WO2020170997A1 - 硬化性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、半導体デバイス、及び、熱塩基発生剤 - Google Patents
硬化性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、半導体デバイス、及び、熱塩基発生剤 Download PDFInfo
- Publication number
- WO2020170997A1 WO2020170997A1 PCT/JP2020/005967 JP2020005967W WO2020170997A1 WO 2020170997 A1 WO2020170997 A1 WO 2020170997A1 JP 2020005967 W JP2020005967 W JP 2020005967W WO 2020170997 A1 WO2020170997 A1 WO 2020170997A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- curable resin
- resin composition
- formula
- cured film
- Prior art date
Links
- 239000011342 resin composition Substances 0.000 title claims abstract description 158
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 35
- 239000004065 semiconductor Substances 0.000 title claims abstract description 19
- 150000001875 compounds Chemical class 0.000 claims abstract description 211
- 239000002243 precursor Substances 0.000 claims abstract description 110
- 125000000962 organic group Chemical group 0.000 claims abstract description 59
- 229920000642 polymer Polymers 0.000 claims abstract description 50
- 125000000623 heterocyclic group Chemical group 0.000 claims abstract description 38
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 27
- 125000005647 linker group Chemical group 0.000 claims abstract description 14
- 239000010410 layer Substances 0.000 claims description 109
- -1 1,2-ethylene group Chemical group 0.000 claims description 92
- 125000004432 carbon atom Chemical group C* 0.000 claims description 69
- 229920001721 polyimide Polymers 0.000 claims description 58
- 239000004642 Polyimide Substances 0.000 claims description 56
- 238000010438 heat treatment Methods 0.000 claims description 48
- 229910052751 metal Inorganic materials 0.000 claims description 46
- 239000002184 metal Substances 0.000 claims description 46
- 125000000217 alkyl group Chemical group 0.000 claims description 34
- 125000003118 aryl group Chemical group 0.000 claims description 28
- 239000000203 mixture Substances 0.000 claims description 28
- 229920005989 resin Polymers 0.000 claims description 28
- 239000011347 resin Substances 0.000 claims description 28
- 239000000758 substrate Substances 0.000 claims description 24
- 229920002577 polybenzoxazole Polymers 0.000 claims description 18
- 239000003505 polymerization initiator Substances 0.000 claims description 12
- 239000011229 interlayer Substances 0.000 claims description 10
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 9
- 125000004429 atom Chemical group 0.000 claims description 8
- 125000002030 1,2-phenylene group Chemical group [H]C1=C([H])C([*:1])=C([*:2])C([H])=C1[H] 0.000 claims description 6
- 125000001183 hydrocarbyl group Chemical group 0.000 claims 1
- 239000010408 film Substances 0.000 description 116
- 239000002585 base Substances 0.000 description 76
- 238000000034 method Methods 0.000 description 47
- 239000002253 acid Substances 0.000 description 31
- 239000000463 material Substances 0.000 description 28
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 27
- 230000015572 biosynthetic process Effects 0.000 description 23
- 238000003786 synthesis reaction Methods 0.000 description 23
- 125000001931 aliphatic group Chemical group 0.000 description 21
- 239000003112 inhibitor Substances 0.000 description 21
- 239000002904 solvent Substances 0.000 description 21
- YEJRWHAVMIAJKC-UHFFFAOYSA-N 4-Butyrolactone Chemical compound O=C1CCCO1 YEJRWHAVMIAJKC-UHFFFAOYSA-N 0.000 description 20
- 239000007787 solid Substances 0.000 description 19
- 238000003860 storage Methods 0.000 description 19
- GTDPSWPPOUPBNX-UHFFFAOYSA-N ac1mqpva Chemical compound CC12C(=O)OC(=O)C1(C)C1(C)C2(C)C(=O)OC1=O GTDPSWPPOUPBNX-UHFFFAOYSA-N 0.000 description 18
- 239000007870 radical polymerization initiator Substances 0.000 description 18
- 229910052802 copper Inorganic materials 0.000 description 17
- 239000010949 copper Substances 0.000 description 17
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 17
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 16
- 150000004985 diamines Chemical class 0.000 description 16
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 16
- 238000011156 evaluation Methods 0.000 description 16
- 239000003999 initiator Substances 0.000 description 16
- 239000003960 organic solvent Substances 0.000 description 16
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 16
- 150000003254 radicals Chemical class 0.000 description 16
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 15
- 238000006116 polymerization reaction Methods 0.000 description 15
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 14
- 229910052799 carbon Inorganic materials 0.000 description 14
- 238000001723 curing Methods 0.000 description 14
- BGTOWKSIORTVQH-UHFFFAOYSA-N cyclopentanone Chemical compound O=C1CCCC1 BGTOWKSIORTVQH-UHFFFAOYSA-N 0.000 description 14
- 238000001914 filtration Methods 0.000 description 13
- 239000000126 substance Substances 0.000 description 13
- 239000004094 surface-active agent Substances 0.000 description 13
- FVCSARBUZVPSQF-UHFFFAOYSA-N 5-(2,4-dioxooxolan-3-yl)-7-methyl-3a,4,5,7a-tetrahydro-2-benzofuran-1,3-dione Chemical compound C1C(C(OC2=O)=O)C2C(C)=CC1C1C(=O)COC1=O FVCSARBUZVPSQF-UHFFFAOYSA-N 0.000 description 12
- WOBHKFSMXKNTIM-UHFFFAOYSA-N Hydroxyethyl methacrylate Chemical compound CC(=C)C(=O)OCCO WOBHKFSMXKNTIM-UHFFFAOYSA-N 0.000 description 12
- 230000000052 comparative effect Effects 0.000 description 12
- 230000005012 migration Effects 0.000 description 12
- 238000013508 migration Methods 0.000 description 12
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical group CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 11
- 150000001721 carbon Chemical group 0.000 description 11
- 238000000576 coating method Methods 0.000 description 11
- 150000002148 esters Chemical class 0.000 description 11
- HLBLWEWZXPIGSM-UHFFFAOYSA-N 4-Aminophenyl ether Chemical compound C1=CC(N)=CC=C1OC1=CC=C(N)C=C1 HLBLWEWZXPIGSM-UHFFFAOYSA-N 0.000 description 10
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 10
- 229910052731 fluorine Inorganic materials 0.000 description 10
- 125000001424 substituent group Chemical group 0.000 description 10
- QQGYZOYWNCKGEK-UHFFFAOYSA-N 5-[(1,3-dioxo-2-benzofuran-5-yl)oxy]-2-benzofuran-1,3-dione Chemical compound C1=C2C(=O)OC(=O)C2=CC(OC=2C=C3C(=O)OC(C3=CC=2)=O)=C1 QQGYZOYWNCKGEK-UHFFFAOYSA-N 0.000 description 9
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 9
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 9
- 239000012986 chain transfer agent Substances 0.000 description 9
- 238000006243 chemical reaction Methods 0.000 description 9
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 9
- 125000001153 fluoro group Chemical group F* 0.000 description 9
- 150000002430 hydrocarbons Chemical group 0.000 description 9
- 238000010030 laminating Methods 0.000 description 9
- 230000008569 process Effects 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- 239000004593 Epoxy Substances 0.000 description 8
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 8
- 125000004122 cyclic group Chemical group 0.000 description 8
- 238000001035 drying Methods 0.000 description 8
- LZCLXQDLBQLTDK-UHFFFAOYSA-N ethyl 2-hydroxypropanoate Chemical compound CCOC(=O)C(C)O LZCLXQDLBQLTDK-UHFFFAOYSA-N 0.000 description 8
- 239000011541 reaction mixture Substances 0.000 description 8
- 238000007363 ring formation reaction Methods 0.000 description 8
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 7
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 7
- 150000001408 amides Chemical class 0.000 description 7
- 238000000354 decomposition reaction Methods 0.000 description 7
- 238000011161 development Methods 0.000 description 7
- 235000014113 dietary fatty acids Nutrition 0.000 description 7
- 230000000694 effects Effects 0.000 description 7
- 235000019441 ethanol Nutrition 0.000 description 7
- 229930195729 fatty acid Natural products 0.000 description 7
- 239000000194 fatty acid Substances 0.000 description 7
- 150000004665 fatty acids Chemical class 0.000 description 7
- 229910052710 silicon Inorganic materials 0.000 description 7
- 239000010703 silicon Substances 0.000 description 7
- 238000004528 spin coating Methods 0.000 description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- VLDPXPPHXDGHEW-UHFFFAOYSA-N 1-chloro-2-dichlorophosphoryloxybenzene Chemical compound ClC1=CC=CC=C1OP(Cl)(Cl)=O VLDPXPPHXDGHEW-UHFFFAOYSA-N 0.000 description 6
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 6
- TXBCBTDQIULDIA-UHFFFAOYSA-N 2-[[3-hydroxy-2,2-bis(hydroxymethyl)propoxy]methyl]-2-(hydroxymethyl)propane-1,3-diol Chemical compound OCC(CO)(CO)COCC(CO)(CO)CO TXBCBTDQIULDIA-UHFFFAOYSA-N 0.000 description 6
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 6
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 6
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical group C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 6
- 125000003647 acryloyl group Chemical group O=C([*])C([H])=C([H])[H] 0.000 description 6
- 239000003463 adsorbent Substances 0.000 description 6
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 6
- 125000006165 cyclic alkyl group Chemical group 0.000 description 6
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 6
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 6
- 150000005690 diesters Chemical class 0.000 description 6
- 239000003822 epoxy resin Substances 0.000 description 6
- CATSNJVOTSVZJV-UHFFFAOYSA-N heptan-2-one Chemical compound CCCCCC(C)=O CATSNJVOTSVZJV-UHFFFAOYSA-N 0.000 description 6
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 description 6
- 229920000647 polyepoxide Polymers 0.000 description 6
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 235000012431 wafers Nutrition 0.000 description 6
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 5
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical group CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 5
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 5
- 125000002947 alkylene group Chemical group 0.000 description 5
- DKPFZGUDAPQIHT-UHFFFAOYSA-N butyl acetate Chemical compound CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 5
- 239000007795 chemical reaction product Substances 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 239000011248 coating agent Substances 0.000 description 5
- 238000004132 cross linking Methods 0.000 description 5
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 5
- 230000003993 interaction Effects 0.000 description 5
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 5
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 5
- 239000002994 raw material Substances 0.000 description 5
- 238000005507 spraying Methods 0.000 description 5
- 125000006158 tetracarboxylic acid group Chemical group 0.000 description 5
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 description 4
- ARXJGSRGQADJSQ-UHFFFAOYSA-N 1-methoxypropan-2-ol Chemical compound COCC(C)O ARXJGSRGQADJSQ-UHFFFAOYSA-N 0.000 description 4
- HEQOJEGTZCTHCF-UHFFFAOYSA-N 2-amino-1-phenylethanone Chemical class NCC(=O)C1=CC=CC=C1 HEQOJEGTZCTHCF-UHFFFAOYSA-N 0.000 description 4
- XLLIQLLCWZCATF-UHFFFAOYSA-N 2-methoxyethyl acetate Chemical compound COCCOC(C)=O XLLIQLLCWZCATF-UHFFFAOYSA-N 0.000 description 4
- OZJPLYNZGCXSJM-UHFFFAOYSA-N 5-valerolactone Chemical compound O=C1CCCCO1 OZJPLYNZGCXSJM-UHFFFAOYSA-N 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- FFOPEPMHKILNIT-UHFFFAOYSA-N Isopropyl butyrate Chemical compound CCCC(=O)OC(C)C FFOPEPMHKILNIT-UHFFFAOYSA-N 0.000 description 4
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 4
- 239000006087 Silane Coupling Agent Substances 0.000 description 4
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 4
- 239000000654 additive Substances 0.000 description 4
- 125000005907 alkyl ester group Chemical group 0.000 description 4
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 4
- 230000001588 bifunctional effect Effects 0.000 description 4
- PXKLMJQFEQBVLD-UHFFFAOYSA-N bisphenol F Chemical compound C1=CC(O)=CC=C1CC1=CC=C(O)C=C1 PXKLMJQFEQBVLD-UHFFFAOYSA-N 0.000 description 4
- XUPYJHCZDLZNFP-UHFFFAOYSA-N butyl butanoate Chemical compound CCCCOC(=O)CCC XUPYJHCZDLZNFP-UHFFFAOYSA-N 0.000 description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 4
- 239000000470 constituent Substances 0.000 description 4
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 4
- ISAOCJYIOMOJEB-UHFFFAOYSA-N desyl alcohol Natural products C=1C=CC=CC=1C(O)C(=O)C1=CC=CC=C1 ISAOCJYIOMOJEB-UHFFFAOYSA-N 0.000 description 4
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical group C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 4
- KZTYYGOKRVBIMI-UHFFFAOYSA-N diphenyl sulfone Chemical compound C=1C=CC=CC=1S(=O)(=O)C1=CC=CC=C1 KZTYYGOKRVBIMI-UHFFFAOYSA-N 0.000 description 4
- 125000003700 epoxy group Chemical group 0.000 description 4
- 238000005530 etching Methods 0.000 description 4
- 150000002170 ethers Chemical class 0.000 description 4
- 229940116333 ethyl lactate Drugs 0.000 description 4
- 238000005227 gel permeation chromatography Methods 0.000 description 4
- 125000005843 halogen group Chemical group 0.000 description 4
- NGAZZOYFWWSOGK-UHFFFAOYSA-N heptan-3-one Chemical compound CCCCC(=O)CC NGAZZOYFWWSOGK-UHFFFAOYSA-N 0.000 description 4
- 239000012535 impurity Substances 0.000 description 4
- XEEYBQQBJWHFJM-UHFFFAOYSA-N iron Substances [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 4
- MLFHJEHSLIIPHL-UHFFFAOYSA-N isoamyl acetate Chemical compound CC(C)CCOC(C)=O MLFHJEHSLIIPHL-UHFFFAOYSA-N 0.000 description 4
- XMGQYMWWDOXHJM-UHFFFAOYSA-N limonene Chemical compound CC(=C)C1CCC(C)=CC1 XMGQYMWWDOXHJM-UHFFFAOYSA-N 0.000 description 4
- CYIDZMCFTVVTJO-UHFFFAOYSA-N pyromellitic acid Chemical compound OC(=O)C1=CC(C(O)=O)=C(C(O)=O)C=C1C(O)=O CYIDZMCFTVVTJO-UHFFFAOYSA-N 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- 125000006219 1-ethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 3
- SVONRAPFKPVNKG-UHFFFAOYSA-N 2-ethoxyethyl acetate Chemical compound CCOCCOC(C)=O SVONRAPFKPVNKG-UHFFFAOYSA-N 0.000 description 3
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 3
- CMLFRMDBDNHMRA-UHFFFAOYSA-N 2h-1,2-benzoxazine Chemical compound C1=CC=C2C=CNOC2=C1 CMLFRMDBDNHMRA-UHFFFAOYSA-N 0.000 description 3
- OSUWBBMPVXVSOA-UHFFFAOYSA-N 4-(4-carbonochloridoylphenoxy)benzoyl chloride Chemical compound C1=CC(C(=O)Cl)=CC=C1OC1=CC=C(C(Cl)=O)C=C1 OSUWBBMPVXVSOA-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical group OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical class C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 125000005042 acyloxymethyl group Chemical class 0.000 description 3
- 238000007259 addition reaction Methods 0.000 description 3
- 125000004450 alkenylene group Chemical group 0.000 description 3
- 125000003545 alkoxy group Chemical group 0.000 description 3
- 125000004849 alkoxymethyl group Chemical class 0.000 description 3
- 150000001412 amines Chemical group 0.000 description 3
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 150000008366 benzophenones Chemical class 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- 238000009835 boiling Methods 0.000 description 3
- 150000001733 carboxylic acid esters Chemical class 0.000 description 3
- 150000001735 carboxylic acids Chemical class 0.000 description 3
- 229910052804 chromium Inorganic materials 0.000 description 3
- 239000011651 chromium Substances 0.000 description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 3
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 3
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 3
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 238000006297 dehydration reaction Methods 0.000 description 3
- XXJWXESWEXIICW-UHFFFAOYSA-N diethylene glycol monoethyl ether Chemical compound CCOCCOCCO XXJWXESWEXIICW-UHFFFAOYSA-N 0.000 description 3
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 3
- BHXIWUJLHYHGSJ-UHFFFAOYSA-N ethyl 3-ethoxypropanoate Chemical compound CCOCCC(=O)OCC BHXIWUJLHYHGSJ-UHFFFAOYSA-N 0.000 description 3
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 3
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 3
- 125000003055 glycidyl group Chemical group C(C1CO1)* 0.000 description 3
- 229910052736 halogen Inorganic materials 0.000 description 3
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 3
- 229910052742 iron Inorganic materials 0.000 description 3
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 3
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 3
- 229910052753 mercury Inorganic materials 0.000 description 3
- BDJSOPWXYLFTNW-UHFFFAOYSA-N methyl 3-methoxypropanoate Chemical compound COCCC(=O)OC BDJSOPWXYLFTNW-UHFFFAOYSA-N 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 229910052759 nickel Inorganic materials 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 150000002923 oximes Chemical class 0.000 description 3
- 125000005702 oxyalkylene group Chemical group 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- NWVVVBRKAWDGAB-UHFFFAOYSA-N p-methoxyphenol Chemical compound COC1=CC=C(O)C=C1 NWVVVBRKAWDGAB-UHFFFAOYSA-N 0.000 description 3
- WXZMFSXDPGVJKK-UHFFFAOYSA-N pentaerythritol Chemical compound OCC(CO)(CO)CO WXZMFSXDPGVJKK-UHFFFAOYSA-N 0.000 description 3
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 3
- 238000000206 photolithography Methods 0.000 description 3
- 125000003386 piperidinyl group Chemical group 0.000 description 3
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 3
- 239000004810 polytetrafluoroethylene Substances 0.000 description 3
- 239000011148 porous material Substances 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- LLHKCFNBLRBOGN-UHFFFAOYSA-N propylene glycol methyl ether acetate Chemical compound COCC(C)OC(C)=O LLHKCFNBLRBOGN-UHFFFAOYSA-N 0.000 description 3
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 238000010008 shearing Methods 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 150000003462 sulfoxides Chemical class 0.000 description 3
- 150000000000 tetracarboxylic acids Chemical group 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- 125000003258 trimethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])[*:1] 0.000 description 3
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical group C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 2
- ZFPGARUNNKGOBB-UHFFFAOYSA-N 1-Ethyl-2-pyrrolidinone Chemical compound CCN1CCCC1=O ZFPGARUNNKGOBB-UHFFFAOYSA-N 0.000 description 2
- IANQTJSKSUMEQM-UHFFFAOYSA-N 1-benzofuran Chemical compound C1=CC=C2OC=CC2=C1 IANQTJSKSUMEQM-UHFFFAOYSA-N 0.000 description 2
- HNAGHMKIPMKKBB-UHFFFAOYSA-N 1-benzylpyrrolidine-3-carboxamide Chemical compound C1C(C(=O)N)CCN1CC1=CC=CC=C1 HNAGHMKIPMKKBB-UHFFFAOYSA-N 0.000 description 2
- LIPRQQHINVWJCH-UHFFFAOYSA-N 1-ethoxypropan-2-yl acetate Chemical compound CCOCC(C)OC(C)=O LIPRQQHINVWJCH-UHFFFAOYSA-N 0.000 description 2
- DMFAHCVITRDZQB-UHFFFAOYSA-N 1-propoxypropan-2-yl acetate Chemical compound CCCOCC(C)OC(C)=O DMFAHCVITRDZQB-UHFFFAOYSA-N 0.000 description 2
- HFZLSTDPRQSZCQ-UHFFFAOYSA-N 1-pyrrolidin-3-ylpyrrolidine Chemical compound C1CCCN1C1CNCC1 HFZLSTDPRQSZCQ-UHFFFAOYSA-N 0.000 description 2
- KJUGUADJHNHALS-UHFFFAOYSA-N 1H-tetrazole Chemical compound C=1N=NNN=1 KJUGUADJHNHALS-UHFFFAOYSA-N 0.000 description 2
- SBASXUCJHJRPEV-UHFFFAOYSA-N 2-(2-methoxyethoxy)ethanol Chemical compound COCCOCCO SBASXUCJHJRPEV-UHFFFAOYSA-N 0.000 description 2
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 2
- LTHJXDSHSVNJKG-UHFFFAOYSA-N 2-[2-[2-[2-(2-methylprop-2-enoyloxy)ethoxy]ethoxy]ethoxy]ethyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCCOCCOCCOCCOC(=O)C(C)=C LTHJXDSHSVNJKG-UHFFFAOYSA-N 0.000 description 2
- OJPDDQSCZGTACX-UHFFFAOYSA-N 2-[n-(2-hydroxyethyl)anilino]ethanol Chemical compound OCCN(CCO)C1=CC=CC=C1 OJPDDQSCZGTACX-UHFFFAOYSA-N 0.000 description 2
- UHFFVFAKEGKNAQ-UHFFFAOYSA-N 2-benzyl-2-(dimethylamino)-1-(4-morpholin-4-ylphenyl)butan-1-one Chemical compound C=1C=C(N2CCOCC2)C=CC=1C(=O)C(CC)(N(C)C)CC1=CC=CC=C1 UHFFVFAKEGKNAQ-UHFFFAOYSA-N 0.000 description 2
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 2
- ICPWFHKNYYRBSZ-UHFFFAOYSA-M 2-methoxypropanoate Chemical compound COC(C)C([O-])=O ICPWFHKNYYRBSZ-UHFFFAOYSA-M 0.000 description 2
- SYUYTOYKQOAVDW-UHFFFAOYSA-N 2-nitrosonaphthalen-1-ol Chemical compound C1=CC=C2C(O)=C(N=O)C=CC2=C1 SYUYTOYKQOAVDW-UHFFFAOYSA-N 0.000 description 2
- XOUQAVYLRNOXDO-UHFFFAOYSA-N 2-tert-butyl-5-methylphenol Chemical compound CC1=CC=C(C(C)(C)C)C(O)=C1 XOUQAVYLRNOXDO-UHFFFAOYSA-N 0.000 description 2
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical group C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 description 2
- NUIURNJTPRWVAP-UHFFFAOYSA-N 3,3'-Dimethylbenzidine Chemical group C1=C(N)C(C)=CC(C=2C=C(C)C(N)=CC=2)=C1 NUIURNJTPRWVAP-UHFFFAOYSA-N 0.000 description 2
- LMIOYAVXLAOXJI-UHFFFAOYSA-N 3-ethyl-3-[[4-[(3-ethyloxetan-3-yl)methoxymethyl]phenyl]methoxymethyl]oxetane Chemical compound C=1C=C(COCC2(CC)COC2)C=CC=1COCC1(CC)COC1 LMIOYAVXLAOXJI-UHFFFAOYSA-N 0.000 description 2
- VVBLNCFGVYUYGU-UHFFFAOYSA-N 4,4'-Bis(dimethylamino)benzophenone Chemical compound C1=CC(N(C)C)=CC=C1C(=O)C1=CC=C(N(C)C)C=C1 VVBLNCFGVYUYGU-UHFFFAOYSA-N 0.000 description 2
- AIVVXPSKEVWKMY-UHFFFAOYSA-N 4-(3,4-dicarboxyphenoxy)phthalic acid Chemical compound C1=C(C(O)=O)C(C(=O)O)=CC=C1OC1=CC=C(C(O)=O)C(C(O)=O)=C1 AIVVXPSKEVWKMY-UHFFFAOYSA-N 0.000 description 2
- FWOLORXQTIGHFX-UHFFFAOYSA-N 4-(4-amino-2,3,5,6-tetrafluorophenyl)-2,3,5,6-tetrafluoroaniline Chemical group FC1=C(F)C(N)=C(F)C(F)=C1C1=C(F)C(F)=C(N)C(F)=C1F FWOLORXQTIGHFX-UHFFFAOYSA-N 0.000 description 2
- QYIMZXITLDTULQ-UHFFFAOYSA-N 4-(4-amino-2-methylphenyl)-3-methylaniline Chemical group CC1=CC(N)=CC=C1C1=CC=C(N)C=C1C QYIMZXITLDTULQ-UHFFFAOYSA-N 0.000 description 2
- NVKGJHAQGWCWDI-UHFFFAOYSA-N 4-[4-amino-2-(trifluoromethyl)phenyl]-3-(trifluoromethyl)aniline Chemical group FC(F)(F)C1=CC(N)=CC=C1C1=CC=C(N)C=C1C(F)(F)F NVKGJHAQGWCWDI-UHFFFAOYSA-N 0.000 description 2
- ACQVEWFMUBXEMR-UHFFFAOYSA-N 4-bromo-2-fluoro-6-nitrophenol Chemical compound OC1=C(F)C=C(Br)C=C1[N+]([O-])=O ACQVEWFMUBXEMR-UHFFFAOYSA-N 0.000 description 2
- CQMIJLIXKMKFQW-UHFFFAOYSA-N 4-phenylbenzene-1,2,3,5-tetracarboxylic acid Chemical compound OC(=O)C1=C(C(O)=O)C(C(=O)O)=CC(C(O)=O)=C1C1=CC=CC=C1 CQMIJLIXKMKFQW-UHFFFAOYSA-N 0.000 description 2
- LPEKGGXMPWTOCB-UHFFFAOYSA-N 8beta-(2,3-epoxy-2-methylbutyryloxy)-14-acetoxytithifolin Natural products COC(=O)C(C)O LPEKGGXMPWTOCB-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 2
- MDNWOSOZYLHTCG-UHFFFAOYSA-N Dichlorophen Chemical compound OC1=CC=C(Cl)C=C1CC1=CC(Cl)=CC=C1O MDNWOSOZYLHTCG-UHFFFAOYSA-N 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 2
- WRQNANDWMGAFTP-UHFFFAOYSA-N Methylacetoacetic acid Chemical compound COC(=O)CC(C)=O WRQNANDWMGAFTP-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical group C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 2
- DIQMPQMYFZXDAX-UHFFFAOYSA-N Pentyl formate Chemical compound CCCCCOC=O DIQMPQMYFZXDAX-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical compound CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 description 2
- 244000028419 Styrax benzoin Species 0.000 description 2
- 235000000126 Styrax benzoin Nutrition 0.000 description 2
- 235000008411 Sumatra benzointree Nutrition 0.000 description 2
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical group C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical group C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- MPIAGWXWVAHQBB-UHFFFAOYSA-N [3-prop-2-enoyloxy-2-[[3-prop-2-enoyloxy-2,2-bis(prop-2-enoyloxymethyl)propoxy]methyl]-2-(prop-2-enoyloxymethyl)propyl] prop-2-enoate Chemical compound C=CC(=O)OCC(COC(=O)C=C)(COC(=O)C=C)COCC(COC(=O)C=C)(COC(=O)C=C)COC(=O)C=C MPIAGWXWVAHQBB-UHFFFAOYSA-N 0.000 description 2
- 150000008062 acetophenones Chemical class 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 150000001450 anions Chemical class 0.000 description 2
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 2
- CUFNKYGDVFVPHO-UHFFFAOYSA-N azulene Chemical compound C1=CC=CC2=CC=CC2=C1 CUFNKYGDVFVPHO-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 229960002130 benzoin Drugs 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- 230000008033 biological extinction Effects 0.000 description 2
- 235000010290 biphenyl Nutrition 0.000 description 2
- 239000004305 biphenyl Substances 0.000 description 2
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical compound C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 description 2
- OBNCKNCVKJNDBV-UHFFFAOYSA-N butanoic acid ethyl ester Natural products CCCC(=O)OCC OBNCKNCVKJNDBV-UHFFFAOYSA-N 0.000 description 2
- IWPATTDMSUYMJV-UHFFFAOYSA-N butyl 2-methoxyacetate Chemical compound CCCCOC(=O)COC IWPATTDMSUYMJV-UHFFFAOYSA-N 0.000 description 2
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 2
- 239000012295 chemical reaction liquid Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 238000006482 condensation reaction Methods 0.000 description 2
- 238000011109 contamination Methods 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 238000005260 corrosion Methods 0.000 description 2
- 230000007797 corrosion Effects 0.000 description 2
- 230000018044 dehydration Effects 0.000 description 2
- 125000004427 diamine group Chemical group 0.000 description 2
- 125000006159 dianhydride group Chemical group 0.000 description 2
- 150000001990 dicarboxylic acid derivatives Chemical class 0.000 description 2
- 229940028356 diethylene glycol monobutyl ether Drugs 0.000 description 2
- 229940075557 diethylene glycol monoethyl ether Drugs 0.000 description 2
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 2
- 239000000539 dimer Substances 0.000 description 2
- UKMSUNONTOPOIO-UHFFFAOYSA-N docosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCCCC(O)=O UKMSUNONTOPOIO-UHFFFAOYSA-N 0.000 description 2
- ODQWQRRAPPTVAG-GZTJUZNOSA-N doxepin Chemical compound C1OC2=CC=CC=C2C(=C/CCN(C)C)/C2=CC=CC=C21 ODQWQRRAPPTVAG-GZTJUZNOSA-N 0.000 description 2
- 238000010894 electron beam technology Methods 0.000 description 2
- 238000009713 electroplating Methods 0.000 description 2
- WNIHNYUROPJCLW-UHFFFAOYSA-N ethyl 2-ethoxy-2-methylpropanoate Chemical compound CCOC(=O)C(C)(C)OCC WNIHNYUROPJCLW-UHFFFAOYSA-N 0.000 description 2
- CKSRFHWWBKRUKA-UHFFFAOYSA-N ethyl 2-ethoxyacetate Chemical compound CCOCC(=O)OCC CKSRFHWWBKRUKA-UHFFFAOYSA-N 0.000 description 2
- JLEKJZUYWFJPMB-UHFFFAOYSA-N ethyl 2-methoxyacetate Chemical compound CCOC(=O)COC JLEKJZUYWFJPMB-UHFFFAOYSA-N 0.000 description 2
- WHRLOJCOIKOQGL-UHFFFAOYSA-N ethyl 2-methoxypropanoate Chemical compound CCOC(=O)C(C)OC WHRLOJCOIKOQGL-UHFFFAOYSA-N 0.000 description 2
- FJAKCEHATXBFJT-UHFFFAOYSA-N ethyl 2-oxobutanoate Chemical compound CCOC(=O)C(=O)CC FJAKCEHATXBFJT-UHFFFAOYSA-N 0.000 description 2
- XYIBRDXRRQCHLP-UHFFFAOYSA-N ethyl acetoacetate Chemical compound CCOC(=O)CC(C)=O XYIBRDXRRQCHLP-UHFFFAOYSA-N 0.000 description 2
- 230000005281 excited state Effects 0.000 description 2
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 235000019382 gum benzoic Nutrition 0.000 description 2
- 230000002140 halogenating effect Effects 0.000 description 2
- 230000020169 heat generation Effects 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- GJRQTCIYDGXPES-UHFFFAOYSA-N iso-butyl acetate Natural products CC(C)COC(C)=O GJRQTCIYDGXPES-UHFFFAOYSA-N 0.000 description 2
- 229940117955 isoamyl acetate Drugs 0.000 description 2
- FGKJLKRYENPLQH-UHFFFAOYSA-M isocaproate Chemical compound CC(C)CCC([O-])=O FGKJLKRYENPLQH-UHFFFAOYSA-M 0.000 description 2
- OQAGVSWESNCJJT-UHFFFAOYSA-N isovaleric acid methyl ester Natural products COC(=O)CC(C)C OQAGVSWESNCJJT-UHFFFAOYSA-N 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 229940087305 limonene Drugs 0.000 description 2
- 235000001510 limonene Nutrition 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 2
- PPFNAOBWGRMDLL-UHFFFAOYSA-N methyl 2-ethoxyacetate Chemical compound CCOCC(=O)OC PPFNAOBWGRMDLL-UHFFFAOYSA-N 0.000 description 2
- YVWPDYFVVMNWDT-UHFFFAOYSA-N methyl 2-ethoxypropanoate Chemical compound CCOC(C)C(=O)OC YVWPDYFVVMNWDT-UHFFFAOYSA-N 0.000 description 2
- AKWHOGIYEOZALP-UHFFFAOYSA-N methyl 2-methoxy-2-methylpropanoate Chemical compound COC(=O)C(C)(C)OC AKWHOGIYEOZALP-UHFFFAOYSA-N 0.000 description 2
- XPIWVCAMONZQCP-UHFFFAOYSA-N methyl 2-oxobutanoate Chemical compound CCC(=O)C(=O)OC XPIWVCAMONZQCP-UHFFFAOYSA-N 0.000 description 2
- HSDFKDZBJMDHFF-UHFFFAOYSA-N methyl 3-ethoxypropanoate Chemical compound CCOCCC(=O)OC HSDFKDZBJMDHFF-UHFFFAOYSA-N 0.000 description 2
- 229940057867 methyl lactate Drugs 0.000 description 2
- CWKLZLBVOJRSOM-UHFFFAOYSA-N methyl pyruvate Chemical compound COC(=O)C(C)=O CWKLZLBVOJRSOM-UHFFFAOYSA-N 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- 239000000178 monomer Substances 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- 125000002971 oxazolyl group Chemical group 0.000 description 2
- JCGNDDUYTRNOFT-UHFFFAOYSA-N oxolane-2,4-dione Chemical compound O=C1COC(=O)C1 JCGNDDUYTRNOFT-UHFFFAOYSA-N 0.000 description 2
- 239000000123 paper Substances 0.000 description 2
- 238000000059 patterning Methods 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- 125000003367 polycyclic group Chemical group 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 239000009719 polyimide resin Substances 0.000 description 2
- 229920001451 polypropylene glycol Polymers 0.000 description 2
- 229920001296 polysiloxane Polymers 0.000 description 2
- CYIRLFJPTCUCJB-UHFFFAOYSA-N propyl 2-methoxypropanoate Chemical compound CCCOC(=O)C(C)OC CYIRLFJPTCUCJB-UHFFFAOYSA-N 0.000 description 2
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 2
- 230000001681 protective effect Effects 0.000 description 2
- KIDHWZJUCRJVML-UHFFFAOYSA-N putrescine Chemical compound NCCCCN KIDHWZJUCRJVML-UHFFFAOYSA-N 0.000 description 2
- 125000003373 pyrazinyl group Chemical group 0.000 description 2
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical group C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- WQGWDDDVZFFDIG-UHFFFAOYSA-N pyrogallol Chemical compound OC1=CC=CC(O)=C1O WQGWDDDVZFFDIG-UHFFFAOYSA-N 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 238000006798 ring closing metathesis reaction Methods 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 150000003457 sulfones Chemical class 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 238000012719 thermal polymerization Methods 0.000 description 2
- 125000003396 thiol group Chemical group [H]S* 0.000 description 2
- 150000003573 thiols Chemical group 0.000 description 2
- LDHQCZJRKDOVOX-UHFFFAOYSA-N trans-crotonic acid Natural products CC=CC(O)=O LDHQCZJRKDOVOX-UHFFFAOYSA-N 0.000 description 2
- 238000002834 transmittance Methods 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- XFNJVJPLKCPIBV-UHFFFAOYSA-N trimethylenediamine Chemical compound NCCCN XFNJVJPLKCPIBV-UHFFFAOYSA-N 0.000 description 2
- QXJQHYBHAIHNGG-UHFFFAOYSA-N trimethylolethane Chemical compound OCC(C)(CO)CO QXJQHYBHAIHNGG-UHFFFAOYSA-N 0.000 description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 2
- 239000008096 xylene Substances 0.000 description 2
- PAPBSGBWRJIAAV-UHFFFAOYSA-N ε-Caprolactone Chemical compound O=C1CCCCCO1 PAPBSGBWRJIAAV-UHFFFAOYSA-N 0.000 description 2
- DTGKSKDOIYIVQL-WEDXCCLWSA-N (+)-borneol Chemical group C1C[C@@]2(C)[C@@H](O)C[C@@H]1C2(C)C DTGKSKDOIYIVQL-WEDXCCLWSA-N 0.000 description 1
- QNODIIQQMGDSEF-UHFFFAOYSA-N (1-hydroxycyclohexyl)-phenylmethanone Chemical compound C=1C=CC=CC=1C(=O)C1(O)CCCCC1 QNODIIQQMGDSEF-UHFFFAOYSA-N 0.000 description 1
- UNMJLQGKEDTEKJ-UHFFFAOYSA-N (3-ethyloxetan-3-yl)methanol Chemical compound CCC1(CO)COC1 UNMJLQGKEDTEKJ-UHFFFAOYSA-N 0.000 description 1
- PGSHZSDDBWTHDU-UHFFFAOYSA-N (3-oxobutan-2-ylideneamino) 4-methylbenzenesulfonate Chemical compound CC(=O)C(C)=NOS(=O)(=O)C1=CC=C(C)C=C1 PGSHZSDDBWTHDU-UHFFFAOYSA-N 0.000 description 1
- MRUDPRFGMHTPFJ-UHFFFAOYSA-N (3-oxobutan-2-ylideneamino) acetate Chemical compound CC(=O)ON=C(C)C(C)=O MRUDPRFGMHTPFJ-UHFFFAOYSA-N 0.000 description 1
- SKCKRZWCOYHBEC-UHFFFAOYSA-N (3-oxobutan-2-ylideneamino) benzoate Chemical compound CC(=O)C(C)=NOC(=O)C1=CC=CC=C1 SKCKRZWCOYHBEC-UHFFFAOYSA-N 0.000 description 1
- JFFCVOSCPLKMLG-UHFFFAOYSA-N (3-oxobutan-2-ylideneamino) propanoate Chemical compound CCC(=O)ON=C(C)C(C)=O JFFCVOSCPLKMLG-UHFFFAOYSA-N 0.000 description 1
- 125000004642 (C1-C12) alkoxy group Chemical group 0.000 description 1
- 125000003837 (C1-C20) alkyl group Chemical group 0.000 description 1
- 125000006710 (C2-C12) alkenyl group Chemical group 0.000 description 1
- NSGXIBWMJZWTPY-UHFFFAOYSA-N 1,1,1,3,3,3-hexafluoropropane Chemical compound FC(F)(F)CC(F)(F)F NSGXIBWMJZWTPY-UHFFFAOYSA-N 0.000 description 1
- MYWOJODOMFBVCB-UHFFFAOYSA-N 1,2,6-trimethylphenanthrene Chemical compound CC1=CC=C2C3=CC(C)=CC=C3C=CC2=C1C MYWOJODOMFBVCB-UHFFFAOYSA-N 0.000 description 1
- WZCQRUWWHSTZEM-UHFFFAOYSA-N 1,3-phenylenediamine Chemical compound NC1=CC=CC(N)=C1 WZCQRUWWHSTZEM-UHFFFAOYSA-N 0.000 description 1
- 229940005561 1,4-benzoquinone Drugs 0.000 description 1
- FBMQNRKSAWNXBT-UHFFFAOYSA-N 1,4-diaminoanthracene-9,10-dione Chemical compound O=C1C2=CC=CC=C2C(=O)C2=C1C(N)=CC=C2N FBMQNRKSAWNXBT-UHFFFAOYSA-N 0.000 description 1
- CBCKQZAAMUWICA-UHFFFAOYSA-N 1,4-phenylenediamine Chemical compound NC1=CC=C(N)C=C1 CBCKQZAAMUWICA-UHFFFAOYSA-N 0.000 description 1
- VWBVCOPVKXNMMZ-UHFFFAOYSA-N 1,5-diaminoanthracene-9,10-dione Chemical compound O=C1C2=C(N)C=CC=C2C(=O)C2=C1C=CC=C2N VWBVCOPVKXNMMZ-UHFFFAOYSA-N 0.000 description 1
- FLBAYUMRQUHISI-UHFFFAOYSA-N 1,8-naphthyridine Chemical group N1=CC=CC2=CC=CN=C21 FLBAYUMRQUHISI-UHFFFAOYSA-N 0.000 description 1
- RDSVPKGMYHANML-UHFFFAOYSA-N 1-[2-[1-(2-aminopropoxy)propan-2-yloxy]ethoxy]propan-2-amine Chemical compound CC(N)COCCOC(C)COCC(C)N RDSVPKGMYHANML-UHFFFAOYSA-N 0.000 description 1
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical group C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 description 1
- JWYVGKFDLWWQJX-UHFFFAOYSA-N 1-ethenylazepan-2-one Chemical compound C=CN1CCCCCC1=O JWYVGKFDLWWQJX-UHFFFAOYSA-N 0.000 description 1
- BBPMQQKHHDHOMI-UHFFFAOYSA-N 1-iminopentan-3-one Chemical compound CCC(=O)CC=N BBPMQQKHHDHOMI-UHFFFAOYSA-N 0.000 description 1
- YXAOOTNFFAQIPZ-UHFFFAOYSA-N 1-nitrosonaphthalen-2-ol Chemical compound C1=CC=CC2=C(N=O)C(O)=CC=C21 YXAOOTNFFAQIPZ-UHFFFAOYSA-N 0.000 description 1
- WJFKNYWRSNBZNX-UHFFFAOYSA-N 10H-phenothiazine Chemical compound C1=CC=C2NC3=CC=CC=C3SC2=C1 WJFKNYWRSNBZNX-UHFFFAOYSA-N 0.000 description 1
- KGRVJHAUYBGFFP-UHFFFAOYSA-N 2,2'-Methylenebis(4-methyl-6-tert-butylphenol) Chemical compound CC(C)(C)C1=CC(C)=CC(CC=2C(=C(C=C(C)C=2)C(C)(C)C)O)=C1O KGRVJHAUYBGFFP-UHFFFAOYSA-N 0.000 description 1
- WCZNKVPCIFMXEQ-UHFFFAOYSA-N 2,3,5,6-tetramethylbenzene-1,4-diamine Chemical compound CC1=C(C)C(N)=C(C)C(C)=C1N WCZNKVPCIFMXEQ-UHFFFAOYSA-N 0.000 description 1
- YDYSEBSNAKCEQU-UHFFFAOYSA-N 2,3-diamino-n-phenylbenzamide Chemical compound NC1=CC=CC(C(=O)NC=2C=CC=CC=2)=C1N YDYSEBSNAKCEQU-UHFFFAOYSA-N 0.000 description 1
- KKTUQAYCCLMNOA-UHFFFAOYSA-N 2,3-diaminobenzoic acid Chemical compound NC1=CC=CC(C(O)=O)=C1N KKTUQAYCCLMNOA-UHFFFAOYSA-N 0.000 description 1
- VEPOHXYIFQMVHW-XOZOLZJESA-N 2,3-dihydroxybutanedioic acid (2S,3S)-3,4-dimethyl-2-phenylmorpholine Chemical group OC(C(O)C(O)=O)C(O)=O.C[C@H]1[C@@H](OCCN1C)c1ccccc1 VEPOHXYIFQMVHW-XOZOLZJESA-N 0.000 description 1
- ZVDSMYGTJDFNHN-UHFFFAOYSA-N 2,4,6-trimethylbenzene-1,3-diamine Chemical compound CC1=CC(C)=C(N)C(C)=C1N ZVDSMYGTJDFNHN-UHFFFAOYSA-N 0.000 description 1
- BTJPUDCSZVCXFQ-UHFFFAOYSA-N 2,4-diethylthioxanthen-9-one Chemical compound C1=CC=C2C(=O)C3=CC(CC)=CC(CC)=C3SC2=C1 BTJPUDCSZVCXFQ-UHFFFAOYSA-N 0.000 description 1
- BWAPJIHJXDYDPW-UHFFFAOYSA-N 2,5-dimethyl-p-phenylenediamine Chemical compound CC1=CC(N)=C(C)C=C1N BWAPJIHJXDYDPW-UHFFFAOYSA-N 0.000 description 1
- QYXHDJJYVDLECA-UHFFFAOYSA-N 2,5-diphenylcyclohexa-2,5-diene-1,4-dione Chemical compound O=C1C=C(C=2C=CC=CC=2)C(=O)C=C1C1=CC=CC=C1 QYXHDJJYVDLECA-UHFFFAOYSA-N 0.000 description 1
- VRMKFAFIVOVETG-UHFFFAOYSA-N 2,6-bis(methoxymethyl)-4-methylphenol Chemical compound COCC1=CC(C)=CC(COC)=C1O VRMKFAFIVOVETG-UHFFFAOYSA-N 0.000 description 1
- STMDPCBYJCIZOD-UHFFFAOYSA-N 2-(2,4-dinitroanilino)-4-methylpentanoic acid Chemical compound CC(C)CC(C(O)=O)NC1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O STMDPCBYJCIZOD-UHFFFAOYSA-N 0.000 description 1
- VXQBJTKSVGFQOL-UHFFFAOYSA-N 2-(2-butoxyethoxy)ethyl acetate Chemical compound CCCCOCCOCCOC(C)=O VXQBJTKSVGFQOL-UHFFFAOYSA-N 0.000 description 1
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 1
- BZRAULNYWZRKMB-UHFFFAOYSA-N 2-(2-methylprop-2-enoyloxy)ethyl 3,5-diaminobenzoate Chemical compound CC(=C)C(=O)OCCOC(=O)C1=CC(N)=CC(N)=C1 BZRAULNYWZRKMB-UHFFFAOYSA-N 0.000 description 1
- JAHNSTQSQJOJLO-UHFFFAOYSA-N 2-(3-fluorophenyl)-1h-imidazole Chemical compound FC1=CC=CC(C=2NC=CN=2)=C1 JAHNSTQSQJOJLO-UHFFFAOYSA-N 0.000 description 1
- GJKGAPPUXSSCFI-UHFFFAOYSA-N 2-Hydroxy-4'-(2-hydroxyethoxy)-2-methylpropiophenone Chemical compound CC(C)(O)C(=O)C1=CC=C(OCCO)C=C1 GJKGAPPUXSSCFI-UHFFFAOYSA-N 0.000 description 1
- HDPLHDGYGLENEI-UHFFFAOYSA-N 2-[1-(oxiran-2-ylmethoxy)propan-2-yloxymethyl]oxirane Chemical compound C1OC1COC(C)COCC1CO1 HDPLHDGYGLENEI-UHFFFAOYSA-N 0.000 description 1
- AOBIOSPNXBMOAT-UHFFFAOYSA-N 2-[2-(oxiran-2-ylmethoxy)ethoxymethyl]oxirane Chemical compound C1OC1COCCOCC1CO1 AOBIOSPNXBMOAT-UHFFFAOYSA-N 0.000 description 1
- YUZSJKBFHATJHV-UHFFFAOYSA-N 2-[4-[2-[4-(2-aminophenoxy)phenyl]-1,1,1,3,3,3-hexafluoropropan-2-yl]phenoxy]aniline Chemical compound NC1=CC=CC=C1OC1=CC=C(C(C=2C=CC(OC=3C(=CC=CC=3)N)=CC=2)(C(F)(F)F)C(F)(F)F)C=C1 YUZSJKBFHATJHV-UHFFFAOYSA-N 0.000 description 1
- FJYCAYKHNVQCJW-UHFFFAOYSA-N 2-[4-[4-(2-aminophenoxy)phenyl]sulfonylphenoxy]aniline Chemical compound NC1=CC=CC=C1OC1=CC=C(S(=O)(=O)C=2C=CC(OC=3C(=CC=CC=3)N)=CC=2)C=C1 FJYCAYKHNVQCJW-UHFFFAOYSA-N 0.000 description 1
- BXVXPASMKWYBFD-UHFFFAOYSA-N 2-[[2-hydroxy-3-(hydroxymethyl)-5-methylphenyl]methyl]-6-(hydroxymethyl)-4-methylphenol Chemical compound CC1=CC(CO)=C(O)C(CC=2C(=C(CO)C=C(C)C=2)O)=C1 BXVXPASMKWYBFD-UHFFFAOYSA-N 0.000 description 1
- MSTZGVRUOMBULC-UHFFFAOYSA-N 2-amino-4-[2-(3-amino-4-hydroxyphenyl)-1,1,1,3,3,3-hexafluoropropan-2-yl]phenol Chemical compound C1=C(O)C(N)=CC(C(C=2C=C(N)C(O)=CC=2)(C(F)(F)F)C(F)(F)F)=C1 MSTZGVRUOMBULC-UHFFFAOYSA-N 0.000 description 1
- UHIDYCYNRPVZCK-UHFFFAOYSA-N 2-amino-4-[2-(3-amino-4-hydroxyphenyl)propan-2-yl]phenol Chemical compound C=1C=C(O)C(N)=CC=1C(C)(C)C1=CC=C(O)C(N)=C1 UHIDYCYNRPVZCK-UHFFFAOYSA-N 0.000 description 1
- KHAFBBNQUOEYHB-UHFFFAOYSA-N 2-amino-5-(4-amino-3-hydroxyphenyl)sulfonylphenol Chemical compound C1=C(O)C(N)=CC=C1S(=O)(=O)C1=CC=C(N)C(O)=C1 KHAFBBNQUOEYHB-UHFFFAOYSA-N 0.000 description 1
- NPUFCLGAODQFLG-UHFFFAOYSA-N 2-amino-5-(4-aminophenyl)cyclohexa-2,4-diene-1,1-diol Chemical group C1C(O)(O)C(N)=CC=C1C1=CC=C(N)C=C1 NPUFCLGAODQFLG-UHFFFAOYSA-N 0.000 description 1
- JDFAWEKPFLGRAK-UHFFFAOYSA-N 2-amino-5-[2-(4-amino-3-hydroxyphenyl)propan-2-yl]phenol Chemical compound C=1C=C(N)C(O)=CC=1C(C)(C)C1=CC=C(N)C(O)=C1 JDFAWEKPFLGRAK-UHFFFAOYSA-N 0.000 description 1
- UXGVMFHEKMGWMA-UHFFFAOYSA-N 2-benzofuran Chemical group C1=CC=CC2=COC=C21 UXGVMFHEKMGWMA-UHFFFAOYSA-N 0.000 description 1
- NLGDWWCZQDIASO-UHFFFAOYSA-N 2-hydroxy-1-(7-oxabicyclo[4.1.0]hepta-1,3,5-trien-2-yl)-2-phenylethanone Chemical class OC(C(=O)c1cccc2Oc12)c1ccccc1 NLGDWWCZQDIASO-UHFFFAOYSA-N 0.000 description 1
- PCKZAVNWRLEHIP-UHFFFAOYSA-N 2-hydroxy-1-[4-[[4-(2-hydroxy-2-methylpropanoyl)phenyl]methyl]phenyl]-2-methylpropan-1-one Chemical compound C1=CC(C(=O)C(C)(O)C)=CC=C1CC1=CC=C(C(=O)C(C)(C)O)C=C1 PCKZAVNWRLEHIP-UHFFFAOYSA-N 0.000 description 1
- ZWVHTXAYIKBMEE-UHFFFAOYSA-N 2-hydroxyacetophenone Chemical class OCC(=O)C1=CC=CC=C1 ZWVHTXAYIKBMEE-UHFFFAOYSA-N 0.000 description 1
- 125000006290 2-hydroxybenzyl group Chemical group [H]OC1=C(C([H])=C([H])C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- LWRBVKNFOYUCNP-UHFFFAOYSA-N 2-methyl-1-(4-methylsulfanylphenyl)-2-morpholin-4-ylpropan-1-one Chemical compound C1=CC(SC)=CC=C1C(=O)C(C)(C)N1CCOCC1 LWRBVKNFOYUCNP-UHFFFAOYSA-N 0.000 description 1
- RIWRBSMFKVOJMN-UHFFFAOYSA-N 2-methyl-1-phenylpropan-2-ol Chemical compound CC(C)(O)CC1=CC=CC=C1 RIWRBSMFKVOJMN-UHFFFAOYSA-N 0.000 description 1
- WNYJRJRHKRZXEO-UHFFFAOYSA-N 2-propan-2-ylbenzene-1,4-diamine Chemical compound CC(C)C1=CC(N)=CC=C1N WNYJRJRHKRZXEO-UHFFFAOYSA-N 0.000 description 1
- UAIUNKRWKOVEES-UHFFFAOYSA-N 3,3',5,5'-tetramethylbenzidine Chemical group CC1=C(N)C(C)=CC(C=2C=C(C)C(N)=C(C)C=2)=C1 UAIUNKRWKOVEES-UHFFFAOYSA-N 0.000 description 1
- JRBJSXQPQWSCCF-UHFFFAOYSA-N 3,3'-Dimethoxybenzidine Chemical group C1=C(N)C(OC)=CC(C=2C=C(OC)C(N)=CC=2)=C1 JRBJSXQPQWSCCF-UHFFFAOYSA-N 0.000 description 1
- HSTOKWSFWGCZMH-UHFFFAOYSA-N 3,3'-diaminobenzidine Chemical group C1=C(N)C(N)=CC=C1C1=CC=C(N)C(N)=C1 HSTOKWSFWGCZMH-UHFFFAOYSA-N 0.000 description 1
- NBAUUNCGSMAPFM-UHFFFAOYSA-N 3-(3,4-dicarboxyphenyl)phthalic acid Chemical compound C1=C(C(O)=O)C(C(=O)O)=CC=C1C1=CC=CC(C(O)=O)=C1C(O)=O NBAUUNCGSMAPFM-UHFFFAOYSA-N 0.000 description 1
- NDXGRHCEHPFUSU-UHFFFAOYSA-N 3-(3-aminophenyl)aniline Chemical group NC1=CC=CC(C=2C=C(N)C=CC=2)=C1 NDXGRHCEHPFUSU-UHFFFAOYSA-N 0.000 description 1
- LJGHYPLBDBRCRZ-UHFFFAOYSA-N 3-(3-aminophenyl)sulfonylaniline Chemical compound NC1=CC=CC(S(=O)(=O)C=2C=C(N)C=CC=2)=C1 LJGHYPLBDBRCRZ-UHFFFAOYSA-N 0.000 description 1
- PWEHHKOWZUPWBI-UHFFFAOYSA-N 3-(3-aminopropyl-methyl-trimethylsilyloxysilyl)propan-1-amine Chemical compound NCCC[Si](C)(O[Si](C)(C)C)CCCN PWEHHKOWZUPWBI-UHFFFAOYSA-N 0.000 description 1
- RNLHGQLZWXBQNY-UHFFFAOYSA-N 3-(aminomethyl)-3,5,5-trimethylcyclohexan-1-amine Chemical compound CC1(C)CC(N)CC(C)(CN)C1 RNLHGQLZWXBQNY-UHFFFAOYSA-N 0.000 description 1
- BQGXXEGJJMEZMZ-UHFFFAOYSA-N 3-(n-ethyl-3-hydroxy-4-nitrosoanilino)propane-1-sulfonic acid Chemical compound OS(=O)(=O)CCCN(CC)C1=CC=C(N=O)C(O)=C1 BQGXXEGJJMEZMZ-UHFFFAOYSA-N 0.000 description 1
- ZZYZUFNSKAIYCJ-UHFFFAOYSA-N 3-[(3-aminocyclohexyl)methyl]cyclohexan-1-amine Chemical compound C1C(N)CCCC1CC1CC(N)CCC1 ZZYZUFNSKAIYCJ-UHFFFAOYSA-N 0.000 description 1
- CKOFBUUFHALZGK-UHFFFAOYSA-N 3-[(3-aminophenyl)methyl]aniline Chemical compound NC1=CC=CC(CC=2C=C(N)C=CC=2)=C1 CKOFBUUFHALZGK-UHFFFAOYSA-N 0.000 description 1
- UCFMKTNJZCYBBJ-UHFFFAOYSA-N 3-[1-(2,3-dicarboxyphenyl)ethyl]phthalic acid Chemical compound C=1C=CC(C(O)=O)=C(C(O)=O)C=1C(C)C1=CC=CC(C(O)=O)=C1C(O)=O UCFMKTNJZCYBBJ-UHFFFAOYSA-N 0.000 description 1
- DKKYOQYISDAQER-UHFFFAOYSA-N 3-[3-(3-aminophenoxy)phenoxy]aniline Chemical compound NC1=CC=CC(OC=2C=C(OC=3C=C(N)C=CC=3)C=CC=2)=C1 DKKYOQYISDAQER-UHFFFAOYSA-N 0.000 description 1
- MFTFTIALAXXIMU-UHFFFAOYSA-N 3-[4-[2-[4-(3-aminophenoxy)phenyl]-1,1,1,3,3,3-hexafluoropropan-2-yl]phenoxy]aniline Chemical compound NC1=CC=CC(OC=2C=CC(=CC=2)C(C=2C=CC(OC=3C=C(N)C=CC=3)=CC=2)(C(F)(F)F)C(F)(F)F)=C1 MFTFTIALAXXIMU-UHFFFAOYSA-N 0.000 description 1
- WCXGOVYROJJXHA-UHFFFAOYSA-N 3-[4-[4-(3-aminophenoxy)phenyl]sulfonylphenoxy]aniline Chemical compound NC1=CC=CC(OC=2C=CC(=CC=2)S(=O)(=O)C=2C=CC(OC=3C=C(N)C=CC=3)=CC=2)=C1 WCXGOVYROJJXHA-UHFFFAOYSA-N 0.000 description 1
- GPXCORHXFPYJEH-UHFFFAOYSA-N 3-[[3-aminopropyl(dimethyl)silyl]oxy-dimethylsilyl]propan-1-amine Chemical compound NCCC[Si](C)(C)O[Si](C)(C)CCCN GPXCORHXFPYJEH-UHFFFAOYSA-N 0.000 description 1
- KNWLMUXHNAQOJM-UHFFFAOYSA-N 3-ethyl-3-(3-ethylheptyl)oxetane Chemical compound CCCCC(CC)CCC1(CC)COC1 KNWLMUXHNAQOJM-UHFFFAOYSA-N 0.000 description 1
- FNYWFRSQRHGKJT-UHFFFAOYSA-N 3-ethyl-3-[(3-ethyloxetan-3-yl)methoxymethyl]oxetane Chemical compound C1OCC1(CC)COCC1(CC)COC1 FNYWFRSQRHGKJT-UHFFFAOYSA-N 0.000 description 1
- 125000006291 3-hydroxybenzyl group Chemical group [H]OC1=C([H])C([H])=C([H])C(=C1[H])C([H])([H])* 0.000 description 1
- HTNUUDFQRYBJPH-UHFFFAOYSA-N 3-methoxypropanehydrazide Chemical compound COCCC(=O)NN HTNUUDFQRYBJPH-UHFFFAOYSA-N 0.000 description 1
- ATVJXMYDOSMEPO-UHFFFAOYSA-N 3-prop-2-enoxyprop-1-ene Chemical compound C=CCOCC=C ATVJXMYDOSMEPO-UHFFFAOYSA-N 0.000 description 1
- TXFPEBPIARQUIG-UHFFFAOYSA-N 4'-hydroxyacetophenone Chemical compound CC(=O)C1=CC=C(O)C=C1 TXFPEBPIARQUIG-UHFFFAOYSA-N 0.000 description 1
- WECDUOXQLAIPQW-UHFFFAOYSA-N 4,4'-Methylene bis(2-methylaniline) Chemical compound C1=C(N)C(C)=CC(CC=2C=C(C)C(N)=CC=2)=C1 WECDUOXQLAIPQW-UHFFFAOYSA-N 0.000 description 1
- ZSSJQYMNCUJSBR-UHFFFAOYSA-N 4,5-dimethoxy-1,3-bis(methoxymethyl)imidazolidin-2-one Chemical compound COCN1C(OC)C(OC)N(COC)C1=O ZSSJQYMNCUJSBR-UHFFFAOYSA-N 0.000 description 1
- RQBIGPMJQUKYAH-UHFFFAOYSA-N 4-(3,4-diaminophenoxy)benzene-1,2-diamine Chemical compound C1=C(N)C(N)=CC=C1OC1=CC=C(N)C(N)=C1 RQBIGPMJQUKYAH-UHFFFAOYSA-N 0.000 description 1
- VWCCEIUKGFGDDS-UHFFFAOYSA-N 4-(4-amino-2-methylphenyl)-3-methylaniline Chemical group CC1=C(C=CC(=C1)N)C1=C(C=C(N)C=C1)C.CC1=C(C=CC(=C1)N)C1=C(C=C(C=C1)N)C VWCCEIUKGFGDDS-UHFFFAOYSA-N 0.000 description 1
- QQWWWAQUMVHHQN-UHFFFAOYSA-N 4-(4-amino-4-phenylcyclohexa-1,5-dien-1-yl)aniline Chemical group C1=CC(N)=CC=C1C1=CCC(N)(C=2C=CC=CC=2)C=C1 QQWWWAQUMVHHQN-UHFFFAOYSA-N 0.000 description 1
- BDBMKUGEVIQCGQ-UHFFFAOYSA-N 4-(4-aminophenyl)-2,3-dimethylaniline Chemical group C1=C(N)C(C)=C(C)C(C=2C=CC(N)=CC=2)=C1 BDBMKUGEVIQCGQ-UHFFFAOYSA-N 0.000 description 1
- QNJWXYRVQVNUQC-UHFFFAOYSA-N 4-(4-aminophenyl)-3-(2-phenylphenyl)aniline Chemical group C1(=C(C=CC=C1)C1=C(C=CC(=C1)N)C1=CC=C(N)C=C1)C1=CC=CC=C1 QNJWXYRVQVNUQC-UHFFFAOYSA-N 0.000 description 1
- OMHOXRVODFQGCA-UHFFFAOYSA-N 4-[(4-amino-3,5-dimethylphenyl)methyl]-2,6-dimethylaniline Chemical compound CC1=C(N)C(C)=CC(CC=2C=C(C)C(N)=C(C)C=2)=C1 OMHOXRVODFQGCA-UHFFFAOYSA-N 0.000 description 1
- DZIHTWJGPDVSGE-UHFFFAOYSA-N 4-[(4-aminocyclohexyl)methyl]cyclohexan-1-amine Chemical compound C1CC(N)CCC1CC1CCC(N)CC1 DZIHTWJGPDVSGE-UHFFFAOYSA-N 0.000 description 1
- SGPJBCCVSFQQED-UHFFFAOYSA-N 4-[(4-aminophenyl)-[[dimethyl(trimethylsilyloxy)silyl]oxy-dimethylsilyl]oxy-methylsilyl]oxysilylaniline Chemical compound C=1C=C(N)C=CC=1[Si](C)(O[Si](C)(C)O[Si](C)(C)O[Si](C)(C)C)O[SiH2]C1=CC=C(N)C=C1 SGPJBCCVSFQQED-UHFFFAOYSA-N 0.000 description 1
- IJJNNSUCZDJDLP-UHFFFAOYSA-N 4-[1-(3,4-dicarboxyphenyl)ethyl]phthalic acid Chemical compound C=1C=C(C(O)=O)C(C(O)=O)=CC=1C(C)C1=CC=C(C(O)=O)C(C(O)=O)=C1 IJJNNSUCZDJDLP-UHFFFAOYSA-N 0.000 description 1
- ASNOFHCTUSIHOM-UHFFFAOYSA-N 4-[10-(4-aminophenyl)anthracen-9-yl]aniline Chemical compound C1=CC(N)=CC=C1C(C1=CC=CC=C11)=C(C=CC=C2)C2=C1C1=CC=C(N)C=C1 ASNOFHCTUSIHOM-UHFFFAOYSA-N 0.000 description 1
- APXJLYIVOFARRM-UHFFFAOYSA-N 4-[2-(3,4-dicarboxyphenyl)-1,1,1,3,3,3-hexafluoropropan-2-yl]phthalic acid Chemical compound C1=C(C(O)=O)C(C(=O)O)=CC=C1C(C(F)(F)F)(C(F)(F)F)C1=CC=C(C(O)=O)C(C(O)=O)=C1 APXJLYIVOFARRM-UHFFFAOYSA-N 0.000 description 1
- GEYAGBVEAJGCFB-UHFFFAOYSA-N 4-[2-(3,4-dicarboxyphenyl)propan-2-yl]phthalic acid Chemical compound C=1C=C(C(O)=O)C(C(O)=O)=CC=1C(C)(C)C1=CC=C(C(O)=O)C(C(O)=O)=C1 GEYAGBVEAJGCFB-UHFFFAOYSA-N 0.000 description 1
- BEKFRNOZJSYWKZ-UHFFFAOYSA-N 4-[2-(4-aminophenyl)-1,1,1,3,3,3-hexafluoropropan-2-yl]aniline Chemical compound C1=CC(N)=CC=C1C(C(F)(F)F)(C(F)(F)F)C1=CC=C(N)C=C1 BEKFRNOZJSYWKZ-UHFFFAOYSA-N 0.000 description 1
- UHNUHZHQLCGZDA-UHFFFAOYSA-N 4-[2-(4-aminophenyl)ethyl]aniline Chemical compound C1=CC(N)=CC=C1CCC1=CC=C(N)C=C1 UHNUHZHQLCGZDA-UHFFFAOYSA-N 0.000 description 1
- WUPRYUDHUFLKFL-UHFFFAOYSA-N 4-[3-(4-aminophenoxy)phenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC1=CC=CC(OC=2C=CC(N)=CC=2)=C1 WUPRYUDHUFLKFL-UHFFFAOYSA-N 0.000 description 1
- BOVVHULZWVFIOX-UHFFFAOYSA-N 4-[3-(4-aminophenyl)phenyl]aniline Chemical compound C1=CC(N)=CC=C1C1=CC=CC(C=2C=CC(N)=CC=2)=C1 BOVVHULZWVFIOX-UHFFFAOYSA-N 0.000 description 1
- JCRRFJIVUPSNTA-UHFFFAOYSA-N 4-[4-(4-aminophenoxy)phenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC(C=C1)=CC=C1OC1=CC=C(N)C=C1 JCRRFJIVUPSNTA-UHFFFAOYSA-N 0.000 description 1
- GACNZKUFHGKSEX-UHFFFAOYSA-N 4-[4-(4-aminophenyl)-1,1,2,2,3,3,4,4-octafluorobutyl]aniline Chemical compound C1=CC(N)=CC=C1C(F)(F)C(F)(F)C(F)(F)C(F)(F)C1=CC=C(N)C=C1 GACNZKUFHGKSEX-UHFFFAOYSA-N 0.000 description 1
- HBLYIUPUXAWDMA-UHFFFAOYSA-N 4-[4-[2-[4-(4-aminophenoxy)-3,5-bis(trifluoromethyl)phenyl]-1,1,1,3,3,3-hexafluoropropan-2-yl]-2,6-bis(trifluoromethyl)phenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC1=C(C(F)(F)F)C=C(C(C=2C=C(C(OC=3C=CC(N)=CC=3)=C(C=2)C(F)(F)F)C(F)(F)F)(C(F)(F)F)C(F)(F)F)C=C1C(F)(F)F HBLYIUPUXAWDMA-UHFFFAOYSA-N 0.000 description 1
- VCFYKCXKADGLPS-UHFFFAOYSA-N 4-[4-[2-[4-(4-aminophenoxy)-3,5-dimethylphenyl]-1,1,1,3,3,3-hexafluoropropan-2-yl]-2,6-dimethylphenoxy]aniline Chemical compound CC1=CC(C(C=2C=C(C)C(OC=3C=CC(N)=CC=3)=C(C)C=2)(C(F)(F)F)C(F)(F)F)=CC(C)=C1OC1=CC=C(N)C=C1 VCFYKCXKADGLPS-UHFFFAOYSA-N 0.000 description 1
- HHLMWQDRYZAENA-UHFFFAOYSA-N 4-[4-[2-[4-(4-aminophenoxy)phenyl]-1,1,1,3,3,3-hexafluoropropan-2-yl]phenoxy]aniline Chemical compound C1=CC(N)=CC=C1OC1=CC=C(C(C=2C=CC(OC=3C=CC(N)=CC=3)=CC=2)(C(F)(F)F)C(F)(F)F)C=C1 HHLMWQDRYZAENA-UHFFFAOYSA-N 0.000 description 1
- KMKWGXGSGPYISJ-UHFFFAOYSA-N 4-[4-[2-[4-(4-aminophenoxy)phenyl]propan-2-yl]phenoxy]aniline Chemical compound C=1C=C(OC=2C=CC(N)=CC=2)C=CC=1C(C)(C)C(C=C1)=CC=C1OC1=CC=C(N)C=C1 KMKWGXGSGPYISJ-UHFFFAOYSA-N 0.000 description 1
- HYDATEKARGDBKU-UHFFFAOYSA-N 4-[4-[4-(4-aminophenoxy)phenyl]phenoxy]aniline Chemical group C1=CC(N)=CC=C1OC1=CC=C(C=2C=CC(OC=3C=CC(N)=CC=3)=CC=2)C=C1 HYDATEKARGDBKU-UHFFFAOYSA-N 0.000 description 1
- IWFSADBGACLBMH-UHFFFAOYSA-N 4-[4-[4-[4-amino-2-(trifluoromethyl)phenoxy]phenyl]phenoxy]-3-(trifluoromethyl)aniline Chemical group FC(F)(F)C1=CC(N)=CC=C1OC1=CC=C(C=2C=CC(OC=3C(=CC(N)=CC=3)C(F)(F)F)=CC=2)C=C1 IWFSADBGACLBMH-UHFFFAOYSA-N 0.000 description 1
- DPCDFSDBIWVMJC-UHFFFAOYSA-N 4-[4-[4-[4-amino-3-(trifluoromethyl)phenoxy]phenyl]phenoxy]-2-(trifluoromethyl)aniline Chemical group C1=C(C(F)(F)F)C(N)=CC=C1OC1=CC=C(C=2C=CC(OC=3C=C(C(N)=CC=3)C(F)(F)F)=CC=2)C=C1 DPCDFSDBIWVMJC-UHFFFAOYSA-N 0.000 description 1
- FPAKXKXRPMFMLR-UHFFFAOYSA-N 4-[5-(4-aminophenyl)-1,1,2,2,3,3,4,4,5,5-decafluoropentyl]aniline Chemical compound C1=CC(N)=CC=C1C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C1=CC=C(N)C=C1 FPAKXKXRPMFMLR-UHFFFAOYSA-N 0.000 description 1
- BOTFKTVSBXEXKF-UHFFFAOYSA-N 4-[7-(4-aminophenyl)-1,1,2,2,3,3,4,4,5,5,6,6,7,7-tetradecafluoroheptyl]aniline Chemical compound C1=CC(N)=CC=C1C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C1=CC=C(N)C=C1 BOTFKTVSBXEXKF-UHFFFAOYSA-N 0.000 description 1
- 125000003143 4-hydroxybenzyl group Chemical group [H]C([*])([H])C1=C([H])C([H])=C(O[H])C([H])=C1[H] 0.000 description 1
- SHDBJOYDLRRTRA-UHFFFAOYSA-N 4-tert-butyl-2,6-bis(methoxymethyl)phenol Chemical compound COCC1=CC(C(C)(C)C)=CC(COC)=C1O SHDBJOYDLRRTRA-UHFFFAOYSA-N 0.000 description 1
- XESZUVZBAMCAEJ-UHFFFAOYSA-N 4-tert-butylcatechol Chemical compound CC(C)(C)C1=CC=C(O)C(O)=C1 XESZUVZBAMCAEJ-UHFFFAOYSA-N 0.000 description 1
- NSPMIYGKQJPBQR-UHFFFAOYSA-N 4H-1,2,4-triazole Chemical compound C=1N=CNN=1 NSPMIYGKQJPBQR-UHFFFAOYSA-N 0.000 description 1
- VQVIHDPBMFABCQ-UHFFFAOYSA-N 5-(1,3-dioxo-2-benzofuran-5-carbonyl)-2-benzofuran-1,3-dione Chemical compound C1=C2C(=O)OC(=O)C2=CC(C(C=2C=C3C(=O)OC(=O)C3=CC=2)=O)=C1 VQVIHDPBMFABCQ-UHFFFAOYSA-N 0.000 description 1
- ZHBXLZQQVCDGPA-UHFFFAOYSA-N 5-[(1,3-dioxo-2-benzofuran-5-yl)sulfonyl]-2-benzofuran-1,3-dione Chemical compound C1=C2C(=O)OC(=O)C2=CC(S(=O)(=O)C=2C=C3C(=O)OC(C3=CC=2)=O)=C1 ZHBXLZQQVCDGPA-UHFFFAOYSA-N 0.000 description 1
- RZWRYPGAUIOOMK-UHFFFAOYSA-N 5-nitroso-8-quinolinol Chemical compound C1=CN=C2C(O)=CC=C(N=O)C2=C1 RZWRYPGAUIOOMK-UHFFFAOYSA-N 0.000 description 1
- MARUHZGHZWCEQU-UHFFFAOYSA-N 5-phenyl-2h-tetrazole Chemical compound C1=CC=CC=C1C1=NNN=N1 MARUHZGHZWCEQU-UHFFFAOYSA-N 0.000 description 1
- SNCJAJRILVFXAE-UHFFFAOYSA-N 9h-fluorene-2,7-diamine Chemical compound NC1=CC=C2C3=CC=C(N)C=C3CC2=C1 SNCJAJRILVFXAE-UHFFFAOYSA-N 0.000 description 1
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 235000021357 Behenic acid Nutrition 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- FCKYPQBAHLOOJQ-UHFFFAOYSA-N Cyclohexane-1,2-diaminetetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)C1CCCCC1N(CC(O)=O)CC(O)=O FCKYPQBAHLOOJQ-UHFFFAOYSA-N 0.000 description 1
- 239000009261 D 400 Substances 0.000 description 1
- 239000004641 Diallyl-phthalate Substances 0.000 description 1
- ORAWFNKFUWGRJG-UHFFFAOYSA-N Docosanamide Chemical compound CCCCCCCCCCCCCCCCCCCCCC(N)=O ORAWFNKFUWGRJG-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 239000004386 Erythritol Substances 0.000 description 1
- UNXHWFMMPAWVPI-UHFFFAOYSA-N Erythritol Natural products OCC(O)C(O)CO UNXHWFMMPAWVPI-UHFFFAOYSA-N 0.000 description 1
- XXRCUYVCPSWGCC-UHFFFAOYSA-N Ethyl pyruvate Chemical compound CCOC(=O)C(C)=O XXRCUYVCPSWGCC-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- UBUCNCOMADRQHX-UHFFFAOYSA-N N-Nitrosodiphenylamine Chemical compound C=1C=CC=CC=1N(N=O)C1=CC=CC=C1 UBUCNCOMADRQHX-UHFFFAOYSA-N 0.000 description 1
- XQVWYOYUZDUNRW-UHFFFAOYSA-N N-Phenyl-1-naphthylamine Chemical compound C=1C=CC2=CC=CC=C2C=1NC1=CC=CC=C1 XQVWYOYUZDUNRW-UHFFFAOYSA-N 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- SLMPOMXBUNUQKZ-UHFFFAOYSA-N NC1=CC=C(C=C1)C(C(C(C1=CC=C(C=C1)N)(F)F)(F)F)(F)F.NC=1C(=C(C=CC1)C(F)(F)F)N Chemical compound NC1=CC=C(C=C1)C(C(C(C1=CC=C(C=C1)N)(F)F)(F)F)(F)F.NC=1C(=C(C=CC1)C(F)(F)F)N SLMPOMXBUNUQKZ-UHFFFAOYSA-N 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Chemical group C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical group OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 1
- 206010034972 Photosensitivity reaction Diseases 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-IGMARMGPSA-N Protium Chemical compound [1H] YZCKVEUIGOORGS-IGMARMGPSA-N 0.000 description 1
- 239000005700 Putrescine Substances 0.000 description 1
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical group C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 1
- 229910052581 Si3N4 Inorganic materials 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- ZJCCRDAZUWHFQH-UHFFFAOYSA-N Trimethylolpropane Chemical compound CCC(CO)(CO)CO ZJCCRDAZUWHFQH-UHFFFAOYSA-N 0.000 description 1
- QYKIQEUNHZKYBP-UHFFFAOYSA-N Vinyl ether Chemical compound C=COC=C QYKIQEUNHZKYBP-UHFFFAOYSA-N 0.000 description 1
- 229910021536 Zeolite Inorganic materials 0.000 description 1
- DPQRMIPRAHPPNE-UHFFFAOYSA-N [(1-oxo-1-phenylpropan-2-ylidene)amino] acetate Chemical compound CC(=O)ON=C(C)C(=O)C1=CC=CC=C1 DPQRMIPRAHPPNE-UHFFFAOYSA-N 0.000 description 1
- ZNZDJSGUWLGTLA-UHFFFAOYSA-N [(1-oxo-1-phenylpropan-2-ylidene)amino] benzoate Chemical compound C=1C=CC=CC=1C(=O)C(C)=NOC(=O)C1=CC=CC=C1 ZNZDJSGUWLGTLA-UHFFFAOYSA-N 0.000 description 1
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical group C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 1
- YPCHGLDQZXOZFW-UHFFFAOYSA-N [2-[[4-methyl-3-[[3-prop-2-enoyloxy-2,2-bis(prop-2-enoyloxymethyl)propoxy]carbonylamino]phenyl]carbamoyloxymethyl]-3-prop-2-enoyloxy-2-(prop-2-enoyloxymethyl)propyl] prop-2-enoate Chemical compound CC1=CC=C(NC(=O)OCC(COC(=O)C=C)(COC(=O)C=C)COC(=O)C=C)C=C1NC(=O)OCC(COC(=O)C=C)(COC(=O)C=C)COC(=O)C=C YPCHGLDQZXOZFW-UHFFFAOYSA-N 0.000 description 1
- JOFSEHHMYPDWFR-UHFFFAOYSA-N [3-(acetyloxymethyl)-2-hydroxy-5-methylphenyl]methyl acetate Chemical compound CC(=O)OCC1=CC(C)=CC(COC(C)=O)=C1O JOFSEHHMYPDWFR-UHFFFAOYSA-N 0.000 description 1
- HIVQCJOGAHNXBO-UHFFFAOYSA-N [3-prop-2-enoyloxy-2-[[3-prop-2-enoyloxy-2,2-bis(prop-2-enoyloxymethyl)propoxy]methyl]-2-(prop-2-enoyloxymethyl)propyl] propanoate Chemical compound CCC(=O)OCC(COC(=O)C=C)(COC(=O)C=C)COCC(COC(=O)C=C)(COC(=O)C=C)COC(=O)C=C HIVQCJOGAHNXBO-UHFFFAOYSA-N 0.000 description 1
- OXIKYYJDTWKERT-UHFFFAOYSA-N [4-(aminomethyl)cyclohexyl]methanamine Chemical compound NCC1CCC(CN)CC1 OXIKYYJDTWKERT-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 125000004062 acenaphthenyl group Chemical group C1(CC2=CC=CC3=CC=CC1=C23)* 0.000 description 1
- TUVYSBJZBYRDHP-UHFFFAOYSA-N acetic acid;methoxymethane Chemical compound COC.CC(O)=O TUVYSBJZBYRDHP-UHFFFAOYSA-N 0.000 description 1
- NJYZCEFQAIUHSD-UHFFFAOYSA-N acetoguanamine Chemical compound CC1=NC(N)=NC(N)=N1 NJYZCEFQAIUHSD-UHFFFAOYSA-N 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 229920006397 acrylic thermoplastic Polymers 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 238000005054 agglomeration Methods 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000007754 air knife coating Methods 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 229910021417 amorphous silicon Inorganic materials 0.000 description 1
- 239000003945 anionic surfactant Substances 0.000 description 1
- 125000005577 anthracene group Chemical group 0.000 description 1
- 229940053200 antiepileptics fatty acid derivative Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 150000004984 aromatic diamines Chemical class 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 150000008365 aromatic ketones Chemical class 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 125000000732 arylene group Chemical group 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- PAQKRBLMHQTWAS-UHFFFAOYSA-N azane;n-hydroxy-n-naphthalen-1-ylnitrous amide Chemical compound N.C1=CC=C2C(N(N=O)O)=CC=CC2=C1 PAQKRBLMHQTWAS-UHFFFAOYSA-N 0.000 description 1
- 125000000751 azo group Chemical group [*]N=N[*] 0.000 description 1
- 238000007611 bar coating method Methods 0.000 description 1
- 229940116226 behenic acid Drugs 0.000 description 1
- GCAIEATUVJFSMC-UHFFFAOYSA-N benzene-1,2,3,4-tetracarboxylic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C(C(O)=O)=C1C(O)=O GCAIEATUVJFSMC-UHFFFAOYSA-N 0.000 description 1
- JGFLAAWSLCPCDY-UHFFFAOYSA-N benzene;cyclopenta-1,3-diene;iron Chemical class [Fe].C1C=CC=C1.C1=CC=CC=C1 JGFLAAWSLCPCDY-UHFFFAOYSA-N 0.000 description 1
- RWCCWEUUXYIKHB-UHFFFAOYSA-N benzophenone Chemical compound C=1C=CC=CC=1C(=O)C1=CC=CC=C1 RWCCWEUUXYIKHB-UHFFFAOYSA-N 0.000 description 1
- 239000012965 benzophenone Substances 0.000 description 1
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical class C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 1
- QRUDEWIWKLJBPS-UHFFFAOYSA-N benzotriazole Chemical compound C1=CC=C2N[N][N]C2=C1 QRUDEWIWKLJBPS-UHFFFAOYSA-N 0.000 description 1
- 239000012964 benzotriazole Substances 0.000 description 1
- 150000005130 benzoxazines Chemical class 0.000 description 1
- TUQQUUXMCKXGDI-UHFFFAOYSA-N bis(3-aminophenyl)methanone Chemical compound NC1=CC=CC(C(=O)C=2C=C(N)C=CC=2)=C1 TUQQUUXMCKXGDI-UHFFFAOYSA-N 0.000 description 1
- QUDWYFHPNIMBFC-UHFFFAOYSA-N bis(prop-2-enyl) benzene-1,2-dicarboxylate Chemical compound C=CCOC(=O)C1=CC=CC=C1C(=O)OCC=C QUDWYFHPNIMBFC-UHFFFAOYSA-N 0.000 description 1
- HEYYMASVKJXRDV-UHFFFAOYSA-N bis[(3-ethyloxetan-3-yl)methyl] benzene-1,4-dicarboxylate Chemical compound C=1C=C(C(=O)OCC2(CC)COC2)C=CC=1C(=O)OCC1(CC)COC1 HEYYMASVKJXRDV-UHFFFAOYSA-N 0.000 description 1
- 150000001639 boron compounds Chemical class 0.000 description 1
- WKDNYTOXBCRNPV-UHFFFAOYSA-N bpda Chemical compound C1=C2C(=O)OC(=O)C2=CC(C=2C=C3C(=O)OC(C3=CC=2)=O)=C1 WKDNYTOXBCRNPV-UHFFFAOYSA-N 0.000 description 1
- 125000006226 butoxyethyl group Chemical group 0.000 description 1
- 229930188620 butyrolactone Natural products 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 239000003093 cationic surfactant Substances 0.000 description 1
- 229910010293 ceramic material Inorganic materials 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- QZHPTGXQGDFGEN-UHFFFAOYSA-N chromene Chemical group C1=CC=C2C=C[CH]OC2=C1 QZHPTGXQGDFGEN-UHFFFAOYSA-N 0.000 description 1
- 125000005578 chrysene group Chemical group 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- 125000000332 coumarinyl group Chemical class O1C(=O)C(=CC2=CC=CC=C12)* 0.000 description 1
- LDHQCZJRKDOVOX-NSCUHMNNSA-N crotonic acid Chemical compound C\C=C\C(O)=O LDHQCZJRKDOVOX-NSCUHMNNSA-N 0.000 description 1
- 238000007766 curtain coating Methods 0.000 description 1
- 150000004292 cyclic ethers Chemical group 0.000 description 1
- VKIRRGRTJUUZHS-UHFFFAOYSA-N cyclohexane-1,4-diamine Chemical compound NC1CCC(N)CC1 VKIRRGRTJUUZHS-UHFFFAOYSA-N 0.000 description 1
- ZQWRZCZEOLZBQF-UHFFFAOYSA-N cyclopentane-1,3-diamine Chemical compound NC1CCC(N)C1 ZQWRZCZEOLZBQF-UHFFFAOYSA-N 0.000 description 1
- 238000000151 deposition Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 229940043279 diisopropylamine Drugs 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- HNPSIPDUKPIQMN-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical compound O=[Si]=O.O=[Al]O[Al]=O HNPSIPDUKPIQMN-UHFFFAOYSA-N 0.000 description 1
- 238000003618 dip coating Methods 0.000 description 1
- VFHVQBAGLAREND-UHFFFAOYSA-N diphenylphosphoryl-(2,4,6-trimethylphenyl)methanone Chemical compound CC1=CC(C)=CC(C)=C1C(=O)P(=O)(C=1C=CC=CC=1)C1=CC=CC=C1 VFHVQBAGLAREND-UHFFFAOYSA-N 0.000 description 1
- 238000007598 dipping method Methods 0.000 description 1
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical compound [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 208000018459 dissociative disease Diseases 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- BXKDSDJJOVIHMX-UHFFFAOYSA-N edrophonium chloride Chemical compound [Cl-].CC[N+](C)(C)C1=CC=CC(O)=C1 BXKDSDJJOVIHMX-UHFFFAOYSA-N 0.000 description 1
- 238000007772 electroless plating Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- UNXHWFMMPAWVPI-ZXZARUISSA-N erythritol Chemical group OC[C@H](O)[C@H](O)CO UNXHWFMMPAWVPI-ZXZARUISSA-N 0.000 description 1
- 235000019414 erythritol Nutrition 0.000 description 1
- 229940009714 erythritol Drugs 0.000 description 1
- 125000005678 ethenylene group Chemical group [H]C([*:1])=C([H])[*:2] 0.000 description 1
- IJUHLFUALMUWOM-UHFFFAOYSA-N ethyl 3-methoxypropanoate Chemical compound CCOC(=O)CCOC IJUHLFUALMUWOM-UHFFFAOYSA-N 0.000 description 1
- YDMWUMUNUXUYKT-UHFFFAOYSA-N ethyl [(1-oxo-1-phenylpropan-2-ylidene)amino] carbonate Chemical compound CCOC(=O)ON=C(C)C(=O)C1=CC=CC=C1 YDMWUMUNUXUYKT-UHFFFAOYSA-N 0.000 description 1
- UHESRSKEBRADOO-UHFFFAOYSA-N ethyl carbamate;prop-2-enoic acid Chemical class OC(=O)C=C.CCOC(N)=O UHESRSKEBRADOO-UHFFFAOYSA-N 0.000 description 1
- 229940117360 ethyl pyruvate Drugs 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 238000007765 extrusion coating Methods 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 238000007756 gravure coating Methods 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- 229910052734 helium Inorganic materials 0.000 description 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 1
- 125000002192 heptalenyl group Chemical group 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 125000004836 hexamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- ACCCMOQWYVYDOT-UHFFFAOYSA-N hexane-1,1-diol Chemical compound CCCCCC(O)O ACCCMOQWYVYDOT-UHFFFAOYSA-N 0.000 description 1
- NAQMVNRVTILPCV-UHFFFAOYSA-N hexane-1,6-diamine Chemical compound NCCCCCCN NAQMVNRVTILPCV-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 125000003427 indacenyl group Chemical group 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 239000010954 inorganic particle Substances 0.000 description 1
- 238000005040 ion trap Methods 0.000 description 1
- 238000010884 ion-beam technique Methods 0.000 description 1
- JEIPFZHSYJVQDO-UHFFFAOYSA-N iron(III) oxide Inorganic materials O=[Fe]O[Fe]=O JEIPFZHSYJVQDO-UHFFFAOYSA-N 0.000 description 1
- 230000001678 irradiating effect Effects 0.000 description 1
- BHIWKHZACMWKOJ-UHFFFAOYSA-N isobutyric acid methyl ester Natural products COC(=O)C(C)C BHIWKHZACMWKOJ-UHFFFAOYSA-N 0.000 description 1
- LDHQCZJRKDOVOX-IHWYPQMZSA-N isocrotonic acid Chemical compound C\C=C/C(O)=O LDHQCZJRKDOVOX-IHWYPQMZSA-N 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- IQPQWNKOIGAROB-UHFFFAOYSA-N isocyanate group Chemical group [N-]=C=O IQPQWNKOIGAROB-UHFFFAOYSA-N 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- ZFSLODLOARCGLH-UHFFFAOYSA-N isocyanuric acid Chemical compound OC1=NC(O)=NC(O)=N1 ZFSLODLOARCGLH-UHFFFAOYSA-N 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical group C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 239000004922 lacquer Substances 0.000 description 1
- 238000003475 lamination Methods 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- JDSHMPZPIAZGSV-UHFFFAOYSA-N melamine powder Natural products NC1=NC(N)=NC(N)=N1 JDSHMPZPIAZGSV-UHFFFAOYSA-N 0.000 description 1
- 150000002734 metacrylic acid derivatives Chemical class 0.000 description 1
- 229910001507 metal halide Inorganic materials 0.000 description 1
- 150000005309 metal halides Chemical class 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 239000007769 metal material Substances 0.000 description 1
- 125000005395 methacrylic acid group Chemical group 0.000 description 1
- LVHBHZANLOWSRM-UHFFFAOYSA-N methylenebutanedioic acid Natural products OC(=O)CC(=C)C(O)=O LVHBHZANLOWSRM-UHFFFAOYSA-N 0.000 description 1
- 238000004377 microelectronic Methods 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- KADGVXXDDWDKBX-UHFFFAOYSA-N naphthalene-1,2,4,5-tetracarboxylic acid Chemical compound OC(=O)C1=CC=CC2=C(C(O)=O)C(C(=O)O)=CC(C(O)=O)=C21 KADGVXXDDWDKBX-UHFFFAOYSA-N 0.000 description 1
- HDLRSIQOZFLEPK-UHFFFAOYSA-N naphthalene-1,2,5,8-tetracarboxylic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C2=C(C(O)=O)C(C(=O)O)=CC=C21 HDLRSIQOZFLEPK-UHFFFAOYSA-N 0.000 description 1
- DSCIZKMHZPGBNI-UHFFFAOYSA-N naphthalene-1,3,5,8-tetracarboxylic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C2=CC(C(=O)O)=CC(C(O)=O)=C21 DSCIZKMHZPGBNI-UHFFFAOYSA-N 0.000 description 1
- KQSABULTKYLFEV-UHFFFAOYSA-N naphthalene-1,5-diamine Chemical compound C1=CC=C2C(N)=CC=CC2=C1N KQSABULTKYLFEV-UHFFFAOYSA-N 0.000 description 1
- DOBFTMLCEYUAQC-UHFFFAOYSA-N naphthalene-2,3,6,7-tetracarboxylic acid Chemical compound OC(=O)C1=C(C(O)=O)C=C2C=C(C(O)=O)C(C(=O)O)=CC2=C1 DOBFTMLCEYUAQC-UHFFFAOYSA-N 0.000 description 1
- YTVNOVQHSGMMOV-UHFFFAOYSA-N naphthalenetetracarboxylic dianhydride Chemical compound C1=CC(C(=O)OC2=O)=C3C2=CC=C2C(=O)OC(=O)C1=C32 YTVNOVQHSGMMOV-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- SLCVBVWXLSEKPL-UHFFFAOYSA-N neopentyl glycol Chemical compound OCC(C)(C)CO SLCVBVWXLSEKPL-UHFFFAOYSA-N 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- 229920003986 novolac Polymers 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- 238000007645 offset printing Methods 0.000 description 1
- 239000012788 optical film Substances 0.000 description 1
- 150000001451 organic peroxides Chemical class 0.000 description 1
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical group C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 description 1
- 150000004893 oxazines Chemical class 0.000 description 1
- 150000002921 oxetanes Chemical class 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- MPQXHAGKBWFSNV-UHFFFAOYSA-N oxidophosphanium Chemical class [PH3]=O MPQXHAGKBWFSNV-UHFFFAOYSA-N 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 125000005582 pentacene group Chemical group 0.000 description 1
- GUVXZFRDPCKWEM-UHFFFAOYSA-N pentalene group Chemical group C1=CC=C2C=CC=C12 GUVXZFRDPCKWEM-UHFFFAOYSA-N 0.000 description 1
- 125000004817 pentamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- 125000005010 perfluoroalkyl group Chemical group 0.000 description 1
- 125000002080 perylenyl group Chemical group C1(=CC=C2C=CC=C3C4=CC=CC5=CC=CC(C1=C23)=C45)* 0.000 description 1
- YNPNZTXNASCQKK-UHFFFAOYSA-N phenanthrene Chemical group C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 description 1
- JGGWKXMPICYBKC-UHFFFAOYSA-N phenanthrene-1,8,9,10-tetracarboxylic acid Chemical compound C1=CC=C(C(O)=O)C2=C(C(O)=O)C(C(O)=O)=C3C(C(=O)O)=CC=CC3=C21 JGGWKXMPICYBKC-UHFFFAOYSA-N 0.000 description 1
- RDOWQLZANAYVLL-UHFFFAOYSA-N phenanthridine Chemical group C1=CC=C2C3=CC=CC=C3C=NC2=C1 RDOWQLZANAYVLL-UHFFFAOYSA-N 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- 229950000688 phenothiazine Drugs 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- GJSGGHOYGKMUPT-UHFFFAOYSA-N phenoxathiine Chemical group C1=CC=C2OC3=CC=CC=C3SC2=C1 GJSGGHOYGKMUPT-UHFFFAOYSA-N 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- DYFXGORUJGZJCA-UHFFFAOYSA-N phenylmethanediamine Chemical compound NC(N)C1=CC=CC=C1 DYFXGORUJGZJCA-UHFFFAOYSA-N 0.000 description 1
- 150000003009 phosphonic acids Chemical class 0.000 description 1
- 238000000016 photochemical curing Methods 0.000 description 1
- 230000036211 photosensitivity Effects 0.000 description 1
- LFSXCDWNBUNEEM-UHFFFAOYSA-N phthalazine Chemical group C1=NN=CC2=CC=CC=C21 LFSXCDWNBUNEEM-UHFFFAOYSA-N 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- CLYVDMAATCIVBF-UHFFFAOYSA-N pigment red 224 Chemical compound C=12C3=CC=C(C(OC4=O)=O)C2=C4C=CC=1C1=CC=C2C(=O)OC(=O)C4=CC=C3C1=C42 CLYVDMAATCIVBF-UHFFFAOYSA-N 0.000 description 1
- 238000009832 plasma treatment Methods 0.000 description 1
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 1
- 229920001515 polyalkylene glycol Polymers 0.000 description 1
- 229910021420 polycrystalline silicon Inorganic materials 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920000193 polymethacrylate Polymers 0.000 description 1
- 229920005591 polysilicon Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 230000001376 precipitating effect Effects 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 238000007639 printing Methods 0.000 description 1
- CDQXHVDVGLVACE-UHFFFAOYSA-N propan-2-amine Chemical compound [CH2]C(C)N CDQXHVDVGLVACE-UHFFFAOYSA-N 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- ILPVOWZUBFRIAX-UHFFFAOYSA-N propyl 2-oxopropanoate Chemical compound CCCOC(=O)C(C)=O ILPVOWZUBFRIAX-UHFFFAOYSA-N 0.000 description 1
- AOHJOMMDDJHIJH-UHFFFAOYSA-N propylenediamine Chemical compound CC(N)CN AOHJOMMDDJHIJH-UHFFFAOYSA-N 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- MIROPXUFDXCYLG-UHFFFAOYSA-N pyridine-2,5-diamine Chemical compound NC1=CC=C(N)N=C1 MIROPXUFDXCYLG-UHFFFAOYSA-N 0.000 description 1
- 229940079877 pyrogallol Drugs 0.000 description 1
- 239000010453 quartz Substances 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 150000004053 quinones Chemical class 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000027756 respiratory electron transport chain Effects 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- 230000000630 rising effect Effects 0.000 description 1
- 150000003872 salicylic acid derivatives Chemical class 0.000 description 1
- 238000007650 screen-printing Methods 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 239000003566 sealing material Substances 0.000 description 1
- 230000001235 sensitizing effect Effects 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- HQVNEWCFYHHQES-UHFFFAOYSA-N silicon nitride Chemical compound N12[Si]34N5[Si]62N3[Si]51N64 HQVNEWCFYHHQES-UHFFFAOYSA-N 0.000 description 1
- 229910052814 silicon oxide Inorganic materials 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000004544 sputter deposition Methods 0.000 description 1
- 125000004079 stearyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 150000005846 sugar alcohols Chemical class 0.000 description 1
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- ISXSCDLOGDJUNJ-UHFFFAOYSA-N tert-butyl prop-2-enoate Chemical compound CC(C)(C)OC(=O)C=C ISXSCDLOGDJUNJ-UHFFFAOYSA-N 0.000 description 1
- 125000001935 tetracenyl group Chemical group C1(=CC=CC2=CC3=CC4=CC=CC=C4C=C3C=C12)* 0.000 description 1
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 1
- 150000003536 tetrazoles Chemical group 0.000 description 1
- 229920001187 thermosetting polymer Polymers 0.000 description 1
- GVIJJXMXTUZIOD-UHFFFAOYSA-N thianthrene Chemical group C1=CC=C2SC3=CC=CC=C3SC2=C1 GVIJJXMXTUZIOD-UHFFFAOYSA-N 0.000 description 1
- 239000010409 thin film Substances 0.000 description 1
- 125000005000 thioaryl group Chemical group 0.000 description 1
- 125000000101 thioether group Chemical group 0.000 description 1
- 150000003568 thioethers Chemical class 0.000 description 1
- 150000003585 thioureas Chemical class 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 125000005424 tosyloxy group Chemical group S(=O)(=O)(C1=CC=C(C)C=C1)O* 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 125000004953 trihalomethyl group Chemical group 0.000 description 1
- 125000005580 triphenylene group Chemical group 0.000 description 1
- GRPURDFRFHUDSP-UHFFFAOYSA-N tris(prop-2-enyl) benzene-1,2,4-tricarboxylate Chemical compound C=CCOC(=O)C1=CC=C(C(=O)OCC=C)C(C(=O)OCC=C)=C1 GRPURDFRFHUDSP-UHFFFAOYSA-N 0.000 description 1
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 239000010937 tungsten Substances 0.000 description 1
- 239000006097 ultraviolet radiation absorber Substances 0.000 description 1
- 238000000870 ultraviolet spectroscopy Methods 0.000 description 1
- 150000003673 urethanes Chemical class 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- GPPXJZIENCGNKB-UHFFFAOYSA-N vanadium Chemical compound [V]#[V] GPPXJZIENCGNKB-UHFFFAOYSA-N 0.000 description 1
- 238000007740 vapor deposition Methods 0.000 description 1
- ZTWTYVWXUKTLCP-UHFFFAOYSA-N vinylphosphonic acid Chemical compound OP(O)(=O)C=C ZTWTYVWXUKTLCP-UHFFFAOYSA-N 0.000 description 1
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 1
- 239000010457 zeolite Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F290/00—Macromolecular compounds obtained by polymerising monomers on to polymers modified by introduction of aliphatic unsaturated end or side groups
- C08F290/08—Macromolecular compounds obtained by polymerising monomers on to polymers modified by introduction of aliphatic unsaturated end or side groups on to polymers modified by introduction of unsaturated side groups
- C08F290/14—Polymers provided for in subclass C08G
- C08F290/145—Polyamides; Polyesteramides; Polyimides
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B15/00—Layered products comprising a layer of metal
- B32B15/04—Layered products comprising a layer of metal comprising metal as the main or only constituent of a layer, which is next to another layer of the same or of a different material
- B32B15/08—Layered products comprising a layer of metal comprising metal as the main or only constituent of a layer, which is next to another layer of the same or of a different material of synthetic resin
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/38—Polymerisation using regulators, e.g. chain terminating agents, e.g. telomerisation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/46—Polymerisation initiated by wave energy or particle radiation
- C08F2/48—Polymerisation initiated by wave energy or particle radiation by ultraviolet or visible light
- C08F2/50—Polymerisation initiated by wave energy or particle radiation by ultraviolet or visible light with sensitising agents
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F290/00—Macromolecular compounds obtained by polymerising monomers on to polymers modified by introduction of aliphatic unsaturated end or side groups
- C08F290/08—Macromolecular compounds obtained by polymerising monomers on to polymers modified by introduction of aliphatic unsaturated end or side groups on to polymers modified by introduction of unsaturated side groups
- C08F290/14—Polymers provided for in subclass C08G
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/10—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L79/00—Compositions of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing nitrogen with or without oxygen or carbon only, not provided for in groups C08L61/00 - C08L77/00
- C08L79/04—Polycondensates having nitrogen-containing heterocyclic rings in the main chain; Polyhydrazides; Polyamide acids or similar polyimide precursors
- C08L79/08—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/027—Non-macromolecular photopolymerisable compounds having carbon-to-carbon double bonds, e.g. ethylenic compounds
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/027—Non-macromolecular photopolymerisable compounds having carbon-to-carbon double bonds, e.g. ethylenic compounds
- G03F7/028—Non-macromolecular photopolymerisable compounds having carbon-to-carbon double bonds, e.g. ethylenic compounds with photosensitivity-increasing substances, e.g. photoinitiators
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/027—Non-macromolecular photopolymerisable compounds having carbon-to-carbon double bonds, e.g. ethylenic compounds
- G03F7/032—Non-macromolecular photopolymerisable compounds having carbon-to-carbon double bonds, e.g. ethylenic compounds with binders
- G03F7/037—Non-macromolecular photopolymerisable compounds having carbon-to-carbon double bonds, e.g. ethylenic compounds with binders the binders being polyamides or polyimides
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/09—Photosensitive materials characterised by structural details, e.g. supports, auxiliary layers
- G03F7/095—Photosensitive materials characterised by structural details, e.g. supports, auxiliary layers having more than one photosensitive layer
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/20—Exposure; Apparatus therefor
Definitions
- the present invention relates to a curable resin composition containing a heterocycle-containing polymer precursor, a cured film, a laminate, a method for producing a cured film, a semiconductor device, and a thermal base generator.
- Resins obtained by cyclizing and curing a precursor of a polymer containing a heterocycle such as a polyimide resin and a polybenzoxazole resin have various heat resistance and insulating properties, and thus are various. It is applied to various uses.
- the use is not particularly limited, but when the semiconductor device for mounting is taken as an example, it may be used as a material of an insulating film or a sealing material or as a protective film. It is also used as a base film or coverlay for flexible substrates.
- the heterocycle-containing polymer precursor is used as a curable resin composition containing the heterocycle-containing polymer precursor.
- the curable resin composition can be applied by a known coating method or the like, for example, the shape, size, and application position of the curable resin composition to be applied have a high degree of freedom in designing, etc. It can be said that it has excellent adaptability.
- the curable resin composition containing the heterocycle-containing polymer precursor is expected to be industrially applied and developed from the viewpoint of such excellent manufacturing adaptability.
- Patent Document 1 an acidic compound that generates a base when heated to 40° C. or higher, and a thermal base generator containing at least one selected from ammonium salts having an anion having a pKa1 of 0 to 4 and an ammonium cation, And a thermosetting resin, are described.
- Patent Document 2 describes a curable resin composition containing a polyimide precursor having a specific structural unit and a compound that generates a radical upon irradiation with an actinic ray.
- cyclization does not easily proceed during storage of the curable resin composition (for example, storage at room temperature (25° C., the same applies below)). Further, it is desired to provide a curable resin composition having excellent elongation at break of the obtained cured product.
- the fact that the cyclization does not easily progress during storage of the curable resin composition is also referred to as “excellent storage stability of the curable resin composition”.
- One embodiment of the present invention is excellent in storage stability, and a curable resin composition having excellent elongation at break of the obtained cured film, a cured film obtained by curing the curable resin composition, and the cured film.
- An object of the present invention is to provide a laminated body including the cured film, a method for producing the cured film, and a semiconductor device including the cured film or the laminated body.
- Another object of the present invention is to provide a novel thermal base generator.
- ⁇ 1> a heterocycle-containing polymer precursor, and Including a compound represented by the following formula (1-1): Curable resin composition.
- R 1 and R 2 each independently represent a monovalent organic group
- L 1 represents a divalent linking group
- R 3 represents a hydrogen atom or a monovalent organic group.
- R 1 and R 2 may combine to form a ring structure.
- R 1 and R 2 are each independently a hydrocarbon group.
- L 1 is a 1,2-ethylene group, a 1,3-propanediyl group, a 1,2-cyclohexanediyl group, a cisvinylene group, a 1,2-phenylene group, a 1,2-phenylenemethylene group, or
- R 3 is a hydrogen atom, an alkyl group, or an aryl group.
- ⁇ 6> The curable resin composition according to any one of ⁇ 1> to ⁇ 5>, wherein the compound represented by the formula (1-1) has a molecular weight of 100 or more and 2,000 or less.
- the heterocycle-containing polymer precursor contains at least one precursor selected from the group consisting of a polyimide precursor and a polybenzoxazole precursor.
- a 1 and A 2 each independently represent an oxygen atom or —NH—
- R 111 represents a divalent organic group
- R 115 represents a tetravalent organic group
- R 1 113 and R 114 each independently represent a hydrogen atom or a monovalent organic group.
- ⁇ 11> The curable resin composition according to ⁇ 10>, wherein at least one of R 113 and R 114 in the above formula (1) contains a radically polymerizable group.
- ⁇ 12> The curable resin composition according to any one of ⁇ 1> to ⁇ 11>, which is used for forming an interlayer insulating film for a rewiring layer.
- ⁇ 13> A cured film obtained by curing the curable resin composition according to any one of ⁇ 1> to ⁇ 12>.
- ⁇ 14> A laminate including two or more layers of the cured film according to ⁇ 13> and a metal layer between any of the cured films.
- ⁇ 15> A method for producing a cured film, comprising a film forming step of applying the curable resin composition according to any one of ⁇ 1> to ⁇ 12> to a substrate to form a film.
- ⁇ 17> A semiconductor device comprising the cured film according to ⁇ 13> or the laminate according to ⁇ 14>.
- ⁇ 18> A thermal base generator represented by the following formula (1-2).
- R 21 and R 22 each independently represent a monovalent organic group
- L 21 represents a divalent linking group
- atoms at both ends of L 21 are carbon atoms.
- R 23 represents a hydrogen atom or a monovalent organic group.
- a curable resin composition having excellent storage stability and excellent elongation at break of a cured film obtained, a cured film obtained by curing the curable resin composition, and the above cured film
- a laminated body including a film, a method for producing the cured film, and a semiconductor device including the cured film or the laminated body.
- a novel thermal base generator is provided.
- the numerical range represented by using the symbol “to” means a range including the numerical values before and after "to” as the lower limit value and the upper limit value, respectively.
- the term “process” is meant to include not only an independent process but also a process that cannot be clearly distinguished from other processes as long as the intended action of the process can be achieved.
- the group (atomic group) in the present specification the notation in which substitution and non-substitution are not included includes a group (atomic group) having no substituent and a group (atomic group) having a substituent.
- the “alkyl group” includes not only an alkyl group having no substituent (unsubstituted alkyl group) but also an alkyl group having a substituent (substituted alkyl group).
- the term “exposure” as used herein includes not only exposure using light but also exposure using particle beams such as electron beams and ion beams. Examples of the light used for exposure include a bright line spectrum of a mercury lamp, far ultraviolet rays represented by an excimer laser, extreme ultraviolet rays (EUV light), X-rays, active rays such as electron rays, or radiation.
- (meth)acrylate means both “acrylate” and “methacrylate”, or either
- (meth)acrylic means both “acrylic” and “methacrylic”, or
- (meth)acryloyl means both or either of “acryloyl” and “methacryloyl”.
- Me in the structural formula represents a methyl group
- Et represents an ethyl group
- Bu represents a butyl group
- Ph represents a phenyl group.
- the total solid content refers to the total mass of components excluding the solvent from all components of the composition.
- the solid content concentration is a mass percentage of the other components excluding the solvent with respect to the total mass of the composition.
- the weight average molecular weight (Mw) and the number average molecular weight (Mn) are defined as polystyrene equivalent values according to gel permeation chromatography (GPC measurement), unless otherwise specified.
- the weight average molecular weight (Mw) and the number average molecular weight (Mn) are, for example, HLC-8220GPC (manufactured by Tosoh Corporation), and the columns are guard columns HZ-L, TSKgel Super HZM-M, and TSKgel.
- a third layer or element may be further interposed between the reference layer and the other layer, and the reference layer and the other layer do not have to be in contact with each other.
- the direction in which the layers are stacked on the substrate is referred to as "upper", or, if there is a photosensitive layer, the direction from the substrate to the photosensitive layer is referred to as "upper”, The opposite direction is called "down". It should be noted that such setting of the vertical direction is for convenience in the present specification, and in an actual aspect, the “up” direction in the present specification may be different from the vertical upward direction.
- the composition may include, as each component included in the composition, two or more kinds of compounds corresponding to the component.
- the content of each component in the composition means the total content of all compounds corresponding to the component.
- the physical property values are values under the conditions of a temperature of 23° C. and an atmospheric pressure of 101,325 Pa (1 atmospheric pressure), unless otherwise specified.
- a combination of the preferred embodiments is a more preferred embodiment.
- the curable resin composition of the present invention (hereinafter, also simply referred to as “the composition of the present invention”) contains a heterocycle-containing polymer precursor and a compound represented by the formula (1-1). Further, the curable resin composition of the present invention preferably further contains a photo radical polymerization initiator described below and a radical polymerizable compound described below.
- the curable resin composition of the present invention is excellent in storage stability and the elongation at break of the obtained cured film.
- the mechanism by which the above effects are obtained is unknown, but is presumed as follows.
- the compound represented by the formula (1-1) contained in the curable resin composition of the present invention is excellent in storage stability because it is difficult to decompose at storage temperature such as room temperature, and is hardened by, for example, heating. It is considered that since the generation efficiency of the base is excellent at this time, cyclization of the heterocycle-containing polymer precursor is likely to proceed and the resulting cured film has an excellent elongation at break.
- the curable resin composition contains the compound represented by the formula (1-1), the cyclization of the heterocycle-containing polymer precursor is likely to proceed due to heating or the like as described above, so that the base material such as copper is used. It is thought that it is also easy to have excellent adhesion with. That is, it is considered that by using the curable resin composition of the present invention, a laminate having excellent adhesion between layers can be easily obtained.
- Patent Documents 1 and 2 do not describe or suggest a curable resin composition containing a compound represented by the formula (1-1).
- the components contained in the curable resin composition of the present invention will be described in detail.
- the curable resin composition of the present invention contains a heterocycle-containing polymer precursor.
- the curable resin composition of the present invention preferably contains, as the heterocycle-containing polymer precursor, at least one precursor selected from the group consisting of a polyimide precursor and a polybenzoxazole precursor. More preferably.
- the polyimide precursor preferably has a repeating unit represented by the following formula (1).
- a 1 and A 2 each independently represent an oxygen atom or —NH—
- R 111 represents a divalent organic group
- R 115 represents a tetravalent organic group
- R 1 113 and R 114 each independently represent a hydrogen atom or a monovalent organic group.
- a -A 1 and A 2- A 1 and A 2 in formula (1) each independently represent an oxygen atom or —NH—, and an oxygen atom is preferable.
- -R 111- R 111 in formula (1) represents a divalent organic group.
- the divalent organic group include a linear or branched aliphatic group, a cyclic aliphatic group, and an aromatic group, a heteroaromatic group, or a group in which two or more thereof are combined. 2 to 20 linear aliphatic group, 3 to 20 carbon branched aliphatic group, 3 to 20 carbon cyclic aliphatic group, 6 to 20 carbon aromatic group, or a combination thereof
- the group consisting of is preferable, and an aromatic group having 6 to 20 carbon atoms is more preferable.
- R 111 in formula (1) is preferably derived from a diamine.
- the diamine used for producing the polyimide precursor include linear or branched aliphatic, cyclic aliphatic or aromatic diamine. Only one diamine may be used, or two or more diamines may be used.
- the diamine is a linear aliphatic group having 2 to 20 carbon atoms, a branched or cyclic aliphatic group having 3 to 20 carbon atoms, an aromatic group having 6 to 20 carbon atoms, or these
- a diamine containing a group composed of a combination is preferable, and a diamine containing an aromatic group having 6 to 20 carbon atoms is more preferable.
- the following are mentioned as an example of an aromatic group.
- A is a single bond or an aliphatic hydrocarbon group having 1 to 10 carbon atoms which may be substituted with a fluorine atom, —O—, —C( ⁇ O)—, —S—, —S A group selected from ( ⁇ O) 2 —, —NHC( ⁇ O)— and a combination thereof is preferable, and a single bond or an alkylene group having 1 to 3 carbon atoms which may be substituted with a fluorine atom.
- —O—, —C( ⁇ O)—, —S— and S( ⁇ O) 2 — are more preferred, and —CH 2 —, —O—, —S—, — A divalent group selected from the group consisting of S( ⁇ O) 2 —, —C(CF 3 ) 2 —, and —C(CH 3 ) 2 — is more preferred.
- diamine examples include 1,2-diaminoethane, 1,2-diaminopropane, 1,3-diaminopropane, 1,4-diaminobutane, 1,6-diaminohexane; 1,2- or 1 ,3-Diaminocyclopentane, 1,2-, 1,3- or 1,4-diaminocyclohexane, 1,2-, 1,3- or 1,4-bis(aminomethyl)cyclohexane, bis-(4- Aminocyclohexyl)methane, bis-(3-aminocyclohexyl)methane, 4,4'-diamino-3,3'-dimethylcyclohexylmethane or isophoronediamine; meta or paraphenylenediamine, diaminotoluene, 4,4'- or 3 ,3'-diaminobiphenyl, 4,4'-diaminodiphenyl ether
- the diamines (DA-1) to (DA-18) shown below are also preferable.
- a diamine having at least two alkylene glycol units in the main chain is also mentioned as a preferable example.
- diamines containing two or more of ethylene glycol chain and/or propylene glycol chain in one molecule more preferably diamines containing no aromatic ring.
- Specific examples include Jeffarmin (registered trademark) KH-511, Jeffarmin (registered trademark) ED-600, Jeffermin (registered trademark) ED-900, Jeffermin (registered trademark) ED-2003, and Jeffermin (registered trademark).
- x, y, and z are arithmetic average values.
- R 111 in formula (1) is preferably represented by —Ar 0 —L 0 —Ar 0 — from the viewpoint of flexibility of the cured film to be obtained.
- Each Ar 0 independently represents an aromatic hydrocarbon group (preferably having 6 to 22 carbon atoms, more preferably 6 to 18 carbon atoms, particularly preferably 6 to 10 carbon atoms), and preferably a phenylene group.
- L 0 is a single bond, an aliphatic hydrocarbon group having 1 to 10 carbon atoms which may be substituted with a fluorine atom, —O—, —C( ⁇ O)—, —S—, —S( ⁇ O) It represents a group selected from the group consisting of 2-, -NHCO-, and groups each combining two or more thereof.
- the preferable range of L 0 has the same meaning as A above.
- R 111 in the formula (1) is preferably a divalent organic group represented by the following formula (51) or formula (61) from the viewpoint of i-ray transmittance.
- a divalent organic group represented by the formula (61) is more preferable from the viewpoints of i-ray transmittance and availability.
- R 50 to R 57 are each independently a hydrogen atom, a fluorine atom or a monovalent organic group, and at least one of R 50 to R 57 is a fluorine atom, a methyl group or a fluoromethyl group, It is a difluoromethyl group or a trifluoromethyl group.
- the monovalent organic group represented by R 50 to R 57 is an unsubstituted alkyl group having 1 to 10 carbon atoms (preferably 1 to 6 carbon atoms), a monovalent organic group having 1 to 10 carbon atoms (preferably 1 to 6 carbon atoms), Examples thereof include fluorinated alkyl groups.
- R 58 and R 59 are each independently a fluorine atom, a fluoromethyl group, a difluoromethyl group, or a trifluoromethyl group.
- Examples of the diamine compound giving the structure of formula (51) or (61) include dimethyl-4,4′-diaminobiphenyl, 2,2′-bis(trifluoromethyl)-4,4′-diaminobiphenyl, 2,2 Examples thereof include'-bis(fluoro)-4,4'-diaminobiphenyl and 4,4'-diaminooctafluorobiphenyl. You may use these 1 type or may use it in combination of 2 or more type.
- -R 115- R 115 in the formula (1) represents a tetravalent organic group.
- a tetravalent organic group containing an aromatic ring is preferable, and a group represented by the following formula (5) or formula (6) is more preferable.
- R 112 has the same meaning as A, and the preferred range is also the same.
- tetravalent organic group represented by R 115 in the formula (1) include a tetracarboxylic acid residue remaining after removing the acid dianhydride group from the tetracarboxylic dianhydride.
- the tetracarboxylic dianhydride may be used alone or in combination of two or more.
- the tetracarboxylic dianhydride is preferably a compound represented by the following formula (7).
- R 115 represents a tetravalent organic group.
- R 115 has the same meaning as R 115 in formula (1).
- tetracarboxylic dianhydride examples include pyromellitic acid, pyromellitic dianhydride (PMDA), 3,3′,4,4′-biphenyltetracarboxylic dianhydride, 3,3′,4.
- DAA-1 to DAA-5 shown below are also preferred examples.
- R 113 and R 114- R 113 and R 114 in formula (1) each independently represent a hydrogen atom or a monovalent organic group. At least one of R 113 and R 114 preferably contains a radically polymerizable group, and more preferably both contain a radically polymerizable group.
- the radically polymerizable group is a group capable of undergoing a crosslinking reaction by the action of a radical, and a preferable example thereof is a group having an ethylenically unsaturated bond.
- Examples of the group having an ethylenically unsaturated bond include a vinyl group, an allyl group, a (meth)acryloyl group and a group represented by the following formula (III).
- R 200 represents a hydrogen atom or a methyl group, and preferably a methyl group.
- R 201 is an alkylene group having 2 to 12 carbon atoms, —CH 2 CH(OH)CH 2 — or a (poly)oxyalkylene group having 4 to 30 carbon atoms (the alkylene group has 1 carbon atoms.
- the number of repetitions is preferably 1 to 12, more preferably 1 to 6, and particularly preferably 1 to 3; the number of repeats is preferably 1 to 12, more preferably 1 to 6, and particularly preferably 1 to 3).
- the (poly)oxyalkylene group means an oxyalkylene group or a polyoxyalkylene group.
- R 201 examples include ethylene group, propylene group, trimethylene group, tetramethylene group, 1,2-butanediyl group, 1,3-butanediyl group, pentamethylene group, hexamethylene group, octamethylene group, dodecamethylene group. , —CH 2 CH(OH)CH 2 —, and an ethylene group, a propylene group, a trimethylene group, and —CH 2 CH(OH)CH 2 — are more preferable.
- R 200 is a methyl group and R 201 is an ethylene group.
- An alkyl group etc. are mentioned. Specific examples thereof include an aromatic group having 6 to 20 carbon atoms having an acid group and an arylalkyl group having 7 to 25 carbon atoms having an acid group. More specific examples include a phenyl group having an acid group and a benzyl group having an acid group.
- the acid group is preferably a hydroxy group. That is, R 113 or R 114 is preferably a group having a hydroxy group.
- a substituent that improves the solubility of the developer is preferably used.
- R 113 or R 114 is a hydrogen atom, 2-hydroxybenzyl, 3-hydroxybenzyl and 4-hydroxybenzyl from the viewpoint of solubility in an aqueous developer.
- R 113 or R 114 is preferably a monovalent organic group.
- the monovalent organic group a linear or branched alkyl group, a cyclic alkyl group or an aromatic group is preferable, and an alkyl group substituted with an aromatic group is more preferable.
- the alkyl group preferably has 1 to 30 carbon atoms (3 or more when it is cyclic).
- the alkyl group may be linear, branched or cyclic.
- As the linear or branched alkyl group for example, methyl group, ethyl group, propyl group, butyl group, pentyl group, hexyl group, heptyl group, octyl group, nonyl group, decyl group, dodecyl group, tetradecyl group, octadecyl group , Isopropyl group, isobutyl group, sec-butyl group, t-butyl group, 1-ethylpentyl group, and 2-ethylhexyl group.
- the cyclic alkyl group may be either a monocyclic cyclic alkyl group or a polycyclic cyclic alkyl group.
- Examples of the monocyclic cyclic alkyl group include a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, a cycloheptyl group and a cyclooctyl group.
- Examples of the polycyclic cyclic alkyl group include an adamantyl group, a norbornyl group, a bornyl group, a camphenyl group, a decahydronaphthyl group, a tricyclodecanyl group, a tetracyclodecanyl group, a camphoroyl group, a dicyclohexyl group and a pinenyl group.
- As the alkyl group substituted with an aromatic group a linear alkyl group substituted with an aromatic group described below is preferable.
- aromatic group examples include a substituted or unsubstituted aromatic hydrocarbon group (the cyclic structure constituting the group includes a benzene ring, a naphthalene ring, a biphenyl ring, a fluorene ring, a pentalene ring, an indene ring, and an azulene.
- the cyclic structure constituting the group includes a benzene ring, a naphthalene ring, a biphenyl ring, a fluorene ring, a pentalene ring, an indene ring, and an azulene.
- the cyclic structure constituting the group includes a fluorene ring, a pyrrole ring, a furan ring, a thiophene ring, an imidazole ring, an oxazole ring, a thiazole ring, a pyridine ring, a pyrazine ring, a pyrimidine ring, a pyridazine ring, an indolizine ring, an indole ring, and benzofuran.
- Ring benzothiophene ring, isobenzofuran ring, quinolidine ring, quinoline ring, phthalazine ring, naphthyridine ring, quinoxaline ring, quinoxazoline ring, isoquinoline ring, carbazole ring, phenanthridine ring, acridine ring, phenanthroline ring, thianthrene ring, chromene ring , Xanthene ring, phenoxathiin ring, phenothiazine ring or phenazine ring).
- the polyimide precursor preferably has a fluorine atom in the repeating unit.
- the fluorine atom content in the polyimide precursor is preferably 10% by mass or more, more preferably 20% by mass or more. There is no particular upper limit, but 50% by mass or less is practical.
- an aliphatic group having a siloxane structure may be copolymerized with the repeating unit represented by the formula (1) for the purpose of improving the adhesion to the base material.
- the diamine component include bis(3-aminopropyl)tetramethyldisiloxane and bis(paraaminophenyl)octamethylpentasiloxane.
- the repeating unit represented by the formula (1) is preferably a repeating unit represented by the formula (1-A) or (1-B).
- a 11 and A 12 represent an oxygen atom or —NH—
- R 111 and R 112 each independently represent a divalent organic group
- R 113 and R 114 each independently represent a hydrogen atom or 1 It represents a valent organic group
- at least one of R 113 and R 114 is preferably a group containing a radically polymerizable group, and more preferably a radically polymerizable group.
- a 11 , A 12 , R 111 , R 113, and R 114 are the same as the preferred ranges of A 1 , A 2 , R 111 , R 113, and R 114 in formula (1), respectively.
- R 112 has the same meaning as R 112 in formula (5), and more preferably among others oxygen atoms.
- the bonding position of the carbonyl group in the formula to the benzene ring is preferably 4,5,3',4' in formula (1-A).
- 1,2,4,5 are preferable.
- the repeating unit represented by the formula (1) may be one type or two or more types. Further, the repeating unit represented by the formula (1) may contain a structural isomer. Further, the polyimide precursor may contain other types of repeating units in addition to the repeating unit of the above formula (1).
- a polyimide precursor in which 50 mol% or more, further 70 mol% or more, particularly 90 mol% or more of all repeating units are repeating units represented by the formula (1) is exemplified.
- the upper limit is practically 100 mol% or less.
- the weight average molecular weight (Mw) of the polyimide precursor is preferably 2,000 to 500,000, more preferably 5,000 to 100,000, and further preferably 10,000 to 50,000.
- the number average molecular weight (Mn) is preferably 800 to 250,000, more preferably 2,000 to 50,000, and further preferably 4,000 to 25,000.
- the molecular weight dispersity of the polyimide precursor is preferably 1.5 to 3.5, more preferably 2 to 3.
- the polydispersity of the molecular weight means a value obtained by dividing the weight average molecular weight by the number average molecular weight (weight average molecular weight/number average molecular weight).
- the polyimide precursor is obtained by reacting a dicarboxylic acid or a dicarboxylic acid derivative with a diamine.
- a dicarboxylic acid or a dicarboxylic acid derivative is obtained by halogenating a dicarboxylic acid or a dicarboxylic acid derivative with a halogenating agent and then reacting it with a diamine.
- the organic solvent may be one kind or two or more kinds.
- the organic solvent can be appropriately determined according to the raw material, and examples thereof include pyridine, diethylene glycol dimethyl ether (diglyme), N-methylpyrrolidone and N-ethylpyrrolidone.
- a step of depositing a solid is included in the production of the polyimide precursor.
- solid precipitation can be performed by precipitating the polyimide precursor in the reaction liquid in water and dissolving it in a solvent in which the polyimide precursor is soluble, such as tetrahydrofuran.
- the polybenzoxazole precursor preferably contains a repeating unit represented by the following formula (2).
- R 121 represents a divalent organic group
- R 122 represents a tetravalent organic group
- R 123 and R 124 each independently represent a hydrogen atom or a monovalent organic group.
- R 121 represents a divalent organic group.
- the divalent organic group an aliphatic group (preferably having 1 to 24 carbon atoms, more preferably 1 to 12 and particularly preferably 1 to 6) and an aromatic group (preferably having 6 to 22 carbon atoms, 6 to 14) Is more preferable, and 6 to 12 is particularly preferable).
- the aromatic group that constitutes R 121 include the examples of R 111 in the above formula (1).
- a linear aliphatic group is preferable.
- R 121 is preferably derived from 4,4′-oxydibenzoyl chloride.
- R 122 represents a tetravalent organic group.
- the tetravalent organic group has the same meaning as R 115 in the above formula (1), and the preferred range is also the same.
- R 122 is preferably derived from 2,2′-bis(3-amino-4-hydroxyphenyl)hexafluoropropane.
- R -R 123 and R 124- R 123 and R 124 each independently represent a hydrogen atom or a monovalent organic group, have the same meaning as R 113 and R 114 in the above formula (1), and the preferred ranges are also the same.
- the polybenzoxazole precursor may contain other types of repeating units in addition to the repeating unit of the above formula (2).
- the polybenzoxazole precursor preferably further contains a diamine residue represented by the following formula (SL) as another type of repeating unit from the viewpoint of suppressing the occurrence of warpage of the cured film due to ring closure.
- SL diamine residue represented by the following formula (SL) as another type of repeating unit from the viewpoint of suppressing the occurrence of warpage of the cured film due to ring closure.
- Z has a structure and b structure
- R 1s is a hydrogen atom or a hydrocarbon group having 1 to 10 carbon atoms (preferably having 1 to 6 carbon atoms, more preferably 1 to 3 carbon atoms)
- R 2s Is a hydrocarbon group having 1 to 10 carbon atoms (preferably having 1 to 6 carbon atoms, more preferably 1 to 3 carbon atoms)
- at least one of R 3s , R 4s , R 5s , and R 6s is aromatic.
- a group (preferably having 6 to 22 carbon atoms, more preferably 6 to 18 carbon atoms, particularly preferably 6 to 10 carbon atoms), the rest being hydrogen atoms or 1 to 30 carbon atoms (preferably 1 to 18 carbon atoms, more preferably It is preferably an organic group having 1 to 12 carbon atoms, particularly preferably 1 to 6 carbon atoms, and they may be the same or different.
- the polymerization of the a structure and the b structure may be block polymerization or random polymerization. In the Z portion, the a structure is preferably 5 to 95 mol%, the b structure is 95 to 5 mol%, and a+b is 100 mol%.
- preferable Z includes those in which R 5s and R 6s in the b structure are phenyl groups.
- the molecular weight of the structure represented by the formula (SL) is preferably 400 to 4,000, more preferably 500 to 3,000.
- the molecular weight can be determined by commonly used gel permeation chromatography. When the molecular weight is in the above range, the elastic modulus of the polybenzoxazole precursor after dehydration ring closure can be lowered, and the effect of suppressing warpage and the effect of improving solubility can be made compatible.
- the tetracarboxylic acid dianhydride is further added in that the alkali solubility of the curable resin composition is improved. It is preferable to include a tetracarboxylic acid residue remaining after the removal of the acid dianhydride group from the compound as a repeating unit. Examples of such a tetracarboxylic acid residue include the example of R 115 in the formula (1).
- the weight average molecular weight (Mw) of the polybenzoxazole precursor is preferably 2,000 to 500,000, more preferably 5,000 to 100,000, and further preferably 10,000 to 50,000. is there.
- the number average molecular weight (Mn) is preferably 800 to 250,000, more preferably 2,000 to 50,000, and further preferably 4,000 to 25,000.
- the dispersity of the molecular weight of the polybenzoxazole precursor is preferably 1.5 to 3.5, more preferably 2 to 3.
- the content of the heterocyclic ring-containing polymer precursor in the curable resin composition of the present invention is preferably 20% by mass or more, more preferably 30% by mass or more, based on the total solid content of the composition. It is more preferably 40% by mass or more, further preferably 50% by mass or more, even more preferably 60% by mass or more, and even more preferably 70% by mass or more.
- the content of the heterocyclic ring-containing polymer precursor in the curable resin composition of the present invention is preferably 99.5% by mass or less and is 99% by mass or less based on the total solid content of the composition. It is more preferably 98% by mass or less, further preferably 97% by mass or less, still more preferably 95% by mass or less.
- the curable resin composition of the present invention may contain only one kind of the heterocycle-containing polymer precursor, or may contain two or more kinds thereof. When two or more kinds are contained, the total amount is preferably within the above range.
- the curable resin composition of the present invention contains a compound represented by formula (1-1).
- R 1 and R 2 each independently represent a monovalent organic group
- L 1 represents a divalent linking group
- R 3 represents a hydrogen atom or a monovalent organic group.
- R 1 and R 2 may combine to form a ring structure.
- the compound represented by the formula (1-1) is preferably a compound which decomposes to generate a base.
- the compound represented by the formula (1-1) is more preferably a compound that decomposes by heat to generate a base. That is, the compound represented by formula (1-1) is preferably a base generator, and more preferably a thermal base generator.
- the compound represented by the following formula (B-1) is decomposed by heating, for example, as shown in the following reaction formula, and the generated diisopropylamine acts as a base, so that the complex in the curable resin composition is It is believed to promote cyclization of the ring-containing polymer precursor.
- R 1 and R 2 are each independently preferably a hydrocarbon group, more preferably a hydrocarbon group having 1 to 20 carbon atoms, and a carbon group having 1 to 10 carbon atoms. It is more preferably a hydrogen group.
- the hydrocarbon group is preferably an alkyl group or an aryl group, more preferably an alkyl group having 1 to 10 carbon atoms, or a phenyl group, further preferably an alkyl group having 1 to 10 carbon atoms.
- 1-ethylpentyl group, 2-ethylhexyl group, cyclopropyl group, cyclobutyl group, cyclopentyl group, cyclohexyl group, cycloheptyl group, cyclooctyl group, or phenyl group is preferred, and methyl group, ethyl group, propyl group, butyl group Group, pentyl group, hexyl group, heptyl group, octyl group, nonyl group, decyl group, isopropyl group, isobutyl group, sec-butyl group, t-butyl group, 1-ethylpentyl group, 2-ethylhexyl group, cyclopropyl group , A cyclobutyl group, a cyclopentyl group, a cyclohexyl group, a cycloheptyl group, or a cycloo
- R 1 and R 2 may combine to form a ring structure, and the ring structure formed is preferably an aliphatic ring structure having 5 or 6 ring members, such as a pyrrolidine ring, a piperidine ring, a piperazine ring, A morpholine ring structure is more preferred.
- the formula weight of R 1 and R 2 (sum of the atomic weights of the atoms contained in R 1 or R 2 ) is preferably 29 to 300, more preferably 57 to 282, and 57 to 200, respectively. More preferably, Further, the generated bases R 1 and R 2 have a high basicity of the generated base, and the interaction of the generated base with a base material such as a copper base material is small.
- R 1 and R 2 are each independently an alkyl group having 1 to 10 carbon atoms, or R 1 and R 2 are bonded to each other to have 5 or 6 ring members. It is preferable that R 1 and R 2 are each independently an ethyl group or an isopropyl group, or it is more preferable that R 1 and R 2 are bonded to each other to form a piperidine ring structure.
- the structures of R 1 and R 2 are, for example, the basic strength of the generated base, the strength of the interaction between the generated base and the base material, the mobility of the generated base, the generated base in the film. It can be selected in consideration of interaction with other materials.
- L 1 is preferably a divalent linking group having a linking chain length of 1 to 5, and more preferably a divalent linking group having a linking chain length of 2 or 3.
- the connecting chain length of L 1, an atom contained in L 1, and the following formula (1 -1 ') by two in the compound represented by the carbon atom C 1 and C 2 The smallest number among the number of atoms existing between them.
- the following formula (1-1′) describes the symbols C 1 and C 2 on the carbon atoms in the above formula (1-1), respectively, and R 1 and R in the formula (1-1′) are represented.
- 2 , R 3 and L 1 have the same meanings as R 1 , R 2 , R 3 and L 1 in formula (1-1), respectively.
- L 1 in the compound represented by the above formula (B-1) is a 1,2-phenylene group, and the connecting chain length is 2.
- All of the atoms at the bonding positions at both ends of L 1 are preferably carbon atoms.
- the bonding positions at both ends of L 1 refer to two positions in the above formula (1-1′), that is, the bonding site between L 1 and carbon atom C 1 and the bonding site between L 1 and carbon atom C 2.
- the structure existing between the carbon atom C 1 and the carbon atom C 2 in L 1 is a part of an aromatic ring structure, or an unsaturated aliphatic hydrocarbon structure. Is preferred, and a phenylene group (particularly preferably a 1,2-phenylene group) or a cisvinylene group is more preferred.
- L 1 may further include a base generation site in a structure other than the structure existing between the carbon atom C 1 and the carbon atom C 2 .
- the base generation site include a structure excluding L 1 in the above formula (1-1) (a combination of two structures described in the following formula (1-1A)).
- Examples of the compound further containing a base generation site in a structure other than the structure in which L 1 exists between the carbon atom C 1 and the carbon atom C 2 include compounds represented by the formulas (B-16, B-17, B-18) described later. ) And the like.
- the structure of L 1 can be selected, for example, in consideration of the generation efficiency of bases.
- the two structures in formula (1-1A) are preferably linked by a linking group having a linking chain length of 1 to 5, and are linked by a linking group having a linking chain length of 2 or 3. Is preferred.
- the structure existing between the two structures in the formula (1-1A) is preferably a part of the aromatic ring structure or an unsaturated aliphatic hydrocarbon structure.
- L 1 is a hydrocarbon group.
- the hydrocarbon group an alkylene group having 1 to 5 carbon atoms, an alkenylene group having 2 to 5 carbon atoms, or an arylene group is preferable, and an alkylene group having 2 to 3 carbon atoms, an alkenylene group having 2 to 3 carbon atoms, Alternatively, a phenylene group is more preferable, an alkenylene group having 2 to 3 carbon atoms is more preferable, and a phenylene group is more preferable, and a vinylene group or a phenylene group is more preferable.
- L 1 is 1,2-ethylene group, 1,3-propanediyl group (trimethylene group), 1,2-cyclohexanediyl group, cisvinylene group, 1,2-phenylene group, 1,2-phenylenemethylene group Or a 1,2-ethyleneoxy-1,2-ethylene group is preferable, and a cisvinylene group or a 1,2-phenylene group is more preferable.
- R 3 is preferably a hydrogen atom or a hydrocarbon group, more preferably a hydrogen atom, an alkyl group or an aryl group, and is an alkyl group having 1 to 20 carbon atoms or 6 to 20 carbon atoms.
- An aryl group is more preferable, an alkyl group having 2 to 8 carbon atoms or a phenyl group is further preferable, and the resulting base has a small interaction with a base material such as a copper base material, and the resulting curing is achieved.
- an alkyl group having 2 to 4 carbon atoms or a phenyl group is more preferable.
- R 3 When R 3 is an alkyl group, the alkyl group may have either a linear or branched structure or a structure having a cyclic structure, but from the viewpoint of base generation efficiency. Is preferably linear.
- the structure of R 3 may be selected in consideration of, for example, the generation efficiency of a base, the strength of interaction of a compound after decomposition with a base material such as a copper base material, and the strength of electron donating property. it can.
- the molecular weight of the compound represented by the formula (1-1) may be determined in consideration of, for example, the decomposition temperature and volatility, but is preferably 100 or more, more preferably 150 or more, and 200 More preferably, it is more preferably 250 or more.
- the upper limit is not particularly limited, but it is preferably 1,000 or less.
- the decomposition temperature is preferably 50° C. or higher, more preferably 80° C. or higher, and 120° C. or higher. More preferably, it is more preferably 140° C. or higher.
- the upper limit is more preferably 450° C. or lower, further preferably 350° C. or lower, and further preferably 250° C. or lower.
- the energy required for curing the curable resin composition can be reduced by using a compound that decomposes at a lower temperature.
- the decomposition temperature is determined as the peak temperature of the lowest exothermic peak when the compound represented by the formula (1-1) is heated to 500° C. at 5° C./minute in a pressure resistant capsule.
- These compounds can be synthesized, for example, by reacting an acid anhydride compound with an amine compound to synthesize an amic acid compound, and then further conducting a condensation reaction between the amic acid compound and the amine compound.
- the acid anhydride compound and the amine compound may be appropriately selected in consideration of the structure of the finally desired compound. Specifically, it can be synthesized using the method described in Organic Chemistry International (2014), 576715/1-576715/10, 10pp and the like.
- the pKa of the conjugate acid of the generated base is preferably 8 or more, more preferably 9 or more, and further preferably 10 or more. There is no particular upper limit, but it is practical that the upper limit is 14 or less.
- the generated base can efficiently progress the cyclization reaction of the heterocycle-containing polymer precursor, and the elongation at break of the cured film at low temperature can be increased. It is preferable because it can be increased.
- pKa is a value specified by the following method.
- pKa represents the equilibrium constant Ka of the negative common logarithm pKa of the dissociation reaction in which hydrogen ions are released from an acid. A smaller pKa indicates a stronger acid. Unless otherwise specified, pKa is a value calculated by ACD/ChemSketch (registered trademark). Alternatively, you may refer to the values listed in “Revised 5th Edition Chemical Handbook Basic Edition” edited by The Chemical Society of Japan.
- the content of the compound represented by the formula (1-1) is based on the total solid content of the curable resin composition. On the other hand, it is preferably 0.005 to 50 mass %.
- the lower limit is more preferably 0.05% by mass or more, further preferably 0.5% by mass or more, and particularly preferably 1% by mass or more.
- the upper limit is more preferably 20% by mass or less, further preferably 10% by mass or less, and particularly preferably 5% by mass or less, from the viewpoint of corrosion resistance of metal (eg, copper used for wiring).
- the content of the compound represented by the formula (1-1) relative to 100 parts by mass of the heterocycle-containing polymer precursor is It is preferably 0.005 parts by mass or more, more preferably 0.06 parts by mass or more, further preferably 0.5 parts by mass or more, and further preferably 1 part by mass or more.
- the upper limit is, for example, preferably 20 parts by mass or less, more preferably 15 parts by mass or less, and more preferably 10 parts by mass from the viewpoint of corrosion resistance of a metal (for example, copper used for wiring).
- the amount is more preferably the following, and particularly preferably 7.5 parts by mass or less.
- the compound represented by the formula (1-1) may be used alone or in combination of two or more. When two or more kinds are used, the total amount is preferably within the above range.
- the curable resin composition of the present invention may contain another base generator.
- the other base generator does not include the compound represented by the above formula (1-1).
- Other base generators include thermal base generators and photobase generators.
- the content of the other thermal base generator is 5% by mass or less of the content of the compound represented by the formula (1-1). It means that the content is preferably 3% by mass or less, and more preferably 1% by mass.
- the curable resin composition of the present invention preferably contains a photopolymerization initiator.
- the photopolymerization initiator is preferably a photoradical polymerization initiator.
- the photo radical polymerization initiator is not particularly limited and can be appropriately selected from known photo radical polymerization initiators.
- a photoradical polymerization initiator having photosensitivity to light rays in the ultraviolet region to the visible region is preferable.
- it may be an activator which produces an active radical by causing some action with the photoexcited sensitizer.
- the photo-radical polymerization initiator contains at least one compound having a molar extinction coefficient of at least about 50 L ⁇ mol ⁇ 1 ⁇ cm ⁇ 1 within a range of about 300 to 800 nm (preferably 330 to 500 nm). Is preferred.
- the molar extinction coefficient of a compound can be measured using a known method. For example, it is preferable to measure with an ultraviolet-visible spectrophotometer (Cary-5 spectrophotometer manufactured by Varian) using an ethyl acetate solvent at a concentration of 0.01 g/L.
- any known compound can be used.
- halogenated hydrocarbon derivatives for example, compounds having a triazine skeleton, compounds having an oxadiazole skeleton, compounds having a trihalomethyl group, etc.
- acylphosphine compounds such as acylphosphine oxide, hexaarylbiimidazole, oxime derivatives, etc.
- the description in paragraphs 0165 to 0182 of JP-A-2016-027357 and paragraphs 0138 to 0151 of International Publication No. 2015/199219 can be referred to, and the contents thereof are incorporated herein.
- Examples of the ketone compound include compounds described in paragraph 0087 of JP-A-2005-087611, the contents of which are incorporated herein.
- Kayacure DETX manufactured by Nippon Kayaku Co., Ltd.
- Nippon Kayaku Co., Ltd. is also preferably used.
- a hydroxyacetophenone compound, an aminoacetophenone compound, and an acylphosphine compound can also be preferably used. More specifically, for example, the aminoacetophenone type initiator described in JP-A-10-291969 and the acylphosphine oxide type initiator described in Japanese Patent No. 4225898 can be used.
- IRGACURE 184 (IRGACURE is a registered trademark)
- DAROCUR 1173 As the hydroxyacetophenone initiator, IRGACURE 184 (IRGACURE is a registered trademark), DAROCUR 1173, IRGACURE 500, IRGACURE-2959, IRGACURE 127 (trade name: all manufactured by BASF) can be used.
- aminoacetophenone initiator commercially available products such as IRGACURE 907, IRGACURE 369, and IRGACURE 379 (trade names: all manufactured by BASF) can be used.
- the compound described in JP-A-2009-191179 in which the absorption maximum wavelength is matched with a wavelength light source such as 365 nm or 405 nm can also be used.
- acylphosphine-based initiator examples include 2,4,6-trimethylbenzoyl-diphenyl-phosphine oxide.
- commercially available products such as IRGACURE-819 and IRGACURE-TPO (trade names: all manufactured by BASF) can be used.
- metallocene compounds examples include IRGACURE-784 (manufactured by BASF).
- the photo-radical polymerization initiator is an oxime compound.
- the exposure latitude can be more effectively improved.
- Oxime compounds are particularly preferable because they have a wide exposure latitude (exposure margin) and also act as a photocuring accelerator.
- the compounds described in JP 2001-233842 A the compounds described in JP 2000-080068 A, and the compounds described in JP 2006-342166 A can be used.
- Preferred oxime compounds include, for example, compounds having the following structures, 3-benzoyloxyiminobutan-2-one, 3-acetoxyiminobutan-2-one, 3-propionyloxyiminobutan-2-one, and 2-acetoxy.
- Iminopentan-3-one 2-acetoxyimino-1-phenylpropan-1-one, 2-benzoyloxyimino-1-phenylpropan-1-one, 3-(4-toluenesulfonyloxy)iminobutan-2-one , And 2-ethoxycarbonyloxyimino-1-phenylpropan-1-one.
- an oxime compound (oxime-based photopolymerization initiator) as a photoradical polymerization initiator.
- IRGACURE OXE 01, IRGACURE OXE 02, IRGACURE OXE 03, IRGACURE OXE 04 (above, manufactured by BASF), Adeka Optimer N-1919 (manufactured by ADEKA, Inc., JP 2012-014052 A)
- the radical polymerization initiator 2 is also preferably used.
- TR-PBG-304 manufactured by Changzhou Power Electronics Co., Ltd.
- ADEKA ARKUL'S NCI-831 and ADEKA ARKRUZ NCI-930 can also be used.
- DFI-091 (manufactured by Daito Chemix Co., Ltd.) can be used.
- an oxime compound having a fluorine atom examples include the compounds described in JP 2010-262028 A, the compounds 24, 36 to 40 described in paragraph 0345 of JP-A-2014-500852, and JP 2013-2013 A.
- the compound (C-3) described in paragraph 0101 of JP-A-164471 can be mentioned.
- Examples of the most preferable oxime compound include an oxime compound having a specific substituent shown in JP-A 2007-269779 and an oxime compound having a thioaryl group shown in JP-A 2009-191061.
- the photoradical polymerization initiator is a trihalomethyltriazine compound, a benzyldimethylketal compound, an ⁇ -hydroxyketone compound, an ⁇ -aminoketone compound, an acylphosphine compound, a phosphine oxide compound, a metallocene compound, an oxime compound, a triaryl, from the viewpoint of exposure sensitivity.
- More preferred photoradical polymerization initiators are trihalomethyltriazine compounds, ⁇ -aminoketone compounds, acylphosphine compounds, phosphine oxide compounds, metallocene compounds, oxime compounds, triarylimidazole dimers, onium salt compounds, benzophenone compounds, acetophenone compounds, At least one compound selected from the group consisting of trihalomethyltriazine compounds, ⁇ -aminoketone compounds, oxime compounds, triarylimidazole dimers and benzophenone compounds is more preferable, metallocene compounds or oxime compounds are even more preferable, and oxime compounds are preferable. Is even more preferable.
- the photoradical polymerization initiator is N,N′-tetraalkyl-4,4′-diaminobenzophenone, 2-benzyl such as benzophenone, N,N′-tetramethyl-4,4′-diaminobenzophenone (Michler's ketone) or the like.
- Aromatic ketones such as -2-dimethylamino-1-(4-morpholinophenyl)-butanone-1,2-methyl-1-[4-(methylthio)phenyl]-2-morpholino-propanone-1 and alkylanthraquinones It is also possible to use quinones condensed with the aromatic ring, benzoin ether compounds such as benzoin alkyl ether, benzoin compounds such as benzoin and alkylbenzoin, and benzyl derivatives such as benzyldimethylketal. Further, a compound represented by the following formula (I) can also be used.
- R I00 represents an alkyl group having 1 to 20 carbon atoms, an alkyl group having 2 to 20 carbon atoms interrupted by one or more oxygen atoms, an alkoxy group having 1 to 12 carbon atoms, a phenyl group, C1-C20 alkyl group, C1-C12 alkoxy group, halogen atom, cyclopentyl group, cyclohexyl group, C2-C12 alkenyl group, C2-C interrupted by one or more oxygen atoms 18 alkyl group and at least one substituted phenyl group of the alkyl group having 1 to 4 carbon atoms, or biphenyl
- R I01 is a group represented by formula (II), the same as R I00 R I02 to R I04 each independently represent alkyl having 1 to 12 carbons, alkoxy group having 1 to 12 carbons, or halogen.
- R I05 to R I07 are the same as R I02 to R I04 in the above formula (I).
- radical photopolymerization initiator compounds described in paragraphs 0048 to 0055 of WO 2015/125469 can also be used.
- the photopolymerization initiator When the photopolymerization initiator is contained, its content is preferably 0.1 to 30% by mass, more preferably 0.1 to 20% by mass, based on the total solid content of the curable resin composition of the present invention. Is more preferable, 0.5 to 15% by mass is more preferable, and 1.0 to 10% by mass is more preferable.
- the photopolymerization initiator may contain only one type, or may contain two or more types. When two or more photopolymerization initiators are contained, the total amount is preferably within the above range.
- the curable resin composition of the present invention may contain a thermal polymerization initiator as a polymerization initiator, and particularly may contain a thermal radical polymerization initiator.
- the thermal radical polymerization initiator is a compound that generates radicals by the energy of heat and initiates or accelerates the polymerization reaction of the polymerizable compound. By adding the thermal radical polymerization initiator, the polymerization reaction of the heterocycle-containing polymer precursor can be advanced together with the cyclization of the heterocycle-containing polymer precursor, so that higher heat resistance can be achieved.
- thermal radical polymerization initiator examples include compounds described in paragraphs 0074 to 0118 of JP 2008-063554 A.
- thermal radical polymerization initiator When the thermal radical polymerization initiator is contained, its content is preferably 0.1 to 30% by mass, more preferably 0.1 to 20% by mass, based on the total solid content of the curable resin composition of the present invention. %, and more preferably 5 to 15% by mass.
- the thermal radical polymerization initiator may contain only one type, or may contain two or more types. When two or more thermal radical polymerization initiators are contained, the total amount is preferably within the above range.
- the curable resin composition of the present invention preferably contains a polymerizable compound.
- a radically polymerizable compound can be used as the polymerizable compound.
- the radically polymerizable compound is a compound having a radically polymerizable group. Examples of the radically polymerizable group include groups having an ethylenically unsaturated bond such as vinyl group, allyl group, vinylphenyl group and (meth)acryloyl group.
- the radically polymerizable group is preferably a (meth)acryloyl group.
- the number of radically polymerizable groups contained in the radically polymerizable compound may be one or two or more, but the radically polymerizable compound preferably has two or more radically polymerizable groups, and preferably three or more. More preferable.
- the upper limit is preferably 15 or less, more preferably 10 or less, and further preferably 8 or less.
- the molecular weight of the radically polymerizable compound is preferably 2,000 or less, more preferably 1,500 or less, even more preferably 900 or less.
- the lower limit of the molecular weight of the radically polymerizable compound is preferably 100 or more.
- the curable resin composition of the present invention preferably contains at least one bifunctional or more radically polymerizable compound containing two or more radically polymerizable groups, and preferably a trifunctional or more radically polymerizable compound. It is more preferable to contain at least one. Further, it may be a mixture of a bifunctional radically polymerizable compound and a trifunctional or higher functional radically polymerizable compound.
- the number of functional groups of a bifunctional or higher polymerizable monomer means that the number of radical polymerizable groups in one molecule is 2 or more.
- the radically polymerizable compound examples include unsaturated carboxylic acids (for example, acrylic acid, methacrylic acid, itaconic acid, crotonic acid, isocrotonic acid, maleic acid, etc.) and their esters and amides, and preferably, They are esters of unsaturated carboxylic acids and polyhydric alcohol compounds, and amides of unsaturated carboxylic acids and polyhydric amine compounds.
- unsaturated carboxylic acids for example, acrylic acid, methacrylic acid, itaconic acid, crotonic acid, isocrotonic acid, maleic acid, etc.
- esters and amides and preferably, They are esters of unsaturated carboxylic acids and polyhydric alcohol compounds, and amides of unsaturated carboxylic acids and polyhydric amine compounds.
- a dehydration condensation reaction product with a functional carboxylic acid is also preferably used.
- an unsaturated carboxylic acid ester or amide having an electrophilic substituent such as an isocyanate group or an epoxy group, an addition reaction product of a monofunctional or polyfunctional alcohol, amines, thiols, and a halogeno group.
- a substitution reaction product of an unsaturated carboxylic acid ester or amide having a leaving substituent such as or tosyloxy group with a monofunctional or polyfunctional alcohol, amine, or thiol is also suitable.
- the description in paragraphs 0113 to 0122 of JP-A-2016-027357 can be referred to, and the contents thereof are incorporated in the present specification.
- the radically polymerizable compound is preferably a compound having a boiling point of 100° C. or higher under normal pressure.
- examples thereof include polyethylene glycol di(meth)acrylate, trimethylolethane tri(meth)acrylate, neopentyl glycol di(meth)acrylate, pentaerythritol tri(meth)acrylate, pentaerythritol tetra(meth)acrylate, dipentaerythritol.
- Such urethane (meth)acrylates polyester acrylates described in JP-A-48-064183, JP-B-49-043191 and JP-B-52-030490, epoxy resins and (meth)acrylics. Mention may be made of polyfunctional acrylates and methacrylates such as epoxy acrylates, which are reaction products with acids, and mixtures thereof. Further, the compounds described in paragraphs 0254 to 0257 of JP-A 2008-292970 are also suitable. Further, a polyfunctional (meth)acrylate obtained by reacting a polyfunctional carboxylic acid with a compound having a cyclic ether group such as glycidyl (meth)acrylate and an ethylenically unsaturated bond can also be used.
- radically polymerizable compounds other than the above there are fluorene rings and ethylenically unsaturated bonds described in JP 2010-160418 A, JP 2010-129825 A, JP 4364216 A, etc. It is also possible to use a compound having two or more groups having a or a cardo resin.
- Radical-polymerizable compounds include dipentaerythritol triacrylate (commercially available KAYARAD D-330; manufactured by Nippon Kayaku Co., Ltd.), dipentaerythritol tetraacrylate (commercially available KAYARAD D-320; Nippon Kayaku Co., Ltd., A-TMMT: Shin-Nakamura Chemical Co., Ltd., dipentaerythritol penta(meth)acrylate (commercially available KAYARAD D-310; manufactured by Nippon Kayaku Co., Ltd.), dipentaerythritol hexa (meta).
- dipentaerythritol triacrylate commercially available KAYARAD D-330; manufactured by Nippon Kayaku Co., Ltd.
- dipentaerythritol tetraacrylate commercially available KAYARAD D-320; Nippon Kayaku Co., Ltd., A-TMMT: Shin-Nakamura Chemical Co.,
- radically polymerizable compounds examples include SR-494, which is a tetrafunctional acrylate having four ethyleneoxy chains manufactured by Sartomer, and SR-209, which is a bifunctional methacrylate having four ethyleneoxy chains, manufactured by Sartomer. 231, 239, DPCA-60, which is a hexafunctional acrylate having six pentyleneoxy chains, manufactured by Nippon Kayaku Co., Ltd., TPA-330, which is a trifunctional acrylate having three isobutyleneoxy chains, and urethane oligomer UAS-.
- Examples of the radically polymerizable compound include urethane acrylates such as those described in JP-B-48-041708, JP-A-51-037193, JP-B-02-032293, and JP-B-02-016765.
- the urethane compounds having an ethylene oxide skeleton described in JP-B-58-049860, JP-B-56-017654, JP-B-62-039417 and JP-B-62-039418 are also suitable.
- compounds having an amino structure or a sulfide structure in the molecule which are described in JP-A-63-277653, JP-A-63-260909 and JP-A-01-105238, can be used. It can also be used.
- the radically polymerizable compound may be a radically polymerizable compound having an acid group such as a carboxy group and a phosphoric acid group.
- the radically polymerizable compound having an acid group is preferably an ester of an aliphatic polyhydroxy compound and an unsaturated carboxylic acid, and an unreacted hydroxy group of the aliphatic polyhydroxy compound is reacted with a non-aromatic carboxylic acid anhydride to form an acid.
- a radically polymerizable compound having a group is more preferable.
- the aliphatic polyhydroxy compound in which an unreacted hydroxy group of the aliphatic polyhydroxy compound is reacted with a non-aromatic carboxylic acid anhydride to have an acid group, is pentaerythritol or dipenta A compound that is erythritol.
- examples of commercially available products include M-510 and M-520 as polybasic acid-modified acrylic oligomer manufactured by Toagosei Co., Ltd.
- the preferable acid value of the radically polymerizable compound having an acid group is 0.1 to 40 mgKOH/g, particularly preferably 5 to 30 mgKOH/g.
- the acid value of the radically polymerizable compound is within the above range, the handleability in production is excellent, and further the developability is excellent. Also, the polymerizability is good.
- the acid value is measured according to the description of JIS K0070:1992.
- a monofunctional radical-polymerizable compound can be preferably used as the radical-polymerizable compound from the viewpoint of suppressing warpage associated with controlling the elastic modulus of the cured film.
- the monofunctional radically polymerizable compound include n-butyl (meth)acrylate, 2-ethylhexyl (meth)acrylate, 2-hydroxyethyl (meth)acrylate, butoxyethyl (meth)acrylate, carbitol (meth)acrylate, cyclohexyl ( (Meth)acrylate, benzyl (meth)acrylate, phenoxyethyl (meth)acrylate, N-methylol (meth)acrylamide, glycidyl (meth)acrylate, polyethylene glycol mono (meth)acrylate, polypropylene glycol mono (meth)acrylate, etc.
- Acrylic acid derivatives such as N-vinyl compounds such as N-vinylpyrrolidone and N-vinylcaprolactam, and allyl compounds such as allyl glycidyl ether, diallyl phthalate and triallyl trimellitate are preferably used.
- a compound having a boiling point of 100° C. or higher under normal pressure is also preferable in order to suppress volatilization before exposure.
- the curable resin composition of the present invention may further contain a polymerizable compound other than the radically polymerizable compound described above.
- a polymerizable compound other than the above radically polymerizable compound include compounds having a hydroxymethyl group, an alkoxymethyl group or an acyloxymethyl group; epoxy compounds; oxetane compounds; and benzoxazine compounds.
- the compound having a hydroxymethyl group, an alkoxymethyl group or an acyloxymethyl group is preferably a compound represented by the following formula (AM1), (AM4) or (AM5).
- R 104 represents a t-valent organic group having 1 to 200 carbon atoms
- R 105 represents a group represented by —OR 106 or —OCO—R 107
- R 106 represents a hydrogen atom or an organic group having 1 to 10 carbon atoms
- R 107 represents an organic group having 1 to 10 carbon atoms.
- R 404 represents a divalent organic group having 1 to 200 carbon atoms
- R 405 represents a group represented by —OR 406 or —OCO—R 407
- R 406 represents a hydrogen atom or a carbon atom.
- R 407 represents an organic group having 1 to 10 carbon atoms.
- u represents an integer of 3 to 8
- R 504 represents a u-valent organic group having 1 to 200 carbon atoms
- R 505 represents a group represented by —OR 506 or —OCO—R 507.
- R 506 represents a hydrogen atom or an organic group having 1 to 10 carbon atoms
- R 507 represents an organic group having 1 to 10 carbon atoms.
- Specific examples of the compound represented by the formula (AM4) include 46DMOC, 46DMOEP (these are trade names, manufactured by Asahi Organic Materials Co., Ltd.), DML-MBPC, DML-MBOC, DML-OCHP, DML-PCHP, DML.
- specific examples of the compound represented by the formula (AM5) include TriML-P, TriML-35XL, TML-HQ, TML-BP, TML-pp-BPF, TML-BPA, TMOM-BP, HML-TPPHBA, HML-TPHAP, HMOM-TPPHBA, HMOM-TPHAP (trade names, manufactured by Honshu Chemical Industry Co., Ltd.), TM-BIP-A (trade name, manufactured by Asahi Organic Materials Co., Ltd.), NIKALAC MX-280, Examples include NIKALAC MX-270 and NIKALAC MW-100LM (trade names, manufactured by Sanwa Chemical Co., Ltd.).
- the epoxy compound is preferably a compound having two or more epoxy groups in one molecule.
- the epoxy group undergoes a cross-linking reaction at 200° C. or lower, and a dehydration reaction due to the cross-linking does not occur, so that the film shrinkage hardly occurs. Therefore, the inclusion of the epoxy compound is effective in curing the composition at low temperature and suppressing warpage.
- the epoxy compound preferably contains a polyethylene oxide group.
- the polyethylene oxide group means that the number of repeating units of ethylene oxide is 2 or more, and the number of repeating units is preferably 2 to 15.
- epoxy compound examples include bisphenol A type epoxy resin; bisphenol F type epoxy resin; alkylene glycol type epoxy resin such as propylene glycol diglycidyl ether; polyalkylene glycol type epoxy resin such as polypropylene glycol diglycidyl ether; polymethyl (glycidyl Examples include, but are not limited to, epoxy group-containing silicones such as loxypropyl)siloxane.
- Epicron (registered trademark) 850-S Epiclon (registered trademark) HP-4032, Epiclon (registered trademark) HP-7200, Epiclon (registered trademark) HP-820, Epiclon (registered trademark) HP-4700, Epicron (registered trademark) EXA-4710, epicuron (registered trademark) HP-4770, epiclon (registered trademark) EXA-859CRP, epiclon (registered trademark) EXA-1514, epiclon (registered trademark) EXA-4880, epiclon (registered trademark) EXA-4850-150, Epiclon EXA-4850-1000, Epiclon (registered trademark) EXA-4816, Epicron (registered trademark) EXA-4822 (these product names, manufactured by DIC Corporation), Licaredin (registered trademark) BEO-60E (Trade name, Shin Nihon Rika Co., Ltd.), EP-4003S, EP-4000S (the above
- an epoxy resin containing a polyethylene oxide group is preferable in terms of suppression of warpage and excellent heat resistance.
- Epiclon (registered trademark) EXA-4880, Epiclon (registered trademark) EXA-4822, and Lycaledin (registered trademark) BEO-60E are preferable because they contain a polyethylene oxide group.
- oxetane compound compound having oxetanyl group
- oxetane compound a compound having two or more oxetane rings in one molecule, 3-ethyl-3-hydroxymethyloxetane, 1,4-bis ⁇ [(3-ethyl-3-oxetanyl)methoxy]methyl ⁇ benzene, Examples thereof include 3-ethyl-3-(2-ethylhexylmethyl)oxetane and 1,4-benzenedicarboxylic acid-bis[(3-ethyl-3-oxetanyl)methyl] ester.
- the Aron oxetane series manufactured by Toagosei Co., Ltd. (for example, OXT-121, OXT-221, OXT-191, OXT-223) can be preferably used, and these can be used alone or You may mix 2 or more types.
- the benzoxazine compound is preferable because it does not generate degas during curing because it is a cross-linking reaction derived from a ring-opening addition reaction, and further reduces heat shrinkage to suppress warpage.
- benzoxazine compound examples include Ba type benzoxazine, Bm type benzoxazine (these are trade names, manufactured by Shikoku Chemicals Co., Ltd.), a benzoxazine adduct of polyhydroxystyrene resin, and phenol novolac type dihydrobenzo.
- examples include oxazine compounds. These may be used alone or in combination of two or more.
- the polymerizable compound When the polymerizable compound is contained, its content is preferably more than 0% by mass and 60% by mass or less based on the total solid content of the curable resin composition of the present invention.
- the lower limit is more preferably 5% by mass or more.
- the upper limit is more preferably 50% by mass or less, further preferably 30% by mass or less.
- the polymerizable compounds may be used alone or in a mixture of two or more. When two or more kinds are used in combination, the total amount is preferably within the above range.
- the curable resin composition of the present invention preferably contains a solvent. Any known solvent can be used as the solvent.
- the solvent is preferably an organic solvent. Examples of the organic solvent include compounds such as esters, ethers, ketones, aromatic hydrocarbons, sulfoxides and amides.
- esters examples include ethyl acetate, n-butyl acetate, isobutyl acetate, amyl formate, isoamyl acetate, butyl propionate, isopropyl butyrate, ethyl butyrate, butyl butyrate, methyl lactate, ethyl lactate, ⁇ -butyrolactone, ⁇ -caprolactone.
- alkyl alkyloxyacetate eg methyl alkyloxyacetate, ethyl alkyloxyacetate, butyl alkyloxyacetate (eg methyl methoxyacetate, ethyl methoxyacetate, butyl methoxyacetate, methyl ethoxyacetate, ethyl ethoxyacetate etc. )
- 3-alkyloxypropionic acid alkyl esters eg, methyl 3-alkyloxypropionate, ethyl 3-alkyloxypropionate, etc.
- ethers include diethylene glycol dimethyl ether, tetrahydrofuran, ethylene glycol monomethyl ether, ethylene glycol monoethyl ether, methyl cellosolve acetate, ethyl cellosolve acetate, diethylene glycol monomethyl ether, diethylene glycol monoethyl ether, diethylene glycol monobutyl ether, propylene glycol monomethyl ether, propylene glycol.
- Suitable examples include monomethyl ether acetate, propylene glycol monoethyl ether acetate, and propylene glycol monopropyl ether acetate.
- ketones include methyl ethyl ketone, cyclohexanone, cyclopentanone, 2-heptanone, 3-heptanone and the like.
- aromatic hydrocarbons include toluene, xylene, anisole, limonene and the like.
- Preferred examples of sulfoxides include dimethyl sulfoxide.
- Preferred amides include N-methyl-2-pyrrolidone, N-ethyl-2-pyrrolidone, N,N-dimethylacetamide, N,N-dimethylformamide and the like.
- the combined use of dimethyl sulfoxide and ⁇ -butyrolactone is particularly preferred.
- the content of the solvent is preferably such that the total solid content concentration of the curable resin composition of the present invention is 5 to 80% by mass, and preferably 5 to 75% by mass. It is more preferable that the amount is 10 to 70% by mass, further preferably 40 to 70% by mass.
- the solvent content may be adjusted according to the desired thickness and coating method.
- Only one type of solvent may be contained, or two or more types may be contained.
- the total amount is preferably within the above range.
- the curable resin composition of the present invention preferably further contains a migration inhibitor.
- a migration inhibitor By including the migration inhibitor, it is possible to effectively suppress the migration of metal ions derived from the metal layer (metal wiring) into the curable resin composition layer.
- the migration inhibitor is not particularly limited, but a heterocycle (pyrrole ring, furan ring, thiophene ring, imidazole ring, oxazole ring, thiazole ring, pyrazole ring, isoxazole ring, isothiazole ring, tetrazole ring, pyridine ring, Compounds having pyridazine ring, pyrimidine ring, pyrazine ring, piperidine ring, piperazine ring, morpholine ring, 2H-pyran ring and 6H-pyran ring, triazine ring), compounds having thioureas and sulfanyl groups, hindered phenol compounds , Salicylic acid derivative-based compounds, and hydrazide derivative-based compounds.
- triazole compounds such as 1,2,4-triazole and benzotriazole can be preferably used, and tetrazole compounds such as 1H-tetrazole
- an ion trap agent that traps anions such as halogen ions can be used.
- the rust preventives described in paragraph 0094 of JP2013-015701A As other migration inhibitors, the rust preventives described in paragraph 0094 of JP2013-015701A, the compounds described in paragraphs 0073 to 0076 of JP2009-283711, and JP2011-059656A are described.
- the compounds described in paragraph 0052, the compounds described in paragraphs 0114, 0116 and 0118 of JP 2012-194520 A, the compounds described in paragraph 0166 of WO 2015/199219 and the like can be used.
- the content of the migration inhibitor is preferably 0.01 to 5.0 mass% with respect to the total solid content of the curable resin composition, and 0 It is more preferably from 0.05 to 2.0% by mass, further preferably from 0.1 to 1.0% by mass.
- the migration inhibitor may be only one kind or two or more kinds. When two or more migration inhibitors are used, it is preferable that the total amount thereof is within the above range.
- the curable resin composition of the present invention preferably contains a polymerization inhibitor.
- polymerization inhibitor examples include hydroquinone, p-methoxyphenol, di-tert-butyl-p-cresol, pyrogallol, p-tert-butylcatechol, 1,4-benzoquinone, diphenyl-p-benzoquinone, 4,4′.
- the content of the polymerization inhibitor is 0.01 to 5% by mass based on the total solid content of the curable resin composition of the present invention. Is preferable, 0.02 to 3% by mass is more preferable, and 0.05 to 2.5% by mass is further preferable.
- the polymerization inhibitor may be only one kind or two or more kinds. When two or more polymerization inhibitors are used, the total amount is preferably within the above range.
- the curable resin composition of the present invention preferably contains a metal adhesion improver for improving the adhesion to metal materials used for electrodes and wiring.
- a metal adhesion improver for improving the adhesion to metal materials used for electrodes and wiring.
- the metal adhesion improver include a silane coupling agent and the like.
- silane coupling agent examples include compounds described in paragraph 0167 of WO 2015/199219, compounds described in paragraphs 0062 to 0073 of JP2014-191002A, paragraphs of WO2011/080992.
- the compound as described in paragraph 0055 is mentioned. It is also preferable to use two or more different silane coupling agents as described in paragraphs 0050 to 0058 of JP2011-128358A. Further, it is also preferable to use the following compounds as the silane coupling agent.
- Et represents an ethyl group.
- the content of the metal adhesion improver is preferably 0.1 to 30 parts by mass, more preferably 0.5 to 15 parts by mass, based on 100 parts by mass of the heterocycle-containing polymer precursor. It is preferably in the range of 0.5 to 5 parts by mass. When it is at least the above lower limit, the adhesiveness between the cured film and the metal layer after the curing step will be good, and when it is at most the above upper limit, the heat resistance and mechanical properties of the cured film after the curing step will be good.
- the metal adhesion improver may be only one kind or two or more kinds. When two or more kinds are used, it is preferable that the total is within the above range.
- the curable resin composition of the present invention contains various additives, if necessary, such as a thermal acid generator, a sensitizer such as N-phenyldiethanolamine, and a chain transfer agent, as long as the effects of the present invention are not impaired.
- a surfactant, a higher fatty acid derivative, inorganic particles, a curing agent, a curing catalyst, a filler, an antioxidant, an ultraviolet absorber, an agglomeration inhibitor and the like can be added.
- the total blending amount is preferably 3% by mass or less of the solid content of the composition.
- the curable resin composition of the present invention may contain a sensitizer.
- the sensitizer absorbs specific actinic radiation to be in an electronically excited state.
- the agent in the electronically excited state comes into contact with a thermal base generator, a thermal radical polymerization initiator, a photoradical polymerization initiator, etc., and effects such as electron transfer, energy transfer, and heat generation.
- the thermal base generator, thermal radical polymerization initiator, and photo radical polymerization initiator undergo a chemical change and decompose to generate a radical, an acid, or a base.
- the description of the sensitizing dyes described in paragraphs 0161 to 0163 of JP-A-2016-027357 can be referred to, and the contents thereof are incorporated in the present specification.
- the content of the sensitizer is 0.01 to 20% by mass based on the total solid content of the curable resin composition of the present invention. It is preferably 0.1 to 15% by mass, and more preferably 0.5 to 10% by mass.
- the sensitizers may be used alone or in combination of two or more.
- the curable resin composition of the present invention may contain a chain transfer agent.
- the chain transfer agent is defined, for example, in Polymer Dictionary, Third Edition (edited by The Polymer Society of Japan, 2005), pages 683-684.
- As the chain transfer agent for example, a group of compounds having SH, PH, SiH, and GeH in the molecule is used. These can generate a radical by donating hydrogen to a low-activity radical, or can generate a radical by deprotonating after being oxidized.
- a thiol compound can be preferably used.
- the content of the chain transfer agent is 0.01 to 20 parts by mass based on 100 parts by mass of the total solid content of the curable resin composition of the present invention. It is preferably 1 to 10 parts by mass, more preferably 1 to 5 parts by mass.
- the chain transfer agent may be only one kind or two or more kinds. When two or more chain transfer agents are used, the total is preferably within the above range.
- each type of surfactant may be added to the curable resin composition of the present invention.
- the surfactant various kinds of surfactants such as a fluorine-based surfactant, a nonionic surfactant, a cationic surfactant, an anionic surfactant, and a silicone-based surfactant can be used.
- the following surfactants are also preferable.
- the parentheses indicating the constituent units of the main chain represent the content (mol %) of each constituent unit
- the parentheses indicating the constituent units of the side chain represent the number of repetitions of each constituent unit.
- the surfactant the compounds described in paragraphs 0159 to 0165 of WO 2015/199219 can also be used.
- the content of the surfactant is 0.001 to 2.0% by mass based on the total solid content of the curable resin composition of the present invention. It is preferably present, and more preferably 0.005 to 1.0% by mass.
- the surfactant may be only one kind or two or more kinds. When two or more kinds of surfactants are used, it is preferable that the total amount is within the above range.
- the curable resin composition of the present invention contains a higher fatty acid derivative such as behenic acid or behenic acid amide in order to prevent polymerization inhibition caused by oxygen, and the surface of the composition is dried in the course of drying after coating. May be unevenly distributed.
- the content of the higher fatty acid derivative is 0.1 to 10% by mass based on the total solid content of the curable resin composition of the present invention. Is preferred. Only one kind of higher fatty acid derivative may be used, or two or more kinds thereof may be used. When two or more higher fatty acid derivatives are used, the total is preferably within the above range.
- the water content of the curable resin composition of the present invention is preferably less than 5% by mass, more preferably less than 1% by mass, and even more preferably less than 0.6% by mass from the viewpoint of the properties of the coated surface.
- the metal content of the curable resin composition of the present invention is preferably less than 5 mass ppm (parts per million), more preferably less than 1 mass ppm, and even more preferably less than 0.5 mass ppm from the viewpoint of insulating properties.
- the metal include sodium, potassium, magnesium, calcium, iron, chromium and nickel. When a plurality of metals are contained, the total of these metals is preferably within the above range.
- a raw material having a low metal content is selected as a raw material constituting the curable resin composition of the present invention.
- Examples include a method of performing filter filtration on the raw materials constituting the curable resin composition of the invention and a method of performing distillation under conditions in which the inside of the apparatus is lined with polytetrafluoroethylene or the like to suppress contamination as much as possible. be able to.
- the curable resin composition of the present invention has a halogen atom content of preferably less than 500 mass ppm, more preferably less than 300 mass ppm, and more than 200 mass ppm from the viewpoint of wiring corrosivity. Less than ppm is even more preferred. Among them, those existing in the state of halogen ions are preferably less than 5 mass ppm, more preferably less than 1 mass ppm, still more preferably less than 0.5 mass ppm.
- the halogen atom include chlorine atom and bromine atom. It is preferable that the total of chlorine atom and bromine atom, or the total of chlorine ion and bromine ion be within the above ranges.
- a conventionally known container can be used as a container for the curable resin composition of the present invention.
- a multi-layer bottle in which the inner wall of the container is composed of 6 kinds of 6 layers of resin, or 6 kinds of resin of 7 layers structure is used for the purpose of suppressing contamination of impurities into raw materials and compositions. It is also preferable to use a bottle.
- a container for example, the container described in JP-A-2015-123351 can be mentioned.
- the curable resin composition of the present invention can be prepared by mixing the above components.
- the mixing method is not particularly limited and may be a conventionally known method.
- the pore size of the filter is preferably 1 ⁇ m or less, more preferably 0.5 ⁇ m or less, still more preferably 0.1 ⁇ m or less.
- the material of the filter is preferably polytetrafluoroethylene, polyethylene or nylon.
- the filter may be washed with an organic solvent in advance. In the filter filtration step, plural kinds of filters may be connected in series or in parallel and used. When using a plurality of types of filters, filters having different pore diameters or materials may be used in combination. Also, various materials may be filtered multiple times. When filtration is performed a plurality of times, circulation filtration may be used.
- the pressurizing pressure is preferably 0.05 MPa or more and 0.3 MPa or less.
- removal treatment of impurities using an adsorbent may be performed. Filter filtration and impurity removal treatment using an adsorbent may be combined.
- a known adsorbent can be used. Examples thereof include inorganic adsorbents such as silica gel and zeolite, and organic adsorbents such as activated carbon.
- the curable resin composition of the present invention is preferably used for forming an interlayer insulating film for a rewiring layer. In addition, it can also be used for forming an insulating film of a semiconductor device or forming a stress buffer film.
- the cured film of the present invention is obtained by curing the curable resin composition of the present invention.
- the thickness of the cured film of the present invention can be, for example, 0.5 ⁇ m or more, and can be 1 ⁇ m or more. Further, the upper limit value may be 100 ⁇ m or less, and may be 30 ⁇ m or less.
- the laminate of the present invention includes two or more cured films and a metal layer between any of the cured films.
- a metal layer is preferably used as a metal wiring such as a rewiring layer.
- the fields to which the cured film of the present invention can be applied include insulating films of semiconductor devices, interlayer insulating films for rewiring layers, stress buffer films, and the like.
- a sealing film, a substrate material (a base film or a coverlay of a flexible printed circuit board, an interlayer insulating film), or an insulating film for mounting as described above may be patterned by etching.
- the cured film of the present invention can also be used for manufacturing plate surfaces such as offset printing plates or screen printing plates, for use in etching molded parts, and for manufacturing protective lacquers and dielectric layers in electronics, especially microelectronics.
- the method for producing a cured film of the present invention includes a film forming step of applying the curable resin composition of the present invention to a substrate to form a film. Is preferred. Further, the method for producing a cured film of the present invention more preferably includes the film forming step and further includes a heating step of heating the film to decompose the compound represented by the formula (1-1). preferable. Specifically, it is also preferable to include the following steps (a) to (d).
- a film forming step of applying a curable resin composition to a substrate to form a film (curable resin composition layer) (b) An exposure step of exposing the film after the film forming step (c) An exposure step A developing step of performing a developing treatment on the above film (d) A heating step of heating the developed film to decompose the compound represented by the formula (1-1), and a heating step in the heating step.
- the resin layer cured by exposure can be further cured. In this heating step, for example, the compound represented by the above formula (1-1) is decomposed and sufficient curability is obtained.
- a method for producing a laminate according to a preferred embodiment of the present invention includes the method for producing a cured film of the present invention.
- the step (a) or the steps (a) to (c), or (a) is performed again.
- )-(D) are performed.
- the manufacturing method according to a preferred embodiment of the present invention includes a film forming step (layer forming step) of applying a curable resin composition to a substrate to form a film (layer).
- the type of the base material can be appropriately determined according to the application, but is a semiconductor manufacturing base material such as silicon, silicon nitride, polysilicon, silicon oxide, and amorphous silicon, quartz, glass, an optical film, a ceramic material, a vapor deposition film, A magnetic film, a reflective film, a metal substrate such as Ni, Cu, Cr, and Fe, paper, SOG (Spin On Glass), a TFT (thin film transistor) array substrate, an electrode plate of a plasma display panel (PDP), etc. are not particularly limited.
- a semiconductor manufacturing base material is particularly preferable, and a silicon base material is more preferable.
- the base material for example, a plate-shaped base material (substrate) is used.
- the resin layer or the metal layer serves as the base material.
- Coating is preferred as the means for applying the curable resin composition to the substrate.
- a dip coating method, an air knife coating method, a curtain coating method, a wire bar coating method, a gravure coating method, an extrusion coating method, a spray coating method, a spin coating method, a slit coating method, The inkjet method and the like are exemplified. From the viewpoint of the uniformity of the thickness of the curable resin composition layer, the spin coating method, the slit coating method, the spray coating method, and the inkjet method are more preferable. A resin layer having a desired thickness can be obtained by adjusting an appropriate solid content concentration and coating conditions according to the method.
- the coating method can be appropriately selected depending on the shape of the base material, and spin coating method, spray coating method, inkjet method or the like is preferable for circular base materials such as wafers, and slit coating method or spray coating for rectangular base materials. Method, inkjet method and the like are preferable.
- spin coating for example, it can be applied at a rotation speed of 500 to 2,000 rpm for about 10 seconds to 1 minute. It is also possible to apply a method in which a coating film previously formed on the temporary support by the above-mentioned application method is transferred onto the substrate.
- the production method of the present invention may include a step of drying the film (curable resin composition layer) after forming the film and then, after the film forming step (layer forming step), to remove the solvent.
- the preferred drying temperature is 50 to 150°C, more preferably 70°C to 130°C, even more preferably 90°C to 110°C.
- the drying time is, for example, 30 seconds to 20 minutes, preferably 1 minute to 10 minutes, more preferably 3 minutes to 7 minutes.
- the production method of the present invention may include an exposure step of exposing the above film (curable resin composition layer).
- the amount of exposure is not particularly limited as long as the curable resin composition can be cured, but, for example, 100 to 10,000 mJ/cm 2 in terms of exposure energy at a wavelength of 365 nm is preferable, and 200 to 8,000 mJ/cm 2 is preferable. More preferably, it is irradiated with cm 2 .
- the exposure wavelength can be appropriately set in the range of 190 to 1,000 nm, and preferably 240 to 550 nm.
- the exposure wavelength is (1) semiconductor laser (wavelength 830 nm, 532 nm, 488 nm, 405 nm etc.), (2) metal halide lamp, (3) high pressure mercury lamp, g line (wavelength 436 nm), h Line (wavelength 405 nm), i line (wavelength 365 nm), broad (3 wavelengths of g, h and i lines), (4) excimer laser, KrF excimer laser (wavelength 248 nm), ArF excimer laser (wavelength 193 nm), F2 excimer Laser (wavelength: 157 nm), (5) extreme ultraviolet rays; EUV (wavelength: 13.6 nm), (6) electron beam, etc.
- exposure with a high pressure mercury lamp is particularly preferable, and exposure with i-line is particularly preferable. Thereby, a particularly high exposure sensitivity can be obtained.
- the production method of the present invention may include a development treatment step of performing development treatment on the exposed film (curable resin composition layer). By developing, the unexposed portion (non-exposed portion) is removed.
- the developing method is not particularly limited as long as a desired pattern can be formed, and for example, a developing method such as paddle, spraying, dipping, ultrasonic wave or the like can be adopted.
- the developer can be used without particular limitation as long as the unexposed portion (non-exposed portion) is removed.
- the developer preferably contains an organic solvent, and more preferably the developer contains 90% or more of the organic solvent.
- the developer preferably contains an organic solvent having a ClogP value of -1 to 5, and more preferably an organic solvent having a ClogP value of 0 to 3.
- the ClogP value can be obtained as a calculated value by inputting a structural formula in ChemBioDraw.
- organic solvent examples include esters such as ethyl acetate, n-butyl acetate, amyl formate, isoamyl acetate, isobutyl acetate, butyl propionate, isopropyl butyrate, ethyl butyrate, butyl butyrate, methyl lactate, ethyl lactate, ⁇ -butyrolactone.
- esters such as ethyl acetate, n-butyl acetate, amyl formate, isoamyl acetate, isobutyl acetate, butyl propionate, isopropyl butyrate, ethyl butyrate, butyl butyrate, methyl lactate, ethyl lactate, ⁇ -butyrolactone.
- alkyl alkyloxyacetate eg, methyl alkyloxyacetate, ethyl alkyloxyacetate, butyl alkyloxyacetate (for example, methyl methoxyacetate, ethyl methoxyacetate, butyl methoxyacetate, methyl ethoxyacetate, Ethyl ethoxyacetate, etc.
- alkyl alkyloxyacetate eg, methyl alkyloxyacetate, ethyl alkyloxyacetate, butyl alkyloxyacetate (for example, methyl methoxyacetate, ethyl methoxyacetate, butyl methoxyacetate, methyl ethoxyacetate, Ethyl ethoxyacetate, etc.
- 3-alkyloxypropionic acid alkyl esters eg, methyl 3-alkyloxypropionate, ethyl 3-alkyloxypropionate, etc.
- cyclopentanone and ⁇ -butyrolactone are particularly preferable, and cyclopentanone is more preferable.
- the developer preferably has 50% by mass or more of an organic solvent, more preferably 70% by mass or more of an organic solvent, and further preferably 90% by mass or more of an organic solvent. Further, 100% by mass of the developer may be an organic solvent.
- the temperature of the developing solution at the time of development is not particularly limited, but it can usually be 20 to 40°C.
- Rinsing may be further performed after the treatment with the developing solution. Rinsing is preferably performed in a solvent different from the developing solution. For example, rinsing can be performed using the solvent contained in the curable resin composition.
- the rinse time is preferably 5 seconds to 1 minute.
- the production method of the present invention preferably includes a step (heating step) of heating the film to decompose the compound represented by the formula (1-1).
- the heating step is preferably included after the film forming step (layer forming step), the drying step, and the developing step.
- the heating step for example, the compound represented by the formula (1-1) is decomposed to generate a base, and the cyclization reaction of the heterocycle-containing polymer precursor proceeds.
- the composition of the present invention may contain a radical polymerizable compound other than the heterocyclic ring-containing polymer precursor, but curing of the radically polymerizable compound other than the unreacted heterocyclic ring-containing polymer precursor is also performed in this step. You can proceed.
- the heating temperature (maximum heating temperature) of the layer in the heating step is preferably 50°C or higher, more preferably 80°C or higher, even more preferably 140°C or higher, and 150°C or higher. Is more preferred, 160°C or higher is even more preferred, and 170°C or higher is even more preferred.
- the upper limit is preferably 500°C or lower, more preferably 450°C or lower, further preferably 350°C or lower, further preferably 250°C or lower, and 220°C or lower. Even more preferable.
- the heating is preferably performed at a temperature rising rate of 1 to 12° C./minute from the temperature at the start of heating to the maximum heating temperature, more preferably 2 to 10° C./minute, further preferably 3 to 10° C./minute.
- a temperature rising rate of 1 to 12° C./minute from the temperature at the start of heating to the maximum heating temperature, more preferably 2 to 10° C./minute, further preferably 3 to 10° C./minute.
- the temperature at the start of heating is preferably 20°C to 150°C, more preferably 20°C to 130°C, further preferably 25°C to 120°C.
- the temperature at the start of heating refers to the temperature at which the step of heating to the maximum heating temperature is started.
- the temperature of the film (layer) after the drying is, for example, higher than the boiling point of the solvent contained in the curable resin composition, It is preferable to gradually raise the temperature from 30 to 200° C. lower.
- the heating time (heating time at the maximum heating temperature) is preferably 10 to 360 minutes, more preferably 20 to 300 minutes, and further preferably 30 to 240 minutes.
- the heating temperature is preferably 180°C to 320°C, more preferably 180°C to 260°C. The reason for this is not clear, but it is considered that at this temperature, the ethynyl groups of the inter-layer heterocycle-containing polymer precursors are undergoing a crosslinking reaction.
- ⁇ Heating may be done in stages.
- the temperature is raised from 25° C. to 180° C. at 3° C./min, held at 180° C. for 60 minutes, raised from 180° C. to 200° C. at 2° C./min, and held at 200° C. for 120 minutes.
- a pretreatment step such as, may be performed.
- the heating temperature in the pretreatment step is preferably 100 to 200°C, more preferably 110 to 190°C, and further preferably 120 to 185°C.
- the pretreatment step may be performed for a short time of about 10 seconds to 2 hours, more preferably 15 seconds to 30 minutes.
- the pretreatment may be performed in two or more steps.
- the pretreatment step 1 may be performed in the range of 100 to 150°C
- the pretreatment step 2 may be performed thereafter in the range of 150 to 200°C.
- the cooling rate in this case is preferably 1 to 5° C./minute.
- the heating step is preferably performed in an atmosphere of low oxygen concentration by flowing an inert gas such as nitrogen, helium, or argon from the viewpoint of preventing decomposition of the heterocycle-containing polymer precursor.
- the oxygen concentration is preferably 50 ppm (volume ratio) or less, more preferably 20 ppm (volume ratio) or less.
- the production method of the present invention preferably includes a metal layer forming step of forming a metal layer on the surface of the film (curable resin composition layer) after the development treatment.
- the metal layer is not particularly limited, and existing metal species can be used, and copper, aluminum, nickel, vanadium, titanium, chromium, cobalt, gold, and tungsten are exemplified, and copper and aluminum are more preferable, and copper is More preferable.
- the method for forming the metal layer is not particularly limited, and existing methods can be applied.
- the methods described in JP-A-2007-157879, JP-A-2001-521288, JP-A-2004-214501 and JP-A-2004-101850 can be used.
- photolithography, lift-off, electrolytic plating, electroless plating, etching, printing, and a method combining these can be considered.
- a patterning method that combines sputtering, photolithography and etching, and a patterning method that combines photolithography and electrolytic plating can be mentioned.
- the thickness of the metal layer is preferably 0.1 to 50 ⁇ m, and more preferably 1 to 10 ⁇ m in the thickest part.
- the manufacturing method of the present invention preferably further includes a laminating step.
- the laminating step means, again on the surface of the cured film (resin layer) or the metal layer, (a) film forming step (layer forming step), (b) exposure step, (c) development processing step, (d) heating step. Is carried out in this order. However, the mode may be such that only the film forming step (a) is repeated.
- the heating step (d) may be performed collectively at the end or the middle of the stacking. That is, the steps (a) to (c) may be repeated a predetermined number of times, and then the step (d) may be heated to collectively cure the laminated curable resin composition layers. Further, (c) the developing step may include (e) the metal layer forming step.
- the laminating step may further include the above-mentioned drying step, heating step and the like as appropriate.
- a surface activation treatment step may be further performed after the heating step, the exposure step, or the metal layer forming step.
- a plasma treatment is exemplified as the surface activation treatment.
- the above laminating step is preferably performed 2 to 5 times, more preferably 3 to 5 times.
- a resin layer/metal layer/resin layer/metal layer/resin layer/metal layer structure having 3 to 7 layers is preferable, and 3 to 5 layers is more preferable.
- a cured film (resin layer) of the curable resin composition is further formed so as to cover the metal layer.
- the steps include (b) exposing step, (c) developing step, (e) metal layer forming step, and repeating (d) heating step at the end or in the middle.
- the present invention also discloses a semiconductor device having the cured film or laminate of the present invention.
- the semiconductor device in which the curable resin composition of the present invention is used for forming the interlayer insulating film for the rewiring layer the description in paragraphs 0213 to 0218 of JP-A-2016-027357 and the description in FIG. 1 are referred to. Yes, and their contents are incorporated herein.
- the thermal base generator of the present invention is a thermal base generator represented by the following formula (1-2).
- R 21 and R 22 each independently represent a monovalent organic group
- L 21 represents a divalent linking group
- atoms at both ends of L 21 at the bonding positions are both It is a carbon atom
- R 23 represents a hydrogen atom or a monovalent organic group.
- R 21 , R 22 and R 23 in formula (1-2) have the same meanings as R 1 , R 2 and R 3 in formula (1-1), respectively, and the preferred embodiments are also the same.
- L 21 in formula (1-2) has the same meaning as L 1 in formula (1-1), except that all the atoms at the bonding positions at both ends are limited to carbon atoms, and the preferred embodiments are also the same. is there.
- the use of the thermal base generator of the present invention is not particularly limited, but for example, a curable resin composition can be formed by using it together with the above-mentioned heterocycle-containing polymer precursor. Further, the thermal base generator of the present invention is considered to be, for example, difficult to decompose at room temperature and excellent in base generation efficiency during heating. Therefore, in various applications in which a known thermal base generator is used, the thermal base generator of the present invention is used instead of the conventional thermal base generator or in combination with the conventional thermal base generator. Are considered useful.
- reaction mixture was cooled to room temperature and 21.43 g (270.9 mmol) pyridine and 90 mL N-methylpyrrolidone were added.
- the reaction mixture was then cooled to ⁇ 10° C. and 16.12 g (135.5 mmol) SOCl 2 was added over 10 minutes keeping the temperature at ⁇ 10 ⁇ 4° C. The viscosity increased during the addition of SOCl 2 .
- the reaction mixture was stirred at room temperature for 2 hours.
- the polybenzoxazole precursor was then precipitated in 6 liters of water and the water-polybenzoxazole precursor mixture was stirred for 15 minutes at a speed of 5000 rpm.
- the polybenzoxazole precursor was filtered off, stirred again in 6 liters of water for 30 minutes and filtered again. Then, the obtained polybenzoxazole precursor was dried under reduced pressure at 45° C. for 3 days.
- the weight average molecular weight of this polybenzoxazole precursor was 15,000.
- ⁇ Synthesis example 7> [Synthesis of polyimide precursor (A-6: polyimide precursor having radically polymerizable group) from 4,4'-oxydiphthalic anhydride, 4,4'-diaminodiphenyl ether, and 2-hydroxyethyl methacrylate] 155.1 g of 4,4′-oxydiphthalic anhydride (ODPA) was placed in a separable flask having a capacity of 2 liters, and 134.0 g of 2-hydroxyethyl methacrylate (HEMA) and 400 ml of ⁇ -butyrolactone were added. A reaction mixture was obtained by adding 79.1 g of pyridine with stirring at room temperature. After the heat generation due to the reaction was completed, the mixture was allowed to cool to room temperature and left to stand for 16 hours.
- ODPA 4,4′-oxydiphthalic anhydride
- HEMA 2-hydroxyethyl methacrylate
- the obtained reaction solution was added to 3 liters of ethyl alcohol to form a precipitate composed of a crude polymer.
- the produced crude polymer was collected by filtration and dissolved in 1.5 liter of tetrahydrofuran to obtain a crude polymer solution.
- the obtained crude polymer solution was dropped into 28 liters of water to precipitate a polymer, and the obtained precipitate was collected by filtration and dried in vacuum to obtain a powdery polymer A-6.
- the weight average molecular weight (Mw) of this polymer A-6 was measured and found to be 20,000.
- Synthesis Example 8 [3,3′4,4′-biphenyltetracarboxylic dianhydride, 4,4′-diaminodiphenyl ether, and polyimide precursor from 2-hydroxyethyl methacrylate (A-7: polyimide precursor having radically polymerizable group) Body)] Synthesis Example 7 was repeated except that 147.1 g of 3,3′4,4′-biphenyltetracarboxylic dianhydride was used in place of 155.1 g of 4,4′-oxydiphthalic anhydride in Synthesis Example 7. Polymer A-7 was obtained by carrying out the reaction in the same manner as described. The weight average molecular weight (Mw) of this polymer A-7 was measured and found to be 22,000.
- Examples 1 to 40 and Comparative Examples C1 and C2> In each of the Examples and Comparative Examples, the components shown in Table 1 or Table 2 below were mixed to obtain curable resin compositions. The resulting curable resin composition was pressure filtered through a polytetrafluoroethylene filter having a pore width of 0.45 ⁇ m.
- Table 1 or Table 2 the numerical value in the column of "part by mass” indicates the content (parts by mass) of each component. Further, in Table 1 or Table 2, the description of "-" indicates that the corresponding component is not contained.
- Viscosity fluctuation rate (%)
- each curable resin composition was applied (applied) in a layer form on a silicon wafer by a spin coating method to form a curable resin composition layer.
- the obtained silicon wafer to which the curable resin composition layer was applied was dried on a hot plate at 100° C. for 5 minutes to form a uniform curable resin composition layer having a thickness of 20 ⁇ m on the silicon wafer.
- the curable resin composition layer on the silicon wafer was exposed with a stepper (Nikon NSR 2005 i9C) at an exposure energy of 500 mJ/cm 2 .
- the exposed curable resin composition layer (resin layer) was heated in a nitrogen atmosphere at a heating rate of 10° C./min to 180° C., and then this temperature was maintained for 3 hours.
- the cured resin layer was immersed in a 4.9% hydrofluoric acid solution, and the resin layer was peeled off from the silicon wafer to obtain a resin film 1.
- the elongation at break of the resin film 1 was measured using a tensile tester (Tensilon) with a crosshead speed of 300 mm/min, a sample width of 10 mm, and a sample length of 50 mm in the longitudinal direction of the film and in the width direction at 25° C. and 65% relative humidity.
- the elongation at break was measured in the (RH) environment in accordance with JIS-K6251:2017.
- the elongation at break in the longitudinal direction was measured 5 times, and the average value was used. The evaluation was performed according to the following evaluation criteria. The evaluation results are shown in the column of "elongation at break" in Table 3. -Evaluation criteria- A: Elongation at break exceeded 60%. B: The elongation at break was more than 55% and 60% or less. C: The elongation at break was more than 40% and 55% or less. D: Elongation at break was 40% or less.
- each curable resin composition was applied in layers on a copper substrate by a spin coating method, dried at 100° C. for 5 minutes, and a curable resin composition layer having a film thickness of 20 ⁇ m was formed. Was formed.
- the obtained copper substrate to which the curable resin composition layer was applied was dried on a hot plate at 100° C. for 5 minutes to form a uniform curable resin composition layer having a thickness of 20 ⁇ m on the copper substrate.
- the curable resin composition layer on the copper substrate was exposed with a stepper (Nikon NSR 2005 i9C) at an exposure energy of 500 mJ/cm 2 using a photomask having a square-shaped unmasked portion of 100 ⁇ m square. Then, it was developed with cyclopentanone for 60 seconds to obtain a 100 ⁇ m square resin layer. Furthermore, in a nitrogen atmosphere, the temperature was raised at a heating rate of 10° C./min, and after reaching 180° C., this temperature was maintained for 3 hours.
- the shear force was measured for a 100 ⁇ m square resin layer on a copper substrate in a 25° C., 65% relative humidity (RH) environment using a bond tester (Condition Sigma, XYZTEC). The larger the shearing force, the larger the adhesion and the more preferable the result.
- the evaluation was performed according to the following evaluation criteria. The evaluation results are shown in the column of "Adhesion" in Table 3. -Evaluation criteria- A: Shear force exceeded 40 gf. B: The shearing force exceeded 25 gf and was 40 gf or less. C: The shearing force was 25 gf or less. However, 1 gf is set to 9.80665 ⁇ 10 ⁇ 3 N (Newton).
- the curable resin composition containing the heterocycle-containing polymer precursor according to the present invention and the compound represented by the formula (1-1) is excellent in storage stability and is a cured product obtained. It can be seen that the elongation at break of the film is excellent.
- the curable resin composition according to Comparative Example 1 did not contain the compound represented by the formula (1-1) but contained the compound represented by the formula (RB-1) as a thermal base generator. It can be seen that the curable resin composition according to Comparative Example 1 is inferior in storage stability.
- the curable resin composition according to Comparative Example 2 does not contain the compound represented by the formula (1-1). It can be seen that the curable resin composition according to Comparative Example 2 is inferior in elongation at break.
- Example 101 The curable resin composition described in Example 1 was applied in layers on the surface of a resin substrate on which a thin copper layer was formed by a spin coating method and dried at 100° C. for 5 minutes to give a curable film having a thickness of 20 ⁇ m. After forming the resin composition layer, exposure was performed using a stepper (manufactured by Nikon, NSR1505 i6). The exposure was performed at a wavelength of 365 nm through a mask (a binary mask having a pattern of 1:1 line and space and a line width of 10 ⁇ m). After exposure, it was developed with cyclopentanone for 30 seconds and rinsed with PGMEA for 20 seconds to obtain a pattern. Then, it was heated at 230° C.
- the interlayer insulating film for the redistribution layer had excellent insulating properties. Moreover, when a semiconductor device was manufactured using these interlayer insulating films for rewiring layers, it was confirmed that the device operates without problems.
Landscapes
- Chemical & Material Sciences (AREA)
- Physics & Mathematics (AREA)
- Medicinal Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Physics & Mathematics (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Engineering & Computer Science (AREA)
- Architecture (AREA)
- Structural Engineering (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
- Laminated Bodies (AREA)
- Macromonomer-Based Addition Polymer (AREA)
Abstract
Description
本明細書において、硬化性樹脂組成物の保管時において環化が進行しにくいことを、「硬化性樹脂組成物の保存安定性に優れる」ともいう。
また、本発明の別の一つの態様は、新規な熱塩基発生剤を提供することを目的とする。
下記式(1-1)で表される化合物を含む、
硬化性樹脂組成物。

<2> 上記L1が連結鎖長1~5の2価の連結基である、<1>に記載の硬化性樹脂組成物。
<3> 上記R1及び上記R2がそれぞれ独立に、炭化水素基である、<1>又は<2>に記載の硬化性樹脂組成物。
<4> 上記L1が、1,2-エチレン基、1,3-プロパンジイル基、1,2-シクロヘキサンジイル基、シスビニレン基、1,2-フェニレン基、1,2-フェニレンメチレン基、又は、1,2-エチレンオキシ-1,2-エチレン基である、<1>~<3>のいずれか1つに記載の硬化性樹脂組成物。
<5> 上記R3が、水素原子、アルキル基、又は、アリール基である、<1>~<4>のいずれか1つに記載の硬化性樹脂組成物。
<6> 上記式(1-1)で表される化合物の分子量が100以上2,000以下である、<1>~<5>のいずれか1つに記載の硬化性樹脂組成物。
<7> 光ラジカル重合開始剤及びラジカル重合性化合物を更に含む、<1>~<6>のいずれか1つに記載の硬化性樹脂組成物。
<8> 上記複素環含有ポリマー前駆体として、ポリイミド前駆体及びポリベンゾオキサゾール前駆体よりなる群から選ばれた少なくとも1種の前駆体を含む、<1>~<7>のいずれか1つに記載の硬化性樹脂組成物。
<9> 上記複素環含有ポリマー前駆体として、ポリイミド前駆体を含む、<1>~<8>のいずれか1つに記載の硬化性樹脂組成物。
<10> 上記ポリイミド前駆体が下記式(1)で表される繰返し単位を有する、<9>に記載の硬化性樹脂組成物;

<11> 上記式(1)におけるR113及びR114の少なくとも一方がラジカル重合性基を含む、<10>に記載の硬化性樹脂組成物。
<12> 再配線層用層間絶縁膜の形成に用いられる、<1>~<11>のいずれか1つに記載の硬化性樹脂組成物。
<13> <1>~<12>のいずれか1つに記載の硬化性樹脂組成物を硬化してなる硬化膜。
<14> <13>に記載の硬化膜を2層以上含み、上記硬化膜同士のいずれかの間に金属層を含む積層体。
<15> <1>~<12>のいずれか1つに記載の硬化性樹脂組成物を基材に適用して膜を形成する膜形成工程を含む、硬化膜の製造方法。
<16> 上記膜を加熱して上記式(1-1)により表される化合物を分解する加熱工程を更に含む、<15>に記載の硬化膜の製造方法。
<17> <13>に記載の硬化膜又は<14>に記載の積層体を含む、半導体デバイス。
<18> 下記式(1-2)により表される熱塩基発生剤。

また、本発明の別の一つの態様によれば、新規な熱塩基発生剤が提供される。
本明細書において「~」という記号を用いて表される数値範囲は、「~」の前後に記載される数値をそれぞれ下限値及び上限値として含む範囲を意味する。
本明細書において「工程」との語は、独立した工程だけではなく、その工程の所期の作用が達成できる限りにおいて、他の工程と明確に区別できない工程も含む意味である。
本明細書における基(原子団)の表記において、置換及び無置換を記していない表記は、置換基を有しない基(原子団)と共に置換基を有する基(原子団)をも包含する。例えば、「アルキル基」とは、置換基を有しないアルキル基(無置換アルキル基)のみならず、置換基を有するアルキル基(置換アルキル基)をも包含する。
本明細書において「露光」とは、特に断らない限り、光を用いた露光のみならず、電子線、イオンビーム等の粒子線を用いた露光も含む。また、露光に用いられる光としては、水銀灯の輝線スペクトル、エキシマレーザーに代表される遠紫外線、極紫外線(EUV光)、X線、電子線等の活性光線又は放射線が挙げられる。
本明細書において、「(メタ)アクリレート」は、「アクリレート」及び「メタクリレート」の両方、又は、いずれかを意味し、「(メタ)アクリル」は、「アクリル」及び「メタクリル」の両方、又は、いずれかを意味し、「(メタ)アクリロイル」は、「アクリロイル」及び「メタクリロイル」の両方、又は、いずれかを意味する。
本明細書において、構造式中のMeはメチル基を表し、Etはエチル基を表し、Buはブチル基を表し、Phはフェニル基を表す。
本明細書において、全固形分とは、組成物の全成分から溶剤を除いた成分の総質量をいう。また本明細書において、固形分濃度とは、組成物の総質量に対する、溶剤を除く他の成分の質量百分率である。
本明細書において、重量平均分子量(Mw)及び数平均分子量(Mn)は、特に述べない限り、ゲル浸透クロマトグラフィ(GPC測定)に従い、ポリスチレン換算値として定義される。本明細書において、重量平均分子量(Mw)及び数平均分子量(Mn)は、例えば、HLC-8220GPC(東ソー(株)製)を用い、カラムとしてガードカラムHZ-L、TSKgel Super HZM-M、TSKgel Super HZ4000、TSKgel Super HZ3000、TSKgel Super HZ2000(東ソー(株)製)を用いることによって求めることができる。それらの分子量は特に述べない限り、溶離液としてTHF(テトラヒドロフラン)を用いて測定したものとする。また、GPC測定における検出は特に述べない限り、UV線(紫外線)の波長254nm検出器を使用したものとする。
本明細書において、積層体を構成する各層の位置関係について、「上」又は「下」と記載したときには、注目している複数の層のうち基準となる層の上側又は下側に他の層があればよい。すなわち、基準となる層と上記他の層の間に、更に第3の層や要素が介在していてもよく、基準となる層と上記他の層は接している必要はない。また、特に断らない限り、基材に対し層が積み重なっていく方向を「上」と称し、又は、感光層がある場合には、基材から感光層へ向かう方向を「上」と称し、その反対方向を「下」と称する。なお、このような上下方向の設定は、本明細書中における便宜のためであり、実際の態様においては、本明細書における「上」方向は、鉛直上向きと異なることもありうる。
本明細書において、特段の記載がない限り、組成物は、組成物に含まれる各成分として、その成分に該当する2種以上の化合物を含んでもよい。また、特段の記載がない限り、組成物における各成分の含有量とは、その成分に該当する全ての化合物の合計含有量を意味する。
本明細書において、物性値は、特に述べない限り、温度23℃及び気圧101,325Pa(1気圧)の条件下での値である。
本明細書において、好ましい態様の組み合わせは、より好ましい態様である。
本発明の硬化性樹脂組成物(以下、単に、「本発明の組成物」ともいう。)は、複素環含有ポリマー前駆体、及び、式(1-1)で表される化合物を含む。
また、本発明の硬化性樹脂組成物は、後述する光ラジカル重合開始剤及び後述するラジカル重合性化合物を更に含むことが好ましい。
上記効果が得られるメカニズムは不明であるが、下記のように推測される。
本発明の硬化性樹脂組成物に含まれる式(1-1)で表される化合物は、室温等の保管時の温度においては分解しにくいため保存安定性に優れ、また、例えば加熱等による硬化時においては塩基の発生効率に優れるため、複素環含有ポリマー前駆体の環化が進行しやすく、得られる硬化膜の破断伸び率に優れると考えられる。
また、硬化性樹脂組成物が式(1-1)で表される化合物を含むことにより、上述の通り加熱等によって複素環含有ポリマー前駆体の環化が進行しやすいため、銅等の基材との密着性にも優れやすいと考えられる。すなわち、本発明の硬化性樹脂組成物を用いることにより、層間の密着性に優れた積層体が得られやすいと考えられる。
以下、本発明の硬化性樹脂組成物に含まれる成分について詳細に説明する。
本発明の硬化性樹脂組成物は、複素環含有ポリマー前駆体を含む。
本発明の硬化性樹脂組成物は、上記複素環含有ポリマー前駆体として、ポリイミド前駆体及びポリベンゾオキサゾール前駆体よりなる群から選ばれた少なくとも1種の前駆体を含むことが好ましく、ポリイミド前駆体を含むことがより好ましい。
得られる硬化膜の膜強度の観点からは、ポリイミド前駆体は、下記式(1)で表される繰返し単位を有することが好ましい。

式(1)におけるA1及びA2は、それぞれ独立に、酸素原子又は-NH-を表し、酸素原子が好ましい。
式(1)におけるR111は、2価の有機基を表す。2価の有機基としては、直鎖状又は分岐鎖状の脂肪族基、環状の脂肪族基、及び芳香族基、複素芳香族基、又はこれらを2以上組み合わせた基が例示され、炭素数2~20の直鎖の脂肪族基、炭素数3~20の分岐の脂肪族基、炭素数3~20の環状の脂肪族基、炭素数6~20の芳香族基、又は、これらの組み合わせからなる基が好ましく、炭素数6~20の芳香族基がより好ましい。



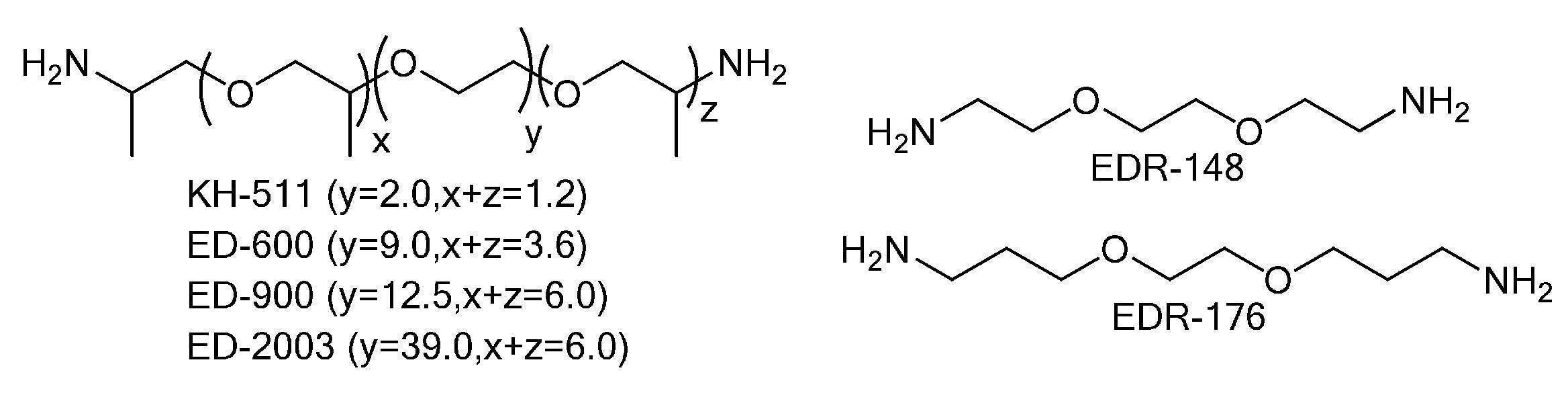


式(1)におけるR115は、4価の有機基を表す。4価の有機基としては、芳香環を含む4価の有機基が好ましく、下記式(5)又は式(6)で表される基がより好ましい。



式(1)におけるR113及びR114はそれぞれ独立に、水素原子又は1価の有機基を表す。R113及びR114の少なくとも一方がラジカル重合性基を含むことが好ましく、両方がラジカル重合性基を含むことがより好ましい。ラジカル重合性基としては、ラジカルの作用により、架橋反応することが可能な基であって、好ましい例として、エチレン性不飽和結合を有する基が挙げられる。
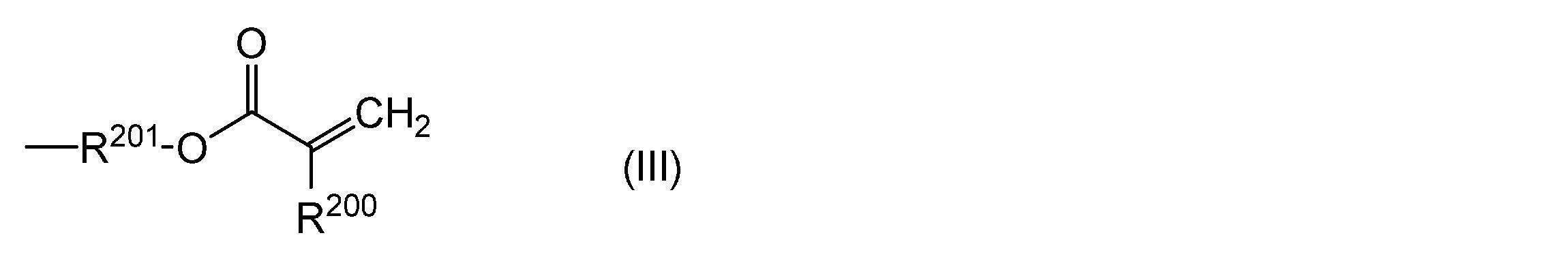
好適なR201の例は、エチレン基、プロピレン基、トリメチレン基、テトラメチレン基、1,2-ブタンジイル基、1,3-ブタンジイル基、ペンタメチレン基、ヘキサメチレン基、オクタメチレン基、ドデカメチレン基、-CH2CH(OH)CH2-が挙げられ、エチレン基、プロピレン基、トリメチレン基、-CH2CH(OH)CH2-がより好ましい。
特に好ましくは、R200がメチル基で、R201がエチレン基である。
本発明におけるポリイミド前駆体の好ましい実施形態として、R113又はR114の1価の有機基として、1、2又は3つの、好ましくは1つの酸基を有する、脂肪族基、芳香族基及びアリールアルキル基などが挙げられる。具体的には、酸基を有する炭素数6~20の芳香族基、酸基を有する炭素数7~25のアリールアルキル基が挙げられる。より具体的には、酸基を有するフェニル基及び酸基を有するベンジル基が挙げられる。酸基は、ヒドロキシ基が好ましい。すなわち、R113又はR114はヒドロキシ基を有する基であることが好ましい。
R113又はR114が表す1価の有機基としては、現像液の溶解度を向上させる置換基が好ましく用いられる。

本明細書において、分子量の分散度とは、重量平均分子量を数平均分子量により除した値(重量平均分子量/数平均分子量)をいう。
ポリベンゾオキサゾール前駆体は、下記式(2)で表される繰返し単位を含むことが好ましい。

式(2)中、R121は、2価の有機基を表す。2価の有機基としては、脂肪族基(炭素数1~24が好ましく、1~12がより好ましく、1~6が特に好ましい)及び芳香族基(炭素数6~22が好ましく、6~14がより好ましく、6~12が特に好ましい)の少なくとも一方を含む基が好ましい。R121を構成する芳香族基としては、上記式(1)のR111の例が挙げられる。上記脂肪族基としては、直鎖の脂肪族基が好ましい。R121は、4,4’-オキシジベンゾイルクロリドに由来することが好ましい。
式(2)中、R122は、4価の有機基を表す。4価の有機基としては、上記式(1)におけるR115と同義であり、好ましい範囲も同様である。R122は、2,2'-ビス(3-アミノ-4-ヒドロキシフェニル)ヘキサフルオロプロパンに由来することが好ましい。
R123及びR124は、それぞれ独立に、水素原子又は1価の有機基を表し、上記式(1)におけるR113及びR114と同義であり、好ましい範囲も同様である。

本発明の硬化性樹脂組成物は式(1-1)で表される化合物を含む。

例えば、下記式(B-1)により表される化合物は、加熱により、例えば下記反応式に記載のように分解し、発生したジイソプロピルアミンが塩基として働くことにより、硬化性樹脂組成物中の複素環含有ポリマー前駆体の環化を促進すると考えられる。

式(1-1)中、R1及びR2はそれぞれ独立に、炭化水素基であることが好ましく、炭素数1~20の炭化水素基であることがより好ましく、炭素数1~10の炭化水素基であることがより好ましい。上記炭化水素基としては、アルキル基又はアリール基が好ましく、炭素数1~10のアルキル基、又は、フェニル基がより好ましく、炭素数1~10のアルキル基が更に好ましい。具体的には、メチル基、エチル基、プロピル基、ブチル基、ペンチル基、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、イソプロピル基、イソブチル基、sec-ブチル基、t-ブチル基、1-エチルペンチル基、2-エチルヘキシル基、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、シクロヘプチル基、シクロオクチル基、又は、フェニル基が好ましく、メチル基、エチル基、プロピル基、ブチル基、ペンチル基、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、イソプロピル基、イソブチル基、sec-ブチル基、t-ブチル基、1-エチルペンチル基、2-エチルヘキシル基、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、シクロヘプチル基、又は、シクロオクチル基がさらに好ましく、メチル基、エチル基、イソプロピル基、又は、シクロへキシル基が最も好ましい。
また、R1とR2は結合して環構造を形成してもよく、形成される環構造としては、環員数5又は6の脂肪族環構造が好ましく、ピロリジン環、ピペリジン環、ピペラジン環、モルホリン環構造がより好ましい。
R1及びR2の式量(R1又はR2に含まれる原子の原子量の総和)は、それぞれ、29~300であることが好ましく、57~282であることがより好ましく、57~200であることが更に好ましい。
また、発生する塩基のR1及びR2は、発生する塩基の塩基性が高くなり、かつ、発生する塩基の銅基材等の基材との相互作用が小さく、得られる硬化膜と基材との密着性が向上する観点からは、R1及びR2がそれぞれ独立に炭素数1~10のアルキル基であるか、又は、R1及びR2が結合して環員数5若しくは6の脂肪族環構造を形成することが好ましく、R1及びR2がそれぞれ独立にエチル基若しくはイソプロピル基であるか、又は、R1及びR2が結合してピペリジン環構造を形成することがより好ましい。
その他、R1及びR2の構造は、例えば、発生する塩基の塩基性の強さ、発生する塩基と基材との相互作用の強さ、発生する塩基の運動性、発生する塩基の膜中の他素材との相互作用等を考慮して選択することができる。
式(1-1)中、L1は、連結鎖長1~5の2価の連結基であることが好ましく、連結鎖長2又は3の2価の連結基であることがより好ましい。
本明細書において、L1の連結鎖長とは、L1に含まれる原子であり、かつ、下記式(1ー1’)により表される化合物における2つの炭素原子C1とC2との間に存在する原子の数のうち、最小の数をいう。
下記式(1ー1’)は、上述の式(1-1)における炭素原子にそれぞれC1及びC2と記号を記載したものであり、式(1ー1’)中のR1、R2、R3及びL1はそれぞれ、式(1ー1)中のR1、R2、R3及びL1と同義である。
例えば、上述の式(B-1)で表される化合物におけるL1は1,2-フェニレン基であり、連結鎖長は2である。

塩基の発生効率の観点からは、L1における、炭素原子C1と炭素原子C2との間に存在する構造は、芳香環構造の一部であるか、又は、不飽和脂肪族炭化水素構造であることが好ましく、フェニレン基(特に好ましくは1,2-フェニレン基)又はシスビニレン基がより好ましい。上記フェニレン基又はシスビニレン基は、公知の置換基を更に有していてもよい。
また、L1は、炭素原子C1と炭素原子C2との間に存在する構造以外の構造に塩基発生部位を更に含んでもよい。塩基発生部位としては、例えば、上述の式(1-1)におけるL1を除いた構造(下記式(1-1A)に記載の2つの構造の組み合わせ)が挙げられる。L1が炭素原子C1と炭素原子C2との間に存在する構造以外の構造に塩基発生部位を更に含む化合物の例としては、後述する式(B-16、B-17、B-18)で表される化合物等が挙げられる。
その他、L1の構造は、例えば、塩基の発生効率を考慮して選択することができる。

R3は、水素原子又は炭化水素基であることが好ましく、水素原子、アルキル基、又は、アリール基であることがより好ましく、炭素数1~20のアルキル基、又は、炭素数6~20のアリール基であることがより好ましく、炭素数2~8のアルキル基、又は、フェニル基であることが更に好ましく、発生する塩基の銅基材等の基材との相互作用が小さく、得られる硬化膜と基材との密着性が向上する観点からは、炭素数2~4のアルキル基、又は、フェニル基がより好ましい。
R3がアルキル基である場合、上記アルキル基は直鎖状、分岐鎖状のいずれの構造であってもよいし、環状構造を有する構造であってもよいが、塩基の発生効率の観点からは、直鎖状であることが好ましい。
その他、R3の構造は、例えば、塩基の発生効率、分解後の化合物の銅基材等の基材との相互作用の強さ、電子供与性の強さ等を考慮して選択することができる。
式(1-1)で表される化合物の分子量は、例えば分解温度、揮発性等を考慮して決定すればよいが、100以上であることが好ましく、150以上であることがより好ましく、200以上であることがより好ましく、250以上であることが更に好ましい。上限としては、特に限定されないが、1,000以下であることが好ましい。
式(1-1)で表される化合物が熱塩基発生剤である場合、分解温度は、50℃以上であることが好ましく、80℃以上であることがより好ましく、120℃以上であることが更に好ましく、140℃以上であることが一層好ましい。上限としては、450℃以下であることがより一層好ましく、350℃以下であることがより好ましく、250℃以下であることが更に好ましい。
より低温で分解する化合物とすることにより、硬化性樹脂組成物の硬化に必要なエネルギーを減少することができる。
一方、分解温度を上記下限値以上とすることで、室温等での保管時に不用意に塩基を発生し効果を促進することが抑制され、硬化性樹脂組成物の保存安定性が向上する。
上記分解温度は、式(1-1)で表される化合物を耐圧カプセル中5℃/分で500℃まで加熱した場合に、最も温度が低い発熱ピークのピーク温度として求められる。


本明細書でいうpKaとは、酸から水素イオンが放出される解離反応を考え、その平衡定数Kaをその負の常用対数pKaによって表したものである。pKaが小さいほど強い酸であることを示す。pKaは、特に断らない限り、ACD/ChemSketch(登録商標)による計算値とする。又は、日本化学会編「改定5版 化学便覧 基礎編」に掲載の値を参照してもよい。
式(1-1)で表される化合物は、1種又は2種以上を用いることができる。2種以上を用いる場合は、合計量が上記範囲であることが好ましい。
本発明の硬化性樹脂組成物は、他の塩基発生剤を含んでいてもよい。
他の塩基発生剤には、上述の式(1-1)で表される化合物は含まれないものとする。
他の塩基発生剤としては、熱塩基発生剤及び光塩基発生剤が挙げられる。
他の熱塩基発生剤を含む場合、国際公開第2015/199219号及び国際公開第2015/199220号に記載のものが例示され、これらの内容は本明細書に組み込まれる。また、本発明では、他の熱塩基発生剤を実質的に含まない構成とすることもできる。実質的に含まないとは、本発明の硬化性樹脂組成物において、他の熱塩基発生剤の含有量が、式(1-1)で表される化合物の含有量の5質量%以下であることをいい、好ましくは3質量%以下、より好ましくは1質量%であることをいう。
本発明の硬化性樹脂組成物は、光重合開始剤を含むことが好ましい。
光重合開始剤は、光ラジカル重合開始剤であることが好ましい。光ラジカル重合開始剤としては、特に制限はなく、公知の光ラジカル重合開始剤の中から適宜選択することができる。例えば、紫外線領域から可視領域の光線に対して感光性を有する光ラジカル重合開始剤が好ましい。また、光励起された増感剤と何らかの作用を生じ、活性ラジカルを生成する活性剤であってもよい。

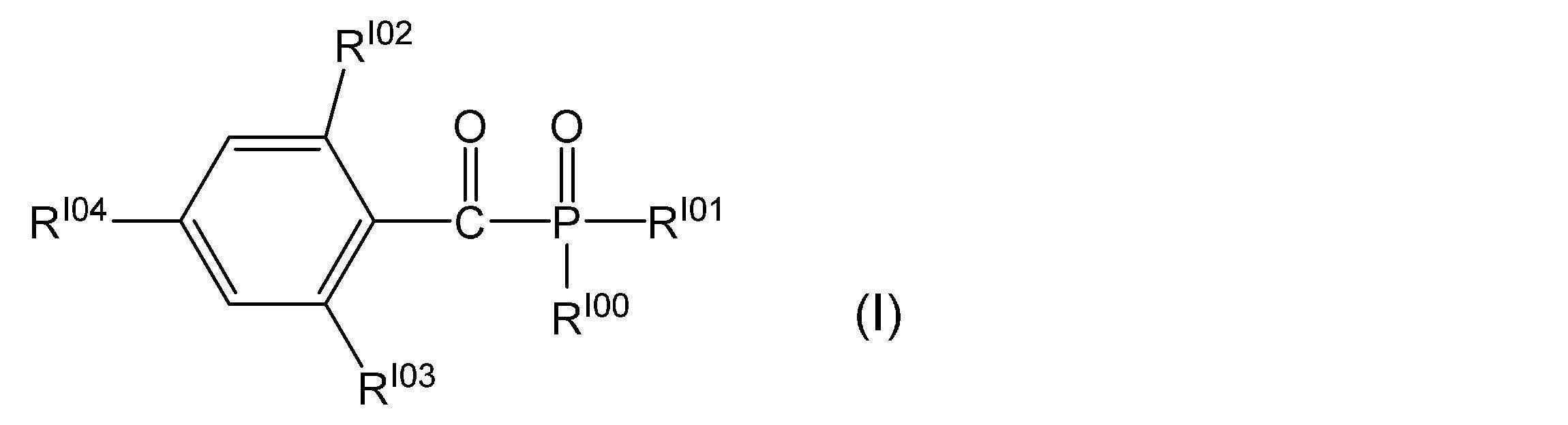

本発明の硬化性樹脂組成物は、重合開始剤として熱重合開始剤を含んでもよく、とくに熱ラジカル重合開始剤を含んでもよい。熱ラジカル重合開始剤は、熱のエネルギーによってラジカルを発生し、重合性を有する化合物の重合反応を開始又は促進させる化合物である。熱ラジカル重合開始剤を添加することによって、複素環含有ポリマー前駆体の環化と共に、複素環含有ポリマー前駆体の重合反応を進行させることもできるので、より高度な耐熱化が達成できることとなる。
〔ラジカル重合性化合物〕
本発明の硬化性樹脂組成物は重合性化合物を含むことが好ましい。重合性化合物としては、ラジカル重合性化合物を用いることができる。ラジカル重合性化合物は、ラジカル重合性基を有する化合物である。ラジカル重合性基としては、ビニル基、アリル基、ビニルフェニル基、(メタ)アクリロイル基などのエチレン性不飽和結合を有する基が挙げられる。ラジカル重合性基は、(メタ)アクリロイル基が好ましい。
本発明の硬化性樹脂組成物は、上述したラジカル重合性化合物以外の重合性化合物を更に含むことができる。上述したラジカル重合性化合物以外の重合性化合物としては、ヒドロキシメチル基、アルコキシメチル基又はアシルオキシメチル基を有する化合物;エポキシ化合物;オキセタン化合物;ベンゾオキサジン化合物が挙げられる。
ヒドロキシメチル基、アルコキシメチル基又はアシルオキシメチル基を有する化合物としては、下記式(AM1)、(AM4)又は(AM5)で示される化合物が好ましい。



エポキシ化合物としては、一分子中にエポキシ基を2以上有する化合物であることが好ましい。エポキシ基は、200℃以下で架橋反応し、かつ、架橋に由来する脱水反応が起こらないため膜収縮が起きにくい。このため、エポキシ化合物を含有することは、組成物の低温硬化及び反りの抑制に効果的である。
オキセタン化合物としては、一分子中にオキセタン環を2つ以上有する化合物、3-エチル-3-ヒドロキシメチルオキセタン、1,4-ビス{[(3-エチル-3-オキセタニル)メトキシ]メチル}ベンゼン、3-エチル-3-(2-エチルヘキシルメチル)オキセタン、1,4-ベンゼンジカルボン酸-ビス[(3-エチル-3-オキセタニル)メチル]エステル等を挙げることができる。具体的な例としては、東亞合成株式会社製のアロンオキセタンシリーズ(例えば、OXT-121、OXT-221、OXT-191、OXT-223)が好適に使用することができ、これらは単独で、又は2種以上混合してもよい。
ベンゾオキサジン化合物は、開環付加反応に由来する架橋反応のため、硬化時に脱ガスが発生せず、更に熱収縮を小さくして反りの発生が抑えられることから好ましい。
本発明の硬化性樹脂組成物は、溶剤を含有することが好ましい。溶剤は、公知の溶剤を任意に使用できる。溶剤は有機溶剤が好ましい。有機溶剤としては、エステル類、エーテル類、ケトン類、芳香族炭化水素類、スルホキシド類、アミド類などの化合物が挙げられる。
本発明の硬化性樹脂組成物は、更にマイグレーション抑制剤を含むことが好ましい。マイグレーション抑制剤を含むことにより、金属層(金属配線)由来の金属イオンが硬化性樹脂組成物層内へ移動することを効果的に抑制可能となる。

本発明の硬化性樹脂組成物は、重合禁止剤を含むことが好ましい。

本発明の硬化性樹脂組成物は、電極や配線などに用いられる金属材料との接着性を向上させるための金属接着性改良剤を含んでいることが好ましい。金属接着性改良剤としては、シランカップリング剤などが挙げられる。

本発明の硬化性樹脂組成物は、本発明の効果を損なわない範囲で、必要に応じて、各種の添加物、例えば、熱酸発生剤、N-フェニルジエタノールアミンなどの増感剤、連鎖移動剤、界面活性剤、高級脂肪酸誘導体、無機粒子、硬化剤、硬化触媒、充填剤、酸化防止剤、紫外線吸収剤、凝集防止剤等を配合することができる。これらの添加剤を配合する場合、その合計配合量は組成物の固形分の3質量%以下とすることが好ましい。
本発明の硬化性樹脂組成物は、増感剤を含んでいてもよい。増感剤は、特定の活性放射線を吸収して電子励起状態となる。電子励起状態となった剤は、熱塩基発生剤、熱ラジカル重合開始剤、光ラジカル重合開始剤などと接触して、電子移動、エネルギー移動、発熱などの作用が生じる。これにより、熱塩基発生剤、熱ラジカル重合開始剤、光ラジカル重合開始剤は化学変化を起こして分解し、ラジカル、酸又は塩基を生成する。増感剤の詳細については、特開2016-027357号公報の段落0161~0163に記載の増感色素の記載を参酌でき、この内容は本明細書に組み込まれる。
本発明の硬化性樹脂組成物は、連鎖移動剤を含有してもよい。連鎖移動剤は、例えば高分子辞典第三版(高分子学会編、2005年)683-684頁に定義されている。連鎖移動剤としては、例えば、分子内にSH、PH、SiH、及びGeHを有する化合物群が用いられる。これらは、低活性のラジカルに水素を供与して、ラジカルを生成するか、若しくは、酸化された後、脱プロトンすることによりラジカルを生成しうる。特に、チオール化合物を好ましく用いることができる。
本発明の硬化性樹脂組成物には、塗布性をより向上させる観点から、各種類の界面活性剤を添加してもよい。界面活性剤としては、フッ素系界面活性剤、ノニオン系界面活性剤、カチオン系界面活性剤、アニオン系界面活性剤、シリコーン系界面活性剤などの各種類の界面活性剤を使用できる。また、下記界面活性剤も好ましい。下記式中、主鎖の構成単位を示す括弧は各構成単位の含有量(モル%)を、側鎖の構成単位を示す括弧は各構成単位の繰り返し数をそれぞれ表す。

本発明の硬化性樹脂組成物は、酸素に起因する重合阻害を防止するために、ベヘン酸やベヘン酸アミドのような高級脂肪酸誘導体を添加して、塗布後の乾燥の過程で組成物の表面に偏在させてもよい。
本発明の硬化性樹脂組成物の水分含有量は、塗布面性状の観点から、5質量%未満が好ましく、1質量%未満がより好ましく、0.6質量%未満が更に好ましい。
本発明の硬化性樹脂組成物は、上記各成分を混合して調製することができる。混合方法は特に限定はなく、従来公知の方法で行うことができる。
フィルターを用いたろ過の他、吸着材を用いた不純物の除去処理を行ってもよい。フィルターろ過と吸着材を用いた不純物除去処理とを組み合わせてもよい。吸着材としては、公知の吸着材を用いることができる。例えば、シリカゲル、ゼオライトなどの無機系吸着材、活性炭などの有機系吸着材が挙げられる。
本発明の硬化性樹脂組成物は、再配線層用層間絶縁膜の形成に用いられることが好ましい。
また、その他、半導体デバイスの絶縁膜の形成、又は、ストレスバッファ膜の形成等にも用いることができる。
次に、硬化膜、積層体、半導体デバイス、及びそれらの製造方法について説明する。
具体的には、以下の(a)~(d)の工程を含むことも好ましい。
(a)硬化性樹脂組成物を基材に適用して膜(硬化性樹脂組成物層)を形成する膜形成工程
(b)膜形成工程の後、膜を露光する露光工程
(c)露光された上記膜に対して、現像処理を行う現像工程
(d)現像された上記膜を加熱して上記式(1-1)により表される化合物を分解する加熱工程
上記加熱工程において加熱することにより、露光で硬化した樹脂層を更に硬化させることができる。この加熱工程で、例えば上記式(1-1)により表される化合物が分解し、十分な硬化性が得られる。
<膜形成工程(層形成工程)>
本発明の好ましい実施形態に係る製造方法は、硬化性樹脂組成物を基材に適用して膜(層状)にする、膜形成工程(層形成工程)を含む。
また、基材としては、例えば板状の基材(基板)が用いられる。
また、あらかじめ仮支持体上に上記付与方法によって付与して形成した塗膜を、基材上に転写する方法を適用することもできる。
転写方法に関しては特開2006-023696号公報の段落0023、0036~0051や、特開2006-047592号公報の段落0096~0108に記載の作製方法を本発明においても好適に用いることができる。
本発明の製造方法は、上記膜(硬化性樹脂組成物層)を形成後、膜形成工程(層形成工程)の後に、溶剤を除去するために乾燥する工程を含んでいてもよい。好ましい乾燥温度は50~150℃で、70℃~130℃がより好ましく、90℃~110℃が更に好ましい。乾燥時間としては、30秒~20分が例示され、1分~10分が好ましく、3分~7分がより好ましい。
本発明の製造方法は、上記膜(硬化性樹脂組成物層)を露光する露光工程を含んでもよい。露光量は、硬化性樹脂組成物を硬化できる限り特に定めるものではないが、例えば、波長365nmでの露光エネルギー換算で100~10,000mJ/cm2照射することが好ましく、200~8,000mJ/cm2照射することがより好ましい。
本発明の製造方法は、露光された膜(硬化性樹脂組成物層)に対して、現像処理を行う現像処理工程を含んでもよい。現像を行うことにより、露光されていない部分(非露光部)が除去される。現像方法は、所望のパターンを形成できれば特に制限は無く、例えば、パドル、スプレー、浸漬、超音波等の現像方法が採用可能である。
本発明の製造方法は、上記膜を加熱して上記式(1-1)により表される化合物を分解する工程(加熱工程)を含むことが好ましい。
加熱工程は、膜形成工程(層形成工程)、乾燥工程、及び現像工程の後に含まれることが好ましい。加熱工程では、例えば上記式(1-1)により表される化合物が分解することにより塩基が発生し、複素環含有ポリマー前駆体の環化反応が進行する。また、本発明の組成物は複素環含有ポリマー前駆体以外のラジカル重合性化合物を含んでいてもよいが、未反応の複素環含有ポリマー前駆体以外のラジカル重合性化合物の硬化などもこの工程で進行させることができる。加熱工程における層の加熱温度(最高加熱温度)としては、50℃以上であることが好ましく、80℃以上であることがより好ましく、140℃以上であることが更に好ましく、150℃以上であることが一層好ましく、160℃以上であることがより一層好ましく、170℃以上であることが更に一層好ましい。上限としては、500℃以下であることが好ましく、450℃以下であることがより好ましく、350℃以下であることが更に好ましく、250℃以下であることが一層好ましく、220℃以下であることがより一層好ましい。
本発明の製造方法は、現像処理後の膜(硬化性樹脂組成物層)の表面に金属層を形成する金属層形成工程を含んでいることが好ましい。
<積層工程>
本発明の製造方法は、更に、積層工程を含むことが好ましい。
本発明の熱塩基発生剤は、下記式(1-2)により表される熱塩基発生剤である。

式(1-2)におけるL21は、両末端の結合位置における原子がいずれも炭素原子に限定されている以外は、式(1-1)におけるL1と同義であり、好ましい態様も同様である。
本発明の熱塩基発生剤の用途としては、特に限定されないが、例えば、上述の複素環含有ポリマー前駆体と併用することにより、硬化性樹脂組成物を構成することができる。
また、本発明の熱塩基発生剤は、例えば、室温において分解しにくく、加熱時の塩基発生効率に優れると考えられる。そのため、公知の熱塩基発生剤が用いられている様々な用途において、従来の熱塩基発生剤の代わりに、又は、従来の熱塩基発生剤と併用して、本発明の熱塩基発生剤を用いることが有用であると考えられる。
〔ピロメリット酸二無水物、4,4’-ジアミノジフェニルエーテル及びベンジルアルコールからのポリイミド前駆体(A-1:ラジカル重合性基を有さないポリイミド前駆体)の合成〕
14.06g(64.5ミリモル)のピロメリット酸二無水物(140℃で12時間乾燥)と、14.22g(131.58ミリモル)のベンジルアルコールを、50mLのN-メチルピロリドンに懸濁させ、モレキュラーシーブで乾燥させた。懸濁液を100℃で3時間加熱した。反応混合物を室温に冷却し、21.43g(270.9ミリモル)のピリジン及び90mLのN-メチルピロリドンを加えた。次いで、反応混合物を-10℃に冷却し、温度を-10±4℃に保ちながら16.12g(135.5ミリモル)のSOCl2を10分かけて加えた。SOCl2を加えている間、粘度が増加した。50mLのN-メチルピロリドンで希釈した後、反応混合物を室温で2時間撹拌した。次いで、100mLのN-メチルピロリドンに11.08g(58.7ミリモル)の4,4’-ジアミノジフェニルエーテルを溶解させた溶液を、-5~0℃で20分かけて反応混合物に滴下した。次いで、反応混合物を0℃で1時間反応させたのち、エタノールを70g加えて、室温で1晩撹拌した。次いで、5リットルの水の中でポリイミド前駆体を沈殿させ、水-ポリイミド前駆体混合物を5,000rpmの速度で15分間撹拌した。ポリイミド前駆体をろ過して除き、4リットルの水の中で再度30分間撹拌し再びろ過した。次いで、得られたポリイミド前駆体を減圧下で、45℃で3日間乾燥した。このポリイミド前駆体の重量平均分子量は、18,000であった。

〔ピロメリット酸二無水物、4,4’-ジアミノジフェニルエーテル及び2-ヒドロキシエチルメタクリレートからのポリイミド前駆体(A-2:ラジカル重合性基を有するポリイミド前駆体)の合成〕
14.06g(64.5ミリモル)のピロメリット酸二無水物(140℃で12時間乾燥した)と、16.8g(129ミリモル)の2-ヒドロキシエチルメタクリレートと、0.05gのハイドロキノンと、20.4g(258ミリモル)のピリジンと、100gのダイグライム(ジエチレングリコールジメチルエーテル)を混合し、60℃の温度で18時間撹拌して、ピロメリット酸と2-ヒドロキシエチルメタクリレートのジエステルを製造した。次いで、得られたジエステルをSOCl2により塩素化した後、合成例1と同様の方法で4,4’-ジアミノジフェニルエーテルでポリイミド前駆体に変換し、合成例1と同様の方法でポリイミド前駆体を得た。このポリイミド前駆体の重量平均分子量は、19,000であった。

〔4,4’-オキシジフタル酸無水物、4,4’-ジアミノジフェニルエーテル及び2-ヒドロキシエチルメタクリレートからのポリイミド前駆体(A-3:ラジカル重合性基を有するポリイミド前駆体)の合成〕
20.0g(64.5ミリモル)の4,4’-オキシジフタル酸無水物(140℃で12時間乾燥した)と、16.8g(129ミリモル)の2-ヒドロキシエチルメタクリレートと、0.05gのハイドロキノンと、20.4g(258ミリモル)のピリジンと、100gのダイグライムとを混合し、60℃の温度で18時間撹拌して、4,4’-オキシジフタル酸と2-ヒドロキシエチルメタクリレートのジエステルを製造した。次いで、得られたジエステルをSOCl2により塩素化した後、合成例1と同様の方法で4,4’-ジアミノジフェニルエーテルでポリイミド前駆体に変換し、合成例1と同様の方法でポリイミド前駆体を得た。このポリイミド前駆体の重量平均分子量は、18,000であった。

〔4,4’-オキシジフタル酸無水物、4,4’-ジアミノ-2,2’-ジメチルビフェニル(オルトトリジン)及び2-ヒドロキシエチルメタクリレートからのポリイミド前駆体(A-4:ラジカル重合性基を有するポリイミド前駆体)の合成〕
20.0g(64.5ミリモル)の4,4’-オキシジフタル酸無水物(140℃で12時間乾燥した)と、16.8g(129ミリモル)の2-ヒドロキシエチルメタクリレートと、0.05gのハイドロキノンと、20.4g(258ミリモル)のピリジンと、100gのダイグライムとを混合し、60℃の温度で18時間撹拌して、4,4’-オキシジフタル酸と2-ヒドロキシエチルメタクリレートのジエステルを製造した。次いで、得られたジエステルをSOCl2により塩素化した後、合成例1と同様の方法で4,4’-ジアミノ-2,2’-ジメチルビフェニルでポリイミド前駆体に変換し、合成例1と同様の方法でポリイミド前駆体を得た。このポリイミド前駆体の重量平均分子量は、19,000であった。

〔2,2'-ビス(3-アミノ-4-ヒドロキシフェニル)ヘキサフルオロプロパン及び4,4'-オキシジベンゾイルクロリドからのポリベンゾオキサゾール前駆体(A-5)の合成〕
N-メチル-2-ピロリドン100mLに、2,2'-ビス(3-アミノ-4-ヒドロキシフェニル)ヘキサフルオロプロパン13.92gを添加し、撹拌溶解した。続いて、温度を0~5℃に保ちながら、11.21gの4,4'-オキシジベンゾイルクロリドを10分間で滴下した後、60分間撹拌を続けた。次いで、6リットルの水の中でポリベンゾオキサゾール前駆体を沈殿させ、水-ポリベンゾオキサゾール前駆体混合物を5000rpmの速度で15分間撹拌した。ポリベンゾオキサゾール前駆体を濾過して除き、6リットルの水の中で再度30分間撹拌し再び濾過した。次いで、得られたポリベンゾオキサゾール前駆体を減圧下で、45℃で3日間乾燥した。このポリベンゾオキサゾール前駆体の重量平均分子量は、15,000であった。

〔化合物B-1~B-18の合成〕
Organic Chemistry International(2014), 576715/1-576715/10, 10ppに記載の方法を参考に化合物B-1~B-18を合成した。
〔4,4’-オキシジフタル酸無水物、4,4’-ジアミノジフェニルエーテル、及び2-ヒドロキシエチルメタクリレートからのポリイミド前駆体(A-6:ラジカル重合性基を有するポリイミド前駆体)の合成〕
4,4’-オキシジフタル酸無水物(ODPA)155.1gを2リットル容量のセパラブルフラスコに入れ、2-ヒドロキシエチルメタクリレート(HEMA)134.0g及びγ―ブチロラクトン400mlを加えた。室温下で撹拌しながら、ピリジン79.1gを加えることにより、反応混合物を得た。反応による発熱の終了後、室温まで放冷し、更に16時間静置した。
〔3,3’4,4’-ビフェニルテトラカルボン酸二無水物、4,4’-ジアミノジフェニルエーテル、及び2-ヒドロキシエチルメタクリレートからのポリイミド前駆体(A-7:ラジカル重合性基を有するポリイミド前駆体)の合成〕
合成例7において、4,4’-オキシジフタル酸無水物155.1gに代えて、3,3’4,4’-ビフェニルテトラカルボン酸二無水物147.1gを用いた以外は、合成例7に記載の方法と同様にして反応を行うことにより、ポリマーA-7を得た。このポリマーA-7の重量平均分子量(Mw)を測定したところ、22,000であった。
各実施例及び比較例において、それぞれ、下記表1又は表2に記載の成分を混合し、各硬化性樹脂組成物を得た。得られた硬化性樹脂組成物を、細孔の幅が0.45μmのポリテトラフルオロエチレン製フィルターを通して加圧ろ過した。
表1又は表2中、「質量部」の欄の数値は各成分の含有量(質量部)を示している。
また、表1又は表2中、「-」の記載は該当する成分を含有していないことを示している。


〔複素環含有ポリマー前駆体〕
・A-1~A-5 :上記で合成したA-1~A-5
〔式(1-1)で表される化合物〕
・B-1~B-18:上記の例示化合物B-1~B-18
・比較例のための化合物RB-1:下記式(RB-1)により表される化合物

・C-1:IRGACURE OXE 01(BASF社製)
・C-2:IRGACURE OXE 02(BASF社製)
・C-3:IRGACURE 369(BASF社製)
〔ラジカル重合性化合物〕
・D-1:A-DPH(新中村化学工業(株)製、ジペンタエリスリトールヘキサアクリレート)
・D-2:SR-209(サートマー社製、下記化合物))

〔重合禁止剤〕
・E-1:2,6-ジ-tert-ブチル-4-メチルフェノール(東京化成工業社製)
・E-2:パラベンゾキノン(東京化成工業(株)製)
・E-3:パラメトキシフェノール(東京化成工業(株)製)
・E-4:2-ニトロソ-1-ナフト-ル(東京化成工業(株)製)
〔マイグレーション抑制剤〕
・F-1~F-4:下記式(F-1)~式(F-4)で表される化合物

・G-1~G-3:下記式(G-1)~式(G-4)で表される化合物

I-1:γ-ブチロラクトン(三和油化社製)
I-2:ジメチルスルホキシド(富士フイルム和光純薬(株)製)
I-3:N-メチル-2-ピロリドン(Ashland社製)
I-4:乳酸エチル(東京化成工業(株)製)
〔添加剤〕
H-1:N-フェニルジエタノールアミン(東京化成工業(株)製)
各実施例及び比較例において、それぞれ、表3に記載の硬化性樹脂組成物を用いて、保存安定性、破断伸び率、及び、密着性の評価を行った。
各評価における評価方法の詳細を下記に記載する。
各実施例及び比較例において、それぞれ、E型粘度計を用いて各硬化性樹脂組成物の粘度(0日)を測定した。遮光の密閉容器中、25℃で14日間、硬化性樹脂組成物を静置した後、再度E型粘度計を用いて上記静置後の各硬化性樹脂組成物の粘度(14日)を測定した。各実施例及び各比較例において、それぞれ、上記粘度(0日)及び上記粘度(14日)の値を用いて、以下の式から、粘度変動率を算出した。粘度変動率が低ければ低い程、保存安定性が高いことを表す。評価は下記評価基準に従い行った。評価結果は表3の「保存安定性」の欄に記載した。
粘度の測定は25℃で行うこととし、その他はJIS Z 8803:2011に準拠することとした。
-評価基準-
A:粘度変動率が5%以下であった。
B:粘度変動率が5%を超えて20%以下であった。
C:粘度変動率が20%を超えた。
各実施例及び比較例において、それぞれ、各硬化性樹脂組成物を、シリコンウェハ上にスピンコート法により層状に適用(塗布)して、硬化性樹脂組成物層を形成した。得られた硬化性樹脂組成物層を適用したシリコンウェハをホットプレート上で、100℃で5分間乾燥し、シリコンウェハ上に20μmの厚さの均一な硬化性樹脂組成物層とした。シリコンウェハ上の硬化性樹脂組成物層を、ステッパー(Nikon NSR 2005 i9C)を用いて、500mJ/cm2の露光エネルギーで露光した。上記露光した硬化性樹脂組成物層(樹脂層)を、窒素雰囲気下で、10℃/分の昇温速度で昇温し、180℃に達した後、この温度を3時間維持した。硬化後の樹脂層を4.9%フッ化水素酸溶液に浸漬し、シリコンウェハから樹脂層を剥離し、樹脂膜1を得た。
-評価基準-
A:破断伸び率が60%を超えた。
B:破断伸び率が55%を超えて60%以下であった。
C:破断伸び率が40%を超えて55%以下であった。
D:破断伸び率が40%以下であった。
各実施例及び比較例において、それぞれ、各硬化性樹脂組成物を、銅基板上にスピンコート法により層状に適用して、100℃で5分間乾燥し、膜厚20μmの硬化性樹脂組成物層を形成した。得られた硬化性樹脂組成物層を適用した銅基板をホットプレート上で、100℃で5分間乾燥し、銅基板上に20μmの厚さの均一な硬化性樹脂組成物層を形成した。銅基板上の硬化性樹脂組成物層を、ステッパー(Nikon NSR 2005 i9C)を用いて、500mJ/cm2の露光エネルギーで100μm四方の正方形状の非マスク部を有するフォトマスクを使用して露光し、その後シクロペンタノンで60秒間現像して、100μm四方形の樹脂層を得た。さらに、窒素雰囲気下で、10℃/分の昇温速度で昇温し、180℃に達した後、この温度を3時間維持した。
-評価基準-
A:せん断力が40gfを超えた。
B:せん断力が25gfを超えて40gf以下であった。
C:せん断力が25gf以下であった。
ただし、1gfは9.80665×10-3N(ニュートン)とする。

比較例1に係る硬化性樹脂組成物は、式(1-1)で表される化合物を含有せず、式(RB-1)で表される化合物を熱塩基発生剤として含有する。この比較例1に係る硬化性樹脂組成物は、保存安定性に劣ることが分かる。
比較例2に係る硬化性樹脂組成物は、式(1-1)で表される化合物を含有しない。この比較例2係る硬化性樹脂組成物は、破断伸び率に劣ることが分かる。
実施例1に記載の硬化性樹脂組成物を、銅薄層が形成された樹脂基材の表面にスピンコート法により層状に適用して、100℃で5分間乾燥し、膜厚20μmの硬化性樹脂組成物層を形成した後、ステッパー(ニコン製、NSR1505 i6)を用いて露光した。露光はマスク(パターンが1:1ラインアンドスペースであり、線幅が10μmであるバイナリマスク)を介して、波長365nmで行った。露光の後、シクロペンタノンで30秒間現像し、PGMEAで20秒間リンスし、パターンを得た。
次いで、230℃で3時間加熱し、再配線層用層間絶縁膜を形成した。この再配線層用層間絶縁膜は、絶縁性に優れていた。
また、これらの再配線層用層間絶縁膜を使用して半導体デバイスを製造したところ、問題なく動作することを確認した。
Claims (18)
- 複素環含有ポリマー前駆体、及び、
下記式(1-1)で表される化合物を含む、
硬化性樹脂組成物。

- 前記L1が連結鎖長1~5の2価の連結基である、請求項1に記載の硬化性樹脂組成物。
- 前記R1及び前記R2がそれぞれ独立に、炭化水素基である、請求項1又は2に記載の硬化性樹脂組成物。
- 前記L1が、1,2-エチレン基、1,3-プロパンジイル基、1,2-シクロヘキサンジイル基、シスビニレン基、1,2-フェニレン基、1,2-フェニレンメチレン基、又は、1,2-エチレンオキシ-1,2-エチレン基である、請求項1~3のいずれか1項に記載の硬化性樹脂組成物。
- 前記R3が、水素原子、アルキル基、又は、アリール基である、請求項1~4のいずれか1項に記載の硬化性樹脂組成物。
- 前記式(1-1)で表される化合物の分子量が100以上2,000以下である、請求項1~5のいずれか1項に記載の硬化性樹脂組成物。
- 光ラジカル重合開始剤及びラジカル重合性化合物を更に含む、請求項1~6のいずれか1項に記載の硬化性樹脂組成物。
- 前記複素環含有ポリマー前駆体として、ポリイミド前駆体及びポリベンゾオキサゾール前駆体よりなる群から選ばれた少なくとも1種の前駆体を含む、請求項1~7のいずれか1項に記載の硬化性樹脂組成物。
- 前記複素環含有ポリマー前駆体として、ポリイミド前駆体を含む、請求項1~8のいずれか1項に記載の硬化性樹脂組成物。
- 前記ポリイミド前駆体が下記式(1)で表される繰返し単位を有する、請求項9に記載の硬化性樹脂組成物;

- 前記式(1)におけるR113及びR114の少なくとも一方がラジカル重合性基を含む、請求項10に記載の硬化性樹脂組成物。
- 再配線層用層間絶縁膜の形成に用いられる、請求項1~11のいずれか1項に記載の硬化性樹脂組成物。
- 請求項1~12のいずれか1項に記載の硬化性樹脂組成物を硬化してなる硬化膜。
- 請求項13に記載の硬化膜を2層以上含み、前記硬化膜同士のいずれかの間に金属層を含む積層体。
- 請求項1~12のいずれか1項に記載の硬化性樹脂組成物を基材に適用して膜を形成する膜形成工程を含む、硬化膜の製造方法。
- 前記膜を加熱して前記式(1-1)により表される化合物を分解する加熱工程を更に含む、請求項15に記載の硬化膜の製造方法。
- 請求項13に記載の硬化膜又は請求項14に記載の積層体を含む、半導体デバイス。
- 下記式(1-2)により表される熱塩基発生剤。

Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020217025744A KR102555592B1 (ko) | 2019-02-22 | 2020-02-17 | 경화성 수지 조성물, 경화막, 적층체, 경화막의 제조 방법, 반도체 디바이스, 및 열염기 발생제 |
| JP2021501965A JP7351896B2 (ja) | 2019-02-22 | 2020-02-17 | 硬化性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、半導体デバイス、及び、熱塩基発生剤 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019030183 | 2019-02-22 | ||
| JP2019-030183 | 2019-02-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020170997A1 true WO2020170997A1 (ja) | 2020-08-27 |
Family
ID=72144978
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/005967 WO2020170997A1 (ja) | 2019-02-22 | 2020-02-17 | 硬化性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、半導体デバイス、及び、熱塩基発生剤 |
Country Status (4)
| Country | Link |
|---|---|
| JP (1) | JP7351896B2 (ja) |
| KR (1) | KR102555592B1 (ja) |
| TW (1) | TW202104511A (ja) |
| WO (1) | WO2020170997A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022050278A1 (ja) * | 2020-09-01 | 2022-03-10 | 富士フイルム株式会社 | 硬化性樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス、並びに、光塩基発生剤 |
| WO2023120035A1 (ja) * | 2021-12-23 | 2023-06-29 | 富士フイルム株式会社 | 樹脂組成物、硬化物、積層体、硬化物の製造方法、積層体の製造方法、半導体デバイスの製造方法、及び、半導体デバイス |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009080452A (ja) * | 2007-09-03 | 2009-04-16 | Tokyo Univ Of Science | 感光性樹脂組成物 |
| JP2012093746A (ja) * | 2010-09-30 | 2012-05-17 | Dainippon Printing Co Ltd | 感光性樹脂組成物、パターン形成用材料及びパターン形成方法 |
| WO2018025738A1 (ja) * | 2016-08-01 | 2018-02-08 | 富士フイルム株式会社 | 感光性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、積層体の製造方法および半導体デバイス |
| WO2018151195A1 (ja) * | 2017-02-20 | 2018-08-23 | 富士フイルム株式会社 | 感光性樹脂組成物、複素環含有ポリマー前駆体、硬化膜、積層体、硬化膜の製造方法、および半導体デバイス |
| WO2018181182A1 (ja) * | 2017-03-29 | 2018-10-04 | 富士フイルム株式会社 | 感光性樹脂組成物、硬化膜、積層体、硬化膜の製造方法および半導体デバイス |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004198678A (ja) * | 2002-12-18 | 2004-07-15 | Toray Ind Inc | ポジ型感光性樹脂前駆体組成物 |
| JPWO2010001779A1 (ja) * | 2008-07-01 | 2011-12-22 | 日本電気株式会社 | 感光性絶縁樹脂組成物及びそれを用いたパターン形成方法 |
| JP5795748B2 (ja) * | 2012-04-16 | 2015-10-14 | 富士フイルム株式会社 | 感光性樹脂組成物、硬化膜の製造方法、硬化膜、有機el表示装置および液晶表示装置 |
| JP6136486B2 (ja) | 2013-04-08 | 2017-05-31 | 日立化成デュポンマイクロシステムズ株式会社 | 樹脂組成物及びそれを用いたパターン形成方法 |
| TWI671343B (zh) | 2014-06-27 | 2019-09-11 | 日商富士軟片股份有限公司 | 熱硬化性樹脂組成物、硬化膜、硬化膜的製造方法以及半導體裝置 |
| KR102500455B1 (ko) * | 2018-09-27 | 2023-02-16 | 후지필름 가부시키가이샤 | 감광성 수지 조성물, 경화막, 적층체, 경화막의 제조 방법, 반도체 디바이스 |
-
2020
- 2020-02-17 WO PCT/JP2020/005967 patent/WO2020170997A1/ja active Application Filing
- 2020-02-17 KR KR1020217025744A patent/KR102555592B1/ko active IP Right Grant
- 2020-02-17 JP JP2021501965A patent/JP7351896B2/ja active Active
- 2020-02-18 TW TW109105066A patent/TW202104511A/zh unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009080452A (ja) * | 2007-09-03 | 2009-04-16 | Tokyo Univ Of Science | 感光性樹脂組成物 |
| JP2012093746A (ja) * | 2010-09-30 | 2012-05-17 | Dainippon Printing Co Ltd | 感光性樹脂組成物、パターン形成用材料及びパターン形成方法 |
| WO2018025738A1 (ja) * | 2016-08-01 | 2018-02-08 | 富士フイルム株式会社 | 感光性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、積層体の製造方法および半導体デバイス |
| WO2018151195A1 (ja) * | 2017-02-20 | 2018-08-23 | 富士フイルム株式会社 | 感光性樹脂組成物、複素環含有ポリマー前駆体、硬化膜、積層体、硬化膜の製造方法、および半導体デバイス |
| WO2018181182A1 (ja) * | 2017-03-29 | 2018-10-04 | 富士フイルム株式会社 | 感光性樹脂組成物、硬化膜、積層体、硬化膜の製造方法および半導体デバイス |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022050278A1 (ja) * | 2020-09-01 | 2022-03-10 | 富士フイルム株式会社 | 硬化性樹脂組成物、硬化物、積層体、硬化物の製造方法、及び、半導体デバイス、並びに、光塩基発生剤 |
| WO2023120035A1 (ja) * | 2021-12-23 | 2023-06-29 | 富士フイルム株式会社 | 樹脂組成物、硬化物、積層体、硬化物の製造方法、積層体の製造方法、半導体デバイスの製造方法、及び、半導体デバイス |
Also Published As
| Publication number | Publication date |
|---|---|
| KR102555592B1 (ko) | 2023-07-14 |
| TW202104511A (zh) | 2021-02-01 |
| JP7351896B2 (ja) | 2023-09-27 |
| JPWO2020170997A1 (ja) | 2021-11-25 |
| KR20210116541A (ko) | 2021-09-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN112639616A (zh) | 感光性树脂组合物、固化膜、层叠体、固化膜的制造方法及半导体器件 | |
| CN110692018B (zh) | 感光性树脂组合物、聚合物前体、固化膜、层叠体、固化膜的制造方法及半导体器件 | |
| JP7281533B2 (ja) | 硬化性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、及び、半導体デバイス | |
| JP7277572B2 (ja) | 硬化性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、及び、半導体デバイス | |
| JP7333383B2 (ja) | 硬化性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、及び、半導体デバイス | |
| JP7237978B2 (ja) | 樹脂組成物、硬化膜、積層体、硬化膜の製造方法、および半導体デバイス | |
| WO2020054226A1 (ja) | 感光性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、および半導体デバイス | |
| JP2020154205A (ja) | パターン形成方法、硬化性樹脂組成物、膜、硬化膜、積層体、及び、半導体デバイス | |
| JP7477579B2 (ja) | 硬化性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、半導体デバイス、及び、ポリマー前駆体 | |
| JP7277573B2 (ja) | 硬化性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、及び、半導体デバイス | |
| WO2020255859A1 (ja) | 硬化性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、半導体デバイス、及び、ポリイミド、又は、ポリイミド前駆体 | |
| JP7023379B2 (ja) | 樹脂組成物、硬化膜、積層体、硬化膜の製造方法、および半導体デバイス | |
| JP2023003421A (ja) | 硬化膜の製造方法、樹脂組成物、硬化膜、積層体の製造方法および半導体デバイスの製造方法 | |
| JP7086882B2 (ja) | 硬化性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、及び、半導体デバイス | |
| JP7351896B2 (ja) | 硬化性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、半導体デバイス、及び、熱塩基発生剤 | |
| JP7078744B2 (ja) | 樹脂組成物、硬化膜、積層体、硬化膜の製造方法、および半導体デバイス | |
| WO2019189327A1 (ja) | 感光性樹脂組成物、硬化膜、積層体およびこれらの応用 | |
| JP7334248B2 (ja) | 硬化性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、及び、半導体デバイス | |
| JP7426375B2 (ja) | 硬化性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、半導体デバイス、及び、熱塩基発生剤 | |
| WO2020066244A1 (ja) | 感光性樹脂組成物、硬化膜、積層体、硬化膜の製造方法、半導体デバイス、および熱塩基発生剤 | |
| JPWO2019013241A1 (ja) | 熱硬化性樹脂組成物、およびその硬化膜、積層体、半導体デバイス、ならびにそれらの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20758941 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2021501965 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20217025744 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 20758941 Country of ref document: EP Kind code of ref document: A1 |