WO2022075107A1 - ポリアセタール樹脂組成物及び自動車部品 - Google Patents
ポリアセタール樹脂組成物及び自動車部品 Download PDFInfo
- Publication number
- WO2022075107A1 WO2022075107A1 PCT/JP2021/035360 JP2021035360W WO2022075107A1 WO 2022075107 A1 WO2022075107 A1 WO 2022075107A1 JP 2021035360 W JP2021035360 W JP 2021035360W WO 2022075107 A1 WO2022075107 A1 WO 2022075107A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mass
- parts
- carbon
- resin composition
- polyacetal resin
- Prior art date
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L59/00—Compositions of polyacetals; Compositions of derivatives of polyacetals
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L59/00—Compositions of polyacetals; Compositions of derivatives of polyacetals
- C08L59/04—Copolyoxymethylenes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/02—Elements
- C08K3/04—Carbon
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/02—Elements
- C08K3/04—Carbon
- C08K3/041—Carbon nanotubes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/18—Oxygen-containing compounds, e.g. metal carbonyls
- C08K3/20—Oxides; Hydroxides
- C08K3/22—Oxides; Hydroxides of metals
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/0008—Organic ingredients according to more than one of the "one dot" groups of C08K5/01 - C08K5/59
- C08K5/005—Stabilisers against oxidation, heat, light, ozone
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/13—Phenols; Phenolates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L71/00—Compositions of polyethers obtained by reactions forming an ether link in the main chain; Compositions of derivatives of such polymers
- C08L71/02—Polyalkylene oxides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/18—Oxygen-containing compounds, e.g. metal carbonyls
- C08K3/20—Oxides; Hydroxides
- C08K3/22—Oxides; Hydroxides of metals
- C08K2003/2217—Oxides; Hydroxides of metals of magnesium
- C08K2003/222—Magnesia, i.e. magnesium oxide
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/18—Oxygen-containing compounds, e.g. metal carbonyls
- C08K3/20—Oxides; Hydroxides
- C08K3/22—Oxides; Hydroxides of metals
- C08K2003/2296—Oxides; Hydroxides of metals of zinc
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K2201/00—Specific properties of additives
- C08K2201/001—Conductive additives
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K2201/00—Specific properties of additives
- C08K2201/002—Physical properties
- C08K2201/003—Additives being defined by their diameter
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K2201/00—Specific properties of additives
- C08K2201/002—Physical properties
- C08K2201/006—Additives being defined by their surface area
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K2201/00—Specific properties of additives
- C08K2201/011—Nanostructured additives
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K2201/00—Specific properties of additives
- C08K2201/014—Additives containing two or more different additives of the same subgroup in C08K
Definitions
- the present invention relates to a method for improving resistance to an acid component while imparting conductivity to a polyacetal resin composition, an automobile part formed by molding the polyacetal resin composition, and a polyacetal resin molded product.
- POM resins Polyacetal resins or polyacetal copolymers (hereinafter, these are also referred to as "POM resins") are widely used as engineering plastics because they are excellent in various physical and mechanical properties, chemical resistance, and slidability. There is.
- POM resin has excellent resistance to hydrocarbon fuels such as gasoline, and is therefore used for flanges and case-shaped molded products around fuel pumps of automobiles.
- the toughness is significantly reduced. There is a problem of doing. That is, in the POM resin composition, if the antistatic effect and the durability against the acid component are simultaneously realized, the toughness will be significantly reduced.
- the present invention has been made in view of the above-mentioned conventional problems, and the problems thereof are POM resin compositions and automobile parts, which are endowed with durability against acid components and antistatic effects without significantly reducing toughness.
- Another object of the present invention is to provide a method for imparting an antistatic effect to a POM resin molded product and improving resistance to an acid component.
- A With respect to 100 parts by mass of the polyacetal copolymer resin having a hemiformal terminal group amount of 0.8 mmol / kg or less.
- B 0.2 to 2.0 parts by mass of a hindered phenolic antioxidant,
- C At least one of magnesium oxide and zinc oxide is contained in an amount of more than 2.0 parts by mass and 20 parts by mass or less.
- D A carbon-based conductive additive is blended in an amount of 0.3 to 2.5 parts by mass, and
- E a polyalkylene glycol is blended in an amount of 0.5 to 3.0 parts by mass.
- One of the (D) carbon-based conductive additives selected from only (D1) carbon nanostructures and a combination of (D1) carbon nanostructures and (D2) carbon black having a BET specific surface area of 300 m 2 / g or more. Is a polyacetal resin composition.
- the POM resin composition and automobile parts to which the durability against the acid component and the antistatic effect are imparted, and the POM resin molded product are imparted with the antistatic effect without significantly reducing the toughness, and the antistatic effect is imparted. It is possible to provide a method for improving resistance to an acid component.
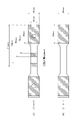
- FIG. 1 is a diagram schematically showing the states of the carbon nanostructure before (A) melt-kneading, (B) immediately after the start of melt-kneading, and (C) after melt-kneading.
- FIG. 2 is a top view (A) and a back view (B) of the test piece used for measuring the surface resistivity and the volume resistivity in the embodiment.
- the POM resin composition of the present embodiment contains (A) 100 parts by mass of a polyacetal copolymer resin having a hemiformal terminal group amount of 0.8 mmol / kg or less, and (B) a hindered phenol-based antioxidant. 2 to 2.0 parts by mass, (C) at least one of magnesium oxide and zinc oxide is more than 2.0 parts by mass and 20 parts by mass or less, and (D) carbon-based conductive additive is 0.3 to 2.5. It is made by blending 0.5 to 3.0 parts by mass of (E) polyalkylene glycol.
- the (D) carbon-based conductive additive is selected from only (D1) carbon nanostructures and a combination of (D1) carbon nanostructures and (D2) carbon black having a BET specific surface area of 300 m 2 / g or more. It is one.
- durability against an acid component can be imparted by blending at least one of (C) magnesium oxide and zinc oxide with a predetermined POM resin. Further, by blending (D) a carbon-based conductive additive, conductivity can be imparted and an antistatic effect can be exhibited.
- (C) magnesium oxide and zinc oxide with a predetermined POM resin.
- (D) a carbon-based conductive additive conductivity can be imparted and an antistatic effect can be exhibited.
- carbon black or the like is added in order to exhibit the antistatic effect, it causes a significant decrease in toughness in combination with magnesium oxide or the like.
- the (D) carbon-based conductive additive imparts conductivity, a significant decrease in toughness can be suppressed. The mechanism will be described later.
- each component of the POM resin composition of the present embodiment will be described.
- the (A) polyacetal copolymer having specific terminal characteristics is used as the substrate resin.
- the polyacetal copolymer is a resin having an oxymethylene group (-OCH 2- ) as a main constituent unit and having other comonomer units in addition to the oximethylene unit. Further, it is generally produced by copolymerizing formaldehyde or a cyclic oligomer of formaldehyde as a main monomer and a compound selected from cyclic ether or cyclic formal as a comonomer. Then, usually, the unstable portion at the end is removed and stabilized by hydrolysis.
- trioxane which is a cyclic trimer of formaldehyde
- Trioxane is generally obtained by reacting an aqueous formaldehyde solution in the presence of an acidic catalyst, and is purified by a method such as distillation before use.
- the trioxane used for the polymerization preferably contains as little impurities as possible, such as water, methanol, and formic acid.
- Examples of the comonomer cyclic ether and cyclic formal include ethylene oxide, propylene oxide, butylene oxide, cyclohexene oxide, oxetane, tetrahydrofuran, trioxepan, 1,3-dioxane, 1,3-dioxolane, propylene glycol formal, diethylene glycol formal, and tri. Examples thereof include ethylene glycol formal, 1,4-butanediol formal, and 1,6-hexanediol formal.
- a compound capable of forming a branched structure or a crosslinked structure can be used as a comonomer (or a tar monomer), and such compounds include methyl glycidyl ether, ethyl glycidyl ether, butyl glycidyl ether, and 2-ethyl-hexyl.
- alkyl or aryl glycidyl ethers such as glycidyl ether and phenyl glycidyl ether
- alkylene glycols such as ethylene glycol diglycidyl ether, triethylene glycol diglycidyl ether and butanediol diglycidyl ether
- diglycidyl ethers of polyalkylene glycol examples thereof include alkyl or aryl glycidyl ethers such as glycidyl ether and phenyl glycidyl ether, alkylene glycols such as ethylene glycol diglycidyl ether, triethylene glycol diglycidyl ether and butanediol diglycidyl ether, and diglycidyl ethers of polyalkylene glycol. These comonomer can be used alone or in combination of two or more.
- the polyacetal copolymer as described above can generally be obtained by adding an appropriate amount of a molecular weight adjusting agent and cationically polymerizing using a cationic polymerization catalyst.
- the molecular weight adjuster used, the cationic polymerization catalyst, the polymerization method, the polymerization apparatus, the deactivation treatment of the catalyst after the polymerization, the end stabilization treatment method of the crude polyacetal copolymer obtained by the polymerization, and the like are known from many documents. , Basically any of them can be used.
- the molecular weight of the (A) polyacetal copolymer used in the present embodiment is not particularly limited, but a weight average molecular weight of about 10,000 to 400,000 is preferable.
- the melt mass flow rate (MFR) (measured at 190 ° C. and a load of 2.16 kg according to ISO1133), which is an index of the fluidity of the resin, is preferably 0.1 to 100 g / 10 minutes, more preferably 0. .5-80g / 10 minutes.
- the (A) polyacetal copolymer used in the present embodiment needs to have specific terminal characteristics as described above, and specifically, the hemiformal terminal group amount is 0.8 mmol / kg or less. It is essential to be there.
- the hemiformal terminal group is represented by ⁇ OCH 2 OH, and the amount of such hemiformal terminal group can be determined by 1 H-NMR measurement, and a specific measurement method thereof is disclosed in JP-A-2001-11143. You can refer to the method described in.
- the polyacetal copolymer (A) used in the present embodiment preferably has a hemiformal terminal group amount of 0.6 mmol / kg or less, and more preferably. It is 0.4 mmol / kg or less.
- the lower limit of the amount of hemiformal terminal groups is not particularly limited.
- the (A) polyacetal copolymer having specific terminal characteristics can be produced by reducing impurities contained in the monomer and the comonomer, selecting a production process, optimizing the production conditions thereof, and the like.
- impurities such as water, alcohol (for example, methanol), and acid (for example, formic acid) contained in the monomer and comonomer. be.
- a chain transfer agent that does not form an unstable terminal for example, a low molecular weight linear acetal having an alkoxy group at both ends such as methylal, contains an arbitrary amount to adjust the molecular weight of the polyacetal copolymer. be able to.
- the amount of catalyst used during the polymerization reaction is also an important requirement. If the amount of the catalyst is too large, it becomes difficult to properly control the polymerization temperature, the decomposition reaction during the polymerization becomes predominant, and it becomes difficult to obtain a polyacetal copolymer having few unstable terminals. On the other hand, if the amount of the catalyst is too small, the polymerization reaction rate is lowered and the polymerization yield is lowered, which is not preferable.
- any conventionally known method is possible, but a continuous lumpy polymerization method in which a liquid monomer is used to obtain a solid powder lumpy polymer as the polymerization progresses is industrially preferable, and the polymerization temperature is 60 to 60 to. It is desirable to keep it at 105 ° C, especially 65-100 ° C.
- a method such as adding the polymer after polymerization to an aqueous solution containing a basic compound is possible as a method for deactivating the catalyst after polymerization.
- the polymer obtained by the polymerization reaction is pulverized and subdivided into contact with an inactivating agent to promptly deactivate the catalyst.
- a polymer to be deactivated by a catalyst is crushed, and 80% by mass or more, preferably 90% by mass, has a particle size of 1.5 mm or less, and 15% by mass or more, preferably 20% by mass or more is 0. It is desirable that the particles are subdivided to a particle size of 3 mm or less.
- Examples of the basic compound for neutralizing and inactivating the polymerization catalyst include ammonia, amines such as triethylamine, tributylamine, triethanolamine, and tributanolamine, or oxides of alkali metals and alkaline earth metals. , Hydroxides, salts and other known catalytic deactivating agents can be used. These basic compounds are preferably added as an aqueous solution of 0.001 to 0.5% by mass, particularly 0.02 to 0.3% by mass.
- the temperature of the preferable aqueous solution is 10 to 80 ° C, particularly preferably 15 to 60 ° C. Further, after the completion of the polymerization, it is preferable to immediately add the aqueous solution to these aqueous solutions to inactivate the catalyst.
- a polyacetal copolymer having a small amount of unstable terminals can be produced by reducing impurities contained in the monomers and comonomer as described above, selecting a production process, and optimizing the production conditions thereof. Then, it is possible to further reduce the amount of hemiformal terminal groups through the stabilization step.
- Stabilization steps include heating the polyacetal copolymer to a temperature above its melting point and treating it in a molten state to decompose and remove only unstable portions, or maintaining a heterogeneous system in an insoluble liquid medium at 80 ° C.
- Known methods such as decomposing and removing only the unstable end portion by heat treatment at the above temperature can be mentioned.
- (B) Hindered phenolic antioxidant examples include 2,2'-methylenebis (4-methyl-6-t-butylphenol) and hexamethylene-bis [3- (3,5-).
- At least one or two or more selected from these antioxidants can be used.
- the blending amount of the (B) hindered phenolic antioxidant in the present embodiment is 0.2 to 2.0 parts by mass and 0.2 to 1.5 parts by mass with respect to 100 parts by mass of the (A) POM resin. It is more preferable that it is a part.
- the POM resin composition of the present embodiment contains at least one of magnesium oxide and zinc oxide (hereinafter, also referred to as “component (C)”).
- component (C) used in the present embodiment has an excellent balance between improvement in detergent resistance (durability against acid components (hereinafter, also referred to as “acid resistance”)) and performance such as mechanical properties and moldability. It is preferable.
- the BET specific surface area is 100 m 2 / g or more and the average particle size is 1.5 ⁇ m or less. By satisfying these conditions, acid resistance can be obtained while suppressing a decrease in toughness.
- the BET specific surface area of magnesium oxide is preferably 100 to 500 m 2 / g, more preferably 120 to 300 m 2 / g.
- the average particle size of magnesium oxide is preferably 0.2 to 1.3 ⁇ m, more preferably 0.3 to 1.0 ⁇ m.
- the average particle size is determined by the particle size of 50% of the integrated value in the particle size distribution (volume basis) measured by the laser diffraction / scattering method.
- the blending amount of the component (C) in the present embodiment is more than 2.0 parts by mass and 20 parts by mass or less, and 4.0 parts by mass or more and 15 parts by mass or less with respect to 100 parts by mass of (A) POM resin. Is preferable.
- the blending amount of the component (C) exceeds 2.0 parts by mass, it is particularly excellent in acid resistance, stable production is possible within 20 parts by mass, and it is particularly excellent in the balance of mechanical properties within 10 parts by mass. ..
- the amount of the component (C) increased, the decomposition of unstable terminals in the POM resin was sometimes promoted, but the (A) POM resin of the present embodiment can suppress the decomposition.
- (C) It was possible to find the property of improving acid resistance by increasing the amount of the component.
- (D) Carbon-based conductive additive In the POM resin composition of the present embodiment, a predetermined amount of (D) carbon-based conductive additive is blended with respect to (A) POM resin.
- the (D) carbon-based conductive additive is only (D1) carbon nanostructure (hereinafter, also referred to as "CNS"), and (D1) carbon nanostructure and (D2) carbon having a BET specific surface area of 300 m 2 / g or more. It is one selected from the combination with black. Then, by adding the (D) carbon-based conductive additive to the POM resin composition, conductivity is imparted and an antistatic effect is exhibited.
- the CNS used in the present embodiment is a structure containing a plurality of carbon nanotubes in a bonded state, and the carbon nanotubes are bonded to other carbon nanotubes by a branched bond or a crosslinked structure. Details of such CNS are described in US Patent Application Publication No. 2013-0071565, US Pat. No. 9,133,031, US Pat. No. 9,447,259, US Pat. No. 9,111,658. It is described in the specification.
- FIG. 1 schematically shows the CNS used in the present embodiment
- (A) is a state before melt-kneading with a POM resin
- (B) is a state immediately after the start of melt-kneading
- (C) is after melt-kneading. Indicates the state of.
- the CNS 10 before melt-kneading forms a structure in which a large number of branched carbon nanotubes 12 are entangled and bonded.
- the CNS 10 is poured into the POM resin 20 and melt-kneaded, the CNS 10 is divided into a large number as shown in FIG. 1 (B).
- each of the carbon nanotubes 12 is in contact with each other via the contact point 14. That is, in the state of FIG. 1C, in the POM resin, a large number of carbon nanotubes 12 are in contact with each other over a wide range to form a conductive path, so that conductivity is exhibited. Further, it is considered that the carbon nanotubes 12 are randomly entangled to form a three-dimensional network structure, so that the decrease in toughness can be suppressed.
- the CNS shown in FIG. 1 (A) has a predetermined flake shape.
- the flake-shaped CNS shown in FIG. 1 (A) contains a plurality of carbon nanotubes, and the carbon nanotubes are branched, crosslinked, and share a common wall with each other.
- not all of the carbon nanotubes have structural characteristics such as branching, cross-linking, and sharing a common wall, and the carbon nanotubes as a whole have at least one of these structural characteristics. It suffices to have.
- the form shown in FIG. 1 (C) is obtained by melt-kneading.
- the flake-shaped CNS as described above is obtained by growing the CNS on a growth substrate such as a fiber material and extracting the grown CNS from the growth substrate.
- Growth substrates such as fibers, tow, yarn, woven fabrics, non-woven fabrics, sheets, tapes and belts can be used in the CNS growth process. That is, the growth base material can be a fiber material having a size that can be spooled, and the formation of CNS can be continuously performed while the growth base material is conveyed. More specifically, the catalyst can be applied to the growth substrate and the CNS can be grown by the pore CVD process. Then, the growth substrate on which the CNS is formed can be stored and then rolled up to remove the CNS.
- a catalyst containing a plurality of transition metal nanoparticles When growing CNS on a growth substrate, it is preferable to use a catalyst containing a plurality of transition metal nanoparticles.
- the catalyst can be applied onto the growth substrate via particle adsorption, for example, direct catalyst application using vapor deposition with a liquid or colloidal precursor.
- Transition metal nanoparticle catalysts include d-block transition metals or d-block transition metal salts.
- the transition metal salt may be applied to the growth substrate without heat treatment, or the transition metal salt may be converted to a zero-valent transition metal on the growth substrate by heat treatment.
- CNS contains carbon nanotubes in a network having a complex structural morphology, which is a CNS on a growth substrate under the growth conditions of carbon nanotubes produced at a rapid growth rate of several microns per second. It is considered that it is derived from the formation of.
- the CNS that grows on the fiber can be formed by techniques such as microcavities, heat and plasma enhanced CVD techniques, laser ablation, arc discharge, and high pressure carbon monoxide (HiPCO). It is also possible to ionize the acetylene gas to generate a low temperature carbon plasma for synthesizing carbon nanotubes. At this time, the plasma is directed at the fiber material having a catalyst.
- the carbon nanotubes are formed by the size of the carbon nanotube forming catalyst. Further, the synthesis of CNS can be facilitated by heating the sized fiber material to about 550 to 800 ° C.
- a process gas such as argon, helium, or nitrogen
- a carbon-containing gas such as acetylene, ethylene, ethanol, or methane. Then, the carbon nanotube grows at the position of the carbon nanotube forming catalyst.
- the CNS used in this embodiment may be a commercially available product.
- ATHLOS 200, ATHLOS 100, etc. manufactured by CABOT can be used.
- carbon black having a BET specific surface area of 300 m 2 / g or more is used among the carbon blacks.
- the carbon black is not used alone, but in combination with CNS. Since the POM resin composition containing the carbon black has high conductivity, the conductivity can be maintained even when used in combination with CNS. On the contrary, the POM resin composition containing carbon black having a BET specific surface area of less than 300 m 2 / g has low conductivity, and it is necessary to increase the amount to ensure sufficient conductivity, so that the toughness is increased. The decline cannot be suppressed.
- the BET specific surface area is preferably 310 m 2 / g or more, more preferably 350 m 2 / g or more, and the upper limit is not particularly limited, but is about 2000 m 2 / g.
- the BET specific surface area can be measured according to ASTM D4820.
- Specific carbon blacks as described above include Ketjen Black EC300J (BET specific surface area: 800 m 2 / g), Ketjen Black EC600JD (BET specific surface area: 1270 m 2 / g), Lionite EC200L manufactured by Lion Corporation. (BET specific surface area: 377 m 2 / g) and the like.
- the (D) carbon-based conductive additive is blended with respect to 100 parts by mass of the POM resin.
- the blending amount of the CNS is preferably 0.5 to 2.0 parts by mass, more preferably 0.6 to 1.8 parts by mass, still more preferably 0.8 to 1.5 parts by mass.
- the mass ratio ((D2) / (D1)) of (D1) CNS and (D2) carbon black is preferably 10 or less. , It is more preferable that it is more than 0 and 5 or less. When the mass ratio is 10 or less, the balance between conductivity and toughness can be ensured. Further, the closer the value of D2 / D1 is to 0, the more the CNS is blended in an excessive amount, but the CNS may be blended in an excessive amount as such. However, considering that CNS is expensive, the lower limit of the value of D2 / D1 is preferably 0.1 from the viewpoint of cost effectiveness.
- (E) Polyalkylene Glycol In the POM resin composition of the present embodiment, a predetermined amount of (E) polyalkylene glycol is blended with respect to (A) POM resin.
- These types are not particularly limited, but from the viewpoint of affinity with the POM resin, those containing polyethylene glycol or polypropylene glycol are preferable, and those containing polyethylene glycol are more preferable.
- the number average molecular weight (Mn) of the polyalkylene glycol is not particularly limited, but is preferably 1,000 or more and 50,000 or less, and 5,000 or more and 30,000 or less, from the viewpoint of dispersibility in the polyacetal resin. It is more preferable to have.
- the number average molecular weight is assumed to be a polystyrene-equivalent number average molecular weight determined by size exclusion chromatography (SEC) using tetrahydrofuran (THF) as a solvent.
- the blending amount of (E) polyalkylene glycol in the present embodiment is 0.5 to 3.0 parts by mass and 0.8 to 2.5 parts by mass with respect to 100 parts by mass of (A) POM resin. Is preferable.
- the upper limit of the blending amount is selected in consideration of the balance with the mechanical properties of the molded product. These may be used by mixing two or more kinds.
- the POM resin composition of the present embodiment may contain other components, if necessary.
- One or more known stabilizers for the POM resin composition can be added as long as the purpose and effect of the POM resin composition of the present embodiment are not impaired.
- the method for producing a molded product using the POM resin composition of the present embodiment is not particularly limited, and a known method can be adopted.
- the POM resin composition of the present embodiment may be put into an extruder, melt-kneaded and pelletized, and the pellets may be put into an injection molding machine equipped with a predetermined mold and injection-molded. can.
- the above-mentioned POM resin composition of the present embodiment can be used as an automobile part described later, or can be a molded product having an antistatic function and resistance to an acid component.
- the automobile parts of the present embodiment are molded by a conventional molding method, for example, injection molding, extrusion molding, compression molding, blow molding, vacuum molding, foam molding, rotary molding, etc., using the POM resin composition. Can be obtained by Even if the automobile parts of the present embodiment come into contact with a strong acid cleaning agent having a pH of 2 or less, deterioration is suppressed and a good surface appearance of the molded product can be maintained.
- a strong acid cleaning agent having a pH of 2 or less, deterioration is suppressed and a good surface appearance of the molded product can be maintained.
- the above-mentioned POM resin composition of the present embodiment is used as a method for imparting an antistatic effect to the polyacetal resin molded product of the present embodiment and improving resistance to an acid component.
- the molded product obtained by molding the POM resin composition of the present embodiment is imparted with durability against an acid component and an antistatic effect without significantly reducing the toughness. That is, by using the POM resin composition of the present embodiment, the durability and antistatic effect of the POM resin composition with respect to the acid component can be exhibited, and the toughness is not significantly reduced.
- the acid component one derived from an acidic detergent can be used.
- each component with respect to the POM resin, a preferable content thereof, and other components are as described in the above-mentioned POM resin composition of the present embodiment.
- Examples 1 to 15, Comparative Examples 1 to 12 In each Example / Comparative Example, the raw material components shown in Tables 1 and 2 were dry-blended, then put into a twin-screw extruder having a cylinder temperature of 200 ° C., melt-kneaded, and pelletized. In Tables 1 and 2, the numerical values of each component indicate parts by mass. The details of each raw material component used are shown below.
- POM resin Polyacetal copolymer resin
- A-1 Polyacetal copolymer resin
- POM resin with hemiformal terminal group amount 0.7 mmol / kg
- POM resin with hemiformal terminal group amount 1.0 mmol / kg
- ISO1133 the MFR measured at 190 ° C. and a load of 2.16 kg is Both A-1 and A-2 were 9 g / 10 minutes.
- the polyacetal copolymers A-1 to A-2 were obtained as follows.
- A-1 A mixture of 96.7% by mass of trioxane and 3.3% by mass of 1,3-dioxolane was continuously supplied to a biaxial paddle type continuous polymerizer, and 10 ppm of boron trifluoride was added as a catalyst. Polymerization was performed. The mixture of trioxane and 1,3-dioxolane to be subjected to polymerization contained 10 ppm of water, 3.5 ppm of methanol and 5 ppm of formic acid as impurities.
- the polymer discharged from the discharge port of the polymer is immediately added with an aqueous solution containing 1000 ppm of triethylamine, pulverized and stirred to deactivate the catalyst, and then centrifuged and dried to obtain both crude polyoxymethylene. A polymer was obtained.
- this crude polyoxymethylene copolymer was supplied to a twin-screw extruder having a vent port.
- 0.4% of a 0.3% aqueous solution of triethylamine was added to the crude polyoxymethylene copolymer, and the mixture was melt-kneaded at a resin temperature of about 220 ° C. to decompose unstable ends and a decomposition product.
- the volatile matter containing the above was devolatile from the vent port under reduced pressure.
- the polymer taken out from the die of the extruder was cooled and shredded to obtain a pellet-shaped polyacetal copolymer A-1 from which the unstable end portion was removed.
- a mixture of 96.7% by mass of trioxane and 3.3% by mass of 1,3-dioxolane was continuously supplied to a twin-screw paddle type continuous polymerizer, and 15 ppm of boron trifluoride was added as a catalyst. Polymerization was performed.
- the mixture of trioxane and 1,3-dioxolane to be subjected to polymerization contained 10 ppm of water, 3.5 ppm of methanol and 5 ppm of formic acid as impurities. Then, the polymer discharged from the discharge port of the polymerizer was subjected to the same treatment as in A-1 to obtain a pellet-shaped polyacetal copolymer A-2.
- Hindered Phenolic Antioxidant B-1 Tetrakis [methylene-3- (3,5-di-t-butyl-4-hydroxyphenyl) propionate] methane (product name: Irganox1010, manufactured by BASF)
- C Magnesium oxide, etc.
- C-1 Magnesium oxide, specific surface area 135 m 2 / g, average particle size 0.9 ⁇ m (Kyowa Chemical Industry Co., Ltd., Kyowa Mag MF150)
- C-2 Magnesium oxide, specific surface area 30 m 2 / g, average particle size 0.6 ⁇ m (Kyowa Chemical Industry Co., Ltd., Kyowa Mag MF30)
- C-3 Magnesium oxide, specific surface area 155 m 2 / g, average particle size 7 ⁇ m (Kyowa Chemical Industry Co., Ltd., Kyowa Mag 150)
- C-4 Zinc oxide (BET specific surface area 60-90 m 2 / g) (manufactured by Shodo Chemical Industry Co., Ltd., active zinc flower AZO)
- D Carbon Nanostructure, Carbon Black
- D-1 Carbon Nanostructure (manufactured by CABOT, ATHLOS 200)
- D-2 Carbon black (manufactured by Lion Corporation, Ketjen Black EC300J, BET specific surface area: 800 m 2 / g)
- D-3 Carbon black (manufactured by Lion Corporation, Lionite EC200L, BET specific surface area: 377m 2 / g)
- D-4 Carbon black (manufactured by Denka Co., Ltd., Denka black, BET specific surface area: 65 m 2 / g)
- the following acidic cleaning agents were used as the acidic cleaning agents.
- Detergent Sulfuric acid: 1.5%
- Hydrofluoric acid 1.5%
- Phosphoric acid 10%
- Spray of acid detergent-Leave the multipurpose test piece at 60 ° C for 4 hours-Leave the multipurpose test piece at 23 ° C and 50% RH for 4 hours-Spray the acid cleaner again-Leave the multipurpose test piece at 23 ° C and 50% RH for 16 hours One cycle was set, and each time this one cycle was completed, the state of crack generation on the surface of the dumbbell test piece was visually observed. Then, based on the number of cycles in which cracks were confirmed, evaluation was performed according to the following evaluation criteria. The evaluation results are shown in Tables 1 and 2. [Evaluation criteria] A; Number of cycles: 10 or more B; Number of cycles: 4-9 C; Number of cycles: 3 or less
- FIG. 2 (A) shows the front surface
- FIG. 2 (B) shows the back surface
- a conductive paint Dotite D500, manufactured by Fujikura Kasei Co., Ltd.
- a predetermined region hatchched region in FIG. 2 on each surface of the test piece and dried.
- DIGITAL MULTIMETER R6450 manufactured by Advantest
- Mold deposit A mold deposit test piece (disk type) was molded under the following conditions using the POM resin compositions prepared in Examples and Comparative Examples. [Evaluation methods] After 3000 shot molding, the surface of the cavity portion on the mold moving side was visually observed, and the amount of deposits was determined according to the following criteria. A: No deposits or slight deposits are confirmed. B: A large amount of deposits are confirmed * Molding machine: FANUC ROBOSHOT S-2000i 50B (FANUC Corporation) * Molding conditions: Cylinder temperature (° C) Nozzle-C1-C2-C3 205 215 205 185 ° C Injection pressure: 40 (MPa) Injection speed: 1.5 (m / min) Mold temperature: 60 (° C)
- Comparative Example 1 is different from Example 1 in that the components (C) to (E) are not blended, and is inferior in acid resistance and conductivity.
- Comparative Example 2 is inferior in conductivity to Example 1 in that the components (D) and (E) are not blended.
- Comparative Example 3 is different from Example 1 in that the component (D) is not blended, and is inferior in conductivity.
- Comparative Example 4 is inferior in acid resistance to Example 1 in that the components (C) and (E) are not blended.
- Comparative Examples 5 and 6 are different from Example 1 in that the component (C) is not blended or is insufficient, respectively, and both are inferior in acid resistance.
- Comparative Example 7 is inferior in acid resistance unlike Example 1 in that the component (C) is excessive.
- Comparative Example 8 is inferior in conductivity to Example 1 in that the component (D) is too small.
- Comparative Example 9 is different from Example 1 in that the component (D) is excessive, and is inferior in toughness.
- Comparative Example 10 is different from Examples 11 and 13 in that the carbon black of the component (D) having a BET specific surface area of less than 300 m 2 / g is inferior in conductivity.
- Comparative Example 11 is different from Example 1 in that a POM resin having an excessive amount of hemiformal terminal groups is used, and is inferior in moldability.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Nanotechnology (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Description
(1)(A)ヘミホルマール末端基量が0.8mmol/kg以下であるポリアセタール共重合体樹脂100質量部に対し、
(B)ヒンダードフェノール系酸化防止剤を0.2~2.0質量部、
(C)酸化マグネシウム及び酸化亜鉛のうちの少なくとも1種を2.0質量部超20質量部以下、
(D)炭素系導電添加剤を0.3~2.5質量部、及び
(E)ポリアルキレングリコールを0.5~3.0質量部、を配合してなり、
前記(D)炭素系導電添加剤が、(D1)カーボンナノストラクチャーのみ、及び(D1)カーボンナノストラクチャーと(D2)BET比表面積が300m2/g以上のカーボンブラックとの組合せから選ばれる一つである、ポリアセタール樹脂組成物。
本実施形態のPOM樹脂組成物は、(A)ヘミホルマール末端基量が0.8mmol/kg以下であるポリアセタール共重合体樹脂100質量部に対し、(B)ヒンダードフェノール系酸化防止剤を0.2~2.0質量部、(C)酸化マグネシウム及び酸化亜鉛のうちの少なくとも1種を2.0質量部超20質量部以下、(D)炭素系導電添加剤を0.3~2.5質量部、及び(E)ポリアルキレングリコールを0.5~3.0質量部、を配合してなる。そして、(D)炭素系導電添加剤が、(D1)カーボンナノストラクチャーのみ、及び(D1)カーボンナノストラクチャーと(D2)BET比表面積が300m2/g以上のカーボンブラックとの組合せから選ばれる一つである。
以下、本実施形態のPOM樹脂組成物の各成分について説明する。
本実施形態においては、基体樹脂として特定の末端特性を有する(A)ポリアセタール共重合体が用いられる。ポリアセタール共重合体は、オキシメチレン基(-OCH2-)を主たる構成単位とし、オキシメチレン単位以外に他のコモノマー単位を有する樹脂である。また、一般的にはホルムアルデヒド又はホルムアルデヒドの環状オリゴマーを主モノマーとし、環状エーテルや環状ホルマールから選ばれた化合物をコモノマーとして共重合させることによって製造される。そして、通常、加水分解によって末端の不安定部分を除去して安定化される。
本実施形態において使用される(B)ヒンダードフェノール系酸化防止剤としては、2,2’-メチレンビス(4-メチル-6-t-ブチルフェノール)、ヘキサメチレン-ビス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオネート]、テトラキス[メチレン-3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオネート]メタン、トリエチレングリコール-ビス[3-(3-t-ブチル-4-ヒドロキシ-5-メチルフェニル)プロピオネート]、1,3,5-トリメチル-2,4,6-トリス(3’,5’-ジ-t-ブチル-4-ヒドロキシ-ベンジル)ベンゼン、n-オクタデシル-3-(4’-ヒドロキシ-3’,5’-ジ-t-ブチルフェニル)プロピオネート、4,4’-メチレンビス(2,6-ジ-t-ブチルフェノール)、4,4’-ブチリデンビス(6-t-ブチル-3-メチル-フェノール)、ジステアリル(3,5-ジ-t-ブチル-4-ヒドロキシベンジル)ホスホネート、2-t-ブチル-6-(3-t-ブチル-5-メチル-2-ヒドロキシベンジル)-4-メチルフェニルアクリレート、3,9-ビス{2-〔3-(3-t-ブチル-4-ヒドロキシ-5-メチルフェニル)プロピオニルオキシ〕-1,1-ジメチルエチル}-2,4,8,10-テトラオキサスピロ〔5,5〕ウンデカン等が例示され、中でも、テトラキス[メチレン-3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオネート]メタン、ヘキサメチレン-ビス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオネート]が好ましい。
本実施形態のPOM樹脂組成物には、酸化マグネシウム及び酸化亜鉛のうちの少なくとも1種(以下、「(C)成分」とも呼ぶ。)が配合される。本実施形態において使用される(C)成分は、耐洗浄剤性(酸成分に対する耐久性(以下、「耐酸性」とも呼ぶ。))の改善と機械物性や成形性等の性能のバランスが優れており好ましい。
本実施形態のPOM樹脂組成物には、(A)POM樹脂に対して所定量の(D)炭素系導電添加剤が配合される。(D)炭素系導電添加剤は、(D1)カーボンナノストラクチャー(以下、「CNS」とも呼ぶ。)のみ、及び(D1)カーボンナノストラクチャーと(D2)BET比表面積が300m2/g以上のカーボンブラックとの組合せから選ばれる一つである。そして、POM樹脂組成物に(D)炭素系導電添加剤を添加することにより、導電性が付与され、帯電防止効果が発揮される。また、カーボンブラックを単独で添加すると、得られる成形品の靭性が低下するが、(D)炭素系導電添加剤の添加によっては靭性の低下を抑えることができる。
以下に、(D1)カーボンナノストラクチャー及び(D2)BET比表面積が300m2/g以上のカーボンブラックのそれぞれについて説明する。
本実施形態で使用するCNSは、複数のカーボンナノチューブが結合した状態で含む構造体であり、カーボンナノチューブは分岐結合や架橋構造で他のカーボンナノチューブと結合している。このようなCNSの詳細は、米国特許出願公開第2013-0071565号明細書、米国特許第9,133,031号明細書、同第9,447,259号明細書、同第9,111,658号明細書に記載されている。
より具体的には、触媒を成長基材に塗布し、細孔CVDプロセスによりCNSの成長を図ることができる。そして、CNSを形成した成長基材を保存して、その後、CNSを取り出すために巻き取ることができる。
本実施形態においては、カーボンブラックの中でも、BET比表面積が300m2/g以上のカーボンブラックを用いる。ただし、当該カーボンブラックは単独で用いるのではなく、CNSと組合せて用いる。当該カーボンブラックが配合されたPOM樹脂組成物は導電性が高いため、CNSと併用しても導電性を保持することができる。逆に、BET比表面積が300m2/g未満のカーボンブラックが配合されたPOM樹脂組成物は導電性が低く、導電性を十分に確保するには配合量を増加させる必要があり、そうすると靭性の低下を抑えることができない。当該BET比表面積は、310m2/g以上が好ましく、350m2/g以上がより好ましく、上限としては特に限定はないが、2000m2/g程度である。
なお、BET比表面積は、ASTM D4820に準拠して測定することができる。
本実施形態のPOM樹脂組成物には、(A)POM樹脂に対して所定量の(E)ポリアルキレングリコールが配合される。これらの種類は特に限定されないが、POM樹脂との親和性の観点から、ポリエチレングリコール又はポリプロピレングリコールを含有するものが好ましく、ポリエチレングリコールを含有するものがより好ましい。
本実施形態のPOM樹脂組成物は、必要に応じて他の成分を含有するものであってもよい。本実施形態のPOM樹脂組成物の目的・効果を阻害しない限り、POM樹脂組成物に対する公知の安定剤を1種又は2種以上添加することができる。
本実施形態の自動車部品は、上述の本実施形態のPOM樹脂組成物の成形品からなる。従って、本実施形態の自動車部品は、靭性が大きく低下することなく、酸成分に対する耐久性と帯電防止効果が付与される。従って、自動車の燃料ポンプ周辺のフランジやケース状の成形品に使用されることが好適である。すなわち、帯電防止効果に優れるため、静電気による燃料の引火を防止することができるし、酸成分に対する耐久性に優れるため、強酸性(pH=1程度)のクリーナーが付着した場合であっても、表面の劣化を防止することができる。すなわち、本実施形態の自動車部品は、酸性洗浄剤に接触し得る環境下で用いることができる。
本実施形態の自動車部品は、上記POM樹脂組成物を用いて、慣用の成形方法、例えば、射出成形、押出成形、圧縮成形、ブロー成形、真空成形、発泡成形、回転成形等の方法で成形することにより得ることができる。
本実施形態の自動車部品は、例えばpH2以下の強酸性洗浄剤に接触したとしても、劣化が抑制され、良好な成形品表面外観を保持できる。
本実施形態のポリアセタール樹脂成形品に帯電防止効果を付与し、かつ、酸成分に対する耐性を向上させる方法は、上述の本実施形態のPOM樹脂組成物を用いる。
上述の通り、本実施形態のPOM樹脂組成物を成形して得られる成形品は、靭性が大きく低下することなく、酸成分に対する耐久性と帯電防止効果が付与される。つまり、本実施形態のPOM樹脂組成物を用いることにより、POM樹脂組成物の酸成分に対する耐久性と帯電防止効果を発現することができ、かつ、靭性が大きく低下することもない。酸成分としては、酸性洗浄剤由来のものを用いることができる。
本実施形態の方法において、POM樹脂に対する各成分及びその好ましい含有量、及び他の成分は上述の本実施形態のPOM樹脂組成物で説明した通りである。
各実施例・比較例において、表1及び表2に示す各原料成分をドライブレンドした後、シリンダー温度200℃の二軸押出機に投入して、溶融混練し、ペレット化した。なお、表1、表2において、各成分の数値は質量部を示す。
また、使用した各原料成分の詳細を以下に示す。
(A)ポリアセタール共重合体樹脂(POM樹脂)
A-1;ヘミホルマール末端基量:0.7mmol/kgのPOM樹脂
A-2;ヘミホルマール末端基量:1.0mmol/kgのPOM樹脂
ISO1133に準じ、190℃、荷重2.16kgで測定したMFRはA-1、A-2ともに9g/10分であった。
ポリアセタール共重合体A-1~A-2は、次のようにして得た。
B-1:テトラキス[メチレン-3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオネート]メタン(製品名:Irganox1010,BASF社製)
C-1:酸化マグネシウム、比表面積135m2/g、平均粒径0.9μm(協和化学工業(株)製、キョーワマグMF150)
C-2:酸化マグネシウム、比表面積30m2/g、平均粒径0.6μm(協和化学工業(株)製、キョーワマグMF30)
C-3:酸化マグネシウム、比表面積155m2/g、平均粒径7μm(協和化学工業(株)製、キョーワマグ150)
C-4:酸化亜鉛(BET比表面積 60~90m2/g)(正同化学工業(株)製、活性亜鉛華AZO)
(株)堀場製作所製、レーザー回折/散乱式粒度分布測定装置LA-920を用いて、以下の測定条件の下、レーザー回折/散乱法により粒度分布を測定し、積算値50%の平均粒径(50%d)を求めた。
~測定条件~
・循環速度:5
・レーザー光源:632.8nmHe-Neレーザー1mW、タングステンランプ50W・検出器:リング状75分割シリコンフォトダイオード×1、シリコンフォトダイオード×12
・分散媒:蒸留水
・超音波:有り
・透過率:75~90%
・水との相対屈折率:1.32
・粒子径基準:体積
D-1:カーボンナノストラクチャー(CABOT社製、ATHLOS 200)
D-2:カーボンブラック(ライオン(株)製、ケッチェンブラックEC300J、BET比表面積:800m2/g)
D-3:カーボンブラック(ライオン(株)製、ライオナイトEC200L、BET比表面積:377m2/g)
D-4:カーボンブラック(デンカ(株)製、デンカブラック、BET比表面積:65m2/g)
E-1:ポリエチレングリコール(三洋化成工業(株)製、PEG6000S)
実施例及び比較例で調製したPOM樹脂組成物を用い、ISO294-1に記載の多目的試験片を、ISO9988-1,2に準じた条件で射出成形機(EC40,東芝機械(株)製)にて射出成形により作製し、以下の(1)から(3)の評価に用いた。
POM樹脂組成物の酸性洗浄剤への耐性を評価するため、上記多目的試験片の両端を固定し、負荷歪み:2.0%の割合で湾曲させた。そして、多目的試験片の表面に酸性洗浄剤をスプレーし、スプレー後の多目的試験片を60℃の条件下で4時間放置した。その後、多目的試験片を23℃50%RHの条件下で4時間放置した。その後、酸性洗浄剤を再度スプレーし、23℃50%RHの条件下で16時間放置した。
洗浄剤:硫酸:1.5%、フッ化水素酸:1.5%、リン酸:10%
酸性洗浄剤のスプレー-多目的試験片の60℃4時間の放置-多目的試験片の23℃50%RH4時間の放置-再度酸性洗浄剤のスプレー-多目的試験片の23℃50%RH16時間の放置を1サイクルとし、この1サイクルが終了する毎に、ダンベル試験片表面のクラック発生状況を目視で観察した。そして、クラックが確認されたサイクル数を基に、以下の評価基準に従い評価した。評価結果を表1及び表2に示す。
[評価基準]
A;サイクル数:10以上
B;サイクル数:4~9
C;サイクル数:3以下
上記多目的試験片を用いてISO527-1、2に準拠した引張破壊呼び歪の測定を実施し、以下の評価基準に従い評価した。評価結果を表1及び表2に示す。
[評価基準]
A:10%以上
B:6~9%
C:5%未満
上記多目的試験片を用いて以下の評価をした。
(表面抵抗率・体積抵抗率)
上記のようにして得た多目的試験片の外観を図2に示す。図2(A)は表面を示し、図2(B)は裏面を示す。当該試験片の各面の所定領域(図2のハッチング領域)に導電塗料(ドータイトD500、藤倉化成(株)製)を塗布して乾燥した。その後、低抵抗率測定装置(DIGITAL MULTIMETER R6450、アドバンテスト製)を使用し、図2(A)のA-B間の抵抗を測定し、これを表面抵抗率とした。また、図2のC-D間の抵抗を測定し、これを体積抵抗率とした。表面抵抗率及び体積抵抗率のそれぞれについて、以下の評価基準に従い評価した。評価結果を表1及び表2に示す。なお、表面抵抗率の測定上限は5.0×109Ω/□であり、体積抵抗率の測定上限は1.8×1011Ω・cmである。
[表面抵抗率の評価基準]
A:1.0×104Ω/□以下
B:1.0×104Ω/□超1.0×109Ω/□以下
C:1.0×109Ω/□超
[体積抵抗率の評価基準]
A:1.0×104Ω・cm以下
B:1.0×104Ω・cm超1.0×109Ω・cm以下
C:1.0×109Ω・cm超
実施例及び比較例で調製したPOM樹脂組成物を用い、下記条件でモールドデポジット試験片(円盤型)を成形した。
[評価方法]
3000shot成形した後、金型移動側のCavity部表面を目視にて観察し、以下の基準に従って付着物量を判定した。
A:付着物は確認されないもしくはわずかに確認される。
B:多量の付着物が確認される
*成形機: FANUC ROBOSHOT S-2000i 50B(ファナック(株))
*成形条件:シリンダー温度(℃) ノズル-C1-C2-C3
205 215 205 185℃
射出圧力:40(MPa)
射出速度:1.5(m/min)
金型温度:60(℃)


比較例1は、(C)~(E)成分が配合されていない点で実施例1と異なり、耐酸性及び導電性に劣る。比較例2は、(D)及び(E)成分が配合されていない点で実施例1と異なり、導電性に劣る。比較例3は、(D)成分が配合されていない点で実施例1と異なり、導電性に劣る。比較例4は、(C)及び(E)成分が配合されていない点で実施例1と異なり、耐酸性に劣る。比較例5及び6は、それぞれ、(C)成分が配合されていないか過少である点で実施例1と異なり、いずれも耐酸性に劣る。比較例7は、(C)成分が過剰である点で実施例1と異なり耐酸性に劣る。比較例8は、(D)成分が過少である点で実施例1と異なり、導電性に劣る。比較例9は、(D)成分が過剰である点で実施例1と異なり、靭性に劣る。比較例10は、(D)成分のカーボンブラックとしてBET比表面積が300m2/gに満たないものを用いた点で実施例11及び13とは異なり、導電性に劣る。比較例11は、ヘミホルマール末端基量が過剰のPOM樹脂を用いている点で実施例1と異なり、成形性に劣る。
Claims (7)
- (A)ヘミホルマール末端基量が0.8mmol/kg以下であるポリアセタール共重合体樹脂100質量部に対し、
(B)ヒンダードフェノール系酸化防止剤を0.2~2.0質量部、
(C)酸化マグネシウム及び酸化亜鉛のうちの少なくとも1種を2.0質量部超20質量部以下、
(D)炭素系導電添加剤を0.3~2.5質量部、及び
(E)ポリアルキレングリコールを0.5~3.0質量部、を配合してなり、
前記(D)炭素系導電添加剤が、(D1)カーボンナノストラクチャーのみ、及び(D1)カーボンナノストラクチャーと(D2)BET比表面積が300m2/g以上のカーボンブラックとの組合せから選ばれる一つである、ポリアセタール樹脂組成物。 - 前記(D1)カーボンナノストラクチャーと前記(D2)カーボンブラックとの質量比((D2)/(D1))が10以下である、請求項1に記載のポリアセタール樹脂組成物。
- 前記酸化マグネシウムのBET比表面積が100m2/g以上であり、平均粒径が1.5μm以下である、請求項1又は2に記載のポリアセタール樹脂組成物。
- 請求項1~3のいずれか1項に記載のポリアセタール樹脂組成物の成形品からなる自動車部品。
- 酸性洗浄剤と接触し得る環境下で用いられる、請求項4に記載の自動車部品。
- 請求項1~3のいずれか1項に記載のポリアセタール樹脂組成物を用いる、ポリアセタール樹脂成形品に帯電防止効果を付与し、かつ、酸成分に対する耐性を向上させる方法。
- 前記酸成分が、酸性洗浄剤由来である、請求項6に記載の方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022517888A JP7217384B2 (ja) | 2020-10-09 | 2021-09-27 | ポリアセタール樹脂組成物及び自動車部品 |
| US18/030,572 US12037487B2 (en) | 2020-10-09 | 2021-09-27 | Polyacetal resin composition and automobile part |
| MX2023004042A MX2023004042A (es) | 2020-10-09 | 2021-09-27 | Composición de resina de poliacetal y parte de automóvil. |
| KR1020237012741A KR102587117B1 (ko) | 2020-10-09 | 2021-09-27 | 폴리아세탈 수지 조성물 및 자동차 부품 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2020171176 | 2020-10-09 | ||
| JP2020-171176 | 2020-10-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022075107A1 true WO2022075107A1 (ja) | 2022-04-14 |
Family
ID=81126726
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2021/035360 WO2022075107A1 (ja) | 2020-10-09 | 2021-09-27 | ポリアセタール樹脂組成物及び自動車部品 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US12037487B2 (ja) |
| JP (1) | JP7217384B2 (ja) |
| KR (1) | KR102587117B1 (ja) |
| MX (1) | MX2023004042A (ja) |
| WO (1) | WO2022075107A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2022137998A1 (ja) * | 2020-12-23 | 2022-06-30 |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007032081A1 (ja) * | 2005-09-16 | 2007-03-22 | Asahi Kasei Chemicals Corporation | マスターバッチおよびそれを配合した組成物 |
| JP2007084604A (ja) * | 2005-09-20 | 2007-04-05 | Asahi Kasei Chemicals Corp | ポリオキシメチレン樹脂製ハードディスクランプ |
| JP2008527128A (ja) * | 2005-01-14 | 2008-07-24 | ビーエーエスエフ ソシエタス・ヨーロピア | 導電性ポリオキシメチレン組成物 |
| JP2008189891A (ja) * | 2007-02-08 | 2008-08-21 | Misuzu Kogyo:Kk | ポリアセタール樹脂コンポジット材、ポリアセタール樹脂コンポジット材からなる平面カム、及びその平面カムの製造方法 |
| JP2010144112A (ja) * | 2008-12-22 | 2010-07-01 | Polyplastics Co | 燃料用部品 |
| JP2011132370A (ja) * | 2009-12-24 | 2011-07-07 | Polyplastics Co | ポリアセタール樹脂組成物の製造方法 |
| JP2012140482A (ja) * | 2010-12-28 | 2012-07-26 | Hodogaya Chem Co Ltd | ポリアセタール樹脂/カーボンナノチューブ導電性樹脂複合材料 |
| CN102634162A (zh) * | 2012-05-09 | 2012-08-15 | 四川大学 | 一种导热聚甲醛复合材料及其制备方法 |
| CN102675818A (zh) * | 2012-05-24 | 2012-09-19 | 兖矿鲁南化肥厂 | 一种增强增韧聚甲醛及其制备方法 |
| JP2013028737A (ja) * | 2011-07-29 | 2013-02-07 | Showa Denko Kk | ポリオキシメチレン樹脂組成物及びそれからなる成形品 |
| CN103897331A (zh) * | 2014-04-21 | 2014-07-02 | 四川大学 | 一种导热聚甲醛复合材料及其制备方法 |
| JP2014122264A (ja) * | 2012-12-20 | 2014-07-03 | Asahi Kasei Chemicals Corp | 導電性ポリアセタール樹脂組成物のペレット及びその製造方法 |
| JP2018172456A (ja) * | 2017-03-31 | 2018-11-08 | ポリプラスチックス株式会社 | ポリアセタール樹脂組成物 |
| JP2019218442A (ja) * | 2018-06-19 | 2019-12-26 | ポリプラスチックス株式会社 | ポリアセタール樹脂組成物 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH072891B2 (ja) | 1986-05-16 | 1995-01-18 | ポリプラスチックス株式会社 | ポリアセタ−ル樹脂組成物及びその製造方法 |
| JPS6386124U (ja) | 1986-11-20 | 1988-06-06 | ||
| TW289021B (ja) | 1993-05-08 | 1996-10-21 | Hoechst Ag | |
| US6730401B2 (en) | 2001-03-16 | 2004-05-04 | Eastman Chemical Company | Multilayered packaging materials for electrostatic applications |
| CA3055734A1 (en) * | 2017-03-07 | 2018-09-13 | Esprix Technologies, LP. | Aliphatic polyketone modified with carbon nanostructures |
| CN115885017B (zh) | 2020-06-30 | 2024-08-23 | 宝理塑料株式会社 | 热塑性树脂组合物、部件及其制造方法以及热塑性树脂组合物的导电性表达方法 |
| CN115916905B (zh) | 2020-06-30 | 2024-08-23 | 宝理塑料株式会社 | 热塑性树脂组合物、部件及其制造方法以及热塑性树脂组合物的导电性表达方法 |
| JP2022015904A (ja) | 2020-07-10 | 2022-01-21 | ポリプラスチックス株式会社 | 熱可塑性樹脂組成物及び部材、並びに熱可塑性樹脂組成物からなる部材の製造方法及び機械強度の向上方法 |
| CN116783245A (zh) | 2020-12-23 | 2023-09-19 | 宝理塑料株式会社 | 聚缩醛树脂组合物以及燃料接触体 |
| WO2022210013A1 (ja) * | 2021-04-02 | 2022-10-06 | 三菱エンジニアリングプラスチックス株式会社 | 樹脂組成物および成形体 |
| US20240026126A1 (en) * | 2022-04-07 | 2024-01-25 | Molecular Rebar Design, Llc | Compositions with carbon nanotubes for low hysteresis elastomers |
-
2021
- 2021-09-27 MX MX2023004042A patent/MX2023004042A/es unknown
- 2021-09-27 JP JP2022517888A patent/JP7217384B2/ja active Active
- 2021-09-27 US US18/030,572 patent/US12037487B2/en active Active
- 2021-09-27 KR KR1020237012741A patent/KR102587117B1/ko active IP Right Grant
- 2021-09-27 WO PCT/JP2021/035360 patent/WO2022075107A1/ja active Application Filing
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008527128A (ja) * | 2005-01-14 | 2008-07-24 | ビーエーエスエフ ソシエタス・ヨーロピア | 導電性ポリオキシメチレン組成物 |
| WO2007032081A1 (ja) * | 2005-09-16 | 2007-03-22 | Asahi Kasei Chemicals Corporation | マスターバッチおよびそれを配合した組成物 |
| JP2007084604A (ja) * | 2005-09-20 | 2007-04-05 | Asahi Kasei Chemicals Corp | ポリオキシメチレン樹脂製ハードディスクランプ |
| JP2008189891A (ja) * | 2007-02-08 | 2008-08-21 | Misuzu Kogyo:Kk | ポリアセタール樹脂コンポジット材、ポリアセタール樹脂コンポジット材からなる平面カム、及びその平面カムの製造方法 |
| JP2010144112A (ja) * | 2008-12-22 | 2010-07-01 | Polyplastics Co | 燃料用部品 |
| JP2011132370A (ja) * | 2009-12-24 | 2011-07-07 | Polyplastics Co | ポリアセタール樹脂組成物の製造方法 |
| JP2012140482A (ja) * | 2010-12-28 | 2012-07-26 | Hodogaya Chem Co Ltd | ポリアセタール樹脂/カーボンナノチューブ導電性樹脂複合材料 |
| JP2013028737A (ja) * | 2011-07-29 | 2013-02-07 | Showa Denko Kk | ポリオキシメチレン樹脂組成物及びそれからなる成形品 |
| CN102634162A (zh) * | 2012-05-09 | 2012-08-15 | 四川大学 | 一种导热聚甲醛复合材料及其制备方法 |
| CN102675818A (zh) * | 2012-05-24 | 2012-09-19 | 兖矿鲁南化肥厂 | 一种增强增韧聚甲醛及其制备方法 |
| JP2014122264A (ja) * | 2012-12-20 | 2014-07-03 | Asahi Kasei Chemicals Corp | 導電性ポリアセタール樹脂組成物のペレット及びその製造方法 |
| CN103897331A (zh) * | 2014-04-21 | 2014-07-02 | 四川大学 | 一种导热聚甲醛复合材料及其制备方法 |
| JP2018172456A (ja) * | 2017-03-31 | 2018-11-08 | ポリプラスチックス株式会社 | ポリアセタール樹脂組成物 |
| JP2019218442A (ja) * | 2018-06-19 | 2019-12-26 | ポリプラスチックス株式会社 | ポリアセタール樹脂組成物 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2022137998A1 (ja) * | 2020-12-23 | 2022-06-30 | ||
| JP7217385B2 (ja) | 2020-12-23 | 2023-02-02 | ポリプラスチックス株式会社 | ポリアセタール樹脂組成物及び燃料接触体 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR102587117B1 (ko) | 2023-10-10 |
| US12037487B2 (en) | 2024-07-16 |
| US20230331977A1 (en) | 2023-10-19 |
| JP7217384B2 (ja) | 2023-02-02 |
| JPWO2022075107A1 (ja) | 2022-04-14 |
| KR20230069981A (ko) | 2023-05-19 |
| MX2023004042A (es) | 2023-08-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11634576B2 (en) | Polyacetal resin composition | |
| US10844191B2 (en) | Polyacetal resin composition | |
| WO2022075107A1 (ja) | ポリアセタール樹脂組成物及び自動車部品 | |
| JP6318237B2 (ja) | ポリアセタール樹脂組成物及びその成形体 | |
| WO2022075004A1 (ja) | ポリアセタール樹脂組成物及び自動車部品 | |
| JP7217385B2 (ja) | ポリアセタール樹脂組成物及び燃料接触体 | |
| TW202323341A (zh) | 甲醛(Oxymethylene)共聚物之製造方法及成形品之製造方法 | |
| EP4032944B1 (en) | Method for producing oxymethylene copolymer resin composition, and oxymethylene copolymer resin composition | |
| TWI765800B (zh) | 氧化亞甲基共聚物樹脂組成物之製造方法及氧化亞甲基共聚物樹脂組成物 | |
| JP5783685B2 (ja) | ポリオキシメチレン製スライド部品 | |
| JP2000230026A (ja) | ポリアセタール樹脂の連続製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2022517888 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21877408 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20237012741 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 21877408 Country of ref document: EP Kind code of ref document: A1 |