CN1057770C - 19,11β-桥甾族化合物在制药中的应用 - Google Patents
19,11β-桥甾族化合物在制药中的应用 Download PDFInfo
- Publication number
- CN1057770C CN1057770C CN96100917A CN96100917A CN1057770C CN 1057770 C CN1057770 C CN 1057770C CN 96100917 A CN96100917 A CN 96100917A CN 96100917 A CN96100917 A CN 96100917A CN 1057770 C CN1057770 C CN 1057770C
- Authority
- CN
- China
- Prior art keywords
- ketone
- phenylene
- group
- compound
- steroid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J71/00—Steroids in which the cyclopenta(a)hydrophenanthrene skeleton is condensed with a heterocyclic ring
- C07J71/0036—Nitrogen-containing hetero ring
- C07J71/0057—Nitrogen and oxygen
- C07J71/0063—Nitrogen and oxygen at position 2(3)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/38—Drugs for disorders of the endocrine system of the suprarenal hormones
- A61P5/44—Glucocorticosteroids; Drugs increasing or potentiating the activity of glucocorticosteroids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J21/00—Normal steroids containing carbon, hydrogen, halogen or oxygen having an oxygen-containing hetero ring spiro-condensed with the cyclopenta(a)hydrophenanthrene skeleton
- C07J21/005—Ketals
- C07J21/006—Ketals at position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J31/00—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring
- C07J31/006—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring not covered by C07J31/003
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J41/00—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring
- C07J41/0033—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005
- C07J41/0038—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005 with an androstane skeleton, including 18- or 19-substituted derivatives, 18-nor derivatives and also derivatives where position 17-beta is substituted by a carbon atom not directly bonded to a further carbon atom and not being part of an amide group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J53/00—Steroids in which the cyclopenta(a)hydrophenanthrene skeleton has been modified by condensation with a carbocyclic rings or by formation of an additional ring by means of a direct link between two ring carbon atoms, including carboxyclic rings fused to the cyclopenta(a)hydrophenanthrene skeleton are included in this class
- C07J53/002—Carbocyclic rings fused
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J71/00—Steroids in which the cyclopenta(a)hydrophenanthrene skeleton is condensed with a heterocyclic ring
- C07J71/0005—Oxygen-containing hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J71/00—Steroids in which the cyclopenta(a)hydrophenanthrene skeleton is condensed with a heterocyclic ring
- C07J71/0036—Nitrogen-containing hetero ring
- C07J71/0042—Nitrogen only
- C07J71/0047—Nitrogen only at position 2(3)
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Animal Behavior & Ethology (AREA)
- Endocrinology (AREA)
- Public Health (AREA)
- Diabetes (AREA)
- Steroid Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- External Artificial Organs (AREA)
- Separation By Low-Temperature Treatments (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
描述了通式I的新的19,11β-桥接甾族化合物以及其在医药上容许的酸加成盐,这些新的化合物具有药理学上有价值的特性。下式中,各基团的定义见说明书。
Description
本发明涉及专利权利要求中表示的主题内容,即新的19,11β-桥甾族化合物,这些化合物的生产方法,含有这些化合物的药物制剂,它们在药物制品生产中的应用以及为此目的所需要的新的中间产物。
通过下述通式I描述本发明19,11β-桥甾族化合物,以及其药学上可允许的酸加成盐, 其中:R1代表甲基或乙基,R2代表氢、氯原子、C1~C4-烷基,B和G可相同或不同,分别代表氢原子、C1~C4-烷基,或一起代表碳原子6和7之间的第二个键,B和R2一起代表亚甲基或亚乙基,Z代表5元或6元环基,它们可以被取代,以及可以是不饱和的,V代表可被取代的碳环或杂环芳基,A环代表:
其中:R1代表甲基或乙基,R2代表氢、氯原子、C1~C4-烷基,B和G可相同或不同,分别代表氢原子、C1~C4-烷基,或一起代表碳原子6和7之间的第二个键,B和R2一起代表亚甲基或亚乙基,Z代表5元或6元环基,它们可以被取代,以及可以是不饱和的,V代表可被取代的碳环或杂环芳基,A环代表: M和N一起代表第二个键,或M为氢原子,N为羟基,那么B、R2、G、R3、D和E为氢原子,
M和N一起代表第二个键,或M为氢原子,N为羟基,那么B、R2、G、R3、D和E为氢原子,
X为氧原子、两个氢原子或一个肟基N~OH,
R3和D可相同或不同,分别为氢原子、腈基或C1~C4-烷基,或一起代表亚甲基或亚乙基,
E为氢原子、C1~C4-烷基,
D和E一起代表碳原子1和2之间的第二个键,或一起代表亚甲基,
或 或其中R11代表氢原子或C1~C8-烷基,芳基V可以是被取代和未被取代的碳环或杂环基,例如,苯基、萘基、呋喃基、噻吩基、吡啶基、咪唑基、吡咯基、嘧啶基、噁唑基、哒嗪基、吡嗪基。
或其中R11代表氢原子或C1~C8-烷基,芳基V可以是被取代和未被取代的碳环或杂环基,例如,苯基、萘基、呋喃基、噻吩基、吡啶基、咪唑基、吡咯基、嘧啶基、噁唑基、哒嗪基、吡嗪基。
本发明尤其涉及其中V代表下式苯环基的通式I化合物,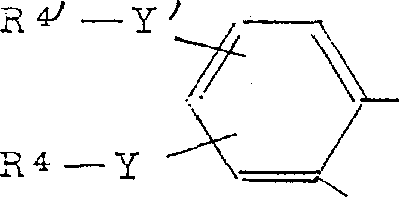 其中:R4和R4′可相同或不同,分别为氢原子、氰基、-OR11-、-S(O)kR11-、N(O)nR11R12-、-O-SO2R13--P(O)(OR14)2-、SiR3 14-或SnR3 14-基,K为0、1或2,n为0或1,R11为氢原子或C1~C8-烷基,R12为R11、氰基或C1~C10-酰基,R13为全氟化的C1~C4-烷基,R14为C1~C4-烷基,
其中:R4和R4′可相同或不同,分别为氢原子、氰基、-OR11-、-S(O)kR11-、N(O)nR11R12-、-O-SO2R13--P(O)(OR14)2-、SiR3 14-或SnR3 14-基,K为0、1或2,n为0或1,R11为氢原子或C1~C8-烷基,R12为R11、氰基或C1~C10-酰基,R13为全氟化的C1~C4-烷基,R14为C1~C4-烷基,
在-N(O)nR11R12基中的R11和R12包括氮一起代表5元或6元杂环,在环中也可以含有另外的杂原子N、O或S。
Y和Y′可相同或不同,分别为直接键、直链或支链的亚烷基,其可含一个或多个双键或三键,碳原子数多至20个。根据具体情况,其亚烷基可被一个或多个桥氧基、C1~C10-酰氧基、-OR11、-S(O)kR11和/或-N(O)nR11R12基取代,或可能被芳基取代,或
R4-Y和R4′-Y′一起可代表带有0至2个氧原子、硫原子和/或NR11基的被取代的、饱和的、不饱和的或芳族的5元或6元环基,附有条件是,如果R11为C1~C8-烷基,k和n仅大于0。
如果Y-R4=H,Y1是在1位上用桥氧基取代的亚乙基,并R4=H,那么,Y′-R4是在本发明范围内起择优作用的乙酰基。
在苯环的取代基中,优先选择在3、4和5位上的单取代和在4和5或3和4位上的双取代,并形成被稠合的第二个环,例如,环己烯、吡咯、呋喃基、吡咯啉、1,3-二噁环戊烯、吡唑啉、二脱氢吗啉、二脱氢哌啶、二脱氢哌嗪、脱氢吡喃、嘧啶、吡啶、吡嗪、1,4-二噁环己烷的环。
R1和R11,以及R2、R3、B、G和D分别所代表的烷基是,R1为1~2个碳原子,R11为1~8个碳原子,其它的为1~4个碳原子,它们是甲基、乙基、丙基、异丙基、丁基、优先选择的分别是甲基、乙基、丙基。
如果R12为酰基,优先选用的是甲酰基、乙酰基、丙酰基、丁酰基和苯甲酰基。
R11和R12也可以包括氮原子一起代表5员或6员的杂环,该环除了含N和C原子外,还可含O和S原子,其实例为:吡咯、吡咯烷、哌啶、哌嗪、吗啉、噁唑烷、噻唑烷以及噻二唑烷的环。
此外,本发明也特别涉及其中Z为下式环基的通式I化合物, 其中:R1具有权利要求1中的意义,在W上出现的虚线代表可存在双键,W为CH2-,CH-,CH2CH2-或CHCH2-基R5/R6为-OR7/-C≡C-U
其中:R1具有权利要求1中的意义,在W上出现的虚线代表可存在双键,W为CH2-,CH-,CH2CH2-或CHCH2-基R5/R6为-OR7/-C≡C-U

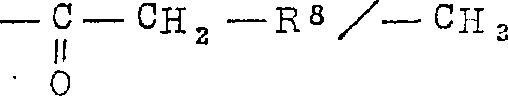
 -OR7/-(CH2)m-CH2-R9-OR7/-CH=CH(CH2)k-CH2-R9-OR10/-H-OR10/-(CH2)k-C≡C-U
-OR7/-(CH2)m-CH2-R9-OR7/-CH=CH(CH2)k-CH2-R9-OR10/-H-OR10/-(CH2)k-C≡C-U
其中:
R7为氢原子或具有1~4个碳原子的酰基,
U为氢原子、在其各自的烷基、酰基中分别具有1~4个碳原子的烷基、羟烷基、烷氧基烷基、酰氧基烷基,
R9为氢原子、羟基、或氰基、以及分别具有1~4个碳原子的O-烷基或O-酰基,
R8为氢原子、羟基、以及分别具有1~4个碳原子的烷基、O-烷基或O-酰基,
R10为氢原子、分别具有1~10个碳原子的烷基或酰基,
m为0、1、2或3,
k为0、1或2。
在通式I中的R5和R6、及R7、R8、R9、R10和U中分别含有的烷基、烷氧基和酰基各自含有1~4个碳原子,优先选用的是甲基、乙基、丙基、异丙基、甲氧基、乙氧基、丙氧基、异丙氧基、甲酰基、乙酰基、丙酰基和异丙酰基。
R6中可以以E或Z构型存在的链烯基可优先选用丙烯基和丁烯基,即,如果R6代表-CH=CH-(CH2)k-CH2-R9,k最好应是0或1。
按着本发明权利要求21中所述的方法,生产通式I化合物。关于通式II的中间产物的生产, 其中:R1是甲基或乙基,1是1或2,k是以缩酮或酮缩硫醇的形式被保护的酮基,V′是在连接点的α位上带有氟、氯、溴或碘原子的可被取代的碳环或杂环芳族化合物的基团,任何羟基、巯基、氨基、桥氧基和/或末端炔基已被保护。
其中:R1是甲基或乙基,1是1或2,k是以缩酮或酮缩硫醇的形式被保护的酮基,V′是在连接点的α位上带有氟、氯、溴或碘原子的可被取代的碳环或杂环芳族化合物的基团,任何羟基、巯基、氨基、桥氧基和/或末端炔基已被保护。
以及特别是关于同样是本发明内容的其中V′为下式苯环基的通式II化合物的生产, 其中:Hal是氟、氯、溴或碘原子,
其中:Hal是氟、氯、溴或碘原子,
R4a、R4′a、Ya和Y′a具有与R4、R4′、Y和Y′相同的意义,同时排除氰基、任何羟基、巯基、氨基、桥氧基和/或末端炔基已被保护。
它可从通式III环氧化合物得到,而通式III环氧化合物是按照公开号为0110434的欧洲专利申请、DE3438484或公开号为0127864的欧洲专利申请中所述的方法得到的。
其中:
R1、1和k具有上述的意义。
将格利雅试剂加入到式V芳甲基卤化物的上述环氧化物中,制得本发明的通式II中间产物(Tetrahedron Letters 1979,2051)。
V′CH2Hal (V)
其中:
Hal是氯、溴或碘原子,
在芳族化合物(碳环或杂环)的2位上带有氟、氯、溴或碘原子,而芳族化合物被连在甲基上。
将可能存在于V′中的官能团保护后,环化通式II的新化合物。
V1和K中的保护了的羟基、巯基和酮基是那些在酸性介质中易于分离的基团,例如,甲氧甲基、乙氧甲基、四氢吡喃基、亚乙二氧缩酮基、亚乙二硫缩酮基或2,2-二甲基亚丙基二氧缩酮基。
氨基和末端炔基的保护基(例如,三甲基甲硅烷基和叔丁基二甲基甲硅烷基)也是专家们所熟知的,并且它们在所需要的反应顺序之后,用文献中所述的方法分离。〔Synthesis 1980,627,J.Org.Chem.46(1986)2280〕
通式II化合物被转化成为下述通式IVa的19,11β-桥甾族化合物, 该化合物同样是本发明的内容,其中R1、k和1具有上述的意义,V″与V意义相同,但存在于V中的任何羟基、巯基、氨基、桥氧基和/或末端炔基是被保护的。如果V′中的α-卤素取代基是溴或碘原子,用已知的方法,通过还原性基环化(TetrahedronLetters 1982,2575;1985,6001;1986,2833;J.Am.Chem.Soc.1982,104,2321;Radicals in Organic Synthesis:Formation ofCarbon-Carbon Bonds,Pergamon Press,1986)。
该化合物同样是本发明的内容,其中R1、k和1具有上述的意义,V″与V意义相同,但存在于V中的任何羟基、巯基、氨基、桥氧基和/或末端炔基是被保护的。如果V′中的α-卤素取代基是溴或碘原子,用已知的方法,通过还原性基环化(TetrahedronLetters 1982,2575;1985,6001;1986,2833;J.Am.Chem.Soc.1982,104,2321;Radicals in Organic Synthesis:Formation ofCarbon-Carbon Bonds,Pergamon Press,1986)。
氟和氯取代的芳族化合物的相应方法,至今还是未知的。现已发现,当在-100和-30℃之间,最好是-78至-60℃,在液态氨草胶中,离析物用电正性金属,如钠、钾、锂或钙处理,并与一种或多种的有机溶剂,例如乙醚、二甲羟基乙烷(DME)、二噁烷或四氢呋喃相混合时,这种环化作用竟意想不到地获得成功,且具有良好的收率。而且氟离子作为离去基团,这种环化作用也是适宜的,必定产生特别意想不到的效果。
这种新方法也适于用溴和碘取代的芳族化合物。
用与文献(例如:J.Fried,J.A.Edwards,“OrganicReactions in Steroid Chemistry”,Van NostrandReinhold Company,1972,Vol.1和2;“Terpenoidsand Steroids”,Specialist Periocical Report,The Chemical Society,London,Vol,1~12)中所述的相似方法,将由此产生的环化产物转化成最后希望的通式I最终产物。
首先进行下述步骤:
a)根据具体情况,氧化C-17羟基,接着
b)根据具体情况,将V′中的带有保护基的羟基,从所说的保护基中释放出来,如果需要,从羟基化合物制备其相应的全氟烷基磺酸酯,可通过进一步的反应,或者直接,或者通过相应的锡三烷基化合物,用锡三烷基替换全氟烷基磺酸酯离去基团,而将该全氟烷基磺酸酯转化成为所示的在V″中具有所希望的取代型式的化合物。
或者首先进行b),然后进行a),接着
c)用所需要的已知方法,使D环进行官能化作用,所得的产物用能够释放出3-桥氧基的脱水剂作用,以便脱去水,同时形成4(5)双键,接着在重新保护了包含在V和/或Z中的反应中被释放的官能团之后,将所希望的A环和B环官能团引入到甾族化合物的结构中,或
d)如此得到的产物用能释放出3-桥氧基脱水剂作用,以便脱去水,同时形成4(5)双键,将所希望的A环和B环官能团引入到甾族化合物的结构中,接着在保护了3-桥氧基之后,用所要求的方法,使D环进行官能化作用。
或者在步骤c)或d)之后,进行步骤a)或b)。
根据具体情况,将如此得到的产物从保护基中释放出来。如果需要,可将含在V中的羟基、巯基和/或氨基烷基化或环化。如果需要,将氰基引入到芳基取代基中。如果需要,氧化可能含在芳基取代基中的氨基和/或硫化物基。如果需要,用羟胺盐酸盐将其转化成X为肟基N~OH的通式I的产物。根据具体情况,制备药学上允许的酸加成盐。
在这些反应过程中,中间再次将保护基引入到中间产物中是非常必要的。例如,对于在Z中含有的官能团,后来的A环和B环的官能化,或者,对于3-酮基,后来的D环的构成。
为生产几乎每一种最终产物所需要的17β-羟基的氧化作用,是使用已知技术完成的,如通过奥盆诺尔氧化作用(OppenauerOxidation)或铬酸试剂〔琼斯试剂(Jones′reagent)或铬酸吡啶〕。
通过用酸或酸离子交换剂处理,在脱水及形成4(5)双键的同时,3-酮基官能团被释放出来。酸处理是用已知方法进行的,通过将相应的5α-羟基-3-缩酮溶于一种与水混溶的溶剂中,例如含水甲醇、乙醇或丙酮,用催化量的无机酸或磺酸,如盐酸、硫酸、磷酸、高氯酸或对甲苯磺酸,或用有机酸,如乙酸处理该溶液,直到任何保护基已被除去,及水已被脱去为止。在温度为0至100℃下发生的转化反应,也可用酸性离子交换剂进行,转化过程后可进行样品分析,如薄层色谱法。
通常,正如在实施例1c)中所述的那样,通过使相应的5α-羟基-3-缩酮、5-烯-缩酮分别在强酸介质中反应一定时间,使保护基的去除和脱水在一步反应中进行。然而,对于本发明,去除保护基和脱水发生在两个单独的反应步聚中进行。根据具体情况,首先在一种适度的酸性介质中,短时间处理相应的5α-羟基-3-缩酮化合物,首先得到并分离出相应的5α-羟基-3-酮化合物。然后,与酸进一步反应,5α-羟基-3-酮化合物便转化成3-酮-4-烯化合物,同时水被脱去。
本发明的一个非常特别的优点表现在大谱带宽度的取代基可以被引入到碳环或杂环芳基V中(M.Pereyre,J.-P.Quintard,A.Rahm,Tin in Organic Synthesis;Butterworths,1987)。例如,通过式V,即V′CH2Hal的芳甲基卤化物的偶合,直接引入分别存在于最终产物中的取代基R4-Y、R4′-Y′。通过适当的通式III5α,10α-环氧化合物的格利雅反应,并用已述的生成中间产物通式II化合物的方法,将相应的该芳甲基卤化物取代到芳基中。
用这种方法能够生产在V中被取代的化合物的数量相对来说是有限的。因为并非在最终产物中所希望的取代基都能经受格利雅反应的条件而不受损害,格利雅反应发生在V′CH2Hal上须优先于与各自的5α,10α-环氧化物III的偶合,并且,特别是不能经受将中间产物II环化成通式IV的19,11β-桥甾族化合物过程中的还原条件。
然而,在本发明另一种可实施的方法中,在环化作用后,即在A、B和D环结构形式之前、同时或仅此之后,有可能在很大范围内,仅仅通过引入取代基来改变在芳基V中的取代基。为此目的,将至少一个分别存在于基团V″、V中的波保护的羟基从它的保护基中释放出来,并且用已知的方法〔P.J.Stang,M.Hanack and L.R。Subramanian,Synthesis 85(1982)〕。用全氟烷基磺酸酐(烷基=C1~C4),从游离的OH-基化合物转化生成相应的全氟烷基磺酸酯化合物。
在这一点上,以这样的方式进行反应是很必要的,即在过渡金属(最好为pd°)催化的反应中,或者用所希望的取代基或它的前一步来取代全氟过渡基,实际上,取代作用的发生几乎是同时进行的(J.E.McMurry and S.Mohanraj,TetrahedronLetters,24,No.27,P.2723-2726,1983;X.Lu and J.Zhu,Communications,P.726-727,1987;Q.-Y.Chen and Z.-Q.Yang,TetrahedronLetters 27,No.10,P.1171-1174,1986;S.Cacchi,P.G.Ciattini,E.Morera and G.Ortar,Tetrahedron Letters,27,No.33,P.3931-3934,1986;A.M.Echavarren and J.K.Stille,J.Am.Chem.Soc.1987,109,P.5478-5486);或者通过过渡金属的催化作用,在反应中由全氟烷基磺酸酯化合物生成相应的三-有机甲烷锡基化合物,最好生成三-正-烷基甲烷锡基化合物〔J.K.Stille,Angew.Chem.98(1986),P.504-519〕。在单釜反应中,用卤素取代的,最好用溴或碘取代的碳环或杂环芳族化合物〔Y.Yamamoto,Y.Azuma,H.Mitoh,Communications,P.564-565,1986;T.J.Bailey,Tetrahedron Letters,27,No.37,P.4407-4410,1986〕,根据具体情况。它也能够带有更多的取代基,转化成19,11β-桥甾族化合物,在其中的芳基V、V″分别含有所希望的取代基或取代前的基团。
反应中存在的三-正-烷基甲锡烷基化合物也可作为单一物质被分离出来,对此,实施例39a)、α)中就11β,19-(4-三-正-丁基甲锡烷基-O-亚苯基)-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-5α,17β-二醇进行了说明。
除了3,4双键之外,用相似的方法引入1,2和/或6,7双键,例如,用脱水剂,如二氧化硒、氯醌、三乙酸铊或二氯二氯苯醌(DDQ),分别通过烯丙基或二烯醇醚的溴化作用,然后将溴化氢分离〔J.Fried,J.A.Edwards,Organic Reactionsin Steroid Chemistry,Van Nostrand ReinholdCompany,1972,P.265-374),1;Tetrahedron 42,(1986)2971〕。
例如,于一种溶剂中,在一种基团形成物如二苯甲酰过氧化物存在下,用N-溴丁二酰亚胺、N-溴代乙酰胺、1,3-二溴-5,5-二甲基乙内酰脲或二溴四氯乙烷进行烯丙基的溴化作用。可用的溶剂是非质子溶剂,象二噁烷和氯代烃,如四氯化碳、氯仿或四氯乙烯。在0℃和溶剂的沸点温度之间发生转化。
例如,用相似于Steroids I,233中所述的方法,进行二烯醇醚的溴化作用。
在50~120℃的温度下,在非质子溶剂如二甲基甲酰胺中,使用碱性试剂,最好用溴化锂和碳酸锂或用溴化锂和碳酸钙,通过加热6-溴化合物,分离出溴化氢,并形成Δ6双键。分离HBr的另一种可能的方法是在可力丁或二甲基吡啶中,加热6-溴化合物。
从构成A环出发,可以同时在1,2和4,5位上引入双键,例如通过溴化作用形成2,4-二溴-3-酮,在二甲基甲酰胺中,使用例如碳酸锂或钙和溴化锂,使二溴化物脱去溴化氢。
通过下述方法可引入6-亚甲基,例如,在乙醇溶液中,用甲醛水溶液,由3-氨基-3(4),5(6)-二烯衍生物转化成6α-羟甲基〔Helv.Chim.Acta.56(1973),2396〕。接着,如在二噁烷/水中,用盐酸进行水的酸离析;或者,由3-烷氧基-3(4),5(6)-二烯衍生物,用相似于USP4544555中所述的方法;或者,直接由3-桥氧基-4(5)-烯衍生物,用相似于Synthesis(1982)34中所述的方法。
用二甲基·亚甲基硫氧将6-亚甲基化合物甲基化,形成6,6-亚乙基化合物。为此目的,将6-亚甲基甾族化合物加入到矿物油和二甲基亚砜中的含有氢化钠的三甲基氧化锍碘化物的悬浮液中,或加入到含有三甲基氧化锍碘化物和氢氧化钠的二甲基亚砜溶液中,在20-40℃下,经15至60分钟后,完成反应(J.Am.Chem.Soc.84(1962)866;European Patentapplication 0150157)。
用与A.J.Manson和D.Wood〔J.Org.Chem.32(1967)3434〕相似的方法或用文本提到的方法引入2-亚甲基,。
用与6-亚甲基化合物进行甲基化的相似方法〔cf.alsoChem.Ber.98(1965)1470〕,使2-亚甲基化合物甲基化,形成2,2-亚乙基化合物。
例如,用与L.Nedelec,Tetrahedron 30(1974)3263所述的相似方法,分别得到在2位上的单烷基化合物和二烷基化合物。
用相似的方法〔J.Fried,J.A.Edwards:OrganicReactions in Steroid Chemistry,Van NostrandReinhold Company,1972,PP75~82,2;andJ.Am.Chem.Soc.99(1977)1673〕,将1,4-亚甲基化合物、1,6-亚甲基化合物分别加入到相应的烯酮中,得到1位上、7位上分别烷基化的化合物。
例如,通过断开相应的5α,6α-环氧化物,接着进行反应(J.Fried,J.A.Edwards:Organic Reactions inSteroid Chemistry,Van Nostrand ReinholdCompany,1972,PP82~86,2),可得到在6位上烷基化的化合物。
通过相应的烯丙基醇的Simmons-Smith反应(J.Fried,J.A.Edwards;Reactions in Steroid Chemistry ,Van Nostrand Reinhold Company,1972,PP100-126;Rev.Soc.Quim.Mex.(1969)171A;Chem.Ber.101(1986)935;Chem.Ber.99(1966)1118;Zeitschr.f.Naturf,196(1964)944),将重氮甲烷或二甲基·亚甲基硫氧添加到相应的烯酮中,可得到1α,2α-、6α,7α-、6β,7β-亚甲基化合物,或1α、2α-亚甲基结构元素与两个6,7-亚甲基结构元素相结合的化合物。
生成被稠合到2和3位上的异噁烷环是通过合成2-二羟亚甲基化合物〔Steroids 6(1962)178;J.Amer.Chem.Soc.83(1961)1478〕,并用羟基胺将其转化〔J.Med.Chem.6(1963)1〕。
对于合成2-氰基-甾族化合物,异噁烷也是良好的起始原料〔J.Med.Chem.6(1963)1〕。
生成被稠合到2和3位上的吡唑环是通过用R11-取代肼转化2-羟基亚甲基-3-桥氧基的离析物(US专利3704295)。
例如,分别用与德国专利说明书1158966、US专利4544555和US专利4196203相同的方法,分别通过6,7-环氧化物、6-亚甲基衍生物,以及在酸性条件下,用二氯二氰基苯醌(DDQ)氧化6-氯代-3,5-二烯醇醚〔比利时专利621197(1962)〕,将氯、甲基取代基分别引入到甾族化合物结构中的C-6上。
根据DOS 2805490所述的方法,例如,通过酮缩硫醇作用,并接着进行还原分离,可脱出3-桥氧基,形成X为两个氢原子的通式I最终产物。
例如,用与已经公开的在Australian J.Chem 8(1955),519和“Organic Reactions in Steroid Chemistry”Vol.2,388中所述的相似方法,通过Tifteneau重排,也可得到具有D-高甾族化合物结构的离析物。用氨草胶断开17,20-螺环氧化物,或者也可通过乙酰化的17β-羟基-17α-氰基化合物的锂-铝还原,便可得到所需要的17α-氨甲基-17β-羟基化合物。
在二甲基甲酰胺中,用二甲基·亚甲基硫氧转化相应的17-酮,得到螺环氧化物〔Journal f.Prakt.Chemie 314(1972),667-668〕。按着已知方法(例如,AustralianJ.Chem.8(1955),519)将氢氰酸加入到相应的17-酮中,接着进行乙酰化作用,产生乙酰化氰基醇。
例如,通过17-酮的相应的烯醇化合物的改进了的Saegusa氧化作用(Tetrahedron 42(1986)2971),可得到具有不饱和D环的离析物。例如,在四氢呋喃中,用二异丙基酰胺锂,将17-酮转化成相应的烯醇酯,并用三甲基氯硅烷回收(Synthesis 1983,1),得到三甲基甲硅烷基烯醇醚。
用通常的方法,利用已构成的C-17侧键,将亲核试剂加入到17-酮中,该酮通过C-17羟基的奥盆诺尔氧化作用得到,接着进行系列反应(“Terpenoids and Steroids”,Specialist Periodical Report,The ChemicalSociety,London,Vol.1-12)来引入取代基R5和R6。
借助于式MC≡C-U′化合物,引入取代基-C≡C-U作为R6,其中U为上述意义,而式MC≡C-U′中的U′是被保护的U基,例如可被三甲基甲硅烷基或叔丁基二甲基甲硅烷基保护,或者,如果U是1~4个碳原子的烷基,那么,U′本身就是基团U。
也可就地形成有机金属化合物,并使之与17-酮反应。于是,例如,在一种适合的溶剂中,在有醇或氨草胶的存在下,可使17-酮与乙炔基和碱金属,特别是钾、钠或锂反应。例如,碱金属可以甲基锂或丁基锂的形式进行反应。适合的溶剂特别是二烷基醚、四氢呋喃、二噁烷、苯和甲苯。
通过用炔丙醇(3-羟基丙炔)的二价阴离子,如用就地生成的炔丙醇二钾盐,转化17-酮,生成17α-(3-羟基丙-1-炔基)-17β-羟基衍生物,或者用3-羟基丙炔的金属衍生物,如1-锂-3-(四氢吡喃-2′-基-氧代)-丙-1-炔基转化17-酮,生成17-〔3-(四氢吡喃-2′-基-氧代)-丙-1-炔基〕-17β羟基衍生物,然后将上述化合物水合,分别形成17-(3-羟丙基)-17β-羟基化合物和17-(3-羟丙烯基)-17β-羟基化合物,这样可分别将3-羟基丙炔、3-羟基丙烯、3-羟基丙烷引入到17位上。例如,在室温和常压下,在溶剂如甲醇、乙醇、丙醇、四氢呋喃(THF)或乙酸乙酯中,加入贵金属催化剂,象铂或钯,通过水合作用,完成上述内容。
以相似的方法,用炔丙醇同系物引入羟基炔基、羟基烯基和羟基烷基同系物。
用非活化的贵金属催化剂水合炔三键,便得到在羟丙烯基中具有Z构型双键的化合物(J.Fried,J.A.Edwards:OrganicReactions in Steroid Chemistry,Van NostrandReinhold Company,1972,P.134;and H.O.House:Modern Synthetic Reactions,1972,P.19)。可用的非活化贵金属催化剂是例如,在胺存在下,10%的钯硫酸钡,或5%的钯碳酸钙,另外加入醋酸铅(II)。一当量氢被吸收后,停止水合作用。
用已知的方法,通过还原炔三键,得到在羟基丙烯基中具有E构型双键的化合物。在文献中,叙述了大量将炔烃转化成交叉结构烯烃的方法。例如,在液态氨草胶中,用钠还原(J.Am.Chem.Soc.63(1941)216),在液态氨草胶中,用氨基钠还原(J.Chem.Soc。1955,3558),在低分子胺中,用锂还原〔J.Am.Chem.Soc.77(1955)3378〕,用硼烷还原(J.Am.Chem.Soc。93(1971)3395和94(1972)6560),用氢化二异丁基铝和甲基锂还原(J.Am.Chem.Soc.89(1967)5085),以及特别是用氢化铝锂/醇化物还原(J.Am.Chem.Soc.89(1967)4245),另一种可使用的方法是,在微酸性介质中,在有水或二甲基甲酰胺存在下,用硫酸铬还原三键(J.Am.Chem.Soc.86(1964)4358),以及通常使用的方法是,随着氧化级数的变化,通过与过渡金属化合物反应来还原。
通过添加相应的金属羟烯烃化合物,如1-锂-3-(四氢吡喃-2′-基氧代)-丙-1(E)-烯(J.Org.Chen.40,2265)或1-锂-3-(四氢吡喃-2′-基氧代)-丙-1(Z)-烯(Synthesis 1981,999),也可直接引入羟基烯烃。同样,用这种方法,也可引入同系物。
在17-位上引入3-羟基丙烷可如下进行,用3-卤代-丙醇的金属衍生物-其中羟基是以醇化物形式(TetrahedronLetters 1978,3013),或以被保护官能团的形式(J.Org.Chem.37,1947)存在于敷金属阶段-转化17-酮,分别形成17-(3-羟丙基)-17β-羟基化合物、在末端羟基上被保护的化合物。可保护的基是如乙氧乙基、四氢吡喃基和甲氧甲基。
如果希望具有R5/R6意义如下的通式I的最终产物,
那么,可用已知的方法,例如用琼斯试剂,二氧化锰、重铬酸吡啶锡、氯化铬酸吡啶鎓、铬酸吡啶、或Fetizon试剂碳酸银/硅藻土(Compt.rend.267〔1968〕900)氧化17-(3-羟丙基)-化合物。
如果希望具有R5/R6意义如下的通式I最终产物,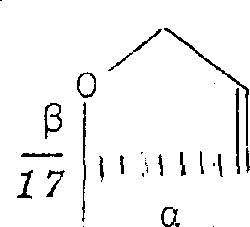
那么,可以通过相应的17-(3-羟丙-1(Z)-烯基)-17β-羟基离析物的闭环反应来得到。
由17-酮,用已知的方法,如用17-螺环氧化物,并按照Z.Chem.18(1978)259-260的方法,用HCN分离该螺环氧化物,来构成17-氰甲基侧键。
用已知的方法,例如按着J.Org.Chem.47(1982),2993-2995,Chem。Ber.113(1984),1184,或US专利4600538中所述的方法,也可引入17-羟乙酰基侧键。导入基团
用对甲苯磺酰基甲基异氰化物(Chem Ind.1972,213)把17-酮转化成17-腈化合物(Tetrahedron,31(1975),2151),该腈化物可用甲基锂溴化物或甲基镁溴化物直接转化成17-乙酰基化合物。在四氢呋喃中用叔丁醇钾烯醇化并且与碘甲烷反应之后17-乙酰基化合物生成所需要的17α-甲基-17β-酰基基团。这种包括把甲基加到腈上以及随后烷基化的程序也可以反向进行。
在Z为V中分别存在游离羟基,羟基、巯基和/或氨基,可以使用已知的方法烷基化或乙酰化。
在V中含有的硫化物和/或二烷基胺可以用适当的氧化剂(例如过氧化氢或过酸)分别转化为所需要的亚砜(n=1)、N-氧化物(n=1)〔例如参见Kontakte(Darmstadt)1986,3,P.12〕和砜(n=2)。
在V中带有二烷基取代基的化合物在质子溶剂中,例如在二噁烷、苯或甲苯中,升温的条件下(用Brauns方法分离胺)与溴化氰反应,能够转化成相应的高收率的(N-氰基-N-烷基氨基芳基)衍生物。使用的方法类似于在Org.Reactions 7,198(1953),K.W.Bentley.Techniques of Organic Chemistry11,773(1963)和Houben-Weyl,5/4,151(1960)中的详细说明。在最终产品中取决于R12的定义。用已知方法将N-氰基-N-烷基氨基芳基衍生物还原成相应的二烷基胺化合物(例如在甲苯中,用二异丁基氢化铝反应生成N-甲酰-N-烷基氨基苯基中向产品,随后再与氢化铝锂反应)、N-H-N-烷基化合物(例如在液态氨中与氢化铝锂反应)。如果需要的话,用文献中的已知方法随后将N-H-N-烷基化合物进行乙酰化,情况可能是在已知方法中随后用氢化铝锂还原,产生新的二烷基胺衍生物(参考DE3623038)。
若需要的话,在X定义为氧子所得到的通式I的化合物,在叔胺存在下,温度-20℃到40℃之间,通过用羟基胺氢氯化物,能转化成肟(在X定义为肟基N~OH基团的通式I,羟基可能是在顺位或反位)。适宜的叔碱是:例如:三甲胺、三乙胺、吡啶,N,N-二甲氨基吡啶,1,5-二氮杂双环〔4,3,0 〕壬烯-5(DBN)和1,5-二氮杂双环〔5,4,0〕十一碳烯-5(DBU),最好是吡啶。
通式I的新化合物和其与医药上容许的酸加成盐是有用的药物。它们对妊娠受体有极其大的亲和性并且具有令人吃惊的广范围妊娠的、抗妊娠的、抗糖皮质激素、抗盐皮质激素和抗雄性激素的特性。这些重要的生物作用能够用于药物。
由于它们代替来自受体维持妊娠所需要的孕甾酮,带有标记抗妊娠活性的这类有效成分适于用来引发流产。因此,在有关交媾后控制生育方面,它们是有价值的和有意的。
它们也可用于抗激素失调、诱发月经和引起分娩。
另外,它们能治疗与激素有关的癌症。
通式I的化合物和其与医药上容许的酸加成盐也表现出抗糖皮质激素的活性,因此可以用作药物,治疗引起类皮质激素失调(青光眼),防治在长期使用糖皮质激素治疗时出现的付作用(Cushing′s综合症)。因此这些药物可以用来治糖皮质激素过量失调而引起的各种疾病。最重要的是肥胖症、动脉硬化、高血压、骨质疏松、糖尿症和失眠症。表现出妊娠活性的通式I的化合物和其与医药上容许的酸加成盐可以用于治疗停经、经痛、月经过多和黄体机能不足,并且可以使用在治疗醛甾酮过多症中抗盐皮质激素特性的疾病。
呈现出抗产生男性特征活性的通式I化合物和其与医药上容许的酸加成盐能用来治疗前列腺肥大症及前列腺癌症。而且这些药物容许特效治疗女性上产生男性特征作用的多种症状:如多毛症中毛发病态生长者、产生男性特征的脱发和在粉刺中皮酯腺增强,并且它能有利地影响皮脂溢出。
因而本发明也涉及通式I的医药上可接受的化合物为基础的药物。即,按剂量使用的非毒性的化合物以及其医药上容许的酸加成盐,它们也带有常规的辅剂和载体(赋形剂)。
依照本发明的化合物及其盐能用已知的galen氏方法加工产生适于肠内的、经皮的、肠胃外的或局部施用的医药制剂。这些药物能以片剂、肠溶衣、胶囊、粒剂、栓剂、植入物、可注射的无菌消毒水剂或油剂、悬浮剂或乳剂、软膏、乳膏和凝胶等形式施用。
与此有关,有效成分(或多种有效成分)能与galen氏常用的辅剂混合,例如辅剂为阿拉伯树胶、滑石、淀粉、甘露糖醇、甲基纤维素、乳糖、表面活性剂如Tweens(R)或Myrj(R)、硬酸镁、水或非水的载体(赋形剂)、石蜡衍生物、润湿剂、分散剂、乳化剂、防腐剂,还有调味用的各种物质(如芳香油)。
因此,本发明也涉及到医药上的化合物,这类化合物至少含有依据本发明用作有效组分的一种化合物或含有其与医学上容许的酸加成盐。
尤其应该提到的是氢氯酸和甲磺酸盐,它们用作本发明产品与酸的加成盐。一剂量单位含有将近1-100毫克的一种(或多种组分)组分。
依照本发明的化合物剂量,用于人体大致每天是1-1000毫克。
选择流产作用以确定抗妊娠作用。
在重量约200克的雌鼠上进行实验。雌鼠交媾后,通过在阴道垢涂片显示精子存在的方法确定妊娠的开始,以有精子存在的这一天视为妊娠的第一天(=dl P.C)。
从妊娠后的第五天到第七天,胚胞着床后用各种需要进行实验的物质或溶剂对这些雌鼠进行处理。妊娠后第九天杀死这些鼠,检测其子宫内的植入物和吸收点。所有子宫要进行拍摄照片。把没有植入物、着床的病理出血点或其它不正常的吸收点视为一次流产。
试验物质溶解在苯甲酸苄酯和蓖麻油的混合物中(1+4的比例)。每个单体剂量的载体容量总计达到0.2毫升,以皮下方式处理。
依照本发明化合物的优点是可以通过比较下列化合物的流产作用表明:17α-(丙-1-炔基)-17β-羟基-11β,19-(4-二甲基氨基-邻-亚苯基)-4-雄(甾)烯-3-酮(A),17α-(丙-1-基)-17β-羟基-11β,19-(4-乙酰基-邻-亚苯基)-4-雄(甾)烯-3-酮(B),17α-(3-羟基)丙-1(Z)-烯基)-17β-羟基-11β,19-(4-二甲基氨基-邻-亚苯基)-4-雄(甾)烯-3-酮(C)和17α-(3-羟基丙-1(Z)-烯基)-17β-羟基-11β,19-(4-甲基硫代-邻-亚苯基)-4-雄(甾)烯-3-酮(D)与和一种在欧州专利号0057115上描述的11β-(4-二甲基氨基苯基)-17β-羟基-17α-(丙-1-炔基)-4,9(10)-雌(甾)二烯-3-酮(E)和在欧州专利号84730147.0上列出的11β-(4-二甲基氨基苯基)-17β-羟基-17α-(3-羟基丙-1(Z)-烯基)-4,9(10)-雌(甾)二烯-3-酮(F)。
从表1可以看出:依照本发明,只有化合物A-D和化合物F,这些也是本人申请的专利,以1.0毫克/d·s·c的剂量施用仍然具有完全的流产药效。以此剂量对照物E仅表现出50%的效果。按照本发明的化合物B)即使以0.3毫克/d·s·c剂量使用仍然显示出完全的效果,而F以此剂量使用不再有效。
表1
在早期怀孕的鼠体内,抗孕前化合物的流产活性
——妊娠5-7天处理,第9天尸体剖检化合物 剂量 流产率
毫克/鼠/d·s·c 流产鼠数/处理过的鼠数 (%)
3.0 4/4 (100)A 1.0 4/4 (100)
0.3 1/4 (25)
3.0 4/4 (100)B 1.0 4/4 (100)
0.3 4/4 (100)
3.0 4/4 (100)C 1.0 4/4 (100)
0.3 0/4 (00)
3.0 4/4 (100)D 1.0 4/4 (100)
0.3 1/4 (25)
3.0 4/4 (100)E
1.0 2/4 (50)
3.0 4/4 (100)F 1.0 4/4 (100)
0.3 0/4 (0)载体对照 - 0/5 (0)0.2毫升苯甲酸苄酯+蓖麻油(1+4)
为了估测抗糖皮质激素的活性,在鼠上以17α-(3-羟基丙-1(Z)-烯基)-17β-羟基-11β,19-(4-二甲基氨基-邻-亚苯基)-4-雄(甾)烯-3-酮(C)作为通式I中所有化合物的代表。所得结果依次与对照物E和F进行比较。
在糖皮质激素的影响下,鼠的胸腺重量大大下降(=抑制胸腺细胞的作用)。如果在同时服用抗糖皮质激素的几种物质,可望能补偿糖皮质激素引起的胸腺抑制。
在重量100到130克切除了肾上腺的雄性幼鼠上进行试验。幼鼠生存条件保持在:常规的光照节律:10小时黑暗/14小时灯照,平均温度20±2℃,标准的进食(颗粒饲料),通过分开的饮水瓶供应自来水和0.9%Nacl溶液。
为了皮下使用,把化合物溶于苯甲酸苄酯和蓖麻油(比例为1+4)的混合物中,每日分别以0.2毫升的载体(赋形剂)容量注射。选用的剂量参见表2。
所用的标准糖皮质激素物质是剂量0.01毫克/鼠/d·s·c的地塞米松。这种剂量—与对照溶剂相关—在胸腺重量上大致降低75%。溶剂;苯甲酸苄酯/蓖麻油(1+4),每日载体(赋形剂)容量:0.2毫升。
在开始上述处置前的5天,被试验的鼠在醚麻醉下切除肾上腺。而后将这些鼠随意分成不同的试验组,随机试验的程度范围见表2。
处理的组别:地塞米松对照
溶剂对照
试验—物质剂量+地塞米松
处理持续4天(1-4天)。在第五天用CO2窒死动物。测定胸腺的重量并转换成毫克/鼠体重。
为了评价一种物质的抗糖皮质激素作用,把对照溶剂和地塞米松(0.01毫克/鼠/d·s·c)之间的差异规定为100%。
以平均百分比的抗糖皮质激素的作用(用%表示地塞米松引起的胸腺抑制的补偿)是以下列公式平均随机取样值为基础加以计算: 在此
如表2所示:化合物C仅用最大的试验量30.0毫克/d·s·c才产生地塞米松引起的胸腺抑制补偿。以较低的剂量(3.0;10.0毫克/d·s·c)不可能确定其有任何抗糖皮质激素的作用。
与化合物E(图2)和化合物F(图1)比较,化合物C的抗糖皮质激素的活性明显降低。
确实由法国专利申请86400057.5中已知在位置10和9(11)双键上有一个与取代芳基结构上类似的甾族化合物;但这些已知的化合物在17α位置上总有一个烷基、链烯基或炔基基团。然而这些化合物表现出相当大的抗糖皮质激素的活性,而在黄体酮接受体方面它们的活性可以不计,因此产生抗妊娠的效果。
依照本发明的化合物可以制备一些药剂,这些药剂与最接近的现有技术相比,具有新的效率,即具有比较高的抗妊娠效果并仅带有适度的抗糖皮质激素的活性。
表2
抗糖皮质激素的胸腺溶解试验
地塞米松引起的胸腺抑制的补偿
| 地塞 化合物 数量 相对胸腺米松 C n 重量 补偿(mg/ds.c) (mg/100g体重)平均±偏差 |
| a)- - 7 361.0±51.5 |
| 0.01 - 7 77.4±7.8 |
| 0.01 3.0 7 72.4±10.9 -1.8(-12.1-7.5) |
| 0.01 10.0 7 76.9±6.6 -0.2(-10.4-9.1) |
| 0.01 30.0 7 125.0±16.4 16.8(7.5-25.5) |
表2续(1)
| 地塞 化合物 数量 相对胸腺米松 F n 重量 补偿(mg/ds.c) (mg/100g体重)平均±偏差 |
| a)- - 12 419.8±61.9 |
| 0.01 - 12 91.2±16.6 |
| 0.01 3.0 6 110.6±16.3 5.9(-4.1-15.4) |
| 0.01 10.0 6 194.4±27.9 31.4(22.1-40.5) |
| 0.01 30.0 6 197.3±49.6 32.3(23.0-41.3) |
表2续(2)
a)对照组:苯甲酸苄酯+蓖麻油(1+4)0.4ml/ds.c( )=%补偿的95%置信区间n=每组鼠数
| 地塞 化合物 数量 相对胸腺米松 E n 重量 补偿(mg/ds.c) (mg/100g体重)平均±偏差 |
| a)- - 24 385.4±51.3 |
| 0.01 - 18 87.2±13.2 |
| 0.01 3.0 6 125.6±19.9 12.9(0.5-24.6) |
| 0.01 10.0 6 178.1±44.8 30.5(18.6-42.0) |
| 0.01 30.0 6 264.7±41.0 59.5(48.0-71.0) |
在下述实施例中,除非另有说明,都用乙酸酯和己烷的混合物进行色层分离。
实施例1
17β-羟基-11β,19-(0-亚苯基)-4-雄甾烯-3-酮
a)。19-(2-氯代苯基)-3,3-(2,2-二甲基亚丙基二氧基)-9(11)-雄甾烯-5α,17β-二醇
在惰性气体下,将4.9g镁屑加入到40ml无水乙醚中,然后与0.5ml2-氯代苄基氯混合,然后小心地与0.4ml的1,2-二溴甲烷混合。反应开始后,在40分钟内,向其中滴加溶于135ml无水乙醚中的其余量(18.4ml)的2-氯代苄基氯,而反应容器内的温度不高于30℃。在格利雅试剂形成之后,反应混合物冷却到0℃,並向其中缓慢滴加溶在75ml无水四氢呋喃中的5α,10α-环氧-3,3-(2,2-二甲基亚丙基二氧,基)-9(11)-雄甾烯-17β-醇(13.6g)。接着在水浴中搅拌后,反应混合物缓慢加热一夜至室温,然后倒入稀的氯化铵溶液中,水相用醋酸酯萃取几次,合并的有机相用氯化钠溶液洗涤至中性,用硫酸钠干燥,並在真空下浓缩,剩余物经氧化铝(中性,三级)色层分离,得到14.8g的上述化合物。
〔α〕D 22=-2°(CHCL3;C=0.51)
闪点:188-191℃(乙酸乙酯)
b).11β,19-(0-亚苯基)-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-5α,17β-二醇
600ml无水氨草胶在反应烧瓶中于-65℃下进行浓缩,除去所有水份,並与970mg新切的锂屑混合,立刻出现特有的兰色以后,将14g按照a)得到的产物溶液,以反应溶液在无色和兰色之间交替变化的方式,滴加到450ml无水四氢呋喃中。过量的锂用滴加乙醇的方法除去后,大多数的氨草胶通过蒸发除去。该反应混合物倾入水中,水相用醋酸乙酯萃取,合并的有机相用氯化钠溶液洗涤至中性,用硫酸钠干燥,並在真空下浓缩,经氧化铝(中性,三级)色层分离,得到10.3g的上述化合物。
〔α)D 22=+13°(CHCL3;C=0.52)
闪点:=164-167℃(乙酸乙酯)
1H-NMR(CDCL3)〔δ〕:7.0~7.45(4H,m,芳族质子);3.13(1H,d,J=16Hz,C-19上的质子);2。68(1H,d,J=16Hz,C-19上的质子);0.98(3H,S,缩酮基甲基的质子);0.95(3H,S,缩酮基甲基的质子);0.25(3H,S,C-18上的质子)。
在b)中表示的标题化合物也能用下述方法制备:
α).19-(2-溴代苯基)-3,3-(2,2-二甲基亚丙基二氧基)-9(11)-雄甾烯-5α,17β-二醇
用与实施例1a)相似的方法,将5g5α,10α-环氧-3,3-(2,2-二甲基亚丙基二氧基)-9(11)-雌甾烯-17β-醇与26.7g2-溴代苄基溴反应。色层分离后,得到5.9g白色泡沫状的上述化合物。
1H-NMR(CDCL3)〔δ〕:6.95~7.55(4H,m,芳族化合物上的质子);5.45(1H,S,宽峰,C-11上的质子);3.7-3.82(1H,m,C-17上的质子);3.4-3.6(4H,m,缩酮亚甲基的质子);3.16和3.07(每个〔1H,d,J=15Hz〕,C-19上的A、B系统的质子);0.98(3H,S,缩酮甲基的质子); 0.9(3H,S,缩酮甲基的质子);0.55(3H,S,C-18上的质子)。
β).11β,19-(0-亚苯基)-3,3-(2,2-二甲基亚丙基二氧基)雄甾烷-5α,17β-二醇
2.5g按照α)得到的化合物溶于250ml无水甲苯中,与2.25ml三丁基氢化锡和250mgα,α-偶氮异丁腈混合,回流加热3小时,然后在真空下除去溶剂,剩余物溶于四氢呋喃中,与50ml的氟化钾饱和水溶液一起搅拌1小时,然后水相用醋酸乙酯萃取并分离,合并有机相,经硫酸钠干燥並在真空下浓缩,剩余物经氧化铝(中性、三级)色层分离,得到1.75g的标题化合物。
在b)中表示的标题化合物,也能由下述化合物制备:
r).19-(2-氟代苯基)-3,3-(2,2-二甲基亚丙基二氧基)-9(11)-雄甾烯-5α,17β-二醇
用与实施例1a)相似的方法,使750mg5α,10α-环氧-3,3-(2,2-二甲基亚丙基二氧基)-9(11)雌甾烯-17β-醇与2g2-氟代苄基氯反应,色层分离后,得到798mg白色泡沫状的上述化合物。
1H-NMR(CD2CL2)〔δ〕:6.92-7.33(4H,m,芳族化合物上的质子);5.09(1H,m,C-11上的质子);3.62-3.72(1H,m,C-17上的质子);3.45-3.58(4H,m,缩酮亚甲基的质子);2.97和2.9(每个〔1H,d,J=15Hz〕,C-19上的A、B系统的质子);0.99(3H,S,缩酮甲基上的质子);0.9(3H,S,缩酮甲基的质子);0.61(3H,S,C-18上的质子)。
δ)11β,19-(0-亚苯基)-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-5α,17β-二醇
用与实施例1b)相似的方法,使750mg19-(2-氟代苯基)-3,3-(2,2-二甲基亚丙基二氧基)-9(11)-雄甾烯-5α,17β-二醇与60mg锂反应,经色层分离后,得到585mg白色泡沫状的上述化合物。
c)17β-羟基-11β,19-(0-亚苯基)-4-雄甾烯-3-酮
2g按照b)得到的产物溶于100ml丙酮中,並与5ml 4N盐酸混合,在室温下搅拌4小时后,反应混合物倾入到碳酸氢钠饱和溶液中,水相用二氯甲烷萃取,合并的有机相用硫酸钠干燥,並在真空下浓缩。剩余物经硅胶色层分离,得到1.13g上述化合物。
〔α〕D 22=+84°(CHCL3;C=0.5)
1H-NMR(CDCL3)〔δ〕:7~7.5(4H,m,芳族化合物上的质子);5.88(1H,S,C-4上的质子);3.68(1H,tr,J=9Hz,C-17上的质子);3.3(1H,m,G-11上的质子);3.26(1H,d,J=17Hz,C-19上的质子);2.74(1H,d,J1=17Hz,C-19上的质子);0.29(3H,S,C-18上的质子)。
实施例2
17β-羟基-17-(丙-1-炔基)-11β,19-(0-亚苯基)-4-雄甾烯-3-酮
a)11β,19-(0-亚苯基)-5α-羟基-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-17-酮
将11.28g三氧化铬在0℃下分几份加入到34.3ml的吡啶和287ml二氯甲烷的混合物中,接着将实施例1b)得到的甾族化合物(8g)溶于50ml二氯甲烷中,将其在相同的温度下缓慢滴加到反应混合物中,后者在冰浴温度下另外搅拌2小时,搅拌完成后,将反应的固体成份沉淀,倾出上层清液,该沉淀物用二氯甲烷彻底洗涤几次,合并的有机相通过用0.5M氢氧化钾水溶液洗涤,使其从剩余的无机成份中分离出来,用水洗涤至中性,用硫酸钠干燥,並在真空下浓缩,分离出7g11β,19(0-亚苯基)-5α-羟基-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-17-酮粗产物,该纯度对进一步的反应是合适的(见下面),为分析目的,用氧化铝(中性,三级)色层分离提纯500mg,得到432mg目的产物。
〔α〕D 22=+31°(CHCL3,C=0.505)
闪点=206-210℃(乙酸乙酯)
b)17-(丙-1-炔基)-11β,19-(0-亚苯基)-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-5α,17β-二醇
在0℃下,将225ml无水四氢呋喃用丙炔饱和,接着将1.6M正丁基锂溶液(己烷)(27.7ml)缓慢地滴加到该溶液中,温度没有明显升高,继之搅拌15分钟后,将溶于45ml无水四氢呋喃中的2g按照a)得到的粗产物溶液缓慢地滴加到上述反应溶液中,接着搅拌30分钟,然后将反应混合物倾入到水中,水相用乙酸乙酯萃取,合并的有机相用氯化钠溶液洗涤,经硫酸钠干燥,並在真空下浓缩,得到2.44g粗产物,经氧化铝(中性,三级)色层分离,得到1.8g上述的化合物。
IR(KBr):2230cm-1三键
c)17β-羟基-17-(丙-1-炔基)-11β,19-(0-亚苯基)-4-雄甾烯-3-酮
用相似于在实施例1c)中所描述的酸裂解方法,将1.5g由b)得到的产物转化,得到738mg标题化合物。
实施例3
17β-羟基-17-(丙-1-炔基)-11β,19-(0-亚苯基)-4,6-雄甾二烯-3-酮
a)11β,19-(0-亚苯基)-4-雄甾烯-3,17-二酮
用相似于实施例1c)所述的方法,将20g通过实施例1a)、实施例1b)和实施例2a)的反应步骤得到的产物分离,得到8.69g标题化合物。
〔α〕D 22=+116°(CHCL3;C=0.51)
闪点=284~288℃
b)11β,19-(0-亚苯基)-3-乙氧基-3,5-雄甾二烯-17-酮
将8g按照a)得到的产物置于85ml无水二氯甲烷、25ml乙醇和6.7ml原甲酸三乙酯的混合物中,並在0℃下,与170mg P-甲苯磺酸(一水合物)混合,接着该混合物在冰浴温度下搅拌一夜,然后与过量的碳酸氢钠溶液混合,水相用二氯甲烷萃取,合并的有机相用氯化钠饱和溶液洗涤,经硫酸钠干燥並在真空下浓缩,得到11.3g含杂质的粗产物。由乙醇(与几滴吡啶混合)结晶,得到5.43g晶形标题化合物。
闪点=182-186℃
〔α〕D 22=+39°(CHCL3;C=0.5)
c)17-(丙-1-炔基)-11β,19-(0-亚苯基)-3-乙氧基-3,5-雄甾二烯-17β-醇
用相似于实施例2b)的方法,将5g按照b)得到的粗产物转化成5.4g粗产品,该纯度足以满足进一步反应的要求,用乙醇结晶400mg粗产物,得到268mg晶形标题化合物。
闪点=203~207℃
〔α〕D 22=-91°(CHCL3;C=0.5)
d)17-(丙-1-炔基)-17β-羟基-11β,19-(0-亚苯基)-4,6-雄甾二烯-3-酮
将5g由c)得到的粗产物悬浮于50ml80%二噁烷的水溶液和24ml 10%醋酸钠水溶液的混合物中,在0℃下,将1.6g1,3-二溴-5,5-二甲基乙内酰脲分几份加入到该悬浮液中,该甾族化合物缓慢地进入到溶液中,反应二小时后,该反应混合物倾入水中,水相用二氯甲烷萃取,合并的有机相用硫代硫酸钠饱和水溶液洗涤,用硫酸钠干燥,並在真空下浓缩。将由此得到的17-(丙-1-炔基)-17β-羟基-11β,19-(0-亚苯基)-6β-溴代-4-雄甾烯-3-酮粗产物溶于48ml无水二甲基甲酰胺中。在惰性气体下,将其与2.4g溴化锂和1.65g碳酸锂混合,并在100℃下搅拌1小时,反应混合物冷却到室温后倾入水中,水相用4N盐酸中和,冷至冰浴温度,接着在此温度下搅拌1小时,得到沉淀的甾族化合物,如此得到4.14g含有微量杂质的粗产物,该纯度足以满足进一步反应的需要,用1.14g粗产物开始,用二异丙醚结晶得到638mg上述化合物。
〔α〕D 22=+80°(CHCL3;C=0.5)
闪点=215-217℃
实施例4
17-(丙-1-炔基)-17β-羟基-11β,19-(4-甲氧基-0-亚苯基)-4-雄甾烯-3-酮
a)19-(2-氯代-5-甲氧基苯基)-3,3-(2,2-二甲基亚丙基二氧基)-9(11)-雄甾烯-5α,17β-二醇
用相似于实施例1a)所述的方法,当使15g5α,10α-环氧-3,3-(2,2-二甲基亚丙基二氧基)-9(11)-雌甾烯-17β-醇与2-氯代-5-甲氧基苄基氯反应时,得到15.5g的上述化合物。
b)11β,19-(4-甲氧基-0-亚苯基)-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-5α,17β-二醇
用相似于实施例1b)所述的方法,15g由a)得到的化合物生成11.6g白色泡沫状标题化合物。
〔α〕D 22=+21.1°(CHCL3;C=0.52)
闪点=223-224℃(二异丙醚)
标题化合物b)也能用下述方法合成:
α)19-(2-溴代-5-甲氧基苯基)-3,3-(2,2-二甲基亚丙基二氧基)-9(11)-雄甾烯-5α,17β-二醇
在惰性气体中,将150g2-溴代-5-甲氧基苄基溴悬浮于11无水乙醚中,並与13.3g镁屑混合,在格利雅反应开始后,反应温度通过冷却保持在30℃以下,在完全形成格利雅试剂后,在搅拌的同时,向其中滴加溶于330ml无水四氢呋喃中的50g5α,10α-环氧-3,3-(2,2-二甲基亚丙基二氧基)-9(11)-雌甾烯-5α,17β-二醇,接着将反应混合物搅拌1.5小时,並按实施例1a)所述的过程进行。经色层分离后,得到66.5g白色泡沫状的上述化合物。
闪点=128-130℃(二异丙醚/己烷)
β)11β,19-(4-甲氧基-0-亚苯基-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-5α,17β-二醇
用相似于实施例1β)的方法,通过使用660mg2,2-偶氮异丁腈作为起始基团,使在1.7l无水甲苯中的66g19-(2-溴代-5-甲氧基苯基)-3,3-(2,2-二甲基亚丙基二氧基)-9(11)-雄甾烯-5α,17β-二醇与34ml三丁基氢化锡反应。完全反应后,在真空下蒸出溶剂,剩余物用二异丙醚结晶,得到49g晶形上述化合物。
c)11β,19-(4-甲氧基-0-亚苯基)-5α-羟基-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-17-酮
用相似于实施例2a)的方法,将11g由b)得到的化合物转化为相应的酮化合物,得到9.53g白色泡沫状上述化合物。
〔α〕D 22=+33°(CHCL3;C=0.55)
闪点=235-238℃
d)17-(丙-1-炔基)-11β,19-(4-甲氧基-0-亚苯基)-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-5α,17β-二醇
用相似于实施例2b)的方法,将4g由c)得到的化合物转化为相应的17α-丙炔基化合物,经色谱分离后,得到3.3g白色泡沫状的上述化合物。
IR(KBr):2230cm-1三键
e)17-(丙-1-炔基)-17β-羟基-11β,19-(4-甲氧基-0-亚苯基)-4-雄甾烯-3-酮
用相似于实施例1c)的方法,将3g由d)得到的化合物转化为相应的4-烯-酮化合物,得到1.5g白色泡沫状的标题化合物。
〔α〕D 22=+18°(CHCL3;C=0.465)
实施例5
17-(3-羟基丙-1-(Z)-烯基)-17β-羟基-11β,19-(4-甲氧基-0-亚苯基)-4-雄甾烯-3-酮
a)17-〔3-(四氢吡喃-2-基氧代)-丙-1-炔基〕-11β,19-(4-甲氧基-0-亚苯基)-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-5α,17β-二醇
在0℃和惰性气体下,将28.3ml 15%的n-丁基锂的己烷溶液缓慢滴加到溶有5.7g3-(四氢吡喃-2-基氧代)-丙-1-炔的100ml无水四氢呋喃溶液中。接着,该混合物在0℃下搅拌15分钟,然后在0~+5℃下向其中滴加含有4g按照实施例4c)得到的产物的60ml无水四氢呋喃溶液。接着,该混合物在室温下搅拌3小时,然后倾入冰水中,並用醋酸乙酯萃取,有机相用硫酸钠干燥,並在真空下浓缩后,粗产物经氧化铝(中性,三级)色层分离,得到4.36g白色泡沫状的上述化合物。
闪点=150-153℃(二异丙醚〔把1∶1的差向异构体的混合物看作四氢吡喃醚〕)
b)17-〔3-(四氢吡喃-2-基氧代)-丙-1(Z)-烯基〕-11β,19-(4-甲氧基-0-亚苯基)-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-5α,17β-二醇。
在室温和常压下,在添加5ml吡啶和400mg钯/硫酸钡(10%pd)后,溶于75ml四氢呋喃的4g按照a)得到的产物被氢化。当水不再被析出时,从催化剂中过滤出该混合物,並将其滤液浓缩,得到3.91g淡黄色泡沫状的上述化合物。
c)17-〔3-羟丙基-1(Z)-烯基〕-17β-羟基-11β,19-(4-甲氧基-0-亚苯基)-4-雄甾烯-3-酮
用相似于实施例1c)的方法,3.5g按照b)得到的化合物被分解,得到1.5g白色泡沫状的标题化合物。
〔α〕D 22=+60°(CHCl3;C=0.5)
实施例6
17-(氰甲基)-17β-羟基-11β,19-(4-甲氧基-0-亚苯基)-4-雄甾烯-3-酮
a)11β,19-(4-甲氧基-0-亚苯基)-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-〔17(β-1′)-螺-3′〕-环氧乙烷-5α-醇
在惰性气体下,将1g按照4c)所述的方法得到的酮化合物溶于20ml无水二甲基甲酰胺中,在0℃下,首先与2.04g三甲基碘化硫混合,然后与1.40g叔丁酸钾混合,接着,将该混合物缓慢加热一夜至室温,並伴随连续搅拌,然后将其倾入到氯化铵饱和溶液中,其水相用乙酸乙酯萃取几次,合并的有机相用硫酸钠干燥,并在真空下浓缩,其剩余物经氧化铝(中性,三级)色层分离,得到895mg白色泡沫状的上述化合物。
b)17-氰甲基-11β,19-(4-甲氧基-0-亚苯基)-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-5α,17β-二醇
在惰性气体下,将850mg由a)得到的环氧化物溶于17ml的无水乙醇中,然后与含有1.7g氰化钾的3.4ml水的溶液混合。接着,将该反应混合物加热一夜至50℃,然后倾入冰水中。水相用乙酸乙酯萃取几次,合并的有机相用硫酸钠干燥,並在真空下浓缩至干,其剩余物经氧化铝(中性,三级)色层分离,得到815mg上述的化合物。
IR(KBr):2250cm-1 C≡N三键
c)17-氰甲基-17β-羟基-11β,19-(4-甲氧基-0-亚苯基)-4-雄甾烯-3-酮
用相似于实施例1c)的方法,将800mg由b)得到的化合物转化成相应的4-烯-3-酮化合物。得到575mg的标题化合物。
〔α〕D 22=59°(CHCL3;C=0.505)
闪点=155-156℃(乙酸乙酯/己烷)
实施例7
17-(丙-1-炔基)-17β-羟基-11β,19-(4-甲氧基-0-亚苯基)-4,6-雄甾二烯-3-酮
a)11β,19-(4-甲氧基-0-亚苯基)-4-雄甾烯-3,17-二酮
用相似于实施例1c)的方法,将11.2g由实施例4c)中得到的物质转化成相应的4-烯-3-酮化合物,得到7.6g上述化合物。
〔α〕D 22=130°(CHCL3;C=0.5)
闪点=184~187℃(乙酸乙酯)
b)11β,19-(4-甲氧基-0-亚苯基)-3-乙氧基-3,5-雄甾二烯-17-酮
用相似于实施例3b)的方法,使5g由a)得到的物质与乙醇反应,得到2.45g晶形的标题化合物。
〔α〕D 22=57°(CHCL3;C=0.5)
闪点=174~176℃
c)17-(丙-1-炔基)-11β,19-(4-甲氧基-0-亚苯基-3-乙氧基-3,5-雄甾二烯-17β-醇
用相似于实施例3c)的方法,转化2.4g由b)得到的酮化合物,得到2.45g粗产物。
(α〕D 22=-86°(CHCL3;C=0.505)
闪点=168~171℃(乙醇)
d)17-(丙-1-炔基)-17β-羟基-11β,19-(4-甲氧基-0-亚苯基)-4,6-雄甾二烯-3-酮
用相似于实施例3d)的方法,转化2.35g由c)得到的粗产物,成为相应的4,6-二烯-3-酮化合物,经硅胶色层分离得到1.43g标题化合物。
〔α〕D 22=132°(CHCL3;C=0.5)
闪点=237~242℃(乙酸酯)
实施例8
17-(3-羟基-1(Z)-烯基)-17β-羟基-11β,19-(4-甲硫基-0-亚苯基)-4-雄甾烯-3-酮
a)19-(2-氯代-5-甲硫基苯基)-3,3-(2,2-二甲基亚丙基二氧基)-9(11)-雄甾烯-5α,17β-二醇
用相似于实施例1a)的方法,使3.4g5α,10α-环氧-3,3-(2,2-二甲基亚丙基二氧基)-9(11)-雄甾烯-17β-醇与94.5g2-氯代-5-甲硫苄基氯反应,经色层分离后,得到43.2g的白色泡沫状上述化合物。
b)11β,19-(4-甲硫基-0-亚苯基)-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-5α,17β-二醇
将40g按照a)得到的物质溶于750ml无水四氢呋喃中,并在-78℃下,向其中滴加3.79g锂和3.41液态氨的混合物。搅拌45分钟后,在相同温度下,缓慢地一滴一滴地加入200ml甲醇、200ml四氢呋喃和4.6ml甲基碘的混合物,滴加完成后,用相似于实施例1b)的方法处理该混合物。由粗产物结晶出18.37g纯的标题化合物。
闪点=173-176℃(乙酸酯)
c)11β,19-(4-甲硫基-0-亚苯基)-5α-羟基-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-17-酮
用相似于实施例13c)的方法,使18g由b)得到的化合物与溶于780ml无水甲苯中的21.47g三异丁酸铝和156ml环己酮反应,生成相应的17-酮化合物,经氧化铝(中性,三级)色层分离后,得到13.98g白色泡沫状的上述化合物。
〔α〕D 22=41.8°(CHCL3;C=0.5)
闪点=224-225℃(乙酸酯/己烷)
d)17-〔3-(四氢吡喃-2-基氧代)-丙-1-炔基〕-11β,19-(4-甲硫基-0-亚苯基)-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-5α,17β-二醇
用相似于实施例5a)的方法,使13.8g由c)得到的物质与19g3-(四氢吡喃-2-基氧代)-丙-1-炔反应,经色层分离后,得到15.65g白色泡沫状的上述化合物。
IR(KBr):2230cm-1三键
e)17-〔3-(四氢吡喃-2-基氧代)-丙-1(Z)-烯基〕-11β,19-(4-甲硫基-0-亚苯基)-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-5α,17β-二醇
用相似于实施例5b)的方法,使用被18.9ml吡啶抑制的1.51g硫酸钡钯(10%pd)作为催化剂,用氢使15.5g由d)得到的物质氢化,色层分离后,得到14.15g白色泡沫状的标题化合物。
f)17-(3-羟基丙-1(Z)-烯基)-17β-羟基-11β,19-(4-甲硫基-0-亚苯基)-4-雄甾烯-3-酮
用相似于实施例1c)的方法,使5.5g由e)得到的烯烃与200ml丙酮中的5ml 4N含水盐酸反应,生成4-烯-3-酮化合物。经硅胶色层分离后,得到2.34g白色泡沫状的标题化合物。
〔α〕D 22=86°(CHCL3;C=0.51)
闪点=146-148℃(乙酸酯/己烷)
实施例9
17-(3-羟基丙-1(Z)-烯基)-5α,17β-二羟基-11β,19-(4-甲硫基-0-亚苯基)-雄甾烷-3-酮
a)17-(3-羟基丙-1(Z)-烯基)-5α、17β-二羟基-11β,19-(4-甲硫基-0-亚苯基)-雄甾烷-3-酮
在室温下,在50ml 70%乙酸中,将5g按照实施例8e)得到的物质转化成所希望的3-酮化合物。接着,该反应混合物用水稀释,水相用二氯甲烷萃取,合并的有机相依次用饱和硫酸氢钠和饱和食盐溶液洗涤。接着用硫酸钠干燥,真空蒸发溶剂后,其剩余物经硅胶色层分离,得到2.66g白色泡沫状的上述化合物。
〔α〕D 22=-5°(CHCL3;C=0.5)
闪点=193-195℃(乙酸酯)
实施例10
17-(3-羟基丙-1(Z)-烯基)-17β-羟基-11β,19-(4-甲基亚磺酰基-0-亚苯基)-4-雄甾烯-3-酮
a)17-〔3-(四氢吡喃-2-基氧代)-丙-1(Z)-烯基〕-11β,19-(4-甲基亚磺酰基-0-亚苯基〕-3,3-(2,2-二甲基亚丙基二氧基)-雄甾烷-5α,17β-二醇
将5g由实施例8e)得到的物质溶于45ml四氢呋喃、45ml甲醇和10ml水的混合物中,并用5.1g高碘酸钠处理。在室温下,将反应混合物搅拌一夜,经硅藻土过滤,其滤液用乙酸酯稀释,有机相用硫酸氢钠饱和溶液洗涤,用硫酸钠干燥,并在真空下浓缩,其剩余物经氧化铝(中性,三级)色层分离,得到3.94g白色泡沫状的上述化合物。
b)17-(3-羟基丙-1(Z)-烯基)-17β-羟基-11β,19-(4-甲基亚磺酰基-0-亚苯基-4-雄甾烯-3-酮
用相似于实施例1c)的方法,将3.8g按着a)得到的物质转化成为4-烯-3-酮化合物。经硅胶色层分离后,得到1.66g的标题化合物。
〔α〕D 22=51°(CHCL3;C=0.5)
实施例11
17-(3-羟基丙-1-炔基)-17β-羟基-11β,19-(4-甲硫基-0-亚苯基)-4-雄甾烯-3-酮。
a)17-(3-羟基丙-1-炔基)-17β-羟基-11β,19-(4-甲硫基-0-亚苯基)-4-雄甾烯-3-酮
用相似于实施例1c)的方法,将2.5g由实施例8d)得到的物质分解成相应的4-烯-3-醇化合物。经硅胶色层分离后,得到1.13g白色泡沫状的标题化合物。
实施例12
17-(3-羟基丙-1(Z)-烯基)-17β-羟基-11β,19-(4-乙硫基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)11β,19-(4-乙硫基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷- 5α,17β-二醇
类似于例8b),依据例8a)产生的10克物质与620毫克锂和14.7毫升碘乙烷进行反应,不使用碘甲烷。用醋酸酯/己烷处理,从该粗产品中得到4.62克上述结晶产品的化合物。
〔α〕D 22=39°(CHCL3;C=0.5)
FP.=164℃
b)11β,19-(4-乙硫基-邻-亚苯基)-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
类似于例13c),把依据例a)所生成的4.5克物质转化成相应的17-酮化合物。色谱法分析后,离析得到3.4克上面所述的化合物。
〔α〕D 22=44°(CHCL3;C=0.505);
IR(KBr):1740cm-1五环酮
c)17-〔3-(四氢吡喃-2-基氧)-丙-1-炔基〕-11β,19-(4-乙硫基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
类似例5a),依据b)产生的3.2克酮使其与4.31克3-(四氢吡喃-2-基氧)-丙炔-1进行反应。色谱法分析后,离析得到3.25克上面所述的白色泡沫状化合物
IR(KBr):2230cm-1三键
d)17-〔3-(四氢吡喃-2-基氧)-丙-1(Z)-烯基〕-11β,19-(4-乙硫基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
类似例5b),依据c)所得3.1克物质与在硫酸钡(10%钯)上附有钯的300毫克催化剂进行加氢反应。用3.75毫升吡啶抑制,经色谱法分析后,离析得到2.85克白色泡沫状标题化合物。
e)17-(3-羟基丙-1(Z)-烯基)-17β-羟基-11β,19-(4-乙硫基-邻-亚甲基)-4-雄(甾)烯-3-酮
根据d)所得2.7克烯与2.5毫升4N盐酸水溶液在100毫升丙酮中以类似于例1c)的方式转化为相应的4-烯-3-酮化合物。在硅胶上色谱分析后,离析得到1.35克浅黄色泡沫状的上述标题化合物。
〔α〕D 22=85°(CHCL3;C=0.5)
实施例13
17-(丙-1-炔基)-17β-羟基-11β,19-(4-二甲氨基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)19-(2-氯-5-二甲氨基苯基)-3,3-(2,2-二甲基三亚甲基二氧)-9(11)-雄(甾)烯-5α,17β-二醇
类似例1a)的方法,从15克5α,10α-环氧-3,3-(2,2-二甲基三亚甲基二氧)-9(11)-estrene-17β-醇起始,用2-氯-5-二甲基氨基苯甲基氯进行反应,得到14.39克上面所述的化合物。
b)11β,19-(4-二甲基氨基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
用类似例1b)的方法,从依据a)所得14克化合物为起始物,制得9.9克上面所述白色泡沫状化合物。
c)11β,19-(4-二甲基氨基-邻-亚苯基)-5-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
依据b)所生成的11.5克化合物溶于500毫升甲苯中,依次用13.8克三异丙醇铝和100毫升环己酮处理。然后,在迴流下加热反应混合物,蒸出大约1/3溶剂。冷却后,把混合物倒入冰水中。在硅藻土上过滤生成的乳胶,用乙酸乙酯充分洗涤滤渣,分离滤液的有机相,在硫酸钠上干燥并在真空下浓缩。在氧化铝上(中性III级)将剩余物色谱法分析后,得到8.13克标题化合物。
从醋酸酯中结晶130毫克这类化合物,得到67毫克晶体状的11β,19-(4-二甲基氨基-邻-亚苯基)-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
FP.=264-267℃
〔α〕D 22=28°(CHCL3;C=0.5)
d)17-(丙-1-炔基)-11β,19-(4-二甲基氨基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
类似例2b)的方法,把依据c)所得的3克化合物转化成相应的17α-丙炔化合物。色谱法分析后,离析得到2.6克浅黄色泡沫状上面所述的化合物。
IR(KBr):2235cm-1三键
e)17-(丙-1-炔基)-17β-羟基-11β,19-(4-二甲基氨基-邻-亚苯基)-4-雄(甾)烯-3-酮
类似例1c)的方法,把依据d)所得的2.5克产品转化成相应的4-烯-3-酮化合物。离析得到1.58克白色泡沫状上面所述的化合物。
〔α〕D 22=+28°(CHCL3;C=0.51)
FP。=231-234℃(醋酸乙酯)
以下述方法制造反应步骤7a)所需的2-氯-5-二甲基氨基苄基氯:
α)2-氯-5-二甲基氨基苄基氯
在惰性气氛下,在0℃的3升四氢呋喃中放入300克氢化锂铝,用500克5-氨基-2-氯苯甲酸(工业用85%)按份进行处理。然后把反应混合物缓慢加热到室温,在该室温下搅拌过夜。进一步做如下处理:反应混合物冷却至0℃,用饱和氯化铵溶液小心地分解过量的氢化铝锂。从沉积物中分离出有机相并且用醋酸酯和二氯甲烷洗涤沉淀物数次。用氯化钠饱和溶液洗涤合并的有机相直至有机相成中性,硫酸钠上干燥并在真空下浓缩。得到242克粗2-氯-5-氨基苯甲醇,它的纯度足以适于进行后面的反应。
1H-NMR(CDCL3)〔δ〕:6.3-7.15(3H,m芳香质子);
4.55(2H,S,苄基质子)
β)2-氯-5-二甲基氨基苯甲醇
在30克2-氯-5-氨基苯甲醇和1升四氢呋喃的混合物中悬浮51.8克硼氢化钠,冷却下滴加到搅拌着的235毫升2M硫酸和88毫升38%福尔马林溶液的混合物中,温度维持在-10℃和20℃之间。滴加完成后,用固体氢氧化钠使反应混合物成为明显的碱性并用水处理。分离出有机相,用二氯甲烷将水相萃取数次,并用饱和氯化钠溶液洗涤合并的有机相直至有机相呈中性。其后在硫酸钠上干燥并在真空下浓缩,得到油状的24克2-氯-5-二甲基氨基苯甲醇。
1H-NMR(CDCL3)〔δ〕:6.4-7.25(3H,m,芳族质子);
4.67(3H,S,苄基质子);
2.92(6H,S,二个甲基基团质子)。
r)2-氯-5-二甲基氨基苄氯
把600毫升无水二氯甲烷中有23.8克N-氯代琥珀酰亚胺的溶液冷却至0℃,并与15.6毫升二甲硫缓慢混合。其后,把由此生成的悬浮液冷却至-30℃,小心地与20克2-氯-5-二甲基氨基苄醇混合。将该反应混合物温热到0℃并在此温度下搅拌3小时。其后用二氯甲烷稀释混合物并倒入冰水中。分离有机相,用饱和氯化钠溶液洗涤,在硫酸钠上干燥并且在真空下浓缩。在氯化铝上(中性III级)色谱法用己烷离析残余物。得到17.2克2-氯-5-二甲基氨基苄基氯
1H-NMR(CDCL3)〔δ〕:6.4-7.3(3H,m,芳族质子);
4.61(2H,S,苄基质子);
2.92(6H,S,二个甲基基团的质子)。
实施例14
17β-羟基-17-甲氧基甲基-11β,19-(4-二甲基氨基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)11β,19-(4-二甲基氨基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-〔17(β-1′)-螺-3′〕-环氧乙烷-5-醇
把依据例13c)制得的2.5克化合物在惰性气氛下溶于50毫升无水二甲基甲酰胺中并冷却至0℃。依次用5克碘化三甲基锍和3.4克叔丁醇钾处理该溶液。搅拌反应混合物一直到反应完成为止(DC检测)。其后把反应混合物倒入冰水中,用醋酸酯萃取水相,用氯化钠水溶液洗涤有机相,并在硫酸钠上干燥。溶剂蒸发后,在氧化铝上(中性III级)色谱法离析剩余物。得2.1克上面所述白色泡沫状化合物。
b)17-甲氧基甲基-11β,19-(4-二甲基氨基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5,17β-二醇
依据a)制得的2克化合物溶于40毫升3m甲醇的甲醇钠溶液中,在惰性气氛下迥流加热5小时。冷却后将反应混合物倒入冰水中,用二氯甲烷萃取水相并用氯化钠溶液洗涤有机相。在硫酸钠上干燥有机相后,真空浓缩。在氢氧化铝上(中性III级)色谱法分析剩余物。离析得到1.41克上面所述白色泡沫状化合物。
c)17β-羟基-17-甲氧基甲基-11β,19-(4-二甲基氨基-邻-亚甲基)-4-雄(甾)烯-3-酮
类似例1c)的方法,把依据b)所得的1.3克产品转化为相应的4-烯-3-酮化合物。离析得到0.75克白色泡沫状的标题化合物。
〔α〕D 22=80°(CHCL3;C=0.505)
FP=124-127℃
实施例15
17-氰甲基-17β-羟基-11β,19-(4-二甲基氨基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)17-氰甲基-11β,19-(4-二甲基氨基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
类似例6)的方法,把依据例14a)所制得的2.2克环氧化物在42毫升乙醇中与在8.4毫升水有4.22克氰化钾的溶液进行反应。色谱法分析后,离析得到1.95克上面提到的化合物。
IR(KBr):2245cm-1C≡N三键
b)17-氰基甲基-17β-羟基-11β,19-(4-二甲基氨基-邻-亚苯基)-4-雄(甾)烯-3-酮
类似例1c)的方法,把依据a)所得的1.9克氰化物转化成相应的4-烯-3-酮化合物。从粗产品中直接结晶1.23克标题化合物。色谱分析母液,进而产生138毫克所需的化合物。
〔α〕D 22=77°(CHCL3;C=0.5)
FP.=172-176℃
实施例16
17-(3-羟基丙-1-炔基)-17β-羟基-11β,19-(4-二甲基氨基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)17-〔3-(四氢吡喃-2-基氧)-丙-1-炔基〕-11β,19-(4-二甲基氨基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
类似例5a)的方法,把依据例13c)所制得的10克酮化合物与55.3克3-(四氢吡喃-2-基氧)-丙炔和247毫升在己烷中有15%丁基锂的溶液进行反应。色谱法分析后,得到11.74克上面所述的白色泡沫状化合物。
IR(KBr):2230cm-1三键
b)17-(3-羟基丙-1-炔基)-17β-羟基-11β,19-(4-二甲基氨基-邻-亚苯基)-4-雄(甾)烯-3-酮
类似例1c)的方法,把依据a)所制得的11.74克化合物转化成6.97克标题化合物。
〔α〕D 22=25.6°(CHCL3;C=0.5)
FP.=251-253℃(醋酸酯)
实施例17
17-(3-羟基丙-1(Z)-烯基)-17β-羟基-11β,19-(4-二甲基氨基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)17-(3-羟基丙-1(Z)-烯基)-17β-羟基-11β,19-(4-二甲基氨基-邻-亚苯基)-4-雄(甾)烯-3-酮
类似例5b)的方法,把依据例16b)所制得的6.5克乙炔化合物转化成相应的Z-烯烃。在硅胶上色谱法分析后,离析得到4.76克上面所述白色泡沫状标题化合物。
〔α〕D 22=71°(CHCL3;C=0.5)
实施例18
17β-羟基-11β,19-(4-羟基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)11β,19-(4-羟基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
以类似例4β)的方式得到的50克甲氧基化合物溶于500毫升无水的二甲基甲酰胺中,然后在惰性气氛下用28.2克甲硫醇钠处理。在此惰性气体条件下,把反应混合物迥流加热3小时,冷至室温,然后倒入8升冰水中。在室温下搅拌该混合物直至粗产品成为固态物质沉淀。取出该产品,用水洗涤并且在真空中干燥。离析得到49.2克白色固体标题粗化合物,其质量足以适于进行下一步反应。
〔α〕D 22=21°(CHCL3;C=0.5)
FP.=267-270℃(醋酸酯)
b)17β-羟基-11β,19-(4-羟基-邻-亚苯基)-4-雄(甾)烯-3-酮
依据a)所得的2克苯酚,以类似例1c)的方式在60毫升丙酮中与3毫升4N氯化氢水溶液反应,转化为4-烯-3-酮化合物。离析得到1.05克上面所述白色泡沫状化合物。
FP.=242-245℃(醋酸酯)
实施例19
17β-羟基-11β,19-(4-三氟甲基-磺酰基氧-邻-亚苯基)-4-雄(甾)烯-3-酮
a)17β-羟基-11β,19-(4-三氟甲基-磺酰基氧-邻-亚苯基)-4-雄(甾)烯-3-酮
类似例18b)的方式制得的10克苯酚溶于250毫升无水二氯甲烷中并用17.3克4-二甲基氨基-吡啶处理。在惰性气氛中将溶液冷至-50℃,并慢慢地把在30毫升无水二氯甲烷中溶解有4.76毫升三氟甲烷磺酸酐的溶液滴加处理。在-50℃搅拌15分钟后,将反应混合物倒入饱和碳酸氢钠溶液中并且用二氯甲烷萃取水相。用饱和氯化钠溶液洗涤合并的有机相,在硫酸钠上干燥并且在真空下浓缩。在硅酸上对剩余物色谱法分析后,得到11.37克浅黄色泡沫状标题化合物。
FP.=204-205℃(二异丙基醚)
实施例20
17β-羟基-11β,19-〔4-(2-三甲基-甲硅烷基-乙炔基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
a)17β-羟基-11β,19-〔4-(2-三甲基-甲硅烷基-乙炔基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
依据例19a)制得的1克三氟甲烷磺酸盐化合物溶于10毫升无水二甲基甲酰胺中,在惰性气体下用三乙基胺,1.39毫升三甲基硅乙炔和49毫克四个三苯膦钯进行处理。其后反应混合物在110℃加热一小时,冷却至室温并且用醋酸酯稀释。在硅藻土上过滤后,用饱和氯化钠溶液洗涤滤渣数次。分离有机相,在硫酸钠上干燥并且在真空中浓缩。在硅胶上对剩余物色谱分析,得到656毫克白色泡沫状标题化合物。
FP.=267-271℃(二异丙醚)
实施例21
17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(2-三甲基甲硅烷基-乙炔基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
a)11β,19-(4-三氟甲基-磺酰基氧-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依照例18a)所制得的40克苯酚,用类似例19a)的方法使其与14.93毫升三氟甲烷磺酸酐反应。色谱法分析后,离析得到37.3克白色泡沫状的上述化合物
b)11β,19-〔4-(2-三甲基甲硅烷基乙炔基)-邻-亚苯基〕-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据例20a)制得15克化合物,使其与17.3毫升三甲基硅乙炔进行反应。在氧化铝上(中性III级)色谱分析后,离析得到11.7克白色泡沫状的上述化合物。
IR(KBr):2150cm-1芳醇盐中的三键
c)11β,19-〔4-(三甲基甲硅烷基乙炔基)-邻-亚苯基〕-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
依据b)所得到的11.2克物质,用类似例2a)的方式使其与11.69克三氧化铬进行氧化反应生成相应的17-酮化合物。在氧化铝上(中性III级)色谱法分析产生9.05克白色泡沫状上述的化合物。
〔α〕D 22=51°(CHCL3;C=0.5)
FP.=245-248℃(二异丙醚)
d)17-(丙-1-炔基)-11β,19-〔4-(2-三甲基甲硅烷基乙炔基)-邻-亚苯基〕-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据c)制得1克酮,用类似例2b)的方式;用丙炔处理。在氧化铝上(中性III级)色谱法分析后,离析得到956毫克白色泡沫状产物。
IR(KBr):2250cm-1芳醇盐中的三键
2235cm-1三键
e)17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(2-三甲基甲硅烷基乙炔基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
依据d)制得900毫克化合物,用类似例1c)方式分裂成相应的4-烯-3-酮化合物。在硅胶上色谱法分析后,离析得到471毫克白色泡沫状标题化合物。
〔α〕D 22=59°(CHCL3;C=0.505)
实施例22
17-(丙-1-炔基)-17β-羟基-11β,19-(4-乙炔基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)11β,19-(4-乙炔基-邻-亚苯基)-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
依据例21c)所制得的1.5克酮溶于26毫升无水甲醇中,并和1.1克无水碳酸钾混合。在惰性气氛中,将反应混合物搅拌3小时,然后倒入饱和氯化钠溶液中。用二氯甲烷萃取水相数次。用饱和氯化钠溶液洗涤合并的有机相,在硫酸钠上干燥并在真空中浓缩。粗产品(1.31克)质量足以适用于进行后面的反应。在氧化铝上(中性III级)对100毫克粗产品色谱法分析,得到67毫克白色泡沫状纯净的标题化合物。
b)17-(丙-1-炔基)-11β,19-(4-乙炔基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇。
依据a)所得1.2克产品,用类似例2b)的方式使其与丙炔反应。在氧化铝上(中性,III级)色谱法分析后,离析得到1.12克上面所述的白色泡沫状化合物。
IR(KBr):2110cm-1芳醇盐的三键
2235cm-1三键
c)17-(丙-1-炔基)-17β-羟基-11β,19-(4-乙炔基-邻-亚苯基)-4-雄(甾)烯-3-酮
依据b)制得1.1克物质,用类似例1c)的方法使其转化成相应的4-烯-3-酮化合物。在硅胶上色谱法分析后,离析得到612毫克白色泡沫状标题化合物。
〔α〕D 22=41°(CHCL3;C=0.5)
实施例23
17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(2-三甲基甲硅烷基乙基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
a)11β,19-〔4-(2-三甲基甲硅烷基乙基)-邻-亚苯基〕-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
依据例21c)制得1克酮,溶于10毫升无水乙醇中,用100毫克碳上附着钯(10%)作催化剂,在常压下进行加氢反应。反应吸收二当量氢后,在硅藻土上抽滤反应混合物。用醋酸酯洗涤过滤后的剩余物并且在真空中浓缩滤液。在氧化铝上(中性,III级)色谱法分析浓缩后残渣,离析得到884毫克白色泡沫状上述的化合物。
b)17-(丙-1-炔基)-11β,19-〔4-(2-三甲基甲硅烷基乙基)-邻-亚苯基〕-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇。
依据a)制得850毫克酮,用类似例2b)的方法使其与丙炔反应。在氧化铝上(中性,III级)色谱法分析后,离析得到845毫克白色泡沫状上面所述的化合物。
IR(KBr):2235cm-1三键
c)17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(2-三甲基甲硅烷基乙基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
依据b)制得800毫克物质,用类似例1c)的方法,使其转化成相应的4-烯-3-酮化合物。在硅胶上色谱法分析后,离析得到512毫克白色泡沫状的上面所述化合物。
〔α〕D 22=23°(CHCL3;C=0.505)
实施例24
17-(丙-1-炔基)-17β-羟基-11β,19-(4-乙基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)11β,19-(4-乙基-邻-亚苯基)-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
依据例22a)所得到的4克乙酰基化合物,用类似例23a)的方法,使其加氢生成相应的乙基化合物。在氧化铝上(中性,III级)色谱法分析后,离析得到3.63克白色泡沫状的上面化合物。
b)17-(丙-1-炔基)-11β,19-(4-乙基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据a)制得1.5克化合物,用类似例2b)的方法,使其与丙炔反应。在氧化铝上(中性,III级)色谱法分析后,得到1.43克上面所述的白色泡沫状化合物。
IR(KBr):2240cm-1三键
c)17-(丙-1-炔基)-17β-羟基-11β,19-(4-乙基-邻-亚苯基)-4-雄(甾)烯-3-酮
依据b)所得到的1.3克物质,用类似例1c)的方式可以使其转化成相应的4-烯-3-酮化合物。在硅胶上色谱法分析后,得到879毫克上面所述的白色泡沫状化合物。
〔α〕D 22=18°(CHCL3;C=0.5)
FP.=283-285℃(醋酸酯)
实施例25
17-(3-羟基-丙-1(Z)-烯基)-17β-羟基-11β,19-(4-乙基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)17-〔3-(四氢吡喃-2-基氧)-丙-1-炔基〕-11β,19-(4-乙基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据例24a)制得2克乙基化合物,用类似例5a)说明的方法,使其与3-(四氢吡喃-2-基氧)-丙炔反应。在氧化铝上色谱法分析后,离析得到2.29克白色泡沫状的标题化合物。
IR(KBr):2240cm-1三键
b)17-〔3-(四氢吡喃-2-基氧)-丙-1(Z)-烯基〕-11β,19-(4-乙基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据a)所得2.2克乙酰基化合物,用类似例5b)的方法,使其氢化成Z-烯烃。色谱法分析后(中性,III级),离析得到1.95克白色泡沫状的标题化合物
c)17-(3-羟基-丙-1(Z)-烯基)-17β-羟基-11β,19-(4-乙基-邻-亚苯基)-4-雄(甾)烯-3-酮。
依据b)制得1.9克化合物,用类似例1c)的方法,使其分裂成相应的4-烯-3-酮化合物。在硅胶上色谱法分析后,离析得到706毫克上面提到的白色泡沫状标题化合物。
〔α〕D 22=62°(CHCL3;C=0.505)
FP.=127-129℃(醋酸酯)
实施例26
17-(丙-1-炔基)-17β-羟基-11β,19-(4-乙烯基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)11β,19-(4-羟基-邻-亚苯基)-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
依据例4)制得20克11β,19-(4-甲氧基-邻-亚苯基)-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮,用类似例18a)的方法,使其转化成相应的苯酚。离析得到16.8克粗产品。其产品纯度足以适于进行后面的反应。在氧化铝上色谱法分析500毫克粗产品,得到412毫克标题化合物。
b)11β,19-(4-三氟甲基磺酰基氧-邻-亚苯基)-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)雄(甾)烷-17-酮。
依据a)所得的16.3克酚,用类似例19a)的方法,使其转化成相应的三氟甲烷磺酸盐。在硅胶上色谱法分析后,离析得到15.1克白色泡沫状的标题化合物。
IR(KBr):1740cm-1五环酮
1215和1420cm-1三氟甲烷磺酸盐
c)11β,19-(4-乙烯基-邻-亚苄基)-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
依据b)制得的1.5克物质溶于18毫升无水二甲基甲酰胺中,然后在惰性气体下用146毫克四个三苯膦钯和207毫克氯化锂处理。搅拌5分钟后,用0.89毫升三正丁基乙烯基锡处理反应混合物并且把它加热到110℃。一小时后,反应混合物冷却到室温,用醋酸酯稀释。在硅胶上抽滤,用饱和的氯化钠液洗涤滤液。在硫酸钠上干燥有机相并且在真空中浓缩。在氧化铝上(中性,III级)对剩余物色谱分析,得到1.1克白色泡沫状标题化合物。
d)17-(丙-1-炔基)-11β,19-(4-乙烯基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇。
依据例a)制得1克酮,用类似例2b)的方法使其与丙炔反应。在氧化铝上(中性,III级)色谱法分析后,离析得到912毫克白色泡沫状的标题化合物。
IR(KBr):2240cm-1三键
e)17-(丙-1-炔基)-17β-羟基-11β,19-(4-乙烯基-邻-亚苯基)-4-雄(甾)烯-3-酮
依据d)所得到的850克化合物,用类似例1e)的方法,使其转化或相应的4-烯-3-酮化合物。在硅胶上色谱法分析后,离析得到485毫克白色泡沫状的标题化合物。
〔α〕D 22=50°(CHCL3;C=0.505)
FP。=243-245℃(二异丙醚)
实施例27
17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(2-丙烯基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
a)11β,19-〔4-(2-丙烯基)-邻-亚苯基〕-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)-17-酮
依据26b)制得1.5克物质,用类似例26c)的方法,使其与0.36毫升四烯丙基锡反应。在硅胶上色谱法分析后,离析得到1.06克白色泡沫状的标题化合物。
b)17-(丙-1-炔基)-11β,19-〔4-(2-丙烯基)-邻-亚苯基〕-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-酮
依据a)所得的1克物质,用类似例2b)的方法,使其与丙炔反应。在氧化铝上(中性,III级)色谱法分析后,离析得到942毫克白色泡沫状的标题化合物。
IR(KBr):2240cm-1三键
c)17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(2-丙烯基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
依据b)制得900毫克物质,用类似例1c)的方法,使其转化成相应的4-烯-3-酮化合物。在硅胶上色谱分析,得到397毫克白色泡沫状的标题化合物。
〔α)D 22=18°(CHCL3;C=0.5)
FP.=275-277℃(二氯甲烷)
实施例28
17β-羟基-11β,19-(4-乙酰基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)11β,19-(4-乙酰基-邻-亚苯基)-8,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据21a)制得30克三氟甲烷磺酸盐,用类似例26c)的方法,用22.06克1-乙氧基-乙烯基-三丁基锡处理。在硅胶上色谱分析后,离析得到18.75克白色泡沫状的标题化合物。
FP。=179-181℃(二异丙醚)
〔α〕D 22=146°(CHCL3;C=0.5)
b)17β-羟基-11β,19-(4-乙酰基-邻-亚苯基)-4-雄(甾)烯-3-酮
依据a)所得的18克物质,用类似例1c)的方法使其转化为相应的4-烯-3-酮化合物。在硅胶上色谱分析后,离析得到10.8克浅黄色泡沫状的标题化合物。
FP.=135-138℃(醋酸酯/己烷)
实施例29
17-(丙-1-炔基)-17β-羟基-11β,19-(4-乙酰基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)11β,19-{4-〔1,1-(2,2-二甲基三亚甲基二氧)-乙基〕-邻-亚苯基}-3,3-(2,2-二甲基三亚甲基二氧)-5-雄(甾)烯-17β-醇
依据28b)所得的10克物质,溶于250毫升无水甲苯中,依次用25.7克1,3-二甲基丙二醇和1.86克对甲苯磺酸吡啶鎓处理。反应混合物在迴流下加热4小时同时用共沸法去除产生的水。将混合物冷却至室温,倒入冰冷的5%氢氧化钠水溶液中。用醋酸酯萃取水相,用饱和的氯化钠溶液洗涤合并的有机相,在硫酸钠上干燥和在真空中浓缩。在氧化铝上(中性,III级)对剩余物色谱法分析,得到10.7克白色泡沫状的标题化合物。
b)11β,19-{4-〔1,1-(2,2-二甲基三亚甲基二氧)乙基〕-邻-亚苯基}-3,3-(2,2-二甲基三亚甲基二氧)-5-雄(甾)烯-17-酮
依据a)所得的10.5克物质,用类似例2a)的方法,使其转化成17-酮化合物。离析得到10.2克粗产品,其纯度足以适于进行后面的反应。在氧化铝上(中性,III级)将500毫克产品色谱分析,得到443毫克白色泡沫状的标题化合物。
〔α〕D 22=43°(CHCL3;C=0.5)
FP.=244-266℃(醋酸酯)
c)17-(丙-1-炔基)-11β,19-{4-〔1,1-(2,2-二甲基三亚甲基二氧)-乙基〕-邻-亚苯基}-3,3-(2,2-二甲基三亚甲基二氧)-5-雄(甾)烯-17β-醇
依据b)所得的1.5克粗产品,用类似例2b)的方法,使其与丙炔反应。在氧化铝上(中性,III级),用醋酸酯/己烷的混合物色谱法分析后,得到1.35克白色泡沫状的标题化合物。
IR(KBr):2240cm-1三键
d)17-(丙-1-炔基)-17β-羟基-11β,19-(4-乙酰基-邻-亚苯基)-4-雄(甾)烯-3酮
依据c)制得1.3克物质,用类似例1c)的方法分裂成相应的4-烯-3-酮化合物。在硅胶上色谱法分析后,离析得到747毫克浅黄色泡沫状的标题化合物。
〔α〕D 22=36°(CHCL3;C=0.5)
FP.=186-187℃(醋酸酯)
实施例30
17-(丙-2-炔基)-17β-羟基-11β,19-(4-乙酰基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)17-(3-三甲基甲硅烷基丙-2-炔基)-11β,.19-{4-〔1,1-(2,2-二甲基三亚甲基二氧)-乙基〕-邻-亚苯基}-3,3-(2,2-二甲基三亚甲基二氧-5-雄(甾)烯-17β-醇
依据例29b)制得的1.5克化合物,用类似例5a)的方法,使其与2.3毫升1-三甲基甲硅烷基-丙-1-炔反应。在氧化铝上(中性,III级)色谱法分析后,离析得到1.31克白色泡沫状的标题化合物。
IR(KBr):2170cm-1三键
b)17-(丙-2-炔基)-17β-羟基-11β,19-(4-乙酰基-邻-亚苯基)-4-雄(甾)烯-3-酮
依据a)制得的1.2克物质,在类似例1c)叙述的条件下,使其转化成相应的4-烯-3-酮化合物。在硅胶上色谱法分析后,离析得到547毫克白色泡沫状的标题化合物。
〔α〕D 22=91°(CHCL3;C=0.5)
FP.=257-259℃
实施例31
17-(丙-1-炔基)-17β-羟基-11β,19-(4-乙酰基-邻-亚苯基)-4,15-雄(甾)二烯-3-酮
a)11β,19-{4-〔1,1-(2,2-二甲基三亚甲基二氧)-乙基〕-邻-亚苯基}-3,3-(2,2-二甲基三亚甲基二氧)-5,15-雄(甾)二烯-17-酮
依据例29b)制得的1.89克化合物溶于20毫升无水四氢呋喃中,在0℃,惰性气氛下滴加到在40毫升无水四氢呋喃中含9.9毫摩尔的二异丙酰胺锂的溶液中。随后将三甲基氯硅烷(2.39毫升)滴加到该反应混合物中。搅拌30分钟后,把反应溶液倒入冰冷的饱和碳酸氢钠溶液中。然后,用醋酸酯萃取水相,用饱和氯化铵水溶液洗涤有机相。在硫酸钠上干燥后,在真空中浓缩有机相。在粗制阶段离析得到浅黄色泡沫状的1.96克17-三甲基甲硅烷基氧-11β,19-{4-〔1,1-(2,2-二甲基三亚甲基二氧)-乙基〕-邻-亚苯基}-3,3-(2,2-二甲基三亚甲基二氧)-5,16-雄(甾)二烯。粗产品悬浮在23毫升的无水乙腈中,用1克醋酸钯(II)进行处理。在室温搅拌2小时后,在硅藻土上抽滤反应混合物。用醋酸酯洗涤过滤后的剩余物,在真空中浓缩滤液。在氧化铝上(中性,III级),色谱法分析浓缩后剩余物,得到1.33克白色泡沫状的标题化合物。
IR(KBr):1710cm-1不饱和五环酮
b)17-(丙-1-炔基)-11β,19-{4-〔1,1-(2,2-二甲基三亚甲基二氧)-乙基〕-邻-亚苯基}-3,3-(2,2-二甲基三亚甲基二氧)-5,15-雄(甾)-烯-17β-醇
依据a)制得的1.3克物质,用类似例2b)的方法,使其与丙炔反应。在氧化铝上(中性,III级)色谱法分析后,离析得到1.23克白色泡沫状的标题化合物。
IR(KBr):2230cm-1三键
c)17-(丙-1-炔基)-17β-羟基-11β,19-(4-乙酰基-邻-亚苯基)-4,15-雄(甾)二烯-3-酮
依据b)制得的1.1克物质,用类似例1c)的方法使其分裂成相应的4-烯-3-酮化合物。在硅胶上色谱分析后,离析得到617毫克浅黄色泡沫状的标题化合物。
〔α〕D 22=114°(CHCL3;C=0.5)
FP.=189-191℃(醋酸酯)
实施例32
17-甲氧基甲基-17β-羟基-11β,19-(4-乙酰基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)11β,19-{4-〔1,1-(2,2-二甲基三亚甲基二氧)-乙基〕-邻-亚苯基}-3,3-(2,2-二甲基三亚甲基二氧)-5-雄(甾)烯-〔17(β-1′)-螺-3′〕-环氧乙烷
依据例29b)所制得的4克化合物,用类似例14a)的方法,使其与7.13克碘化三甲硫鎓反应。在氧化铝上(中性,III级)色谱分析后,离析得到3.76克白色泡沫状标题化合物。
b)17-甲氧基甲基-11β,19-{4-〔1,1-(2,2-二甲基三亚甲基二氧)-乙基〕-邻-亚苯基}-3,3-(2,2-二甲基三亚甲基二氧)-5-雄(甾)烯-17β-醇
依据a)制得的1.8克物质,用类似例14b)的方法,使其与甲醇钠进行反应。在氧化铝上(中性,III级)色谱分析后,离析得到1.55克白色泡沫状的标题化合物。
c)17-甲氧基甲基-17β-羟基-11β,19-(4-乙酰基-邻-亚苯基)-4-雄(甾)烯-3-酮
依据b)所得到的1.5克物质,用类似例1c)的方法,使其分裂成相应的4-烯-3-酮化合物。在硅胶上色谱分析后,离析得到561毫克浅黄色泡沫状的标题化合物。
〔α〕D 22=76°(CHCL3;C=0.5)
实施例33
17-氰甲基-17β-羟基-11β,19-(4-乙酰基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)17-氰甲基-11β,19-{4-〔1,1-(2,2-二甲基三亚甲基二氧)-乙基〕-邻-亚苯基}-3,3-(2,2-二甲基三亚甲基二氧)-5-雄(甾)烯-17β-醇
依据例32a)所得的1.8克物质,用类似例6b)的方法,使其与氰化钾反应。在氧化铝上(中性,III级)色谱分析后。离析得到1.67克白色泡沫状的标题化合物。
IR(KBr):2250cm-1 C≡N三键
b)17-氰甲基-17β-羟基-11β,19-(4-乙酰基-邻-亚苯基)-4-雄(甾)烯-3-酮
依据a)所得的1.5克物质,用类似例1c)的方法,使其分裂成相应的4-烯-3-酮化合物。在硅胶上色谱分析后,离析得到732毫克白色泡沫状的标题化合物。
〔α〕D 22=83°(CHCL3;C=0.5)
FP.=184-185℃(二氯甲烷)
实施例34
17-(丙-1-炔基)-17β-羟基-11β,19-(4-异丙烯基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)11β,19-( 4-异丙烯基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
在惰性气氛中,将1.94克氢化钠在20毫升无水的二甲基亚砜中70℃温热,直到检测不出多余气体产生为止。将溶液冷却到0℃,把61毫升无水二甲亚砜中溶解溴甲基三苯膦的溶液滴状加入。在室温搅拌后,把依据例28a)制得的10.3克化合物溶于10毫升无水二甲基亚砜中,滴入,并将反应混合物搅拌3小时。随后将反应混合物倒入饱和的冷碳酸氢钠溶液中。用醋酸酯萃取水相,用饱和氯化钠溶液洗涤有机相。在硫酸钠上干燥合并的有机相并在真空中浓缩。在氧化铝上(中性,III级)色谱分析残余物,得到8.3克白色泡沫状的标题化合物。
FP。=155-157℃(二异丙醚)
b)11β,19-(4-异丙烯基-邻-亚苯基)-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
依据a)制得的8.1克物质,用类似例2a)的方法,使其用三氧化铬氧化。在氧化铝上(中性,III级)色谱分析后,离析得到7.8克白色泡沫状的标题化合物。
FP.=238-240℃(二异丙基醚)
c)17-(丙-1-炔基)-11β,19-(4-异丙烯基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇。
依据b)所得到的1.5克物质,用类似例2b)的方法,使其与丙炔反应。在氧化铝上(中性,III级)色谱分析后,离析得到1.34克白色泡沫状的标题化合物。
IR(KBr):2240cm-1三键
d)17-(丙-1-炔基)-17β-羟基-11β,19-(4-异丙烯基-邻-亚苯基)-4-雄(甾)烯-3-酮
依据c)所得的1.3克化合物,用类似例1c)的方法,使其转化成4-烯-3-酮化合物。在硅胶上色谱分析后,离析得到706毫克白色泡沫状的标题化合物。
〔α〕D 22=41°(CHCL3;C=0.5)
FP.=247-250℃(二异丙基醚)
离析作为副产品的白色泡沫状化合物17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(1-甲基-羟基乙基)-邻-亚苯基〕-4-雄(甾)烯-3-酮(354毫克)
〔α〕D 22=17°(CHCL3;C=0.5)
FP.=222-224℃
实施例35
17-(丙-1-炔基)-17β-(羟基-11β,19-(4-异丙基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)11β,19-(4-异丙基-邻-亚苯基)-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
依据例34b)所得到的2克物质,如例23a)中叙述的方法,钯作为催化剂进行加氢反应,但反应只进行到吸收-当量的氢。在氧化铝上(中性,III级)色谱分析后,离析得到1.83克白色泡沫状的标题化合物。
b)17-(丙-1-炔基)-11β,19-(4-异丙烯基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烯-5α,17β-二醇
根据a)制得的1.8克物质,用类似例2b)的方法,使其与丙炔反应。在氧化铝上(中性,III级)色谱分析后,离析得到1.81克白色泡沫状的标题化合物。
c)17-(丙-1-炔基)-17β-羟基-11β,19-(4-异丙烯基-邻-亚苯基)-4-雄(甾)烯-3-酮
根据b)制得的1.7克物质,用类似例1c)的方法,使其分裂成4-烯-3-酮化合物。在硅胶上色谱分析后,离析得到932毫克白色泡沫状的标题化合物。
〔α〕D 22=21°(CHCL3;C=0.505)
FP.=240-243℃(醋酸酯)
实施例36
17-(3-羟基丙-1(Z)-丙烯基)-17β-羟基-11β,19-(4-异丙烯基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)17-〔3-(四氢吡喃-2-基氧)-丙-1-炔基〕-11β,19-(4-异丙烯基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据例34b)所得的2克物质,用类似例5a)的方法,使其与3-(四氢吡喃-2-基氧)-丙炔反应。在氧化铝上(中性,III级)色谱分析后,离析得到2.1克白色泡沫状的标题化合物。
b)17-〔3-(四氢呋喃-2-基氧)-丙-1(Z)-烯基〕-11β,19-(4-异丙烯基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据a)所得的2克化合物,用类似例5b)的方法,使其用Lindlar′s催化剂进行加氢反应。在氧化铝上(中性,III级)色谱分析后,离析得到1.78克白色泡沫状的标题化合物。
c)17-(3-羟基丙-1(Z)-烯基)-17β-羟基-11β,19-(4-异丙烯基-邻-亚苯基)-4-雄(甾)烯-3-酮
依据b)所得的1.7克化合物,用类似例1c)的方法,使其分裂成4-烯-3-酮化合物。在硅胶上色谱分析后,离析得到567毫克白色泡沫状的标题化合物。
〔α〕D 22=79°(CHCL3;C=0.5)
FP.=143-145℃(醋酸酯)
离析作为副产品的白色泡沫状化合物为178毫克17-(3-羟基丙-1(Z)-烯基)-17β-羟基-11β,19-〔4-(1-甲基-1-羟基乙基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
〔α〕D 22=61°(CHCL3;C=0.5)
FP.=208-211℃(醋酸酯)
实施例37
17-(4-羟基丁-1(Z)-烯基)-17β-羟基-11β,19(4-异丙烯基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)17-(4-羟基丁-1-炔基)-11β,19-(4-异丙烯基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据例34b)所得的2克物质,溶于60毫升无水四氢呋喃中,在惰性气氛中依次用2.27毫升正丁炔-4-醇和4.09克的甲醇钠处理。在加料和随后14小时搅拌期间,反应混合物保持在0℃。把反应混合物倒入水中并且用醋酸酯萃取水相。用饱和的氯化钠溶液洗涤合并的有机相,在硫酸钠上干燥并且在真空中浓缩。在氧化铝上(中性,III级)色谱分析浓缩后的剩余物,离析得到1.2克白色泡沫状的标题化合物。
红外(KBr):2240cm-1三键
b)17-(4-羟基丁-1(Z)-烯基)-11β,19-(4-异丙烯基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇。
依据a)所得的1克物质,用类似例5b)的方法,使其用Lindlar′催化剂加氢反应。在氧化铝上(中性,III级)色谱分析后,得到836毫克白色泡沫状的标题化合物。
c)17-(4-羟基丁-1(Z)-烯基)-17β-羟基-11β,19-(4-异丙烯基-邻-亚苯基)-4-雄(甾)烯-3-酮
依据b)所得的800毫克物质,用类似例1c)的方法,使其分裂成4-烯-3-酮化合物。在硅胶上色谱分析后,离析得到306毫克白色泡沫状的标题化合物。
〔α〕D 22=78°(CHCL3;C=0.515)
实施例38
17-(丙-1-炔基)-17β-羟基-11β,19-14-二乙氧基磷酰基-邻-亚苯基)-4-雄(甾)烯-3-酮
a)11β,19-(4-二乙氧基磷酰基-邻-亚苯基)-5α-羟基-3.3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
依据例26b)所得的1.5克三氟甲烷磺酸盐,溶于10毫升无水三乙胺中。在惰性气氛中用122毫克四个三苯膦钯和0.46毫升亚磷酸二乙酯处理。随后反应混合物迴流加热1.5小时。再加100毫克四个三苯膦钯和0.46毫升亚磷酸二乙酯后,反应混合物迴流加热1.5小时。反应混合物冷却到室温并且在真空中浓缩。在硅胶上色谱分析浓缩后的剩余物,得到1.05克白色泡沫状的标题化合物。
b)17-(丙-1-炔基)-11β,19-(4-二乙氧基磷酰基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据a)制得的1克物质,用类似例2b)的方法,使其与丙炔反应。在氧化铝上(中性,III级)色谱分析后,得到832毫克白色泡沫状的标题化合物。
IR(KBr):2240cm-1三键
c)17-(丙-1-炔基)-17β-羟基-11β,19-(4-二乙氧基磷酰基-邻-亚苯基)-4-雄(甾)烯-3-酮
依据b)制得的800毫克物质,用类似例1c)的方式,使其分裂成4-烯-3-酮化合物。在硅胶上色谱分析后,离析得到440毫克白色泡沫状的标题化合物。
〔α〕D 22=17°(CHCL3;C=0.5)
实施例39
17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(2-噻吩基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
a)11β,19-〔4-(2-噻吩基)-邻-亚苯基〕-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据例21a)制得的1.26克物质,溶于36毫升无水二噁烷中。在惰性气氛中,用3.04毫升六-正-丁基-二锡和254毫克氯化锂和100毫克四个三苯膦钯处理。随后在加1.94毫升2-溴(代)噻吩之前,将反应混合物加热到110℃,维持温度1小时。然后在110℃再搅拌18小时。将反应混合物冷却到室温并且在硅藻土上过滤。滤液过滤后,在硅胶上色谱分析剩余物,得到545毫克浅黄色泡沫状的标题化合物。
作为这类偶合的例子,离析以下锡化合物。
α)11β,19-(4-三-正-丁基甲锡烷基-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据例21a)制得的1.26克物质,用类似a)叙述的条件,使其与3.04毫升六-正-丁基二锡反应。在110℃加热1小时后,以一般方式进一步处理反应混合物。在硅胶上色谱分析,得到625毫克白色泡沫状的标题化合物。
〔α〕D 22=25°(CHCL3;C=0.5)
FP.=137-139℃(二异丙醚)
b)11β,19-〔4-(2-噻吩基)-邻-亚苯基〕-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
将400毫克N-氯代琥珀酰亚胺置于5毫升0℃无水的二氯甲烷中。把0.3毫升二甲硫滴加完后,在0℃搅拌混合物30分钟。随后依据a)所得的510毫克物质溶于5毫升无水二氯甲烷中,慢慢地滴加。排斥潮气搅拌2小时后,将0.6毫升三乙胺滴加到反应混合物中,然后倒入冰水中。用二氯甲烷萃取有机相并且用饱和氯化钠溶液洗涤有机相。随后在硫酸钠上干燥并且在真空中浓缩。在氧化铝上(中性,III级)色谱分析剩余物后,离析得出387毫克浅黄色泡沫状的标题化合物。
IR(KBr):1740cm-1五环酮
c)17-(丙-1-炔基)-11β,19-〔4-(2-噻吩基)-邻-亚苯基〕-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据b)所得的350毫克物质,用类似例2b)的方法,使其与丙炔进行反应。在氧化铝上(中性,III级)色谱分析后,离析得到343毫克浅黄色泡沫状的标题化合物。
IR(KBr):2240cm-1三键
d)17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(2-噻吩基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
依据d)所得的320毫克化合物,用类似例1c)的方法,使其分裂成4-烯-3-酮化合物。在硅胶上色谱分析后,离析出234毫克浅黄色泡沫状的标题化合物。
〔α〕=65°(CHCL3;C=0.5)
实施例40
17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(3-噻吩基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
a)11β,19-〔4-(3-噻吩基)-邻-亚苯基〕-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
依据例26b)所得的1.26克物质,用类似例39a)叙述的条件,使其与2毫升3-溴(代)噻吩反应。在氧化铝上(中性,III级)色谱分析后,离析得到546毫克浅黄色泡沫状的物质。
b)17-(丙-1-炔基)-11β,19-〔4-(3-噻吩基)-邻-亚苯基〕-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据a)制得的520毫克物质,用类似例2b)的方法,使其与丙炔反应。在氧化铝上(中性,III级)色谱分析后,离析出462毫克浅黄色泡沫状的标题化合物。
IR(KBr):2250cm-1三键
c)17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(3-噻吩基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
依据b)所得的410毫克物质,用类似例1c)的方法,使其分裂成4-烯-3-酮化合物。在硅胶上色谱分析后,离析得到287毫克浅黄色泡沫状的标题化合物。
〔α〕D 22=61°(CHCL3;C=0.51)
实施例41
17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(3-呋喃基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
a)11β,19-〔4-(3-呋喃基)-邻-亚苯基〕-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
依据例26b)制备的1.26克物质,用类似例39a)的方法,使其与1.8毫升3-溴(代)呋喃进行反应。在硅胶上色谱分析后,离析得到660毫克白色泡沫状的标题化合物。
FP.=240-243℃(醋酸酯)
b)17-(丙-1-炔基)-11β,19-〔4-(3-呋喃基)-邻-亚苯基〕-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据a)所得的630毫克物质,用类似例2b)的方法,使其与丙炔反应。在氧化铝上(中性,III级)色谱分析后,离析得到615毫克白色泡沫状的标题化合物。
IR(KBr):2240cm-1三键
c)17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(3-呋喃基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
依据b)所得的590毫克物质,用类似例1c)的方法,使其反应生成4-烯-3-酮化合物。在硅胶上色谱分析后,离析得到289毫克白色泡沫状的标题化合物。
〔α〕D 22=49°(CHCL3;C=0.51)
实施例42
17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(3-甲氧基苯基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
a)11β,19-〔4-(3-甲氧基苯基)-邻-亚苯基〕-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
依据例26b)制得的1.26克物质,用类似例39a)的方法,使其与2.53毫升3-溴(代)苯甲醚反应。在硅胶上色谱分析后,离析得到685毫克白色泡沫状的标题化合物。
b)17-(丙-1-炔基)-11β,19-〔4-(3-甲氧基苯基)-邻-亚苯基〕-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据a)制得的650毫克物质,用类似例2b)的方法,使其与与丙炔反应。在氧化铝上(中性,III级)色谱分析后,离析得到635毫克白色泡沫状的标题化合物。
IR(KBr):2230cm-1三键
c)17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(3-甲氧基苯基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
依据b)制备的600毫克物质,用类似例1c)所叙述的条件,使其转化成4-烯-3-酮化合物。在硅胶上色谱分析后,离析得到366毫克白色泡沫状的标题化合物。
〔α〕D 22=66°(CHCL3;C=0.5)
FP.=158-162℃(乙酸酯/己烷)
实施例43
17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(4-甲氧基苯基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
a)11β,19-〔4-(4-甲氧基苯基)-邻-亚苯基〕-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
依据例26b)制得的1.26克物质,用类似例39a)的方法,使其与2.53毫升4-溴(代)苯甲醚反应。在硅胶上色谱分析后,离析得到522毫克白色泡沫状的标题化合物。
FP.=171-173℃(醋酸酯)
〔α〕D 22=48°(CHCL3;C=0.5)
b)17-(丙-1-炔基)-11β,19-〔4-(4-甲氧基苯基)-邻-亚苯基〕-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据a)制得的500毫克物质,用类似例2b)的方法,使其与丙炔反应。在氧化铝上(中性,III级)色谱分析后,离析得到498毫克白色泡沫状的标题化合物。
IR(KBr):2240cm-1三键
c)17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(4-甲氧基苯基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
依据b)制得的450毫克物质,用类似例1c)的方法,使其转化成4-烯-3-酮化合物。在硅胶上色谱分析后,离析得到276毫克白色泡沫状的标题化合物。
〔α〕D 22=70°(CHCL3;C=0.505)
FP.=165-169℃(醋酸酯/己烷)
实施例44
17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(2-甲氧基苯基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
a)11β,19-〔4-(2-甲氧基苯基)-邻-亚苯基-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮
依据例26b)所得的1.26克物质,用类似例39a)的方法,使其与2.53毫升2-溴(代)苯甲醚反应。在硅胶上色谱分析后,离析得到448毫克白色泡沫状的标题化合物。
b)17-(丙-1-炔基)-11β,19-〔4-(2-甲氧基苯基)-邻-亚苯基〕-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇。
依据a)制得的410毫克物质,用类似例2b)的方法,使其与丙炔反应。在氧化铝上(中性,III级)色谱分析后,离析得到405毫克白色泡沫状的标题化合物。
IR(KBr):2240cm-1三键
c)17-(丙-1-炔基)-17β-羟基-11β,19-〔4-(2-甲氧基苯基)-邻-亚苯基〕-4-雄(甾)烯-3-酮
依据b)制得的380毫克物质,用类似例1c)的方法,使其转化成4-烯-3-酮化合物。在硅胶上色谱分析后,离析得到205毫克白色泡沫状的标题化合物。
〔α〕D 22=49°(CHCL3;C=0.51)
实施例45
17-(丙-1-炔基)-17β-羟基-11β,19-(4,5-亚甲基二氧)-邻-亚苯基)-4-雄(甾)烯-3-酮
α)19-(2-氯-4,5-亚甲基二氧苯基)-3,3-(2,2-二甲基三亚甲基二氧)-9(11)-雄(甾)烯-5α,17β-二醇
从10克5α,10α-桥氧基-3,3-(2,2-二甲基三亚甲基二氧)-9(11)-雌(甾)烯-17β-醇,用类似例1a)的方法,使其与6-氯代-3,4-亚甲二氧苯甲基氯反应,得到10.3克19-(2-氯-4,5-亚甲基二氧苯基)-3,3-(2,2-二甲基三亚甲基二氧)-9(11)-雄(甾)烯-5α,17β-二醇
b)11β,19-(4,5-亚甲基二氧-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇。
根据a)所得的10克化合物,用类似例1b)的方法,生成5.9克白色泡沫状的11β,19-(4,5-亚甲基二氧-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇。
c)11β,19-(4,5-亚甲基二氧-邻-亚苯基)-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮。
依据b)所得的5.5克化合物,用类似例2a)的方法,使其转化成相应的酮化合物,得到4.2克白色泡沫状的11β,19-(4,5-亚甲基二氧-邻-亚苯基)-5α-羟基-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-17-酮。
〔α〕D 22=+45°(CHCL3;C=0.525)
FP.=219-222℃(醋酸乙酯)
d)17-(丙-1-炔基)-11β,19-(4,5-亚甲基二氧-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇
依据c)所得的4克化合物,用类似例2b)的方法,使其转化成相应的17α-丙炔基化合物。色谱分析后,离析得到3.5克白色泡沫状的17-(丙-1-炔基)-11β,19-(4,5-亚甲基二氧-邻-亚苯基)-3,3-(2,2-二甲基三亚甲基二氧)-雄(甾)烷-5α,17β-二醇。
IR(KBr):2230cm-1三键
e)17-(丙-1-炔基)-17β-羟基-11β,19-(4,5-亚甲基二氧-邻-亚苯基)-4-雄(甾)烯-3-酮
依据d)所得的3克化合物,用类似例1c)的方法,使其转化成相应的4-烯-3-酮化合物,离析得到1.36克17-(丙-1-炔基)-17β-羟基-11β,19-(4,5-亚甲基二氧-邻-亚苯基)-4-雄(甾)烯-3-酮。
〔α)D 22=+2°(CHCL3;C=0.485)
Claims (14)
1.通式I的19,11β-桥甾族化合物以及它们的药学上可允许的酸加成盐在制药中的应用, 其中:R1代表甲基或乙基,R2代表氢、氯原子、C1-C4-烷基,B和G可相同或不同,分别代表氢原子、C1-C4-烷基,或一起代表碳原子6和7之间的第二个键,
其中:R1代表甲基或乙基,R2代表氢、氯原子、C1-C4-烷基,B和G可相同或不同,分别代表氢原子、C1-C4-烷基,或一起代表碳原子6和7之间的第二个键,
B和R2一起代表亚甲基或亚乙基,
Z代表5元或6元环基,它们可以被取代以及可以不饱和,
V代表可被取代的碳环或杂环芳基,
A环代表:
M和N一起代表第二键或M为氢原子,N为羟基,B、R2、G、R3、D和E为氢原子,以及
X为氧原子、两个氢原子或一个肟基N~OH,
R3和D可相同或不同,分别为氢原子、腈基或C1-C4-烷基,或一起代表亚甲基或亚乙基,
E为氢原子、C1-C4-烷基,
D和E一起代表碳原子1和2之间的第二键,或一起代表亚甲基,或
 或
或
其中R11代表氢原子或C1-C8-烷基。
2.根据权利要求1的应用,其特征在于所述化合物中V代表下式的苯环基团:
其中:
R4和R4′可相同或不同,分别为氢原子、氰基、-OR11-、-S(O)kR11-、N(O)nR11R12-、-O-SO2R13-、P(O)(OR14)2-、SiR3 14-或SnR3 14-基团,
其中k为0、1或2,n为0或1,
R11为氢原子或C1-C8-烷基,
R12为R11、氰基或C1-C10-酰基,
R13为全氟化的C1-C4-烷基,
R14为C1-C4-烷基,或
在-N(O)nR11R12-基团中的R11和R12包括N一起代表5或6元的杂环,在环中也可以含有另外的杂原子N、O或S,
Y和Y′可相同或不同,分别代表直接的键、直链或支链的亚烷基,它可含有一个或多个双键或叁键,碳原子数多至20个。根据具体情况,该亚烷基可被一个或多个氧代、C1-C10-酰氧基、-OR11-、-S(O)kR11-和/或-N(O)nR11R12-基团所取代,或者可以被芳基取代,或
R4-Y和R4′-Y′一起代表具有0至2个氧原子、硫原子和/或NR11基的可被取代的、饱和的、不饱和的或芳族的5元或6元环基团,附加条件是,如果R11为C1-C8的烷基,k和n仅大于0。
3.根据权利要求1的应用,其特征在于所述化合物中V代表下式的具有1至2个N、O或S原子的5元或6元杂环芳族基团:
其中:
R4和R4′以及Y和Y′具有与权利要求2相同的意义。
4.根据权利要求1的应用,其特征在于在所述化合物中Z代表下式环基: 其中:R1的意义同权利要求1,在W上出现的虚线代表可能存在双键,W为CH2-,CH-,CH2CH2-或CHCH2-基R5/R6为-OR7/-C≡C-U
其中:R1的意义同权利要求1,在W上出现的虚线代表可能存在双键,W为CH2-,CH-,CH2CH2-或CHCH2-基R5/R6为-OR7/-C≡C-U 

 -OR7/-(CH2)m-CH2-R9-OR7/-CH=CH(CH2)k-CH2-R9-OR10/-H-OR10/-(CH2)k-C≡C-U
-OR7/-(CH2)m-CH2-R9-OR7/-CH=CH(CH2)k-CH2-R9-OR10/-H-OR10/-(CH2)k-C≡C-U
其中,R7是氢原子或具有1~4个碳原子的酰基,
U是氢原子,在其烷基、酰基部分分别带有1~4个碳原子的烷基、羟烷基、烷氧基烷基、酰氧基烷基、或者卤素原子,
R8为氢原子、羟基、分别带有1~4个碳原子的烷基、O-烷基或O-酰基,
R9为氢原子、羟基或氰基,分别带有1~4个碳原子的O-烷基或O-酰基,
R10为氢原子,分别具有1~10个碳原子的烷基或酰基,
m为0、1、2或3,
k为0、1或2。
5.根据权利要求1的应用,其特征在于在所述化合物中R2、B和G分别为氢原子。
6.根据权利要求1的应用,其特征在于在所述化合物中B和G一起代表第二个键,且R2代表氢原子。
7.根据权利要求2或3的应用,其特征在于在所述化合物中Y和Y’分别为直接的键,以及R4和R4’分别为氢原子。
8.根据权利要求2或3的应用,其特征在于在所述化合物中A和A’分别为直接的键,R4为氢原子,R4’为被两个C1~C8-烷基取代的N原子。
9.根据权利要求2或3的应用,其特征在于在所述化合物中Y和Y’分别为直接的键,R4为氢原子,以及R4’为C1~C8-烷氧基。
10.根据权利要求2或3的应用,其特征在于在所述化合物中Y为直接的键,R4和R4’分别为氢原子,以及Y’是直链或支链的亚烷基,可带有一个或多个双键和/或叁键,碳原子数多至20个,且被氧代或OR11-基取代,其中R11是氢原子或C1~C8-烷基。
11.根据权利要求2或3的应用,其特征在于所述化合物中R4-Y和R4’-Y’一起代表具有0至2个氧原子、硫原子和/或NR11一基的饱和的、不饱和的或芳族的5元或6元环基,其中R11是氢原子或C1~C8-烷基。
12.根据权利要求2或3的应用,其特征在于所述化合物中Y’-R4’是氢原子,Y-R4是乙基、乙烯基、异丙基、异丙烯基、丙-1(Z)-烯基、丙-1(E)-烯基、丙-2-烯基、乙炔基、丙炔基、丙-2-炔基、甲氧基、硫甲基、硫乙基、1-羟乙基或二乙氧磷酰基、被取代或未被取代的碳环或杂环芳基或三氟甲基磺酸酯基。
13.根据权利要求12的应用,其特征在于在所述化合物中芳基是苯基、萘基、2-甲氧苯基、3-甲氧苯基、4-甲氧苯基、2-甲苯基、3--甲苯基、4-甲苯基、2-二甲氨基、3-二甲氨基、4-二甲氨基、呋喃-2-基、呋喃-3-基、噻吩-2-基、噻吩-3-基、吡啶-2-基、吡啶-3-基、吡啶-4-基、噻唑基、咪唑基。
14.根据权利要求3的应用,其特征在于所述化合物中5元或6元杂环是呋喃基、噻吩基、吡啶基、吡唑基、吡咯基、嘧啶基、噁唑基、哒嗪基、吡嗪基。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19873708942 DE3708942A1 (de) | 1987-03-18 | 1987-03-18 | 19,11ss-ueberbrueckte steroide, deren herstellung und diese enthaltende pharmazeutische praeparate |
| DEP3708942.0 | 1987-03-18 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN88102162A Division CN1033033C (zh) | 1987-03-18 | 1988-03-18 | 具有孕甾酮拮抗活性的11β,19桥接甾族化合物的制备方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1135488A CN1135488A (zh) | 1996-11-13 |
| CN1057770C true CN1057770C (zh) | 2000-10-25 |
Family
ID=6323462
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN88102162A Expired - Fee Related CN1033033C (zh) | 1987-03-18 | 1988-03-18 | 具有孕甾酮拮抗活性的11β,19桥接甾族化合物的制备方法 |
| CN96100917A Expired - Fee Related CN1057770C (zh) | 1987-03-18 | 1996-01-17 | 19,11β-桥甾族化合物在制药中的应用 |
| CN96102566A Expired - Fee Related CN1080267C (zh) | 1987-03-18 | 1996-02-01 | 19,11β-桥甾族化合物的中间体的制备 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN88102162A Expired - Fee Related CN1033033C (zh) | 1987-03-18 | 1988-03-18 | 具有孕甾酮拮抗活性的11β,19桥接甾族化合物的制备方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN96102566A Expired - Fee Related CN1080267C (zh) | 1987-03-18 | 1996-02-01 | 19,11β-桥甾族化合物的中间体的制备 |
Country Status (19)
| Country | Link |
|---|---|
| US (1) | US5095129A (zh) |
| EP (1) | EP0283428B1 (zh) |
| JP (1) | JP2588267B2 (zh) |
| CN (3) | CN1033033C (zh) |
| AT (1) | ATE69055T1 (zh) |
| AU (1) | AU628015B2 (zh) |
| CA (1) | CA1326015C (zh) |
| DD (1) | DD273262A5 (zh) |
| DE (2) | DE3708942A1 (zh) |
| ES (1) | ES2040373T3 (zh) |
| FI (1) | FI101883B (zh) |
| GR (1) | GR3003362T3 (zh) |
| HU (2) | HU206368B (zh) |
| IE (1) | IE62480B1 (zh) |
| IL (1) | IL85773A (zh) |
| NZ (1) | NZ223924A (zh) |
| PT (1) | PT87007B (zh) |
| WO (1) | WO1988007051A1 (zh) |
| ZA (1) | ZA881962B (zh) |
Families Citing this family (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5446178A (en) * | 1987-03-18 | 1995-08-29 | Schering Aktiengesellschaft | Process for preparing 19,11β-bridged steroids |
| DE3832303A1 (de) * | 1988-09-20 | 1990-04-12 | Schering Ag | 11ss-phenyl-14ssh-steroide |
| DE3917274A1 (de) * | 1989-05-24 | 1990-11-29 | Schering Ag | 9(alpha)-hydroxy-19, 11(beta)-ueberbrueckte steroide, deren herstellung und diese enthaltende pharmazeutische praeparate |
| DE3921059A1 (de) * | 1989-06-23 | 1991-01-10 | Schering Ag | 11(beta)-aryl-4-estrene, verfahren zu ihrer herstellung sowie deren verwendung als arzneimittel |
| DD289541A5 (de) * | 1989-08-04 | 1991-05-02 | ��@�K@�������������@�K@��������������@��������k�� | Verfahren zur herstellung von 11beta-aryl-16 alpha, 17 alph-cyclohexanoestra-4,9-dienen |
| DE4038128A1 (de) * | 1990-11-27 | 1992-06-04 | Schering Ag | 8-en-19, 11(beta)-ueberbrueckte steroide, deren herstellung und diese enthaltende pharmazeutische praeparate |
| US5439913A (en) * | 1992-05-12 | 1995-08-08 | Schering Aktiengesellschaft | Contraception method using competitive progesterone antagonists and novel compounds useful therein |
| DE4235220A1 (de) * | 1992-10-13 | 1994-06-16 | Schering Ag | Gestagen wirksame 19,11ß-überbrückte 4-Estrene |
| DE4413184A1 (de) * | 1994-04-12 | 1995-10-19 | Schering Ag | 10,11-C¶3¶-überbrückte Steroide |
| DE4413185A1 (de) * | 1994-04-12 | 1995-10-19 | Schering Ag | 11beta,19-überbrückte 13alpha-Alkylsteroide |
| DE4417880A1 (de) * | 1994-05-18 | 1995-11-23 | Schering Ag | 19,11beta-Überbrückte 18-Nor-Steroide, Verfahren zu ihrer Herstellung, diese Steroide enthaltende Arzneimittel sowie deren Verwendung zur Herstellung von Arzneimitteln |
| US5639598A (en) * | 1994-05-19 | 1997-06-17 | The Trustees Of The University Of Pennsylvania | Method and kit for identification of antiviral agents capable of abrogating HIV Vpr-Rip-1 binding interactions |
| US5780220A (en) * | 1994-05-19 | 1998-07-14 | Trustees Of The University Of Pennsylvania | Methods and compositions for inhibiting HIV replication |
| DE4424222C1 (de) * | 1994-07-09 | 1995-09-14 | Metallgesellschaft Ag | Gelöstes Methyllithium enthaltendes Synthesemittel sowie Verfahren zu deren Herstellung |
| DE4434488A1 (de) * | 1994-09-14 | 1996-03-21 | Schering Ag | Steroidester und -amide, Verfahren zu ihrer Herstellung und ihre pharmazeutische Verwendung |
| US5521166A (en) * | 1994-12-19 | 1996-05-28 | Ortho Pharmaceitical Corporation | Antiprogestin cyclophasic hormonal regimen |
| ZA9510926B (en) | 1994-12-23 | 1996-07-03 | Schering Ag | Compounds with progesterone-antagonistic and antiestrogenic action to be used together for female contraception |
| IL118974A (en) * | 1995-08-17 | 2001-09-13 | Akzo Nobel Nv | History 11 - (Transformed Phenyl) - Astra - 4, 9 - Diane, their preparation and pharmaceutical preparations containing them |
| DE19652408C2 (de) * | 1996-12-06 | 2002-04-18 | Schering Ag | Steroidester, Verfahren zu ihrer Herstellung und ihre Verwendung zur Herstellung von Arzneimitteln |
| US6090798A (en) * | 1997-12-19 | 2000-07-18 | Alcon Laboratories, Inc. | Treatment of GLC1A glaucoma with glucocorticoid antagonists |
| GB0000313D0 (en) | 2000-01-10 | 2000-03-01 | Astrazeneca Uk Ltd | Formulation |
| EP3693376A1 (en) | 2006-10-24 | 2020-08-12 | Allergan Pharmaceuticals International Limited | Compositions for suppressing endometrial proliferation |
| TWI539953B (zh) | 2008-04-28 | 2016-07-01 | 瑞波若斯治療學公司 | 用於治療乳癌之組成物和方法 |
| EP2550288A1 (en) | 2010-03-22 | 2013-01-30 | Repros Therapeutics Inc. | Compositions and methods for non-toxic delivery of antiprogestins |
| US11103514B2 (en) | 2010-05-26 | 2021-08-31 | Corcept Therapeutics, Inc. | Treatment of muscular dystrophy |
| CN103555806B (zh) * | 2013-11-16 | 2015-05-13 | 江南大学 | 一种高效利用亚麻刺盘孢霉合成7α-羟基-雄甾烯酮的方法 |
| JP6821582B2 (ja) | 2015-03-02 | 2021-01-27 | コーセプト セラピューティクス, インコーポレイテッド | Acth分泌腫瘍を処置するための糖質コルチコイドレセプターアンタゴニストおよびソマトスタチンアナログの使用 |
| EP3277281B1 (en) | 2015-03-30 | 2022-01-05 | Corcept Therapeutics, Inc. | Use of glucocorticoid receptor antagonists in combination with glucocorticoids to treat adrenal insufficiency |
| WO2017027851A1 (en) | 2015-08-13 | 2017-02-16 | Corcept Therapeutics, Inc. | Method for differentially diagnosing acth-dependent cushing's syndrome |
| WO2017127448A1 (en) | 2016-01-19 | 2017-07-27 | Corcept Therapeutics, Inc. | Differential diagnosis of ectopic cushing's syndrome |
| AU2018244928B2 (en) | 2017-03-31 | 2023-10-19 | Corcept Therapeutics, Inc. | Glucocorticoid receptor modulators to treat cervical cancer |
| US11213526B2 (en) | 2017-06-20 | 2022-01-04 | Corcept Therapeutics, Inc. | Methods of treating neuroepithelial tumors using selective glucocorticoid receptor modulators |
| EP4263807A2 (en) | 2020-12-18 | 2023-10-25 | Instil Bio (Uk) Limited | Processing of tumor infiltrating lymphocytes |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1983003099A1 (en) * | 1982-03-01 | 1983-09-15 | Roussel Uclaf | 3-keto-delta 4,9-19-norsteroids |
| US4447424A (en) * | 1981-01-09 | 1984-05-08 | Roussel Uclaf | Steroid derivatives |
| EP0188396A2 (fr) * | 1985-01-14 | 1986-07-23 | Roussel-Uclaf | Nouveaux stéroides substitués en position 10, leur procédé et les intermédiaires de préparation, leur application comme médicaments, les compositions pharmaceutiques les contenant |
-
1987
- 1987-03-18 DE DE19873708942 patent/DE3708942A1/de not_active Withdrawn
-
1988
- 1988-03-11 WO PCT/DE1988/000150 patent/WO1988007051A1/de active IP Right Grant
- 1988-03-11 AT AT88730061T patent/ATE69055T1/de not_active IP Right Cessation
- 1988-03-11 ES ES198888730061T patent/ES2040373T3/es not_active Expired - Lifetime
- 1988-03-11 JP JP63502319A patent/JP2588267B2/ja not_active Expired - Fee Related
- 1988-03-11 HU HU911518A patent/HU206368B/hu not_active IP Right Cessation
- 1988-03-11 EP EP88730061A patent/EP0283428B1/de not_active Expired - Lifetime
- 1988-03-11 DE DE88730061T patent/DE3865863D1/de not_active Expired - Lifetime
- 1988-03-11 AU AU14209/88A patent/AU628015B2/en not_active Ceased
- 1988-03-17 DD DD88313750A patent/DD273262A5/de not_active IP Right Cessation
- 1988-03-17 CA CA000561771A patent/CA1326015C/en not_active Expired - Fee Related
- 1988-03-17 NZ NZ223924A patent/NZ223924A/en unknown
- 1988-03-17 IL IL8577388A patent/IL85773A/en not_active IP Right Cessation
- 1988-03-18 PT PT87007A patent/PT87007B/pt not_active IP Right Cessation
- 1988-03-18 IE IE79388A patent/IE62480B1/en not_active IP Right Cessation
- 1988-03-18 ZA ZA881962A patent/ZA881962B/xx unknown
- 1988-03-18 CN CN88102162A patent/CN1033033C/zh not_active Expired - Fee Related
- 1988-12-09 US US07/283,632 patent/US5095129A/en not_active Expired - Fee Related
-
1989
- 1989-09-15 FI FI894364A patent/FI101883B/fi not_active IP Right Cessation
-
1991
- 1991-12-17 GR GR91401682T patent/GR3003362T3/el unknown
-
1994
- 1994-07-11 HU HU94P/P00010P patent/HU210532A9/hu unknown
-
1996
- 1996-01-17 CN CN96100917A patent/CN1057770C/zh not_active Expired - Fee Related
- 1996-02-01 CN CN96102566A patent/CN1080267C/zh not_active Expired - Fee Related
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4447424A (en) * | 1981-01-09 | 1984-05-08 | Roussel Uclaf | Steroid derivatives |
| WO1983003099A1 (en) * | 1982-03-01 | 1983-09-15 | Roussel Uclaf | 3-keto-delta 4,9-19-norsteroids |
| EP0188396A2 (fr) * | 1985-01-14 | 1986-07-23 | Roussel-Uclaf | Nouveaux stéroides substitués en position 10, leur procédé et les intermédiaires de préparation, leur application comme médicaments, les compositions pharmaceutiques les contenant |
Also Published As
| Publication number | Publication date |
|---|---|
| CN1080267C (zh) | 2002-03-06 |
| US5095129A (en) | 1992-03-10 |
| AU1420988A (en) | 1988-10-10 |
| CN1141926A (zh) | 1997-02-05 |
| FI894364A0 (fi) | 1989-09-15 |
| IE62480B1 (en) | 1995-02-08 |
| EP0283428A1 (de) | 1988-09-21 |
| JP2588267B2 (ja) | 1997-03-05 |
| PT87007B (pt) | 1992-06-30 |
| FI101883B1 (fi) | 1998-09-15 |
| HU210532A9 (en) | 1995-04-28 |
| ZA881962B (en) | 1988-09-08 |
| ATE69055T1 (de) | 1991-11-15 |
| DD273262A5 (de) | 1989-11-08 |
| PT87007A (pt) | 1988-04-01 |
| EP0283428B1 (de) | 1991-10-30 |
| FI101883B (fi) | 1998-09-15 |
| NZ223924A (en) | 1991-12-23 |
| HU911518D0 (en) | 1991-11-28 |
| WO1988007051A1 (en) | 1988-09-22 |
| DE3708942A1 (de) | 1988-09-29 |
| CA1326015C (en) | 1994-01-11 |
| CN88102162A (zh) | 1988-11-23 |
| GR3003362T3 (en) | 1993-02-17 |
| AU628015B2 (en) | 1992-09-10 |
| CN1033033C (zh) | 1996-10-16 |
| IE880793L (en) | 1988-09-18 |
| IL85773A0 (en) | 1988-09-30 |
| CN1135488A (zh) | 1996-11-13 |
| IL85773A (en) | 1994-01-01 |
| JPH02503194A (ja) | 1990-10-04 |
| DE3865863D1 (zh) | 1991-12-05 |
| ES2040373T3 (es) | 1993-10-16 |
| HU206368B (en) | 1992-10-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1057770C (zh) | 19,11β-桥甾族化合物在制药中的应用 | |
| CN1036342C (zh) | 9,11-脱氢甾类化合物及9-α-羟基-16,17-脱氢甾类化合物的制备方法 | |
| CN1061052C (zh) | 11β-芳基-4-雌烯及其药物上适宜的酸加成盐的制药应用 | |
| CN1214039C (zh) | 3-氮-6,7-二氧甾族化合物及其应用 | |
| CN1026236C (zh) | 甾类5-α-还原酶抑制剂的制备方法 | |
| CN1294142C (zh) | 作为抗炎剂的17β-硫代羧酸酯17α-芳基羰氧基雄甾烷衍生物 | |
| CN1031942C (zh) | 新的药用甾体的制备方法 | |
| CN1231670A (zh) | 7α-(ξ-氨基烷基)-雌三烯、其制备方法、含有7α-(ξ-氨基烷基)-雌三烯的药剂及其用于制备药物的应用 | |
| CN1158297C (zh) | 用于治疗的具有c-17烷基侧链和芳香a-环的甾族化合物 | |
| CN87107371A (zh) | 螺-取代的戊二酰胺利尿剂 | |
| CN1085561A (zh) | 治疗血胆甾醇过多的甾族苷类 | |
| CN1087092A (zh) | 用作5α-还原酶抑制剂的新的7β-取代的-4-氮杂-5α-雄烷-3-酮类 | |
| CN87103193A (zh) | 一种在位置17含有螺环的新的甾族化合物,它的生产方法及中间产物,它作为药剂的应用,以及含有该化合物的医药用组合物 | |
| CN1075722A (zh) | 新的甾族化合物及其制法,中间体和应用 | |
| CN101061133A (zh) | 制备17-羟基-6β,7β;15β,16β-双亚甲基-3-氧代-17α-孕甾-4-烯-21-羧酸γ-内酯的工业方法和用于该方法的关键中间体 | |
| CN1046166A (zh) | 含有11β-羟酸链的新的19-去甲甾体化合物的制备方法 | |
| CN1029683C (zh) | 在十位以含有双键或叁键的基团取代的新的甾体化合物的制备方法 | |
| CN1066271A (zh) | 14β-H-14-和15-烯键-11β-芳基-4-雌烯 | |
| CN1065070A (zh) | 6,7-变性的11β-芳基-4-雌烯 | |
| CN1633445A (zh) | 制备依匹乐酮的方法 | |
| CN1878786A (zh) | 新的I型17β-羟化类固醇脱氢酶抑制剂 | |
| CN1326461A (zh) | 减数分裂调控化合物 | |
| CN1052238C (zh) | 有促孕素作用的19,11-架桥的雌烯 | |
| CN1101914A (zh) | 新的甾族化合物及其中间体的制法和用途 | |
| CN1729200A (zh) | 大环内酯类 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C06 | Publication | ||
| PB01 | Publication | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |