.1-12h。
优选地,所述结构式(1)所示的化合物的制备方法包括以下步骤:将结构式(3)所示的化合物与结构式(4)所示的化合物在叔胺存在下于有机溶剂B中进行反应而得到所述结构式(1)所示的化合物;
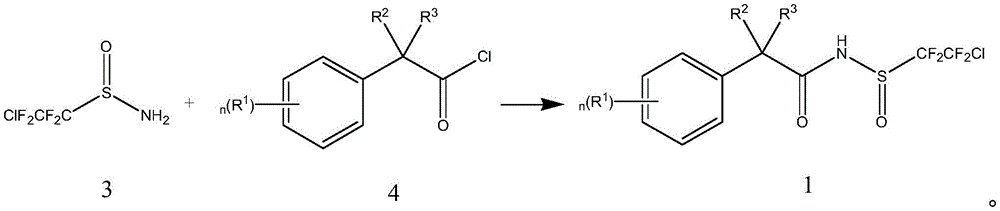
叔胺在上述反应中充当缚酸剂,与反应中生成的盐酸结合生成叔胺盐酸盐。
优选地,所述叔胺为三乙胺。
优选地,所述有机溶剂B为二氯甲烷。
优选地,所述结构式(3)所示的化合物与结构式(4)所示的化合物反应时,结构式(3)所示的化合物、结构式(4)所示的化合物以及叔胺的摩尔比为结构式(3)所示的化合物:结构式(4)所示的化合物:叔胺=1:0.9-2:5-20,反应温度为0℃至室温,反应时间为1-3h。
优选地,所述结构式(3)所示的化合物的制备方法包括以下步骤:
(1)将1-碘-2-氯四氟乙烷在连二亚硫酸钠以及碳酸氢钠存在下于溶剂C中进行搅拌反应,反应结束后,得到反应混合液;
(2)采用有机溶剂对步骤(1)所得反应混合液进行萃取,收集有机相并除去溶剂,再加入硫酸,混合均匀后进行热过滤,收集滤液并浓缩,得到浓缩液;
(3)在步骤(2)所得浓缩液中加入二氯亚砜进行反应,即得结构式(3)所示的化合物。
优选地,所述1-碘-2-氯四氟乙烷、连二亚硫酸钠、碳酸氢钠以及二氯亚砜的摩尔比为1-碘-2-氯四氟乙烷:连二亚硫酸钠:碳酸氢钠:二氯亚砜=1:1-2:1-2:1-5;所述步骤(1)中的反应温度为室温,反应时间为5-10h,溶剂C为水和乙腈的混合液;所述步骤(3)中的反应温度为0℃至室温,反应时间为1-5h。
本发明所涉及的室温,均是指室内温度,不一定为25℃。
与现有技术相比,本发明具有如下优点:本发明以氟烷基亚磺酰基为无痕导向基团,烯烃为偶联试剂,在过渡金属催化剂催化下进行邻位碳氢键烯基化反应,制得单烯基化芳香族化合物,反应过程中,氟烷基亚磺酰基导向基团直接从酰胺底物中脱除,故反应后无需再进行脱除导向基团的处理,大大节省了反应步骤,提高了步骤经济性,且底物兼容性好,能以较高产率得到相应的单烯基化产物。
附图说明
图1为实施例7所涉及的反应、相应的产物芳香族烯基化合物及其收率;
图2为实施例8所涉及的反应、相应的产物芳香族烯基化合物及其收率。
具体实施方式
为更好的说明本发明的目的,技术方案和优点,本发明通过下列实施例进一步说明。显然,下列实施例仅是本发明的一部分实施例,而不是全部的实施例。应理解,本发明实施例仅用于说明本发明的技术效果,而非用于限制本发明的保护范围。
本发明提供了一种通过氟烷基亚磺酰基制备芳香族烯基化合物的方法,所述芳香族烯基化合物的结构式如式(5)所示,所述方法包括以下步骤:将结构式(1)所示的化合物与结构式(2)所示的化合物在过渡金属催化剂、氧化剂以及碱的存在下于有机溶剂A中进行反应而得所述芳香族烯基化合物;

其中,R1为氢原子、卤原子、烷基、芳基、烷氧基中的一种,n=1或2;R2以及R3独立地为烷基或者R2与R3共同形成环烷基;R4为烷基、环烷基、芳基、苄基或任选具有取代基的烷基;所述氧化剂为醋酸盐、氯化盐中的至少一种;所述碱为钠盐、钾盐中的至少一种。
作为由R1表示的卤原子,可举出氯原子、溴原子等。
作为由R1表示的烷基,可举出甲基、乙基、叔丁基等。
作为由R1表示的烷氧基,可举出甲氧基。
作为由R2以及R3各自独立地表示的烷基,可举出甲基、乙基等。
作为由R2与R3共同形成的环烷基,可举出环丙基、环丁基、环戊基、环己基等。
作为由R4表示的烷基,可以是直链烷基或支链烷基,如甲基、乙基、正丁基、异丁基等。
作为由R4表示的环烷基,可以是环己基等。
作为由R4表示的具有取代基的烷基,可以是羟基烷基、烷氧基烷基、杂环基烷基等,其中“羟基烷基”是指含有至少有一个羟基的烷基;“烷氧基”是通过氧原子与母体分子部分连接的烷基,如甲氧基,“烷氧基烷基”是如上所定义的烷氧基和烷基的组合基团,如2-甲氧基乙基;“杂环基”是指含有至少一个杂原子的饱和杂环,如四氢呋喃基,“杂环基烷基”是如上所定义的杂环基和烷基的组合基团,如(四氢呋喃基)烷基。
结构式(2)所示的化合物的用量,相对于1mol结构式(1)所示的化合物,通常为2-5mol。
过渡金属催化剂优选使用钯金属催化剂。
钯金属催化剂优选使用醋酸钯。
过渡金属催化剂的用量,相对于1mol结构式(1)所示的化合物,通常为0.05-0.2mol。
氧化剂优选使用醋酸银。研究过程中发现在醋酸盐、氯化盐等氧化剂存在下,结构式(1)所示的化合物与结构式(2)所示的化合物能反应生成结构式(5)所示的化合物,如氯化铜、醋酸铜、醋酸银等,尤其是在醋酸银存在下,结构式(5)所示的化合物的收率非常高,而在氧化银、碳酸银或三氟醋酸银存在下,均检测不到目标产物(结构式(5)所示的化合物)的生成。
氧化剂的用量,相对于1mol结构式(1)所示的化合物,通常为2-5mol。
碱优选使用碳酸钠。研究过程中发现当碱为碳酸锂时,得不到结构如式(5)所示的化合物;当碱为碳酸钠时产率最高,碱为碳酸钾时产率次之,碱为磷酸钾、磷酸氢钾、醋酸钠或三氟醋酸钠时产率再次之。
碱的用量,相对于1mol结构式(1)所示的化合物,通常为1-3mol。
有机溶剂A可选为含氟有机试剂,如六氟异丙醇、三氟乙醇等,尤其以六氟异丙醇作为溶剂时,结构式(5)所示的化合物的收率非常高,另外,当有机溶剂A为1,4-二氧六环、二氯甲烷、叔丁醇或乙腈时,检测不到目标产物(结构式(5)所示的化合物)的生成。
有机溶剂A的用量,相对于1mol结构式(1)所示的化合物,通常为(0.8-1.5)*104mL。
上述结构式(1)所示的化合物与结构式(2)所示的化合物发生的反应在密闭、非密闭条件下均可进行,优选在密闭条件下进行。
上述结构式(1)所示的化合物与结构式(2)所示的化合物发生的反应在空气氛围中即可进行。
上述结构式(1)所示的化合物与结构式(2)所示的化合物发生的反应的温度通常为60-120℃。
上述结构式(1)所示的化合物与结构式(2)所示的化合物发生的反应的时间通常在0.1~12h范围内。
经过对上述结构式(1)所示的化合物与结构式(2)所示的化合物发生的反应的反应条件包括氧化剂、溶剂、碱、温度和时间等反应变量进行筛选,确定最佳合成条件为:碱为碳酸钠,氧化剂为醋酸银,有机溶剂A为六氟异丙醇,结构式(1)所示的化合物、结构式(2)所示的化合物、过渡金属催化剂、氧化剂、碱以及有机溶剂A的比例为结构式(1)所示的化合物:结构式(2)所示的化合物:过渡金属催化剂:氧化剂:碱:有机溶剂A=1mol:3mol:0.1mol:2mol:2mol:1*104mL,该反应在空气氛围中进行,反应温度为80℃,反应时间为10h。
上述结构式(1)所示的化合物与结构式(2)所示的化合物发生的反应结束后,例如可以在反应混合液冷却至室温后,用水进行淬灭反应,然后采用有机溶剂萃取,收集有机相,干燥除水后,蒸馏除去有机溶剂,再采用柱层析等进行纯化。这样所得结构如式(5)所示的化合物不仅能应用于有机化学领域,还能应用于医药领域。
所述结构式(1)所示的化合物可通过包括以下步骤的方法制备而成:将结构式(3)所示的化合物与结构式(4)所示的化合物在叔胺存在下于有机溶剂B中进行反应而得到所述结构式(1)所示的化合物;

结构式(4)所示的化合物的用量,相对于1mol结构式(3)所示的化合物,通常为0.9-2mol。
叔胺优选为三乙胺。
叔胺的用量,相对于1mol结构式(3)所示的化合物,通常为5-20mol。
有机溶剂B优选为二氯甲烷。
有机溶剂B的用量,相对于1mol结构式(3)所示的化合物,通常为(10-20)*103mL。
上述结构式(3)所示的化合物与结构式(4)所示的化合物发生的反应的温度通常为0℃至室温。
上述结构式(3)所示的化合物与结构式(4)所示的化合物发生的反应的时间通常为1-3h。
经过对上述结构式(3)所示的化合物与结构式(4)所示的化合物发生的反应的反应条件包括叔胺、溶剂、温度和时间等反应变量进行筛选,确定最佳合成条件为:结构式(3)所示的化合物与结构式(4)所示的化合物反应时,结构式(3)所示的化合物、结构式(4)所示的化合物以及叔胺的摩尔比为结构式(3)所示的化合物:结构式(4)所示的化合物:叔胺=1:1.5:5,反应温度为0℃至室温,反应时间为2h,所述有机溶剂B为二氯甲烷。
上述结构式(3)所示的化合物与结构式(4)所示的化合物发生的反应结束后,例如可以在反应混合液冷却至室温后,采用有机溶剂萃取,收集有机相,干燥除水后,蒸馏除去有机溶剂,再采用柱层析等进行纯化,其中柱层析采用的流动相可选为石油醚和乙酸乙酯的混合液(石油醚/乙酸乙酯=5-20:1,v/v)等。
所述结构式(3)所示的化合物可通过包括以下步骤的方法制备而成:
(1)将1-碘-2-氯四氟乙烷在连二亚硫酸钠以及碳酸氢钠存在下于溶剂C中进行搅拌反应,反应结束后,得到反应混合液;
(2)采用有机溶剂对步骤(1)所得反应混合液进行萃取,收集有机相并除去溶剂,再加入硫酸,混合均匀后进行热过滤,收集滤液并浓缩,得到浓缩液;
(3)在步骤(2)所得浓缩液中加入二氯亚砜进行反应,即得结构式(3)所示的化合物。
连二亚硫酸钠的用量,相对于1mol 1-碘-2-氯四氟乙烷,通常为1-2mol。
碳酸氢钠的用量,相对于1mol 1-碘-2-氯四氟乙烷,通常为1-2mol。
溶剂C优选为水和乙腈的混合液。
溶剂C的用量,相对于1mol 1-碘-2-氯四氟乙烷,通常为500-3000mL。
上述步骤(1)中的反应在室温下即可进行。
上述步骤(1)中的反应的时间通常为5-10h。
二氯亚砜的用量,相对于1mol 1-碘-2-氯四氟乙烷,通常为1-5mol。
上述步骤(3)中的反应的反应温度通常为0℃至室温,反应时间通常为1-5h。
经过对1-碘-2-氯四氟乙烷制备结构式(3)所示的化合物的反应条件进行筛选,确定优选反应条件为:1-碘-2-氯四氟乙烷、连二亚硫酸钠、碳酸氢钠以及二氯亚砜的摩尔比为1-碘-2-氯四氟乙烷:连二亚硫酸钠:碳酸氢钠:二氯亚砜=1:2:2:2;步骤(1)中的反应温度为室温,反应时间为12h,溶剂C为水和乙腈的混合液;步骤(3)中的反应程序为先在冰浴中反应1h,再在室温下反应2h。
实施例1
本实施例为本发明通过氟烷基亚磺酰基制备芳香族烯基化合物的方法的一种实施方式,该方法包括以下几个工艺:
1、结构式(3)所示的化合物的制备
以廉价的工业副产品1-碘-2-氯四氟乙烷为原料,利用亚磺化脱卤反应,制备相应的亚磺酰胺,制备路线如下所示:

具体的制备方法包括以下步骤:
步骤A:将160mmol 1-碘-2-氯四氟乙烷、320mmol连亚硫酸钠以及320mmol碳酸氢钠加入反应容器中,然后加入150-300mL水和100-200mL乙腈作为溶剂,在室温搅拌12h,反应结束后,得到反应混合物;
步骤B:采用50mL乙酸乙酯对步骤A所得反应混合物中萃取2次,收集上层有机相并旋干抽净残余水,然后加入70mL98%硫酸混合均匀后热过滤,取滤液减压蒸馏,得浓缩液;
步骤C:待步骤B所得浓缩液冷却后,加入320mmol二氯亚砜,先于冰浴中搅拌反应1h,再在室温下反应2h,然后常压蒸馏,取馏出液,即得结构式(3)所示的化合物,称重为15g。
2、亚磺酰胺底物的制备
本实施例中亚磺酰胺底物为N-((2-氯-1,1,2,2-四氟乙基)亚砜基)-2-甲基-2-苯丙酰胺,即结构式(101)所示的化合物,制备路线如下所示:

具体的制备方法包括以下步骤:
步骤D:向80mmol上述所得结构式(3)所示的化合物中慢慢加入120mmol2-甲基-2-苯基丙酰氯(结构式(401)所示的化合物)、400mmol三乙胺(Et3N)以及1200mL的二氯甲烷(DCM),在0℃至室温温度范围内反应2h,得到反应混合物;
步骤E:待步骤D所得反应混合物冷却至室温后,采用乙醚萃取3次,收集上层有机相并用无水硫酸钠干燥除水分后过滤,得到粗品;
步骤F:将步骤E所得粗品减压蒸发除溶剂后,采用硅胶柱层析进行纯化(流动相为石油醚和乙酸乙酯的混合物,其中石油醚/乙酸乙酯=15:1,v/v),得到白色固体,即得所述亚磺酰胺底物。该亚磺酰胺底物为1.6g,产率为77%,用NMR和HRMS对其进行表征,结果为:(1)1H NMR(400MHz,CDCl3)δ:1.63(s,3H),1.66(s,3H),7.33-7.45(m,5H);19F NMR(376MHz,CDCl3)δ-67.93–-67.91(m,2F),-117.0(d,J=233.1Hz,1F),-120.1(d,J=233.1Hz,1F);13CNMR(100MHz,CDCl3):26.0,26.1,48.3,116.7(tt,JC–F=310Hz,JC–F=40Hz),121.5(tt,J1=300Hz,J2=30Hz),126.1,128.0,129.2,141.8,176.9;(2)该产物高分辨质谱(ESI-TOF)分子量计算值为[M+Na]+368.0106,实测值为[M+Na]+368.0404。
3、芳香族烯基化合物的制备
本实施例中芳香族烯基化合物为2-(4,4-二甲基-3-氧-1,2,3,4-四氢异喹啉-1-基)乙酸乙酯,即结构式(501)所示的化合物,制备路线如下所示:

具体的制备方法包括以下步骤:
步骤G:于35mL的螺旋式密封管中,加入上述制得的亚磺酰胺底物(0.2mmol)、丙烯酸乙酯(0.6mmol,结构式(201)所示的化合物)、过渡金属催化剂醋酸钯(0.02mmol)、氧化剂醋酸银(0.4mmol)以及碱碳酸钠(0.4mmol),最后加入2mL六氟异丙醇(HFIP),密封后置于80℃油浴锅中反应10h,得到反应混合物;
步骤H:待反应混合物冷却至室温,用10mL的蒸馏水淬灭反应,再采用20mL乙酸乙酯萃取3次;
步骤I:将步骤H萃取得到的有机相合并,用无水硫酸钠干燥除水分后过滤,然后依次经减压蒸馏、柱层析纯化(流动相为石油醚和丙酮的混合物,其中石油醚/丙酮=3:1,v/v),得到白色固体,即得芳香族烯基化合物。该芳香族烯基化合物为38mg,产率为73%,用NMR和HRMS对其进行表征,结果为:(1)1H NMR(400MHz,CDCl3)δ:2.65(dd,J1=17.2Hz,J2=10.8Hz,1H),2.88(dd,J1=17.2Hz,J2=2.8Hz,1H),4.21(q,J=7.2Hz,2H),5.00(dt,J1=10.8Hz,J2=2.8Hz,1H),6.66(br,1H),7.11(d,J=7.6Hz,1H),7.24(t,J=6.4Hz,1H),7.32(t,J=11.2Hz,1H);13C NMR(100MHz,CDCl3):7.38(d,41.8,45.7,52.6,62.2,126.4,127.2,127.6,129.1,132.1,142.1,172.0,176.7;(2)该产物高分辨质谱(ESI-TOF)分子量计算值为[M+H]+262.1438,实测值为[M+H]+262.1437。
实施例2
本实施例为本发明通过氟烷基亚磺酰基制备芳香族烯基化合物的方法的一种实施方式,该方法包括以下几个工艺:
1、结构式(3)所示的化合物的制备
方法步骤以及工艺参数同实施例1。
2、亚磺酰胺底物的制备
本实施例中亚磺酰胺底物为N-((2-氯-1,1,2,2-四氟乙基)亚砜基)-2-(4-甲氧基苯基)-2-甲基丙酰胺,即结构式(105)所示的化合物,制备路线如下所示:

具体的制备方法包括以下步骤:除用结构式(405)所示的化合物替代2-甲基-2-苯基丙酰氯外,其他方法步骤及工艺参数均同实施例1。所得亚磺酰胺底物为白色固体,质量为1.3g,产率为59%,用NMR和HRMS对其进行表征,结果为:(1)1H NMR(400MHz,CDCl3)δ:1.60(s,3H),1.62(s,3H),3.83(s,3H),6.94(d,J=8.8Hz,2H),7.26(m,2H);19F NMR(376MHz,CDCl3)δ-69.76–-69.80(m,2F),-118.8(dt,J1=233.1Hz,J2=3.76Hz,1F),-122.0(dt,J1=229.4Hz,J2=3.76Hz,1F);13C NMR(100MHz,CDCl3):26.3,30.9,47.7,55.3,114.7,127.4,133.6,159.3,177.3;(2)该产物高分辨质谱(ESI-TOF)分子量计算值为[M+H]+376.0392,实测值为[M+H]+376.0387。
3、芳香族烯基化合物的制备
本实施例中芳香族烯基化合物为2-(7-甲氧基-4,4-二甲基-3-氧-1,2,3,4-四氢异喹啉-1-基)乙酸乙酯,即结构式(505)所示的化合物,制备路线如下所示:

具体的制备方法包括以下步骤:除亚磺酰胺底物为本实施例制得的亚磺酰胺底物外,其他方法步骤及工艺参数均同实施例1。所得芳香族烯基化合物为白色固体,质量为26mg,产率为44%,用NMR和HRMS对其进行表征,结果为:1H NMR(400MHz,CDCl3)δ:1.30(t,J=7.2Hz,3H),1.49(s,3H),1.58(s,1H),2.66(dd,J1=17.6Hz,J2=10.8Hz,1H),2.88(dd,J1=17.6Hz,J2=2.8Hz,1H),3.80(s,3H),4.22(q,J=7.2Hz,2H),4.96(dt,J1=10.8Hz,J2=2.8Hz,1H),6.56(br,1H),6.60(d,J=2.4Hz,1H),6.89(dd,J1=8.4Hz,J2=2.4Hz,1H),7.30(d,J=8.8Hz,1H);13C NMR(100MHz,CDCl3)δ:15.2,28.0,32.2,41.4,45.7,52.6,56.4,62.3,111.1,115.4,128.6,133.3,134.2,159.0,172.1,176.9。
实施例3
本实施例为本发明通过氟烷基亚磺酰基制备芳香族烯基化合物的方法的一种实施方式,该方法包括以下几个工艺:
1、结构式(3)所示的化合物的制备
方法步骤以及工艺参数同实施例1。
2、亚磺酰胺底物的制备
本实施例中亚磺酰胺底物为N-((2-氯-1,1,2,2-四氟乙基)亚砜基)-1-苯基环戊烷-1-甲酰胺,即结构式(113)所示的化合物,制备路线如下所示:

具体的制备方法包括以下步骤:除用结构式(413)所示的化合物替代2-甲基-2-苯基丙酰氯外,其他方法步骤以及工艺参数均同实施例1。所得亚磺酰胺底物为白色固体,质量为1.6g,产率为70%,用NMR和HRMS对其进行表征,结果为:(1)1H NMR(400MHz,CDCl3)δ:(1.76-1.89)(m,4H),(2.06-2.13)(m,2H),(2.44-2.57)(m,2H),(7.31-7.38)(m,3H),7.40(t,J=7.6Hz,3H);19F NMR(376MHz,CDCl3)δ-67.31–-67.30(m,2F),-115.96(dt,J1=229.3Hz,J2=3.76Hz,1F),-119.51(dt,J1=229.36Hz,J2=3.76,1F);13C NMR(100MHz,CDCl3)δ:23.9,36.2,36.3,60.3,116.8(tt,JC–F=293.2Hz,JC–F=36.2Hz),121.5(tt,J1=298.6Hz,J2=32.6Hz),126.6,128.0,129.3,141.0,175.9;(2)该产物高分辨质谱(ESI-TOF)分子量计算值为[M+Na]+382.0262,实测值为[M+Na]+382.0259。
3、芳香族烯基化合物的制备
本实施例中芳香族烯基化合物为2-(3'-氧-2',3'-二氢-1'-氢-螺环[环戊烷-1,4'-异喹啉]-1'-基)乙酸乙酯,即结构式(513)所示的化合物,制备路线如下所示:

具体的制备方法包括以下步骤:除亚磺酰胺底物为本实施例制得的亚磺酰胺底物外,其他方法步骤以及工艺参数均同实施例1。所得芳香族烯基化合物为白色固体,质量为29mg,产率为51%,用NMR和HRMS对其进行表征,结果为:1H NMR(400MHz,CDCl3)δ:1.27(t,J=7.2Hz,3H),(1.71-2.09)(m,7H),2.65(dd,J1=7.2Hz,J2=10.8Hz,1H),(2.71-2.76)(m,1H),2.82(dd,J1=17.2Hz,J2=2.8Hz,1H),4.20(q,J=6.8Hz,2H),4.98(dt,J1=10.8Hz,J2=2.8Hz,1H),6.58(br,1H),7.08(d,J=3.6Hz,1H),(7.19-7.30)(m,3H);13C NMR(100MHz,CDCl3)δ:15.2,28.4,29.0,39.2,45.6,46.2,52.3,52.9,62.2,126.3,127.3,127.3,129.2,132.8,143.5,172.1,177.7。
实施例4
本实施例为本发明通过氟烷基亚磺酰基制备芳香族烯基化合物的方法的一种实施方式,该方法包括以下几个工艺:
1、结构式(3)所示的化合物的制备
方法步骤以及工艺参数均同实施例1。
2、亚磺酰胺底物的制备
本实施例中亚磺酰胺底物为1-(4-溴苯基)-N-((2-氯-1,1,2,2-四氟乙基)亚砜基)环戊烷-1-甲酰胺,即结构式(116)所示的化合物,制备路线如下所示:

具体的制备方法包括以下步骤:除用结构式(416)所示的化合物替代2-甲基-2-苯基丙酰氯外,其他方法步骤以及工艺参数均同实施例1。所得亚磺酰胺底物为白色固体,质量为1.9g,产率为71%,用NMR和HRMS对其进行表征,结果为:(1)1H NMR(400MHz,CDCl3)δ:1.74–1.79(m,4H),1.96–2.01(m,2H),2.42-2.50(m,2H),7.16(d,J=8.4Hz,2H),7.48(d,J=8.4Hz,2H),7.78(br,1H);19F NMR(376MHz,CDCl3)δ-67.36(s,2F),-116.1(d,J1=229.4Hz,1F),-119.3(dd,J=229.4Hz,1F);13C NMR(100MHz,CDCl3)δ:23.7,23.8,36.2,36.3,60.0,116.8(tt,JC–F=300Hz,JC–F=30Hz),121.5(tt,JC-F=300Hz,JC-F=30Hz),128.4,132.3,132.4,140.0,175.2;(2)该产物高分辨质谱(ESI-TOF)分子量计算值为[M+Na]+471.9367,实测值为[M+Na]+471.9365。
3、芳香族烯基化合物的制备
本实施例中芳香族烯基化合物为2-(7'-溴-3'-氧-2',3'-二氢-1'-氢螺环戊烷-1,4'-异喹啉]-1'-基)乙酸乙酯,即结构式(516)所示的化合物,制备路线如下所示:

具体的制备方法包括以下步骤:除亚磺酰胺底物为本实施例制得的亚磺酰胺底物外,其他方法步骤以及工艺参数均同实施例1。所得芳香族烯基化合物为白色固体,质量为27mg,产率为44%,用NMR和HRMS对其进行表征,结果为:1H NMR(400MHz,CDCl3)δ:1.30(t,J=7.2Hz,3H),(1.63-1.75)(m,1H),(1.82-2.00)(m,4H),(2.06-2.11)(m,1H),(2.27-2.33)(m,1H),2.66(dd,J1=17.2Hz,J2=10.8Hz,1H),(2.72-2.78)(m,1H),2.83(dd,J1=17.2Hz,J2=3.2Hz,1H),4.22(q,J=7.2Hz,2H),4.94(dt,J1=10.8Hz,J2=3.2Hz,1H),6.53(br,1H),7.16(d,J=8.4Hz,4H),7.25(s,1H),7.42(d,J=8.8Hz,1H);13C NMR(100MHz,CDCl3)δ:15.2,28.4,28.9,39.3,45.4,46.1,52.1,52.5,62.4,121.0,129.2,129.2,132.3,135.0,142.6,171.8,177.2。
实施例5
本实施例为本发明通过氟烷基亚磺酰基制备芳香族烯基化合物的方法的一种实施方式,本实施例中芳香族烯基化合物为2-(4,4-二甲基-3-氧-1,2,3,4-四氢异喹啉-1-基)乙酸环己酯(即结构式(522)所示的化合物),该方法制备路线如下所示:

具体的制备方法包括以下步骤:除采用结构式(222)所示的化合物替换丙烯酸乙酯外,其他方法步骤以及工艺参数均同实施例1。所得芳香族烯基化合物为白色固体,质量为50mg,产率为80%,用NMR和HRMS对其进行表征,结果为:1H NMR(400MHz,CDCl3)δ:(1.25-1.48)(m,5H),1.52(s,3H),1.60(s,3H),(1.69-1.89)(m,5H),2.64(dd,J1=17.2Hz,J2=10.8Hz,1H),2.87(dd,J1=17.2Hz,J2=2.8Hz,1H),(4.81-4.86)(m,1H),(4.98-5.01)(m,1H),6.63(br,1H),7.12(d,J=7.6Hz,1H),7.24(d,J=7.2Hz,1H),7.32(d,J=7.2Hz,1H),7.38(d,J=7.6Hz,1H);13C NMR(100MHz,CDCl3)δ:24.7,26.2,27.9,31.9,32.6,41.8,45.9,52.6,74.8,126.5,127.2,127.5,129.1,132.1,142.1,171.5,176.6。
实施例6
本实施例为本发明通过氟烷基亚磺酰基制备芳香族烯基化合物的方法的一种实施方式,本实施例中芳香族烯基化合物为物2-甲氧基乙基2-(4,4-二甲基-3-氧-1,2,3,4-四氢异喹啉-1-基)乙酸酯(即结构式(524)所示的化合物),该方法制备路线如下所示:

具体的制备方法包括以下步骤:除采用结构式(224)所示的化合物替换丙烯酸乙酯外,其他方法步骤以及工艺参数均同实施例1。所得芳香族烯基化合物为白色固体,质量为43mg,产率为74%,用NMR和HRMS对其进行表征,结果为:1H NMR(400MHz,CDCl3)δ:1.84(s,3H),1.94(s,3H),3.03(dd,J1=16.8Hz,J2=10.8Hz,1H),3.27(dd,J1=17.2Hz,J2=2.8Hz,1H),3.74(s,3H),3.96(t,J=4.4Hz,2H),(4.59-4.60)(m,1H),(4.69-4.74)(m,1H),5.34(dt,J1=10.8Hz,J2=2.8Hz,1H),7.10(br,1H),7.44(d,J=7.6Hz,1H),7.58(t,J=8.8Hz,1H),7.66(t,J=7.6Hz,1H),7.72(d,J=7.6Hz,1H);13C NMR(100MHz,CDCl3)δ:27.7,32.1,41.8,45.7,52.7,60.0,64.9,71.1,126.5,127.3,127.6,129.2,132.0,142.1,172.0,176.6。
实施例7
发明人研究了结构式(1)所示的化合物为不同结构时,与丙烯酸乙酯(即结构式(201)所示的化合物)进行反应纯化,具体步骤以及工艺参数同实施例1中“3、芳香族烯基化合物的制备”,所得产物与收率如图1所示。
由图1结果可知,当芳环上带有给电子取代基如甲基、叔丁基和甲氧基(502-506)时,可得到较好的产率,最高达74%。而芳环上带有缺电子取代基如氯、溴(507-508)时,反应效果较差,最高仅有52%的产率。其次,我们发现该反应具有明显的偕二取代基效应,在苄位为环状取代基时,如三元环(511),四元环(512)时,可得到不错的产率。当羰基α位环增大至五元环和六元环时,其产率有下降趋势(513,514),但有意思的是当苯环上带有Br、Cl等吸电子基时(515,516)时,反而有近70%的产率。研究苯环上的立体位阻效应时,发现苯环的间位(503)和对位(502,504)的给电子基团对产率几乎无影响(与501产率相比较),分别是73%、71%、74%,但甲基在邻位时,得不到目标产物,结果显示该反应更倾向于在位阻小的位置进行偶联反应。联苯类底物(517,518)在此反应中也表现出中等的反应产率(分别为55%,67%)。当苯环上有双给电子取代基时,以56%的产率得到目标产物506,明显比单富电子取代基(505)的产率高(44%)。
实施例8
发明人对不同取代基的丙烯酸酯底物进行普适性考察,具体为研究了结构式(2)所示的化合物为不同结构时,与结构式(101)所示的化合物进行反应纯化,具体步骤以及工艺参数同实施例1中“3、芳香族烯基化合物的制备”,所得产物与收率如图2所示。
由图2反应结果可知,丙烯酸酯类烯烃在该反应体系中是一个很好的偶联试剂。富电子烷氧基取代的绝大多数丙烯酸酯类底物合成得到的烯基化目标产物的产率较理想,包括正丁基(520,产率69%),异丁基(521,产率60%),环己基(522,产率80%),4-羟基丁基(523,产率74%),2-甲氧基乙基(524,产率74%),四氢-2-呋喃基甲基(525,产率67%)等烷基取代基。然而,实验过程发明人发现叔丁基取代的丙烯酸酯类底物只能得到痕量的目标产物,可能是由于反应中对邻位产生较大的空间位阻,抑制了反应的进行。同理,可解释为何苄基取代(取代基苯环可旋转)的丙烯酸酯类烯烃(526)能获得86%的理想产率,而芳基取代(平面结构,取代基苯环无法自由旋转,对邻位产生较大位阻)的丙烯酸酯类烯烃(527)只得到33%的目标产物。
文中,亚磺酰胺底物(即结构式(1)所示的化合物)的产率以及芳香族烯基化合物(即结构式(5)所示的化合物)的产率的计算公式如下:
亚磺酰胺底物的产率=亚磺酰胺底物的摩尔量/结构式(3)所示的化合物的摩尔量×100%;
芳香族烯基化合物的产率=芳香族烯基化合物的摩尔量/亚磺酰胺底物的摩尔量×100%。
最后应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
Claims (10)
1.一种通过氟烷基亚磺酰基制备芳香族烯基化合物的方法,其特征在于,所述芳香族烯基化合物的结构式如式(5)所示,所述方法包括以下步骤:将结构式(1)所示的化合物与结构式(2)所示的化合物在过渡金属催化剂、氧化剂以及碱的存在下于有机溶剂A中进行反应而得所述芳香族烯基化合物;

其中,R1为氢原子、卤原子、烷基、芳基、烷氧基中的一种;R2以及R3各自独立地为烷基或者R2与R3共同形成环烷基;R4为烷基、环烷基、芳基、苄基或任选具有取代基的烷基,所述具有取代基的烷基为羟基烷基、烷氧基烷基、杂环基烷基中的一种;所述氧化剂为醋酸银;所述碱为碳酸钠、碳酸钾中的至少一种;所述有机溶剂A为六氟异丙醇、三氟乙醇中的至少一种;所述过渡金属催化剂为醋酸钯。
2.一种通过氟烷基亚磺酰基制备芳香族烯基化合物的方法,其特征在于,所述方法包括以下步骤:将结构式(1)所示的化合物与结构式(2)所示的化合物在过渡金属催化剂、氧化剂以及碱的存在下于有机溶剂A中进行反应而得所述芳香族烯基化合物;

所述芳香族烯基化合物的结构式如式(5)所示,所述式(5)化合物为:

其中,所述氧化剂为醋酸银;所述碱为碳酸钠、碳酸钾中的至少一种;所述有机溶剂A为六氟异丙醇、三氟乙醇中的至少一种;所述过渡金属催化剂为醋酸钯。
3.根据权利要求1所述的方法,其特征在于,所述R4表示的烷基为直链烷基或支链烷基。
4.根据权利要求3所述的方法,其特征在于,所述R1为氢原子、氯原子、溴原子、甲基、乙基、叔丁基、芳基、甲氧基中的一种;所述R2与R3各自独立地为甲基、乙基中的一种,或者所述R2与R3共同形成环丙基、环丁基、环戊基、环己基中的一种;所述R4为甲基、乙基、正丁基、异丁基、环己基、芳基、苄基、4-羟基丁基、2-甲氧基乙基、四氢-2-呋喃基甲基中的一种。
5.根据权利要求1-4任一项所述的方法,其特征在于,所述结构式(1)所示的化合物、结构式(2)所示的化合物、过渡金属催化剂、氧化剂、碱以及有机溶剂A的比例为结构式(1)所示的化合物:结构式(2)所示的化合物:过渡金属催化剂:氧化剂:碱:有机溶剂A=1mol:2-5mol:0.05-0.2mol:2-5mol:1-3mol:(0.8-1.5)*104mL;所述反应温度为60-120℃,反应时间为0.1-12h。
6.根据权利要求1-4任一项所述的方法,其特征在于,所述结构式(1)所示的化合物的制备方法包括以下步骤:将结构式(3)所示的化合物与结构式(4)所示的化合物在叔胺存在下于有机溶剂B中进行反应而得到所述结构式(1)所示的化合物;

7.根据权利要求6所述的方法,其特征在于,所述叔胺为三乙胺,所述有机溶剂B为二氯甲烷。
8.根据权利要求6所述的方法,其特征在于,所述结构式(3)所示的化合物与结构式(4)所示的化合物反应时,结构式(3)所示的化合物、结构式(4)所示的化合物以及叔胺的摩尔比为结构式(3)所示的化合物:结构式(4)所示的化合物:叔胺=1:0.9-2:5-20,反应温度为0℃至室温,反应时间为1-3h。
9.根据权利要求6所述的方法,其特征在于,所述结构式(3)所示的化合物的制备方法包括以下步骤:
(1)将1-碘-2-氯四氟乙烷在连二亚硫酸钠以及碳酸氢钠存在下于溶剂C中进行搅拌反应,反应结束后,得到反应混合液;
(2)采用有机溶剂对步骤(1)所得反应混合液进行萃取,收集有机相并除去溶剂,再加入硫酸,混合均匀后进行热过滤,收集滤液并浓缩,得到浓缩液;
(3)在步骤(2)所得浓缩液中加入二氯亚砜进行反应,即得结构式(3)所示的化合物。
10.根据权利要求9所述的方法,其特征在于,所述1-碘-2-氯四氟乙烷、连二亚硫酸钠、碳酸氢钠以及二氯亚砜的摩尔比为1-碘-2-氯四氟乙烷:连二亚硫酸钠:碳酸氢钠:二氯亚砜=1:1-2:1-2:1-5;所述步骤(1)中的反应温度为室温,反应时间为5-10h,溶剂C为水和乙腈的混合液;所述步骤(3)中的反应温度为0℃至室温,反应时间为1-5h。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010411494.8A CN111635359B (zh) | 2020-05-14 | 2020-05-14 | 一种通过氟烷基亚磺酰基制备芳香族烯基化合物的方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010411494.8A CN111635359B (zh) | 2020-05-14 | 2020-05-14 | 一种通过氟烷基亚磺酰基制备芳香族烯基化合物的方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN111635359A CN111635359A (zh) | 2020-09-08 |
| CN111635359B true CN111635359B (zh) | 2022-08-23 |
Family
ID=72325498
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202010411494.8A Active CN111635359B (zh) | 2020-05-14 | 2020-05-14 | 一种通过氟烷基亚磺酰基制备芳香族烯基化合物的方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN111635359B (zh) |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111393364B (zh) * | 2020-04-14 | 2021-07-13 | 五邑大学 | 一种含C(sp2)-S键芳基砜化合物的合成方法 |
-
2020
- 2020-05-14 CN CN202010411494.8A patent/CN111635359B/zh active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CN111635359A (zh) | 2020-09-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN108947894B (zh) | 联芳结构手性n-甲基吡哆醛催化剂及其合成和应用 | |
| Nie et al. | Chiral bifunctional thiourea-catalyzed enantioselective aldol reaction of trifluoroacetaldehyde hemiacetal with aromatic ketones | |
| Li et al. | An efficient enantioselective synthesis of florfenicol via a vanadium-catalyzed asymmetric epoxidation | |
| CN109956870A (zh) | 一种罗沙司他的合成方法及其中间体化合物 | |
| CN112442008B (zh) | 一种温度调控单质硫与活泼内炔制备1,4-二噻烯和噻吩类化合物的方法及其转化反应 | |
| CN111718372B (zh) | 一种轴手性膦-烯配体及其制备方法与应用 | |
| JP7464234B2 (ja) | 軸不斉を有する高光学活性アレンカルボン酸系化合物を製造する方法 | |
| CN111635359B (zh) | 一种通过氟烷基亚磺酰基制备芳香族烯基化合物的方法 | |
| CN102863371B (zh) | 氟代二氢吡咯或氟代吡咯 | |
| CN114163445B (zh) | 拉罗替尼中间体及其制备方法 | |
| CN109265385B (zh) | 一种手性催化剂的合成工艺 | |
| JP2008503453A (ja) | 無水アルキルホスホン酸類を用いた、アルコール類から水の脱離によるアルケン類の製造方法 | |
| CN107513056A (zh) | 一种含四氢呋喃基团的喹啉类化合物的合成方法 | |
| JP4425654B2 (ja) | 水溶性遷移金属−ジアミン錯体、及びその製造方法、並びにその用途 | |
| CN114082446A (zh) | 用于制备手性α-羟基-β-酮酸酯化合物的手性锆催化剂及其制备方法 | |
| CN107531662B (zh) | 奈必洛尔的合成方法及其中间体化合物 | |
| CN107325025A (zh) | 一种手性α‑氨基酸衍生物及其制备方法 | |
| CN114805017B (zh) | 一种2-氟-1,5-己二烯类化合物的制备方法 | |
| CN115232163B (zh) | 一种硅中心手性分子化合物及其制备方法与应用 | |
| CN112442042B (zh) | 一种螺环吲哚化合物的制备方法 | |
| CN114057717B (zh) | 一种喹啉取代的双噁唑啉配体、其合成方法及其应用 | |
| JP4860510B2 (ja) | β位に不斉点を有するカルボン酸の製造及び求核剤 | |
| JP2006206550A (ja) | δ−イミノマロン酸誘導体の製造方法、及びそのための触媒 | |
| JP2005247715A (ja) | 配位子の合成方法 | |
| CN106278968B (zh) | 一种合成硫代氨基酸衍生物的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |