WO2020179931A1 - ハイスループット遺伝子編集技術 - Google Patents
ハイスループット遺伝子編集技術 Download PDFInfo
- Publication number
- WO2020179931A1 WO2020179931A1 PCT/JP2020/009987 JP2020009987W WO2020179931A1 WO 2020179931 A1 WO2020179931 A1 WO 2020179931A1 JP 2020009987 W JP2020009987 W JP 2020009987W WO 2020179931 A1 WO2020179931 A1 WO 2020179931A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- gene
- tissue stem
- stem cells
- edited
- target gene
- Prior art date
Links
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/90—Stable introduction of foreign DNA into chromosome
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/90—Stable introduction of foreign DNA into chromosome
- C12N15/902—Stable introduction of foreign DNA into chromosome using homologous recombination
- C12N15/907—Stable introduction of foreign DNA into chromosome using homologous recombination in mammalian cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0647—Haematopoietic stem cells; Uncommitted or multipotent progenitors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0693—Tumour cells; Cancer cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPRs]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2750/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssDNA viruses
- C12N2750/00011—Details
- C12N2750/14011—Parvoviridae
- C12N2750/14111—Dependovirus, e.g. adenoassociated viruses
- C12N2750/14141—Use of virus, viral particle or viral elements as a vector
- C12N2750/14143—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
Abstract
本発明の目的の一つは、遺伝子編集のされた未分化状態の組織幹細胞を短期間の内に製造する方法を提供することである。また、本発明の目的の一つは、遺伝子編集のされた未分化状態の組織幹細胞を用いた疾患の治療および/または予防方法を提供することである。 組織幹細胞中の標的遺伝子の遺伝子配列を編集する工程、および遺伝子配列の編集された組織幹細胞を選別する工程を含み、場合により、標的遺伝子の遺伝子配列にタグを付加する工程、および/または組織幹細胞における標的遺伝子の遺伝子発現を活性化する工程をさらに含む方法により、遺伝子編集のされた組織幹細胞を製造する。そして、そのようにして製造された組織幹細胞を疾患の治療/予防に利用する。
Description
本発明は、遺伝子編集方法に関し、詳細には、組織幹細胞の遺伝子編集を短期間に行う方法に関する。
ゲノム編集は、部位特異的なヌクレアーゼを利用して、標的遺伝子を改変する技術である。ヌクレアーゼとしては、ZFN、TALEN、CRISPR/Cas9などが用いられている。従来の遺伝子工学、遺伝子治療と比較して、非常に応用範囲が広く、ゲノム編集技術を応用した治療法の開発も進められている(非特許文献1)。
例えば、CRISPR Therapeutics社(スイス)は、ゲノム編集を利用した鎌状赤血球貧血の治療法(CTX001)の開発を行っている(非特許文献2および3)。鎌状赤血球貧血は、酸素を運搬する赤血球内の分子であるヘモグロビンを作るための指示を与えるHBB遺伝子の変異によって引き起こされる。鎌状赤血球貧血では、この変異の結果、ヘモグロビンの欠損が生じ、赤血球の酸素運搬機能が低下する。
CTX001は、遺伝子編集技術を利用して、遺伝子を変化させ、患者の赤血球中の胎児ヘモグロビン(HbF)の産生を増加させることを狙っている。胎児ヘモグロビンは、新生児に天然に存在するヘモグロビンの一種で、後に成人型のヘモグロビンに置き換えられる。しかしながら、時として、胎児ヘモグロビンが成人においても存続することがあり、鎌状赤血球貧血やβ-サラセミアから人々を守っている。
治療のためには、血液を構成する赤血球および白血球を生じさせる骨髄由来の細胞である造血幹細胞が患者から採取され、それから、高レベルの胎児ヘモグロビンを産生することができるように遺伝子が改変される。より具体的には、HbFの転写リプレッサーであるBCL11A遺伝子を破壊する遺伝子改変が行われる。このような処理を施された細胞はそれから、患者の体に戻され、患者の体内で胎児ヘモグロビンを含有する赤血球を大量に産生し、それにより、疾患によって引き起こされるヘモグロビン欠乏症を克服できると考えられている。
しかしながら、血液・免疫・癌などの疾患治療においては、組織幹細胞への遺伝子編集の効率の問題、細胞選択の困難さなど多くの問題点が残っており、未だ、遺伝子編集技術の医療応用には不十分な点も多い。
Dunbar et al., Gene therapy comes of age. Science. 2018;359:175-184. DOI: 10.1126/science.aan4672
Antony et al., CRISPR/Cas9 system: A promising technology for the treatment of inherited and neoplastic hematological diseases, Adv Cell Gene Ther. 2018;1:e10. DOI: 10.1002/acg2.10
Lin MI, et al. CRISPR/Cas9 Genome Editing to Treat Sickle Cell Disease and B-Thalassemia: Re-Creating Genetic Variants to Upregulate Fetal Hemoglobin Appear Well-Tolerated, Effective and Durable. Blood. 2017;130:284.
本発明は、遺伝子編集のされた未分化状態の組織幹細胞を短期間の内に製造する方法を提供することを目的の一つとする。また、本発明は、遺伝子編集のされた未分化状態の組織幹細胞を用いた疾患の治療および/または予防方法を提供することも目的の一つとする。
本発明者らは、ゲノム編集技術を応用することにより、遺伝子編集のされた未分化状態の組織幹細胞を短期間の内に製造するための手法を開発した。本手法を用いることで、遺伝子編集のされた未分化状態の組織幹細胞を用いた疾患の治療および/または予防方法を提供することができる。本発明は、より具体的には以下の態様を包含する。
[態様1]
遺伝子編集のされた組織幹細胞の製造方法であって、組織幹細胞中の標的遺伝子の遺伝子配列を編集する工程、および遺伝子配列の編集された組織幹細胞を選別する工程を含む、方法。
[態様2]
標的遺伝子の遺伝子配列にタグを付加する工程、および/または組織幹細胞における標的遺伝子の遺伝子発現を活性化する工程をさらに含む、態様1記載の方法。
[態様3]
組織幹細胞が患者から単離された組織幹細胞である、態様1または2記載の方法。
[態様4]
遺伝子配列の編集がex vivoで行われる、態様1~3のいずれか記載の方法。
[態様5]
遺伝子配列の編集がCRISPR/Cas系またはTALEN系を用いて行われる、態様1~4のいずれか記載の方法。
[態様6]
標的遺伝子が組織幹細胞中では定常発現しない遺伝子である、態様1~5のいずれか記載の方法。
[態様7]
遺伝子発現の活性化がCRISPRa系またはTALENエフェクター系を用いて行われる、態様2~6のいずれか記載の方法。
[態様8]
遺伝子配列の編集された組織幹細胞を遺伝子配列に付加したタグを利用して選別する、態様2~7のいずれか記載の方法。
[態様9]
組織幹細胞中の標的遺伝子の遺伝子配列を編集する工程と、標的遺伝子の遺伝子配列にタグを付加する工程とが同時に行われる、態様2~8のいずれか記載の方法。
[態様10]
遺伝子配列の編集された組織幹細胞を該細胞を患者から単離してから24時間以内に選別する、態様1~9のいずれか記載の方法。
[態様11]
選別した組織幹細胞が未分化状態を維持している、態様1~10のいずれか記載の方法。
[態様12]
選別した組織幹細胞を増殖させる工程をさらに含む、態様1~11のいずれか記載の方法。
[態様13]
選別した組織幹細胞を患者に移植する工程をさらに含む、態様1~12のいずれか記載の方法。
[態様14]
遺伝子配列の編集された組織幹細胞が疾患の治療に用いるための細胞である、態様1~13のいずれか記載の方法。
[態様15]
疾患が血液疾患または免疫疾患である、態様14記載の方法。
[態様16]
血液疾患または免疫疾患が、ADA欠損症、X連鎖性重症複合免疫不全症(SCID)、その他のSCID、Wiskott-Aldrich症候群、慢性肉芽腫症、白血球粘着不全症、家族性血球貪食症候群、X連鎖性 Hyper IgM症候群、X連鎖性リンパ増殖性疾患、X連鎖性無γグロブリン血症、Hyper-IgE症候群、鎌状赤血球症、およびβ-サラセミアからなる群より選択される、態様15記載の方法。
[態様17]
標的遺伝子が、ADA、IL2RG、WAS、CYBB、INTGB2、UNC13D、CD40L、SAP/SH2D1A、BTK、STAT3、およびヘモグロビンからなる群より選択される、態様1~16のいずれか記載の方法。
[態様18]
標的遺伝子の遺伝子配列が編集された、生体から単離後48時間以内の造血幹細胞を含む、血液疾患または免疫疾患の治療剤。
[態様19]
血液疾患または免疫疾患が、ADA欠損症、X連鎖性重症複合免疫不全症(SCID)、その他のSCID、Wiskott-Aldrich症候群、慢性肉芽腫症、白血球粘着不全症、家族性血球貪食症候群、X連鎖性 Hyper IgM症候群、X連鎖性リンパ増殖性疾患、X連鎖性無γグロブリン血症、Hyper-IgE症候群、鎌状赤血球症、およびβ-サラセミアからなる群より選択される、態様18記載の治療剤。
[態様20]
標的遺伝子が、ADA、IL2RG、WAS、CYBB、INTGB2、UNC13D、CD40L、SAP/SH2D1A、BTK、STAT3、およびヘモグロビンからなる群より選択される、態様18または19記載の治療剤。
[態様21]
遺伝子編集のされた組織幹細胞の製造方法であって、
組織幹細胞中の標的遺伝子の遺伝子配列を編集する工程、
標的遺伝子の遺伝子配列にタグを付加する工程、
組織幹細胞における標的遺伝子の遺伝子発現を活性化する工程、および
遺伝子配列の編集された組織幹細胞を選別する工程
を含み、
組織幹細胞中の標的遺伝子の遺伝子配列を編集する工程と、標的遺伝子の遺伝子配列にタグを付加する工程とが、CRISPR/Cas系またはTALEN系を用いて同時に行われる、方法。
[態様22]
標的遺伝子が組織幹細胞中では定常発現しない遺伝子であり、
標的遺伝子の遺伝子配列の編集とタグの付加が行われた細胞においてのみ、標的遺伝子の遺伝子発現を活性化する工程によってタグで標識された目的タンパク質が産生し、該タグを用いて遺伝子配列の編集された組織幹細胞を選別する、態様21に記載の方法。
[態様23]
組織細胞が患者から単離された組織幹細胞であり、遺伝子配列の編集された組織幹細胞を該細胞を患者から単離してから48時間以内に選別する、態様21または22に記載の方法。
遺伝子編集のされた組織幹細胞の製造方法であって、組織幹細胞中の標的遺伝子の遺伝子配列を編集する工程、および遺伝子配列の編集された組織幹細胞を選別する工程を含む、方法。
[態様2]
標的遺伝子の遺伝子配列にタグを付加する工程、および/または組織幹細胞における標的遺伝子の遺伝子発現を活性化する工程をさらに含む、態様1記載の方法。
[態様3]
組織幹細胞が患者から単離された組織幹細胞である、態様1または2記載の方法。
[態様4]
遺伝子配列の編集がex vivoで行われる、態様1~3のいずれか記載の方法。
[態様5]
遺伝子配列の編集がCRISPR/Cas系またはTALEN系を用いて行われる、態様1~4のいずれか記載の方法。
[態様6]
標的遺伝子が組織幹細胞中では定常発現しない遺伝子である、態様1~5のいずれか記載の方法。
[態様7]
遺伝子発現の活性化がCRISPRa系またはTALENエフェクター系を用いて行われる、態様2~6のいずれか記載の方法。
[態様8]
遺伝子配列の編集された組織幹細胞を遺伝子配列に付加したタグを利用して選別する、態様2~7のいずれか記載の方法。
[態様9]
組織幹細胞中の標的遺伝子の遺伝子配列を編集する工程と、標的遺伝子の遺伝子配列にタグを付加する工程とが同時に行われる、態様2~8のいずれか記載の方法。
[態様10]
遺伝子配列の編集された組織幹細胞を該細胞を患者から単離してから24時間以内に選別する、態様1~9のいずれか記載の方法。
[態様11]
選別した組織幹細胞が未分化状態を維持している、態様1~10のいずれか記載の方法。
[態様12]
選別した組織幹細胞を増殖させる工程をさらに含む、態様1~11のいずれか記載の方法。
[態様13]
選別した組織幹細胞を患者に移植する工程をさらに含む、態様1~12のいずれか記載の方法。
[態様14]
遺伝子配列の編集された組織幹細胞が疾患の治療に用いるための細胞である、態様1~13のいずれか記載の方法。
[態様15]
疾患が血液疾患または免疫疾患である、態様14記載の方法。
[態様16]
血液疾患または免疫疾患が、ADA欠損症、X連鎖性重症複合免疫不全症(SCID)、その他のSCID、Wiskott-Aldrich症候群、慢性肉芽腫症、白血球粘着不全症、家族性血球貪食症候群、X連鎖性 Hyper IgM症候群、X連鎖性リンパ増殖性疾患、X連鎖性無γグロブリン血症、Hyper-IgE症候群、鎌状赤血球症、およびβ-サラセミアからなる群より選択される、態様15記載の方法。
[態様17]
標的遺伝子が、ADA、IL2RG、WAS、CYBB、INTGB2、UNC13D、CD40L、SAP/SH2D1A、BTK、STAT3、およびヘモグロビンからなる群より選択される、態様1~16のいずれか記載の方法。
[態様18]
標的遺伝子の遺伝子配列が編集された、生体から単離後48時間以内の造血幹細胞を含む、血液疾患または免疫疾患の治療剤。
[態様19]
血液疾患または免疫疾患が、ADA欠損症、X連鎖性重症複合免疫不全症(SCID)、その他のSCID、Wiskott-Aldrich症候群、慢性肉芽腫症、白血球粘着不全症、家族性血球貪食症候群、X連鎖性 Hyper IgM症候群、X連鎖性リンパ増殖性疾患、X連鎖性無γグロブリン血症、Hyper-IgE症候群、鎌状赤血球症、およびβ-サラセミアからなる群より選択される、態様18記載の治療剤。
[態様20]
標的遺伝子が、ADA、IL2RG、WAS、CYBB、INTGB2、UNC13D、CD40L、SAP/SH2D1A、BTK、STAT3、およびヘモグロビンからなる群より選択される、態様18または19記載の治療剤。
[態様21]
遺伝子編集のされた組織幹細胞の製造方法であって、
組織幹細胞中の標的遺伝子の遺伝子配列を編集する工程、
標的遺伝子の遺伝子配列にタグを付加する工程、
組織幹細胞における標的遺伝子の遺伝子発現を活性化する工程、および
遺伝子配列の編集された組織幹細胞を選別する工程
を含み、
組織幹細胞中の標的遺伝子の遺伝子配列を編集する工程と、標的遺伝子の遺伝子配列にタグを付加する工程とが、CRISPR/Cas系またはTALEN系を用いて同時に行われる、方法。
[態様22]
標的遺伝子が組織幹細胞中では定常発現しない遺伝子であり、
標的遺伝子の遺伝子配列の編集とタグの付加が行われた細胞においてのみ、標的遺伝子の遺伝子発現を活性化する工程によってタグで標識された目的タンパク質が産生し、該タグを用いて遺伝子配列の編集された組織幹細胞を選別する、態様21に記載の方法。
[態様23]
組織細胞が患者から単離された組織幹細胞であり、遺伝子配列の編集された組織幹細胞を該細胞を患者から単離してから48時間以内に選別する、態様21または22に記載の方法。
本発明によれば、遺伝子編集のされた未分化状態の組織幹細胞を短期間の内に製造することができる。そして、そのような組織幹細胞を用いて、これまで介入の困難であった疾患の治療および/または予防を行うことも可能となる。
本発明の一部の実施形態は、遺伝子編集のされた組織幹細胞を、短期間で一細胞レベルで正確に選択することが可能な遺伝子編集のされた組織幹細胞の製造方法に関する。本発明の方法は、組織幹細胞中の標的遺伝子の遺伝子配列を編集する工程、遺伝子配列の編集された組織幹細胞を選別する工程を含み、さらに場合により、標的遺伝子の遺伝子配列にタグを付加する工程、および/または組織幹細胞における標的遺伝子の遺伝子発現を活性化する工程を含む。各工程について、以下に詳細に説明する。
[組織幹細胞中の標的遺伝子の遺伝子配列を編集する工程]
ゲノム編集とはゲノムの標的部位を特異的に切断できる人工制限酵素を用いて任意のゲノム配列に変異(置換、挿入、欠失)を導入する技術である。制限酵素により標的部位の二重鎖切断が生じると、鋳型DNAの非存在下では、細胞の修復機構であるNHEJ(Non-Homologous End Joining)によるゲノム修復の際、末端同士の連結時のエラーにより、塩基対の欠損、挿入が生じる。その結果フレームシフト等により最終的に標的遺伝子がKnock-outされる。一方、鋳型DNAの存在下では修復機構であるHDR(Homology Dependent Repair)によって標的部位への鋳型配列の置換・挿入が生じる(Knock-in)。
ゲノム編集とはゲノムの標的部位を特異的に切断できる人工制限酵素を用いて任意のゲノム配列に変異(置換、挿入、欠失)を導入する技術である。制限酵素により標的部位の二重鎖切断が生じると、鋳型DNAの非存在下では、細胞の修復機構であるNHEJ(Non-Homologous End Joining)によるゲノム修復の際、末端同士の連結時のエラーにより、塩基対の欠損、挿入が生じる。その結果フレームシフト等により最終的に標的遺伝子がKnock-outされる。一方、鋳型DNAの存在下では修復機構であるHDR(Homology Dependent Repair)によって標的部位への鋳型配列の置換・挿入が生じる(Knock-in)。
本発明に係る方法において、遺伝子配列の編集は、遺伝子を破壊するノックアウト(Knock-out)または導入・改変を行いたい配列を置換・挿入するノックイン(Knock-in)のいずれでもあり得るが、好ましくは、所望の鋳型配列を用いたKnock-inであり、それにより、例えば、疾患等の原因となる配列変異が正常な配列または無害な配列に訂正される。
本発明に係る方法において、標的遺伝子の遺伝子配列の編集に利用できる主な編集ツール(Correction tool)としては、例えば、ZFN系(Zinc Finger Nucleases)、TALEN系(Transcription Activator Like Effector Nucleases)、及びCRISPR/Cas系(Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR Associated Protein 9)等が挙げられる。これらの系は、ゲノムのDNA二重鎖の切断を担うヌクレアーゼと、該ヌクレアーゼをゲノム上の標的部位に誘導するガイド分子と、場合により鋳型DNAと、から構成される、特異性の高いゲノム編集ツールである。鋳型DNAは一本鎖DNAであっても、二本鎖DNAであっても良い。
ZFN系(例えば、Kim, Y. G., et al., 1996, Proc. Natl. Acad. Sci. USA 93:1156-1160.を参照)は、簡単には、多くの転写因子のDNA結合ドメインに存在するDNA結合モチーフの一つであるZinc-Fingerと、FokIヌクレアーゼドメインという2つの機能ドメインで構成された人工キメラタンパク質であり、標的配列を挟み込む様に設計された一対のZFNsを用いてゲノム編集を行う。ZFN系では、DNA結合ドメインであるZinc-Fingerが標的遺伝子上の特異的な遺伝子配列を認識し、それによりDNA切断ドメインであるFokIヌクレアーゼが高い特異性で二重鎖切断を引き起こす。
TALEN系(例えば、Cermak T, et al., 2011, Nucleic Acids Res 39: e82を参照)も、ZFNと同様、任意の塩基配列を認識するDNA結合ドメイン(TALエフェクター(TALE))にFokIヌクレアーゼドメインを融合させた人工キメラタンパク質である。TALENのDNA結合ドメインは15~20塩基を認識し、FolIの標的配列となる14~20塩基のスペーサーを挟み込むようにセンス鎖、アンチセンス鎖それぞれに対して設計された一対のTALENsを用いて二重鎖切断を行う。TALEN系によれば、ZFNより特異的な標的配列の切断が可能となり、理論上任意のゲノム配列を標的とすることができる。TALEN系はオフターゲット変異の抑制という点でCRISPR/CAS系よりも優れているとも言われている。
CRISPR/CAS9を中心とするCRISPR/CAS系(例えばCong, L. et al., 2013, Science, 339, 819-823.を参照)は、前述のZFN及びTALENが標的配列の認識にタンパク質とDNAの相互作用を用いているのに対し、RNAとDNAの相補的結合を標的配列の認識に用いる。II型CRISPR/CAS系では、簡単には、guideRNA(crRNAとtracrRNAの複合体またはそれらを一本鎖化したsgRNA)によって、二重鎖切断を担うCas9ヌクレアーゼが標的配列に誘導され、CAS9-RNA複合体がguide RNAと相補的なDNA配列を切断する。V型CRISPR/CAS系では、RNA依存性DNAヌクレアーゼCpf1が用いられる。V型CRISPR/CAS系のguideRNAはcrRNAのみから構成される。CRISPR/Cas系は、guideRNAとヌクレアーゼとが別々に存在して機能するため、複数のguideRNAの導入による多重変異の導入が可能であるという点で利便性に優れる。例えば、Integrated DNA Technologies(IDT)社から、CRISPR/CAS系において利用されるコンポーネント(Cas9 gRNA、Cas9タンパク質、Cas9プラスミド、Cas9 キャリアDNA、バッファー等)およびCRISPR/Cas12a系において利用されるコンポーネント(Cas12a(Cpf1)gRNA、Cas12a(Cpf1)タンパク質、Cas12a(Cpf1)キャリアDNA、バッファー等)を購入することができる。sgRNAの設計ツールとしては、例えばCRISPRdirect(http://crispr.dbcls.jp)を用いることができる。RNAの合成及び精製のためには、例えば、mMessage mMachine(商標) T7 UTRA Transcription Kit(Ambion, AM1345)、MEGAclear(商標) Transcription Clean-Up Kit(Ambion, AM1908)等を用いることができる。
例えば、Alt-RTM(IDT社)を用いてguideRNAを作成する場合、標的配列を含むAlt-RTM crRNA(100μM、IDT社)、tracrRNA(100μM、IDT社、1072532)、Nuclease-free Duplex Buffer(IDT社、11-01-03-01)をそれぞれ3.58μl、3.58μl、7.84μlで混和し、95℃下で5分おき、室温で冷却して使用することができる。gRNA vector(Addgene No.41824)を用いたsgRNAの作成においては、インバースPCR法により標的配列を導入し、gRNA鋳型DNAを作成する。mMessage mMachine(商標) T7 UTRA Transcription Kit(Ambion, AM1345)にてRNAを合成し、MEGAclear(商標) Transcription Clean-Up Kit(Ambion, AM1908)を用いて精製し、100μMとすることができる。組換えタンパク質としてヌクレアーゼを用いる場合、SpCas9(IDT社、1074182)、cpf1(IDT社、1081069)を10mg/mlに希釈し、使用することができる。RNAとしてヌクレアーゼを用いる場合は、例えば、文献(Nishimasu H, et al. Science. 2018;361:1259-1262. Esvelt KM, et al. Nat Methods. 2013;10(11):1116-1121.)記載の方法を参考として、ベクター上のSpCas9、SpCas-9-NG、dCas9、M-NMn-VP64(Addgene # 80425、#41816、#48676)を鋳型DNAとしてmMessage mMachine(商標) T7 UTRA Transcription Kit(Ambion, AM1345)にてRNAを合成し、エタノール沈殿法により精製することができる。
以上、ZFN系、TALEN系、及びCRISPR/Cas系について簡単に記載したが、これらの系の改良型や、その他の任意のゲノム編集ツールも本発明に用いることができる。特異性の観点では、遺伝子配列の編集は、CRISPR/Cas系またはTALEN系を用いて行われることが好ましい。
これらのゲノム編集ツールを用いる際には、ガイド分子-ヌクレアーゼ複合体、又はguide RNA及び/若しくはヌクレアーゼの発現プラスミド又は該発現プラスミドがパッケージされたウイルスを標的細胞に導入して、標的細胞内でガイド分子-ヌクレアーゼ複合体、又はguide RNA及び/若しくはヌクレアーゼを発現してもよいし、あるいはヌクレアーゼ又はガイド分子-ヌクレアーゼ複合体をmRNAまたはタンパク質として、またguide RNAをRNAとして、標的細胞にそれぞれ導入してもよい。発現プラスミド等を導入する場合は発現を誘導する作用物質を標的細胞に作用させてもよい。プラスミド導入によりヌクレアーゼを発現し続けることによるオフターゲットリスクや、転写・翻訳にかかるタイムラグ、変異導入の効率化の観点では、ヌクレアーゼ又はガイド分子-ヌクレアーゼ複合体をmRNAまたはタンパク質として、またguide RNAをRNAとして導入することが好ましい。これらのRNA、タンパク質、プラスミド、又はパッケージウイルスの導入方法は特に限定されず当業者に知られている方法を用いることができるが、非限定的な例としては、リン酸カルシウム沈殿、リポフェクション、ポリマーカプセル、微粒子銃、マイクロインジェクション、エレクトロポレーション等が挙げられる。簡便性と導入効率のバランスから、エレクトロポレーションが一般的である。
細胞にRNA、タンパク質、DNAを導入するためのエレクトロポレーションは例えば、Nepa21 pulse generator(Nepa Gene)を用いて行うことができる。送電は、例えば、2つの矩形電気パルス(225V、2ms幅、50ms間隔)、続いて5パルス(30V、50ms幅、50ms間隔)を行う。エレクトロポレーションを行なった混合液に2mlのHBSS+培地を加え、800rpmで3分間遠心し洗浄することができる。この細胞を造血幹細胞採取の際に用いた培地を用いて24時間程度培養してもよい。更に、GFPシグナルなどの一細胞レベルでの検出によって、遺伝子編集が的確に行われた細胞を選別することができる。
組織幹細胞を取り扱う際には、滅菌下でRNA及びタンパク質を用いるのに適した作業スペース、動物実験施設としてSPF区を有していることが望ましい。また、変異が導入された細胞を効率的に抽出するための手段としては、発光性タグを検出して、選別するための機器が好適に利用され得る。
[遺伝子配列の編集された組織幹細胞を選別する工程]
本発明に係る方法は、標的細胞における標的遺伝子の編集後、組織幹細胞のプールから標的遺伝子が編集されている細胞を一細胞レベルで選別する工程を含む。標的遺伝子が編集されている細胞を選別する方法としては、編集された標的遺伝子の核酸配列に基づく方法や編集された標的遺伝子の発現に基づく方法が挙げられる。編集された標的遺伝子を正常に発現可能な細胞を一細胞レベルで正確に選択する観点では、標的遺伝子の発現に基づく方法、例えば一過性に活性化された標的遺伝子の発現を検出することにより行うことが好ましい。活性化された標的遺伝子の発現の検出には、例えば蛍光または燐光の検出を利用する系を用いることができる。
本発明に係る方法は、標的細胞における標的遺伝子の編集後、組織幹細胞のプールから標的遺伝子が編集されている細胞を一細胞レベルで選別する工程を含む。標的遺伝子が編集されている細胞を選別する方法としては、編集された標的遺伝子の核酸配列に基づく方法や編集された標的遺伝子の発現に基づく方法が挙げられる。編集された標的遺伝子を正常に発現可能な細胞を一細胞レベルで正確に選択する観点では、標的遺伝子の発現に基づく方法、例えば一過性に活性化された標的遺伝子の発現を検出することにより行うことが好ましい。活性化された標的遺伝子の発現の検出には、例えば蛍光または燐光の検出を利用する系を用いることができる。
[組織幹細胞における標的遺伝子の遺伝子発現を活性化する工程]
標的遺伝子は、多くの場合、組織幹細胞が対応する各組織細胞に分化して様々な機能を発揮する際に発現する遺伝子である。すなわち、多くの場合、標的遺伝子は組織幹細胞中では定常発現しない遺伝子である。このため、標的遺伝子の発現に基づき標的遺伝子の編集された細胞を選択するためには、標的遺伝子の発現を一過的に活性化することが求められる。したがって、一実施形態では、本発明の方法は、組織幹細胞における標的遺伝子の遺伝子発現を活性化する工程をさらに含み得る。このような遺伝子発現の活性化は、一過的なものであり得る。この工程により、標的遺伝子が正常に発現し得るように編集されている細胞のみを一細胞レベルで選別することができる。この工程により、上記遺伝子配列を編集する工程において意図しない編集、例えばオフターゲット変異等が生じた細胞を除外することもできる。
標的遺伝子は、多くの場合、組織幹細胞が対応する各組織細胞に分化して様々な機能を発揮する際に発現する遺伝子である。すなわち、多くの場合、標的遺伝子は組織幹細胞中では定常発現しない遺伝子である。このため、標的遺伝子の発現に基づき標的遺伝子の編集された細胞を選択するためには、標的遺伝子の発現を一過的に活性化することが求められる。したがって、一実施形態では、本発明の方法は、組織幹細胞における標的遺伝子の遺伝子発現を活性化する工程をさらに含み得る。このような遺伝子発現の活性化は、一過的なものであり得る。この工程により、標的遺伝子が正常に発現し得るように編集されている細胞のみを一細胞レベルで選別することができる。この工程により、上記遺伝子配列を編集する工程において意図しない編集、例えばオフターゲット変異等が生じた細胞を除外することもできる。
標的遺伝子を一過的に活性化する活性化ツール(activation tool)としては、例えば、標的遺伝子の上流に存在するプロモーター領域に転写活性化因子(VP64など)を作用させる方法が挙げられる。一実施形態では、上述の標的遺伝子を編集する工程、又は後述の標的遺伝子をタグ化する工程と実質的に同時に行うことが可能な方法が好ましい。
このような活性化ツールとしては、CRISPRa(CRISPR activation)系、TALENエフェクター系が挙げられる。
CRISPRa系(例えば、Silvana Konermann et al. Nature. 2015 Jan 29;6(7536):583-588を参照)は、簡単には、DNA切断活性を欠失させたdeadCas9(dCas9)ヌクレアーゼと転写活性因子(VP16、VP64等)を融合させたdCas9-転写活性因子複合体と、標的遺伝子のプロモーター部位を標的とするguide RNAとを導入することにより、標的遺伝子の転写開始点にdCas9-転写活性因子複合体を誘導し、標的遺伝子の発現を活性化させるシステムである。CRISPRa系としては、dCas9-転写活性因子複合体及びguide RNAの発現ベクター、又は該発現ベクターをパッケージしたウイルス粒子等を標的細胞に導入してもよいし、dCas9-転写活性因子複合体をタンパク質として、guide RNAをRNAとしてそれぞれ標的細胞に導入してもよいが、本実施形態では、迅速な活性化と安全性の観点で、dCas9-転写活性因子複合体をタンパク質として、guide RNAをRNAとしてそれぞれ標的細胞に導入することがより好ましい。
また、標的遺伝子のプロモーター領域を標的とするTAL DNA結合ドメインを転写活性因子(VP16、VP64等)に融合させたTALエフェクター系等を用いることも好ましい。TALエフェクター系の設計ツールとしては、例えば http://tale-nt.cac.cornell.edu/node/add/single-tale を用いることができる。例えば、文献(Matsubara,Y, et al. Sci Rep, 4 : 5043, 2014)記載の方法を参考に、TALエフェクターをTAL Effector Nucleotide Targeter 2.0(https://tale-nt.cac.cornell.edu/node/add/single-tale)を参考にして、Golden Gate法によって哺乳類発現ベクター(pcDNA-TAL-VP-64(Addgene、47107))に導入し作成する。CRISPRa系を活性化ツールとして用いる場合、上記編集ツールと同様の方法により設計することができる。
これらの活性化ツールは、上記編集ツールと同様の手法で標的細胞に導入することができる。
[標的遺伝子の遺伝子配列にタグを付加する工程]
標的遺伝子の発現を検出する方法は、標的遺伝子の特性によって適宜選択することができ、例えば膜タンパク質に反応性の抗体を用いた方法、分泌タンパク質をその酵素活性により検出する方法等が挙げられる。
標的遺伝子の発現を検出する方法は、標的遺伝子の特性によって適宜選択することができ、例えば膜タンパク質に反応性の抗体を用いた方法、分泌タンパク質をその酵素活性により検出する方法等が挙げられる。
一実施形態では、遺伝子配列の編集された組織幹細胞を遺伝子配列に付加したタグを利用して選別することができる。例えば、標的遺伝子に、標的遺伝子が目的とする正常遺伝子に編集された場合にのみ発現するタグ配列を付加することにより、標的遺伝子を正常に発現する細胞のみを一細胞レベルで正確に選択することができる。このようなタグ配列としては、特に限定されるものでないが、ペプチドもしくはポリペプチドタグ(FLAG、HA、His、Myc、V5、S、Trx等)、レポーター遺伝子(Luciferase等)、蛍光タンパク質(GFP、RFP、Venus等)等が挙げられる。Venus配列は、文献(Matsubara,Y, et al. Sci Rep, 4 : 5043, 2014)記載の方法を参考とし、pVenus vectorに標的遺伝子の相同遺伝子を導入することで作成し、鋳型DNAとして使用することができる。このようなタグを、標識抗体や蛍光もしくは発光等を用いて検出することができる。本発明では、生細胞検出や変異導入効率の観点から、細胞へのダメージの少ないペプチド等の小分子(FLAG、HA等)や後述のHiBiTシステム(Promega)等が好ましい。
一実施形態において、標的細胞における不測の影響を抑える観点では、タグがペプチド等、なるべく小分子であることが好ましい場合がある。このようなタグの非限定的な例としては、HiBiTシステム(Promega)等が挙げられる。このシステムは、タグとして11アミノ酸のペプチドタグ(HiBiT)を用い、検出に該タグに結合するルシフェラーゼ断片(LgBiT)を用いるため、標的細胞における影響が最小限であり、かつ、標的細胞を高感度に検出することができる。
このようなタグ配列を標的遺伝子配列に付加するためのタグ化ツール(Tagging tool)は特に限定されず、例えば、上述の編集ツールと同じであってもよい。一実施形態では、タグ化ツールは、標的遺伝子を標的とするguide RNA、Casヌクレアーゼ、及びタグ配列挿入用鋳型DNAを含む。このようなタグ化ツールも、上記編集ツール及び活性化ツールと同様の手法で標的細胞に導入することができる。なお、タグを導入する位置は、標的遺伝子のN末端、C末端、中央部分の任意の部位であり得る。標的遺伝子のインタクトなタンパク質の発現を検出し得るという観点ではC末端側に導入することも好ましい。
[ACTゲノム編集]
一部の実施形態では、上記の組織幹細胞中の標的遺伝子の遺伝子配列を編集する工程と、組織幹細胞における標的遺伝子の遺伝子発現を活性化する工程と、標的遺伝子の遺伝子配列にタグを付加する工程とを組み合わせて行う。本明細書において「組み合わせて行う」とは、標的の組織幹細胞を体外で維持し得る短期間の間に編集から選択までの工程を行うことを意味する。
一部の実施形態では、上記の組織幹細胞中の標的遺伝子の遺伝子配列を編集する工程と、組織幹細胞における標的遺伝子の遺伝子発現を活性化する工程と、標的遺伝子の遺伝子配列にタグを付加する工程とを組み合わせて行う。本明細書において「組み合わせて行う」とは、標的の組織幹細胞を体外で維持し得る短期間の間に編集から選択までの工程を行うことを意味する。
このために、一実施形態では、活性化ツール(activation tool)と、編集ツール(correction tool)と、タグ化ツール(tagging tool)とを実質的に同時に標的細胞に導入することができる。このように活性化ツール(activation tool)、編集ツール(correction tool)およびタグ化ツール(tagging tool)を組み合わせたゲノム編集技術を本願では「ACTゲノム編集」と呼ぶ。ACTゲノム編集では、例えば、標的遺伝子の編集部位を標的とするZFN、TALEN、またはCRISPR/Cas系の編集ツールと、標的遺伝子の翻訳領域(C末端側もしくはN末端、または翻訳領域内)を標的とするZFN、TALEN、またはCRISPR/Cas系のタグ化ツールと、標的遺伝子のプロモーター部位を標的としたZFN、TALEN、またはCRISPR/Cas系の活性化ツールとを実質的に同時に標的細胞に導入する。ここで、編集ツールとタグ化ツールとは同じ系を使用してもよい。このようにすることにより、多重変異の導入(標的遺伝子の編集とタグ化)と、編集された標的遺伝子の発現とを極めて短時間に行うことができる。その結果、標的遺伝子が的確に編集された組織幹細胞を短期間の間に正確に選択することができる。
ここで「短期間」とは、標的の組織幹細胞を生体外で維持し得る期間であればよいが、好ましくは1週間以下、より好ましくは3日以下(72時間以下)、例えば2日以下(48時間以下、36時間以下、24時間以下、または18時間以下)である。また、「実質的に同時」とは数時間以内(例えば2もしくは3時間以内)を意味し、作業の簡便さを考慮すれば活性化ツールと編集ツールとタグ化ツールを同時が導入することが好ましいが、遺伝子編集・遺伝子発現の効率・正確性を向上させるために導入する順番を変更したり、時間をずらしたりしてもよい。例えば、活性化ツールと編集ツールとタグ化ツールに用いられる全てのコンポーネントを1回のエレクトロポレーションで細胞に導入してもよいし、編集ツールとタグ化ツールに用いられるコンポーネントを導入した後に時間を空けて活性化ツールに用いられるコンポーネントを導入してもよい。
従来のゲノム編集技術では、CRISPR/Cas系等を用いて遺伝子編集を行った場合であっても、遺伝子編集後に一細胞クローニングを行い、各クローンについてPCR等によりジェノタイピングを行って目的の遺伝子が編集された細胞を選別する必要があり、目的の細胞を得るまで数日から数週間程度の期間が必要であった。また、目的の遺伝子内に薬剤耐性遺伝子等を挿入すれば薬剤耐性株の選別により簡便に目的細胞を取得できるが、薬剤耐性遺伝子等の外来DNAの挿入をゲノムDNAとドナーベクターとの相同組換えによって行う場合、一般的に極めて低効率である等の問題もあった。しかし本実施形態に係る方法によれば、編集・タグ化・活性化を実質的に同時に行うことにより、目的の遺伝子が編集された細胞のみを、組織幹細胞を生体外で維持できる期間内に極めて短時間で高効率に選別することが可能となる。
一実施形態では、本発明の方法によれば、遺伝子配列の編集された組織幹細胞を該細胞を患者から単離してから2日以内、好ましくは24時間以内に選別することが可能である。
遺伝子編集のされた組織幹細胞を一細胞レベルで選択する方法としては、限界希釈法、セルソーター法等が挙げられるが、これらに限定されない。遺伝子配列の編集された組織幹細胞は、遺伝子配列に付加したタグを利用して選別されてもよい。一部の実施形態において、タグは蛍光または燐光を発する系の一部または全部を構成していてもよい。タグには、例えば、上述のように、プロメガ社(米国ウィスコンシン州)のHiBiTシステムを利用することができる。
[組織幹細胞]
本発明に係る方法において、遺伝子配列の編集は、通常単離した組織幹細胞においてex vivoで行われる。
本発明に係る方法において、遺伝子配列の編集は、通常単離した組織幹細胞においてex vivoで行われる。
組織幹細胞(組織特異的幹細胞または体性幹細胞とも呼ばれる)は細胞が由来する部位に基づき、例えば、皮膚系(表皮幹細胞、毛包幹細胞等)、消化器系(膵(共通)幹細胞、肝幹細胞等)、骨髄系(造血幹細胞、間葉系幹細胞(筋サテライト細胞等を含む)等)、神経系(神経幹細胞、網膜幹細胞等)等に分類することができる。本発明に係る方法を適用し得る組織幹細胞は特に限定されないが、疾患の病態、確立された移植方法、さらに移植細胞の組織定着性等の理由から、骨髄系、特に造血幹細胞において、特に有効である。
組織幹細胞は、由来する各組織の細胞から単離して取得しても、ES細胞、iPS細胞等のより未分化な多能性幹細胞から分化誘導して取得してもよいが、本発明に係る方法においては、安全性等の観点から、由来する各組織の細胞から単離することが好ましい。例えば、造血幹細胞は、骨髄、臍帯血、末梢血から単離して得ることができる。このような単離方法は特に限定されず、当業者に知られている方法を用いることができる。造血幹細胞の取得方法は、例えば、文献(Bak RO, et al. Nat Protoc. 2018;13(2):358-376、Forraz N, et al. Stem Cells. 2004;22(1):100-108.)記載の方法を参考とすることができる。具体的には、例えば、骨髄液を遠心(1500rpm、5分、4℃)により上精を除去し、赤血球溶解培地(Takara Bio、786-649)1mL と RPMI-1640培地(富士フィルム和光純薬、189-02025)9mlを加え、さらに遠心(1500rpm、5分、4℃)を行うことで赤血球を除去し、HBSS+培地により洗浄した細胞を、抗体を用いてlineage negative selectionにより選別する。Lineage negative selectionには、MagniSort(商標) Human Hematopoietic Lineage Depletion Kit(Thermofisher Scientific、8804-6836-74)を用いることができる。
マウス造血幹細胞の抽出は、例えば、文献(Ema H, et al. Nat Protoc.2006;1(6):2979-87、Gundry MC, et al. Cell Rep.2016;17:1453-61、Hetzel M, et al. Blood.2018;131(5):533-545)記載の方法を参考として行うことができる。具体的には、マウスの両側大腿骨と脛骨を採取し、6ml程度の2%ウシ胎児血清入りHBSS+培地(富士フィルム和光純薬、082-09365)を入れた6cmディッシュへ回収する。両側骨端を切離し、切断断片に25Gの針を入れて、HBSS+培地で押し出し、70μm nylonセルストレイナー(FALCON、352350)を通し細胞を回収する。遠心(1500rpm、5分、4℃)により上精を除去し、赤血球溶解培地(Takara Bio、786-649)1mLとRPMI-1640培地(富士フィルム和光純薬、189-02025)、10%ウシ胎児血清9mlを加え、さらに遠心(1500rpm、5分、4℃)を行うことで赤血球を除去する。HBSS+培地により洗浄した細胞を、抗体を用いてlineage negative selectionを行う。抗体は、1.0×107細胞当たり、以下の抗体を2μlずつ投与する(Biotin anti-mouse Ter-119/Erythroid Antibody(BioLegend、79748)、Biotin anti-mouse CD11b Antibody(BioLegend、79749)、Biotin anti-mouse Ly-6G/Ly-6C(Gr-1) Antibody(BioLegend、79750)、Biotin anti-mouse NK-1.1 Antibody(BioLegend、108703)、Biotin anti-mouse CD45R/B220 Antibody(BioLegend、79752)、Biotin anti-mouse CD127(IL-7Rα) Antibody(BioLegend、135005)、Biotin anti-mouse CD3 Antibody(BioLegend、79751))。氷上で60分抗体反応し、洗浄後、1.0×107細胞当たり、500μlの0.5% ウシ胎児血清+1% PBS溶液で懸濁し、50μlのDynabeads(商標) M-280 Streptavidin(Invitrogen、11205D)を追加する。さらに氷上で60分反応させ、磁気スタンドを用いて抗体の結合していない陰性細胞を回収する。HBSS+培地にて洗浄を行い、1×106細胞当たり100μlの培養液で懸濁し、37℃、5%CO2下で培養(1時間から24時間)する。培養液は、StemSpan(商標) SFEM培地(STEM CELL Technologies、09600)を用いて、L-グルタミン(富士フィルム和光純薬、073-05391)、マウス幹細胞因子(富士フィルム和光純薬、196-15581)、トロンボポエチン(R&D、488-TO-005)、インスリン様成長因子-2(コスモバイオ、100-14)、線維芽細胞成長因子(富士フィルム和光純薬、062-06041)をそれぞれ200mmol/L、100ng/ml、50μg/ml、100ng/ml、100ng/ml、1mMの最終濃度となる様に調整する。
ヒト造血幹細胞の抽出は、例えば、文献(Bak RO, et al. Nat Protoc. 2018;13(2):358-376、Forraz N, et al. Stem Cells. 2004;22(1):100-108.)記載の方法を参考として行うことができる。具体的には、例えば、ヒト骨髄液を遠心(1500rpm、5分、4℃)により上精を除去し、赤血球溶解培地(Takara Bio、786-649)1mLとRPMI-1640培地(富士フィルム和光純薬、189-02025)、10%ウシ胎児血清9mlを加え、さらに遠心(1500rpm、5分、4℃)を行うことで赤血球を除去する。HBSS+培地により洗浄した細胞を、抗体を用いてlineage negative selectionを行う。Lineage negative selectionは、MagniSort(商標) Human Hematopoietic Lineage Depletion Kit(Thermofisher Scientific、8804-6836-74)を用いる。HBSS+培地にて洗浄を行い、1×106細胞当たり100μlの培養液で懸濁し、37℃、5%CO2下で培養する(1時間から24時間)。培養液は、StemSpan(商標) SFEM II培地(StemCell Technologies、9655)を用いて、IL-6(PeproTech、200-06)、StemRegenin1(CellagenTech、C7710)、UM171(StemCell Technologies、72914)、Flt3L(PeproTech、300-19)、トロンボポエチン(PeproTech、300-18)、ヒト幹細胞因子(PeproTech、300-07)をそれぞれ100ng/ml、0.75μM、35nM、100ng/ml、100ng/ml、100ng/mlの最終濃度となる様に調整する。
特に、組織幹細胞は、患者から単離された組織幹細胞であることが好ましい。患者由来の組織幹細胞を用いることにより、本発明の方法により遺伝子の編集された組織幹細胞を患者の治療に用いる場合も、ドナーの有無や移植によるGVHDの問題を回避することができる。また、前処置が軽度であるため患者への負担が少ないという利点もある。
本発明に係る方法は、選別した組織幹細胞を必要により増殖させる工程をさらに含んでもよい。
組織幹細胞の培養・増殖には、細胞の未分化状態維持及び増殖のために当技術分野で知られている成分及び条件を用いることができる。例えば、基本培地(無機塩、炭水化物、ホルモン、必須アミノ酸、非必須アミノ酸、ビタミン、脂肪酸等を含む)としては、SFEM培地、SFEM II、D-MEM、MEM、RPMI 1640、BME、D-MEM/F-12、Glasgow MEM、ハンクス液、mTeSR1等が挙げられる。必要に応じて、培地に、幹細胞因子、塩基性線維芽細胞増殖因子(bFGF)、白血球遊走阻止因子(LIF)、インターロイキン、インスリン様成長因子、トランスフェリン、ヘパリン、ヘパラン硫酸、コラーゲン、フィブロネクチン、プロゲステロン、セレナイト、B27-サプリメント、N2-サプリメント、ITS-サプリメント、抗生物質等を添加してもよい。また、培地には、血清又は血漿を添加してもよい。各成分は、患者への移植に適合するグレードのものを選択することが好ましい。
一実施形態では、本発明に係る方法により選別し、必要により増殖させた遺伝子配列の編集された組織幹細胞を患者に移植することができる。特に、本発明によれば、患者から単離された組織幹細胞に遺伝子編集を施し極めて短時間で患者に移植することが可能となるため、様々な組織幹細胞、中でも肝細胞や骨格筋の内在性組織幹細胞(サテライト細胞)等の生体外で未分化な状態での維持が困難な細胞にも適用することができる。また、ストレスにより標的遺伝子発現量の増加が必要となる等、患者の状態変化に応答した迅速な治療が可能になり得る等の利点もある。
[治療剤]
本発明の一態様は、疾患の治療に用いるための標的遺伝子の遺伝子配列が編集された組織幹細胞を含む治療剤または予防剤にも関する。
本発明の一態様は、疾患の治療に用いるための標的遺伝子の遺伝子配列が編集された組織幹細胞を含む治療剤または予防剤にも関する。
本発明に係る治療・予防剤は、疾患の治療に有効な量の遺伝子配列の編集された組織幹細胞を含む。遺伝子配列の編集された組織幹細胞は特に、生体から単離後72時間以内、48時間以内、36時間以内、24時間、または18時間以内のものであり得る。治療効果を得るための組織幹細胞の量は、適切な治療応答を得るために、投与経路、投与形態、標的となる疾患、治療対象、治療標的部位等を考慮して、当業者が適宜設定することができる。
投与レジメンの非限定的な例としては、患者体重kgあたり1日用量1×105~2×106細胞を投与する。投与は、単回投与もしくは複数回投与であってもよく、または連続注入であってもよい。投与は、低用量から開始し、治療効果を考慮して標的用量まで漸増してもよい。また、当然ながら、治療対象の状態及び緊急性に応じて、上記の範囲外の量に変更してもよい。投与経路は、局所投与であっても全身投与であってもよく、例えば、静脈内、門脈内、筋肉内、腹腔内、標的組織内、皮下、若しくは皮内投与等の非経口送達が挙げられる。投与形態としては、注射剤、懸濁剤、点滴剤、医療用ハイドロゲル等の非経口剤形が挙げられる。
本実施形態に係る治療剤は、遺伝子配列の編集された組織幹細胞に加えて、任意の追加成分を含んでもよい。非限定的な追加成分の例としては、薬学的に許容可能な希釈剤、担体、そのほかの添加剤等、例えば、生理食塩水、等張液、緩衝剤、無痛化剤、安定剤、保存剤、酸化防止剤等が挙げられる。
本実施形態に係る治療剤は、単独で用いてもよいし、標的疾患の治療に有効な他の薬剤と組み合わせて用いてもよい。
本実施形態において、遺伝子配列の編集された組織幹細胞は、造血幹細胞、間葉系幹細胞、または肝幹細胞であり得る。
また、別の観点からは、本発明の一態様は、処置を必要とする患者に対して、標的遺伝子の遺伝子配列が編集された組織幹細胞を投与することを含む、疾患の治療方法または予防方法に関する。さらに、本発明の一態様は、疾患の治療方法または予防方法に用いるための医薬の製造における、遺伝子配列が編集された組織幹細胞の使用に関する。これらの組織幹細胞の特徴は上に述べたとおりである。
[標的となり得る疾患群]
本発明に係る方法を用いたゲノム編集技術は、組織幹細胞由来の細胞における標的遺伝子発現の調節が関与する疾患であれば、遺伝子疾患に限らず、幅広く適用することができる。本発明の方法は常染色体優性遺伝、常染色体劣性遺伝、X連鎖性劣性遺伝、X連鎖性優性遺伝を問わず、全ての遺伝形式に有効であり、特に、従来の遺伝子導入では治療効果が乏しい常染色体優性遺伝病への適用に適している。
本発明に係る方法を用いたゲノム編集技術は、組織幹細胞由来の細胞における標的遺伝子発現の調節が関与する疾患であれば、遺伝子疾患に限らず、幅広く適用することができる。本発明の方法は常染色体優性遺伝、常染色体劣性遺伝、X連鎖性劣性遺伝、X連鎖性優性遺伝を問わず、全ての遺伝形式に有効であり、特に、従来の遺伝子導入では治療効果が乏しい常染色体優性遺伝病への適用に適している。
目的遺伝子の補充・付加を行う従来の遺伝子治療においては、異常遺伝子が残ること、正常遺伝子の組み込み部位が制御不能であること、導入遺伝子の発現調節が困難であること等の課題があり、適用し得る疾患も限られていた。しかし、本発明の方法は、異常遺伝子や特定遺伝子の機能消失、異常遺伝子の変異の修復、がん化を生じない安全な部位への遺伝子導入、遺伝子の発現調節等が可能となる。さらに標的遺伝子が正常に編集された細胞を患者の生体内で安定的に維持できるため、治療効果の長期的な維持が期待される。
中でも、本発明の方法は、単一遺伝子疾患、特には欠失変異/フレームシフトや、それに準じる遺伝子変異に起因する疾患に特に有効であり得る。
本発明に係る方法または治療剤の標的となり得る疾患としては、
- 免疫不全症候群:例えば、ADA欠損症、X連鎖性重症複合免疫不全症(SCID)、その他のSCID、Wiskott-Aldrich症候群、慢性肉芽腫症、白血球粘着不全症、家族性血球貪食症候群、X連鎖性Hyper IgM症候群、X連鎖性リンパ増殖性疾患、X連鎖性無γグロブリン血症、Hyper-IgE症候群等
- 異常Hb症:例えば、鎌状赤血球症、β-サラセミア等
- 代謝疾患:例えば、ゴーシェ病、ムコ多糖症、X連鎖性副腎白質ジストロフィー、異染性白質ジストロフィー、大理石骨病等
- その他、ファンコニ貧血、Schwachman-Diamond症候群、Kostmann症候群
等が挙げられる(例えば、Richard A. Morgan et al., 2017, Cell Stem Cell, 21, 574-590を参照)。
- 免疫不全症候群:例えば、ADA欠損症、X連鎖性重症複合免疫不全症(SCID)、その他のSCID、Wiskott-Aldrich症候群、慢性肉芽腫症、白血球粘着不全症、家族性血球貪食症候群、X連鎖性Hyper IgM症候群、X連鎖性リンパ増殖性疾患、X連鎖性無γグロブリン血症、Hyper-IgE症候群等
- 異常Hb症:例えば、鎌状赤血球症、β-サラセミア等
- 代謝疾患:例えば、ゴーシェ病、ムコ多糖症、X連鎖性副腎白質ジストロフィー、異染性白質ジストロフィー、大理石骨病等
- その他、ファンコニ貧血、Schwachman-Diamond症候群、Kostmann症候群
等が挙げられる(例えば、Richard A. Morgan et al., 2017, Cell Stem Cell, 21, 574-590を参照)。
本発明に係る方法または治療剤の標的となり得る疾患及びその標的遺伝子の一例を下記表1に記載する。

なかでも、本発明に係る方法は、極めて短期間で遺伝子編集のされた組織幹細胞を得ることができるため、長期間培養によって増殖能の高い細胞が濃縮された非生理的な細胞集団となる可能性や、分化が進むことによって定着性が低下する可能性、培養による細菌・ウイルス・マイコプラズマ増殖の可能性をそれぞれ減ずることで、より効率的かつ安全に変異導入細胞の組織定着率を上昇させることが期待される。これらの理由から、現在まで治療法が確立されていない血液疾患・免疫疾患、例えば、長期間生着が有用と考えられる慢性肉芽腫症やWiskott-Aldrich症候群、さらにはHyper-IgE症候群のようにドミナントネガティブによる発症をきたす疾患に適用することが好ましい。
以下に、本発明を実施例に基づいて具体的に説明するが、本発明はこれらの実施例に何ら限定されるものではない。
例1:マウス造血幹細胞への遺伝子変異導入
野生型マウス大腿骨より抽出したマウス造血幹細胞に対して、CRISPR/Cas9系を用いて標的遺伝子部位への変異導入を行った。マウス造血幹細胞の採取は、文献(Ema H, et al. Nat Protoc.2006;1(6):2979-87、Gundry MC, et al. Cell Rep.2016;17:1453-61、Hetzel M, et al. Blood.2018;131(5):533-545)記載の方法を参考にして行った。具体的には、マウスの両側大腿骨と脛骨を採取し、6ml程度の2%ウシ胎児血清入りHBSS+培地(富士フィルム和光純薬、082-09365)を入れた6cmディッシュへ回収した。両側骨端を切離し、切断断片に25Gの針を入れて、HBSS+培地で押し出し、70μm nylonセルストレイナー(FALCON、352350)を通し細胞を回収した。遠心(1500rpm、5分、4℃)により上清を除去し、赤血球溶解培地(Takara Bio、786-649)1mLとRPMI-1640培地(富士フィルム和光純薬、189-02025)、10%ウシ胎児血清9mlを加え、さらに遠心(1500rpm、5分、4℃)を行うことで赤血球を除去した。HBSS+培地により洗浄した細胞に対し、抗体を用いてlineage negative selectionを行った。抗体は、1.0×107細胞当たり、以下の抗体を2μlずつ投与した(Biotin anti-mouse Ter-119/Erythroid Antibody(BioLegend、79748)、Biotin anti-mouse CD11b Antibody(BioLegend、79749)、Biotin anti-mouse Ly-6G/Ly-6C(Gr-1) Antibody(BioLegend、79750)、Biotin anti-mouse NK-1.1 Antibody(BioLegend、108703)、Biotin anti-mouse CD45R/B220 Antibody(BioLegend、79752)、Biotin anti-mouse CD127(IL-7Rα) Antibody(BioLegend、135005)、Biotin anti-mouse CD3 Antibody(BioLegend、79751))。氷上で60分抗体を反応させ、洗浄後、1.0×107細胞当たり、500μlの0.5%ウシ胎児血清+1% PBS溶液で懸濁し、50μlのDynabeads(商標) M-280 Streptavidin(Invitrogen、11205D)を追加した。さらに氷上で60分反応させ、磁気スタンドを用いて抗体の結合していない陰性細胞を回収した。
野生型マウス大腿骨より抽出したマウス造血幹細胞に対して、CRISPR/Cas9系を用いて標的遺伝子部位への変異導入を行った。マウス造血幹細胞の採取は、文献(Ema H, et al. Nat Protoc.2006;1(6):2979-87、Gundry MC, et al. Cell Rep.2016;17:1453-61、Hetzel M, et al. Blood.2018;131(5):533-545)記載の方法を参考にして行った。具体的には、マウスの両側大腿骨と脛骨を採取し、6ml程度の2%ウシ胎児血清入りHBSS+培地(富士フィルム和光純薬、082-09365)を入れた6cmディッシュへ回収した。両側骨端を切離し、切断断片に25Gの針を入れて、HBSS+培地で押し出し、70μm nylonセルストレイナー(FALCON、352350)を通し細胞を回収した。遠心(1500rpm、5分、4℃)により上清を除去し、赤血球溶解培地(Takara Bio、786-649)1mLとRPMI-1640培地(富士フィルム和光純薬、189-02025)、10%ウシ胎児血清9mlを加え、さらに遠心(1500rpm、5分、4℃)を行うことで赤血球を除去した。HBSS+培地により洗浄した細胞に対し、抗体を用いてlineage negative selectionを行った。抗体は、1.0×107細胞当たり、以下の抗体を2μlずつ投与した(Biotin anti-mouse Ter-119/Erythroid Antibody(BioLegend、79748)、Biotin anti-mouse CD11b Antibody(BioLegend、79749)、Biotin anti-mouse Ly-6G/Ly-6C(Gr-1) Antibody(BioLegend、79750)、Biotin anti-mouse NK-1.1 Antibody(BioLegend、108703)、Biotin anti-mouse CD45R/B220 Antibody(BioLegend、79752)、Biotin anti-mouse CD127(IL-7Rα) Antibody(BioLegend、135005)、Biotin anti-mouse CD3 Antibody(BioLegend、79751))。氷上で60分抗体を反応させ、洗浄後、1.0×107細胞当たり、500μlの0.5%ウシ胎児血清+1% PBS溶液で懸濁し、50μlのDynabeads(商標) M-280 Streptavidin(Invitrogen、11205D)を追加した。さらに氷上で60分反応させ、磁気スタンドを用いて抗体の結合していない陰性細胞を回収した。
標的遺伝子はIL2RGとし、sgRNA混合液、Cas9組換えタンパク質をエレクトロポレーションによって導入した。インターロイキン2受容体γ鎖(Il2rg)は、X連鎖重症複合免疫不全症(X-SCID)の原因遺伝子である。sgRNAは、Alt-RTM(IDT社)を用いて作成した。標的配列(aggattgatgttcaggcttc;配列番号1)を含むAlt-RTM crRNA(100μM、IDT社、カスタム合成品(https://sg.idtdna.com/jp/site/))、tracrRNA(100μM、IDT社、1072532)、Nuclease-free Duplex Buffer(IDT社、11-01-03-01)をそれぞれ3.58μl、3.58μl、7.84μlで混和し、95℃下で5分おき、室温で冷却した。Cas9組換えタンパク質には、SpCas9(IDT社、1074182)を10mg/mlに希釈して使用した。細胞培養とRNAおよびタンパク質の導入方法の詳細としては、sgRNAのRNA混合液(100μM)に、ヌクレアーゼ組換えタンパク質(10mg/mlでは3.69μl)を加え、室温で20分おいた。これに1.0×106細胞のマウス造血幹細胞と鋳型DNA(1mg/ml)を7.7μl混和し、OptiMEM培地(ThermoFisher Scientific、31985070)で希釈し、100μlとした。この細胞、RNA、タンパク質、オリゴDNA混合液に、Nepa21 pulse generator(Nepa Gene)を用いてエレクトロポレーションを行なった。送電は、2つの矩形電気パルス(225V、2ms幅、50ms間隔)、続いて5パルス(30V、50ms幅、50ms間隔)を行った。エレクトロポレーションを行なった混合液に2mlのHBSS+培地を加え、800rpmで3分遠心し洗浄した。
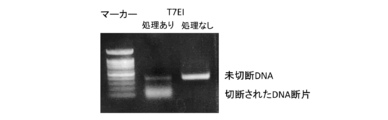
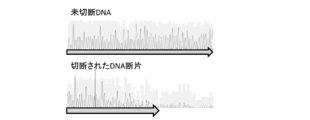
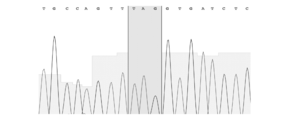
変異導入24時間後にゲノムDNAを抽出し(DNeasy Blood & Tissue Kit、Qiagen、69504)、標的部位を挟んだ500bp程度の長さのゲノムDNA配列についてPCR増幅を行なった(Quick Taq(登録商標) HS DyeMix、Toyobo、DTM-101)。Forwardプライマー(gagctatctgtctttaggcctggag;配列番号2)、Reverseプライマー(caacctggcctacatagtgagctc;配列番号3)を用いた。PCRの条件は、94℃ 2分の後、94℃ 30秒/60℃ 30秒/68℃ 30秒を30サイクル行なった。このPCR産物を精製し、NEBuffer2(New England BioLab、B7002)内で95℃ 5分処理して変性を行った。この後に室温にて緩徐にアニーリングを行うことで、変異導入部位が存在するところでDNAミスマッチが形成される。これを、ミスマッチを含むDNAのみを認識して切断するT7エンドヌクレアーゼI(T7EI)(New England BioLab、M0302S)で37℃ 15分処理することで、DNAミスマッチ切断を行った。図1はこのT7エンドヌクレアーゼIで処理したPCR産物を1%アガロースゲルにて電気泳動したものであり、標的部位でのDNAミスマッチの形成、即ち変異導入が得られたことを示している。図2は切断されたPCR産物のシークエンス結果を示す(FASMAC、Big Dye terminator v3.1、3130xl Genetic Analyzer)。シークエンス時のプライマーは塩基配列:gagctatctgtctttaggcctggag(配列番号4)を用いた。切断後のPCR産物(図2下)は切断前のPCR産物(図2上)と異なり、標的部位以降の配列が判読困難となっており、同部位で切断されていることを示している。
例2:標的遺伝子へのタグ配列の導入
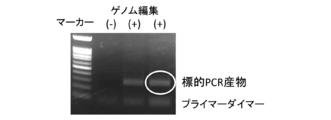
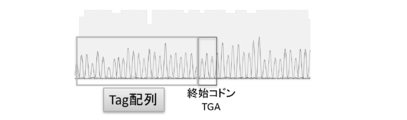
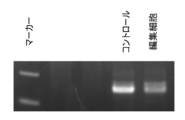
野生型マウス大腿骨より抽出したマウス造血幹細胞に対して、CRISPR/Cas9系を用いて標的遺伝子部位へのタグ配列変異導入を行った。標的遺伝子はIL2RGとし、標的配列(aggattgatgttcaggcttc;配列番号5)を含むsgRNA混合液、Cas9組換えタンパク質、タグ配列を導入するための鋳型DNA(tgcatagcccttactggcctcccccatgttattctctgaagccggaagccgtgagcggctggcggctgttcaagaagattagctgaacatcaatcctttgatggaacctcaaagtcctatagtcctaagtgac;配列番号6)をエレクトロポレーションによって導入した。変異導入24時間後にゲノムDNAを抽出し(DNeasy Blood & Tissue Kit、Qiagen、69504)、タグ配列内にForwardプライマー(gtgagcggctggcggctgtt;配列番号7)を、300bp程度下流にReverseプライマー(caacctggcctacatagtgagctc;配列番号8)を設計し、PCR増幅を行なった(Quick Taq(登録商標) HS DyeMix、Toyobo、DTM-101)。PCRの条件は、94℃ 2分の後、94℃ 30秒/60℃ 30秒/68℃ 30秒を30サイクル行なった。図3はこのPCR産物を1%アガロースゲルにて電気泳動したものである。図4はこのPCR産物のシークエンス結果を示す。シークエンス時のプライマーは塩基配列:caacctggcctacatagtgagctc(配列番号9)を用いた。これらは標的部位にタグ配列が導入されたことを示している。
野生型マウス大腿骨より抽出したマウス造血幹細胞に対して、CRISPR/Cas9系を用いて標的遺伝子部位へのタグ配列変異導入を行った。標的遺伝子はIL2RGとし、標的配列(aggattgatgttcaggcttc;配列番号5)を含むsgRNA混合液、Cas9組換えタンパク質、タグ配列を導入するための鋳型DNA(tgcatagcccttactggcctcccccatgttattctctgaagccggaagccgtgagcggctggcggctgttcaagaagattagctgaacatcaatcctttgatggaacctcaaagtcctatagtcctaagtgac;配列番号6)をエレクトロポレーションによって導入した。変異導入24時間後にゲノムDNAを抽出し(DNeasy Blood & Tissue Kit、Qiagen、69504)、タグ配列内にForwardプライマー(gtgagcggctggcggctgtt;配列番号7)を、300bp程度下流にReverseプライマー(caacctggcctacatagtgagctc;配列番号8)を設計し、PCR増幅を行なった(Quick Taq(登録商標) HS DyeMix、Toyobo、DTM-101)。PCRの条件は、94℃ 2分の後、94℃ 30秒/60℃ 30秒/68℃ 30秒を30サイクル行なった。図3はこのPCR産物を1%アガロースゲルにて電気泳動したものである。図4はこのPCR産物のシークエンス結果を示す。シークエンス時のプライマーは塩基配列:caacctggcctacatagtgagctc(配列番号9)を用いた。これらは標的部位にタグ配列が導入されたことを示している。
例3:ACTゲノム編集
Activationにおいては、文献(Matsubara,Y, et al. Sci Rep, 4 : 5043, 2014)記載の方法を参考に、TALエフェクターをTAL Effector Nucleotide Targeter 2.0(https://tale-nt.cac.cornell.edu/node/add/single-tale)を参考にして、Golden Gate法によって哺乳類発現ベクター(pcDNA-TAL-VP-64(Addgene、47107))に導入し作成する。
Activationにおいては、文献(Matsubara,Y, et al. Sci Rep, 4 : 5043, 2014)記載の方法を参考に、TALエフェクターをTAL Effector Nucleotide Targeter 2.0(https://tale-nt.cac.cornell.edu/node/add/single-tale)を参考にして、Golden Gate法によって哺乳類発現ベクター(pcDNA-TAL-VP-64(Addgene、47107))に導入し作成する。
CorrectionとTaggingにおいては、Alt-RTM(IDT社)を用いてsgRNAを作成する場合、標的配列を含むAlt-RTM crRNA(100μM、IDT社、カスタム合成品(https://sg.idtdna.com/jp/site/))、tracrRNA(100μM、IDT社、1072532)、Nuclease-free Duplex Buffer(IDT社、11-01-03-01)をそれぞれ3.58μl、3.58μl、7.84μlで混和し、95℃下で5分おき、室温で冷却する。
gRNA vector(Addgene No.41824)を用いたsgRNAの作成においては、インバースPCR法により標的配列を導入し、gRNA鋳型DNAを作成する。mMessage mMachine(商標) T7 UTRA Transcription Kit(Ambion, AM1345)にてRNAを合成し、MEGAclear(商標) Transcription Clean-Up Kit(Ambion, AM1908)を用いて精製し、100μMとする。組換えタンパク質としてヌクレアーゼを用いる場合、SpCas9(IDT社、1074182)、cpf1(IDT社、1081069)を10mg/mlに希釈し、使用する。
RNAとしてヌクレアーゼを用いる場合、文献(Nishimasu H, et al. Science. 2018;361:1259-1262. Esvelt KM, et al. Nat Methods. 2013;10(11):1116-1121.)記載の方法を参考として、ベクター上のSpCas9、SpCas-9-NG、dCas9、M-NMn-VP64(Addgene #80425、#41816、#48676)を鋳型DNAとしてmMessage mMachine(商標) T7 UTRA Transcription Kit(Ambion, AM1345)にてRNAを合成し、エタノール沈殿法により精製する。
次に、以上の、Activation、Correction、TaggingのRNAおよび組換えタンパク質のツールをエレクトロポレーションによって同時に細胞に導入することで、標的遺伝子の編集修復が行われた細胞を選別する。即ち、Correctionとして標的遺伝子の標的変異部位を標的としたsgRNAとSpCas9、及び鋳型DNAを用いて変異導入を行う。さらに、Taggingとしてタンパク質翻訳領域のC末端側を標的としたsgRNAとSpCas9あるいはSpCas9-NG及び鋳型DNAを用いて標識配列の導入を行なう。これらがうまく作動した場合に限り、標的遺伝子のプロモーター領域を標的としたsgRNAによりM-NMn-VP64あるいはTALエフェクターによって誘導された、標的遺伝子の活性化により標的遺伝子の変異部位が的確に編集され、タンパク質として産生できるようになった細胞が、標識配列のシグナルに基づき選別される。標識配列として、前述のHiBit配列、HA配列、GFP配列、またはVenus配列等を用いる。Venus配列は、文献(Matsubara,Y, et al. Sci Rep, 4 : 5043, 2014)記載の方法を参考とし、pVenus vectorに標的遺伝子の相同遺伝子を導入することで作成し、鋳型DNAとして使用する。
細胞培養とRNAおよびタンパク質の導入方法の詳細として、sgRNAのRNA混合液(100μM)に、ヌクレアーゼ(組換えタンパク質10mg/mlでは3.69μl、RNAでは250ng/μlで2μl)を加え、室温で20分おく。これに1.0×106細胞の組織幹細胞と鋳型DNA(1mg/ml)を7.7μl混和し、OptiMEM培地(ThermoFisher Scientific、31985070)で希釈し、100μlとする。この細胞、RNA、タンパク質、オリゴDNA混合液に、Nepa21 pulse generator(Nepa Gene)を用いてエレクトロポレーションを行なう。送電は、2つの矩形電気パルス(225V、2ms幅、50ms間隔)、続いて5パルス(30V、50ms幅、50ms間隔)を行う。エレクトロポレーションを行なった混合液に2mlのHBSS+培地を加え、800rpm 3分で遠心し洗浄する。この細胞を組織幹細胞採取の際に用いた培地を用いて24時間培養する。更に、GFPシグナルなどの一細胞レベルでの検出によって、遺伝子編集が的確に行われた細胞を選別する。
例4:ヒト胎児腎癌細胞への目的遺伝子変異導入(Correction)とタグ標識挿入(Tagging)
ヒト胎児腎癌細胞株(293FTcell細胞(ThermofisherScientific))に対して、CRISPR/Cas9系を用いて標的遺伝子部位への目的とする変異の導入とタグ配列としてHiBIT(Flag付加)配列の導入を行った。標的遺伝子はRBP(TruB1)とし、標的配列(CTGCGCTGTCTAGAGTCCCT;配列番号10、CAAAAGTATGGCCGCTTCTG:配列番号11)を含むsgRNA混合液と、Cas9組換えタンパク質、タグ配列を導入するための鋳型DNA(GACCAAGAGGAAAAAGCAGACTTTGAAAATTGGGCATGGAGGGACTCTAGTGAGCGCAGCCCGAGGAGTTCTGGTTGTTGGAATTGGAAGCGGAACAAAAA;配列番号12、CAGCGTGCACCTCCACGATGAAACAGGTCTGGGCTACAAAAGTATGGCCGCTTCTGTGAGCGGCTGGCGGCTGTTCAAGAAGATTAGCGACTACAAAGACCATGACGGTGATTATAAAGATCATGACATCGATTACAAGGATGACGATGACAAGGCCGCTTCTGAGGCGGCGGTGGTGTCTTCGCCGTCTTTGAAAACAG;配列番号13)をエレクトロポレーションによって導入した。
ヒト胎児腎癌細胞株(293FTcell細胞(ThermofisherScientific))に対して、CRISPR/Cas9系を用いて標的遺伝子部位への目的とする変異の導入とタグ配列としてHiBIT(Flag付加)配列の導入を行った。標的遺伝子はRBP(TruB1)とし、標的配列(CTGCGCTGTCTAGAGTCCCT;配列番号10、CAAAAGTATGGCCGCTTCTG:配列番号11)を含むsgRNA混合液と、Cas9組換えタンパク質、タグ配列を導入するための鋳型DNA(GACCAAGAGGAAAAAGCAGACTTTGAAAATTGGGCATGGAGGGACTCTAGTGAGCGCAGCCCGAGGAGTTCTGGTTGTTGGAATTGGAAGCGGAACAAAAA;配列番号12、CAGCGTGCACCTCCACGATGAAACAGGTCTGGGCTACAAAAGTATGGCCGCTTCTGTGAGCGGCTGGCGGCTGTTCAAGAAGATTAGCGACTACAAAGACCATGACGGTGATTATAAAGATCATGACATCGATTACAAGGATGACGATGACAAGGCCGCTTCTGAGGCGGCGGTGGTGTCTTCGCCGTCTTTGAAAACAG;配列番号13)をエレクトロポレーションによって導入した。
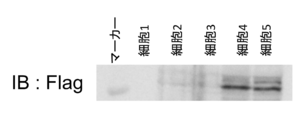
変異導入後、Nano-Glo(登録商標) HiBiT Lytic Detection System(Promega N3030)を用いてNano-Gloの発光を検出し、陽性となった細胞のみ培養を継続することで濃縮を行なった。これらの代表的な細胞からタンパク質を抽出(RIPA buffer、Nacalaitesk 16488-34)し、Flag抗体(MBL, FLA-1)を用いてウェスタンブロットを行なった。図5はFlag抗体によるウェスタンブロットの結果を示す。一部の細胞においてよくタグ配列が挿入され、ペプチドタグとして機能していることを示す。
この細胞からクローニングを行い、ゲノムDNAを抽出し(DNeasy Blood & Tissue Kit、Qiagen、69504)、変異導入部位上流200bp程度上流にForwardプライマー(GTTTTGAAAATGCCATCCCC;配列番号14)を、300bp程度下流にReverseプライマー(AGAAATAGCTACTTTTATGT;配列番号15)を設計し、PCR増幅を行なった(Quick Taq(登録商標) HS DyeMix、Toyobo、DTM-101)。PCRの条件は、94℃ 2分の後、94℃ 30秒/60℃ 30秒/68℃ 30秒を30サイクル行なった。図6はそのうちの一つのPCR産物のシークエンス結果を示す(FASMAC、Big Dye terminator v3.1、3130xl Genetic Analyzer)。シークエンス時のプライマーはForwardプライマー(配列番号14)を用いた。標的部位において目的の配列へ変異が導入されていることを示す。
例5:ヒト胎児腎癌細胞への目的遺伝子変異導入(Correction)とタグ標識挿入(Tagging)
ヒト胎児腎癌細胞株(293FTcell細胞(ThermofisherScientific))に対して、CRISPR/Cas9系を用いて標的遺伝子部位への目的とする変異の導入(exon2の除去)とタグ配列としてHiBIT配列の導入し、直後に終止コドンの導入を行った。標的遺伝子はRBP(TruB1)とし、標的配列(TCCCCTTTTCCTCCCAAGTT;配列番号16、TTTCTCTCATAGAAGCTGGA:配列番号17)の相補鎖を含むsgRNA混合液と、Cas9組換えタンパク質、タグ配列を導入するための鋳型DNA(CTAGTAATGAGGTCATAGTCTCTTAACATGTAAAGTTTGTATAATACTTTGTGAGCGGCTGGCGGCTGTTCAAGAAGATTAGCTTAATAGAAGCTGGAATGCCTTCTCCAGAATGGACCAAGAGGAAAAAGCAGAC;配列番号18)をエレクトロポレーションによって導入した。
ヒト胎児腎癌細胞株(293FTcell細胞(ThermofisherScientific))に対して、CRISPR/Cas9系を用いて標的遺伝子部位への目的とする変異の導入(exon2の除去)とタグ配列としてHiBIT配列の導入し、直後に終止コドンの導入を行った。標的遺伝子はRBP(TruB1)とし、標的配列(TCCCCTTTTCCTCCCAAGTT;配列番号16、TTTCTCTCATAGAAGCTGGA:配列番号17)の相補鎖を含むsgRNA混合液と、Cas9組換えタンパク質、タグ配列を導入するための鋳型DNA(CTAGTAATGAGGTCATAGTCTCTTAACATGTAAAGTTTGTATAATACTTTGTGAGCGGCTGGCGGCTGTTCAAGAAGATTAGCTTAATAGAAGCTGGAATGCCTTCTCCAGAATGGACCAAGAGGAAAAAGCAGAC;配列番号18)をエレクトロポレーションによって導入した。
変異導入後、Nano-Glo(登録商標) HiBiT Lytic Detection System(Promega N3030)を用いてNano-Gloの発光検出を行なった。図7はLytic Detectionの結果を示す。ノックインした細胞においてLuciferase活性が上昇していることを示している。
例6:ヒト胎児腎癌細胞への目的遺伝子変異導入(Correction)とタグ標識挿入(Tagging)と活性化(Activation)
ヒト胎児腎癌細胞株(293FTcell細胞(ThermofisherScientific))に対して、CRISPR/Cas9系を用いて標的遺伝子部位への目的とする変異の導入とタグ配列としてHiBIT配列の導入し、その後に活性化を行なった。標的遺伝子はSTAT3とし、はじめに変異型STAT3の細胞株を樹立した。この際、標的配列(GTTGTGGTGATCTCCAACAT;配列番号19)の相補鎖を含むsgRNA混合液と、Cas9組換えタンパク質、変異を導入するための鋳型DNA(CCCAGCTCAGTCCCCACTCCCTCCGCAGACCCACTCCTTGCCAGTTGTGTAGATCTCCAACATCTGTCAGATGCCAAATGCCTGGGCGTCCATCCTGTGGTA;配列番号20)をエレクトロポレーションによって導入した。
ヒト胎児腎癌細胞株(293FTcell細胞(ThermofisherScientific))に対して、CRISPR/Cas9系を用いて標的遺伝子部位への目的とする変異の導入とタグ配列としてHiBIT配列の導入し、その後に活性化を行なった。標的遺伝子はSTAT3とし、はじめに変異型STAT3の細胞株を樹立した。この際、標的配列(GTTGTGGTGATCTCCAACAT;配列番号19)の相補鎖を含むsgRNA混合液と、Cas9組換えタンパク質、変異を導入するための鋳型DNA(CCCAGCTCAGTCCCCACTCCCTCCGCAGACCCACTCCTTGCCAGTTGTGTAGATCTCCAACATCTGTCAGATGCCAAATGCCTGGGCGTCCATCCTGTGGTA;配列番号20)をエレクトロポレーションによって導入した。
変異導入後、限界希釈法によって変異導入された細胞をクローニングした。クローニングした変異導入細胞のゲノムDNAを抽出し、シークエンスによって変異導入を確認した。図8はこの際のシークエンス結果である。この際のPCRにおいては、Forwardプライマー(GCAGCAGGTGTGGTTTATGG;配列番号21)、Reverseプライマー(ACCATCCCTCATCTAAACAA;配列番号22)を用い、PCRの条件は、94℃ 2分の後、94℃ 30秒/60℃ 30秒/68℃ 30秒を30サイクル行なった。Forwardプライマーを用いてシークエンスを行なった。
ここで得られた変異細胞に対して、変異を元に戻すためのsgRNA(配列番号19と同様)とタグ配列付加のためのsgRNA(標的配列としてTGCGCTACCTCCCCCATGTG;配列番号23を含む)と、Cas9組換えタンパク質、変異を戻すための鋳型DNA(CCCAGCTCAGTCCCCACTCCCTCCGCAGACCCACTCCTTGCCAGTTGTGGTGATCTCCAACATCTGTCAGATGCCAAATGCCTGGGCGTCCATCCTGTGG;配列番号24)とタグ配列を導入するための鋳型DNA(CCCTCACCTTTGACATGGAGTTGACCTCGGAGTGCGCTACCTCCCCCATGGTGAGCGGCTGGCGGCTGTTCAAGAAGATTAGCTGAGGAGCTGAGAACGGAAGCTGCAGAAAGATACGACTGAGGCGCCTACC;配列番号25)をエレクトロポレーションによって導入した。変異導入後、STAT3遺伝子の活性化を行なった。STAT3遺伝子の転写開始因子より上流1000bp、500bp、200bpの部位をそれぞれ標的とするsgRNA(それぞれUUGUUGAGUAGGAGAAUCUC;配列番号26、UUUAAAAAAUGAGUGUGGCA;配列番号27、UCAAGGCCACCCUGGGCAAC;配列番号28)とdCas9-VP64 vector(addgene, #47107)から得られたmRNAをFugene HD Transfection reagent(プロメガ, E2311)を用いて導入し、24時間後にNano-Glo(登録商標) HiBiT Lytic Detection System(Promega N3030)を用いてNano-Gloの発光検出を行なった。mRNAは、dCas9-VP64 vectorを鋳型とし、mMESSAGE mMACHINE T7 Ultra(Invitrogen, AM1345)を用いてRNAを合成し、MEGA Clear Transcription clean kit(Invitrogen, AM1908)を用いて精製した。図9はLytic Detectionの結果を示す。活性化によってHiBITのLuciferase活性が上昇しており、変異導入がなされ、遺伝子の活性化によってその変化が強調されていることを示す。
さらに、この細胞をForwardプライマー(GCAGCAGGTGTGGTTTATGG;配列番号21)、Reverseプライマー(ACCATCCCTCATCTAAACAA;配列番号22)を用いてPCR法を行い、その産物を、Forwardプライマーを用いてシークエンスすることで、変異を戻す効率を調べた。図10はこの際のシークエンス結果である。目的配列に波形の重なりがあり、変異を戻した細胞が含まれていることを示す。
例7:マウス造血幹細胞の遺伝子変異修復とのタグ配列の導入
Prkdc遺伝子変異を有するC.B-17/Icr-SCIDマウス(日本クレア)の大腿骨より抽出したSCIDマウス造血幹細胞に対して、CRISPR/Cas9系を用いて標的遺伝子変異の修復と、標的遺伝子へのタグ配列導入を行った。標的遺伝子はPrkdcとし、変異を修復するめのsgRNA(gcuuagcguauuuuauguug;配列番号29を含む)とタグ配列付加のためのsgRNA(acaccacagacuuuacaucc;配列番号30を含む)と、Cas9組換えタンパク質、変異を修復するための鋳型DNA(gatcatggattcaagaaataaatgtaacggaaaagaattggtatccacaacataaaatacgctatgctaagagaaagttagcaggggccaacccagctgt;配列番号31)とタグ配列を導入するための鋳型DNA(cagaccccaatatccttggcaggacttgggaaggatgggagccctggatgCGGGACCACATGGTGCTGCACGAGTACGTGAACGCCGCCGGCATCACATAAtaaagtctgtggtgtcaccaatcataaagcattctgtctccgagaggacc;配列番号32)をエレクトロポレーションによって導入した。タグ配列として、GFP-11を用いた。
Prkdc遺伝子変異を有するC.B-17/Icr-SCIDマウス(日本クレア)の大腿骨より抽出したSCIDマウス造血幹細胞に対して、CRISPR/Cas9系を用いて標的遺伝子変異の修復と、標的遺伝子へのタグ配列導入を行った。標的遺伝子はPrkdcとし、変異を修復するめのsgRNA(gcuuagcguauuuuauguug;配列番号29を含む)とタグ配列付加のためのsgRNA(acaccacagacuuuacaucc;配列番号30を含む)と、Cas9組換えタンパク質、変異を修復するための鋳型DNA(gatcatggattcaagaaataaatgtaacggaaaagaattggtatccacaacataaaatacgctatgctaagagaaagttagcaggggccaacccagctgt;配列番号31)とタグ配列を導入するための鋳型DNA(cagaccccaatatccttggcaggacttgggaaggatgggagccctggatgCGGGACCACATGGTGCTGCACGAGTACGTGAACGCCGCCGGCATCACATAAtaaagtctgtggtgtcaccaatcataaagcattctgtctccgagaggacc;配列番号32)をエレクトロポレーションによって導入した。タグ配列として、GFP-11を用いた。
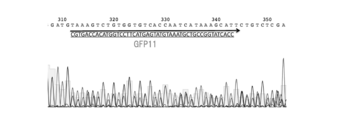
変異導入48時間後にゲノムDNAを抽出し(DNeasy Blood & Tissue Kit、Qiagen、69504)、Forwardプライマー(CAAACTTTGAATTCACAGTCATGAGTGAC;配列番号33)、Reverseプライマー(GAGGTCCTCTCGGAGACAGAATG;配列番号34)を用いてPCR増幅を行なった(Quick Taq(登録商標) HS DyeMix、Toyobo、DTM-101)。PCRの条件は、94℃ 2分の後、94℃ 30秒/60℃ 30秒/68℃ 30秒を30サイクル行なった。図11はこのPCR産物を1%アガロースゲルにて電気泳動したものである。図12はこのPCR産物のシークエンス結果を示す。シークエンス時のプライマーはForwardプライマー(配列番号33)を用いた。これらは標的部位に目的のタグ配列の重なりを認めており、約50%程度の効率でタグ配列が導入されたことを示している。
さらに、変異導入後48時間後の細胞に対して、アデノ随伴ウイルスを用いてGFP1-10を導入し、24時間後に細胞の観察を行った。アデノ随伴ウイルスの作成及び導入は、AAVpro(商標登録) Helper Free System (Takara, 6673)を用いてpAAV-CMV(Takara, 6673)へGFP1-10(配列番号35)を導入して使用した。図13はこの細胞の蛍光を示す。細胞内にGFP-11が導入され、AAV由来のGFP1-10と結合することでGFPの蛍光を得た細胞が存在することを示す。
本明細書には、本発明の好ましい実施態様を示してあるが、そのような実施態様が単に例示の目的で提供されていることは、当業者には明らかであり、当業者であれば、本発明から逸脱することなく、様々な変形、変更、置換を加えることが可能であろう。本明細書に記載されている発明の様々な代替的実施形態が、本発明を実施する際に使用されうることが理解されるべきである。また、本明細書中において参照している特許および特許出願書類を含む、全ての刊行物に記載の内容は、その引用によって、本明細書中に明記された内容と同様に取り込まれていると解釈すべきである。
本発明は、今まで治療法が十分なかった血液、免疫疾患などの根本的治療法を早期に可能にするものである。
Claims (23)
- 遺伝子編集のされた組織幹細胞の製造方法であって、
組織幹細胞中の標的遺伝子の遺伝子配列を編集する工程、および
遺伝子配列の編集された組織幹細胞を選別する工程
を含む、方法。 - 標的遺伝子の遺伝子配列にタグを付加する工程、および/または
組織幹細胞における標的遺伝子の遺伝子発現を活性化する工程
をさらに含む、請求項1記載の方法。 - 組織幹細胞が患者から単離された組織幹細胞である、請求項1または2記載の方法。
- 遺伝子配列の編集がex vivoで行われる、請求項1~3のいずれか一項記載の方法。
- 遺伝子配列の編集がCRISPR/Cas系またはTALEN系を用いて行われる、請求項1~4のいずれか一項記載の方法。
- 標的遺伝子が組織幹細胞中では定常発現しない遺伝子である、請求項1~5のいずれか一項記載の方法。
- 遺伝子発現の活性化がCRISPRa系またはTALENエフェクター系を用いて行われる、請求項2~6のいずれか一項記載の方法。
- 遺伝子配列の編集された組織幹細胞を遺伝子配列に付加したタグを利用して選別する、請求項2~7のいずれか一項記載の方法。
- 組織幹細胞中の標的遺伝子の遺伝子配列を編集する工程と、標的遺伝子の遺伝子配列にタグを付加する工程とが同時に行われる、請求項2~8のいずれか一項記載の方法。
- 遺伝子配列の編集された組織幹細胞を該細胞を患者から単離してから24時間以内に選別する、請求項1~9のいずれか一項記載の方法。
- 選別した組織幹細胞が未分化状態を維持している、請求項1~10のいずれか一項記載の方法。
- 選別した組織幹細胞を増殖させる工程をさらに含む、請求項1~11のいずれか一項記載の方法。
- 選別した組織幹細胞を患者に移植する工程をさらに含む、請求項1~12のいずれか一項記載の方法。
- 遺伝子配列の編集された組織幹細胞が疾患の治療に用いるための細胞である、請求項1~13のいずれか一項記載の方法。
- 疾患が血液疾患または免疫疾患である、請求項14記載の方法。
- 血液疾患または免疫疾患が、ADA欠損症、X連鎖性重症複合免疫不全症(SCID)、その他のSCID、Wiskott-Aldrich症候群、慢性肉芽腫症、白血球粘着不全症、家族性血球貪食症候群、X連鎖性 Hyper IgM症候群、X連鎖性リンパ増殖性疾患、X連鎖性無γグロブリン血症、Hyper-IgE症候群、鎌状赤血球症、およびβ-サラセミアからなる群より選択される、請求項15記載の方法。
- 標的遺伝子が、ADA、IL2RG、WAS、CYBB、INTGB2、UNC13D、CD40L、SAP/SH2D1A、BTK、STAT3、およびヘモグロビンからなる群より選択される、請求項1~16のいずれか一項記載の方法。
- 標的遺伝子の遺伝子配列が編集された、生体から単離後48時間以内の造血幹細胞を含む、血液疾患または免疫疾患の治療剤。
- 血液疾患または免疫疾患が、ADA欠損症、X連鎖性重症複合免疫不全症(SCID)、その他のSCID、Wiskott-Aldrich症候群、慢性肉芽腫症、白血球粘着不全症、家族性血球貪食症候群、X連鎖性 Hyper IgM症候群、X連鎖性リンパ増殖性疾患、X連鎖性無γグロブリン血症、Hyper-IgE症候群、鎌状赤血球症、およびβ-サラセミアからなる群より選択される、請求項18記載の治療剤。
- 標的遺伝子が、ADA、IL2RG、WAS、CYBB、INTGB2、UNC13D、CD40L、SAP/SH2D1A、BTK、STAT3、およびヘモグロビンからなる群より選択される、請求項18または19記載の治療剤。
- 遺伝子編集のされた組織幹細胞の製造方法であって、
組織幹細胞中の標的遺伝子の遺伝子配列を編集する工程、
標的遺伝子の遺伝子配列にタグを付加する工程、
組織幹細胞における標的遺伝子の遺伝子発現を活性化する工程、および
遺伝子配列の編集された組織幹細胞を選別する工程
を含み、
組織幹細胞中の標的遺伝子の遺伝子配列を編集する工程と、標的遺伝子の遺伝子配列にタグを付加する工程とが、CRISPR/Cas系またはTALEN系を用いて同時に行われる、方法。 - 標的遺伝子が組織幹細胞中では定常発現しない遺伝子であり、
標的遺伝子の遺伝子配列の編集とタグの付加が行われた細胞においてのみ、標的遺伝子の遺伝子発現を活性化する工程によってタグで標識された目的タンパク質が産生し、該タグを用いて遺伝子配列の編集された組織幹細胞を選別する、請求項21に記載の方法。 - 組織細胞が患者から単離された組織幹細胞であり、遺伝子配列の編集された組織幹細胞を該細胞を患者から単離してから48時間以内に選別する、請求項21または22に記載の方法。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US17/436,208 US20220177922A1 (en) | 2019-03-07 | 2020-03-09 | High-throughput gene-editing technique |
| JP2021503678A JPWO2020179931A1 (ja) | 2019-03-07 | 2020-03-09 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019-041177 | 2019-03-07 | ||
| JP2019041177 | 2019-03-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020179931A1 true WO2020179931A1 (ja) | 2020-09-10 |
Family
ID=72337498
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/009987 WO2020179931A1 (ja) | 2019-03-07 | 2020-03-09 | ハイスループット遺伝子編集技術 |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US20220177922A1 (ja) |
| JP (1) | JPWO2020179931A1 (ja) |
| WO (1) | WO2020179931A1 (ja) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016044416A1 (en) * | 2014-09-16 | 2016-03-24 | Sangamo Biosciences, Inc. | Methods and compositions for nuclease-mediated genome engineering and correction in hematopoietic stem cells |
| WO2018170239A1 (en) * | 2017-03-15 | 2018-09-20 | The Regents Of The University Of California | Methods of treating lysosomal disorders |
| WO2019239361A1 (en) * | 2018-06-14 | 2019-12-19 | Novartis Ag | Method for sequence insertion using crispr |
-
2020
- 2020-03-09 US US17/436,208 patent/US20220177922A1/en active Pending
- 2020-03-09 WO PCT/JP2020/009987 patent/WO2020179931A1/ja active Application Filing
- 2020-03-09 JP JP2021503678A patent/JPWO2020179931A1/ja active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016044416A1 (en) * | 2014-09-16 | 2016-03-24 | Sangamo Biosciences, Inc. | Methods and compositions for nuclease-mediated genome engineering and correction in hematopoietic stem cells |
| WO2018170239A1 (en) * | 2017-03-15 | 2018-09-20 | The Regents Of The University Of California | Methods of treating lysosomal disorders |
| WO2019239361A1 (en) * | 2018-06-14 | 2019-12-19 | Novartis Ag | Method for sequence insertion using crispr |
Non-Patent Citations (2)
| Title |
|---|
| KONERMANN, S. ET AL.: "Genome-scale transcriptional activation by an engineered CRISPR- Cas9 complex", NATURE, vol. 517, 2015, pages 583 - 588, XP055585957, DOI: 10.1038/nature14136 * |
| MADSEN, R. R. ET AL.: "Luminescent peptide tagging enables efficient screening for CRISPR-mediated knock-in in human induced pluripotent stem cells", WELLCOME OPEN RESEARCH, vol. 4, no. 37, 2019 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2020179931A1 (ja) | 2020-09-10 |
| US20220177922A1 (en) | 2022-06-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2021106611A (ja) | ヌクレアーゼ介在性遺伝子発現調節 | |
| JP2020530307A (ja) | 遺伝子治療のためのアデノ随伴ウイルスベクター | |
| JP2020518256A (ja) | Crispr/cpf1を用いる、t細胞における遺伝子編集のための組成物および方法 | |
| WO2019210042A1 (en) | Expression of foxp3 in edited cd34+ cells | |
| JP7198823B2 (ja) | 全身においてタンパク質を発現させるためのプラットフォームとしての遺伝子工学操作された造血幹細胞 | |
| JP7428712B2 (ja) | 低/最小操作による遺伝子改変細胞の製造 | |
| KR102338993B1 (ko) | 인위적으로 조작된 조작면역세포 | |
| US20210139935A1 (en) | Methods of manufacturing car-t cells | |
| US20210253652A1 (en) | Expression of human foxp3 in gene edited t cells | |
| JP2021524233A (ja) | 薬物耐性免疫細胞及びその用途 | |
| US20230081343A1 (en) | Crispr-based foxp3 gene engineered t cells and hematopoietic stem cell precursors to treat ipex syndrome patients | |
| JP2021522837A (ja) | 造血幹細胞移植のための組成物及び方法 | |
| JP2022519070A (ja) | 免疫療法の改善のための遺伝子調節組成物及び遺伝子調節方法 | |
| WO2020149395A1 (ja) | 栄養障害型表皮水疱症治療薬 | |
| WO2020179931A1 (ja) | ハイスループット遺伝子編集技術 | |
| JP7457302B2 (ja) | Hla遺伝子のrnaガイドゲノム編集を使用して関節リウマチを処置する方法 | |
| US20180066253A1 (en) | Methods and compositions for modifying endothelial cells | |
| CN115960204B (zh) | Kras_g12v突变抗原特异性tcr及其与cd8共表达重定向cd4 t细胞 | |
| WO2021172583A1 (ja) | 遺伝子改変巨核球、改変血小板及びそれらの製造方法 | |
| WO2024002279A1 (zh) | 免疫兼容型人多能干细胞、其制备方法及应用 | |
| WO2024059618A2 (en) | Immune cells having co-expressed tgfbr shrnas | |
| KR20230131816A (ko) | 저면역원성 줄기세포, 줄기세포로부터 분화되거나 유래된 저면역원성 세포및 이의 제조방법 | |
| Lee | Genome Editing with Crispr/Cas9 to Study and Treat Primary Immune Regulatory Disorders | |
| WO2023122099A2 (en) | Crispr-based gene editing to preserve splicing and expression of foxp3 isoforms 1 and 2 | |
| CN118056014A (zh) | 单碱基编辑修复hba2基因突变的方法及其应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20765623 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2021503678 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 20765623 Country of ref document: EP Kind code of ref document: A1 |