BR112015004274B1 - Método para a produção de dímeros, trímeros, oligômeros e polímeros a partir da oxidação de um átomo de hidrogênio alfa para um anel aromático - Google Patents
Método para a produção de dímeros, trímeros, oligômeros e polímeros a partir da oxidação de um átomo de hidrogênio alfa para um anel aromático Download PDFInfo
- Publication number
- BR112015004274B1 BR112015004274B1 BR112015004274-0A BR112015004274A BR112015004274B1 BR 112015004274 B1 BR112015004274 B1 BR 112015004274B1 BR 112015004274 A BR112015004274 A BR 112015004274A BR 112015004274 B1 BR112015004274 B1 BR 112015004274B1
- Authority
- BR
- Brazil
- Prior art keywords
- reaction
- gas
- aromatic ring
- nitrous oxide
- starting material
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 84
- 229920000642 polymer Polymers 0.000 title claims abstract description 29
- 239000000539 dimer Substances 0.000 title claims description 20
- 238000004519 manufacturing process Methods 0.000 title claims description 9
- 239000013638 trimer Substances 0.000 title claims description 5
- 230000003647 oxidation Effects 0.000 title claims description 4
- 238000007254 oxidation reaction Methods 0.000 title claims description 4
- YZCKVEUIGOORGS-UHFFFAOYSA-N Hydrogen atom Chemical compound [H] YZCKVEUIGOORGS-UHFFFAOYSA-N 0.000 title 1
- 238000006243 chemical reaction Methods 0.000 claims abstract description 107
- GQPLMRYTRLFLPF-UHFFFAOYSA-N Nitrous Oxide Chemical compound [O-][N+]#N GQPLMRYTRLFLPF-UHFFFAOYSA-N 0.000 claims abstract description 70
- 239000007789 gas Substances 0.000 claims abstract description 55
- 239000001272 nitrous oxide Substances 0.000 claims abstract description 35
- 150000001875 compounds Chemical class 0.000 claims abstract description 29
- 230000015572 biosynthetic process Effects 0.000 claims abstract description 21
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 claims abstract description 15
- 239000008096 xylene Substances 0.000 claims abstract description 15
- 239000007858 starting material Substances 0.000 claims description 34
- 239000007795 chemical reaction product Substances 0.000 claims description 27
- 239000000178 monomer Substances 0.000 claims description 23
- 125000003118 aryl group Chemical group 0.000 claims description 21
- 238000009833 condensation Methods 0.000 claims description 21
- 230000005494 condensation Effects 0.000 claims description 21
- 230000005291 magnetic effect Effects 0.000 claims description 21
- 230000005684 electric field Effects 0.000 claims description 17
- 239000011261 inert gas Substances 0.000 claims description 15
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 12
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 claims description 9
- 239000001257 hydrogen Substances 0.000 claims description 5
- 229910052739 hydrogen Inorganic materials 0.000 claims description 5
- 239000011541 reaction mixture Substances 0.000 claims description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 4
- 238000002156 mixing Methods 0.000 claims description 3
- 238000010438 heat treatment Methods 0.000 claims description 2
- 125000004429 atom Chemical group 0.000 claims 1
- 230000008569 process Effects 0.000 abstract description 32
- 239000012048 reactive intermediate Substances 0.000 abstract description 21
- OOLUVSIJOMLOCB-UHFFFAOYSA-N 1633-22-3 Chemical compound C1CC(C=C2)=CC=C2CCC2=CC=C1C=C2 OOLUVSIJOMLOCB-UHFFFAOYSA-N 0.000 abstract description 11
- 239000000543 intermediate Substances 0.000 abstract description 11
- 238000000197 pyrolysis Methods 0.000 abstract description 11
- 230000008021 deposition Effects 0.000 abstract description 8
- 239000012159 carrier gas Substances 0.000 abstract description 5
- 238000007429 general method Methods 0.000 abstract description 4
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 27
- 239000001301 oxygen Substances 0.000 description 27
- 229910052760 oxygen Inorganic materials 0.000 description 27
- 239000000463 material Substances 0.000 description 26
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 24
- 238000006116 polymerization reaction Methods 0.000 description 21
- NRNFFDZCBYOZJY-UHFFFAOYSA-N p-quinodimethane Chemical group C=C1C=CC(=C)C=C1 NRNFFDZCBYOZJY-UHFFFAOYSA-N 0.000 description 18
- 239000000047 product Substances 0.000 description 18
- 239000007787 solid Substances 0.000 description 15
- URLKBWYHVLBVBO-UHFFFAOYSA-N Para-Xylene Chemical group CC1=CC=C(C)C=C1 URLKBWYHVLBVBO-UHFFFAOYSA-N 0.000 description 12
- 229910052786 argon Inorganic materials 0.000 description 12
- 239000000203 mixture Substances 0.000 description 12
- 229920000052 poly(p-xylylene) Polymers 0.000 description 12
- 238000001816 cooling Methods 0.000 description 11
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 10
- 230000006872 improvement Effects 0.000 description 9
- 239000007788 liquid Substances 0.000 description 9
- 238000003786 synthesis reaction Methods 0.000 description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 8
- 238000000354 decomposition reaction Methods 0.000 description 8
- 230000004048 modification Effects 0.000 description 8
- 238000012986 modification Methods 0.000 description 8
- 238000000151 deposition Methods 0.000 description 7
- 150000003839 salts Chemical class 0.000 description 7
- 125000001424 substituent group Chemical group 0.000 description 7
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- 229910001882 dioxygen Inorganic materials 0.000 description 6
- 230000007246 mechanism Effects 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 230000000704 physical effect Effects 0.000 description 6
- -1 poly(p-xylylene) Polymers 0.000 description 6
- 230000004913 activation Effects 0.000 description 5
- 238000000576 coating method Methods 0.000 description 5
- 238000001704 evaporation Methods 0.000 description 5
- 230000008020 evaporation Effects 0.000 description 5
- 239000011521 glass Substances 0.000 description 5
- 230000009257 reactivity Effects 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- 239000000758 substrate Substances 0.000 description 5
- 238000009834 vaporization Methods 0.000 description 5
- 230000008016 vaporization Effects 0.000 description 5
- 125000006839 xylylene group Chemical group 0.000 description 5
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 230000037361 pathway Effects 0.000 description 4
- 238000005086 pumping Methods 0.000 description 4
- 239000007790 solid phase Substances 0.000 description 4
- 238000007711 solidification Methods 0.000 description 4
- 230000008023 solidification Effects 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 239000011248 coating agent Substances 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 235000021056 liquid food Nutrition 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 235000005985 organic acids Nutrition 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 235000021055 solid food Nutrition 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 2
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 2
- 229910000990 Ni alloy Inorganic materials 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- 239000006227 byproduct Substances 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 238000006555 catalytic reaction Methods 0.000 description 2
- 239000007805 chemical reaction reactant Substances 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 239000004020 conductor Substances 0.000 description 2
- 239000000112 cooling gas Substances 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 239000003989 dielectric material Substances 0.000 description 2
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 2
- 230000005284 excitation Effects 0.000 description 2
- 230000001747 exhibiting effect Effects 0.000 description 2
- 230000004907 flux Effects 0.000 description 2
- 238000004817 gas chromatography Methods 0.000 description 2
- 238000010574 gas phase reaction Methods 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 229910001026 inconel Inorganic materials 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 150000002688 maleic acid derivatives Chemical class 0.000 description 2
- 229910044991 metal oxide Inorganic materials 0.000 description 2
- 150000004706 metal oxides Chemical class 0.000 description 2
- 229910003455 mixed metal oxide Inorganic materials 0.000 description 2
- 229910001172 neodymium magnet Inorganic materials 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 2
- 230000006641 stabilisation Effects 0.000 description 2
- 238000011105 stabilization Methods 0.000 description 2
- 239000010935 stainless steel Substances 0.000 description 2
- 229910001220 stainless steel Inorganic materials 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 238000005979 thermal decomposition reaction Methods 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- 238000005985 Hofmann elimination reaction Methods 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 239000007772 electrode material Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000013461 intermediate chemical Substances 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 239000010453 quartz Substances 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N silicon dioxide Inorganic materials O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 238000001771 vacuum deposition Methods 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2/00—Preparation of hydrocarbons from hydrocarbons containing a smaller number of carbon atoms
- C07C2/02—Preparation of hydrocarbons from hydrocarbons containing a smaller number of carbon atoms by addition between unsaturated hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2/00—Preparation of hydrocarbons from hydrocarbons containing a smaller number of carbon atoms
- C07C2/76—Preparation of hydrocarbons from hydrocarbons containing a smaller number of carbon atoms by condensation of hydrocarbons with partial elimination of hydrogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C15/00—Cyclic hydrocarbons containing only six-membered aromatic rings as cyclic parts
- C07C15/12—Polycyclic non-condensed hydrocarbons
- C07C15/18—Polycyclic non-condensed hydrocarbons containing at least one group with formula
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/92—Systems containing at least three condensed rings with a condensed ring system consisting of at least two mutually uncondensed aromatic ring systems, linked by an annular structure formed by carbon chains on non-adjacent positions of the aromatic system, e.g. cyclophanes
Abstract
formação de [2,2]paraciclofano e os compostos relacionados e métodos para a formação de polímeros a partir de ciclofanos. a presente invenção se refere a um processo melhorado e a um método para a formação de ciclofanos intermediários estáveis. a invenção apresenta um método geral para a produção de ciclofanos substituídos e não substituídos. os componentes da invenção são um tubo de reação de pirólise que pode ser aquecido de maneira elétrica, em que um fluxo de óxido nitroso com vapor de xileno em um gás transportador inerte opcional à pressão atmosférica. o gás de saída é condensado resultante da deposição de [2,2']paraciclofano. além disso, um processo e um método pelos quais os intermediários reativos da reação descrita acima podem ser depositados e polimerizados de maneira direta a pressões atmosféricas ou em torno disso são descritos.
Description
[001] A presente invenção refere-se ao campo da síntese deciclofanos e ao seu método de aplicação e utilidade como precursores de polímeros.
[002] Os ciclofanos são um subconjunto das estruturasorgânicas, que são bem conhecidas e caracterizadas. Várias revisões e livros excelentes foram publicados que abrangem os métodos e aplicações muito bem.
[003] De forma resumida, os ciclofanos e outros compostosrelacionados ao benzocicloide são moléculas orgânicas que possuem as estruturas em que um carbono cíclico ou cadeia substituída por heteroátomo está ligado a duas ou mais posições de um anel aromático. O termo ciclofanos é usado para descrever os compostos que têm uma relação ou uma estrutura que se encaixa amplamente nessa categoria estrutural.
[004] Um dos compostos de ciclofano mais pesquisados é aestrutura paraciclofano. Em um conjunto de compostos de ciclofano (como mostrado nas estruturas de [n]metaciclofanos (I), [n]paraciclofanos (II) e [n, n’'] ciclofanos abaixo), é vista estrutura III como uma estrutura geral para paraciclofanos.
[005] Nesse padrão de substituição, nota-se que o membro maissimples da série é onde n = 1. Nesse caso, a molécula é denominada [2,2']paraciclofano. Essa molécula e os seus derivados são uma importante classe de compostos que são capazes de formar uma variedade de estruturas de polímero. Por esta razão, eles são intermediários orgânicos altamente desejáveis que foram utilizados como precursores de revestimentos isolantes para numerosas aplicações. Nessas aplicações, a molécula mostrada em III (n = 1) é normalmente aquecida em vácuo para produzir uma pressão de vapor significativa e para forçar uma dissociação da molécula em um intermediário altamente reativo. Essa clivagem pirolítica do [2,2']paraciclofano resulta em duas moléculas do p-xilileno intermediário reativo (mostrado abaixo).Pirólise, 600 a 700°C, 0,1 a 0,2 torr [2,2'] paraciclofano, "dímero" p-xilileno, "monômero"
[2,2'] paraciclofano, "dímero" p-xilileno, "monômero"
[006] Além disso, o p-xilileno intermediário reativo pode serformado a partir do "dímero" pela utilização de uma descarga de pirólise sob pressão atmosférica reduzida. (Ref. Gorham patente U.S. n° 3.342.754). Esse procedimento tem sido comumente chamado de "processo de Gorham".
[007] Como a estrutura indicada, o p-xilileno intermediário reativoé uma espécie intermediária de longa vida que tem a capacidade de reagir para formar um polímero altamente desejável. Em particular, esse polímero é um revestimento isolante, que tem a capacidade de revestir as superfícies em camadas relativamente uniformes que são altamente resistentes aos solventes químicos, gases e de ataque biológico. O p-xilileno é depositado no vácuo sobre uma superfície- alvo para o revestimento isolante. Na superfície, ele sofre uma reforma para uma unidade de repetição do poli(p-xilileno), também conhecido como parileno. No caso de não haver substituintes suplementares nos anéis aromáticos ou cadeias laterais alifáticas além do hidrogênio, esse composto é denominado polímero parileno-N.Deposição de vácuo (0,1 torr) p-xilileno parileno-N
p-xilileno parileno-N
[008] O Para-xilileno, como um valioso intermediário reativo, tevea sua síntese, principalmente através da pirólise de [2,2']paraciclofano. Assim, a síntese de [2,2']paraciclofano é um intermediário estável crítico na utilização de p-xilileno e o parileno de polímero.
[009] A síntese de [2,2’]paraciclofano é através da via daeliminação de 1,6-Hofmann de sais de amônio quaternário.
[0010] Essa rota que passa por um sal de amônio quaternário,embora amplamente utilizada, apresenta várias desvantagens. O paraciclofano é, em geral, produzido em baixos rendimentos com o uso de um processo de múltiplas etapas.
[0011] Além disso, devido ao baixo rendimento e grandequantidade de produtos secundários, uma purificação extensiva do dímero resultante é um procedimento de processo adicional.
[0012] Até o presente, a restrição do processo de vácuo (Gorham)conhecido tanto para a pirólise do dímero quanto para a deposição do monômero para produzir o polímero aumentaram o custo e limitaram a utilidade de aplicações do polímero. Um método pelo qual a necessidade de uma pressão reduzida durante o processo e durante a aplicação dos materiais é altamente desejado.
[0013] Uma variedade de substituições para os vários átomos dehidrogênio e a substituição de heteroátomos no lugar dos vários átomos de carbono nos anéis e cadeias têm sido feita. Seria desejável dispor de um método eficiente e eficaz para a formação desses compostos, tanto conhecidos como desconhecidos.
[0014] Assim, existe uma necessidade de uma síntese melhoradado dímero intermediário estável de xilileno ([2,2’]paraciclofano) e derivados relacionados a esse composto e estrutura geral. Além disso, um método geral para a formação de ciclofanos e de compostos relacionados a vários substituintes, por meio de um método de baixo custo, é desejado. Também necessário é um método melhorado para aplicar os monômeros de xilileno (ou xilileno substituído) para a produção de revestimentos e outros produtos de polímeros derivados desse intermediário reativo. Ainda mais desejável seria um método para modificar as propriedades físicas dos polímeros resultantes para aplicações e extensões de métodos adicionais. Na discussão que se segue, o exemplo de p-xilileno será usado para ilustrar os métodos e processos que são ensinados. O método e os processos podem ser ampliados a outras moléculas semelhantes pelos versados na técnica da presente invenção, e as moléculas não serão, portanto, expressamente discutidas de maneira exaustiva.
[0015] Por coseguinte, é o objetivo da presente invenção aliviar oscustos e os problemas descritos nos processos descritos acima. Primeiro, um método geral para a formação do p-xilileno intermediário reativo de exemplo é mostrado. Em segundo lugar, um método em que a formação do composto químico intermediário estável, tal como [2,2’]paraciclofano ("dímero"), e as estruturas relacionadas, é mostrado para utilidade nos processos de fabricação existentes. Em terceiro lugar, também é mostrado um método direto e econômico para a aplicação do monômero para o alvo, sem a necessidade de uma pressão reduzida, ou qualquer mudança de pressão para isso. Em quarto lugar, será ensinado um método através do qual as propriedades físicas dos produtos resultantes das reações podem ser modificadas e controladas para fins específicos para a melhoria de outros processos.
[0016] Uma vez que os produtos derivados dos métodos que seseguem podem ser altamente controlados, tanto para a pureza quanto o controle das propriedades físicas, a marca Puralene® foi utilizada para os produtos produzidos pelos métodos aqui descritos. O desvio substancial sob controle do operador do processo normal de produção de parileno permite que Puralene® apresente as propriedades únicas e inovadoras normalmente não associadas aos produtos de parileno tradicionais.
[0017] Em uma modalidade preferencial da invenção, um aparelhoe o método são apresentados para formar o p-xilileno intermediário reativo de exemplo, através da utilização de um tubo de reação de pirólise aquecido, dentro do qual um fluxo de uma mistura de gás inerte e óxido nitroso com vapor de xileno em gás de veículo inerte à pressão atmosférica com os produtos de reação de gases de saída não voláteis são condensados para um recipiente arrefecido.
[0018] Em uma outra modalidade de um método através do qualum intermediário reativo de maneira química é formado quer a partir de óxido nitroso ou diretamente a partir de oxigênio, que permite a formação seletiva de p-xilileno na fase gasosa em pressões atmosféricas.
[0019] Em uma outra modalidade, um aparelho e um método sãoapresentados para misturar os gases não reativos resfriados dentro do fluxo de reação quente, que resulta no arrefecimento da temperatura elevada dos gases da reação e, assim, melhora a capacidade dos intermediários reativos de condensar e aderir às superfícies.
[0020] Em uma outra modalidade, um aparelho e um método sãoapresentados para que a reação prossiga em uma pressão aumentada e um valor de expansão na saída do tubo de reação de pirólise aquecido para fornecer um arrefecimento de expansão dos gases quentes abaixo das suas temperaturas de inversão pelo efeito de Joule-Thomson.
[0021] Em uma outra modalidade, um aparelho e um método sãoapresentados com o uso de materiais de partida orgânicos com substituintes que incluem cloro, dicloro, metóxi e metila.
[0022] Em uma outra modalidade, um aparelho e um método sãoapresentados para a utilização de materiais de partida orgânicos com orientação meta e/ou orto dos substituintes nos anéis aromáticos.
[0023] Em uma outra modalidade, um aparelho e um método sãoapresentados para a formação indireta radical e/ou ionização do material de partida através da reação do material de partida (por exemplo, p-xileno) com uma fonte de aquecimento por plasma e/ou por combinação.
[0024] Em uma outra modalidade, a substituição de nitrogênio porargônio e/ou outros gases inertes é essencialmente descrita.
[0025] Uma modificação adicional dos métodos descritos acima éintroduzida através do qual as propriedades físicas do polímero resultante ou outros produtos de reação podem ser modificadas através da utilização de campos magnéticos e/ou elétricos para proporcionar as propriedades físicas controladas.
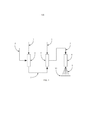
[0026] Os aspectos, objetivos, características e vantagensanteriores ou outros da invenção serão melhor compreendidos com referência à descrição a seguir, as reivindicações anexas e desenhos anexos, em que:A figura 1 é um desenho esquemático do aparelho básico para a produção de uma reação;a figura 2 é um desenho esquemático do aparelho básico para a produção de reação 2; ea figura 3 é um fluxograma de alto nível que ilustra um método de exemplo para produzir um material de permitividade aumentada de acordo com os princípios da invenção.
[0027] Agora, com referência à figura 1, onde a alimentação domaterial de partida 1 é introduzida dentro da câmara 4 através da utilização de um mecanismo de bombeamento (não mostrado) para alimentos líquidos ou sólidos. Tipicamente, a câmara 4 seria um tubo aquecido ou outro dispositivo de evaporação para volatilizar a alimentação do material de partida 1. A alimentação do material de partida 1 é evaporada e misturada com o gás inerte 2 na câmara 4. O gás inerte 2 pode ser qualquer um de um grupo de gases inertes, tais como, mas não limitado a argônio. A mistura volátil resultante 3 é transportada para a câmara 6 e, subsequentemente, misturada com o óxido nitroso 5, para produzir a mistura de reação química 7. A câmara de reação 8 é tipicamente aquecida a cerca de 450 °C a 800 °C para permitir a reação e permitir a vaporização do produtos de reação a serem expulsos como produtos 9, na superfície de coleta 10.
[0028] Agora, com referência à figura 2, onde a alimentação do material de partida 1 é introduzida dentro da câmara 4 através da utilização de um mecanismo de bombeamento (não mostrado) para a alimentos líquidos ou sólidos. Tipicamente, a câmara 4 seria um tubo aquecido ou outro dispositivo de evaporação para volatilizar a alimentação do material de partida 1. A alimentação do material de partida 1 é evaporada e misturada com o gás inerte 2 na câmara 4. O gás inerte 2 pode ser qualquer um de um grupo de gases inertes, tais como, mas não limitado a argônio. A mistura volátil resultante 3 é transportada para a câmara 6. A formação do plasma gasoso 5, é a partir da câmara 12, que está conectada de maneira elétrica através do condutor 14 ao gerador de plasma elétrico 13. A alimentação de gás 11 é uma alimentação de um gás adequado para a conversão de plasma gasoso 5 mediante a indução para dentro da câmara 12, que resulta em plasma gasoso 5. A mistura volátil 3 é subsequentemente misturada com o plasma gasoso 5 na câmara 6 para produzir a mistura de reação química 7, que é transportada para a câmara de reação 8. A câmara de reação 8 é tipicamente aquecida em aproximadamente 450 °C a 800°C para permitir a reação e para permitir a vaporização dos produtos da reação a serem expelidos como produtos 9, para a superfície de coleta 10.
[0029] Agora, com referência à figura 1, a alimentação do materialde partida 1 é introduzida dentro da câmara 4 através da utilização de um mecanismo de bombeamento (não mostrado) para alimentos líquidos ou sólidos. Tipicamente, a câmara 4 seria um tubo aquecido ou outro dispositivo de evaporação para volatilizar a alimentação do material de partida 1. A alimentação do material de partida 1 é aquecida e na câmara 4. A mistura volátil resultante 3 é transportada para a câmara 6 e, subsequentemente, misturada com o óxido nitroso 5, para produzir a mistura de reação química 7. A câmara de reação 8 é tipicamente aquecida a cerca de 450 °C a 800 °C para permitir a reação e permitir a vaporização dos produtos a serem expelidos como os produtos de reação 9, para a superfície de coleta 10.
[0030] Agora, com referência à figura 2, em que a alimentação domaterial de partida 1 é introduzida dentro da câmara 4 através da utilização de um mecanismo de bombeamento (não mostrado) para a alimentação de líquidos ou sólidos. Tipicamente, a câmara 4 seria um tubo aquecido ou outro dispositivo de evaporação para volatilizar a alimentação do material de partida 1. A alimentação do material de partida 1 é evaporada na câmara 4. A mistura volátil resultante 3 é transportada para a câmara 6. A formação do plasma gasoso 5, é a partir de câmara 12, que está conectada de maneira elétrica através do condutor 14 ao gerador de plasma elétrico 13. A alimentação de gás 11 é uma alimentação de um gás adequado para a conversão de plasma gasoso 5 mediante a indução para dentro da câmara 12, que resulta no plasma gasoso 5. A mistura volátil 3 é subsequentemente misturada com o plasma gasoso 5 na câmara 6 para produzir a mistura de reação química 7, que é transportada para a câmara de reação 8. A câmara de reação 8 é tipicamente aquecida a cerca de 450 °C a 800 °C para permitir a reação e para permitir a vaporização dos produtos a serem expelidos como produtos da reação 9, para a superfície de coleta 10.
[0031] Para formar o p-xilileno intermediário reativo, um tubo dereação de pirólise foi construído. O principal elemento na área aquecida foi um tubo de Inconel (liga de níquel 600) (0,826 cm (0,325") de diâmetro externo X 0,703 cm (0,277") de diâmetro interno X 152,4 cm (60") de comprimento, Grainger # 3ACP8). O tubo foi aquecido até as temperaturas indicadas. Uma corrente de fluxo de mistura de gás de argônio composta por óxido nitroso (Airgas # UM1070) com vapor de xileno (Aldrich # 134449-4L) no gás veículo de argônio (Airgas # UM1006) foi introduzida ao tubo com uma taxa de fluxo total de 20 a 100 mL/minuto a uma temperatura de 450 °C a 630 °C e à pressão atmosférica. A proporção de gases é ajustada para fornecer as proporções estequiométricas molares de aproximadamente 1:1 (em xileno ao óxido nitroso). O gás de saída foi composto por um fluxo incolor límpido de gás reativo. A condensação do gás em um recipiente de vidro arrefecido resultou na deposição de um sólido de incolor a cor creme. Esse sólido é parcialmente solúvel em etanol a 95%. O sólido foi comparado a uma amostra de [2,2’]paraciclofano (Aldrich # P225-5G-A), por análise de GC (SRI # 310, 15m, coluna megabore, detector FID) e mostrou resultar em tempos de retenção idênticos. Em temperaturas mais elevadas (650 °C a 800 °C), a saída do tubo de reação é quente o suficiente para manter o p-xilileno monomérico na forma monomérica. O arrefecimento rápido do monômero para uma superfície resulta em um líquido de condensação do monômero e rápida polimerização do monômero em um polímero. A comparação da película assim produzida parece ser idêntica à película de parileno produzida pelo processo de Gorham. (Ambas as permitividades medidas sendo 3, as resistências de ruptura elétrica são idênticas em 100V/mícron, e a solubilidade em ambos os solventes quentes e frios está abaixo dos níveis detectáveis).
[0032] Nessa reação, presume-se, mas não é provado, que ointermediário reativo de p-xilileno reativo é formado e subsequentemente dimerizado no tubo de reação ou durante a condensação no recipiente de vidro. Essa reação usada para sintetizar o dímero, em comparação com o "processo de Gorham" conhecido, resulta em uma grande melhoria no rendimento geral de síntese do dímero e também resulta em uma grande melhoria na pureza do dímero diretamente a partir da reação. Entende-se que a variação nas quantidades estequiométricas dos reagentes pode ser ajustada para proporcionar maior ou menor rendimento com purezas variáveis associadas para proporcionar um processo mais econômico ou com melhor eficiência geral de produção, substancialmente sem se desviar do âmbito da presente invenção. As purificações posteriores dos materiais a partir dessa reação podem ser realizadas nesse material de uma maneira que é muito mais fácil de realizar do que com os processos previamente ensinados.
[0033] A reação é mostrada abaixo.Pirólise, 450° a 630°C, 1 atm;fluxo de gás de argônio
[0034] À medida que a temperatura da reação é aumentada para>650 °C, a deposição do monômero de xilileno pode prosseguir diretamente para um substrato sólido alvo sem a necessidade de isolamento do dímero intermediário. A deposição do gás de saída acima da temperatura de reação de 650 °C sobre uma placa de vidro resfriado que resultou na formação de uma substância insolúvel em etanol, que exibe características de um polímero de parileno. No entanto, as características de solubilidade mostram claramente que o material é insolúvel em todos os solventes comuns (isto é, hexano, xileno, acetato de etila, etanol, água).
[0035] O aumento da quantidade de óxido nitroso resultou naoxidação parcial e/ou total de xileno com formação reduzida dos ciclofanos desejados ou o seu polímero. O controle fechado da estequiometria é desejado nessa reação da fase gasosa.
[0036] Propõe-se que o mecanismo de reação prossiga através de uma rota que envolve a decomposição anterior de óxido nitroso. O óxido nitroso é a molécula energeticamente instável que pode ser decomposta de maneira térmica a temperaturas elevadas. Os produtos da reação são o nitrogênio diatômico e oxigênio monoatômico. O oxigênio monoatômico é capaz de reagir com ele próprio para formar o oxigênio diatômico, mas essa reação é relativamente lenta. As estimativas variam na determinação da temperatura que a decomposição térmica pura ocorre, mas as estimativas de 1.100 °C são frequentemente citadas. A catálise dessa reação, como mostrado abaixo na equação 1 é conhecida por ocorrer com uma variedade de óxidos de metais e óxidos de metal mistos. Algumas temperaturas utilizadas para a decomposição de óxido nitroso com certos catalisadores são tão baixos quanto 350 °C.
[0037] A espécie reativa para o processo é muito provável para o oxigênio monoatômico produzido a partir da decomposição do óxido nitroso. Nesse sentido, o óxido nitroso pode ser visto como um veículo conveniente para a liberação de oxigênio monoatômico intermediário reativo. Seria então de se pensar que o oxigênio diatômico poderia ser usado para realizar a mesma reação. No entanto, o uso de oxigênio diatômico sob as mesmas condições que o óxido nitroso não produz a reação. Com a ativação da reação com uma faísca de plasma quente, a ignição do xileno ocorre com subprodutos altamente coloridos, bem como diminuiu substancialmente o rendimento do produto desejado.
[0038] De uma forma semelhante à reação de óxido nitroso, o oxigênio puro diatômico pode ser utilizado como um reagente. No entanto, para produzir rendimentos substanciais dos produtos desejados, a ativação do oxigênio é necessária. Presume-se, mas não é provado, que a ativação do oxigênio deve-se à excitação da molécula de oxigênio para produzir o oxigênio monoatômico como mostrado na Equação 3.
[0039] A reação com o oxigênio monoatômico produzido dessemodo na Equação 2 prossegue assim, de um modo semelhante ao da via de decomposição de óxido nitroso.
[0040] As explicações mecanicistas da reação química oferecidasnessa divulgação são para um quadro proposto para discussão dos resultados, e de forma alguma limitam o efeito dos resultados reais dos procedimentos da invenção aqui descrita. As explicações alternativas podem descrever completamente alguns resultados aqui contidos e podem ser adequadas para discussão intelectual, mas não prejudicam em qualquer forma a invenção descrita na presente memória descritiva ou a análise descrita.
[0041] O arrefecimento dos gases de temperatura elevada quesaem do tubo de reação é necessário. Se o gás de reação estiver em uma temperatura muito elevada, a capacidade do intermediário reativo condensar e aderir a uma superfície é muito reduzida. Para esse fim, um dispositivo foi desenvolvido para misturar os gases não reativos frescos na corrente de reação quente. Além disso, a reação pode prosseguir em uma pressão aumentada (acima da pressão atmosférica), e uma válvula de expansão pode ser utilizada na saída da reação para proporcionar o efeito Joule-Thomson de arrefecimento do gás quente quando o gás está abaixo da temperatura de inversão. Os métodos para fazer ambos são bem conhecidos, e podem ser opcionalmente incorporados nos procedimentos.
[0042] O método pode ser estendido a outros substratos, taiscomo os que são apresentados abaixo.
[0043] Deve-se observar que osindicados acima (cloro, dicloro, metóxi e metila) não são os únicos substituintes aromáticos que são capazes de ser modificados por esse processo em intermediários reativos e seus polímeros subsequentes. Além disso, os paraciclofanos e os compostos derivados dos mesmos não são exclusivos para esse processo. A orientação meta e orto dos substituintes nos anéis aromáticos é também material de partida de reação viável. A reação pode ser generalizada para incluir todos os compostos que são capazes de reação com óxido nitroso produtos de reação intermediária e também contêm hidrogênio estabilizados pela presença de um anelTipicamente, esses átomos de hidrogênio estão localizados em uma posição alfa em relação a um anel de fenila (posição benzílica). As estruturas de Michael removidas das posições do anel aromático alfa são conhecidas para dar reatividade semelhante ao alfa de hidrogênio para a posição do anel aromático tal como é bem conhecido pelos versados na síntese orgânica. No entanto, a reatividade de tais átomos de hidrogênio não está limitada às posições alfa e/ou de Michael de um anel aromático ou o anel aromático, tal como benzeno. Outra estabilização aromática é conhecida para muitos anéis diferentes, anéis fundidos e sistemas de não anéis, como é conhecido para os versados na técnica da química orgânica. Tais materiais de partida podem, de preferência, ter a presença de dois átomos de hidrogênio que são capazes de ser removidos para formar os materiais de partida parcialmente oxidados. Esses materiais preferenciais podemopcionalmente ter a capacidade de dimerizar, trimerizar, oligiomerizar ou polimerizar. O exemplo utilizado na presente invenção é p-xileno.
[0044] Em alternativa, um gás de plasma pode ser usado com osmateriais de partida mencionados acima para formar os produtos intermediários oxidados que podem, subsequentemente, reagir para formar os produtos de reação que são formas dos materiais de partida que podem ser monômeros, dímeros, trímeros, oligômeros ou polímeros oxidados.
[0045] O tratamento das superfícies reativas que podem ficar comcontato com os produtos dessas reações com o uso de plasma com limpeza da superfície antes da exposição ao intermediário reativo é bem conhecido. No entanto, esse processo é incidental para esse método de formação dos compostos químicos necessários para o revestimento ou polímero.
[0046] Um método para a formação de plasma é bemdocumentado e é conhecido para os que estão familiarizados na técnica de formação de plasma. Um exemplo, Reação 2, de uma tal reação de plasma com o uso do método semelhante ao descrito na reação 1, é uma outra modalidade para esse método geral.
[0047] Uma vez que a reação é semelhante para todos oscompostos reivindicados por esse método, a utilização de p-xileno será utilizada para fins de discussão. Fica claro para os versados na técnica da química que a utilização de compostos semelhantes daria resultados semelhantes e, portanto, a discussão exaustiva das diferenças estruturais na reatividade adicionaria pouco, se algo, a partir dos ensinamentos dessa descoberta.
[0048] A um tubo de quartzo de 952,5 cm (3/8") de diâmetro e 30,5cm (12") de comprimento liga-se um tubo de aço inoxidável 316 de 0,15875 cm (1/16") conectado a uma fonte gasosa (tal como argônio ou nitrogênio). O tubo de aço inoxidável é posicionado de tal modo que ele está a uma curta distância a partir de um eletrodo ligado à terra (cerca de 5 mm a 15 mm). Um gerador de plasma (InfoUnlimited PVM- 400, de 20 a 50 kHz, 0 a 6000 V) é conectado ao tubo de 0,15875 cm (1/16") e o eletrodo conectado à terra. O eletrodo conectado à terra é localizado e conectado de tal modo que os gases, depois de terem passado pelo eletrodo conectado à terra, são misturados com uma mistura de argônio e p-xileno. A mistura resultante é deixada passar através de um tubo de um 30,5 cm (12") de 0,9525 cm diâmetro de (3/8") X a uma temperatura que varia desde a temperatura ambiente até 800 °C. A energia elétrica é fornecida ao gerador suficiente para permitir que a reação do xileno ou outros materiais de partida prossigam principalmente para a conclusão. A reação completa do material de partida não é necessária. Em temperaturas mais baixas (temperatura ambiente até 500 °C), um sólido é formado no tubo de saída. Em altas temperaturas (500 a 650 °C), o tubo de saída pode ser misturado com os gases de arrefecimento e/ou direcionado para um alvo sólido ou líquido arrefecido para condensar o dímero. Em temperaturas ainda mais elevadas (650 a 800 °C), o tubo de saída pode ser misturado com os gases de arrefecimento e/ou direcionado para um alvo sólido ou líquido arrefecido para condensar o monômero. A polimerização subsequente do monômero condensado é provável que ocorra de maneira rápida.
[0049] A substituição de nitrogênio por argônio e/ou outros gasesinertes, essencialmente, é possível sem se desviar substancialmente a partir desse procedimento. Além disso, a modificação da polaridade do eletrodo, dos materiais de eletrodo, do material de contenção e das temperaturas é possível sem um desvio significativo do âmbito da presente invenção.
[0050] Uma vez que a condensação de "monômero" de p-xileno édifícil devido às altas temperaturas de reação, é vantajoso adicionar os gases inertes frescos aos produtos de reação. Os métodos para fazer isso são muito simples e bem conhecidos.
[0051] No lugar do método de arrefecimento de gás para oarrefecimento dos produtos de reação, há alguma vantagem de permitir que a reação prossiga a uma pressão mais elevada e permitir que os produtos de reação se expandam para um ambiente de pressão inferior. O arrefecimento de Joule-Thomson ocorre e os produtos da reação são arrefecidos de maneira muito rápida. A condensação subsequente no alvo pode ocorrer, em seguida, com uma carga térmica inferior no alvo.
[0052] As alterações das reações descritas acima podem serrealizadas para introduzir as variações nas propriedades físicas dos polímeros resultantes e produtos de reação. O método para fazer isso está descrito a seguir.Modificação de reação 1: controle de permitividade
[0053] Para a emissão das reações descritas acima, a correntegasosa de produto de reação é direcionada para uma superfície sólida resfriada. O alvo da superfície está imerso em um campo magnético, como o previsto por um ímã de neodímio (S84, K&J Magnetics). A condensação do monômero e a polimerização subsequente podem prosseguir de maneira rápida, enquanto no campo magnético. Se o alvo e o ímã tiverem a mesma orientação relativa durante o processo de polimerização, em seguida, um aumento da linha de base na permitividade elétrica vai ocorrer. Se a orientação da relação de campo magnético para o alvo for girada durante o processo de polimerização ou a condensação em fase sólida, em seguida, a permitividade resultante irá diminuir.
[0054] Quando a reação é realizada como mencionado acima, autilização do monômero de p-xilileno como a molécula de polimerização, mas sem a presença do campo magnético, a permitividade relativa do material depositado é de cerca de 3. Quando o material é executado como descrito com uma densidade de fluxo magnético de cerca de 0,02 a 0,2 teslas (200 a 2.000 gauss), a permitividade relativa é de cerca de 7. Assim, o campo magnético aumentou substancialmente a permitividade por mais de um fator de 2 vezes. De um modo semelhante, outros sais, dipolos e os sais de ácidos orgânicos podem ser orientados de modo eletrópico durante a solidificação ou polimerizações para produzir os materiais de alta permitividade melhorada. As melhorias na permitividade variam de 10 a mais de 1.000%.Modificação de reação 2: controle de orientação de empacotamento eletrostático
[0055] Para a emissão das reações descritas acima, a correntegasosa de produto de reação é direcionado para uma superfície sólida arrefecida. O alvo da superfície está imerso em um campo elétrico, como o previsto por uma fonte de alimentação de alta tensão (G40, Emco, espaçamento de chumbo de 5,08 cm (2") em 4.000V). A condensação do monômero e a polimerização subsequente podem prosseguir de maneira rápida, enquanto no campo elétrico. Se o alvo e o campo elétrico tiverem a mesma orientação relativa durante o processo de polimerização, em seguida, um aumento da linha de base na permitividade elétrica vai ocorrer. Se a orientação da relação de campo elétrico para o alvo for girada durante o processo de polimerização ou a condensação em fase sólida, em seguida, a permitividade resultante irá diminuir.
[0056] Quando a reação é realizada como mencionado acima, ouso do sal do ácido maleico com guanidina como o material alto dielétrico, mas sem a presença do campo elétrico, a permitividade relativa do material depositado é de cerca de 500. Quando o material é executado como descrito com uma densidade de campo elétrico de 10.000 a 30.000 V/m, a permitividade relativa é de cerca de 25.000 a 40.000. Assim, o campo elétrico aumentou substancialmente a permitividade por pelo menos um fator de 25 no caso particular. De um modo semelhante outros sais dipolos e sais de ácidos orgânicos podem ser orientados de modo entrópico durante a solidificação ou polimerizações para produzir os materiais de alta permitividade melhorada. As melhorias de permitividade variam de 50 a mais de 10.000%.
[0057] O uso de campos elétricos e/ou magnéticos durante oprocesso de condensação irá modificar a resistência mecânica. O material pode ser não anisotrópico após condensação em campos fortes. Assim, esse método é um modo de controlar as propriedades mecânicas dos produtos de reação feitas por esse processo.
[0058] Agora, com referência à figura 3, um fluxograma de altonível é mostrado, que ilustra um processo de exemplo para produzir um material de permitividade aumentada de acordo com princípios da presente invenção. As seções, chamadas de câmaras, podem compreender reservatórios que têm uma entrada e uma saída ou estruturas tubulares com uma entrada e uma saída. A seção 210 é um tubo aquecido ou outro dispositivo de evaporação para volatilizar a alimentação do material de partida 200. A alimentação do material de partida 200 é evaporada e misturada com o gás inerte 205 na câmara 210. O gás inerte 205 pode ser qualquer de um grupo de gases inertes, tais como, mas não se limitando a, argônio. A substituição de nitrogênio por argônio e/ou outros gases inertes, essencialmente, é possível. As bombas e as válvulas podem ser utilizadas para impulsionar e controlar o fluxo de fluidos a partir de uma estação para outra.
[0059] A título de exemplo e não de limitação, a câmara 210 podecompreender um tubo de reação de pirólise aquecido de maneira elétrica Inconel (liga de níquel 600). O tubo é aquecido a uma temperatura de cerca de 450 °C a 630 °C à pressão atmosférica. Uma corrente de fluxo de gás argônio por si só, ou com um composto reativo tal como óxido nitroso, é fornecido para o tubo de reação de pirólise. A alimentação do material de partida 200 pode ser vapor xileno (Aldrich # 134449-4L). Se o gás de transporte 205 incluir um composto reativo (por exemplo, N2O), a proporção de gases é ajustada para fornecer as relações estequiométricas aproximadamente molares de 1:1 (xileno para óxido nitroso).
[0060] O material de partida aquecido 200 na mistura volátil comum gás inerte reage com oxigênio monatômico na câmara de reação 215. Sendo muito reativo e transiente, o oxigênio monatômico deve estar disponível para reagir com a mistura volátil na câmara de reação 215. Como discutido acima, a fonte de oxigênio monatômico pode ser um composto gasoso fornecido com o gás de suporte 205, ou um composto gasoso fornecido separadamente 240, ou de outra fonte, tal como um gerador de plasma 235.
[0061] O plasma de oxigênio monatômico pode ser criado atravésda exposição de oxigênio (O2) em um gás de baixa pressão a uma fonte de energia de alta potência, tal como uma descarga de RF, que ioniza o gás. Em alternativa, um composto tal como óxido nitroso (N2O) pode fornecer o oxigênio monatômico para a reação. Assim, um gerador de plasma de oxigênio monatômico 235, ou uma alimentação de composto químico de oxigênio monatômico (por exemplo, N2O) 240, ou outra fonte adequada de oxigênio monatômico é fornecido.
[0062] Um gás de plasma pode ser usado com os materiais departida acima mencionados para formar os produtos intermediários oxidados que podem subsequentemente reagem para formar os produtos de reação que são formas de materiais de partida que podem ser monômeros, dímeros, trímeros, oligômeros ou polímeros oxidados. O gerador de plasma 235 inclui uma alimentação de gás 230 que fornece o gás para uma câmara de reação de plasma 220. Um acionador de plasma 225 fornece energia de alta potência para ionizar o gás.
[0063] A proporção de gases é ajustada para fornecer relaçõesestequiométricas aproximadamente molares de 1:1 (xileno para óxido nitroso ou xileno ao plasma de oxigênio monatômico). De maneirailustrativa, o aumento da quantidade de óxido nitroso em resultado deoxidação parcial e/ou total de xileno com formação reduzida deciclofanos desejados ou o seu polímero. O controle próximo daestequiometria é desejado nessa reação da fase gasosa.
[0064] Os produtos da reação são fornecidos para uma câmara dereação 235, que é aquecida a aproximadamente 450 °C a 800 °C para facilitar a vaporização dos produtos da reação. Os produtos de reação vaporizados 245 são expelidos para uma superfície de coleta de baixa temperatura 250, onde os produtos de reação condensam e formam um sólido. Em temperaturas mais elevadas (650 °C a 800 °C), a saída da câmara de reação 235 é suficientemente quente o suficiente para manter o p-xilileno monomérico na forma monomérica.
[0065] A condensação do gás para um recipiente de vidroarrefecida resultou na deposição de um líquido incolor de sólido de cor creme. Esse sólido é parcialmente solúvel em etanol a 95%. O sólido foi comparado com uma amostra de [2,2’]paraciclofano (Aldrich # P225-5G-A), por análise de GC (SRI # 310, 15m, coluna megabore, detector FID) e mostrou resultar tempos de retenção idênticos.
[0066] O arrefecimento rápido do monômero para uma superfície250 resulta em um líquido de condensação do monômero e rápida polimerização do monômero em um polímero. A comparação da película assim produzida parece ser idêntica à película de parileno produzida pelo processo de Gorham. Sem aumento, a permitividade do produto solidificado é de cerca de 3, os pontos fortes de degradação elétricos são idênticos sobre 100 V/mícron, e solubilidade em ambos os solventes quentes e frios estão abaixo dos níveis detectáveis.
[0067] Nessa reação, acredita-se que o intermediário reativo de p-xilileno reativo é formado e subsequentemente dimerizado no tubo de reação 235 ou durante a condensação 245 na superfície do substrato 250. Essa reação usada para sintetizar o dímero, em comparação com o conhecido "processo de Gorham", resulta em uma grande melhoria no rendimento geral de síntese do dímero e também resulta em uma grande melhoria na pureza do dímero diretamente a partir da reação. Entende-se que a variação nas quantidades estequiométricas dos reagentes pode ser ajustada para proporcionar maior ou menor rendimento com purezas variáveis associadas a proporcionar um processo mais econômico ou com melhor eficiência geral de produção substancialmente sem se desviar do âmbito da presente invenção. As purificações posteriores dos materiais a partir dessa reação podem ser realizadas nesse material de uma maneira que é muito mais fácil de realizar do que com os processos previamente ensinados. A reação é mostrada abaixo.
[0068] À medida que a temperatura da reação na estação 235 éaumentada para > 650 °C, a deposição do monômero de xilileno pode prosseguir diretamente para um alvo de substrato sólido, sem necessidade de isolamento do dímero intermediário. A deposição do gás de saída acima de 650 °C de temperatura de reação sobre uma placa de vidro resfriado resultou na formação de uma substância insolúvel em etanol, que exibe características de um polímero de parileno. No entanto, as características de solubilidade mostram claramente que o material é insolúvel em todos os solventes comuns (isto é, hexano, xileno, acetato de etilo, etanol, água).
[0069] Acredita-se que o mecanismo de reação prossiga por umavia que envolve a decomposição antes de óxido nitroso. O óxido nitroso é a molécula instável de maneira energética que pode ser decomposta de modo térmico a temperaturas elevadas. Os produtos da reação são o nitrogênio diatômico e o oxigênio monoatômico. O oxigênio monoatômico é capaz de reagir com ele próprio para formar o oxigênio diatômico, mas esta reação é relativamente lenta. As estimativas variam determinam a temperatura em que a decomposição térmica pura ocorre, mas as estimativas de 1.100 °C são frequentemente citadas. A catálise dessa reação, como mostrado abaixo na equação 1, é conhecida por ocorrer com uma variedade de óxidos de metais e óxidos de metal mistos. Algumas temperaturas utilizadas para a decomposição de óxido nitroso com certos catalisadores são tão baixas como 350 °C.
[0070] A espécie reativa para o processo é muito provável para ooxigênio monoatômico produzido a partir da decomposição do óxido nitroso. Nesse sentido, o óxido nitroso pode ser visto como um veículo conveniente para a liberação de oxigênio monoatômico intermediário reativo.
[0071] De uma forma semelhante à reação de óxido nitroso, ooxigênio diatômico puro pode ser utilizado como um reagente. No entanto, para produzir rendimentos substanciais de produtos desejados, a ativação do oxigênio é necessária. Acredita-se que a ativação do oxigênio deve-se à excitação da molécula de oxigênio para produzir o oxigênio monoatômico como mostrado na Equação 3.
[0072] A reação com o oxigênio monoatômico produzido desse modo prossegue assim, de um modo semelhante ao da via de decomposição de óxido nitroso.
[0073] O arrefecimento dos gases de temperatura elevada 245 que saem do tubo de reação 235 é necessário. Se o gás de reação estiver em uma temperatura muito elevada, a capacidade do intermediário reativo condensar e aderir a uma superfície é muito reduzida. Para esse fim, um dispositivo foi desenvolvido para misturar os gases não reativos frescos na corrente de reação quente. A reação pode prosseguir em uma pressão aumentada (acima da pressão atmosférica). Em conformidade, uma válvula de expansão pode ser utilizada na saída do tubo de reação 235 para proporcionar o efeito Joule-Thomson de arrefecimento do gás quente quando o gás está abaixo da temperatura de inversão.
[0074] O método pode ser estendido a outros substratos, taiscomo os que são apresentados abaixo.
[0075] Os substituintes, tais como os indicados acima (cloro,dicloro, metóxi e metila) não são os únicos substituintes aromáticos que são capazes de ser modificados por esse processo em intermediários reativos e seus polímeros subsequentes. Além disso, os paraciclofanos e os compostos derivados dos mesmos não são exclusivos para esse processo. A orientação meta e orto dos substituintes nos anéis aromáticos é também material de partida de reação viável. A reação pode ser generalizada para incluir todos os compostos que são capazes de reação com óxido nitroso ou os seus produtos de reação intermediária e também contêm átomos de hidrogênio estabilizados pela presença de um anel aromático. Tipicamente, esses átomos de hidrogênio estão localizados em uma posição alfa em relação a um anel de fenila (posição benzílica). As estruturas de Michael removidas das posições do anel aromático alfa são conhecidas para dar reatividade semelhante ao alfa de hidrogênio para a posição do anel aromático tal como é bem conhecido pelos versados na síntese orgânica. No entanto, a reatividade de tais átomos de hidrogênio não está limitada às posições alfa e/ou de Michael de um anel aromático ou o anel aromático, tal como benzeno. Outra estabilização aromática é conhecida para muitos anéis diferentes, anéis fundidos e sistemas de não anéis, como é conhecido para os versados na técnica da química orgânica. Tais materiais de partida podem, de preferência, ter a presença de dois átomos de hidrogênio que são capazes de ser removidos para formar os materiais de partida parcialmente oxidados. Esses materiais preferenciais podem opcionalmente ter a capacidade de dimerizar, trimerizar, oligiomerizar ou polimerizar. O exemplo utilizado na presente invenção é p-xileno.
[0076] Uma implementação preferencial da presente invençãoaumenta a permitividade do polímero, exibindo os produtos de reação de condensação 245 a um campo magnético ou elétrico. Para a produção das reações descritas acima, a corrente gasosa de produto da reação de 245 é direcionada para uma superfície sólida arrefecida 250. De modo ilustrativo, a superfície alvo 250 pode ser imersa em um campo magnético 255, como fornecida por um ímã de neodímio (S84, K&J Magnetics). Outras fontes do campo magnético podem ser utilizadas e têm a intenção de ser abrangidas pelo âmbito da invenção. A condensação do monômero e a polimerização subsequente podem prosseguir de maneira rápida, enquanto no campo magnético 255. Se o alvo e o ímã tiverem a mesma orientação relativa durante o processo de polimerização, em seguida, um aumento da linha de base na permitividade elétrica vai ocorrer. Se a orientação do campo magnético 255 em relação ao alvo for girada durante o processo de polimerização ou de condensação em fase sólida, em seguida, a permitividade resultante vai diminuir.
[0077] Quando a reação é realizada como mencionado acima, ouso de o monômero de p-xilileno como a molécula de polimerização, mas sem a presença do campo magnético, a permitividade relativa do material depositado é de cerca de 3. Quando o material é executado como descrito com um fluxo magnético 255, a densidade de cerca de 0,02 a 0,2 teslas (200-2000 gauss), a permitividade relativa é de cerca de 7. Assim, o campo magnético aumenta substancialmente a permitividade em mais de um fator de 2 vezes. De um modo semelhante outros sais dipolos e os sais de ácidos orgânicos podem ser orientados de modo entrópico durante a solidificação ou polimerizações para produzir os materiais de elevada permitividade melhoradas. As melhorias na permitividade que variam de 10 a mais de 1.000% podem ser alcançadas.
[0078] Em outra aplicação, o alvo de superfície 250 está imersoem um campo elétrico 255, como fornecido por uma fonte de alimentação de alta tensão (G40, Emco, espaçamento de chumbo de 5,08 cm (2") em 4.000V). A condensação do monômero e a subsequente polimerização podem prosseguir de maneira rápida, enquanto no campo elétrico. Se o alvo e o campo elétrico tiverem a mesma orientação relativa durante o processo de polimerização, em seguida, um aumento da linha de base na permitividade elétrica vai ocorrer. Se a orientação da relação de campo elétrico para o alvo for girada durante o processo de polimerização ou condensação em fase sólida, em seguida, a permitividade resultante será inferior.
[0079] A condensação de produtos de reação dielétrica napresença de um campo elétrico e/ou magnético aumenta a permitividade dielétrica do condensado. Essa etapa pode ser aplicada a outros compostos além dos polímeros de parileno.
[0080] Quando a reação é realizada como mencionado acima, ouso do sal do ácido maleico com guanidina como o material alto dielétrico, mas sem a presença do campo elétrico, a permitividade relativa do material depositado é de cerca de 500. Quando o material é executado como descrito com uma densidade de campo elétrico de 10.000 a 30.000 V/m, a permitividade relativa é de cerca de 25.000 a 40.000. Assim, o campo elétrico aumentou substancialmente a permitividade por pelo menos um fator de 25 no caso particular. De um modo semelhante outros sais dipolos e sais de ácidos orgânicos podem ser orientados de modo entrópico durante a solidificação ou polimerizações para produzir os materiais de alta permitividade melhorada. As melhorias de permitividade variam de 50 a mais de 10.000%.
[0081] O uso de campos elétricos e/ou magnéticos durante oprocesso de condensação irá modificar a resistência mecânica. O material pode ser não anisotrópico após condensação em campos fortes. Assim, esse método é um modo de controlar as propriedades mecânicas dos produtos de reação feitas por esse processo.
[0082] Embora uma modalidade de exemplo da invenção tenhasido descrita, deve ser evidente que as modificações e as variações das mesmas são possíveis, todas as quais ficam dentro do verdadeiro espírito e âmbito da invenção. No que diz respeito à descrição acima, em seguida, deve ser compreendido que as relações ideais para os componentes e as etapas da invenção, que incluem as variações em ordem, de forma, de conteúdo, função e modo de operação, são consideradas de maneira rápida, evidente e óbvia para o versado na técnica, e todas as relações equivalentes àquelas ilustradas nos desenhos e descritas na especificação pretendem ser abrangidas pela presente invenção. A descrição acima e os desenhos são ilustrativos das modificações que podem ser feitas sem se afastar da presente invenção, cujo âmbito deve ser apenas limitado pelas reivindicações a seguir. Portanto, o que precede é considerado apenas como ilustrativo dos princípios da invenção. Além disso, uma vez que numerosas modificações e alterações ocorrerão facilmente aos versados na técnica, não é desejado limitar a invenção à construção e operação exatas, mostradas e descritas e, em conformidade, todas as modificações apropriadas e equivalentes destinam-se a ficar dentro do âmbito da invenção tal como reivindicado.
Claims (9)
1. Método para a produção de dímeros, trímeros, oligômeros e polímeros a partir da oxidação de um átomo de hidrogênio alfa para um anel aromático, caracterizado pelo fato de que compreende:a introdução de um material de partida orgânico que contém pelo menos um átomo de hidrogênio alfa para um anel aromático em uma corrente de fluxo de gás;a mistura do dito material de partida orgânico no fluxo de gás com óxido nitroso;a reação do dito átomo de hidrogênio alfa com o anel aromático para formar um produto da reação; eo aquecimento do dito fluxo de corrente de gás com o produto da reação a uma temperatura de reação para permitir a oxidação do átomo de hidrogênio alfa e a subsequente formação de um composto de saída a partir do grupo que consiste em monômeros, dímeros, trímeros, oligômeros e polímeros, de acordo com a dita temperatura de reação.
2. Método, de acordo com a reivindicação 1, caracterizado pelo fato de que o material de partida orgânico compreende xileno.
3. Método, de acordo com a reivindicação 1, caracterizado pelo fato de que há dois átomos de hidrogênio alfa em relação ao mesmo anel aromático.
4. Método, de acordo com a reivindicação 1,caracterizado pelo fato de que o anel aromático é benzeno.
5. Método, de acordo com a reivindicação 1,caracterizado pelo fato de que o composto de saída é misturado com um gás inerte fresco.
6. Método, de acordo com a reivindicação 1, caracterizado pelo fato de que a pressão da corrente de fluxo de gás com o produto da reação sendo aquecido a uma temperatura de reação é aumentada mediante a aplicação de um dispositivo de pressão de retorno.
7. Método, de acordo com a reivindicação 1, caracterizado pelo fato de que compreende ainda uma etapa de mistura de um gás de plasma com o dito fluxo de corrente de gás e o material de partida orgânico e óxido nitroso, formando uma mistura de reação.
8. Método, de acordo com a reivindicação 1, caracterizado pelo fato de que compreende ainda uma etapa de expor o composto de saída a um campo magnético durante a condensação.
9. Método, de acordo com a reivindicação 1, caracterizado pelo fato de que compreende ainda uma etapa de expor o composto de saída a um campo elétrico durante a condensação.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13/599,996 | 2012-08-30 | ||
| US13/599,996 US8633289B2 (en) | 2011-08-31 | 2012-08-30 | Formation of [2,2]paracyclophane and related compounds and methods for the formation of polymers from cyclophanes |
| PCT/US2012/072335 WO2014035456A2 (en) | 2012-08-30 | 2012-12-31 | Formation of [2,2] paracyclophane and related compounds and methods for the formation of polymers from cyclophanes |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| BR112015004274A2 BR112015004274A2 (pt) | 2017-07-04 |
| BR112015004274B1 true BR112015004274B1 (pt) | 2021-11-09 |
Family
ID=48173049
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BR112015004274-0A BR112015004274B1 (pt) | 2012-08-30 | 2012-12-31 | Método para a produção de dímeros, trímeros, oligômeros e polímeros a partir da oxidação de um átomo de hidrogênio alfa para um anel aromático |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US8633289B2 (pt) |
| EP (1) | EP2890665B1 (pt) |
| JP (1) | JP6195926B2 (pt) |
| KR (1) | KR101717278B1 (pt) |
| CN (1) | CN105026352B (pt) |
| AU (1) | AU2012388717B2 (pt) |
| BR (1) | BR112015004274B1 (pt) |
| CA (1) | CA2883372C (pt) |
| HK (1) | HK1212322A1 (pt) |
| IN (1) | IN2015DN01925A (pt) |
| MX (1) | MX371156B (pt) |
| MY (1) | MY171391A (pt) |
| SG (1) | SG11201501399WA (pt) |
| WO (1) | WO2014035456A2 (pt) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9011627B2 (en) | 2007-10-05 | 2015-04-21 | Carver Scientific, Inc. | Method of manufacturing high permittivity low leakage capacitor and energy storing device |
| WO2009046341A1 (en) | 2007-10-05 | 2009-04-09 | David Carver | High permittivity low leakage capacitor and energy storing device and method for forming the same |
| US8940850B2 (en) | 2012-08-30 | 2015-01-27 | Carver Scientific, Inc. | Energy storage device |
| US9214281B2 (en) | 2008-10-03 | 2015-12-15 | Carver Scientific, Inc. | Very thin dielectrics for high permittivity and very low leakage capacitors and energy storing devices |
| US9214280B2 (en) | 2008-10-03 | 2015-12-15 | Carver Scientific, Inc. | Very thin dielectrics for high permittivity and very low leakage capacitors and energy storing devices |
| US10227432B2 (en) | 2011-08-31 | 2019-03-12 | Carver Scientific, Inc. | Formation of xylylene type copolymers, block polymers, and mixed composition materials |
| US9899846B2 (en) | 2012-08-30 | 2018-02-20 | Carver Scientific, Inc. | Entropic energy transfer methods and circuits |
| US10199165B2 (en) | 2012-08-30 | 2019-02-05 | Carver Scientific, Inc. | Energy storage device |
| US9805869B2 (en) | 2012-11-07 | 2017-10-31 | Carver Scientific, Inc. | High energy density electrostatic capacitor |
| KR102414054B1 (ko) * | 2014-09-26 | 2022-06-28 | 카버 싸이언티픽, 아이엔씨. | 자일릴렌 유형의 코폴리머, 블록 폴리머, 및 혼합 조성물 재료의 형성 |
| KR102533118B1 (ko) * | 2014-12-17 | 2023-05-16 | 카버 싸이언티픽, 아이엔씨. | 엔트로픽 에너지 전달 방법 및 회로 |
| WO2016100260A2 (en) | 2014-12-17 | 2016-06-23 | Carver Scientific, Inc. | Energy storage device |
| JP6869239B2 (ja) | 2015-11-06 | 2021-05-12 | カーバー サイエンティフィック インコーポレイテッドCarver Scientific, Inc. | 電子エントロピー・メモリデバイス |
| US10249409B2 (en) | 2016-06-21 | 2019-04-02 | Schlumberger Technology Corporation | Coated conductors |
| SG10201912363TA (en) | 2016-12-02 | 2020-02-27 | Carver Scientific Inc | Memory device and capacitive energy storage device |
Family Cites Families (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3149175A (en) * | 1960-08-22 | 1964-09-15 | Union Carbide Corp | Preparation of di-para-xylylenes |
| US3198844A (en) * | 1961-07-12 | 1965-08-03 | Minnesota Mining & Mfg | Cyclic products of para-xylylene |
| US3271417A (en) | 1963-02-06 | 1966-09-06 | Eastman Kodak Co | 1, 2-thiazetidine-3-one-1-oxides and their preparation |
| US3271471A (en) * | 1965-03-23 | 1966-09-06 | Union Carbide Corp | Dimerization process |
| US3342754A (en) | 1966-02-18 | 1967-09-19 | Union Carbide Corp | Para-xylylene polymers |
| US3616314A (en) | 1969-11-24 | 1971-10-26 | Dow Chemical Co | Electrolytic process for preparing(2.2)-paracyclophane |
| US3907748A (en) | 1974-06-17 | 1975-09-23 | University Patents Inc | Paracyclophane polymers and the production thereof |
| FR2457709A1 (fr) | 1979-05-29 | 1980-12-26 | Anvar | Nouveaux agents d'adsorption de gaz utiles en particulier pour separer de l'hydrogene d'une phase le renfermant |
| DE3240303C1 (de) | 1982-10-30 | 1984-01-19 | Merck Patent Gmbh, 6100 Darmstadt | Verfahren zur Herstellung von [2,2]-Paracyclophan |
| US4500562A (en) | 1983-03-02 | 1985-02-19 | The United States Of America As Represented By The United States Department Of Energy | Di-p-xylylene polymer and method for making the same |
| IT1191632B (it) | 1985-10-30 | 1988-03-23 | Montedison Spa | Processo per la preparazione di (2,2)-paraciolofano |
| IT1191645B (it) | 1985-12-19 | 1988-03-23 | Montedison Spa | Processo per la preparazione di (2,2)-paraciclofano e suoi derivati |
| IT1190350B (it) | 1986-06-27 | 1988-02-16 | Montedison Spa | Processo per la preparazione di (2,2)-paraciclofano e suoi derivati |
| IT1190647B (it) | 1986-06-27 | 1988-02-16 | Montedison Spa | Processo per la preparazione di (2,2)-paraciclofano e suoi derivati |
| IT1203876B (it) | 1987-04-10 | 1989-02-23 | Montedison Spa | Processo per la preparazione di (2,2)-paraciclofano e suoi derivati |
| US4849559A (en) | 1987-05-15 | 1989-07-18 | Union Carbide Corporation | Process for the preparation of dichloro-[2,2]paracyclophane |
| US4806702A (en) | 1987-05-26 | 1989-02-21 | Union Carbide Corporation | Process for the preparation of the parylene dimer |
| US4769505A (en) | 1987-07-17 | 1988-09-06 | Union Carbide Corporation | Process for the preparation of the parylene dimer |
| IT1222683B (it) | 1987-09-18 | 1990-09-12 | Montedison Spa | Processo per la preparazione del triciclo 8.2.2.2. esadeca 4,6,10,12,13,15 esaene clorurato nei nuclei benzenici |
| US5110903A (en) | 1990-12-20 | 1992-05-05 | Union Carbide Chemicals & Plastics Technology Corporation | Process for the preparation of mixed parylene dimers free of alpha-halogens |
| US5324702A (en) | 1991-11-22 | 1994-06-28 | Amoco Corporation | Catalytic oxidation and oxidative dehydrogenation using metal-compound-loaded, deboronated hams-1b crystalline borosilicate molecular sieve compositions |
| US5266291A (en) * | 1992-05-05 | 1993-11-30 | Praxair Technology, Inc. | Packed bed arrangement for oxidation reaction system |
| US6107184A (en) * | 1998-12-09 | 2000-08-22 | Applied Materials, Inc. | Nano-porous copolymer films having low dielectric constants |
-
2012
- 2012-08-30 US US13/599,996 patent/US8633289B2/en active Active
- 2012-12-31 MY MYPI2015700600A patent/MY171391A/en unknown
- 2012-12-31 JP JP2015529779A patent/JP6195926B2/ja active Active
- 2012-12-31 WO PCT/US2012/072335 patent/WO2014035456A2/en active Application Filing
- 2012-12-31 BR BR112015004274-0A patent/BR112015004274B1/pt active IP Right Grant
- 2012-12-31 KR KR1020157007263A patent/KR101717278B1/ko active IP Right Grant
- 2012-12-31 SG SG11201501399WA patent/SG11201501399WA/en unknown
- 2012-12-31 CN CN201280076532.2A patent/CN105026352B/zh active Active
- 2012-12-31 MX MX2015002577A patent/MX371156B/es active IP Right Grant
- 2012-12-31 IN IN1925DEN2015 patent/IN2015DN01925A/en unknown
- 2012-12-31 EP EP12883727.5A patent/EP2890665B1/en active Active
- 2012-12-31 CA CA2883372A patent/CA2883372C/en active Active
- 2012-12-31 AU AU2012388717A patent/AU2012388717B2/en active Active
-
2016
- 2016-01-07 HK HK16100103.6A patent/HK1212322A1/xx unknown
Also Published As
| Publication number | Publication date |
|---|---|
| IN2015DN01925A (pt) | 2015-08-07 |
| US20130109827A1 (en) | 2013-05-02 |
| AU2012388717B2 (en) | 2017-03-09 |
| HK1212322A1 (en) | 2016-06-10 |
| MY171391A (en) | 2019-10-10 |
| MX2015002577A (es) | 2015-06-10 |
| WO2014035456A3 (en) | 2015-06-25 |
| KR20150044955A (ko) | 2015-04-27 |
| BR112015004274A2 (pt) | 2017-07-04 |
| EP2890665B1 (en) | 2019-05-22 |
| JP6195926B2 (ja) | 2017-09-13 |
| MX371156B (es) | 2020-01-20 |
| JP2015529205A (ja) | 2015-10-05 |
| SG11201501399WA (en) | 2015-03-30 |
| EP2890665A4 (en) | 2016-05-25 |
| CN105026352B (zh) | 2018-03-16 |
| CA2883372C (en) | 2019-10-15 |
| CN105026352A (zh) | 2015-11-04 |
| CA2883372A1 (en) | 2014-03-06 |
| AU2012388717A1 (en) | 2015-03-19 |
| EP2890665A2 (en) | 2015-07-08 |
| KR101717278B1 (ko) | 2017-03-16 |
| US8633289B2 (en) | 2014-01-21 |
| WO2014035456A2 (en) | 2014-03-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| BR112015004274B1 (pt) | Método para a produção de dímeros, trímeros, oligômeros e polímeros a partir da oxidação de um átomo de hidrogênio alfa para um anel aromático | |
| US10227432B2 (en) | Formation of xylylene type copolymers, block polymers, and mixed composition materials | |
| CN104619678B (zh) | 通过至少一种分子活性化合物催化热解聚‑3‑羟基丙酸酯而制备丙烯酸的方法 | |
| Ferguson et al. | Strong enthalpically driven complexation of neutral benzene guests in aqueous solution | |
| JP2018026581A (ja) | 高エネルギー密度静電キャパシタ | |
| US20110137081A1 (en) | Continuous Method For Producing Amides Of Low Aliphatic Carboxylic Acids | |
| Gaines et al. | High melting precision sulfone polyethylenes synthesized by ADMET chemistry | |
| Fondren et al. | Effects of solution conditions on polymorph development in 2, 4, 6-trinitrotoluene | |
| Brancart et al. | Substituent effect on the thermophysical properties and thermal dissociation behaviour of 9-substituted anthracene derivatives | |
| Florin et al. | γ-IRRADIATION OF SMALL MOLECULES AT 4 AND 77° K. | |
| Lian et al. | Determination and correlation solubility of 4-nitroimidazole in twelve pure solvents from 278.15 K to 323.15 K | |
| TWI720948B (zh) | 伸二甲苯類型共聚物、嵌段聚合物及混合組成物材料之形成 | |
| Gorbachuk et al. | Molybdenum‐Mediated Insertion of Ketones into the P− P bond of cyclo‐(P5Ph5) and Formation of Trinuclear Molybdenum Complexes | |
| Nasirmahale et al. | Introduction of TiO 2-[bip]-NH 2+ C (NO 2) 3− as an effective nanocatalyst for the Hantzsch reactions | |
| Couturier-Tamburelli et al. | Cyanoacetylenic complexes as pre-reactional species leading to the HC7N synthesis. Part II: Experimental and theoretical identifications of the HC5N: C2H2 complex | |
| Baggi et al. | Flow‐Integrated Preparation of Norbornadiene Precursors for Solar Thermal Energy Storage | |
| Abdelkader et al. | Cycloaddition and polymerization reactions of N-ethyl-3-vinylcarbazole with electron-poor olefins | |
| Kichigina et al. | Kinetics and mechanism of radiation telomerization of tetrafluoroethylene in hexafluoropropan-2-ol and trifluoroethanol | |
| Lagow et al. | Synthesis of Linear Acetylenic Carbon The Fourth Carbon Allotrope | |
| Fiilop et al. | Production of C59N: C6o Solid Solution | |
| Fülöp et al. | Production of C 59 N: C 60 solid solution | |
| JPH03151332A (ja) | パラシクロファンの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B06U | Preliminary requirement: requests with searches performed by other patent offices: procedure suspended [chapter 6.21 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 31/12/2012, OBSERVADAS AS CONDICOES LEGAIS. |