BRPI0410488B1 - Composição farmacêutica líquida estabilizada e livre de hsa, método para preparação de composição farmacêutica líquida estabilizada e livre de hsa, e recipiente vedado hermeticamente - Google Patents
Composição farmacêutica líquida estabilizada e livre de hsa, método para preparação de composição farmacêutica líquida estabilizada e livre de hsa, e recipiente vedado hermeticamente Download PDFInfo
- Publication number
- BRPI0410488B1 BRPI0410488B1 BRPI0410488-9A BRPI0410488A BRPI0410488B1 BR PI0410488 B1 BRPI0410488 B1 BR PI0410488B1 BR PI0410488 A BRPI0410488 A BR PI0410488A BR PI0410488 B1 BRPI0410488 B1 BR PI0410488B1
- Authority
- BR
- Brazil
- Prior art keywords
- ifn
- liquid pharmaceutical
- formulation
- pharmaceutical composition
- interferon
- Prior art date
Links
Images













Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/08—Solutions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
- A61K38/21—Interferons [IFN]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
- A61K38/21—Interferons [IFN]
- A61K38/215—IFN-beta
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/10—Alcohols; Phenols; Salts thereof, e.g. glycerol; Polyethylene glycols [PEG]; Poloxamers; PEG/POE alkyl ethers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/20—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing sulfur, e.g. dimethyl sulfoxide [DMSO], docusate, sodium lauryl sulfate or aminosulfonic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/12—Keratolytics, e.g. wart or anti-corn preparations
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/12—Carboxylic acids; Salts or anhydrides thereof
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Immunology (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Gastroenterology & Hepatology (AREA)
- Zoology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Dermatology (AREA)
- Oncology (AREA)
- Virology (AREA)
- Molecular Biology (AREA)
- Communicable Diseases (AREA)
- Biochemistry (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Hematology (AREA)
- Biotechnology (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
Abstract
composição farmacêutica líquida estabilizada livre de hsa, método para preparação de composição farmacêutica líquida estabilizada livre de hsa, recipiente vedado hermeticamente e kit para administração de múltiplas doses de composição farmacêutica líquida estabilizada livre de hsa, descreve uma composição farmacêutica líquida estabilizada livre de hsa, que é composta de um interferon (ifn), caracterizada pelo fato de que a referida formulação seja uma solução que seja composta de um tampão, um surfactante, um agente de isotonicidade e um antioxidante. preferivelmente, o interferon é um recombinante humano ifn-beta.
Description
[001] A presente patente de privilégio de invenção se refere, de modo geral, a composições farmacêuticas contendo um interferon, mais especificamente a formulações estabilizadas de interferon-beta que sejam livres de albumina sérica humana como excipiente farmacêutica adicional.
[002] Os interferons são citocinas, ou seja, proteínas solúveis que transmitem mensagens entre as células e desempenham um papel essencial no sistema imunológico, ajudando a destruir microorganismos que causam infecção e consertando os danos resultantes. Os interferons são naturalmente secretados pelas células infectadas e foram identificados pela primeira vez em 1957. Seu nome se deriva do fato de que eles “interferem” na replicação e produção viral.
[003] Os interferons exibem atividade antiviral e antiproliferativa. Com base nas propriedades bioquímicas e imunológicas, os interferons humanos que ocorrem naturalmente são agrupados em três classes principais: interferon-alfa (leucócito), interferon-beta (fibroblasto) e interferon-gama (imunológico). O alfa-interferon é atualmente aprovado para os Estados Unidos e outros países para o tratamento de leucemia de células pilosas, verrugas genitais, Sarcoma de Kaposi (um câncer que normalmente afeta pacientes que sofrem de Síndrome de imunodeficiência Adquirida - AIDS), e hepatite crônica não- A, não-B.
[004] Mais além, interferons (IFNs) são glicoproteínas produzidas pelo corpo em resposta a uma infecção viral. Eles inibem a multiplicação de vírus em células protegidas. Consistindo de uma proteína de peso molecular menor, os IFNs são notadamente não-específicos em sua ação, ou seja, IFN induzido por um vírus é eficaz contra uma ampla variedade de outros vírus. Eles são, entretanto, específicos por espécie, ou seja, IFN produzido por uma espécie irá somente estimular a atividade antiviral em células da mesma espécie ou de uma espécie próxima. Os IFNs foram o primeiro grupo de citocinas a serem exploradas por suas potenciais atividades antitumorais e antivirais.
[005] Os três maiores IFNs são referidos como IFN-α, IFN-β e IFN-Y. Esses principais tipos de IFNs foram inicialmente classificados de acordo com suas células de origem (leucócito, fibroblasto ou célula T). Entretanto, ficou claro que vários tipos podem ser produzidos por uma célula. Então o leucócito IFN é agora chamado de IFN-α, o fibroblasto IFN é IFN-β e a célula T IFN é IFN-y. Há também um quarto tipo de IFN, o linfoblastóide IFN, produzido na linha celular “Namalwa” (derivada do linfoma de Burkitt), que parece produzir uma mistura de leucócito e fibroblasto IFN.
[006] A unidade de interferon ou unidade internacional de interferon (U ou UI, de unidade internacional) tem sido relatada como uma medida de atividade de IFN definida como a quantidade necessária para proteger 50% das células contra danos virais. O ensaio que pode ser usado para medir a bioatividade é o ensaio de inibição de efeito citopático conforme descrito (Rubinstein et al. 1981; Familletti, P. C. et al., 1981). Nesse ensaio antiviral de interferon cerca de 1 unidade/ml de interferon é a quantidade necessária para produzir um efeito citopático de 50%. As unidades são determinadas em relação ao padrão internacional de referência para Hu-IFN-beta fornecido pelo National Institutes of Health (Pestka, S. 1986).
[007] Cada classe de IFn contém vários tipos distintos. IFN-β e IFN-y são cada um o produto de um único gene.
[008] As proteínas classificadas como IFNs-α são o grupo mais diverso, contendo cerca de 15 tipos. Há um grupo de genes IFN-α no cromossomo 9, contendo no mínimo 23 membros, dos quais 15 são ativos e transcritos. Os IFNs-α maduros não são glicosilados.
[009] IFNs-α e IFN-β são todos do mesmo comprimento (165 ou 166 aminoácidos) com atividades biológicas similares. IFNs—Y são 146 aminoácidos em comprimento, e assemelham as classes α e β de forma menos próxima. Somente os IFNs—Y podem ativar os macrófagos ou induzir a maturação de células T assassinas. Esses novos tipos de agentes terapêuticos podem ser às vezes chamados de modificadores biológicos de resposta (BRMs), porque eles têm um efeito sobre a resposta do organismo ao tumor, afetando o reconhecimento por meio de imunomodulação.
[0010] O interferon de fibroblasto humano (IFN- β) tem atividade antiviral e também pode estimular as células assassinas naturais contra as células neoplásticas. É um polipeptídio de cerca de 20.000Da induzido por vírus e RNAs de cepa dupla. Da sequência de nucleotídeo do gene de interferon de fibroblasto, clonado por tecnologia de DNA recombinante, (Derynk et al. 1980) deduziu o aminoácido completo da sequência da proteína. Tem o comprimento de 166 aminoácidos.
[0011] Shepard et al. (1981) descreveu uma mutação em base 842 (Cys ^ Tyr na posição 141) que aboliu sua atividade antiviral, e um clone variante com uma deleção dos nucleotídeos 1119-1121.
[0012] Mark et al. (1984) inseriu uma mutação artificial substituindo a base 469 (T) por (A) causando uma troca de aminoácido de Cys ^ Ser na posição 17. O IFN-β resultante foi relatado como sendo o IFN-β “nativo” e estável durante o armazenamento em longo prazo (-70°C).
[0013] Rebif® (Serono - interferon-β humano de recombinante), o mais recente desenvolvimento em terapia de interferon para esclerose múltipla (EM), é interferon (IFN)- beta-1a, produzido de linhas celulares de mamíferos. Seu “Nome Não-Proprietário Internacional” (INN) é “Interferon beta-1a”.
[0014] Como em todos os produtos farmacêuticos à base de proteínas, um principal obstáculo, que pode ser superado no uso de IFN-beta como agente terapêutico, é a perda de utilidade farmacêutica que pode resultar de sua instabilidade em formulações farmacêuticas.
[0015] As instabilidades físicas que ameaçam a atividade e eficácia de polipeptídios em formulações farmacêuticas incluem a desnaturação e formação de agregados solúveis e insolúveis incluem hidrólise, formação de imida, oxidação, racemização e desarnidação. Algumas dessas mudanças são conhecidas por levar à perda ou redução da atividade farmacêutica da proteína de interesse. Em outros casos, os efeitos precisos dessas mudanças são desconhecidos, mas os produtos degradativos são ainda considerados como farmaceuticamente inaceitáveis em função do potencial de efeitos colaterais indesejáveis.
[0016] A estabilização de polipeptídios em composições farmacêuticas permanece uma área em que a tentativa e o erro desempenham um papel importante (analisado por Wang (1999) Int. J. Pharm. 185:129-188; Wang and Hanson (1988) J. Parenteral Sci. Tech. 42: S3-S26). Os excipientes que são adicionados às formulações farmacêuticas de polipeptídios para aumentar sua estabilidade incluem tampões, açúcares, surfactantes, aminoácidos, glicóis de polietileno e polímeros, mas os efeitos estabilizantes desses aditivos químicos variam dependendo da proteína.
[0017] As formulações de IFN-beta empregam o uso de HSA como um agente aprimorador de solubilidade de IFN- beta. Entretanto, o uso de HSA tem alguns prejuízos. HSA é um produto do sangue humano e deve, portanto, ser retirado de pacientes humanos. Enquanto as etapas são tomadas para reduzir o risco, o uso de produtos de sangue humano como HSA carrega em si o potencial de introdução de vírus humanos como HIV e HCV.
[0018] Consequentemente, há uma necessidade de composições farmacêuticas IFN-beta adicionais compostas de estabilizantes fisiologicamente compatíveis que aprimoram a solubilidade dessa proteína e estabilizam a proteína contra a formação de agregados, com isso aumentando sua utilidade farmacêutica.
[0019] A presente invenção é voltada para composições farmacêuticas estabilizadas que sejam compostas de interferon (IFN) e métodos para sua preparação. Essas composições são preparadas na ausência de albumina sérica humana (HSA) e, portanto, são livres desse excipiente farmacêutico. Tais composições são aqui referidas como composições farmacêuticas de IFN “livres de HSA” e elas são compostas de interferon (IFN) ou uma isoforma, muteína, proteína fundida, derivado funcional, fração ativa ou sal dos mesmos, caracterizado pelo fato de que a referida composição seja uma solução que seja composta de um tampão, um surfactante, um agente de isotonicidade e um antioxidante.
[0020] De acordo com uma configuração da presente invenção, as composições também são compostas de um agente bacteriostático.
[0021] O termo “interferon”, ou “IFN”, conforme aqui utilizado, tem a intenção de incluir quaisquer moléculas definida como tal na literatura, compreendendo por exemplo quaisquer tipos de IFNs mencionados na seção “Histórico da Invenção” acima. Em específico, IFN-α, IFN-β e IFN-y são incluídos na definição acima, IFN-β é o IFN preferido de acordo com a presente invenção. O IFN-β adequado de acordo com a presente invenção é comercialmente disponível como por exemplo Rebif® (Serono), Avonex® (Biogen) ou Betaferon® (Schering). O uso de interferons de origem humano também é preferido de acordo com a presente invenção. O termo interferon, conforme aqui empregado, tem a intenção de englobar uma isoforma, uma muteína, uma proteína fundida, um derivado funcional, uma fração ativa ou um sal dos mesmos.
[0022] O termo “interferon-beta (IFN-beta ou IFN-β)”, conforme aqui empregado, tem a intenção de incluir interferon de fibroblasto, em específico de origem humana, conforme obtido por isolação de fluidos biológicos ou conforme obtido por técnicas recombinantes de DNA de células hospedeiras procarióticas ou eucarióticas, assim como seus sais, derivados funcionais, variantes, análogos e fragmentos ativos. Preferivelmente, o termo IFN-beta significa Interferon beta- 1a.
[0023] Conforme aqui empregado, o termo “muteínas” se refere a análogos de IFN em que um ou mais resíduos de aminoácidos de um IFN natural são substituídos por resíduos de aminoácidos diferentes, ou um ou mais resíduos de aminoácidos são adicionados à sequência natural de IFN, sem mudar consideravelmente a atividade dos produtos resultantes se comparado ao tipo IFN selvagem. Essas muteínas são preparadas por síntese conhecida e/ou por técnicas de mutagênese voltadas para o local, ou qualquer outra técnica conhecida adequada. As muteínas preferidas incluem, por exemplo, as descritas por Shepard et al. (1981) ou Mark et al. (1984).
[0024] Qualquer uma dessas muteínas tem preferivelmente uma sequência de aminoácidos suficientemente duplicativa da sequência do IFN, de modo a ter atividade substancialmente similar ou até melhor a de IFN. A função biológica de interferon é bem familiar àqueles com experiência na técnica, e os padrões biológicos são estabelecidos e estão disponíveis, por exemplo, pelo National Institute for Biological Standards and Control (Instituto Nacional para Padrões e Controles Biológicos) (http://immunology.org/links/NIBSC).
[0025] Foram descritos os bioensaios para determinação de atividade de IFN. Um ensaio de IFN pode ser conduzido, por exemplo, conforme descrito por Rubinstein et al., 1981. Assim, pode ser determinado se qualquer muteína tem uma atividade substancialmente similar, ou até melhor, do que IFN, por meio de experimentação de rotina.
[0026] As muteínas de IFN, que podem ser usadas de acordo com a presente invenção, ou codificação de ácido nucléico das mesmas, incluem um conjunto finito de sequências substancialmente correspondentes, como peptídeos de substituição ou polinucleotídeos que podem ser obtidos de forma rotineira por pessoas com conhecimento da técnica, sem experimentação indevida, com base nos ensinamentos e diretrizes aqui presentes.
[0027] As mudanças preferidas das muteínas de acordo com a presente invenção são conhecidas como substituições “conservadoras”. As substituições conservadoras de aminoácidos de polipeptídios ou proteínas da invenção podem incluir aminoácidos sinônimos dentro de um grupo, que podem ter propriedades físico-químicas suficientemente similares que a substituição entre os membros do grupo irá preservar a função biológica da molécula. É evidente que as inserções e deleções de aminoácidos também podem ser feitas nas sequências acima definidas sem alterar sua função, especificamente se as inserções ou deleções somente envolverem alguns aminoácidos como, por exemplo, abaixo de trinta, e preferivelmente abaixo de dez, e não removerem ou mudarem de lugar aminoácidos que sejam importantes para uma conformação funcional, como resíduos de cisteína. As proteínas e muteínas produzidas por tais deleções e/ou inserções que caem dentro da competência da presente invenção.
[0028] Preferivelmente, os grupos de aminoácidos sinônimos são aqueles definidos na Tabela I. Mais preferivelmente, os grupos de aminoácidos sinônimos são aqueles definidos na Tabela II; e mais preferivelmente os grupos de aminoácidos sinônimos são aqueles definidos na Tabela III. Tabela I - Grupos Preferidos de Aminoácidos Sinônimos.

[0029] Os exemplos de produção de substituições de aminoácido em proteínas que podem ser usados para se obter muteínas de IFN, para uso na presente invenção, incluem quaisquer etapas de método conhecidas, como as apresentadas nas patentes americanas 4.959.314, 4.588.585 e 4.737.462, para Mark et al; 5.116.943 para Koths et al., 4.965.195 para Namen et al; 4.879.111 para Chong et al; e 5.017.691 para Lee et al.; e proteínas substituídas por lisina, apresentadas na patente americana N° 4.904.584 (Shaw et al). As muteínas específicas de IFN-beta foram descritas, por exemplo, por Mark et al, 1984.
[0030] O termo “proteína fundida” se refere a um polipeptídio composto de um IFN, ou uma muteína do mesmo, fundido a outra proteína que, por exemplo, tenha sido um tempo de residência estendido nos fluidos corporais. Um IFN pode, portanto, ser fundida a outra proteína, polipeptídio ou outros, como por exemplo uma imunoglobulina ou fragmento do mesmo.
[0031] O termo “derivados funcionais” conforme, aqui empregado, cobre derivados de IFN e suas muteínas e proteínas fundidas, que podem ser preparados dos grupos funcionais que ocorrem como cadeias laterais nos resíduos ou os grupos terminais N- ou C-, por meios conhecidos na técnica, e são incluídos na invenção, uma vez que eles permanecem farmaceuticamente aceitável, ou seja, eles não destroem a atividade da proteína que é substancialmente similar à atividade IFN, e não conferem propriedades tóxicas a composições que o contém. Esses derivados podem, por exemplo, incluir cadeias laterais de glicol polietileno, que podem mascarar locais antigênicos e estender a permanência de IFN em fluidos corporais. Outros derivados incluem ésteres alifáticos dos grupos de carboxil, amidas dos grupos de carboxil por reação com amônia ou com aminas primárias ou secundárias, derivados de N-acil de grupos de amino livres dos resíduos de aminoácidos formados com porções de acil (como grupos alcanoil ou aroil carbocíclicos) ou derivados de O-acil de grupos de hidroxil livres (por exemplo os de resíduos de seril ou treonil formados com porções de acil.
[0032] As “frações ativas” de IFN, ou muteínas e proteínas fundidas, a presente invenção cobre qualquer fragmento ou precursor da cadeia de polipeptídios da cadeia de polipeptídios da molécula de proteína sozinha ou em conjunto com moléculas associadas ou resíduos a eles ligados, como por exemplo resíduos de açúcar ou fosfato, ou agregados da molécula de proteína ou os resíduos de açúcar sozinhos, em que a referida fração não tenha atividade significantemente reduzida se comparada ao IFN correspondente.
[0033] O termo “sais” aqui empregado se refere tanto a sais de grupos de carboxil como a sais de adição de ácido de grupos de amino das proteínas acima descritas ou de análogos das mesmas. Sais de um grupo carboxil podem ser formados por meios conhecidos na técnica e incluem sais inorgânicos, por exemplo, sódio, cálcio, amônio, sais férricos ou de zinco, entre outros, e sais com bases orgânicas como aqueles formados, por exemplo, com aminas, como trietanolamina, arginina ou lisina, piperidina, procaína, entre outros. Sais de adição ácida incluem, por exemplo, sais com ácidos minerais, como, por exemplo, ácido clorídrico ou ácido sulfúrico, e sais com ácidos orgânicos, como por exemplo, ácido acético ou ácido oxálico. Logicamente, todos esses sais devem manter a atividade biológica das proteínas (IFN) relevantes para a presente invenção, ou seja, a capacidade de ligar-se ao receptor correspondente e iniciar a sinalização do receptor.
[0034] De acordo com a presente invenção, o uso de IFN-beta recombinante humano e os compostos da invenção é mais especificamente preferido.
[0035] Um tipo especial de variante de interferon foi descrito recentemente. Os chamados “interferons de consenso” são variantes que não ocorrem naturalmente de IFN (patente americana 6.013.253). De acordo com uma configuração preferida da invenção, os compostos da invenção são usados em combinação com um interferon de consenso.
[0036] Conforme aqui empregado, o termo “interferon de consenso humano” (IFN-con) significa um polipeptídio que ocorre naturalmente, que inclui predominantemente os resíduos de aminoácidos que são comuns a um subconjunto de representante de IFN-alfa da maioria das sequências de subtipo de interferon de leucócito humano que ocorre naturalmente e que inclui, em uma ou mais dessas posições em que há aminoácido comum a todos os subtipos, um aminoácido que ocorre predominantemente nessa posição e em nenhum evento inclui resíduo de aminoácido que não seja existente nessa posição em no mínimo um subtipo que ocorre naturalmente. IFN-con engloba mas não se limita às sequências de aminoácidos chamadas de IFN-con1, IFN-con2 e IFN-con3 que são revelados nas patentes americanas 4.695.623, 4.897.471 e 5.541.293. As sequências de DNA que codificam IFN-con podem ser produzidas conforme descrito nas patentes acima mencionadas, ou por outros métodos padrão.
[0037] Em uma outra configuração preferida, a proteína fundida é composta de uma fusão de Ig. A fusão pode ser direta ou por meio de um peptídeo ligador curto que pode ter o comprimento de apenas 1 a 3 resíduos de aminoácidos. O referido ligador pode ser um tripeptídeo da sequência E-F-M (Glu-Phe-Met), por exemplo, ou uma sequência ligadora de 13 aminoácidos composta de Glu-Phe-Gly-Ala-Gly-Leu-Val-Leu-Gly- Gly-Gln-Phe-Met introduzida entre a sequência de IFN e a sequência de imunoglobulina. A proteína de fusão resultante pode ter propriedades aprimoradas, como um tempo de permanência estendido nos fluidos corporais (meia-vida), atividade específica aumentada, nível de expressão aumentado ou a purificação da proteína de fusão é facilitada.
[0038] Em uma outra configuração preferida, IFN é usado para a região constante de uma molécula de Ig. Preferivelmente, é fundida para regiões de cadeia pesada, como os domínios de CH2 e CH3 de IgG1 humana, por exemplo. Outras isoformas de moléculas de Ig são também adequados para a geração de proteínas de fusão de acordo com a presente invenção, como isoformas IgG2, IgG3, IgG4 ou outras classes de Ig, como IgM ou IgA, por exemplo. As proteínas de fusão podem ser monoméricas ou multiméricas, hétero ou homomultiméricas.
[0039] Em uma outra configuração preferida, o derivado funcional é composto de no mínimo uma porção ligada a um ou mais grupos funcionais, que ocorrem como uma ou mais cadeias nos resíduos de aminoácidos. Preferivelmente, a porção é uma porção de polietileno (PEG). A PEGilação pode ser conduzida por métodos conduzidos, como os métodos descritos em WO99/55377, por exemplo.
[0040] A dosagem administrada, como dose única ou múltipla a um indivíduo, irá variar dependendo de uma variedade de fatores, incluindo propriedades farmacocinéticas, a rota de administração, condições e características do paciente (sexo, idade, peso, saúde, altura), extensão dos sintomas, tratamentos concomitantes, frequência de tratamento e o efeito desejado.
[0041] As dosagens padrão de IFN-beta humano variam de 80.000IU/kg e 200.000IU/kg por dia ou 6MIU (milhões de unidades internacionais) e 12MIU por pessoa por dia ou 22 a 44μg (micrograma) por pessoa. De acordo com a presente invenção, o IFN pode preferivelmente ser administrado a uma dosagem de cerca de 1 a 50μg, mais preferivelmente de cerca de 10 a 30μg ou cerca de 10 a 20μg por pessoa por dia.
[0042] A administração de ingredientes ativos de acordo com a presente invenção pode ser por via intravenosa, intramuscular ou rota subcutânea. A rota de administração preferida de IFN é a rota subcutânea.
[0043] A IFN também pode ser administrada diariamente ou dia sim dia não, ou com menos frequência. Preferivelmente, o IFN é administrado uma, duas ou três vezes por semana.
[0044] A rota de administração preferida é a administração subcutânea, administrada por exemplo três vezes por semana. Uma rota de administração também preferida é a rota intramuscular, que pode ser aplicada, por exemplo, uma vez por semana.
[0045] Preferivelmente, 22 a 44μg ou 6MIU a 12MIU de IFN-beta é administrada três vezes por semana por injeção subcutânea.
[0046] IFN-beta pode ser administrado por via subcutânea, em uma dosagem de 25 a 30μg ou 8MIU a 9,6MIU, dia sim dia não. 30μg ou 6MIU de IFN-beta podem ainda ser administrados via intramuscular uma vez por semana.
[0047] O termo “estabilidade” se refere à estabilidade física, química e de conformação de formulações de interferon da presente invenção (incluindo manutenção de potência biológica). A instabilidade de uma formulação de proteína pode ser causada pela degradação química ou agregação de moléculas de proteínas para formar polímeros de ordem maior, deglicosilação, modificação de glicosilação, oxidação ou qualquer outra modificação estrutural que reduza no mínimo uma atividade biológica de um polipeptídio de interferon incluído na presente invenção.
[0048] Uma solução ou formulação “estável”, é uma em que o grau de degradação, modificação, agregação, perda de atividade biológica e outros, das proteínas, é aceitavelmente controlada e não se torna inaceitável com o tempo. Preferivelmente, a formulação mantém no mínimo ou cerca de 60%, mais preferivelmente no mínimo ou cerca de 70%, mais preferivelmente no mínimo ou cerca de 80%, da atividade rotulada de interferon por um período de 12 a 24 meses. As composições de IFN livres de HSA estabilizadas da invenção têm preferivelmente uma vida de prateleira de no mínimo cerca de 6 meses, 12 meses, 18 meses, mais preferivelmente no mínimo 20 meses, mais preferivelmente ainda no mínimo cerca de 22 meses, mais preferivelmente ainda no mínimo 24 meses quando armazenadas a 2 a 8°C.
[0049] Métodos para monitoração da estabilidade das composições farmacêuticas de IFN livres de HSA estão disponíveis na técnica, incluindo os métodos descritos nos exemplos aqui revelados. Assim, a formação de agregado de IFN durante o armazenamento de uma composição farmacêutica líquida da invenção pode ser facilmente determinada medindo-se a mudança no IFN solúvel em solução com o tempo. A quantidade de polipeptídio solúvel em solução pode ser quantificada por um número de ensaios analíticos adaptados para a detecção de IFN. Tais ensaios incluem, por exemplo, HPLC de fase reversa (RP) e espectroscopia de absorção de UV, conforme descrito nos Exemplos abaixo.
[0050] A determinação de ambos agregados solúveis e insolúveis durante o armazenamento em formulações líquidas pode ser atingida, por exemplo, com o uso de ultracentrifugação analítica conforme observado nos Exemplos a seguir para distinguir entre a parte de polipeptídio solúvel que está presente na forma de agregados solúveis e a parte que está presente na forma molecular não-agregada, biologicamente ativa.
[0051] A expressão “uso de múltiplas doses” pretende incluir o uso de um único frasco, ampola ou cartucho de uma formulação de interferon para mais de uma injeção, por exemplo 2, 3, 4, 5, 6 ou mais injeções. As injeções são preferivelmente dadas por um período de no mínimo ou cerca de a cada 12 horas, 24 horas, 48 horas, etc., preferivelmente até um período de cerca de 12 dias. As injeções podem ser espaçadas no tempo, por exemplo, por um período de 6, 12, 24, 48 ou 72 horas.
[0052] O termo “tampão” ou “tampão fisiologicamente aceitável” se refere a soluções de compostos que sejam conhecidos como seguros para uso farmacêutico ou veterinário em formulações e que têm o efeito de manter ou controlar o pH da formulação na faixa de pH desejada para a formulação. Os tampões aceitáveis para controle de pH em um pH moderadamente ácido a um pH moderadamente básico incluem, mas não se limitam a compostos como fosfato, acetato, citrato, arginina, TRIS e histidina. “TRIS” se refere a 2-amino-2- hidroximetil-1,3-propanodiol e a qualquer sal farmacologicamente aceitável do mesmo. Os tampões preferíveis são tampões de acetato com salina ou um sal aceitável.
[0053] O “agente de isotonicidade” é um composto que seja fisiologicamente tolerado e confira uma tonicidade adequada a uma formulação para prevenir que o fluxo líquido de água pela membrana que esteja em contato com a formulação. Compostos como glicerina são comumente usados para tais fins em concentrações conhecidas. Outros agentes de isotonicidade adequados incluem, mas não se limitam a aminoácidos ou proteínas (como glicina ou albumina), sais (como cloreto de sódio) e açúcares (como dextrose, manitol, sacarose e lactose). Preferivelmente, o agente de isotonicidade é manitol.
[0054] O termo “antioxidante” se refere a um composto que previne que o oxigênio ou radicais livres derivados de oxigênio interajam com outras substâncias. Os antioxidantes estão entre um número de excipientes comumente adicionados a sistemas farmacêuticos para aumentar a estabilidade física e química. Os antioxidantes são adicionados para minimizar ou retardar processos oxidantes que ocorrem com algumas drogas ou excipientes mediante a exposição ao oxigênio ou na presença de radicais livres. Esses processos podem frequentemente ser catalisadas por luz, temperatura, hidrogênio em concentração, presença de traços de metal ou peróxidos. Sulfitos, bissulfetos, tiouréia, metionina, sais de ácido etilenodiaminatetra acético (EDTA), butil hidroxi anisol (BHT) e butil hidroxi anisola (BHA) são frequentemente usados como antioxidantes em drogas. Descobriu-se que o EDTA de sódio aumenta a atividade de antioxidantes por íons metálicos quelantes que de outra forma catalisariam a reação de oxidação. O antioxidante mais preferido é metionina.
[0055] O termo “bacteriostático” se refere a um composto ou composições adicionados a uma formulação para agir como agente antibacteriano. Uma formulação preservada contendo interferon preferivelmente atende às diretrizes de estatutos e regulamentos para eficácia de preservativos para que seja um produto multiuso comercialmente viável. Exemplos de bacteriostáticos incluem fenol, m-cresol, p-cresol, o-cresol, clorocresol, benzil álcool, alquilparabeno (metil, etil, propil, butil e outros), cloreto de benzalcônio, cloreto de benzetônio, deidroacetato de sódio e timerosal. Preferivelmente, o agente bacteriostático é benzil álcool.
[0056] O termo “surfactante” se refere a um composto solúvel que reduz a tensão de superfície de líquidos ou reduz a tensão interfacial entre dois líquidos ou um líquido e um sólido, em que a tensão de superfície seja a força que age sobre a superfície de um líquido, tendendo a minimizar a área da superfície. Os surfactantes por vezes são usados em formulações farmacêuticas, incluindo o fornecimento de drogas de massa molecular baixa e polipeptídios, a fim de modificar a absorção da droga ou seu fornecimento para os tecidos desejados. Surfactantes bem conhecidos incluem polisorbatos (derivados de polioxietileno; Tween) assim como Pluronic.
[0057] De acordo com uma configuração preferida da invenção, foi descoberto que se formulando interferon com um surfactante selecionado de Pluronic® F77, Pluronic F87, Pluronic F88 e Pluronic® F68, em específico preferivelmente Pluronic F68 (BASF, Pluronic F68 também é conhecido como Poloxamer 188) eles obtêm formulações estáveis que minimizam a perda de princípio ativo causado pela adsorção das superfícies do frasco e/ou dispositivo de entrega (como seringa, bomba, cateter, etc.). Foi também descoberto que pela formulação de interferon com surfactante selecionado de Pluronic® F77, Pluronic F87, Pluronic F88 e Pluronic® F68, em específico preferivelmente Pluronic F68 (BASF, Pluronic F68 também é conhecido como Poloxamer 188) eles obtêm uma formulação estável, que é mais resistente à oxidação e à formação de agregados de proteínas.
[0058] Os surfactantes Pluronic são copolímeros de bloco de óxido de etileno (OE) e óxido de propileno (OP). O bloco de óxido de propileno (OP) é ensanduichado entre dois blocos de óxido de etileno (OE).
[0059] Surfactantes Pluronic são sintetizados em um processo de duas etapas: 1. Um hidrófobo de peso molecular desejado é criado pela adição controlada de óxido de propileno aos dois grupos de hidroxil do propileno glicol; e 2. O óxido de etileno é adicionado para ensanduichar o hidrófobo entre os grupos hidrofílicos. No Pluronic® F77, a porcentagem de polioxietileno (hidrófilo) é de 70% e o peso molecular do hidrófobo (polioxipropileno) é de aproximadamente 2.306 Da.
[0060] No Pluronic F87, a porcentagem de polioxietileno (hidrófilo) é de 70% e o peso molecular do hidrófobo (polioxipropileno) é de aproximadamente 2.644Da.
[0061] No Pluronic F88, a porcentagem de polioxietileno (hidrófilo) é de 80% e o peso molecular do hidrófobo (polioxipropileno) é de aproximadamente 2.644Da.
[0062] No Pluronic F68, a porcentagem de polioxietileno (hidrófilo) é de 80% e o peso molecular do hidrófobo (polioxipropileno) é de aproximadamente 1.967Da.
[0063] As propriedades típicas do Pluronic F77 são listadas a seguir: • Peso Molecular Médio: 6600; • Ponto de Fusão/precipitação: 48°C; • Forma Física a 20°C: sólido; • Viscosidade (Brookfield) cps: 480 [líquido a 25°C, pastoso a 60°C e sólido a 77°C]; • Tensão de superfície, dinas/cm a 25°C; • 0,1% Conc.: 47,0 • 0,01% Conc.: 49,3 • 0,001% Conc.: 52,8 • Tensão interfacial, dinas/cm a 25°C vs. Nujol; • 0,1% Conc.: 17,7 • 0,01% Conc.: 20,8 • 0,01% Conc.: 25,5 • Molhadura de Draves, Segundos a 25°C • 1,0% Conc.: > 360 • 0,1% Conc.: > 360 • Altura da Espuma • Milhas Ross, 0,1%, mm a 50°C: 100 • Milhas Ross, 0,1%, mm a 26°C: 47 • Dinâmica, 0,1%, mm a 400 ml/min: > 600 • Ponto de evaporação em solução aquosa, °C • 1% Conc.: > 100 • 10% Conc.: > 100 • HLB (equilíbrio hidrófilo-lipófilo): 25
[0064] As propriedades típicas de Pluronic F87 são listadas a seguir: • Peso Molecular Médio: 7700; • Ponto de Fusão/Precipitação: 49°C; • Forma física a 20°C: sólida; • Cps de viscosidade (Brookfield): 700 [líquido a 25°C, pastoso a 60°C e sólido a 77°]; • Tensão de superfície, dinas/cm a 25°; • 0,1% Conc.: 44,0 • 0,01% Conc.: 47,0 • 0,001% Conc.: 50,2 • Tensão interfacial, dinas/cm a 25°C vs. Nujol; • 0,1% Conc.: 17,4 • 0,01% Conc.: 20,3 • 0,001% Conc.: 23,3 • Molhadura de Draves, Segundos a 25°C • 1,0% Conc.: > 360 • 0,1% Conc.: > 360 • Altura da Espuma • Milhas Ross, 0,1%, mm a 50°C: 80 • Milhas Ross, 0,1%, mm a 26°C: 37 • Dinâmica, 0,1%, mm a 400 ml/min: > 600 • Ponto de evaporação em solução aquosa, °C • 1% Conc.: > 100 • 10% Conc.: > 100 • HLB (equilíbrio hidrófilo-lipófilo): 24
[0065] As propriedades típicas de Pluronic F88 são listadas a seguir: • Peso Molecular Médio: 11400; • Ponto de Fusão/Precipitação: 54°C; • Forma física a 20°C: sólida; • Cps de viscosidade (Brookfield): 2300 [líquido a 25°C, pastoso a 60°C e sólido a 77°]; • Tensão de superfície, dinas/cm a 25°; • 0,1% Conc.: 48,5 • 0,01% Conc.: 52,6 • 0,001% Conc.: 55,7 • Tensão interfacial, dinas/cm a 25°C vs. Nujol; • 0,1% Conc.: 20,5 • 0,01% Conc.: 23,3 • 0,001% Conc.: 27,0 • Molhadura de Draves, Segundos a 25°C • 1,0% Conc.: > 360 • 0,1% Conc.: > 360 • Altura da Espuma • Milhas Ross, 0,1%, mm a 50°C: 80 • Milhas Ross, 0,1%, mm a 26°C: 37 • Dinâmica, 0,1%, mm a 400 ml/min: > 600 • Ponto de evaporação em solução aquosa, °C • 1% Conc.: > 100 • 10% Conc.: > 100 • HLB (equilíbrio hidrófilo-lipófilo): 28
[0066] As propriedades típicas de Pluronic F68 são listadas a seguir: • Peso Molecular Médio: 8400; • Ponto de Fusão/Precipitação: 52°C; • Forma física a 20°C: sólida; • Cps de viscosidade (Brookfield): 1000 [líquido a 25°C, pastoso a 60°C e sólido a 77°]; • Tensão de superfície, dinas/cm a 25°; • 0,1% Conc.: 50,3 • 0,01% Conc.: 51,2 • 0,001% Conc.: 53,6 • Tensão interfacial, dinas/cm a 25°C vs. Nujol; • 0,1% Conc.: 19,8 • 0,01% Conc.: 24,0 • 0,001% Conc.: 26,0 • Molhadura de Draves, Segundos a 25°C • 1,0% Conc.: > 360 • 0,1% Conc.: > 360 • Altura da Espuma • Milhas Ross, 0,1%, mm a 50°C: 35 • Milhas Ross, 0,1%, mm a 26°C: 40 • Dinâmica, 0,1%, mm a 400 ml/min: > 600 • Ponto de evaporação em solução aquosa, °C • 1% Conc.: > 100 • 10% Conc.: > 100 • HLB (equilíbrio hidrófilo-lipófilo): 29
[0067] Outros polímeros com propriedades similares às acima listadas também podem ser usados nas formulações da invenção. O surfactante preferido é o Pluronic F68 e surfactantes com propriedades similares.
[0068] O Pluronic, especialmente o Pluronic F68, é preferivelmente presente na formulação a uma concentração que seja suficiente para manter estabilidade de FSH e/ou LH ao longo do período de armazenamento desejado (por exemplo, 12 a 24 meses) e também a uma concentração que seja suficiente para prevenir as perdas de proteína em função da adsorção nas superfícies, como o frasco, ampola ou cartucho ou seringa.
[0069] Preferivelmente a concentração de Pluronic, especificamente o Pluronic F68, em formulações líquidas é de cerca de 0,01mg/ml ou cerca de 10mg/ml, mais preferivelmente a cerca de 0,05mg/ml até cerca de ,5,0mg/ml, mais especificamente preferivelmente a cerca de 0,1mg/ml a cerca de 2mg/ml, mais preferivelmente a cerca de 0,1mg/ml.
[0070] Preferivelmente a concentração de IFN- beta na formulação é de cerca de 10μg/ml a cerca de 800μg/ml, mais preferivelmente a cerca de 20μg/ml até cerca de 500μg/ml, mais especificamente preferivelmente a cerca de 30 até a cerca de 300, mais preferivelmente a cerca de 22, 44, 88 ou 264μg/ml.
[0071] Preferivelmente, as formulações de FSH da presente invenção têm pH entre cerca de 3,0 e cerca de 5,0, mais preferivelmente de cerca de 3,7 ou 4,7. Um tampão preferido é o acetato, com contraíons preferidos sendo de sódio ou potássio. Os tampões de salina de acetato são bem conhecidos na técnica. As concentrações de tampão na solução total podem variar entre 5mM, 9,5mM, 10mM, 50mM, 100mM, 150mM, 200mM, 250mM e 500mM. Preferivelmente, a concentração de tampão é de cerca de 10mM. Especificamente, o tampão preferido é de 10mM em íons de acetato com pH de 3,5±0,2 ou 4,5±0,2.
[0072] Preferivelmente, na composição da invenção o antioxidante, por exemplo metionina, está presente a uma concentração de cerca de 0,01 a cerca de 5,0mg/ml, mais preferivelmente de cerca de 0,05 até cerca de 0,3mg/ml, mais preferivelmente cerca de 0,1mg/ml.
[0073] Preferivelmente, a concentração do agente de isotonicidade (por exemplo, manitol) nas formulações líquidas é de cerca de 0,5mg/ml a cerca de 500mg/ml, mais preferivelmente a cerca de 1mg/ml até cerca de 250mg/ml, mais especificamente preferível a cerca de 10mg/ml até cerca de 100mg/ml, mais preferivelmente a cerca de 55mg/ml.
[0074] A invenção inclui formulações líquidas. O solvente preferido é água para injeção.
[0075] As formulações líquidas podem ser para dose única ou múltiplas doses. As formulações de interferon líquidas da invenção que são para uso em doses múltiplas são compostas preferivelmente de um bacteriostático, como fenol, m-cresol, p-cresol, o-cresol, clorocresol, benzil álcool, alquilparabeno (metil, etil, propil, butil e outros), cloreto de benzalcônio, cloreto de benzetônio, deidroacetato de sódio e timerosal. Os particularmente preferidos são fenol, benzil álcool e m-cresol, o mais preferido é benzil álcool. O agente bacteriostático é usado em uma quantidade que produza uma concentração que seja eficaz para manter a formulação essencialmente livre de bactérias (adequada para injeção) pelo período das múltiplas doses, que pode ser cerca de 12 ou 24 horas até 12 dias, preferivelmente cerca de 6 até cerca de 12 dias. O bacteriostático é preferivelmente presente em uma concentração de cerca de 0,1% (massa bacteriostático/massa de solvente) até cerca de 2,0%, mais preferivelmente cerca de 0,2% a cerca de 1,0%. No caso do benzil álcool, são especificamente preferidas as concentrações de 0,2% ou 0,3%. Entretanto, o uso de preservativo, como benzil álcool, não se limita a formulações de doses múltiplas, mas também pode se usar em formulações para dose única.
[0076] A faixa de interferon nas formulações da invenção inclui quantidades produzidas mediante reconstituição, concentrações de cerca de 1,0μg/ml a cerca de 50mg/ml, embora concentrações menores e maiores sejam operáveis e sejam dependentes do veículo desejado, como formulações de solução diferem de adesivo transdérmico, métodos pulmonares, transmucosais, bomba osmótica ou micrombomba. A concentração de interferon é preferivelmente de cerca de 5,0μg/ml até cerca de 2 mg/ml, mais preferivelmente de cerca de 10μg até cerca de 1mg/ml, mais preferivelmente de cerca de 30μg/ml até cerca de 100μg/ml.
[0077] Preferivelmente, as formulações da invenção mantêm no mínimo ou cerca de 60%, mais preferivelmente no mínimo ou cerca de 70%, mais preferivelmente no mínimo ou cerca de 80% da atividade de interferon no momento da embalagem, por um período de 24 meses.
[0078] Em uma outra configuração preferida, a invenção fornece um método para fabricação de uma composição farmacêutica líquida conforme descrito anteriormente.
[0079] Em ainda outra configuração preferida, a invenção fornece um método para fabricação de uma composição farmacêutica embalada com uma solução composta do ingrediente ativo e excipientes conforme descrito anteriormente.
[0080] Em ainda outra configuração preferida, a invenção fornece um artigo de fabricação para uso farmacêutico humano, composto de um frasco com as composições farmacêuticas conforme descrito anteriormente, e material escrito declarando que tal solução pode ser mantida por um período de cerca de 24 horas ou mais após o primeiro uso. Preferivelmente o material escrito declare que a solução pode ser mantida por até ou cerca de 12 dias.
[0081] Após o primeiro uso de uma formulação de múltiplas doses, ela pode ser mantida e utilizada por no mínimo ou cerca de 24 horas, preferivelmente no mínimo ou cerca de 4, 5 ou 6 dias, mais preferivelmente por até 12 dias. Após o primeiro uso a formulação é preferivelmente armazenada abaixo da temperatura ambiente (ou seja, abaixo de ou a cerca de 25°C), mais preferivelmente abaixo de ou a cerca de 10°C, mais preferivelmente a cerca de 2 a 8°C, mais preferivelmente a cerca de 4 a 6°C.
[0082] As formulações líquidas da presente invenção podem ser preparadas por um processo que compreende a adição das quantidades calculadas dos excipientes à solução com tampão e em seguida a adição do interferon.
[0083] A solução resultante é então colocada em frascos, ampolas ou cartuchos. As variações desse processo seriam reconhecidas por alguém com conhecimento básico na técnica. Por exemplo, a ordem em que os componentes são adicionados, se mais aditivos são usados, a temperatura e pH em que a formulação é preparada, são todos os fatores que podem ser otimizados para a concentração e meio de administração usados.
[0084] No caso de uma formulação de uso em múltiplas doses, o agente bacteriostático pode ser adicionado à solução contendo o ingrediente ativo (interferon) ou em vez disso, pode ser mantido em um frasco ou cartucho separado e em seguida misturado à solução contendo o ingrediente ativo no momento do uso.
[0085] As formulações da invenção podem ser administradas usando-se dispositivos reconhecidos. Os exemplos compreendendo esses sistemas de frasco único incluem auto- injetores ou dispositivos de caneta de injeção para administração de uma solução, como Rebiject®.
[0086] Os produtos neste reivindicados incluem material de embalagem. O material de embalagem fornece, em adição às informações solicitadas pelas agências regulamentares, as condições sob as quais o produto pode ser usado. O material de embalagem da presente invenção fornece instruções ao paciente, se necessário, para preparar a solução final e usar essa solução final por um período de vinte e quatro horas ou mais para o produto de dois frascos, molhado/seco. Para um frasco único, produto em solução, a etiqueta indica que tal solução pode ser armazenada após o primeiro uso por um período de vinte e quatro horas ou mais. Os produtos neste reivindicados são úteis para uso farmacêutico humano.
[0087] As formulações preservadas estáveis podem ser fornecidas a pacientes como soluções transparentes. A solução pode ser para dose única ou tempos múltiplos de reuso e pode ser o bastante para ciclos únicos ou múltiplos de tratamento de pacientes e assim fornece um regime de tratamento mais conveniente do que os atualmente disponíveis.
[0088] O interferon, seja nas formulações ou soluções estáveis ou preservadas aqui descritas, pode ser administrado a um paciente de acordo com a presente invenção por uma variedade de métodos de administração incluindo injeção via subcutânea ou intramuscular; transdérmico, pulmonar, transmucosal, implante, bomba osmótica, cartucho, microbomba, oral ou outros meios apreciados por aqueles com experiência na técnica, conforme é conhecido pela técnica.
[0089] O termo “frasco” se refere de modo amplo a um recipiente adequado para manter o interferon em forma sólida ou líquida em um estado estéril contido. Exemplos de um frasco conforme aqui utilizado incluem ampolas, cartuchos, blisters ou outros recipientes adequados para administração ao paciente via seringa, bomba (incluindo a bomba osmótica), cateter, adesivo transdérmico, spray pulmonar ou transmucosal. Os frascos adequados para embalar produtos para administração parenteral, pulmonar, transmucosal ou transdérmica são bem conhecidos e reconhecidos na técnica.
[0090] O termo “tratamento” dentro do contexto se refere a qualquer efeito benéfico na progressão da doença, incluindo atenuação, redução, diminuição ou cessão do desenvolvimento patológico após a doença se instalar.
[0091] As composições farmacêuticas da invenção compostas de IFN ou uma isoforma, muteína, proteína fundida, derivado funcional, fração ativa ou sal são úteis no diagnóstico, prevenção e tratamento (local ou sistêmico) de indicações clínicas que respondem à terapia com esse polipeptídio. Tais indicações clínicas incluem, por exemplo, distúrbios ou doenças do sistema nervoso central (SNC), cérebro e/ou coluna vertebral, incluindo esclerose múltipla; doenças autoimunes, incluindo artrite reumatóide, psoríase, doença de Crohn; e cânceres, incluindo os cânceres de mama, próstata, bexiga, rim e de cólon.
[0092] Todas as referências aqui citadas, incluindo artigos ou resumos de revistas especializadas, pedidos de patente americana ou estrangeira publicados ou não, patentes americanas ou estrangeiras emitidas ou quaisquer outras referências, são totalmente incorporadas aqui por referência, incluindo todos os dados, tabelas, figuras e texto apresentados nas referências citadas. Além disso, todo o conteúdo das referências aqui citadas é totalmente incorporado por referência.
[0093] Referências a etapas de método conhecido, etapas de métodos convencionais, métodos conhecidos ou convencional não significam de forma alguma uma admissão de que qualquer aspecto, descrição ou configuração da presente invenção é revelado, ensinado ou sugerido na técnica relevante.
[0094] A descrição acima das configurações específicas irá revelar a natureza geral da invenção de forma tão completa que terceiros poderão, pela aplicação dentro das capacidades da técnica (incluindo o conteúdo das referências aqui citadas), modificá-la e/ou adaptá-la facilmente para várias configurações específicas, sem experimentação indevida, sem fugir do conceito geral da presente invenção. Portanto, tais adaptações pretendem estar dentro do significado de uma faixa de equivalência das configurações reveladas, com base no ensinamento e na diretriz aqui apresentada. Deve ser entendido que a fraseologia ou terminologia aqui empregada é para fins de descrição e não de limitação, de modo que a terminologia ou fraseologia da presente especificação deve ser interpretada pela pessoa com experiência na técnica à luz dos ensinamentos e diretrizes neste apresentados, em combinação com o conhecimento de uma pessoa com conhecimento básico da técnica. DESCRIÇÃO DA FIGURAS
[0095] A presente patente é mais precisamente descrita com base nas figuras abaixo relacionadas, nas quais: • A figura 1 relata a porcentagem de formas oxidadas, que estão presentes nas formulações de múltiplas doses de Interferon beta-1a com diferentes concentrações de benzil álcool após armazenamento a 40°C; • A figura 2 relata a porcentagem de formas oxidadas, que estão presentes nas formulações de múltiplas doses de Interferon beta-1a com diferentes concentrações de benzil álcool após armazenamento a 25°C; • A figura 3 relata a porcentagem de formas oxidadas, que estão presentes nas formulações de múltiplas doses de Interferon beta-1a com diferentes concentrações de benzil álcool após armazenamento de 2 a 8°C; • A figura 4 relata a porcentagem total de agregados, que estão presentes nas formulações de múltiplas doses de Interferon beta-1a com diferentes concentrações de benzil álcool após armazenamento a 40°C; • A figura 5 relata a porcentagem total de agregados, que estão presentes nas formulações de múltiplas doses de Interferon beta-1a com diferentes concentrações de benzil álcool após armazenamento a 25°C; • A figura 6 relata a porcentagem total de agregados, que estão presentes nas formulações de múltiplas doses de Interferon beta-1a com diferentes concentrações de benzil álcool após armazenamento de 2 a 8°C; • A figura 7 mostra a porcentagem de formas oxidadas, que estão presentes nas formulações de múltiplas doses de Interferon beta-1a com agentes bacteriostáticos alternativos após armazenamento a 25°C; • A figura 8 relata a porcentagem de formas oxidadas, que estão presentes nas formulações de múltiplas doses de Interferon beta-1a com agentes bacteriostáticos alternativos após armazenamento de 2 a 8°C; • A figura 9 relata a porcentagem total de agregados, que estão presentes nas formulações de múltiplas doses de Interferon beta-1a com agentes bacteriostáticos alternativos; • A figura 10 mostra a porcentagem de formas oxidadas, que estão presentes nas formulações de múltiplas doses de Interferon beta-1a com EDTA após armazenamento a 25°C; • A figura 11 mostra a porcentagem total de agregados, que estão presentes nas formulações de múltiplas doses de Interferon beta-1a com EDTA após armazenamento a 25°C; • A figura 12 mostra a eficácia de 0,012% de L-Metionina como antioxidante (2 a 8°C); e • A figura 13 mostra a eficácia de 0,012% de L-Metionina como antioxidante (a 25±2°C). EXEMPLOS Exemplo 1 - Formulação líquida de Interferon beta-1a livre de HSA de dose única em ampolas de injeção 1.1 Estudos Preliminares de Compatibilidade
[0096] Os experimentos foram conduzidos para verificar o efeito protetor mostrado por alguns excipientes como antioxidantes e surfactantes, uma vez que se espera que a eliminação da albumina sérica humana (HSA) do produto atual possa ter afetado o produto em termos de oxidação, formação de agregados e adsorção às superfícies.
[0097] O Interferon beta-1a foi formulado em concentrações de 44mcg/ml e 88mcg/nl em tampão de acetato de sódio contendo 54,6mg/ml de manitol em combinação com vários excipientes como 0,4% HSA, 0,012% de L-Metionina, Tween 20 (0,005%, 0,007%, 0,01%), Poloxamer 188 (a 0,05%, 0,1%, 0,5%). As diferentes combinações foram expostas a condições estressantes (armazenamento a 40°C ou turbilhonamento) e testadas quanto à oxidação (por HPLC de fase reversa) e agregação (por HPLC de exclusão molecular).
[0098] As Tabelas DEP-1 e 2 resumem os níveis de oxidação e agregação após 2 semanas de armazenamento a 40°C. A combinação de Interferon beta-1a com os dois surfactantes testados (Tween 20 e Poloxamer 188) resultou em um aumento no nível de oxidação que, para cada tipo de surfactante, é dependente de concentração (Tabela DEP-1, combinações #4-6 e #7-9); em um nível de 0,5% de Poloxamer 188 (também mencionado como Pluronic F-68 ou F-68), a substância da droga é completamente degradada (Tabela DEP-1, #9). Uma taxa de degradação mais alta é observada em Tween 20, conforme esperado, em função de espécies oxidantes (como peróxidos) que podem estar presentes na forma de resíduos de sínteses.
[0099] Ambos os surfactantes nas diferentes concentrações testadas não influenciam o nível de agregação mediante armazenamento a 40°C (Tabela DEP-2). Tabela DEP-1 Formas oxidadas (%) por HPLC de fase reversa após armazenamento a 40°C
[00100] A Tabela DEP-3 mostra que ambos os surfactantes, Tween 20 e Poloxamer 188 (F-68), usados em sua Concentração Crítica de Micela (CMC), ajudam na prevenção da agregação induzida pelo turbilhonamento por 5 minutos. Tabela DEP-3 Total de agregados (%) por HPLC de exclusão molecular após turbilhonamento por 5 minutos
[00101] As características físico-químicas que são conhecidas como críticas para a qualidade do produto da droga são as quantidades de oxidação e de dímeros/agregados. Essas características são consideradas nos estudos de compatibilidade, resumidas acima. 1.2 Excipientes 1.2.1 10mM de tampão de acetato de sódio, pH 3,5
[00102] Um tampão de acetato de sódio com pH 3,5 contendo 54,6mg/ml de manitol como agente de isotonicidade estabiliza o produto, conforme mostrado durante o desenvolvimento prévio do produto atualmente comercializado (Rebif®) e conforme descrito na patente européia 759.775. 1.2.2. Poloxamer 188
[00103] O Poloxamer 188 (ou Pluronic F-68) está incluído na formulação em um nível de 0,1% (Concentração Crítica de Micela), a fim de prevenir a adsorção da substância da droga pela superfície dos recipientes durante o processo de fabricação; concentrações maiores podem afetar de forma negativa a estabilidade do produto (oxidação maior); concentrações menores podem ser menos eficazes na limitação da adsorção.
[00104] A eficácia do Poloxamer 188 em prevenir a adsorção da substância da droga durante o processo de fabricação foi demonstrado pelo estudo a seguir: soluções contendo 44mcg/ml de Interferon beta-1a foram combinadas com 3 concentrações diferentes de um surfactante (Tween 20 ou Poloxamer 188) ou HSA, imitando o processo de fabricação; amostras foram retiradas em diferentes etapas (composição, filtração asséptica, preenchimento) e testadas por um método de HPLC de fase reversa quantitativa.
[00105] As amostras a seguir foram retiradas: • Antes da filtração (AF) • Após 1a filtração (AF1) • Após 2a filtração (AF2) • Após preenchimento (produto acabado a T = 0)
[00106] Os resultados foram relatados na Tabela DEP-4 e são expressos na forma de recuperação (%) versus o valor inicial (ou seja, a solução composta antes da filtração): Poloxamer 188 é mais eficaz do que Tween 20, mas tão eficaz quanto HSA na prevenção da adsorção da substância de droga durante a fabricação. Tabela DEP-4 % de recuperação de Interferon beta-1a durante a fabricação
[00107] Diferentes graus de Poloxamer 188 obtidos de diferentes fornecedores foram investigados em termos de produtos da oxidação mediante condições aceleradas (2 semanas a 40°C) para definir a qualidade a ser usada: Poloxamer 188 da BASF foi escolhido, pois ele fornece um nível menor de oxidação e é fornecido como grau farmacêutico. Os resultados estão resumidos na Tabela DEP-5: Tabela DEP-5 % de formas oxidadas detectadas em formulações contendo 0,1% de Poloxamer 188 (diferentes qualidades e fornecedores)
[00108] A L-Metionina (L-Met) está incluída na formulação a um nível de 0,012% para limitar a oxidação. A eficácia dessa concentração é mostrada pela comparação com uma formulação que não contém L-Metionina; concentrações maiores (0,05%, 0,1%) de L-Metionina mostram um efeito comparável sobre a estabilidade. Os produtos de oxidação detectados mediante armazenamento a 40°C são mostrados na Tabela DEP-6. Tabela DEP-6 % de formas oxidadas em formulações de Interferon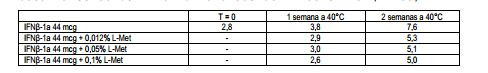
[00109] Durante o desenvolvimento da formulação, a eficácia de L-Metionina como um antioxidante foi confirmada pelos dados de estabilidade de três meses a 2-8°C e 25±2°C, gerada em formulações com L-Metionina em combinação com surfactantes: L-Metionina é eficaz como antioxidante em um nível de 0,012% e podem garantir uma estabilidade comparável à observada pelo produto atual (ver Figuras 12 e 13). 1.3 Produto da Droga 1.3.2 Desenvolvimento da Formulação
[00110] O desenvolvimento da nova formulação livre de HSA de Interferon beta-1a focado na confirmação dos resultados das investigações preliminares (eficácia de L- Metionina como antioxidante, inclusão de Poloxamer 188 para prevenir perdas durante a fabricacao) no recipiente final.
[00111] Soluções de Interferon beta-1a a 44mcg/ml e 88mcg/ml contendo 54,6mg/ml de manitol em 10mM de tampão de acetato de sódio a pH 3,5 foram preparadas e os seguintes excipientes incluídos: • Tween 20 (0,003%, 0,007%, 0,02%) • Poloxamer 188 (0,05%, 0,1%, 0,2%) • L-Metionina (0%, 0,012%) • HSA (0,4%, formulação atual, designada como “ref”)
[00112] A composição das formulações que foram investigadas é mostrada na Tabela DEP-7. Tabela DEP-7 Composição de formulações contendo Interferon beta-1a
[00113] As formulações foram fabricadas de acordo com o procedimento descrito a seguir:
[00114] 90ml de cada formulação foram fabricadas sob condições assépticas, por composto da quantidade necessária de excipientes dissolvidas em água para injeção com a substância de droga (Interferon beta-1a); as formulações foram então filtradas através de uma membrana de 0,22μm (filtradas duas vezes por meio de dois filtros de membrana) e 0,5ml de cada solução preenchida em seringas de vidro Hypak de 1ml. O tamanho de lote era de cerca de 180 seringas.
[00115] As formulações foram então armazenadas a 2-8°C, 25±2°C e 40±2°C e testadas quanto à estabilidade até 12 semanas (até 6 semanas em amostras armazenadas a 40±2°C).
[00116] Os testes analíticos e métodos a seguir foram usados durante o desenvolvimento (para mais detalhes sobre esses ensaios veja o Exemplo 2): • bioatividade (ensaio de CPE) • ensaio (método de HPLC de fase reversa) • produtos de oxidação (método de HPLC de fase reversa) • dímeros/agregados (método de HPLC de exclusão molecular e SDS-PAGE) • pH (método potenciométrico) • osmolalidade (medição crioscópica)
[00117] Os resultados e sua avaliação estão resumidos nas Tabelas DEP-8 a DEP-17. Tabela DEP-8 Bioidentidade (MIU/ml)

[00118] Os desvios calculados por análise de regressão linear e resumidos na Tabela DEP-9 mostram uma diminuição na atividade biológica de todas as formulações contendo Tween 20 (#1, 2, 3, 9) e armazenados a 40°C; uma diminuição na bioatividade é também observada nas formulações #3 e 6 (mais alta concentração de surfactantes) após o armazenamento a 25°C e 2-8°C. Tabela DEP-9 Análise de regressão linear para bioidentidade (desvios expressos como MIU/ml x semana)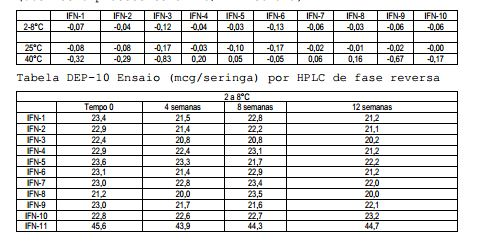
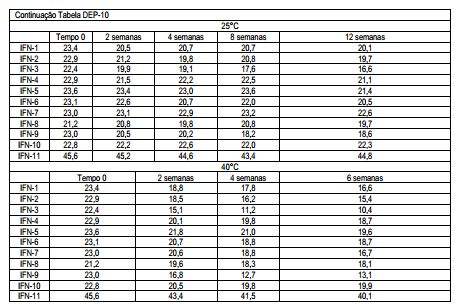
[00119] Os desvios calculados por análise de regressão linear e resumidos na Tabela DEP-11 mostram uma perda maior do conteúdo de proteína das formulações contendo Tween 20 (#1, 2, 3, 9) e armazenados a 40°C; a mesma tendência é observada a 25°C assim como nas formulações #5 e 6. Uma diminuição significante no conteúdo de proteína ocorre a 2-8°C nas formulações #1, 2, 3 (com Tween 20), 4, 5 (com Poloxamer 188) e 9 (com Tween 20 e L-Metionina). Tabela DEP-11 Análise de regressão linear para o ensaio (desvios expressos como mcg/seringa x semana)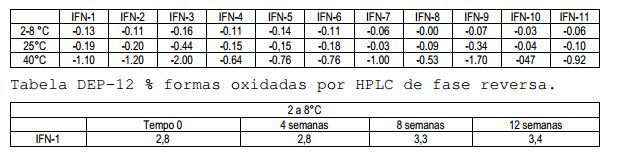
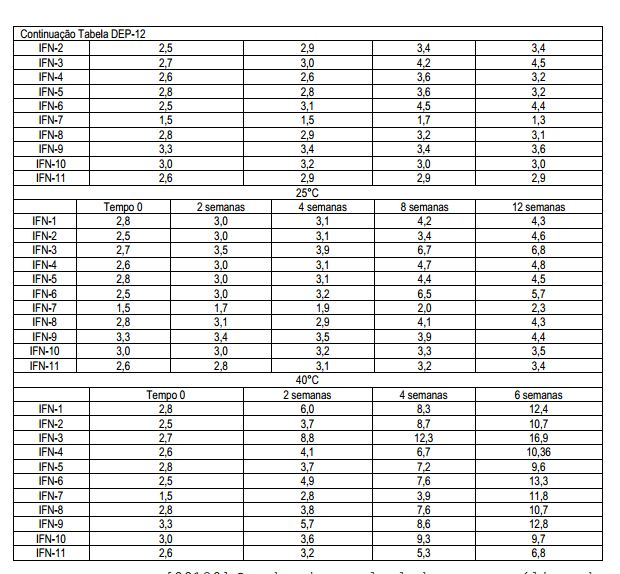
[00120] Os desvios calculados por análise de regressão linear e resumidos na Tabela DEP-13 mostram que as formulações contendo Tween 20 têm uma taxa de oxidação maior quando comparadas às formulações contendo Poloxamer 188 e um nível de oxidação dependente da concentração de Tween 20.
[00121] A eficácia de L-Metionina na limitação da oxidação nas diferentes temperaturas testadas é demonstrada também pela comparação da formulação #9 (Tween 20 + L- Metionina) com #3 (Tween 20) e formulação #10 (Poloxamer 188 + L-Metionina) com #6 (Poloxamer 188). Tabela DEP-13 Análise de regressão linear das formas oxidadas
[00122] Os desvios calculados por análise de regressão linear e resumidos na Tabela DEP-15 mostram que não ocorre nenhum aumento significante no total de conteúdo de agregados com o armazenamento em diferentes temperaturas. Tabela DEP-15 Análise de regressão linear de total de agregados

[00123] Nenhuma troca de pH é observada com o armazenamento. Tabela DEP-17 Osmolalidade (OSM/kg)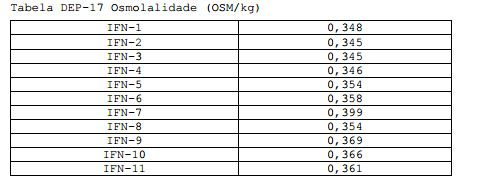
[00124] A osmolalidade das formulações testadas é adequada.
[00125] Com base nos resultados do desenvolvimento da formulação, foi selecionada a formulação livre de HSA a seguir: • 44 ou 88mcg/ml de Interferon beta-1a em tampão de acetato de sódio de pH 3,5 contendo • 54,6mg/ml manitol, • 1mg/ml Poloxamer 188 • 0,12mg/ml de L-Metionina 1.3.2.Envelhecimento
[00126] Não se aplica nenhum envelhecimento. 1.3.3 Propriedades Físico-químicas e Biológicas
[00127] Essas características foram consideradas nos estudos de desenvolvimento de formulação, conforme descrito acima. 1.4 Desenvolvimento do Processo de Fabricação 1.4.1 Desenvolvimento do Processo de Fabricação
[00128] O atual processo de fabricação foi adaptado à preparação dos lotes em escala de laboratório da nova formulação: a substância da droga foi composta diretamente com ingredientes; em seguida uma filtração foi feita para imitar o processo em escala industrial que prevê uma filtração asséptica seguida por uma filtração em linha antes do preenchimento de seringa. As seringas foram então preenchidas manualmente com a solução estéril final.
[00129] As duas etapas de filtração e o preenchimento de seringa foram feitos sob fluxo laminar.
[00130] Uma descrição de cada etapa do processo é feita a seguir. 1.4.2. Cálculos Preliminares
[00131] A quantidade de substância da droga Interferon beta-1a D(mg) necessária para se obter uma solução de 44mcg/ml: D(mg) = 44mcg/ml x 90ml = 3960mcg = 3,96mg
[00132] Volume de substância da droga Interferon beta-1a B(ml) correspondente à quantidade D(mg): B(ml) = 3,96mg: titulação de volume (mg/ml)
[00133] Volume de solução de excipiente V(ml) necessária para se obter 90 ml de uma solução de 44mcg/ml: V(ml) = 90ml - B(ml)* *(d [símbolo] 1g/ml) 1.4.3 Preparação de 1M de solução de hidróxido de sódio
[00134] Foi preparado 1M de hidróxido de sódio em água de injeção. 1.4.4 Preparação de 0,01M de tampão de acetato de sódio pH 3,5
[00135] Uma quantidade adequada de ácido acético glacial foi adicionada à água de injeção e o pH foi ajustado para 3,5±0,2 usando 1M NaOH ou 50% de ácido acético diluído. A solução foi preenchida até o volume final usando água de injeção. 1.4.5 Preparação da solução excipiente
[00136] A quantidade calculada de excipientes (manitol, Tween 20 ou Poloxamer 188, L-Metionina) foi pesada e dissolvida na quantidade necessária de 0,01M de tampão de acetato de sódio pH 3,5; o pH é então checado e ajustado, se necessário, para 3,5±0,2 com 1M de NaOH ou 50% de ácido acético diluído; a solução é então preenchida até o peso final com 0,01M de tampão de acetato de sódio. 1.4.6 Composição da solução da substância da droga
[00137] A quantidade necessária B(g) da substância de droga Interferon beta-1a é adicionada à quantidade necessária da solução excipiente V(g) e gentilmente mexida para se obter homogeneidade. 1.4.7 1a Filtração da solução da substância da droga
[00138] A solução composta é então filtrada através de uma membrana de náilon de 0,2μm (Ultipor N66 0,2μm, 0 2,5cm, Pall), montada sobre um suporte de aço inoxidável, sob pressão de nitrogênio (1bar máx.) e coletada em um béquer de vidro. 1.4.8 2a Filtração da solução da substância da droga
[00139] A solução da filtração anterior é então filtrada através de uma nova membrana de náilon de 0,2μm sob as mesmas condições. 1.4.9 Preenchimento de seringas
[00140] Seringas de vidro de 1ml foram assepticamente preenchidas com 0,5ml da solução final. 1.4.10 Temperatura durante o processo
[00141] Durante todo o processo a temperatura é mantida o mais próximo possível a condições refrigeradas, usando-se água de injeção refrigerada e armazenando a solução de excipiente e as soluções compostas a 2-8°C. Exemplo 2 - Formulação líquida de Interferon beta-1a livre de HSA de múltiplas doses em cartuchos adequados para um auto- injetor.
[00142] A necessidade de se desenvolver um produto de doses múltiplas em cartuchos surgiu durante o desenvolvimento de uma nova formulação livre de HSA que visava a eliminação de HSA do produto atualmente comercializado em seringas. A formulação em doses múltiplas aumentaria a conveniência ao permitir a auto-administração por meio de um auto-injetor.
[00143] Os agentes bacteriostáticos mais comumente usados (0,3% de m-cresol, 0,5% de fenol e 0,9% de benzil álcool) foram inicialmente estudados em combinação com a substância ativa e comparados à formula de dose única em seringa selecionada no quadro de desenvolvimento de dose única (44 ou 88mcg/ml de Interferon beta-1a, 54,6 mg/ml de manitol, 1mg/ml de Poloxamer 188, 0,12mg/ml de L-metionina em 10mM de tampão de acetato de sódio a pH 3,5); foram observados os seguintes dados: • A inclusão de cada um dos agentes bacteriostáticos, nas concentrações comumente usadas para prevenir a contaminação microbiana, determinou um aumento nas formas oxidadas e promoveu um dramático aumento na agregação; • 0,3% de m-cresol e a combinação de 0,5% de fenol com 0,1% de Poloxamer 188 determinou um aumento dramático em agregação.
[00144] Com base nas informações obtidas durante a pré-formulação, o desenvolvimento da formulação focou inicialmente em benzil álcool e fenol (sem Poloxamer 188) assim como em agentes bacteriostáticos adicionais (clorobutanol, feniletanol); o EDTA foi também investigado em combinação com benzil álcool: a oxidação e agregação da droga ativa foram as principais trilhas de degradação observadas; mostrou-se que a redução da quantidade de benzil álcool na formulação aumenta a vida de prateleira do produto.
[00145] Todos os outros agentes preservativos investigados durante esta fase não resultaram em um aprimoramento significante da estabilidade do produto.
[00146] No fim do desenvolvimento da formulação as formulações de dose múltipla candidatas a seguir foram identificadas: • Formulação B - 264mcg de Interferon beta-1a, 163,8mg de manitol, 3mg de Poloxamer 188, 0,36 mg de L-metionina, 6mg de benzil álcool em 3ml de 10mM de tampão de acetato de sódio pH 3,5 • Formulação A - 264mcg de Interferon beta-1a, 163,8mg de manitol, 3mg de Poloxamer 188, 0,36 mg de L-metionina em 2,7ml de 11mM tampão de acetato de sódio ph 3,5 são misturados com 0,3ml de 3% de benzil álcool em água de injeção obtendo-se assim a formulação de múltipla dose final.
[00147] Três lotes de escala em labotório foram fabricados para cada formulação candidata e testados quanto à estabilidade indicado métodos até 6 meses: nenhuma degradação significante ocorreu nas duas formulações candidatas mediante armazenamento a 2-8°C; a principal degradação a ocorrer mediante condições aceleradas (25°C) é a oxidação.
[00148] Ao final do estudo, duas formulações de múltiplas doses candidatas foram identificadas com um perfil de estabilidade comparável: • A Formulação B é uma formulação de dose múltipla pronta para uso contendo 0,2% de benzil álcool; • A Formulação A é uma formulação de dose múltipla contendo 0,3% de benzil álcool que é obtida após misturar-se o conteúdo de 2 cartuchos (um contendo o princípio ativo e os excipientes e um contendo a quantidade necessária de benzil álcool para se alcançar a apresentação final). 2.1 Objetivo do estudo
[00149] O objetivo desse estudo era desenvolver uma formulação livre de HSA de doses múltiplas de Interferon beta-1a a 264mcg em cartuchos de 3ml para permitir a administração por um auto-injetor. 2.2 Peça Experimental 2.2.1 Materiais • Interferon beta-1a (Serono S.A.) • Manitol DAB, Ph Eur, Farmacopéia Americana, FCC, E421 (Merck) • Ácido acético glacial 100% GR (Merck) • Pellets de hidróxido de sódio GR (Merck) • Poloxamer 188 (Lutrol F 68 DAC, Farmacopéia Americana /NF, BASF) • L-Metionina para bioquímica (Merck) • M-Cresol para síntese (Merck) • Fenol para síntese (Merck) • Benzil álcool Ph Eur, BP, NF (Merck) • Clorobutanol (Aldrich) • Feniletanol (Sigma) • Metilparabeno de sódio BP, Farmacopéia Americana /NF (Formenti) • Propilparabeno de sódio BP, Farmacopéia Americana /NF (Formenti) • Sal dissódico EDTA (Fluka) • 1,2-Propanodiol extrapuro DAB, Ph Eur, BP, Farmacopéia Americana (Merck) • Acetonitrila (Merck) • Ácido trifluoroacético (Baker) • Ácido heptafluorobutírico (Pierce) 2.2.2 Equipamentos • Sistemas de HPLC (Waters) • Software Millenium 32 (Waters) • Osmômetro (Osmomat 030-D, Gonotec) • Medidor de pH (mod. 654, Metrohm) • Pipetas calibradas (Gilson) • Membranas de náilon de 0,2μm Ultipor N66, FTKNF, 0 4,7cm (Pall) • Membranas de náilon de 0,2μm Ultipor N66, NR 14225, 0 14,2cm (Pall) • Suportes de aço inoxidável, 0 4,7cm e 0 10cm (Sartorius) • Tanque de aço inoxidável (Sartorius) • Coluna C4 5μm (0,46 x 25cm) (Baker) • Coluna C4, Supelcosil LC-304 5μm (0,46 x 25cm) (Supelco) • Coluna TSK, G2000SWXL (0,46 x 25cm) (TosoHaas) 2.3 Estudo Pré-Formulação
[00150] Os agentes bacteriostáticos mais comumente usados (0,3% de m-cresol, 0,5% de fenol e 0,9% de benzil álcool) foram inicialmente estudados em combinação com a substância ativa e diferentes misturas de excipiente no recipiente final (cartuchos de 3ml): tampão de acetato, tampão de acetato/manitol, tampão de acetato/manitol/L-Met/Poloxamer 188. A compatibilidade da substância ativa nos diferentes ambientes foi investigada em termos de oxidação (por HPLC de fase reversa) e agregação (por HPLC de exclusão molecular) mediante armazenamento a 40°C. Um resumo das formulações investigadas durante as diferentes etapas dessa fase preliminar é dado na Tabela 1.
[00151] O efeito da inclusão de cada agente bacteriostático foi comparado à fórmula de dose única (referência) selecionada no quadro de um desenvolvimento de dose única na seringa (44 ou 88mcg/ml de Interferon beta-1a, 54,6mg/ml de manitol, 1mg/ml de Poloxamer 188, 0,12mg/ml de L- metionina em 10mM de tampão de acetato de sódio a pH 3,5. Tabela 1: Composições das formulações de doses múltiplas de Interferon beta-1a (pré-formulação) Ace = 10mM de tampão de acetato de sódio pH 3,5; Man = 54,6mg/ml; Plu = 1mg/ml de Poloxamer 188; Met1 = 0,12mg/ml de L-metionina; Met2 = 0,24 de mg/ml de L-metionina; CR = 3mg/ml de m-cresol; PH = 5mg/ml de fenol; BA = 9mg/ml de benzil álcool 2.4 Desenvolvimento da Formulação
Ace = 10mM de tampão de acetato de sódio pH 3,5; Man = 54,6mg/ml; Plu = 1mg/ml de Poloxamer 188; Met1 = 0,12mg/ml de L-metionina; Met2 = 0,24 de mg/ml de L-metionina; CR = 3mg/ml de m-cresol; PH = 5mg/ml de fenol; BA = 9mg/ml de benzil álcool 2.4 Desenvolvimento da Formulação
[00152] Com base nas informações obtidas durante a pré-formulação, o desenvolvimento da formulação focou inicialmente em benzil álcool e fenol; agentes preservativos adicionais (clorobutanol, feniletanol) e EDTA combinados ao benzil álcool também foram investigados. Uma formulação comparativa (MS-3), correspondente à nova formulação livre de HSA da dose única de Interferon beta-1a, também foi preparada em cartuchos e usada como referência.
[00153] A composição (em mg/ml) das formulações fabricadas durante essa fase é relatada nas Tabelas 2 a 5: Tabela 2. Formulações de doses múltiplas de Interferon beta-1a contendo benzil álcool (composição em mg/ml)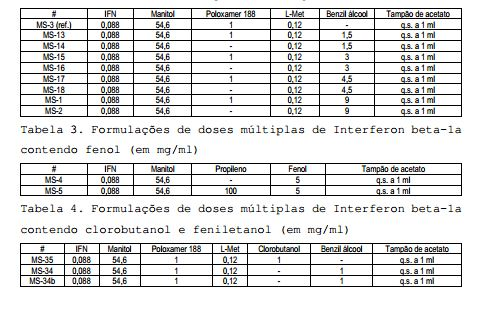
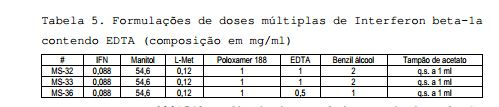
[00154] No fim do desenvolvimento da formulação as formulações de dose múltipla candidatas a seguir foram identificadas: • Formulação B - 264mcg de Interferon beta-1a em 3ml de tampão de acetato de sódio a pH 3,5 contendo 54,6mg/ml de manitol, 1 mg/ml de Poloxamer 188 e 0,12mg/ml de L-Metionina e 2mg/ml de benzil álcool (0,2% de benzil álcool) • Formulação A - 264mcg de Interferon beta-1a em 2,7ml de tampão de acetato de sódio a pH 3,5 contendo 54,6mg/ml de manitol, 1mg/ml de Poloxamer 188 e 0,12mg/ml de L-Metionina a serem misturados com 0,3ml de 3% de benzil álcool em água de injeção obtendo-se assim a formulação de múltipla dose final (0,3% de benzil álcool). 2.5 Teste de eficácia do preservativo
[00155] As formulações de múltiplas doses contendo diferentes concentrações de benzil álcool (0,2% a 0,9%) passaram por triagem para o teste de eficácia de preservativo de acordo com a Farmacopéia Européia e a Farmacopéia Americana.
[00156] Os testes preliminares foram feitos selecionando-se como indicadores o Staphylococcus aureus, Aspergillus niger e Candida albicans: a evidência de que o pH ácido da formulação tem em si um efeito bacteriostático sobre as bactérias (Staphylococcus aureus) e o fato de que algumas cepas de Candida albicans serem relatadas como sobreviventes mesmo a valores baixos de pH (pH por volta de 2) levou à seleção de Candida albicans como o indicador crítico do teste. Os critérios de aceitação do teste de eficácia de preservativo descrito na Farmacopéia Européia e na Farmacopéia Americana foram aplicados. 2.6 Formulações Candidatas 2.6.1 Formulação A
[00157] Três lotes de escala em laboratório de cerca de 140 cartuchos/lote foram fabricados; a fórmula é dada na Tabela 6: Tabela 6: Composição de Formulação A de doses múltiplas de Interferon beta-1a.
[00158] O ingrediente ativo foi composto com a solução de excipientes e então filtrado através de uma membrana de náilon de 0,22μm; os cartuchos foram preenchidos com 2,7ml de solução final. As amostras foram tiradas antes, após a filtração e após o preenchimento para monitoramento de perdas do princípio ativo durante o processo.
[00159] As amostras foram armazenadas e testadas quanto à estabilidade a 2-8°C (6 meses), 25±2°C (3 meses) e 40±2°C (1 mês).
[00160] Seis lotes de escala de laboratório de 3% de benzil álcool em água para injeção foram fabricados para serem misturados com os cartuchos contendo o ingrediente ativo; a composição dos lotes é dada na Tabela 7: Tabela 7 Composição de 3% de benzil álcool em água para injeção
[00161] A quantidade necessária de benzil álcool foi adicionada à água para injeção e então filtrada através de uma membrana Durapore de 0,22μm; os cartuchos foram então preenchidos com 0,5ml e por fim esterilizados por autoclave.
[00162] As amostras foram armazenadas a 25±2°C e testadas quanto ao conteúdo de benzil álcool e pH até 1 mês. 2.6.2 Formulação B
[00163] Três lotes de escala em laboratório de cerca de 500 cartuchos/lote foram fabricados; a fórmula é dada na Tabela 8: Tabela 8 Composição de Formulação B de doses múltiplas de Interferon beta-1a
[00164] O ingrediente ativo foi composto com a solução de excipientes e então filtrado através de uma membrana de náilon de 0,22μm; os cartuchos foram preenchidos com 3ml de solução final. As amostras foram tiradas antes, após a filtração e após o preenchimento para monitoramento de perdas do princípio ativo durante o processo.
[00165] As amostras foram armazenadas e testadas quanto à estabilidade a 2-8°C (6 meses), 25±2°C (6 meses) e 40±2°C (3 semanas). 2.6.3 Teste de eficácia do preservativo
[00166] Ambas as formulações candidatas foram testadas quanto à eficácia de preservativo de acordo com a Farmacopéia Européia e Farmacopéia Americana. 2.7 Testes e Métodos Analíticos
[00167] Os testes e métodos analíticos descritos a seguir foram usados para monitorar a estabilidade das formulações em escala de laboratório: pH (medição potenciométrico). Ensaio Quantificação de Proteína (HPLC de fase reversa).
[00168] A quantificação de proteína é feita em uma coluna de 5μm, C4, Wide Pore Butyl (Baker); o comprimento de onda é estabelecido a 214nm e a eluição é feita a 1ml/min usando a seguinte fase móvel e gradiente: • A = água/ácido trifluoroacético 0,1% • B = acetonitrila /ácido trifluoroacético 0,1% • C = acetonitrila Tabela G1 Gradiente:
[00169] As amostras são analisadas pela injeção de 100μl de uma amostra da forma como é (amostras de 44mcg/ml) ou com diluição (1:1) em um placebo equivalente para amostras a 88mcg/ml.
[00170] A quantificação das amostras é feita versus uma curva padrão na faixa de 0,0125mg/ml a 0,2mg/ml preparadas por um material de padrão de referência. Formas oxidadas (HPLC de fase reversa).
[00171] A quantificação das formas oxidadas é feita em uma coluna C4, Supelcosil LC-304 (Supelco) com termostato a 40°C; o comprimento de onda é estabelecido a 208nm e a eluição é feita a 1ml/min usando a seguinte fase móvel e gradiente:
[00172] As amostras são analisadas da forma como são por injeção de 200μl (amostras de 88mcg/ml) ou 400μl (amostras de 44mcg/ml). Total de agregados (HPLC de fase reversa).
[00173] A detecção do total de conteúdo de agregados é feito em uma coluna TSK G2000SWXL (TosoHaas); a eluição é feita no modo isocrático a 0,5ml/min usando acetonitrila:água (30:70) + 0,2% de ácido trifluoroacético; o comprimento de onda é estabelecido a 214nm. O tempo de execução é 20 min.
[00174] As amostras são analisadas da forma como são por injeção de 100μl (amostras de 88mcg/ml) ou 200μl (amostras de 44mcg/ml). Atividade biológica (bioensaio in vitro).
[00175] A atividade biológica é medida por um ensaio antiviral com base na proteção induzida de IFN-β de células (células WISH - tecido amniótico humano) contra o efeito citopático de um vírus (Vírus da Estomatite Vesicular). Osmolalidade (medição crioscópica).
[00176] A determinação de osmolalidade é feita por uma medição crioscópica com base na depressão de ponto de congelamento observada para a solução testada. Ensaio de benzil álcool (GC).
[00177] O método GC para detecção de benzil álcool usa a abordagem de calibração de ponto único, usando como referência o benzil álcool conforme fornecido por Merck. Além disso, um padrão interno (feniletil álcool) é usado para normalizar as áreas de pico das duas amostras de teste, solução de amostra de controle e da solução padrão. O método é conduzido em uma coluna de aço de 6pés x 2mm ID com 10% Carbowax 20M em malha supelcoport 80/100. O detector é um FID (detector de ionização de chama).
[00178] Os resultados são expressos como mg de benzil álcool por ml. 2.7.1 Resultados 2.7.1.1 Pré-formulação
[00179] Os níveis de formas oxidadas e total de agregados detectados mediante condições estressados (40°C) são mostrados nas Tabelas 9 e 10: Tabela 9: formas oxidadas nas formulações de doses múltiplas de Interferon beta-1a mediante armazenamento a 40°C (por HPLC de fase reversa).
 Ace = 10mM de tampão de acetato de sódio pH 3,5; Man = 54,6mg/ml; Plu = 1mg/ml de Poloxamer 188; Met1 = 0,12mg/ml de L-metionina; Met2 = 0,24 de mg/ml de L-metionina; CR = 3mg/ml de m-cresol; PH = 5mg/ml de fenol; BA = 9mg/ml de benzil álcool.
Ace = 10mM de tampão de acetato de sódio pH 3,5; Man = 54,6mg/ml; Plu = 1mg/ml de Poloxamer 188; Met1 = 0,12mg/ml de L-metionina; Met2 = 0,24 de mg/ml de L-metionina; CR = 3mg/ml de m-cresol; PH = 5mg/ml de fenol; BA = 9mg/ml de benzil álcool.
[00180] A inclusão de cada um dos agentes bacteriostáticos, nas concentrações comumente usadas para prevenir a contaminação microbiana, determinou um aumento nas formas oxidadas e promoveu um dramático aumento na agregação em comparação com a formulação de dose única (Referência).
[00181] Ocorreu a combinação de 0,5% fenol e 0,1% de Poloxamer 188 afetou de forma negativa a estabilidade do produto desde cerca de 40% de agregação (formulações P-Q- R). 0,3% de m-cresol foi excluído para posterior desenvolvimento uma vez que afeta de forma negativa a estabilidade do produto em função de um aumento na agregação (formulação I). 2.7.1.2 Desenvolvimento de Formulação
[00182] Todos os dados de estabilidade (dados brutos) são coletados na seção Tabelas; a análise de regressão linear foi usada para avaliar os dados. 2.7.1.2.1 Formulações Contendo Benzil Álcool
[00183] Uma alta concentração de benzil álcool (0,9) afetou negativamente a estabilidade do produto em termos de conteúdo de formas e agregados oxidados, conforme mostrado nas figuras 1 a 6: • Mediante condições estressadas (40°C), o aumento em oxidação é mais alto nas formulações contendo 0,9% de benzil álcool (MS-1 e MS-2), conforme mostrado na Figura 1; • Mediante condições aceleradas (25°C), o aumento em oxidação é mais alto nas formulações contendo benzil álcool comparado à formulação sem benzil álcool (MS-3, referência), conforme mostrado na Figura 2; • Mediante armazenamento em longo prazo (2-8°C); foram observadas taxas de oxidação mais altas nas formulações contendo 0,9% de benzil álcool (MS-1 e MS-2); foram detectadas taxas de degradação comparáveis nas formulações contendo benzil álcool abaixo de 0,45% (Figura 3).
[00184] Quanto ao nível de agregados foi observado o seguinte: • Mediante condições estressadas (40°C), o aumento em agregação é observado nas formulações contendo 0,9% de benzil álcool (MS-1 e MS-2), conforme mostrado nas Figuras 5 e 6; • Uma diminuição na bioatividade foi observada nas formulações com 0,9% benzil álcool (MS-1 e MS-2) a 40°C, isso não é observado nas mesmas amostras armazenadas a 25°C e 2-8°C. • Não foi observada diminuição da titulação mediante armazenamento em todas as temperaturas. • Nenhuma troca de pH ocorreu mediante armazenamento em todas as temperaturas. 2.7.1.2.2 Formulações contendo agentes bacteriostáticos alternativos (fenol, clorobutanol, feniletanol)
[00185] O fenol (MS-4 e MS-5) afetou negativamente a estabilidade do produto em termos de conteúdo de formas oxidadas enquanto o clorobutanol (MS-35) e feniletanol (MS-34 e MS-34b) mostrou uma estabilidade comparável à solução de referência (MS-3, sem bacteriostático), conforme mostrado nas figuras 7 e 8:
[00186] Quanto ao nível total de agregados, foi observado um aumento dramático na agregação mediante condições estressadas (40°C) nas formulações contendo fenol (MS-4 e MS- 5) conforme mostrado na Figura 9; não houve aumento da agregação nas temperaturas menores (25°C e 2-8°C). 2.7.1.2.3 Formulações contendo EDTA
[00187] A adição de EDTA a formulações contendo 0,1% a 0,2% de benzil álcool (MS-32, MS-33, MS-36) não diminuiu o nível de oxidação (Figura 10); um aumento no nível de agregados foi observado mediante condições aceleradas nas formulações contendo 0,1% EDTA e 0,2% de benzil álcool (MS-32, MS-33) (Figura 11); foi observada degradação comparável a 28°C. 2.7.1.2.4 Resultados de eficácia de preservativos.
[00188] Os resultados do estudo de triagem mostraram que os critérios de Farmacopéia Americana e Européia são atendidos no mínimo em concentrações de benzil álcool que são iguais ou maiores que 0,3% (para Candida albicans). 2.8 Formulações Candidatas
[00189] A avaliação de todos os dados foi feita da seguinte forma: uma análise de regressão linear foi feita em cada lote, seguida por análise de covariância (valor P > 0,25) para avaliar a variabilidade lote-a-lote; nenhuma análise estatística formal foi feita quando foi observada nenhuma variabilidade mediante armazenamento. 2.8.1 Candidato A 2.8.1.1 Recuperação durante a fabricação.
[00190] A recuperação de Interferon beta-1a nas amostras em processo (antes e depois da filtração, produto acabado) coletadas durante a fabricação do candidato A está resumida na Tabela 11: nenhuma perda significante de ingrediente ativo foi registrada. Tabela 11: % de recuperação de interferon beta-1a durante fabricação do candidato A
[00191] Foram calculados um desvio comum e intercepção no nível das formas oxidadas nos três lotes armazenados nas diferentes temperaturas; os resultados da análise estatística são mostrados na Tabela 12: Tabela 12: resultados das análises estatísticas de candidato A de Interferon beta-1a (formas oxidadas) 0,063 2,73 0,322 2,67 2,206 2,51
0,063 2,73 0,322 2,67 2,206 2,51
[00192] Nenhuma variabilidade foi observada no nível de total de agregados e no ensaio; portanto, não foi feita nenhuma análise estatística. O nível diferente nos agregados nos três lotes em função do nível diferente nos volumes usados na fabricação. 2.8.2 Candidato B 2.8.2.1 Recuperação durante a fabricação
[00193] A recuperação de Interferon beta-1a nas amostras em processo (antes e depois da filtração, produto acabado) coletadas durante a fabricação do candidato B está resumida na Tabela 15: nenhuma perda significante de ingrediente ativo foi registrada. Tabela 15: % de recuperação de interferon beta-1a durante fabricação do candidato B
[00194] A análise estatística feita no nível de formas oxidadas mostrou o seguinte: • Um desvio comum e intercepção foram calculados nos 3 lotes a 40°C: o desvio acumulado é igual a 1,42%/semana e a intercepção acumulada é igual a 2,72%; • Nenhum desvio comum pôde ser calculado nos 3 lotes a 25°C (valor P = 0,095); portanto, foi usada a abordagem de pior caso (lote MS-03): foi observado um aumento de 1,17% nas formas oxidadas após 1 mês de armazenamento (0,27%/semana); • Nenhum desvio comum pôde ser calculado nos 3 lotes a 2-8°C (valor P = 0,016); portanto, foi usada a abordagem de pior caso (lote MS-03): foi observado um aumento de 0,2% nas formas oxidadas após 1 mês de armazenamento (0,047%/semana);
[00195] Nenhuma variabilidade significante foi observada dos dados de estabilidade no nível de total de agregados e o ensaio; portanto, não foi feita análise estatística formal.
[00196] Não foi observada diminuição na bioatividade com o armazenamento, mesmo mediante condições estressadas (40°C). 2.9 Conclusões
[00197] Ao final do estudo, duas formulações de múltiplas doses foram identificadas com um perfil de estabilidade comparável: • A Formulação Candidata B é uma formulação de múltiplas doses pronta para uso contendo 0,2% de benzil álcool; • A Formulação Candidata A é uma formulação de dose múltipla contendo 0,3% de benzil álcool que é obtida após misturar-se o conteúdo de 2 cartuchos (um contendo o princípio ativo e os excipientes e um contendo a quantidade necessária de benzil álcool para se alcançar a apresentação final).
[00198] - Essas soluções candidatas também foram testadas a um pH maior (4,5±0,2) e não foram mostradas mudanças no perfil de estabilidade. O Requerente descobriu agora que esse pH levemente maior das formulações de IFN aumenta a tolerabilidade local de injeções subcutâneas. Portanto, as 2 soluções candidatas acima a um pH de 4,5 ou 4,7 poderiam oferecer mais vantagens em termos de conformidade com o paciente. Exemplo 3: Método de Fabricação para Múltiplas Doses - Formulação Candidata A
[00199] Primeiramente, a solução de hidróxido de sódio 1N foi preparada pela dissolução de 20g de pellets de hidróxido de sódio em 500g de água para injeção.
[00200] Em seguida o tampão de acetato de sódio com pH a 3,5±0,2 foi preparado pela adição de 1,32g de ácido acético glacial a cerca de 1800g de água de injeção. O pH da solução a 3,5±0,2 foi ajustado com 1N NaOH. Então a solução foi levada ao peso final de 2000g. O pH foi então ajustado novamente a 3,5±0,2 com 1N NaOH ou 50% de ácido acético diluído. A solução é então levada ao peso final de 2000g.
[00201] A solução final foi preparada conforme segue.
[00202] A quantidade calculada de excipientes foi pesada e dissolvida na quantidade necessária de 11mM de tampão de acetato de sódio a pH 3,5±0,2 e o pH da solução checado e ajustado (se necessário); então é adicionada a quantidade necessária de r-h Interferon beta-1a (produzido de forma recombinante a partir de células de ovário de hamster); o peso final é alcançado pela adição de 11mM de tampão de acetato de sódio a pH 3,5±0,2.
[00203] 1ml dessa solução final foi retirado para amostra por HPLC de fase reversa quantitativa (amostra AF = Antes da 1a Filtração).
[00204] A solução final foi então filtrada através de uma membrana de 0,2μm montada sobre um suporte de aço inoxidável e a solução coletada para o béquer de vidro.
[00205] 1ml dessa solução final foi retirado para amostra para ser testado por HPLC de fase reversa.
[00206] A solução final filtrada da forma descrita no ponto anterior foi filtrada através de uma 2a membrana de 0,2μm montada sobre um suporte de aço inoxidável.
[00207] 1ml dessa solução final foi retirado para amostra para ser testado por HPLC quantitativa de fase reversa.
[00208] Cartuchos de vidro de 3ml foram preenchidos com 2,7ml da solução final e foram tampados.
[00209] Essa solução está pronta para ser misturada com um cartucho contendo a solução de benzil álcool em água para injeção. Exemplo 4: Método de Fabricação para Múltiplas Doses - Formulação Candidata B.
[00210] Primeiramente, a solução de hidróxido de sódio 1N foi preparada pela dissolução de 20g de pellets de hidróxido de sódio em 500g de água para injeção.
[00211] Em seguida 0,011M de tampão de acetato de sódio com pH a 3,5±0,2 foi preparado pela adição de 1,32g de ácido acético glacial a cerca de 1800g de água de injeção. O pH da solução a 3,5±0,2 foi ajustado com 1N de NaOH. Então a solução foi levada ao peso final de 2000g. O pH foi então ajustado novamente a 3,5±0,2 com 1N de NaOH ou 50% de ácido acético diluído. A solução é então levada ao peso final de 2000g.
[00212] A solução final foi preparada conforme segue.
[00213] A quantidade calculada de excipientes foi pesada e dissolvida na quantidade necessária de 10mM de tampão de acetato de sódio a pH 3,5±0,2 e o pH da solução checado e ajustado (se necessário); então é adicionada a quantidade necessária de r-h Interferon beta-1a (produzido de forma recombinante a partir de células de ovário de hamster); o peso final é alcançado pela adição de 10mM de tampão de acetato de sódio a pH 35±0,2.
[00214] 1ml dessa solução final foi retirado para amostra por HPLC de fase reversa quantitativa.
[00215] A solução final foi então filtrada através de uma membrana de 0,2μm montada sobre um suporte de aço inoxidavel e a solucao coletada para o béquer de vidro.
[00216] 1ml dessa solução final foi retirado para amostra para ser testado por HPLC quantitativa de fase reversa.
[00217] A solução final filtrada da forma descrita no ponto anterior foi filtrada através de uma 2a membrana de 0,2μm montada sobre um suporte de aço inoxidável.
[00218] 1ml dessa solução final foi retirado para amostra para ser testado por HPLC quantitativa de fase reversa.
[00219] Cartuchos de vidro de 3ml foram preenchidos com 3ml da solução final e foram tampados. REFERÊNCIAS 1. Grupo de Estudo. The Lancet 1998; 352, 1498-1504. 2. Clegg e Bryant, Exp. Opin. Parmacother 2001; 2(4): 623-639 3. Derynk R. et al., Nature 1980; 285, 542-547. 4. Familetti, P. C. Rubinstein, S., e Pestka, S. 1981 “Um Ensaio Conveniente e Rápido de Inibição de Efeito Citopático de Interferon”, em Methods in Enzymology, Vol. 78 (S. Pestka, ed.), Academic Press, Nova Iorque, 387-394; 5. Hultgren C., Milich DR, Weiland O., Sallberg M. (1998). “O composto antiviral modula o ajudante T (Th) 1/Th2 equilíbrio de subconjunto nas respostas imunológicas específicas de vírus de hepatite B e C”. J Gen Virol 1998; 79: 2381-2391. 6. McCormick JB, King IJ, Webb PA, Scribner CL, Craven RB, Johnson KM, Elliott LH, Belmont-Williams R. “Febre Lassa. Terapia eficaz com ribavirina”. N Engl J Méd. 1986 2 de janeiro; 314(1): 20-6. 7. Mark D. F. et al., Proc. Natl. Acad. Sci. EUA 81 (18) 56625666 (1984). 8. Pestka, S. (1986) “Padrões de interferon e abreviações gerais” in Methods in Enzymology (S. Pestka, et.) Academic Press, Nova Iorque 119, 14-23. 9. Rubinstein, S., Familetti, P.C. e Pestka, S. ”Convenient Assay for Interferons.“ J Virol 1981; 37, 755-758. 10. Shepard H. M. et al., Nature 1981; 294, 563-565.
Claims (11)
1. “COMPOSIÇÃO FARMACÊUTICA LÍQUIDA ESTABILIZADA E LIVRE DE HSA”, compreendendo um interferon beta (IFN-beta), caracterizada por a referida formulação ser uma solução aquosa de Interferon-β recombinante humano presente em uma concentração de 10μg/ml a 800μg/ml, que compreende um tampão de acetato presente em uma concentração de 5mM a 500mM, preferivelmente 10mM; um surfactante, poloxâmero 188, presente em uma concentração de 0,01mg/ml a 10mg/ml, preferivelmente 1mg/ml; um agente bacteriostático, álcool benzílico, presente em uma concentração de 0,1% a 2,0%, preferivelmente 0,2 a 0,3%; um agente de isotonicidade, manitol, presente em uma concentração de 0,5mg/ml a 500mg/ml, preferivelmente 55mg/ml; e um antioxidante, metionina, presente em uma concentração de 0,01mg/ml a 5,0mg/ml, preferivelmente 0,1mg/ml.
2. “COMPOSIÇÃO FARMACÊUTICA LÍQUIDA ESTABILIZADA E LIVRE DE HSA”, de acordo com a reivindicação 1, caracterizada por o tampão de acetato ajustar o pH da composição dentro de ±0,5 unidades de pH especificado, entre 3,0 e 5,0.
3. “COMPOSIÇÃO FARMACÊUTICA LÍQUIDA ESTABILIZADA E LIVRE DE HSA”, de acordo com a reivindicação 2, caracterizada por o pH ser 3,5±0,2.
4. “COMPOSIÇÃO FARMACÊUTICA LÍQUIDA ESTABILIZADA E LIVRE DE HSA”, de acordo com a reivindicação 3, caracterizada por o pH ser 4,5±0,2.
5. “COMPOSIÇÃO FARMACÊUTICA LÍQUIDA ESTABILIZADA E LIVRE DE HSA”, de acordo com qualquer uma das reivindicações anteriores, caracterizada por o interferon beta recombinante humano estar presente, preferivelmente, em uma concentração de 22, 44, 88 ou 264μg/ml.
6. “MÉTODO PARA PREPARAÇÃO DE COMPOSIÇÃO FARMACÊUTICA LÍQUIDA ESTABILIZADA E LIVRE DE HSA”, para preparação da composição definida nas reivindicações de 1 a 5, caracterizado por o referido método compreender a adição de uma quantidade calculada de surfactante poloxâmero 188, antioxidante metionina e agente de isotonicidade à solução tamponada e, em seguida, a adição do interferon beta (IFN- beta).
7. “RECIPIENTE VEDADO HERMETICAMENTE”, em condições estéreis e adequado para armazenamento antes do uso, caracterizado por compreender a formulação farmacêutica líquida definida nas reivindicações de 1 a 5.
8. “RECIPIENTE VEDADO HERMETICAMENTE”, de acordo com a reivindicação 7, caracterizado por o referido recipiente ser uma seringa pré-preenchida para administração de dose única.
9. “RECIPIENTE VEDADO HERMETICAMENTE”, de acordo com a reivindicação 7, caracterizado por o referido recipiente ser um frasco.
10. “RECIPIENTE VEDADO HERMETICAMENTE”, de acordo com a reivindicação 7, caracterizado por o referido recipiente ser um cartucho para um autoinjetor.
11. “RECIPIENTE VEDADO HERMETICAMENTE”, de acordo com qualquer uma das reivindicações 8 ou 9, caracterizado por o referido recipiente servir para administração de dose única ou de doses múltiplas.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP03101210.7 | 2003-05-01 | ||
| EP03101210 | 2003-05-01 | ||
| US53016903P | 2003-12-17 | 2003-12-17 | |
| US60/530,169 | 2003-12-17 | ||
| PCT/EP2004/004806 WO2004096263A2 (en) | 2003-05-01 | 2004-04-29 | Human serum albumin-free stabilized interferon liquid formulations |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| BRPI0410488A BRPI0410488A (pt) | 2006-06-13 |
| BRPI0410488B1 true BRPI0410488B1 (pt) | 2021-06-22 |
| BRPI0410488B8 BRPI0410488B8 (pt) | 2021-09-08 |
Family
ID=36908159
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0410488A BRPI0410488B8 (pt) | 2003-05-01 | 2004-04-29 | Composição farmacêutica líquida estabilizada e livre de hsa, método para preparação de composição farmacêutica líquida estabilizada e livre de hsa, e recipiente vedado hermeticamente |
Country Status (29)
| Country | Link |
|---|---|
| US (1) | US8309069B2 (pt) |
| EP (1) | EP1617861B1 (pt) |
| JP (2) | JP4870550B2 (pt) |
| KR (1) | KR101116058B1 (pt) |
| CN (1) | CN1816347B (pt) |
| AR (1) | AR044147A1 (pt) |
| AU (2) | AU2004233603B2 (pt) |
| BR (1) | BRPI0410488B8 (pt) |
| CA (1) | CA2521560A1 (pt) |
| DK (1) | DK1617861T3 (pt) |
| EA (2) | EA200800327A1 (pt) |
| ES (1) | ES2417061T3 (pt) |
| HK (1) | HK1088847A1 (pt) |
| HR (1) | HRP20130512T1 (pt) |
| IL (1) | IL171709A (pt) |
| ME (1) | ME00404B (pt) |
| MX (1) | MXPA05011718A (pt) |
| MY (1) | MY151121A (pt) |
| NO (1) | NO335674B1 (pt) |
| NZ (2) | NZ542912A (pt) |
| PL (1) | PL1617861T3 (pt) |
| PT (1) | PT1617861E (pt) |
| RS (1) | RS52869B (pt) |
| SG (1) | SG159389A1 (pt) |
| SI (1) | SI1617861T1 (pt) |
| TW (1) | TWI272948B (pt) |
| UA (2) | UA80331C2 (pt) |
| WO (1) | WO2004096263A2 (pt) |
| ZA (2) | ZA200508124B (pt) |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE526032T1 (de) * | 2003-12-11 | 2011-10-15 | Ares Trading Sa | Stabilisierte flüssige interferon-formulierungen |
| CN1984673A (zh) * | 2004-05-17 | 2007-06-20 | 阿雷斯贸易股份有限公司 | 干扰素水凝胶制剂 |
| EA010979B1 (ru) * | 2004-06-01 | 2008-12-30 | Арес Трейдинг С.А. | Стабилизированные жидкие препаративные формы интерферона |
| BRPI0510527A (pt) * | 2004-06-01 | 2007-10-30 | Ares Trading Sa | método de estabilização de proteìnas |
| UA95461C2 (en) * | 2005-09-14 | 2011-08-10 | Арес Трейдинг С.А. | Method for quantitative determination of poloxamers |
| AU2007253254B2 (en) | 2006-05-24 | 2013-01-17 | Merck Serono Sa | Cladribine regimen for treating multiple sclerosis |
| WO2008066322A1 (en) * | 2006-11-28 | 2008-06-05 | Daewoong Co., Ltd. | A stable hsa-free and antioxidant-free pharmaceutical composition comprising interferon-beta |
| WO2008065752A1 (fr) * | 2006-11-30 | 2008-06-05 | National University Corporation Hokkaido University | Agent immunothérapeutique contenant un arndi en tant que principe actif |
| PL2134353T3 (pl) * | 2007-03-30 | 2017-06-30 | Helix Biopharma Corp. | Dwufazowa kompozycja pęcherzyków lipidowych i sposób leczenia dysplazji szyjki macicy przez podawanie dopochwowe |
| WO2008145323A1 (en) * | 2007-05-31 | 2008-12-04 | F. Hoffmann-La Roche Ag | Pharmaceutical formulation for interferons |
| RS52417B (en) | 2007-12-20 | 2013-02-28 | Merck Serono S.A. | PEG-INTERFERON-BETA FORMULATIONS |
| KR20150074167A (ko) | 2012-10-26 | 2015-07-01 | 루핀 리미티드 | Peg 인터페론 알파-2b의 안정한 약학 조성물 |
| US8986732B2 (en) | 2013-08-12 | 2015-03-24 | Helix Biopharma Corporation | Biphasic lipid-vesicle compositions and methods for treating cervical dysplasia by intravaginal delivery |
| US10159646B2 (en) | 2013-08-12 | 2018-12-25 | Altum-Avro Pharma Partnership | Biphasic lipid-vesicle compositions and methods for treating cervical dysplasia by intravaginal delivery |
| WO2016046101A1 (en) * | 2014-09-23 | 2016-03-31 | F. Hoffmann-La Roche Ag | Stable, benzyl alcohol-free aqueous solution formulations containing alpha-type interferon |
| US9937223B2 (en) | 2015-01-30 | 2018-04-10 | Par Pharmaceutical, Inc. | Vasopressin formulations for use in treatment of hypotension |
| US9744209B2 (en) | 2015-01-30 | 2017-08-29 | Par Pharmaceutical, Inc. | Vasopressin formulations for use in treatment of hypotension |
| US9687526B2 (en) | 2015-01-30 | 2017-06-27 | Par Pharmaceutical, Inc. | Vasopressin formulations for use in treatment of hypotension |
| US9744239B2 (en) | 2015-01-30 | 2017-08-29 | Par Pharmaceutical, Inc. | Vasopressin formulations for use in treatment of hypotension |
| US9925233B2 (en) | 2015-01-30 | 2018-03-27 | Par Pharmaceutical, Inc. | Vasopressin formulations for use in treatment of hypotension |
| US9750785B2 (en) | 2015-01-30 | 2017-09-05 | Par Pharmaceutical, Inc. | Vasopressin formulations for use in treatment of hypotension |
| KR101943160B1 (ko) | 2016-10-06 | 2019-01-30 | 에이비온 주식회사 | 인터페론 베타 변이체의 안정화 제제 |
Family Cites Families (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6936694B1 (en) | 1982-05-06 | 2005-08-30 | Intermune, Inc. | Manufacture and expression of large structural genes |
| US4588585A (en) | 1982-10-19 | 1986-05-13 | Cetus Corporation | Human recombinant cysteine depleted interferon-β muteins |
| US4737462A (en) | 1982-10-19 | 1988-04-12 | Cetus Corporation | Structural genes, plasmids and transformed cells for producing cysteine depleted muteins of interferon-β |
| US4959314A (en) | 1984-11-09 | 1990-09-25 | Cetus Corporation | Cysteine-depleted muteins of biologically active proteins |
| US5116943A (en) | 1985-01-18 | 1992-05-26 | Cetus Corporation | Oxidation-resistant muteins of Il-2 and other protein |
| US5017691A (en) | 1986-07-03 | 1991-05-21 | Schering Corporation | Mammalian interleukin-4 |
| US4879111A (en) | 1986-04-17 | 1989-11-07 | Cetus Corporation | Treatment of infections with lymphokines |
| US4965195A (en) | 1987-10-26 | 1990-10-23 | Immunex Corp. | Interleukin-7 |
| US4904584A (en) | 1987-12-23 | 1990-02-27 | Genetics Institute, Inc. | Site-specific homogeneous modification of polypeptides |
| IT1272252B (it) * | 1994-05-16 | 1997-06-16 | Applied Research Systems | Formulazioni liquide di interferone beta |
| JP2758154B2 (ja) * | 1995-04-06 | 1998-05-28 | エフ・ホフマン−ラ ロシユ アーゲー | インターフェロンを含む液体製剤 |
| ZA9610374B (en) * | 1995-12-11 | 1997-06-23 | Elan Med Tech | Cartridge-based drug delivery device |
| JP4878664B2 (ja) | 1996-12-24 | 2012-02-15 | バイオジェン・アイデック・エムエイ・インコーポレイテッド | 安定な液体インターフェロン処方物 |
| US6013253A (en) | 1997-08-15 | 2000-01-11 | Amgen, Inc. | Treatment of multiple sclerosis using consensus interferon and IL-1 receptor antagonist |
| CA2304808C (en) * | 1997-09-23 | 2011-03-22 | Michael Tschope | Liquid interferon-.beta. formulations |
| US6299869B1 (en) * | 1997-12-08 | 2001-10-09 | Genentech, Inc. | Human interferon-epsilon: a type I interferon |
| ES2224649T3 (es) | 1998-04-28 | 2005-03-01 | Applied Research Systems Ars Holding N.V. | Conjugados de poliol-ifn-beta. |
| CA2331816C (en) * | 1998-05-11 | 2010-11-16 | Basf Aktiengesellschaft | Method for producing isoxazoline-3-yl-acyl benzene |
| CN1175901C (zh) * | 1999-12-06 | 2004-11-17 | 天津华立达生物工程有限公司 | 一种稳定的干扰素水溶液 |
| US6465425B1 (en) * | 2000-02-10 | 2002-10-15 | Alkermes Controlled Therapeutics, Inc. | Microencapsulation and sustained release of biologically active acid-stable or free sulfhydryl-containing proteins |
| US6887462B2 (en) | 2001-04-09 | 2005-05-03 | Chiron Corporation | HSA-free formulations of interferon-beta |
| UY27373A1 (es) * | 2001-07-09 | 2003-02-28 | Schering Ag | Formulaciones de interferón beta-humano |
| ATE526032T1 (de) | 2003-12-11 | 2011-10-15 | Ares Trading Sa | Stabilisierte flüssige interferon-formulierungen |
| CN1984673A (zh) | 2004-05-17 | 2007-06-20 | 阿雷斯贸易股份有限公司 | 干扰素水凝胶制剂 |
| BRPI0510527A (pt) | 2004-06-01 | 2007-10-30 | Ares Trading Sa | método de estabilização de proteìnas |
-
2004
- 2004-04-23 TW TW093111368A patent/TWI272948B/zh active
- 2004-04-29 PL PL04730264T patent/PL1617861T3/pl unknown
- 2004-04-29 PT PT47302641T patent/PT1617861E/pt unknown
- 2004-04-29 AU AU2004233603A patent/AU2004233603B2/en not_active Expired
- 2004-04-29 CA CA002521560A patent/CA2521560A1/en not_active Withdrawn
- 2004-04-29 EA EA200800327A patent/EA200800327A1/ru unknown
- 2004-04-29 SG SG200705206-1A patent/SG159389A1/en unknown
- 2004-04-29 RS YU20050821A patent/RS52869B/sr unknown
- 2004-04-29 EA EA200501699A patent/EA009995B1/ru not_active IP Right Cessation
- 2004-04-29 KR KR1020057020444A patent/KR101116058B1/ko active IP Right Grant
- 2004-04-29 UA UAA200509798A patent/UA80331C2/uk unknown
- 2004-04-29 JP JP2006505383A patent/JP4870550B2/ja not_active Expired - Lifetime
- 2004-04-29 CN CN2004800188759A patent/CN1816347B/zh not_active Expired - Lifetime
- 2004-04-29 SI SI200432066T patent/SI1617861T1/sl unknown
- 2004-04-29 NZ NZ542912A patent/NZ542912A/en unknown
- 2004-04-29 MY MYPI20041605 patent/MY151121A/en unknown
- 2004-04-29 NZ NZ566625A patent/NZ566625A/en unknown
- 2004-04-29 US US10/554,602 patent/US8309069B2/en active Active
- 2004-04-29 EP EP04730264.1A patent/EP1617861B1/en not_active Expired - Lifetime
- 2004-04-29 UA UAA200704143A patent/UA94032C2/ru unknown
- 2004-04-29 ME MEP-2008-604A patent/ME00404B/me unknown
- 2004-04-29 BR BRPI0410488A patent/BRPI0410488B8/pt active IP Right Grant
- 2004-04-29 WO PCT/EP2004/004806 patent/WO2004096263A2/en active Application Filing
- 2004-04-29 MX MXPA05011718A patent/MXPA05011718A/es active IP Right Grant
- 2004-04-29 ZA ZA200508124A patent/ZA200508124B/xx unknown
- 2004-04-29 ES ES04730264T patent/ES2417061T3/es not_active Expired - Lifetime
- 2004-04-29 DK DK04730264.1T patent/DK1617861T3/da active
- 2004-04-30 AR ARP040101486A patent/AR044147A1/es not_active Application Discontinuation
-
2005
- 2005-11-01 IL IL171709A patent/IL171709A/en active IP Right Grant
- 2005-11-30 NO NO20055666A patent/NO335674B1/no unknown
-
2006
- 2006-10-03 HK HK06110914.6A patent/HK1088847A1/xx not_active IP Right Cessation
-
2007
- 2007-10-04 ZA ZA200708484A patent/ZA200708484B/xx unknown
-
2009
- 2009-03-31 AU AU2009201261A patent/AU2009201261A1/en not_active Abandoned
-
2011
- 2011-07-15 JP JP2011156524A patent/JP5346065B2/ja not_active Expired - Lifetime
-
2013
- 2013-06-11 HR HRP20130512TT patent/HRP20130512T1/hr unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5346065B2 (ja) | Hsaを含まない安定なインターフェロン液体製剤 | |
| EP1750751B1 (en) | Stabilized interferon liquid formulations | |
| US7879320B2 (en) | Hydrogel interferon formulations | |
| US7846427B2 (en) | Stabilized interferon liquid formulations | |
| KR101191536B1 (ko) | 안정화된 인터페론 액상 제제 | |
| MXPA06006579A (en) | Stabilized interferon liquid formulations | |
| MXPA06013308A (es) | Formulaciones de hidrogel que contienen interferon. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] | ||
| B07E | Notification of approval relating to section 229 industrial property law [chapter 7.5 patent gazette] |
Free format text: NOTIFICACAO DE ANUENCIA RELACIONADA COM O ART 229 DA LPI |
|
| B07A | Application suspended after technical examination (opinion) [chapter 7.1 patent gazette] | ||
| B06A | Patent application procedure suspended [chapter 6.1 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 29/04/2004, OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF, , QUE DETERMINA A ALTERACAO DO PRAZO DE CONCESSAO. |
|
| B16C | Correction of notification of the grant [chapter 16.3 patent gazette] |
Free format text: REF. RPI 2633 DE 22/06/2021 QUANTO AOS DESENHOS. |