WO2022265086A1 - ヒト多能性幹細胞由来大脳皮質細胞製剤の製造方法 - Google Patents
ヒト多能性幹細胞由来大脳皮質細胞製剤の製造方法 Download PDFInfo
- Publication number
- WO2022265086A1 WO2022265086A1 PCT/JP2022/024226 JP2022024226W WO2022265086A1 WO 2022265086 A1 WO2022265086 A1 WO 2022265086A1 JP 2022024226 W JP2022024226 W JP 2022024226W WO 2022265086 A1 WO2022265086 A1 WO 2022265086A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cerebral
- cells
- cell
- cell mass
- culture
- Prior art date
Links
- 210000004027 cell Anatomy 0.000 title claims abstract description 1116
- 230000002490 cerebral effect Effects 0.000 title claims abstract description 515
- 230000001054 cortical effect Effects 0.000 title claims abstract description 221
- 210000001778 pluripotent stem cell Anatomy 0.000 title claims abstract description 100
- 238000004519 manufacturing process Methods 0.000 title claims description 76
- 238000002360 preparation method Methods 0.000 title description 17
- 210000002220 organoid Anatomy 0.000 claims abstract description 356
- 238000000034 method Methods 0.000 claims abstract description 214
- 230000004069 differentiation Effects 0.000 claims abstract description 112
- 210000002569 neuron Anatomy 0.000 claims abstract description 85
- 238000012258 culturing Methods 0.000 claims abstract description 49
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 claims abstract description 39
- 102100024785 Fibroblast growth factor 2 Human genes 0.000 claims abstract description 29
- 230000001939 inductive effect Effects 0.000 claims abstract description 8
- 239000002609 medium Substances 0.000 claims description 181
- 239000003112 inhibitor Substances 0.000 claims description 156
- 210000003710 cerebral cortex Anatomy 0.000 claims description 151
- 230000014509 gene expression Effects 0.000 claims description 146
- 239000003550 marker Substances 0.000 claims description 131
- 108090000623 proteins and genes Proteins 0.000 claims description 130
- DWJXYEABWRJFSP-XOBRGWDASA-N DAPT Chemical compound N([C@@H](C)C(=O)N[C@H](C(=O)OC(C)(C)C)C=1C=CC=CC=1)C(=O)CC1=CC(F)=CC(F)=C1 DWJXYEABWRJFSP-XOBRGWDASA-N 0.000 claims description 107
- 238000011977 dual antiplatelet therapy Methods 0.000 claims description 107
- 239000001963 growth medium Substances 0.000 claims description 89
- 102000004887 Transforming Growth Factor beta Human genes 0.000 claims description 84
- 108090001012 Transforming Growth Factor beta Proteins 0.000 claims description 84
- 102000014736 Notch Human genes 0.000 claims description 68
- 108010070047 Notch Receptors Proteins 0.000 claims description 68
- 230000006698 induction Effects 0.000 claims description 57
- 210000001519 tissue Anatomy 0.000 claims description 48
- 230000001537 neural effect Effects 0.000 claims description 41
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 claims description 40
- 238000004114 suspension culture Methods 0.000 claims description 40
- 230000002062 proliferating effect Effects 0.000 claims description 39
- 238000002054 transplantation Methods 0.000 claims description 39
- 230000011664 signaling Effects 0.000 claims description 37
- 210000004129 prosencephalon Anatomy 0.000 claims description 34
- 238000004113 cell culture Methods 0.000 claims description 33
- -1 NEUROD2 Proteins 0.000 claims description 31
- 102100038039 Vesicular glutamate transporter 1 Human genes 0.000 claims description 31
- 108050003627 Wnt Proteins 0.000 claims description 31
- 102000013814 Wnt Human genes 0.000 claims description 31
- 102100023823 Homeobox protein EMX1 Human genes 0.000 claims description 28
- 101001048956 Homo sapiens Homeobox protein EMX1 Proteins 0.000 claims description 28
- 108091006283 SLC17A7 Proteins 0.000 claims description 28
- 101150006690 NEUROD6 gene Proteins 0.000 claims description 26
- 102100030589 Neurogenic differentiation factor 6 Human genes 0.000 claims description 26
- 239000003540 gamma secretase inhibitor Substances 0.000 claims description 25
- 108010025020 Nerve Growth Factor Proteins 0.000 claims description 24
- 101150026630 FOXG1 gene Proteins 0.000 claims description 23
- FHYUGAJXYORMHI-UHFFFAOYSA-N SB 431542 Chemical compound C1=CC(C(=O)N)=CC=C1C1=NC(C=2C=C3OCOC3=CC=2)=C(C=2N=CC=CC=2)N1 FHYUGAJXYORMHI-UHFFFAOYSA-N 0.000 claims description 23
- 229940125373 Gamma-Secretase Inhibitor Drugs 0.000 claims description 22
- 102000007072 Nerve Growth Factors Human genes 0.000 claims description 21
- 239000003900 neurotrophic factor Substances 0.000 claims description 21
- 102100033601 Collagen alpha-1(I) chain Human genes 0.000 claims description 20
- 102100032062 Neurogenic differentiation factor 2 Human genes 0.000 claims description 20
- 108010029483 alpha 1 Chain Collagen Type I Proteins 0.000 claims description 20
- 235000010323 ascorbic acid Nutrition 0.000 claims description 20
- 229960005070 ascorbic acid Drugs 0.000 claims description 20
- 239000011668 ascorbic acid Substances 0.000 claims description 20
- 210000004263 induced pluripotent stem cell Anatomy 0.000 claims description 20
- 102000004169 proteins and genes Human genes 0.000 claims description 19
- 101000873786 Homo sapiens Glutamate decarboxylase 2 Proteins 0.000 claims description 18
- 101000595526 Homo sapiens T-box brain protein 1 Proteins 0.000 claims description 18
- 102100036083 T-box brain protein 1 Human genes 0.000 claims description 18
- 208000026106 cerebrovascular disease Diseases 0.000 claims description 18
- 102100035857 Glutamate decarboxylase 2 Human genes 0.000 claims description 17
- 101150079937 NEUROD1 gene Proteins 0.000 claims description 16
- 239000008194 pharmaceutical composition Substances 0.000 claims description 16
- 101150004658 BHLHE22 gene Proteins 0.000 claims description 15
- 102100026204 Class E basic helix-loop-helix protein 22 Human genes 0.000 claims description 15
- 101001108436 Homo sapiens Neurexin-1 Proteins 0.000 claims description 15
- 101001108433 Homo sapiens Neurexin-1-beta Proteins 0.000 claims description 15
- 101000829127 Homo sapiens Somatostatin receptor type 2 Proteins 0.000 claims description 15
- 102100021582 Neurexin-1-beta Human genes 0.000 claims description 15
- 108700020297 NeuroD Proteins 0.000 claims description 15
- 102100032063 Neurogenic differentiation factor 1 Human genes 0.000 claims description 15
- 102100023802 Somatostatin receptor type 2 Human genes 0.000 claims description 15
- 210000001671 embryonic stem cell Anatomy 0.000 claims description 15
- 102100036817 Ankyrin-3 Human genes 0.000 claims description 14
- 102100024316 Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1A Human genes 0.000 claims description 14
- 102100021849 Calretinin Human genes 0.000 claims description 14
- 102100024175 Centriole, cilia and spindle-associated protein Human genes 0.000 claims description 14
- 101150025643 Epha5 gene Proteins 0.000 claims description 14
- 102100021605 Ephrin type-A receptor 5 Human genes 0.000 claims description 14
- 102100021888 Helix-loop-helix protein 1 Human genes 0.000 claims description 14
- 102100021889 Helix-loop-helix protein 2 Human genes 0.000 claims description 14
- 102100039542 Homeobox protein Hox-A2 Human genes 0.000 claims description 14
- 101000897856 Homo sapiens Adenylyl cyclase-associated protein 2 Proteins 0.000 claims description 14
- 101000928342 Homo sapiens Ankyrin-3 Proteins 0.000 claims description 14
- 101001117044 Homo sapiens Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1A Proteins 0.000 claims description 14
- 101000898072 Homo sapiens Calretinin Proteins 0.000 claims description 14
- 101000980796 Homo sapiens Centriole, cilia and spindle-associated protein Proteins 0.000 claims description 14
- 101000616435 Homo sapiens Gamma-sarcoglycan Proteins 0.000 claims description 14
- 101000897691 Homo sapiens Helix-loop-helix protein 1 Proteins 0.000 claims description 14
- 101000897700 Homo sapiens Helix-loop-helix protein 2 Proteins 0.000 claims description 14
- 101000962636 Homo sapiens Homeobox protein Hox-A2 Proteins 0.000 claims description 14
- 101001003169 Homo sapiens Insulin-like growth factor-binding protein-like 1 Proteins 0.000 claims description 14
- 101001050607 Homo sapiens KH domain-containing, RNA-binding, signal transduction-associated protein 3 Proteins 0.000 claims description 14
- 101000979909 Homo sapiens NMDA receptor synaptonuclear signaling and neuronal migration factor Proteins 0.000 claims description 14
- 101000577555 Homo sapiens Neuritin Proteins 0.000 claims description 14
- 101000596532 Homo sapiens Neuronal vesicle trafficking-associated protein 2 Proteins 0.000 claims description 14
- 101000602176 Homo sapiens Neurotensin/neuromedin N Proteins 0.000 claims description 14
- 101000995264 Homo sapiens Protein kinase C-binding protein NELL2 Proteins 0.000 claims description 14
- 101000735368 Homo sapiens Protocadherin-9 Proteins 0.000 claims description 14
- 101000823237 Homo sapiens Reticulon-1 Proteins 0.000 claims description 14
- 101000873658 Homo sapiens Secretogranin-3 Proteins 0.000 claims description 14
- 101000829203 Homo sapiens Serine/arginine repetitive matrix protein 4 Proteins 0.000 claims description 14
- 101000836079 Homo sapiens Serpin B8 Proteins 0.000 claims description 14
- 101000891867 Homo sapiens Synaptotagmin-4 Proteins 0.000 claims description 14
- 101000654935 Homo sapiens Thrombospondin type-1 domain-containing protein 7A Proteins 0.000 claims description 14
- 101000798702 Homo sapiens Transmembrane protease serine 4 Proteins 0.000 claims description 14
- 101000987017 Homo sapiens Tumor protein p53-inducible protein 11 Proteins 0.000 claims description 14
- 101000964479 Homo sapiens Zinc finger and BTB domain-containing protein 18 Proteins 0.000 claims description 14
- 101000931371 Homo sapiens Zinc finger protein ZFPM2 Proteins 0.000 claims description 14
- 102100020781 Insulin-like growth factor-binding protein-like 1 Human genes 0.000 claims description 14
- 102100023428 KH domain-containing, RNA-binding, signal transduction-associated protein 3 Human genes 0.000 claims description 14
- 102100024546 NMDA receptor synaptonuclear signaling and neuronal migration factor Human genes 0.000 claims description 14
- 102100028749 Neuritin Human genes 0.000 claims description 14
- 102100035069 Neuronal vesicle trafficking-associated protein 2 Human genes 0.000 claims description 14
- 102100037590 Neurotensin/neuromedin N Human genes 0.000 claims description 14
- 102100034433 Protein kinase C-binding protein NELL2 Human genes 0.000 claims description 14
- 102100034957 Protocadherin-9 Human genes 0.000 claims description 14
- 102100022647 Reticulon-1 Human genes 0.000 claims description 14
- 102100023663 Serine/arginine repetitive matrix protein 4 Human genes 0.000 claims description 14
- 102100040817 Synaptotagmin-4 Human genes 0.000 claims description 14
- 102100032612 Thrombospondin type-1 domain-containing protein 7A Human genes 0.000 claims description 14
- 102100027873 Tumor protein p53-inducible protein 11 Human genes 0.000 claims description 14
- 102100040762 Zinc finger and BTB domain-containing protein 18 Human genes 0.000 claims description 14
- 102100020996 Zinc finger protein ZFPM2 Human genes 0.000 claims description 14
- 239000012190 activator Substances 0.000 claims description 14
- 239000000470 constituent Substances 0.000 claims description 14
- 102100023046 Band 4.1-like protein 3 Human genes 0.000 claims description 13
- 101001049975 Homo sapiens Band 4.1-like protein 3 Proteins 0.000 claims description 13
- 101000661816 Homo sapiens Suppression of tumorigenicity 18 protein Proteins 0.000 claims description 13
- 102100031623 Myelin transcription factor 1-like protein Human genes 0.000 claims description 13
- 101150059596 Myt1l gene Proteins 0.000 claims description 13
- 102100037943 Suppression of tumorigenicity 18 protein Human genes 0.000 claims description 13
- 230000019491 signal transduction Effects 0.000 claims description 13
- 102100036466 Delta-like protein 3 Human genes 0.000 claims description 12
- 102100020913 Follistatin-related protein 5 Human genes 0.000 claims description 12
- 102100021260 Galactosylgalactosylxylosylprotein 3-beta-glucuronosyltransferase 1 Human genes 0.000 claims description 12
- 101000928513 Homo sapiens Delta-like protein 3 Proteins 0.000 claims description 12
- 101000931687 Homo sapiens Follistatin-related protein 5 Proteins 0.000 claims description 12
- 101000894906 Homo sapiens Galactosylgalactosylxylosylprotein 3-beta-glucuronosyltransferase 1 Proteins 0.000 claims description 12
- 101001007738 Homo sapiens Neurexophilin-4 Proteins 0.000 claims description 12
- 101000604058 Homo sapiens Neuronal pentraxin-1 Proteins 0.000 claims description 12
- 102100027531 Neurexophilin-4 Human genes 0.000 claims description 12
- 102100038436 Neuronal pentraxin-1 Human genes 0.000 claims description 12
- 229940127276 delta-like ligand 3 Drugs 0.000 claims description 12
- 239000004017 serum-free culture medium Substances 0.000 claims description 11
- 102100025488 CUGBP Elav-like family member 4 Human genes 0.000 claims description 10
- 101000914306 Homo sapiens CUGBP Elav-like family member 4 Proteins 0.000 claims description 10
- 239000000203 mixture Substances 0.000 claims description 10
- 230000008093 supporting effect Effects 0.000 claims description 10
- 102100041021 Coronin-2B Human genes 0.000 claims description 9
- 101000748863 Homo sapiens Coronin-2B Proteins 0.000 claims description 9
- 101001124900 Homo sapiens PR domain zinc finger protein 8 Proteins 0.000 claims description 9
- 102100029128 PR domain zinc finger protein 8 Human genes 0.000 claims description 9
- 102100020871 Forkhead box protein G1 Human genes 0.000 claims description 8
- 239000004480 active ingredient Substances 0.000 claims description 8
- 230000019612 pigmentation Effects 0.000 claims description 8
- 101000603698 Homo sapiens Neurogenin-2 Proteins 0.000 claims description 7
- 102100038554 Neurogenin-2 Human genes 0.000 claims description 7
- 230000000977 initiatory effect Effects 0.000 claims description 7
- 206010011732 Cyst Diseases 0.000 claims description 6
- 101000978570 Homo sapiens Noelin Proteins 0.000 claims description 6
- 102100023731 Noelin Human genes 0.000 claims description 6
- JNGZXGGOCLZBFB-IVCQMTBJSA-N compound E Chemical compound N([C@@H](C)C(=O)N[C@@H]1C(N(C)C2=CC=CC=C2C(C=2C=CC=CC=2)=N1)=O)C(=O)CC1=CC(F)=CC(F)=C1 JNGZXGGOCLZBFB-IVCQMTBJSA-N 0.000 claims description 6
- 208000031513 cyst Diseases 0.000 claims description 6
- 239000012634 fragment Substances 0.000 claims description 6
- 230000008569 process Effects 0.000 claims description 6
- HIJMSZGHKQPPJS-UHFFFAOYSA-N 3-(6-methylpyridin-2-yl)-n-phenyl-4-quinolin-4-ylpyrazole-1-carbothioamide Chemical compound CC1=CC=CC(C=2C(=CN(N=2)C(=S)NC=2C=CC=CC=2)C=2C3=CC=CC=C3N=CC=2)=N1 HIJMSZGHKQPPJS-UHFFFAOYSA-N 0.000 claims description 5
- KLGQSVMIPOVQAX-UHFFFAOYSA-N XAV939 Chemical compound N=1C=2CCSCC=2C(O)=NC=1C1=CC=C(C(F)(F)F)C=C1 KLGQSVMIPOVQAX-UHFFFAOYSA-N 0.000 claims description 4
- 238000005259 measurement Methods 0.000 claims description 4
- 210000004227 basal ganglia Anatomy 0.000 claims description 3
- 102100021879 Adenylyl cyclase-associated protein 2 Human genes 0.000 claims 4
- 102100021792 Gamma-sarcoglycan Human genes 0.000 claims 4
- 229940126585 therapeutic drug Drugs 0.000 claims 1
- 238000002560 therapeutic procedure Methods 0.000 abstract description 7
- 230000001172 regenerating effect Effects 0.000 abstract description 3
- 238000009630 liquid culture Methods 0.000 abstract 1
- 210000003172 sustentacular cell Anatomy 0.000 abstract 1
- 239000010410 layer Substances 0.000 description 74
- 239000000126 substance Substances 0.000 description 41
- 210000002966 serum Anatomy 0.000 description 40
- 210000000130 stem cell Anatomy 0.000 description 36
- 230000015572 biosynthetic process Effects 0.000 description 33
- 238000004458 analytical method Methods 0.000 description 30
- 230000000694 effects Effects 0.000 description 28
- 108010085895 Laminin Proteins 0.000 description 26
- 102000007547 Laminin Human genes 0.000 description 26
- 102000007354 PAX6 Transcription Factor Human genes 0.000 description 26
- 101150081664 PAX6 gene Proteins 0.000 description 26
- 210000001178 neural stem cell Anatomy 0.000 description 26
- 239000000243 solution Substances 0.000 description 24
- 102000000568 rho-Associated Kinases Human genes 0.000 description 23
- 108010041788 rho-Associated Kinases Proteins 0.000 description 23
- 238000012744 immunostaining Methods 0.000 description 22
- 238000012423 maintenance Methods 0.000 description 21
- 210000004498 neuroglial cell Anatomy 0.000 description 20
- 238000012360 testing method Methods 0.000 description 20
- 210000002950 fibroblast Anatomy 0.000 description 19
- 230000001965 increasing effect Effects 0.000 description 19
- 210000003618 cortical neuron Anatomy 0.000 description 18
- 230000003247 decreasing effect Effects 0.000 description 18
- 108010071690 Prealbumin Proteins 0.000 description 17
- 102000009190 Transthyretin Human genes 0.000 description 17
- 210000002987 choroid plexus Anatomy 0.000 description 17
- 238000010195 expression analysis Methods 0.000 description 17
- 238000000684 flow cytometry Methods 0.000 description 17
- 210000001362 glutamatergic neuron Anatomy 0.000 description 17
- 239000002771 cell marker Substances 0.000 description 16
- 235000018102 proteins Nutrition 0.000 description 16
- 210000005155 neural progenitor cell Anatomy 0.000 description 15
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 15
- 238000011529 RT qPCR Methods 0.000 description 14
- 210000002744 extracellular matrix Anatomy 0.000 description 14
- 150000002632 lipids Chemical class 0.000 description 14
- 238000003762 quantitative reverse transcription PCR Methods 0.000 description 14
- 210000004556 brain Anatomy 0.000 description 13
- 239000003814 drug Substances 0.000 description 13
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 12
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 12
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 12
- IDDDVXIUIXWAGJ-LJDSMOQUSA-N chembl1605605 Chemical compound Cl.Cl.C1C[C@@H]([C@H](N)C)CC[C@@H]1C(=O)NC1=CC=NC=C1 IDDDVXIUIXWAGJ-LJDSMOQUSA-N 0.000 description 12
- 235000021186 dishes Nutrition 0.000 description 12
- 210000001222 gaba-ergic neuron Anatomy 0.000 description 12
- 101000687905 Homo sapiens Transcription factor SOX-2 Proteins 0.000 description 11
- 239000012580 N-2 Supplement Substances 0.000 description 11
- 108010032788 PAX6 Transcription Factor Proteins 0.000 description 11
- 102100037506 Paired box protein Pax-6 Human genes 0.000 description 11
- 102100024270 Transcription factor SOX-2 Human genes 0.000 description 11
- 230000002401 inhibitory effect Effects 0.000 description 11
- 229910052760 oxygen Inorganic materials 0.000 description 11
- 230000002441 reversible effect Effects 0.000 description 11
- 102000003974 Fibroblast growth factor 2 Human genes 0.000 description 10
- 101100310657 Mus musculus Sox1 gene Proteins 0.000 description 10
- 102100035897 Secretogranin-3 Human genes 0.000 description 10
- 102100032471 Transmembrane protease serine 4 Human genes 0.000 description 10
- 239000000853 adhesive Substances 0.000 description 10
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 10
- 238000011156 evaluation Methods 0.000 description 10
- 210000001982 neural crest cell Anatomy 0.000 description 10
- 239000001301 oxygen Substances 0.000 description 10
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 10
- CJGYSWNGNKCJSB-YVLZZHOMSA-N bucladesine Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](OC(=O)CCC)[C@@H]2N1C(N=CN=C2NC(=O)CCC)=C2N=C1 CJGYSWNGNKCJSB-YVLZZHOMSA-N 0.000 description 9
- 229960005263 bucladesine Drugs 0.000 description 9
- 210000003169 central nervous system Anatomy 0.000 description 9
- 239000012141 concentrate Substances 0.000 description 9
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 9
- 238000002073 fluorescence micrograph Methods 0.000 description 9
- 210000005036 nerve Anatomy 0.000 description 9
- 210000000933 neural crest Anatomy 0.000 description 9
- 230000036961 partial effect Effects 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- 210000003556 vascular endothelial cell Anatomy 0.000 description 9
- 102000004219 Brain-derived neurotrophic factor Human genes 0.000 description 8
- 108090000715 Brain-derived neurotrophic factor Proteins 0.000 description 8
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 8
- 102100035423 POU domain, class 5, transcription factor 1 Human genes 0.000 description 8
- 230000001070 adhesive effect Effects 0.000 description 8
- 229940077737 brain-derived neurotrophic factor Drugs 0.000 description 8
- 210000004438 cajal-retzius cell Anatomy 0.000 description 8
- 201000010099 disease Diseases 0.000 description 8
- 230000007705 epithelial mesenchymal transition Effects 0.000 description 8
- 230000007774 longterm Effects 0.000 description 8
- 230000000877 morphologic effect Effects 0.000 description 8
- 230000002829 reductive effect Effects 0.000 description 8
- 230000003068 static effect Effects 0.000 description 8
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 7
- 239000000654 additive Substances 0.000 description 7
- 210000004102 animal cell Anatomy 0.000 description 7
- 239000006185 dispersion Substances 0.000 description 7
- 239000003102 growth factor Substances 0.000 description 7
- 229960002897 heparin Drugs 0.000 description 7
- 229920000669 heparin Polymers 0.000 description 7
- 230000008672 reprogramming Effects 0.000 description 7
- 210000001082 somatic cell Anatomy 0.000 description 7
- 230000002861 ventricular Effects 0.000 description 7
- 239000012099 Alexa Fluor family Substances 0.000 description 6
- 241000283074 Equus asinus Species 0.000 description 6
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 6
- 108091007433 antigens Proteins 0.000 description 6
- 102000036639 antigens Human genes 0.000 description 6
- 229910002091 carbon monoxide Inorganic materials 0.000 description 6
- 150000001875 compounds Chemical class 0.000 description 6
- 238000005516 engineering process Methods 0.000 description 6
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 6
- 108010082117 matrigel Proteins 0.000 description 6
- 230000035755 proliferation Effects 0.000 description 6
- 239000012679 serum free medium Substances 0.000 description 6
- 239000013589 supplement Substances 0.000 description 6
- 108010043324 Amyloid Precursor Protein Secretases Proteins 0.000 description 5
- 102000002659 Amyloid Precursor Protein Secretases Human genes 0.000 description 5
- 101001094700 Homo sapiens POU domain, class 5, transcription factor 1 Proteins 0.000 description 5
- 101001116302 Homo sapiens Platelet endothelial cell adhesion molecule Proteins 0.000 description 5
- 101000713575 Homo sapiens Tubulin beta-3 chain Proteins 0.000 description 5
- 229930182555 Penicillin Natural products 0.000 description 5
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 5
- 102100024616 Platelet endothelial cell adhesion molecule Human genes 0.000 description 5
- 102000043322 Reelin Human genes 0.000 description 5
- 108700038365 Reelin Proteins 0.000 description 5
- 102100036790 Tubulin beta-3 chain Human genes 0.000 description 5
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 description 5
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 5
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 5
- 238000004115 adherent culture Methods 0.000 description 5
- 230000001464 adherent effect Effects 0.000 description 5
- 150000001413 amino acids Chemical class 0.000 description 5
- 239000000427 antigen Substances 0.000 description 5
- 239000007640 basal medium Substances 0.000 description 5
- 239000006285 cell suspension Substances 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 108020004999 messenger RNA Proteins 0.000 description 5
- 229940049954 penicillin Drugs 0.000 description 5
- 239000000049 pigment Substances 0.000 description 5
- 238000003757 reverse transcription PCR Methods 0.000 description 5
- 229960005322 streptomycin Drugs 0.000 description 5
- 229940124597 therapeutic agent Drugs 0.000 description 5
- KHZOJCQBHJUJFY-UHFFFAOYSA-N 2-[4-(2-methylpyridin-4-yl)phenyl]-n-(4-pyridin-3-ylphenyl)acetamide Chemical compound C1=NC(C)=CC(C=2C=CC(CC(=O)NC=3C=CC(=CC=3)C=3C=NC=CC=3)=CC=2)=C1 KHZOJCQBHJUJFY-UHFFFAOYSA-N 0.000 description 4
- 102000009027 Albumins Human genes 0.000 description 4
- 108010088751 Albumins Proteins 0.000 description 4
- 229920001342 Bakelite® Polymers 0.000 description 4
- 102100031181 Glyceraldehyde-3-phosphate dehydrogenase Human genes 0.000 description 4
- 102100032530 Glypican-3 Human genes 0.000 description 4
- 206010019196 Head injury Diseases 0.000 description 4
- 102100022377 Homeobox protein DLX-2 Human genes 0.000 description 4
- 101001014668 Homo sapiens Glypican-3 Proteins 0.000 description 4
- 101000901635 Homo sapiens Homeobox protein DLX-2 Proteins 0.000 description 4
- 101001069727 Homo sapiens Paired mesoderm homeobox protein 1 Proteins 0.000 description 4
- 101000945496 Homo sapiens Proliferation marker protein Ki-67 Proteins 0.000 description 4
- 101000652332 Homo sapiens Transcription factor SOX-1 Proteins 0.000 description 4
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 4
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 4
- 229930182816 L-glutamine Natural products 0.000 description 4
- 108010012255 Neural Cell Adhesion Molecule L1 Proteins 0.000 description 4
- 102100024964 Neural cell adhesion molecule L1 Human genes 0.000 description 4
- 102100033786 Paired mesoderm homeobox protein 1 Human genes 0.000 description 4
- 102100034836 Proliferation marker protein Ki-67 Human genes 0.000 description 4
- 238000003559 RNA-seq method Methods 0.000 description 4
- 102000004338 Transferrin Human genes 0.000 description 4
- 108090000901 Transferrin Proteins 0.000 description 4
- 108010083162 Twist-Related Protein 1 Proteins 0.000 description 4
- 102100030398 Twist-related protein 1 Human genes 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 230000004956 cell adhesive effect Effects 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- 239000011248 coating agent Substances 0.000 description 4
- 239000003636 conditioned culture medium Substances 0.000 description 4
- 238000001218 confocal laser scanning microscopy Methods 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 210000000981 epithelium Anatomy 0.000 description 4
- 108020004445 glyceraldehyde-3-phosphate dehydrogenase Proteins 0.000 description 4
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 4
- 238000002493 microarray Methods 0.000 description 4
- 230000037361 pathway Effects 0.000 description 4
- 102000005962 receptors Human genes 0.000 description 4
- 108020003175 receptors Proteins 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 239000012581 transferrin Substances 0.000 description 4
- 229940088594 vitamin Drugs 0.000 description 4
- 229930003231 vitamin Natural products 0.000 description 4
- 235000013343 vitamin Nutrition 0.000 description 4
- 239000011782 vitamin Substances 0.000 description 4
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 3
- 102100034134 Activin receptor type-1B Human genes 0.000 description 3
- 102100034135 Activin receptor type-1C Human genes 0.000 description 3
- 108010059616 Activins Proteins 0.000 description 3
- HJCMDXDYPOUFDY-WHFBIAKZSA-N Ala-Gln Chemical compound C[C@H](N)C(=O)N[C@H](C(O)=O)CCC(N)=O HJCMDXDYPOUFDY-WHFBIAKZSA-N 0.000 description 3
- 102100022983 B-cell lymphoma/leukemia 11B Human genes 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 238000002965 ELISA Methods 0.000 description 3
- 108010001498 Galectin 1 Proteins 0.000 description 3
- 102100021736 Galectin-1 Human genes 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- 101150092640 HES1 gene Proteins 0.000 description 3
- 239000012981 Hank's balanced salt solution Substances 0.000 description 3
- 102100022373 Homeobox protein DLX-5 Human genes 0.000 description 3
- 102100035349 Homeobox protein DLX-6 Human genes 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 101000799189 Homo sapiens Activin receptor type-1B Proteins 0.000 description 3
- 101000799193 Homo sapiens Activin receptor type-1C Proteins 0.000 description 3
- 101000903697 Homo sapiens B-cell lymphoma/leukemia 11B Proteins 0.000 description 3
- 101000901627 Homo sapiens Homeobox protein DLX-5 Proteins 0.000 description 3
- 101000804582 Homo sapiens Homeobox protein DLX-6 Proteins 0.000 description 3
- 101001139134 Homo sapiens Krueppel-like factor 4 Proteins 0.000 description 3
- 101000979001 Homo sapiens Methionine aminopeptidase 2 Proteins 0.000 description 3
- 101000969087 Homo sapiens Microtubule-associated protein 2 Proteins 0.000 description 3
- 101000713275 Homo sapiens Solute carrier family 22 member 3 Proteins 0.000 description 3
- 101000800571 Homo sapiens T-box transcription factor T Proteins 0.000 description 3
- 101000757378 Homo sapiens Transcription factor AP-2-alpha Proteins 0.000 description 3
- 101000976653 Homo sapiens Zinc finger protein ZIC 1 Proteins 0.000 description 3
- ZGSXEXBYLJIOGF-ALFLXDJESA-N IWR-1-endo Chemical compound C=1C=CC2=CC=CN=C2C=1NC(=O)C(C=C1)=CC=C1N1C(=O)[C@@H]2[C@H](C=C3)C[C@H]3[C@@H]2C1=O ZGSXEXBYLJIOGF-ALFLXDJESA-N 0.000 description 3
- 102100026818 Inhibin beta E chain Human genes 0.000 description 3
- 108090001061 Insulin Proteins 0.000 description 3
- 102000004877 Insulin Human genes 0.000 description 3
- 102100020677 Krueppel-like factor 4 Human genes 0.000 description 3
- MIJPAVRNWPDMOR-ZAFYKAAXSA-N L-ascorbic acid 2-phosphate Chemical compound OC[C@H](O)[C@H]1OC(=O)C(OP(O)(O)=O)=C1O MIJPAVRNWPDMOR-ZAFYKAAXSA-N 0.000 description 3
- XXYGTCZJJLTAGH-UHFFFAOYSA-N LGK974 Chemical compound C1=NC(C)=CC(C=2C(=CC(CC(=O)NC=3N=CC(=CC=3)C=3N=CC=NC=3)=CN=2)C)=C1 XXYGTCZJJLTAGH-UHFFFAOYSA-N 0.000 description 3
- 102100023174 Methionine aminopeptidase 2 Human genes 0.000 description 3
- 102000015336 Nerve Growth Factor Human genes 0.000 description 3
- 102100037369 Nidogen-1 Human genes 0.000 description 3
- 101710126211 POU domain, class 5, transcription factor 1 Proteins 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 101100247004 Rattus norvegicus Qsox1 gene Proteins 0.000 description 3
- 102100033130 T-box transcription factor T Human genes 0.000 description 3
- 108091005735 TGF-beta receptors Proteins 0.000 description 3
- 102100022972 Transcription factor AP-2-alpha Human genes 0.000 description 3
- 102100030248 Transcription factor SOX-1 Human genes 0.000 description 3
- 102000016715 Transforming Growth Factor beta Receptors Human genes 0.000 description 3
- 101710107964 Vesicular glutamate transporter 1 Proteins 0.000 description 3
- 239000000488 activin Substances 0.000 description 3
- 230000002411 adverse Effects 0.000 description 3
- 230000000735 allogeneic effect Effects 0.000 description 3
- APKFDSVGJQXUKY-INPOYWNPSA-N amphotericin B Chemical compound O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1/C=C/C=C/C=C/C=C/C=C/C=C/C=C/[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-INPOYWNPSA-N 0.000 description 3
- 239000004637 bakelite Substances 0.000 description 3
- 210000002469 basement membrane Anatomy 0.000 description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 3
- 239000007853 buffer solution Substances 0.000 description 3
- 239000003638 chemical reducing agent Substances 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- 238000010494 dissociation reaction Methods 0.000 description 3
- 230000005593 dissociations Effects 0.000 description 3
- 230000002255 enzymatic effect Effects 0.000 description 3
- 230000001747 exhibiting effect Effects 0.000 description 3
- 238000007667 floating Methods 0.000 description 3
- 230000003371 gabaergic effect Effects 0.000 description 3
- 239000008103 glucose Substances 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- 238000002513 implantation Methods 0.000 description 3
- 230000001976 improved effect Effects 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 229940125396 insulin Drugs 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 230000035800 maturation Effects 0.000 description 3
- 238000010208 microarray analysis Methods 0.000 description 3
- 208000022084 motor paralysis Diseases 0.000 description 3
- 229940053128 nerve growth factor Drugs 0.000 description 3
- 210000003061 neural cell Anatomy 0.000 description 3
- 108010008217 nidogen Proteins 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 238000000513 principal component analysis Methods 0.000 description 3
- 229940107700 pyruvic acid Drugs 0.000 description 3
- 238000013441 quality evaluation Methods 0.000 description 3
- 210000003124 radial glial cell Anatomy 0.000 description 3
- 235000000346 sugar Nutrition 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 230000001988 toxicity Effects 0.000 description 3
- 231100000419 toxicity Toxicity 0.000 description 3
- 230000000007 visual effect Effects 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- GDVRVPIXWXOKQO-UHFFFAOYSA-N 1-[(3-hydroxyphenyl)methyl]-3-(4-pyridin-4-yl-1,3-thiazol-2-yl)urea Chemical compound OC1=CC=CC(CNC(=O)NC=2SC=C(N=2)C=2C=CN=CC=2)=C1 GDVRVPIXWXOKQO-UHFFFAOYSA-N 0.000 description 2
- BJHCYTJNPVGSBZ-YXSASFKJSA-N 1-[4-[6-amino-5-[(Z)-methoxyiminomethyl]pyrimidin-4-yl]oxy-2-chlorophenyl]-3-ethylurea Chemical compound CCNC(=O)Nc1ccc(Oc2ncnc(N)c2\C=N/OC)cc1Cl BJHCYTJNPVGSBZ-YXSASFKJSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- PMYDPQQPEAYXKD-UHFFFAOYSA-N 3-hydroxy-n-naphthalen-2-ylnaphthalene-2-carboxamide Chemical compound C1=CC=CC2=CC(NC(=O)C3=CC4=CC=CC=C4C=C3O)=CC=C21 PMYDPQQPEAYXKD-UHFFFAOYSA-N 0.000 description 2
- JTVBXQAYBIJXRP-SNVBAGLBSA-N 4-[(1R)-1-aminoethyl]-N-(1H-pyrrolo[2,3-b]pyridin-4-yl)benzamide Chemical compound C1=CC([C@H](N)C)=CC=C1C(=O)NC1=CC=NC2=C1C=CN2 JTVBXQAYBIJXRP-SNVBAGLBSA-N 0.000 description 2
- LLSFXOUAPAZFIV-SNVBAGLBSA-N 4-[(1r)-1-aminoethyl]-n-pyridin-4-ylbenzamide Chemical compound C1=CC([C@H](N)C)=CC=C1C(=O)NC1=CC=NC=C1 LLSFXOUAPAZFIV-SNVBAGLBSA-N 0.000 description 2
- UZOVYGYOLBIAJR-UHFFFAOYSA-N 4-isocyanato-4'-methyldiphenylmethane Chemical compound C1=CC(C)=CC=C1CC1=CC=C(N=C=O)C=C1 UZOVYGYOLBIAJR-UHFFFAOYSA-N 0.000 description 2
- LCGTWRLJTMHIQZ-UHFFFAOYSA-N 5H-dibenzo[b,f]azepine Chemical compound C1=CC2=CC=CC=C2NC2=CC=CC=C21 LCGTWRLJTMHIQZ-UHFFFAOYSA-N 0.000 description 2
- WHIWGRCYMQLLAO-UHFFFAOYSA-N 6-(4-tert-butylphenoxy)pyridin-3-amine Chemical compound C1=CC(C(C)(C)C)=CC=C1OC1=CC=C(N)C=N1 WHIWGRCYMQLLAO-UHFFFAOYSA-N 0.000 description 2
- NRSGWEVTVGZDFC-UHFFFAOYSA-N 6-chloro-4-n-[3,5-difluoro-4-[(3-methyl-1h-pyrrolo[2,3-b]pyridin-4-yl)oxy]phenyl]pyrimidine-2,4-diamine Chemical compound C=12C(C)=CNC2=NC=CC=1OC(C(=C1)F)=C(F)C=C1NC1=CC(Cl)=NC(N)=N1 NRSGWEVTVGZDFC-UHFFFAOYSA-N 0.000 description 2
- 102100022142 Achaete-scute homolog 1 Human genes 0.000 description 2
- APKFDSVGJQXUKY-KKGHZKTASA-N Amphotericin-B Natural products O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1C=CC=CC=CC=CC=CC=CC=C[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-KKGHZKTASA-N 0.000 description 2
- 102000004888 Aquaporin 1 Human genes 0.000 description 2
- 108090001004 Aquaporin 1 Proteins 0.000 description 2
- 239000012583 B-27 Supplement Substances 0.000 description 2
- 108060000903 Beta-catenin Proteins 0.000 description 2
- 102000015735 Beta-catenin Human genes 0.000 description 2
- 102000004506 Blood Proteins Human genes 0.000 description 2
- 108010017384 Blood Proteins Proteins 0.000 description 2
- 241000282693 Cercopithecidae Species 0.000 description 2
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 2
- 108010005939 Ciliary Neurotrophic Factor Proteins 0.000 description 2
- 102100031614 Ciliary neurotrophic factor Human genes 0.000 description 2
- 102000008186 Collagen Human genes 0.000 description 2
- 108010035532 Collagen Proteins 0.000 description 2
- 102000004266 Collagen Type IV Human genes 0.000 description 2
- 108010042086 Collagen Type IV Proteins 0.000 description 2
- 102000004127 Cytokines Human genes 0.000 description 2
- 108090000695 Cytokines Proteins 0.000 description 2
- 102100032883 DNA-binding protein SATB2 Human genes 0.000 description 2
- 102100030074 Dickkopf-related protein 1 Human genes 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- OHCQJHSOBUTRHG-KGGHGJDLSA-N FORSKOLIN Chemical compound O=C([C@@]12O)C[C@](C)(C=C)O[C@]1(C)[C@@H](OC(=O)C)[C@@H](O)[C@@H]1[C@]2(C)[C@@H](O)CCC1(C)C OHCQJHSOBUTRHG-KGGHGJDLSA-N 0.000 description 2
- 102000018233 Fibroblast Growth Factor Human genes 0.000 description 2
- 108050007372 Fibroblast Growth Factor Proteins 0.000 description 2
- 229930182566 Gentamicin Natural products 0.000 description 2
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 2
- 239000012571 GlutaMAX medium Substances 0.000 description 2
- 239000007995 HEPES buffer Substances 0.000 description 2
- 102000008055 Heparan Sulfate Proteoglycans Human genes 0.000 description 2
- 229920002971 Heparan sulfate Polymers 0.000 description 2
- 102100033636 Histone H3.2 Human genes 0.000 description 2
- 108010033040 Histones Proteins 0.000 description 2
- 108700005087 Homeobox Genes Proteins 0.000 description 2
- 102100023830 Homeobox protein EMX2 Human genes 0.000 description 2
- 102100025110 Homeobox protein Hox-A5 Human genes 0.000 description 2
- 102100029240 Homeobox protein Hox-B5 Human genes 0.000 description 2
- 101000901099 Homo sapiens Achaete-scute homolog 1 Proteins 0.000 description 2
- 101000655236 Homo sapiens DNA-binding protein SATB2 Proteins 0.000 description 2
- 101001048970 Homo sapiens Homeobox protein EMX2 Proteins 0.000 description 2
- 101001077568 Homo sapiens Homeobox protein Hox-A5 Proteins 0.000 description 2
- 101000840553 Homo sapiens Homeobox protein Hox-B5 Proteins 0.000 description 2
- 101000599858 Homo sapiens Intercellular adhesion molecule 2 Proteins 0.000 description 2
- 101001020544 Homo sapiens LIM/homeobox protein Lhx2 Proteins 0.000 description 2
- 101001030211 Homo sapiens Myc proto-oncogene protein Proteins 0.000 description 2
- 101000984042 Homo sapiens Protein lin-28 homolog A Proteins 0.000 description 2
- 101000664703 Homo sapiens Transcription factor SOX-10 Proteins 0.000 description 2
- 101000652324 Homo sapiens Transcription factor SOX-17 Proteins 0.000 description 2
- 101000851018 Homo sapiens Vascular endothelial growth factor receptor 1 Proteins 0.000 description 2
- 102100037872 Intercellular adhesion molecule 2 Human genes 0.000 description 2
- 102100036132 LIM/homeobox protein Lhx2 Human genes 0.000 description 2
- 101150009249 MAP2 gene Proteins 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 102100038895 Myc proto-oncogene protein Human genes 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- 102000004230 Neurotrophin 3 Human genes 0.000 description 2
- 108090000742 Neurotrophin 3 Proteins 0.000 description 2
- 108090000099 Neurotrophin-4 Proteins 0.000 description 2
- 102100033857 Neurotrophin-4 Human genes 0.000 description 2
- 108090000095 Neurotrophin-6 Proteins 0.000 description 2
- 240000007594 Oryza sativa Species 0.000 description 2
- 235000007164 Oryza sativa Nutrition 0.000 description 2
- 229930040373 Paraformaldehyde Natural products 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- RJKFOVLPORLFTN-LEKSSAKUSA-N Progesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RJKFOVLPORLFTN-LEKSSAKUSA-N 0.000 description 2
- 102100025460 Protein lin-28 homolog A Human genes 0.000 description 2
- 101150057388 Reln gene Proteins 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- 238000012300 Sequence Analysis Methods 0.000 description 2
- 108090000054 Syndecan-2 Proteins 0.000 description 2
- 102000003620 TRPM3 Human genes 0.000 description 2
- 108060008547 TRPM3 Proteins 0.000 description 2
- 102100038808 Transcription factor SOX-10 Human genes 0.000 description 2
- 102100030243 Transcription factor SOX-17 Human genes 0.000 description 2
- 102100033178 Vascular endothelial growth factor receptor 1 Human genes 0.000 description 2
- 235000010724 Wisteria floribunda Nutrition 0.000 description 2
- 102100036973 X-ray repair cross-complementing protein 5 Human genes 0.000 description 2
- 101710124921 X-ray repair cross-complementing protein 5 Proteins 0.000 description 2
- 101000928515 Xenopus laevis Homeobox protein DLL-1 Proteins 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 230000004931 aggregating effect Effects 0.000 description 2
- 238000013019 agitation Methods 0.000 description 2
- 229960003942 amphotericin b Drugs 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 239000003125 aqueous solvent Substances 0.000 description 2
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 2
- 210000003050 axon Anatomy 0.000 description 2
- 230000003376 axonal effect Effects 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 230000009992 cAMP activation Effects 0.000 description 2
- 108010007877 calpain inhibitor III Proteins 0.000 description 2
- 230000030833 cell death Effects 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 229910052804 chromium Inorganic materials 0.000 description 2
- 239000011651 chromium Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 229920001436 collagen Polymers 0.000 description 2
- 238000011109 contamination Methods 0.000 description 2
- 238000005138 cryopreservation Methods 0.000 description 2
- 229940068840 d-biotin Drugs 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 210000001900 endoderm Anatomy 0.000 description 2
- 229960002435 fasudil Drugs 0.000 description 2
- LFVPBERIVUNMGV-UHFFFAOYSA-N fasudil hydrochloride Chemical compound Cl.C=1C=CC2=CN=CC=C2C=1S(=O)(=O)N1CCCNCC1 LFVPBERIVUNMGV-UHFFFAOYSA-N 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- 229940126864 fibroblast growth factor Drugs 0.000 description 2
- 238000010304 firing Methods 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 229960002518 gentamicin Drugs 0.000 description 2
- 210000004602 germ cell Anatomy 0.000 description 2
- 210000001654 germ layer Anatomy 0.000 description 2
- 230000000848 glutamatergic effect Effects 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- ZAVGJDAFCZAWSZ-UHFFFAOYSA-N hydroxyfasudil Chemical compound C1=CC=C2C(O)=NC=CC2=C1S(=O)(=O)N1CCCNCC1 ZAVGJDAFCZAWSZ-UHFFFAOYSA-N 0.000 description 2
- 238000010191 image analysis Methods 0.000 description 2
- 239000007943 implant Substances 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 229910052500 inorganic mineral Inorganic materials 0.000 description 2
- 229910052742 iron Inorganic materials 0.000 description 2
- 239000004310 lactic acid Substances 0.000 description 2
- 235000014655 lactic acid Nutrition 0.000 description 2
- 108010038862 laminin 10 Proteins 0.000 description 2
- 210000002752 melanocyte Anatomy 0.000 description 2
- 210000002901 mesenchymal stem cell Anatomy 0.000 description 2
- 210000003716 mesoderm Anatomy 0.000 description 2
- 235000010755 mineral Nutrition 0.000 description 2
- 239000011707 mineral Substances 0.000 description 2
- 230000000394 mitotic effect Effects 0.000 description 2
- 230000007659 motor function Effects 0.000 description 2
- 210000002161 motor neuron Anatomy 0.000 description 2
- 210000002894 multi-fate stem cell Anatomy 0.000 description 2
- 210000001020 neural plate Anatomy 0.000 description 2
- 230000004031 neuronal differentiation Effects 0.000 description 2
- 229940032018 neurotrophin 3 Drugs 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- SECPZKHBENQXJG-FPLPWBNLSA-N palmitoleic acid Chemical compound CCCCCC\C=C/CCCCCCCC(O)=O SECPZKHBENQXJG-FPLPWBNLSA-N 0.000 description 2
- 229920002866 paraformaldehyde Polymers 0.000 description 2
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 2
- 239000008363 phosphate buffer Substances 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- 230000001766 physiological effect Effects 0.000 description 2
- 239000002504 physiological saline solution Substances 0.000 description 2
- 102000040430 polynucleotide Human genes 0.000 description 2
- 108091033319 polynucleotide Proteins 0.000 description 2
- 239000002157 polynucleotide Substances 0.000 description 2
- KIDHWZJUCRJVML-UHFFFAOYSA-N putrescine Chemical compound NCCCCN KIDHWZJUCRJVML-UHFFFAOYSA-N 0.000 description 2
- 230000007261 regionalization Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 235000009566 rice Nutrition 0.000 description 2
- QSKQVZWVLOIIEV-NSHDSACASA-N ripasudil Chemical compound C[C@H]1CNCCCN1S(=O)(=O)C1=CC=CC2=CN=CC(F)=C12 QSKQVZWVLOIIEV-NSHDSACASA-N 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 238000005070 sampling Methods 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- 239000011655 sodium selenate Substances 0.000 description 2
- 235000018716 sodium selenate Nutrition 0.000 description 2
- 229960001881 sodium selenate Drugs 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 238000010186 staining Methods 0.000 description 2
- 238000013513 substance screening Methods 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 231100000820 toxicity test Toxicity 0.000 description 2
- 238000013518 transcription Methods 0.000 description 2
- 230000035897 transcription Effects 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- 210000002444 unipotent stem cell Anatomy 0.000 description 2
- 238000001262 western blot Methods 0.000 description 2
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 1
- HQWTUOLCGKIECB-XZWHSSHBSA-N (6S,9aS)-6-[(4-hydroxyphenyl)methyl]-8-(1-naphthalenylmethyl)-4,7-dioxo-N-(phenylmethyl)-3,6,9,9a-tetrahydro-2H-pyrazino[1,2-a]pyrimidine-1-carboxamide Chemical compound C1=CC(O)=CC=C1C[C@H]1C(=O)N(CC=2C3=CC=CC=C3C=CC=2)C[C@H]2N1C(=O)CCN2C(=O)NCC1=CC=CC=C1 HQWTUOLCGKIECB-XZWHSSHBSA-N 0.000 description 1
- OYHQOLUKZRVURQ-NTGFUMLPSA-N (9Z,12Z)-9,10,12,13-tetratritiooctadeca-9,12-dienoic acid Chemical compound C(CCCCCCC\C(=C(/C\C(=C(/CCCCC)\[3H])\[3H])\[3H])\[3H])(=O)O OYHQOLUKZRVURQ-NTGFUMLPSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- BFOPDSJOLUQULZ-GXKRWWSZSA-N (S)-2-methyl-1-(4-methylisoquinoline-5-sulfonyl)-1,4-diazepane dihydrochloride Chemical compound Cl.Cl.C[C@H]1CNCCCN1S(=O)(=O)C1=CC=CC2=CN=CC(C)=C12 BFOPDSJOLUQULZ-GXKRWWSZSA-N 0.000 description 1
- LBPKYPYHDKKRFS-UHFFFAOYSA-N 1,5-naphthyridine, 2-[3-(6-methyl-2-pyridinyl)-1h-pyrazol-4-yl]- Chemical compound CC1=CC=CC(C2=C(C=NN2)C=2N=C3C=CC=NC3=CC=2)=N1 LBPKYPYHDKKRFS-UHFFFAOYSA-N 0.000 description 1
- 108020004463 18S ribosomal RNA Proteins 0.000 description 1
- ZSZRUEAFVQITHH-UHFFFAOYSA-N 2-(2-methylprop-2-enoyloxy)ethyl 2-(trimethylazaniumyl)ethyl phosphate Chemical compound CC(=C)C(=O)OCCOP([O-])(=O)OCC[N+](C)(C)C ZSZRUEAFVQITHH-UHFFFAOYSA-N 0.000 description 1
- QESQGTFWEQMCMH-UHFFFAOYSA-N 2-[(4-oxo-3-phenyl-6,7-dihydrothieno[3,2-d]pyrimidin-2-yl)sulfanyl]-n-(5-phenylpyridin-2-yl)acetamide Chemical compound C=1C=C(C=2C=CC=CC=2)C=NC=1NC(=O)CSC1=NC=2CCSC=2C(=O)N1C1=CC=CC=C1 QESQGTFWEQMCMH-UHFFFAOYSA-N 0.000 description 1
- DVBJNSKWHSGTDK-YHYXMXQVSA-N 2-[2-methoxy-4-[(z)-(4-oxo-2-sulfanylidene-1,3-thiazolidin-5-ylidene)methyl]phenoxy]acetic acid Chemical compound C1=C(OCC(O)=O)C(OC)=CC(\C=C/2C(NC(=S)S\2)=O)=C1 DVBJNSKWHSGTDK-YHYXMXQVSA-N 0.000 description 1
- RNUXIZKXJOGYQP-UHFFFAOYSA-N 2-[3-[[4-(4-methoxyphenyl)-5-pyridin-4-yl-1,2,4-triazol-3-yl]sulfanyl]propyl]benzo[de]isoquinoline-1,3-dione Chemical compound C1=CC(OC)=CC=C1N1C(C=2C=CN=CC=2)=NN=C1SCCCN(C1=O)C(=O)C2=C3C1=CC=CC3=CC=C2 RNUXIZKXJOGYQP-UHFFFAOYSA-N 0.000 description 1
- PNPFMWIDAKQFPY-UHFFFAOYSA-N 2-[4-[[4-[1-cyclopropyl-3-(oxan-4-yl)pyrazol-4-yl]oxypyridin-2-yl]amino]pyridin-2-yl]propan-2-ol Chemical compound C1(CC1)N1N=C(C(=C1)OC1=CC(=NC=C1)NC1=CC(=NC=C1)C(C)(C)O)C1CCOCC1 PNPFMWIDAKQFPY-UHFFFAOYSA-N 0.000 description 1
- RHUJMHOIQBDFQR-UHFFFAOYSA-N 2-[[3-(2-methoxyphenyl)-4-oxo-6,7-dihydrothieno[3,2-d]pyrimidin-2-yl]sulfanyl]-n-(6-methyl-1,3-benzothiazol-2-yl)acetamide Chemical compound COC1=CC=CC=C1N1C(=O)C(SCC2)=C2N=C1SCC(=O)NC1=NC2=CC=C(C)C=C2S1 RHUJMHOIQBDFQR-UHFFFAOYSA-N 0.000 description 1
- XVMHQSDMKWQNBK-UHFFFAOYSA-N 2-[[3-(4-fluorophenyl)-4-oxo-6,7-dihydrothieno[3,2-d]pyrimidin-2-yl]sulfanyl]-n-(6-methyl-1,3-benzothiazol-2-yl)acetamide Chemical compound S1C2=CC(C)=CC=C2N=C1NC(=O)CSC1=NC=2CCSC=2C(=O)N1C1=CC=C(F)C=C1 XVMHQSDMKWQNBK-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 1
- IDDDVXIUIXWAGJ-DDSAHXNVSA-N 4-[(1r)-1-aminoethyl]-n-pyridin-4-ylcyclohexane-1-carboxamide;dihydrochloride Chemical compound Cl.Cl.C1CC([C@H](N)C)CCC1C(=O)NC1=CC=NC=C1 IDDDVXIUIXWAGJ-DDSAHXNVSA-N 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- 102100024959 5-hydroxytryptamine receptor 2C Human genes 0.000 description 1
- DKPQHFZUICCZHF-UHFFFAOYSA-N 6-[2-tert-butyl-5-(6-methyl-2-pyridinyl)-1H-imidazol-4-yl]quinoxaline Chemical compound CC1=CC=CC(C2=C(N=C(N2)C(C)(C)C)C=2C=C3N=CC=NC3=CC=2)=N1 DKPQHFZUICCZHF-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 1
- 108091023037 Aptamer Proteins 0.000 description 1
- 208000020925 Bipolar disease Diseases 0.000 description 1
- 102100024505 Bone morphogenetic protein 4 Human genes 0.000 description 1
- 102100022544 Bone morphogenetic protein 7 Human genes 0.000 description 1
- 108091016585 CD44 antigen Proteins 0.000 description 1
- 101150008656 COL1A1 gene Proteins 0.000 description 1
- 102100029761 Cadherin-5 Human genes 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 102000005403 Casein Kinases Human genes 0.000 description 1
- 108010031425 Casein Kinases Proteins 0.000 description 1
- 102100025745 Cerberus Human genes 0.000 description 1
- 101710010675 Cerberus Proteins 0.000 description 1
- 241000202252 Cerberus Species 0.000 description 1
- 206010008190 Cerebrovascular accident Diseases 0.000 description 1
- 108091006146 Channels Proteins 0.000 description 1
- 102100023506 Chloride intracellular channel protein 6 Human genes 0.000 description 1
- 241000725101 Clea Species 0.000 description 1
- 108060005980 Collagenase Proteins 0.000 description 1
- 102000029816 Collagenase Human genes 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- 235000001815 DL-alpha-tocopherol Nutrition 0.000 description 1
- 239000011627 DL-alpha-tocopherol Substances 0.000 description 1
- 108020001019 DNA Primers Proteins 0.000 description 1
- 239000003155 DNA primer Substances 0.000 description 1
- 102100033587 DNA topoisomerase 2-alpha Human genes 0.000 description 1
- 206010012289 Dementia Diseases 0.000 description 1
- SUZLHDUTVMZSEV-UHFFFAOYSA-N Deoxycoleonol Natural products C12C(=O)CC(C)(C=C)OC2(C)C(OC(=O)C)C(O)C2C1(C)C(O)CCC2(C)C SUZLHDUTVMZSEV-UHFFFAOYSA-N 0.000 description 1
- 102000007260 Deoxyribonuclease I Human genes 0.000 description 1
- 108010008532 Deoxyribonuclease I Proteins 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 101710099518 Dickkopf-related protein 1 Proteins 0.000 description 1
- 102100030751 Eomesodermin homolog Human genes 0.000 description 1
- 108700039887 Essential Genes Proteins 0.000 description 1
- 208000009331 Experimental Sarcoma Diseases 0.000 description 1
- 102100030456 Follistatin-related protein 4 Human genes 0.000 description 1
- 102000027587 GPCRs class F Human genes 0.000 description 1
- 108091008884 GPCRs class F Proteins 0.000 description 1
- 101000625722 Gallus gallus Tubulin beta-4 chain Proteins 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 108090000031 Hedgehog Proteins Proteins 0.000 description 1
- 102000003693 Hedgehog Proteins Human genes 0.000 description 1
- 102100030087 Homeobox protein DLX-1 Human genes 0.000 description 1
- 102100028707 Homeobox protein MSX-1 Human genes 0.000 description 1
- 102100030634 Homeobox protein OTX2 Human genes 0.000 description 1
- 102100027345 Homeobox protein SIX3 Human genes 0.000 description 1
- 101000761348 Homo sapiens 5-hydroxytryptamine receptor 2C Proteins 0.000 description 1
- 101000762379 Homo sapiens Bone morphogenetic protein 4 Proteins 0.000 description 1
- 101000899361 Homo sapiens Bone morphogenetic protein 7 Proteins 0.000 description 1
- 101000794587 Homo sapiens Cadherin-5 Proteins 0.000 description 1
- 101000906631 Homo sapiens Chloride intracellular channel protein 6 Proteins 0.000 description 1
- 101000864646 Homo sapiens Dickkopf-related protein 1 Proteins 0.000 description 1
- 101001064167 Homo sapiens Eomesodermin homolog Proteins 0.000 description 1
- 101001062597 Homo sapiens Follistatin-related protein 4 Proteins 0.000 description 1
- 101001002170 Homo sapiens Glutamine amidotransferase-like class 1 domain-containing protein 3, mitochondrial Proteins 0.000 description 1
- 101000864690 Homo sapiens Homeobox protein DLX-1 Proteins 0.000 description 1
- 101000985653 Homo sapiens Homeobox protein MSX-1 Proteins 0.000 description 1
- 101000584400 Homo sapiens Homeobox protein OTX2 Proteins 0.000 description 1
- 101000651928 Homo sapiens Homeobox protein SIX3 Proteins 0.000 description 1
- 101000998011 Homo sapiens Keratin, type I cytoskeletal 19 Proteins 0.000 description 1
- 101000975496 Homo sapiens Keratin, type II cytoskeletal 8 Proteins 0.000 description 1
- 101000831616 Homo sapiens Protachykinin-1 Proteins 0.000 description 1
- 101000928535 Homo sapiens Protein delta homolog 1 Proteins 0.000 description 1
- 101001123986 Homo sapiens Protein-serine O-palmitoleoyltransferase porcupine Proteins 0.000 description 1
- 101000825960 Homo sapiens R-spondin-3 Proteins 0.000 description 1
- 101000740178 Homo sapiens Sal-like protein 4 Proteins 0.000 description 1
- 101000911790 Homo sapiens Sister chromatid cohesion protein DCC1 Proteins 0.000 description 1
- 101000829138 Homo sapiens Somatostatin receptor type 3 Proteins 0.000 description 1
- 101000851696 Homo sapiens Steroid hormone receptor ERR2 Proteins 0.000 description 1
- 101000909641 Homo sapiens Transcription factor COE2 Proteins 0.000 description 1
- 101000843556 Homo sapiens Transcription factor HES-1 Proteins 0.000 description 1
- 101000635938 Homo sapiens Transforming growth factor beta-1 proprotein Proteins 0.000 description 1
- 101000857270 Homo sapiens Zinc finger protein GLIS1 Proteins 0.000 description 1
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 1
- 102000004218 Insulin-Like Growth Factor I Human genes 0.000 description 1
- 102000012411 Intermediate Filament Proteins Human genes 0.000 description 1
- 108010061998 Intermediate Filament Proteins Proteins 0.000 description 1
- 102100033420 Keratin, type I cytoskeletal 19 Human genes 0.000 description 1
- 102100023972 Keratin, type II cytoskeletal 8 Human genes 0.000 description 1
- IVRXNBXKWIJUQB-UHFFFAOYSA-N LY-2157299 Chemical compound CC1=CC=CC(C=2C(=C3CCCN3N=2)C=2C3=CC(=CC=C3N=CC=2)C(N)=O)=N1 IVRXNBXKWIJUQB-UHFFFAOYSA-N 0.000 description 1
- 101150107475 MEF2C gene Proteins 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 108010006035 Metalloproteases Proteins 0.000 description 1
- 102000005741 Metalloproteases Human genes 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- 101100445099 Mus musculus Emx1 gene Proteins 0.000 description 1
- 101100012007 Mus musculus Etv1 gene Proteins 0.000 description 1
- OLIIUAHHAZEXEX-UHFFFAOYSA-N N-(6-fluoro-1H-indazol-5-yl)-6-methyl-2-oxo-4-[4-(trifluoromethyl)phenyl]-3,4-dihydro-1H-pyridine-5-carboxamide Chemical compound C1C(=O)NC(C)=C(C(=O)NC=2C(=CC=3NN=CC=3C=2)F)C1C1=CC=C(C(F)(F)F)C=C1 OLIIUAHHAZEXEX-UHFFFAOYSA-N 0.000 description 1
- WRKPZSMRWPJJDH-UHFFFAOYSA-N N-(6-methyl-1,3-benzothiazol-2-yl)-2-[(4-oxo-3-phenyl-6,7-dihydrothieno[3,2-d]pyrimidin-2-yl)thio]acetamide Chemical compound S1C2=CC(C)=CC=C2N=C1NC(=O)CSC1=NC=2CCSC=2C(=O)N1C1=CC=CC=C1 WRKPZSMRWPJJDH-UHFFFAOYSA-N 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 108010088225 Nestin Proteins 0.000 description 1
- 102000008730 Nestin Human genes 0.000 description 1
- 102000003683 Neurotrophin-4 Human genes 0.000 description 1
- 108091005461 Nucleic proteins Proteins 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 235000021319 Palmitoleic acid Nutrition 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 206010057249 Phagocytosis Diseases 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 108010039918 Polylysine Proteins 0.000 description 1
- 102100024304 Protachykinin-1 Human genes 0.000 description 1
- 102100036467 Protein delta homolog 1 Human genes 0.000 description 1
- 102100028119 Protein-serine O-palmitoleoyltransferase porcupine Human genes 0.000 description 1
- 239000005700 Putrescine Substances 0.000 description 1
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 description 1
- 102100022766 R-spondin-3 Human genes 0.000 description 1
- 101150105130 RORB gene Proteins 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 101150086694 SLC22A3 gene Proteins 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 102100037192 Sal-like protein 4 Human genes 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 102100034201 Sclerostin Human genes 0.000 description 1
- 108050006698 Sclerostin Proteins 0.000 description 1
- 108091058545 Secretory proteins Proteins 0.000 description 1
- 102000040739 Secretory proteins Human genes 0.000 description 1
- 102000005806 Serine Peptidase Inhibitor Kazal-Type 5 Human genes 0.000 description 1
- 108010005020 Serine Peptidase Inhibitor Kazal-Type 5 Proteins 0.000 description 1
- 102100027040 Sister chromatid cohesion protein DCC1 Human genes 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- 108050001286 Somatostatin Receptor Proteins 0.000 description 1
- 102000011096 Somatostatin receptor Human genes 0.000 description 1
- 102100023803 Somatostatin receptor type 3 Human genes 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 102100036831 Steroid hormone receptor ERR2 Human genes 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 102000003978 Tissue Plasminogen Activator Human genes 0.000 description 1
- 108090000373 Tissue Plasminogen Activator Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102100024204 Transcription factor COE2 Human genes 0.000 description 1
- 102100030798 Transcription factor HES-1 Human genes 0.000 description 1
- 102100030742 Transforming growth factor beta-1 proprotein Human genes 0.000 description 1
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 108010046308 Type II DNA Topoisomerases Proteins 0.000 description 1
- 102000008790 VE-cadherin Human genes 0.000 description 1
- 102100035071 Vimentin Human genes 0.000 description 1
- 108010065472 Vimentin Proteins 0.000 description 1
- 108010031318 Vitronectin Proteins 0.000 description 1
- 102100035140 Vitronectin Human genes 0.000 description 1
- 102000006757 Wnt Receptors Human genes 0.000 description 1
- 108010047118 Wnt Receptors Proteins 0.000 description 1
- 102000052549 Wnt-3 Human genes 0.000 description 1
- 108700020985 Wnt-3 Proteins 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- NGBKFLTYGSREKK-PMACEKPBSA-N Z-Val-Phe-H Chemical compound N([C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C=O)C(=O)OCC1=CC=CC=C1 NGBKFLTYGSREKK-PMACEKPBSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 102100025883 Zinc finger protein GLIS1 Human genes 0.000 description 1
- 102100023497 Zinc finger protein ZIC 1 Human genes 0.000 description 1
- SMNRFWMNPDABKZ-WVALLCKVSA-N [[(2R,3S,4R,5S)-5-(2,6-dioxo-3H-pyridin-3-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4S,5R,6R)-4-fluoro-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound OC[C@H]1O[C@H](OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@H](O)[C@@H]2O)C2C=CC(=O)NC2=O)[C@H](O)[C@@H](F)[C@@H]1O SMNRFWMNPDABKZ-WVALLCKVSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 108010076089 accutase Proteins 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 230000004523 agglutinating effect Effects 0.000 description 1
- 230000004520 agglutination Effects 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 1
- DTOSIQBPPRVQHS-PDBXOOCHSA-N alpha-linolenic acid Chemical compound CC\C=C/C\C=C/C\C=C/CCCCCCCC(O)=O DTOSIQBPPRVQHS-PDBXOOCHSA-N 0.000 description 1
- 235000020661 alpha-linolenic acid Nutrition 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 235000021342 arachidonic acid Nutrition 0.000 description 1
- 229940114079 arachidonic acid Drugs 0.000 description 1
- 210000000576 arachnoid Anatomy 0.000 description 1
- 238000013528 artificial neural network Methods 0.000 description 1
- 208000029560 autism spectrum disease Diseases 0.000 description 1
- 239000003855 balanced salt solution Substances 0.000 description 1
- 239000000090 biomarker Substances 0.000 description 1
- 210000003969 blast cell Anatomy 0.000 description 1
- 210000002459 blastocyst Anatomy 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 230000004641 brain development Effects 0.000 description 1
- 230000003925 brain function Effects 0.000 description 1
- DNSISZSEWVHGLH-UHFFFAOYSA-N butanamide Chemical compound CCCC(N)=O DNSISZSEWVHGLH-UHFFFAOYSA-N 0.000 description 1
- 108010018828 cadherin 5 Proteins 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 210000005056 cell body Anatomy 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 210000004720 cerebrum Anatomy 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- SECPZKHBENQXJG-UHFFFAOYSA-N cis-palmitoleic acid Natural products CCCCCCC=CCCCCCCCC(O)=O SECPZKHBENQXJG-UHFFFAOYSA-N 0.000 description 1
- OHCQJHSOBUTRHG-UHFFFAOYSA-N colforsin Natural products OC12C(=O)CC(C)(C=C)OC1(C)C(OC(=O)C)C(O)C1C2(C)C(O)CCC1(C)C OHCQJHSOBUTRHG-UHFFFAOYSA-N 0.000 description 1
- 229960002424 collagenase Drugs 0.000 description 1
- 238000010835 comparative analysis Methods 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 239000002577 cryoprotective agent Substances 0.000 description 1
- 238000012136 culture method Methods 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- GVJHHUAWPYXKBD-UHFFFAOYSA-N d-alpha-tocopherol Natural products OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 238000013135 deep learning Methods 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 210000001787 dendrite Anatomy 0.000 description 1
- 238000000151 deposition Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- BVTBRVFYZUCAKH-UHFFFAOYSA-L disodium selenite Chemical compound [Na+].[Na+].[O-][Se]([O-])=O BVTBRVFYZUCAKH-UHFFFAOYSA-L 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 108010007093 dispase Proteins 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 230000000857 drug effect Effects 0.000 description 1
- 210000003981 ectoderm Anatomy 0.000 description 1
- 235000013601 eggs Nutrition 0.000 description 1
- 210000002242 embryoid body Anatomy 0.000 description 1
- 210000002257 embryonic structure Anatomy 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 210000003038 endothelium Anatomy 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 239000003797 essential amino acid Substances 0.000 description 1
- 235000020776 essential amino acid Nutrition 0.000 description 1
- QHPJWPQRZMBKTG-KPKJPENVSA-N ethyl 2-[2-methoxy-4-[(e)-(4-oxo-2-sulfanylidene-1,3-thiazolidin-5-ylidene)methyl]phenoxy]acetate Chemical compound C1=C(OC)C(OCC(=O)OCC)=CC=C1\C=C\1C(=O)NC(=S)S/1 QHPJWPQRZMBKTG-KPKJPENVSA-N 0.000 description 1
- 230000002964 excitative effect Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- NGOGFTYYXHNFQH-UHFFFAOYSA-N fasudil Chemical compound C=1C=CC2=CN=CC=C2C=1S(=O)(=O)N1CCCNCC1 NGOGFTYYXHNFQH-UHFFFAOYSA-N 0.000 description 1
- 230000004720 fertilization Effects 0.000 description 1
- 210000004700 fetal blood Anatomy 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 1
- 101150022222 foxp1 gene Proteins 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000011990 functional testing Methods 0.000 description 1
- 230000005251 gamma ray Effects 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 108091008053 gene clusters Proteins 0.000 description 1
- 210000004907 gland Anatomy 0.000 description 1
- 230000000285 glutaminergic effect Effects 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000005484 gravity Effects 0.000 description 1
- 239000003966 growth inhibitor Substances 0.000 description 1
- 238000012835 hanging drop method Methods 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 238000002649 immunization Methods 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- ZGSXEXBYLJIOGF-UHFFFAOYSA-N iwr-1-endo Chemical compound C=1C=CC2=CC=CN=C2C=1NC(=O)C(C=C1)=CC=C1N1C(=O)C2C(C=C3)CC3C2C1=O ZGSXEXBYLJIOGF-UHFFFAOYSA-N 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 229960004488 linolenic acid Drugs 0.000 description 1
- KQQKGWQCNNTQJW-UHFFFAOYSA-N linolenic acid Natural products CC=CCCC=CCC=CCCCCCCCC(O)=O KQQKGWQCNNTQJW-UHFFFAOYSA-N 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 238000000691 measurement method Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 208000004141 microcephaly Diseases 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 238000000465 moulding Methods 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 229940028444 muse Drugs 0.000 description 1
- SAGZIBJAQGBRQA-UHFFFAOYSA-N n-(oxan-4-yl)-4-[4-(5-pyridin-2-yl-1h-pyrazol-4-yl)pyridin-2-yl]benzamide Chemical compound C=1C=C(C=2N=CC=C(C=2)C2=C(NN=C2)C=2N=CC=CC=2)C=CC=1C(=O)NC1CCOCC1 SAGZIBJAQGBRQA-UHFFFAOYSA-N 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- JJEDWBQZCRESJL-DEDYPNTBSA-N n-[(e)-(5-methylfuran-2-yl)methylideneamino]-2-phenoxybenzamide Chemical compound O1C(C)=CC=C1\C=N\NC(=O)C1=CC=CC=C1OC1=CC=CC=C1 JJEDWBQZCRESJL-DEDYPNTBSA-N 0.000 description 1
- DOBKQCZBPPCLEG-UHFFFAOYSA-N n-benzyl-2-(pyrimidin-4-ylamino)-1,3-thiazole-4-carboxamide Chemical compound C=1SC(NC=2N=CN=CC=2)=NC=1C(=O)NCC1=CC=CC=C1 DOBKQCZBPPCLEG-UHFFFAOYSA-N 0.000 description 1
- 210000000944 nerve tissue Anatomy 0.000 description 1
- 210000005055 nestin Anatomy 0.000 description 1
- 210000002241 neurite Anatomy 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 229940097998 neurotrophin 4 Drugs 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 238000012758 nuclear staining Methods 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 235000021313 oleic acid Nutrition 0.000 description 1
- 238000001543 one-way ANOVA Methods 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 238000000059 patterning Methods 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 230000008782 phagocytosis Effects 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 210000004694 pigment cell Anatomy 0.000 description 1
- 229920001992 poloxamer 407 Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920002714 polyornithine Polymers 0.000 description 1
- 108010055896 polyornithine Proteins 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 239000013615 primer Substances 0.000 description 1
- 239000000186 progesterone Substances 0.000 description 1
- 229960003387 progesterone Drugs 0.000 description 1
- 210000001176 projection neuron Anatomy 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 230000020978 protein processing Effects 0.000 description 1
- 208000020016 psychiatric disease Diseases 0.000 description 1
- 229940076788 pyruvate Drugs 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 238000003753 real-time PCR Methods 0.000 description 1
- 108091006082 receptor inhibitors Proteins 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 210000001202 rhombencephalon Anatomy 0.000 description 1
- 229950007455 ripasudil Drugs 0.000 description 1
- 238000011076 safety test Methods 0.000 description 1
- 201000000980 schizophrenia Diseases 0.000 description 1
- 230000000862 serotonergic effect Effects 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 229960001471 sodium selenite Drugs 0.000 description 1
- 239000011781 sodium selenite Substances 0.000 description 1
- 235000015921 sodium selenite Nutrition 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 101150077014 sox10 gene Proteins 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000023895 stem cell maintenance Effects 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 210000002536 stromal cell Anatomy 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 230000009469 supplementation Effects 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 210000000225 synapse Anatomy 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 210000001587 telencephalon Anatomy 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- TUNFSRHWOTWDNC-HKGQFRNVSA-N tetradecanoic acid Chemical compound CCCCCCCCCCCCC[14C](O)=O TUNFSRHWOTWDNC-HKGQFRNVSA-N 0.000 description 1
- 238000010257 thawing Methods 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 229960000187 tissue plasminogen activator Drugs 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 229960000984 tocofersolan Drugs 0.000 description 1
- 239000011573 trace mineral Substances 0.000 description 1
- 235000013619 trace mineral Nutrition 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 238000009281 ultraviolet germicidal irradiation Methods 0.000 description 1
- 210000005048 vimentin Anatomy 0.000 description 1
- 239000011534 wash buffer Substances 0.000 description 1
- 229920003169 water-soluble polymer Polymers 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 1
Images




























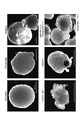














Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0618—Cells of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/30—Nerves; Brain; Eyes; Corneal cells; Cerebrospinal fluid; Neuronal stem cells; Neuronal precursor cells; Glial cells; Oligodendrocytes; Schwann cells; Astroglia; Astrocytes; Choroid plexus; Spinal cord tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/30—Organic components
- C12N2500/38—Vitamins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/01—Modulators of cAMP or cGMP, e.g. non-hydrolysable analogs, phosphodiesterase inhibitors, cholera toxin
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/115—Basic fibroblast growth factor (bFGF, FGF-2)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/13—Nerve growth factor [NGF]; Brain-derived neurotrophic factor [BDNF]; Cilliary neurotrophic factor [CNTF]; Glial-derived neurotrophic factor [GDNF]; Neurotrophins [NT]; Neuregulins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/15—Transforming growth factor beta (TGF-β)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/40—Regulators of development
- C12N2501/415—Wnt; Frizzeled
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/40—Regulators of development
- C12N2501/42—Notch; Delta; Jagged; Serrate
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/70—Enzymes
- C12N2501/72—Transferases (EC 2.)
- C12N2501/727—Kinases (EC 2.7.)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/02—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from embryonic cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/45—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from artificially induced pluripotent stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2513/00—3D culture
Definitions
- the present invention relates to cerebral organoids or cerebral cortical cell clusters derived from pluripotent stem cells, methods for producing them, and the like.
- cell transplantation therapy is expected by transplanting nerve cells differentiated from human pluripotent stem cells, especially human induced pluripotent stem cells (iPS cells). It is possible to transplanting nerve cells differentiated from human pluripotent stem cells, especially human induced pluripotent stem cells (iPS cells). It is possible to transplanting nerve cells differentiated from human pluripotent stem cells, especially human induced pluripotent stem cells (iPS cells). It is possible to transplanting nerve cells differentiated from human pluripotent stem cells, especially human induced pluripotent stem cells (iPS cells). It is
- ES cells human embryonic stem cells
- support cells sometimes referred to as feeder cells
- MEFs mouse embryonic fibroblasts
- Non-Patent Document 3 When human pluripotent stem cells maintained in the absence of support cells are induced to differentiate by the SFEBq method, there is a problem that the efficiency of producing cerebral organoids is low (Non-Patent Document 3).
- the purpose of the present invention is to provide human pluripotent stem cell-derived cerebral organoids, cell aggregates containing cerebral cortical cells, and methods for producing these, which are useful for regenerative medicine. More specifically, the object is to provide a method for producing cerebral organoids from pluripotent stem cells in the absence of support cells and products obtained by the method.
- the present inventors cultured pluripotent stem cells in a culture medium that does not substantially contain bFGF and does not substantially induce TGF ⁇ signals in the absence of feeder cells. It was found that cerebral organoids can be produced efficiently by inducing the differentiation of cells from cerebral cortex into neurons. I found that it can be manufactured well.
- Step (2) is (2a) a step of suspension culture of the cells obtained in step (1) in a culture medium containing a TGF ⁇ signaling inhibitor and a Wnt signaling inhibitor to obtain a cell mass; [ 1].
- [4] The method according to [2] or [3], wherein the suspension culture in step (2b) is shaking culture.
- [5] The method according to any one of [1] to [4], wherein the culture period in step (1) is less than 3 days.
- [6] The method according to any one of [1] to [5], wherein the culture period in step (1) is 12 hours or more and 2 days or less.
- the culture medium of step (1) contains a TGF ⁇ signal inhibitor selected from the group consisting of SB431542, A-83-01, and XAV-939, according to any one of [1] to [6].
- Method. [8] The method according to any one of [2] to [7], wherein the culture medium in steps (1), (2a), and (2b) is a serum-free culture medium.
- the pluripotent stem cells are human induced pluripotent stem cells or human embryonic stem cells.
- the method according to any one of [1] to [9] further comprising the step of selecting cerebral organoids.
- the cerebral organoid further comprises the following (1) to (5): (1) be a spherical cell mass, (2) having a cerebral cortex-like structure inside the cell mass; (3) having no pigmentation on the surface; (4) having neither a cyst-like shape, a protrusion shape, or a balloon-like shape in a part of the cell mass, and (5) NEUROD6, NEUROD2, SSTR2, TBR1, ZBTB18, NHLH1, IGFBPL1, NRN1, RTN1, THSD7A, NRXN1, BHLHE22, CALB2, KHDRBS3, CCSAP, PDE1A, NEUROD1, NPTX1, NXPH4, NTS, NEUROD2, COROMOG2, , TP53I11, ZFPM2, PCDH9, NELL2, SRRM4, SCG3, DCC, EPB41L3, SLC17A7, ST18, NSG2,
- the number of proliferative marker-positive cells is 10% or less of the total number of cells
- the number of cells positive for one or more markers selected from the group consisting of neuronal markers, cortical V/VI layer markers, and forebrain markers is 70% or more of the total number of cells
- a cerebral cortex cell mass characterized by being substantially free of neuroepithelial or cerebral cortex-like structures.
- the proliferative marker of (a) is Ki67
- the neuronal marker of (b) is ⁇ III-Tubulin
- the cortical V/VI layer marker is Ctip2
- the forebrain marker is FOXG1
- a method for producing a cerebral cortical cell mass from pluripotent stem cells in the absence of supporting cells comprising: (i) obtaining cerebral organoids from pluripotent stem cells; (ii) culturing the cerebral organoid obtained in step (i) in a culture medium containing a Notch signal inhibitor to obtain a cerebral cortex cell mass. [20] The method of [19], wherein in step (i), cerebral organoids are obtained from pluripotent stem cells by the method of any one of [1] to [10].
- (A) the number of proliferative marker-positive cells is 5% or less of the total number of cells
- (B) One or more marker-positive cells selected from neuronal markers, cortical V/VI layer markers, and forebrain markers are 70% or more of the total number of cells
- the proliferative marker of (A) is Ki67
- the neuronal marker of (B) is ⁇ III-Tubulin
- the cortical V/VI layer marker is Ctip2
- the forebrain marker is FOXG1
- the highly purified cerebral cortex cell mass according to [21].
- a method for producing highly purified cerebral cortical cell clusters from pluripotent stem cells in the absence of supporting cells comprising: (i) obtaining cerebral organoids from pluripotent stem cells; (ii) culturing the cerebral organoids obtained in step (i) in a culture medium; (iii) dispersing the cell culture obtained in step (ii) into a single cell or a cell clump of 2 to 5 cells; (iv) culturing the cell culture obtained in step (ii) or the cell population obtained in step (iii) in a medium comprising one or more neurotrophic factors, ascorbic acid, and a cAMP activator; and obtaining a cell mass, wherein the culture medium in step (ii) and/or the culture
- step (i) The method of [26], wherein in step (i), cerebral organoids are obtained from pluripotent stem cells by the method of any one of [1] to [10].
- step (i) Any of [19], [20], [26] and [27], wherein the cerebral organoids given in step (ii) are cerebral organoids 28 to 44 days after initiation of induction of differentiation into neurons. The method described in Crab.
- step (ii) The method according to any one of [19], [20], and [26] to [28], wherein the culture period in step (ii) is 2 to 6 days.
- the culture period in step (iv) is 2 to 14 days.
- [31] The method according to any one of [19], [20], and [26] to [30], wherein the Notch signaling inhibitor is a ⁇ -secretase inhibitor.
- the ⁇ -secretase inhibitor is N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) or Compound E. .
- DAPT N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester
- Compound E. Compound E.
- a cell population comprising the highly purified cerebral cortical cell clusters according to any one of [21] to [25], wherein the highly purified cerebral cortical cell clusters are uniform in size, shape or constituent cell composition. cell population.
- step (aa) measuring the expression level of at least one gene selected from the group consisting of GAD2, COL1A1, TYR, TTR and HOXA2 or a protein encoded by the gene or a fragment thereof in a cerebral organoid or cerebral cortex cell mass and (bb) based on the measurement result of step (aa), when the expression level of the gene is below the reference value, the amount of non-target cells contained in the cerebral organoid or the cerebral cortical cell mass is the reference.
- evaluating to be A method for evaluating the quality of cerebral organoids or cerebral cortical cell masses comprising: [39] (AA) NEUROD6, NEUROD2, SSTR2, TBR1, ZBTB18, NHLH1, IGFBPL1, NRN1, RTN1, THSD7A, NRXN1, BHLHE22, CALB2, KHDRBS3, CCSAP, PDE1A, NEUROD1, NPTX1 in cerebral organoids or cerebral cortical cell masses , NXPH4, NTS, NEUROG2, OLFM1, PRDM8, CORO2B, TP53I11, ZFPM2, PCDH9, NELL2, SRRM4, SCG3, DCC, EPB41L3, SLC17A7, ST18, NSG2, EMX1, CAP2, SYT4, NSMF, ANK3, MYT1L, FSTL4, , B3GAT1, EPHA5, NHLH2, and a step of measuring the expression level of at least one gene selected from the group consisting of DLL3; A step of
- cerebral organoids that can be used as materials for cerebral cortex cell preparations can be produced from human pluripotent stem cells with high efficiency.
- the cerebral cortical cell mass or the like derived from the cerebral organoid of the present invention is useful as a therapeutic agent or a transplant material for treating cerebrovascular disorders and the like.
- FIG. 10 shows images of the morphology of cell aggregates after induction of differentiation into neurons in preliminary test 1.
- FIG. 2 shows the results of microarray expression analysis of FGF2 and TGF ⁇ pathway-related genes in preliminary test 2.
- FIG. An example of a differentiation induction scheme in Example 1 is shown. 1 shows a bright field image (A) and a confocal fluorescence microscope image (B) of cell aggregates after induction of differentiation in 1-1 of Example 1.
- FIG. An example of a differentiation induction scheme in Example 1 is shown. 1 shows a confocal fluorescence microscopy image of a cerebral cortex cell mass after differentiation induction in 1-1 of Example 1.
- FIG. FIG. 2 shows confocal fluorescence microscope images of cerebral organoids on Day 35 after induction of differentiation in 1-2 of Example 1.
- FIG. 2 shows representative bright-field images of cultures on Day 18, Day 27, and Day 34 after induction of differentiation in Example 2.
- FIG. 2 shows the formation efficiency (%) of cerebral organoids under each condition after induction of differentiation in Example 2.
- FIG. 4 shows the results of expression analysis of various marker genes after step (1) (Day 0) in Example 3.
- Representative confocal fluorescence microscope images of immunostaining in 4-1 of Example 4 are shown.
- the differentiation induction scheme in 4-2 of Example 4 is shown.
- 4 shows the analysis results of marker gene expression by flow cytometry in 4-2 of Example 4.
- FIG. 4 shows the analysis results of marker gene expression by flow cytometry in 4-2 of Example 4.
- Example 4 The scheme of the DAPT method in 4-3 of Example 4 (A) and the results of RT-qPCR analysis of changes in gene expression in the obtained cell mass (B) are shown.
- the differentiation induction scheme (A) in Example 5 and the results (B) of immunostaining of cerebral organoids on Day 28 (4wk), Day 42 (6wk), and Day 75 (10wk) are shown.
- 4 shows the analysis results of relative expression levels of various markers of cerebral organoids on Day 28 (4wk), Day 35 (5wk), Day 42 (6wk), and Day 75 (10wk) in Example 5.
- FIG. Representative confocal fluorescence microscopy images of immunostained grafts in Example 6 are shown.
- FIG. 10 shows the volumetric analysis results of immunostained grafts in Example 6.
- FIG. 1 shows a scheme of the method of the preferred embodiment; Representative confocal fluorescence microscope images of immunostaining in Example 7 are shown.
- 2 shows the analysis results of flow cytometry in Example 7.
- FIG. The schemes of the two methods in 8-1 of Example 8 are shown.
- FIG. 10 shows the results of RT-qPCR analysis of gene expression levels over time for each marker in cell masses obtained by the single-cell DAPT method in 8-1 of Example 8.
- FIG. 10 shows the results of RT-qPCR analysis of gene expression levels over time for each marker of cell clusters obtained by the single-cell DAPT method and the organoid method in 8-1 of Example 8.
- FIG. 10 shows flow cytometry analysis results of gene expression levels on day 10 for each marker of cell aggregates obtained by the single-cell DAPT method and the organoid method in 8-1 of Example 8.
- FIG. The scheme of the single-cell DAPT method in 8-2 of Example 8 is shown. 8 shows the results of flow cytometry analysis of gene expression in cell masses obtained by the single-cell DAPT method in 8-2 of Example 8.
- FIG. A representative bright-field image of the organoid morphology in 9-1 of Example 9 is shown. Representative bright field images of the seven morphological groups in 9-1 of Example 9 are shown. 10 shows a bar graph of the ratio of organoids of each form in 9-1 of Example 9.
- FIG. 10 shows bright-field images of nine organoids obtained by three rounds of differentiation induction in 9-2 of Example 9.
- FIG. 9 shows the results of UMAP display of data of single-cell RNA-seq analysis for nine organoids in 9-2 of Example 9.
- FIG. 9 shows the results of UMAP display of data of single-cell RNA-seq analysis for nine organoids in 9-2 of Example 9.
- FIG. The result of identifying the cell type of each cluster based on the gene expression of each cluster in 9-2 of Example 9 is shown.
- the characteristic gene for each cluster and the expression profile of known marker genes are shown by dot plots.
- the characteristic gene for each cluster and the expression profile of known marker genes are shown by dot plots.
- the percentage of each cell type in nine organoids in 9-2 of Example 9 (A) and the percentage of neural crest cells at each differentiation stage (B) are shown.
- Immunostaining results of representative marker proteins for each group of organoids in 9-3 of Example 9 are shown.
- Bright field images of three organoids for each group in 9-4 of Example 9 are shown.
- FIG. 10 shows the result of analysis of marker gene expression by RT-qPCR method for each organoid in 9-4 of Example 9.
- FIG. FIG. 10 shows the result of analysis of marker gene expression by RT-qPCR method for each organoid in 9-4 of Example 9.
- stem cell means an undifferentiated cell having differentiation potential and proliferation potential (especially self-renewal potential) while maintaining differentiation potential.
- Stem cells include subpopulations such as pluripotent stem cells, multipotent stem cells, and unipotent stem cells, depending on their differentiation potential.
- Pluripotent stem cells are capable of being cultured in vitro and capable of differentiating into all cell lineages belonging to the three germ layers (ectoderm, mesoderm, endoderm) and/or extra-embryonic tissues (pluripotent stem cells).
- stem cells with pluripotency refer to stem cells that have the ability to differentiate into multiple, but not all, types of tissues and cells.
- a unipotent stem cell means a stem cell that has the ability to differentiate into a specific tissue or cell.
- Pluripotent stem cells can be derived from fertilized eggs, cloned embryos, germ stem cells, tissue stem cells, somatic cells, etc.
- pluripotent stem cells include embryonic stem cells (ES cells: Embryonic Stem cells), EG cells (Embryonic Germ cells), induced pluripotent stem cells (iPS cells: induced Pluripotent Stem cells), and the like.
- Muse cells Multi-lineage differentiating stress enduring cells obtained from mesenchymal stem cells (MSCs), and mGS cells produced from germ cells (e.g., testis) are also included in pluripotent stem cells.
- Embryonic stem cells Human embryonic stem cells were established in 1998 and are being used in regenerative medicine. Embryonic stem cells can be produced by culturing the inner cell mass at the blastocyst stage, ie within 14 days after fertilization, on feeder cells or in a medium containing FGF2. Methods for producing embryonic stem cells are described, for example, in WO96/22362, WO02/101057, US5,843,780, US6,200,806, US6,280,718 and the like. Embryonic stem cells can be obtained from designated institutions or can be purchased commercially. For example, human embryonic stem cells KhES-1, KhES-2 and KhES-3 are available from Institute for Frontier Medical Sciences, Kyoto University.
- induced pluripotent stem cells refers to cells in which pluripotency is induced by reprogramming somatic cells by a known method or the like.
- Induced pluripotent stem cells were established in mouse cells by Yamanaka et al. in 2006 (Cell, 2006, 126(4), pp.663-676). Induced pluripotent stem cells were also established in human fibroblasts in 2007, and have pluripotency and self-renewal ability like embryonic stem cells (Cell, 2007, 131(5), pp.861-872; Science , 2007, 318(5858), pp.1917-1920; Nat. Biotechnol., 2008, 26(1), pp.101-106).
- induced pluripotent stem cells are OCT3/4, SOX2, KLF4, MYC (c-MYC, N-MYC, L-MYC), fibroblasts, peripheral blood mononuclear cells, and other differentiated somatic cells.
- Cells in which pluripotency is induced by reprogramming by forcibly expressing any combination of a plurality of genes selected from the reprogramming gene group including GLIS1, NANOG, SALL4, LIN28, ESRRB, etc., can be mentioned.
- Preferred combinations of reprogramming factors include (1) OCT3/4, SOX2, KLF4 and MYC (c-MYC or L-MYC), (2) OCT3/4, SOX2, KLF4, LIN28 and L-MYC (Stem Cells, 2013; 31: 458-466).
- induced pluripotent stem cells in addition to the method of producing by reprogramming induction by gene expression, induced pluripotent stem cells can also be induced by adding compounds to somatic cells (Science, 2013, 341, pp.651 -654; Nature, 2022, 605, pp.325-331).
- iPS cells can be produced by reprogramming somatic cells such as peripheral blood/cord blood-derived hematopoietic progenitor cells and fibroblasts using reprogramming factors.
- S2WCB1, S2WCB3 used herein were established from adult peripheral blood mononuclear cells using CytoTuneTM-2.0 (ID Pharma).
- pluripotent stem cells are preferably embryonic stem cells or induced pluripotent stem cells, more preferably induced pluripotent stem cells.
- pluripotent stem cells are mammalian pluripotent stem cells, preferably rodent (e.g., mouse, rat) or primate (e.g., human, monkey) pluripotent stem cells, Human pluripotent stem cells are more preferred, and human induced pluripotent stem cells (iPS cells) or human embryonic stem cells (ES cells) are even more preferred.
- rodent e.g., mouse, rat
- primate e.g., human, monkey
- iPS cells human induced pluripotent stem cells
- ES cells human embryonic stem cells
- Pluripotent stem cells such as human iPS cells can be subjected to maintenance culture and expansion culture by methods well known to those skilled in the art.
- markers means a substance present in a cell and capable of identifying or distinguishing the type or property of the cell based on its presence or amount. Specific examples of markers include mRNAs, proteins encoded by the mRNAs, sugar chains, and fragments thereof.
- nerve cells refer to nerve units composed of cell bodies, dendrites and axons, and are also called neurons. Nerve cells have the function of transmitting stimuli from other nerve cells or stimuli-receiving cells to other nerve cells, muscles or gland cells. Although classified into serotonergic nerves, GABAergic nerves, glutamatergic nerves, etc., the type of neurotransmitter is not particularly limited in the present specification. Neurons can be identified by significantly expressed markers, such as ⁇ III-Tubulin, MAP2, and the like.
- neural stem cell refers to a stem cell that is destined to differentiate into neural cells but has the ability to differentiate into multiple neural cells and maintains proliferative properties, and neural progenitor cells. It is a cell that has both the ability to differentiate into cerebral cortex cells and the ability to differentiate into cerebral cortical cells. They can be identified by markers of primitive neuroectoderm and neural stem cells such as intermediate filament proteins (nestin, vimentin, etc.), transcription factors SOX1, SOX2, PAX6, and the like. Neural stem cells herein include radial glia.
- immature cells that are generated from neural stem cells and are capable of differentiating into multiple nerve cells are also called neural progenitor cells.
- cell culture refers to products obtained by culturing cells, regardless of specific composition. Cell cultures can be aggregates of multiple cell masses, and may further include single cells and aggregates of 2 to 5 cells as described below.
- a cell culture may or may not contain a medium or suspending medium and, as used herein, refers to one that does not contain a medium or suspending medium unless otherwise specified.
- Cerebral organoid means a spherical cell aggregate (cell mass) containing one or more, preferably multiple, cerebral cortex-like structures with a neuronal cell layer on the outside of the neuroepithelium.
- cortex-like structures are rosette-like structures with neuronal layers outside the neuroepithelium.
- cortex-like structures may include neuroepithelial-like structures
- cerebral organoids of the present application may include neuroepithelial-like structures.
- neural epithelium is a layered structure whose main constituent cells are neural stem cells and/or neural progenitor cells, and can also be said to be a localized area of neural stem cells and/or neural progenitor cells.
- the "neuronal layer” is a layered structure containing neurons.
- the nerve cells are not particularly limited as long as they are nerve cells that can be generated at the differentiation stage of cerebral organoids.
- the neuronal cell layer preferably contains two or more of neuronal marker-positive cells, forebrain marker-positive cells, and cerebral cortical neuronal cells or their progenitor cell marker-positive cells, or all of them.
- the neuronal layer can also be said to be a localized area of neurons (cerebral cortical neurons) generated from the neuroepithelium.
- One aspect of the cerebral organoid in the present specification is a cerebral organoid obtained by inducing differentiation of pluripotent stem cells.
- spherical refers to neither a rod-like shape nor a flat plate-like shape (plate-like), and preferably means "a three-dimensional structure close to a sphere". Examples include those that exhibit, for example, a circular or elliptical shape when projected onto a two-dimensional surface. However, it is not necessary to show a smooth curve, and even if unevenness is partially observed, if the entire cell mass can be recognized as having a shape close to a sphere, it corresponds to "spherical”.
- the term “spherical” in the present specification does not necessarily require a high sphericity, but an example is a structure having a sphericity of 0.7 or more, 0.8 or more, preferably 0.9 or more. be done.
- marker genes for neural stem cells or neural progenitor cells include, for example, SOX1, SOX2, PAX6, and the like.
- neuronal marker genes examples include ⁇ III-Tubulin, MAP2, and the like.
- Forebrain marker genes include, for example, FOXG1 (also called BF1), SIX3, and EMX1.
- the cerebral cortex neuron marker genes include markers for layers I to VI, which will be described later.
- marker genes for various cells include the genes listed in Table 6 below, as described later.
- Cerebral cortical cell is also referred to as cerebral cortical neuron or cerebral cortical nerve cell, and refers to nerve cells that constitute the cerebral cortex.
- the nerve cell layer may be a single layer, or may be divided into multiple cell layers.
- the cerebral cortex VI Tbr1, Tbr2
- V Ctip2, Er81, Fezf2
- IV Rorb
- III/II Foxp1, Mef2c, Satb2
- Marker genes frequently used for identification of each layer are shown in parentheses.
- cerebral cortical cells are preferably cells positive for the forebrain marker FOXG1.
- FOXG1 includes polynucleotides represented by NCBI Accession No. NM_005249 and proteins encoded by these.
- cerebral cortical cells may include neurons in the motor area of the cerebral cortex or upper motor neurons, i.e., neurons anterior to the cerebral cortex; and/or layer VI neurons.
- neurons in layer V or layer VI are cell populations characterized by being positive for Ctip2.
- Ctip2 includes polynucleotides represented by NCBI accession numbers NM_001282237, NM_001282238, NM_022898 or NM_138576 and proteins encoded by these.
- the cerebral cortical cells may also include layer I, layer II/III, and layer IV neurons.
- the cerebral cortical cells herein may be produced as a cell population containing other cell types, and the cell population containing cerebral cortical cells is, for example, 15% or more, 20% or more, It may contain 30% or more, 40% or more, or 50% or more cerebral cortical cells.
- Cerebral cortical cells is a concept including “cerebral cortical progenitor cells”
- the target cells of the “cerebral organoid” of the present invention include cerebral cortical progenitor cells.
- the target cells of the "cerebral cortical cell mass (including highly purified cerebral cortical cell mass)" of the present invention which are at a more advanced stage of differentiation, contain almost no cerebral progenitor cells, or contain only a minimal number of cerebral progenitor cells. can be included to some extent.
- cell aggregate As used herein, the term "cell aggregate” (also referred to as cell aggregate) is not particularly limited as long as a plurality of cells adhere to each other to form a three-dimensional structure. It refers to a mass formed by aggregation of cells dispersed in a medium, a mass of cells formed through cell division, or the like. A cell mass also includes a case where a specific tissue is formed. Embryoid bodies, spheres, and spheroids are included in the cell mass.
- the cell aggregates may have any shape, and examples thereof include spherical cell aggregates and layered cell aggregates.
- a "cell population” is a population containing a plurality of cells, and is a cell cluster (spherical cell cluster, layered cell cluster), or a cluster of 2 to 5 cells (Cell clump) good too.
- a “cell population” may also refer to a population of multiple cell clumps, a population of multiple dispersed single cells, or a population of multiple cell clumps of 2-5 cells, or a population of any combination thereof. may be
- the high purity of the cell mass means that the content of the target cells contained in the cell mass is high.
- the specific content rate depends on the type of cells, for example, when the content rate of the target cells is 70% or more, preferably 80% or more, and 90% or more with respect to the total number of cells in the cell cluster, the purity is high. It can be said that there is
- the high-purity cell mass means that the content of non-target cells is 30% or less, preferably 20% or less, 10% or less with respect to the total number of cells in the cell mass. be able to. More preferably, the content of proliferative cells in the cell mass is 10% or less, 5% or less, preferably 3% or less, and more preferably 2% or less relative to the total number of cells in the cell mass. .
- reaggregated cell cluster refers to a cell cluster that is dispersed into single cells or aggregates of 2 to 5 cells, and then the dispersed single cells and aggregates of cells are reassembled and formed. Refers to cell clusters (eg, reaggregated cerebral cortical cell clusters).
- the reaggregated cerebral cortex cell mass contains at least a highly purified cerebral cortex cell mass.
- the "culture medium may be any culture medium (medium) that is commonly used for culturing animal cells, and is not particularly limited as long as animal cells can sustain life, but is preferably It provides an environment in which target cells can proliferate.
- the culture solution (medium) may be prepared by the patient or a commercially available medium may be purchased and used.
- basal media examples include BME medium, BGJb medium, CMRL 1066 medium, Glasgow MEM (GMEM) medium, Improved MEM Zinc Option medium, IMDM medium, Medium 199 medium, Eagle MEM medium, ⁇ MEM medium, DMEM medium, F-12 Medium, DMEM/F-12 medium, IMDM/F12 medium, Ham's medium, RPMI 1640 medium, Fischer's medium, or a mixed medium of these mediums, which can be used for culturing animal cells.
- These basal media contain carbohydrates such as glucose, carbon sources such as amino acids, vitamins, inorganic salts and the like.
- Any component normally used for culturing animal cells may be added to the basal medium as long as it does not adversely affect the desired induction of differentiation.
- the medium used in the present invention is preferably a serum-free culture medium from the viewpoint of producing cell clusters suitable for transplantation.
- serum-free culture medium in the present invention means a medium substantially free of unadjusted or unpurified serum.
- serum-free culture even if the medium is contaminated with purified blood-derived components or animal tissue-derived components (e.g., growth factors), as long as it does not contain unadjusted or unpurified serum. contained in the liquid.
- the serum-free culture medium optionally contains fatty acids or lipids, amino acids (e.g., non-essential amino acids), vitamins, growth factors, cytokines, antioxidants, 2-mercaptoethanol, pyruvic acid, buffers, inorganic salts, and the like. good too.
- the medium used in the present invention is preferably a xeno-free medium.
- xeno-free means conditions under which components derived from a biological species different from the biological species of cells to be cultured (heterologous components, also referred to as Xenogenic factors) are eliminated.
- a portion of the serum-free medium may be xeno-free medium.
- the medium used in the present invention may contain serum replacement.
- serum substitutes include those containing albumin, transferrin, fatty acids, collagen precursors, trace elements, 2-mercaptoethanol, 3' thiol glycerol, or equivalents thereof as appropriate.
- serum substitutes can be prepared, for example, by the method described in WO98/30679. Commercially available products may be used as serum substitutes.
- Such commercially available serum replacements include, for example, Knockout Serum Replacement (manufactured by Thermo Fisher Scientific; hereinafter sometimes referred to as KSR), “StemSure (registered trademark) Serum Replacement (SSR)" Chemically-defined Lipid concentrated (manufactured by Thermo Fisher Scientific), B27 supplement (manufactured by Thermo Fisher Scientific), N2 supplement (manufactured by Thermo Fisher Scientific), ITS supplement (manufactured by Thermo Fisher Scientific), N2 supplements or B27 supplements are preferred.
- support cells are also referred to as feeder cells, and refer to cells other than stem cells that coexist when stem cells such as pluripotent stem cells are cultured.
- Supporting cells include, for example, mouse fibroblasts (MEFs, etc.), human fibroblasts, SNL cells, STO cells, and the like.
- the feeder cells may be antiproliferatively treated feeder cells.
- the growth inhibitory treatment includes growth inhibitor (eg, mitomycin C) treatment, gamma ray irradiation, UV irradiation, or the like.
- feeder-free means culturing in the absence of feeder cells.
- the absence of feeder cells means, for example, conditions in which feeder cells are not added, or substantially free of feeder cells (e.g., the ratio of feeder cells to the total number of cells is 3% or less, preferably 1% below).
- the term "suspension culture” refers to allowing cells to survive while suspended in a medium, or culturing cells by forming aggregates (also referred to as spheres) in a non-adherent state to a culture vessel. means In the present specification, cells are cultured in suspension in the state of a single cell or a cluster of a plurality of cells (cell mass or cell population).
- Culture vessels used for suspension culture are not particularly limited, but examples include flasks, tissue culture flasks, dishes, petri dishes, tissue culture dishes, multidishes, microplates, microwell plates, micropores, multiplates, Examples include multiwell plates, chamber slides, petri dishes, tubes, trays, culture bags, bioreactors, and roller bottles.
- the incubator is preferably cell non-adhesive in order to allow culture under non-adhesive conditions.
- the cell non-adhesive culture vessel is a culture vessel whose surface is not artificially treated (for example, coating treatment with extracellular matrix etc.) for the purpose of improving adhesion to cells, or an artificial A treatment that effectively inhibits adhesion (e.g., polyhydroxyethyl methacrylic acid (poly-HEMA), a nonionic surfactant polyol (Pluronic F-127, etc.) or a phospholipid-like structure (e.g., 2-methacryloyloxyethylphosphorylcholine) It can be carried out by using a culture vessel coated with a water-soluble polymer (Lipidure (registered trademark)) having a constituent unit PrimeSurface (registered trademark) as a culture vessel used in suspension culture, especially in the SFEBq method. (low protein adsorption treatment 96-well plate, manufactured by Sumito
- suspension culture may be static culture, shaking culture, orbital culture, or agitation culture.
- static culture refers to a culture method in which cell clusters are cultured in a state in which they are not intentionally moved. That is, for example, the medium convects with a change in local medium temperature, and the flow may move the cell aggregates. In the present invention, it is referred to as static culture, including the case.
- bFGF means basic fibroblast growth factor and is a protein also called FGF2.
- TGF ⁇ signal inhibitor refers to a substance that inhibits signal transduction from binding of TGF ⁇ to a receptor to SMAD, and a substance that inhibits binding to the ALK family of receptors, or an inhibitor of the ALK family and substances that inhibit the phosphorylation of SMAD by .
- the TGF ⁇ signal inhibitor is not particularly limited as long as it can suppress signal transduction mediated by TGF ⁇ , and may be any of nucleic acids, proteins, and low-molecular-weight organic compounds.
- TGF ⁇ signal inhibitors include substances that act directly on TGF ⁇ (e.g., proteins, antibodies, aptamers, etc.), substances that suppress the expression of genes encoding TGF ⁇ (e.g., antisense oligonucleotides, siRNA, etc.), and TGF ⁇ receptors. Substances that inhibit binding of TGF ⁇ and substances that inhibit physiological activity caused by signal transduction by TGF ⁇ receptors (for example, inhibitors of TGF ⁇ receptors, etc.) can be mentioned.
- the TGF ⁇ signal inhibitor includes a substance that inhibits the binding to the ALK family receptor, or a substance that inhibits the phosphorylation of SMAD by the ALK family.
- the ALK family specifically includes ALK4, ALK5 and ALK7.
- TGF ⁇ signal inhibitors examples include Lefty-1 (as NCBI Accession No., mouse: NM_010094, human: NM_020997), Lefty-2 (as NCBI Accession No., mouse: NM_177099, human: NM_003240 and NM_001172425 is exemplified), SB431542 (4-[4-(1,3-benzodioxol-5-yl)-5-(2-pyridinyl)-1H-imidazol-2-yl]-benzamide), SB202190 (above, R.K.
- the TGF ⁇ signaling inhibitor used in the present invention is preferably SB431542 or A-83-01.
- a Wnt signaling inhibitor is a substance that suppresses the production of Wnt (e.g., Wnt3), or a substance that inhibits signal transduction from binding of Wnt to its receptor to accumulation of ⁇ -catenin, Examples thereof include substances that inhibit binding to Frizzled family receptors, and substances that promote degradation of ⁇ -catenin.
- Wnt signaling inhibitors examples include PORCN involved in Wnt protein processing (in humans, proteins represented by NCBI accession numbers: NP_001269096, NP_073736, NP_982299, NP_982300, or NP_982301). ), DKK1 protein (for example, NCBI accession number for humans: NM_012242), sclerostin (for example, for humans, NCBI accession number: NM_025237), Cerberus protein, Wnt receptor inhibitor , soluble Wnt receptors, anti-Wnt antibodies, casein kinase inhibitors, dominant-negative Wnt proteins and the like, but are not limited to these, and one or more substances may be used in combination.
- PORCN involved in Wnt protein processing
- DKK1 protein for example, NCBI accession number for humans: NM_012242
- sclerostin for example, for humans, NCBI accession number: NM_025237
- Cerberus protein for example
- IWR-1-endo ((4-[(3aR,4S,7R,7aS)-1,3,3a,4,7,7a-hexahydro-1,3-dioxo- 4,7-methano-2H-isoindol-2-yl]-N-8-quinolinyl-benzamide), Merck Millipore), IWP-2 (Sigma-Aldrich), IWP-3 (Sigma-Aldrich), IWP-4 ( Sigma-Aldrich), IWP-L6 (EMD Millipore), C59 (or Wnt-C59) (Cellagen technology), ICG-001 (Cellagen Technology), LGK-974 (or NVP-LGK-974) (Cellagen Technology) , FH535 (Sigma-Aldrich), WIKI4 (Sigma-Aldrich), KYO2111 (Minami I, et al, Cell Rep.2:1448-1460, 2012), P
- Notch signal inhibitors are not limited as long as they are substances capable of suppressing signal transduction by Notch.
- Notch signal inhibitors include ⁇ -secretase inhibitors, Notch transcription complex inhibitors such as MAML-1 inhibitors, and the like.
- the ⁇ -secretase inhibitor is not limited as long as it is a substance capable of inhibiting the enzymatic activity of ⁇ -secretase.
- DAPT N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester
- DBZ dibenzazepine
- MDL28170 Calpain Inhibitor III
- Compound E N -[(1S)-2-[[(3S)-2,3-Dihydro-1-methyl-2-oxo-5-phenyl-1H-1,4-benzodiazepin-3-yl]amino]-1-methyl -2-oxoethyl]-3,5-difluorobenzeneacetamide
- Compound 34 ((2S,3R)-3-(3,4-Difluorophenyl)-2-(4-fluorophenyl)-4-hydroxy-N-((3
- Notch transcription complex inhibitors include CB-103 (6-[4-(1,1-dimethylethyl)phenoxy]-3-pyridinamine), IMR-1 (2-Methoxy-4-(4-oxo-2-thioxo -thiazolidin-5-ylidenemethyl)-phenoxy]-acetic acid ethyl ester) and the like.
- ROCK inhibitor is an inhibitor of Rho-associated coiled-coil kinase (ROCK), and is not particularly limited as long as it is a substance that suppresses the function of ROCK.
- ROCK inhibitors include Y-27632 ((+)-(R)-trans-4-(1-aminoethyl)-N-(4-pyridyl)cyclohexanecarboxamide dihydrochloride), H-1152 ((S)-4-Methyl -5-((2-methyl-1,4-diazepan-1-yl)sulfonyl)isoquinoline dihydrochloride), Fasudil (HA-1077; 1-(5-Isoquinolinesulfonyl)homopiperazine Hydrochloride), Wf-536 (4-[( 1R)-1-aminoethyl]-N-(pyridin-4-yl)benzamide), Thiazovivin (N-Benzyl-2-(pyrimidin-4-
- neurotrophic factor is a general term for secretory proteins having activities to promote survival of nerve cells, extension of neurites and axons, formation of synapses, and the like.
- neurotrophic factors Nerve Growth Factor (NGF), Brain-derived Neurotrophic Factor (BDNF), Neurotrophin 3 (NT-3), Neurotrophin 4/5 (NT-4/5), Neurotrophin 6 (NT-6), Glia Cell line-derived Neurotrophic Factor (GDNF), Ciliary Neurotrophic Factor (CNTF) and the like.
- a preferred neurotrophic factor in the present invention is a factor selected from the group consisting of GDNF and BDNF.
- Neurotrophic factors are commercially available from, for example, Wako, R&D Systems, etc. and can be easily used, but may be obtained by forced expression in cells by methods known to those skilled in the art.
- cAMP activators include cAMP, dibutyryl-cAMP, forskolin, and the like.
- One embodiment of the method for producing cerebral organoids of the present invention is a method for producing cerebral organoids from pluripotent stem cells in the absence of support cells, including the following steps (1) and (2). are mentioned. “In the absence of feeder cells” is as described above, and both step (1) and step (2) are performed “in the absence of feeder cells”.
- Step of culturing pluripotent stem cells in a culture medium that is substantially free of bFGF and does not substantially induce TGF ⁇ signals (2) Cells obtained in step (1) are transformed into nerve cells and the step of inducing differentiation ⁇ Step (1)>
- culturing "in the presence of substance X” means culturing in "a medium containing substance X", and the substance X is contained as an original component in the medium. or may be added exogenously.
- an endogenous substance X that can be expressed, secreted or produced by a cell or tissue during the culture is distinguished from exogenous substance X, and a medium that does not contain exogenous substance X is endogenous Even if it contains substance X, it is understood that it does not fall under the category of "medium containing substance X".
- a "medium containing a TGF ⁇ signal inhibitor” is a medium supplemented with a TGF ⁇ signal inhibitor or a medium containing a TGF ⁇ signal inhibitor as an original component.
- the culture medium "substantially free of bFGF” does not deny the existence of bFGF expressed by the cells themselves, but does not originally contain bFGF, or does not exogenously add bFGF. Means culture medium.
- the present application also encompasses culturing using a medium containing a low dose of bFGF to the extent that the efficiency of neuronal differentiation in the present invention is not affected.
- the culture medium that "substantially does not induce TGF ⁇ signals” does not deny the existence of TGF ⁇ expressed by the cells themselves. It means that the culture medium is not Alternatively, a culture solution containing an exogenously added TGF ⁇ or a culture solution containing no exogenously added TGF ⁇ , to which an effective amount of a TGF ⁇ signal inhibitor is added, is also referred to as “substantially inhibiting the TGF ⁇ signal. It is in the category of "culture solution that does not induce
- the above-mentioned TGF ⁇ signal inhibitor can be appropriately selected so as not to substantially induce TGF ⁇ signals.
- the concentration of the TGF ⁇ signal inhibitor can be appropriately adjusted according to the intensity of TGF ⁇ signal activity in the culture medium. That is, the concentration of the TGF ⁇ signal inhibitor in the culture medium is not particularly limited as long as it inhibits the activity of ALK4, ALK5 or ALK7 signaling.
- the TGF ⁇ signal inhibitor added to the culture medium in step (1) may be any of the above TGF ⁇ signal inhibitors, preferably selected from the group consisting of SB431542 and A-83-01.
- As the culture solution in step (1) for example, a commercially available medium for the purpose of culturing pluripotent stem cells such as ES cells or iPS cells while maintaining their pluripotency, and which does not contain bFGF is used.
- the concentration of the TGF ⁇ signal inhibitor is 100 nM to 1 mM, 100 nM to 500 ⁇ M, 100 nM to 100 ⁇ M, 100 nM to 50 ⁇ M, 100 nM to 40 ⁇ M, 100 nM to 30 ⁇ M, 100 nM to 25 ⁇ M when SB431542 is used as the TGF ⁇ signal inhibitor. .
- the concentration that exhibits ALK inhibitory activity or TGF ⁇ inhibitory activity equivalent to that of SB431542 at the concentration described above can be appropriately set.
- the concentration equivalent to the concentration when SB431542 is used means a concentration that has an inhibitory effect on the TGF ⁇ signaling pathway equivalent to the concentration of SB431542 (e.g., an effect of inhibiting the activity of ALK4, ALK5 or ALK7 signaling). means The concentration can be easily set by those skilled in the art.
- a TGF ⁇ signal inhibitor can be added to a commercially available pluripotent stem cell culture medium that does not contain bFGF.
- a medium that can maintain and grow pluripotent stem cells e.g., Essential 8
- a commercially available culture medium eg, Essential 6 can also be used.
- the culture solution used in step (1) is to the extent that it does not affect the culture, that is, to the extent that it does not substantially affect the formation of cerebral organoids obtained in steps (1) and (2).
- other substances may be contained, preferably a substance that enhances or inhibits signal transduction that affects differentiation induction of pluripotent stem cells, such as BMP signal, Sonic hedge hog signal, etc., is added externally. preferably not.
- the culture medium used in step (1) may contain a Wnt signaling inhibitor, but desirably no substances that enhance Wnt signaling are externally added.
- the pluripotent stem cells to be subjected to step (1) are preferably human induced pluripotent stem cells (iPS cells) or human embryonic stem cells (ES cells), more preferably human induced pluripotent stem cells.
- iPS cells human induced pluripotent stem cells
- ES cells human embryonic stem cells
- pluripotent stem cells are cultured in the absence of feeder cells.
- the absence of feeder cells is also referred to as feeder-free, and refers to a state in which no feeder cells are present in the medium.
- the culture conditions in the absence of feeder cells herein specifically include culture conditions in which feeder cells such as fibroblasts, SNL cells, and STO cells are not added.
- the culture medium used in step (1) is preferably a serum-free culture medium that does not substantially contain serum, and if necessary, a serum-free culture medium supplemented with a serum substitute may be used.
- serum substitutes include those described above, preferably KSR, preferably 1 to 30% KSR, or the like can be used.
- the culture medium used in step (1) is not particularly limited as long as it is substantially free of bFGF and does not substantially induce TGF ⁇ signals, preferably serum-free. It is desirable that the medium contains components necessary for culturing pluripotent stem cells while maintaining their pluripotency, except for substances that induce signals.
- pluripotent stem cell media including Essential 8 (manufactured by Thermo Fisher Scientific), S-medium (DS Pharma Biomedical Co.), StemPro (Thermo Fisher Scientific), hESF9 (Proc Natl Acad Sci USA.
- the pluripotent stem cells to be subjected to step (1) may be cryopreserved pluripotent stem cells immediately after thawing, but are preferably expanded while maintaining the pluripotency of the pluripotent stem cells. It can be cultured and subcultured in advance in a medium suitable for The passage number of the pluripotent stem cells to be subjected to step (1) is not particularly limited, but it is desirable to pass 2 to 8 passages.
- the culture period in step (1) is less than 5 days, less than 4 days, preferably less than 3 days, more preferably 12 hours or more and 48 hours or less, 18 hours or more and 48 hours or less, more preferably 24 hours or more and 48 hours or less, 24 hours or more and 36 hours or less, or 18 hours or more and 36 hours or less, most preferably about one day. Note that the culture period does not include a period of subculture of pluripotent stem cells as a preparatory stage.
- the culture in step (1) may be performed under either suspension culture or adherent culture, but is preferably performed by adherent culture.
- the incubator used for adherent culture is not particularly limited as long as it is capable of "adherent culture", but cell-adhesive incubators are preferred.
- Examples of cell-adhesive incubators include those in which the surface of the incubator is artificially treated for the purpose of improving adhesion to cells. Specifically, the interior is coated with the aforementioned coating agent. and an incubator.
- Coating agents include, for example, laminin [laminin ⁇ 5 ⁇ 1 ⁇ 1 (hereinafter, laminin 511), laminin ⁇ 1 ⁇ 1 ⁇ 1 (hereinafter, laminin 111), laminin ⁇ 1 ⁇ 1 ⁇ 2 (laminin 112), laminin ⁇ 2 ⁇ 1 ⁇ 1 (laminin 211), laminin ⁇ 2 ⁇ 1 ⁇ 2 (laminin 212), laminin ⁇ 2 ⁇ 2 ⁇ 1 (laminin 221), laminin ⁇ 2 ⁇ 2 ⁇ 2 (laminin 222), laminin ⁇ 5 ⁇ 1 ⁇ 2 (laminin 512), etc., and laminin fragments (laminin 511E8, etc.)], entactin, collagen, gelatin, vitronectin, synthmax (Corning), Examples include extracellular matrices such as matrigel, and polymers such as polylysine and polyornithine.
- a culture vessel that has been surface-treated such as a positive charge treatment, can also be used.
- Laminin is preferred, and laminin 511E8 is more preferred.
- Laminin 511E8 can be purchased commercially (eg iMatrix-511, Nippi).
- Culture conditions such as culture temperature and CO 2 concentration in step (1) can be appropriately set.
- the culture temperature is, for example, about 30°C to about 40°C, preferably about 37°C.
- the CO 2 concentration is, for example, about 1% to about 10%, preferably about 5%.
- Step (1) is a maintenance culture step of culturing pluripotent stem cells while maintaining their pluripotency in the absence of support cells, and is performed before step (2) (differentiation induction step).
- This is the neural-differentiation preparation step.
- This neural differentiation preparatory step is a step of preparation before induction of neural differentiation or before introduction of neural differentiation, and the pluripotent stem cells cultured in step (1) are TGF ⁇ -related When gene expression is suppressed and the pluripotent stem cells are thereby induced to differentiate into nerve cells, the formation efficiency of cerebral organoids is high.
- Step (2)> The cells obtained in step (1) can be induced to differentiate into nerve cells by methods well known to those skilled in the art, thereby obtaining cerebral organoids.
- a differentiation induction method capable of differentiating pluripotent stem cells into cell populations constituting cerebral organoids can be appropriately selected.
- the method is well known, for example, WO2015/076388, WO2016/167372, Sakaguchi et al., Stem Cell Reports 2019 Vol 13 458-473, Kitahara, et al., Stem Cell Reports 2020 Vol. 15, 467-481 (Non-Patent Document 1) and Kadoshima et al., 2013, PNAS, vol.110, No. 50, 20284-20289, etc. can be used.
- the differentiation induction method performed in step (2) includes the following steps (2a) and (2b): (2a) the cells obtained in step (1) are subjected to suspension culture, preferably stationary culture, in a culture medium containing a TGF ⁇ signal inhibitor and a Wnt signal transduction inhibitor to obtain a cell mass (2b) The cell mass obtained in step (2a) is placed in a culture medium substantially free of TGF ⁇ signal inhibitors or Wnt signal transduction inhibitors, preferably neither TGF ⁇ signal inhibitors nor Wnt signal transduction inhibitors. A step of obtaining cerebral organoids by floating culture in a culture medium.
- the cells obtained in step (1) are dispersed in a culture medium, preferably in a serum-free medium substantially free of serum (unadjusted or unpurified serum), and cultured under non-adhesive conditions ( (i.e., suspension culture), and aggregate a plurality of cells to form a cell cluster.
- a serum-free culture medium containing a serum substitute may be used as the culture medium used for agglutination.
- the incubator used for this cell cluster formation is not particularly limited, but examples include flasks, tissue culture flasks, dishes, Petri dishes, tissue culture dishes, multidishes, microplates, microwell plates, micropores, and multiplates. , multiwell plates, chamber slides, Petri dishes, tubes, trays, culture bags, bioreactors, and roller bottles.
- a method of forming a cell mass by embedding in a gel such as alginic acid hydrogel can also be used.
- the incubator is preferably cell non-adherent so as to allow culture under non-adherent conditions.
- Non-cell-adhesive culture vessels include those in which the surface of the culture vessel is artificially treated to make it non-adhesive to cells, and those artificially treated for the purpose of improving adhesion to cells (e.g., Those that have not been coated with an extracellular matrix or the like) can be used.
- the cells obtained in step (1) are recovered from the subculture and dispersed to a single cell or a state close to it. This dispersal is performed using a suitable cell dissociation solution.
- a suitable cell dissociation solution for example, a chelating agent such as EDTA, a proteolytic enzyme such as trypsin, collagenase IV, metalloprotease, and the like can be used alone or in appropriate combination. Among them, those with low cytotoxicity are preferred, and commercially available products such as dispase (Edia), TrypLE (manufactured by Gibco) and Accutase (MILLIPORE) are available as such cell dissociation solutions.
- the dispersed cells are suspended in a medium obtained by adding Y-27632 to the above medium (serum-free medium, ie, basal medium used in step (2a) described below).
- an inhibitor of Rho-associated coiled-coil kinase was added from the start of culture. It is preferably added (WO2008/035110, Watanabe, K. et al., Nature Biotechnology, 2007, vol.25, No.6, page 681-686).
- ROCK inhibitors examples include those mentioned above, preferably Y-27632 and the like.
- the ROCK inhibitor is added, for example, within 20 days, within 15 days, preferably within 10 days, more preferably within 6 days from the start of culture.
- the concentration of ROCK inhibitor may be constant or tapered.
- culturing in the presence of the ROCK inhibitor for 10 to 20 days, preferably 15 to 20 days, even more preferably about 17 to 19 days, during which the concentration of the ROCK inhibitor is gradually decreased. can be done.
- culturing in the presence of a ROCK inhibitor for 10 to 25 days, preferably 12 to 25 days, 10 to 20 days, even more preferably about 15 to 20 days, or about 17 to 19 days, and the concentration of the ROCK inhibitor may be tapered in the meantime.
- the concentration of the ROCK inhibitor used for suspension culture is a concentration that can suppress cell death of pluripotent stem cells induced by dispersion.
- the concentration corresponds to about 0.1 to 200 ⁇ M, preferably about 2 to 100 ⁇ M, more preferably about 30 to 100 ⁇ M.
- the concentration of the ROCK inhibitor may be varied during the period of addition.
- the concentration may be halved in the second half of the period, or the concentration may be gradually reduced from the start of step (2a).
- the cell suspension obtained in step (1) is sown in the culture vessel and cultured under non-adhesive conditions to aggregate a plurality of cells to form a cell mass.
- SFEBq method it is preferable to rapidly aggregate dispersed cells to form one cell cluster in one culture compartment.
- Methods for rapidly aggregating dispersed cells include, for example, the following methods: 1) A method in which dispersed cells are confined in a culture compartment of relatively small volume (e.g., 1 ml or less, 500 ⁇ l or less, 200 ⁇ l or less, 100 ⁇ l or less) to form a single cell cluster in the compartment, or 2). A method of placing dispersed cells in a centrifugation tube, centrifuging it, and depositing the cells in one place to form a single cell mass in the tube.
- the culture compartment is allowed to stand after the dispersed cells are confined.
- culture compartments include wells in multiwell plates (384 wells, 192 wells, 96 wells, 48 wells, 24 wells, etc.), micropores, chamber slides, tubes, medium droplets in the hanging drop method, and the like. can be, but are not limited to: Dispersed cells confined in the compartments are sedimented in one place by gravity, or the cells adhere to each other to form one cell mass per culture compartment.
- the shape of the bottom of multiwell plates, micropores, chamber slides, tubes, etc. is preferably U-bottom or V-bottom so that dispersed cells can easily settle in one place.
- the number of cells to be seeded in one culture compartment is such that one cell cluster is formed per culture compartment, and if the cell cluster can be induced to differentiate into forebrain cells by the method of the present invention,
- the number of cells obtained in step (1) is usually about 1 ⁇ 10 3 to about 5 ⁇ 10 4 , preferably about 1 ⁇ 10 3 to about 2 ⁇ 10 4 per culture compartment, More preferably, about 2 ⁇ 10 3 to about 1.2 ⁇ 10 4 seeds are sown.
- the cells usually about 1 ⁇ 10 3 to about 5 ⁇ 10 4 cells, preferably about 1 ⁇ 10 3 to about 2 ⁇ 10 4 cells, more preferably about 1 ⁇ 10 3 to about 5 ⁇ 10 4 cells per culture compartment A single cluster of 2 ⁇ 10 3 to about 1.2 ⁇ 10 4 cells is formed.
- the cells obtained in step (1) are usually about 1 ⁇ 10 3 to about 5 ⁇ 10 5 , preferably about 1 ⁇ 10 3 to about 2 ⁇ 10 5 , more preferably about 1 ⁇ 10 3 to about 2 ⁇ 10 5 cells per culture compartment. About 2 ⁇ 10 3 to about 1 ⁇ 10 5 seeds are sown. Then, by rapidly agglutinating the cells, usually 5 ⁇ 10 2 to about 5 ⁇ 10 5 cells, preferably about 5 ⁇ 10 2 to about 2 ⁇ 10 5 cells, more preferably about 1 ⁇ 10 5 cells per culture compartment are obtained. A single cell cluster of ⁇ 10 3 to about 1 ⁇ 10 5 cells is formed.
- the time to cell cluster formation can be determined as appropriate as long as one cell cluster is formed per compartment and the cell cluster can be induced to differentiate into cerebral organoids, but is preferably 24 hours.
- Cell clusters are formed within, more preferably within 12 hours.
- the time to form cell clusters is preferably within 48 hours, more preferably within 24 hours.
- culture temperature is not particularly limited, but is, for example, about 30 to 40°C, preferably about 37°C.
- the CO 2 concentration is, for example, about 1-10%, preferably about 5%.
- a qualitatively uniform population of cell masses can be obtained.
- the qualitative uniformity of the cell aggregates is determined by the size and number of cells, macroscopic morphology, microscopic morphology and uniformity thereof by tissue staining analysis, expression of differentiation and undifferentiated markers and their uniformity, differentiation It can be evaluated based on the regulation of marker expression and its synchrony, reproducibility of differentiation efficiency between cell clusters, and the like.
- the term "homogeneous" cell mass population means that 80% or more of the cell mass population is within the range of the average value of the parameter ⁇ 20% in the cell mass population, preferably It means within the range of mean value ⁇ 10%, more preferably mean value ⁇ 5%.
- the culture medium used in step (2a) contains a TGF ⁇ signal inhibitor and a Wnt signaling inhibitor.
- TGF ⁇ signal inhibitors can be used, preferably SB431542, A-83-01, or XAV-939.
- Wnt signaling inhibitors those described above can be used, and preferred examples include IWR-1-end, C59, LGK-974, and DKK-1 (protein).
- Wnt signaling inhibitor and TGF ⁇ signaling inhibitor is IWR-1-endo and SB431542.
- the concentration of the Wnt signaling inhibitor in the medium can be appropriately set within a range in which cell masses can be induced to differentiate into forebrain cells. , 0.1 to 50 ⁇ M, preferably 0.3 to 10 ⁇ M, more preferably 0.3 to 5 ⁇ M.
- the concentration of the TGF ⁇ signal inhibitor in the medium can be appropriately set within a range in which cell masses can be induced to differentiate into forebrain cells. , preferably 1 to 50 ⁇ M, more preferably 1 to 10 ⁇ M, which exhibits TGF ⁇ signal inhibitory activity.
- the culture solution used in step (2a), i.e., the medium used for forming the cell mass and for suspension culture of the cell mass, is not particularly limited as long as it can be used for culturing animal cells, and is defined as above.
- the medium used for culturing animal cells can be prepared from the basal medium.
- basal medium used in step (2a) examples include Glasgow MEM medium, DMEM medium, F-12 medium (also referred to as F12 medium) and DMEM/F12 medium. Glasgow MEM medium is preferably used.
- the medium used for cell cluster formation may contain a serum substitute.
- the above-mentioned serum substitutes can be used, and examples thereof include KSR (knockout serum replacement) (manufactured by Invitrogen), Chemically-defined Lipid concentrated (manufactured by Gibco), and Glutamax (manufactured by Gibco).
- a medium containing about 10-30% serum replacement eg, KSR
- KSR serum replacement
- the medium used during cell cluster formation may contain a serum replacement.
- the above-mentioned serum substitutes can be used, and examples thereof include KSR (manufactured by Gibco), Chemically-defined Lipid concentrated (manufactured by Gibco), and Glutamax (manufactured by Gibco).
- a medium containing an appropriate amount of serum replacement eg, 1-30% KSR
- an appropriate amount of serum replacement eg, 1-30% KSR
- the medium used for suspension culture of cell masses can contain other additives to the extent that they do not adversely affect the induction of differentiation into forebrain cells.
- Additives include, for example, insulin, iron sources (e.g., transferrin), minerals (e.g., sodium selenate, etc.), sugars (e.g., glucose, etc.), organic acids (e.g., pyruvic acid, lactic acid, etc.), serum proteins (e.g., albumin, etc.). ), amino acids (e.g. L-glutamine etc.), reducing agents (e.g. 2-mercaptoethanol etc.), vitamins (e.g. ascorbic acid, d-biotin etc.), antibiotics (e.g. streptomycin, penicillin, gentamicin etc.), buffers ( For example, HEPES, etc.) and the like, but are not limited to these.
- iron sources e.g., transferrin
- minerals e.g., sodium selenate, etc.
- the medium used for the suspension culture of the cell mass contains patterning factors such as Fgf, Wnt, Nodal, Notch, and Shh; It preferably does not contain growth factors such as albumin.
- culture temperature is, for example, about 30-40°C, preferably about 37°C.
- the CO 2 concentration is for example about 1-10%, preferably about 5%.
- the O2 concentration is for example about 20%.
- the suspension culture in step (2a) may be static culture, shaking culture, orbital culture, or stirring culture, but static culture is preferred. Alternatively, static culture may be performed only for a part of the period of step (2a).
- Step (2a) is carried out for a period of time sufficient for the differentiation direction to the forebrain region to be determined and forebrain marker-positive cell clusters (eg, Foxg1-positive cell clusters) to be induced. That is, a cell mass containing forebrain marker-positive cells can be obtained by step (2a).
- forebrain marker-positive cell clusters eg, Foxg1-positive cell clusters
- step (2a) is performed until at least one forebrain marker-positive cell is generated.
- step (2a) is performed, for example, until 50% or more, preferably 70% or more of the cell masses in culture become positive for the forebrain marker.
- step (2a) is performed until 30% or more, preferably 50% or more, more preferably 70% or more of the cells contained in the cell mass generate a forebrain marker-positive cell mass. .
- the culture period in step (2a) may vary depending on the type of Wnt signaling inhibitor and TGF ⁇ signaling inhibitor, and the culture conditions. is used, it is 7 to 30 days, preferably 15 to 20 days (eg, 18 days).
- step (2b) the cell mass obtained in step (2a) is subjected to suspension culture in a culture medium substantially free of TGF ⁇ signal inhibitors and Wnt signal transduction inhibitors to obtain cerebral organoids.
- the suspension culture in step (2b) may be performed under high oxygen partial pressure conditions.
- a high oxygen partial pressure condition means an oxygen partial pressure condition exceeding the oxygen partial pressure in air (20%).
- the oxygen partial pressure in step (2b) is, for example, 30-60%, preferably 35-60%, more preferably 38-60%.
- the medium used in step (2b) is not particularly limited as long as it is a medium used for culturing animal cells, and the medium defined above is prepared as a basal medium. can do.
- basal media examples include Glasgow MEM medium, DMEM medium, F-12 medium, and DMEM/F12 medium.
- DMEM/F12 medium is used.
- the medium used in step (2b) is preferably a serum-free medium, and may contain serum replacement if necessary.
- the above-mentioned serum substitutes can be used, and examples thereof include N2 supplement, B27 supplement, Neurocult SM1 Neuronal supplement, KSR and the like.
- the concentration of the serum substitute may be adjusted as appropriate. Specifically, for example, N2 supplements are about 0.1 to 3%, preferably about 1%, and B27 supplements are about 0.1%. A medium containing ⁇ 10%, preferably 2%, and for KSR, preferably about 1-30% serum replacement can be used.
- step (2b) the Wnt signaling inhibitor and TGF ⁇ signaling inhibitor used in step (2a) are unnecessary.
- the medium used in step (2b) is free of Wnt signaling inhibitors and TGF ⁇ signaling inhibitors.
- the culture medium in step (2b) is preferably a serum-free culture medium substantially free of serum, more preferably a serum-free culture medium containing a serum substitute.
- the medium used in step (2b) preferably contains an N2 supplement as a serum substitute in order to promote differentiation induction into cerebral cortical cells.
- N2 supplement is a known serum-replacement composition containing insulin, transferrin, progesterone, putrescine and sodium selenite, available from Gibco/Thermo Fisher Scientific and others.
- the amount of the N2 supplement added is such that differentiation induction into forebrain tissue or its progenitor tissue, or differentiation induction into nerve cells, and/or differentiation induction into cells constituting the cerebral cortex or their progenitor cells is promoted. can be set as appropriate.
- the medium used in step (2b) preferably contains a chemically defined lipid concentrate for long-term maintenance and culture of the ventricular zone.
- Chemically Defined Lipid Concentrate is a lipid mixture containing cholesterol, DL- ⁇ -tocopherol, arachidonic acid, linolenic acid, linoleic acid, myristic acid, oleic acid, palmitic acid, palmitoleic acid, and stearic acid, each purified.
- a commercially available chemically defined lipid concentrate can be used, for example, it can be purchased from Gibco/Thermo Fisher Scientific.
- the medium used for suspension culture of cell clusters can contain other additives to the extent that they do not adversely affect differentiation induction into cerebral cortical cells.
- Additives include, for example, insulin, iron sources (e.g., transferrin), minerals (e.g., sodium selenate, etc.), sugars (e.g., glucose, etc.), organic acids (e.g., pyruvic acid, lactic acid, etc.), serum proteins (e.g., albumin, etc.). ), amino acids (e.g. L-glutamine etc.), reducing agents (e.g. 2-mercaptoethanol etc.), vitamins (e.g. ascorbic acid, d-biotin etc.), antibiotics (e.g. streptomycin, penicillin, gentamicin etc.), buffers ( For example, HEPES, etc.) and the like, but are not limited to these.
- iron sources e.g., transferrin
- minerals e.g., sodium selenate, etc.
- the medium in step (2b) may contain serum.
- Serum can contribute to long-term maintenance culture of the ventricular zone.
- Serum includes, but is not limited to, FBS and the like.
- the serum is inactivated.
- the serum concentration in the medium can be appropriately adjusted within a range that can contribute to long-term maintenance culture of the ventricular zone, but is usually 1-20% (v/v).
- the medium in step (2b) may contain heparin.
- Heparin may contribute to long-term maintenance of the ventricular zone.
- the heparin concentration in the medium can be appropriately adjusted within a range that can contribute to long-term maintenance culture of the ventricular zone, but is usually 0.5 to 50 ⁇ g/ml, preferably 1 to 10 ⁇ g/ml (eg, 5 ⁇ g/ml). /ml).
- the medium in step (2b) may contain extracellular matrix components.
- Extracellular matrix may contribute to long-term maintenance culture of the ventricular zone.
- Extracellular matrix components refer to various components normally found in extracellular matrices.
- basement membrane components are used in the methods of the present invention. Main components of the basement membrane include, for example, type IV collagen, laminin, heparan sulfate proteoglycan, and entactin.
- Commercially available extracellular matrix components to be added to the medium include, for example, Matrigel (BD Bioscience) and human laminin (Sigma-Aldrich). Matrigel is a basement membrane preparation from Engelbreth Holm Swarn (EHS) mouse sarcoma.
- EHS Engelbreth Holm Swarn
- Matrigel The main components of Matrigel are type IV collagen, laminin, heparan sulfate proteoglycans, and entactin, in addition to TGF ⁇ , fibroblast growth factor (FGF), tissue plasminogen activator, a growth factor naturally produced by EHS tumors. is included.
- FGF fibroblast growth factor
- Matrigel's growth factor reduced products have lower concentrations of growth factors than regular Matrigel, with typical concentrations of ⁇ 0.5 ng/ml EGF, ⁇ 0.2 ng/ml NGF, and ⁇ 5 pg/ml PDGF. , IGF-1 at 5 ng/ml and TGF ⁇ at 1.7 ng/ml.
- the use of growth factor reduced products is preferred in the method of the present invention.
- the concentration of extracellular matrix components in the medium can be appropriately adjusted within a range that can contribute to long-term maintenance and culture of the ventricular zone. volume, more preferably 1/100 volume.
- the medium in step (2b) contains serum and heparin in addition to N2 supplement and Chemically Defined Lipid Concentrate.
- the medium may further contain an extracellular matrix.
- the culture medium of this embodiment is suitable for observing long-term differentiation induction of the telencephalon or its partial tissue, or its progenitor tissue.
- a medium containing N2 supplement, Chemically Defined Lipid Concentrate, serum and heparin (optionally further extracellular matrix) may be used over the entire extent of step (2b), but only for a part of the period medium may be used.
- a medium containing N2 supplement and Chemically Defined Lipid Concentrate and containing no serum, heparin and extracellular matrix is used, and from the middle (for example, in Foxg1 positive aggregates, brain ventricle Neuroepithelial-like structure with similar cavities (multi-columnar columnar epithelium), for example, after the stage of formation of multi-columnar columnar epithelium with multiple hemispherical or spherical cavities), N2 supplement, Chemically Defined Lipid Concentrate, serum, Media may be switched to containing heparin (optionally extracellular matrix).
- culture temperature and CO 2 concentration in step (2b) can be appropriately set.
- the culture temperature is, for example, about 30-40°C, preferably about 37°C.
- the CO 2 concentration is for example about 1-10%, preferably about 5%.
- Step (2b) is carried out for at least a period of time sufficient for forebrain marker (eg, Foxg1)-positive cells to be generated and a cerebral cortex-like structure to be formed.
- forebrain marker eg, Foxg1
- the cerebral cortex-like structure can be confirmed by microscopic observation.
- the culture period in step (2b) may vary depending on the types of Wnt signaling inhibitor and TGF ⁇ signaling inhibitor in step (2a), etc., and thus cannot be specified unconditionally.
- sex stem cells it is at least 10 days, or 10 to 40 days, preferably 10 to 31 days, more preferably 15 to 24 days.
- step (2b) is preferably continued until reaching the desired stage of differentiation, ie, the formation of cerebral organoids comprising a plurality of cerebral cortex-like structures.
- the culture vessel used for suspension culture of the cell mass is not particularly limited, but examples include flasks, tissue culture flasks, dishes, petri dishes, tissue culture dishes, multidishes, microplates, and microwells. Plates, micropores, multiplates, multiwell plates, chamber slides, petri dishes, tubes, trays, culture bags, bioreactors, and roller bottles.
- the incubator is preferably cell non-adherent so as to allow culture under non-adherent conditions.
- Non-cell-adhesive incubators include those whose surface has been artificially treated to make it non-adhesive to cells, and those that have been artificially treated for the purpose of improving adhesion to cells (e.g. , coating treatment with an extracellular matrix, etc.) can be used.
- an oxygen-permeable culture vessel may be used as the culture vessel used for floating culture of the cell mass in step (2b).
- Oxygen supply to the cell mass is improved by using an oxygen permeable incubator.
- an oxygen-permeable incubator may be used in order to avoid the risk that the cell aggregates grow large and the cells in the cell aggregates are not supplied with sufficient oxygen.
- the cell mass may be statically cultured, or the aggregate may be intentionally moved by rotating culture or shaking culture.
- Static culture may be performed throughout the entire period of step (2b), or static culture may be performed only for a part of the period.
- the suspension culture in step (2b) is stationary culture. At this time, it is preferable to culture under the high oxygen partial pressure conditions described above.
- the suspension culture in step (2b) is shaking culture. At this time, culture under high oxygen partial pressure conditions is unnecessary.
- a forebrain marker-positive cerebral cortex-like structure is formed in the cell mass by step (2b).
- 70% or more of the cells contained in the cell mass containing cerebral cortex-like structures are forebrain marker-positive (eg, Foxg1-positive).
- the cerebral cortex-like structure formed in the cell mass in step (2b) is a rosette-like structure with a neuronal layer outside the neuroepithelium.
- the cerebral cortex-like structure has a neural stem cell marker-positive, specifically PAX6 and/or SOX2-positive cell layer on the luminal side, and a phosphorylated Histone H3-positive cell layer on the luminal side. Contains mitotic cells.
- ⁇ III-Tubulin ⁇ TubIII, TUBB3 or TUJ1
- Ctip2 an early cortical plate marker for the cerebral cortex
- the aggregate obtained by the production method of the present invention contains cerebral cortex precursor tissue.
- the method for producing cerebral organoids of the present invention may further include step (3), which is a step of selecting cerebral organoids.
- step (3) from the plurality of cell clusters obtained in step (2), one or more selected from the group consisting of cell cluster shape, internal structure, size, surface color or pattern, and gene expression are used as indicators. , the process of selecting the desired cerebral organoids.
- the indicators it is preferable to adopt 2 or more, 3 or more, or 4 or more, and the indicators are preferably the shape of the cell mass, the internal structure, the surface color or pattern, and/or gene expression.
- cell mass shape, internal structure and/or gene expression are more preferred, and cell mass shape is particularly preferred.
- the shape of the cell mass is spherical (spherical), it can be selected as a desired cerebral organoid.
- Each index for selecting cerebral organoids preferably conforms to the general definition of cerebral organoids, for example, at least one, at least two, at least three of the following (1) to (5), or Cell clusters that fill four may be determined to be cerebral organoids.
- the cerebral organoid preferably expresses one or more genes selected from the group consisting of SLC17A7, NEUROD6, and EMX1, preferably all genes.
- At least one, at least two, at least three or more, or at least five or more of the genes expressed in the non-target cells listed in Table 6 below are not substantially expressed. Furthermore, it is preferable that one or more genes selected from the group consisting of GAD2, COL1A1, TYR, TTR and HOXA2 are not substantially expressed, and 2 or more, 3 or more, 4 or more or all of these markers are It is more preferable not to express.
- the term "substantially not expressed" of a gene means that the expression level of the protein, which is the expression product of the gene, is below the level at which physiological functions are exhibited.
- the state of "substantially not expressed” specifically refers to any control gene that is constantly expressed in cells (e.g., GAPDH, ACTB as a constantly expressed gene). , B2M, 18S ribosomal RNA, etc.) is 1/10 or less of the expression level of the control gene.
- the spherical structure in (1) above and the cerebral cortex-like structure in (2) above are as described above.
- representative examples of cell clusters that are spherical but do not have cerebral cortex-like structures are the cell clusters called “Potato-like” or “Jelly-like” in FIG. is.
- Pigmentation in (3) above means that a part of the cell mass has a black or brown area.
- a representative example of the cell mass with pigmentation is the cell mass called "Pigment" in FIG.
- a projection shape, or a balloon-like shape in part of the cell mass in (4) above means that the cell mass has a highly transparent bag-like structure, a fibrous epithelial structure, Or it means that there is no protrusion-like structure.
- a representative example of the cystic shape is the shape called "Transparent” in FIG.
- a representative example of the projection shape is a shape called "Cotton-like” in FIG.
- a representative example of the balloon-like shape is the shape called "Balloon” in FIG.
- the features (1) to (4) above can be determined and removed by visual observation after the cell mass is enlarged as the case may be.
- the shape may be determined by an apparatus using software capable of image analysis based on an enlarged image of a microscope. In this case, a method such as deep learning may be used to improve the accuracy of shape discrimination.
- the cerebral organoid may be a spherical cell mass with a diameter (equivalent circle diameter) of about 100 ⁇ m to 10000 ⁇ m, preferably about 500 ⁇ m to 5000 ⁇ m.
- the cerebral cortex-like structure contained inside the cell mass may have a diameter (equivalent circle diameter) of about 10 ⁇ m to 1000 ⁇ m, preferably about 50 ⁇ m to 500 ⁇ m.
- the expression of a gene can be determined by its expression level.
- the expression level of a gene can be evaluated by the amount of expression product (mRNA or protein or fragment thereof) of the gene, preferably the expression level of mRNA. Specifically, it can be determined by quantitative RT-PCR, RT-PCR, next-generation sequence analysis, microarray analysis, Western blotting, ELISA, immunostaining, flow cytometry, and the like.
- the determination may be made for each cell mass, or in the case of the same lot, a certain number of cell masses (eg, 1 or more, 3 or more, 5 or more) may be sampled for determination.
- the shape, internal structure, size, and surface color or pattern of a cell mass can be determined by visual observation under an inverted microscope, preferably for each cell mass.
- Gene expression is preferably determined by sampling rather than by cell mass.
- quantitative RT-PCR method, RT-PCR method, next-generation sequence analysis, microarray analysis, Western blotting method, ELISA method, immunostaining method , flow cytometry, and the like are examples of cell masses selected by shape and / or internal structure.
- step (3) cell clusters determined to be cerebral organoids by the determination method described above are collected and selected as cerebral organoids.
- the cerebral organoids sorted in step (3) are enriched in cerebral organoids compared to when they are not sorted. That is, through step (3), the ratio of cerebral organoids in the population of cell masses can be further increased.
- the cell culture of the present invention is a cell culture produced using the method for producing a cerebral organoid comprising steps (1) and (2) described above, and contains a plurality of spherical cell aggregates.
- the ratio can be calculated by sampling a cell culture and calculating the number of cerebral organoids in the total number of spherical cell aggregates in the sample.
- a cerebral organoid is a spherical cell mass that encloses one or more, preferably multiple, cerebral cortex-like structures with a neuronal layer on the outside of the neuroepithelium.
- the determination method of cerebral organoids conforms to the method of step (3) above.
- the proportion of cerebral cortex-like structures in individual cerebral organoids may be 20% or more, preferably 40% or more, and more preferably 50% or more.
- the ratio of cerebral cortex-like structures to the cerebral organoid is the ratio of the cerebral cortex-like structures to the area or volume of the entire cerebral organoid, and from the viewpoint of easy evaluation, it is preferably the ratio of the area.
- the ratio may be the average value of a plurality (eg, 2 to 20, preferably 5 or 10) of cerebral organoids.
- the ratio may be evaluated, for example, by the ratio of cerebral cortex-like structures to the cross-sectional area of a section containing the central part of the cerebral organoid.
- the central part means the central part of the cross section having the maximum diameter of the cerebral organoid.
- immunostaining is performed on cerebral organoid sections, and cells that are positive for neural stem cell markers and / or neural progenitor cell markers, and neuronal markers are determined as cerebral cortex-like structures, and cerebral cortex-like It may be performed by calculating the ratio of the area of the structure to the area of the section. Alternatively, it may be calculated as the ratio of the volume of the cerebral cortex-like structure to the volume of the cerebral organoid.
- the neural stem cell and/or neural progenitor cell marker includes Pax6, and the neuronal cell marker includes the forebrain marker Foxg1, the cerebral cortex marker Ctip2, and the like.
- 20% or more, preferably 40% or more, preferably 70% or more of the total number of cells are Foxg1-positive cells.
- the cerebral cortex-like structure has a neural stem cell marker: PAX6 and/or SOX2-positive cell layer on the luminal side, and contains phosphorylated Histone H3-positive mitotic cells in the most luminal part. .
- ⁇ III-Tubulin a marker for postmitotic neurons
- ⁇ TubIII, TUBB3 or TUJ1 a marker for postmitotic neurons
- ⁇ TubIII, TUBB3 or TUJ1 a marker for postmitotic neurons
- ⁇ TubIII, TUBB3 or TUJ1 a marker for postmitotic neurons
- Ctip2 and/or Tbr1 are included. They contain Reelin-positive Cajal-Retius cells, the first layer of neurons in the cerebral cortex, and may have a Laminin-rich layer near the surface.
- the cerebral organoid may be a cerebral organoid produced using the method for producing a cerebral organoid, which includes steps (1) and (2) above.
- a cerebral organoid of one embodiment may be a cerebral organoid selected by the above step (3), or may be a cell mass having the following characteristics (1) to (5).
- the cerebral organoids express one or more, two or more selected from the group consisting of SLC17A7, NEUROD6, and EMX1, or all of them. Furthermore, it is preferable that one or more genes selected from the group consisting of GAD2, COL1A1, TYR, TTR and HOXA2 are not substantially expressed, and 2 or more, 3 or more, 4 or more or all of these markers are It is more preferable not to express.
- the cerebral organoid is as described above, and the number of cells per cell mass is about 5 ⁇ 10 3 to 5 ⁇ 10 6 , preferably about 1 ⁇ 10 4 to 3 ⁇ 10 6 .
- Method 1 for producing cerebral cortical cell mass includes a method for producing a cerebral cortex cell mass from pluripotent stem cells in the absence of feeder cells, including the following steps (i) and (ii). (i) obtaining cerebral organoids from pluripotent stem cells; (ii) culturing the cerebral organoids obtained in step (i) in a culture medium containing a Notch signal inhibitor, preferably a ⁇ -secretase inhibitor, to obtain a cerebral cortex cell mass;
- a Notch signal inhibitor preferably a ⁇ -secretase inhibitor
- the method for obtaining cerebral organoids from pluripotent stem cells is not limited, and they can be prepared by methods well known to those skilled in the art. Examples of such a well-known method include step (2) in the method for producing cerebral organoids in 2 above.
- Method 1 for producing cerebral cortical cell mass comprises obtaining cerebral organoids from pluripotent stem cells by the method for producing cerebral organoids described in 2 above in step (i). That is, Method 1 for producing a cerebral cortical cell mass includes treating the cerebral organoids obtained by the method for producing a cerebral organoid described in 2 above in the following steps (i) and (ii). A method for producing cerebral cortical cell masses from pluripotent stem cells in the presence.
- step (i) obtaining cerebral organoids by the method for producing cerebral organoids according to 2 above (that is, including the method according to any one of claims 1 to 10); (ii) culturing the cerebral organoids obtained in step (i) in a culture medium containing a Notch signal inhibitor, preferably a ⁇ -secretase inhibitor, to obtain a cerebral cortex cell mass;
- a Notch signal inhibitor preferably a ⁇ -secretase inhibitor
- Step (i) is as described in step (1), step (2a) and step (2b) in 2 above.
- Step (i) may further include step (3) in 2 above.
- step (2b) is performed for about 7-31 days, preferably about 7-21 days.
- step (ii) is performed by optionally performing selection according to step (3) and replacing the medium with a medium containing a Notch inhibitor, preferably a ⁇ -secretase inhibitor. be able to.
- step (ii) may be performed after step (2b) described above, followed by screening in step (3).
- Notch signal inhibitors used in step (ii) preferably include ⁇ -secretase inhibitors.
- ⁇ -secretase inhibitors include N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) or Compound E.
- the culture medium used may be the same as the culture medium used in step (i), that is, step (2b) of the production method of 2, except that it contains a Notch signal inhibitor. can.
- a culture medium different from the culture medium used in step (2b) may be appropriately selected from the culture conditions described in step (2b) of the production method in 2 above.
- step (ii) may conform to step (2b) as appropriate.
- the concentration of the Notch signal inhibitor, preferably the ⁇ -secretase inhibitor, in the culture medium can be appropriately set within a range where proliferative cells such as neural stem cells that may be contained in the cell mass can be suppressed.
- DAPT a ⁇ -secretase inhibitor
- ⁇ -secretase activity corresponding to 0.1 to 1000 ⁇ M, 1 to 100 ⁇ M, preferably 1 to 30 ⁇ M, more preferably 5 to 20 ⁇ M or the ⁇ - Concentrations exhibiting Notch signaling inhibitory activity based on secretase activity can be mentioned.
- Step (ii) is performed about 28 to 49 days, preferably about 28 to 44 days after the start of suspension culture in step (i) (start of induction of differentiation into nerve cells).
- the culture period of step (ii) is 1 to 7 days, preferably 2 to 6 days, more preferably 2 to 4 days.
- Method 2 for producing cerebral cortical cell mass includes a method for producing cerebral cortical cell clusters from pluripotent stem cells in the absence of supporting cells, the method comprising (i) obtaining cerebral organoids from pluripotent stem cells; (ii) culturing the cerebral organoids obtained in step (i) in a culture medium; (iii) dispersing the cell culture obtained in step (ii) into a single cell or a cell clump of 2 to 5 cells; (iv) culturing the cell culture obtained in step (ii) or the cell population obtained in step (iii) in a medium containing one or more neurotrophic factors, ascorbic acid, and a cAMP activator;
- the step of dispersing into a single cell or a cell clump of 2 to 5 cells in the step (iii) is an optional step, and the culture medium and / or the step in the step (ii)
- the culture solution (iv) contains a Notch signal inhibitor.
- step (i) the method for obtaining cerebral organoids from pluripotent stem cells is not limited, and they can be prepared by methods well known to those skilled in the art.
- cerebral organoids are obtained from pluripotent stem cells by the method for producing cerebral organoids described in 2 above, particularly the production method including steps (1), (2a) and (2b).
- the culture solution in step (ii) and/or the culture solution in step (iv) may contain a Notch signal inhibitor during part or all of the steps.
- a culture medium not containing the Notch signal inhibitor is used when exchanging the culture medium in step (ii) or the culture medium in step (iv).
- Part of the period is not particularly limited as long as the effect of treatment with a Notch signal inhibitor is obtained, and specifically several hours to several days during the culture period of the step (ii) or the step (iv) may be 1-7 days, preferably 2-6 days, more preferably 2-4 days. It may also be repeated for very short treatment periods (eg, 4 hours, 12 hours, 24 hours, 2 days).
- a period containing a Notch signal inhibitor and a period not containing a Notch signal inhibitor are alternately performed, or combinations using Notch signal inhibitors at different concentrations are also within the scope of the present application.
- the description of step (ii) in Method 1 for producing cerebral cortical aggregates can be referred to.
- One aspect of the present invention includes “Method 2-1 for producing cerebral cortical cell mass”, in which step (i) is performed according to the method for producing cerebral organoids described in 2 above, and step (iii) is included. That is, the method 2-1 for producing a cerebral cortex cell mass includes the following step (iii) and step ( A method for producing a highly purified cerebral cortical cell mass, comprising treatment with iv-1). This is a highly purified cerebral cortical cell mass that has been dispersed and reaggregated.
- the cerebral organoids obtained in step (i) are cultured in a medium containing a Notch signal inhibitor, preferably a ⁇ -secretase inhibitor.
- the step (ii) may be performed in the same manner as the step (ii) in the method 1 for producing a cerebral cortical cell cluster in 4 above.
- (iii) dispersing the cerebral organoids obtained in step (ii) into single cells or cell clumps of 2 to 5 cells;
- the cell population obtained in step (iii) is cultured in a serum-free medium containing one or more neurotrophic factors, ascorbic acid and a cAMP activator (eg, dibutyryl cAMP (dbcAMP)). to obtain a cell mass (consisting of, for example, 20 or more cells).
- a cAMP activator eg, dibutyryl cAMP (dbcAMP)
- the cerebral organoids obtained in step (ii) can be divided into single cells or clusters of 2 to 5 cells (cell clumps) by methods well known to those skilled in the art, such as physically by pipetting or by enzymatic treatment. can be dispersed.
- the cell population obtained by dispersion may be a single cell and/or a population of 2 to 5 cell clumps. That is, in step (iii), the cerebral organoids obtained in step (ii) may be dispersed into single cells or clusters of 2 to 5 cells (cell clumps) by methods well known to those skilled in the art.
- the culture medium used in step (iv-1) is a culture medium containing one or more neurotrophic factors, ascorbic acid or a derivative thereof, and a cAMP activator.
- neurotrophic factor 1 to 4, 1 to 3, preferably 1 or 2 factors may be selected from those defined above.
- neurotrophic factors preferably BDNF and/or GDNF can be mentioned.
- the concentration of BDNF includes 1-100 ng/mL, preferably 10-30 ng/mL.
- the concentration of GDNF includes 1-100 ng/mL, preferably 5-20 ng/mL.
- the concentration of ascorbic acid or a derivative thereof includes, in the case of ascorbic acid, a concentration corresponding to 10 to 500 ⁇ M, preferably 50 to 300 ⁇ M, and similar concentrations for derivatives. be done.
- the concentration of the cAMP activator includes, for example, a concentration capable of activating cAMP equivalent to 10 to 1000 ⁇ M, preferably 100 to 600 ⁇ M, more preferably 300 to 500 ⁇ M when dbcAMP is used.
- the culture period of step (iv-1) is 1 to 21 days, preferably 2 to 15 days, 2 to 10 days, 2 to 8 days, 2 to 6 days, more preferably about 4 days.
- the culture period in step (iv-1) is 1 to 40 days, preferably 2 to 28 days, more preferably 2 to 14 days.
- the suspension culture in step (iv-1) can be performed by referring to the method described in step (2b) of 2 above.
- the same medium as in step (2b) of 2 above may be added with one or more neurotrophic factors, ascorbic acid or a derivative thereof, and a cAMP activator to prepare and use the medium.
- the culture medium used in step (iv-1) may optionally contain a ROCK inhibitor.
- a ROCK inhibitor those defined above can be appropriately used, preferably Y-27632 and the like.
- the concentration of the ROCK inhibitor used here corresponds to, for example, about 0.1 to 200 ⁇ M, preferably about 2 to 100 ⁇ M, more preferably about 30 to 100 ⁇ M when Y-27632 is used as the ROCK inhibitor. concentration.
- the cerebral cortical cell aggregates obtained through step (iii) are highly purified cerebral cortical cell aggregates through the steps of dispersion-reaggregation.
- step (iii) is not included in the method 2-1 for producing a cerebral cortex cell mass described in 5 above. That is, in the method 1 for producing a cerebral cortex cell mass described in 4 above, in addition to the steps (i) and (ii), the following step (iv-2) is performed to produce a cerebral cortex cell mass. manufacturing methods.
- step (ii) the cerebral organoids obtained in step (i) are cultured in a medium containing a Notch signal inhibitor, preferably a ⁇ -secretase inhibitor.
- step (iv-2) The cell culture containing the cerebral organoids obtained in step (ii) is treated with one or more neurotrophic factors, ascorbic acid or a derivative thereof (eg, ascorbic acid-2-phosphate), and cAMP activation.
- a step of obtaining a cerebral cortex cell mass by suspension culture in a culture medium containing an agent for example, dibutyryl cAMP (dbcAMP)).
- a cell mass containing cerebral cortical cells (cerebral cortical cell mass) can be obtained by culturing the cell culture containing the cerebral organoids obtained in step (ii) in a suspension culture.
- the culture medium used in step (iv-2) includes the culture medium described in step (iv-1) of 5 above. ).
- Methods for producing cerebral cortical cell clusters 3-1 to 3-7 includes “Method 3-1 for producing cerebral cortex cell mass”, wherein the culture medium in step (ii) contains a Notch signal inhibitor. That is, the method 3-1 for producing a cerebral cortex cell mass includes a method for producing a cerebral cortex cell mass including the following step (I).
- step (I) A step of culturing pluripotent stem cell-derived cerebral organoids in a medium containing a Notch inhibitor, preferably a ⁇ -secretase inhibitor.
- step (I) may be performed in the same manner as step (ii) in method 1 for producing a cerebral cortex cell mass.
- An aspect of the present invention includes “Method 3-2 for producing cerebral cortical cell clusters”, which includes the following steps in addition to the above step (I).
- (II) a step of dispersing the cell culture containing the cerebral organoids obtained in step (I) into a single cell or a cell clump of 2 to 5 cells, and (III) in step (II) A step of culturing the resulting cell population in a culture medium containing one or more neurotrophic factors, ascorbic acid or a derivative thereof, and a cAMP activator to obtain a cerebral cortical cell mass.
- step (I) may be performed in the same manner as step (ii) in method 1 for producing a cerebral cortex cell mass
- step (II) and step (III) are the steps ( iii) and step (iv-1) may be performed in the same manner.
- the cerebral cortex cell aggregates obtained through step (III) have undergone the dispersion-reaggregation process, and are high-purity cerebral cortex cell aggregates.
- One aspect of the present invention includes an aspect that does not include the step (II). That is, in addition to the step (I), there is a “Method 3-3 for producing a cerebral cortex cell mass” including the following steps. (III-2) The cell culture containing the cerebral organoids obtained in step (I) was treated with one or more neurotrophic factors, ascorbic acid or a derivative thereof (eg, ascorbic acid-2-phosphate), and cAMP activation. A step of obtaining a cerebral cortex cell mass by suspension culture in a culture medium containing an agent (for example, dibutyryl cAMP (dbcAMP)).
- an agent for example, dibutyryl cAMP (dbcAMP)
- step (I) may be performed in the same manner as step (ii) in method 1 for producing a cerebral cortex cell mass
- step (III-2) is step (iv-1) in method 2 for producing a cerebral cortex cell mass. can be done in the same way as
- One aspect of the present invention includes “Method 3-4 for producing cerebral cortical cell mass”, which includes the following steps (I-2), (II) and (III-3).
- (I-2) a step of culturing cerebral organoids derived from pluripotent stem cells in a culture medium;
- (II) a step of dispersing the cell culture containing the cerebral organoids obtained in step (I-2) into a single cell or a cell clump of 2 to 5 cells, and
- the culture medium in step (I-2) does not contain a Notch signal inhibitor, and the culture medium in step (III-3) contains a Notch signal inhibitor.
- step (I-2) may be performed in the same manner as step (2b) in the method for producing cerebral organoids described in 2 above.
- the step (I-2) may be a culture corresponding to a part of the steps for producing pluripotent stem cell-derived cerebral organoids, and the culture period is not particularly limited.
- Step (II) may be performed in the same manner as step (iii) in method 2-1 for producing a cerebral cortex cell mass described in 5 above.
- the culture medium in step (III-3) may contain a Notch signal inhibitor during part or all of the step. If the treatment period with the Notch signal inhibitor is only part of the period, a culture medium that does not contain the Notch signal inhibitor is used when exchanging the culture medium in step (III-3).
- Part of the period is not particularly limited as long as the effect of treatment with a Notch signal inhibitor is obtained, and several hours to several days in the culture period of the above step (III-3), specifically 1 to It may be 7 days, preferably 2-6 days, more preferably 2-4 days. It may also be repeated for very short treatment periods (eg, 4 hours, 12 hours, 24 hours, 2 days).
- a period containing a Notch signal inhibitor and a period not containing a Notch signal inhibitor are alternately performed, or combinations using Notch signal inhibitors at different concentrations are also within the scope of the present application.
- the culture medium of steps (III-4) and (III-5) below may also contain a Notch signal inhibitor during part or all of the steps, like the culture medium of step (III-3).
- Step (III-3)> The cell population obtained in step (II) is reaggregated by suspension culture to obtain a cell mass containing highly purified cerebral cortical cells (highly purified cerebral cortical cell mass).
- Examples of the culture medium used in step (III-3) include the culture medium described in step (iv-1) of method 2 for producing a cerebral cortical cell mass described in 5 above.
- the period also includes those described in step (iv-1) of 5 above.
- the culture solution contains a Notch signal inhibitor, preferably a ⁇ -secretase inhibitor, during part or all of the suspension culture period.
- a Notch signal inhibitor preferably a ⁇ -secretase inhibitor
- the Notch signal inhibitor used here those described in the definition can be appropriately selected and used, but the Notch signal inhibitor preferably includes a ⁇ -secretase inhibitor.
- the ⁇ -secretase inhibitor those described in the definition can be appropriately selected and used.
- DAPT ⁇ -S-phenylglycine t-butylester
- the concentration of the Notch signal inhibitor, preferably the ⁇ -secretase inhibitor, in the culture medium can be appropriately set within a range that can suppress proliferating cells that may be contained in the generated cell mass.
- ⁇ -secretase activity corresponding to 0.1 to 1000 ⁇ M, 1 to 100 ⁇ M, preferably 1 to 30 ⁇ M, more preferably 5 to 20 ⁇ M or the ⁇ - Concentrations exhibiting Notch signaling inhibitory activity based on secretase activity can be mentioned.
- step (III-3) there is no particular limitation on the time to start culturing in the presence of a Notch signal inhibitor, preferably a ⁇ -secretase inhibitor, but for example, 28 to 42 days after initiation of differentiation can be mentioned.
- the culture period is preferably about 1 day to about 20 days, more preferably 2 days to 8 days.
- the cerebral cortical cell aggregates obtained through step (III-3) have undergone the steps of dispersion-reaggregation, and are high-purity cerebral cortical cell aggregates.
- step (I), the step (II) and the step (III-4) are included, and both the culture solution of the step (I) and the culture solution of the step (III-4) Examples include an embodiment containing a Notch signal inhibitor (Method 3-5 for producing cerebral cortex cell mass).
- step (I) may be performed in the same manner as step (ii) in method 1 for producing a cerebral cortex cell mass described in 4 above
- step (II) is the method for producing a cerebral cortex cell mass described in 5 above.
- step (III-4) may be performed in the same manner as step (III-3) in method 3-4 for producing a cerebral cortical cell mass.
- the cerebral cortex cell mass obtained through step (III-4) has undergone the steps of dispersion-reaggregation and is a highly purified cerebral cortex cell aggregate.
- step (I) and step (III-5) are included, and the culture medium in step (III) contains a Notch signal inhibitor (Method 3-6 for producing cerebral cortical cell mass ).
- step (I) may be performed in the same manner as step (ii) in method 1 for producing a cerebral cortex cell mass described in 4 above
- step (III-5) is the method for producing a cerebral cortex cell mass 3- It may be carried out in the same manner as the step (III-3) in 4.
- step (I) and/or step (III-5) culturing in a culture medium containing a Notch signal inhibitor means part of step (I) and/or step (III-5)
- the medium used during the culture period or the entire culture period is meant to contain a Notch signaling inhibitor.
- the culture medium not containing the Notch signal inhibitor at the time of exchanging the culture medium in step (I) or the culture medium in step (III-5) Use This partial period is not particularly limited as long as the effect of treatment with the Notch signal inhibitor is obtained, and several hours to several days during the culture period of the step (I) or the step (III-5), Specifically, it may be 1 to 7 days, preferably 2 to 6 days, more preferably 2 to 4 days. Alternatively, short treatment periods (eg, 4 hours, 12 hours, 24 hours, 2 days) may be repeated. In addition, a period containing a Notch signal inhibitor and a period not containing a Notch signal inhibitor are alternately performed, or combinations using Notch signal inhibitors at different concentrations are also within the scope of the present application.
- One aspect of the present invention includes an aspect (method 3-7 for producing a cerebral cortex cell mass) in which the step (I) of the method 3-6 for producing a cerebral cortex cell mass is changed to the step (I-2). .
- the production method 3-6 of the cerebral cortex cell mass is followed except that the step (I) is changed to the step (I-2).
- the culture medium of step (III-5) further contains a Notch signaling inhibitor in the medium containing one or more neurotrophic factors, ascorbic acid or a derivative thereof, and a cAMP activator.
- the method for producing the cerebral organoids used in step (I) or (I-2) is not particularly limited, and can be prepared by methods well known to those skilled in the art. can be done. For example, it can be produced according to the method for producing cerebral organoids described in 2 above. Alternatively, without performing step (1) of the method for producing cerebral organoids described in 2 above, using pluripotent stem cells instead of "cells obtained in step (1)" in step (2) (2) ) should be implemented.
- step 2 (2b) instead of the treatment in steps (III) to (III-5), or nerve cells may be cultured in a medium that allows them to survive.
- addition or non-addition of a Notch signal inhibitor can be selected.
- the above 4 method 1 for producing a cerebral cortex cell mass the above 5 method 2 for producing a cerebral cortex cell mass, the above 6 method 2-2 for producing a cerebral cortex cell mass, and the above 7 cerebral cortex
- the above 4 method 1 for producing a cerebral cortex cell mass the above 5 method 2 for producing a cerebral cortex cell mass
- the above 6 method 2-2 for producing a cerebral cortex cell mass
- the above 7 cerebral cortex In the absence of supporting cells, including the step of collecting the cerebral cortex cell mass obtained by cell mass production methods 3-1 to 3-7 and preparing a cerebral cortex cell preparation containing the cell mass and medium, A method of producing a cerebral cortex cell preparation from pluripotent stem cells is included.
- media for preparing cerebral cortex cell preparations include non-frozen media such as physiological saline, phosphate buffer (PBS(-)), Hank's balanced salt solution (HBSS), Earl's balanced salt solution, Solutions such as art celebrities can be mentioned.
- non-frozen media such as physiological saline, phosphate buffer (PBS(-)), Hank's balanced salt solution (HBSS), Earl's balanced salt solution, Solutions such as art celebrities can be mentioned.
- media for preparing cerebral cortex cell preparations include, in the case of freezing, cryoprotective agents such as DMSO, glycerol, polyethylene glycol, propylene glycol, glycerin, polyvinylpyrrolidone, sorbitol, dextran, Solutions containing trehalose and the like, and commercially available cryopreservation solutions such as Cell banker, Stem cell banker, Bum banker and the like can be mentioned.
- cryoprotective agents such as DMSO, glycerol, polyethylene glycol, propylene glycol, glycerin, polyvinylpyrrolidone, sorbitol, dextran, Solutions containing trehalose and the like
- cryoprotective agents such as DMSO, glycerol, polyethylene glycol, propylene glycol, glycerin, polyvinylpyrrolidone, sorbitol, dextran, Solutions containing trehalose and the like
- Cerebral Cortex Cell Clusters One aspect of the present invention includes cerebral cortex cell clusters having the following properties.
- the cerebral cortex cell mass can be produced by the method 1 for producing a cerebral cortex cell mass described in 4 above.
- one aspect of the present invention includes a cerebral cortical cell cluster having the following characteristics (a) to (c).
- the number of proliferative marker-positive cells is 10% or less of the total number of cells;
- the number of cells positive for one or more markers selected from the group consisting of neuronal markers, cortical V/VI layer markers, and forebrain markers is 60% or more, preferably 70% or more of the total number of cells; more preferably 80% or more,
- Ki67 is an example of the proliferative marker.
- the neuronal cell marker includes ⁇ III-Tubulin ( ⁇ TubIII).
- the cortical layer V/VI markers include Ctip2.
- the forebrain markers include Foxg1.
- One aspect includes a cerebral cortical cell mass having the following characteristics: (a') the number of Ki67-positive cells is 5% or less of the total number of cells; (b') ⁇ III-Tubulin ( ⁇ TubIII), Ctip2, and Foxg1-positive cells account for 60% or more, preferably 70% or more, and more preferably 80% or more of the total number of cells; (c) substantially free of neuroepithelial or cerebral cortex-like structures;
- Neuroepithelial and cerebral cortex-like structures in cerebral organoids can be visually confirmed under an inverted microscope.
- substantially no neuroepithelium- or cerebral cortex-like structures include the fact that the above-mentioned neuroepithelium- or cerebral cortex-like structures cannot be visually identified by observation with an inverted microscope or the like.
- the cerebral cortical cell mass further comprises (d) NEUROD6, NEUROD2, SSTR2, TBR1, ZBTB18, NHLH1, IGFBPL1, NRN1, RTN1, THSD7A, NRXN1, BHLHE22, CALB2, KHDRBS3, CCSAP, PDE1A, NEUROD1, NPTX1 , NXPH4, NTS, NEUROG2, OLFM1, PRDM8, CORO2B, TP53I11, ZFPM2, PCDH9, NELL2, SRRM4, SCG3, DCC, EPB41L3, SLC17A7, ST18, NSG2, EMX1, CAP2, SYT4, NSMF, ANK3, MYT1L, FSTL5, , B3GAT1, EPHA5, NHLH2, and DLL3.
- the cerebral cortex cell mass preferably expresses one or more genes selected from the group consisting of SLC17A7, NEUROD6, and EMX1, and more preferably one or more selected from the group consisting of GAD2, COL1A1, TYR, TTR, and HOXA2. gene is not substantially expressed.
- the cerebral cortex cell mass further includes (e) neural stem cell marker-positive cells of 10% or less of the total number of cells,
- the neural stem cell markers include Pax6, Sox1 or Sox2.
- the cerebral cortical cell mass is further (f) negative for pluripotency markers.
- Pluripotent marker-negative means that pluripotent stem cells are not substantially detected, specifically pluripotent marker-positive cells are 1% or less of the total number of cells. Oct4 etc. are mentioned as said pluripotency marker.
- the cerebral cortex cell mass may further contain (g) cortex II to IV layer marker (SATB2)-positive cells in some cases.
- SATB2 cortex II to IV layer marker
- One aspect of the present invention includes a cell population containing 10% or more, preferably 20% or more, more preferably 40% or more, and even more preferably 50% or more of the total cell mass, the cerebral cortex cell mass.
- the cerebral cortical cell mass is: It may be a globular-like cell mass with a diameter (equivalent circle diameter) of about 100 ⁇ m to 1000 ⁇ m, preferably about 300 ⁇ m to 600 ⁇ m.
- the number of cells per cell clump may be about 1 ⁇ 10 3 to 5 ⁇ 10 4 , preferably about 5 ⁇ 10 3 to 2 ⁇ 10 4 .
- the cerebral cortex cell mass may be a spherical cell mass with a diameter (equivalent circle diameter) of about 100 ⁇ m to 5000 ⁇ m, preferably about 300 ⁇ m to 2000 ⁇ m.
- the number of cells per cell clump may be about 5 ⁇ 10 3 to 5 ⁇ 10 6 , preferably about 1 ⁇ 10 4 to 3 ⁇ 10 6 .
- the cell population of the cerebral cortical cell mass has an average diameter (equivalent circle diameter) of about 300 ⁇ m to 2000 ⁇ m.
- the cerebral cortex cell mass and the cell population thereof are characterized in that, when transplanted into the brain in vivo, they engraft to the transplant site and suppress proliferation of the transplant-derived cells. and
- the volume of the graft 3 months after transplantation of the cerebral cortical cell mass at 5 weeks of differentiation induction is 2% to 50% compared to the cerebral organoid graft at the same time. Suppressed.
- cerebral cortical cell clusters and cell populations thereof are useful for cell transplantation therapy for patients suffering from cerebrovascular disease or head injuries, as described later.
- a high-purity cerebral cortical cell cluster having the following properties, in which proliferative marker-positive cells are suppressed and the target nerve cell content is improved, is the method for producing a cerebral cortical cell cluster according to 5 above, 2-1. , method 2-2 for producing a cerebral cortex cell mass described in 6 above, or method 3-1 to 3-6 for producing a cerebral cortex cell mass described in 7 above.
- one aspect of the present invention includes a highly purified cerebral cortical cell mass having the following characteristics.
- the number of proliferative marker-positive cells is 5% or less, preferably 3% or less, more preferably 1% or less of the total number of cells;
- One or more marker-positive cells selected from neuronal markers, cortical V/VI layer markers, and forebrain markers account for 60% or more, preferably 70% or more, more preferably 80% or more of the total number of cells to be
- C be substantially free of neuroepithelial or cerebral cortex-like structures;
- Ki67 is an example of the proliferative marker.
- the neuronal cell marker includes ⁇ III-Tubulin ( ⁇ TubIII).
- the cortical layer V/VI markers include Ctip2.
- the forebrain markers include Foxg1.
- One embodiment includes highly purified cerebral cortical cell clusters having the following characteristics: (A) the number of Ki67-positive cells is 5% or less of the total number of cells; (B) ⁇ III-Tubulin ( ⁇ TubIII), Ctip2, and Foxg1-positive cells account for 60% or more, preferably 70% or more, and more preferably 80% or more of the total number of cells; (C) be substantially free of neuroepithelial or cerebral cortex-like structures;
- substantially no neuroepithelium- or cerebral cortex-like structures include the fact that the above-mentioned neuroepithelium- or cerebral cortex-like structures cannot be visually identified by observation with an inverted microscope or the like.
- a method for obtaining a highly purified cerebral cortical cell mass that is, an effective step for increasing the content of cerebral cortical cells in the cerebral cortical cell mass and decreasing the content of unintended cells such as proliferative cells is , the steps described in step (iv) or step (III) above (the Notch signal inhibitor may be present but not essential), more preferably in addition to the steps described above ( iii) or a step comprising dispersing and reaggregating the cerebral cortical cell clusters described in step (II).
- the highly purified cerebral cortical cell mass further includes (D) NEUROD6, NEUROD2, SSTR2, TBR1, ZBTB18, NHLH1, IGFBPL1, NRN1, RTN1, THSD7A, NRXN1, BHLHE22, CALB2, KHDRBS3, CCSAP, PDE1A, NEUROD1 , NPTX1, NXPH4, NTS, NEUROG2, OLFM1, PRDM8, CORO2B, TP53I11, ZFPM2, PCDH9, NELL2, SRRM4, SCG3, DCC, EPB41L3, SLC17A7, ST18, NSG2, EMX1, CAP2, SYT4, NSMF, ANK3, MYT5L, FSTLT5L , CELF4, B3GAT1, EPHA5, NHLH2, and DLL3.
- D NEUROD6, NEUROD2, SSTR2, TBR1, ZBTB18, NHLH1, IGFBPL1, NRN1, RTN1, THSD7A, NRXN1,
- the cerebral cortical cell mass preferably expresses one or more genes selected from the group consisting of SLC17A7, NEUROD6, and EMX1, or all genes, and more preferably the group consisting of GAD2, COL1A1, TYR, TTR, and HOXA2. does not substantially express one or more genes selected from
- the high-purity cerebral cortical cell mass further includes (E) neural stem cell marker-positive cells of 10% or less, preferably 5% or less, more preferably 3% or less of the total number of cells,
- the neural stem cell markers include Pax6, Sox1 or Sox2.
- the high-purity cerebral cortical cell mass is further (F) negative for pluripotent markers.
- Pluripotent marker-negative means that pluripotent stem cells are not substantially detected, specifically pluripotent marker-positive cells are 1% or less of the total number of cells.
- the above pluripotency markers include Oct4 and the like.
- the cerebral cortex cell mass may further contain (G) cortex II to IV layer marker (SATB2)-positive cells in some cases.
- G cortex II to IV layer marker
- One aspect of the present invention includes a cell population containing 10% or more, preferably 20% or more, more preferably 40% or more, and even more preferably 50% or more of the total cell mass, the cerebral cortex cell mass.
- the highly purified cerebral cortical cell mass may be a spherical cell mass with a diameter (equivalent circle diameter) of about 100 ⁇ m to 10000 ⁇ m, preferably about 200 ⁇ m to 3000 ⁇ m.
- the number of cells per cell clump may be about 1 ⁇ 10 3 to 5 ⁇ 10 6 , preferably about 5 ⁇ 10 3 to 3 ⁇ 10 6 .
- the cell population of the high-purity cerebral cortical cell mass has an average diameter (equivalent circle diameter) of about 300 ⁇ m to 500 ⁇ m.
- the highly purified cerebral cortex cell mass and the cell population obtained therefrom when transplanted into the brain in vivo, engraft to the transplant site and suppress proliferation of the transplant-derived cells.
- the volume of the implant at 5 weeks post-implantation can be 2% to 50% of that immediately after implantation.
- the high-purity cerebral cortex cell mass and the cell population obtained therefrom are useful for cell transplantation therapy for patients suffering from cerebrovascular disorders, as described later.
- one aspect of the present invention includes a cell population of highly purified cerebral cortex cell clusters that are uniform in size, constituent cell composition, or shape.
- the cell population can be produced by methods 2-1, 2-2, and 3-1 to 3-6 for producing a cerebral cortical cell mass described in 5 to 7 above. That is, according to the production method, cerebral organoids can be dispersed and reaggregated to produce a highly purified cerebral cortical cell mass uniform in size, cell composition, or shape.
- homogeneity is defined as, for example, variations in numerical values such as marker expression levels, sizes, and the number of cells contained in one cell mass of highly purified cerebral cortex cell masses of ⁇ 20% or less, preferably ⁇ 10%. % or less, more preferably ⁇ 5% or less, can be used as an index for identification.
- the high-purity cerebral cortex cell mass and its cell population are useful for cell transplantation therapy for patients suffering from cerebrovascular disease or head injuries, as described later.
- compositions As an aspect of the present invention, a cerebral organoid obtained by the method for producing a cerebral organoid described in 2 above, or the cerebral organoid described in 3 above, a cell population containing cerebral cortical cells obtained from these cerebral organoids, a cerebral cortical cell mass obtained by the method for producing a cerebral cortical cell mass according to 4 to 7 above, a cerebral cortical cell mass according to 8 or 9 above, or a cell population containing cerebral cortical cells obtained from these cell masses
- a pharmaceutical composition is included as an active ingredient.
- the cerebral cortical cell mass obtained by the method for producing a cortical cell mass, the cerebral cortical cell mass according to 8 or 9 above, or the cell mass containing cerebral cortical cells obtained from these cell masses is a cell mass for transplantation (implant It can be used as an active ingredient for cell transplantation therapy as a cell/tissue for transplantation.
- the effective amount of the active ingredient varies depending on the purpose of administration, administration method, and conditions of the administration subject (sex, age, body weight, medical condition, etc.), but for example, the number of cells is 1 ⁇ 10 4 to 1 ⁇ 10 10 . , 1 ⁇ 10 5 to 1 ⁇ 10 9 , 1 ⁇ 10 6 to 1 ⁇ 10 7 , 3 ⁇ 10 6 to 3 ⁇ 10 7 , or 1 ⁇ 10 6 to 1 ⁇ 10 9 be able to.
- the pharmaceutical composition, cell mass for transplantation, cell for transplantation, tissue for transplantation may contain a pharmaceutically acceptable carrier in addition to an effective amount of the above-mentioned active ingredients.
- a pharmaceutically acceptable carrier a physiological aqueous solvent (physiological saline, buffer solution, serum-free culture medium, etc.) can be used.
- the pharmaceutical composition and the like may contain preservatives, stabilizers, reducing agents, tonicity agents, etc., which are commonly used in medicines containing cell aggregates or cells for transplantation, if necessary.
- a cell suspension for transplantation can be produced by suspending a cell mass for transplantation, a cell for transplantation, or a tissue for transplantation in an appropriate physiological aqueous solvent. If necessary, the cell population for transplantation is cryopreserved by adding a cryopreservation agent, thawed at the time of use, washed with a buffer solution, or stored under low temperature conditions, washed with a buffer solution at the time of use, It may be used for transplantation medicine.
- all cerebral organoids or cell clusters are collected and, if necessary, used.
- the collected cell mass can be washed with the medium, other medium, phosphate buffer, or the like, and then suspended in the medium used in the pharmaceutical composition.
- the pharmaceutical composition, etc. of the present invention may be a suspension obtained by dispersing or suspending cell aggregates, or may be a suspension or sheet preparation obtained by dispersing cells from the cell aggregates.
- the pharmaceutical composition or the like obtained by the production method of the present invention is cultured on a plate after carrying out the above 2 production method for cerebral organoids or the above 4 to 7 production methods for cerebral cortical cell clusters. It is also possible to form a cell tissue structure that is a two-dimensional tissue by molding it into a sheet, or to form a cell tissue structure that is a three-dimensional tissue by culturing it on a scaffold.
- the pharmaceutical composition or the like containing a cell mass or cell population containing cerebral cortical cells of the present invention is administered to the cerebral cortex of an animal model of cerebrovascular disease (e.g., a mouse), and the damaged nerves at the site of injury. It brings about remarkable effects such as restoration of the system and recovery of motor function (for example, improvement of symptoms such as motor paralysis). Therefore, the pharmaceutical composition, etc. of the present invention is useful as a therapeutic agent for cerebrovascular disorders or head trauma. In addition, the pharmaceutical composition, etc. of the present invention is useful as a motor function-improving drug for patients suffering from cerebrovascular disorders.
- an animal model of cerebrovascular disease e.g., a mouse
- the cell clusters or cell populations containing cerebral cortical cells which are contained in the pharmaceutical compositions and the like described herein, are produced from established pluripotent stem cells, identified by markers and the like, and subjected to quality control. Therefore, a large amount of cell population for transplantation with stable quality can be produced and used for transplantation. In addition, since the cell population for transplantation can be preserved, the cell population for transplantation can be prepared according to the time of transplantation for the patient.
- the cerebral organoid obtained by the method for producing a cerebral organoid described in 2 above, or the cerebral organoid described in 3 above, and cells containing cerebral cortical cells obtained from these cerebral organoids A population, a cerebral cortical cell mass obtained by the method for producing a cerebral cortical cell mass according to 4 to 7 above, a cerebral cortical cell mass according to 8 or 9 above, a cell mass containing cerebral cortical cells obtained from these cell masses , or diseases requiring supplementation or functional improvement of cerebral cortical cells, including transplantation (administration) of an effective amount of the pharmaceutical composition according to 10 above to a subject in need of transplantation, specifically cerebrovascular Methods of treating disorders are included.
- the administration (implantation) site may be the cerebral cortex or the basal ganglia.
- the subject may be a mammal, preferably a rodent (eg mouse, rat) or a primate (eg human, monkey), more preferably a human.
- the cerebrovascular accident may also be a head injury.
- the treatment method is a concept that includes a method for improving motor function (for example, improvement of symptoms such as motor paralysis) in patients with cerebrovascular disease, a method for replenishing cerebral cortical cells in patients with cerebrovascular disease, and the like.
- pluripotent stem cells eg, induced pluripotent stem cells
- pluripotent stem cells eg, induced pluripotent stem cells
- pluripotent stem cells e.g., induced pluripotent stem cells
- somatic cells of others whose immunity is compatible with the recipient (e.g., HLA type or MHC type is compatible)
- allo (allogeneic) A cell mass (cell population) may be produced, and the cell mass (cell population) or a cell mass for transplantation containing cells obtained from the cell mass (cell population) may be transplanted into the recipient.
- iPS cells in which the expression of histocompatibility antigens (e.g., antigen proteins constituting HLA Class I and HLA Class II) or factors required for the expression of the antigens are suppressed are used to obtain the cell clusters of the present invention. ), rejection can be avoided even in allogeneic cell transplantation.
- histocompatibility antigens e.g., antigen proteins constituting HLA Class I and HLA Class II
- factors required for the expression of the antigens are suppressed are used to obtain the cell clusters of the present invention.
- the aforementioned pharmaceutical composition can be used as a therapeutic agent to be administered or transplanted to a patient or recipient in the therapeutic method of the present invention.
- One aspect of the present invention includes use of cell masses or cell populations comprising the cerebral cortical cells of the present invention for use in treating cerebrovascular disorders.
- the cerebral organoid obtained by the method for producing a cerebral organoid described in 2 above, or the cerebral organoid described in 3 above, or cerebral cortical cells obtained from these cerebral organoids is included.
- a cell population, a cerebral cortex cell mass obtained by the method for producing a cerebral cortex cell mass according to 4 to 7 above, a cerebral cortex cell mass according to 8 or 9 above, or cells obtained by dispersing these cell masses Examples include methods for evaluating toxicity or efficacy of a test substance, including contacting a population with a test substance and detecting or quantifying the effect of the test substance on the cell mass or cell population.
- Cerebral organoid obtained by the method for producing a cerebral organoid described in 2 above, or the cerebral organoid described in 3 above, or a cell population containing cerebral cortical cells obtained from these cerebral organoids, and the cerebral cortex described in 4 to 7 above The cerebral cortex cell mass obtained by the method for producing a cell mass, the cerebral cortex cell mass according to 8 or 9 above, or the cell mass obtained by dispersing these cell masses are used as disease model cells for cerebrovascular disease. It can be used for screening and efficacy evaluation of therapeutic agents for diseases associated with or preventive agents thereof.
- the cerebral cortex cell mass obtained by the method for producing a cerebral cortex cell mass, the cerebral cortex cell mass described in 8 or 9 above, or the cell mass obtained by dispersing these cell masses are used as healthy model cells, chemically It can be used for safety tests, stress tests, toxicity tests, side effect tests, infection or contamination tests of substances, etc.
- the cell mass or cell population of the present invention contains cells of cortical layer V/VI tissue, functional tests of nerve tissue (e.g., cerebral cortex) containing these cells, specifically glutamatergic nerves It is also possible to use it for functional evaluation such as cerebral cortical cell proliferative ability and differentiation ability evaluation.
- nerve tissue e.g., cerebral cortex
- glutamatergic nerves it is also possible to use it for functional evaluation such as cerebral cortical cell proliferative ability and differentiation ability evaluation.
- test to evaluate the effects of chemical substances on differentiation, axonal outgrowth ability, and firing ability of cerebral cortical cells RT-PCR of various gene markers, Cytokine expression protein analysis by ELISA or the like, phagocytosis test, patch clamp method, electrophysiological analysis using multi-electrode array (MEA) or the like), and the like.
- RT-PCR of various gene markers
- Cytokine expression protein analysis by ELISA or the like phagocytosis test
- patch clamp method electrophysiological analysis using multi-electrode array (MEA) or the like
- search for compounds that promote or inhibit neural differentiation, axonal outgrowth ability, and firing ability, and rescue disease-specific phenotypes of cells differentiated from patient-derived iPS cells suffering from cerebrovascular disease It can be used for searching for compounds, proteins, and the like.
- the cells of the cell mass of the present invention are dispersed, seeded and adhered to a plate, a cell suspension, or a sheet or molded body thereof can be provided.
- the cerebral cortex cell mass obtained by the method for producing a cerebral cortex cell mass, the cerebral cortex cell mass described in 8 or 9 above, or the cell mass obtained by dispersing these cell masses are outside human and animal tests. It can be used as an insertion test.
- a method for evaluating the quality of a cerebral organoid or a cerebral cortical cell mass comprising the following steps (aa) and (bb): A quality evaluation method can be mentioned.
- the cerebral cortex cell mass includes the highly purified cerebral cortex cell mass of the present invention.
- step (aa) measuring the expression levels of at least one or all genes selected from the group consisting of GAD2, COL1A1, TYR, TTR and HOXA2 in cerebral organoids or cerebral cortical cell masses; (bb) based on the measurement result of step (aa), when the expression level of the gene is below the reference value, the amount of non-target cells contained in the cerebral organoid or the cerebral cortex cell mass is below the reference value; process to evaluate.
- Another aspect of the present invention includes a method for evaluating the quality of cerebral organoids or cerebral cortex cell clusters, including the following steps (AA) and (BB).
- (AA) NEUROD6, NEUROD2, SSTR2, TBR1, ZBTB18, NHLH1, IGFBPL1, NRN1, RTN1, THSD7A, NRXN1, BHLHE22, CALB2, KHDRBS3, CCSAP, PDE1A, NEUROD1, NPTX41, NXPHH in cerebral organoids or cerebral cortical cell masses NTS, NEUROG2, OLFM1, PRDM8, CORO2B, TP53I11, ZFPM2, PCDH9, NELL2, SRRM4, SCG3, DCC, EPB41L3, SLC17A7, ST18, NSG2, EMX1, CAP2, SYT4, NSMF, ANK3, MYT1L, FSTL5, CATELF14, B3G measuring the expression levels of at least one, at least two, at least three, or at least five genes selected
- the amount of gene expression (that is, the amount of mRNA or protein expression) and its measurement method are as described above.
- Examples of the reference value for the expression level of each gene that serves as a marker for non-target cells, such as the gene described in (aa) above, include values measured in cerebral organoids or cerebral cortical aggregates that serve as samples. , equal to or lower than the reference value can be used as an index for evaluation.
- the reference value of each gene that serves as a target cell marker such as the gene described in (AA) above, includes, for example, the value measured in a cerebral organoid or cerebral cortex aggregate that serves as a standard. It can be evaluated as an index that it is equal to or higher than the value.
- the cerebral cortex cell mass obtained by the method for producing a cerebral cortex cell mass described in 4 to 7 above a part of the cell population of the cerebral cortex cell mass is sampled, and the cerebral organoid or cerebral cortex is obtained.
- a population of cerebral cortical cell clusters with non-target cells below the reference value and/or target cells above the reference value based on the evaluation results evaluated by the cell cluster quality evaluation method can be used for transplantation. It can be selected (identified) as a possible population.
- One aspect of the present invention includes a method for producing a cerebral cortex cell mass, which includes the method for producing a cerebral cortex cell mass described in 4 to 7 above, and a method for evaluating the quality of the cerebral organoid or cerebral cortex cell mass.
- a method for producing a cerebral cortex cell mass comprising the following steps: (1) a step of producing a cerebral cortex cell mass by the production method according to 4 to 7; (2) a step of evaluating that the amount of target cells is equal to or greater than the standard and/or that the amount of non-target cells is equal to or less than the standard, by the above quality evaluation method; (3) A step of selecting or identifying a cerebral cortex cell cluster that can be used for transplantation based on the evaluation result of (2).
- hiPSCs Maintenance culture of human induced pluripotent stem cells (hiPSC)
- hiPSC human induced pluripotent stem cells
- hiPSCs were added to a 6-well plate with iMatrix-511 (Nippi Co., Ltd.), an E8 fragment of laminin 511, at 0.5 ⁇ g/cm 2 and grown in StemFit (registered trademark) AK02N medium or AK03N on a coded plate.
- a medium hereinafter sometimes referred to as “StemFit”; manufactured by Ajinomoto Healthy Supply Co., Ltd.
- TransFit manufactured by Ajinomoto Healthy Supply Co., Ltd.
- Passaging was performed every 7 days.
- Organoids were fixed with 4% paraformaldehyde for 30 minutes, dehydrated with 30% (w/v) sucrose in PBS, and O.D. C. T. It was embedded using a compound (Sakura Finetek). Cryosections were made with a cryostat (CM1850, Leica Biosystems) at a thickness of 16 ⁇ m. Cells were permeabilized with 0.3% or 2% (v/v) Triton-X100 and antigen retrieval reactions were performed as needed. Blocking was performed with Block ACE (KAC) followed by double or triple label staining. The primary and secondary antibodies used for immunostaining were used at recommended dilution concentrations.
- KAC Block ACE
- L1CAM 554273 (BD) Ctip2: ab18465 (Abcam)
- Pax6 EPR15858 (Abcam), 561482 (BD)
- Foxg1 M227 (Takara) Ki67: NCL (Leica) Emx1: M196 (Takara) Gad65: 559931 (BD)
- Col1a1 AF6220 (R&D) TTR: A0002 (DAKO)
- TYR MA5-14177 (Thermo Fisher) Alexa Fluor® 488 donkey anti-mouse IgG (H+L): A21202 (Thermo Fisher) Alexa Fluor® 647 donkey anti-mouse IgG (H+L): 1900251 (Thermo Fisher) Alexa Fluor® 594 donkey anti-rat IgG (H+L): 1979379 (Thermo Fisher) Alexa Fluor® 488 donkey anti-rabbit IgG (H+L)
- Oligo DNA primer POU5F1 Forward: AGACCATCTGCCGCTTTGAG (sequence number 1) Reverse: GCAAGGGCCGCAGCTT (SEQ ID NO: 2)
- NANOG Forward: GGCTCTGTTTTGCTATATCCCCTAA (sequence number 3) Reverse: CATTACGATGCAGCAAATACGAGA (SEQ ID NO: 4)
- BMP4 Forward: ATGATTCCTGGTAACCGAATGC (sequence number 5) Reverse: CCCCGTCTCAGGTATCAAAACT (SEQ ID NO: 6)
- NODAL Forward: TGAGCCAACAAGAGGATCTG (sequence number 7) Reverse: TGGAAAATCTCAATGGCAAG (SEQ ID NO: 8)
- TGFB1 Forward: TACCTGAACCCGTGTTGCTCTCTC (SEQ ID NO: 9) Reverse: GTTGCTGAGGTATCGCCAGGAA (SEQ ID NO: 10) SOX
- hESC Human embryonic stem cells
- hiPSC human iPS cell lines
- Human ES KhES-1 and KhES-2 were obtained from the Institute for Frontier Medical Sciences, Kyoto University, and human iPS cell lines 201B7 cells, 1231A3 cells and Ff-I01s04 cells were obtained from Kyoto University.
- mouse embryonic fibroblasts (MEF, Oriental Yeast) were used as feeder cells.
- Various stem cell lines such as KhES-1, were seeded at a density of 1.2 ⁇ 10 6 cells per 90 mm dish and treated with 1% (v/v) NEAA, 1% (v/v) in the presence of MEFs.
- the cells after maintenance culture were induced to differentiate into cerebral organoids by a method partially modified from the method described in Non-Patent Document 1.
- various pluripotent stem cells were treated with 0.5 ⁇ TrypLE Select and dispersed into single cells. After that, replace with a differentiation medium (described later) supplemented with 50 ⁇ M Y-27632 (Fuji Film Wako Pure Chemical Industries, Ltd.), and transfer to a non-cell-adsorbing V-bottom 96-well plate (Sumitomo Bakelite) at 9,000 cells/well. sown.
- Differentiation medium was DMEM/F-12 GlutaMAX medium (Gibco) supplemented with 20% (v/v) KSR, 5 ⁇ M SB431542 (TGF ⁇ inhibitor; TOCRIS), and 3 ⁇ M IWR1e (Wnt inhibitor; Calbiochem). From the 3rd day to the 15th day of differentiation, the differentiation medium was used, and half the medium was replaced every 3 days.
- the aggregated cells were transferred to a 90 mm suspension culture dish (Sumitomo Bakelite), and 1% (v/v) N2-supplement (Gibco) until 35 days after the initiation of differentiation induction (Day35), 1% (v/v) DMEM/F-12 GlutaMAX (Gibco) supplemented with Chemically defined lipid concentrate (CDLC; Gibco), 0.25 ⁇ g/ml Amphotericin B (Gibco), 100 U/ml penicillin, and 100 ⁇ g/ml streptomycin cultured. Up to Day35, the medium was replaced every 3 days.
- the denominator represents the number of experiments in which differentiation was performed, and the numerator represents the number of times cerebral organoids were formed. For example, "1/3" indicates that differentiation was induced three times and cerebral organoids were formed once. In any ES or iPS cell line, it was confirmed that the formation efficiency of cerebral organoids was lower in the case of feeder-free than in the case of on-feeder.
- KhES-1, 201B7, Ff-I01s04 and 1231A3 cell lines were cultured under three conditions, on feeder, feeder-free, and feeder conditioned medium, and differentiated. induced.
- Feeder conditioned medium conditions are MEF, 1% (v / v) NEAA, 1% (v / v) L-glutamine, 1% (v / v) 2-Mercaptoethanol, 20% (v / v) KSR
- the expression of FGF2 and TGF ⁇ pathway-related genes (FGF2, LEFTY, NODAL, TGF ⁇ 1, Activin) in these obtained cells before and after induction of differentiation after culture was analyzed by microarray.
- Microarray analysis was performed using TaKaRa's NucleoSpin RNA Plus XS from four types of iPS cell lines (KhES1, 201B7, 1231A3, Ff-I01s04) cultured under three conditions: on-feeder, feeder-free, and MEF conditioned medium. According to the protocol, RNA was extracted and gene expression analysis was performed using a Clariom S (Applied Biosystems) microarray. Analysis of gene expression levels was performed using Transcriptome viewer (Kurabo).
- FIG. 2 shows the results after culture and before induction of differentiation.
- gene expression are comparatively analyzed using a microarray (n means the type of cell line).
- n means the type of cell line.
- FGF2 and TGF ⁇ pathway-related genes FGF2, LEFTY, NODAL, TGF ⁇ 1, Activin was more than twice as high in the on-feeder culture group as compared to the feeder-free culture group.
- FIG. 2 shows that these cell lines were differentiated into iPS cells under three conditions: on feeder, feeder-free, and feeder conditioned medium, using four types of cell lines according to preliminary test 1.
- n is the number of cerebral organoids means the number of conditions in which the formation efficiency was high.
- FGF2 and TGF ⁇ pathway-related genes FGF2, LEFTY, NODAL, TGF ⁇ 1, Activin
- bFGF- and TGF ⁇ -related genes were found to be expressed at high levels.
- compositions of known human pluripotent stem cell maintenance media are shown in Table 2 below. It can be seen that the feeder-free maintenance medium is usually supplemented with high concentrations of bFGF and TGF ⁇ . It was suggested that ES/iPS cells cultured in these media show lower expression of bFGF- and TGF ⁇ -related genes compared to cultured on feeder. Therefore, it was considered possible that the difference in the expression levels of bFGF- and TGF ⁇ -related genes causes the difference in cerebral organoid formation efficiency.
- the present inventors cultured ES/iPS cells in the presence of various compounds, then induced differentiation into neurons, and analyzed the formation efficiency of cerebral organoids, which may affect the formation efficiency. searched for compounds.
- iPS cells are cultured in "bFGF-free and TGF ⁇ -free medium or supplemented with a TGF ⁇ signaling inhibitor" (step (1)) to induce differentiation into nerve cells.
- Step (2) the formation efficiency of cerebral organoids was found to be significantly increased.
- Step (1) Effect of step (1) on organoid formation efficiency 1-1.
- Maintenance Medium in Step (1) Steps (1) and (2) were carried out according to the scheme shown in FIG. 3 or FIG.
- Human iPS cells S2WCB1 strain, S2WCB3 strain
- StemFit medium iMatrix-511-coated 6-well plates (feeder-free).
- StemFit C solution-free medium i.e., bFGF-free medium
- 5 ⁇ M SB431542 FGF-free medium
- Essential 6 medium i.e., bFGF-free, TGF ⁇ -free medium
- the human iPS cells cultured in step (1) were dispersed into single cells by enzymatic treatment.
- Human iPS cells dispersed into single cells were seeded in a non-cell-adhesive 96-well culture plate (PrimeSurface 96V bottom plate, manufactured by Sumitomo Bakelite Co., Ltd.) (9,000 cells/well/100 ⁇ L), and 0.1 mM non-essential Glasgow MEM (GMEM; Thermo (Fisher) medium supplemented with amino acids, 1 mM pyruvate, 0.1 mM 2-mercaptoethanol, 20% (v/v) KnockOutTM serum replacement, 100 U/mL penicillin, and 100 ⁇ g/ml streptomycin) hereinafter also referred to as 20GMK), 5 ⁇ M SB431542 (TGF ⁇ signal inhibitor; Tocris), 3 ⁇
- Step (2b) On Day18, transfer the obtained cell mass to a 90 mm dish, add 1 ⁇ N2 supplement (Thermo Fisher), Chemically Defined Lipid Concentrate (Invitro-gen), 0.25 mg/mL fungizone (Gibco), 1% penicillin In DMEM/F12/GlutaMAX medium supplemented with /streptomycin and 0.1% amphotericin B, shaking culture was performed by Orbital Shaker from Day 35 to Day 42 at 37° C., 20% O 2 , 5% CO 2 . Medium changes were performed every 3-4 days.
- Fig. 4 (A) The cell culture on Day 35 was observed with an inverted microscope, and a representative image is shown in Fig. 4 (A).
- the cell culture was subjected to fluorescent immunostaining for each marker (L1CAM, CTIP2, PAX6). Fluorescent immunostaining used the primary antibodies described above and their corresponding fluorescently labeled secondary antibodies.
- nuclear staining was performed with DAPI.
- a representative fluorescence-stained image is shown in FIG. 4(B).
- step (1) in the case of maintenance culture (StemFit-bFGF+SB) in StemFit medium containing no bFGF to which SB431542 (hereinafter sometimes referred to as SB) is added, cell clusters having a plurality of distinct rosette structures ( Rightmost of (B)) was confirmed.
- a Pax6-positive neural progenitor cell layer radial glial cells
- a neuronal cell layer expressing Ctip2 and L1CAM was seen outside.
- step (1) maintenance culture (step (1)) in a bFGF-free medium (“StemFit-bFGF” or “StemFit-bFGF + SB”) showed a formation efficiency of cerebral organoids ( ⁇ 10% for “StemFit-bFGF”). %, about 40%-50% for “StemFit ⁇ bFGF +SB”) was significantly elevated compared to maintenance cultures (StemFit, ⁇ 1%) on bFGF-containing medium.
- essential 6 (E6) medium supplemented with SB431542 was used as a medium for maintenance culture, and differentiation was induced in the same manner as described above. Immunostaining was performed in the same manner as above, and confocal fluorescence microscope images of representative cell clusters are shown in FIG.
- cerebral organoids were obtained in the case of using the E6 medium supplemented with SB431542, similarly to the StemFit medium supplemented with SB431542.
- step (1) of the scheme in FIG. to evaluate the formation efficiency of cerebral organoids The results of immunostaining are shown in FIG. 7, and the formation efficiency of cerebral organoids is shown in Table 3. "None" in FIG. 7 means no additive.
- Example 1 From the results of Example 1, when human pluripotent stem cells were cultured in "a medium containing no bFGF and no TGF ⁇ or supplemented with a TGF ⁇ signaling inhibitor" and then induced to differentiate into neurons, cerebral organoids It was shown that the formation efficiency of Therefore, when pluripotent stem cells are cultured in a “culture medium that does not substantially contain bFGF and do not substantially induce TGF ⁇ signals” and then induce differentiation into neurons, the formation efficiency of cerebral organoids is remarkable. was found to rise to
- Example 2 Culture period of step (1) The period for culturing the iPS cells in the SB431542-free/SB431542-added medium was as shown in the table below.
- FIG. 8 shows representative bright field images of cultures on Day 18, Day 27 and Day 34.
- FIG. 9 shows the results of analyzing the formation efficiency of cerebral organoids on Day 35.
- the formation efficiency of cerebral organoids was about 30% to 40% when treated with the “bFGF-free/SB431542-added medium” for 1 day or 2 days. In the above cases, almost no formation of cerebral organoids was observed. Under the conditions of treatment with the "bFGF-free, SB431542-added medium” for 6 days, the proliferation of iPS cells was remarkably reduced, and a sufficient number of cells could not be obtained for induction of differentiation. Therefore, it was clarified that the culture period in step (1) is preferably less than 3 days, more preferably 12 hours or more and 2 days or less, and most preferably about 1 day.
- Example 3 Influence of step (1) on gene expression
- S2WCB1 human iPS cells
- FIG. 10 shows the results of gene analysis.
- Each point represents a sample of each time, the organoids (Control) differentiated from iPS cells cultured in StemFit medium (bFGF-containing medium), which is a normal maintenance medium, are indicated by black circles, and StemFit (-bFGF, + TGF ⁇ inhibitor) is indicated by black circles.
- StemFit -bFGF, + TGF ⁇ inhibitor
- Organoids induced to differentiate after culturing are indicated by open circles.
- the vertical axis represents the relative expression level corrected for one lot of GAPDH and Control by the ⁇ Ct method.
- TGF ⁇ i in the figure means TGF ⁇ inhibitor.
- step (1) may change iPS cells into a state that facilitates differentiation into three germ layers. That is, it is suggested that pluripotent stem cells, when cultured for an appropriate period of time in a “culture medium that does not substantially contain bFGF and do not substantially induce TGF ⁇ signals”, tend to differentiate into trigerm layers. was done.
- Step (2) Effect 1 of Steps (ii)-(iv) of Step (2): Increase in Nerve Cells and Decrease in Proliferative Cells in Layer V/VI 4-1. With dispersion and reaggregation -Cultivation was performed in the same manner as in Example 1 from Day 7 to Day 35.
- step (1) (1) StemFit C solution-free medium (bFGF-free medium) to which 5 ⁇ M SB431542 was added was used.
- step (ii) cerebral organoids were selected on day 33 (Day33) after the initiation of differentiation induction, and the medium was the medium in step (2b) with 10 ⁇ M DAPT added. , and suspended culture was carried out at 37° C. under 5% CO 2 for 3 days. Cerebral cortical cell clusters were thus obtained.
- step (iii) the cerebral cortex cell mass obtained in step (ii) above is incubated in a 0.5 ⁇ TrypLE Select, 0.25 mM EDTA solution at 37° C. for 20 minutes, and further step (2b) The medium was incubated at 37°C for 10 minutes in a solution containing 25 U/mL DNase I. Then, after pipetting to disperse into single cells, the dispersed cells were added to the medium of step (2b) with 10 ng/mL GDNF, 20 ng/mL BDNF, 200 ⁇ M ascorbic acid, 400 ⁇ M dibutyryl-cAMP, 50 ⁇ M Y-27632. was suspended in medium supplemented with
- step (iv) the cell suspension obtained in step (iii) was dispensed into a non-cell-adhesive 96-well plate at 30,000 cells/well and suspended for 4 days at 37°C under 5% CO2 . did. This yielded a highly pure cerebral cortical cell mass. Bright-field images of the cerebral cortical cell mass (Day 36) and the highly purified cerebral cortical cell mass (Day 40) are shown in FIG. 11(B).
- cortical V/VI layer progenitor cell (Deep Layer) marker Ctip2, telencephalic marker Foxg1, neural stem cell (Radial Glia) Immunostaining was performed for the marker Pax6/Ki67 and the proliferative cell marker Ki67.
- FIG. Fig. 12 shows immunization of cerebral cortical cell clusters on Day 36 without DAPT in the upper row, and cerebral cortical cell clusters on Day 36 with DAPT in the lower row (both obtained through steps (i)-(ii)).
- FIG. 12B shows an immunostained image of Day 40 highly purified cerebral cortical cell clusters (obtained through steps (i)-(iv)).
- the Pax6/Ki67-positive neural stem cell (Radial Glia) layer which is characteristic of cerebral organoids
- the Ctip2/Foxg1 double-positive cortex outside it.
- a large number of cerebral cortex-like layered structures with neuronal layers of layers V/VI were confirmed. Almost no expression was observed, and no cerebral cortex-like layered structure was observed.
- Ctip2/Foxg1-positive cells were observed throughout the cell mass. Therefore, it was revealed that the cerebral cortex-like layered structure disappeared in the cerebral cortex cell mass, and Ctip2/Foxg1 double-positive cortical layer V/VI neurons were widely distributed inside.
- step (ii) the cell culture immediately after DAPT treatment (immediately after step (ii)) was analyzed for the proportion of constituent cells using flow cytometry.
- a differentiation induction scheme is shown in FIG. Cerebral organoids on Day 40 were subjected to DAPT treatment for 3 days, and then the expression of marker genes was analyzed by flow cytometry.
- FIG. 14 shows the results of expression analysis of the marker Ki67.
- Each point in FIG. 14 represents a cell.
- the left and center panels show the results of displaying all viable cells, and the right panel shows the results of displaying only Ki67-positive cells.
- the percentage of Ctip2/ ⁇ III-Tubulin double-positive cells (left panel) was 36.4% in the DAPT-untreated group (top), but increased to 86.1% in the DAPT-treated group (bottom).
- the ratio of Pax6/Ki67 double-positive neural stem cells (Radial Glia) (middle panel) was 46.7% in the DAPT-untreated group (upper row), but 1.0% in the DAPT-treated group (lower row). It was down to 5%.
- the ratio of Pax6-positive/Sox1-positive/Ki67-positive cells was 0.68% in the DAPT-untreated group (upper), but 0.01% in the DAPT-treated group (lower). decreased to
- Ki67, Sox1, Pax6 triple positive cells were 0.68% in “Ctrl”, but decreased to 0.01% in “DAPT3d” and 0% in “DAPT3d + Release4d” (from right 1st column). In ⁇ DAPT 3d + Release 4d'', the ratio of Pax6-positive, Sox1-positive, and Ki67 triple-positive cells is even lower than in ⁇ DAPT 3d''. I found out.
- Organoid DAPT method and single-cell DAPT method (2) In order to analyze the effect of the DAPT treatment period on differentiation, according to the method scheme (organoid DAPT method) shown in FIG. Changes in expression were analyzed by RT-qPCR and the results are shown in FIG. 16(B). The vertical axis represents the relative expression level to GAPDH.
- Fig. 17(A) shows a scheme of induction of differentiation.
- Organoids on Day25, Day32, Day39, and Day72 (differentiation induction periods are approximately 4 weeks, 5 weeks, 6 weeks, 10 weeks) were cultured for 3 days in the presence or absence of DAPT, Day28 (4wk), Organoids on Day 42 (6 wk) and Day 75 (10 wk) were analyzed by immunostaining for various markers.
- the results of immunostaining (confocal fluorescence microscope image) are shown in FIG. 17(B).
- Satb2 is a cortical II-IV layer progenitor cell (Upper Layer) marker. From FIG.
- step (ii) is preferably about 20 to 44 days (Day 20 to Day 44) after the initiation of differentiation induction, more preferably Day 25 to Day 39. It was also found that the period of step (ii) is preferably about 2 to 4 days, more preferably about 3 days (for example, 60 to 84 hours).
- Fig. 20 shows the analysis results of the volume of the stained graft.
- the volume of the graft was calculated by observing the immunostained brain sections at intervals of 350 ⁇ m, calculating the area of the graft from the Ku80-stained image for each section, and integrating the area and the section interval. did. Tests were performed by one-way ANOVA (Tukey Multiple Test) (***p ⁇ 0.0001, **p ⁇ 0.01).
- the cerebral cortical cell clusters obtained by performing step (ii) about 5 weeks or 6 weeks after the initiation of differentiation induction are the same as those obtained without performing step (ii). Compared to , the engraftment rate after transplantation into the brain was high, and the increase in volume was very small.
- cerebral organoids differentiated from pluripotent stem cells are cultured in the presence of DAPT, and then cultured in a culture medium containing one or more neurotrophic factors, ascorbic acid, and a cAMP activator. It was revealed that the cerebral cortical cell clusters in the brain have a high engraftment rate after transplantation into the brain and are difficult to increase (cell proliferation is difficult).
- a preferred method of producing cerebral organoids from pluripotent stem cells in the absence of feeder cells comprises the following steps. Step (1): culturing pluripotent stem cells in a culture medium that is substantially free of bFGF and does not substantially induce TGF ⁇ signals; Step (2): A step of inducing differentiation of the cells obtained in step (1) into cerebral organoids.
- Step (2) includes the following steps.
- Step (2a) A step of suspension culture of the cells obtained in step (1) in a culture medium containing a TGF ⁇ signaling inhibitor and a Wnt signaling inhibitor to obtain a cell mass
- Step (2b) A step of suspension culture of the cell mass obtained in step (2a) in a culture medium substantially free of TGF ⁇ signal inhibitors or Wnt signal transduction inhibitors to obtain cerebral organoids.
- a preferred method for producing a cerebral cortex cell mass and a highly purified cerebral cortex cell mass from pluripotent stem cells in the absence of feeder cells includes the following steps.
- DAPT Notch signal inhibitor
- a high-purity cerebral cortex cell mass can be obtained by skipping step (iii) and culturing the cell culture that has undergone step (ii) in the culture medium of step (iv).
- Example 7 Investigation of the effect of DAPT on organoids induced with preparation for neural differentiation - From Day 7 to Day 35, culture was performed by the same method as in the scheme of Fig. 3 of Example 1.
- cerebral cortical cell clusters were induced by the method described in step (ii) of Example 4, and evaluated by immunostaining.
- dispersing into single cells by the method described in steps (iii) and (iv) of Example 4 they were reaggregated to induce high-density cerebral cortical cell clusters, which were analyzed by flow cytometry.
- FIG. 1 A representative confocal fluorescence microscope image of immunostaining is shown in FIG.
- a cerebral cortex-like structure with a Pax6-positive neuroepithelium and an outer Ctip2-positive cortical V/VI neuronal layer was formed, albeit at a lower efficiency. rice field.
- this organoid was treated with DAPT, Ctip2-positive cells were found throughout the cell mass, as in the preparation for neural differentiation.
- Fig. 23 shows the analysis results of flow cytometry. From FIG. 23 , the percentage of Ctip2 ⁇ TubIII double-positive cells (left panel) increased from 22.5% to 85.9% with DAPT treatment even without neural differentiation preparation. In addition, the ratio of Pax6/Ki67 double-positive neural stem cells (Radial Glia) (middle panel) decreased from 59.5% to 1.9% by DAPT treatment. Furthermore, the percentage of proliferating cells, Pax6-positive/Sox1-positive/Ki67-positive cells (right panel), was reduced from 14.5% to 0% by DAPT treatment.
- Organoid DAPT method and single-cell DAPT method (1) Cultured in the same manner as in Example 1 from Day 7 to Day 35.
- step (ii) "Organoid DAPT method (when DAPT treatment is performed in step (ii))" cerebral organoids were selected 35 days after the start of differentiation induction, and the medium was added to the medium in step (2b) with 10 ⁇ M DAPT. was added, and suspension culture was carried out for 3 days at 37° C. under 5% CO 2 .
- the resulting cerebral cortex cell mass was dispersed into single cells by the method described in steps (iii) and (iv) in Example 4, and then reaggregated to obtain a highly purified cerebral cortex cell mass.
- step (B) Single-cell DAPT method (when DAPT treatment is performed in step (iv))
- cerebral organoids were selected 38 days after the start of differentiation induction, and described in step (iii) of Example 4.
- the cells are added to the medium of step (2b) with 10 ng/mL GDNF, 20 ng/mL BDNF, 200 ⁇ M Ascorbic Acid, 400 ⁇ M dibutyl-cAMP, and 30 ⁇ M Y-27632, DAPT was added at concentrations of 0 ⁇ M, 0.1 ⁇ M, 1 ⁇ M, or 10 ⁇ M and suspended.
- the cells were reaggregated by the method described in step (iv) of Example 4, and cultured for 1, 2, or 3 days.
- the medium was switched to the medium in step (2b) supplemented with 10 ng/mL GDNF, 20 ng/mL BDNF, 200 ⁇ M ascorbic acid, and 400 ⁇ M dibutylyl-cAMP, and culture was continued until day 10 from the start of reaggregation.
- Cells on day 3, 4, or 10 after the start of reaggregation were analyzed by RT-qPCR, and cells on day 10 were analyzed by flow cytometry, and the results are shown in FIGS.
- the DAPT concentration was 10 ⁇ M, and the DAPT treatment period was 1 day, 2 days, and 3 days.
- the results when analyzed by qPCR are shown in FIG. In FIG. 25, “day” represents the number of days of step (iv), “Conc.” represents the concentration of DAPT, and “Treatment” represents the number of days of DAPT treatment.
- Hes1 whose expression is activated downstream of the Notch signal, decreased in expression immediately after DAPT treatment regardless of the DAPT treatment period. It was found that the expression increased again with culture. On the other hand, in the case of DAPT treatment for 3 days, Hes1 expression was suppressed for 10 days. At this time, the expression of the proliferative marker Ki67 and the expression of the neural stem cell marker Pax6 were also suppressed. On the other hand, the expression of Map2, which is a neuromaturation marker, and the expression of Ctip2, which is a neuronal layer marker of cortical layer V/VI, increased with the culture in step (iv). Therefore, it was found that DAPT treatment for at least 3 days can suppress proliferation of proliferative neural stem cells and increase the ratio of neurons in cortical layers V/VI.
- DAPT treatment at 10 ⁇ M for 3 days could suppress the proliferation of proliferative neural stem cells, and the ratio of neurons in cortical layers V/VI could be reduced. is increased, and the same or better effect can be obtained as compared with the case where the DAPT treatment is performed in the step (ii).
- step (iv) When DAPT treatment is performed in step (iv), the DAPT concentration is 0 ⁇ M, 0.1 ⁇ M, 1 ⁇ M, 10 ⁇ M, the DAPT treatment period is 3 days, and each marker is cultured for 10 days in step (iv).
- FIG. 27 shows the results of analyzing the expression levels by flow cytometry. For comparison, the case where the DAPT concentration was 10 ⁇ M and the DAPT treatment period was 3 days in step (ii) is also shown (organoid DAPT method).
- step (iv) when DAPT treatment was performed in step (iv), the percentage of Ctip2/ ⁇ TubIII (Tuj1) double-positive cells (upper panel) increased from 32.2% to 89% as the concentration of DAPT treatment increased. It was found to have increased to .6%. In addition, the ratio of Pax6-positive/Sox1-positive/Ki67-positive cells (bottom panel), which are proliferative cells, decreased from 35.0% to 0.8% by DAPT treatment. Therefore, even when DAPT treatment was performed in step (iv), as in the case of step (ii), the Ctip2/ ⁇ TubIII double-positive cortical layers V/VI increased with an increase in the DAPT treatment concentration. It was found that the percentage of nerve cells increased and the number of proliferating cells decreased.
- Example 9 Organoid morphology and expression profile analysis 9-1. Morphological analysis of organoids - Cultured in the same manner as in Example 1 from Day 7 to Day 35. Bright-field images of cerebral organoids obtained from 12 induction differentiation lots (Lot1 to Lot12) were taken using an inverted microscope (Leica DMS1000), and representative bright-field images are shown in FIG. 30 (scale bar: 5 mm). ). From FIG. 30, it was found that there was a difference in the morphology of the induced organoids depending on the lot.
- Table 5 shows the percentage of organoids in each form. Organoids were classified according to the type of morphology contained within one organoid. In rows 1-7 from the top, each column indicates the morphology contained in individual organoids. The 1st to 12th rows from the bottom show the results of morphological classification of the organoids induced in the 12 differentiation induction lots.
- the numerical values in Table 5 indicate the ratio of the organoids of each form to the total organoids in the lot as a percentage, and the shade of color corresponds to the magnitude of the numerical value.
- FIG. 32 shows a bar graph of the proportion of organoids in each form.
- organoid groups that develop varies greatly among differentiation lots, with lots containing both “rosette structures,” “balloon or flocculent tissue,” and “transparent tissue,” Lots of "cotton-like tissue” and mostly “potato-like tissue” were found ( Figure 32, Table 5). In this way, it was found that the ratio of organoid types generated by differentiation-inducing rounds varied from lot to lot. Some organoids had characteristics belonging to more than one of the above seven morphological classifications. In addition, these organoids can be visually selected by observing them under magnification.
- the organoids were separated into single cells using a nerve cell dispersion (Fujifilm Wako Pure Chemical Industries, Ltd.) according to the recommended conditions.
- the dispersed cells were resuspended in HBSS supplemented with 10% (v/v) KSR and 10 ⁇ M Y-27632 (Fuji Film Wako Pure Chemical Industries) at a density of 1,000 cells/ ⁇ L.
- Approximately 4,670 cells per channel were loaded onto the ChromiumNextGEM Chip G (2000177 10X Genomics) and processed with the Chromium controller to acquire Gel Beads-in-Emulsion (GEM).
- Libraries were generated using Chromium Next GEM Single Cell 3' Reagent Kits v3.1 (1000121; 10x Genomics) according to the manufacturer's protocol (CG000204 RevC). Library sequences were analyzed by Novaseq 6000 (Illumina).
- the Cell Ranger pipeline was used for single-cell RNA-seq sequence mapping.
- the GRCh38 human genome sequence was used as the reference genome sequence.
- the UMI count value obtained by analysis with the next-generation sequencer was analyzed using the Seurat R package.
- PCA principal component analysis
- Figures 34 and 35 show the results of single-cell RNA-seq analysis data displayed by the UMAP method for the cells contained in the nine analyzed organoids. From FIG. 34, it was considered that the cells contained in the nine organoids were divided into 10 or more different clusters, and contained cell populations exhibiting 10 or more characteristic gene expression patterns. In addition, the plot patterns differed depending on the organoid (Fig. 35), suggesting that the cell types contained differed depending on the organoid.
- each cluster In order to identify the cell types contained in each cluster, they were statistically classified into 14 clusters by the Nearest Neighbors method. Genes characteristically expressed in each cluster were extracted based on the percentage of expressing cells and the average expression level, and are shown in Table 6. The top 50 genes in each cell in Table 6 are listed in order from the top. In addition, based on literature information, annotation of cell types was performed, and the cell type of each cluster was identified based on the gene expression of each cluster (Fig. 36). Each dot represents an individual cell and the shade of dots represents a cluster.
- Cortical Neuron (Radial Glia (RG): cerebral cortical neural progenitor cells (Radial Glia (RG)), Cortical neuron (CR): Cajal Retzius cell (CR), Cortical Neuron (Glutamatergic neuron (GN)): Cerebral cortical neuron (Glutamatergic Neuron (GN)), GABAergic neuron: GABAergic neuron, Choroid plexus (ChP): Choroid plexus, CNS fibroblast: Center Neural fibroblasts, Neural Crest: neural crest cells, Vascular Endothelial cells: vascular endothelial cells, Caudal Neurons: considered to be caudal nerve cells.
- Figures 37 and 38 show in dot plots the expression profile of the known marker genes, the genes characteristic of each cluster, in the single-cell gene expression data.
- the vertical axis represents clusters, and the horizontal axis represents genes.
- the circle size represents the percentage of expressing cells to all cells in the cluster.
- the shade of color represents the average gene expression level of the cells within the cluster.
- MAP2 and TUBB3 were analyzed as neuronal markers.
- KRT19 and KRT8 were analyzed as markers of epithelial cells.
- MKI67 and TOP2A were analyzed as markers of proliferating cells.
- PAX6, SOX2, and HES1 were analyzed as markers of radial glia (RG).
- EMX1 was analyzed as a marker for the dorsal forebrain area.
- NEUROD6 and SLC17A7 were analyzed as markers of glutamatergic neurons.
- SSTR3 is a somatostatin receptor and is known to be expressed in glutamatergic neurons.
- TBR1 and BCL11B were analyzed as markers of Deep Layer, which is a precursor of layers V and VI of the cerebral cortex.
- RELN was analyzed as a marker for Cajal Retzius cells.
- ASCL1, DLX1, DLX2, DLX5, and DLX6 were analyzed as markers whose expression is observed during the differentiation process of GABAergic neurons.
- GAD2 was analyzed as a marker of GABAergic neurons.
- SST and TAC1 were analyzed as markers expressed in subtypes of GABAergic neurons.
- TTR, RSPO3, CLIC6, HTR2C, and TRPM3 were analyzed as markers whose expression is observed during the developmental process of the choroid plexus.
- COL1A1 was analyzed as a CNS Fibroblast marker.
- DCN, LUM, and DLK1 are genes expressed in stromal cells.
- AQP1 has been reported to be expressed in the choroid plexus and arachnoid granules.
- OTX2 and EMX2 are region-specific markers and are known to be expressed in the telencephalic choroid plexus.
- HOXA5 and HOXB5 were analyzed as caudal neural markers.
- ZIC1, MSX1, LGALS1, TWIST1, PRRX1, GPC3, SOX10, and TFAP2A were analyzed as markers whose expression is observed during the differentiation process of Neural Crest.
- LGALS1, TWIST1, PRRX1, and GPC3 are genes involved in epithelial-mesenchymal transition (EMT), and SOX10 and TFAP2A are markers expressed in mature neural crest.
- PECAM1, KDR, FLT1 and ICAM2 were analyzed as markers of vascular endothelial cells.
- EMX1 Clusters of cortical neurons expressed EMX1, a forebrain marker.
- “Radial Glia (RG)” expressed SOX2 and PAX6, which are essential genes for the regulation of LHX2 and Radial Glia.
- Glutamatergic neuron (GN) is a transcription factor characteristic of deep layer neuron and pyramidal neuron TBR1 (Robert F. Hevner, Tbr1 Regulates Differentiation of the Preplate and Layer 6, Neuron, 2001), NEUROD6 (Tukova al., The Role of Neurod Genes in Brain Development, Function, and Disease, Frontiers in Molecular Neuroscience, 2021), glutamate transporters SLC17A7 (VGluT1) and BCL11B (Ctip2).
- Cajal Retzius cell (CR) expressed Cajal Retzius cell marker genes such as RELN and TBR1.
- the GABAergic neuron cluster commonly expressed the glutamate decarboxylase GAD2.
- GAD2 glutamate decarboxylase 2
- high expression of MKI67 (Ki67) which is a proliferative marker, and ASCL1, whose expression is observed in the early stage of differentiation (VZ)
- ASCL1 a proliferative marker
- ASCL1 whose expression is observed in the early stage of differentiation (VZ)
- DLX5 and DLX6 which are expressed after the middle stage of differentiation (SVZ) were expressed.
- TTR Transthyretin
- COL1A1 a marker for fibroblasts associated with the central nervous system, was highly expressed.
- expression of AQP1 and the like was also observed, suggesting the possibility that some of these cells are fibroblasts associated with the choroid plexus.
- Neural Crest cluster expression of various genes observed during the differentiation process of Neural Crest Cell (NCC) was observed (Simoes-Costa M et al., Establishing neural crest identity: a gene regulatory recipe, Development, 2015).
- NCC Neural Crest Cell
- HOX genes In the Caudal Neuron cluster, expression of numerous HOX genes was observed, including TUBB3, a neuronal marker, and HOXA5 and HOXB5, which control caudal regionalization.
- Fig. 39 (A) shows the proportion of each cell type in the nine organoids
- Fig. 39 (B) shows the proportion of neural crest cells in each differentiation stage in the neural crest.
- Organoid9 containing pigment cells had a higher percentage of Neural Crest than other organoids.
- Neural Crest-2 which is a cell population that expresses a group of genes involved in EMT, has a high proportion, and contains many actively migrating Neural Crests.
- the forebrain marker EMX1 was specifically expressed in the area where the rosette structure was seen (rosette).
- GABAergic neuron marker GAD65 GAD65 (GAD2) was found to be expressed in the region of "potato-like tissue".
- GAD65 GABAergic neuron marker
- COL1A1 an ECM Fibroblast marker, was observed in balloon-like tissue, cotton-like tissue, and areas surrounding the pigment. Choroid plexus-specific markers were expressed in transparent tissue.
- TYR a melanocyte marker, was found to be expressed in the organoid "pigment” where pigmentation was observed.
- VGluT1 and EMX1 which were identified as markers of cortical neurons, was observed in Rosettes organoids.
- DLX2 and GAD2 which were identified as GABAergic neuron markers, were highly expressed in potato-like organoids.
- TYR which is a melanocyte marker, was highly expressed in Pigment organoids.
- COL1A1 a CNS Fibroblast marker, was found to be expressed in balloon-like tissue and cotton-like tissue.
- HOXA2 a marker of Caural Neuron, was found to be expressed in the jelly-like tissue.
- organoids Cerebral organoids obtained under the DAPT-condition were divided into two clusters and contained two types of cell populations (Fig. 44 (B)). It was found that the organoids (cerebral cortical cell clusters) obtained under the DAPT+ condition aggregated into one cluster and consisted of one type of cell population. Since the distribution of the cells in the three manufacturing lots overlapped, it was found that the difference due to the manufacturing lot was small, the reproducibility was good, and similar cell populations were obtained.
- Cerebral organoids are divided into two clusters. In the right cluster, SLC17A7 and NEUROD6, which are markers of glutamatergic neurons, and Ctip2, which is a marker of deep layer neurons, are highly expressed. It was thought to contain its progenitor cells. In the cluster on the left, PAX6 and MKI67, markers of radial glia, were highly expressed, suggesting that the cluster was highly proliferative radial glia. Almost no expression of Satb2, a marker of Upper Neuron, was observed, and most of them were thought to be progenitor cells of cerebral cortex layers V and VI.
- TTR, COL1A1, TYR, and PECAM1 which are markers of non-target cells identified this time, were hardly detected. From this, it was found that cerebral cortical cells can be obtained by selecting organoids having a rosette structure as a whole or by excluding organoids of other morphologies, and contamination with unintended cells can be reduced. .
- DAPT+ cerebral cortical cell mass
- SLC17A7 VGluT1
- NEUROD6 NEUROD6
- Ctip2 the cell population expressing Pax6 and MKI67, which are markers of non-target cells, is significantly increased. decreased to Moreover, TTR, COL1A1, TYR and PECAM1 were hardly detected. From this, it was found that DAPT treatment yields highly pure cerebral cortical cell clusters containing glutaminergic neurons.
- Each of the genes listed in Table 6 corresponds to cerebral cortical neural progenitor cells (in a broad sense, it is included in cerebral cortical neurons, but corresponds to radial glia, radial Glia (RG)), cerebral cortical neurons (glutamatergic neurons (GN), and Cajal-Retius cells (CR)), GABAergic neurons, choroid plexus (ChP), central nervous system fibroblasts, neural crest cells, vascular endothelial cells, or caudal neurons It can also be used as a marker.
- markers for cerebral cortical neural progenitor cells and cerebral cortical neurons can be used as markers for cerebral organoids because these cells are contained in cerebral organoids.
- genes expressed in cerebral cortical neurons can be used as markers for the cerebral cortical cell clusters of the present invention and the highly purified cerebral cortical cell clusters of the present invention. can.
- NEUROD6, NEUROD2, SSTR2, TBR1, NRXN1, BHLHE22, NEUROD1, NEUROG2, SLC17A7, and EMX1 are located in the dorsal forebrain region, which is the region where the cerebral cortex is generated, and layer II of the cerebral cortex. ⁇ Planned region of layer VI) (Cortical Plate), and glutamatergic neurons are known to be expressed, but the other 37 genes were newly identified as markers of cerebral cortical neurons. It is a thing.
- cerebral cortical neurons which are target cells
- highly purified cerebral cortical cell clusters defined in the present application means that NEUROD6, NEUROD2, SSTR2, TBR1, ZBTB18, NHLH1, IGFBPL1, NRN1, RTN1, THSD7A, NRXN1, BHLHE22, CALB2, KHDRBS3, CCSAP, PDE1A, NEUROD1, NPTX1, NXPH4, NTS, NEUROG2, OLFM1, PRDM8, CORO2B, TP53I11, ZFPM2, PCDH9, NELL2, SRRM4, SCG3, DCC1CA7, STB41L3, SCB41L3, DCC1, STB41L3 at least 1, at least 2, at least 3, or at least 5 genes selected from the group consisting of NSG2, EMX1, CAP2, SYT4, NSMF, ANK3, MYT1L, FSTL5, CELF4, B3GAT1, EPHA5, NHLH2,
- cerebral cortical neural progenitor cells radial glia (RG)
- GABAergic neurons choroid plexus (ChP)
- central nervous system fibers described in Table 6
- CNS fibroblast central nervous system fibroblast
- neural crest cells Neurovascular Endothelial cells
- Vascular Endothelial cells vascular endothelial cells
- Caudal nerve cells Cerudal Neuron
- the expression level of one, at least two, or at least three genes is substantially not expressed, or is below the reference level, and can be evaluated as an index.
- LHX2, SOX2, and PAX6 identified as markers of radial glia, GAD2DLX1, DLX2, DLX5, and DLX6 identified as markers of GABAergic neurons, TTR identified as a marker of the choroid plexus, TRPM3, COL1A1 identified as a marker for central nervous system fibroblasts, ZIC gene group identified as a marker for neural crest cells, Msx1, ID gene group downstream of Smad signal, Epithelial-Mesenchymal Transition (EMT)-related gene (TWIST1 , PRRX1, GPC3, Sox10, and TFAP2, KDR (VEGFR-2) identified as a marker for vascular endothelial cells, PECAM1 and CDH5 (VE-Cadherin), as well as numerous vascular endothelial cell markers, and caudal nerve
- TUBB3 which has been identified as a cell marker, at least one, at least two, or at least three genes selected from a
- one or more genes selected from the group consisting of GAD2, COL1A1, TYR, TTR and HOXA2 in Table 6 are not substantially expressed, or are below the standard value.
- the cerebral organoids produced by the production method specified in the present invention are not necessarily limited in use, and may be used for the following purposes.
- ⁇ Cerebral application (substance screening)>
- Low-molecular-weight compounds, antibodies, nucleic acids, and other substances may be added to the cerebral organoids produced by the production method of the present invention, and their effects may be examined. That is, cerebral organoids can be used for the purpose of clarifying the effects, toxicity, and usefulness of low-molecular-weight compounds, antibodies, and other substances. In this case, the effects of organized neurons, organized neural networks formed in cerebral organoids can be investigated.
- the method for producing cerebral organoids and the method for selecting cerebral organoids defined in the present invention make it possible to produce a large amount of cerebral organoids having relatively uniform expression profiles, shapes, etc., or tissue structures.
- the method for evaluating the quality of the cerebral organoids described in 13 above may be used.
- cerebral organoids with relatively uniform expression profiles, shapes, etc., or tissue structures By using cerebral organoids with relatively uniform expression profiles, shapes, etc., or tissue structures, multiple tests can be easily performed, and reproducibility, which has been difficult in the past, can be easily achieved. .
- cerebral organoids induced from pluripotent stem cells derived from specific genotype cells may be produced, and the above substance screening may be applied to specific diseases.
- the optimum process can be appropriately adjusted according to the properties of the derived pluripotent stem cells.
Landscapes
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biomedical Technology (AREA)
- Chemical & Material Sciences (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Cell Biology (AREA)
- Zoology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Organic Chemistry (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- Medicinal Chemistry (AREA)
- Immunology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- Developmental Biology & Embryology (AREA)
- Urology & Nephrology (AREA)
- Molecular Biology (AREA)
- Pharmacology & Pharmacy (AREA)
- Hematology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- General Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Food Science & Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Epidemiology (AREA)
- Virology (AREA)
- Ophthalmology & Optometry (AREA)
- General Chemical & Material Sciences (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
Abstract
Description
[1] 支持細胞の非存在下、多能性幹細胞から大脳オルガノイドを製造する方法であって、
(1)多能性幹細胞を、bFGFが実質的に含まれず、かつTGFβシグナルを実質的に惹起しない培養液中で培養する工程と、
(2)工程(1)で得られた細胞を、神経細胞へと分化誘導する工程と
を含む、方法。
[2] 工程(2)が、
(2a)工程(1)で得られた細胞を、TGFβシグナル阻害剤、及びWntシグナル伝達阻害剤を含む培養液中で浮遊培養して、細胞塊を得る工程と、
(2b)工程(2a)で得られた細胞塊を、TGFβシグナル阻害剤及びWntシグナル伝達阻害剤を実質的に含まない培養液中で浮遊培養して、大脳オルガノイドを得る工程と
を含む、[1]に記載の方法。
[3] 工程(2a)の浮遊培養が静置培養である、[2]に記載の方法。
[4] 工程(2b)の浮遊培養が振とう培養である、[2]又は[3]に記載の方法。
[5] 工程(1)の培養期間が3日間未満である、[1]~[4]のいずれかに記載の方法。
[6] 工程(1)の培養期間が12時間以上2日間以下である、[1]~[5]のいずれかに記載の方法。
[7] 工程(1)の培養液が、SB431542、A-83-01、及びXAV-939からなる群から選ばれるTGFβシグナル阻害剤を含む、[1]~[6]のいずれかに記載の方法。
[8] 工程(1)、工程(2a)、及び工程(2b)における培養液が、無血清培養液である、[2]~[7]のいずれかに記載の方法。
[9] 前記多能性幹細胞が、ヒト人工多能性幹細胞又はヒト胚性幹細胞である、[1]~[8]のいずれかに記載の方法。
[10] (3)工程(2)で得られた複数の細胞塊から、細胞塊の形状、内部構造、サイズ、表面の色彩若しくは模様、及び遺伝子発現からなる群から選ばれる1以上を指標とし、大脳オルガノイドを選別する工程をさらに含む、[1]~[9]のいずれかに記載の方法。
[11] [1]~[9]のいずれかに記載の方法を用いて製造される細胞培養物であって、
複数の球状の細胞塊を含み、
前記複数の球状の細胞塊に占める大脳オルガノイドの割合が40%以上である、細胞培養物。
[12] 前記大脳オルガノイドに占める大脳皮質様構造物の割合が40%以上である、[11]に記載の細胞培養物。
[13] 大脳オルガノイドが、更に以下の(1)~(5):
(1)球状の細胞塊であること、
(2)細胞塊の内部に、大脳皮質様構造物を有すること、
(3)表面に色素沈着を有さないこと、
(4)細胞塊の一部に嚢胞状形状、突起形状、及びバルーン状形状のいずれも有さないこと、並びに、
(5)NEUROD6、NEUROD2、SSTR2、TBR1、ZBTB18、NHLH1、IGFBPL1、NRN1、RTN1、THSD7A、NRXN1、BHLHE22、CALB2、KHDRBS3、CCSAP、PDE1A、NEUROD1、NPTX1、NXPH4、NTS、NEUROG2、OLFM1、PRDM8、CORO2B、TP53I11、ZFPM2、PCDH9、NELL2、SRRM4、SCG3、DCC、EPB41L3、SLC17A7、ST18、NSG2、EMX1、CAP2、SYT4、NSMF、ANK3、MYT1L、FSTL5、CELF4、B3GAT1、EPHA5、NHLH2、及びDLL3からなる群から選ばれる少なくとも1つのマーカーを発現していること
から選択される、1以上の特徴を有する細胞塊である、[12]に記載の細胞培養物。
[14] (a)増殖性マーカー陽性細胞数が全細胞数の10%以下であり、
(b)神経細胞マーカー、皮質V/VI層マーカー、及び前脳マーカーからなる群から選ばれる1つ以上のマーカーが陽性である細胞数が全細胞数の70%以上であり、且つ
(c)神経上皮又は大脳皮質様構造を実質的に含まないことを特徴とする、大脳皮質細胞塊。
[15] 前記(a)の増殖性マーカーがKi67であり、
前記(b)の神経細胞マーカーがβIII-Tubulinであり、皮質V/VI層マーカーがCtip2であり、前脳マーカーがFOXG1である、
[14]に記載の大脳皮質細胞塊。
[16] (d)NEUROD6、NEUROD2、SSTR2、TBR1、ZBTB18、NHLH1、IGFBPL1、NRN1、RTN1、THSD7A、NRXN1、BHLHE22、CALB2、KHDRBS3、CCSAP、PDE1A、NEUROD1、NPTX1、NXPH4、NTS、NEUROG2、OLFM1、PRDM8、CORO2B、TP53I11、ZFPM2、PCDH9、NELL2、SRRM4、SCG3、DCC、EPB41L3、SLC17A7、ST18、NSG2、EMX1、CAP2、SYT4、NSMF、ANK3、MYT1L、FSTL5、CELF4、B3GAT1、EPHA5、NHLH2、及びDLL3からなる群から選ばれる少なくとも1つのマーカーをさらに発現している、[14]又は[15]に記載の大脳皮質細胞塊。
[17] SLC17A7を発現している、[16]に記載の大脳皮質細胞塊。
[18] GAD2、COL1A1、TYR、TTR及びHOXA2からなる群から選ばれる1以上の遺伝子を実質的に発現していない、[15]~[17]のいずれかに記載の大脳皮質細胞塊。
[19] 支持細胞の非存在下で、多能性幹細胞から大脳皮質細胞塊を製造する方法であって、
(i)多能性幹細胞から大脳オルガノイドを得る工程と、
(ii)工程(i)で得られた大脳オルガノイドを、Notchシグナル阻害剤を含む培養液中で培養して、大脳皮質細胞塊を得る工程と
を含む、方法。
[20] 前記工程(i)において、[1]~[10]のいずれかに記載の方法により多能性幹細胞から大脳オルガノイドを得る、[19]に記載の方法。
[21] (A)増殖性マーカー陽性細胞数が全細胞数の5%以下であり、
(B)神経細胞マーカー、皮質V/VI層マーカー、及び前脳マーカーから選ばれる1つ以上のマーカー陽性細胞数が全細胞数の70%以上であり、且つ
(C)神経上皮又は大脳皮質様構造を実質的に含まない
ことを特徴とする、高純度大脳皮質細胞塊。
[22] 前記(A)の増殖性マーカーがKi67であり、
前記(B)の神経細胞マーカーがβIII-Tubulinであり、皮質V/VI層マーカーがCtip2であり、前脳マーカーがFOXG1である、
[21]に記載の高純度大脳皮質細胞塊。
[23] (D)NEUROD6、NEUROD2、SSTR2、TBR1、ZBTB18、NHLH1、IGFBPL1、NRN1、RTN1、THSD7A、NRXN1、BHLHE22、CALB2、KHDRBS3、CCSAP、PDE1A、NEUROD1、NPTX1、NXPH4、NTS、NEUROG2、OLFM1、PRDM8、CORO2B、TP53I11、ZFPM2、PCDH9、NELL2、SRRM4、SCG3、DCC、EPB41L3、SLC17A7、ST18、NSG2、EMX1、CAP2、SYT4、NSMF、ANK3、MYT1L、FSTL5、CELF4、B3GAT1、EPHA5、NHLH2、及びDLL3からなる群から選ばれる少なくとも1つの遺伝子を発現している、[21]又は[22]に記載の高純度大脳皮質細胞塊。
[24] SLC17A7、NEUROD6、及びEMX1からなる群から選ばれる1以上の遺伝子を発現している、[23]に記載の高純度大脳皮質細胞塊。
[25] GAD2、COL1A1、TYR、TTR及びHOXA2からなる群から選ばれる1以上の遺伝子を実質的に発現していない、[21]~[24]のいずれかに記載の高純度大脳皮質細胞塊。
[26] 支持細胞の非存在下で、多能性幹細胞から高純度大脳皮質細胞塊を製造する方法であって、
(i)多能性幹細胞から大脳オルガノイドを得る工程と、
(ii)工程(i)で得られた大脳オルガノイドを、培養液中で培養する工程と、
(iii)工程(ii)で得られた細胞培養物を、単細胞又は2~5個の細胞の集合体(Cell clump)にまで分散させる工程と、
(iv)工程(ii)で得られた細胞培養物又は工程(iii)で得られた細胞集団を、1以上の神経栄養因子、アスコルビン酸、及びcAMP活性化剤を含む培養液中で培養して、細胞塊を得る工程と
を含み、前記工程(ii)の培養液及び/又は前記工程(iv)の培養液は、Notchシグナル阻害剤を含む、方法。
[27] 前記工程(i)において、[1]~[10]のいずれかに記載の方法により多能性幹細胞から大脳オルガノイドを得る、[26]に記載の方法。
[28] 工程(ii)に付与する大脳オルガノイドが、神経細胞への分化誘導開始から28日後~44日後の大脳オルガノイドである、[19]、[20]、[26]及び[27]のいずれかに記載の方法。
[29] 工程(ii)の培養期間が2~6日間である、[19]、[20]、及び[26]~[28]のいずれかに記載の方法。
[30] 工程(iv)の培養期間が2~14日間である、[19]、[20]、及び[26]~[29]のいずれかに記載の方法。
[31] 前記Notchシグナル阻害剤は、γセクレターゼ阻害剤である、[19]、[20]、及び[26]~[30]のいずれかに記載の方法。
[32] 前記γセクレターゼ阻害剤がN-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester(DAPT)又はCompound Eである、[31]に記載の方法。
[33] [21]~[25]のいずれかに記載の高純度大脳皮質細胞塊を含む細胞集団であって、前記高純度大脳皮質細胞塊のサイズ、形状又は構成細胞組成が均一である、細胞集団。
[34] [14]~[18]のいずれかに記載の大脳皮質細胞塊、[21]~[25]のいずれかに記載の高純度大脳皮質細胞塊、若しくは[33]に記載の細胞集団、又はそれらを構成細胞にまで分散させて得られる細胞集団を有効成分として含む医薬組成物。
[35] [14]~[18]のいずれかに記載の大脳皮質細胞塊、[21]~[25]のいずれかに記載の高純度大脳皮質細胞塊、若しくは[33]に記載の細胞集団、又はそれらを構成細胞にまで分散させて得られる細胞集団を含む、移植用組織。
[36] [14]~[18]のいずれかに記載の大脳皮質細胞塊、[21]~[25]のいずれかに記載の高純度大脳皮質細胞塊、若しくは[33]に記載の細胞集団、又はそれらを構成細胞にまで分散させて得られる細胞集団を有効成分として含む、脳血管障害治療薬。
[37] [14]~[18]のいずれかに記載の大脳皮質細胞塊、[21]~[25]のいずれかに記載の高純度大脳皮質細胞塊、若しくは[33]に記載の細胞集団、又はそれらを構成細胞にまで分散させて得られる細胞集団を、これを必要とする対象の大脳皮質又は大脳基底核に投与又は移植することを含む、脳血管障害の治療方法。
[38] (aa)大脳オルガノイド又は大脳皮質細胞塊における、GAD2、COL1A1、TYR、TTR及びHOXA2からなる群から選ばれる少なくとも1つの遺伝子又は該遺伝子によりコードされるタンパク質もしくはその断片の発現量を測定する工程、及び
(bb)工程(aa)の測定結果に基づき、上記遺伝子の発現量が基準値以下である場合に、上記大脳オルガノイド又は上記大脳皮質細胞塊に含まれる目的外細胞の量が基準以下であると評価する工程、
を含む、大脳オルガノイド又は大脳皮質細胞塊の品質評価方法。
[39] (AA)大脳オルガノイド又は大脳皮質細胞塊における、NEUROD6、NEUROD2、SSTR2、TBR1、ZBTB18、NHLH1、IGFBPL1、NRN1、RTN1、THSD7A、NRXN1、BHLHE22、CALB2、KHDRBS3、CCSAP、PDE1A、NEUROD1、NPTX1、NXPH4、NTS、NEUROG2、OLFM1、PRDM8、CORO2B、TP53I11、ZFPM2、PCDH9、NELL2、SRRM4、SCG3、DCC、EPB41L3、SLC17A7、ST18、NSG2、EMX1、CAP2、SYT4、NSMF、ANK3、MYT1L、FSTL5、CELF4、B3GAT1、EPHA5、NHLH2、及びDLL3からなる群から選ばれる少なくとも1つの遺伝子の発現量を測定する工程、及び
(BB)工程(AA)の測定結果に基づき、上記遺伝子の発現量が基準値以上である場合に、上記大脳オルガノイド又は上記大脳皮質細胞塊に含まれる目的細胞の量が基準以上であると評価する工程、
を含む、大脳オルガノイド又は大脳皮質細胞塊の品質評価方法。
〔幹細胞〕
本明細書において「幹細胞」とは、分化能及び分化能を維持した増殖能(特に自己複製能)を有する未分化細胞を意味する。幹細胞には、分化能力に応じて、多能性幹細胞(pluripotent stem cell)、複能性幹細胞(multipotent stem cell)、単能性幹細胞(unipotent stem cell)等の亜集団が含まれる。
本明細書において「マーカー」は、細胞に存在する物質であって、その存在又は存在量に基づいて、当該細胞の種類若しくは性質等を同定若しくは判別することができる物質を意味する。マーカーとして、具体的には、mRNA、当該mRNAによりコードされるタンパク質、及び糖鎖、並びにこれらの断片が挙げられる。
本明細書において神経細胞とは、細胞体、樹状突起及び軸索から構成される神経単位のことを言い、ニューロンとも呼ばれる。神経細胞は、他の神経細胞又は刺激受容細胞からの刺激を別の神経細胞、筋又は腺細胞に刺激を伝える機能を有し、神経細胞が産生する神経伝達物質の違いにより、ドパミン作動性神経、セロトニン作動性神経、GABA作動性神経、グルタミン酸作動性神経等のように分類されるが、本明細書においては神経伝達物質の種類については特に限定しない。神経細胞は、有意に発現するマーカーによって同定することができ、当該マーカーとしては、例えばβIII-Tubulin、MAP2等が挙げられる。
本明細書において「神経幹細胞」は、神経系細胞への分化が運命づけられているが、複数の神経系細胞へ分化する能力を有し増殖性を保っている幹細胞を意味し、神経前駆細胞への分化能、及び大脳皮質細胞への分化能を共に有する細胞である。中間体フィラメントタンパク質(ネスチン、ビメンチン等)、転写因子SOX1,SOX2、PAX6等の原始的神経外胚葉及び神経幹細胞のマーカー等によって同定されうる。本明細書における神経幹細胞には、放射状グリア(Radial Gila)が含まれる。
本明細書において、「細胞培養物」は、特定の組成を有するものに限らず、細胞を培養することで得られる産物をさす用語である。細胞培養物は、複数の細胞塊の集合体であり得、さらに単細胞及び後述の2~5個の細胞の集合体を含んでもよい。細胞培養物は、培地又は懸濁媒体を含んでいても含まなくてもよく、本明細書において、特に明記しない限り、培地又は懸濁媒体を含まないものを指す。
本明細書において、「大脳皮質細胞」は、大脳皮質ニューロン又は大脳皮質神経細胞ともいい、大脳皮質を構成する神経細胞を指す。
本明細書において「細胞塊」(Cell Aggregate;細胞凝集体ともいう)とは、複数の細胞同士が接着して立体構造を形成しているものであれば特に限定されず、例えば、培地等の媒体中に分散していた細胞が集合して形成する塊、又は細胞分裂を経て形成される細胞の塊等をいう。細胞塊には、特定の組織を形成している場合も含まれる。胚様体(Embryoid body)、スフェア(Sphere)、スフェロイド(Spheroid)は、細胞塊に包含される。細胞塊は、任意の形状を有してよく、球状の細胞塊、層状の細胞塊などが挙げられる。
本明細書において「培養液(培地とも称する)」は、動物細胞の培養に通常用いられる培養液(培地)であればよく、動物細胞が生命を維持することができれば特に限定されないが、好ましくは目的細胞に増殖可能な環境を提供するものである。培養液(培地)は自ら調製してもよく市販培地を購入して用いてもよい。
本発明で使用される培地には、血清代替物が含まれていてもよい。血清代替物として、例えば、アルブミン、トランスフェリン、脂肪酸、コラーゲン前駆体、微量元素、2-メルカプトエタノール又は3’チオールグリセロール、或いはこれらの均等物等を適宜含有するものを挙げることができる。かかる血清代替物は、例えば、WO98/30679に記載の方法により調製することができる。血清代替物としては市販品を利用してもよい。かかる市販の血清代替物としては、例えば、Knockout Serum Replacement(サーモフィッシャーサイエンティフィック社製;以下、KSRと記す場合がある。)、「StemSure(登録商標)Serum Replacement(SSR)」Chemically-defined Lipid concentrated(サーモフィッシャーサイエンティフィック社製)、B27サプリメント(サーモフィッシャーサイエンティフィック社製)、N2サプリメント(サーモフィッシャーサイエンティフィック社製)、ITSサプリメント(サーモフィッシャーサイエンティフィック社製)が挙げられ、好ましくはN2サプリメント又はB27サプリメントが挙げられる。
本明細書において、支持細胞とは、フィーダー細胞ともいい、多能性幹細胞等の幹細胞を培養するときに共存させる当該幹細胞以外の細胞をいう。支持細胞としては、例えば、マウス線維芽細胞(MEF等)、ヒト線維芽細胞、SNL細胞、STO細胞等が挙げられる。支持細胞は、増殖抑制処理された支持細胞であってもよい。ここで、増殖抑制処理としては、増殖抑制剤(例えば、マイトマイシンC)処理又はガンマ線照射若しくはUV照射等による処理が挙げられる。
本発明において「浮遊培養」とは、細胞を培地中に浮遊させた状態で生存させること、又は、細胞を培養容器へ非接着の状態で凝集体(スフェアとも言う)を形成させて培養することを意味する。本明細書中では、細胞は、単細胞又は複数の細胞が集まった塊(細胞塊又は細胞集団)の状態で浮遊培養される。
本明細書において、bFGFは、塩基性線維芽細胞成長因子(basic fibroblast growth factor)を意味し、FGF2とも称されるタンパク質である。
本明細書において、TGFβシグナル阻害剤とは、TGFβの受容体への結合からSMADへと続くシグナル伝達を阻害する物質であり、受容体であるALKファミリーへの結合を阻害する物質、又はALKファミリーによるSMADのリン酸化を阻害する物質が挙げられる。
本明細書において、Wntシグナル伝達阻害剤とは、Wnt(例えばWnt3)の産生を抑制する物質、又はWntの受容体への結合からβカテニンの蓄積へと続くシグナル伝達を阻害する物質であり、受容体であるFrizzledファミリーへの結合を阻害する物質、又はβカテニンの分解を促進する物質などが挙げられる。
Notchシグナル阻害剤は、Notchによるシグナル伝達を抑制し得る物質である限り限定されない。Notchシグナル阻害剤としては、γ-セクレターゼ阻害剤、Notch転写複合体阻害剤、例えばMAML-1阻害剤等が挙げられる。
ROCK阻害剤は、Rho-associated coiled-coilキナーゼ(ROCK)の阻害剤であり、ROCKの機能を抑制する物質である限り特に限定されない。ROCK阻害剤としては、Y-27632((+)-(R)-trans-4-(1-aminoethyl)-N-(4-pyridyl)cyclohexanecarboxamide dihydrochloride)、H-1152((S)-4-Methyl-5-((2-methyl-1,4-diazepan-1-yl)sulfonyl)isoquinoline dihydrochloride)、Fasudil(HA-1077;1-(5-Isoquinolinesulfonyl)homopiperazine Hydrochloride)、Wf-536(4-[(1R)-1-aminoethyl]-N-(pyridin-4-yl)benzamide)、Thiazovivin(N-Benzyl-2-(pyrimidin-4-ylamino)thiazole-4-carboxamide)、Ripasudil(4-Fluoro-5-[[(2S)-hexahydro-2-methyl-1H-1,4-diazepin-1-yl]sulfonyl]isoquinoline)、GSK42928(4-[4-(Trifluoromethyl)phenyl]-N-(6-Fluoro-1H-indazol-5-yl)-2-methyl-6-oxo-1,4,5,6-tetrahydro-3-pyridinecarboxamide)6、RKI-1447(N-[(3-Hydroxyphenyl)methyl]-N'-[4-(4-pyridinyl)-2-thiazolyl]urea)、Azaindole1(6-chloro-N4-[3,5-difluoro-4-[(3-methyl-1H-pyrrolo[2,3-b]pyridin-4-yl)oxy]phenyl]pyrimidine-2,4-diamine)、HA-1100(1-[(1,2-Dihydro-1-oxo-5-isoquinolinyl)sulfonyl]hexahydro-1H-1,4-diazepine)、Y-39983(4-[(1R)-1-Aminoethyl]-N-1H-pyrrolo[2,3-b]pyridin-4-ylbenzamide)等が挙げられる。ROCK阻害剤として、好ましくはY-27632が挙げられる。
本明細書において、神経栄養因子(Neurotrophic Factor)とは、神経細胞の生存、神経突起や軸索の伸展、シナプス形成等を促進する活性を有する分泌タンパク質の総称である。神経栄養因子として、Nerve Growth Factor(NGF)、Brain-derived Neurotrophic Factor(BDNF)、Neurotrophin 3(NT-3)、Neurotrophin 4/5(NT-4/5)、Neurotrophin 6(NT-6)、Glia cell line-derived Neurotrophic Factor(GDNF)、及びCiliary Neurotrophic Factor(CNTF)などが挙げられる。本発明において好ましい神経栄養因子は、GDNF、及びBDNFから成るグループより選択される因子である。神経栄養因子は、例えばWako社やR&D systems社等から市販されており容易に利用することが可能であるが、当業者に公知の方法によって細胞へ強制発現させることによって得てもよい。
本明細書において、cAMP活性化剤としては、cAMP、dibutyryl-cAMP又はフォルスコリン等が挙げられる。
本発明の大脳オルガノイドの製造方法の一態様としては、下記工程(1)及び工程(2)を含む、支持細胞の非存在下、多能性幹細胞から大脳オルガノイドを製造する方法が挙げられる。「支持細胞の非存在下」は、上述のとおりであり、工程(1)及び工程(2)は共に、「支持細胞の非存在下」で行われる。
(1)多能性幹細胞を、bFGFが実質的に含まれず、かつTGFβシグナルを実質的に惹起しない培養液中で培養する工程
(2)工程(1)で得られた細胞を、神経細胞へと分化誘導する工程
<工程(1)>
本明細書において、「物質Xの存在下」での培養とは、「物質Xを含む培地」中で培養することを意味し、上記物質Xは上記培地に本来の成分として含まれているものであってもよく、外来性(exogenous)に添加されたものであってもよい。すなわち、当該培養中に細胞又は組織によって発現、分泌若しくは産生され得る内在性(endogenous)の物質Xは外来性の物質Xとは区別され、外来性の物質Xを含んでいない培地は内在性の物質Xを含んでいても「物質Xを含む培地」の範疇には該当しないと解する。
工程(1)で得られる細胞は、当業者に周知の方法で、神経細胞へ分化誘導することができ、これにより、大脳オルガノイドを得ることができる。
(2a)工程(1)で得られた細胞を、TGFβシグナル阻害剤、及びWntシグナル伝達阻害剤を含む培養液中で浮遊培養、好ましくは静置培養して、細胞塊を得る工程
(2b)工程(2a)で得られた細胞塊を、TGFβシグナル阻害剤又はWntシグナル伝達阻害剤を実質的に含まない培養液中、好ましくは、TGFβシグナル阻害剤及びWntシグナル伝達阻害剤のいずれも含まない培養液中で浮遊培養して、大脳オルガノイドを得る工程。
工程(1)で得られる細胞は、培養液中、好ましくは血清(無調整又は未精製の血清)を実質的に含まない無血清培養液中に分散され、非接着性の条件下で培養(即ち浮遊培養)し、複数の細胞を集合させて細胞塊を形成させる。凝集時に用いられる培養液としては、血清代替物を含む無血清培養液を用いてもよい。
1)比較的小さな体積(例えば、1ml以下、500μl以下、200μl以下、100μl以下)の培養コンパートメント中に、分散した細胞を閉じ込め、該コンパートメント中に1個の細胞塊を形成する方法、又は
2)遠心チューブに分散した細胞を入れ、これを遠心し、1箇所に細胞を沈殿させることにより、該チューブ中に1個の細胞塊を形成する方法。
工程(2b)においては、工程(2a)で得られた細胞塊を、更にTGFβシグナル阻害剤及びWntシグナル伝達阻害剤を実質的に含まない培養液中で浮遊培養することにより、大脳オルガノイドを得る。
本発明の大脳オルガノイドの製造方法は、さらに大脳オルガノイドを選別する工程である工程(3)を含んでよい。工程(3)は、工程(2)で得られた複数の細胞塊から、細胞塊の形状、内部構造、サイズ、表面の色彩若しくは模様、及び遺伝子発現からなる群から選ばれる1以上を指標として、所望の大脳オルガノイドを選別する工程である。これらの指標のうち、2以上、3以上又は4以上を採用することが好ましく、また、指標として、細胞塊の形状、内部構造、表面の色彩若しくは模様、及び/又は遺伝子発現であることが好ましく、細胞塊の形状、内部構造、及び/又は遺伝子発現であることがより好ましく、細胞塊の形状であることが特に好ましい。例えば、細胞塊の形状が球状(スフェア状)である場合、所望の大脳オルガノイドとして選別することができる。
(1)球状の細胞塊であること、
(2)細胞塊の内部に、大脳皮質様構造物を有すること、
(3)細胞塊の表面に色素沈着を有しないこと、
(4)細胞塊の一部に嚢胞状形状、突起形状、バルーン状形状を有しないこと、並びに、
(5)細胞塊が、NEUROD6、NEUROD2、SSTR2、TBR1、ZBTB18、NHLH1、IGFBPL1、NRN1、RTN1、THSD7A、NRXN1、BHLHE22、CALB2、KHDRBS3、CCSAP、PDE1A、NEUROD1、NPTX1、NXPH4、NTS、NEUROG2、OLFM1、PRDM8、CORO2B、TP53I11、ZFPM2、PCDH9、NELL2、SRRM4、SCG3、DCC、EPB41L3、SLC17A7、ST18、NSG2、EMX1、CAP2、SYT4、NSMF、ANK3、MYT1L、FSTL5、CELF4、B3GAT1、EPHA5、NHLH2、及びDLL3からなる群から選ばれる少なくとも少なくとも1つ、少なくとも2つ、少なくとも3つ、又は少なくとも5つの遺伝子を発現していること。
。すなわち、工程(3)を経ることにより、細胞塊の集団における大脳オルガノイドの割合を更に向上させることができる。
本発明の細胞培養物は、上述の工程(1)及び工程(2)を含む大脳オルガノイドの製造方法を用いて製造される細胞培養物であって、複数の球状の細胞凝集体を含み、該複数の球状の細胞凝集体に占める大脳オルガノイドの割合が40%以上、好ましくは50%以上、更に好ましくは60%以上である細胞培養物である。ここで当該割合は、細胞培養物をサンプリングして、該サンプルにおける、球状の細胞凝集体の全数に占める大脳オルガノイドの数で算出することができる。
(1)球状の細胞塊であること、
(2)細胞塊の内部に、大脳皮質様構造物を有すること、
(3)表面に色素沈着を有しないこと、
(4)細胞塊の一部に嚢胞状形状、突起形状、バルーン状形状を有しないこと、並びに、
(5)NEUROD6、NEUROD2、SSTR2、TBR1、ZBTB18、NHLH1、IGFBPL1、NRN1、RTN1、THSD7A、NRXN1、BHLHE22、CALB2、KHDRBS3、CCSAP、PDE1A、NEUROD1、NPTX1、NXPH4、NTS、NEUROG2、OLFM1、PRDM8、CORO2B、TP53I11、ZFPM2、PCDH9、NELL2、SRRM4、SCG3、DCC、EPB41L3、SLC17A7、ST18、NSG2、EMX1、CAP2、SYT4、NSMF、ANK3、MYT1L、FSTL5、CELF4、B3GAT1、EPHA5、NHLH2、及びDLL3からなる群から選ばれる少なくとも1つ、少なくとも2つ、少なくとも3つ、又は少なくとも5つの遺伝子を発現していること。
本発明の一態様として、下記の工程(i)及び工程(ii)を含む、支持細胞の非存在下で、多能性幹細胞から大脳皮質細胞塊を製造する方法が挙げられる。
(i)多能性幹細胞から大脳オルガノイドを得る工程、
(ii)工程(i)で得られた大脳オルガノイドを、Notchシグナル阻害剤、好ましくはγ-セクレターゼ阻害剤を含む培養液中で培養して、大脳皮質細胞塊を得る工程。
(i)上記2に記載の大脳オルガノイドの製造方法(即ち、請求項1~10のいずれか1項に記載の方法を含む)により、大脳オルガノイドを得る工程、
(ii)工程(i)で得られた大脳オルガノイドを、Notchシグナル阻害剤、好ましくはγ-セクレターゼ阻害剤を含む培養液中で培養して、大脳皮質細胞塊を得る工程。
工程(i)は上記2における工程(1)、工程(2a)及び工程(2b)に記載のとおりである。また、工程(i)は上記2における工程(3)をさらに含んでもよい。
上述の工程(2b)を行った後、場合により工程(3)による選別を行い、Notch阻害剤、好ましくはγ-セクレターゼ阻害剤を含む培地に培地交換を行うことにより、工程(ii)を行うことができる。あるいは、上述の工程(2b)を行った後工程(ii)を実施し、次いで工程(3)の選別を行ってもよい。
本発明の一態様として、支持細胞の非存在下で、多能性幹細胞から大脳皮質細胞塊を製造する方法が挙げられ、当該方法が、
(i)多能性幹細胞から大脳オルガノイドを得る工程と、
(ii)工程(i)で得られた大脳オルガノイドを、培養液中で培養する工程と、
(iii)工程(ii)で得られた細胞培養物を、単細胞又は2~5個の細胞の集合体(Cell clump)にまで分散させる工程と、
(iv)工程(ii)で得られた細胞培養物又は工程(iii)で得られた細胞集団を、1以上の神経栄養因子、アスコルビン酸、及びcAMP活性化剤を含む培養液中で培養する工程と
を含み、上記工程(iii)の単細胞又は2~5個の細胞の集合体(Cell clump)への分散工程は任意の工程であり、上記工程(ii)の培養液及び/又は上記工程(iv)の培養液は、Notchシグナル阻害剤を含む。
(iii)工程(ii)で得られた大脳オルガノイドを、単細胞又は2~5個の細胞の集合体(Cell clump)にまで分散する工程、
(iv-1)工程(iii)で得られた細胞集団を、1以上の神経栄養因子、アスコルビン酸及びcAMP活性化剤(例えば、dibutyryl cAMP(dbcAMP))を含む無血清培養液中で培養して、(例えば20細胞以上の細胞からなる)細胞塊を得る工程。
工程(ii)で得られた大脳オルガノイドは、当業者に周知の方法、例えばピペッティング等により物理的に、又は酵素処理により、単細胞又は2~5個の細胞の集合体(Cell clump)にまで分散させることができる。ここで、分散して得られた細胞集団は、単細胞及び/又は2~5個の細胞の集合体(Cell Clump)の集団であり得る。すなわち、工程(iii)において、当業者に周知の方法によって、工程(ii)で得られた大脳オルガノイドを単細胞、又は2~5個の細胞の集合体(Cell Clump)にまで分散してよい。
工程(iii)で得られる細胞集団を浮遊培養することにより再凝集させ、高純度の、大脳皮質細胞を含む細胞塊(高純度大脳皮質細胞塊)を得ることができる。
本発明の一態様として、上記5に記載の大脳皮質細胞塊の製造方法2-1において、工程(iii)を含まない態様が挙げられる。すなわち、上記4に記載の大脳皮質細胞塊の製造方法1において、工程(i)及び工程(ii)に加えて、以下の工程(iv-2)で処理することを含む、大脳皮質細胞塊の製造方法が挙げられる。ここで、工程(ii)において、工程(i)で得られた大脳オルガノイドを、Notchシグナル阻害剤、好ましくはγ-セクレターゼ阻害剤を含む培養液中で培養する。
(iv-2)工程(ii)で得られた大脳オルガノイドを含む細胞培養物を、1以上の神経栄養因子、アスコルビン酸若しくはその誘導体(例えば、アスコルビン酸-2-リン酸)、及びcAMP活性化剤(例えば、dibutyryl cAMP(dbcAMP))を含む培養液中で浮遊培養して、大脳皮質細胞塊を得る工程。
工程(ii)で得られる大脳オルガノイドを含む細胞培養物を浮遊培養することにより、大脳皮質細胞を含む細胞塊(大脳皮質細胞塊)を得ることができる。
本発明の一態様として、工程(ii)の培養液が、Notchシグナル阻害剤を含む、「大脳皮質細胞塊の製造方法3-1」が挙げられる。すなわち、大脳皮質細胞塊の製造方法3-1としては、以下の工程(I)を含む、大脳皮質細胞塊の製造方法が挙げられる。
(I)多能性幹細胞由来の大脳オルガノイドを、Notch阻害剤、好ましくはγ-セクレターゼ阻害剤を含む培養液中で培養する工程。
ここで、工程(I)は大脳皮質細胞塊の製造方法1における工程(ii)と同様に行えばよい。
(II)工程(I)で得られた大脳オルガノイドを含む細胞培養物を、単細胞又は2~5個の細胞の集合体(Cell clump)にまで分散する工程、及び
(III)工程(II)で得られた細胞集団を、1以上の神経栄養因子、アスコルビン酸若しくはその誘導体、及びcAMP活性化剤を含む培養液中で培養して、大脳皮質細胞塊を得る工程。
ここで、工程(I)は大脳皮質細胞塊の製造方法1における工程(ii)と同様に行えばよく、工程(II)及び工程(III)は、大脳皮質細胞塊の製造方法2における工程(iii)及び工程(iv-1)と同様に行えばよい。また、工程(III)を経て得られる大脳皮質細胞塊は、分散-再凝集の工程を経ており、高純度大脳皮質細胞凝集塊である。
(III-2)工程(I)で得られた大脳オルガノイドを含む細胞培養物を、1以上の神経栄養因子、アスコルビン酸若しくはその誘導体(例えば、アスコルビン酸-2-リン酸)、及びcAMP活性化剤(例えば、dibutyryl cAMP(dbcAMP))を含む培養液中で浮遊培養して、大脳皮質細胞塊を得る工程。
ここで、工程(I)は大脳皮質細胞塊の製造方法1における工程(ii)と同様に行えばよく、工程(III-2)は大脳皮質細胞塊の製造方法2における工程(iv-1)と同様に行えばよい。
(I-2)多能性幹細胞由来の大脳オルガノイドを培養液中で培養する工程、
(II)工程(I―2)で得られた大脳オルガノイドを含む細胞培養物を、単細胞又は2~5個の細胞の集合体(Cell clump)にまで分散する工程、及び
(III-3)工程(II)で得られた細胞集団を、1以上の神経栄養因子、アスコルビン酸若しくはその誘導体、及びcAMP活性化剤を含む培養液中で培養して、大脳皮質細胞塊を得る工程を含み、ここで、工程(I-2)の培養液はNotchシグナル阻害剤を含まず、上記工程(III-3)の培養液はNotchシグナル阻害剤を含む。ここで、工程(I-2)は上記2に記載の大脳オルガノイドの製造方法における工程(2b)と同様に行えばよい。工程(I-2)は多能性幹細胞由来の大脳オルガノイドを製造する工程の一部に相当する培養であってもよく、培養期間に特に限定はない。工程(II)は上記5に記載の大脳皮質細胞塊の製造方法2-1における工程(iii)と同様に行えばよい。
工程(III-3)の培養液は、当該工程の一部又は全ての期間においてNotchシグナル阻害剤を含む態様でもよい。Notchシグナル阻害剤の処理期間が一部の期間の場合は、上記工程(III-3)の培養液の交換時にNotchシグナル阻害剤を含まない培養液を用いる。一部の期間は、Notchシグナル阻害剤による処理の効果が得られる限り、特に限定されず、上記工程(III-3)のの培養期間のうちの数時間~数日、具体的には1~7日間、好ましくは2~6日間、より好ましくは2~4日間でもよい。また、極めて短い処理期間(例えば、4時間、12時間、24時間、2日間)の繰り返しでもよい。また、Notchシグナル阻害剤を含む期間と、Notchシグナル阻害剤を含まない期間を交互に行ったり、濃度を変えたNotchシグナル阻害剤を用いた組み合わせも本願の範疇である。下記工程(III-4)及び工程(III-5)の培養液も工程(III-3)の培養液と同様に当該工程の一部又は全ての期間においてNotchシグナル阻害剤を含む態様でもよい。
工程(II)で得られる細胞集団を浮遊培養することにより再凝集させ、高純度の大脳皮質細胞を含む細胞塊(高純度大脳皮質細胞塊)を得ることができる。
本発明の一態様として、以下の性質を有する大脳皮質細胞塊が挙げられる。当該大脳皮質細胞塊は、上記4に記載の大脳皮質細胞塊の製造方法1により製造できる。
(a)増殖性マーカー陽性細胞数が全細胞数の10%以下であること、
(b)神経細胞マーカー、皮質V/VI層マーカー、及び前脳マーカーからなる群から選ばれる1つ以上のマーカーが陽性である細胞数が全細胞数の60%以上、好ましくは70%以上、より好ましくは80%以上であること、
(c)神経上皮又は大脳皮質様構造を実質的に含まないこと。
(a’)Ki67陽性細胞数が全細胞数の5%以下であること、
(b’)βIII-Tubulin(βTubIII)、Ctip2、及びFoxg1がいずれも陽性の細胞数が全細胞数の60%以上、好ましくは70%以上、より好ましくは80%以上であること、
(c)神経上皮又は大脳皮質様構造を実質的に含まないこと。
(d)NEUROD6、NEUROD2、SSTR2、TBR1、ZBTB18、NHLH1、IGFBPL1、NRN1、RTN1、THSD7A、NRXN1、BHLHE22、CALB2、KHDRBS3、CCSAP、PDE1A、NEUROD1、NPTX1、NXPH4、NTS、NEUROG2、OLFM1、PRDM8、CORO2B、TP53I11、ZFPM2、PCDH9、NELL2、SRRM4、SCG3、DCC、EPB41L3、SLC17A7、ST18、NSG2、EMX1、CAP2、SYT4、NSMF、ANK3、MYT1L、FSTL5、CELF4、B3GAT1、EPHA5、NHLH2、及びDLL3からなる群から選ばれる少なくとも1つの遺伝子をさらに発現している。上記大脳皮質細胞塊は好ましくはSLC17A7、NEUROD6、及びEMX1からなる群から選ばれる1以上の遺伝子を発現しており、より好ましくはGAD2、COL1A1、TYR、TTR及びHOXA2からなる群から選ばれる1以上の遺伝子を実質的に発現していない。
(e)神経幹細胞マーカー陽性細胞が全細胞数の10%以下であり、
上記神経幹細胞マーカーとしては、Pax6、Sox1又はSox2が挙げられる。
(f)多能性マーカー陰性である。
本発明の一態様として、以下の性質を有する高純度大脳皮質細胞塊が挙げられる。以下の性質を有し、増殖性マーカー陽性細胞が抑制され、目的とする神経細胞の含有率が向上した高純度大脳皮質細胞塊は、上記5に記載の大脳皮質細胞塊の製造方法2-1、上記6に記載の大脳皮質細胞塊の製造方法2-2、又は、上記7に記載の大脳皮質細胞塊の製造方法3-1乃至3-6により製造できる。
(A)増殖性マーカー陽性細胞数が全細胞数の5%以下、好ましくは3%以下、更に好ましくは1%以下であること、
(B)神経細胞マーカー、皮質V/VI層マーカー、及び前脳マーカーから選ばれる1つ以上のマーカー陽性細胞数が全細胞数の60%以上、好ましくは70%以上、より好ましくは80%以上であること、
(C)神経上皮又は大脳皮質様構造を実質的に含まないこと。
(A)Ki67陽性細胞数が全細胞数の5%以下であること、
(B)βIII-Tubulin(βTubIII)、Ctip2、及びFoxg1がいずれも陽性の細胞数が全細胞数の60%以上、好ましくは70%以上、より好ましくは80%以上であること、
(C)神経上皮又は大脳皮質様構造を実質的に含まないこと。
(D)NEUROD6、NEUROD2、SSTR2、TBR1、ZBTB18、NHLH1、IGFBPL1、NRN1、RTN1、THSD7A、NRXN1、BHLHE22、CALB2、KHDRBS3、CCSAP、PDE1A、NEUROD1、NPTX1、NXPH4、NTS、NEUROG2、OLFM1、PRDM8、CORO2B、TP53I11、ZFPM2、PCDH9、NELL2、SRRM4、SCG3、DCC、EPB41L3、SLC17A7、ST18、NSG2、EMX1、CAP2、SYT4、NSMF、ANK3、MYT1L、FSTL5、CELF4、B3GAT1、EPHA5、NHLH2、及びDLL3からなる群から選ばれる少なくとも少なくとも1つ、少なくとも2つ、少なくとも3つ、又は少なくとも5つの遺伝子をさらに発現している。上記大脳皮質細胞塊は好ましくはSLC17A7、NEUROD6、及びEMX1からなる群から選ばれる1以上の遺伝子、又は全ての遺伝子を発現しており、より好ましくはGAD2、COL1A1、TYR、TTR及びHOXA2からなる群から選ばれる1以上の遺伝子を実質的に発現していない。
(E)神経幹細胞マーカー陽性細胞が全細胞数の10%以下、好ましくは5%以下、更に好ましくは3%以下であり、
上記神経幹細胞マーカーとしては、Pax6、Sox1又はSox2が挙げられる。
(F)多能性マーカー陰性である。
本発明の一態様として、上記2に記載の大脳オルガノイドの製造方法により得られる大脳オルガノイド、若しくは上記3に記載の大脳オルガノイド、これらの大脳オルガノイドから得られる大脳皮質細胞を含む細胞集団、上記4~7に記載の大脳皮質細胞塊の製造方法で得られる大脳皮質細胞塊、上記8若しくは9に記載の大脳皮質細胞塊、又はこれらの細胞塊から得られる大脳皮質細胞を含む細胞集団を有効成分とする、医薬組成物が挙げられる。すなわち、上記2に記載の大脳オルガノイドの製造方法により得られる大脳オルガノイド、若しくは上記3に記載の大脳オルガノイド、これらの大脳オルガノイドから得られる大脳皮質細胞を含む細胞集団、上記4~7に記載の大脳皮質細胞塊の製造方法で得られる大脳皮質細胞塊、上記8若しくは9に記載の大脳皮質細胞塊、又はこれらの細胞塊から得られる大脳皮質細胞を含む細胞集団は、移植用細胞塊(移植片ともいう)又は移植用細胞・移植用組織として、細胞移植治療のための有効成分として利用できる。
本発明の一態様として、本発明の上記2に記載の大脳オルガノイドの製造方法で得られる大脳オルガノイド、若しくは上記3に記載の大脳オルガノイド、これらの大脳オルガノイドから得られる大脳皮質細胞を含む細胞集団、上記4~7に記載の大脳皮質細胞塊の製造方法で得られる大脳皮質細胞塊、上記8若しくは9に記載の大脳皮質細胞塊、これらの細胞塊から得られる大脳皮質細胞を含む細胞集団、又は上記10に記載の医薬組成物の有効量を、移植を必要とする対象に移植(投与)することを含む、大脳皮質細胞の補充又は機能改善が必要な疾患、具体的には脳血管障害の治療方法が挙げられる。ここで、投与(移植)部位は大脳皮質又は大脳基底核であってよい。対象は、哺乳動物あってよく、好ましくはげっ歯類(例、マウス、ラット)又は霊長類(例、ヒト、サル)であり、より好ましくはヒトである。また、脳血管障害は、頭部外傷であってもよい。また、当該治療方法は、脳血管障害の患者における運動機能(例えば運動麻痺等の症状の改善)の改善方法、脳血管障害の患者における大脳皮質細胞の補充方法等を含む概念である。
本発明の一態様として、上記2に記載の大脳オルガノイドの製造方法で得られる大脳オルガノイド、若しくは上記3に記載の大脳オルガノイド、又はこれらの大脳オルガノイドから得られる大脳皮質細胞を含む細胞集団、上記4~7に記載の大脳皮質細胞塊の製造方法で得られる大脳皮質細胞塊、上記8若しくは9に記載の大脳皮質細胞塊、又は、これらの細胞塊を分散させて得られる細胞集団に被験物質を接触させ、被検物質が該細胞塊又は該細胞集団に及ぼす影響を検出又は定量することを含む、被験物質の毒性又は薬効を評価するための方法が挙げられる。
本発明の一態様として、以下の工程(aa)及び工程(bb)を含む、大脳オルガノイド又は大脳皮質細胞塊の品質評価方法が挙げられる。ここにおいて大脳皮質細胞塊には、本発明の高純度大脳皮質細胞塊が包含される。
(aa)大脳オルガノイド又は大脳皮質細胞塊における、GAD2、COL1A1、TYR、TTR及びHOXA2からなる群から選ばれる少なくとも1つ、又は全ての遺伝子の発現量を測定する工程、
(bb)工程(aa)の測定結果に基づき、上記遺伝子の発現量が基準値以下である場合に、上記大脳オルガノイド又は上記大脳皮質細胞塊に含まれる目的外細胞の量が基準以下であると評価する工程。
(AA)大脳オルガノイド又は大脳皮質細胞塊における、NEUROD6、NEUROD2、SSTR2、TBR1、ZBTB18、NHLH1、IGFBPL1、NRN1、RTN1、THSD7A、NRXN1、BHLHE22、CALB2、KHDRBS3、CCSAP、PDE1A、NEUROD1、NPTX1、NXPH4、NTS、NEUROG2、OLFM1、PRDM8、CORO2B、TP53I11、ZFPM2、PCDH9、NELL2、SRRM4、SCG3、DCC、EPB41L3、SLC17A7、ST18、NSG2、EMX1、CAP2、SYT4、NSMF、ANK3、MYT1L、FSTL5、CELF4、B3GAT1、EPHA5、NHLH2、及びDLL3からなる群から選ばれる少なくとも1つ、少なくとも2つ、少なくとも3つ、又は少なくとも5つの遺伝子の発現量を測定する工程、
(BB)工程(AA)の測定結果に基づき、上記遺伝子の発現量が基準値以上である場合に、上記大脳オルガノイド又は上記大脳皮質細胞塊に含まれる目的細胞の量が基準以上であると評価する工程。
(1)4~7に記載の製造方法で、大脳皮質細胞塊を製造する工程、
(2)上記品質評価方法により、目的細胞の量が基準以上であること、及び/又は、目的外細胞の量が基準以下であることを評価する工程、
(3)(2)の評価結果に基づき、移植に用いることが可能な大脳皮質細胞塊を選択又は同定する工程。
hiPSCは、ラミニン511のE8フラグメントであるiMatrix-511(株式会社ニッピ)を0.5μg/cm2になるように6ウェルプレートに添加しコーディングしたプレート上にてStemFit(登録商標)AK02N培地又はAK03N培地で(以下、「StemFit」という場合がある;味の素ヘルシーサプライ社製)を用いて維持培養した。継代は、hiPSCを0.5×Tryple Selectを用いて、細胞を37℃で8分間処理し、単細胞に分離後、1~1.5×104細胞の細胞密度で6ウェルプレートに播種した。継代は7日ごとに行った。
オルガノイドを4%パラホルムアルデヒドで30分間固定し、PBS中の30%(w/v)スクロースで脱水反応を行い、O.C.T.コンパウンド(Sakura Finetek)を用いて包埋した。凍結切片は、クリオスタット(CM1850、ライカバイオシステムズ)を用いて16μmの厚さで作成した。0.3%又は2%(v/v)Triton-X100で透過処理し、必要に応じて抗原賦活化反応を実施した。ブロックACE(KAC)でブロッキングを実施した後、二重又は三重標識染色を行った。免疫染色に使用した一次抗体及び二次抗体は、推奨希釈濃度にて使用した。
L1CAM:554273 (BD)
Ctip2:ab18465 (Abcam)
Pax6:EPR15858 (Abcam),561482 (BD)
Foxg1:M227 (Takara)
Ki67:NCL (Leica)
Emx1:M196 (Takara)
Gad65:559931 (BD)
Col1a1:AF6220(R&D)
TTR:A0002(DAKO)
TYR:MA5-14177(Thermo Fisher)
Alexa Fluor(登録商標) 488 donkey anti-mouse IgG (H+L):A21202 (Thermo Fisher)
Alexa Fluor(登録商標) 647 donkey anti-mouse IgG (H+L):1900251 (Thermo Fisher)
Alexa Fluor(登録商標) 594 donkey anti-rat IgG (H+L):1979379 (Thermo Fisher)
Alexa Fluor(登録商標) 488 donkey anti-rabbit IgG (H+L):A21206 (Thermo Fisher)
Alexa Fluor(登録商標) 647 donkey anti-rabbit IgG (H+L):A32795 (Thermo Fisher)
Alexa Fluor(登録商標) 594 donkey anti-sheep IgG (H+L):A11016 (Thermo Fisher)
培養中のオルガノイドの画像(明視野像)は、デジタル倒立顕微鏡(Leica Biosystems、DMS1000)で撮影した。共焦点蛍光顕微鏡画像はLSM800共焦点顕微鏡(Zeiss)で撮影し、ZENBlue画像処理ソフトウェアで解析した。
大脳オルガノイド、大脳皮質細胞塊、及び高純度大脳皮質細胞塊は、神経細胞用分散液(富士フィルム和光純薬)を用いて単細胞に分散し、Fixation Buffer(BD Biosciences)を用いて4℃で30分間固定した後、Perm/Wash buffer(BD Biosciences)で室温15分間処理し、二重又は三重染色を行い、AriaIIIを用いて解析した。解析ソフトウェアは、FACSDiva software (BD)を用いた。使用した一次抗体、二次抗体は下記に示す。
PerCP-Cy5.5 Mouse IgG1 k isotype control: 550795 (BD Biosciences)
Alexa 647 Mouse Anti-human Oct3/4 antigen: 560329 (BD Biosciences)
Tra-2-49/6E-FITC antibody : FCMAB133F (Merck)
Alexa647 mouse anti β-tubulin Class III: 560394 (BD)
PerCP-Cy5.5 Mouse anti-Human Sox1: 561549 (BD)
Alexa647 mouse anti-Human Pax6: 561165(BD)
Alexa488 mouse anti-Ki67: 562249 (BD)
Alexa488 rat anti-Ctip2 antibody: ab123449 (Abcam)
全RNAはRNeasyMicroKit(QIAGEN)によって取得し、SuperScript III First-Strand Synthesis System(QIAGEN)によってメーカーのプロトコルに従って逆転写反応を行った。定量的PCRは、TaqMan(商標)Gene Expression Master Mix(Thermo Fisher)、若しくはSYBR Green Master Mix(Thermo Fisher)を使用して、製造元の指示に従って実行した。遺伝子の発現量は、ΔΔCt法を使用してGAPDHで標準化した。
オリゴDNAプライマー
POU5F1:
Forward: AGACCATCTGCCGCTTTGAG (配列番号1)
Reverse: GCAAGGGCCGCAGCTT (配列番号2)
NANOG:
Forward: GGCTCTGTTTTGCTATATCCCCTAA (配列番号3)
Reverse: CATTACGATGCAGCAAATACGAGA (配列番号4)
BMP4:
Forward: ATGATTCCTGGTAACCGAATGC (配列番号5)
Reverse: CCCCGTCTCAGGTATCAAACT (配列番号6)
NODAL:
Forward: TGAGCCAACAAGAGGATCTG (配列番号7)
Reverse: TGGAAAATCTCAATGGCAAG (配列番号8)
TGFB1:
Forward: TACCTGAACCCGTGTTGCTCTC (配列番号9)
Reverse: GTTGCTGAGGTATCGCCAGGAA (配列番号10)
SOX1:
Forward: GCGGAGCTCGTCGCATT (配列番号11)
Reverse: GCGGTAACAACTACAAAAAACTTGTAA (配列番号12)
PAX6:
Forward: CTGGCTAGCGAAAAGCAACAG (配列番号13)
Reverse: CCCGTTCAACATCCTTAGTTTATCA (配列番号14)
T (TBXT):
Forward: ATGGAGGAACCCGGAGACA (配列番号15)
Reverse: TGAGGATTTGCAGGTGGACA (配列番号16)
SOX17:
Forward: CGCTTTCATGGTGTGGGCTAAGGACG (配列番号17)
Reverse: TAGTTGGGGTGGTCCTGCATGTGCTG (配列番号18)
hCGalpha:
Forward: ACCGCCCTGAACACATCCTGC (配列番号19)
Reverse: GCGTGCATTCTGGGCAATCCTGC (配列番号20)
SOX2:
Forward: GCCGAGTGGAAACTTTTGTCG (配列番号21)
Reverse: GGCAGCGTGTACTTATCCTTCT (配列番号22)
Taqman probe
SLC17A7: Hs01574213
EMX1: Hs00417957
GAD2: Hs00609536
DLX2: Hs00269993
TTR: Hs00174914
COL1A1: Hs00164004
TYR: Hs00165976
HOXA2: Hs00534579
ヒト胚性幹細胞(hESC)又はヒトiPS細胞株(hiPSC)を、支持細胞の存在下(on feeder)又は非存在下(feeder-free)で神経細胞へ分化誘導し、生じた球状の細胞凝集体を解析し、大脳オルガノイドへの分化誘導の効率を比較した。

予備試験1と同様にKhES-1、201B7、Ff-I01s04及び1231A3細胞株を、on feeder、feeder-free、及び、feeder conditioned mediumの3条件で培養し、分化誘導した。feeder conditioned mediumの条件は、MEFを、1%(v/v)NEAA、1%(v/v)L-Glutamine、1%(v/v)2-Mercaptoethanol、20%(v/v)KSRを含むDMEM/F12培地で24時間培養して培地上清を回収し、フィルトレーションして、フィーダーフリーの細胞の培養に用いた。得られたこれらの細胞における、培養後分化誘導前及び分化誘導後の、FGF2及びTGFβ経路関連遺伝子(FGF2、LEFTY、NODAL、TGFβ1、Activin)の発現をマイクロアレイで解析した。

1-1.工程(1)の維持培地について
図3又は図5に示すスキームにしたがって、工程(1)及び工程(2)を行った。ヒトiPS細胞(S2WCB1株、S2WCB3株)の維持培養は、iMatrix-511でコーティングした6ウェルプレート上(フィーダーフリー)、StemFit培地中で行った。
(1)StemFitのC液非添加培地(すなわち、bFGF不含培地)に5μM SB431542を添加したもの(図3)、又は、
(2)Essential 6培地(すなわち、bFGF不含、TGFβ不含培地)(図5)
に交換し、1日間接着培養した。

工程(1)において、培地として、SB431542添加したEssential 6(E6)培地を用いて維持培養して、上記と同様に分化誘導した。上記と同様に免疫染色し、代表的な細胞塊の共焦点蛍光顕微鏡画像を図6に示す。図6から分かるように、SB431542を添加したE6培地を用いた場合において、SB431542を添加したStemFit培地と同様に、大脳オルガノイドが得られた。また、大脳オルガノイドの形成効率(%)=(22/39)×100%=56.9%であった。
図3のスキームの工程(1)において、-Day1~Day0の期間に下記添加剤を添加した培地で培養し、Day0で神経細胞に分化誘導し、Day35で大脳オルガノイドの形成効率を評価した。免疫染色の結果を図7に示し、大脳オルガノイドの形成効率を表3に示す。なお、図7中の「無し」は添加剤なしを意味する。

実施例1で(1)StemFitのC液非添加培地に5μM SB431542を添加した「bFGF不含・SB431542添加培地」を使用する実験系において、「bFGF不含・SB431542添加培地」でiPS細胞を培養する期間を下表の通りに行った。図8はDay18、Day27、Day34の培養物の代表的な明視野像を示す。図9はDay35における大脳オルガノイドの形成効率を解析した結果を示す。

実施例1で(1)StemFitのC液非添加培地に5μM SB431542を添加した「bFGF不含・SB431542添加培地」を使用する実験系において、Day0におけるヒトiPS細胞(S2WCB1)の遺伝子発現解析を行った。Controlは、通常の維持培地であるStemFit培地(bFGF含有培地)で培養したiPS細胞であった。
4-1.分散及び再凝集あり
-Day7~Day35までは、実施例1と同じ方法で培養した。工程(1)には、(1)StemFitのC液非添加培地(bFGF不含培地)に5μM SB431542を添加したものを使用した。
次に、DAPT処理直後(工程(ii)を経た直後)の細胞培養物について、フローサイトメトリーを用いて構成細胞の割合を解析した。分化誘導スキームを図13に示す。Day40の大脳オルガノイドに対してDAPT処理を3日間行った後、フローサイトメトリーでマーカー遺伝子の発現を解析した。
DAPT処理期間が分化に与える影響を解析するため、図16(A)の示す方法スキーム(オルガノイドDAPT法)に従って、分化誘導7週目の大脳オルガノイドを10μMのDAPTで3~7日間処理し、遺伝子発現の変化をRT-qPCRで解析し、その結果を図16(B)に示す。縦軸は、GAPDHに対する相対発現量を表す。
図17の(A)は分化誘導のスキームを示す。Day25、Day32、Day39、及びDay72(それぞれの分化誘導期間は、凡そ4週間、5週間、6週間、10週間)のオルガノイドを3日間DAPT存在下・非存在下で培養し、Day28(4wk)、Day42(6wk)、及びDay75(10wk)のオルガノイドを各種マーカーに対して免疫染色で解析した。免疫染色の結果(共焦点蛍光顕微鏡画像)を図17の(B)に示す。なお、Satb2は、皮質II-IV層前駆細胞(Upper Layer)マーカーである。図17の(B)から、4週間から6週間のオルガノイドにおいて、DAPT非存在下では、Ctip2、Bf1共陽性細胞とKi67陽性細胞からなるロゼット構造が認められるが、DAPT処理後にはロゼット構造が見られなくなり、Ctip2、Bf1共陽性の細胞が増加し、Ki67陽性の細胞が減少することが分かった。10週のオルガノイドでは、DAPT処理後にロゼット構造は不明瞭になったが、Ctip2陽性細胞、Satb2陽性細胞の割合に顕著な変化は見られなかった。
図17の(A)の分化誘導スキームで得られた大脳皮質細胞塊をScidマウス(日本クレア)の脳へ定位脳移植法によって1.5×105細胞を移植し、移植3か月後に、4%パラホルムアルデヒドによって脳を還流固定し、35μm厚の凍結切片を作成した。作成した脳切片を用いて、Ku80の免疫染色でヒト細胞の生着を評価した。免疫染色した移植片の代表的な共焦点蛍光顕微鏡画像を図19に示す。
好ましい、支持細胞の非存在下で、多能性幹細胞から大脳オルガノイドを製造する方法は、下記の工程を含む。
工程(1):多能性幹細胞を、bFGFが実質的に含まれず、かつTGFβシグナルを実質的に惹起しない培養液中で培養する工程、
工程(2):工程(1)で得られた細胞を、大脳オルガノイドへと分化誘導する工程。
工程(2a):工程(1)で得られた細胞を、TGFβシグナル阻害剤、及びWntシグナル伝達阻害剤を含む培養液中で浮遊培養して、細胞塊を得る工程、
工程(2b):工程(2a)で得られた細胞塊を、TGFβシグナル阻害剤又はWntシグナル伝達阻害剤を実質的に含まない培養液中で浮遊培養して、大脳オルガノイドを得る工程。
工程(i):工程(2)を含む方法、又は、工程(1)及び工程(2)を含む方法により、大脳オルガノイドを得る工程、
工程(ii):工程(i)で得られた大脳オルガノイドを、Notchシグナル阻害剤(DAPT等)を含む培養液中で培養して、大脳皮質細胞塊を得る工程、
工程(iii):工程(ii)を経た細胞培養物を、単細胞又は2~5個の細胞の集合体(Cell clump)にまで分散させる工程、
工程(iv):工程(iii)で得られた細胞を、1以上の神経栄養因子、アスコルビン酸、及びcAMP活性化剤を含む培養液中で培養して、高純度大脳皮質細胞塊を得る工程。
-Day7~Day35までは、実施例1の図3のスキームと同じ方法で培養した。「神経分化準備あり」(工程(1)あり)では、工程(1)にStemFitのC液非添加培地(=bFGF不含培地)に5μMSB431542を添加したものを使用し、「神経分化準備入なし」(工程(1)なし)では、StemFit培地を用いた。その後、実施例4の工程(ii)に記載の方法で大脳皮質細胞塊を誘導し、免疫染色で評価を行った。また、実施例4の工程(iii)、工程(iv)に記載の方法で単細胞に分散後、再凝集して、高密度大脳皮質細胞塊を誘導し、フローサイトメトリーで解析を行った。
8-1.オルガノイドDAPT法及びシングルセルDAPT法(1)
-Day7~Day35までは、実施例1と同じ方法で培養した。工程(1)には、StemFitのC液非添加培地(=bFGF不含培地)に5μMSB431542を添加したものを使用した。
図28に示す方法スキームに従って、分化誘導5週目の大脳オルガノイドを、工程(II)で単細胞に分散後、工程(III)で10μMのDAPTで3日間処理を行った。その後、DAPTを含まない培地条件下において、4日間(DAPT除去後1日)及び14日間(DAPT除去後11日)培養を継続し、得られた細胞塊に対してフローサイトメトリーでマーカー発現を解析した。解析の結果を図29に示す。
9-1.オルガノイドの形態解析
-Day7~Day35まで実施例1と同じ方法で培養した。12回の誘導分化ロット(Lot1~Lot12)で得られた大脳オルガノイドの明視野像を倒立顕微鏡(Leica DMS1000)を用いて撮影し、代表的な明視野像を図30に示す(スケールバー:5mm)。図30から、ロットによって、誘導されるオルガノイドの形態に違いがあったことが分かった。

各形態グループのオルガノイドに含まれる細胞種を同定するため、シングルセル遺伝子発現解析を行った。解析には、3回の分化誘導回で得られた9つのオルガノイド(図33)を用いた。各オルガノイドに含まれる組織の形態を各写真の上部に示す。シングルセル遺伝子発現解析は、以下のとおりに行った。



各グループのオルガノイドに対し、代表的なマーカータンパク質の免疫染色を行った。結果を図40に示す。
-Day7~Day35まで実施例1と同じ方法で培養した。各グループにつき3個のオルガノイドを取得し、倒立顕微鏡(Leica DMS1000)を用いて明視野像を撮影した(図41)。各オルガノイドに対し、RT-qPCR法でマーカー遺伝子の発現を解析した。解析結果を図42及び43に示す。
-Day7~Day35まで実施例1と同じ方法で培養した3回の分化誘導回で得られたオルガノイドから、Rosettesのオルガノイドを目視で選別した。その後、実施例4の工程(ii)に記載の方法で、大脳オルガノイドをDAPTで3日間処理し、大脳皮質細胞塊を誘導した(“DAPT+”)。対照群として、工程(ii)でDAPT非添加の群を作成した(大脳オルガノイド:“DAPT-”)。
これらの細胞を用いて、9-2と同じ方法でシングルセル遺伝子発現解析を行った。
低分子化合物、抗体、核酸、その他の物質を、本発明の製造方法で作製した大脳オルガノイドに添加し、その影響を調べてもよい。すなわち、低分子化合物、抗体、その他の物質の影響、毒性、及び有用性を明らかにする目的で、大脳オルガノイドを用いることができる。この場合、大脳オルガノイドにおいて形成された、組織化された神経細胞、組織化された神経ネットワークの影響を調べることができる。特に本発明に規定する大脳オルガノイドの製造方法、及び、選別方法により、発現プロファイル又は形状等又は組織構成が比較的均一な大脳オルガノイドを大量に作成することが可能である。この際に、大脳オルガノイドの品質を客観的に評価するために、上記13に記載の大脳オルガノイドの品質評価方法を用いてもよい。これらの発現プロファイル又は形状等又は組織構成が比較的均一な大脳オルガノイドを用いることにより、容易に複数回の試験を行うことができ、従来は困難であった再現性を容易に達成することができる。
また、特定の遺伝子型細胞由来の多能性幹細胞から誘導した大脳オルガノイドを作製し、特定の疾患に対して上記物質スクリーニングを適用してもよい。この場合、上記2に記載の大脳オルガノイドの製造方法を用いて大脳オルガノイドを作製する際に、由来する多能性幹細胞の性質に応じて最適な工程は適宜調整できる。特に、統合失調症、双極性障害、自閉症スペクトラム障害、アルツハイマー病、認知症、小頭症、等の患者から採取した細胞由来の多能性幹細胞から誘導した大脳オルガノイドを作製し(精神疾患大脳オルガノイド)、薬剤の効果を調べる他、疾患のメカニズムの解明に用いてもよい。より具体的には、興奮性神経細胞のグルタミン酸作動性神経等、抑制性神経細胞のGABA作動性神経等、グリア細胞等を健常者と比較することや、遺伝子発現、タンパク質発現、代謝状態、電気生理学的性質等の細胞特性を解析することが含まれる。
Claims (39)
- 支持細胞の非存在下、多能性幹細胞から大脳オルガノイドを製造する方法であって、
(1)多能性幹細胞を、bFGFが実質的に含まれず、かつTGFβシグナルを実質的に惹起しない培養液中で培養する工程と、
(2)工程(1)で得られた細胞を、神経細胞へと分化誘導する工程と
を含む、方法。 - 工程(2)が、
(2a)工程(1)で得られた細胞を、TGFβシグナル阻害剤、及びWntシグナル伝達阻害剤を含む培養液中で浮遊培養して、細胞塊を得る工程と、
(2b)工程(2a)で得られた細胞塊を、TGFβシグナル阻害剤及びWntシグナル伝達阻害剤を実質的に含まない培養液中で浮遊培養して、大脳オルガノイドを得る工程と
を含む、請求項1に記載の方法。 - 工程(2a)の浮遊培養が静置培養である、請求項2に記載の方法。
- 工程(2b)の浮遊培養が振とう培養である、請求項2又は3に記載の方法。
- 工程(1)の培養期間が3日間未満である、請求項1~4のいずれか1項に記載の方法。
- 工程(1)の培養期間が12時間以上2日間以下である、請求項1~5のいずれか1項に記載の方法。
- 工程(1)の培養液が、SB431542、A-83-01、及びXAV-939からなる群から選ばれるTGFβシグナル阻害剤を含む、請求項1~6のいずれか1項に記載の方法。
- 工程(1)、工程(2a)、及び工程(2b)における培養液が、無血清培養液である、請求項2~7のいずれか1項に記載の方法。
- 前記多能性幹細胞が、ヒト人工多能性幹細胞又はヒト胚性幹細胞である、請求項1~8のいずれか1項に記載の方法。
- (3)工程(2)で得られた複数の細胞塊から、細胞塊の形状、内部構造、サイズ、表面の色彩若しくは模様、及び遺伝子発現からなる群から選ばれる1以上を指標とし、大脳オルガノイドを選別する工程をさらに含む、請求項1~9のいずれか1項に記載の方法。
- 請求項1~9のいずれか1項に記載の方法を用いて製造される細胞培養物であって、
複数の球状の細胞塊を含み、
前記複数の球状の細胞塊に占める大脳オルガノイドの割合が40%以上である、細胞培養物。 - 前記大脳オルガノイドに占める大脳皮質様構造物の割合が40%以上である、請求項11に記載の細胞培養物。
- 大脳オルガノイドが、更に以下の(1)~(5):
(1)球状の細胞塊であること、
(2)細胞塊の内部に、大脳皮質様構造物を有すること、
(3)表面に色素沈着を有さないこと、
(4)細胞塊の一部に嚢胞状形状、突起形状、及びバルーン状形状のいずれも有さないこと、並びに、
(5)NEUROD6、NEUROD2、SSTR2、TBR1、ZBTB18、NHLH1、IGFBPL1、NRN1、RTN1、THSD7A、NRXN1、BHLHE22、CALB2、KHDRBS3、CCSAP、PDE1A、NEUROD1、NPTX1、NXPH4、NTS、NEUROG2、OLFM1、PRDM8、CORO2B、TP53I11、ZFPM2、PCDH9、NELL2、SRRM4、SCG3、DCC、EPB41L3、SLC17A7、ST18、NSG2、EMX1、CAP2、SYT4、NSMF、ANK3、MYT1L、FSTL5、CELF4、B3GAT1、EPHA5、NHLH2、及びDLL3からなる群から選ばれる少なくとも1つのマーカーを発現していること
から選択される、1以上の特徴を有する細胞塊である、請求項12に記載の細胞培養物。 - (a)増殖性マーカー陽性細胞数が全細胞数の10%以下であり、
(b)神経細胞マーカー、皮質V/VI層マーカー、及び前脳マーカーからなる群から選ばれる1つ以上のマーカーが陽性である細胞数が全細胞数の70%以上であり、且つ
(c)神経上皮又は大脳皮質様構造を実質的に含まないことを特徴とする、大脳皮質細胞塊。 - 前記(a)の増殖性マーカーがKi67であり、
前記(b)の神経細胞マーカーがβIII-Tubulinであり、皮質V/VI層マーカーがCtip2であり、前脳マーカーがFOXG1である、
請求項14に記載の大脳皮質細胞塊。 - (d)NEUROD6、NEUROD2、SSTR2、TBR1、ZBTB18、NHLH1、IGFBPL1、NRN1、RTN1、THSD7A、NRXN1、BHLHE22、CALB2、KHDRBS3、CCSAP、PDE1A、NEUROD1、NPTX1、NXPH4、NTS、NEUROG2、OLFM1、PRDM8、CORO2B、TP53I11、ZFPM2、PCDH9、NELL2、SRRM4、SCG3、DCC、EPB41L3、SLC17A7、ST18、NSG2、EMX1、CAP2、SYT4、NSMF、ANK3、MYT1L、FSTL5、CELF4、B3GAT1、EPHA5、NHLH2、及びDLL3からなる群から選ばれる少なくとも1つのマーカーをさらに発現している、請求項14又は15に記載の大脳皮質細胞塊。
- SLC17A7を発現している、請求項16に記載の大脳皮質細胞塊。
- GAD2、COL1A1、TYR、TTR及びHOXA2からなる群から選ばれる1以上の遺伝子を実質的に発現していない、請求項15~17のいずれか1項に記載の大脳皮質細胞塊。
- 支持細胞の非存在下で、多能性幹細胞から大脳皮質細胞塊を製造する方法であって、
(i)多能性幹細胞から大脳オルガノイドを得る工程と、
(ii)工程(i)で得られた大脳オルガノイドを、Notchシグナル阻害剤を含む培養液中で培養して、大脳皮質細胞塊を得る工程と
を含む、方法。 - 前記工程(i)において、請求項1~10のいずれか1項に記載の方法により多能性幹細胞から大脳オルガノイドを得る、請求項19に記載の方法。
- (A)増殖性マーカー陽性細胞数が全細胞数の5%以下であり、
(B)神経細胞マーカー、皮質V/VI層マーカー、及び前脳マーカーから選ばれる1つ以上のマーカー陽性細胞数が全細胞数の70%以上であり、且つ
(C)神経上皮又は大脳皮質様構造を実質的に含まない
ことを特徴とする、高純度大脳皮質細胞塊。 - 前記(A)の増殖性マーカーがKi67であり、
前記(B)の神経細胞マーカーがβIII-Tubulinであり、皮質V/VI層マーカーがCtip2であり、前脳マーカーがFOXG1である、
請求項21に記載の高純度大脳皮質細胞塊。 - (D)NEUROD6、NEUROD2、SSTR2、TBR1、ZBTB18、NHLH1、IGFBPL1、NRN1、RTN1、THSD7A、NRXN1、BHLHE22、CALB2、KHDRBS3、CCSAP、PDE1A、NEUROD1、NPTX1、NXPH4、NTS、NEUROG2、OLFM1、PRDM8、CORO2B、TP53I11、ZFPM2、PCDH9、NELL2、SRRM4、SCG3、DCC、EPB41L3、SLC17A7、ST18、NSG2、EMX1、CAP2、SYT4、NSMF、ANK3、MYT1L、FSTL5、CELF4、B3GAT1、EPHA5、NHLH2、及びDLL3からなる群から選ばれる少なくとも1つの遺伝子を発現している、請求項21又は22に記載の高純度大脳皮質細胞塊。
- SLC17A7、NEUROD6、及びEMX1からなる群から選ばれる1以上の遺伝子を発現している、請求項23に記載の高純度大脳皮質細胞塊。
- GAD2、COL1A1、TYR、TTR及びHOXA2からなる群から選ばれる1以上の遺伝子を実質的に発現していない、請求項21~24のいずれか1項に記載の高純度大脳皮質細胞塊。
- 支持細胞の非存在下で、多能性幹細胞から高純度大脳皮質細胞塊を製造する方法であって、
(i)多能性幹細胞から大脳オルガノイドを得る工程と、
(ii)工程(i)で得られた大脳オルガノイドを、培養液中で培養する工程と、
(iii)工程(ii)で得られた細胞培養物を、単細胞又は2~5個の細胞の集合体(Cell clump)にまで分散させる工程と、
(iv)工程(ii)で得られた細胞培養物又は工程(iii)で得られた細胞集団を、1以上の神経栄養因子、アスコルビン酸、及びcAMP活性化剤を含む培養液中で培養して、細胞塊を得る工程と
を含み、前記工程(ii)の培養液及び/又は前記工程(iv)の培養液は、Notchシグナル阻害剤を含む、方法。 - 前記工程(i)において、請求項1~10のいずれか1項に記載の方法により多能性幹細胞から大脳オルガノイドを得る、請求項26に記載の方法。
- 工程(ii)に付与する大脳オルガノイドが、神経細胞への分化誘導開始から28日後~44日後の大脳オルガノイドである、請求項19、20、26及び27のいずれか1項に記載の方法。
- 工程(ii)の培養期間が2~6日間である、請求項19、20、及び26~28のいずれか1項に記載の方法。
- 工程(iv)の培養期間が2~14日間である、請求項19、20、及び26~29のいずれか1項に記載の方法。
- 前記Notchシグナル阻害剤は、γセクレターゼ阻害剤である、請求項19、20、及び26~30のいずれか1項に記載の方法。
- 前記γセクレターゼ阻害剤がN-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT)又はCompound Eである、請求項31に記載の方法。
- 請求項21~25のいずれか1項に記載の高純度大脳皮質細胞塊を含む細胞集団であって、前記高純度大脳皮質細胞塊のサイズ、形状又は構成細胞組成が均一である、細胞集団。
- 請求項14~18のいずれか1項に記載の大脳皮質細胞塊、請求項21~25のいずれか1項に記載の高純度大脳皮質細胞塊、若しくは請求項33に記載の細胞集団、又はそれらを構成細胞にまで分散させて得られる細胞集団を有効成分として含む医薬組成物。
- 請求項14~18のいずれか1項に記載の大脳皮質細胞塊、請求項21~25のいずれか1項に記載の高純度大脳皮質細胞塊、若しくは請求項33に記載の細胞集団、又はそれらを構成細胞にまで分散させて得られる細胞集団を含む、移植用組織。
- 請求項14~18のいずれか1項に記載の大脳皮質細胞塊、請求項21~25のいずれか1項に記載の高純度大脳皮質細胞塊、若しくは請求項33に記載の細胞集団、又はそれらを構成細胞にまで分散させて得られる細胞集団を有効成分として含む、脳血管障害治療薬。
- 請求項14~18のいずれか1項に記載の大脳皮質細胞塊、請求項21~25のいずれか1項に記載の高純度大脳皮質細胞塊、若しくは請求項33に記載の細胞集団、又はそれらを構成細胞にまで分散させて得られる細胞集団を、これを必要とする対象の大脳皮質又は大脳基底核に投与又は移植することを含む、脳血管障害の治療方法。
- (aa)大脳オルガノイド又は大脳皮質細胞塊における、GAD2、COL1A1、TYR、TTR及びHOXA2からなる群から選ばれる少なくとも1つの遺伝子又は該遺伝子によりコードされるタンパク質もしくはその断片の発現量を測定する工程、及び
(bb)工程(aa)の測定結果に基づき、前記遺伝子の発現量が基準値以下である場合に、前記大脳オルガノイド又は前記大脳皮質細胞塊に含まれる目的外細胞の量が基準以下であると評価する工程、
を含む、大脳オルガノイド又は大脳皮質細胞塊の品質評価方法。 - (AA)大脳オルガノイド又は大脳皮質細胞塊における、NEUROD6、NEUROD2、SSTR2、TBR1、ZBTB18、NHLH1、IGFBPL1、NRN1、RTN1、THSD7A、NRXN1、BHLHE22、CALB2、KHDRBS3、CCSAP、PDE1A、NEUROD1、NPTX1、NXPH4、NTS、NEUROG2、OLFM1、PRDM8、CORO2B、TP53I11、ZFPM2、PCDH9、NELL2、SRRM4、SCG3、DCC、EPB41L3、SLC17A7、ST18、NSG2、EMX1、CAP2、SYT4、NSMF、ANK3、MYT1L、FSTL5、CELF4、B3GAT1、EPHA5、NHLH2、及びDLL3からなる群から選ばれる少なくとも1つの遺伝子の発現量を測定する工程、及び
(BB)工程(AA)の測定結果に基づき、前記遺伝子の発現量が基準値以上である場合に、前記大脳オルガノイド又は前記大脳皮質細胞塊に含まれる目的細胞の量が基準以上であると評価する工程、
を含む、大脳オルガノイド又は大脳皮質細胞塊の品質評価方法。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA3224178A CA3224178A1 (en) | 2021-06-17 | 2022-06-16 | Method for producing cerebral cortical cell preparation derived from human pluripotent stem cells |
| KR1020247001757A KR20240034191A (ko) | 2021-06-17 | 2022-06-16 | 인간 다능성 줄기세포 유래 대뇌 피질 세포 제제의 제조 방법 |
| EP22825074.2A EP4357449A1 (en) | 2021-06-17 | 2022-06-16 | Method for producing cerebral cortical cell preparation derived from human pluripotent stem cells |
| IL309344A IL309344A (en) | 2021-06-17 | 2022-06-16 | A method for producing the preparation of cortical cells derived from human pluripotent stem cells |
| CN202280042648.8A CN117769591A (zh) | 2021-06-17 | 2022-06-16 | 源自人多能干细胞的大脑皮质细胞制剂的制造方法 |
| AU2022294563A AU2022294563A1 (en) | 2021-06-17 | 2022-06-16 | Method for producing cerebral cortical cell preparation derived from human pluripotent stem cells |
| JP2023530419A JPWO2022265086A1 (ja) | 2021-06-17 | 2022-06-16 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202163211622P | 2021-06-17 | 2021-06-17 | |
| US63/211,622 | 2021-06-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022265086A1 true WO2022265086A1 (ja) | 2022-12-22 |
Family
ID=84527112
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/024226 WO2022265086A1 (ja) | 2021-06-17 | 2022-06-16 | ヒト多能性幹細胞由来大脳皮質細胞製剤の製造方法 |
Country Status (8)
| Country | Link |
|---|---|
| EP (1) | EP4357449A1 (ja) |
| JP (1) | JPWO2022265086A1 (ja) |
| KR (1) | KR20240034191A (ja) |
| CN (1) | CN117769591A (ja) |
| AU (1) | AU2022294563A1 (ja) |
| CA (1) | CA3224178A1 (ja) |
| IL (1) | IL309344A (ja) |
| WO (1) | WO2022265086A1 (ja) |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996022362A1 (en) | 1995-01-20 | 1996-07-25 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| WO1998030679A1 (en) | 1997-01-10 | 1998-07-16 | Life Technologies, Inc. | Embryonic stem cell serum replacement |
| US6280718B1 (en) | 1999-11-08 | 2001-08-28 | Wisconsin Alumni Reasearch Foundation | Hematopoietic differentiation of human pluripotent embryonic stem cells |
| WO2002101057A1 (fr) | 2001-06-08 | 2002-12-19 | Dnavec Research Inc. | Transfert genique dans des cellules souches embryonnaires de primate a l'aide d'un virus de l'immunodeficience simienne de pseudo type vsv-g utilise comme vecteur |
| WO2008035110A1 (en) | 2006-09-22 | 2008-03-27 | Riken | Stem cell culture medium and method |
| WO2009146408A1 (en) | 2008-05-30 | 2009-12-03 | Summa Health Systems Llc | Methods for using tgf-b receptor inhibitors or activin-like kinase (alk) 5 inhibitors a-83-01 and sb-431542 to treat eye disease and wound healing conditions |
| WO2015076388A1 (ja) | 2013-11-22 | 2015-05-28 | 国立研究開発法人理化学研究所 | 終脳又はその前駆組織の製造方法 |
| WO2016063985A1 (ja) | 2014-10-24 | 2016-04-28 | 大日本住友製薬株式会社 | 神経組織の製造方法 |
| WO2016167372A1 (ja) | 2015-04-14 | 2016-10-20 | 国立大学法人京都大学 | 大脳皮質ニューロンの誘導方法 |
| US20200208090A1 (en) * | 2015-11-13 | 2020-07-02 | The Johns Hopkins University | Cell culture system and method of use thereof |
-
2022
- 2022-06-16 WO PCT/JP2022/024226 patent/WO2022265086A1/ja active Application Filing
- 2022-06-16 JP JP2023530419A patent/JPWO2022265086A1/ja active Pending
- 2022-06-16 EP EP22825074.2A patent/EP4357449A1/en active Pending
- 2022-06-16 KR KR1020247001757A patent/KR20240034191A/ko unknown
- 2022-06-16 AU AU2022294563A patent/AU2022294563A1/en active Pending
- 2022-06-16 IL IL309344A patent/IL309344A/en unknown
- 2022-06-16 CA CA3224178A patent/CA3224178A1/en active Pending
- 2022-06-16 CN CN202280042648.8A patent/CN117769591A/zh active Pending
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996022362A1 (en) | 1995-01-20 | 1996-07-25 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| US5843780A (en) | 1995-01-20 | 1998-12-01 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| US6200806B1 (en) | 1995-01-20 | 2001-03-13 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| WO1998030679A1 (en) | 1997-01-10 | 1998-07-16 | Life Technologies, Inc. | Embryonic stem cell serum replacement |
| US6280718B1 (en) | 1999-11-08 | 2001-08-28 | Wisconsin Alumni Reasearch Foundation | Hematopoietic differentiation of human pluripotent embryonic stem cells |
| WO2002101057A1 (fr) | 2001-06-08 | 2002-12-19 | Dnavec Research Inc. | Transfert genique dans des cellules souches embryonnaires de primate a l'aide d'un virus de l'immunodeficience simienne de pseudo type vsv-g utilise comme vecteur |
| WO2008035110A1 (en) | 2006-09-22 | 2008-03-27 | Riken | Stem cell culture medium and method |
| WO2009146408A1 (en) | 2008-05-30 | 2009-12-03 | Summa Health Systems Llc | Methods for using tgf-b receptor inhibitors or activin-like kinase (alk) 5 inhibitors a-83-01 and sb-431542 to treat eye disease and wound healing conditions |
| WO2015076388A1 (ja) | 2013-11-22 | 2015-05-28 | 国立研究開発法人理化学研究所 | 終脳又はその前駆組織の製造方法 |
| WO2016063985A1 (ja) | 2014-10-24 | 2016-04-28 | 大日本住友製薬株式会社 | 神経組織の製造方法 |
| WO2016167372A1 (ja) | 2015-04-14 | 2016-10-20 | 国立大学法人京都大学 | 大脳皮質ニューロンの誘導方法 |
| US20200208090A1 (en) * | 2015-11-13 | 2020-07-02 | The Johns Hopkins University | Cell culture system and method of use thereof |
Non-Patent Citations (31)
| Title |
|---|
| "NCBI", Database accession no. NM-00 1172425 |
| "NM-177099", Database accession no. NM-003240 |
| ALBANESE ALEXANDRE, SWANEY JUSTIN M., YUN DAE HEE, EVANS NICHOLAS B., ANTONUCCI JENNA M., VELASCO SILVIA, SOHN CHANG HO, ARLOTTA P: "Multiscale 3D phenotyping of human cerebral organoids", SCIENTIFIC REPORTS, vol. 10, no. 1, XP093016062, DOI: 10.1038/s41598-020-78130-7 * |
| BEJOY JULIE, BIJONOWSKI BRENT, MARZANO MARK, JESKE RICHARD, MA TENG, LI YAN: "Wnt-Notch Signaling Interactions During Neural and Astroglial Patterning of Human Stem Cells", TISSUE ENGINEERING PART A, MARY ANN LIEBERT, US, vol. 26, no. 7-8, 1 April 2020 (2020-04-01), US , pages 419 - 431, XP093016079, ISSN: 1937-3341, DOI: 10.1089/ten.tea.2019.0202 * |
| CELL, vol. 131, no. 5, 2007, pages 861 - 872 |
| EIRAKU, M ET AL., CELL STEM CELL, vol. 3, 2008, pages 519 - 532 |
| FAZILATY H ET AL., A GENE REGULATORY NETWORK TO CONTROL EMT PROGRAMS IN DEVELOPMENT AND DISEASE, 2019 |
| GONCHALOV ET AL.: "Markers and Biomarkers of Endothelium: When Something Is Rotten in the State", OXIDATIVE MEDICINE AND CELLULAR LONGEVITY, 2017 |
| HASAN MD FAYAD, BERDICHEVSKY YEVGENY: "Neuron and astrocyte aggregation and sorting in three-dimensional neuronal constructs", COMMUNICATIONS BIOLOGY, vol. 4, no. 1, XP093016075, DOI: 10.1038/s42003-021-02104-2 * |
| IZSAK JULIA, VIZLIN-HODZIC DZENETA, ILJIN MARGARITA, STRANDBERG JOAKIM, JADASZ JANUSZ, OLSSON BONTELL THOMAS, THEISS STEPHAN, HANS: "TGF-β1 Suppresses Proliferation and Induces Differentiation in Human iPSC Neural in vitro Models", FRONTIERS IN CELL AND DEVELOPMENTAL BIOLOGY, vol. 8, XP093016076, DOI: 10.3389/fcell.2020.571332 * |
| KADOSHIMA ET AL., PNAS, vol. 110, no. 50, 2013, pages 20284 - 20289 |
| KITAHARA ET AL., STEM CELL REPORTS, vol. 15, 2020, pages 467 - 481 |
| KUWAHARA ET AL., SCIENTIFIC REPORTS, vol. 9, 2019, pages 18936 |
| LIQING SONG, YUAN XUEGANG, JONES ZACHARY, GRIFFIN KYLE, ZHOU YI, MA TENG, LI YAN: "Assembly of Human Stem Cell-Derived Cortical Spheroids and Vascular Spheroids to Model 3-D Brain-like Tissues", SCIENTIFIC REPORTS, NATURE PUBLISHING GROUP, vol. 9, no. 1, XP055605380, DOI: 10.1038/s41598-019-42439-9 * |
| LOPEZ-BENDITO G., STURGESS KATHERINE, ERDÉLYI FERENC, SZABÓ GÁBOR, MOLNÁR ZOLTÁN, PAULSEN OLE: "Preferential Origin and Layer Destination of GAD65-GFP Cortical Interneurons", CEREBRAL CORTEX, vol. 14, no. 10, 31 October 2004 (2004-10-31), pages 1122 - 1133, XP093016081, DOI: 10.1093/cercor/bhh072 * |
| MINAMI I. ET AL., CELL REP., vol. 2, 2012, pages 1448 - 1460 |
| NAT. BIOTECHNOL., vol. 26, no. 1, 2008, pages 101 - 106 |
| NATURE BIOTECHNOLOGY, vol. 37, 2019, pages 38 - 44 |
| NATURE, vol. 605, 2022, pages 325 - 331 |
| OYAMA HIROSHI, TAKAHASHI KOJI, TANAKA YOSHIKAZU, TAKEMOTO HIROSHI, HAGA HISASHI: "Long-term Culture of Human iPS Cell-derived Telencephalic Neuron Aggregates on Collagen Gel", CELL STRUCTURE AND FUNCTION., JAPAN SOCIETY FOR CELL BIOLOGY (JSCB), KYOTO., JP, vol. 43, no. 1, 1 January 2018 (2018-01-01), JP , pages 85 - 94, XP093016066, ISSN: 0386-7196, DOI: 10.1247/csf.18002 * |
| PROC NATL ACAD SCI USA., vol. 105, no. 36, 9 September 2008 (2008-09-09), pages 13409 - 14 |
| R. K. LINDEMANN ET AL., MOL. CANCER, vol. 2, 2003, pages 20 |
| SAKAGUCHI ET AL., STEM CELL REPORTS, vol. 13, 2019, pages 458 - 473 |
| SAKAGUCHI HIDEYA, OZAKI YUKI, ASHIDA TOMOKA, MATSUBARA TAKAYOSHI, OISHI NAOTAKA, KIHARA SHUNSUKE, TAKAHASHI JUN: "Self-Organized Synchronous Calcium Transients in a Cultured Human Neural Network Derived from Cerebral Organoids", STEM CELL REPORTS, CELL PRESS, UNITED STATES, vol. 13, no. 3, 1 September 2019 (2019-09-01), United States , pages 458 - 473, XP093016065, ISSN: 2213-6711, DOI: 10.1016/j.stemcr.2019.05.029 * |
| SCIENCE, vol. 318, no. 5858, 2007, pages 1917 - 1920 |
| SCIENCE, vol. 341, 2013, pages 651 - 654 |
| SIMOES-COSTA M ET AL.: "Establishing neural crest identity: a gene regulatory recipe", DEVELOPMENT, 2015 |
| STEM CELLS, vol. 31, 2013, pages 458 - 466 |
| TUTUKOVA S ET AL.: "The Role of Neurod Genes in Brain Development, Function, and Disease, Frontiers", MOLECULAR NEUROSCIENCE, 2021 |
| WATANABE, K. ET AL., NATURE BIOTECHNOLOGY, vol. 25, no. 6, 2007, pages 681 - 686 |
| YAMANAKA ET AL., CELL, vol. 126, no. 4, 2006, pages 663 - 676 |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2022294563A1 (en) | 2024-01-18 |
| CA3224178A1 (en) | 2022-12-22 |
| EP4357449A1 (en) | 2024-04-24 |
| CN117769591A (zh) | 2024-03-26 |
| IL309344A (en) | 2024-02-01 |
| KR20240034191A (ko) | 2024-03-13 |
| JPWO2022265086A1 (ja) | 2022-12-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7075556B2 (ja) | 神経組織の製造方法 | |
| JP7088496B2 (ja) | 網膜組織の製造方法 | |
| JP7116964B2 (ja) | 終脳又はその前駆組織の製造方法 | |
| JP7360583B2 (ja) | 網膜組織の製造方法 | |
| CN110396499B (zh) | 一种诱导神经干细胞的方法及其应用 | |
| US20180258388A1 (en) | Method for producing retinal pigment epithelial cells | |
| JP2008099662A (ja) | 幹細胞の培養方法 | |
| CN112601814A (zh) | 包含嗅神经细胞或其前体细胞的细胞团块及其制备方法 | |
| Kumar et al. | Generation of an expandable intermediate mesoderm restricted progenitor cell line from human pluripotent stem cells | |
| CN113684182A (zh) | TGF-β抑制剂在诱导神经干细胞及类器官形成中的应用 | |
| JP2023169391A (ja) | 細胞凝集体、細胞凝集体の混合物及びそれらの製造方法 | |
| EP4011449A1 (en) | Method for producing cell aggregate including glial progenitor cells | |
| WO2022265086A1 (ja) | ヒト多能性幹細胞由来大脳皮質細胞製剤の製造方法 | |
| JPWO2020100481A1 (ja) | 脳オルガノイドの製造方法 | |
| CN115485364A (zh) | 分离垂体激素产生细胞及其祖细胞的方法 | |
| CN114736864A (zh) | 一种神经干细胞诱导分化培养基及诱导分化方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22825074 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 309344 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202280042648.8 Country of ref document: CN Ref document number: 3224178 Country of ref document: CA Ref document number: 2301008170 Country of ref document: TH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023530419 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022294563 Country of ref document: AU Ref document number: AU2022294563 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 20247001757 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020247001757 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022825074 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2022294563 Country of ref document: AU Date of ref document: 20220616 Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2022825074 Country of ref document: EP Effective date: 20240117 |