WO2023080024A1 - 電荷輸送性組成物 - Google Patents
電荷輸送性組成物 Download PDFInfo
- Publication number
- WO2023080024A1 WO2023080024A1 PCT/JP2022/039830 JP2022039830W WO2023080024A1 WO 2023080024 A1 WO2023080024 A1 WO 2023080024A1 JP 2022039830 W JP2022039830 W JP 2022039830W WO 2023080024 A1 WO2023080024 A1 WO 2023080024A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- charge
- transporting
- carbon atoms
- sulfonic acid
- Prior art date
Links
- 239000000203 mixture Substances 0.000 title claims abstract description 130
- 229920000123 polythiophene Polymers 0.000 claims abstract description 71
- 239000002904 solvent Substances 0.000 claims abstract description 60
- 125000000542 sulfonic acid group Chemical group 0.000 claims abstract description 57
- 239000010409 thin film Substances 0.000 claims abstract description 57
- 238000006243 chemical reaction Methods 0.000 claims abstract description 48
- 239000000126 substance Substances 0.000 claims abstract description 44
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 37
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 30
- 239000003960 organic solvent Substances 0.000 claims abstract description 29
- 239000002253 acid Substances 0.000 claims abstract description 25
- 239000002019 doping agent Substances 0.000 claims abstract description 22
- 125000001273 sulfonato group Chemical group [O-]S(*)(=O)=O 0.000 claims abstract description 9
- 125000002947 alkylene group Chemical group 0.000 claims abstract description 8
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims abstract description 8
- 150000007513 acids Chemical class 0.000 claims abstract description 5
- 125000001624 naphthyl group Chemical group 0.000 claims abstract description 5
- ONUFSRWQCKNVSL-UHFFFAOYSA-N 1,2,3,4,5-pentafluoro-6-(2,3,4,5,6-pentafluorophenyl)benzene Chemical group FC1=C(F)C(F)=C(F)C(F)=C1C1=C(F)C(F)=C(F)C(F)=C1F ONUFSRWQCKNVSL-UHFFFAOYSA-N 0.000 claims abstract description 4
- 125000004432 carbon atom Chemical group C* 0.000 claims description 104
- 229920000642 polymer Polymers 0.000 claims description 25
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 24
- 229910052731 fluorine Inorganic materials 0.000 claims description 23
- 125000001153 fluoro group Chemical group F* 0.000 claims description 19
- 125000003709 fluoroalkyl group Chemical group 0.000 claims description 19
- 125000003118 aryl group Chemical group 0.000 claims description 18
- 125000003545 alkoxy group Chemical group 0.000 claims description 17
- 239000004094 surface-active agent Substances 0.000 claims description 15
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical group C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 claims description 10
- XMWRBQBLMFGWIX-UHFFFAOYSA-N C60 fullerene Chemical compound C12=C3C(C4=C56)=C7C8=C5C5=C9C%10=C6C6=C4C1=C1C4=C6C6=C%10C%10=C9C9=C%11C5=C8C5=C8C7=C3C3=C7C2=C1C1=C2C4=C6C4=C%10C6=C9C9=C%11C5=C5C8=C3C3=C7C1=C1C2=C4C6=C2C9=C5C3=C12 XMWRBQBLMFGWIX-UHFFFAOYSA-N 0.000 claims description 9
- 239000011964 heteropoly acid Substances 0.000 claims description 9
- 229910003472 fullerene Inorganic materials 0.000 claims description 8
- 238000003475 lamination Methods 0.000 claims description 8
- 229910052799 carbon Inorganic materials 0.000 claims description 5
- CGFYHILWFSGVJS-UHFFFAOYSA-N silicic acid;trioxotungsten Chemical compound O[Si](O)(O)O.O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1.O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1.O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1.O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1 CGFYHILWFSGVJS-UHFFFAOYSA-N 0.000 claims description 5
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 4
- 125000004428 fluoroalkoxy group Chemical group 0.000 claims description 4
- 229910052708 sodium Inorganic materials 0.000 claims description 4
- 229910052783 alkali metal Inorganic materials 0.000 claims description 3
- 150000001340 alkali metals Chemical class 0.000 claims description 3
- 125000005577 anthracene group Chemical group 0.000 claims description 3
- 229910052744 lithium Inorganic materials 0.000 claims description 3
- 229910052700 potassium Inorganic materials 0.000 claims description 3
- 125000000547 substituted alkyl group Chemical group 0.000 claims description 3
- 125000004429 atom Chemical group 0.000 claims description 2
- 229910052736 halogen Inorganic materials 0.000 claims description 2
- 150000002367 halogens Chemical class 0.000 claims description 2
- 125000005156 substituted alkylene group Chemical group 0.000 claims description 2
- -1 —O—[ZO] p —R e Chemical group 0.000 description 214
- 238000000034 method Methods 0.000 description 61
- 239000000243 solution Substances 0.000 description 52
- 239000000463 material Substances 0.000 description 50
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 37
- 230000000052 comparative effect Effects 0.000 description 26
- 239000004065 semiconductor Substances 0.000 description 24
- 150000001412 amines Chemical class 0.000 description 23
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 21
- 150000001875 compounds Chemical class 0.000 description 19
- 239000000758 substrate Substances 0.000 description 19
- 239000002105 nanoparticle Substances 0.000 description 18
- 239000011148 porous material Substances 0.000 description 17
- 229910044991 metal oxide Inorganic materials 0.000 description 15
- 150000004706 metal oxides Chemical class 0.000 description 15
- 230000008569 process Effects 0.000 description 15
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 14
- 230000015572 biosynthetic process Effects 0.000 description 13
- 238000004519 manufacturing process Methods 0.000 description 13
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- 238000000576 coating method Methods 0.000 description 11
- 239000010408 film Substances 0.000 description 11
- 239000011159 matrix material Substances 0.000 description 10
- 229910052751 metal Inorganic materials 0.000 description 10
- 239000002184 metal Substances 0.000 description 10
- 239000007787 solid Substances 0.000 description 10
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 9
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 9
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 9
- 239000010405 anode material Substances 0.000 description 9
- 230000000903 blocking effect Effects 0.000 description 9
- 239000011737 fluorine Substances 0.000 description 9
- 125000005843 halogen group Chemical group 0.000 description 9
- 150000002739 metals Chemical class 0.000 description 9
- HMUNWXXNJPVALC-UHFFFAOYSA-N 1-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]-2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)C(CN1CC2=C(CC1)NN=N2)=O HMUNWXXNJPVALC-UHFFFAOYSA-N 0.000 description 8
- 239000000178 monomer Substances 0.000 description 8
- 239000000047 product Substances 0.000 description 8
- 230000009467 reduction Effects 0.000 description 8
- 239000000377 silicon dioxide Substances 0.000 description 8
- 239000003638 chemical reducing agent Substances 0.000 description 7
- 238000001035 drying Methods 0.000 description 7
- 125000005010 perfluoroalkyl group Chemical group 0.000 description 7
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 7
- XOLBLPGZBRYERU-UHFFFAOYSA-N tin dioxide Chemical compound O=[Sn]=O XOLBLPGZBRYERU-UHFFFAOYSA-N 0.000 description 7
- LDXJRKWFNNFDSA-UHFFFAOYSA-N 2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-1-[4-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]piperazin-1-yl]ethanone Chemical compound C1CN(CC2=NNN=C21)CC(=O)N3CCN(CC3)C4=CN=C(N=C4)NCC5=CC(=CC=C5)OC(F)(F)F LDXJRKWFNNFDSA-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 239000011248 coating agent Substances 0.000 description 6
- 229910052809 inorganic oxide Inorganic materials 0.000 description 6
- 239000002736 nonionic surfactant Substances 0.000 description 6
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 238000000137 annealing Methods 0.000 description 5
- 239000012298 atmosphere Substances 0.000 description 5
- 229910052801 chlorine Inorganic materials 0.000 description 5
- 239000006185 dispersion Substances 0.000 description 5
- 238000011156 evaluation Methods 0.000 description 5
- 239000011261 inert gas Substances 0.000 description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 5
- 235000012239 silicon dioxide Nutrition 0.000 description 5
- HZNVUJQVZSTENZ-UHFFFAOYSA-N 2,3-dichloro-5,6-dicyano-1,4-benzoquinone Chemical compound ClC1=C(Cl)C(=O)C(C#N)=C(C#N)C1=O HZNVUJQVZSTENZ-UHFFFAOYSA-N 0.000 description 4
- STTGYIUESPWXOW-UHFFFAOYSA-N 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline Chemical compound C=12C=CC3=C(C=4C=CC=CC=4)C=C(C)N=C3C2=NC(C)=CC=1C1=CC=CC=C1 STTGYIUESPWXOW-UHFFFAOYSA-N 0.000 description 4
- ZCSHACFHMFHFKK-UHFFFAOYSA-N 2-methyl-1,3,5-trinitrobenzene;2,4,6-trinitro-1,3,5-triazinane Chemical compound [O-][N+](=O)C1NC([N+]([O-])=O)NC([N+]([O-])=O)N1.CC1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O ZCSHACFHMFHFKK-UHFFFAOYSA-N 0.000 description 4
- DHDHJYNTEFLIHY-UHFFFAOYSA-N 4,7-diphenyl-1,10-phenanthroline Chemical compound C1=CC=CC=C1C1=CC=NC2=C1C=CC1=C(C=3C=CC=CC=3)C=CN=C21 DHDHJYNTEFLIHY-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 229920002873 Polyethylenimine Polymers 0.000 description 4
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 4
- COHDHYZHOPQOFD-UHFFFAOYSA-N arsenic pentoxide Chemical compound O=[As](=O)O[As](=O)=O COHDHYZHOPQOFD-UHFFFAOYSA-N 0.000 description 4
- 125000003710 aryl alkyl group Chemical group 0.000 description 4
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 4
- 239000010406 cathode material Substances 0.000 description 4
- 229910052681 coesite Inorganic materials 0.000 description 4
- ORTQZVOHEJQUHG-UHFFFAOYSA-L copper(II) chloride Chemical compound Cl[Cu]Cl ORTQZVOHEJQUHG-UHFFFAOYSA-L 0.000 description 4
- 229910052906 cristobalite Inorganic materials 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- YBMRDBCBODYGJE-UHFFFAOYSA-N germanium dioxide Chemical compound O=[Ge]=O YBMRDBCBODYGJE-UHFFFAOYSA-N 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 230000007774 longterm Effects 0.000 description 4
- 239000010955 niobium Substances 0.000 description 4
- LIVNPJMFVYWSIS-UHFFFAOYSA-N silicon monoxide Chemical compound [Si-]#[O+] LIVNPJMFVYWSIS-UHFFFAOYSA-N 0.000 description 4
- 229910052709 silver Inorganic materials 0.000 description 4
- 239000004332 silver Substances 0.000 description 4
- 238000004528 spin coating Methods 0.000 description 4
- 229910052682 stishovite Inorganic materials 0.000 description 4
- 239000010936 titanium Substances 0.000 description 4
- 229910052905 tridymite Inorganic materials 0.000 description 4
- 239000011787 zinc oxide Substances 0.000 description 4
- ARXJGSRGQADJSQ-UHFFFAOYSA-N 1-methoxypropan-2-ol Chemical compound COCC(C)O ARXJGSRGQADJSQ-UHFFFAOYSA-N 0.000 description 3
- VZSRBBMJRBPUNF-UHFFFAOYSA-N 2-(2,3-dihydro-1H-inden-2-ylamino)-N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]pyrimidine-5-carboxamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C(=O)NCCC(N1CC2=C(CC1)NN=N2)=O VZSRBBMJRBPUNF-UHFFFAOYSA-N 0.000 description 3
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 229910052782 aluminium Inorganic materials 0.000 description 3
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 3
- 125000001309 chloro group Chemical group Cl* 0.000 description 3
- 229920000547 conjugated polymer Polymers 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 230000006866 deterioration Effects 0.000 description 3
- 238000010586 diagram Methods 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- 239000002612 dispersion medium Substances 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 239000011344 liquid material Substances 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 239000011368 organic material Substances 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 239000011164 primary particle Substances 0.000 description 3
- 239000011347 resin Substances 0.000 description 3
- 229920005989 resin Polymers 0.000 description 3
- 238000007789 sealing Methods 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- 238000004381 surface treatment Methods 0.000 description 3
- 229910052715 tantalum Inorganic materials 0.000 description 3
- 125000005309 thioalkoxy group Chemical group 0.000 description 3
- 229930192474 thiophene Natural products 0.000 description 3
- 229910001887 tin oxide Inorganic materials 0.000 description 3
- 125000003944 tolyl group Chemical group 0.000 description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 3
- 238000001771 vacuum deposition Methods 0.000 description 3
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical compound C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 description 2
- PAMIQIKDUOTOBW-UHFFFAOYSA-N 1-methylpiperidine Chemical compound CN1CCCCC1 PAMIQIKDUOTOBW-UHFFFAOYSA-N 0.000 description 2
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 2
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 2
- RKMGAJGJIURJSJ-UHFFFAOYSA-N 2,2,6,6-tetramethylpiperidine Chemical compound CC1(C)CCCC(C)(C)N1 RKMGAJGJIURJSJ-UHFFFAOYSA-N 0.000 description 2
- OZDGMOYKSFPLSE-UHFFFAOYSA-N 2-Methylaziridine Chemical compound CC1CN1 OZDGMOYKSFPLSE-UHFFFAOYSA-N 0.000 description 2
- SVTBMSDMJJWYQN-UHFFFAOYSA-N 2-methylpentane-2,4-diol Chemical compound CC(O)CC(C)(C)O SVTBMSDMJJWYQN-UHFFFAOYSA-N 0.000 description 2
- KDSNLYIMUZNERS-UHFFFAOYSA-N 2-methylpropanamine Chemical compound CC(C)CN KDSNLYIMUZNERS-UHFFFAOYSA-N 0.000 description 2
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 2
- YEJRWHAVMIAJKC-UHFFFAOYSA-N 4-Butyrolactone Chemical compound O=C1CCCO1 YEJRWHAVMIAJKC-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- GOLCXWYRSKYTSP-UHFFFAOYSA-N Arsenious Acid Chemical compound O1[As]2O[As]1O2 GOLCXWYRSKYTSP-UHFFFAOYSA-N 0.000 description 2
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 2
- 229910011255 B2O3 Inorganic materials 0.000 description 2
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- MKYBYDHXWVHEJW-UHFFFAOYSA-N N-[1-oxo-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propan-2-yl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(C(C)NC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 MKYBYDHXWVHEJW-UHFFFAOYSA-N 0.000 description 2
- NIPNSKYNPDTRPC-UHFFFAOYSA-N N-[2-oxo-2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 NIPNSKYNPDTRPC-UHFFFAOYSA-N 0.000 description 2
- AFCARXCZXQIEQB-UHFFFAOYSA-N N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CCNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 AFCARXCZXQIEQB-UHFFFAOYSA-N 0.000 description 2
- QCOGKXLOEWLIDC-UHFFFAOYSA-N N-methylbutylamine Chemical compound CCCCNC QCOGKXLOEWLIDC-UHFFFAOYSA-N 0.000 description 2
- ATHHXGZTWNVVOU-UHFFFAOYSA-N N-methylformamide Chemical compound CNC=O ATHHXGZTWNVVOU-UHFFFAOYSA-N 0.000 description 2
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 229920001609 Poly(3,4-ethylenedioxythiophene) Polymers 0.000 description 2
- 229920001167 Poly(triaryl amine) Polymers 0.000 description 2
- 229910006095 SO2F Inorganic materials 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 229910003069 TeO2 Inorganic materials 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 2
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 2
- MCMNRKCIXSYSNV-UHFFFAOYSA-N Zirconium dioxide Chemical compound O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- 125000003342 alkenyl group Chemical group 0.000 description 2
- 125000000304 alkynyl group Chemical group 0.000 description 2
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- 125000002078 anthracen-1-yl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C3C([*])=C([H])C([H])=C([H])C3=C([H])C2=C1[H] 0.000 description 2
- 125000000748 anthracen-2-yl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C3C([H])=C([*])C([H])=C([H])C3=C([H])C2=C1[H] 0.000 description 2
- ADCOVFLJGNWWNZ-UHFFFAOYSA-N antimony trioxide Inorganic materials O=[Sb]O[Sb]=O ADCOVFLJGNWWNZ-UHFFFAOYSA-N 0.000 description 2
- LZYIDMKXGSDQMT-UHFFFAOYSA-N arsenic dioxide Inorganic materials [O][As]=O LZYIDMKXGSDQMT-UHFFFAOYSA-N 0.000 description 2
- 125000004104 aryloxy group Chemical group 0.000 description 2
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 2
- 229920001400 block copolymer Polymers 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- MOWNZPNSYMGTMD-UHFFFAOYSA-N boron monoxide Inorganic materials O=[B] MOWNZPNSYMGTMD-UHFFFAOYSA-N 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- WERYXYBDKMZEQL-UHFFFAOYSA-N butane-1,4-diol Chemical compound OCCCCO WERYXYBDKMZEQL-UHFFFAOYSA-N 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- UUAGAQFQZIEFAH-UHFFFAOYSA-N chlorotrifluoroethylene Chemical group FC(F)=C(F)Cl UUAGAQFQZIEFAH-UHFFFAOYSA-N 0.000 description 2
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 2
- SWXVUIWOUIDPGS-UHFFFAOYSA-N diacetone alcohol Chemical compound CC(=O)CC(C)(C)O SWXVUIWOUIDPGS-UHFFFAOYSA-N 0.000 description 2
- IYYZUPMFVPLQIF-UHFFFAOYSA-N dibenzothiophene Chemical compound C1=CC=C2C3=CC=CC=C3SC2=C1 IYYZUPMFVPLQIF-UHFFFAOYSA-N 0.000 description 2
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 2
- DMBHHRLKUKUOEG-UHFFFAOYSA-N diphenylamine Chemical compound C=1C=CC=CC=1NC1=CC=CC=C1 DMBHHRLKUKUOEG-UHFFFAOYSA-N 0.000 description 2
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical class [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 2
- LZCLXQDLBQLTDK-UHFFFAOYSA-N ethyl 2-hydroxypropanoate Chemical compound CCOC(=O)C(C)O LZCLXQDLBQLTDK-UHFFFAOYSA-N 0.000 description 2
- 239000010419 fine particle Substances 0.000 description 2
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 2
- PVADDRMAFCOOPC-UHFFFAOYSA-N germanium monoxide Inorganic materials [Ge]=O PVADDRMAFCOOPC-UHFFFAOYSA-N 0.000 description 2
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 2
- 229910052737 gold Inorganic materials 0.000 description 2
- 239000010931 gold Substances 0.000 description 2
- 125000001188 haloalkyl group Chemical group 0.000 description 2
- 125000001072 heteroaryl group Chemical group 0.000 description 2
- AOGQPLXWSUTHQB-UHFFFAOYSA-N hexyl acetate Chemical compound CCCCCCOC(C)=O AOGQPLXWSUTHQB-UHFFFAOYSA-N 0.000 description 2
- 229920001519 homopolymer Polymers 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 230000001771 impaired effect Effects 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- FBAFATDZDUQKNH-UHFFFAOYSA-M iron chloride Chemical compound [Cl-].[Fe] FBAFATDZDUQKNH-UHFFFAOYSA-M 0.000 description 2
- 238000010030 laminating Methods 0.000 description 2
- 238000004768 lowest unoccupied molecular orbital Methods 0.000 description 2
- 239000000395 magnesium oxide Substances 0.000 description 2
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 2
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 2
- 229910052752 metalloid Inorganic materials 0.000 description 2
- 150000002738 metalloids Chemical class 0.000 description 2
- IBHBKWKFFTZAHE-UHFFFAOYSA-N n-[4-[4-(n-naphthalen-1-ylanilino)phenyl]phenyl]-n-phenylnaphthalen-1-amine Chemical compound C1=CC=CC=C1N(C=1C2=CC=CC=C2C=CC=1)C1=CC=C(C=2C=CC(=CC=2)N(C=2C=CC=CC=2)C=2C3=CC=CC=C3C=CC=2)C=C1 IBHBKWKFFTZAHE-UHFFFAOYSA-N 0.000 description 2
- GVWISOJSERXQBM-UHFFFAOYSA-N n-methylpropan-1-amine Chemical compound CCCNC GVWISOJSERXQBM-UHFFFAOYSA-N 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 229910052758 niobium Inorganic materials 0.000 description 2
- GUCVJGMIXFAOAE-UHFFFAOYSA-N niobium atom Chemical compound [Nb] GUCVJGMIXFAOAE-UHFFFAOYSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 150000002894 organic compounds Chemical class 0.000 description 2
- 125000000962 organic group Chemical group 0.000 description 2
- 239000007800 oxidant agent Substances 0.000 description 2
- DPBLXKKOBLCELK-UHFFFAOYSA-N pentan-1-amine Chemical compound CCCCCN DPBLXKKOBLCELK-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- DHRLEVQXOMLTIM-UHFFFAOYSA-N phosphoric acid;trioxomolybdenum Chemical compound O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.OP(O)(O)=O DHRLEVQXOMLTIM-UHFFFAOYSA-N 0.000 description 2
- IYDGMDWEHDFVQI-UHFFFAOYSA-N phosphoric acid;trioxotungsten Chemical compound O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.OP(O)(O)=O IYDGMDWEHDFVQI-UHFFFAOYSA-N 0.000 description 2
- 229920000301 poly(3-hexylthiophene-2,5-diyl) polymer Polymers 0.000 description 2
- 229920000767 polyaniline Polymers 0.000 description 2
- 238000006116 polymerization reaction Methods 0.000 description 2
- 229920001296 polysiloxane Polymers 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical compound CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 2
- LLHKCFNBLRBOGN-UHFFFAOYSA-N propylene glycol methyl ether acetate Chemical compound COCC(C)OC(C)=O LLHKCFNBLRBOGN-UHFFFAOYSA-N 0.000 description 2
- VSZWPYCFIRKVQL-UHFFFAOYSA-N selanylidenegallium;selenium Chemical compound [Se].[Se]=[Ga].[Se]=[Ga] VSZWPYCFIRKVQL-UHFFFAOYSA-N 0.000 description 2
- 238000004544 sputter deposition Methods 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 238000000859 sublimation Methods 0.000 description 2
- 230000008022 sublimation Effects 0.000 description 2
- 238000006277 sulfonation reaction Methods 0.000 description 2
- GUVRBAGPIYLISA-UHFFFAOYSA-N tantalum atom Chemical compound [Ta] GUVRBAGPIYLISA-UHFFFAOYSA-N 0.000 description 2
- JBQYATWDVHIOAR-UHFFFAOYSA-N tellanylidenegermanium Chemical compound [Te]=[Ge] JBQYATWDVHIOAR-UHFFFAOYSA-N 0.000 description 2
- LAJZODKXOMJMPK-UHFFFAOYSA-N tellurium dioxide Chemical compound O=[Te]=O LAJZODKXOMJMPK-UHFFFAOYSA-N 0.000 description 2
- YEAUATLBSVJFOY-UHFFFAOYSA-N tetraantimony hexaoxide Chemical compound O1[Sb](O2)O[Sb]3O[Sb]1O[Sb]2O3 YEAUATLBSVJFOY-UHFFFAOYSA-N 0.000 description 2
- BFKJFAAPBSQJPD-UHFFFAOYSA-N tetrafluoroethene Chemical group FC(F)=C(F)F BFKJFAAPBSQJPD-UHFFFAOYSA-N 0.000 description 2
- VJYJJHQEVLEOFL-UHFFFAOYSA-N thieno[3,2-b]thiophene Chemical group S1C=CC2=C1C=CS2 VJYJJHQEVLEOFL-UHFFFAOYSA-N 0.000 description 2
- 150000003577 thiophenes Chemical class 0.000 description 2
- 229910052719 titanium Inorganic materials 0.000 description 2
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 2
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 2
- 229910052721 tungsten Inorganic materials 0.000 description 2
- 239000010937 tungsten Substances 0.000 description 2
- DCTMXCOHGKSXIZ-UHFFFAOYSA-N (R)-1,3-Octanediol Chemical compound CCCCCC(O)CCO DCTMXCOHGKSXIZ-UHFFFAOYSA-N 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- OBDUMNZXAIUUTH-HWKANZROSA-N (e)-tetradec-2-ene Chemical group CCCCCCCCCCC\C=C\C OBDUMNZXAIUUTH-HWKANZROSA-N 0.000 description 1
- 125000006013 1,1-difluoroethoxy group Chemical group 0.000 description 1
- 125000006002 1,1-difluoroethyl group Chemical group 0.000 description 1
- 125000004317 1,3,5-triazin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=N1 0.000 description 1
- CYSGHNMQYZDMIA-UHFFFAOYSA-N 1,3-Dimethyl-2-imidazolidinon Chemical compound CN1CCN(C)C1=O CYSGHNMQYZDMIA-UHFFFAOYSA-N 0.000 description 1
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 description 1
- YIWGJFPJRAEKMK-UHFFFAOYSA-N 1-(2H-benzotriazol-5-yl)-3-methyl-8-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carbonyl]-1,3,8-triazaspiro[4.5]decane-2,4-dione Chemical compound CN1C(=O)N(c2ccc3n[nH]nc3c2)C2(CCN(CC2)C(=O)c2cnc(NCc3cccc(OC(F)(F)F)c3)nc2)C1=O YIWGJFPJRAEKMK-UHFFFAOYSA-N 0.000 description 1
- MNCMBBIFTVWHIP-UHFFFAOYSA-N 1-anthracen-9-yl-2,2,2-trifluoroethanone Chemical group C1=CC=C2C(C(=O)C(F)(F)F)=C(C=CC=C3)C3=CC2=C1 MNCMBBIFTVWHIP-UHFFFAOYSA-N 0.000 description 1
- RRQYJINTUHWNHW-UHFFFAOYSA-N 1-ethoxy-2-(2-ethoxyethoxy)ethane Chemical compound CCOCCOCCOCC RRQYJINTUHWNHW-UHFFFAOYSA-N 0.000 description 1
- 125000004790 1-fluoroethoxy group Chemical group FC(C)O* 0.000 description 1
- 125000004776 1-fluoroethyl group Chemical group [H]C([H])([H])C([H])(F)* 0.000 description 1
- BMVXCPBXGZKUPN-UHFFFAOYSA-N 1-hexanamine Chemical compound CCCCCCN BMVXCPBXGZKUPN-UHFFFAOYSA-N 0.000 description 1
- 125000006019 1-methyl-1-propenyl group Chemical group 0.000 description 1
- 125000006021 1-methyl-2-propenyl group Chemical group 0.000 description 1
- 125000006018 1-methyl-ethenyl group Chemical group 0.000 description 1
- AVFZOVWCLRSYKC-UHFFFAOYSA-N 1-methylpyrrolidine Chemical compound CN1CCCC1 AVFZOVWCLRSYKC-UHFFFAOYSA-N 0.000 description 1
- KWKAKUADMBZCLK-UHFFFAOYSA-N 1-octene Chemical group CCCCCCC=C KWKAKUADMBZCLK-UHFFFAOYSA-N 0.000 description 1
- 125000004793 2,2,2-trifluoroethoxy group Chemical group FC(CO*)(F)F 0.000 description 1
- 125000004778 2,2-difluoroethyl group Chemical group [H]C([H])(*)C([H])(F)F 0.000 description 1
- FGRJGEWVJCCOJJ-UHFFFAOYSA-N 2,2-dimethylaziridine Chemical compound CC1(C)CN1 FGRJGEWVJCCOJJ-UHFFFAOYSA-N 0.000 description 1
- LXOFYPKXCSULTL-UHFFFAOYSA-N 2,4,7,9-tetramethyldec-5-yne-4,7-diol Chemical compound CC(C)CC(C)(O)C#CC(C)(O)CC(C)C LXOFYPKXCSULTL-UHFFFAOYSA-N 0.000 description 1
- ZEBFPAXSQXIPNF-UHFFFAOYSA-N 2,5-dimethylpyrrolidine Chemical compound CC1CCC(C)N1 ZEBFPAXSQXIPNF-UHFFFAOYSA-N 0.000 description 1
- SDGKUVSVPIIUCF-UHFFFAOYSA-N 2,6-dimethylpiperidine Chemical compound CC1CCCC(C)N1 SDGKUVSVPIIUCF-UHFFFAOYSA-N 0.000 description 1
- VXQBJTKSVGFQOL-UHFFFAOYSA-N 2-(2-butoxyethoxy)ethyl acetate Chemical compound CCCCOCCOCCOC(C)=O VXQBJTKSVGFQOL-UHFFFAOYSA-N 0.000 description 1
- FPZWZCWUIYYYBU-UHFFFAOYSA-N 2-(2-ethoxyethoxy)ethyl acetate Chemical compound CCOCCOCCOC(C)=O FPZWZCWUIYYYBU-UHFFFAOYSA-N 0.000 description 1
- LCZVSXRMYJUNFX-UHFFFAOYSA-N 2-[2-(2-hydroxypropoxy)propoxy]propan-1-ol Chemical compound CC(O)COC(C)COC(C)CO LCZVSXRMYJUNFX-UHFFFAOYSA-N 0.000 description 1
- POAOYUHQDCAZBD-UHFFFAOYSA-N 2-butoxyethanol Chemical compound CCCCOCCO POAOYUHQDCAZBD-UHFFFAOYSA-N 0.000 description 1
- WBIQQQGBSDOWNP-UHFFFAOYSA-N 2-dodecylbenzenesulfonic acid Chemical compound CCCCCCCCCCCCC1=CC=CC=C1S(O)(=O)=O WBIQQQGBSDOWNP-UHFFFAOYSA-N 0.000 description 1
- 125000004791 2-fluoroethoxy group Chemical group FCCO* 0.000 description 1
- 125000004777 2-fluoroethyl group Chemical group [H]C([H])(F)C([H])([H])* 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- IULJSGIJJZZUMF-UHFFFAOYSA-N 2-hydroxybenzenesulfonic acid Chemical compound OC1=CC=CC=C1S(O)(=O)=O IULJSGIJJZZUMF-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- 125000006020 2-methyl-1-propenyl group Chemical group 0.000 description 1
- 125000006022 2-methyl-2-propenyl group Chemical group 0.000 description 1
- DLBWPRNUXWYLRN-UHFFFAOYSA-N 2-methylazetidine Chemical compound CC1CCN1 DLBWPRNUXWYLRN-UHFFFAOYSA-N 0.000 description 1
- RGHPCLZJAFCTIK-UHFFFAOYSA-N 2-methylpyrrolidine Chemical compound CC1CCCN1 RGHPCLZJAFCTIK-UHFFFAOYSA-N 0.000 description 1
- YEYKMVJDLWJFOA-UHFFFAOYSA-N 2-propoxyethanol Chemical compound CCCOCCO YEYKMVJDLWJFOA-UHFFFAOYSA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- IDWRJRPUIXRFRX-UHFFFAOYSA-N 3,5-dimethylpiperidine Chemical compound CC1CNCC(C)C1 IDWRJRPUIXRFRX-UHFFFAOYSA-N 0.000 description 1
- QCAHUFWKIQLBNB-UHFFFAOYSA-N 3-(3-methoxypropoxy)propan-1-ol Chemical compound COCCCOCCCO QCAHUFWKIQLBNB-UHFFFAOYSA-N 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- KYINPWAJIVTFBW-UHFFFAOYSA-N 3-methylpyrrolidine Chemical compound CC1CCNC1 KYINPWAJIVTFBW-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- GZEFZLXJPGMRSP-UHFFFAOYSA-N 37,38,39,40-tetrazanonacyclo[28.6.1.13,10.112,19.121,28.04,9.013,18.022,27.031,36]tetraconta-1(37),2,4,6,8,10,12(39),13,15,17,19,21,23,25,27,29,31,33,35-nonadecaene Chemical compound c1ccc2c3cc4[nH]c(cc5nc(cc6[nH]c(cc(n3)c2c1)c1ccccc61)c1ccccc51)c1ccccc41 GZEFZLXJPGMRSP-UHFFFAOYSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- YCPXWRQRBFJBPZ-UHFFFAOYSA-N 5-sulfosalicylic acid Chemical compound OC(=O)C1=CC(S(O)(=O)=O)=CC=C1O YCPXWRQRBFJBPZ-UHFFFAOYSA-N 0.000 description 1
- 229910018072 Al 2 O 3 Inorganic materials 0.000 description 1
- 229920003937 Aquivion® Polymers 0.000 description 1
- 238000004438 BET method Methods 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 1
- AZSFNTBGCTUQFX-UHFFFAOYSA-N C12=C3C(C4=C5C=6C7=C8C9=C(C%10=6)C6=C%11C=%12C%13=C%14C%11=C9C9=C8C8=C%11C%15=C%16C=%17C(C=%18C%19=C4C7=C8C%15=%18)=C4C7=C8C%15=C%18C%20=C(C=%178)C%16=C8C%11=C9C%14=C8C%20=C%13C%18=C8C9=%12)=C%19C4=C2C7=C2C%15=C8C=4C2=C1C12C3=C5C%10=C3C6=C9C=4C32C1(CCCC(=O)OC)C1=CC=CC=C1 Chemical compound C12=C3C(C4=C5C=6C7=C8C9=C(C%10=6)C6=C%11C=%12C%13=C%14C%11=C9C9=C8C8=C%11C%15=C%16C=%17C(C=%18C%19=C4C7=C8C%15=%18)=C4C7=C8C%15=C%18C%20=C(C=%178)C%16=C8C%11=C9C%14=C8C%20=C%13C%18=C8C9=%12)=C%19C4=C2C7=C2C%15=C8C=4C2=C1C12C3=C5C%10=C3C6=C9C=4C32C1(CCCC(=O)OC)C1=CC=CC=C1 AZSFNTBGCTUQFX-UHFFFAOYSA-N 0.000 description 1
- 229940126062 Compound A Drugs 0.000 description 1
- 229910021591 Copper(I) chloride Inorganic materials 0.000 description 1
- 229920003935 Flemion® Polymers 0.000 description 1
- 229920000028 Gradient copolymer Polymers 0.000 description 1
- WJYIASZWHGOTOU-UHFFFAOYSA-N Heptylamine Chemical compound CCCCCCCN WJYIASZWHGOTOU-UHFFFAOYSA-N 0.000 description 1
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 1
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 1
- DJEQZVQFEPKLOY-UHFFFAOYSA-N N,N-dimethylbutylamine Chemical compound CCCCN(C)C DJEQZVQFEPKLOY-UHFFFAOYSA-N 0.000 description 1
- SUAKHGWARZSWIH-UHFFFAOYSA-N N,N‐diethylformamide Chemical compound CCN(CC)C=O SUAKHGWARZSWIH-UHFFFAOYSA-N 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- AHVYPIQETPWLSZ-UHFFFAOYSA-N N-methyl-pyrrolidine Natural products CN1CC=CC1 AHVYPIQETPWLSZ-UHFFFAOYSA-N 0.000 description 1
- OHLUUHNLEMFGTQ-UHFFFAOYSA-N N-methylacetamide Chemical compound CNC(C)=O OHLUUHNLEMFGTQ-UHFFFAOYSA-N 0.000 description 1
- PYFSCIWXNSXGNS-UHFFFAOYSA-N N-methylbutan-2-amine Chemical compound CCC(C)NC PYFSCIWXNSXGNS-UHFFFAOYSA-N 0.000 description 1
- 229920000557 Nafion® Polymers 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical compound C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 description 1
- 239000004695 Polyether sulfone Substances 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 229910004298 SiO 2 Inorganic materials 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- BOTDANWDWHJENH-UHFFFAOYSA-N Tetraethyl orthosilicate Chemical compound CCO[Si](OCC)(OCC)OCC BOTDANWDWHJENH-UHFFFAOYSA-N 0.000 description 1
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 1
- 235000010724 Wisteria floribunda Nutrition 0.000 description 1
- JAWMENYCRQKKJY-UHFFFAOYSA-N [3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-ylmethyl)-1-oxa-2,8-diazaspiro[4.5]dec-2-en-8-yl]-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]methanone Chemical compound N1N=NC=2CN(CCC=21)CC1=NOC2(C1)CCN(CC2)C(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F JAWMENYCRQKKJY-UHFFFAOYSA-N 0.000 description 1
- MCEWYIDBDVPMES-UHFFFAOYSA-N [60]pcbm Chemical compound C123C(C4=C5C6=C7C8=C9C%10=C%11C%12=C%13C%14=C%15C%16=C%17C%18=C(C=%19C=%20C%18=C%18C%16=C%13C%13=C%11C9=C9C7=C(C=%20C9=C%13%18)C(C7=%19)=C96)C6=C%11C%17=C%15C%13=C%15C%14=C%12C%12=C%10C%10=C85)=C9C7=C6C2=C%11C%13=C2C%15=C%12C%10=C4C23C1(CCCC(=O)OC)C1=CC=CC=C1 MCEWYIDBDVPMES-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000005456 alcohol based solvent Substances 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- 150000001448 anilines Chemical class 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 229910052787 antimony Inorganic materials 0.000 description 1
- WATWJIUSRGPENY-UHFFFAOYSA-N antimony atom Chemical compound [Sb] WATWJIUSRGPENY-UHFFFAOYSA-N 0.000 description 1
- VMPVEPPRYRXYNP-UHFFFAOYSA-I antimony(5+);pentachloride Chemical compound Cl[Sb](Cl)(Cl)(Cl)Cl VMPVEPPRYRXYNP-UHFFFAOYSA-I 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 150000004982 aromatic amines Chemical class 0.000 description 1
- 229910052785 arsenic Inorganic materials 0.000 description 1
- RQNWIZPPADIBDY-UHFFFAOYSA-N arsenic atom Chemical compound [As] RQNWIZPPADIBDY-UHFFFAOYSA-N 0.000 description 1
- YBGKQGSCGDNZIB-UHFFFAOYSA-N arsenic pentafluoride Chemical compound F[As](F)(F)(F)F YBGKQGSCGDNZIB-UHFFFAOYSA-N 0.000 description 1
- 125000000732 arylene group Chemical group 0.000 description 1
- ZSIQJIWKELUFRJ-UHFFFAOYSA-N azepane Chemical compound C1CCCNCC1 ZSIQJIWKELUFRJ-UHFFFAOYSA-N 0.000 description 1
- QXNDZONIWRINJR-UHFFFAOYSA-N azocane Chemical compound C1CCCNCCC1 QXNDZONIWRINJR-UHFFFAOYSA-N 0.000 description 1
- NRHDCQLCSOWVTF-UHFFFAOYSA-N azonane Chemical compound C1CCCCNCCC1 NRHDCQLCSOWVTF-UHFFFAOYSA-N 0.000 description 1
- 238000007611 bar coating method Methods 0.000 description 1
- 229910052788 barium Inorganic materials 0.000 description 1
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 125000002511 behenyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- LJOLGGXHRVADAA-UHFFFAOYSA-N benzo[e][1]benzothiole Chemical compound C1=CC=C2C(C=CS3)=C3C=CC2=C1 LJOLGGXHRVADAA-UHFFFAOYSA-N 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- OWBTYPJTUOEWEK-UHFFFAOYSA-N butane-2,3-diol Chemical compound CC(O)C(C)O OWBTYPJTUOEWEK-UHFFFAOYSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 238000005266 casting Methods 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 239000013065 commercial product Substances 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 239000004020 conductor Substances 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 238000007334 copolymerization reaction Methods 0.000 description 1
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical compound [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 description 1
- 229910052593 corundum Inorganic materials 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 238000000151 deposition Methods 0.000 description 1
- 239000003599 detergent Substances 0.000 description 1
- WUOBERCRSABHOT-UHFFFAOYSA-N diantimony Chemical compound [Sb]#[Sb] WUOBERCRSABHOT-UHFFFAOYSA-N 0.000 description 1
- 229920000359 diblock copolymer Polymers 0.000 description 1
- 238000007607 die coating method Methods 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 229940019778 diethylene glycol diethyl ether Drugs 0.000 description 1
- XXJWXESWEXIICW-UHFFFAOYSA-N diethylene glycol monoethyl ether Chemical compound CCOCCOCCO XXJWXESWEXIICW-UHFFFAOYSA-N 0.000 description 1
- 125000004786 difluoromethoxy group Chemical group [H]C(F)(F)O* 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- XUCJHNOBJLKZNU-UHFFFAOYSA-M dilithium;hydroxide Chemical compound [Li+].[Li+].[OH-] XUCJHNOBJLKZNU-UHFFFAOYSA-M 0.000 description 1
- JJQZDUKDJDQPMQ-UHFFFAOYSA-N dimethoxy(dimethyl)silane Chemical compound CO[Si](C)(C)OC JJQZDUKDJDQPMQ-UHFFFAOYSA-N 0.000 description 1
- YYLGKUPAFFKGRQ-UHFFFAOYSA-N dimethyldiethoxysilane Chemical compound CCO[Si](C)(C)OCC YYLGKUPAFFKGRQ-UHFFFAOYSA-N 0.000 description 1
- WDNQRCVBPNOTNV-UHFFFAOYSA-N dinonylnaphthylsulfonic acid Chemical class C1=CC=C2C(S(O)(=O)=O)=C(CCCCCCCCC)C(CCCCCCCCC)=CC2=C1 WDNQRCVBPNOTNV-UHFFFAOYSA-N 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- 238000003618 dip coating Methods 0.000 description 1
- WEHWNAOGRSTTBQ-UHFFFAOYSA-N dipropylamine Chemical compound CCCNCCC WEHWNAOGRSTTBQ-UHFFFAOYSA-N 0.000 description 1
- SZXQTJUDPRGNJN-UHFFFAOYSA-N dipropylene glycol Chemical compound OCCCOCCCO SZXQTJUDPRGNJN-UHFFFAOYSA-N 0.000 description 1
- 229940060296 dodecylbenzenesulfonic acid Drugs 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 229940116333 ethyl lactate Drugs 0.000 description 1
- LIWAQLJGPBVORC-UHFFFAOYSA-N ethylmethylamine Chemical compound CCNC LIWAQLJGPBVORC-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 238000010304 firing Methods 0.000 description 1
- 125000003983 fluorenyl group Chemical class C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 125000004785 fluoromethoxy group Chemical group [H]C([H])(F)O* 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 150000002240 furans Chemical class 0.000 description 1
- 238000005227 gel permeation chromatography Methods 0.000 description 1
- 229910052732 germanium Inorganic materials 0.000 description 1
- GNPVGFCGXDBREM-UHFFFAOYSA-N germanium atom Chemical compound [Ge] GNPVGFCGXDBREM-UHFFFAOYSA-N 0.000 description 1
- 125000006343 heptafluoro propyl group Chemical group 0.000 description 1
- VKYKSIONXSXAKP-UHFFFAOYSA-N hexamethylenetetramine Chemical class C1N(C2)CN3CN1CN2C3 VKYKSIONXSXAKP-UHFFFAOYSA-N 0.000 description 1
- 229940051250 hexylene glycol Drugs 0.000 description 1
- 238000004770 highest occupied molecular orbital Methods 0.000 description 1
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 1
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- AMGQUBHHOARCQH-UHFFFAOYSA-N indium;oxotin Chemical compound [In].[Sn]=O AMGQUBHHOARCQH-UHFFFAOYSA-N 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- PQXKHYXIUOZZFA-UHFFFAOYSA-M lithium fluoride Chemical compound [Li+].[F-] PQXKHYXIUOZZFA-UHFFFAOYSA-M 0.000 description 1
- SKEDXQSRJSUMRP-UHFFFAOYSA-N lithium;quinolin-8-ol Chemical compound [Li].C1=CN=C2C(O)=CC=CC2=C1 SKEDXQSRJSUMRP-UHFFFAOYSA-N 0.000 description 1
- 238000001459 lithography Methods 0.000 description 1
- ORUIBWPALBXDOA-UHFFFAOYSA-L magnesium fluoride Chemical compound [F-].[F-].[Mg+2] ORUIBWPALBXDOA-UHFFFAOYSA-L 0.000 description 1
- 229910001635 magnesium fluoride Inorganic materials 0.000 description 1
- ARYZCSRUUPFYMY-UHFFFAOYSA-N methoxysilane Chemical compound CO[SiH3] ARYZCSRUUPFYMY-UHFFFAOYSA-N 0.000 description 1
- BFXIKLCIZHOAAZ-UHFFFAOYSA-N methyltrimethoxysilane Chemical compound CO[Si](C)(OC)OC BFXIKLCIZHOAAZ-UHFFFAOYSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 229920006030 multiblock copolymer Polymers 0.000 description 1
- 125000001802 myricyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- QKYWADPCTHTJHQ-UHFFFAOYSA-N n,2-dimethylpropan-1-amine Chemical compound CNCC(C)C QKYWADPCTHTJHQ-UHFFFAOYSA-N 0.000 description 1
- ZQGJEUVBUVKZKS-UHFFFAOYSA-N n,2-dimethylpropan-2-amine Chemical compound CNC(C)(C)C ZQGJEUVBUVKZKS-UHFFFAOYSA-N 0.000 description 1
- GDHRQDYGUDOEIZ-UHFFFAOYSA-N n,n,2-trimethylpropan-1-amine Chemical compound CC(C)CN(C)C GDHRQDYGUDOEIZ-UHFFFAOYSA-N 0.000 description 1
- OXQMIXBVXHWDPX-UHFFFAOYSA-N n,n,2-trimethylpropan-2-amine Chemical compound CN(C)C(C)(C)C OXQMIXBVXHWDPX-UHFFFAOYSA-N 0.000 description 1
- ORSUTASIQKBEFU-UHFFFAOYSA-N n,n-diethylbutan-1-amine Chemical compound CCCCN(CC)CC ORSUTASIQKBEFU-UHFFFAOYSA-N 0.000 description 1
- USSPHSVODLAWSA-UHFFFAOYSA-N n,n-dimethylbutan-2-amine Chemical compound CCC(C)N(C)C USSPHSVODLAWSA-UHFFFAOYSA-N 0.000 description 1
- DAZXVJBJRMWXJP-UHFFFAOYSA-N n,n-dimethylethylamine Chemical compound CCN(C)C DAZXVJBJRMWXJP-UHFFFAOYSA-N 0.000 description 1
- ZUHZZVMEUAUWHY-UHFFFAOYSA-N n,n-dimethylpropan-1-amine Chemical compound CCCN(C)C ZUHZZVMEUAUWHY-UHFFFAOYSA-N 0.000 description 1
- VMOWKUTXPNPTEN-UHFFFAOYSA-N n,n-dimethylpropan-2-amine Chemical compound CC(C)N(C)C VMOWKUTXPNPTEN-UHFFFAOYSA-N 0.000 description 1
- 125000006606 n-butoxy group Chemical group 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- BBDGYADAMYMJNO-UHFFFAOYSA-N n-butyl-n-ethylbutan-1-amine Chemical compound CCCCN(CC)CCCC BBDGYADAMYMJNO-UHFFFAOYSA-N 0.000 description 1
- 125000006610 n-decyloxy group Chemical group 0.000 description 1
- XQOIBQBPAXOVGP-UHFFFAOYSA-N n-ethyl-2-methylpropan-2-amine Chemical compound CCNC(C)(C)C XQOIBQBPAXOVGP-UHFFFAOYSA-N 0.000 description 1
- GNVRJGIVDSQCOP-UHFFFAOYSA-N n-ethyl-n-methylethanamine Chemical compound CCN(C)CC GNVRJGIVDSQCOP-UHFFFAOYSA-N 0.000 description 1
- QHCCDDQKNUYGNC-UHFFFAOYSA-N n-ethylbutan-1-amine Chemical compound CCCCNCC QHCCDDQKNUYGNC-UHFFFAOYSA-N 0.000 description 1
- KFYKZKISJBGVMR-UHFFFAOYSA-N n-ethylbutan-2-amine Chemical compound CCNC(C)CC KFYKZKISJBGVMR-UHFFFAOYSA-N 0.000 description 1
- XCVNDBIXFPGMIW-UHFFFAOYSA-N n-ethylpropan-1-amine Chemical compound CCCNCC XCVNDBIXFPGMIW-UHFFFAOYSA-N 0.000 description 1
- RIVIDPPYRINTTH-UHFFFAOYSA-N n-ethylpropan-2-amine Chemical compound CCNC(C)C RIVIDPPYRINTTH-UHFFFAOYSA-N 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001298 n-hexoxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000004718 n-hexylthio group Chemical group C(CCCCC)S* 0.000 description 1
- ISRXMEYARGEVIU-UHFFFAOYSA-N n-methyl-n-propan-2-ylpropan-2-amine Chemical compound CC(C)N(C)C(C)C ISRXMEYARGEVIU-UHFFFAOYSA-N 0.000 description 1
- XHFGWHUWQXTGAT-UHFFFAOYSA-N n-methylpropan-2-amine Chemical compound CNC(C)C XHFGWHUWQXTGAT-UHFFFAOYSA-N 0.000 description 1
- 125000006609 n-nonyloxy group Chemical group 0.000 description 1
- 125000006608 n-octyloxy group Chemical group 0.000 description 1
- 125000003935 n-pentoxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004712 n-pentylthio group Chemical group C(CCCC)S* 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical class C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- PAYSBLPSJQBEJR-UHFFFAOYSA-N naphtho[2,3-e][1]benzothiole Chemical compound C1=CC=C2C=C3C(C=CS4)=C4C=CC3=CC2=C1 PAYSBLPSJQBEJR-UHFFFAOYSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 125000005246 nonafluorobutyl group Chemical group FC(F)(F)C(F)(F)C(F)(F)C(F)(F)* 0.000 description 1
- 229910052755 nonmetal Inorganic materials 0.000 description 1
- 150000002843 nonmetals Chemical class 0.000 description 1
- IOQPZZOEVPZRBK-UHFFFAOYSA-N octan-1-amine Chemical compound CCCCCCCCN IOQPZZOEVPZRBK-UHFFFAOYSA-N 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 125000005003 perfluorobutyl group Chemical group FC(F)(F)C(F)(F)C(F)(F)C(F)(F)* 0.000 description 1
- 125000005004 perfluoroethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 125000005804 perfluoroheptyl group Chemical group FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)* 0.000 description 1
- 125000005005 perfluorohexyl group Chemical group FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)* 0.000 description 1
- 125000005007 perfluorooctyl group Chemical group FC(C(C(C(C(C(C(C(F)(F)F)(F)F)(F)F)(F)F)(F)F)(F)F)(F)F)(F)* 0.000 description 1
- 125000005008 perfluoropentyl group Chemical group FC(C(C(C(C(F)(F)F)(F)F)(F)F)(F)F)(F)* 0.000 description 1
- 125000005009 perfluoropropyl group Chemical group FC(C(C(F)(F)F)(F)F)(F)* 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 description 1
- OBCUTHMOOONNBS-UHFFFAOYSA-N phosphorus pentafluoride Chemical compound FP(F)(F)(F)F OBCUTHMOOONNBS-UHFFFAOYSA-N 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 238000009832 plasma treatment Methods 0.000 description 1
- ONJQDTZCDSESIW-UHFFFAOYSA-N polidocanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO ONJQDTZCDSESIW-UHFFFAOYSA-N 0.000 description 1
- 229920001992 poloxamer 407 Polymers 0.000 description 1
- 229920000172 poly(styrenesulfonic acid) Polymers 0.000 description 1
- 229920006393 polyether sulfone Polymers 0.000 description 1
- 230000000379 polymerizing effect Effects 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229940005642 polystyrene sulfonic acid Drugs 0.000 description 1
- 150000004032 porphyrins Chemical class 0.000 description 1
- 238000007639 printing Methods 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 150000003233 pyrroles Chemical class 0.000 description 1
- ZVJDFJRDKIADMB-UHFFFAOYSA-N quinolin-8-ol;sodium Chemical compound [Na].C1=CN=C2C(O)=CC=CC2=C1 ZVJDFJRDKIADMB-UHFFFAOYSA-N 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 229920005604 random copolymer Polymers 0.000 description 1
- 238000001226 reprecipitation Methods 0.000 description 1
- 238000007650 screen-printing Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- BHRZNVHARXXAHW-UHFFFAOYSA-N sec-butylamine Chemical compound CCC(C)N BHRZNVHARXXAHW-UHFFFAOYSA-N 0.000 description 1
- 150000005082 selenophenes Chemical class 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 239000002210 silicon-based material Substances 0.000 description 1
- PUZPDOWCWNUUKD-UHFFFAOYSA-M sodium fluoride Chemical compound [F-].[Na+] PUZPDOWCWNUUKD-UHFFFAOYSA-M 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 229920006301 statistical copolymer Polymers 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- FVRNDBHWWSPNOM-UHFFFAOYSA-L strontium fluoride Chemical compound [F-].[F-].[Sr+2] FVRNDBHWWSPNOM-UHFFFAOYSA-L 0.000 description 1
- 229910001637 strontium fluoride Inorganic materials 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- OBTWBSRJZRCYQV-UHFFFAOYSA-N sulfuryl difluoride Chemical group FS(F)(=O)=O OBTWBSRJZRCYQV-UHFFFAOYSA-N 0.000 description 1
- 229910052714 tellurium Inorganic materials 0.000 description 1
- PORWMNRCUJJQNO-UHFFFAOYSA-N tellurium atom Chemical compound [Te] PORWMNRCUJJQNO-UHFFFAOYSA-N 0.000 description 1
- 150000005087 tellurophenes Chemical class 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- YBRBMKDOPFTVDT-UHFFFAOYSA-N tert-butylamine Chemical compound CC(C)(C)N YBRBMKDOPFTVDT-UHFFFAOYSA-N 0.000 description 1
- PCCVSPMFGIFTHU-UHFFFAOYSA-N tetracyanoquinodimethane Chemical compound N#CC(C#N)=C1C=CC(=C(C#N)C#N)C=C1 PCCVSPMFGIFTHU-UHFFFAOYSA-N 0.000 description 1
- CRUIOQJBPNKOJG-UHFFFAOYSA-N thieno[3,2-e][1]benzothiole Chemical compound C1=C2SC=CC2=C2C=CSC2=C1 CRUIOQJBPNKOJG-UHFFFAOYSA-N 0.000 description 1
- QHGNHLZPVBIIPX-UHFFFAOYSA-N tin(II) oxide Inorganic materials [Sn]=O QHGNHLZPVBIIPX-UHFFFAOYSA-N 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 125000005270 trialkylamine group Chemical group 0.000 description 1
- 125000005259 triarylamine group Chemical group 0.000 description 1
- 229920000428 triblock copolymer Polymers 0.000 description 1
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 description 1
- CPUDPFPXCZDNGI-UHFFFAOYSA-N triethoxy(methyl)silane Chemical compound CCO[Si](C)(OCC)OCC CPUDPFPXCZDNGI-UHFFFAOYSA-N 0.000 description 1
- ZIBGPFATKBEMQZ-UHFFFAOYSA-N triethylene glycol Chemical compound OCCOCCOCCO ZIBGPFATKBEMQZ-UHFFFAOYSA-N 0.000 description 1
- JLGNHOJUQFHYEZ-UHFFFAOYSA-N trimethoxy(3,3,3-trifluoropropyl)silane Chemical compound CO[Si](OC)(OC)CCC(F)(F)F JLGNHOJUQFHYEZ-UHFFFAOYSA-N 0.000 description 1
- 238000009281 ultraviolet germicidal irradiation Methods 0.000 description 1
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000007740 vapor deposition Methods 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229910001845 yogo sapphire Inorganic materials 0.000 description 1
- YVTHLONGBIQYBO-UHFFFAOYSA-N zinc indium(3+) oxygen(2-) Chemical compound [O--].[Zn++].[In+3] YVTHLONGBIQYBO-UHFFFAOYSA-N 0.000 description 1
Images

Classifications
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K30/00—Organic devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation
- H10K30/50—Photovoltaic [PV] devices
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K30/00—Organic devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation
- H10K30/80—Constructional details
- H10K30/84—Layers having high charge carrier mobility
- H10K30/86—Layers having high hole mobility, e.g. hole-transporting layers or electron-blocking layers
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E10/00—Energy generation through renewable energy sources
- Y02E10/50—Photovoltaic [PV] energy
- Y02E10/549—Organic PV cells
Definitions
- the present invention relates to a charge-transporting composition, and more specifically, to a charge-transporting composition for forming a charge-transporting thin film used in an organic photoelectric conversion device.
- An electronic device is a device that converts light energy into electrical energy using an organic semiconductor, and examples thereof include organic solar cells.
- An organic solar cell is a solar cell element using an organic material for an active layer and a charge-transporting material.
- An organic thin-film solar cell developed by Tang is well known (Non-Patent Documents 1 and 2). Both are lightweight and thin, flexible, and roll-to-roll production is possible. expected to form.
- organic thin-film solar cells exhibit high photoelectric conversion efficiency even at low illuminance compared to photoelectric conversion elements using existing silicon-based materials. Due to its features such as being able to combine the properties of a filter, it is attracting attention not only for solar cell applications but also for optical sensor applications such as image sensors (Patent Documents 1 and 2, Non-Patent Document 3). .
- organic solar cells die-sensitized solar cells and organic thin-film solar cells
- applications such as optical sensors are collectively referred to as organic photoelectric conversion devices (hereinafter sometimes abbreviated as OPV).
- An organic photoelectric conversion element is composed of an active layer (photoelectric conversion layer), a charge (hole, electron) collection layer, electrodes (anode, cathode), and the like.
- the hole collection layer has the role of extracting the holes generated in the active layer to the electrode, and this can be effectively performed by reducing the energy barrier between the active layer and the hole collection layer. can.
- a conjugated compound as an electron-donating organic material p-type organic semiconductor
- a conjugated compound having n-type semiconductor characteristics as an electron-accepting organic material n-type organic semiconductor
- Active layers using fullerenes such as C 60 and fullerene derivatives (hereinafter abbreviated as FA active layers) have been used.
- NFA Non-Fullerene Acceptor
- the NFA active layer exhibits a higher PCE than the FA active layer due to an increase in photocurrent and an improvement in battery voltage.
- Jianhui Hou et al. reported that the use of the NFA active layer showed a PCE of 18% (Non-Patent Document 5).
- these organic photoelectric conversion elements exhibiting a high PCE widely use MoO 3 , which is a vapor-deposited hole-collecting layer that is disadvantageous for mass production, as the hole-collecting layer.
- MoO 3 is a vapor-deposited hole-collecting layer that is disadvantageous for mass production, as the hole-collecting layer.
- Ip the ionization potential of PEDOT/PSS. Therefore, there is a demand for a coating-type hole-collecting material having a deep Ip (Non-Patent Document 6).
- the present invention has been made in view of the above circumstances, and is suitable for forming a charge-transporting thin film of a photoelectric conversion device.
- An object of the present invention is to provide a charge-transporting composition capable of greatly improving the durability of an element that is used.
- an organic A charge-transporting composition having a solvent content adjusted to a specific range is suitable for forming a charge-transporting thin film in an organic photoelectric conversion device, particularly when used as a hole collection layer in an organic photoelectric conversion device.
- the inventors have found that the durability of the resulting device can be greatly improved, and have completed the present invention.
- the present invention provides the following charge-transporting composition.
- a charge-transporting composition for forming a charge-transporting thin film in an organic photoelectric conversion device A charge-transporting substance made of a polythiophene derivative containing a repeating unit represented by the following formula (1), an electron-accepting dopant substance, and a solvent,
- the electron-accepting dopant substance contains at least one selected from the group consisting of arylsulfonic acids and heteropolyacids represented by the following formula (2), and the content of the organic solvent in the solvent is 10% by mass or less.
- R 1 and R 2 are each independently a hydrogen atom, an alkyl group having 1 to 40 carbon atoms, a fluoroalkyl group having 1 to 40 carbon atoms, an alkoxy group having 1 to 40 carbon atoms, an alkoxy group having 1 to 40 carbon atoms, or 1 ⁇ 40 fluoroalkoxy group, C6-C20 aryloxy group, -O-[Z-O] p -R e , sulfonic acid group or sulfonic acid group, or formed by combining R 1 and R 2 -O-Y-O-, Y is an alkylene group having 1 to 40 carbon atoms substituted with a sulfonic acid group or a sulfonate group, which may contain an ether bond, and Z is a halogen an alkylene group having 1 to 40 carbon atoms which may be substituted with atoms, p is an integer of 1 or more, and R e is a hydrogen atom, a fluor
- A represents a naphthalene ring or anthracene ring
- B represents a divalent to tetravalent perfluorobiphenyl group
- l represents the number of sulfonic acid groups bonded to A, satisfying 1 ⁇ l ⁇ 4 is an integer
- q indicates the number of bonds between B and X, and is an integer satisfying 2 to 4.
- R 1 and R 2 above are —O—Y—O— formed by combining R 1 and R 2 , and Y is a sulfonic acid group or a sulfonic acid group, which may contain an ether bond.
- a charge-transporting composition 1 which is a substituted alkylene group having 1 to 40 carbon atoms. 3. 1 or 2, wherein the polythiophene derivative containing a repeating unit represented by formula (1) is a polythiophene derivative containing a repeating unit represented by formula (1-4) or formula (1′-1). Composition. (In the formula, R y represents an alkyl group having 1 to 6 carbon atoms or a fluorine atom.
- M is a hydrogen atom, an alkali metal selected from the group consisting of Li, Na and K. , NH(R S ) 3 or HNC 5 H 5.
- R S independently represents a hydrogen atom or an optionally substituted alkyl group having 1 to 6 carbon atoms.) 4.
- the charge-transporting thin film, wherein the charge-transporting thin film is a hole collecting layer of an organic photoelectric conversion device.
- An electronic device comprising 12 or 13 charge-transporting thin films.
- 14 electronic devices, wherein the electronic devices are organic photoelectric conversion devices.
- 16. 13 An organic photoelectric conversion device having a hole collection layer and a non-fullerene active layer provided in contact therewith. 17.
- 20. 19 organic photoelectric conversion devices with a top anode structure.
- the charge-transporting composition for the organic photoelectric conversion device of the present invention can be produced using a charge-transporting substance composed of a polythiophene derivative that is inexpensively available on the market or can be easily synthesized by a known method.
- a thin film obtained therefrom can greatly improve the durability of the device when used as a hole collection layer in a photoelectric conversion device.
- the deep Ip of the resulting charge-transporting thin film can reduce the energy gap with the NFA active layer, enabling the device to operate at a higher voltage.
- FIG. 4 is a diagram showing JV curves obtained from Examples 3-3 and 3-4.
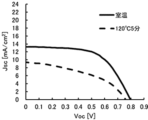
- FIG. 10 is a diagram showing JV curves obtained from Comparative Examples 3-1 and 3-2;
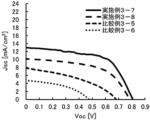
- FIG. 4 is a diagram showing JV curves obtained from Examples 3-7 and 3-8 and Comparative Examples 3-5 and 3-6.
- the charge-transporting composition of the present invention is a charge-transporting composition for forming a charge-transporting thin film in an organic photoelectric conversion device, and comprises a polythiophene derivative containing a repeating unit represented by the following formula (1). a charge-transporting substance and a solvent, wherein the electron-accepting dopant substance contains at least one selected from the group consisting of arylsulfonic acids and heteropolyacids represented by the following formula (2); The content of the organic solvent is 10% by mass or less.
- solid content is a general term for all components of the charge-transporting composition other than the solvent.
- R 1 and R 2 are each independently a hydrogen atom, an alkyl group having 1 to 40 carbon atoms, a fluoroalkyl group having 1 to 40 carbon atoms, an alkoxy group having 1 to 40 carbon atoms, or an alkoxy group having 1 to 40 carbon atoms.
- Y is an alkylene group having 1 to 40 carbon atoms substituted with a sulfonic acid group or a sulfonate group, which may contain an ether bond
- Z is a halogen atom is an alkylene group having 1 to 40 carbon atoms which may be substituted with p is an integer of 1 or more
- R e is a hydrogen atom, a sulfonic acid group or a carbon optionally substituted with a sulfonic acid group an alkyl group having 1 to 40 carbon atoms, a fluoroalkyl group having 1 to 40 carbon atoms optionally substituted with a sulfonic acid group or a sulfonic acid group,
- the alkyl group having 1 to 40 carbon atoms may be linear, branched or cyclic, and specific examples thereof include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl group, s-butyl group, t-butyl group, n-pentyl group, n-hexyl group, n-heptyl group, n-octyl group, n-nonyl group, n-decyl group, n-undecyl group, n- dodecyl group, n-tridecyl group, n-tetradecyl group, n-pentadecyl group, n-hexadecyl group, n-heptadecyl group, n-octadecyl group, n-nonadecyl group, n-eicosanyl group, behenyl group
- the fluoroalkyl group having 1 to 40 carbon atoms is not particularly limited as long as it is an alkyl group having 1 to 40 carbon atoms in which at least one hydrogen atom on the carbon atoms is substituted with a fluorine atom.
- the alkyl group therein may be linear, branched or cyclic. Specific examples thereof include methoxy, ethoxy, n-propoxy, i- propoxy group, c-propoxy group, n-butoxy group, i-butoxy group, s-butoxy group, t-butoxy group, n-pentoxy group, n-hexoxy group, n-heptyloxy group, n-octyloxy group, n-nonyloxy group, n-decyloxy group, n-undecyloxy group, n-dodecyloxy group, n-tridecyloxy group, n-tetradecyloxy group, n-pentadecyloxy group, n-hexadecyloxy group , n-heptadecyloxy group, n-octadecyloxy group, n-
- the fluoroalkoxy group having 1 to 40 carbon atoms is not particularly limited as long as it is an alkoxy group having 1 to 40 carbon atoms in which at least one hydrogen atom on the carbon atoms is substituted with a fluorine atom.
- fluoromethoxy group difluoromethoxy group, perfluoromethoxy group, 1-fluoroethoxy group, 2-fluoroethoxy group, 1,2-difluoroethoxy group, 1,1-difluoroethoxy group, 2,2-difluoroethoxy group, 1,1,2-trifluoroethoxy group, 1,2,2-trifluoroethoxy group, 2,2,2-trifluoroethoxy group, 1,1,2,2-tetrafluoroethoxy group, 1,2, 2,2-tetrafluoroethoxy group, perfluoroethoxy group, 1-fluoropropoxy group, 2-fluoropropoxy group, 3-fluoropropoxy group, 1,1-difluoropropoxy group, 1,2-difluoropropoxy group, 1, 3-difluoropropoxy group, 2,2-difluoropropoxy group, 2,3-difluoropropoxy group, 3,3-difluoropropoxy group, 1,1,2-
- the alkylene group having 1 to 40 carbon atoms may be linear, branched or cyclic, and specific examples include methylene, ethylene, propylene, trimethylene, tetramethylene, pentylene, Hexylene group, heptylene group, octylene group, nonylene group, decylene group, undecylene group, dodecylene group, tridecylene group, tetradecylene group, pentadecylene group, hexadecylene group, heptadecylene group, octadecylene group, nonadecylene group, eicosanylene group and the like.
- aryl group having 6 to 20 carbon atoms include a phenyl group, a tolyl group, a 1-naphthyl group, a 2-naphthyl group, a 1-anthryl group, a 2-anthryl group, a 9-anthryl group, a 1-phenanthryl group, 2-phenanthryl group, 3-phenanthryl group, 4-phenanthryl group, 9-phenanthryl group and the like, with phenyl group, tolyl group and naphthyl group being preferred.
- aryloxy group having 6 to 20 carbon atoms include phenoxy group, anthracenoxy group, naphthoxy group, phenanthreoxy group, fluorenoxy group and the like.
- Halogen atoms include fluorine, chlorine, bromine and iodine atoms.
- sulfonic acid groups and sulfonic acid groups include groups represented by the following formula (S).
- M represents a hydrogen atom, an alkali metal selected from the group consisting of Li, Na and K, NH(R S ) 3 or HNC 5 H 5 .
- R S independently represents a hydrogen atom, or represents an optionally substituted alkyl group having 1 to 6 carbon atoms.
- R S is an alkyl group having a substituent
- substituents include an alkyl group having 1 to 6 carbon atoms, an alkoxy group having 1 to 6 carbon atoms, an aryl group having 6 to 20 carbon atoms, and a hydroxy group. , an amino group, a carboxy group, and the like.
- alkyl group having 1 to 6 carbon atoms include the same groups as those exemplified for the above alkyl group.
- Specific examples of alkoxy groups having 1 to 6 carbon atoms include methoxy, ethoxy, n-propoxy, i-propoxy, n-butoxy, etc.
- aryl groups having 6 to 20 carbon atoms include phenyl, tolyl, 1-naphthyl, 2-naphthyl, 1-anthryl, 2-anthryl, 9-anthryl, 1-phenanthryl, 2-phenanthryl, 3-phenanthryl, 4-phenanthryl, 9-phenanthryl groups and the like.
- a hydroxy group is preferable as the substituent, and specific examples of the alkyl group having a hydroxy group include 2-hydroxyethyl group, 3-hydroxypropyl group, 2-hydroxypropyl group, 2,3-dihydroxypropyl group and the like. mentioned.
- R S is preferably a hydrogen atom or a linear or branched alkyl group having 1 to 3 carbon atoms, more preferably a hydrogen atom or a methyl group.
- R 1 and R 2 are each independently a hydrogen atom, a fluoroalkyl group having 1 to 40 carbon atoms, an alkoxy group having 1 to 40 carbon atoms, —O[C(R a R b )—C(R c R d )—O] p —R e , —OR f , a sulfonate group or a sulfonate group, or —O—Y— formed by combining R 1 and R 2 O- is preferred.
- R a to R d each independently represent a hydrogen atom, an alkyl group having 1 to 40 carbon atoms, a fluoroalkyl group having 1 to 40 carbon atoms, or an aryl group having 6 to 20 carbon atoms. Specific examples of these groups are the same as those listed above. Among them, R a to R d are each independently preferably a hydrogen atom, an alkyl group having 1 to 8 carbon atoms, a fluoroalkyl group having 1 to 8 carbon atoms, or a phenyl group.
- R e represents a hydrogen atom, an alkyl group having 1 to 40 carbon atoms, a fluoroalkyl group having 1 to 40 carbon atoms, or an aryl group having 6 to 20 carbon atoms. Specific examples of these groups are the same as those listed above. Among them, R e is preferably a hydrogen atom, an alkyl group having 1 to 8 carbon atoms, a fluoroalkyl group having 1 to 8 carbon atoms, or a phenyl group, and more preferably a hydrogen atom, a methyl group, a propyl group, or a butyl group. Also, p is preferably 1 to 5, more preferably 1, 2 or 3.
- R f is a hydrogen atom, an alkyl group having 1 to 40 carbon atoms, a fluoroalkyl group having 1 to 40 carbon atoms or an aryl group having 6 to 20 carbon atoms, but a hydrogen atom, an alkyl group having 1 to 8 carbon atoms, A fluoroalkyl group having 1 to 8 carbon atoms or a phenyl group is preferred, and -CH 2 CF 3 is more preferred.
- R 1 is preferably a hydrogen atom, a sulfonic acid group or a sulfonic acid group, more preferably a sulfonic acid group or a sulfonic acid group, and R 2 is preferably an alkoxy group having 1 to 40 carbon atoms.
- a group or -O-[Z-O] p -R e more preferably -O[C(R a R b )-C(R c R d )-O] p -R e or -OR f , more preferably preferably -O[C(R a R b )-C(R c R d )-O] p -R e , -O-CH 2 CH 2 -O-CH 2 CH 2 -O-CH 3 , -O -CH 2 CH 2 -O-CH 2 CH 2 -OH or -O-CH 2 CH 2 -OH, or -O-Y-O- formed by combining R 1 and R 2 together is.
- the polythiophene derivative according to a preferred embodiment of the present invention comprises a repeating unit in which R 1 is a sulfonic acid group or a sulfonate group and R 2 is other than a sulfonic acid group or a sulfonate group, or R 1 and R 2 are combined to form --O--Y--O--.
- R 1 is a sulfonic acid group or a sulfonic acid group
- R 2 is an alkoxy group having 1 to 40 carbon atoms or —O—[ZO] p —R e or repeat units wherein R 1 and R 2 are joined to form -O-Y-O-.
- R 1 is a sulfonic acid group or a sulfonic acid group
- R 2 is —O[C(R a R b )–C(R c R d )–O] p — It contains repeating units that are R e or —OR f .
- R 1 is a sulfonic acid group or a sulfonic acid group
- R 2 is —O[C(R a R b )–C(R c R d )–O] p It includes repeating units that are —R e or —O—Y—O— formed by combining R 1 and R 2 .
- R 1 is a sulfonic acid group or a sulfonic acid group
- R 2 is --O--CH 2 CH 2 --O--CH 2 CH 2 --O--CH 3 , --O-- CH 2 CH 2 —O—CH 2 CH 2 —OH or —O—CH 2 CH 2 —OH
- Y1 Includes repeat units that are represented groups.
- R y represents an alkyl group having 1 to 6 carbon atoms or a fluorine atom. M is the same as above.
- Examples of the alkyl group having 1 to 6 carbon atoms for R y include the same groups as exemplified in the description of R 1 and R 2 .
- An alkyl group having 1 to 3 carbon atoms is preferred, a methyl group and an ethyl group are more preferred, and a methyl group is even more preferred.
- polythiophene derivative examples include, for example, polythiophene containing at least one repeating unit represented by the following formulas (1-1) to (1-4).
- a polythiophene containing a repeating unit represented by the following formula (1-4) is more preferable because it is easily obtainable and has high solubility in a solvent.
- examples of suitable structures of the above polythiophene derivatives include polythiophene derivatives having a structure represented by the following formula (1a).
- each unit may be combined randomly or may be combined as a block polymer.
- M is the same as above.
- polythiophene derivatives may be homopolymers or copolymers (including statistical, random, gradient, and block copolymers).
- block copolymers include, for example, AB diblock copolymers, ABA triblock copolymers, and (AB) m -multiblock copolymers.
- Polythiophenes contain repeat units derived from other types of monomers such as thienothiophenes, selenophenes, pyrroles, furans, tellurophenes, anilines, arylamines, and arylenes such as phenylenes, phenylene vinylenes, and fluorenes. may contain.
- the content of the repeating unit represented by formula (1) in the polythiophene derivative is preferably more than 50 mol%, more preferably 80 mol% or more, more preferably 90 mol% of all repeating units contained in the polythiophene derivative.
- the above is more preferable, 95 mol % or more is more preferable, and 100 mol % is most preferable.
- the content of repeating units having a sulfonic acid group or a sulfonate group is 10% of the repeating units represented by formula (1) in the polythiophene derivative.
- mol % or more is preferred, 30 mol % or more is more preferred, 50 mol % or more is even more preferred, and 100 mol % is even more preferred.
- the polymer formed may contain repeating units derived from impurities, depending on the purity of the starting monomers used for polymerization.
- the term "homopolymer” means a polymer containing repeating units derived from one type of monomer, but may contain repeating units derived from impurities.
- the polythiophene derivative is preferably a polymer in which basically all the repeating units are the repeating units represented by the above formula (1). ) is more preferably a polymer containing at least one repeating unit.
- the polythiophene derivative contains a repeating unit having a sulfonic acid group
- at least part of the sulfonic acid group contained in the polythiophene derivative contains an amine compound from the viewpoint of further improving solubility and dispersibility in solvents.
- An added amine adduct is preferred.
- Amine compounds that can be used to form amine adducts include methylamine, ethylamine, n-propylamine, isopropylamine, n-butylamine, isobutylamine, s-butylamine, t-butylamine, n-pentylamine, n-hexylamine.
- n-heptylamine monoalkylamine compounds such as n-octylamine; aniline, primary amine compounds such as monoarylamine compounds such as tolylamine; N-ethylmethylamine, N-methyl-n-propylamine, N-methyl isopropylamine, N-methyl-n-butylamine, N-methyl-s-butylamine, N-methyl-t-butylamine, N-methylisobutylamine, diethylamine, N-ethyl-n-propylamine, N-ethylisopropylamine, N-ethyl-n-butylamine, N-ethyl-s-butylamine, N-ethyl-t-butylamine, dipropylamine, Nn-propylisopropylamine, Nn-propyl-n-butylamine, Nn- Bropyl-s-butylamine, aziridine (ethyleneimine), 2-
- secondary amine compounds such as alkylarylamine compounds such as diphenylamine and indoline; N,N-dimethylethylamine, N,N-dimethyl-n-propylamine, N,N-dimethylisopropylamine, N,N -dimethyl-n-butylamine, N,N-dimethyl-s-butylamine, N,N-dimethyl-t-butylamine, N,N-dimethylisobutylamine, N,N-diethylmethylamine, N-methyldi(n-propyl ) amine, N-methyldiisopropylamine, triethylamine, N,N-diethyl-n-butylamine, N,N-diisopropylethylamine, N,N-di(n-butyl)ethylamine, 1-methylacetidine, 1-methylpyrrolidine , 1-methylpiperidine and the like, but considering the balance between the solub
- the above polythiophene derivative or its amine adduct may be treated with a reducing agent.
- some of the repeating units constituting them may have an oxidized chemical structure called a "quinoid structure".
- the term "quinoid structure” is used for the term “benzenoid structure”, the latter being a structure containing an aromatic ring, whereas the former is a structure in which the double bond within the aromatic ring moves out of the ring (the As a result, the aromatic ring disappears), meaning a structure in which two exocyclic double bonds conjugated with other double bonds remaining in the ring are formed.
- R 1 and R 2 are as defined in formula (1) above.
- This quinoid structure is generated by a process in which the polythiophene derivative containing the repeating unit represented by the above formula (1) undergoes an oxidation reaction by a dopant, a so-called doping reaction, and imparts charge transport properties to the polythiophene derivative. It forms part of a structure called "bipolaron structure". These structures are known. Introduction of a "polaron structure” and/or a “bipolaron structure” is essential in the production of an organic solar cell element, and in fact, when producing an organic solar cell element, the thin film formed from the charge-transporting composition is baked. Sometimes the doping reactions described above are deliberately allowed to achieve this.
- the reason why the quinoid structure is included in the polythiophene derivative before the doping reaction is that the polythiophene derivative undergoes an unintended oxidation reaction equivalent to the doping reaction during the manufacturing process (especially the sulfonation step therein). This is thought to be due to the
- the polythiophene derivative when the polythiophene derivative is subjected to a reduction treatment using a reducing agent, even if the quinoid structure is excessively introduced into the polythiophene derivative, the quinoid structure is reduced by the reduction, and the solubility and dispersibility of the polythiophene derivative in an organic solvent are improved. is improved, it becomes possible to stably produce a good charge-transporting composition that gives a thin film with excellent uniformity.
- the conditions for the reduction treatment are such that the quinoid structure is reduced to appropriately convert to the non-oxidized structure, that is, the benzenoid structure (for example, in the polythiophene derivative containing the repeating unit represented by the above formula (1),
- the quinoid structure represented by the above formula (1′) is not particularly limited as long as it can be converted to the structure represented by the above formula (1), for example, in the presence of a suitable solvent or
- This treatment can be carried out simply by contacting the polythiophene derivative or amine adduct with a reducing agent in the absence thereof.
- a reducing agent is not particularly limited as long as the reduction is performed properly, but suitable examples include aqueous ammonia, hydrazine, etc., which are readily available on the market.
- the amount of the reducing agent varies depending on the amount of the reducing agent to be used, and cannot be categorically defined. It is 0.1 parts by mass or more and 10 parts by mass or less from the viewpoint of preventing excess reducing
- a polythiophene derivative or an amine adduct is stirred overnight at room temperature in 28% ammonia water.
- the reduction treatment under such relatively mild conditions sufficiently improves the solubility and dispersibility of the polythiophene derivatives and amine adducts in organic solvents.
- the reduction treatment may be performed before or after forming the amine adduct.
- the solubility and dispersibility of the polythiophene derivative or its amine adduct in the solvent change, and as a result, the polythiophene derivative or its amine adduct, which was not dissolved in the reaction system at the start of the treatment, will be removed after the treatment is completed. Sometimes dissolved. In such a case, an organic solvent (acetone, isopropyl alcohol, etc. in the case of sulfonated polythiophene) incompatible with the polythiophene derivative or its amine adduct is added to the reaction system to obtain the polythiophene derivative or its amine adduct.
- the polythiophene derivative or its amine adduct can be recovered by a method such as causing precipitation and filtering.
- Preferred specific examples of the quinoid structure of the above formula (1') include structures represented by the following formula (1'-1).
- the weight-average molecular weight of the polythiophene derivative containing the repeating unit represented by formula (1) or its amine adduct is preferably from about 1,000 to about 1,000,000, more preferably from about 5,000 to about 100,000. Preferably, from about 10,000 to about 50,000 is even more preferred.
- a weight average molecular weight is a polystyrene conversion value by a gel permeation chromatography.
- the polythiophene derivative or its amine adduct contained in the charge-transporting composition of the present invention may be only one kind of polythiophene derivative or its amine adduct containing a repeating unit represented by formula (1), or two kinds. or more.
- a commercially available product or a product obtained by polymerizing a thiophene derivative or the like as a starting material by a known method may be used. It is also preferable to use those purified by methods such as reprecipitation and ion exchange. By using the purified one, the characteristics of the organic solar cell element provided with the thin film obtained from the charge-transporting composition of the present invention can be further enhanced. Examples of commercially available products include SELFTRON (registered trademark) manufactured by Tosoh Corporation.
- conjugated polymers and sulfonated conjugated polymers are described in US Pat. No. 8,017,241 to Seshadri et al. Sulfonated polythiophenes are also described in WO2008/073149 and WO2016/171935.
- At least part of the polythiophene derivative containing the repeating unit represented by formula (1) or its amine adduct contained in the charge-transporting composition is dissolved in an organic solvent.
- a polythiophene derivative containing a repeating unit represented by formula (1) or an amine adduct thereof and a charge-transporting substance other than the polythiophene derivative containing the repeating unit may be used in combination.
- the ionization potential of the hole collection layer is preferably close to the ionization potential of the p-type semiconductor material in the active layer.
- the absolute value of the difference is preferably 0 to 1 eV, more preferably 0 to 0.5 eV, and even more preferably 0 to 0.2 eV. Therefore, the charge-transporting composition of the present invention contains an arylsulfonic acid compound represented by the following formula (2) and a heteropolyacid for the purpose of adjusting the ionization potential of the charge-transporting thin film obtained using the composition.
- A represents a naphthalene ring or anthracene ring
- B represents a divalent to tetravalent perfluorobiphenyl group
- l represents the number of sulfonic acid groups bonded to A, satisfying 1 ⁇ l ⁇ 4 is an integer
- q indicates the number of bonds between B and X, and is an integer satisfying 2 to 4.
- the arylsulfonic acid compound represented by formula (2) can be synthesized by a known method, for example, by the method described in International Publication No. 2006/025342.
- heteropoly acids include heteropolyacids such as phosphomolybdic acid, silicomolybdic acid, phosphotungstic acid, phosphotungstomolybdic acid, silicotungstic acid, sodium phosphomolybdate, and phosphovanadomolybdic acid described in WO 2010/058777.
- heteropolyacids such as phosphomolybdic acid, silicomolybdic acid, phosphotungstic acid, phosphotungstomolybdic acid, silicotungstic acid, sodium phosphomolybdate, and phosphovanadomolybdic acid described in WO 2010/058777.
- examples include inorganic oxidizing agents such as acid compounds.
- silicomolybdic acid and silicotungstic acid are preferred in consideration of solubility in solvents, particularly solubility in water.
- the content of the electron-accepting dopant substance is appropriately set in consideration of the type of the charge-transporting substance and the charge-transporting substance to be expressed. 0.05 to 10, preferably 0.1 to 3.0, more preferably 0.2 to 2.0.
- the mixing ratio of the arylsulfonic acid compound represented by the formula (2) and the heteropolyacid is 10:90 to 90:10 is preferred, and 20:80 to 80:20 is more preferred.
- the charge-transporting composition of the present invention may contain an electron-accepting dopant substance other than the arylsulfonic acid compound represented by formula (2) and the heteropolyacid.
- electron-accepting dopant substances include strong inorganic acids such as hydrogen chloride, sulfuric acid , nitric acid, phosphoric acid; boron chloride (BBr 3 ), boron trifluoride etherate (BF 3 OEt 2 ), iron chloride (III) (FeCl 3 ), copper chloride (II) (CuCl 2 ), antimony pentachloride (V) (SbCl 5 ), arsenic pentafluoride (V) (AsF 5 ), phosphorus pentafluoride (PF 5 ), tris(4-bromophenyl)aluminum hexachloroantimonate (TBPAH); Lewis acids such as benzenesulfonic acid, tosylic acid, hydroxyl Benzenes
- the content thereof is preferably 20% by mass or less, more preferably 10% by mass or less, and still more preferably not contained, based on the total electron-accepting dopant substances.
- the charge-transporting composition of the present invention may contain a surfactant.
- the surfactant is not particularly limited, and fluorine-based surfactants, acetylenic surfactants, alkyl-based surfactants, silicone-based surfactants, etc. can be used. It is preferred to use an active agent.
- the fluorosurfactant used in the present invention is available as a commercial product.
- Such commercial products include Capstone® FS-10, FS-22, FS-30, FS-31, FS-34, FS-35, FS-50, FS- 51, FS-60, FS-61, FS-63, FS-64, FS-65, FS-66, FS-81, FS-83, FS-3100; Daiichi Kogyo Seiyaku Co., Ltd. Neugen FN- 1287; Megafac F-444, F-477, F-559 manufactured by DIC Corporation, but not limited thereto.
- Capstone FS-30, 31, 34, 35 and 3100, Neugen FN-1287 and Megafac F-559, which are nonionic surfactants, are preferred.
- the fluorine-based surfactant is not particularly limited as long as it contains a fluorine atom, and may be cationic, anionic, or nonionic, but fluorine-based nonionic surfactants are preferred. At least one fluorine-based nonionic surfactant selected from the following formulas (A1) and (B1) is particularly preferred.
- R represents a monovalent organic group containing a fluorine atom
- n represents an integer of 1-20.
- organic groups include alkyl groups having 1 to 40 carbon atoms, aryl groups having 6 to 20 carbon atoms, aralkyl groups having 7 to 20 carbon atoms, and heteroaryl groups having 2 to 20 carbon atoms.
- aralkyl groups having 7 to 20 carbon atoms include benzyl group, p-methylphenylmethyl group, m-methylphenylmethyl group, o-ethylphenylmethyl group, m-ethylphenylmethyl group and p-ethylphenylmethyl. group, 2-propylphenylmethyl group, 4-isopropylphenylmethyl group, 4-isobutylphenylmethyl group, ⁇ -naphthylmethyl group and the like.
- heteroaryl groups include 2-thienyl, 3-thienyl, 2-furanyl, 3-furanyl, 2-oxazolyl, 4-oxazolyl, 5-oxazolyl, 3-isoxazolyl, 4 -isoxazolyl group, 5-isoxazolyl group, 2-thiazolyl group, 4-thiazolyl group, 5-thiazolyl group, 3-isothiazolyl group, 4-isothiazolyl group, 5-isothiazolyl group, 2-imidazolyl group, 4-imidazolyl group, 2 -pyridyl group, 3-pyridyl group, 4-pyridyl group, 2-pyrazyl group, 3-pyrazyl group, 5-pyrazyl group, 6-pyrazyl group, 2-pyrimidyl group, 4-pyrimidyl group, 5-pyrimidyl group, 6 -pyrimidyl group, 3-pyridazyl group, 4-pyridazyl group, 5-pyridazyl group
- n is not particularly limited as long as it is an integer of 1-20, but an integer of 1-10 is more preferable.
- n has the same meaning as above.
- a specific example of the perfluoroalkyl group having 1 to 40 carbon atoms is a group in which all the hydrogen atoms of the alkyl group having 1 to 40 carbon atoms are substituted with fluorine atoms.
- acetylenic surfactant a commercially available product can also be used, and specific examples include Surfynol-104, Surfynol-420, Surfynol-440, Surfynol-465, Surfynol-485, Dynol-604, etc. manufactured by Evonik. Examples include, but are not limited to.
- alkyl-based surfactants Commercially available products can also be used as alkyl-based surfactants, and specific examples include Pluronic-10R5, Pluronic-25R5, Pluronic F-127, Pluronic-L44, Genapol X-080 and Genapol manufactured by Sigma-Aldrich. Examples include, but are not limited to, X-100, Genapol C-100, and lgepal C0-430 and lgepal CA-630 manufactured by Solvay.
- a surfactant When a surfactant is included, its content is not particularly limited. About 0.01 to 0.1% by mass of the whole is preferable, 0.02 to 0.08% by mass is more preferable, and 0.03 to 0.06% by mass is most preferable.
- compositions of the present invention may contain one or more metal oxide nanoparticles.
- a nanoparticle means a fine particle having an average primary particle size of the order of nanometers (typically 500 nm or less).
- Metal oxide nanoparticles refer to metal oxides shaped into nanoparticles.
- a nanoparticle means a fine particle having an average primary particle size of the order of nanometers (typically 500 nm or less).
- Metal oxide nanoparticles refer to metal oxides shaped into nanoparticles.
- the primary particle size of the metal oxide nanoparticles used in the present invention is not particularly limited as long as it is nano-sized. 100 nm is more preferred, and 5 to 50 nm is even more preferred.
- the particle size is a measured value using a nitrogen adsorption isotherm by the BET method.
- Metals constituting metal oxide nanoparticles in the present invention include not only metals in the usual sense, but also semimetals.
- Metals in the usual sense include, but are not limited to, tin (Sn), titanium (Ti), aluminum (Al), zirconium (Zr), zinc (Zn), niobium (Nb), tantalum ( It is preferable to use one or more selected from the group consisting of Ta) and W (tungsten).
- metalloids refer to elements whose chemical and/or physical properties are intermediate between those of metals and nonmetals. A universal definition of metalloids has not been established, but in the present invention, a total of six Let the elements be semimetals. These semimetals may be used alone or in combination of two or more, and may also be used in combination with metals in the usual sense.
- Metal oxide nanoparticles used in the present invention include boron (B), silicon (Si), germanium (Ge), arsenic (As), antimony (Sb), tellurium (Te), tin (Sn), titanium (Ti) , aluminum (Al), zirconium (Zr), zinc (Zn), niobium (Nb), tantalum (Ta) and W (tungsten).
- the metal oxide may be a mixture of oxides of individual single metals, or a composite oxide containing a plurality of metals.
- metal oxides include B2O3 , B2O, SiO2 , SiO, GeO2 , GeO, As2O4 , As2O3 , As2O5 , Sb2O3 , Sb2 . O5 , TeO2 , SnO2 , ZrO2 , Al2O3 , ZnO and the like, but also B2O3 , B2O , SiO2 , SiO , GeO2 , GeO, As2O4 , As2 . O3 , As2O5 , SnO2 , SnO, Sb2O3 , TeO2 , and mixtures thereof are preferred , with SiO2 being more preferred.
- the metal oxide nanoparticles may contain one or more organic capping groups.
- This organic capping group may be reactive or non-reactive.
- Examples of reactive organic capping groups include organic capping groups that can be crosslinked by UV light or radical initiators.
- silica sol in which SiO 2 nanoparticles are dispersed in a dispersion medium as the metal oxide nanoparticles.
- the silica sol is not particularly limited, and can be appropriately selected from known silica sols and used. Commercially available silica sols are usually in the form of dispersions.
- SiO2 nanoparticles are mixed with various solvents such as water, methanol, methyl ethyl ketone, methyl isobutyl ketone, N,N-dimethylacetamide, ethylene glycol, isopropanol, methanol, ethylene glycol monopropyl ether, cyclohexanone, acetic acid.
- solvents such as water, methanol, methyl ethyl ketone, methyl isobutyl ketone, N,N-dimethylacetamide, ethylene glycol, isopropanol, methanol, ethylene glycol monopropyl ether, cyclohexanone, acetic acid.
- solvent water-soluble alcohols are preferable, and methanol, 2-propanol and ethylene glycol are more preferable.
- silica sols include Snowtex (registered trademark) ST-O, ST-OS, ST-O-40 and ST-OL manufactured by Nissan Chemical Industries, Ltd., and Silidol 20 manufactured by Nippon Chemical Industries Co., Ltd. , 30, 40, etc.; methanol silica sol manufactured by Nissan Chemical Co., Ltd., MA-ST-M, MA-ST-L, IPA-ST, IPA-ST-L, IPA-ST-ZL, EG- Examples include, but are not limited to, organosilica sols such as ST.
- the solid content concentration of the silica sol is also not particularly limited, but is preferably 5 to 60% by mass, more preferably 10 to 50% by mass, and even more preferably 15 to 30% by mass.
- the content is not particularly limited, but in consideration of sufficient adhesion to the active layer, 50 parts per 100 parts by mass of the charge-transporting substance. ⁇ 95 parts by mass is preferable, 60 to 95 parts by mass is more preferable, and 80 to 95 parts by mass is even more preferable.
- the charge-transporting substance is used as a solution or dispersion, the amount of metal oxide nanoparticles to be added is based on the solid content of the charge-transporting substance.
- the composition of the present invention may contain an alkoxysilane.
- alkoxysilane By containing alkoxysilane, it is possible to improve the solvent resistance and water resistance of the obtained thin film, improve the electron blocking property, and optimize the HOMO level and LUMO level for the active layer.
- the alkoxysilane may be a siloxane-based material.
- any one or more alkoxysilanes selected from tetraalkoxysilane, trialkoxysilane and dialkoxysilane can be used.
- Methoxysilane, methyltriethoxysilane, methyltrimethoxysilane, 3,3,3-trifluoropropyltrimethoxysilane, dimethyldiethoxysilane and dimethyldimethoxysilane are preferred, and tetraethoxysilane is more preferred.
- siloxane-based materials include polysiloxanes such as poly(tetraethoxysilane) and poly(phenylethoxysilane) obtained by reactions such as hydrolysis of the alkoxysilanes.
- alkoxysilane When alkoxysilane is used, its content is not particularly limited as long as it exhibits the above effects. 0.01 to 50 times is more preferred, and 0.05 to 10 times is even more preferred.
- the charge-transporting composition of the present invention may further contain a matrix polymer, if necessary.
- a matrix polymer include matrix polymers containing repeating units represented by the following formula (I) and repeating units represented by the following formula (II).
- R 3 , R 4 , R 5 , R 6 , R 7 , R 8 and R 9 are each independently a hydrogen atom, a halogen atom, a fluoroalkyl group having 1 to 20 carbon atoms, or a a perfluoroalkyl group of 1 to 20,
- Q is -[OC(R h R i )-C(R j R k )] y -O-[CR l R m ] z -SO 3 H
- R h , R i , R j , R k , R l and R m are each independently a hydrogen atom, a halogen atom, a C 1-20 fluoroalkyl group, or a C 1-20 perfluoroalkyl group, y is 0-10, and z is 1-5.
- halogen atom the fluoroalkyl group having 1 to 20 carbon atoms
- perfluoroalkyl group having 1 to 20 carbon atoms are the same as those described above.
- R 3 , R 4 , R 5 and R 6 are preferably a fluorine atom or a chlorine atom, R 3 , R 5 and R 6 are a fluorine atom and R 4 is a chlorine atom is more preferred, and it is even more preferred that all of R 3 , R 4 , R 5 and R 6 are fluorine atoms.
- R 7 , R 8 and R 9 are preferably fluorine atoms.
- R h , R i , R j , R k , R l and R m are preferably a fluorine atom, a C 1-8 fluoroalkyl group, or a C 1-8 perfluoroalkyl group.
- R l and R m are fluorine atoms.
- y is preferably 0, and z is preferably 2.
- R 3 , R 5 and R 6 above are a fluorine atom
- R 4 is a chlorine atom
- each of R 1 and R m is a fluorine atom
- y is 0
- z is preferably two.
- each R3 , R4 , R5 , and R6 is a fluorine atom; and each R1 and Rm is a fluorine atom; y is 0; and z is preferably two.
- the ratio (s:t ratio) between the number "s" of repeating units represented by formula (I) and the number "t” of repeating units represented by formula (II) is not particularly limited.
- the s:t ratio is preferably 9:1 to 1:9, more preferably 8:2 to 2:8.
- the matrix polymer that can be suitably used in the present invention may be synthesized using a known method or may be a commercially available product.
- the polymer containing the repeating unit represented by the formula (I) and the repeating unit represented by the formula (II) is represented by the monomer represented by the following formula (Ia) and the following formula (IIa)
- Monomers can be prepared by copolymerization by known polymerization methods, followed by hydrolysis of the sulfonyl fluoride groups to convert them to sulfonic acid groups.
- TFE tetrafluoroethylene
- CFE chlorotrifluoroethylene
- F 2C CF-[O- CF2 - CR12FO ] y - CF2 - CF2 - SO2F where R12 is F or CF3 and y is 1 to 10
- F 2 C CF-O-CF 2 -CF 2 -CF 2 -SO 2 F
- F 2 C CF-OCF 2 -CF 2 -CF 2 -CF 2 -SO 2 F, etc.
- the matrix polymer means the mass (g/mol) of the matrix polymer per 1 mol of acid groups present in the matrix polymer.
- the equivalent weight of the matrix polymer is preferably from about 400 to about 15,000 g/mol, more preferably from about 500 to about 10,000 g/mol, even more preferably from about 500 to about 8,000 g/mol, even more preferably about 500 to about 2,000 g/mol, most preferably about 600 to about 1,700 g/mol.
- Such matrix polymers are commercially available.
- Commercially available products include, for example, NAFION (registered trademark) manufactured by DuPont, AQUIVION (registered trademark) manufactured by Solvay Specialty Polymers, and FLEMION (registered trademark) manufactured by Asahi Glass Co., Ltd., and the like.
- the matrix polymer is preferably polyethersulfone containing at least one repeating unit containing at least one sulfonic acid residue (--SO 3 H).
- composition of the present invention may contain other additives as long as the object of the present invention can be achieved.
- the type of additive can be appropriately selected from known additives depending on the desired effect.
- a highly soluble solvent that can dissolve the polythiophene derivative and the electron-accepting dopant substance well can be used as the solvent for preparing the charge-transporting composition.
- the highly soluble solvent can be used alone or in combination of two or more, and the amount used can be 5 to 100% by mass of the total solvent used in the composition.
- solvents examples include water; alcohol solvents such as ethanol, 2-propanol, 1-butanol, 2-butanol, s-butanol, t-butanol, 1-methoxy-2-propanol; - amides such as methylformamide, N,N-dimethylformamide, N,N-diethylformamide, N-methylacetamide, N,N-dimethylacetamide, N-methylpyrrolidone, 1,3-dimethyl-2-imidazolidinone
- Organic solvents such as solvents can be mentioned.
- at least one solvent selected from water and alcoholic solvents is preferred, water, ethanol and 2-propanol are more preferred, water and 2-propanol are still more preferred, and water is even more preferred.
- the above highly soluble solvents can be used alone or in combination of two or more.
- the content of the organic solvent in the solvent is 10% by mass or less, preferably 5% by mass or less. (that is, the solvent is only water) is more preferable.
- both the charge-transporting substance and the electron-accepting dopant substance are completely dissolved in the solvent or are in a state of being uniformly dispersed. Considering obtaining a pore-collecting layer with good reproducibility, it is more preferable that these substances are completely dissolved in the solvent.
- the charge-transporting composition of the present invention has a viscosity of 10 to 200 mPa ⁇ s, particularly 35 to 150 mPa ⁇ s at 25° C. in order to improve film-forming properties and ejection properties from a coating apparatus.
- the high-viscosity organic solvent is not particularly limited, and examples include cyclohexanol, ethylene glycol, 1,3-octylene glycol, diethylene glycol, dipropylene glycol, triethylene glycol, tripropylene glycol, and 1,3-butane. diol, 2,3-butanediol, 1,4-butanediol, propylene glycol, hexylene glycol and the like.
- the addition ratio is preferably within a range in which solids do not precipitate, and as long as solids do not precipitate, the total content of the organic solvent exemplified in the above highly soluble solvent , preferably 10% by mass or less in the solvent, more preferably 5% by mass or less, and more preferably not contained (0% by mass).
- solvents examples include butyl cellosolve, diethylene glycol diethyl ether, diethylene glycol dimethyl ether, diethylene glycol monoethyl ether acetate, diethylene glycol monobutyl ether acetate, dipropylene glycol monomethyl ether, propylene glycol monomethyl ether, propylene glycol monomethyl ether acetate, ethyl carbitol, diacetone alcohol, ⁇ -butyrolactone, ethyl lactate, n-hexyl acetate and the like.
- the addition ratio is preferably 1 to 90% by mass, more preferably 1 to 50% by mass, of the total solvent used in the composition.
- the solid content concentration of the composition of the present invention is appropriately set in consideration of the viscosity and surface tension of the composition, the thickness of the thin film to be produced, etc., but is usually 0.1 to 10.0 mass. %, preferably 0.5 to 5.0% by mass, more preferably 1.0 to 3.0% by mass.
- the viscosity of the charge-transporting composition used in the present invention may be appropriately adjusted depending on the coating method, taking into consideration the thickness of the thin film to be produced and the solid content concentration. ⁇ 50 mPa ⁇ s.
- a charge-transporting substance, a surfactant, a metal oxide nanoparticle, an electron-accepting dopant substance, a solvent, and the like are used as long as the solid content is uniformly dissolved or dispersed in the solvent. They can be mixed in any order.
- a method of dissolving a polythiophene derivative in a solvent and then dissolving an electron-accepting dopant substance in the solution a method of dissolving an electron-accepting dopant substance in a solvent and then dissolving a polythiophene derivative in the solution
- Any method of mixing a polythiophene derivative and an electron-accepting dopant substance and then dissolving the mixture in a solvent can be employed as long as the solid content is uniformly dissolved or dispersed in the solvent.
- the order of adding the matrix polymer and the alkoxysilane is also arbitrary.
- the preparation of the composition is carried out under an inert gas atmosphere at normal temperature and normal pressure. bottom) or while heating.
- the composition described above is coated on the anode in the case of a forward stacking type organic thin film solar cell, or on the active layer in the case of a reverse stacking type organic thin film solar cell, and then sintered to obtain the hole collection of the present invention. It can form layers.
- the viscosity and surface tension of the composition, the thickness of the desired thin film, etc. are taken into consideration, and the drop casting method, spin coating method, blade coating method, dip coating method, roll coating method, bar coating method, die coating method, An optimum wet process method such as an inkjet method, a printing method (letterpress, intaglio, lithography, screen printing, etc.) may be adopted. Further, coating is usually carried out in an inert gas atmosphere at normal temperature and pressure. You may carry out, and you may carry out while heating.
- the film thickness is not particularly limited, it is preferably about 0.1 to 800 nm, more preferably about 30 to 500 nm in any case.
- a method for changing the film thickness there are methods such as changing the solid content concentration in the composition and changing the amount of the solution at the time of coating.
- a method for producing an organic thin-film solar cell using the charge-transporting composition of the present invention as a composition for forming a hole-collecting layer will be described below, but the present invention is not limited thereto.
- Laminated organic thin film solar cell A process of forming a layer of anode material on the surface of a transparent substrate to manufacture a transparent electrode.
- Inorganic oxides such as zinc oxide (IZO), metals such as gold, silver and aluminum, and highly charge-transporting organic compounds such as polythiophene derivatives and polyaniline derivatives can be used. Among these, ITO is most preferred.
- the transparent substrate a substrate made of glass or transparent resin can be used as the transparent substrate. The method for forming the anode material layer (anode layer) is appropriately selected according to the properties of the anode material.
- a dry process such as a vacuum deposition method or a sputtering method is selected. Considering the thickness of the thin film, etc., the optimum one is adopted from among the various wet process methods described above.
- a commercially available transparent anode substrate can also be used, and in this case, from the viewpoint of improving the yield of the device, it is preferable to use a substrate that has been smoothed.
- the method for producing an organic thin film solar cell of the present invention does not include the step of forming an anode layer.
- an inorganic oxide such as ITO
- surface treatment such as UV ozone treatment and oxygen-plasma treatment immediately before use.
- the anode material is mainly composed of an organic substance, surface treatment may not be performed.
- a step of forming an active layer on the formed hole collection layer It may be a laminate of layers or a non-laminate thin film made of a mixture of these materials.
- the active layer may be either an FA active layer or an NFA active layer, and any combination can significantly improve the durability of the resulting device.
- the deep Ip of the resulting charge-transporting thin film can reduce the energy gap with the NFA active layer, enabling the device to operate at a higher voltage.
- the NFA active layer means an active layer in which the content of NFA is 70% by mass or more in the n-type semiconductor contained in the active layer in the present invention.
- the FA active layer means an active layer in which the content of FA is 70 mass % or more of the n-type semiconductor contained in the active layer.
- n-type semiconductor materials used for the FA active layer include fullerene, [6,6]-phenyl-C 61 -butyric acid methyl ester (PC 61 BM), [6,6]-phenyl-C 71 -butyric acid methyl ester ( PC 71 BM) and the like.
- n-type semiconductor material used for the NFA active layer examples include compounds represented by the following formulas (3-1) to (3-4).
- Examples of the p-type semiconductor material that can be combined with the n-type semiconductor material include regioregular poly(3-hexylthiophene) (P3HT), PTB7 represented by the following formula (4-1), and the following formula (4-2).
- P3HT regioregular poly(3-hexylthiophene)
- PTB7 represented by the following formula (4-1)
- 4-2 regioregular poly(3-hexylthiophene)
- 4-1 regioregular poly(3-hexylthiophene)
- 4-1 regioregular poly(3-hexylthiophene)
- 4-1 regioregular poly(3-hexylthiophene)
- 4-1 regioregular poly(3-hexylthiophene)
- 4-1 regioregular poly(3-hexylthiophene)
- 4-1 regioregular poly(3-he
- n-type semiconductor material compounds represented by formulas (3-1) and (3-2) are preferable, and among them, BTP-4F in which X is F is more preferable in the former, and BTP-4F in which X is F in the latter ITIC-4F in which both X 1 and X 2 are F is more preferred.
- the p-type semiconductor material polymers containing a thiophene skeleton in the main chain, such as PM6 and PTB7, are preferable.
- thiophene skeleton in the main chain refers to a divalent aromatic ring consisting only of thiophene, or thienothiophene, benzothiophene, dibenzothiophene, benzodithiophene, naphthothiophene, naphthodithiophene, anthrathiophene, anthradithiophene.
- alkyl group having 1 to 20 carbon atoms alkenyl group having 2 to 20 carbon atoms, alkynyl group having 2 to 20 carbon atoms, haloalkyl group having 1 to 20 carbon atoms, aryl group having 6 to 20 carbon atoms, carbon It may be substituted with an aralkyl group having 7 to 20 carbon atoms or an acyl group having 1 to 20 carbon atoms.
- the halogen atom, the alkyl group having 1 to 20 carbon atoms, the alkoxy group having 1 to 20 carbon atoms, the aryl group having 6 to 20 carbon atoms, and the aralkyl group having 7 to 20 carbon atoms are the same as those exemplified above. are mentioned.
- thioalkoxy group having 1 to 20 carbon atoms include groups obtained by substituting the oxygen atom of the above alkoxy group with a sulfur atom.
- thioalkoxy (alkylthio) groups having 1 to 20 carbon atoms include methylthio, ethylthio, n-propylthio, isopropylthio, n-butylthio, isobutylthio, s-butylthio and t-butylthio.
- n-pentylthio group n-hexylthio group, n-heptylthio group, n-octylthio group, n-nonylthio group, n-decylthio group, n-undecylthio group, n-dodecylthio group, n-tridecylthio group, n-tetra decylthio group, n-pentadecylthio group, n-hexadecylthio group, n-heptadecylthio group, n-octadecylthio group, n-nonadecylthio group, n-eicosanylthio group and the like.
- alkenyl groups having 2 to 20 carbon atoms include ethenyl group, n-1-propenyl group, n-2-propenyl group, 1-methylethenyl group, n-1-butenyl group, n-2-butenyl group, n-3-butenyl group, 2-methyl-1-propenyl group, 2-methyl-2-propenyl group, 1-ethylethenyl group, 1-methyl-1-propenyl group, 1-methyl-2-propenyl group, n- 1-pentenyl group, n-1-decenyl group, n-1-eicosenyl group and the like.
- alkynyl groups having 2 to 20 carbon atoms include ethynyl, n-1-propynyl, n-2-propynyl, n-1-butynyl, n-2-butynyl and n-3-butynyl.
- haloalkyl groups having 1 to 20 carbon atoms include groups obtained by substituting at least one hydrogen atom in the above alkyl group with a halogen atom.
- Halogen atoms may be chlorine, bromine, iodine or fluorine atoms. Among them, a fluoroalkyl group is preferred, and a perfluoroalkyl group is more preferred.
- Specific examples thereof include a fluoromethyl group, a difluoromethyl group, a trifluoromethyl group, a pentafluoroethyl group, a 2,2,2-trifluoroethyl group, a heptafluoropropyl group, a 2,2,3,3,3- pentafluoropropyl group, 2,2,3,3-tetrafluoropropyl group, 2,2,2-trifluoro-1-(trifluoromethyl)ethyl group, nonafluorobutyl group, 4,4,4-trifluoro butyl group, undecafluoropentyl group, 2,2,3,3,4,4,5,5,5-nonafluoropentyl group, 2,2,3,3,4,4,5,5-octafluoro pentyl group, tridecafluorohexyl group, 2,2,3,3,4,4,5,5,6,6,6-undecafluorohexyl group, 2,2,3,3,4,4,5 , 5,6,6-
- acyl groups having 1 to 20 carbon atoms include formyl group, acetyl group, propionyl group, butyryl group, isobutyryl group, valeryl group, isovaleryl group, benzoyl group and the like.
- n-type semiconductor material contained in the active layer contains the n-type semiconductor material corresponding to the above NFA as the balance.
- n-type semiconductor materials include BTP-4F and ITIC-4F.
- the n-type semiconductor material corresponding to the above-mentioned FA is used as the balance in the range of less than 30% by mass of the n-type semiconductor material contained in the active layer. may be included.
- the same dry process as described above is selected when the active layer material is a poorly soluble sublimable material, and when it is a solution material or a dispersion liquid material, Considering the viscosity and surface tension of the composition, the desired thickness of the thin film, and the like, the most suitable one is adopted from among the various wet process methods described above.
- An electron collecting layer may be formed.
- Materials for forming the electron collection layer include lithium oxide (Li 2 O), magnesium oxide (MgO), alumina (Al 2 O 3 ), lithium fluoride (LiF), sodium fluoride (NaF), and magnesium fluoride.
- the various dry processes described above are selected. Considering the viscosity, surface tension, desired thickness of the thin film, etc., the optimum wet process method is adopted from among the various wet process methods described above.
- a step of forming a cathode layer on the formed electron collection layer metals such as barium, silver, and gold; inorganic oxides such as indium tin oxide (ITO) and indium zinc oxide (IZO); and highly charge-transporting organic compounds such as polythiophene derivatives and polyaniline derivatives. can be used by laminating or mixing the cathode materials.
- metals such as barium, silver, and gold
- inorganic oxides such as indium tin oxide (ITO) and indium zinc oxide (IZO)
- highly charge-transporting organic compounds such as polythiophene derivatives and polyaniline derivatives.
- the various dry processes described above are selected. Considering the viscosity and surface tension of the film, the desired thickness of the thin film, etc., the optimum one is adopted from among the various wet process methods described above.
- a carrier block layer may be provided between arbitrary layers for the purpose of controlling the rectification of photocurrent.
- a carrier blocking layer When a carrier blocking layer is provided, an electron blocking layer is usually inserted between the active layer and the hole collecting layer or the anode, and a hole blocking layer is inserted between the active layer and the electron collecting layer or the cathode.
- Materials for forming the hole blocking layer include titanium oxide, zinc oxide, tin oxide, bathocuproine (BCP), 4,7-diphenyl-1,10-phenanthroline (BPhen), and the like.
- Materials for forming the electron blocking layer include N,N'-di(1-naphthyl)-N,N'-diphenylbenzidine ( ⁇ -NPD) and triarylamine-based materials such as poly(triarylamine) (PTAA). materials and the like.
- the various dry processes described above are selected. Considering the viscosity and surface tension of the composition, the desired thickness of the thin film, and the like, the most suitable one is adopted from among the various wet process methods described above.
- the dry process described above is selected in the case of the poorly soluble and difficultly dispersible sublimable material, and in the case of the solution material or dispersion liquid material, the viscosity of the composition and the Considering the surface tension, the desired thickness of the thin film, etc., the most suitable wet process method is adopted from among the various wet process methods described above.
- a commercially available transparent cathode substrate can be suitably used, and from the viewpoint of improving the yield of the device, it is preferable to use a substrate that has been smoothed.
- the method for producing an organic thin-film solar cell of the present invention does not include the step of forming a cathode layer.
- an inorganic oxide is used as a cathode material to form a transparent cathode substrate, it may be subjected to the same cleaning treatment and surface treatment as those for the sequentially laminated anode material.
- a trapping layer may be formed.
- Materials for forming the electron collection layer include zinc oxide (ZnO), titanium oxide (TiO), tin oxide (SnO), etc., in addition to the materials exemplified in the above-mentioned forward stacking type materials.
- the above-described dry process is selected in the case of a poorly soluble or difficultly dispersible sublimation material, and in the case of a solution material or a dispersion liquid material, the viscosity and surface tension of the composition are adjusted to the desired value.
- the optimum one is adopted from among the various wet process methods described above.
- a method of forming an inorganic oxide precursor layer on the cathode using a wet process (particularly spin coating or slit coating) and firing to form an inorganic oxide layer can also be employed.
- the active layer consists of an n-layer, which is a thin film made of an n-type semiconductor material, and a p-layer, which is a thin film made of a p-type semiconductor material. or a non-laminated thin film made of a mixture of these materials.
- the n-type and p-type semiconductor materials include the same materials as those exemplified in the above-mentioned forward stacking type semiconductor materials. are preferably polymers containing a thiophene skeleton in the main chain, such as PM6 and PTB7.
- the method for forming the active layer is also the same as the method described for the forward lamination type active layer.
- a step of forming an anode layer on the formed hole collection layer. is the same as that of the positively laminated cathode layer.
- a carrier block layer may be provided between arbitrary layers for the purpose of controlling the rectification of photocurrent.
- Materials for forming the hole blocking layer and materials for forming the electron blocking layer are the same as those described above, and the method for forming the carrier blocking layer is also the same as described above.
- the OPV element manufactured by the method exemplified above is introduced into the glove box again and sealed in an inert gas atmosphere such as nitrogen. It is possible to exhibit the function as a solar cell and to measure the solar cell characteristics.
- a sealing method a concave glass substrate having a UV curable resin attached to the edge is attached to the film forming surface side of the organic thin film solar cell element in an inert gas atmosphere, and the resin is cured by UV irradiation.
- a film-sealing type sealing method using a method such as sputtering under vacuum is a method such as sputtering under vacuum.
- D1 manufactured by Fuji Film Wako Pure Chemical Industries, Ltd.
- D1 Fuji Film Wako Pure Chemical Industries, Ltd.
- D1 was added to prepare a dark blue solution with a concentration of 0.65% by mass.
- the resulting dark blue solution was filtered through a syringe filter with a pore size of 0.45 ⁇ m to obtain a hole-collecting layer composition A1.
- Example 1-2 To a solution of 1.25 g of P1 added with 3.73 g of pure water, 5.00 mg of S1, and 1.00 mg of S2, silicotungstic acid n-hydrate (manufactured by Alfa Aesar Co., Ltd.) (hereinafter: D2) was added. 7.50 mg was added to prepare a dark blue solution with a concentration of 0.65% by mass. The resulting dark blue solution was filtered through a syringe filter with a pore size of 0.45 ⁇ m to obtain a hole-collecting layer composition A2.
- D2 silicotungstic acid n-hydrate
- Example 1-3 7.50 mg of D1 to a solution of 1.25 g of P1, 3.71 g of pure water, 5.00 mg of S1, and 1.00 mg of S2, synthesized based on the description of WO 2006/025342 25.0 mg of the arylsulfonic acid compound A (hereinafter referred to as D3) represented by formula (2-1) was added to prepare a dark blue solution having a concentration of 1.15% by mass. The resulting dark blue solution was filtered through a syringe filter with a pore size of 0.45 ⁇ m to obtain a hole-collecting layer composition A3.
- D3 arylsulfonic acid compound A
- Example 1-4 7.50 mg of D2 and 25.0 mg of D3 were added to a solution in which 3.71 g of pure water, 5.00 mg of S1 and 1.00 mg of S2 were added to 1.25 g of P1 to obtain a concentration of 1.15% by mass. A blue solution was prepared. The resulting dark blue solution was filtered through a syringe filter with a pore size of 0.45 ⁇ m to obtain a hole-collecting layer composition A4.
- Example 1-5 3.21 g of pure water, 0.49 g of isopropanol, 5.00 mg of S1, and 1.00 mg of S2 were added to 1.25 g of P1. A 0.15% by weight dark blue solution was prepared. The resulting dark blue solution was filtered through a syringe filter with a pore size of 0.45 ⁇ m to obtain a hole-collecting layer composition A5.
- Example 1-6 12 molybdo (IV) phosphate n-hydrate (manufactured by Fujifilm Wako Pure Chemical Industries, Ltd.) ) (hereinafter: D4) was added to prepare a dark blue solution having a concentration of 0.65% by mass. The resulting dark blue solution was filtered through a syringe filter with a pore size of 0.45 ⁇ m to obtain a hole-collecting layer composition A6.
- Example 1-7 12 Tungsto (IV) phosphate n-hydrate (manufactured by Fujifilm Wako Pure Chemical Industries, Ltd.) ) (hereinafter referred to as D5) was added to prepare a dark blue solution having a concentration of 0.65% by mass. The resulting dark blue solution was filtered through a syringe filter with a pore size of 0.45 ⁇ m to obtain a hole-collecting layer composition A7.
- Table 1 summarizes the ink compositions of Examples 1-1 to 1-7.
- Table 2 summarizes the ink compositions of Comparative Examples 1-1 to 1-9.
- the film thickness of the active layer was about 100 nm.
- the hole-collecting layer composition A1 prepared in Example 1-1 was applied onto the active layer by a blade coating method to form a hole-collecting layer.
- the film thickness of the hole collection layer was about 100 nm.
- the laminated substrate is placed in a vacuum deposition apparatus, the apparatus is evacuated to a degree of vacuum of 1 ⁇ 10 ⁇ 3 Pa or less, and a silver layer serving as an anode is formed to a thickness of 100 nm by resistance heating.
- a reverse lamination type OPV element having an area of 10 mm ⁇ 10 mm at the intersection of the striped ITO layer and the silver layer was fabricated.
- Example 3-2 After applying the hole-collecting layer composition A1 by a blade coating method, an inverse lamination type OPV element was formed in the same manner as in Example 3-1, except that annealing was performed on a hot plate at 120 ° C. for 5 minutes. was made.
- Example 3-3 A reverse laminated OPV element was fabricated in the same manner as in Example 3-1, except that the hole-collecting layer composition A3 was used instead of the hole-collecting layer composition A1.
- Example 3-4 A reverse laminated OPV element was produced in the same manner as in Example 3-2, except that the hole-collecting layer composition A3 was used instead of the hole-collecting layer composition A1.
- Example 3-5 A reverse laminated OPV element was produced in the same manner as in Example 3-1, except that the hole-collecting layer composition A4 was used instead of the hole-collecting layer composition A1.
- Example 3-6 A reverse laminated OPV element was fabricated in the same manner as in Example 3-2, except that the hole-collecting layer composition A4 was used instead of the hole-collecting layer composition A1.
- Example 3-7 A reverse stacked OPV device was fabricated in the same manner as in Example 3-4, except that the thickness of the hole collection layer was about 300 nm.
- Example 3-8 A reverse laminated OPV element was fabricated in the same manner as in Example 3-7, except that the hole-collecting layer composition A5 was used instead of the hole-collecting layer composition A3.
- Example 3-9 A reverse lamination type OPV element was fabricated in the same manner as in Example 3-1 except that the FA active layer PV-F1062 (manufactured by Merck) was used instead of the NFA active layer PV-X Plus (manufactured by Raynergy tek). .
- Example 3-10 A reverse lamination type OPV element was fabricated in the same manner as in Example 3-5, except that the FA active layer PV-F1062 (manufactured by Merck & Co.) was used instead of the NFA active layer PV-X Plus (manufactured by Raynergy tek).
- Example 3-1 A reverse laminated OPV element was fabricated in the same manner as in Example 3-1, except that the hole-collecting layer composition B3 was used instead of the hole-collecting layer composition A1.
- Example 3-2 A reverse laminated OPV element was fabricated in the same manner as in Example 3-2, except that the hole-collecting layer composition B3 was used instead of the hole-collecting layer composition A1.
- Example 3-3 A reverse laminated OPV element was produced in the same manner as in Example 3-1, except that the hole-collecting layer composition B7 was used instead of the hole-collecting layer composition A1.
- Example 3-4 A reverse laminated OPV element was fabricated in the same manner as in Example 3-2, except that the hole-collecting layer composition B7 was used instead of the hole-collecting layer composition A1.
- Example 3-5 A reverse laminated OPV element was fabricated in the same manner as in Example 3-7, except that the hole-collecting layer composition B5 was used instead of the hole-collecting layer composition A3.
- Example 3-6 A reverse laminated OPV element was fabricated in the same manner as in Example 3-7, except that the hole-collecting layer composition B6 was used instead of the hole-collecting layer composition A3.
- Example 3-7 A reverse laminated OPV element was fabricated in the same manner as in Example 3-9, except that the hole-collecting layer composition B3 was used instead of the hole-collecting layer composition A1.
- Example 3-8 A reverse laminated OPV element was produced in the same manner as in Example 3-9, except that the hole-collecting layer composition B7 was used instead of the hole-collecting layer composition A1.
Landscapes
- Physics & Mathematics (AREA)
- Electromagnetism (AREA)
- Photovoltaic Devices (AREA)
Abstract
有機光電変換素子の電荷輸送性薄膜の形成に好適な電荷輸送性組成物として、式(1)の繰り返し単位を含むポリチオフェン誘導体からなる電荷輸送性物質と電子受容性ドーパント物質と溶媒とを含み、電子受容性ドーパント物質が式(2)で表されるアリールスルホン酸およびヘテロポリ酸からなる群より選ばれる少なくとも1種を含み、溶媒中の有機溶媒の含有量が10質量%以下である電荷輸送性組成物を提供する。 (R1およびR2は、互いに独立して、水素原子、炭素数1~40のアルキル基、またはR1およびR2が結合して形成される-O-Y-O-であり、Yはエーテル結合を含んでいてもよい、スルホン酸基またはスルホン酸塩基で置換されている炭素数1~40のアルキレン基等である。) (Aはナフタレン環等を表し、Bは2~4価のパーフルオロビフェニル基を表し、lは1≦l≦4を満たす整数であり、qは2~4を満たす整数である。)
Description
本発明は、電荷輸送性組成物に関し、更に詳述すると、有機光電変換素子において用いられる電荷輸送性薄膜を形成するための電荷輸送性組成物に関する。
電子素子、特に、有機光電変換素子は、有機半導体を用いて光エネルギーを電気エネルギーに変換するデバイスであり、例えば有機太陽電池が挙げられる。
有機太陽電池は、活性層や電荷輸送性物質に有機物を使用した太陽電池素子であり、M.グレッツェルによって開発された色素増感太陽電池と、C.W.タンによって開発された有機薄膜太陽電池とがよく知られている(非特許文献1、2)。
いずれも軽量・薄膜で、フレキシブル化可能である点、ロール・トゥ・ロールでの生産が可能である点など、現在主流の無機系太陽電池とは異なる特長を持っていることから、新たな市場形成が期待されている。
有機太陽電池は、活性層や電荷輸送性物質に有機物を使用した太陽電池素子であり、M.グレッツェルによって開発された色素増感太陽電池と、C.W.タンによって開発された有機薄膜太陽電池とがよく知られている(非特許文献1、2)。
いずれも軽量・薄膜で、フレキシブル化可能である点、ロール・トゥ・ロールでの生産が可能である点など、現在主流の無機系太陽電池とは異なる特長を持っていることから、新たな市場形成が期待されている。
さらに有機薄膜太陽電池は、既存のシリコン系材料を使用した光電変換素子と比較して、低照度においても高い光電変換効率を示すこと、素子の薄化および画素微細化が可能であること、カラーフィルターの性質を兼ね備えることが可能であること等の特長から、太陽電池用途だけでなく、イメージセンサーをはじめとする光センサー用途としても注目されている(特許文献1、2、非特許文献3)。以下、有機太陽電池(色素増感太陽電池および有機薄膜太陽電池)に加えて、光センサー等の用途を含んで有機光電変換素子(以下OPVと略す場合もある)と総称する。
有機光電変換素子は、活性層(光電変換層)、電荷(正孔、電子)捕集層、および電極(陽極、陰極)等を備えて構成される。これらの中でも、正孔捕集層は活性層で生成した正孔を電極へと抽出する役割があり、活性層-正孔捕集層間のエネルギー障壁を小さくすることで、効果的に行うことができる。
従来の有機光電変換素子の活性層には、電子供与性有機材料(p型有機半導体)として共役系化合物、電子受容性有機材料(n型有機半導体)としてn型の半導体特性を有する共役系化合物C60などのフラーレンやフラーレン誘導体を使用した活性層(以下、FA活性層と略す)が用いられてきた。
これらFA活性層に対しては、上記の観点に加え、量産プロセスを考え、塗布型の正孔捕集層であるPEDOT/PSS等の水分散性高分子有機導電材料が広く用いられており、例えば、Qun Wanらによって、光電変換効率(以下、PCEと略す)が約11%を示すことが報告されている(非特許文献4)。しかしながら、FA活性層はPCEが実用化レベルに到達していないだけでなく、理論限界に近づいていることもあり、活性層の開発が進められてきた。
そこで近年、新規なn型有機半導体として、フラーレンやフラーレン誘導体ではない非フラーレンアクセプター(Non-Fullerene Acceptor:以下、NFAと略す)と呼ばれる新材料が開発され、これを使用したNFA活性層が開発された。NFA活性層は光電流の増加や電池電圧の向上などにより、FA活性層を用いた場合よりも高いPCEを示す。実際、Jianhui Houらによって、NFA活性層を用いることでPCEが18%を示すことが報告されている(非特許文献5)。
一方で、これらの高いPCEを示す有機光電変換素子は正孔捕集層に、量産化に不利とされる蒸着型の正孔捕集層であるMoO3が広く用いられている。これは新規活性層のHOMO-LUMOの準位が従来活性層よりも深く、PEDOT/PSSのイオン化ポテンシャル(以下、Ipと略す)との間にエネルギーギャップが生じたためである。そのため、深いIpを有する塗布型の正孔捕集材料が求められている(非特許文献6)。
このように、有機光電変換素子の性能向上の観点から、PCEを向上させることが多く試みられてきたが、有機光電変換素子が長期的に使用されることを考慮すると、素子の耐久性の向上も重要な課題である。また、生産性向上の観点から、電荷輸送性薄膜を形成するための組成物の保存安定性についても改善が望まれている。
Nature, vol.353, 737-740(1991)
Appl. Phys. Lett., Vol.48, 183-185 (1986)
Scientific Reports, Vol.5:7708, 1-7 (2015)
Adv. Funct. Mater., vol.26, 6635-6640(2016)
Adv. Mater., vol.32, 1908205(2020)
Sol. RRL, 2000749(2021)
本発明は、上記事情に鑑みてなされたものであり、光電変換素子の電荷輸送性薄膜の形成に好適であり、特に、有機光電変換素子の正孔捕集層として用いた場合には、得られる素子の耐久性を大きく向上させ得る電荷輸送性組成物を提供することを目的とする。
本発明者らは、上記目的を達成するために鋭意検討を重ねた結果、所定の繰り返し単位を含有するポリチオフェン誘導体と、特定の電子受容性ドーパント物質と、溶媒とを含み、かつ上記溶媒に対する有機溶媒の含有量が特定範囲に調整された電荷輸送性組成物が、有機光電変換素子における電荷輸送性薄膜の形成に好適であり、特に、有機光電変換素子の正孔捕集層として用いた場合には、得られる素子の耐久性を大きく向上させ得ることを見出し、本発明を完成させた。
すなわち、本発明は、以下の電荷輸送性組成物を提供する。
1. 有機光電変換素子における電荷輸送性薄膜を形成するための電荷輸送性組成物であって、
下記式(1)で表される繰り返し単位を含むポリチオフェン誘導体からなる電荷輸送性物質と、電子受容性ドーパント物質と、溶媒とを含み、
上記電子受容性ドーパント物質が、下記式(2)で表されるアリールスルホン酸およびヘテロポリ酸からなる群より選ばれる少なくとも1種を含み、上記溶媒中の有機溶媒の含有量が10質量%以下である電荷輸送性組成物。

(式中、R1およびR2は、互いに独立して、水素原子、炭素数1~40のアルキル基、炭素数1~40のフルオロアルキル基、炭素数1~40のアルコキシ基、炭素数1~40のフルオロアルコキシ基、炭素数6~20のアリールオキシ基、-O-[Z-O]p-Re、スルホン酸基もしくはスルホン酸塩基、またはR1およびR2が結合して形成される-O-Y-O-であり、Yは、エーテル結合を含んでいてもよい、スルホン酸基またはスルホン酸塩基で置換されている炭素数1~40のアルキレン基であり、Zは、ハロゲン原子で置換されていてもよい炭素数1~40のアルキレン基であり、pは、1以上の整数であり、Reは、水素原子、スルホン酸基またはスルホン酸塩基で置換されていてもよい炭素数1~40のアルキル基、スルホン酸基またはスルホン酸塩基で置換されていてもよい炭素数1~40のフルオロアルキル基、またはスルホン酸基またはスルホン酸塩基で置換されていてもよい炭素数6~20のアリール基である。)

(式中、Aは、ナフタレン環またはアントラセン環を表し、Bは、2~4価のパーフルオロビフェニル基を表し、lは、Aに結合するスルホン酸基数を表し、1≦l≦4を満たす整数であり、qは、BとXとの結合数を示し、2~4を満たす整数である。)
2. 上記R1およびR2が、R1およびR2が結合して形成される-O-Y-O-であり、Yは、エーテル結合を含んでいてもよい、スルホン酸基またはスルホン酸塩基で置換されている炭素数1~40のアルキレン基である1の電荷輸送性組成物。
3. 上記式(1)で表される繰り返し単位を含むポリチオフェン誘導体が、式(1-4)または式(1’-1)で表される繰り返し単位を含むポリチオフェン誘導体である1または2の電荷輸送性組成物。

(式中、Ryは、炭素数1~6のアルキル基、またはフッ素原子を表す。式(1-4)中、Mは、水素原子、Li、NaおよびKからなる群より選ばれるアルカリ金属、NH(RS)3またはHNC5H5を表す。RSは、互いに独立して、水素原子、または置換基を有していてもよい炭素数1~6のアルキル基を表す。)
4. 上記電子受容性ドーパント物質が、上記式(2)で表されるアリールスルホン酸およびヘテロポリ酸を含む1~3のいずれかの電荷輸送性組成物。
5. 上記ヘテロポリ酸が、ケイモリブデン酸およびケイタングステン酸からなる群より選ばれる少なくとも1種を含む1~4のいずれかの電荷輸送性組成物。
6. 上記溶媒中の有機溶媒の含有量が、5質量%以下である1~5のいずれかの電荷輸送性組成物。
7. 上記溶媒が水のみである6の電荷輸送性組成物。
8. さらに、界面活性剤を含む1~7のいずれかの電荷輸送性組成物。
9. 上記界面活性剤が、フッ素系界面活性剤である8の電荷輸送性組成物。
10. 有機光電変換素子の正孔捕集層用である1~9のいずれかの電荷輸送性組成物。
11. 上記有機光電変換素子が、有機薄膜太陽電池、色素増感太陽電池または光センサーである10の電荷輸送性組成物。
12. 1~11のいずれかの電荷輸送性組成物から得られる電荷輸送性薄膜。
13. 上記電荷輸送性薄膜が、有機光電変換素子の正孔捕集層である12の電荷輸送性薄膜。
14. 12または13の電荷輸送性薄膜を備える電子素子。
15. 上記電子素子が、有機光電変換素子である14の電子素子。
16. 13の正孔捕集層と、それに接するように設けられた非フラーレン活性層とを有する有機光電変換素子。
17. 上記非フラーレン活性層が、主鎖にチオフェン骨格を含むポリマーを含む16の有機光電変換素子。
18. 逆積層型である16または17の有機光電変換素子。
19. 上記有機光電変換素子が、有機薄膜太陽電池または光センサーである16~18のいずれかの有機光電変換素子。
20. トップ陽極構造を有する19の有機光電変換素子。
1. 有機光電変換素子における電荷輸送性薄膜を形成するための電荷輸送性組成物であって、
下記式(1)で表される繰り返し単位を含むポリチオフェン誘導体からなる電荷輸送性物質と、電子受容性ドーパント物質と、溶媒とを含み、
上記電子受容性ドーパント物質が、下記式(2)で表されるアリールスルホン酸およびヘテロポリ酸からなる群より選ばれる少なくとも1種を含み、上記溶媒中の有機溶媒の含有量が10質量%以下である電荷輸送性組成物。
2. 上記R1およびR2が、R1およびR2が結合して形成される-O-Y-O-であり、Yは、エーテル結合を含んでいてもよい、スルホン酸基またはスルホン酸塩基で置換されている炭素数1~40のアルキレン基である1の電荷輸送性組成物。
3. 上記式(1)で表される繰り返し単位を含むポリチオフェン誘導体が、式(1-4)または式(1’-1)で表される繰り返し単位を含むポリチオフェン誘導体である1または2の電荷輸送性組成物。
4. 上記電子受容性ドーパント物質が、上記式(2)で表されるアリールスルホン酸およびヘテロポリ酸を含む1~3のいずれかの電荷輸送性組成物。
5. 上記ヘテロポリ酸が、ケイモリブデン酸およびケイタングステン酸からなる群より選ばれる少なくとも1種を含む1~4のいずれかの電荷輸送性組成物。
6. 上記溶媒中の有機溶媒の含有量が、5質量%以下である1~5のいずれかの電荷輸送性組成物。
7. 上記溶媒が水のみである6の電荷輸送性組成物。
8. さらに、界面活性剤を含む1~7のいずれかの電荷輸送性組成物。
9. 上記界面活性剤が、フッ素系界面活性剤である8の電荷輸送性組成物。
10. 有機光電変換素子の正孔捕集層用である1~9のいずれかの電荷輸送性組成物。
11. 上記有機光電変換素子が、有機薄膜太陽電池、色素増感太陽電池または光センサーである10の電荷輸送性組成物。
12. 1~11のいずれかの電荷輸送性組成物から得られる電荷輸送性薄膜。
13. 上記電荷輸送性薄膜が、有機光電変換素子の正孔捕集層である12の電荷輸送性薄膜。
14. 12または13の電荷輸送性薄膜を備える電子素子。
15. 上記電子素子が、有機光電変換素子である14の電子素子。
16. 13の正孔捕集層と、それに接するように設けられた非フラーレン活性層とを有する有機光電変換素子。
17. 上記非フラーレン活性層が、主鎖にチオフェン骨格を含むポリマーを含む16の有機光電変換素子。
18. 逆積層型である16または17の有機光電変換素子。
19. 上記有機光電変換素子が、有機薄膜太陽電池または光センサーである16~18のいずれかの有機光電変換素子。
20. トップ陽極構造を有する19の有機光電変換素子。
本発明の有機光電変換素子の電荷輸送性組成物は、市場で安価に入手可能な、あるいは公知の方法で簡便に合成できるポリチオフェン誘導体からなる電荷輸送性物質を用いて製造可能なだけでなく、それから得られる薄膜は、光電変換素子の正孔捕集層として用いた場合において、素子の耐久性を大きく向上し得る。特にNFA活性層と組み合わせて用いられた場合には、得られる電荷輸送性薄膜が有する深いIpによりNFA活性層とのエネルギーギャップを低減し、素子の高電圧化を実現し得る。
以下、本発明について更に詳しく説明する。
本発明の電荷輸送性組成物は、有機光電変換素子における電荷輸送性薄膜を形成するための電荷輸送性組成物であって、下記式(1)で表される繰り返し単位を含むポリチオフェン誘導体からなる電荷輸送性物質と、溶媒とを含み、上記電子受容性ドーパント物質が、下記式(2)で表されるアリールスルホン酸およびヘテロポリ酸からなる群より選ばれる少なくとも1種を含み、上記溶媒中の有機溶媒の含有量が10質量%以下であることを特徴とする。なお、本発明において、「固形分」とは電荷輸送性組成物の全成分のうち、溶剤以外のものの総称である。
本発明の電荷輸送性組成物は、有機光電変換素子における電荷輸送性薄膜を形成するための電荷輸送性組成物であって、下記式(1)で表される繰り返し単位を含むポリチオフェン誘導体からなる電荷輸送性物質と、溶媒とを含み、上記電子受容性ドーパント物質が、下記式(2)で表されるアリールスルホン酸およびヘテロポリ酸からなる群より選ばれる少なくとも1種を含み、上記溶媒中の有機溶媒の含有量が10質量%以下であることを特徴とする。なお、本発明において、「固形分」とは電荷輸送性組成物の全成分のうち、溶剤以外のものの総称である。

式中、R1およびR2は、互いに独立して、水素原子、炭素数1~40のアルキル基、炭素数1~40のフルオロアルキル基、炭素数1~40のアルコキシ基、炭素数1~40のフルオロアルコキシ基、炭素数6~20のアリールオキシ基、-O-[Z-O]p-Re、スルホン酸基もしくはスルホン酸塩基、またはR1およびR2が結合して形成される-O-Y-O-であり、Yは、エーテル結合を含んでいてもよい、スルホン酸基またはスルホン酸塩基で置換されている炭素数1~40のアルキレン基であり、Zは、ハロゲン原子で置換されていてもよい炭素数1~40のアルキレン基であり、pは、1以上の整数であり、Reは、水素原子、スルホン酸基またはスルホン酸塩基で置換されていてもよい炭素数1~40のアルキル基、スルホン酸基またはスルホン酸塩基で置換されていてもよい炭素数1~40のフルオロアルキル基、またはスルホン酸基またはスルホン酸塩基で置換されていてもよい炭素数6~20のアリール基である。
炭素数1~40のアルキル基としては、直鎖状、分岐鎖状、環状のいずれでもよく、その具体例としては、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基、s-ブチル基、t-ブチル基、n-ペンチル基、n-ヘキシル基、n-ヘプチル基、n-オクチル基、n-ノニル基、n-デシル基、n-ウンデシル基、n-ドデシル基、n-トリデシル基、n-テトラデシル基、n-ペンタデシル基、n-ヘキサデシル基、n-ヘプタデシル基、n-オクタデシル基、n-ノナデシル基、n-エイコサニル基、ベヘニル基、トリアコンチル基、テトラコンチル基等が挙げられる。本発明においては、炭素数1~18のアルキル基が好ましく、炭素数1~8のアルキル基がより好ましい。
炭素数1~40のフルオロアルキル基としては、炭素原子上の少なくとも1個の水素原子がフッ素原子で置換された炭素数1~40のアルキル基であれば特に限定されないが、その具体例としては、フルオロメチル基、ジフルオロメチル基、パーフルオロメチル基、1-フルオロエチル基、2-フルオロエチル基、1,2-ジフルオロエチル基、1,1-ジフルオロエチル基、2,2-ジフルオロエチル基、1,1,2-トリフルオロエチル基、1,2,2-トリフルオロエチル基、2,2,2-トリフルオロエチル基、1,1,2,2-テトラフルオロエチル基、1,2,2,2-テトラフルオロエチル基、パーフルオロエチル基、1-フルオロプロピル基、2-フルオロプロピル基、3-フルオロプロピル基、1,1-ジフルオロプロピル基、1,2-ジフルオロプロピル基、1,3-ジフルオロプロピル基、2,2-ジフルオロプロピル基、2,3-ジフルオロプロピル基、3,3-ジフルオロプロピル基、1,1,2-トリフルオロプロピル基、1,1,3-トリフルオロプロピル基、1,2,3-トリフルオロプロピル基、1,3,3-トリフルオロプロピル基、2,2,3-トリフルオロプロピル基、2,3,3-トリフルオロプロピル基、3,3,3-トリフルオロプロピル基、1,1,2,2-テトラフルオロプロピル基、1,1,2,3-テトラフルオロプロピル基、1,2,2,3-テトラフルオロプロピル基、1,3,3,3-テトラフルオロプロピル基、2,2,3,3-テトラフルオロプロピル基、2,3,3,3-テトラフルオロプロピル基、1,1,2,2,3-ペンタフルオロプロピル基、1,2,2,3,3-ペンタフルオロプロピル基、1,1,3,3,3-ペンタフルオロプロピル基、1,2,3,3,3-ペンタフルオロプロピル基、2,2,3,3,3-ペンタフルオロプロピル基、パーフルオロプロピル基、パーフルオロブチル基、パーフルオロペンチル基、パーフルオロヘキシル基、パーフルオロヘプチル基、パーフルオロオクチル基等が挙げられる。
炭素数1~40のアルコキシ基としては、その中のアルキル基が直鎖状、分岐鎖状、環状のいずれでもよく、その具体例としては、メトキシ基、エトキシ基、n-プロポキシ基、i-プロポキシ基、c-プロポキシ基、n-ブトキシ基、i-ブトキシ基、s-ブトキシ基、t-ブトキシ基、n-ペントキシ基、n-ヘキソキシ基、n-ヘプチルオキシ基、n-オクチルオキシ基、n-ノニルオキシ基、n-デシルオキシ基、n-ウンデシルオキシ基、n-ドデシルオキシ基、n-トリデシルオキシ基、n-テトラデシルオキシ基、n-ペンタデシルオキシ基、n-ヘキサデシルオキシ基、n-ヘプタデシルオキシ基、n-オクタデシルオキシ基、n-ノナデシルオキシ基、n-エイコサニルオキシ基等が挙げられる。
炭素数1~40のフルオロアルコキシ基としては、炭素原子上の少なくとも1個の水素原子がフッ素原子で置換された炭素数1~40のアルコキシ基であれば特に限定されないが、その具体例としては、フルオロメトキシ基、ジフルオロメトキシ基、パーフルオロメトキシ基、1-フルオロエトキシ基、2-フルオロエトキシ基、1,2-ジフルオロエトキシ基、1,1-ジフルオロエトキシ基、2,2-ジフルオロエトキシ基、1,1,2-トリフルオロエトキシ基、1,2,2-トリフルオロエトキシ基、2,2,2-トリフルオロエトキシ基、1,1,2,2-テトラフルオロエトキシ基、1,2,2,2-テトラフルオロエトキシ基、パーフルオロエトキシ基、1-フルオロプロポキシ基、2-フルオロプロポキシ基、3-フルオロプロポキシ基、1,1-ジフルオロプロポキシ基、1,2-ジフルオロプロポキシ基、1,3-ジフルオロプロポキシ基、2,2-ジフルオロプロポキシ基、2,3-ジフルオロプロポキシ基、3,3-ジフルオロプロポキシ基、1,1,2-トリフルオロプロポキシ基、1,1,3-トリフルオロプロポキシ基、1,2,3-トリフルオロプロポキシ基、1,3,3-トリフルオロプロポキシ基、2,2,3-トリフルオロプロポキシ基、2,3,3-トリフルオロプロポキシ基、3,3,3-トリフルオロプロポキシ基、1,1,2,2-テトラフルオロプロポキシ基、1,1,2,3-テトラフルオロプロポキシ基、1,2,2,3-テトラフルオロプロポキシ基、1,3,3,3-テトラフルオロプロポキシ基、2,2,3,3-テトラフルオロプロポキシ基、2,3,3,3-テトラフルオロプロポキシ基、1,1,2,2,3-ペンタフルオロプロポキシ基、1,2,2,3,3-ペンタフルオロプロポキシ基、1,1,3,3,3-ペンタフルオロプロポキシ基、1,2,3,3,3-ペンタフルオロプロポキシ基、2,2,3,3,3-ペンタフルオロプロポキシ基、パーフルオロプロポキシ基等が挙げられる。
炭素数1~40のアルキレン基としては、直鎖状、分岐鎖状、環状のいずれでもよく、その具体例としては、メチレン基、エチレン基、プロピレン基、トリメチレン基、テトラメチレン基、ペンチレン基、へキシレン基、ヘプチレン基、オクチレン基、ノニレン基、デシレン基、ウンデシレン基、ドデシレン基、トリデシレン基、テトラデシレン基、ペンタデシレン基、ヘキサデシレン基、ヘプタデシレン基、オクタデシレン基、ノナデシレン基、エイコサニレン基等が挙げられる。
炭素数6~20のアリール基の具体例としては、フェニル基、トリル基、1-ナフチル基、2-ナフチル基、1-アントリル基、2-アントリル基、9-アントリル基、1-フェナントリル基、2-フェナントリル基、3-フェナントリル基、4-フェナントリル基、9-フェナントリル基等が挙げられ、フェニル基、トリル基およびナフチル基が好ましい。
炭素数6~20のアリールオキシ基の具体例としては、フェノキシ基、アントラセノキシ基、ナフトキシ基、フェナントレノキシ基、フルオレノキシ基等が挙げられる。
ハロゲン原子としては、フッ素原子、塩素原子、臭素原子およびヨウ素原子が挙げられる。
スルホン酸基およびスルホン酸塩基としては、下記式(S)で表される基が挙げられる。
また、RSが置換基を有するアルキル基である場合の当該置換基としては、炭素数1~6のアルキル基、炭素数1~6のアルコキシ基、炭素数6~20のアリール基、ヒドロキシ基、アミノ基、カルボキシ基等が挙げられる。
炭素数1~6のアルキル基としては、上記アルキル基において例示した基と同じものが挙げられる。
炭素数1~6のアルコキシ基の具体例としては、メトキシ、エトキシ、n-プロポキシ、i-プロポキシ、n-ブトキシ等が挙げられ、炭素数6~20のアリール基の具体例としては、フェニル、トリル、1-ナフチル、2-ナフチル、1-アントリル、2-アントリル、9-アントリル、1-フェナントリル、2-フェナントリル、3-フェナントリル、4-フェナントリル、9-フェナントリル基等が挙げられる。
特に、置換基としてはヒドロキシ基が好ましく、ヒドロキシ基を有するアルキル基の具体例としては、2-ヒドロキシエチル基、3-ヒドロキシプロピル基、2-ヒドロキシプロピル基、2,3-ジヒドロキシプロピル基等が挙げられる。
これらの中でも、RSとしては、水素原子、炭素数1~3の直鎖状または分岐鎖状のアルキル基が好ましく、水素原子、メチル基がより好ましい。
炭素数1~6のアルキル基としては、上記アルキル基において例示した基と同じものが挙げられる。
炭素数1~6のアルコキシ基の具体例としては、メトキシ、エトキシ、n-プロポキシ、i-プロポキシ、n-ブトキシ等が挙げられ、炭素数6~20のアリール基の具体例としては、フェニル、トリル、1-ナフチル、2-ナフチル、1-アントリル、2-アントリル、9-アントリル、1-フェナントリル、2-フェナントリル、3-フェナントリル、4-フェナントリル、9-フェナントリル基等が挙げられる。
特に、置換基としてはヒドロキシ基が好ましく、ヒドロキシ基を有するアルキル基の具体例としては、2-ヒドロキシエチル基、3-ヒドロキシプロピル基、2-ヒドロキシプロピル基、2,3-ジヒドロキシプロピル基等が挙げられる。
これらの中でも、RSとしては、水素原子、炭素数1~3の直鎖状または分岐鎖状のアルキル基が好ましく、水素原子、メチル基がより好ましい。
上記式(1)中、R1およびR2は、互いに独立して、水素原子、炭素数1~40のフルオロアルキル基、炭素数1~40のアルコキシ基、-O[C(RaRb)-C(RcRd)-O]p-Re、-ORf、スルホン酸基もしくはスルホン酸塩基であるか、またはR1およびR2が結合して形成される-O-Y-O-が好ましい。
Ra~Rdは、互いに独立して、水素原子、炭素数1~40のアルキル基、炭素数1~40のフルオロアルキル基、または炭素数6~20のアリール基を表す。これらの基の具体例としては上記で挙げたものと同じである。
中でも、Ra~Rdは、互いに独立して、水素原子、炭素数1~8のアルキル基、炭素数1~8のフルオロアルキル基、またはフェニル基が好ましい。
中でも、Ra~Rdは、互いに独立して、水素原子、炭素数1~8のアルキル基、炭素数1~8のフルオロアルキル基、またはフェニル基が好ましい。
Reは、水素原子、炭素数1~40のアルキル基、炭素数1~40のフルオロアルキル基、または炭素数6~20のアリール基を表す。これらの基の具体例としては上記で挙げたものと同じである。
中でも、Reは、水素原子、炭素数1~8のアルキル基、炭素数1~8のフルオロアルキル基、またはフェニル基が好ましく、水素原子、メチル基、プロピル基、またはブチル基がより好ましい。
また、pは、1~5が好ましく、1、2または3がより好ましい。
中でも、Reは、水素原子、炭素数1~8のアルキル基、炭素数1~8のフルオロアルキル基、またはフェニル基が好ましく、水素原子、メチル基、プロピル基、またはブチル基がより好ましい。
また、pは、1~5が好ましく、1、2または3がより好ましい。
Rfは、水素原子、炭素数1~40のアルキル基、炭素数1~40のフルオロアルキル基または炭素数6~20のアリール基であるが、水素原子、炭素数1~8のアルキル基、炭素数1~8のフルオロアルキル基、またはフェニル基が好ましく、-CH2CF3がより好ましい。
本発明においては、R1は、好ましくは水素原子またはスルホン酸基もしくはスルホン酸塩基、より好ましくはスルホン酸基またはスルホン酸塩基であり、かつ、R2は、好ましくは炭素数1~40のアルコキシ基または-O-[Z-O]p-Re、より好ましくは-O[C(RaRb)-C(RcRd)-O]p-Reまたは-ORf、より一層好ましくは-O[C(RaRb)-C(RcRd)-O]p-Re、-O-CH2CH2-O-CH2CH2-O-CH3、-O-CH2CH2-O-CH2CH2-OHまたは-O-CH2CH2-OHであるか、または、R1およびR2が互いに結合して形成される-O-Y-O-である。
例えば、本発明の好ましい態様に係る上記ポリチオフェン誘導体は、R1が、スルホン酸基またはスルホン酸塩基であり、R2が、スルホン酸基またはスルホン酸塩基以外である繰り返し単位を含むか、またはR1およびR2が結合して形成される-O-Y-O-である繰り返し単位を含む。
好ましくは、上記ポリチオフェン誘導体は、R1が、スルホン酸基またはスルホン酸塩基であり、R2が、炭素数1~40のアルコキシ基もしくは-O-[Z-O]p-Reである繰り返し単位を含むか、またはR1およびR2が結合して形成される-O-Y-O-である繰り返し単位を含む。
より好ましくは、上記ポリチオフェン誘導体は、R1が、スルホン酸基またはスルホン酸塩基であり、R2が、-O[C(RaRb)-C(RcRd)-O]p-Reまたは-ORfである繰り返し単位を含む。
より一層好ましくは、上記ポリチオフェン誘導体は、R1が、スルホン酸基またはスルホン酸塩基であり、R2が、-O[C(RaRb)-C(RcRd)-O]p-Reである繰り返し単位を含むか、またはR1およびR2が結合して形成される-O-Y-O-である繰り返し単位を含む。
更に好ましくは、上記ポリチオフェン誘導体は、R1が、スルホン酸基またはスルホン酸塩基であり、R2が、-O-CH2CH2-O-CH2CH2-O-CH3、-O-CH2CH2-O-CH2CH2-OH、もしくは-O-CH2CH2-OHである繰り返し単位を含むか、またはR1およびR2が互いに結合して、下記式(Y1)で表される基である繰り返し単位を含む。
好ましくは、上記ポリチオフェン誘導体は、R1が、スルホン酸基またはスルホン酸塩基であり、R2が、炭素数1~40のアルコキシ基もしくは-O-[Z-O]p-Reである繰り返し単位を含むか、またはR1およびR2が結合して形成される-O-Y-O-である繰り返し単位を含む。
より好ましくは、上記ポリチオフェン誘導体は、R1が、スルホン酸基またはスルホン酸塩基であり、R2が、-O[C(RaRb)-C(RcRd)-O]p-Reまたは-ORfである繰り返し単位を含む。
より一層好ましくは、上記ポリチオフェン誘導体は、R1が、スルホン酸基またはスルホン酸塩基であり、R2が、-O[C(RaRb)-C(RcRd)-O]p-Reである繰り返し単位を含むか、またはR1およびR2が結合して形成される-O-Y-O-である繰り返し単位を含む。
更に好ましくは、上記ポリチオフェン誘導体は、R1が、スルホン酸基またはスルホン酸塩基であり、R2が、-O-CH2CH2-O-CH2CH2-O-CH3、-O-CH2CH2-O-CH2CH2-OH、もしくは-O-CH2CH2-OHである繰り返し単位を含むか、またはR1およびR2が互いに結合して、下記式(Y1)で表される基である繰り返し単位を含む。

上記Ryの炭素数1~6のアルキル基としては、R1およびR2の説明において例示した基と同じものが挙げられる。炭素数1~3のアルキル基が好ましく、メチル基およびエチル基がより好ましく、メチル基がより一層好ましい。
上記ポリチオフェン誘導体の好ましい具体例としては、例えば、下記式(1-1)~(1-4)で示される繰り返し単位の少なくとも一種を含むポリチオフェンを挙げることができ、特に良好な導電性が再現性良く得られ、溶媒に対する溶解性が高い点から、下記式(1-4)で示される繰り返し単位を含むポリチオフェンがより好ましい。

また、上記ポリチオフェン誘導体の好適な構造としては、例えば、下記式(1a)で表される構造を有するポリチオフェン誘導体を挙げることができる。なお、下記式において、各単位はランダムに結合していても、ブロック重合体として結合していてもよい。

式中、a~dは、各単位のモル比を表し、0≦a≦1、0≦b≦1、0<a+b≦1、0≦c<1、0≦d<1、a+b+c+d=1を満足する。Mは、上記と同じである。
さらに、上記ポリチオフェン誘導体は、ホモポリマーまたはコポリマー(統計的、ランダム、勾配、およびブロックコポリマーを含む)であってよい。モノマーAおよびモノマーBを含むポリマーとしては、ブロックコポリマーは、例えば、A-Bジブロックコポリマー、A-B-Aトリブロックコポリマー、および(AB)m-マルチブロックコポリマーを含む。ポリチオフェンは、他のタイプのモノマー(例えば、チエノチオフェン、セレノフェン、ピロール、フラン、テルロフェン、アニリン、アリールアミン、およびアリーレン(例えば、フェニレン、フェニレンビニレン、およびフルオレン等)等)から誘導される繰り返し単位を含んでいてもよい。
本発明において、ポリチオフェン誘導体における式(1)で表される繰り返し単位の含有量は、ポリチオフェン誘導体に含まれる全繰り返し単位中、50モル%超が好ましく、80モル%以上がより好ましく、90モル%以上がより一層好ましく、95モル%以上がさらに好ましく、100モル%が最も好ましい。
また、ポリチオフェン誘導体における式(1)で表される繰り返し単位のうち、スルホン酸基またはスルホン酸塩基を有する繰り返し単位の含有量は、ポリチオフェン誘導体における式(1)で表される繰り返し単位中、10モル%以上が好ましく、30モル%以上がより好ましく、50モル%以上がより一層好ましく、100モル%がさらに好ましい。
本発明において、重合に使用される出発モノマーの純度に応じて、形成されるポリマーは、不純物から誘導される繰り返し単位を含有してもよい。本発明において、上記の「ホモポリマー」という用語は、1つのタイプのモノマーから誘導される繰り返し単位を含むポリマーを意味するものであるが、不純物から誘導される繰り返し単位を含んでいてもよい。本発明において、上記ポリチオフェン誘導体は、基本的に全ての繰り返し単位が、上記式(1)で表される繰り返し単位であるポリマーであることが好ましく、上記式(1-1)~(1-4)で表される繰り返し単位の少なくとも1つを含むポリマーであることがより好ましい。
本発明において、上記ポリチオフェン誘導体が、スルホン酸基を有する繰り返し単位を含む場合、溶媒に対する溶解性や分散性をより向上させる観点から、ポリチオフェン誘導体に含まれるスルホン酸基の少なくとも一部にアミン化合物が付加したアミン付加体とすることが好ましい。
アミン付加体の形成に使用できるアミン化合物としては、メチルアミン、エチルアミン、n-プロピルアミン、イソプロピルアミン、n-ブチルアミン、イソブチルアミン、s-ブチルアミン、t-ブチルアミン、n-ペンチルアミン、n-ヘキシルアミン、n-ヘプチルアミン、n-オクチルアミン等のモノアルキルアミン化合物;アニリン、トリルアミン等のモノアリールアミン化合物等の一級アミン化合物;N-エチルメチルアミン、N-メチル-n-プロピルアミン、N-メチルイソプロピルアミン、N-メチル-n-ブチルアミン、N-メチル-s-ブチルアミン、N-メチル-t-ブチルアミン、N-メチルイソブチルアミン、ジエチルアミン、N-エチル-n-プロピルアミン、N-エチルイソプロピルアミン、N-エチル-n-ブチルアミン、N-エチル-s-ブチルアミン、N-エチル-t-ブチルアミン、ジプロピルアミン、N-n-プロピルイソプロピルアミン、N-n-プロピル-n-ブチルアミン、N-n-ブロピル-s-ブチルアミン、アジリジン(エチレンイミン)、2-メチルアジリジン(プロピレンイミン)、2,2-ジメチルアジリジン、アゼチジン(トリメチレンイミン)、2-メチルアゼチジン、ピロリジン、2-メチルピロリジン、3-メチルピロリジン、2,5-ジメチルピロリジン、ピペリジン、2,6-ジメチルピペリジン、3,5-ジメチルピペリジン,2,2,6,6-テトラメチルピペリジン、ヘキサメチレンイミン、ヘプタメチレンイミン、オクタメチレンイミン等のジアルキルアミン化合物;ジフェニルアミン、インドリン等のアルキルアリールアミン化合物等の二級アミン化合物;N,N-ジメチルエチルアミン、N,N-ジメチル-n-プロピルアミン、N,N-ジメチルイソプロピルアミン、N,N-ジメチル-n-ブチルアミン、N,N-ジメチル-s-ブチルアミン、N,N-ジメチル-t-ブチルアミン、N,N-ジメチルイソブチルアミン、N,N-ジエチルメチルアミン、N-メチルジ(n-プロピル)アミン、N-メチルジイソプロピルアミン、トリエチルアミン、N,N-ジエチル-n-ブチルアミン、N,N-ジイソプロピルエチルアミン、N,N-ジ(n-ブチル)エチルアミン、1-メチルアセチジン、1-メチルピロリジン、1-メチルピペリジン等のトリアルキルアミン化合物が挙げられるが、アミン付加体の溶解性、得られる電荷輸送性薄膜の電荷輸送性等のバランスを考慮すると、三級アミン化合物が好ましく、トリアルキルアミン化合物がより好ましく、トリエチルアミンがより一層好ましい。
アミン付加体は、アミン自体またはその溶液にポリチオフェン誘導体を投入し、よく撹拌することで得ることができる。
アミン付加体は、アミン自体またはその溶液にポリチオフェン誘導体を投入し、よく撹拌することで得ることができる。
本発明においては、上記のポリチオフェン誘導体またはそのアミン付加体は、還元剤で処理したものを用いてもよい。
ポリチオフェン誘導体またはそのアミン付加体では、それらを構成する繰り返し単位の一部において、その化学構造が「キノイド構造」と呼ばれる酸化型の構造となっている場合がある。用語「キノイド構造」は、用語「ベンゼノイド構造」に対して用いられるもので、芳香環を含む構造である後者に対し、前者は、その芳香環内の二重結合が環外に移動し(その結果、芳香環は消失する)、環内に残る他の二重結合と共役する2つの環外二重結合が形成された構造を意味する。当業者にとって、これらの両構造の関係は、ベンゾキノンとヒドロキノンの構造の関係から容易に理解できるものである。種々の共役ポリマーの繰り返し単位についてのキノイド構造は、当業者にとって周知である。一例として、上記式(1)で表される繰り返し単位を含むポリチオフェン誘導体の繰り返し単位に対応するキノイド構造を、下記式(1’)に示す。
ポリチオフェン誘導体またはそのアミン付加体では、それらを構成する繰り返し単位の一部において、その化学構造が「キノイド構造」と呼ばれる酸化型の構造となっている場合がある。用語「キノイド構造」は、用語「ベンゼノイド構造」に対して用いられるもので、芳香環を含む構造である後者に対し、前者は、その芳香環内の二重結合が環外に移動し(その結果、芳香環は消失する)、環内に残る他の二重結合と共役する2つの環外二重結合が形成された構造を意味する。当業者にとって、これらの両構造の関係は、ベンゾキノンとヒドロキノンの構造の関係から容易に理解できるものである。種々の共役ポリマーの繰り返し単位についてのキノイド構造は、当業者にとって周知である。一例として、上記式(1)で表される繰り返し単位を含むポリチオフェン誘導体の繰り返し単位に対応するキノイド構造を、下記式(1’)に示す。

式(1’)中、R1およびR2は、上記式(1)において定義されたとおりである。
このキノイド構造は、上記式(1)で表される繰り返し単位を含むポリチオフェン誘導体がドーパントにより酸化反応を受けるプロセス、いわゆるドーピング反応によって生じ、ポリチオフェン誘導体に電荷輸送性を付与する「ポーラロン構造」および「バイポーラロン構造」と称される構造の一部を成すものである。これらの構造は公知である。有機太陽電池素子の作製において、「ポーラロン構造」および/または「バイポーラロン構造」の導入は必須であり、実際、有機太陽電池素子作成時、電荷輸送性組成物から形成された薄膜を焼成処理するときに、上記のドーピング反応を意図的に起こさせて、これを達成している。このドーピング反応を起こさせる前のポリチオフェン誘導体にキノイド構造が含まれているのは、ポリチオフェン誘導体が、その製造過程(特に、その中のスルホン化工程)において、ドーピング反応と同等の、意図しない酸化反応を起こしたためと考えられる。
上記ポリチオフェン誘導体に含まれるキノイド構造の量と、ポリチオフェン誘導体の有機溶媒に対する溶解性や分散性の間には相関があり、キノイド構造の量が多くなると、その溶解性や分散性は低下する傾向にある。このため、電荷輸送性組成物から薄膜が形成された後でのキノイド構造の導入は問題を生じないが、上記の意図しない酸化反応により、ポリチオフェン誘導体にキノイド構造が過剰に導入されていると、電荷輸送性組成物の製造に支障をきたす場合がある。ポリチオフェン誘導体においては、有機溶媒に対する溶解性や分散性にばらつきがあることが知られているが、その原因の1つは、上記の意図しない酸化反応によりポリチオフェンに導入されたキノイド構造の量が、各々のポリチオフェン誘導体の製造条件の差に応じて変動することであると考えられる。
そこで、上記ポリチオフェン誘導体を、還元剤を用いる還元処理に付すと、ポリチオフェン誘導体にキノイド構造が過剰に導入されていても、還元によりキノイド構造が減少し、ポリチオフェン誘導体の有機溶媒に対する溶解性や分散性が向上するため、均質性に優れた薄膜を与える良好な電荷輸送性組成物を、安定的に製造することが可能になる。
そこで、上記ポリチオフェン誘導体を、還元剤を用いる還元処理に付すと、ポリチオフェン誘導体にキノイド構造が過剰に導入されていても、還元によりキノイド構造が減少し、ポリチオフェン誘導体の有機溶媒に対する溶解性や分散性が向上するため、均質性に優れた薄膜を与える良好な電荷輸送性組成物を、安定的に製造することが可能になる。
還元処理の条件は、上記キノイド構造を還元して非酸化型の構造、すなわち、上記ベンゼノイド構造に適切に変換する(例えば、上記式(1)で表される繰り返し単位を含むポリチオフェン誘導体においては、上記式(1’)で表されるキノイド構造を、上記式(1)で表される構造に変換する)ことができるものである限り特に制限はないが、例えば、適当な溶媒の存在下または非存在下、単にポリチオフェン誘導体やアミン付加体を還元剤と接触させることにより、この処理を行うことができる。
このような還元剤も還元が適切にされる限り特に制限はないが、例えば、市販品で入手が容易であるアンモニア水、ヒドラジン等が適当である。
また、還元剤の量は、用いる還元剤の量に応じて異なるため一概に規定できないが、処理すべきポリチオフェン誘導体やアミン付加体100質量部に対し、通常、還元が適切にされる観点から、0.1質量部以上であり、過剰な還元剤が残存しないようにする観点から、10質量部以下である。
このような還元剤も還元が適切にされる限り特に制限はないが、例えば、市販品で入手が容易であるアンモニア水、ヒドラジン等が適当である。
また、還元剤の量は、用いる還元剤の量に応じて異なるため一概に規定できないが、処理すべきポリチオフェン誘導体やアミン付加体100質量部に対し、通常、還元が適切にされる観点から、0.1質量部以上であり、過剰な還元剤が残存しないようにする観点から、10質量部以下である。
還元処理の具体的な方法の一例としては、ポリチオフェン誘導体やアミン付加体を28%アンモニア水中で、室温にて終夜撹拌する。このような比較的温和な条件下での還元処理により、ポリチオフェン誘導体やアミン付加体の有機溶媒に対する溶解性や分散性は十分に向上する。
本発明の電荷輸送性組成物において、ポリチオフェン誘導体のアミン付加体を使用する場合、上記還元処理は、アミン付加体を形成する前に行っても、アミン付加体を形成した後に行ってもよい。
なお、この還元処理によりポリチオフェン誘導体またはそのアミン付加体の溶媒に対する溶解性や分散性が変化する結果、処理の開始時には反応系に溶解していなかったポリチオフェン誘導体またはそのアミン付加体が、処理の完了時には溶解している場合がある。そのような場合には、ポリチオフェン誘導体またはそのアミン付加体と非相溶性の有機溶媒(スルホン化ポリチオフェンの場合、アセトン、イソプロピルアルコール等)を反応系に添加して、ポリチオフェン誘導体またはそのアミン付加体の沈殿を生じさせ、ろ過する等の方法により、ポリチオフェン誘導体またはそのアミン付加体を回収することができる。
上記式(1’)のキノイド構造の好ましい具体例としては、例えば、下記式(1’-1)で示される構造が挙げられる。

式(1)で表される繰り返し単位を含むポリチオフェン誘導体またはそのアミン付加体の重量平均分子量は、約1,000~約1,000,000が好ましく、約5,000~約100,000がより好ましく、約10,000~約50,000がより一層好ましい。重量平均分子量を下限以上とすることで、良好な導電性が再現性よく得られ、上限以下とすることで、溶媒に対する溶解性が向上する。なお、重量平均分子量は、ゲルパーミエーションクロマトグラフィーによるポリスチレン換算値である。
本発明の電荷輸送性組成物に含まれるポリチオフェン誘導体またはそのアミン付加体は、式(1)で表される繰り返し単位を含むポリチオフェン誘導体またはそのアミン付加体1種のみであってもよく、2種以上であってもよい。
また、式(1)で表される繰り返し単位を含むポリチオフェン誘導体は、市販品を用いても、チオフェン誘導体などを出発原料とした公知の方法によって重合したものを用いてもよいが、いずれの場合も再沈殿やイオン交換等の方法により精製されたものを用いることが好ましい。精製したものを用いることで、本発明の電荷輸送性組成物から得られる薄膜を備えた有機太陽電池素子の特性をより高めることができる。市販品としては、例えば、東ソー(株)製のセルフトロン(SELFTRON,登録商標)等が挙げられる。
また、式(1)で表される繰り返し単位を含むポリチオフェン誘導体は、市販品を用いても、チオフェン誘導体などを出発原料とした公知の方法によって重合したものを用いてもよいが、いずれの場合も再沈殿やイオン交換等の方法により精製されたものを用いることが好ましい。精製したものを用いることで、本発明の電荷輸送性組成物から得られる薄膜を備えた有機太陽電池素子の特性をより高めることができる。市販品としては、例えば、東ソー(株)製のセルフトロン(SELFTRON,登録商標)等が挙げられる。
なお、共役ポリマーのスルホン化およびスルホン化共役ポリマー(スルホン化ポリチオフェンを含む)は、Seshadriらの米国特許第8,017,241号に記載されている。また、スルホン化ポリチオフェンについては、国際公開第2008/073149号および国際公開第2016/171935号に記載されている。
本発明においては、電荷輸送性組成物に含まれる式(1)で表される繰り返し単位を含むポリチオフェン誘導体またはそのアミン付加体の少なくとも一部は、有機溶媒に溶解している。
本発明においては、電荷輸送性物質として、式(1)で表される繰り返し単位を含むポリチオフェン誘導体またはそのアミン付加体と、それ以外の電荷輸送性化合物からなる電荷輸送性物質を併用してよいが、式(1)で表される繰り返し単位を含むポリチオフェン誘導体またはそのアミン付加体のみが含まれることが好ましい。
有機薄膜太陽電池において、正孔捕集層のイオン化ポテンシャルは、活性層中におけるp型半導体材料のイオン化ポテンシャルに近接した値であることが好ましい。その差の絶対値は、0~1eVが好ましく、0~0.5eVがより好ましく、0~0.2eVがより一層好ましい。
したがって、本発明の電荷輸送性組成物には、これを用いて得られる電荷輸送性薄膜のイオン化ポテンシャルを調節することを目的として、下記式(2)で表されるアリールスルホン酸化合物およびヘテロポリ酸からなる群より選ばれる少なくとも1種の電子受容性ドーパント物質を含む。
したがって、本発明の電荷輸送性組成物には、これを用いて得られる電荷輸送性薄膜のイオン化ポテンシャルを調節することを目的として、下記式(2)で表されるアリールスルホン酸化合物およびヘテロポリ酸からなる群より選ばれる少なくとも1種の電子受容性ドーパント物質を含む。

本発明において、好適に用いることができるアリールスルホン酸化合物の例としては、下記式(2-1)で表される化合物が挙げられる。

式(2)で表されるアリールスルホン酸化合物は、公知の方法により合成することができ、例えば、国際公開第2006/025342号に記載の方法により合成することができる。
ヘテロポリ酸としては、国際公開第2010/058777号に記載されているリンモリブデン酸、ケイモリブデン酸、リンタングステン酸、リンタングストモリブデン酸、ケイタングステン酸、リンモリブテン酸ナトリウム、リンバナドモリブテン酸等のヘテロポリ酸化合物等の無機酸化剤が挙げられる。本発明では、溶媒に対する溶解性、特に水に対する溶解性を考慮すると、ケイモリブデン酸およびケイタングステン酸が好ましい。
電子受容性ドーパント物質の含有量は、発現する電荷輸送性、電荷輸送性物質等の種類を考慮して適宜設定されるものではあるが、通常、電荷輸送性物質1に対し、通常、電子受容性ドーパント物質0.05~10、好ましくは0.1~3.0、より好ましくは0.2~2.0である。
また、式(2)で表されるアリールスルホン酸化合物およびヘテロポリ酸を併用することが好ましく、この場合、式(2)で表されるアリールスルホン酸化合物とヘテロポリ酸の混合比率は、質量比で10:90~90:10が好ましく、20:80~80:20がより好ましい。
また、式(2)で表されるアリールスルホン酸化合物およびヘテロポリ酸を併用することが好ましく、この場合、式(2)で表されるアリールスルホン酸化合物とヘテロポリ酸の混合比率は、質量比で10:90~90:10が好ましく、20:80~80:20がより好ましい。
また、本発明の電荷輸送性組成物には、上記の式(2)で表されるアリールスルホン酸化合物およびヘテロポリ酸以外の電子受容性ドーパント物質を含んでいてもよい。その他の電子受容性ドーパント物質の具体例としては、塩化水素、硫酸、硝酸、リン酸等の無機強酸;塩化アルミニウム(III)(AlCl3)、四塩化チタン(IV)(TiCl4)、三臭化ホウ素(BBr3)、三フッ化ホウ素エーテル錯体(BF3・OEt2)、塩化鉄(III)(FeCl3)、塩化銅(II)(CuCl2)、五塩化アンチモン(V)(SbCl5)、五フッ化砒素(V)(AsF5)、五フッ化リン(PF5)、トリス(4-ブロモフェニル)アルミニウムヘキサクロロアンチモナート(TBPAH)等のルイス酸;ベンゼンスルホン酸、トシル酸、ヒドロキシベンゼンスルホン酸、5-スルホサリチル酸、ドデシルベンゼンスルホン酸、ポリスチレンスルホン酸、国際公開第2005/000832号に記載されている1,4-ベンゾジオキサンジスルホン酸化合物、国際公開第2006/025342号に記載されているナフタレンスルホン酸化合物(ただし、式(2)で表されるアリールスルホン酸化合物に包含されるものを除く)、特開2005-108828号公報に記載されているジノニルナフタレンスルホン酸および1,3,6-ナフタレントリスルホン酸等のアリールスルホン酸化合物、ならびにカンファスルホン酸等の有機強酸;7,7,8,8-テトラシアノキノジメタン(TCNQ)、2,3-ジクロロ-5,6-ジシアノ-1,4-ベンゾキノン(DDQ)、ヨウ素等の有機酸化剤等が挙げられる。これらは、1種を単独で、または2種以上を組み合わせて用いることができる。
その他の電子受容性ドーパント物質を含む場合、その含有量は、電子受容性ドーパント物質全体の20質量%以下が好ましく、10質量%以下がより好ましく、含まないことがより一層好ましい。
本発明の電荷輸送性組成物は、成膜性の観点から、界面活性剤を含んでいてもよい。界面活性剤は特に限定されるものではなく、フッ素系界面活性剤、アセチレン系界面活性剤、アルキル系界面活性剤、シリコーン系界面活性剤等を用いることができるが、本発明では、フッ素系界面活性剤を用いることが好ましい。
本発明で用いるフッ素系界面活性剤は、市販品として入手可能である。
そのような市販品としては、デュポン社製のキャップストーン(Capstone,登録商標)FS-10、FS-22、FS-30、FS-31、FS-34、FS-35、FS-50、FS-51、FS-60、FS-61、FS-63、FS-64、FS-65、FS-66、FS-81、FS-83、FS-3100;第一工業製薬(株)製のノイゲンFN-1287;DIC(株)製のメガファックF-444、F-477、F-559等が挙げられるが、これらに限定されるものではない。
特に、ノニオン性界面活性剤である、キャップストーンFS-30、31、34、35、3100、ノイゲンFN-1287、メガファックF-559が好適である。
そのような市販品としては、デュポン社製のキャップストーン(Capstone,登録商標)FS-10、FS-22、FS-30、FS-31、FS-34、FS-35、FS-50、FS-51、FS-60、FS-61、FS-63、FS-64、FS-65、FS-66、FS-81、FS-83、FS-3100;第一工業製薬(株)製のノイゲンFN-1287;DIC(株)製のメガファックF-444、F-477、F-559等が挙げられるが、これらに限定されるものではない。
特に、ノニオン性界面活性剤である、キャップストーンFS-30、31、34、35、3100、ノイゲンFN-1287、メガファックF-559が好適である。
また、フッ素系界面活性剤としては、フッ素原子を含有している限り特に限定されるものではなく、カチオン性、アニオン性、ノニオン性のいずれでもよいが、フッ素系ノニオン性界面活性剤が好適であり、特に、下記式(A1)および(B1)から選ばれる少なくとも1種のフッ素系ノニオン性界面活性剤が好ましい。

上記式中、Rは、フッ素原子を含有する1価の有機基を表し、nは、1~20の整数を表す。
有機基の具体例としては、炭素数1~40のアルキル基、炭素数6~20のアリール基、炭素数7~20のアラルキル基、炭素数2~20のヘテロアリール基等が挙げられる。
有機基の具体例としては、炭素数1~40のアルキル基、炭素数6~20のアリール基、炭素数7~20のアラルキル基、炭素数2~20のヘテロアリール基等が挙げられる。
炭素数7~20のアラルキル基の具体例としては、ベンジル基、p-メチルフェニルメチル基、m-メチルフェニルメチル基、o-エチルフェニルメチル基、m-エチルフェニルメチル基、p-エチルフェニルメチル基、2-プロピルフェニルメチル基、4-イソプロピルフェニルメチル基、4-イソブチルフェニルメチル基、α-ナフチルメチル基等が挙げられる。
ヘテロアリール基の具体例としては、2-チエニル基、3-チエニル基、2-フラニル基、3-フラニル基、2-オキサゾリル基、4-オキサゾリル基、5-オキサゾリル基、3-イソオキサゾリル基、4-イソオキサゾリル基、5-イソオキサゾリル基、2-チアゾリル基、4-チアゾリル基、5-チアゾリル基、3-イソチアゾリル基、4-イソチアゾリル基、5-イソチアゾリル基、2-イミダゾリル基、4-イミダゾリル基、2-ピリジル基、3-ピリジル基、4-ピリジル基、2-ピラジル基、3-ピラジル基、5-ピラジル基、6-ピラジル基、2-ピリミジル基、4-ピリミジル基、5-ピリミジル基、6-ピリミジル基、3-ピリダジル基、4-ピリダジル基、5-ピリダジル基、6-ピリダジル基、1,2,3-トリアジン-4-イル基、1,2,3-トリアジン-5-イル基、1,2,4-トリアジン-3-イル基、1,2,4-トリアジン-5-イル基、1,2,4-トリアジン-6-イル基、1,3,5-トリアジン-2-イル基等が挙げられる。
その他、アルキル基、アリール基の具体例としては、上記と同様のものが挙げられる。
上記nは、1~20の整数であれば、特に限定されるものではないが、1~10の整数がより好ましい。
上記nは、1~20の整数であれば、特に限定されるものではないが、1~10の整数がより好ましい。
これらの中でも、炭素数1~40のパーフルオロアルキル基Rfを有する下記(A2)で示されるパーフルオロアルキルポリオキシエチレンエステルおよび(B2)で示されるパーフルオロアルキルポリオキシエチレンエーテルまたはフッ素テロマーアルコールから選ばれる少なくとも1種のフッ素系ノニオン性界面活性剤がより好ましい。

炭素数1~40のパーフルオロアルキル基の具体例としては、上記炭素数1~40のアルキル基の水素原子が全てフッ素原子に置換された基を挙げることができる。
アセチレン系界面活性剤としては、市販品を用いることもでき、具体例としては、Evonik社製のSurfynol-104、Surfynol-420、Surfynol-440、Surfynol-465、Surfynol-485、Dynol-604等が挙げられるが、これらに限定されるものではない。
アルキル系界面活性剤としては、市販品を用いることもでき、具体例としては、Sigma-Aldrich社製のPluronic-10R5、Pluronic-25R5、Pluronic F-127、Pluronic-L44、Genapol X-080、Genapol X-100、Genapol C-100、ならびにSolvay社製のlgepal C0-430、lgepal CA-630等が挙げられるが、これらに限定されるものではない。
界面活性剤を含む場合、その含有量は特に限定されるものではないが、活性層上での成膜性の向上と、得られる素子の光電変換効率の低下とのバランスを考慮すると、組成物全体の0.01~0.1質量%程度が好ましく、0.02~0.08質量%がより好ましく、0.03~0.06質量%が最も好ましい。
更に、本発明の組成物は、1種以上の金属酸化物ナノ粒子を含んでもよい。ナノ粒子とは、一次粒子についての平均粒子径がナノメートルのオーダー(典型的には500nm以下)である微粒子を意味する。金属酸化物ナノ粒子とは、ナノ粒子に成形された金属酸化物を意味する。
ナノ粒子とは、一次粒子についての平均粒子径がナノメートルのオーダー(典型的には500nm以下)である微粒子を意味する。金属酸化物ナノ粒子とは、ナノ粒子に成形された金属酸化物を意味する。
本発明で用いる金属酸化物ナノ粒子の一次粒子径は、ナノサイズであれば特に限定されるものではないが、活性層に対する密着性をより高めることを考慮すると、2~150nmが好ましく、3~100nmがより好ましく、5~50nmがより一層好ましい。なお、粒子径は、BET法による窒素吸着等温線を用いた測定値である。
ナノ粒子とは、一次粒子についての平均粒子径がナノメートルのオーダー(典型的には500nm以下)である微粒子を意味する。金属酸化物ナノ粒子とは、ナノ粒子に成形された金属酸化物を意味する。
本発明で用いる金属酸化物ナノ粒子の一次粒子径は、ナノサイズであれば特に限定されるものではないが、活性層に対する密着性をより高めることを考慮すると、2~150nmが好ましく、3~100nmがより好ましく、5~50nmがより一層好ましい。なお、粒子径は、BET法による窒素吸着等温線を用いた測定値である。
本発明における金属酸化物ナノ粒子を構成する金属は、通常の意味での金属に加え、半金属も包含する。
通常の意味での金属としては、特に限定されるものではないが、スズ(Sn)、チタン(Ti)、アルミニウム(Al)、ジルコニウム(Zr)、亜鉛(Zn)、ニオブ(Nb)、タンタル(Ta)およびW(タングステン)からなる群より選択される1種または2種以上を用いることが好ましい。
一方、半金属とは、化学的および/または物理的性質が金属と非金属の中間である元素を意味する。半金属の普遍的な定義は確立されていないが、本発明では、ホウ素(B)、ケイ素(Si)、ゲルマニウム(Ge)、ヒ素(As)、アンチモン(Sb)およびテルル(Te)の計6元素を半金属とする。これらの半金属は、単独で用いても、2種以上を組み合わせて用いてもよく、また通常の意味での金属と組み合わせて用いてもよい。
通常の意味での金属としては、特に限定されるものではないが、スズ(Sn)、チタン(Ti)、アルミニウム(Al)、ジルコニウム(Zr)、亜鉛(Zn)、ニオブ(Nb)、タンタル(Ta)およびW(タングステン)からなる群より選択される1種または2種以上を用いることが好ましい。
一方、半金属とは、化学的および/または物理的性質が金属と非金属の中間である元素を意味する。半金属の普遍的な定義は確立されていないが、本発明では、ホウ素(B)、ケイ素(Si)、ゲルマニウム(Ge)、ヒ素(As)、アンチモン(Sb)およびテルル(Te)の計6元素を半金属とする。これらの半金属は、単独で用いても、2種以上を組み合わせて用いてもよく、また通常の意味での金属と組み合わせて用いてもよい。
本発明で用いる金属酸化物ナノ粒子は、ホウ素(B)、ケイ素(Si)、ゲルマニウム(Ge)、ヒ素(As)、アンチモン(Sb)、テルル(Te)、スズ(Sn)、チタン(Ti)、アルミニウム(Al)、ジルコニウム(Zr)、亜鉛(Zn)、ニオブ(Nb)、タンタル(Ta)およびW(タングステン)から選ばれる1種または2種以上の金属の酸化物を含むことが好ましい。なお、金属が2種以上の組み合わせである場合、金属酸化物は、個々の単独の金属の酸化物の混合物であってもよく、複数の金属を含む複合酸化物であってもよい。
金属酸化物の具体例としては、B2O3、B2O、SiO2、SiO、GeO2、GeO、As2O4、As2O3、As2O5、Sb2O3、Sb2O5、TeO2、SnO2、ZrO2、Al2O3、ZnO等が挙げられるが、B2O3、B2O、SiO2、SiO、GeO2、GeO、As2O4、As2O3、As2O5、SnO2、SnO、Sb2O3、TeO2、およびこれらの混合物が好ましく、SiO2がより好ましい。
なお、上記金属酸化物ナノ粒子は、1種以上の有機キャッピング基を含んでもよい。この有機キャッピング基は、反応性であっても非反応性であってもよい。反応性有機キャッピング基の例としては、紫外線またはラジカル開始剤により架橋できる有機キャッピング基が挙げられる。
特に、本発明においては、金属酸化物ナノ粒子として、SiO2ナノ粒子が分散媒に分散したシリカゾルを用いることが好適である。
シリカゾルとしては、特に限定されるものではなく、公知のシリカゾルから適宜選択して用いることができる。
市販のシリカゾルは通常、分散液の形態にある。市販のシリカゾルとしては、SiO2ナノ粒子が種々の溶媒、例えば、水、メタノール、メチルエチルケトン、メチルイソブチルケトン、N,N-ジメチルアセトアミド、エチレングリコール、イソプロパノール、メタノール、エチレングリコールモノプロピルエーテル、シクロヘキサノン、酢酸エチル、トルエン、プロピレングリコールモノメチルエーテルアセタート等に分散したものが挙げられる。
特に、本発明においては、分散媒がアルコール溶媒または水であるシリカゾルが好ましく、分散媒がアルコール溶媒であるシリカゾルがより好ましい。アルコール溶媒としては、水溶性のアルコールが好ましく、メタノール、2-プロパノール、エチレングリコールがより好ましい。
シリカゾルとしては、特に限定されるものではなく、公知のシリカゾルから適宜選択して用いることができる。
市販のシリカゾルは通常、分散液の形態にある。市販のシリカゾルとしては、SiO2ナノ粒子が種々の溶媒、例えば、水、メタノール、メチルエチルケトン、メチルイソブチルケトン、N,N-ジメチルアセトアミド、エチレングリコール、イソプロパノール、メタノール、エチレングリコールモノプロピルエーテル、シクロヘキサノン、酢酸エチル、トルエン、プロピレングリコールモノメチルエーテルアセタート等に分散したものが挙げられる。
特に、本発明においては、分散媒がアルコール溶媒または水であるシリカゾルが好ましく、分散媒がアルコール溶媒であるシリカゾルがより好ましい。アルコール溶媒としては、水溶性のアルコールが好ましく、メタノール、2-プロパノール、エチレングリコールがより好ましい。
市販のシリカゾルの具体例としては日産化学(株)製のスノーテックス(登録商標)ST-O、ST-OS、ST-O-40、ST-OL、日本化学工業(株)製のシリカドール20、30、40等の水分散シリカゾル;日産化学(株)製のメタノールシリカゾル、MA-ST-M、MA-ST-L、IPA-ST、IPA-ST-L、IPA-ST-ZL、EG-ST等のオルガノシリカゾルなどが挙げられるが、これらに限定されるものではない。
また、シリカゾルの固形分濃度も特に限定されるものではないが、5~60質量%が好ましく、10~50質量%がより好ましく、15~30質量%がより一層好ましい。
また、シリカゾルの固形分濃度も特に限定されるものではないが、5~60質量%が好ましく、10~50質量%がより好ましく、15~30質量%がより一層好ましい。
金属酸化物ナノ粒子を使用する場合、その含有量は、特に限定されるものではないが、活性層に対する密着性を十分に発揮させることを考慮すると、電荷輸送性物質100質量部に対し、50~95質量部が好ましく、60~95質量部がより好ましく、80~95質量部がより一層好ましい。
なお、電荷輸送性物質を溶液や分散液として用いる場合、金属酸化物ナノ粒子の添加量は、電荷輸送性物質の固形分量を基準とする。
なお、電荷輸送性物質を溶液や分散液として用いる場合、金属酸化物ナノ粒子の添加量は、電荷輸送性物質の固形分量を基準とする。
更に、本発明の組成物は、アルコキシシランを含んでいてもよい。アルコキシシランを含むことで、得られる薄膜の耐溶剤性および耐水性の向上、電子ブロック性向上、並びにHOMOレベルおよびLUMOレベルを活性層に対して最適な値とすることができる。なお、アルコキシシランは、シロキサン系材料であってもよい。
アルコキシシランとしては、テトラアルコキシシラン、トリアルコキシシラン、ジアルコキシシランの中から任意の1種以上のアルコキシシランを用いることができるが、特にテトラエトキシシラン、テトラメトキシシラン、フェニルトリエトキシシラン、フェニルトリメトキシシラン、メチルトリエトキシシラン、メチルトリメトキシシラン、3,3,3-トリフルオロプロピルトリメトキシシラン、ジメチルジエトキシシラン、ジメチルジメトキシシランが好ましく、テトラエトキシシランがより好ましい。
シロキサン系材料としては、上記アルコキシシランに対して加水分解等の反応により得られる、ポリ(テトラエトキシシラン)、ポリ(フェニルエトキシシラン)等のポリシロキサンが挙げられる。
アルコキシシランを使用する場合、その含有量は、上記の効果が発揮される量であれば特に限定されないが、本発明で用いるポリチオフェン誘導体に対し、質量比で0.0001~100倍が好ましく、0.01~50倍がより好ましく、0.05~10倍がより一層好ましい。
アルコキシシランとしては、テトラアルコキシシラン、トリアルコキシシラン、ジアルコキシシランの中から任意の1種以上のアルコキシシランを用いることができるが、特にテトラエトキシシラン、テトラメトキシシラン、フェニルトリエトキシシラン、フェニルトリメトキシシラン、メチルトリエトキシシラン、メチルトリメトキシシラン、3,3,3-トリフルオロプロピルトリメトキシシラン、ジメチルジエトキシシラン、ジメチルジメトキシシランが好ましく、テトラエトキシシランがより好ましい。
シロキサン系材料としては、上記アルコキシシランに対して加水分解等の反応により得られる、ポリ(テトラエトキシシラン)、ポリ(フェニルエトキシシラン)等のポリシロキサンが挙げられる。
アルコキシシランを使用する場合、その含有量は、上記の効果が発揮される量であれば特に限定されないが、本発明で用いるポリチオフェン誘導体に対し、質量比で0.0001~100倍が好ましく、0.01~50倍がより好ましく、0.05~10倍がより一層好ましい。
本発明の電荷輸送性組成物は、必要に応じて、マトリックス高分子を更に含んでいてもよい。
上記マトリックス高分子の具体例としては、下記式(I)で表される繰り返し単位および下記式(II)で表される繰り返し単位を含むマトリックス高分子を挙げることができる。
上記マトリックス高分子の具体例としては、下記式(I)で表される繰り返し単位および下記式(II)で表される繰り返し単位を含むマトリックス高分子を挙げることができる。

ハロゲン原子、炭素数1~20のフルオロアルキル基および炭素数1~20のパーフルオロアルキル基の具体例としては、上記と同様のものが挙げられる。
上記R3、R4、R5およびR6が、フッ素原子または塩素原子であることが好ましく、R3、R5およびR6が、フッ素原子であり、かつR4が、塩素原子であることがより好ましく、R3、R4、R5およびR6が、全てフッ素原子であることがより一層好ましい。
上記R7、R8およびR9が、全てフッ素原子であることが好ましい。
上記Rh、Ri、Rj、Rk、RlおよびRmが、フッ素原子、炭素数1~8のフルオロアルキル基、または炭素数1~8のパーフルオロアルキル基であることが好ましい。
上記RlおよびRmが、フッ素原子であることがより好ましい。また、yは、0が好ましく、zは、2が好ましい。
また、上記R3、R5、およびR6が、フッ素原子であり、R4が、塩素原子であり、そして各々のRlおよびRmが、フッ素原子であり;yは、0であり;そしてzは、2であることが好ましい。
更に、ある実施態様において、各々のR3、R4、R5、およびR6は、フッ素原子であり;そして各々のRlおよびRmは、フッ素原子であり;yは、0であり;そしてzは、2であることが好ましい。
式(I)で表される繰り返し単位の数「s」と式(II)で表される繰り返し単位の数「t」との比(s:t比)は、特に限定されない。s:t比は、好ましくは9:1~1:9、より好ましくは8:2~2:8である。
本発明において好適に使用できるマトリックス高分子は、公知の方法を用いて合成されたものを用いても、市販品を用いてもよい。例えば、上記式(I)で表される繰り返し単位および上記式(II)で表される繰り返し単位を含むポリマーは、下記式(Ia)で表されるモノマーと下記式(IIa)で表されるモノマーとを、公知の重合方法により共重合し、続いてスルホニルフルオリド基の加水分解によりスルホン酸基に変換することによって製造することができる。

例えば、テトラフルオロエチレン(TFE)またはクロロトリフルオロエチレン(CTFE)は、スルホン酸の前駆体基を含む1種以上のフッ素化モノマー(例えば、F2C=CF-O-CF2-CF2-SO2F;F2C=CF-[O-CF2-CR12F-O]y-CF2-CF2-SO2F(ここで、R12は、FまたはCF3であり、そしてyは、1~10である);F2C=CF-O-CF2-CF2-CF2-SO2F;およびF2C=CF-OCF2-CF2-CF2-CF2-SO2Fなど)と共重合することができる。
本発明において、マトリックス高分子に存在する酸基1モル当たりのマトリックス高分子の質量(g/mol)を意味する。マトリックス高分子の当量は、好ましくは約400~約15,000g/mol、より好ましくは約500~約10,000g/mol、より一層好ましくは約500~約8,000g/mol、更に好ましくは約500~約2,000g/mol、最も好ましくは約600~約1,700g/molである。
このようなマトリックス高分子は、市販品として入手することができる。
市販品としては、例えば、デュポン製のナフィオン(NAFION,登録商標)、Solvay Specialty Polymers製のアクイヴィオン(AQUIVION,登録商標)、および旭硝子(株)製のフレミオン(FLEMION,登録商標)等が挙げられる。
市販品としては、例えば、デュポン製のナフィオン(NAFION,登録商標)、Solvay Specialty Polymers製のアクイヴィオン(AQUIVION,登録商標)、および旭硝子(株)製のフレミオン(FLEMION,登録商標)等が挙げられる。
本発明において、マトリックス高分子は、少なくとも1個のスルホン酸残基(-SO3H)を含む繰り返し単位を1個以上含むポリエーテルスルホンであることが好ましい。
なお、本発明の組成物には、本発明の目的を達成し得る限り、その他の添加剤を配合してもよい。
添加剤の種類としては、所望の効果に応じて公知のものから適宜選択して用いることができる。
添加剤の種類としては、所望の効果に応じて公知のものから適宜選択して用いることができる。
電荷輸送性組成物の調製に用いる溶媒としては、ポリチオフェン誘導体および電子受容性ドーパント物質を良好に溶解し得る高溶解性溶媒を用いることができる。高溶解性溶媒は1種単独で、または2種以上混合して用いることができ、その使用量は、組成物に使用する溶媒全体の5~100質量%とすることができる。
このような高溶解性溶媒としては、例えば、水;エタノール、2-プロパノール、1-ブタノール、2-ブタノール、s-ブタノール、t-ブタノール、1-メトキシ-2-プロパノール等のアルコール系溶媒、N-メチルホルムアミド、N,N-ジメチルホルムアミド、N,N-ジエチルホルムアミド、N-メチルアセトアミド、N,N-ジメチルアセトアミド、N-メチルピロリドン、1,3-ジメチル-2-イミダゾリジノン等のアミド系溶媒などの有機溶媒が挙げられる。
これらの中でも、水およびアルコール系溶媒から選ばれる少なくとも1種が好ましく、水、エタノール、2-プロパノールがより好ましく、水、2-プロパノールがより一層好ましく、水が更に好ましい。
これらの中でも、水およびアルコール系溶媒から選ばれる少なくとも1種が好ましく、水、エタノール、2-プロパノールがより好ましく、水、2-プロパノールがより一層好ましく、水が更に好ましい。
上記の高溶解性溶媒は1種単独で、または2種以上混合して用いることができるが、上記溶媒中の有機溶媒の含有量は10質量%以下であり、5質量%以下が好ましく、含まないこと(すなわち、溶媒が水のみであること)がより好ましい。
電荷輸送性物質および電子受容性ドーパント物質は、いずれも上記溶媒に完全に溶解しているか、均一に分散している状態となっていることが好ましく、高変換効率の有機薄膜太陽電池を与える正孔捕集層を再現性よく得ることを考慮すると、これらの物質は上記溶媒に完全に溶解していることがより好ましい。
また、本発明の電荷輸送性組成物は、成膜性および塗布装置からの吐出性向上のために、25℃で10~200mPa・s、特に35~150mPa・sの粘度を有し、常圧で沸点50~300℃、特に150~250℃の高粘度有機溶媒を、少なくとも1種類含有してもよい。
高粘度有機溶媒としては、特に限定されるものではなく、例えば、シクロヘキサノール、エチレングリコール、1,3-オクチレングリコール、ジエチレングリコール、ジプロピレングリコール、トリエチレングリコール、トリプロピレングリコール、1,3-ブタンジオール、2,3-ブタンジオール、1,4-ブタンジオール、プロピレングリコール、へキシレングリコール等が挙げられる。
高粘度有機溶媒としては、特に限定されるものではなく、例えば、シクロヘキサノール、エチレングリコール、1,3-オクチレングリコール、ジエチレングリコール、ジプロピレングリコール、トリエチレングリコール、トリプロピレングリコール、1,3-ブタンジオール、2,3-ブタンジオール、1,4-ブタンジオール、プロピレングリコール、へキシレングリコール等が挙げられる。
高粘度有機溶媒を使用する場合、その添加割合は、固体が析出しない範囲内であることが好ましく、固体が析出しない限りにおいて、上記高溶解性溶媒で例示した有機溶媒の含有量との合計で、溶媒中10質量%以下となる範囲が好ましく、5質量%以下がより好ましく、含まないこと(0質量%)がより好ましい。
更に、塗布面に対する濡れ性の向上、溶媒の表面張力の調整、極性の調整、沸点の調整等の目的で、熱処理時に膜の平坦性を付与し得るその他の溶媒を含有してもよい。
このような溶媒としては、例えば、ブチルセロソルブ、ジエチレングリコールジエチルエーテル、ジエチレングリコールジメチルエーテル、ジエチレングリコールモノエチルエーテルアセテート、ジエチレングリコールモノブチルエーテルアセテート、ジプロピレングリコールモノメチルエーテル、プロピレングリコールモノメチルエーテル、プロピレングリコールモノメチルエーテルアセテート、エチルカルビトール、ジアセトンアルコール、γ-ブチロラクトン、エチルラクテート、n-ヘキシルアセテート等が挙げられる。
その他の溶媒を使用する場合、その添加割合は、組成物に使用する溶媒全体の1~90質量%であることが好ましく、1~50質量%であることがより好ましい。
本発明の組成物の固形分濃度は、組成物の粘度および表面張力等や、作製する薄膜の厚み等を勘案して適宜設定されるものではあるが、通常、0.1~10.0質量%程度であり、好ましくは0.5~5.0質量%、より好ましくは1.0~3.0質量%である。
そして、本発明において用いる電荷輸送性組成物の粘度は、作製する薄膜の厚み等や固形分濃度を考慮し、塗布方法に応じて適宜調節されるものではあるが、通常25℃で0.1~50mPa・s程度である。
そして、本発明において用いる電荷輸送性組成物の粘度は、作製する薄膜の厚み等や固形分濃度を考慮し、塗布方法に応じて適宜調節されるものではあるが、通常25℃で0.1~50mPa・s程度である。
本発明の電荷輸送性組成物を調製する際、固形分が溶媒に均一に溶解または分散する限り、電荷輸送性物質、界面活性剤、金属酸化物ナノ粒子、電子受容性ドーパント物質、溶媒等を任意の順序で混合することができる。すなわち、例えば、溶媒にポリチオフェン誘導体を溶解させた後、その溶液に電子受容性ドーパント物質を溶解させる方法、溶媒に電子受容性ドーパント物質を溶解させた後、その溶液にポリチオフェン誘導体を溶解させる方法、ポリチオフェン誘導体と電子受容性ドーパント物質とを混合した後、その混合物を溶媒に投入して溶解させる方法のいずれも、固形分が溶媒に均一に溶解または分散する限り、採用することができる。
なお、マトリックス高分子、アルコキシシランの添加順序も任意である。
なお、マトリックス高分子、アルコキシシランの添加順序も任意である。
また、通常、組成物の調製は、常温、常圧の不活性ガス雰囲気下で行われるが、組成物中の化合物が分解したり、組成が大きく変化したりしない限り、大気雰囲気下(酸素存在下)で行ってもよく、加熱しながら行ってもよい。
以上説明した組成物を、順積層型有機薄膜太陽電池の場合は陽極上に、逆積層型有機薄膜太陽電池の場合は活性層上に塗布して焼成することで、本発明の正孔捕集層を形成できる。
塗布にあたっては、組成物の粘度と表面張力、所望する薄膜の厚さ等を考慮し、ドロップキャスト法、スピンコート法、ブレードコート法、ディップコート法、ロールコート法、バーコート法、ダイコート法、インクジェット法、印刷法(凸版、凹版、平版、スクリーン印刷等)等といった各種ウェットプロセス法の中から最適なものを採用すればよい。
また、通常、塗布は、常温、常圧の不活性ガス雰囲気下で行われるが、組成物中の化合物が分解したり、組成が大きく変化したりしない限り、大気雰囲気下(酸素存在下)で行ってもよく、加熱しながら行ってもよい。
塗布にあたっては、組成物の粘度と表面張力、所望する薄膜の厚さ等を考慮し、ドロップキャスト法、スピンコート法、ブレードコート法、ディップコート法、ロールコート法、バーコート法、ダイコート法、インクジェット法、印刷法(凸版、凹版、平版、スクリーン印刷等)等といった各種ウェットプロセス法の中から最適なものを採用すればよい。
また、通常、塗布は、常温、常圧の不活性ガス雰囲気下で行われるが、組成物中の化合物が分解したり、組成が大きく変化したりしない限り、大気雰囲気下(酸素存在下)で行ってもよく、加熱しながら行ってもよい。
膜厚は、特に限定されないが、いずれの場合も0.1~800nm程度が好ましく、更には30~500nm程度が好ましい。膜厚を変化させる方法としては、組成物中の固形分濃度を変化させたり、塗布時の溶液量を変化させたりする等の方法がある。
以下、本発明の電荷輸送性組成物を正孔捕集層形成用組成物として用いた有機薄膜太陽電池の製造方法について説明するが、これらに限定されるものではない。
(1)順積層型有機薄膜太陽電池
[陽極層の形成]:透明基板の表面に陽極材料の層を形成し、透明電極を製造する工程
陽極材料としては、インジウム錫酸化物(ITO)、インジウム亜鉛酸化物(IZO)等の無機酸化物や、金、銀、アルミニウム等の金属、ポリチオフェン誘導体、ポリアニリン誘導体等の高電荷輸送性有機化合物を用いることができる。これらの中ではITOが最も好ましい。また、透明基板としては、ガラスあるいは透明樹脂からなる基板を用いることができる。
陽極材料の層(陽極層)の形成方法は、陽極材料の性質に応じて適宜選択される。通常、難溶性、難分散性昇華性材料の場合には真空蒸着法やスパッタ法等のドライプロセスが選択され、溶液材料あるいは分散液材料の場合には、組成物の粘度と表面張力、所望する薄膜の厚さ等を考慮し、上述した各種ウェットプロセス法の中から最適なものが採用される。
[陽極層の形成]:透明基板の表面に陽極材料の層を形成し、透明電極を製造する工程
陽極材料としては、インジウム錫酸化物(ITO)、インジウム亜鉛酸化物(IZO)等の無機酸化物や、金、銀、アルミニウム等の金属、ポリチオフェン誘導体、ポリアニリン誘導体等の高電荷輸送性有機化合物を用いることができる。これらの中ではITOが最も好ましい。また、透明基板としては、ガラスあるいは透明樹脂からなる基板を用いることができる。
陽極材料の層(陽極層)の形成方法は、陽極材料の性質に応じて適宜選択される。通常、難溶性、難分散性昇華性材料の場合には真空蒸着法やスパッタ法等のドライプロセスが選択され、溶液材料あるいは分散液材料の場合には、組成物の粘度と表面張力、所望する薄膜の厚さ等を考慮し、上述した各種ウェットプロセス法の中から最適なものが採用される。
また、市販の透明陽極基板を用いることもでき、この場合、素子の歩留を向上させる観点からは、平滑化処理がされている基板を用いることが好ましい。市販の透明陽極基板を用いる場合、本発明の有機薄膜太陽電池の製造方法は、陽極層を形成する工程を含まない。
ITO等の無機酸化物を陽極材料として用いて透明陽極基板を形成する場合、上層を積層する前に、洗剤、アルコール、純水等で洗浄してから使用することが好ましい。更に、使用直前にUVオゾン処理、酸素-プラズマ処理等の表面処理を施すことが好ましい。陽極材料が有機物を主成分とする場合、表面処理を行わなくともよい。
ITO等の無機酸化物を陽極材料として用いて透明陽極基板を形成する場合、上層を積層する前に、洗剤、アルコール、純水等で洗浄してから使用することが好ましい。更に、使用直前にUVオゾン処理、酸素-プラズマ処理等の表面処理を施すことが好ましい。陽極材料が有機物を主成分とする場合、表面処理を行わなくともよい。
[正孔捕集層の形成]:形成された陽極材料の層上に正孔捕集層を形成する工程
上記方法に従い、陽極材料の層上に、本発明の組成物を用いて正孔捕集層を形成する。
上記方法に従い、陽極材料の層上に、本発明の組成物を用いて正孔捕集層を形成する。
[活性層の形成]:形成された正孔捕集層上に活性層を形成する工程
活性層は、n型半導体材料からなる薄膜であるn層と、p型半導体材料からなる薄膜であるp層とを積層したものであっても、これら材料の混合物からなる非積層薄膜であってもよい。本発明では、上記活性層は、FA活性層であってもNFA活性層であってもよく、いずれの組み合わせでも得られる素子の耐久性を大きく向上させることができる。特にNFA活性層と組み合わせて用いられた場合には、得られる電荷輸送性薄膜が有する深いIpによりNFA活性層とのエネルギーギャップを低減し、素子の高電圧化を実現し得る。
活性層は、n型半導体材料からなる薄膜であるn層と、p型半導体材料からなる薄膜であるp層とを積層したものであっても、これら材料の混合物からなる非積層薄膜であってもよい。本発明では、上記活性層は、FA活性層であってもNFA活性層であってもよく、いずれの組み合わせでも得られる素子の耐久性を大きく向上させることができる。特にNFA活性層と組み合わせて用いられた場合には、得られる電荷輸送性薄膜が有する深いIpによりNFA活性層とのエネルギーギャップを低減し、素子の高電圧化を実現し得る。
ここで、NFA活性層とは、本発明においてはNFAの含有量が、活性層に含まれるn型半導体のうち70質量%以上である活性層を意味する。また、FA活性層とは、本発明においてはFAの含有量が、活性層に含まれるn型半導体のうち70質量%以上である活性層を意味する。
FA活性層に用いられるn型半導体材料としては、フラーレン、[6,6]-フェニル-C61-酪酸メチルエステル(PC61BM)、[6,6]-フェニル-C71-酪酸メチルエステル(PC71BM)等が挙げられる。
NFA活性層に用いられるn型半導体材料としては、下記式(3-1)~(3-4)で表される化合物等が挙げられる。


上記n型半導体材料に組み合わせられるp型半導体材料としては、レジオレギュラーポリ(3-ヘキシルチオフェン)(P3HT)、下記式(4-1)で表されるPTB7、下記式(4-2)で表されるPM6、特開2009-158921号公報および国際公開第2010/008672号に記載されているようなチエノチオフェンユニット含有ポリマー類等の、主鎖にチオフェン骨格を含むポリマー、CuPC,ZnPC等のフタロシアニン類、テトラベンゾポルフィリン等のポルフィリン類などが挙げられる。

これらの中でも、n型半導体材料としては、式(3-1)および(3-2)で表される化合物が好ましく、その中でも、前者ではXがFであるBTP-4Fがより好ましく、後者ではX1およびX2がともにFであるITIC-4Fがより好ましい。一方、p型半導体材料としては、PM6およびPTB7等の主鎖にチオフェン骨格を含むポリマー類が好ましい。
なお、ここでいう「主鎖にチオフェン骨格」とはチオフェンのみからなる2価の芳香環、またはチエノチオフェン、ベンゾチオフェン、ジベンゾチオフェン、ベンゾジチオフェン、ナフトチオフェン、ナフトジチオフェン、アントラチオフェン、アントラジチオフェン等のような1以上のチオフェンを含む2価の縮合芳香環を表し、これらはハロゲン原子、ニトロ基、シアノ基、スルホン酸基、炭素数1~20のアルコキシ基、炭素数1~20のチオアルコキシ基、炭素数1~20のアルキル基、炭素数2~20のアルケニル基、炭素数2~20のアルキニル基、炭素数1~20のハロアルキル基、炭素数6~20のアリール基、炭素数7~20のアラルキル基、または炭素数1~20のアシル基で置換されていてもよい。
なお、ここでいう「主鎖にチオフェン骨格」とはチオフェンのみからなる2価の芳香環、またはチエノチオフェン、ベンゾチオフェン、ジベンゾチオフェン、ベンゾジチオフェン、ナフトチオフェン、ナフトジチオフェン、アントラチオフェン、アントラジチオフェン等のような1以上のチオフェンを含む2価の縮合芳香環を表し、これらはハロゲン原子、ニトロ基、シアノ基、スルホン酸基、炭素数1~20のアルコキシ基、炭素数1~20のチオアルコキシ基、炭素数1~20のアルキル基、炭素数2~20のアルケニル基、炭素数2~20のアルキニル基、炭素数1~20のハロアルキル基、炭素数6~20のアリール基、炭素数7~20のアラルキル基、または炭素数1~20のアシル基で置換されていてもよい。
ハロゲン原子、炭素数1~20のアルキル基、炭素数1~20のアルコキシ基、炭素数6~20のアリール基、炭素数7~20のアラルキル基としては、上記で例示したものと同様のものが挙げられる。
炭素数1~20のチオアルコキシ基の具体例としては、上記アルコキシ基の酸素原子を硫黄原子で置換した基等が挙げられる。
炭素数1~20のチオアルコキシ(アルキルチオ)基の具体例としては、メチルチオ基、エチルチオ基、n-プロピルチオ基、イソプロピルチオ基、n-ブチルチオ基、イソブチルチオ基、s-ブチルチオ基、t-ブチルチオ基、n-ペンチルチオ基、n-ヘキシルチオ基、n-ヘプチルチオ基、n-オクチルチオ基、n-ノニルチオ基、n-デシルチオ基、n-ウンデシルチオ基、n-ドデシルチオ基、n-トリデシルチオ基、n-テトラデシルチオ基、n-ペンタデシルチオ基、n-ヘキサデシルチオ基、n-ヘプタデシルチオ基、n-オクタデシルチオ基、n-ノナデシルチオ基、n-エイコサニルチオ基等が挙げられる。
炭素数1~20のチオアルコキシ(アルキルチオ)基の具体例としては、メチルチオ基、エチルチオ基、n-プロピルチオ基、イソプロピルチオ基、n-ブチルチオ基、イソブチルチオ基、s-ブチルチオ基、t-ブチルチオ基、n-ペンチルチオ基、n-ヘキシルチオ基、n-ヘプチルチオ基、n-オクチルチオ基、n-ノニルチオ基、n-デシルチオ基、n-ウンデシルチオ基、n-ドデシルチオ基、n-トリデシルチオ基、n-テトラデシルチオ基、n-ペンタデシルチオ基、n-ヘキサデシルチオ基、n-ヘプタデシルチオ基、n-オクタデシルチオ基、n-ノナデシルチオ基、n-エイコサニルチオ基等が挙げられる。
炭素数2~20のアルケニル基の具体例としては、エテニル基、n-1-プロペニル基、n-2-プロペニル基、1-メチルエテニル基、n-1-ブテニル基、n-2-ブテニル基、n-3-ブテニル基、2-メチル-1-プロペニル基、2-メチル-2-プロペニル基、1-エチルエテニル基、1-メチル-1-プロペニル基、1-メチル-2-プロペニル基、n-1-ペンテニル基、n-1-デセニル基、n-1-エイコセニル基等が挙げられる。
炭素数2~20のアルキニル基の具体例としては、エチニル基、n-1-プロピニル基、n-2-プロピニル基、n-1-ブチニル基、n-2-ブチニル基、n-3-ブチニル基、1-メチル-2-プロピニル基、n-1-ペンチニル基、n-2-ペンチニル基、n-3-ペンチニル基、n-4-ペンチニル基、1-メチル-n-ブチニル基、2-メチル-n-ブチニル基、3-メチル-n-ブチニル基、1,1-ジメチル-n-プロピニル基、n-1-ヘキシニル基、n-1-デシニル基、n-1-ペンタデシニル基、n-1-エイコシニル基等が挙げられる。
炭素数1~20のハロアルキル基としては、上記アルキル基中の水素原子の少なくとも1つをハロゲン原子で置換した基等が挙げられる。なお、ハロゲン原子は、塩素、臭素、ヨウ素、フッ素原子のいずれでもよい。中でも、フルオロアルキル基が好ましく、パーフルオロアルキル基がより好ましい。
その具体例としては、フルオロメチル基、ジフルオロメチル基、トリフルオロメチル基、ペンタフルオロエチル基、2,2,2-トリフルオロエチル基、ヘプタフルオロプロピル基、2,2,3,3,3-ペンタフルオロプロピル基、2,2,3,3-テトラフルオロプロピル基、2,2,2-トリフルオロ-1-(トリフルオロメチル)エチル基、ノナフルオロブチル基、4,4,4-トリフルオロブチル基、ウンデカフルオロペンチル基、2,2,3,3,4,4,5,5,5-ノナフルオロペンチル基、2,2,3,3,4,4,5,5-オクタフルオロペンチル基、トリデカフルオロヘキシル基、2,2,3,3,4,4,5,5,6,6,6-ウンデカフルオロヘキシル基、2,2,3,3,4,4,5,5,6,6-デカフルオロヘキシル基、3,3,4,4,5,5,6,6,6-ノナフルオロヘキシル基等が挙げられる。
その具体例としては、フルオロメチル基、ジフルオロメチル基、トリフルオロメチル基、ペンタフルオロエチル基、2,2,2-トリフルオロエチル基、ヘプタフルオロプロピル基、2,2,3,3,3-ペンタフルオロプロピル基、2,2,3,3-テトラフルオロプロピル基、2,2,2-トリフルオロ-1-(トリフルオロメチル)エチル基、ノナフルオロブチル基、4,4,4-トリフルオロブチル基、ウンデカフルオロペンチル基、2,2,3,3,4,4,5,5,5-ノナフルオロペンチル基、2,2,3,3,4,4,5,5-オクタフルオロペンチル基、トリデカフルオロヘキシル基、2,2,3,3,4,4,5,5,6,6,6-ウンデカフルオロヘキシル基、2,2,3,3,4,4,5,5,6,6-デカフルオロヘキシル基、3,3,4,4,5,5,6,6,6-ノナフルオロヘキシル基等が挙げられる。
炭素数1~20のアシル基の具体例としては、ホルミル基、アセチル基、プロピオニル基、ブチリル基、イソブチリル基、バレリル基、イソバレリル基、ベンゾイル基等が挙げられる。
上記FA活性層には、本発明の効果を損なわない限り、活性層に含まれるn型半導体材料のうち30質量%未満範囲において、残部として、上述したNFAに該当するn型半導体材料が含まれていてもよい。そのようなn型半導体材料の具体例としては、BTP-4FやITIC-4F等が挙げられる。
また、上記NFA活性層においても、本発明の効果を損なわない限り、活性層に含まれるn型半導体材料のうち30質量%未満範囲において、残部として、上述したFAに該当するn型半導体材料が含まれていてもよい。
上記FA活性層およびNFA活性層の形成方法も、上記と同様、活性層材料が難溶性昇華性材料の場合には上述した各種ドライプロセスが選択され、溶液材料あるいは分散液材料の場合には、組成物の粘度と表面張力、所望する薄膜の厚さ等を考慮し、上述した各種ウェットプロセス法の中から最適なものが採用される。
[電子捕集層の形成]:形成された活性層上に電子捕集層を形成する工程
必要に応じて、電荷の移動を効率化すること等を目的として、活性層と陰極層の間に電子捕集層を形成してもよい。
電子捕集層を形成する材料としては、酸化リチウム(Li2O)、酸化マグネシウム(MgO)、アルミナ(Al2O3)、フッ化リチウム(LiF)、フッ化ナトリウム(NaF)、フッ化マグネシウム(MgF2)、フッ化ストロンチウム(SrF2)、炭酸セシウム(Cs2CO3)、8-キノリノールリチウム塩(Liq)、8-キノリノールナトリウム塩(Naq)、バソクプロイン(BCP)、4,7-ジフェニル-1,10-フェナントロリン(BPhen)、ポリエチレンイミン(PEI)、エトキシ化ポリエチレンイミン(PEIE)等が挙げられる。
必要に応じて、電荷の移動を効率化すること等を目的として、活性層と陰極層の間に電子捕集層を形成してもよい。
電子捕集層を形成する材料としては、酸化リチウム(Li2O)、酸化マグネシウム(MgO)、アルミナ(Al2O3)、フッ化リチウム(LiF)、フッ化ナトリウム(NaF)、フッ化マグネシウム(MgF2)、フッ化ストロンチウム(SrF2)、炭酸セシウム(Cs2CO3)、8-キノリノールリチウム塩(Liq)、8-キノリノールナトリウム塩(Naq)、バソクプロイン(BCP)、4,7-ジフェニル-1,10-フェナントロリン(BPhen)、ポリエチレンイミン(PEI)、エトキシ化ポリエチレンイミン(PEIE)等が挙げられる。
電子捕集層の形成方法も、上記と同様、電子捕集材料が難溶性昇華性材料の場合には上述した各種ドライプロセスが選択され、溶液材料あるいは分散液材料の場合には、組成物の粘度と表面張力、所望する薄膜の厚さ等を考慮し、上述した各種ウェットプロセス法の中から最適なものが採用される。
[陰極層の形成]:形成された電子捕集層の上に陰極層を形成する工程
陰極材料としては、アルミニウム、マグネシウム-銀合金、アルミニウム-リチウム合金、リチウム、ナトリウム、カリウム、セシウム、カルシウム、バリウム、銀、金等の金属や、インジウム錫酸化物(ITO)、インジウム亜鉛酸化物(IZO)等の無機酸化物や、ポリチオフェン誘導体、ポリアニリン誘導体等の高電荷輸送性有機化合物が挙げられ、複数の陰極材料を積層したり、混合したりして使用することができる。
陰極層の形成方法も、上記と同様、陰極層材料が難溶性、難分散性昇華性材料の場合には上述した各種ドライプロセスが選択され、溶液材料あるいは分散液材料の場合には、組成物の粘度と表面張力、所望する薄膜の厚さ等を考慮し、上述した各種ウェットプロセス法の中から最適なものが採用される。
陰極材料としては、アルミニウム、マグネシウム-銀合金、アルミニウム-リチウム合金、リチウム、ナトリウム、カリウム、セシウム、カルシウム、バリウム、銀、金等の金属や、インジウム錫酸化物(ITO)、インジウム亜鉛酸化物(IZO)等の無機酸化物や、ポリチオフェン誘導体、ポリアニリン誘導体等の高電荷輸送性有機化合物が挙げられ、複数の陰極材料を積層したり、混合したりして使用することができる。
陰極層の形成方法も、上記と同様、陰極層材料が難溶性、難分散性昇華性材料の場合には上述した各種ドライプロセスが選択され、溶液材料あるいは分散液材料の場合には、組成物の粘度と表面張力、所望する薄膜の厚さ等を考慮し、上述した各種ウェットプロセス法の中から最適なものが採用される。
[キャリアブロック層の形成]
必要に応じて、光電流の整流性をコントロールすること等を目的として、任意の層間にキャリアブロック層を設けてもよい。キャリアブロック層を設ける場合、通常、活性層と、正孔捕集層または陽極との間に電子ブロック層を、活性層と、電子捕集層または陰極との間に正孔ブロック層を挿入する場合が多いが、この限りではない。
正孔ブロック層を形成する材料としては、酸化チタン、酸化亜鉛、酸化スズ、バソクプロイン(BCP)、4,7-ジフェニル1,10-フェナントロリン(BPhen)等が挙げられる。
電子ブロック層を形成する材料としては、N,N′-ジ(1-ナフチル)-N,N′-ジフェニルベンジジン(α-NPD)、ポリ(トリアリールアミン)(PTAA)等のトリアリールアミン系材料等が挙げられる。
必要に応じて、光電流の整流性をコントロールすること等を目的として、任意の層間にキャリアブロック層を設けてもよい。キャリアブロック層を設ける場合、通常、活性層と、正孔捕集層または陽極との間に電子ブロック層を、活性層と、電子捕集層または陰極との間に正孔ブロック層を挿入する場合が多いが、この限りではない。
正孔ブロック層を形成する材料としては、酸化チタン、酸化亜鉛、酸化スズ、バソクプロイン(BCP)、4,7-ジフェニル1,10-フェナントロリン(BPhen)等が挙げられる。
電子ブロック層を形成する材料としては、N,N′-ジ(1-ナフチル)-N,N′-ジフェニルベンジジン(α-NPD)、ポリ(トリアリールアミン)(PTAA)等のトリアリールアミン系材料等が挙げられる。
キャリアブロック層の形成方法も、上記と同様、キャリアブロック層材料が難溶性、難分散性昇華性材料の場合には上述した各種ドライプロセスが選択され、溶液材料あるいは分散液材料の場合には、組成物の粘度と表面張力、所望する薄膜の厚さ等を考慮し、上述した各種ウェットプロセス法の中から最適なものが採用される。
(2)逆積層型有機薄膜太陽電池
[陰極層の形成]:透明基板の表面に陰極材料の層を形成し、透明陰極基板を製造する工程
陰極材料としては、上記順積層型の陽極材料で例示したものに加え、フッ素ドープ酸化錫(FTO)が挙げられ、透明基板としては、上記順積層型の陽極材料で例示したものが挙げられる。
陰極材料の層(陰極層)の形成方法も、難溶性、難分散性昇華性材料の場合には上述したドライプロセスが選択され、溶液材料あるいは分散液材料の場合には、組成物の粘度と表面張力、所望する薄膜の厚さ等を考慮し、上述した各種ウェットプロセス法の中から最適なものが採用される。
また、この場合も市販の透明陰極基板を好適に用いることができ、素子の歩留を向上させる観点からは、平滑化処理がされている基板を用いることが好ましい。市販の透明陰極基板を用いる場合、本発明の有機薄膜太陽電池の製造方法は、陰極層を形成する工程を含まない。
無機酸化物を陰極材料として使用して透明陰極基板を形成する場合、順積層型の陽極材料と同様の洗浄処理や、表面処理を施してもよい。
[陰極層の形成]:透明基板の表面に陰極材料の層を形成し、透明陰極基板を製造する工程
陰極材料としては、上記順積層型の陽極材料で例示したものに加え、フッ素ドープ酸化錫(FTO)が挙げられ、透明基板としては、上記順積層型の陽極材料で例示したものが挙げられる。
陰極材料の層(陰極層)の形成方法も、難溶性、難分散性昇華性材料の場合には上述したドライプロセスが選択され、溶液材料あるいは分散液材料の場合には、組成物の粘度と表面張力、所望する薄膜の厚さ等を考慮し、上述した各種ウェットプロセス法の中から最適なものが採用される。
また、この場合も市販の透明陰極基板を好適に用いることができ、素子の歩留を向上させる観点からは、平滑化処理がされている基板を用いることが好ましい。市販の透明陰極基板を用いる場合、本発明の有機薄膜太陽電池の製造方法は、陰極層を形成する工程を含まない。
無機酸化物を陰極材料として使用して透明陰極基板を形成する場合、順積層型の陽極材料と同様の洗浄処理や、表面処理を施してもよい。
[電子捕集層の形成]:形成された陰極上に電子捕集層を形成する工程
必要に応じて、電荷の移動を効率化すること等を目的として、活性層と陰極層の間に電子捕集層を形成してもよい。
電子捕集層を形成する材料としては、上記順積層型の材料で例示したものに加え、酸化亜鉛(ZnO)、酸化チタン(TiO)、酸化スズ(SnO)等が挙げられる。
電子捕集層の形成方法も、難溶性、難分散性昇華性材料の場合には上述したドライプロセスが選択され、溶液材料あるいは分散液材料の場合には、組成物の粘度と表面張力、所望する薄膜の厚さ等を考慮し、上述した各種ウェットプロセス法の中から最適なものが採用される。また、無機酸化物の前駆体層をウェットプロセス(特にスピンコート法かスリットコート法)を用いて陰極上に形成し、焼成して無機酸化物の層を形成する方法を採用することもできる。
必要に応じて、電荷の移動を効率化すること等を目的として、活性層と陰極層の間に電子捕集層を形成してもよい。
電子捕集層を形成する材料としては、上記順積層型の材料で例示したものに加え、酸化亜鉛(ZnO)、酸化チタン(TiO)、酸化スズ(SnO)等が挙げられる。
電子捕集層の形成方法も、難溶性、難分散性昇華性材料の場合には上述したドライプロセスが選択され、溶液材料あるいは分散液材料の場合には、組成物の粘度と表面張力、所望する薄膜の厚さ等を考慮し、上述した各種ウェットプロセス法の中から最適なものが採用される。また、無機酸化物の前駆体層をウェットプロセス(特にスピンコート法かスリットコート法)を用いて陰極上に形成し、焼成して無機酸化物の層を形成する方法を採用することもできる。
[活性層の形成]:形成された電子捕集層上に活性層を形成する工程
活性層は、n型半導体材料からなる薄膜であるn層と、p型半導体材料からなる薄膜であるp層とを積層したものであっても、これら材料の混合物からなる非積層薄膜であってもよい。
n型およびp型半導体材料としては、上記順積層型の半導体材料で例示したものと同様のものが挙げられるが、n型半導体材料としては、BTP-4FおよびITIC-4F、p型半導体材料としては、PM6およびPTB7等の主鎖にチオフェン骨格を含むポリマー類が好ましい。
活性層の形成方法も、上記順積層型の活性層で説明した方法と同様である。
活性層は、n型半導体材料からなる薄膜であるn層と、p型半導体材料からなる薄膜であるp層とを積層したものであっても、これら材料の混合物からなる非積層薄膜であってもよい。
n型およびp型半導体材料としては、上記順積層型の半導体材料で例示したものと同様のものが挙げられるが、n型半導体材料としては、BTP-4FおよびITIC-4F、p型半導体材料としては、PM6およびPTB7等の主鎖にチオフェン骨格を含むポリマー類が好ましい。
活性層の形成方法も、上記順積層型の活性層で説明した方法と同様である。
[正孔捕集層の形成]:形成された活性層材料の層上に正孔捕集層を形成する工程
上記方法に従い、活性層材料の層上に、本発明の組成物を用いて正孔捕集層を形成する。
上記方法に従い、活性層材料の層上に、本発明の組成物を用いて正孔捕集層を形成する。
[陽極層の形成]:形成された正孔捕集層の上に陽極層を形成する工程
陽極材料としては、上記順積層型の陽極材料と同様のものが挙げられ、陽極層の形成方法としても、順積層型の陰極層と同様である。
陽極材料としては、上記順積層型の陽極材料と同様のものが挙げられ、陽極層の形成方法としても、順積層型の陰極層と同様である。
[キャリアブロック層の形成]
順積層型の素子と同様、必要に応じて、光電流の整流性をコントロールすること等を目的として、任意の層間にキャリアブロック層を設けてもよい。
正孔ブロック層を形成する材料および電子ブロック層を形成する材料としては、上記と同様のものが挙げられ、キャリアブロック層の形成方法も上記と同様である。
順積層型の素子と同様、必要に応じて、光電流の整流性をコントロールすること等を目的として、任意の層間にキャリアブロック層を設けてもよい。
正孔ブロック層を形成する材料および電子ブロック層を形成する材料としては、上記と同様のものが挙げられ、キャリアブロック層の形成方法も上記と同様である。
上記で例示した方法によって作製されたOPV素子は、大気による素子劣化を防ぐために、再度グローブボックス内に導入して窒素等の不活性ガス雰囲気下で封止操作を行い、封止された状態で太陽電池としての機能を発揮させたり、太陽電池特性の測定を行ったりすることができる。
封止法としては、端部にUV硬化樹脂を付着させた凹型ガラス基板を、不活性ガス雰囲気下、有機薄膜太陽電池素子の成膜面側に付着させ、UV照射によって樹脂を硬化させる方法や、真空下、スパッタリング等の手法によって膜封止タイプの封止を行う方法などが挙げられる。
封止法としては、端部にUV硬化樹脂を付着させた凹型ガラス基板を、不活性ガス雰囲気下、有機薄膜太陽電池素子の成膜面側に付着させ、UV照射によって樹脂を硬化させる方法や、真空下、スパッタリング等の手法によって膜封止タイプの封止を行う方法などが挙げられる。
以下、実施例および比較例を挙げて、本発明をより具体的に説明するが、本発明は下記の実施例に限定されるものではない。なお、使用した装置は以下のとおりである。
(1)グローブボックス:山八物産(株)製、VACグローブボックスシステム
(2)蒸着装置:アオヤマエンジニアリング(株)製、真空蒸着装置
(3)ソーラーシミュレータ:分光計器(株)製、OTENTOSUN-III、AM1.5Gフィルター、放射強度:100mW/cm2
(4)ソースメジャーユニット:ケースレーインスツルメンツ(株)製、2612A
(1)グローブボックス:山八物産(株)製、VACグローブボックスシステム
(2)蒸着装置:アオヤマエンジニアリング(株)製、真空蒸着装置
(3)ソーラーシミュレータ:分光計器(株)製、OTENTOSUN-III、AM1.5Gフィルター、放射強度:100mW/cm2
(4)ソースメジャーユニット:ケースレーインスツルメンツ(株)製、2612A
[1]正孔捕集層用組成物の製造
[実施例1-1]
SELFTRON S(東ソー(株)製、2.0質量%水溶液)(以下:P1)1.25gに純水を3.73g、フッ素系ノニオン性界面活性剤(FN-1287、第一工業製薬(株)製)(以下:S1)を5.00mg、フッ素系ノニオン性界面活性剤(F-559、DIC(株)製)(以下:S2)を1.00mg加えた溶液に、ケイモリブド酸n水和物(富士フイルム和光純薬(株)製)(以下:D1)を7.50mg加え、濃度0.65質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物A1を得た。
[実施例1-1]
SELFTRON S(東ソー(株)製、2.0質量%水溶液)(以下:P1)1.25gに純水を3.73g、フッ素系ノニオン性界面活性剤(FN-1287、第一工業製薬(株)製)(以下:S1)を5.00mg、フッ素系ノニオン性界面活性剤(F-559、DIC(株)製)(以下:S2)を1.00mg加えた溶液に、ケイモリブド酸n水和物(富士フイルム和光純薬(株)製)(以下:D1)を7.50mg加え、濃度0.65質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物A1を得た。
[実施例1-2]
1.25gのP1に純水を3.73g、S1を5.00mg、S2を1.00mg加えた溶液に、ケイタングステン酸n水和物(Alfa Aesar(株)製)(以下:D2)を7.50mg加え、濃度0.65質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物A2を得た。
1.25gのP1に純水を3.73g、S1を5.00mg、S2を1.00mg加えた溶液に、ケイタングステン酸n水和物(Alfa Aesar(株)製)(以下:D2)を7.50mg加え、濃度0.65質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物A2を得た。
[実施例1-3]
1.25gのP1に純水を3.71g、S1を5.00mg、S2を1.00mg加えた溶液に、D1を7.50mg、国際公開第2006/025342号の記載に基づいて合成した上記式(2-1)で示されるアリールスルホン酸化合物A(以下:D3)を25.0mg加え、濃度1.15質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物A3を得た。
1.25gのP1に純水を3.71g、S1を5.00mg、S2を1.00mg加えた溶液に、D1を7.50mg、国際公開第2006/025342号の記載に基づいて合成した上記式(2-1)で示されるアリールスルホン酸化合物A(以下:D3)を25.0mg加え、濃度1.15質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物A3を得た。
[実施例1-4]
1.25gのP1に純水を3.71g、S1を5.00mg、S2を1.00mg加えた溶液に、D2を7.50mg、D3を25.0mg加え、濃度1.15質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物A4を得た。
1.25gのP1に純水を3.71g、S1を5.00mg、S2を1.00mg加えた溶液に、D2を7.50mg、D3を25.0mg加え、濃度1.15質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物A4を得た。
[実施例1-5]
1.25gのP1に純水を3.21g、イソプロパノールを0.49g、S1を5.00mg、S2を1.00mg加えた溶液に、D1を7.50mg、D3を25.0mg加え、濃度1.15質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物A5を得た。
1.25gのP1に純水を3.21g、イソプロパノールを0.49g、S1を5.00mg、S2を1.00mg加えた溶液に、D1を7.50mg、D3を25.0mg加え、濃度1.15質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物A5を得た。
[実施例1-6]
1.25gのP1に純水を3.71g、S1を5.00mg、S2を1.00mg加えた溶液に、12モリブド(IV)りん酸n水和物(富士フイルム和光純薬(株)製)(以下:D4)を7.50mg加え、濃度0.65質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物A6を得た。
1.25gのP1に純水を3.71g、S1を5.00mg、S2を1.00mg加えた溶液に、12モリブド(IV)りん酸n水和物(富士フイルム和光純薬(株)製)(以下:D4)を7.50mg加え、濃度0.65質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物A6を得た。
[実施例1-7]
1.25gのP1に純水を3.71g、S1を5.00mg、S2を1.00mg加えた溶液に、12タングスト(IV)りん酸n水和物(富士フイルム和光純薬(株)製)(以下:D5)を7.50mg加え、濃度0.65質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物A7を得た。
1.25gのP1に純水を3.71g、S1を5.00mg、S2を1.00mg加えた溶液に、12タングスト(IV)りん酸n水和物(富士フイルム和光純薬(株)製)(以下:D5)を7.50mg加え、濃度0.65質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物A7を得た。
以上、実施例1-1~1-7のインク組成を表1にまとめた。

[比較例1-1]
2.50gのP1にイソプロパノールを2.47g、S2を3.00mg加えた溶液に、D1を15.0mg加え、濃度1.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B1を得た。
2.50gのP1にイソプロパノールを2.47g、S2を3.00mg加えた溶液に、D1を15.0mg加え、濃度1.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B1を得た。
[比較例1-2]
2.50gのP1にイソプロパノールを2.47g、S2を3.00mg加えた溶液に、D2を15.0mg加え、濃度1.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B2を得た。
2.50gのP1にイソプロパノールを2.47g、S2を3.00mg加えた溶液に、D2を15.0mg加え、濃度1.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B2を得た。
[比較例1-3]
2.50gのP1にイソプロパノールを2.44g、S2を3.00mg加えた溶液に、D1を15.0mg、D3を50.0mg加え、濃度2.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B3を得た。
2.50gのP1にイソプロパノールを2.44g、S2を3.00mg加えた溶液に、D1を15.0mg、D3を50.0mg加え、濃度2.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B3を得た。
[比較例1-4]
2.50gのP1にイソプロパノールを2.44g、S2を3.00mg加えた溶液に、D2を15.0mg、D3を50.0mg加え、濃度2.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B4を得た。
2.50gのP1にイソプロパノールを2.44g、S2を3.00mg加えた溶液に、D2を15.0mg、D3を50.0mg加え、濃度2.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B4を得た。
[比較例1-5]
2.50gのP1に純水を0.96g、イソプロパノールを1.46g、S1を5.00mg、S2を1.00mg加えた溶液に、D1を15.0mg、D3を50.0mg加え、濃度2.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B5を得た。
2.50gのP1に純水を0.96g、イソプロパノールを1.46g、S1を5.00mg、S2を1.00mg加えた溶液に、D1を15.0mg、D3を50.0mg加え、濃度2.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B5を得た。
[比較例1-6]
2.50gのP1に純水を1.45g、イソプロパノールを0.98g、S1を5.00mg、S2を1.00mg加えた溶液に、D1を15.0mg、D3を50.0mg加え、濃度2.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B6を得た。
2.50gのP1に純水を1.45g、イソプロパノールを0.98g、S1を5.00mg、S2を1.00mg加えた溶液に、D1を15.0mg、D3を50.0mg加え、濃度2.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B6を得た。
[比較例1-7]
2.50gのP1にエタノールを2.44g、S2を3.00mg加えた溶液に、D1を15.0mg、D3を50.0mg加え、濃度2.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B7を得た。
2.50gのP1にエタノールを2.44g、S2を3.00mg加えた溶液に、D1を15.0mg、D3を50.0mg加え、濃度2.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B7を得た。
[比較例1-8]
2.50gのP1にイソプロパノールを2.47g、S2を3.00mg加えた溶液に、D4を15.0mg加え、濃度1.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B8を得た。
2.50gのP1にイソプロパノールを2.47g、S2を3.00mg加えた溶液に、D4を15.0mg加え、濃度1.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B8を得た。
[比較例1-9]
2.50gのP1にイソプロパノールを2.47g、S2を3.00mg加えた溶液に、D4を15.0mg加え、濃度1.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B9を得た。
2.50gのP1にイソプロパノールを2.47g、S2を3.00mg加えた溶液に、D4を15.0mg加え、濃度1.30質量%の濃青色溶液を調製した。得られた濃青色溶液を、孔径0.45μmのシリンジフィルターでろ過して、正孔捕集層用組成物B9を得た。
以上、比較例1-1~1-9のインク組成を表2にまとめた。

[2]正孔捕集層用組成物の保存安定性の評価
上記実施例1-1~1-7および比較例1-1~1-9の正孔捕集層用組成物を一週間室温で静置し、組成物中の析出物の有無を目視により観察し、下記評価基準に従って評価した。結果を表3に示す。
〈評価基準〉
〇:析出物無し
×:析出物有り
上記実施例1-1~1-7および比較例1-1~1-9の正孔捕集層用組成物を一週間室温で静置し、組成物中の析出物の有無を目視により観察し、下記評価基準に従って評価した。結果を表3に示す。
〈評価基準〉
〇:析出物無し
×:析出物有り

表3の結果より、以下のことが確認された。
・水/有機溶媒(比較例)では、いずれの組成も析出物は確認されなかった。
・溶媒が水のみの実施例では、リンモリブデン酸またはリンタングステン酸を使用した場合において析出が確認された。
・溶媒が水のみの場合では、ケイ酸系のヘテロポリ酸が好適である。
・水/有機溶媒(比較例)では、いずれの組成も析出物は確認されなかった。
・溶媒が水のみの実施例では、リンモリブデン酸またはリンタングステン酸を使用した場合において析出が確認された。
・溶媒が水のみの場合では、ケイ酸系のヘテロポリ酸が好適である。
[3]有機薄膜太陽電池の作製
[実施例3-1]
陰極となるITO透明導電層を10mm×25mmのストライプ状にパターニングした25mm×25mmのガラス基板を15分間UV/オゾン処理した。この基板に、電子捕集層となる酸化亜鉛の溶液(Genes’ Ink製)を滴下し、スピンコート法により成膜した。電子捕集層の膜厚は約30nmであった。その後、不活性ガスにより置換されたグローブボックス中で、形成した電子捕集層上にNFA活性層PV-X Plus(Raynergy tek製)をスピンコート法により成膜し、活性層を形成した。活性層の膜厚は約100nmであった。
次に、この活性層上に実施例1-1で調製した正孔捕集層用組成物A1をブレードコート法により塗布することで、正孔捕集層を形成した。正孔捕集層の膜厚は約100nmであった。
最後に、積層した基板を真空蒸着装置内に設置して、装置内の真空度が1×10-3Pa以下になるまで排気し、抵抗加熱法によって、陽極となる銀層を100nmの厚さに蒸着することで、ストライプ状のITO層と銀層とが交差する部分の面積が10mm×10mmである逆積層型OPV素子を作製した。
[実施例3-1]
陰極となるITO透明導電層を10mm×25mmのストライプ状にパターニングした25mm×25mmのガラス基板を15分間UV/オゾン処理した。この基板に、電子捕集層となる酸化亜鉛の溶液(Genes’ Ink製)を滴下し、スピンコート法により成膜した。電子捕集層の膜厚は約30nmであった。その後、不活性ガスにより置換されたグローブボックス中で、形成した電子捕集層上にNFA活性層PV-X Plus(Raynergy tek製)をスピンコート法により成膜し、活性層を形成した。活性層の膜厚は約100nmであった。
次に、この活性層上に実施例1-1で調製した正孔捕集層用組成物A1をブレードコート法により塗布することで、正孔捕集層を形成した。正孔捕集層の膜厚は約100nmであった。
最後に、積層した基板を真空蒸着装置内に設置して、装置内の真空度が1×10-3Pa以下になるまで排気し、抵抗加熱法によって、陽極となる銀層を100nmの厚さに蒸着することで、ストライプ状のITO層と銀層とが交差する部分の面積が10mm×10mmである逆積層型OPV素子を作製した。
[実施例3-2]
正孔捕集層用組成物A1をブレードコート法により塗布した後、ホットプレート上で120℃、5分のアニール処理を行った以外は実施例3-1と同様の方法で逆積層型OPV素子を作製した。
正孔捕集層用組成物A1をブレードコート法により塗布した後、ホットプレート上で120℃、5分のアニール処理を行った以外は実施例3-1と同様の方法で逆積層型OPV素子を作製した。
[実施例3-3]
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物A3を用いた以外は、実施例3-1と同様の方法で逆積層型OPV素子を作製した。
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物A3を用いた以外は、実施例3-1と同様の方法で逆積層型OPV素子を作製した。
[実施例3-4]
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物A3を用いた以外は、実施例3-2と同様の方法で逆積層型OPV素子を作製した。
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物A3を用いた以外は、実施例3-2と同様の方法で逆積層型OPV素子を作製した。
[実施例3-5]
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物A4を用いた以外は、実施例3-1と同様の方法で逆積層型OPV素子を作製した。
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物A4を用いた以外は、実施例3-1と同様の方法で逆積層型OPV素子を作製した。
[実施例3-6]
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物A4を用いた以外は、実施例3-2と同様の方法で逆積層型OPV素子を作製した。
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物A4を用いた以外は、実施例3-2と同様の方法で逆積層型OPV素子を作製した。
[実施例3-7]
正孔捕集層膜厚が約300nmである以外は、実施例3-4と同様の方法で逆積層型OPV素子を作製した。
正孔捕集層膜厚が約300nmである以外は、実施例3-4と同様の方法で逆積層型OPV素子を作製した。
[実施例3-8]
正孔捕集層用組成物A3の代わりに正孔捕集層用組成物A5を用いた以外は、実施例3-7と同様の方法で逆積層型OPV素子を作製した。
正孔捕集層用組成物A3の代わりに正孔捕集層用組成物A5を用いた以外は、実施例3-7と同様の方法で逆積層型OPV素子を作製した。
[実施例3-9]
NFA活性層PV-X Plus(Raynergy tek製)の代わりにFA活性層PV-F1062(Merck社製)を用いた以外は、実施例3-1と同様の方法で逆積層型OPV素子を作製した。
NFA活性層PV-X Plus(Raynergy tek製)の代わりにFA活性層PV-F1062(Merck社製)を用いた以外は、実施例3-1と同様の方法で逆積層型OPV素子を作製した。
[実施例3-10]
NFA活性層PV-X Plus(Raynergy tek製)の代わりにFA活性層PV-F1062(Merck&Co製)を用いた以外は、実施例3-5と同様の方法で逆積層型OPV素子を作製した。
NFA活性層PV-X Plus(Raynergy tek製)の代わりにFA活性層PV-F1062(Merck&Co製)を用いた以外は、実施例3-5と同様の方法で逆積層型OPV素子を作製した。
[比較例3-1]
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物B3を用いた以外は、実施例3-1と同様の方法で逆積層型OPV素子を作製した。
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物B3を用いた以外は、実施例3-1と同様の方法で逆積層型OPV素子を作製した。
[比較例3-2]
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物B3を用いた以外は、実施例3-2と同様の方法で逆積層型OPV素子を作製した。
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物B3を用いた以外は、実施例3-2と同様の方法で逆積層型OPV素子を作製した。
[比較例3-3]
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物B7を用いた以外は、実施例3-1と同様の方法で逆積層型OPV素子を作製した。
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物B7を用いた以外は、実施例3-1と同様の方法で逆積層型OPV素子を作製した。
[比較例3-4]
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物B7を用いた以外は、実施例3-2と同様の方法で逆積層型OPV素子を作製した。
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物B7を用いた以外は、実施例3-2と同様の方法で逆積層型OPV素子を作製した。
[比較例3-5]
正孔捕集層用組成物A3の代わりに正孔捕集層用組成物B5を用いた以外は、実施例3-7と同様の方法で逆積層型OPV素子を作製した。
正孔捕集層用組成物A3の代わりに正孔捕集層用組成物B5を用いた以外は、実施例3-7と同様の方法で逆積層型OPV素子を作製した。
[比較例3-6]
正孔捕集層用組成物A3の代わりに正孔捕集層用組成物B6を用いた以外は、実施例3-7と同様の方法で逆積層型OPV素子を作製した。
正孔捕集層用組成物A3の代わりに正孔捕集層用組成物B6を用いた以外は、実施例3-7と同様の方法で逆積層型OPV素子を作製した。
[比較例3-7]
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物B3を用いた以外は、実施例3-9と同様の方法で逆積層型OPV素子を作製した。
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物B3を用いた以外は、実施例3-9と同様の方法で逆積層型OPV素子を作製した。
[比較例3-8]
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物B7を用いた以外は、実施例3-9と同様の方法で逆積層型OPV素子を作製した。
正孔捕集層用組成物A1の代わりに正孔捕集層用組成物B7を用いた以外は、実施例3-9と同様の方法で逆積層型OPV素子を作製した。
[4]初期耐熱性評価
上記実施例3-1~3-8および比較例3-1~3-6で作製した各OPV素子について短絡電流密度(Jsc〔mA/cm2〕)、開放電圧(Voc〔V〕)、曲線因子(FF)、およびPCE〔%〕を測定した。結果を表4および表5に示す。また、実施例3-3および3-4より得られたJ-Vカーブを図1に、比較例3-1および3-2より得られたJ-Vカーブを図2に、実施例3-7および3-8、ならびに比較例3-5および3-6より得られたJ-Vカーブを図3にそれぞれ示す。
なおPCE〔%〕は、下式により算出した。
PCE〔%〕=Jsc〔mA/cm2〕×Voc〔V〕×FF÷入射光強度(100〔mW/cm2〕)×100
上記実施例3-1~3-8および比較例3-1~3-6で作製した各OPV素子について短絡電流密度(Jsc〔mA/cm2〕)、開放電圧(Voc〔V〕)、曲線因子(FF)、およびPCE〔%〕を測定した。結果を表4および表5に示す。また、実施例3-3および3-4より得られたJ-Vカーブを図1に、比較例3-1および3-2より得られたJ-Vカーブを図2に、実施例3-7および3-8、ならびに比較例3-5および3-6より得られたJ-Vカーブを図3にそれぞれ示す。
なおPCE〔%〕は、下式により算出した。
PCE〔%〕=Jsc〔mA/cm2〕×Voc〔V〕×FF÷入射光強度(100〔mW/cm2〕)×100


表4および表5、ならびに図1~3の結果より、以下のことが確認された。
・溶媒中の有機溶媒の含有量が10質量%以下である場合、HTL成膜後のアニール処理の温度で特性は変化しなかった。
・組成物の溶媒中の有機溶媒の含有量が50質量%である場合、120℃のアニール処理で大幅に特性が低下した。理由は定かではないが、有機溶媒が活性層中のn型半導体材料が形成したドメインを崩すことで、電荷分離能を低下させたことが要因と推察される。
・組成物の溶媒中の有機溶媒の含有量が20質量%、30質量%の場合でも、特性が著しく低下した。一方、その含有量が10質量%(実施例3-8)である場合、溶媒が水のみの場合(実施例3-7)と同等のVocを示しており、劣化の影響は小さいといえる。
・溶媒中の有機溶媒の含有量が10質量%以下である場合、HTL成膜後のアニール処理の温度で特性は変化しなかった。
・組成物の溶媒中の有機溶媒の含有量が50質量%である場合、120℃のアニール処理で大幅に特性が低下した。理由は定かではないが、有機溶媒が活性層中のn型半導体材料が形成したドメインを崩すことで、電荷分離能を低下させたことが要因と推察される。
・組成物の溶媒中の有機溶媒の含有量が20質量%、30質量%の場合でも、特性が著しく低下した。一方、その含有量が10質量%(実施例3-8)である場合、溶媒が水のみの場合(実施例3-7)と同等のVocを示しており、劣化の影響は小さいといえる。
[5]長期耐熱性評価
上記実施例3-1、3-3、3-5および3-8~3-10、ならびに比較例3-1、3-3、3-7および3-8で作製した各OPV素子を85℃に加熱したホットプレート上に静置することで、長期耐熱性を評価した。所定時間毎に短絡電流密度(Jsc〔mA/cm2〕)、開放電圧(Voc〔V〕)、曲線因子(FF)、およびPCE〔%〕を測定した。表6および表7に、各OPV素子の所定時間におけるPCEを示す。
上記実施例3-1、3-3、3-5および3-8~3-10、ならびに比較例3-1、3-3、3-7および3-8で作製した各OPV素子を85℃に加熱したホットプレート上に静置することで、長期耐熱性を評価した。所定時間毎に短絡電流密度(Jsc〔mA/cm2〕)、開放電圧(Voc〔V〕)、曲線因子(FF)、およびPCE〔%〕を測定した。表6および表7に、各OPV素子の所定時間におけるPCEを示す。


表6および表7の結果より、以下のことが確認された。
・溶媒中の有機溶媒の含有量が10質量%以下である場合、85℃のアニール処理を168時間行っても特性が低下しなかった。
・組成物の溶媒中の有機溶媒の含有量が10質量%である場合も、特性が低下しなかった。本含有量においては長期耐熱性においても劣化の影響は小さいといえる。
・組成物の溶媒中の有機溶媒の含有量が50質量%である場合、85℃のアニール処理で特性が低下した。これは含有する有機溶媒がイソプロパノール、エタノールどちらでも確認された。
・溶媒中の有機溶媒の含有量を10質量%以下とすることによる高い長期耐熱性、および有機溶媒による特性低下は、NFA活性層、FA活性層によらず確認された。
・溶媒中の有機溶媒の含有量が10質量%以下である場合、85℃のアニール処理を168時間行っても特性が低下しなかった。
・組成物の溶媒中の有機溶媒の含有量が10質量%である場合も、特性が低下しなかった。本含有量においては長期耐熱性においても劣化の影響は小さいといえる。
・組成物の溶媒中の有機溶媒の含有量が50質量%である場合、85℃のアニール処理で特性が低下した。これは含有する有機溶媒がイソプロパノール、エタノールどちらでも確認された。
・溶媒中の有機溶媒の含有量を10質量%以下とすることによる高い長期耐熱性、および有機溶媒による特性低下は、NFA活性層、FA活性層によらず確認された。
Claims (20)
- 有機光電変換素子における電荷輸送性薄膜を形成するための電荷輸送性組成物であって、
下記式(1)で表される繰り返し単位を含むポリチオフェン誘導体からなる電荷輸送性物質と、電子受容性ドーパント物質と、溶媒とを含み、
上記電子受容性ドーパント物質が、下記式(2)で表されるアリールスルホン酸およびヘテロポリ酸からなる群より選ばれる少なくとも1種を含み、上記溶媒中の有機溶媒の含有量が10質量%以下である電荷輸送性組成物。


- 上記R1およびR2が、R1およびR2が結合して形成される-O-Y-O-であり、Yは、エーテル結合を含んでいてもよい、スルホン酸基またはスルホン酸塩基で置換されている炭素数1~40のアルキレン基である請求項1記載の電荷輸送性組成物。
- 上記式(1)で表される繰り返し単位を含むポリチオフェン誘導体が、式(1-4)または式(1’-1)で表される繰り返し単位を含むポリチオフェン誘導体である請求項1記載の電荷輸送性組成物。

- 上記電子受容性ドーパント物質が、上記式(2)で表されるアリールスルホン酸およびヘテロポリ酸を含む請求項1記載の電荷輸送性組成物。
- 上記ヘテロポリ酸が、ケイモリブデン酸およびケイタングステン酸からなる群より選ばれる少なくとも1種を含む請求項1記載の電荷輸送性組成物。
- 上記溶媒中の有機溶媒の含有量が、5質量%以下である請求項1記載の電荷輸送性組成物。
- 上記溶媒が水のみである請求項6記載の電荷輸送性組成物。
- さらに、界面活性剤を含む請求項1記載の電荷輸送性組成物。
- 上記界面活性剤が、フッ素系界面活性剤である請求項8記載の電荷輸送性組成物。
- 有機光電変換素子の正孔捕集層用である請求項1記載の電荷輸送性組成物。
- 上記有機光電変換素子が、有機薄膜太陽電池、色素増感太陽電池または光センサーである請求項10記載の電荷輸送性組成物。
- 請求項1記載の電荷輸送性組成物から得られる電荷輸送性薄膜。
- 上記電荷輸送性薄膜が、有機光電変換素子の正孔捕集層である請求項12記載の電荷輸送性薄膜。
- 請求項12記載の電荷輸送性薄膜を備える電子素子。
- 上記電子素子が、有機光電変換素子である請求項14記載の電子素子。
- 請求項13記載の正孔捕集層と、それに接するように設けられた非フラーレン活性層とを有する有機光電変換素子。
- 上記非フラーレン活性層が、主鎖にチオフェン骨格を含むポリマーを含む請求項16記載の有機光電変換素子。
- 逆積層型である請求項16記載の有機光電変換素子。
- 上記有機光電変換素子が、有機薄膜太陽電池または光センサーである請求項16~18のいずれか1項記載の有機光電変換素子。
- トップ陽極構造を有する請求項19記載の有機光電変換素子。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021179827 | 2021-11-02 | ||
| JP2021-179827 | 2021-11-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023080024A1 true WO2023080024A1 (ja) | 2023-05-11 |
Family
ID=86240985
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/039830 WO2023080024A1 (ja) | 2021-11-02 | 2022-10-26 | 電荷輸送性組成物 |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2023080024A1 (ja) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013042623A1 (ja) * | 2011-09-21 | 2013-03-28 | 日産化学工業株式会社 | 電荷輸送性ワニス |
| WO2016208597A1 (ja) * | 2015-06-22 | 2016-12-29 | 住友化学株式会社 | 有機電子素子の製造方法及び有機薄膜の形成方法 |
| WO2019176662A1 (ja) * | 2018-03-15 | 2019-09-19 | 日産化学株式会社 | 電荷輸送性組成物 |
| JP2021520069A (ja) * | 2018-04-03 | 2021-08-12 | 住友化学株式会社 | 近赤外有機光検出器 |
| WO2022202868A1 (ja) * | 2021-03-26 | 2022-09-29 | 日産化学株式会社 | 電荷輸送性組成物 |
-
2022
- 2022-10-26 WO PCT/JP2022/039830 patent/WO2023080024A1/ja unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013042623A1 (ja) * | 2011-09-21 | 2013-03-28 | 日産化学工業株式会社 | 電荷輸送性ワニス |
| WO2016208597A1 (ja) * | 2015-06-22 | 2016-12-29 | 住友化学株式会社 | 有機電子素子の製造方法及び有機薄膜の形成方法 |
| WO2019176662A1 (ja) * | 2018-03-15 | 2019-09-19 | 日産化学株式会社 | 電荷輸送性組成物 |
| JP2021520069A (ja) * | 2018-04-03 | 2021-08-12 | 住友化学株式会社 | 近赤外有機光検出器 |
| WO2022202868A1 (ja) * | 2021-03-26 | 2022-09-29 | 日産化学株式会社 | 電荷輸送性組成物 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7276314B2 (ja) | 電荷輸送性組成物 | |
| JP6696510B2 (ja) | 有機光電変換素子の正孔捕集層用組成物 | |
| JP7180379B2 (ja) | 有機光電変換素子の正孔捕集層用組成物 | |
| JP7243636B2 (ja) | 有機光電変換素子の正孔捕集層用組成物 | |
| WO2022202868A1 (ja) | 電荷輸送性組成物 | |
| WO2022202872A1 (ja) | 電荷輸送性組成物 | |
| WO2020153180A1 (ja) | ペロブスカイト光電変換素子用電荷輸送性組成物 | |
| WO2023080024A1 (ja) | 電荷輸送性組成物 | |
| WO2024071060A1 (ja) | 電荷輸送性組成物 | |
| JP2023149746A (ja) | 電荷輸送性組成物およびその製造方法 | |
| TW202432659A (zh) | 電荷輸送性組成物 | |
| US20230200203A1 (en) | Composition for hole collecting layer of organic photoelectric conversion element | |
| BR112020018774B1 (pt) | Composição de transporte de carga, filme fino, elemento eletrônico e elemento de conversão fotoelétrica orgânica |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22889844 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |