CN109320552B - 具有良好生物活性的葛根素衍生物及其制备方法以及应用 - Google Patents
具有良好生物活性的葛根素衍生物及其制备方法以及应用 Download PDFInfo
- Publication number
- CN109320552B CN109320552B CN201811156463.1A CN201811156463A CN109320552B CN 109320552 B CN109320552 B CN 109320552B CN 201811156463 A CN201811156463 A CN 201811156463A CN 109320552 B CN109320552 B CN 109320552B
- Authority
- CN
- China
- Prior art keywords
- compound
- reaction
- follows
- puerarin
- tertiary amine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- HKEAFJYKMMKDOR-VPRICQMDSA-N puerarin Chemical class O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1C1=C(O)C=CC(C2=O)=C1OC=C2C1=CC=C(O)C=C1 HKEAFJYKMMKDOR-VPRICQMDSA-N 0.000 title claims abstract description 81
- 238000002360 preparation method Methods 0.000 title claims description 32
- 230000004071 biological effect Effects 0.000 title description 5
- 150000001875 compounds Chemical class 0.000 claims abstract description 53
- 238000006243 chemical reaction Methods 0.000 claims abstract description 49
- 150000003512 tertiary amines Chemical class 0.000 claims abstract description 45
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 60
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 33
- 239000007787 solid Substances 0.000 claims description 33
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 32
- 238000000034 method Methods 0.000 claims description 31
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 claims description 24
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 24
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 23
- 238000010828 elution Methods 0.000 claims description 21
- 238000003756 stirring Methods 0.000 claims description 21
- 229940125898 compound 5 Drugs 0.000 claims description 19
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 18
- 230000015572 biosynthetic process Effects 0.000 claims description 18
- 238000003786 synthesis reaction Methods 0.000 claims description 18
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 18
- 229940125782 compound 2 Drugs 0.000 claims description 17
- 229940126214 compound 3 Drugs 0.000 claims description 16
- 238000010992 reflux Methods 0.000 claims description 16
- 239000000843 powder Substances 0.000 claims description 15
- ZVJYSSDFOMJWSE-UHFFFAOYSA-N chloro-bis(phenylmethoxy)phosphane Chemical compound C=1C=CC=CC=1COP(Cl)OCC1=CC=CC=C1 ZVJYSSDFOMJWSE-UHFFFAOYSA-N 0.000 claims description 14
- 229940125904 compound 1 Drugs 0.000 claims description 14
- 239000000243 solution Substances 0.000 claims description 14
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 claims description 12
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 12
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 claims description 10
- 239000003814 drug Substances 0.000 claims description 10
- 235000019441 ethanol Nutrition 0.000 claims description 9
- 239000000706 filtrate Substances 0.000 claims description 9
- 239000000203 mixture Substances 0.000 claims description 9
- 239000012044 organic layer Substances 0.000 claims description 9
- 239000002904 solvent Substances 0.000 claims description 9
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 8
- 238000001035 drying Methods 0.000 claims description 8
- 239000003480 eluent Substances 0.000 claims description 8
- 238000001914 filtration Methods 0.000 claims description 8
- 238000010898 silica gel chromatography Methods 0.000 claims description 8
- 238000005406 washing Methods 0.000 claims description 7
- ULTHEAFYOOPTTB-UHFFFAOYSA-N 1,4-dibromobutane Chemical compound BrCCCCBr ULTHEAFYOOPTTB-UHFFFAOYSA-N 0.000 claims description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 6
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 claims description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 6
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 claims description 6
- 125000000217 alkyl group Chemical group 0.000 claims description 6
- 235000011114 ammonium hydroxide Nutrition 0.000 claims description 6
- 201000007270 liver cancer Diseases 0.000 claims description 6
- 208000014018 liver neoplasm Diseases 0.000 claims description 6
- 201000005202 lung cancer Diseases 0.000 claims description 6
- 208000020816 lung neoplasm Diseases 0.000 claims description 6
- 239000003208 petroleum Substances 0.000 claims description 6
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 6
- 238000004809 thin layer chromatography Methods 0.000 claims description 6
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 claims description 5
- 238000001816 cooling Methods 0.000 claims description 5
- RQKYHDHLEMEVDR-UHFFFAOYSA-N oxo-bis(phenylmethoxy)phosphanium Chemical compound C=1C=CC=CC=1CO[P+](=O)OCC1=CC=CC=C1 RQKYHDHLEMEVDR-UHFFFAOYSA-N 0.000 claims description 5
- 206010008342 Cervix carcinoma Diseases 0.000 claims description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 4
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 claims description 4
- 201000010881 cervical cancer Diseases 0.000 claims description 4
- SRCZQMGIVIYBBJ-UHFFFAOYSA-N ethoxyethane;ethyl acetate Chemical compound CCOCC.CCOC(C)=O SRCZQMGIVIYBBJ-UHFFFAOYSA-N 0.000 claims description 4
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 claims description 3
- WDBQJSCPCGTAFG-QHCPKHFHSA-N 4,4-difluoro-N-[(1S)-3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-pyridin-3-ylpropyl]cyclohexane-1-carboxamide Chemical compound FC1(CCC(CC1)C(=O)N[C@@H](CCN1CCC(CC1)N1C(=NN=C1C)C(C)C)C=1C=NC=CC=1)F WDBQJSCPCGTAFG-QHCPKHFHSA-N 0.000 claims description 3
- BWGRDBSNKQABCB-UHFFFAOYSA-N 4,4-difluoro-N-[3-[3-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]octan-8-yl]-1-thiophen-2-ylpropyl]cyclohexane-1-carboxamide Chemical compound CC(C)C1=NN=C(C)N1C1CC2CCC(C1)N2CCC(NC(=O)C1CCC(F)(F)CC1)C1=CC=CS1 BWGRDBSNKQABCB-UHFFFAOYSA-N 0.000 claims description 3
- LFZAGIJXANFPFN-UHFFFAOYSA-N N-[3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-thiophen-2-ylpropyl]acetamide Chemical compound C(C)(C)C1=NN=C(N1C1CCN(CC1)CCC(C=1SC=CC=1)NC(C)=O)C LFZAGIJXANFPFN-UHFFFAOYSA-N 0.000 claims description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 claims description 3
- 239000007864 aqueous solution Substances 0.000 claims description 3
- 239000003729 cation exchange resin Substances 0.000 claims description 3
- WORJEOGGNQDSOE-UHFFFAOYSA-N chloroform;methanol Chemical compound OC.ClC(Cl)Cl WORJEOGGNQDSOE-UHFFFAOYSA-N 0.000 claims description 3
- 229910052739 hydrogen Inorganic materials 0.000 claims description 3
- 239000001257 hydrogen Substances 0.000 claims description 3
- 239000005457 ice water Substances 0.000 claims description 3
- 229910052757 nitrogen Inorganic materials 0.000 claims description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 claims description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 claims description 3
- 238000000967 suction filtration Methods 0.000 claims description 3
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 claims description 2
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 24
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 10
- RXUWDKBZZLIASQ-UHFFFAOYSA-N Puerarin Natural products OCC1OC(Oc2c(O)cc(O)c3C(=O)C(=COc23)c4ccc(O)cc4)C(O)C(O)C1O RXUWDKBZZLIASQ-UHFFFAOYSA-N 0.000 description 8
- 229940079593 drug Drugs 0.000 description 7
- 230000000694 effects Effects 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- 238000002390 rotary evaporation Methods 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 206010028980 Neoplasm Diseases 0.000 description 3
- 235000010575 Pueraria lobata Nutrition 0.000 description 3
- 230000001093 anti-cancer Effects 0.000 description 3
- 230000000259 anti-tumor effect Effects 0.000 description 3
- 208000019065 cervical carcinoma Diseases 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- AYMUQTNXKPEMLM-UHFFFAOYSA-N 1-bromononane Chemical compound CCCCCCCCCBr AYMUQTNXKPEMLM-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- 241000219781 Pueraria montana var. lobata Species 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 230000000857 drug effect Effects 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- BKIMMITUMNQMOS-UHFFFAOYSA-N nonane Chemical class CCCCCCCCC BKIMMITUMNQMOS-UHFFFAOYSA-N 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- MYMSJFSOOQERIO-UHFFFAOYSA-N 1-bromodecane Chemical compound CCCCCCCCCCBr MYMSJFSOOQERIO-UHFFFAOYSA-N 0.000 description 1
- VMKOFRJSULQZRM-UHFFFAOYSA-N 1-bromooctane Chemical compound CCCCCCCCBr VMKOFRJSULQZRM-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 241000220485 Fabaceae Species 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 244000046146 Pueraria lobata Species 0.000 description 1
- 241000731396 Pueraria montana var. thomsonii Species 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 208000026106 cerebrovascular disease Diseases 0.000 description 1
- 239000013043 chemical agent Substances 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 210000004351 coronary vessel Anatomy 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000000916 dilatatory effect Effects 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 229930182486 flavonoid glycoside Natural products 0.000 description 1
- 150000007955 flavonoid glycosides Chemical class 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- GOMNOOKGLZYEJT-UHFFFAOYSA-N isoflavone Chemical compound C=1OC2=CC=CC=C2C(=O)C=1C1=CC=CC=C1 GOMNOOKGLZYEJT-UHFFFAOYSA-N 0.000 description 1
- CJWQYWQDLBZGPD-UHFFFAOYSA-N isoflavone Natural products C1=C(OC)C(OC)=CC(OC)=C1C1=COC2=C(C=CC(C)(C)O3)C3=C(OC)C=C2C1=O CJWQYWQDLBZGPD-UHFFFAOYSA-N 0.000 description 1
- 235000008696 isoflavones Nutrition 0.000 description 1
- 230000031700 light absorption Effects 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- LFETXMWECUPHJA-UHFFFAOYSA-N methanamine;hydrate Chemical compound O.NC LFETXMWECUPHJA-UHFFFAOYSA-N 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 239000010865 sewage Substances 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65586—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system at least one of the hetero rings does not contain nitrogen as ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
本发明涉及一种葛根素衍生物及其制备方法以及应用,该葛根素衍生物以葛根素为原料先经5步反应制得化合物6,之后化合物6与一些列叔胺反应制得相应的葛根素衍生物,本发明制得的一系列葛根素衍生物具有良好的水溶性,适合做注射剂,另外该葛根素衍生物具有良好的抗肿瘤效果,经实验验证,该葛根素衍生物对肺癌细胞、结肠癌细胞、胃癌细胞、肝癌细胞以及宫颈癌(Hela)细胞均具有良好的抑制作用。
Description
技术领域
本发明涉及一种具有良好生物活性的葛根素衍生物及其制备方法以及应用。
背景技术
葛根素是多年生豆科植物野葛(Pueraria Lobata(Willd.)ohwi)或甘葛藤(又称粉葛)(Pueraria thomsonii Benth)的干燥根中提取的一种黄酮苷。化学名为7,4’-二羟基-8-β-D-葡萄糖异黄酮,分子式为C21H20O9,分子量416.38,CAS号为82373-94-3。作为葛根的特有成份,具有独特的化学结构、生物活性和药理作用。葛根素有扩张冠状动脉和脑血管作用,同时具有降血糖及血脂、抗氧化、抗肿瘤的作用。然而,葛根素在水中溶解度较低,这极大的影响了葛根素的使用以及对其进行更深层次的药理、药效的研究。另外,在葛根素的药效研究方面,大多仅研究其在心脑血管疾病方面上的作用,而对抗肿瘤方面的研究甚少。
发明内容
本发明的目的在于提供一种合成方法简单且集良好水溶性与显著抗肿瘤效果为一体的具有良好生物活性的葛根素衍生物及其制备方法以及应用。
本发明的目的通过如下技术方案实现:一种葛根素衍生物,其结构式如下:

其中,M为Na+或K+中的一种;所述R1为C8-C12的饱和直链烷基;所述R2为C8-C12的饱和直链烷基。
所述的葛根素衍生物的制备方法,它包括以下步骤:
(1)化合物2的合成:将化合物1溶解于吡啶中,再与乙酸酐反应制得化合物2;其中化合物1与化合物2的结构式分别为:

(2)化合物3的合成:将步骤(1)所得的化合物2,在二异丙基乙胺的作用下,与氯代亚磷酸二苄酯反应得化合物3,其中化合物3的结构式为:

(3)化合物4的合成:将步骤(2)所得的化合物3与Pd/C,H2作用,得化合物4,其中化合物4的结构式为:

(4)化合物5的合成:将步骤(3)所得的化合物4与氨水作用,得化合物5,其中化合物5的结构式为:

(5)化合物6的合成:将步骤(4)所得的化合物5与1,4-二溴丁烷以及MOH反应得化合物6;其中MOH为NaOH或KOH中的一种,所述化合物6的结构式为:

(6)葛根素衍生物的合成:将步骤(5)所得的化合物6与一系列叔胺反应,得葛根素衍生物。
所述的葛根素衍生物的应用,在制备治疗肺癌、肝癌、宫颈癌药物中的应用。
较之现有技术而言,本发明的优点在于:1.本发明制得的一系列葛根素衍生物具有良好的水溶性,适合做注射剂,另外,该葛根素衍生物同时具备良好的脂溶性,能够很好的进入细胞,进而最大程度的发挥其药效,由此可知该葛根素衍生物是一种既亲水又亲脂的双亲型化合物。2.该葛根素衍生物具有良好的抗肿瘤效果,经实验验证,该葛根素衍生物对肺癌细胞、结肠癌细胞、胃癌细胞、肝癌细胞以及宫颈癌(Hela)细胞均具有良好的抑制作用,且对Hela细胞的抑制作用尤为显著,可见该葛根素衍生物可以作为潜在的抗癌药物。3.本发明制备葛根素衍生物的制备方法简单,易于葛根素衍生物的合成。
具体实施方式
下面结合实施例对本发明内容进行详细说明:
一种葛根素衍生物,其结构式如下:

其中,M为Na+或K+中的一种;所述R1为C8-C12的饱和直链烷基;所述R2为C8-C12的饱和直链烷基。
所述的葛根素衍生物的制备方法,它包括以下步骤:
(1)化合物2的合成:将化合物1溶解于吡啶中,再与乙酸酐反应制得化合物2;其中化合物1与化合物2的结构式分别为:

(2)化合物3的合成:将步骤(1)所得的化合物2,在二异丙基乙胺的作用下,与氯代亚磷酸二苄酯反应得化合物3,其中化合物3的结构式为:

(3)化合物4的合成:将步骤(2)所得的化合物3与Pd/C,H2作用,得化合物4,其中化合物4的结构式为:

(4)化合物5的合成:将步骤(3)所得的化合物4与氨水作用,得化合物5,其中化合物5的结构式为:

(5)化合物6的合成:将步骤(4)所得的化合物5与1,4-二溴丁烷以及MOH反应得化合物6;其中MOH为NaOH或KOH中的一种,所述化合物6的结构式为:

(6)葛根素衍生物的合成:将步骤(5)所得的化合物6与一系列叔胺反应,得葛根素衍生物。
所述葛根素衍生物的合成路线如下:

其中,步骤(1)的具体操作方法为:在化合物1中加入经干燥后的吡啶,使化合物1完全溶解,之后加入乙酸酐,常温下搅拌25-35min,室温下放置18-24h得混合物A,将混合物A缓慢的倒入冰水中,充分搅拌,析出大量固体,抽滤得固体A,将固体A用二氯甲烷溶解,加入5%的碳酸氢钠水溶液,常温下搅拌45-60min,用分液漏斗分液并收集有机层,有机层减压回收溶剂至干,得固体B,固体B经硅胶柱层析,以石油醚-乙酸乙酯作为洗脱剂进行梯度,收集石油醚与乙酸乙酯体积比为15:1时的洗脱部分,回收溶剂,再利用丙酮重结晶,得化合物2;
其中,化合物1与乙酸酐的摩尔比为1:8-1:10。
步骤(2)的具体操作方法为:
a.氯代亚磷酸二苄酯的制备:将亚磷酸二苄酯与N-氯代丁二酰亚胺反应得氯代亚磷酸二苄酯;
b.将步骤(1)所得的化合物2溶解于二氯甲烷中,加入二异丙基乙胺,然后将反应体系的温度降到0℃,往反应体系中滴加氯代亚磷酸二苄酯的甲苯溶液,滴加完毕后,室温搅拌10-12h,待反应结束后,向反应体系中加入水,然后利用二氯甲烷萃取,饱和食盐水洗涤,无水硫酸钠干燥得到化合物3;
步骤a的操作方法为:将亚磷酸二苄酯用干燥的甲苯溶解后,加入N-氯代丁二酰亚胺,氮气保护下室温搅拌3-5小时,反应结束后,过滤,收集滤液,记得所述氯代亚磷酸二苄酯。
步骤(3)的具体操作方法为:将步骤(2)所得的化合物3用甲醇溶解,加入5%钯碳,在氢气加压下氢化2-3小时,利用TLC检测反应进程,待反应结束后,过滤,收集滤液,即得所述化合物4。
步骤(4)的具体操作方法为:将步骤(3)所得的化合物4溶于甲醇中,之后加入氨水,室温搅拌3-4小时,停止反应,将反应液减压浓缩,固体用水溶解,阳离子交换树脂处理,过滤,滤液减压浓缩,得到化合物5。
步骤(5)的具体操作方法为:将MOH置于反应瓶中,加入无水乙醇,在85-90℃下回流30-45min至MOH溶解,之后加入步骤(4)所得的化合物5,继续回流2-3h至化合物5溶解,接着再加入1,4-二溴丁烷,继续回流反应10-12h,待反应结束后,减压回收溶剂,得淡黄色固体粉末,淡黄色固体粉末利用甲醇重结晶,干燥得到白色粉末状化合物6。
步骤(6)的具体操作方法为:将步骤(5)所得化合物6置于反应瓶中,加入无水乙醇,在85-90℃下回流搅拌30-45min,之后再加入叔胺,继续回流反应20-24h,其中叔胺与化合物6的摩尔比为1:1-1.5;待反应结束后,冷却,减压回收乙醇至干,得淡黄色固体粉末,将该淡黄色固体粉末利用硅胶柱层析分离提纯,以氯仿-甲醇作为洗脱剂,按照氯仿与甲醇的体积比依次为50:1、40:1、30:1、20:1、10:1、8:1的洗脱顺序进行梯度洗脱,收集氯仿与甲醇体积比为8:1时的洗脱部分,收集的洗脱部分经减压除去洗脱剂后得淡黄色固体粉末,即为所述葛根素衍生物。
下面结合实施例对本发明的发明内容作更细致的阐释:
实施例一:化合物6的合成:
(1)化合物2的制备:
在50g化合物1中加入经干燥后的吡啶300mL,使化合物1完全溶解,之后加入乙酸酐100g,常温下搅拌30min,室温下放置24h得混合物A,将混合物A缓慢的倒入冰水中,充分搅拌,析出大量固体,抽滤得固体A,将固体A用二氯甲烷溶解,加入5%的碳酸氢钠水溶液,常温下搅拌60min,用分液漏斗分液并收集有机层,有机层减压回收溶剂至干,得固体B,固体B经硅胶柱层析,以石油醚-乙酸乙酯作为洗脱剂进行梯度,按照石油醚与乙酸乙酯的体积比依次为45:1、35:1、30:1、25:1、15:1的洗脱顺序进行梯度洗脱,收集石油醚与乙酸乙酯体积比为15:1时的洗脱部分,回收溶剂,再利用丙酮重结晶,得化合物2(20g);
其中,化合物2的1H NMR结果为:1H NMR(CDCl3,δ,ppm)8.20(d,1H,J=9.5Hz),7.57(d,2H,J=8.5Hz),7.17(d,2H,8.5Hz),7.01(d,1H,8.5Hz),5.40(m,3H),5.35(m,1H),4.37(dd,1H,J=12.5Hz,3.5Hz),4.21(dd,1H,J=12.5Hz,2Hz),3.99(m,1H),2.32(s,3H),2.14(s,3H),2.09(s,3H),2.02(s,3H),1.67(s,3H)。
(2)化合物3的制备:
a.氯代亚磷酸二苄酯的制备:将亚磷酸二苄酯(160g,0.610mol)用500mL干燥的甲苯溶解后,往里加入N-氯代丁二酰亚胺(115g,0.861mol),氮气保护下室温搅拌4小时,之后过滤,收集滤液,即得氯代亚磷酸二苄酯的甲苯溶液,氯代亚磷酸二苄酯的甲苯溶液直接用于下步反应。
b.将步骤(1)所得的化合物2(60g,95.76mmol)溶解于600mL二氯甲烷中,加入二异丙基乙胺(22.0g,0.169mmol),然后将反应体系的温度降到0℃,往反应体系中滴加80mL步骤a所得的氯代亚磷酸二苄酯的甲苯溶液,滴加完毕后,室温搅拌12h,TLC检测反应进程,待反应结束后,向反应体系中加入水500mL,然后利用二氯甲烷(300mL×3)萃取,饱和食盐水洗涤,无水硫酸钠干燥得到化合物3(60.2g)。
(3)化合物4的制备:取步骤(2)所得的化合物3(55g,61.93mmol)用500mL甲醇溶解,加入5%钯碳(2.7g),氢气加压下氢化2小时,TLC检测反应完全。过滤,收集滤液,即得化合物4的甲醇溶液,化合物4的甲醇溶液直接用于下步反应。
(4)化合物5的制备:往步骤(3)所得的化合物4的甲醇溶液中加入50mL氨水,室温搅拌4小时,HPLC检测显示反应完全。停止反应,将反应液减压浓缩,固体用100mL水溶解,阳离子交换树脂处理,过滤,滤液减压浓缩,得到化合物5(31g)。
(5)化合物6的制备:将NaOH(1.7g,42.5mmol)置于反应瓶中,加入无水800mL乙醇,在90℃下回流30min至NaOH溶解,之后加入步骤(4)所得的化合物5(4.96g,10mmol),继续回流2-3h至化合物5溶解,接着再加入1,4-二溴丁烷(2.591g,12mmol),继续回流反应12h,待反应结束后,减压回收溶剂,得淡黄色固体粉末,淡黄色固体粉末利用甲醇重结晶,干燥得到白色粉末状化合物6。
化合物6的1H NMR结果为:1H NMR(D2O,δ,ppm)8.34(d,1H,J=11.0Hz),8.25(d,1H,J=7.7Hz),7.73(d,2H,J=7.8Hz),7.44(d,2H,6.52Hz),6.97(d,1H,6.5Hz),4.54(d,1H),4.14(t,1H),4.07-4.11(m,3H),3.69(M,1H),3.44-3.47(M,3H),3.23-3.24(m,2H),1.88-2.16(m,4H),1.81-1.84(m,4H)。NaOH也可以用KOH来替代。
实施例二:叔胺的合成:
所述叔胺的结构式为: 叔胺Z1至Z6的碳链均为饱和的直链。其中叔胺Z1、Z2以及Z3均为含有9个碳的长链叔胺,市面上暂无销售,因此叔胺Z1、Z2以及Z3由本发明的发明人合成。其他叔胺Z4、Z5、Z6从国药化学试剂有限公司购置。
叔胺Z1至Z6的碳链均为饱和的直链。其中叔胺Z1、Z2以及Z3均为含有9个碳的长链叔胺,市面上暂无销售,因此叔胺Z1、Z2以及Z3由本发明的发明人合成。其他叔胺Z4、Z5、Z6从国药化学试剂有限公司购置。
叔胺Z1、Z2以及Z3制备路线为:

a.化合物7的合成:
在反应瓶中加入50mL甲胺水溶液、12mL溴代正壬烷以及20mL乙醇,常温搅拌45min后,将反应液用水洗涤(除去过量的甲胺)、氯仿萃取,将氯仿萃取所得的有机相用污水硫酸钠干燥,旋蒸,旋到没有液滴滴下为止,得到淡黄色的粘稠液体即为粗产品化合物7。之后将该淡黄色的粘稠液体进行硅胶柱层析,利用二氯甲烷与乙醇体积比为35:1的混合溶剂洗脱,利用TLC配合碘缸显色检测洗脱进程,洗脱结束后,将收集的流出液进行旋蒸,得无色粘稠液体化合物7。
化合物7的1H NMR结果为:1H NMR(400MHz,CDCl3)δ:3.69(s,1H,-NH),2.61(t,2H,J=6.801Hz,-CH 2-),1.53(m,2H,-NCH2CH 2-),1.26(m,12H,-(CH2)6-),0.87(t,J=6.80Hz,3H,-CH 3),化合物7的ESI-MS m/z 158.20(M+H)+.
b.叔胺Z1的制备:
在反应瓶中加入0.02mol的化合物7、0.02mol的溴代正壬烷C9H19Br、NaOH(0.8g,0.02mol)以及乙醇,回流搅拌5h,之后将反应冷却,用水洗涤除去无机盐,水洗3遍,水洗过程中用大量的氯仿萃取。合并有机相,有机相用无水硫酸钠干燥,旋蒸,得到淡黄色黏稠物质即为粗产品叔胺Z1。粗产品叔胺Z1经硅胶柱层析,利用二氯甲烷洗脱,利用TLC配合碘缸显色检测洗脱进程,洗脱结束后,将收集的流出液进行旋蒸,得无色粘稠液体叔胺Z1。
叔胺Z1的1H NMR结果为:1H NMR(400MHZ,CDCl3)δ:2.33(t,J=7.6Hz,4H,-CH 2-N-CH 2-),2.22(s,3H,N-CH3),1.47(m,4H,2×-NCH2CH 2-),1.27(m,24H,2×-(CH2)6-),0.88(t,J=7.2Hz,6H,2×-CH 3),叔胺Z1的ESI-MS m/z 284.34(M+H)+.
c.叔胺Z2的制备:叔胺Z2的制备方法与叔胺Z1的制备方法相同,仅将原料溴代正壬烷C9H19Br替换成溴代正辛烷C8H17Br。叔胺Z2的1H NMR结果为:1H NMR(400MHZ,CDCl3)δ:2.33(m,4H,-CH 2-N-CH 2-),2.23(s,3H,N-CH3),1.48(m,4H,2×-NCH2CH 2-),1.29(m,22H,-(CH2)5-,-(CH2)6-),0.90(m,6H,2×-CH 3),叔胺Z2的ESI-MS m/z 270.37(M+H)+.
d.叔胺Z3的制备:叔胺Z3的制备方法与叔胺Z1的制备方法相同,仅将原料溴代正壬烷C9H19Br替换成溴代正癸烷C10H21Br。叔胺Z3的1H NMR结果为:1H NMR(400MHz,CDCl3)δ:2.32(t,J=7.6Hz,4H,-CH 2-N-CH 2-),2.22(s,3H,N-CH3),1.47(m,4H,2×-NCH2CH 2-),1.28(m,26H,-(CH2)6-,-(CH2)7-),0.89(t,J=6.8Hz,6H,2×-CH 3),叔胺Z3的ESI-MS m/z 298.39(M+H)+.
实施例三:葛根素衍生物Y1的合成:
取0.250mmol实施例一步骤(5)中所得化合物6置于反应瓶中,加入500mL无水乙醇,在90℃下回流搅拌45min,之后再加入0.252mmol实施例二所得的叔胺Z1,继续回流反应24h,待反应结束后,冷却,减压回收乙醇至干,得淡黄色固体粉末,将该淡黄色固体粉末利用硅胶柱层析分离提纯,以氯仿-甲醇作为洗脱剂,按照氯仿与甲醇的体积比依次为50:1、40:1、30:1、20:1、10:1、8:1的洗脱顺序进行梯度洗脱,收集氯仿与甲醇体积比为8:1时的洗脱部分,收集的洗脱部分经减压除去洗脱剂后得淡黄色固体粉末,即为所述葛根素衍生物Y1。室温下,葛根素衍生物Y1在水中的溶解度约为353mg/mL。
葛根素衍生物Y1的1H NMR结果为:
1H NMR(D2O,δ,ppm):8.44(d,1H,J=11.0Hz),8.35(d,1H,J=7.7Hz),7.63(d,2H,J=7.8Hz),7.51(d,2H,6.52Hz),6.98(d,1H,6.5Hz),4.54(d,1H),4.14(t,2H),3.65-4.07(m,10H),3.47(m,6H,-CH2-N-CH2-),3.34(s,3H,N-CH3),1.88-1.74(m,8H,3×-N-CH2-CH 2-,-O-CH2-CH 2-),1.43-1.26(m,24H,2×-(CH2)6-),0.89(t,J=6.8Hz,6H,2×-CH3)。
实施例四:葛根素衍生物Y2的合成:葛根素衍生物Y2的制备方法与葛根素衍生物Y1的制备方法相同,仅将叔胺Z1替换为实施例二所得的叔胺Z2。室温下,葛根素衍生物Y2在水中的溶解度约为365mg/mL。葛根素衍生物Y2的1H NMR结果为:
1H NMR(D2O,δ,ppm):8.42(d,1H,J=11.0Hz),8.34(d,1H,J=7.7Hz),7.61(d,2H,J=7.8Hz),7.49(d,2H,6.52Hz),7.01(d,1H,6.5Hz),4.52(d,1H),4.13(t,2H),3.63-4.11(m,10H),3.47(m,6H,-CH2-N-CH2-),3.34(s,3H,N-CH3),1.83-1.76(m,8H,3×-N-CH2-CH 2-,-O-CH2-CH 2-),1.42-1.31(m,22H,-(CH2)5-,-(CH2)6-),0.90(t,J=6.8Hz,6H,2×-CH3)。
实施例五:葛根素衍生物Y3的合成:葛根素衍生物Y3的制备方法与葛根素衍生物Y1的制备方法相同,仅将叔胺Z1替换为实施例二所得的叔胺Z3。室温下,葛根素衍生物Y3在水中的溶解度约为372mg/mL。葛根素衍生物Y3的1H NMR结果为:
1H NMR(D2O,δ,ppm):8.31(d,1H,J=11.0Hz),8.35(d,1H,J=7.7Hz),7.59(d,2H,J=7.8Hz),7.50(d,2H,6.52Hz),6.99(d,1H,6.5Hz),4.51(d,1H),4.12(t,2H),3.58-4.03(m,10H),3.47(m,6H,-CH2-N-CH2-),3.34(s,3H,N-CH3),1.86-1.80(m,8H,3×-N-CH2-CH 2-,-O-CH2-CH 2-),1.41-1.27(m,26H,-(CH2)7-,-(CH2)6-),0.89(t,J=6.8Hz,6H,2×-CH3)。
实施例六:葛根素衍生物Y4的合成:葛根素衍生物Y4的制备方法与葛根素衍生物Y1的制备方法相同,仅将叔胺Z1替换为叔胺Z4。室温下,葛根素衍生物Y4在水中的溶解度约为389mg/mL。
葛根素衍生物Y4的1H NMR结果为:
1H NMR(D2O,δ,ppm):8.41(d,1H,J=11.0Hz),8.33(d,1H,J=7.7Hz),7.60(d,2H,J=7.8Hz),7.52(d,2H,6.52Hz),7.02(d,1H,6.5Hz),4.55(d,1H),4.13(t,2H),3.66-4.08(m,10H),3.45(m,6H,-CH2-N-CH2-),3.31(s,3H,N-CH3),1.83-1.76(m,8H,3×-N-CH2-CH 2-,-O-CH2-CH 2-),1.41-1.32(m,20H,2×-(CH2)5-),0.90(t,J=6.8Hz,6H,2×-CH3)。
实施例七:葛根素衍生物Y5的合成:葛根素衍生物Y5的制备方法与葛根素衍生物Y1的制备方法相同,仅将叔胺Z1替换为叔胺Z5。室温下,葛根素衍生物Y5在水中的溶解度约为362mg/mL。葛根素衍生物Y5的1H NMR结果为:
1H NMR(D2O,δ,ppm):8.42(d,1H,J=11.0Hz),8.34(d,1H,J=7.7Hz),7.61(d,2H,J=7.8Hz),7.49(d,2H,6.52Hz),7.01(d,1H,6.5Hz),4.52(d,1H),4.13(t,2H),3.63-4.11(m,10H),3.47(m,6H,-CH2-N-CH2-),3.31(s,3H,N-CH3),1.83-1.78(m,8H,3×-N-CH2-CH 2-,-O-CH2-CH 2-),1.43-1.30(m,28H,2×-(CH2)7-),0.89(t,J=6.8Hz,6H,2×-CH3)。
实施例八:葛根素衍生物Y6的合成:葛根素衍生物Y6的制备方法与葛根素衍生物Y1的制备方法相同,仅将叔胺Z1替换为叔胺Z6。室温下,葛根素衍生物Y6在水中的溶解度约为356mg/mL。葛根素衍生物Y6的1H NMR结果为:
1H NMR(D2O,δ,ppm):8.41(d,1H,J=11.0Hz),8.35(d,1H,J=7.7Hz),7.59(d,2H,J=7.8Hz),7.51(d,2H,6.52Hz),7.01(d,1H,6.5Hz),4.53(d,1H),4.14(t,2H),3.62-4.08(m,10H),3.47(m,6H,-CH2-N-CH2-),3.33(s,3H,N-CH3),1.82-1.78(m,8H,3×-N-CH2-CH 2-,-O-CH2-CH 2-),1.42-1.31(m,36H,2×-(CH2)9-),0.89(t,J=6.8Hz,6H,2×-CH3)。实施例九:葛根素衍生物体外细胞活性测试:
将葛根素衍生物Y1-6分别作为受试药物,用培养基将药物稀释,并分别配置成不同的浓度梯度;
取肺癌细胞H446、肝癌细胞HepG2、人宫颈癌细胞Hela和正常细胞HELF,将其密度调整为1×105个/ml,接种于96孔板,每孔100μl,置37℃、5%CO2培养箱中培养24h;
移去旧的培养基,加入受试药物,每孔100μl,另设空白对照组和葛根素组,每组设3个复孔。药物作用48h后,吸弃含药培养基,于每孔中加入无血清、无酚红1640培养基100μl,再加入MTT溶液10μl,继续孵育4h,终止培养;
小心吸弃96孔板孔内上清液,每孔加入100μl DMSO,振荡10min,在酶标仪上于570nm波长处测定各孔光吸收值(OD值),计算细胞的增殖抑制率:抑制率(%)=(1-用药组的OD平均值÷空白对照组的OD平均值)×100%,使用SPSS13.0软件对数据进行处理,并计算癌细胞半数抑制浓度IC50值。结果如表1所示。
表1处理后癌细胞和正常细胞的活性(IC50,μmol/L)
| 葛根素 | Y<sub>1</sub> | Y<sub>2</sub> | Y<sub>3</sub> | Y<sub>4</sub> | Y<sub>5</sub> | Y<sub>6</sub> | |
| 肺癌细胞H446 | ﹥90 | 8.18 | 5.33 | 7.78 | 9.37 | 9.23 | 9.10 |
| 肝癌细胞HepG2 | ﹥90 | 7.76 | 5.13 | 6.32 | 8.35 | 7.71 | 8.11 |
| 人宫颈癌细胞Hela | ﹥90 | 6.53 | 4.42 | 6.11 | 7.12 | 6.98 | 6.03 |
| 正常细胞HELF | ﹥90 | 30.15 | 30.01 | 28.14 | 32.33 | 29.13 | 31.17 |
实验结果表明,葛根素衍生物Y1-6均显示出对肺癌细胞H446、肝癌细胞HepG2、人宫颈癌细胞Hela的抗癌活性,其中葛根素衍生物Y2对人宫颈癌细胞Hela具有显著的抑制作用,其抗癌活性明显强于比葛根素且其对正常细胞HELF毒性较小,可以作为潜在的抗癌药物。
Claims (10)
1.一种葛根素衍生物,其特征在于:其结构式如下:

其中,M为Na+;所述R1为C9的饱和直链烷基;所述R2为C8的饱和直链烷基。
2.根据权利要求1所述的葛根素衍生物的制备方法,其特征在于:它包括以下步骤:
(1)化合物2的合成:将化合物1溶解于吡啶中,再与乙酸酐反应制得化合物2;其中化合物1与化合物2的结构式分别为:

(2)化合物3的合成:将步骤(1)所得的化合物2,在二异丙基乙胺的作用下,与氯代亚磷酸二苄酯反应得化合物3,其中化合物3的结构式为:

(3)化合物4的合成:将步骤(2)所得的化合物3与Pd/C,H2作用,得化合物4,其中化合物4的结构式为:
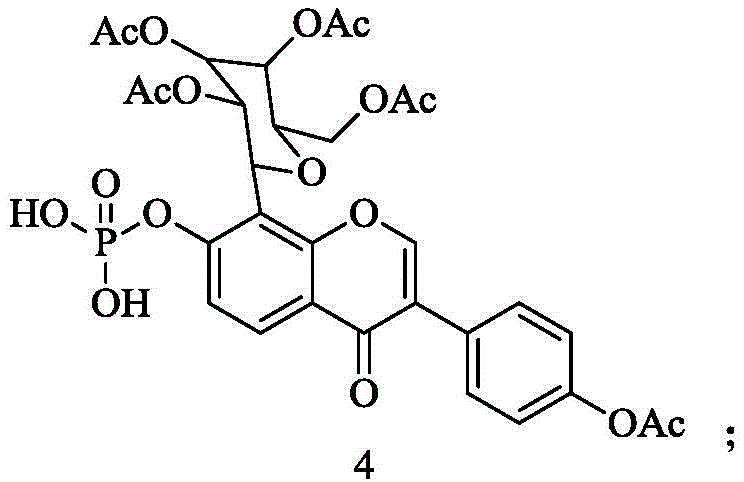
(4)化合物5的合成:将步骤(3)所得的化合物4与氨水作用,得化合物5,其中化合物5的结构式为:

(5)化合物6的合成:将步骤(4)所得的化合物5与1,4-二溴丁烷以及MOH反应得化合物6;其中MOH为NaOH,所述化合物6的结构式为:

其中,M为Na+;
(6)葛根素衍生物的合成:将步骤(5)所得的化合物6与叔胺反应,得葛根素衍生物;其中,所述叔胺为叔胺Z2,其结构式为:

3.根据权利要求2所述的葛根素衍生物的制备方法,其特征在于:步骤(1)的具体操作方法为:在化合物1中加入经干燥后的吡啶,使化合物1完全溶解,之后加入乙酸酐,常温下搅拌25-35min,室温下放置18-24h得混合物A,将混合物A缓慢的倒入冰水中,充分搅拌,析出大量固体,抽滤得固体A,将固体A用二氯甲烷溶解,加入5%的碳酸氢钠水溶液,常温下搅拌45-60min,用分液漏斗分液并收集有机层,有机层减压回收溶剂至干,得固体B,固体B经硅胶柱层析,以石油醚-乙酸乙酯作为洗脱剂进行梯度,收集石油醚与乙酸乙酯体积比为15:1时的洗脱部分,回收溶剂,再利用丙酮重结晶,得化合物2;
其中,化合物1与乙酸酐的摩尔比为1:8-1:10。
4.根据权利要求2所述的葛根素衍生物的制备方法,其特征在于:步骤(2)的具体操作方法为:
a.氯代亚磷酸二苄酯的制备:将亚磷酸二苄酯与N-氯代丁二酰亚胺反应得氯代亚磷酸二苄酯;
b.将步骤(1)所得的化合物2溶解于二氯甲烷中,加入二异丙基乙胺,然后将反应体系的温度降到0℃,往反应体系中滴加氯代亚磷酸二苄酯的甲苯溶液,滴加完毕后,室温搅拌10-12h,待反应结束后,向反应体系中加入水,然后利用二氯甲烷萃取,饱和食盐水洗涤,无水硫酸钠干燥得到化合物3。
5.根据权利要求4所述的葛根素衍生物的制备方法,其特征在于:步骤a的操作方法为:将亚磷酸二苄酯用干燥的甲苯溶解后,加入N-氯代丁二酰亚胺,氮气保护下室温搅拌3-5小时,反应结束后,过滤,收集滤液,即得氯代亚磷酸二苄酯的甲苯溶液。
6.根据权利要求2所述的葛根素衍生物的制备方法,其特征在于:步骤(3)的具体操作方法为:将步骤(2)所得的化合物3用甲醇溶解,加入5%钯碳,在氢气加压下氢化2-3小时,利用TLC检测反应进程,待反应结束后,过滤,收集滤液,可得所述化合物4。
7.根据权利要求2所述的葛根素衍生物的制备方法,其特征在于:步骤(4)的具体操作方法为:
将步骤(3)所得的化合物4溶于甲醇中,之后加入氨水,室温搅拌3-4小时,停止反应,将反应液减压浓缩,固体用水溶解,阳离子交换树脂处理,过滤,滤液减压浓缩,得到化合物5。
8.根据权利要求2所述的葛根素衍生物的制备方法,其特征在于:步骤(5)的具体操作方法为:将NaOH置于反应瓶中,加入无水乙醇,在85-90℃下回流30-45min至MOH溶解,之后加入步骤(4)所得的化合物5,继续回流2-3h至化合物5溶解,接着再加入1,4-二溴丁烷,继续回流反应10-12h,待反应结束后,减压回收溶剂,得淡黄色固体粉末,淡黄色固体粉末利用甲醇重结晶,干燥得到白色粉末状化合物6。
9.根据权利要求2所述的葛根素衍生物的制备方法,其特征在于:步骤(6)的具体操作方法为:将步骤(5)所得化合物6置于反应瓶中,加入无水乙醇,在85-90℃下回流搅拌30-45min,之后再加入叔胺Z2,继续回流反应20-24h,其中叔胺与化合物6的摩尔比为1:1-1.5;待反应结束后,冷却,减压回收乙醇至干,得淡黄色固体粉末,将该淡黄色固体粉末利用硅胶柱层析分离提纯,以氯仿-甲醇作为洗脱剂,按照氯仿与甲醇的体积比依次为50:1、40:1、30:1、20:1、10:1、8:1的洗脱顺序进行梯度洗脱,收集氯仿与甲醇体积比为8:1时的洗脱部分,收集的洗脱部分经减压除去洗脱剂后得淡黄色固体粉末,即为所述葛根素衍生物。
10.根据权利要求1所述的葛根素衍生物的应用,其特征在于:在制备治疗肺癌、肝癌、宫颈癌药物中的应用。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201811156463.1A CN109320552B (zh) | 2018-09-30 | 2018-09-30 | 具有良好生物活性的葛根素衍生物及其制备方法以及应用 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201811156463.1A CN109320552B (zh) | 2018-09-30 | 2018-09-30 | 具有良好生物活性的葛根素衍生物及其制备方法以及应用 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN109320552A CN109320552A (zh) | 2019-02-12 |
| CN109320552B true CN109320552B (zh) | 2020-10-16 |
Family
ID=65264964
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201811156463.1A Active CN109320552B (zh) | 2018-09-30 | 2018-09-30 | 具有良好生物活性的葛根素衍生物及其制备方法以及应用 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN109320552B (zh) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116444378A (zh) * | 2023-02-14 | 2023-07-18 | 广东优康精细化工有限公司 | 一种n-甲基异丙基胺的合成方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1800196A (zh) * | 2006-01-10 | 2006-07-12 | 重庆大学 | 葛根素衍生物及其制备工艺 |
| CN102443027A (zh) * | 2010-10-13 | 2012-05-09 | 南京工业大学 | 果糖基化葛根素及其制备方法与用途 |
| CN103044409A (zh) * | 2012-12-05 | 2013-04-17 | 东南大学 | 葛根素衍生物或其药学上可接受的盐及其应用 |
| CN103288809A (zh) * | 2012-02-24 | 2013-09-11 | 深圳市健元医药科技有限公司 | 葛根素单磷酸盐或单磺酸盐衍生物及其制备方法 |
-
2018
- 2018-09-30 CN CN201811156463.1A patent/CN109320552B/zh active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1800196A (zh) * | 2006-01-10 | 2006-07-12 | 重庆大学 | 葛根素衍生物及其制备工艺 |
| CN102443027A (zh) * | 2010-10-13 | 2012-05-09 | 南京工业大学 | 果糖基化葛根素及其制备方法与用途 |
| CN103288809A (zh) * | 2012-02-24 | 2013-09-11 | 深圳市健元医药科技有限公司 | 葛根素单磷酸盐或单磺酸盐衍生物及其制备方法 |
| CN103044409A (zh) * | 2012-12-05 | 2013-04-17 | 东南大学 | 葛根素衍生物或其药学上可接受的盐及其应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN109320552A (zh) | 2019-02-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN110272342B (zh) | 马齿苋中一种萘酸化合物及其提取分离方法与用途 | |
| CN113845551B (zh) | 一种具有光动力抗三阴乳腺癌活性的Pt(Ⅱ)配合物及其制备方法和应用 | |
| CN111635449B (zh) | 一种羽扇豆醇吡啶季铵盐衍生物及其制备方法与应用 | |
| CN109320552B (zh) | 具有良好生物活性的葛根素衍生物及其制备方法以及应用 | |
| CN112300000A (zh) | 马齿苋中一种具有抗肿瘤和抗胆碱酯酶活性的酯类化合物及其提取分离方法和应用 | |
| CN112409307A (zh) | 马齿苋中化合物Olerafuran A及其提取分离方法和用途 | |
| CN113845549B (zh) | 一种芒柄花黄素衍生物及其制备方法和应用 | |
| CN112300104A (zh) | 马齿苋中一种木脂素类化合物及其提取分离方法和应用 | |
| CN111454241B (zh) | 双异戊烯基黄酮类化合物及其制备方法和应用 | |
| CN104610212B (zh) | 淫羊藿素衍生物及其制备方法和用途 | |
| CN114409670A (zh) | 具有抗肿瘤活性苦木素类化合物及其制备方法与应用 | |
| CN112300185B (zh) | 肝毒性降低的生物碱类化合物及其制备方法和用途 | |
| CN116925018A (zh) | 大黄酸-哌嗪-呋喃酮杂化物及其制备方法和应用 | |
| CN107382944B (zh) | 一类具有抗肿瘤活性的香豆素棉酚衍生物及其合成方法 | |
| CN109824685B (zh) | 马齿苋中化合物oleracone G及其提取分离方法与应用 | |
| CN110964032B (zh) | 毛栲利素硫化氢供体衍生物及其制备方法和用途 | |
| CN113683594A (zh) | 一种喹啉-苯并咪唑盐类化合物及其合成方法和应用 | |
| CN111018780A (zh) | 一种n-羰基-9,10-二氢吖啶类化合物及其应用 | |
| CN107236004B (zh) | 二氢杨梅素环磷酰胺衍生物及其制备方法和应用 | |
| CN114957272B (zh) | 一种色原烷二聚体及其制备方法和应用 | |
| CN114989214B (zh) | 一种紫草素氨基磷酸酯杂合体及其合成方法和应用 | |
| CN118772215B (zh) | 山楂叶中联苯类化合物的制备方法及应用 | |
| CN119101061B (zh) | 环淫羊藿素二氢黄酮类衍生物的合成及其抗肿瘤应用 | |
| CN112920151A (zh) | 异戊烯基黄酮类化合物及其制备方法和应用 | |
| CN109810142B (zh) | 一种含氮芥川芎嗪苦参碱衍生物及其制备方法和应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| TA01 | Transfer of patent application right | ||
| TA01 | Transfer of patent application right |
Effective date of registration: 20200917 Address after: Building D7, No. 555, Chuangye Road, Dayun Town, Jiashan County, Jiashan City, Zhejiang Province Applicant after: Zhejiang Pharmaceutical Garden Biotechnology Co.,Ltd. Address before: 350011 Unit 1004, 6 Rongqiao Yuecheng, 289 Zhuyu Road, Yuefeng Town, Jinan District, Fuzhou City, Fujian Province Applicant before: Yu Daiying |
|
| GR01 | Patent grant | ||
| GR01 | Patent grant |