WO2019163943A1 - 医薬組成物 - Google Patents
医薬組成物 Download PDFInfo
- Publication number
- WO2019163943A1 WO2019163943A1 PCT/JP2019/006780 JP2019006780W WO2019163943A1 WO 2019163943 A1 WO2019163943 A1 WO 2019163943A1 JP 2019006780 W JP2019006780 W JP 2019006780W WO 2019163943 A1 WO2019163943 A1 WO 2019163943A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- polymer
- compound
- cancer
- drug
- Prior art date
Links
- 0 CC(*1)(C1(*)C(*)(**)C(**)(O1)I)C1(C(*)=C)I Chemical compound CC(*1)(C1(*)C(*)(**)C(**)(O1)I)C1(C(*)=C)I 0.000 description 3
- SUDHEDJJFGYYPL-UHFFFAOYSA-N CCOC(N(C)C)=O Chemical compound CCOC(N(C)C)=O SUDHEDJJFGYYPL-UHFFFAOYSA-N 0.000 description 1
- QSCZSBGBDORDGM-UHFFFAOYSA-N COC(c(cc1)cc([N+]([O-])=O)c1S(Cl)(=O)=O)=O Chemical compound COC(c(cc1)cc([N+]([O-])=O)c1S(Cl)(=O)=O)=O QSCZSBGBDORDGM-UHFFFAOYSA-N 0.000 description 1
- OGHLGQSUFGNKDW-DYNHUECUSA-N C[C@@]1(C(C(C2)N(C)S(c(ccc(C(O)=O)c3)c3[N+]([O-])=O)(=O)=O)OC)O[C@H]2[n](c2ccccc22)c3c2c(C(NC2)=O)c2c2c3[n]1c1ccccc21 Chemical compound C[C@@]1(C(C(C2)N(C)S(c(ccc(C(O)=O)c3)c3[N+]([O-])=O)(=O)=O)OC)O[C@H]2[n](c2ccccc22)c3c2c(C(NC2)=O)c2c2c3[n]1c1ccccc21 OGHLGQSUFGNKDW-DYNHUECUSA-N 0.000 description 1
- CCUCCXOZLJSUQE-KWBLKEKNSA-N C[C@@]1(C(C(C2)N(C)S(c(ccc(C(OC)=O)c3)c3[N+]([O-])=O)(=O)=O)OC)O[C@H]2[n](c2ccccc22)c3c2c(C(NC2)=O)c2c2c3[n]1c1ccccc21 Chemical compound C[C@@]1(C(C(C2)N(C)S(c(ccc(C(OC)=O)c3)c3[N+]([O-])=O)(=O)=O)OC)O[C@H]2[n](c2ccccc22)c3c2c(C(NC2)=O)c2c2c3[n]1c1ccccc21 CCUCCXOZLJSUQE-KWBLKEKNSA-N 0.000 description 1
- HKSZLNNOFSGOKW-SKHIGLGASA-N C[C@@]1(C(C(C2)NC)OC)O[C@H]2[n](c2ccccc22)c3c2c(C(NC2)=O)c2c2c3[n]1c1ccccc21 Chemical compound C[C@@]1(C(C(C2)NC)OC)O[C@H]2[n](c2ccccc22)c3c2c(C(NC2)=O)c2c2c3[n]1c1ccccc21 HKSZLNNOFSGOKW-SKHIGLGASA-N 0.000 description 1
Images























































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K33/00—Medicinal preparations containing inorganic active ingredients
- A61K33/24—Heavy metals; Compounds thereof
- A61K33/243—Platinum; Compounds thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/7056—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing five-membered rings with nitrogen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the present invention relates to a pharmaceutical composition.
- the present invention relates to a pharmaceutical composition for treating or preventing cancer resistant to an anticancer drug.
- gefitinib is an anticancer agent that inhibits the tyrosine kinase activity of epidermal growth factor receptor (EGFR) and is used as a therapeutic agent for non-small cell lung cancer.
- Gefitinib is effective in non-small cell lung cancer that expresses EGFR with the L858R mutation, but when the T790M mutation occurs, it becomes a gefitinib resistant cancer.
- Osimertinib is effective for gefitinib-resistant cancers that express EGFR with the T790M mutation. However, when the C797S mutation occurs, the cancer becomes resistant to osmeltinib.
- an effective drug for cancer expressing EGFR having three mutations of L858R, T790M, and C797S has not been developed.
- staurosporine is a natural product isolated from Streptomyces genus actinomycetes (Non-Patent Document 1) and is known to have an antitumor action (for example, Patent Document 1). ).
- Patent Document 1 Non-Patent Document 1
- K252a To date, staurosporine analogues such as K252a have been reported (for example, Patent Document 2).
- X and Y are each independently H, —OH, Cl, propoxy, or ethylthiomethyl; R 6 is H, C 1-3 alkyl, —NH 2 , benzyl,
- R 7 and R 8 are each independently H, —OH, or methoxy, or may be taken together to form O ⁇ ;
- R 9 and R 10 are each independently H, methyl, ⁇ -D-glucopyranosyl, 4-O-methyl- ⁇ -D-glucopyranosyl, cyanoethyl, or
- R 11 is methyl;
- R 12 is H;
- R 13 and R 14 are each independently H, methoxy, —OH, hydroxymethyl, methylcarboxylate, methylamino, methylaminomethyl, propylaminomethyl, dimethylaminomethyl, methoxycarbonyl,
- R 15 and R 16 are each independently H, —OH, or
- R 17 and R 18 are H, —OH, methylamino, dimethylamino, oxime, or a group represented by any one of the following formulae:
- the anticancer agent is at least one anticancer agent selected from the group consisting of gefitinib, afatinib, osmeltinib, dasatinib, erlotinib, gemcitabine, cisplatin, pemetrexed, and midostaurin.
- the pharmaceutical composition as described.
- the kinase is at least one selected from the group consisting of EGFR, ABL1, ALK1, HER2, c-Kit, FGFR1, FGFR2, FGFR3, c-Src, PDGFRa, RET, DDR2, TRKA, and Flt-3.
- the pharmaceutical composition according to [5], wherein the kinase is EGFR having mutations of d746-750, T790M, and C797S.
- the compound (S) is staurosporine, 7-hydroxystautosporin, KT5926, staurosporine aglycone, SF2370, KT5823, 4′-N-benzoylstaurosporine, Go6976, N, N-dimethyl.
- Staurosporine NA 0359, N-ethoxycarbonyl-7-oxostaurosporine, KT-6124, CGP42700, 4'-demethylamino-4 ', 5'-dihydroxystaurosporine, 7-oxostaurosporine, CEP751, NA0346, NA0359, 3′-demethoxy-3′-hydroxystaurosporine, KT 6006, 7-O-methyl-UCN 01, TAN 999, NA 0346, NA 0345, NA 0344, CGP 44171A, SCH 47112, N, N -Jimee Rustaurosporin, TAN 1030A, Restaurtinib, 4′-demethylamino-4′-hydroxystaurosporine, AFN941, edtecalin, becatecarine, K252a, and K252a hydrazide, which is at least one compound selected from the group [1 ] To [6] The pharmaceutical composition according to any one of [6].
- a pharmaceutical composition that can be used for treatment or prevention of cancer resistant to an anticancer drug is provided.
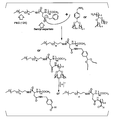
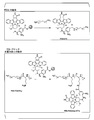
- FIG. 1 is a synthesis scheme of an aromatic aldehyde group-containing polymer and an aliphatic aldehyde group-containing polymer.
- FIG. 2 is a 1 H-NMR spectrum of an acetal group-containing polymer that is an intermediate of an aromatic aldehyde group-containing polymer.
- FIG. 3 is a 1 H-NMR spectrum of an aromatic aldehyde group-containing polymer.
- FIG. 4 is a 1 H-NMR spectrum of an acetal group-containing polymer that is an intermediate of an aliphatic aldehyde group-containing polymer.
- FIG. 5 is a 1 H-NMR spectrum of an aliphatic aldehyde group-containing polymer.
- FIG. 1 is a synthesis scheme of an aromatic aldehyde group-containing polymer and an aliphatic aldehyde group-containing polymer.
- FIG. 2 is a 1 H-NMR spectrum of an acetal group-containing polymer that is an intermediate
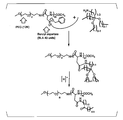
- FIG. 6 is a synthesis scheme of an aromatic ketone group-containing polymer and an aliphatic ketone group-containing polymer.
- FIG. 7 is a 1 H-NMR spectrum of an aliphatic ketone group-containing polymer.
- FIG. 8 is a 1 H-NMR spectrum of K252a and K252a hydrazide (K252a-H).
- FIG. 9 shows the results of HPLC analysis of K252a and K252a-H.
- FIG. 10 is a 1 H-NMR spectrum of a drug complex (K252a-H-bonded aliphatic polymer micelle) in which K252a-H is bound to an aliphatic ketone group-containing polymer.
- FIG. 10 is a 1 H-NMR spectrum of a drug complex (K252a-H-bonded aliphatic polymer micelle) in which K252a-H is bound to an aliphatic ketone group-containing polymer.
- FIG. 11 is a 1 H-NMR spectrum of a drug complex (K252a-H-bonded aromatic polymer micelle) in which K252a-H is bound to an aromatic aldehyde group-containing polymer.
- FIG. 12A shows the analysis results of the micelle size and the dispersion degree (PDI) of the K252a-H-bonded aromatic polymer micelle.
- FIG. 12B is an analysis result of the micelle size and the degree of dispersion (PDI) of K252a-H bonded aliphatic polymer micelles.
- FIG. 13 is an ultraviolet absorption spectrum of a sample prepared by dissolving K252a-H in distilled water or methanol.
- FIG. 14 is a HPLC analysis result of a sample after treatment of K252a-H linked aliphatic polymer micelles with an aqueous buffer at pH 1.
- FIG. 15 shows the results of cytotoxicity tests on lung cancer cells of K252a, K252a-H, K252a-H-linked aliphatic polymer micelles, and K252a-H-linked aromatic polymer micelles (hereinafter also referred to as “K252a class”). It is a graph.
- FIG. 16 is a graph showing the results of a cytotoxicity test for K252a class lung cancer cells.
- FIG. 17 is a graph showing the results of a cytotoxicity test for K252a class pancreatic cancer cells.
- FIG. 18 is a graph showing the results of a cytotoxicity test of K252a class on head and neck cancer cells.
- FIG. 19 is a graph showing the results of a cytotoxicity test for K252a class malignant mesothelioma cells.
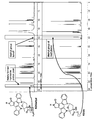
- FIG. 20 is a diagram showing first to third generation tyrosine kinase inhibitors (TKI) and resistance mutations thereof.
- FIG. 21 is a diagram showing the first to third generation TKIs and their resistance mutations.
- FIG. 22 shows the results of K252a-H kinase assay for EGFR mutation.
- FIG. 23A is a diagram showing the results of kinase profiling.
- FIG. 23B is a diagram showing the results of kinase profiling.
- FIG. 23A is a diagram showing the results of kinase profiling.
- FIG. 23C shows the results of kinase profiling.
- FIG. 24 is a diagram showing a list of mutant kinases whose kinase activity is suppressed to 30% or less by K252a-H.
- FIG. 25 is a diagram showing the results of kinase profiling.
- FIG. 26 shows the results of kinase profiling.
- FIG. 27 is a diagram showing the results of kinase profiling.
- FIG. 28 shows the results of kinase profiling.
- FIG. 29 is a diagram showing the results of kinase profiling.
- FIG. 30 shows the results of kinase profiling.
- FIG. 31 shows the results of kinase profiling.
- FIG. 32 shows the results of kinase profiling.
- FIG. 33A shows the results of kinase profiling.
- FIG. 33B shows the results of kinase profiling.
- FIG. 33C shows the results of kinase profiling.
- FIG. 33D shows the results of kinase profiling.
- FIG. 34A shows the results of kinase profiling.
- FIG. 34B shows the results of kinase profiling.
- FIG. 34B shows the results of kinase profiling.
- FIG. 34D shows the results of kinase profiling.
- FIG. 35 shows a list of kinases having an IC50 of staurosporine of 0.2 nM or less.
- the underlined kinase is a kinase that is important as a therapeutic target for cancer (particularly drug-resistant cancer).
- FIG. 36 is a graph showing the results of cytotoxicity tests of staurosporine and K252a class on non-small cell lung cancer.
- FIG. 37 is a graph showing the results of cytotoxicity tests of staurosporine and K252a family against pancreatic cancer.
- FIG. 38 is a graph showing the results of cytotoxicity tests of staurosporine and K252a family on head and neck cancer.
- FIG. 39 is a graph showing the results of cytotoxicity test for staurosporine and K252a malignant mesothelioma.
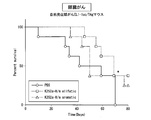
- FIG. 40 is a graph showing the results of an in vivo anti-tumor test for K252a species.
- FIG. 40 is a graph showing the results of an in vivo anti-tumor test for K252a species.
- FIG. 41 is a graph showing the results of an in vivo anti-tumor test for K252a species.
- FIG. 42 is a graph showing the results of an in vivo anti-tumor test for K252a species.
- FIG. 43 is a graph showing the results of cytotoxicity testing of staurosporine and K252a against spontaneous pancreatic cancer.
- FIG. 44 is a graph showing the results of an in vivo anti-tumor test for K252a species.
- FIG. 45 is a graph showing the results of an in vivo antitumor test for K252a species.
- FIG. 46 is a graph showing the results of a cytotoxicity test for K252a class lymphoma.
- FIG. 47 is a graph showing the results of a staurosporine and K252a class inhibitory activity test against MDR-1.
- FIG. 48 is a graph showing the results of a test evaluating the effect of human ⁇ 1-acid glycoprotein (hAGP) on the cytotoxicity of K252a species.
- FIG. 49 is a diagram showing an example of a reaction for binding a linker reagent (L1) to staurosporine.
- FIG. 50 is a diagram showing a 1 H-NMR spectrum of staurosporine bound to a linker reagent (L1).
- FIG. 51 is a diagram illustrating a scheme for preparing a drug complex of staurosporine and a polymer via a linker derived from a linker reagent (L1).
- the present invention provides the compound (S) represented by the following general formula (S), or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable polymer, wherein the compound (S) or the Provided is a pharmaceutical composition for treating or preventing cancer resistant to an anticancer drug, which comprises a drug complex bound with a pharmaceutically acceptable salt.
- composition of the present embodiment comprises the compound (S) represented by the following general formula (S), or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable polymer, the compound (S) or the It includes drug conjugates with pharmaceutically acceptable salts attached.
- X and Y are each independently H, —OH, Cl, propoxy, or ethylthiomethyl; R 6 is H, C 1-3 alkyl, —NH 2 , benzyl,
- R 7 and R 8 are each independently H, —OH, or methoxy, or may be taken together to form O ⁇ ;
- R 9 and R 10 are each independently H, methyl, ⁇ -D-glucopyranosyl, 4-O-methyl- ⁇ -D-glucopyranosyl, cyanoethyl, or
- R 11 is methyl;
- R 12 is H;
- R 13 and R 14 are each independently H, methoxy, —OH, hydroxymethyl, methylcarboxylate, methylamino, methylaminomethyl, propylaminomethyl, dimethylaminomethyl, methoxycarbonyl,
- R 15 and R 16 are each independently H, —OH, or
- R 17 and R 18 are each independently H, —OH, methylamino, dimethylamino, oxime, or a group represented by any one of the following formulae:
- R 19 is a group containing methoxy, —OH, —NH—NH 2 , or an active ester.
- Preferred examples of the compound (S) include compounds (S-1) and (S-2) represented by the following general formulas (S-1) and (S-2), respectively.
- X, Y, and R 6 are preferably H.
- R 13 and R 14 are each independently represented by H, methoxy, —OH, hydroxymethyl, methoxycarbonyl, or the following formula (h1). It is preferably a group.
- R 13 and R 14 in (S-1) are each independently H, methoxycarbonyl, or a group represented by the above formula (h1). More preferably, R 13 and R 14 in (S-2) are each independently H or methoxy.
- R 15 and R 16 are preferably each independently H or —OH.
- R 17 and R 18 are preferably H, —OH, methylamino, dimethylamino, oxime, or a group represented by the following formula (11). More preferred is amino or a group represented by the formula (l1).
- R 19 is methoxy, —OH, —NH—NH 2 , or a group containing an active ester.
- the group containing an active ester in the above formula (l1) includes an active esterifying agent (N-hydroxysuccinimide (NHS), 1-hydroxybenzotriazole (HOBt), 1-hydroxy-7-azabenzotriazole (HOAt). , Pentafluorophenol, p-nitrophenol, etc.).
- an active esterifying agent N-hydroxysuccinimide (NHS), 1-hydroxybenzotriazole (HOBt), 1-hydroxy-7-azabenzotriazole (HOAt).
- Pentafluorophenol p-nitrophenol, etc.
- Preferred examples of the compound (S-1) include a compound (S-1-1) represented by the following general formula (S-1-1).
- a compound (S-2-1) represented by the following general formula (S-2-1) is preferably exemplified.
- R 7 is H or —OH
- R 13 and R 14 are each independently H, methoxy, —OH, hydroxymethyl, methoxycarbonyl, or a group represented by the following formula. ]
- Examples of the compound (S) include the following compounds: staurosporine, 7-hydroxystautosporin, KT5926, staurosporine aglycone, SF2370, KT5823, 4′-N-benzoylstaurosporine, PKC412 , Go6976, N, N-dimethylstaurosporine, NA0359, N-ethoxycarbonyl-7-oxostaurosporine, KT-6124, CGP42700, 4'-demethylamino-4 ', 5'-dihydroxystaurosporine, 7- Oxostaurosporine, CEP751, NA0346, NA0359, 3′-demethoxy-3′-hydroxystaurosporine, KT6006, 7-O-methyl-UCN01, TAN999, NA03 6, NA0345, NA0344, CGP44171A, SCH47112, N, N-dimethylstaurosporine, TAN1030A, restaurtinib,
- Specific examples of the compound (S-1-1) include K252a and K252a hydrazide (hereinafter also referred to as “K252a-H”). Specific examples of the compound (S-2-1) include staurosporine.
- Compound (S) may be in the form of a pharmaceutically acceptable salt.
- the “pharmaceutically acceptable salt” means a salt that does not inhibit the pharmacological action (anticancer activity, kinase inhibitory activity, etc.) of the compound (S).
- the pharmaceutically acceptable salt is preferably one that does not cause side effects when administered to a living body.
- the pharmaceutically acceptable salt is not particularly limited, and for example, a salt with an alkali metal (sodium, potassium, etc.); a salt with an alkaline earth metal (magnesium, calcium, etc.); an organic base (pyridine, triethylamine, etc.) Salt with amine, salt with amine, salt with organic acid (acetic acid, formic acid, propionic acid, fumaric acid, maleic acid, succinic acid, tartaric acid, citric acid, malic acid, oxalic acid, benzoic acid, methanesulfonic acid, etc.) And salts with inorganic acids (hydrochloric acid, phosphoric acid, hydrobromic acid, sulfuric acid, nitric acid, etc.).
- the compound (S) may be a compound (K) or a pharmaceutically acceptable salt of the compound (K) described in [Second aspect] described later.
- Compound (S) may be used alone or in combination of two or more.
- the pharmaceutical composition of this embodiment may include a drug complex in which the compound (S) is bound to a pharmaceutically acceptable polymer.
- the “drug complex” refers to a molecule in which a pharmacological action (drug) is bound to another molecule (for example, a polymer).
- “Pharmaceutically acceptable polymer” means a polymer that does not inhibit the pharmacological action (anticancer activity, kinase inhibitory activity, etc.) of compound (S) when it forms a drug complex with compound (S). .
- a polymer that does not inhibit the pharmacological action of the compound (S) includes a compound in which the drug complex with the compound (S) exhibits a pharmacological action equivalent to or higher than that of the compound (S) in vitro, and in vivo And those having a pharmacological action equivalent to or higher than that of the compound (S).
- the pharmaceutically acceptable polymer is preferably a polymer that does not cause side effects when administered to a living body.
- the polymer that forms the drug complex with the compound (S) is not particularly limited as long as it does not inhibit the pharmacological action of the compound (S).
- the polymer may be a hydrophilic polymer, a hydrophobic polymer, or a copolymer including a hydrophilic polymer segment and a hydrophobic polymer segment.
- hydrophilic polymer segment examples include, for example, polyalkylene glycol, poly (2-oxazoline), polysaccharide, polyvinyl alcohol, polyvinyl pyrrolidone, polyacrylamide, polymethacrylamide, polyacrylic ester, polymethacrylic ester, poly (2-methacryloyloxyethyl phosphorylcholine), poly (N- (2-hydroxypropyl) methacrylamide) (PHPMA) and derivatives thereof.
- polyalkylene glycol and poly (2-oxazoline) are preferable, and polyalkylene glycol is more preferable.
- polyalkylene glycol examples include polyethylene glycol, polypropylene glycol, polyethylene glycol / polypropylene glycol copolymer, and the like, and polyethylene glycol is particularly preferable.
- the hydrophobic polymer segment include a polymer having a repeating unit derived from an amino acid and / or a derivative thereof. More specifically, a polyamino acid or a derivative thereof can be mentioned. Examples of polyamino acids and derivatives thereof include polyaspartic acid, polyglutamic acid, polylysine, poly (benzylaspartic acid), poly (benzylglutamic acid) and the like. Moreover, the hydrophobic polymer segment may contain a repeating unit derived from an amino acid having an alkyl group side chain or an aralkyl group side chain, for example. Examples of amino acids having an alkyl side chain include alanine, valine, leucine, and isoleucine.
- phenylalanine is mentioned as an amino acid which has an aralkyl group side chain.
- aralkyl group side chain When it has a repeating unit derived from two or more alkyl group side chain amino acids and / or aralkyl group side chain amino acids, these side chains may be the same or different.
- the ratio of the repeating unit derived from the alkyl group side chain amino acid or the aralkyl group side chain amino acid with respect to all the repeating units of the hydrophobic polymer segment is not particularly limited, and is, for example, 20% or more, 35% or more, 40% or more, 50 % Or more, 80% or more, 95% or more, 99% or more, or 100%.
- Preferred examples of the polymer that forms the drug complex with the compound (S) include the following polymer (P).
- the polymer (P) is a polymer having a repeating unit (I) represented by the following general formula (I) and a repeating unit (II) represented by the following general formula (II).
- L represents a divalent aromatic hydrocarbon group or a divalent aliphatic hydrocarbon group.
- R 1 represents a hydrogen atom, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- X represents OR x , SR x or NR x1 R x2 .
- R x represents a hydrogen atom, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- R x1 and R x2 each independently represent a hydrogen atom, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- L represents a divalent aromatic hydrocarbon group or a divalent aliphatic hydrocarbon group.
- the divalent aromatic hydrocarbon group for L include a phenylene group and a benzylene group.
- the divalent aromatic hydrocarbon group for L may have a substituent. Examples of the substituent include a methyl group, an ethyl group, a propyl group, an isopropyl group, a tert-butyl group, a nitro group, and a halide.
- Examples of the divalent aliphatic hydrocarbon group for L include an ethylene group, a propylene group, a butylene group, and a pentylene group.
- the divalent aliphatic hydrocarbon group for L may have a substituent.
- the substituent include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, a tert-butyl group, and a halide.
- L is preferably a methylene group, an ethylene group, a propylene group, or a benzylene group, and more preferably a methylene group, an ethylene group, or a benzylene group.
- L is preferably a divalent aliphatic hydrocarbon group, more preferably a methylene group, an ethylene group, or a propylene group, and even more preferably a methylene group or an ethylene group.
- R 1 represents a hydrogen atom, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- the aliphatic hydrocarbon group for R 1 include an ethyl group, a propyl group, a butyl group, and a pentyl group.
- the aliphatic hydrocarbon group for R 1 may have a substituent.
- substituents examples include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, an isobutyl group, a tert-butyl group, a pentyl group, an isopentyl group, a tert-pentyl group, a cyclohexyl group, and a trihalomethyl group.
- the aromatic hydrocarbon group for R 1 includes phenyl, benzyl, pyridyl, naphthyl, hydroxyphenyl, methoxyphenyl, ethoxyphenyl, xylyl, methylphenyl, nitrophenyl, chlorophenyl, fluoro Examples include an orophenyl group, an iodophenyl group, and a bromophenyl group.
- R 1 preferably a hydrogen atom or an aliphatic hydrocarbon group, more preferably a hydrogen atom or a methyl group, further preferably a methyl group.
- X represents OR x , SR x or NR x1 R x2 .
- R x represents a hydrogen atom, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- the aliphatic hydrocarbon group for R x include methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, tert-butyl group, pentyl group, isopentyl group, tert-pentyl group, cyclohexyl group, and trifluoro group.
- a methyl group etc. are mentioned.
- the aromatic hydrocarbon group for R x includes phenyl, benzyl, pyridyl, naphthyl, hydroxyphenyl, methoxyphenyl, ethoxyphenyl, xylyl, methylphenyl, nitrophenyl, chlorophenyl, fluoro, Examples include an orophenyl group, an iodophenyl group, and a bromophenyl group.
- R x1 and R x2 each independently represent a hydrogen atom, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- Examples of the aliphatic hydrocarbon group represented by R x1 and R x2 include methyl group, ethyl group, isopropyl group, butyl group, isobutyl group, tert-butyl group, pentyl group, isopentyl group, tert-pentyl group, cyclohexyl group, and trihalomethyl. Group.
- Examples of the aromatic hydrocarbon group represented by R x1 and R x2 include phenyl group, benzyl group, pyridyl group, naphthyl group, hydroxyphenyl group, methoxyphenyl group, ethoxyphenyl group, xylyl group, methylphenyl group, humanlophenyl group, and chlorophenyl. Group, fluorophenyl group, iodophenyl group, bromophenyl group and the like. Among them, X is preferably OR x and more preferably OH (hydroxy group).
- the polymer (P) may have a repeating unit other than the repeating units (I) and (II) (hereinafter sometimes referred to as “repeating unit (III)”).
- the repeating unit (III) is preferably a hydrophilic repeating unit.
- a repeating unit derived from polyethylene glycol a repeating unit derived from poly (ethylethylene phosphate), a repeating unit derived from polyvinyl alcohol, polyvinyl Examples thereof include a repeating unit derived from pyrrolidone, a repeating unit derived from poly (oxazoline), and a repeating unit derived from poly (N- (2-hydroxypropyl) methacrylamide) (PHPMA).
- the repeating unit (III) is preferably a repeating unit derived from polyethylene glycol.
- the content of the repeating units (I) to (III) is not particularly limited.
- the content of the repeating unit (I) is preferably 5 to 100 mol%, more preferably 10 to 80 mol%, more preferably 20 to 50 mol based on the total (100 mol%) of all repeating units constituting the polymer (P). More preferred is mol%.
- the content of the repeating unit (II) is preferably 0 to 80 mol%, more preferably 10 to 60 mol%, more preferably 20 to 40 mol based on the total (100 mol%) of all the repeating units constituting the polymer (P). More preferred is mol%.
- the content of the repeating unit (III) is preferably from 0 to 95 mol%, more preferably from 20 to 90 mol%, more preferably from 50 to 80, based on the total (100 mol%) of all repeating units constituting the polymer (P). More preferred is mol%.
- the molecular weight of the polymer (P) is preferably from 2,000 to 1,000,000 D, more preferably from 5,000 to 100,000 D, and even more preferably from 10,000 to 40,000 D.
- production method (1) includes the polymer (P1) having the repeating unit (II ′) represented by the following general formula (II ′) and the following general formula (II ′).
- R 2 represents a hydrogen atom, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- L represents a divalent aromatic hydrocarbon group or a divalent aliphatic hydrocarbon group.
- R 1 represents a hydrogen atom, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- Ra 11 and Ra 12 each independently represent a methyl group or an ethyl group, or Ra 11 and Ra 12 are bonded to each other to represent an ethylene group or a propylene group.
- R x represents a hydrogen atom, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- m, L, R 1 and R x are m, L in the general formulas (I) and (II). , R 1 and R x .
- Ra 11 and Ra 12 each independently represent a methyl group or an ethyl group, or Ra 11 and Ra 12 are bonded to each other to represent an ethylene group or a propylene group.
- the compound (1a) becomes a cyclic acetal or a cyclic ketal.
- n1 and n2 are each independently 0 or 1, and 1 is preferable.
- Step (1) of production method (1) is an aminolysis reaction between polymer (P1) and compound (1a).
- the acetal structure or ketal structure of the compound (1a) is introduced into the side chain of the polymer (P1).
- the reaction temperature in the step (1) is not particularly limited as long as the acetal structure or ketal structure of the compound (1a) is introduced into the side chain of the polymer (P1), but is usually 4 ° C. to 100 ° C. ⁇ 40 ° C is preferred.
- the reaction time in the step (1) is not particularly limited as long as the acetal structure or ketal structure of the compound (1a) is introduced into the side chain of the polymer (P1), and the reaction time, the type of the compound (1a) although it can be selected depending on the amount, it is usually 4 hours to 5 days.
- step (2) of production method (1) polymer (P2) is hydrolyzed under neutral or weakly acidic conditions, and the acetal structure of repeating unit (I ′) of polymer (P2) is converted into an aldehyde or ketal. Convert structure to ketone.
- the hydrolysis is not particularly limited as long as the acetal structure of the repeating unit (I ′) of the polymer (P2) can be converted into an aldehyde or a ketal structure into a ketone.
- a method of treating with 0.1N hydrochloric acid for about 30 minutes (i) a method of treating in the presence of acetone and indium (III) trifluoromethanesulfonate (catalyst), and (iii) a catalytic amount in water at 30 ° C.
- a method using 1 to 5 mol% of Er (OTf) 3 in wet nitromethane at room temperature (v) a substantially neutral solution using tetrakis (3,5-trifluoromethylphenyl) borate
- Known methods such as a method using a catalytic amount of cerium (III) triflate in wet nitromethane at room temperature under pH conditions.
- production method (2) includes the polymer (P1) having the repeating unit (II ′) represented by the following general formula (II ′) and the following general formula (II ′).
- the polymer (P3) is hydrolyzed under neutral or weakly acidic conditions, and the repeating unit (I) represented by the following general formula (I) and the repeating unit represented by the following general formula (II) ( (2b) obtaining a polymer (P) having II); including.
- R 2 represents a hydrogen atom, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- L represents a divalent aromatic hydrocarbon group or a divalent aliphatic hydrocarbon group.
- R 1 represents a hydrogen atom, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- Ra 11 and Ra 12 each independently represent a methyl group or an ethyl group, or Ra 11 and Ra 12 are bonded to each other to represent an ethylene group or a propylene group.
- X represents OR x , SR x or NR x1 R x2 .
- R x represents a hydrogen atom, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- R x1 and R x2 each independently represent a hydrogen atom, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- m, L, X, R x , R x1 and R x2 are the same as those in the general formulas (I) and (II). The same as m, L, X, R x , R x1 and R x2 .
- R 1 , Ra 11 , and Ra 12 are the same as described above.
- Step (1) of production method (2) is the same as step (1) of production method (1).
- the side chain of the repeating unit (II ′) is obtained by subjecting the polymer (P2) to a predetermined treatment so that the repeating unit (I ′) is protected with an acetal structure.
- a desired functional group can be introduced into Hydrolysis under alkaline conditions is, for example, a method of treating in a mixture of 0.5N NaOH solution and DMSO (volume ratio: 50/50) at room temperature for 30 minutes, or treating with triethylamine in DMSO at room temperature for 1 hour. And a method of treating with diisopropylethylamine in DMSO for 1 hour at room temperature.
- Aminolysis can introduce an amino functional group by cleaving the ester with, for example, ethylenediamine or diaminopropane. By introducing an amino group, it can be combined with a fluorescent dye. Moreover, it can also attach
- the resulting carboxylic acid is subjected to transesterification or amide coupling using a known coupling agent.
- a known coupling agent for hydrolysis and amide coupling under alkaline conditions, for example, after the ester residue is treated by hydrolysis under alkaline conditions, the resulting carboxylic acid is subjected to transesterification or amide coupling using a known coupling agent. be able to.
- the hydrophilic / hydrophobic balance of the polymer can be made desirable, contributing to self-assembly in polar or non-polar solvents.
- step (2b) of the production method (2) the polymer (P3) is hydrolyzed under weakly acidic conditions to convert the acetal structure of the repeating unit (I ′) of the polymer (P3) into an aldehyde. Hydrolysis conditions are the same as in step (2) of production method (1).
- the bond between the polymer (P) and the compound (S) is a nitrogen atom-containing group capable of forming a Schiff base with an aldehyde group or a ketone group in the compound (S) (hereinafter sometimes referred to as “Schiff base-forming group”).
- the reaction can be performed by reacting the Schiff base-forming group with an aldehyde group or a ketone group contained in the repeating unit (I) of the polymer (P).
- Examples of such a Schiff base include an amino group, an imino group, and a hydrazide group.
- the Schiff base forming group may be introduced into the compound (S). The introduction of the Schiff base forming group can be performed by a known method.
- K252a since there is no Schiff base-forming group in K252a, it can be bonded to the polymer (P) by introducing a hydrazide group to form K252a-H as shown in the examples described later.
- the drug complex of the polymer (P) and K252a include a polymer having the repeating unit (ka-1) and the repeating unit (II) described in [Second embodiment] described later.
- the drug complex of the polymer (P) and the compound (S) has a repeating unit (Ia) represented by the following general formula (Ia) and a repeating unit (II) represented by the following general formula (II).
- a polymer is preferred.
- L represents a divalent aromatic hydrocarbon group or a divalent aliphatic hydrocarbon group.
- R 1 represents a hydrogen atom, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- BM represents a group derived from the compound (S).
- X represents OR x , SR x or NR x1 R x2 .
- R x represents a hydrogen atom, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- R x1 and R x2 each independently represent a hydrogen atom, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- m, L, R 1 , X, R x , R x1 and R x2 are m, L, R 1 in the general formulas (I) and (II), The same as X, R x , R x1 and R x2 .
- BM represents a group derived from the compound (S).
- Preferred examples of the group derived from compound (S) include a group derived from K252a-H.
- the amount of the aldehyde group or ketone group to be introduced (the amount of the repeating unit (I)) can be controlled, and the amount of the drug bonded to the aldehyde group or the ketone group can also be controlled. Therefore, the dose of compound (S) can be appropriately controlled.
- an aldehyde group or a ketone group (a group represented by —LC ( ⁇ O) —R 1 in the above general formula (I)) to be introduced into the repeating unit (I)
- An aromatic aldehyde group (L in the general formula (I) is a divalent aromatic hydrocarbon group, R 1 is a hydrogen atom), an aliphatic aldehyde group (L in the general formula (I) is a divalent aliphatic carbonization
- an aromatic ketone group (L in the general formula (I) is a divalent aromatic hydrocarbon group, R 1 is an aliphatic hydrocarbon group or an aromatic hydrocarbon group; 1 is preferably an aliphatic hydrocarbon group, more preferably a methyl group), an aliphatic ketone group (L in the general formula (I) is a divalent aliphatic hydrocarbon group, R 1 is an aliphatic hydrocarbon group or aromatic R 1 is preferably selected from an
- the repeating unit (I) of the polymer (P) When the aromatic aldehyde group is introduced, the compound (S) is more stably held in the repeating unit (I) of the polymer (P). Therefore, the sustained release property of the drug can be controlled by selecting the type of aldehyde group or ketone group to be introduced into the repeating unit (I) of the polymer (P) according to the disease state and the type of drug.
- the compound (S) is stably maintained and the toxicity is reduced while the compound (S) is held by the polymer (P). Can reduce side effects and enhance the therapeutic effect.
- an aliphatic ketone group is introduced into the repeating unit (I) of the polymer (P) from the viewpoint of the antitumor effect when administered in vivo.
- L is preferably a divalent aliphatic hydrocarbon group (preferably a methylene group or ethylene group)
- R 1 is an aliphatic hydrocarbon group (preferably a methyl group).
- the compound (S) and the polymer (P) may be bonded using a known linker reagent.
- the linker reagent contains a Schiff base forming group
- the compound (S) can be bonded to the polymer (P) using the Schiff base forming group.
- a Schiff base-forming group may be introduced into the compound in which the linker is bound to the compound (S).
- the linker reagent that can be used is not particularly limited.
- a glutathione (GSH) / glutathione-S-transferase (GST) -sensitive linker reagent represented by the following formula (L1) (linker reagent (L1); 2012/039499, Huang CH et al., ChemMedChem. 2017 Jan 5; 12 (1): 19-22 .; Zhang J et al., J Am Chem Soc. 2011 Sep 7; 133 (35): 14109-19 .).
- the compound (SL1) represented by the following general formula (SL1) is a compound obtained by reacting the linker reagent (L1) with the compound (S-2-1).
- the following compound (SL1-H) containing a Schiff base-forming group can be obtained by refluxing the following compound (SL1) without solvent in the presence of anhydrous hydrazine or hydrazine hydrate.
- the compound (SL1-H) is obtained by, for example, converting the compound (SL1) into methanol, ethanol, propanol, isopropanol, butanol, tert-butanol, N, N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), tetrahydrofuran (THF).
- reaction temperature and reaction time are not particularly limited, and examples include room temperature to 50 ° C. for 30 minutes to 15 hours.
- R 7, R 13, and R 14 are the same as R 7, R 13, and R 14 in the general formula (S-2-1). ]
- the drug complex in which the compound (SL1-H) is bound to the polymer (P) is represented by the repeating unit (Ia-S) represented by the following general formula (Ia-S) and the following general formula (II). Having repeating unit (II).
- the drug complex having the repeating unit (Ia-S) and the repeating unit (II) includes a pH-sensitive bond (I) and a GSH / GST-sensitive bond (II). Therefore, when the above drug complex is administered in vivo, first, the bond (I) is cleaved around the tumor tissue in a low pH environment, and the compound (SL1-H) is released. Further, the compound (SL1-H) is taken up by tumor cells, and the bond (II) is cleaved by the action of GSH / GST to release the compound (S-2-1). Therefore, the release of the compound (S) from the polymer (P) in the living body can be controlled in two stages.
- the compound (S) may be a drug complex with a polymer other than the polymer (P).
- the bond between the compound (S) and the polymer can be performed using a known method depending on the functional group contained in the compound (S) and the polymer.
- the compound (S) may be directly bonded to the polymer, or a functional group capable of bonding to the polymer may be introduced into the compound (S) and bonded to the polymer via the functional group.
- a linker reagent is not specifically limited, For example, the said linker reagent (L1) is mentioned.
- an active ester may be introduced by an active esterifying agent and bonded to a polymer having a hydroxy group or an amino group. Conversely, an active ester may be introduced into the polymer.
- the linker reagent (L1) is bound to the compound (S) to obtain the compound (SL1), and then the compound (SL1) is demethylated to produce an active esterifying agent (N-hydroxysuccinimide (NHS)).
- an active esterifying agent N-hydroxysuccinimide (NHS)
- 1-hydroxybenzotriazole (HOBt), 1-hydroxy-7-azabenzotriazole (HOAt), pentafluorophenol, p-nitrophenol, etc.) to give an active ester represented by the following formula (SL2)
- R 7 , R 13 and R 14 are as defined above.
- E represents a group obtained by removing a hydroxy group from an active esterifying agent.
- R 7 , R 13 , R 14 and E are as defined above.
- Px represents an arbitrary polymer, and Ex represents a hydroxy group or an amino group.
- Ex ′ represents an ester bond or an amide bond.
- the drug complex of the compound (S) and polymer contained in the pharmaceutical composition of this embodiment may form a micelle.
- the polymer included in the drug complex preferably has a hydrophilic polymer segment and a hydrophobic polymer segment.
- the micelle of the drug complex of the compound (S) and the polymer can be prepared by a known method. For example, the drug conjugate is dissolved or suspended in a lipophilic or hydrophilic solvent, and the solution or suspension is dropped into the hydrophilic or lipophilic solvent and stirred to prepare a micelle of the drug conjugate. can do.
- the pharmaceutical composition of this embodiment may contain other components in addition to the compound (S).
- other components include pharmaceutically acceptable carriers.
- “Pharmaceutically acceptable carrier” means a carrier that does not inhibit the physiological activity of the active ingredient and does not exhibit substantial toxicity to the administration subject. By “not exhibiting substantial toxicity” is meant that the ingredient is not toxic to the administered subject at the doses normally used.
- the pharmaceutically acceptable carrier those commonly used in the pharmaceutical field can be used without particular limitation. For example, water, physiological saline, phosphate buffer, DMSO, dimethylacetamide, ethanol, glycerol, mineral An oil etc. can be mentioned.
- compositions of the present embodiment may contain an anticancer agent other than the compound (S) or an active ingredient other than the anticancer agent.
- active ingredients other than anticancer agents include, but are not limited to, anti-inflammatory agents, analgesics, antipyretic agents, anti-inflammatory agents, blood circulation promoters, stimulus-relaxing agents, antibiotics, and crude drugs.
- the compound (S) may be encapsulated or supported in known drug delivery particles such as liposomes and micelles. Encapsulation and loading in the drug delivery particles can be performed by a known method.
- the pharmaceutical composition of the present embodiment may also contain such drug delivery particles as an optional component.
- the pharmaceutical composition of this embodiment can be used for treating or preventing cancer resistant to an anticancer drug.
- Anticancer drug resistant cancer means a cancer that is resistant to one or more anticancer drugs.
- Anticancer agent means a compound used for the treatment or prevention of cancer and having an action of suppressing the growth of at least one kind of cancer cell.
- the anticancer agent having resistance to cancer is preferably an anticancer agent developed for the treatment of the cancer. More preferably, the anticancer agent is an anticancer agent approved for the treatment of the cancer. For example, it is an anticancer agent used for standard treatment of the cancer. Cancers that have acquired resistance to anticancer drugs used in such standard treatments are intractable due to ineffective drug therapy.
- the compound (S) exhibits high antitumor activity against cancer resistant to an anticancer drug. Therefore, the pharmaceutical composition of the present embodiment can be effectively used for treating or preventing cancer resistant to an anticancer drug. For example, if a cancer patient is treated with an existing anticancer drug and the effect of the anticancer drug cannot be obtained, or if the effect of the anticancer drug cannot be obtained
- the pharmaceutical composition in the form can be suitably used.
- the pharmaceutical composition of this embodiment can be suitably used as an anticancer agent used for cancer treatment after the second line.
- the pharmaceutical composition of this embodiment may be used for first-line cancer treatment.
- Whether or not a cancer is resistant to anticancer drugs is, for example, compared to the growth of cancer cells in the absence of anticancer drugs, the growth of cancer cells in the presence of anticancer drugs is suppressed. It can be determined by whether or not it is done. Compared with the growth rate of cancer cells in the absence of an anticancer agent, the growth inhibition rate of cancer cells in the presence of an anticancer agent is, for example, 50% or less, preferably 30% or less, more preferably 20%. Hereinafter, when the ratio is more preferably 10% or less, the cancer cell can be determined to be resistant to the anticancer agent.
- the growth rate is 2 times or more, preferably 5 times or more, more preferably 10 times or more, compared to the growth rate of cancer cells that are sensitive to anticancer agents. It can be determined that the drug is resistant to the anticancer drug. Alternatively, whether or not a cancer is resistant to an anticancer agent may be evaluated by, for example, the IC50 of the anticancer agent.
- the IC50 of the anticancer agent against cancer cells is, for example, 10 nM or more, preferably 20 nM or more, more preferably 30 nM or more, and even more preferably 50 nM or more, the cancer cells are against the anticancer agent. Can be determined to be resistant.
- the cancer cell is the anticancer agent.
- cancer resistant to an anticancer drug is a cancer refractory to an anticancer drug. That is, when an anticancer drug is administered to a cancer patient and the cancer patient's cancer does not shrink, the cancer grows, or the cancer growth rate is low, the cancer is It can be determined that the anticancer drug is resistant (refractory).
- the cancer to which the pharmaceutical composition of the present embodiment is applied is not particularly limited as long as it is an anticancer drug resistant cancer.
- cancer include lung cancer, pancreatic cancer, head and neck cancer, mesothelioma, neuroblastoma, liver cancer, malignant melanoma, uterine cancer, bladder cancer, biliary tract cancer, esophageal cancer, Examples include osteosarcoma, testicular tumor, thyroid cancer, acute myeloid leukemia, brain tumor, prostate cancer, squamous cell carcinoma of the head and neck, colon cancer, kidney cancer, ovarian cancer, and breast cancer.
- Preferred examples of cancer to which the pharmaceutical composition of this embodiment is applied include lung cancer, pancreatic cancer, head and neck cancer, and mesothelioma.
- anticancer drug-resistant cancer to which the pharmaceutical composition of the present embodiment can be applied are shown below.
- Cancer showing resistance to at least one anticancer drug selected from the group consisting of gefitinib, afatinib, osmeltinib, dasatinib, erlotinib, gemcitabine, cisplatin, pemetrexed, and midostaurin.
- Lung cancer particularly non-small cell lung cancer
- lung cancer that is resistant to osimertinib.
- lung cancer that is resistant to gefitinib, afitinib, and osimertinib.
- Pancreatic cancer having resistance to at least one anticancer agent selected from the group consisting of gemcitabine and erlotinib. More preferably, pancreatic cancer showing resistance to gemcitabine and erlotinib.
- Head and neck cancer that exhibits resistance to at least one anticancer drug selected from the group consisting of cisplatin, erlotinib, gefitinib, and midostaurin.
- a head and neck cancer resistant to cisplatin and erlotinib or a head and neck cancer resistant to erlotinib, gefitinib, and midostaurin.
- Examples of the molecular target drug include gefitinib, afatinib, osmeltinib, dasatinib, erlotinib, and midostaurin.
- Gatekeeper mutation refers to a mutation at a gatekeeper site.
- Gatekeeper site refers to the site of the amino acid residue located at the back of the ATP binding pocket of the kinase.
- deletion of amino acid residues for example, “d746-750” means deletion of amino acid residues at positions 746 to 750.
- deletion of amino acid residues is expressed as described above.
- the notation in which the one-letter code of amino acid is written on the left and right of the position number of the amino acid indicated by the number indicates the mutation of the amino acid residue of the position number indicated by the number, and the left side of the position number is the original amino acid residue.
- the right side of the position number indicates the amino acid residue after mutation.
- “T790M” indicates that the threonine (T) residue at position 790 is mutated to a methionine (M) residue.
- amino acid residue mutations are represented in the same manner as described above.
- a cancer that expresses at least one c-Kit selected from the group consisting of T670I and V559D (14) A cancer expressing FGFR1 having a V561M mutation. (15) A cancer expressing FGFR2 having a mutation of V564F. (16) A cancer expressing FGFR3 having a mutation of V565M. (17) A cancer expressing c-Src having a T341M mutation. (18) A cancer expressing PDGFRa having a mutation of T674I. (19) A cancer expressing RET having a mutation of V804L. (20) A cancer that expresses DDR2 having a mutation of T654M.
- a cancer that expresses a kinase having at least one mutation described in FIG. A cancer that expresses at least one of the kinases described in Table 3 below.
- AuroraA, AuroraB, CAMK2a, CAMK2d, CyclinC, CyclinA, CyclinA1, CHK1, DDR1, DYRK3, AK3, MEK3, MEK5, PDK1, PIM3, PKCmu, PKC1, and TRK are selected from TRB, TRK, and TRK.
- Cancers that express certain kinases. A cancer that expresses at least one kinase described in FIG.
- the cancer resistant to an anticancer drug preferably has at least one characteristic selected from the group consisting of (1) to (25) above, and from the group consisting of (1) to (25) above More preferably, it has a plurality of selected features.
- the dosage form of the pharmaceutical composition of the present embodiment is not particularly limited, and a known formulation method can be applied.
- Examples of the dosage form of the pharmaceutical composition of the present embodiment include emulsions, emulsions, liquids, gels, capsules, ointments, patches, patches, granules, tablets and the like. It is not limited.
- the administration route of the pharmaceutical composition of this embodiment is not particularly limited, and can be administered by oral or parenteral routes.
- the parenteral route includes all routes of administration other than oral, for example, intravenous, intramuscular, subcutaneous, intranasal, intradermal, ophthalmic, intracerebral, rectal, intravaginal and intraperitoneal. Further, the administration may be local administration or systemic administration.
- the pharmaceutical composition of the present embodiment can be administered in a single dose or multiple doses, and the administration period and interval are the type and condition of the disease, the administration route, the age of the administration subject, the body weight, the sex, etc. Can be selected as appropriate.
- the administration interval examples include, but are not limited to, 1 to 3 times a day, once every 2 to 3 days, 1 to 3 times a week, and once a day.
- the dosage of the pharmaceutical composition of this embodiment, its administration period and interval can be appropriately selected depending on the type of drug, the type and condition of the disease, the administration route, the age, weight and sex of the administration subject.
- the dosage of the pharmaceutical composition of the present embodiment can be a therapeutically effective amount of compound (S).
- “Therapeutically effective amount” means the amount of compound (S) effective for the treatment or prevention of cancer resistant to an anticancer drug.
- the dose of compound (S) per administration can be about 0.01 to 1000 mg, about 0.1 to 100 mg, about 0.5 to 50 mg, and about 1 to 10 mg per kg body weight.
- Rk 1 and Rk 2 are the same or different residues; (a) hydrogen, halogen, substituted or unsubstituted lower alkyl, substituted or unsubstituted lower alkenyl, substituted or unsubstituted lower alkynyl, hydroxy , Lower alkoxy, carboxy, lower alkoxycarbonyl, acyl, nitro, carbamoyl, lower alkylaminocarbonyl, —NR 5 R 6 [where R 5 and R 6 are hydrogen, substituted or unsubstituted lower alkyl, substituted or non-substituted, Substituted lower alkenyl, substituted or unsubstituted lower alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted aralkyl, substituted or unsubstituted lower alkylaminocarbonyl, substituted or unsubstituted Lower aryla
- Rk 3 is hydrogen, halogen, acyl, carbamoyl, substituted or unsubstituted lower alkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted lower alkynyl or amino;
- Wk 1 and Wk 2 are independently hydrogen, hydroxy, or Wk 1 and Wk 2 together represent oxygen.
- lower alkyl when used alone or in combination with other groups is 1 to 6 carbon atoms, preferably 1 to 5, more preferably 1 to 4 and particularly preferably 1 Means a linear or branched lower alkyl group containing ⁇ 3 or 1-2 carbon atoms.
- These groups include in particular methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, amyl, isoamyl, neopentyl, 1-ethylpropyl, hexyl and the like.
- lower alkyl part of “lower alkoxy”, “lower alkoxycarbonyl”, “lower alkylaminocarbonyl”, “lower hydroxyalkyl” and “tri-lower alkylsilyl” has the same meaning as “lower alkyl” as defined above. .
- a “lower alkenyl” group is defined as a C 2 -C 6 alkenyl group, which may be straight or branched and may be Z or E type. Such groups include vinyl, propenyl, 1-butenyl, isobutenyl, 2-butenyl, 1-pentenyl, (Z) -2-pentenyl, (E) -2-pentenyl, (Z) -4-methyl-2 -Pentenyl, (E) -4-methyl-2-pentenyl, pentadienyl, such as 1,3 or 2,4-pentadienyl. More preferred C2-C6-alkenyl groups are C 2 -C 5- , C 2 -C 4 -alkenyl groups, even more preferred C 2 -C 3 -alkenyl groups.
- lower alkynyl refers to a C 2 -C 6 -alkynyl group which may be straight or branched and includes ethynyl, propynyl, 1-butynyl, 2-butynyl, 1-pentynyl, 2-pentynyl, 3 -Methyl-1-pentynyl, 3-pentynyl, 1-hexynyl, 2-hexynyl, 3-hexynyl and the like. More preferred C 2 -C 6 -alkynyl groups are C 2 -C 5- , C 2 -C 4 -alkynyl groups, even more preferred C 2 -C 3 -alkynyl groups.
- aryl group means a C 6 -C 14 -aryl group containing from 6 to 14 cyclic carbon atoms. These groups may be monocyclic, bicyclic, or tricyclic and are fused rings. Preferred aryl groups include phenyl, biphenyl, naphthyl, anthracenyl, phenanthrenyl and the like. The aryl part of the “arylcarbonyl” group and the “arylaminocarbonyl” group has the same meaning as defined above.
- heteroaryl group may contain 1 to 3 heteroatoms independently selected from nitrogen, sulfur or oxygen and refers to a C 3 -C 13 -heteroaryl group. These groups may be monocyclic, bicyclic or tricyclic.
- the C 3 -C 13 heteroaryl groups of the present invention include heteroaromatic and saturated and partially saturated heterocyclic groups. These heterocycles may be monocyclic, bicyclic, or tricyclic.
- Preferred 5- or 6-membered heterocyclic groups are thienyl, furyl, pyrrolyl, pyridyl, pyranyl, monophorinyl, pyrazinyl, methylpyrrolyl, and pyridazinyl.
- C 3 -C 13 -heteroaryl may be a bicyclic heterocyclic group.
- Preferred bicyclic heterocyclic groups are benzofuryl, benzothienyl, indolyl, imidazolyl, and pyrimidinyl.
- the most preferred C 3 -C 13 -heteroaryl is furyl and pyridyl.
- lower alkoxy includes alkoxy groups containing 1 to 6 carbon atoms, preferably 1 to 5, more preferably 1 to 4, particularly preferably 1 to 3 or 1 to 2 carbon atoms. And may be linear or branched. These groups include methoxy, ethoxy, propoxy, butoxy, isopropoxy, tert-butoxy, pentoxy, hexoxy and the like.
- acyl includes lower alkanoyl containing 1 to 6 carbon atoms, preferably 1 to 5, 1 to 4, 1 to 3 or 1 to 2 carbon atoms, linear or It may be a branch. These groups preferably include formyl, acetyl, propionyl, butyryl, isobutyryl, tert-butyryl, pentanoyl and hexanoyl.
- the acyl part of the “acyloxy” group has the same meaning as defined above.
- halogen includes fluoro, chloro, bromo, iodo and the like.
- aralkyl group refers to a C 7 -C 15 -aralkyl in which the alkyl group is substituted by aryl.
- the alkyl group and aryl can be selected from C 1 -C 6 -alkyl groups and C 6 -C 14 -aryl groups as defined above, wherein the total number of carbon atoms is from 7 to 15.
- Preferred C 7 -C 15 -aralkyl groups are benzyl, phenylethyl, phenylpropyl, phenylisopropyl, phenylbutyl, diphenylmethyl, 1,1-diphenylethyl, 1,2-diphenylethyl.
- the aralkyl part of the “aralkyloxy” group has the same meaning as defined above.
- Substituted lower alkyl group, substituted lower alkenyl group, and substituted lower alkynyl group are lower alkyl, hydroxy, lower alkoxy, carboxyl, lower alkoxycarbonyl, nitro, halogen, amino, mono- or di-lower alkylamino, dioxolane, dioxane, dithiolane, And having 1 to 3 independently selected substituents such as dithione.
- the lower alkyl portion of the lower alkylamino substituent has the same meaning as "lower alkyl" defined above.
- the substituted aryl group, substituted heteroaryl group and substituted aralkyl group are each independently selected from lower alkyl, hydroxy, lower alkoxy, carboxy, lower alkoxycarbonyl, nitro, amino, mono- or di-lower alkylamino, and halogen, etc. It has 1 to 3 substituents.
- Heterocyclic groups formed by R 5 and R 6 bonded to a nitrogen atom include pyrrolidinyl, piperidinyl, piperidino, morpholinyl, morpholino, thiomorpholino, N-methylpiperazinyl, indolyl, and isoindolyl.
- the ⁇ -amino acid group includes glycine, alanine, proline, glutamic acid and lysine, and may be L-type, D-type or racemic.
- Rk 1 and Rk 2 are hydrogen, halogen, nitro, —CH 2 OH, — (CH 2 ) k R 14 , —CH ⁇ CH (CH 2 ) m R 16 , —C ⁇ C (CH 2 ) n R 15 , —CO (CH 2 ) j R 4 (where R 4 is —SR 7 ), CH 2 O— (substituted or unsubstituted) lower alkyl (where substituted lower alkyl is preferably Is independently selected from the group consisting of —NR 5 R 6 . More preferably, Rk 1 and Rk 2 are hydrogen.
- residue R 14 is preferably phenyl, pyridyl, imidazolyl, thiazolyl, tetrazolyl, —COOR 15 , —OR 15 (where R 15 is preferably hydrogen, Selected from methyl, ethyl, phenyl or acyl), —SR 7 (wherein R 7 is preferably selected from substituted or unsubstituted lower alkyl, 2-thiazoline and pyridyl) and —NR 5 R 6 wherein R 5 and R 6 are preferably selected from hydrogen, methyl, ethyl, phenyl, carbamoyl and lower alkylaminocarbonyl.
- residue R 16 is preferably hydrogen, methyl, ethyl, phenyl, imidazole, thiazole, tetrazole, —COOR 15 , —OR 15 and —NR 5 R 6 (wherein residues R 15 , R 5 and R 6 is selected from the above preferred meanings).
- residue R 7 is preferably selected from the group consisting of substituted or unsubstituted lower alkyl, substituted or unsubstituted phenyl, pyridyl, pyrimidinyl, thiazole and tetrazole.
- k is preferably 2, 3 or 4
- j is preferably 1 or 2
- m1 and n are independently preferably 0 or 1.
- Rk 3 is hydrogen or acetyl, most preferably hydrogen.
- each Wk 1 and Wk 2 is hydrogen.
- the compound (K) is preferably represented by the following formula (k1-1).
- Compound (K) can be produced, for example, by refluxing a compound represented by the following general formula (k0) without a solvent in the presence of anhydrous hydrazine or hydrazine hydrate. Further, for example, a compound represented by the following general formula (k0) is converted into methanol, ethanol, propanol, isopropanol, butanol, tert-butanol, N, N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), tetrahydrofuran (THF).
- DMF N-dimethylformamide
- DMSO dimethyl sulfoxide
- THF tetrahydrofuran
- reaction temperature and reaction time are not particularly limited.
- the reaction can be performed at room temperature to 50 ° C. for 30 minutes to 15 hours.
- the second aspect of the present invention is a drug complex containing a polymer having a repeating unit (ka) represented by the following general formula (ka) and a repeating unit (II) represented by the following general formula (II) (Hereinafter sometimes referred to as “drug complex (K)”).
- m is preferably 1.
- L is preferably a methylene group, an ethylene group, a propylene group or a benzylene group, and more preferably a methylene group, an ethylene group or a benzylene group.
- R 1 is preferably a hydrogen atom or an aliphatic hydrocarbon group, and more preferably a hydrogen atom or a methyl group.
- Rk 1 and Rk 2 are each independently hydrogen, halogen, nitro, —CH 2 OH, — (CH 2 ) k R 14 , —CH ⁇ CH (CH 2 ) m R 16 , —C ⁇ C (CH 2 ) n R 15 , —CO (CH 2 ) j R 4 (where R 4 is —SR 7 ), CH 2 O— (substituted or unsubstituted) lower alkyl (here The substituted lower alkyl is preferably methoxymethyl, methoxyethyl or ethoxymethyl) or —NR 5 R 6 , more preferably hydrogen.
- Rk 3 is preferably hydrogen or acetyl, and more preferably hydrogen.
- Wk 1 and Wk 2 are preferably hydrogen.
- m is preferably 1.
- X is preferably OR x and more preferably OH (hydroxy group).
- the drug complex (K) contains a polymer having a repeating unit (ka-1) represented by the following general formula (ka-1) and a repeating unit (II) represented by the following general formula (II) It is preferable to do.
- m is preferably 1.
- L is preferably a methylene group, an ethylene group, a propylene group or a benzylene group, more preferably a methylene group, an ethylene group or a benzylene group.
- R 1 is preferably a hydrogen atom or an aliphatic hydrocarbon group, more preferably a hydrogen atom or a methyl group.
- X is preferably OR x and more preferably OH (hydroxy group).
- Compound (K) or drug conjugate (K) shows efficacy against malignant cancer and metastatic cancer for which existing cancer therapeutic agents do not show efficacy. Therefore, the compound (K) and the drug complex (K) are useful as cancer therapeutic agents that are resistant to existing cancer therapeutic agents.
- the sustained release property of the compound (K) can be controlled similarly to the drug complex of the first aspect. While the compound (K) is held in the polymer, the compound (K) is stably maintained and the toxicity is alleviated. Therefore, side effects can be reduced and the therapeutic effect can be enhanced. Further, by preparing micelles containing the drug complex (K), the compound (K) from the drug complex (K) depends on the in vivo pH environment, as in the micelle of the first aspect. The release of can be controlled.
- the micelle containing the drug complex (K) can be prepared by a known method in the same manner as the drug complex micelle of the first aspect.
- the drug conjugate (K) is dissolved or suspended in a lipophilic or hydrophilic solvent, and the solution or suspension is dropped into the hydrophilic or lipophilic solvent and stirred, whereby the drug conjugate ( Micelle containing K) can be prepared.
- the drug conjugate (K) is an embodiment in which the drug is the compound (K) in the drug conjugate of the first aspect, and is included in the drug conjugate of the first aspect. It can also be said that the drug conjugate (K) is an embodiment in which BM is a group derived from the compound (K) in the general formula (Ia) in the first embodiment.
- the compound (K) and the drug complex (K) can be used as the compound (S) and the drug complex in the pharmaceutical composition of the first aspect, respectively.
- the present invention provides a compound (S) or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable salt in the manufacture of a pharmaceutical composition for treating or preventing cancer resistant to an anticancer drug.
- the present invention provides a compound (S) or a pharmaceutically acceptable salt thereof, or a drug in which the compound (S) or a pharmaceutically acceptable salt thereof is bound to a pharmaceutically acceptable polymer.
- a method for treating or preventing cancer resistant to an anticancer drug comprising administering the complex to a subject (for example, a patient suffering from cancer resistant to an anticancer drug).
- the present invention relates to a compound (S) or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable polymer for use in the treatment or prevention of cancer resistant to an anticancer drug.
- a drug conjugate to which compound (S) or a pharmaceutically acceptable salt thereof is bound is bound.
- the present invention provides compound (S) or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable polymer for treating or preventing anticancer drug-resistant cancer, S) or the use of a drug conjugate conjugated with a pharmaceutically acceptable salt thereof.
- K252a-hydrazide K252a-H
- K252a was purchased from BOC Science (US).
- K252a (20 mg) was dissolved in anhydrous methanol (200 ⁇ L) and added to anhydrous hydrazine (300 ⁇ L).
- the reaction mixture was stirred at 40 ° C. for 15 hours.
- the reaction mixture was evaporated to give a dry product.
- toluene was used as the co-evaporation solvent.
- the resulting product was used for the preparation of drug conjugates and micelles without further purification.
- FIG. 8 shows the 1 H-NMR analysis result of the obtained product.
- K252a-H was analyzed by HPLC. The analysis results are shown in FIG. Since K252a-H and K252a have different retention times, it was confirmed that they have different structures.
- the conditions for HPLC analysis are as follows. TSK-GEL ODS-100V column 4.6 ⁇ 150mm, particle size 5 ⁇ m (Tosoh Corporation) Column pressure: 10.7 MPa Column temperature: constant temperature around 40 ° C. Mobile phase: methanol / formate buffer (pH 3.0) mixture (3: 2) Flow rate: 1.2 mL / min, 20-30 minutes UV detection: wavelength 290 nm
- Example 2 Preparation of drug complex of K252a-H and polymer
- Aromatic aldehyde group-containing polymer or aliphatic ketone group-containing polymer and K252a-H were mixed at a mass ratio of 2: 1 and dissolved in DMSO. (Dissolve 15 mg drug / polymer mixture per 1 mL DMSO).
- the coupling reaction between the aromatic aldehyde group-containing polymer and K252a-H was carried out by stirring the reaction mixture at 40 ° C. overnight.
- the coupling reaction between the aliphatic ketone group-containing polymer and K252a-H was performed by stirring the reaction mixture for 96 hours.
- FIG. 10 shows the result of 1 H-NMR analysis confirming the bond between the aliphatic ketone group-containing polymer and K252a-H.
- FIG. 11 shows the results of 1 H-NMR analysis confirming the bond between the aliphatic aldehyde group-containing polymer and K252a-H.
- Example 3 Preparation of K252a-H bonded polymer micelle (Preparation of micelle) The reaction mixture in DMSO was dialyzed against dimethylacetamide (DMAc), and a solvent exchange from DMSO to DMAc was performed (dialysis for 4 hours, dialysis solvent was changed once). In addition, free K252a-H was removed by dialysis. A DMAc solution of the drug complex of polymer and K252a-H obtained as described above was used for the preparation of micelles. The DMAc solution was dropped into water at a ratio of 1 to 10 by volume and vortexed to prepare micelles.
- DMAc dimethylacetamide
- This solution was dialyzed against water for 24 hours in a dialysis bag with a molecular weight cut off (MWCO) of 3500 Da.
- the dialysis medium was changed 5 times during dialysis.
- the solution in the dialysis bag was filtered (0.22 ⁇ m) and concentrated by ultrafiltration using a 100 kDa MWCO filter membrane (Amicon).
- the micelle of the drug complex of K252a-H and the above polymer is sometimes referred to as “K252a-H-bonded polymer micelle”.
- FIGS. 12A and 12B The results are shown in FIGS. 12A and 12B.
- FIG. 12A is a drug complex micelle of an aromatic aldehyde group-containing polymer and K252a-H (hereinafter referred to as “K252a-H-bonded aromatic polymer micelle”)
- FIG. 12B is an aliphatic ketone group-containing polymer.
- K252a-H-linked aliphatic polymer micelle a drug complex micelle of K252a-H (hereinafter referred to as “K252a-H-linked aliphatic polymer micelle”). It was confirmed that the K252a-H-bonded aliphatic polymer micelle has a smaller micelle size than the K252a-H-bonded aromatic polymer micelle. Therefore, K252a-H-bonded aliphatic polymer micelles are expected to have higher cancer accumulation properties than K252a-H-bonded aromatic polymer micelles.
- Example 5 Release of K252a-H from K252a-H-bonded polymer micelles
- K252a-H K252a-H-bonded aromatic polymer micelles and K252a-H-bonded aliphatic polymer micelles
- K252a-H K252a-H-bonded aromatic polymer micelles
- K252a-H-bonded aliphatic polymer micelles Collectively referred to as “K252a”.
- the cell line used was American type. Purchased from Culture Collection (American Type Culture Collection, ATCC) or JCRB Cell Bank (National Research Institute for Pharmaceuticals, Health and Nutrition). PC14 PE6 was distributed by Prof. Hosho (Tottori University). Unless otherwise stated, DMEM, RPMI or E-MEM was used as the medium. Gefetinib, Afatinib, Erlotinib, Cisplatin, Pemetrexed, and Gemicitabine were purchased from Funakoshi Corporation. Osimertinib was purchased from Selleck Chemicals.
- 50 ⁇ L of the drug solution prepared above was added in order of increasing micelle concentration from row 3 to row 12 of a 96-well plate seeded with cancer cells.
- 50 ⁇ L of medium alone was added. Thereafter, it was incubated for 48 hours. After 48 hours, 10 ⁇ L of cell-counting kit-8 solution (DOJINDO) was added to each well. After incubating again, the absorbance at 450 nm was measured after 30 minutes, 1 hour, and 2 hours.
- results are shown in FIG. Gefenitib, Afatinib, and Osimetinib are molecularly targeted therapeutics for lung cancer that are currently approved by the FDA.
- the results shown in FIG. 15 indicate that K252a species (particularly, K252a, K252a-H, and K252a-H-linked aliphatic polymer micelles) exhibit high cytotoxic activity against lung cancer resistant to existing lung cancer therapeutic agents. It was done.
- FIG. 16 shows the extracted PC14 strain and PC14 / PE6 strain in FIG.
- the PC14 / PE6 strain is a cancer cell line obtained by administering the PC14 strain to the tail vein of mice and establishing cancer cells that have metastasized in the thoracic cavity (Yano et al., Oncology Research 9: 573-579 (1997)). Therefore, the PC14 / PE6 strain is a cancer cell line that has extremely high metastatic potential and is malignant. As shown in FIGS. 15 and 16, Gefenitib, Afatinib, and Osimetinib showed high cytotoxic activity against the PC14 strain. However, for existing malignant PC14 / PE6 strains, these existing molecular targeted drugs have significantly reduced cytotoxic activity.
- K252a compounds (particularly K252a-H and K252a-H-linked aliphatic polymer micelles) showed higher cytotoxic activity against the PC14 / PE6 strain. These results indicate that K252a is more effective against malignant metastatic cancer. Of the K252a-H-bonded aromatic polymer micelles and K252a-H-bonded aliphatic polymer micelles, K252a-H-bonded aliphatic polymer micelles tended to have higher cytotoxic activity against cancer cells.
- Example 7 Cytotoxicity test of K252a against pancreatic cancer cells Using various pancreatic cancer cell lines, the cytotoxicity test of K252a was performed in the same manner as described above. The results are shown in FIG.
- the BXPC3-F3 strain is a metastatic strain that has been malignant by repeating the establishment of a cancer cell line that has metastasized to the liver three times after orthotopic transplantation of the BXPC3 strain to mice.
- KP-3L is a metastatic strain established after transplanting human liver metastatic pancreatic cancer into the spleen of a nude mouse and then establishing a cancer cell line that has metastasized to the liver (JCRB, National Institute of Biomedical Innovation, Health and Nutrition) Obtained from cell bank).
- K252a-H-bonded aromatic polymer micelles K252a-H-bonded aliphatic polymer micelles tended to have higher cytotoxic activity against cancer cells.
- Example 8 Cytotoxicity test of K252a against head and neck cancer cells Using various head and neck cancer cell lines, the cytotoxicity test of K252a was performed in the same manner as described above. The results are shown in FIG. CDDP indicates cisplatin, which is a standard treatment for head and neck cancer.
- the SAS strain, FaDu strain, and HSC2 strain are all cisplatin resistant cell lines.
- K252a showed higher cytotoxic activity than any of cisplatin and Erlotinib against any head and neck cancer cell line.
- the FaDu strain the cytotoxic activity of K252a-H-linked aliphatic polymer micelles was high. This result shows that K252a is effective against a cisplatin resistant strain.
- Example 9 Cytotoxicity test of K252a against malignant mesothelioma cells Using the malignant mesothelioma cell line MSTO-211H, a cytotoxicity test of K252a was performed in the same manner as described above. The results are shown in FIG. CDDP indicates cisplatin and is a standard treatment for malignant mesothelioma. Pemetrexed is also a standard treatment for malignant mesothelioma. As shown in FIG. 19, K252a family showed higher cytotoxic activity than cisplatin, Pemetrexed and Midostaurin.
- Hot spot kinase profiling of K252a-H was carried out using the Monthly 202 mutual kinase panel consigned to Rection Biology.
- the conditions for the kinase assay are as follows. K252a was dissolved in DMSO and assayed at 100 nM. Buffer conditions: 20 mM HEPES, pH 7.5, 10 mM MgCl 2 , 1 mM EGTA, (2 mM MnCl 2 if applicable) ATP concentration: 10 ⁇ M Reaction time: 2 hours
- FIGS. 20 and 21 For reference, the first generation, second generation, and third generation tyrosine kinase inhibitors (TKI) and their resistance mutations are shown in FIGS. 20 and 21.
- FIG. 20 and 21 since the first to third generation TKIs are not effective for the C797S mutation, a TKI effective for the C797S mutation is required.
- K252a-H The results of the K252a-H kinase assay for the EGFR mutation are shown in FIG. As shown in FIG. 22, K252a-H is against kinases with gatekeeper mutations (d746-750 / T790M, d746-750 / T790M / C797S, L858R, T790M, etc.) that many TKIs do not show efficacy. In particular, it showed high inhibitory activity.
- Figures 23A-C show the results of kinase profiling of K252a-H.
- FIG. 22 The results of the K252a-H kinase assay for the EGFR mutation are shown in FIG. As shown in FIG. 22, K252a-H is against kinases with gatekeeper mutations (d746-750 / T790M, d746-750 / T790M / C797S, L858R, T790M, etc.) that many TKIs do
- K252a-H shows a list of mutant kinases whose kinase activity was suppressed to 30% or less by K252a-H as a result of kinase profiling for K252a-H.
- K252a-H was confirmed to be a potent inhibitor of the oncogene c-kit / Flt3 / RET.
- FIGS. 25 to 34D The results of kinase profiling using a Monthly Mutant Kinase panel are shown in FIGS. 25 to 34D.
- the kinase surrounded by a solid line has a mutation involved in drug resistance and shows a high inhibitory activity by the tested drug.
- These mutations are summarized in Table 2.
- Staurosporine and K252a were shown to have high inhibitory activity of kinases having the mutations listed in Table 2.
- staurosporine and K252a were shown to have a high inhibitory effect on the triple mutation of EGFR in oximeltinib resistance (d746-750 / T790M / C797S, L858R / T790M / C797S).
- HER2 gatekeeper mutation T798I
- HER2 is an EGFR homologue, and thus was inhibited by staurosporine and K252a in the same manner as the EGFR gatekeeper mutation.
- Figures 33A-D show the results of kinase profiling with 500 nM K252a-H.
- Figures 34A-D show the results of kinase profiling with 20 nM staurosporine.
- the IC50 of K252a-H for each wild type kinase was determined from the results of kinase profiling using the monthly virus type kinase panel. As a result, kinases having an IC50 of K252a-H of 5 nM or less are shown in Table 3. In Table 3, the underlined kinase is a kinase involved in drug resistance.
- IC50 of staurosporine with respect to each wild type kinase was determined from the result of kinase profiling using a monthly wild type kinase panel.
- the kinase in which IC50 of staurosporine was 0.2 nM or less is shown in FIG.
- the underlined kinase is a kinase that is important as a therapeutic target for cancer (particularly drug-resistant cancer).
- Staurosporine showed an especially high inhibitory effect with an IC50 of 0.1 nM or less for CaMK2a, CaMK2b, JAK3, ROS1, RSK4, STK22, and TRKB.
- BxPC3-F3 is a liver metastasis strain in which BxPC3 is malignant.
- BxPC3-F3 was prepared by isotopically transplanting BxPC3 into the pancreas of mice, isolating liver metastasized cells from the liver by trypsin treatment, establishing a cell line, and repeating the work (cell line establishment) three times. did.
- Anticancer agents (Gefetinib, Afetinib, Osimmertinib, Gemicitabine, Cisplatin, Erlotinib, Pemetrexed, Midostaurin) were purchased from Funakoshi.
- the cytotoxicity of the test drug was measured by the following method using cell counting kit 8 (DOJINDO, JAPAN). Cancer cell lines were seeded at 1 to 3 ⁇ 10 3 cells / well. The next day, the medium was changed and 50 ⁇ L of medium was added. Furthermore, a dilution series of the test drug was prepared, and 50 ⁇ L was added to the well and stirred. 72 hours later, 10 ⁇ L of cell counting kit 8 was added, and after 1 hour, absorbance at 450 nm was measured with a microplate reader (TECAN). Based on the measured absorbance, the cell viability at each dilution rate was calculated for each test drug by the following formula (1). Based on the calculated cell viability, the IC50 of the test drug was calculated.
- Non-small cell lung cancer The results of the cytotoxicity test for non-small cell lung cancer cell lines are shown in FIG. The characteristics of each cell line are as shown in Table 1 above.
- Known lung cancer therapies (Gefetinib, Afatinib, Osimertinib, Dasatinib) showed high cytotoxicity against PC14, while Gefetinib, Afatinib, and Osimertinib are against PC14-PE6, a malignant metastatic strain of PC14. The cytotoxicity was low.
- staurosporine, K252a, and K252a-H showed a higher effect on PC14-PE6 than on PC14.
- PC14 was sensitive to known lung cancer therapeutics (Gefetinib, Afatinib, Osimmertinib, Dasatinib), while PC14-PE6, A549, H358, NCI1650, and H520 were resistant to known lung cancer therapeutics. .
- staurosporine, K252a, and K252a-H tended to show high cytotoxicity against these lung cancer therapeutic drug resistant strains.
- pancreatic cancer The result of the cytotoxicity test for the pancreatic cancer cell line is shown in FIG. In FIG. 37, BxPC3-F3 is a malignant liver metastasis strain of BxPC3. KP3L is a metastatic strain that has metastasized to the liver of a mouse after transplanting human liver metastatic pancreatic cancer into the spleen of a nude mouse. Standard treatments for pancreatic cancer (Gemicitabine, Erlotinib) showed high cytotoxicity against BxPC3, but low cytotoxicity against BxPC3-F3 and KP3L. On the other hand, staurosporine, K252a, and K252a-H also showed high cytotoxicity against these pancreatic cancer therapeutic drug resistant strains.
- FIG. 38 The results of the cytotoxicity test against pancreatic cancer cell lines are shown in FIG. In FIG. 38, “CDDP” represents cisplatin.
- staurosporine, K252a, and K252a-H showed higher cytotoxicity than known head and neck cancer therapeutic agents (Midostaurin, cisplatin, Erlotinib, Gefetinib).
- staurosporine, K252a, and K252a-H also showed high cytotoxicity against SAS, which is a Midostaurin resistant strain.
- FIG. 39 The results of the cytotoxicity test on the malignant mesothelioma cell line are shown in FIG. In FIG. 39, “CDDP” represents cisplatin. Staurosporine, K252a, and K252a-H showed higher cytotoxicity than known malignant mesothelioma therapeutics (Midostaurin, cisplatin, Pemetrexed).
- mice In vivo antitumor activity of K252a-H (non-small cell lung cancer) Balb / c nude mice were anesthetized with isoflurane, and 5 ⁇ 10 5 cells / 20% Matrigel 50 ⁇ L of non-small cell lung cancer cell line (PC14-PE6-luc: cells into which luciferase gene was introduced into PC14-PE6) were directly added to the mice. Injected into the lungs. Mark 5mm from the tip of the injection needle, insert the injection needle from the 3rd to 4th position of the rib toward the lower lobe of the ribs, and suspend 20% Matrigel in non-small cell lung cancer cell line 50 ⁇ L of the solution was injected directly into the lung.
- PC14-PE6-luc cells into which luciferase gene was introduced into PC14-PE6
- a human non-small cell lung cancer orthotopic transplant model mouse was prepared. From 5 days after the cancer cell injection, the drug was injected into the human non-small cell lung cancer orthotopic transplantation mouse at a dose of 1 mg / kg body weight twice a week. Twice a week under isoflurane anesthesia, IVIS spectrum (Xwogen Corporation) was used to monitor tumor growth and evaluate the anti-tumor effect of the drug. Statistical processing of tumor size and survival days was performed using Prism7. K252a-H and K252a-H-linked aliphatic polymer micelles (prepared in Example 3) were used as test agents. PBS was used as a negative control.
- FIG. 40 shows tumor growth
- FIG. 41 shows the survival curve of mice by the Kaplan-Meier method.
- K252a-H-linked aliphatic polymer micelles were administered, tumor growth was significantly suppressed and survival time was significantly prolonged compared with PBS administration.
- K252a-H was administered, the survival time tended to be longer than when PBS was administered. From these results, it was confirmed that K252a (especially, K252a-H-bonded aliphatic polymer micelle) exhibits an antitumor effect even in vivo against osimertinib-resistant lung cancer.
- K252a-H, K252a-H linked aliphatic polymer micelles, K252a-H linked aromatic polymer micelles (prepared in Example 3), and gemcitabine were used.
- PBS was used as a negative control.
- the dose per dose of each drug is 3 mg / kg body weight for K252a-H, K252a-H linked aliphatic polymer micelles, and K252a-H linked aromatic polymer micelles, and 5 mg / kg body weight for gemcitabine. It was.
- pancreatic cancer Trial using pancreatic cancer spontaneous mice
- Transgenic mice that spontaneously develop pancreatic cancer Capiller corp
- pancreatic cancer spontaneously developing model mice EL-1-luc / TAg
- the drug administration was started from 13 to 15 weeks of age when pancreatic cancer occurred.
- the drug administration method was the same as the method described in “(Non-small cell lung cancer)”.
- drugs K252a-H-linked aliphatic polymer micelles and K252a-H-linked aromatic polymer micelles were used.
- PBS was used as a negative control.
- the dose per dose of each drug was 1 mg / kg body weight.
- FIG. 43 shows IC50 of staurosporine and K252a family for spontaneous pancreatic cancer EL1-luc / TAg cells taken from mouse pancreas and cell line. IC50 was calculated by the same method as described in Example 12. As shown in FIG. 43, EL-1 / TAg is resistant to gemcitabine and erlotinib. On the other hand, EL-1 / TAg was sensitive to staurosporine and K252a.
- FIG. 44 and 45 are diagrams showing the results of an antitumor effect evaluation test using a pancreatic cancer spontaneous model mouse.
- FIG. 44 shows tumor growth
- FIG. 45 shows the survival curve of mice by Kaplan-Meier method.
- K252a-H-bonded aliphatic polymer micelle or K252a-H-bonded aliphatic polymer micelle was administered, tumor growth was significantly suppressed as compared to PBS.
- the K252a-H-linked aliphatic polymer micelle was administered, the survival time was significantly prolonged as compared with the case where PBS was administered. Even when K252a-H-bonded aromatic polymer micelle was administered, the survival time tended to be longer than when PBS was administered. From these results, it was confirmed that K252a species (particularly, K252a-H-linked aliphatic polymer micelles) exhibited an antitumor effect in vivo against gemcitabine and erlotinib-resistant pancreatic cancer.
- Example 14 Cytotoxicity of K252a class to lymphoma
- the IC50 of K252a class to lymphoma cell line was calculated. The results are shown in FIG. Also for lymphoma cell lines, K252a species (particularly K252a, K252a-H, K252a-H-linked aliphatic polymer micelles) showed high cytotoxicity.
- Example 15 Drug transport inhibitory effect of ABC transporter (MDR-1) Human renal cancer cell line 786-0 is a cell that highly expresses MDR-1 (ABCB-1, p-gp) Strain (data not shown). Therefore, 786-0 was used to evaluate the effects of staurosporine and K252a on the drug excretion activity of MDR-1. The drug excretion activity was evaluated using an eFluxx-ID TM Green Multidrug Resistance Assay kit (Enzo Life Science). After culturing 786-0 cells in a well plate, trypsin / EDTA solution was added and incubated, and the cells were detached from the wells.
- the detached cells were collected and washed with the medium, and then the cells were suspended in the medium at a concentration of 1 ⁇ 10 6 cells / mL to obtain a cell suspension.
- the cell suspension was pre-warmed at 37 ° C. for 10 minutes or more, and a dilution series of drug was added to 250 ⁇ L of the cell suspension (2.5 ⁇ 10 5 cells).
- Staurosporine, K252a, K252a-H, and sunitinib were used as drugs.
- Verapamil final concentration 30 ⁇ M
- an MDR-1 inhibitor was used as a positive control.
- LD50 was calculated as a drug concentration at which the amount of eFluxx-GFP excretion was reduced to half.
- Example 16 Influence of human ⁇ 1-AGP
- human ⁇ 1-AGP acid glycoprotein
- human ⁇ 1-AGP human serum protein
- the cellular attributes of K252a-H and K252a-H linked aliphatic polymer micelles were evaluated. Cytotoxicity was evaluated using cell counting kit 8 (DOJINDO, JAPAN).
- DOJINDO cell counting kit 8
- the lung cancer cell line H460 was seeded in the wells at 1 to 3 ⁇ 10 3 cells / well and cultured. The next day, the medium was changed and hAGP was added to a final concentration of 0.5 mg / mL.
- a medium supplemented with 50 ⁇ L of hAGP-free medium was also prepared.
- a drug K252a-H or K252a-H-linked aliphatic polymer micelle
- a concentration of 1 ⁇ M was added to a concentration of 1 ⁇ M.
- 10 ⁇ L of cell counting kit 8 was added.
- the absorbance at 450 nm was measured with a microplate reader (TECAN). Based on the measured absorbance, the cell viability was calculated by the above formula (1).
- a linker reagent (L1) represented by the following formula (L1) was bound to staurosporine.
- a scheme of the linker binding reaction is shown in FIG. In FIG. 49, “DIC” represents diisopropylcarbodiimide.
- the activated esters in FIG. 49 include N-hydroxysuccinimide (NHS), 1-hydroxybenzotriazole (HOBt), 1-hydroxy-7-azabenzotriazole (HOAt), pentafluorophenol, p-nitrophenol. And active esters derived from these.
- FIG. 50 shows the 1 H NMR analysis result of the organic phase component.
- Staurosporine to which an active ester linker is bound can be bound to a polymer such as polyethylene glycol (PEG) to form a drug complex with the polymer.
- PEG polyethylene glycol
- the upper diagram of FIG. 51 shows a reaction in which polyethylene glycol is bonded to staurosporine via a linker.
- the lower diagram of FIG. 51 shows a reaction in which a PEG-polyamino acid block copolymer is bound to staurosporine via a linker. (Bottom view of FIG. 51).
- the polymer-linker-staurosporine complex reacts with glutathione (GSH) by glutathione transferase (GST) in the cell, and staurosporine is released.
- GSH glutathione
- GST glutathione transferase
- the compound (S′L1-H) can be bound to the polymers synthesized in Polymer Synthesis Examples 1 to 5.
- Examples of drug conjugates in which the compound (S′L1-H) is bound to an aromatic aldehyde group-containing polymer are shown below.
- bond (I) represents a pH-sensitive bond.
- Binding (II) represents a GSH sensitive binding that reacts with GSH in the presence of GST.
- the bond (I) is first cleaved under a low pH environment around the tumor tissue, and the compound (S′L1-H) becomes Released. Next, the compound (S′L1-H) is taken up into the tumor cell, and reacts with GSH by GST, whereby the bond (II) is cleaved to produce staurosporine. The staurosporine kills tumor cells.
Abstract
下記一般式(S)で表される化合物(S)、若しくはその薬学的に許容される塩、又は薬学的に許容されるポリマーに前記化合物(S)若しくはその薬学的に許容される塩が結合した薬物複合体を含む、抗がん剤耐性のがんを治療又は予防するための医薬組成物(X、Y、R6、R7は、H、ヒドロキシ基等であり、R9及びR10は、H、メチル、β-D-グルコピラノシル、4-O-メチル-β-D-グルコピラノシル、シアノエチル、又は下記のいずれかの式で表される基である。)。
Description
本発明は、医薬組成物に関する。特に、抗がん剤耐性のがんを治療又は予防するための医薬組成物に関する。
本願は、2018年2月22日に、日本に出願された特願2018-30208号に基づき優先権を主張し、その内容をここに援用する。
本願は、2018年2月22日に、日本に出願された特願2018-30208号に基づき優先権を主張し、その内容をここに援用する。
抗がん剤による治療を行うと、一時的にはがんの消失が確認できるものの、現在までに開発されている抗がん剤では全てのがん細胞を根絶することは難しい。抗がん剤に耐性を獲得したごく少数のがん細胞が生存することにより、がんが再発・転移することが知られている。
例えば、ゲフィチニブは、上皮成長因子受容体(EGFR)のチロシンキナーゼ活性を阻害する抗がん剤であり、非小細胞肺がん治療薬として用いられている。ゲフィチニブは、L858R変異を有するEGFRを発現する非小細胞肺がんに有効であるが、T790M変異が生じると、ゲフィチニブ耐性がんとなる。T790M変異を有するEGFRを発現するゲフィチニブ耐性がんには、オシメルチニブが有効性を示す。しかし、さらに、C797S変異が生じると、オシメルチニブにも耐性のがんとなる。現在のところ、L858R、T790M、及びC797Sの3つの変異を有するEGFRを発現するがんに有効な薬剤は開発されていない。
一方、スタウロスポリン(Staurosporine)は、ストレプトマイセス属の放線菌から単離された天然物であり(非特許文献1)、抗腫瘍作用を有することが知られている(例えば、特許文献1)。現在までに、K252a等のスタウロスポリン類縁体が報告されている(例えば、特許文献2)。
Omura S, et al., A new alkaloid AM-2282 OF Streptomyces origin. Taxonomy, fermentation, isolation and preliminary characterization. J Antibiot (Tokyo). 1977 Apr;30(4):275-82.
上記のように、遺伝子変異により、がんが抗がん剤耐性を獲得すると、既存の抗がん剤では治療が困難になるという問題がある。そのため、抗がん剤耐性のがんに有効な薬剤の開発が求められている。一方、スタウロスポリン及びその類縁体は、抗腫瘍活性を有することが知られていたが、抗がん剤耐性のがんに対する有効性は示されていなかった。また、安全域が極めて狭いためにがん治療に有効な投与方法が求められている。さらに、スタウロスポリン及びその類縁体は、難溶性であるため静注が困難であった。
本発明は、上記事情に鑑みてなされたものであって、抗がん剤耐性のがんの治療又は予防に利用可能な医薬組成物を提供することを課題とする。
本発明は、上記事情に鑑みてなされたものであって、抗がん剤耐性のがんの治療又は予防に利用可能な医薬組成物を提供することを課題とする。
本発明は以下の態様を含む。
〔1〕下記一般式(S)で表される化合物(S)、若しくはその薬学的に許容される塩、又は薬学的に許容されるポリマーに前記化合物(S)若しくはその薬学的に許容される塩が結合した薬物複合体を含む、抗がん剤耐性のがんを治療又は予防するための医薬組成物。
〔1〕下記一般式(S)で表される化合物(S)、若しくはその薬学的に許容される塩、又は薬学的に許容されるポリマーに前記化合物(S)若しくはその薬学的に許容される塩が結合した薬物複合体を含む、抗がん剤耐性のがんを治療又は予防するための医薬組成物。

X及びYは、それぞれ独立に、H、-OH、Cl、プロポキシ、又はエチルチオメチルであり;
R6は、H、C1-3アルキル、-NH2、ベンジル、


R7及びR8は、それぞれ独立に、H、-OH、又はメトキシであるか、或いは一緒になってO=を形成してもよく;
R9及びR10は、それぞれ独立に、H、メチル、β-D-グルコピラノシル、4-O-メチル-β-D-グルコピラノシル、シアノエチル、又は



R11は、メチルであり;
R12は、Hであり;
R13及びR14は、それぞれ独立に、H、メトキシ、-OH、ヒドロキシメチル、メチルカルボキシレート、メチルアミノ、メチルアミノメチル、プロピルアミノメチル、ジメチルアミノメチル、メトキシカルボニル、

R15及びR16は、それぞれ独立に、H、-OH、又は

R17及びR18は、H、-OH、メチルアミノ、ジメチルアミノ、オキシム、又は下記のいずれかの式で表される基

R19は、メトキシ、-OH、-NH-NH2、又は活性エステルを含む基である。]
〔2〕前記抗がん剤が、ゲフィチニブ、アファチニブ、オシメルチニブ、ダサチニブ、エルロチニブ、ゲムシタビン、シスプラチン、ペメトレキセド、及びミドスタウリンからなる群より選択される少なくとも1種の抗がん剤である、〔1〕に記載の医薬組成物。
〔3〕前記抗がん剤が、キナーゼを標的とする分子標的薬である、〔1〕又は〔2〕に記載の医薬組成物。
〔4〕前記キナーゼが、EGFR、ABL1、ALK1、HER2、c-Kit、FGFR1、FGFR2、FGFR3、c-Src、PDGFRa、RET、DDR2、TRKA、及びFlt-3からなる群より選択される少なくとも1種のキナーゼである、〔3〕に記載の医薬組成物。
〔5〕前記抗がん剤耐性のがんが、ゲートキーパー変異を有するキナーゼを発現するがんである、〔1〕~〔4〕のいずれかに記載の医薬組成物。
〔6〕前記キナーゼが、d746-750、T790M、及びC797Sの変異を有するEGFRである、〔5〕に記載の医薬組成物。
〔7〕前記化合物(S)が、スタウロスポリン、7-ヒドロキシスタウトスポリン、KT5926、スタウロスポリン・アグリコン、SF2370、KT5823、4’-N-ベンゾイルスタウロスポリン、Go6976、N,N-ジメチルスタウロスポリン、NA 0359、N-エトキシカルボニル-7-オキソスタウロスポリン、KT-6124、CGP42700、4’-デメチルアミノ-4’,5’-ジヒドロキシスタウロスポリン、7-オキソスタウロスポリン、CEP751、NA0346、NA0359、3’-デメトキシ-3’-ヒドロキシスタウロスポリン、KT 6006、7-O-メチル-UCN 01、TAN 999、NA 0346、NA 0345、NA 0344、CGP 44171A、SCH 47112、N,N-ジメチルスタウロスポリン、TAN 1030A、レスタウルチニブ、4’-デメチルアミノ-4’-ヒドロキシスタウロスポリン、AFN941、エドテカリン、ベカテカリン、K252a、及びK252aヒドラジドからなる群より選択される少なくとも1種の化合物である、〔1〕~〔6〕のいずれかに記載の医薬組成物。
〔8〕前記化合物(S)が、スタウロスポリン、K252a、及びK252aヒドラジドからなる群より選択される少なくとも1種の化合物である、〔7〕に記載の医薬組成物。
〔9〕前記薬物複合体がミセルを形成している、〔1〕~〔8〕のいずれかに記載の医薬組成物。
本発明によれば、抗がん剤耐性のがんの治療又は予防に利用可能な医薬組成物が提供される。
本明細書及び本特許請求の範囲において、化学式で表される構造によっては不斉炭素が存在し、エナンチオ異性体(enantiomer)やジアステレオ異性体(diastereomer)が存在し得るものがあるが、その場合は一つの式でそれら異性体を代表して表す。それらの異性体は単独で用いてもよいし、混合物として用いてもよい。
[第1の態様]
<医薬組成物>
一実施形態において、本発明は、下記一般式(S)で表される化合物(S)、若しくはその薬学的に許容される塩、又は薬学的に許容されるポリマーに前記化合物(S)若しくはその薬学的に許容される塩が結合した薬物複合体を含む、抗がん剤耐性のがんを治療又は予防するための医薬組成物を提供する。
<医薬組成物>
一実施形態において、本発明は、下記一般式(S)で表される化合物(S)、若しくはその薬学的に許容される塩、又は薬学的に許容されるポリマーに前記化合物(S)若しくはその薬学的に許容される塩が結合した薬物複合体を含む、抗がん剤耐性のがんを治療又は予防するための医薬組成物を提供する。
(化合物(S))
本実施形態の医薬組成物は、下記一般式(S)で表される化合物(S)、若しくはその薬学的に許容される塩、又は薬学的に許容されるポリマーに前記化合物(S)若しくはその薬学的に許容される塩が結合した薬物複合体を含む。
本実施形態の医薬組成物は、下記一般式(S)で表される化合物(S)、若しくはその薬学的に許容される塩、又は薬学的に許容されるポリマーに前記化合物(S)若しくはその薬学的に許容される塩が結合した薬物複合体を含む。

X及びYは、それぞれ独立に、H、-OH、Cl、プロポキシ、又はエチルチオメチルであり;
R6は、H、C1-3アルキル、-NH2、ベンジル、

R7及びR8は、それぞれ独立に、H、-OH、又はメトキシであるか、或いは一緒になってO=を形成してもよく;
R9及びR10は、それぞれ独立に、H、メチル、β-D-グルコピラノシル、4-O-メチル-β-D-グルコピラノシル、シアノエチル、又は



R11は、メチルであり;
R12は、Hであり;
R13及びR14は、それぞれ独立に、H、メトキシ、-OH、ヒドロキシメチル、メチルカルボキシレート、メチルアミノ、メチルアミノメチル、プロピルアミノメチル、ジメチルアミノメチル、メトキシカルボニル、

R15及びR16は、それぞれ独立に、H、-OH、又は

R17及びR18は、それぞれ独立に、H、-OH、メチルアミノ、ジメチルアミノ、オキシム、又は下記のいずれかの式で表される基

R19は、メトキシ、-OH、-NH-NH2、又は活性エステルを含む基である。]
上記化合物(S)の好ましい例としては、下記一般式(S-1)及び(S-2)でそれぞれ表される化合物(S-1)及び化合物(S-2)が挙げられる。

上記一般式(S-1)又は(S-2)中、X、Y、及びR6は、Hであることが好ましい。
上記一般式(S-1)又は(S-2)中、R13及びR14は、それぞれ独立に、H、メトキシ、-OH、ヒドロキシメチル、メトキシカルボニル、又は下記式(h1)で表される基であることが好ましい。

上記(S-1)中のR13及びR14は、それぞれ独立に、H、メトキシカルボニル、又は上記式(h1)で表される基であることがより好ましい。上記(S-2)中のR13及びR14は、それぞれ独立に、H、又はメトキシであることがより好ましい。
上記一般式(S-1)又は(S-2)中、R15及びR16は、それぞれ独立に、H、又は-OHであることが好ましい。
上記一般式(S-2)中、R17及びR18は、H、-OH、メチルアミノ、ジメチルアミノ、オキシム、又は下記式(l1)で表される基であることが好ましく、H、メチルアミノ、又は式(l1)で表される基であることがより好ましい。

上記式(l1)中の活性エステルを含む基は、活性エステル化剤(N-ヒドロキシコハク酸イミド(NHS)、1-ヒドロキシベンゾトリアゾール(HOBt)、1-ヒドロキシ-7-アザベンゾトリアゾール(HOAt)、ペンタフルオロフェノール、p-ニトロフェノール等)を用いて誘導することができる。
上記化合物(S-1)としては、下記一般式(S-1-1)で表される化合物(S-1-1)が好ましく例示される。また、上記化合物(S-2)としては、下記一般式(S-2-1)で表される化合物(S-2-1)が好ましく例示される。

R7はH、又は-OHであり、
R13及びR14は、それぞれ独立に、H、メトキシ、-OH、ヒドロキシメチル、メトキシカルボニル、又は下記式で表される基である。]

上記化合物(S)として、例えば以下の化合物:スタウロスポリン、7-ヒドロキシスタウトスポリン、KT5926、スタウロスポリン・アグリコン(staurosporine aglycone)、SF2370、KT5823、4’-N-ベンゾイルスタウロスポリン、PKC412、Go6976、N,N-ジメチルスタウロスポリン、NA0359、N-エトキシカルボニル-7-オキソスタウロスポリン、KT-6124、CGP42700、4’-デメチルアミノ-4’,5’-ジヒドロキシスタウロスポリン、7-オキソスタウロスポリン、CEP751、NA0346、NA0359、3’-デメトキシ-3’-ヒドロキシスタウロスポリン、KT6006、7-O-メチル-UCN01、TAN999、NA0346、NA0345、NA0344、CGP44171A、SCH47112、N,N-ジメチルスタウロスポリン、TAN1030A、レスタウルチニブ、4’-デメチルアミノ-4’-ヒドロキシスタウロスポリン、AFN941、エドテカリン、ベカテカリン、K252a、及びK252aヒドラジドからなる群より選択され化合物が挙げられる。
上記化合物(S-1-1)の具体例としては、K252a及びK252aヒドラジド(以下、「K252a-H」ともいう。)が例示される。上記化合物(S-2-1)の具体例としては、スタウロスポリンが例示される。

化合物(S)は、薬学的に許容される塩の形態であってもよい。本明細書において、「薬学的に許容される塩」とは、化合物(S)の薬理作用(抗がん活性、キナーゼ阻害活性など)を阻害しない塩を意味する。薬学的に許容される塩は、生体に投与した場合に、副作用を生じないものであることが好ましい。薬学的に許容可能な塩は、特に制限されず、例えば、アルカリ金属(ナトリウム、カリウムなど)との塩;アルカリ土類金属(マグネシウム、カルシウムなど)との塩;有機塩基(ピリジン、トリエチルアミンなど)との塩、アミンとの塩、有機酸(酢酸、ギ酸、プロピオン酸、フマル酸、マレイン酸、コハク酸、酒石酸、クエン酸、リンゴ酸、シュウ酸、安息香酸、メタンスルホン酸など)との塩、及び無機酸(塩酸、リン酸、臭化水素酸、硫酸、硝酸など)との塩等が挙げられる。
また、化合物(S)は、後述する[第2の態様]に記載の化合物(K)又は化合物(K)の薬学的に許容される塩であってもよい。
化合物(S)は、1種を単独で使用してもよく、2種以上を併用してもよい。
(薬物複合体)
本実施形態の医薬組成物は、薬学的に許容されるポリマーに前記化合物(S)が結合した薬物複合体を含んでいてもよい。なお、「薬物複合体」とは、薬理作用を有する分子(薬物)が、他の分子(例えば、ポリマー)に結合したものをいう。「薬学的に許容されるポリマー」とは、化合物(S)と薬物複合体を形成したとき、化合物(S)の薬理作用(抗がん活性、キナーゼ阻害活性など)を阻害しないポリマーを意味する。「化合物(S)の薬理作用を阻害しないポリマー」には、化合物(S)との薬物複合体が、in vitroで化合物(S)と同等程度又はそれ以上の薬理作用を示すもの、及びin vivoで化合物(S)と同等程度又はそれ以上の薬理作用を示すものが包含される。薬学的に許容されるポリマーは、生体に投与した場合に、副作用を生じないものであることが好ましい。
ポリマーとの薬物複合体とすることにより、化合物(S)の安全域を広くすることができる。また、化合物(S)の可溶性を高め、静注等も可能となる。
本実施形態の医薬組成物は、薬学的に許容されるポリマーに前記化合物(S)が結合した薬物複合体を含んでいてもよい。なお、「薬物複合体」とは、薬理作用を有する分子(薬物)が、他の分子(例えば、ポリマー)に結合したものをいう。「薬学的に許容されるポリマー」とは、化合物(S)と薬物複合体を形成したとき、化合物(S)の薬理作用(抗がん活性、キナーゼ阻害活性など)を阻害しないポリマーを意味する。「化合物(S)の薬理作用を阻害しないポリマー」には、化合物(S)との薬物複合体が、in vitroで化合物(S)と同等程度又はそれ以上の薬理作用を示すもの、及びin vivoで化合物(S)と同等程度又はそれ以上の薬理作用を示すものが包含される。薬学的に許容されるポリマーは、生体に投与した場合に、副作用を生じないものであることが好ましい。
ポリマーとの薬物複合体とすることにより、化合物(S)の安全域を広くすることができる。また、化合物(S)の可溶性を高め、静注等も可能となる。
化合物(S)と薬物複合体を形成するポリマーは、化合物(S)の薬理作用を阻害しないものであれば特に限定されない。ポリマーは、親水性ポリマーであってもよく、疎水性ポリマーであってもよく、親水性ポリマーセグメントと疎水性ポリマーセグメントとを含む共重合体であってもよい。
親水性ポリマーセグメントの具体例としては、例えば、ポリアルキレングリコール、ポリ(2-オキサゾリン)、ポリサッカライド、ポリビニルアルコール、ポリビニルピロリドン、ポリアクリルアミド、ポリメタクリルアミド、ポリアクリル酸エステル、ポリメタクリル酸エステル、ポリ(2-メタクロイルオキシエチルホスホリルコリン)、ポリ(N-(2-ヒドロキシプロピル)メタクリルアミド)(PHPMA)及びそれらの誘導体等が挙げられる。中でも、ポリアルキレングリコール、ポリ(2-オキサゾリン)等が好ましく、ポリアルキレングリコールがより好ましい。ポリアルキレングリコールとしては、ポリエチレングリコール、ポリプロピレングリコール、ポリエチレングリコール/ポリプロピレングリコールのコポリマー等が挙げられ、ポリエチレングリコール特に好ましい。
疎水性ポリマーセグメントの具体例としては、例えば、アミノ酸及び/又はその誘導体から誘導される繰り返し単位を有するポリマーが挙げられる。より具体的には、ポリアミノ酸又はその誘導体が挙げられる。ポリアミノ酸及びその誘導体としては、例えば、ポリアスパラギン酸、ポリグルタミン酸、ポリリジン、ポリ(ベンジルアスパラギン酸)、ポリ(ベンジルグルタミン酸)等が挙げられる。
また、疎水性ポリマーセグメントは、例えば、アルキル基側鎖又はアラルキル基側鎖を有するアミノ酸から誘導される繰り返し単位を含んでいてもよい。アルキル基側鎖を有するアミノ酸としては、アラニン、バリン、ロイシン、イソロイシンが挙げられる。また、アラルキル基側鎖を有するアミノ酸としては、フェニルアラニンが挙げられる。2以上のアルキル基側鎖アミノ酸及び/又はアラルキル基側鎖アミノ酸から誘導される繰り返し単位を有する場合、それらの側鎖は同一であってもよく、異なっていてもよい。疎水性ポリマーセグメントの全繰り返し単位に対するアルキル基側鎖アミノ酸又はアラルキル基側鎖アミノ酸から誘導される繰り返し単位の比率は、特に限定されず、例えば、20%以上、35%以上、40%以上、50%以上、80%以上、95%以上、99%以上、又は100%であってよい。
また、疎水性ポリマーセグメントは、例えば、アルキル基側鎖又はアラルキル基側鎖を有するアミノ酸から誘導される繰り返し単位を含んでいてもよい。アルキル基側鎖を有するアミノ酸としては、アラニン、バリン、ロイシン、イソロイシンが挙げられる。また、アラルキル基側鎖を有するアミノ酸としては、フェニルアラニンが挙げられる。2以上のアルキル基側鎖アミノ酸及び/又はアラルキル基側鎖アミノ酸から誘導される繰り返し単位を有する場合、それらの側鎖は同一であってもよく、異なっていてもよい。疎水性ポリマーセグメントの全繰り返し単位に対するアルキル基側鎖アミノ酸又はアラルキル基側鎖アミノ酸から誘導される繰り返し単位の比率は、特に限定されず、例えば、20%以上、35%以上、40%以上、50%以上、80%以上、95%以上、99%以上、又は100%であってよい。
化合物(S)と薬物複合体を形成するポリマーとしては、例えば、下記のポリマー(P)が好ましく例示される。
≪ポリマー(P)≫
ポリマー(P)は、下記一般式(I)で表される繰り返し単位(I)、及び下記一般式(II)で表される繰り返し単位(II)を有するポリマーである。
ポリマー(P)は、下記一般式(I)で表される繰り返し単位(I)、及び下記一般式(II)で表される繰り返し単位(II)を有するポリマーである。

前記一般式(I)及び(II)中、mは1又は2であり、1が好ましい。
前記一般式(I)中、Lは2価の芳香族炭化水素基又は2価の脂肪族炭化水素基を表す。
Lの2価の芳香族炭化水素基としては、フェニレン基、ベンジレン基等が挙げられる。
Lの2価の芳香族炭化水素基は、置換基を有していてもよい。該置換基としては、メチル基、エチル基、プロピル基、イソプロピル基、tert-ブチル基、ニトロ基、ハロゲン化物等が挙げられる。
Lの2価の脂肪族炭化水素基としては、エチレン基、プロピレン基、ブチレン基、ペンチレン基等が挙げられる。Lの2価の脂肪族炭化水素基は、置換基を有していてもよい。
該置換基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert-ブチル基、ハロゲン化物等が挙げられる。
中でも、Lとしては、メチレン基、エチレン基、プロピレン基、ベンジレン基が好ましく、メチレン基、エチレン基又はベンジレン基がより好ましい。Lは、抗腫瘍活性の観点からは、2価の脂肪族炭化水素基が好ましく、メチレン基、エチレン基、又はプロピレン基より好ましく、メチレン基又はエチレン基がさらに好ましい。
前記一般式(I)中、Lは2価の芳香族炭化水素基又は2価の脂肪族炭化水素基を表す。
Lの2価の芳香族炭化水素基としては、フェニレン基、ベンジレン基等が挙げられる。
Lの2価の芳香族炭化水素基は、置換基を有していてもよい。該置換基としては、メチル基、エチル基、プロピル基、イソプロピル基、tert-ブチル基、ニトロ基、ハロゲン化物等が挙げられる。
Lの2価の脂肪族炭化水素基としては、エチレン基、プロピレン基、ブチレン基、ペンチレン基等が挙げられる。Lの2価の脂肪族炭化水素基は、置換基を有していてもよい。
該置換基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert-ブチル基、ハロゲン化物等が挙げられる。
中でも、Lとしては、メチレン基、エチレン基、プロピレン基、ベンジレン基が好ましく、メチレン基、エチレン基又はベンジレン基がより好ましい。Lは、抗腫瘍活性の観点からは、2価の脂肪族炭化水素基が好ましく、メチレン基、エチレン基、又はプロピレン基より好ましく、メチレン基又はエチレン基がさらに好ましい。
前記一般式(I)中、R1は水素原子、脂肪族炭化水素基又は芳香族炭化水素基を表す。
R1の脂肪族炭化水素基としては、エチル基、プロピル基、ブチル基、ペンチル基等が挙げられる。R1の脂肪族炭化水素基は、置換基を有していてもよい。該置換基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、tert-ブチル基、ペンチル基、イソペンチル基、tert-ペンチル基、シクロヘキシル基、トリハロメチル基等が挙げられる。
R1の芳香族炭化水素基としては、フェニル基、ベンジル基、ピリジル基、ナフチル基、ヒドロキシフェニル基、メトキシフェニル基、エトキシフェニル基、キシリル基、メチルフェニル基、ニトロフェニル基、クロロフェニル基、フロオロフェニル基、ヨードフェニル基、ブロモフェニル基等が挙げられる。
なかでも、R1としては、水素原子又は脂肪族炭化水素基が好ましく、水素原子又はメチル基がより好ましく、メチル基がさらに好ましい。
R1の脂肪族炭化水素基としては、エチル基、プロピル基、ブチル基、ペンチル基等が挙げられる。R1の脂肪族炭化水素基は、置換基を有していてもよい。該置換基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、tert-ブチル基、ペンチル基、イソペンチル基、tert-ペンチル基、シクロヘキシル基、トリハロメチル基等が挙げられる。
R1の芳香族炭化水素基としては、フェニル基、ベンジル基、ピリジル基、ナフチル基、ヒドロキシフェニル基、メトキシフェニル基、エトキシフェニル基、キシリル基、メチルフェニル基、ニトロフェニル基、クロロフェニル基、フロオロフェニル基、ヨードフェニル基、ブロモフェニル基等が挙げられる。
なかでも、R1としては、水素原子又は脂肪族炭化水素基が好ましく、水素原子又はメチル基がより好ましく、メチル基がさらに好ましい。
前記一般式(II)中、XはORx、SRx又はNRx1Rx2を表す。
Rxは水素原子、脂肪族炭化水素基又は芳香族炭化水素基を表す。Rxの脂肪族炭化水素基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、tert-ブチル基、ペンチル基、イソペンチル基、tert-ペンチル基、シクロヘキシル基、トリフルオロメチル基等が挙げられる。
Rxの芳香族炭化水素基としては、フェニル基、ベンジル基、ピリジル基、ナフチル基、ヒドロキシフェニル基、メトキシフェニル基、エトキシフェニル基、キシリル基、メチルフェニル基、ニトロフェニル基、クロロフェニル基、フロオロフェニル基、ヨードフェニル基、ブロモフェニル基等が挙げられる。
Rx1及びRx2はそれぞれ独立に水素原子、脂肪族炭化水素基又は芳香族炭化水素基を表す。
Rx1及びRx2の脂肪族炭化水素基としては、メチル基、エチル基、イソプロピル基、ブチル基、イソブチル基、tert-ブチル基、ペンチル基、イソペンチル基、tert-ペンチル基、シクロヘキシル基、トリハロメチル基挙げられる。
Rx1及びRx2の芳香族炭化水素基としては、フェニル基、ベンジル基、ピリジル基、ナフチル基、ヒドロキシフェニル基、メトキシフェニル基、エトキシフェニル基、キシリル基、メチルフェニル基、ヒトロフェニル基、クロロフェニル基、フロオロフェニル基、ヨードフェニル基、ブロモフェニル基等が挙げられる。
中でも、XとしてはORxが好ましく、OH(ヒドロキシ基)がより好ましい。
Rxは水素原子、脂肪族炭化水素基又は芳香族炭化水素基を表す。Rxの脂肪族炭化水素基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、tert-ブチル基、ペンチル基、イソペンチル基、tert-ペンチル基、シクロヘキシル基、トリフルオロメチル基等が挙げられる。
Rxの芳香族炭化水素基としては、フェニル基、ベンジル基、ピリジル基、ナフチル基、ヒドロキシフェニル基、メトキシフェニル基、エトキシフェニル基、キシリル基、メチルフェニル基、ニトロフェニル基、クロロフェニル基、フロオロフェニル基、ヨードフェニル基、ブロモフェニル基等が挙げられる。
Rx1及びRx2はそれぞれ独立に水素原子、脂肪族炭化水素基又は芳香族炭化水素基を表す。
Rx1及びRx2の脂肪族炭化水素基としては、メチル基、エチル基、イソプロピル基、ブチル基、イソブチル基、tert-ブチル基、ペンチル基、イソペンチル基、tert-ペンチル基、シクロヘキシル基、トリハロメチル基挙げられる。
Rx1及びRx2の芳香族炭化水素基としては、フェニル基、ベンジル基、ピリジル基、ナフチル基、ヒドロキシフェニル基、メトキシフェニル基、エトキシフェニル基、キシリル基、メチルフェニル基、ヒトロフェニル基、クロロフェニル基、フロオロフェニル基、ヨードフェニル基、ブロモフェニル基等が挙げられる。
中でも、XとしてはORxが好ましく、OH(ヒドロキシ基)がより好ましい。
ポリマー(P)は、前記繰り返し単位(I)及び(II)以外の他の繰り返し単位(以下、「繰り返し単位(III)」という場合がある)を有していてもよい。
繰り返し単位(III)としては、親水性の繰り返し単位が好ましく、例えば、ポリエチレングリコールから誘導される繰り返し単位、ポリ(エチルエチレンホスフェート)から誘導される繰り返し単位、ポリビニルアルコールから誘導される繰り返し単位、ポリビニルピロリドンから誘導される繰り返し単位、ポリ(オキサゾリン)から誘導される繰り返し単位、ポリ(N-(2-ヒドロキシプロピル)メタクリルアミド)(PHPMA)から誘導される繰り返し単位等が挙げられる。中でも、繰り返し単位(III)としては、ポリエチレングリコールから誘導される繰り返し単位が好ましい。
繰り返し単位(III)としては、親水性の繰り返し単位が好ましく、例えば、ポリエチレングリコールから誘導される繰り返し単位、ポリ(エチルエチレンホスフェート)から誘導される繰り返し単位、ポリビニルアルコールから誘導される繰り返し単位、ポリビニルピロリドンから誘導される繰り返し単位、ポリ(オキサゾリン)から誘導される繰り返し単位、ポリ(N-(2-ヒドロキシプロピル)メタクリルアミド)(PHPMA)から誘導される繰り返し単位等が挙げられる。中でも、繰り返し単位(III)としては、ポリエチレングリコールから誘導される繰り返し単位が好ましい。
ポリマー(P)において、繰り返し単位(I)~(III)の含有量は特に限定されない。
繰り返し単位(I)の含有量は、ポリマー(P)を構成する全繰り返し単位の合計(100モル%)に対し、5~100モル%が好ましく、10~80モル%がより好ましく、20~50モル%が更に好ましい。
繰り返し単位(II)の含有量は、ポリマー(P)を構成する全繰り返し単位の合計(100モル%)に対し、0~80モル%が好ましく、10~60モル%がより好ましく、20~40モル%が更に好ましい。
繰り返し単位(III)の含有量は、ポリマー(P)を構成する全繰り返し単位の合計(100モル%)に対し、0~95モル%が好ましく、20~90モル%がより好ましく、50~80モル%が更に好ましい。
繰り返し単位(I)の含有量は、ポリマー(P)を構成する全繰り返し単位の合計(100モル%)に対し、5~100モル%が好ましく、10~80モル%がより好ましく、20~50モル%が更に好ましい。
繰り返し単位(II)の含有量は、ポリマー(P)を構成する全繰り返し単位の合計(100モル%)に対し、0~80モル%が好ましく、10~60モル%がより好ましく、20~40モル%が更に好ましい。
繰り返し単位(III)の含有量は、ポリマー(P)を構成する全繰り返し単位の合計(100モル%)に対し、0~95モル%が好ましく、20~90モル%がより好ましく、50~80モル%が更に好ましい。
ポリマー(P)の分子量は、2000~1000000Dが好ましく、5000~100000Dがより好ましく、10000~40000Dがさらに好ましい。
〔ポリマー(P)の製造方法(1)〕
ポリマー(P)の製造方法(以下、「製造方法(1)」という場合がある)は、下記一般式(II’)で表される繰り返し単位(II’)を有するポリマー(P1)と下記一般式(1a)で表される化合物(1a)とを反応させて、下記一般式(I’)で表される繰り返し単位及び前記繰り返し単位(II’)を有するポリマー(P2)を得る工程(1)と、前記ポリマー(P2)を弱酸性条件下で加水分解して、下記一般式(I)で表される繰り返し単位(I)及び下記一般式(II-1)で表される繰り返し単位(II-1)を有するポリマー(P)を得る工程(2)と、を含む。
ポリマー(P)の製造方法(以下、「製造方法(1)」という場合がある)は、下記一般式(II’)で表される繰り返し単位(II’)を有するポリマー(P1)と下記一般式(1a)で表される化合物(1a)とを反応させて、下記一般式(I’)で表される繰り返し単位及び前記繰り返し単位(II’)を有するポリマー(P2)を得る工程(1)と、前記ポリマー(P2)を弱酸性条件下で加水分解して、下記一般式(I)で表される繰り返し単位(I)及び下記一般式(II-1)で表される繰り返し単位(II-1)を有するポリマー(P)を得る工程(2)と、を含む。

前記式一般式(I’)、(II’)、(I)及び(II-1)中、m、L、R1及びRxは前記一般式(I)及び(II)中のm、L、R1及びRxと同様である。
前記一般式(1a)及び(I’)中、Ra11及びRa12はそれぞれ独立にメチル基若しくはエチル基を表す、又はRa11及びRa12は相互に結合してエチレン基若しくはプロピレン基を表す。Ra11及びRa12が相互に結合してエチレン基若しくはプロピレン基を表す場合、化合物(1a)は環状アセタール又は環状ケタールとなる。
前記一般式(1a)中、n1及びn2はそれぞれ独立に0又は1であり、1が好ましい。
前記一般式(1a)及び(I’)中、Ra11及びRa12はそれぞれ独立にメチル基若しくはエチル基を表す、又はRa11及びRa12は相互に結合してエチレン基若しくはプロピレン基を表す。Ra11及びRa12が相互に結合してエチレン基若しくはプロピレン基を表す場合、化合物(1a)は環状アセタール又は環状ケタールとなる。
前記一般式(1a)中、n1及びn2はそれぞれ独立に0又は1であり、1が好ましい。
・工程(1)
製造方法(1)の工程(1)は、ポリマー(P1)と化合物(1a)とのアミノリシス反応である。工程(1)により、ポリマー(P1)の側鎖に化合物(1a)のアセタール構造又はケタール構造が導入される。
工程(1)の反応温度は、ポリマー(P1)の側鎖に化合物(1a)のアセタール構造又はケタール構造が導入される条件であれば特に限定されないが、通常4℃~100℃であり、室温~40℃が好ましい。
工程(1)の反応時間は、ポリマー(P1)の側鎖に化合物(1a)のアセタール構造又はケタール構造が導入される条件であれば特に限定されず、反応時間、化合物(1a)の種類や量によって選択できるが、通常4時間~5日間である。
製造方法(1)の工程(1)は、ポリマー(P1)と化合物(1a)とのアミノリシス反応である。工程(1)により、ポリマー(P1)の側鎖に化合物(1a)のアセタール構造又はケタール構造が導入される。
工程(1)の反応温度は、ポリマー(P1)の側鎖に化合物(1a)のアセタール構造又はケタール構造が導入される条件であれば特に限定されないが、通常4℃~100℃であり、室温~40℃が好ましい。
工程(1)の反応時間は、ポリマー(P1)の側鎖に化合物(1a)のアセタール構造又はケタール構造が導入される条件であれば特に限定されず、反応時間、化合物(1a)の種類や量によって選択できるが、通常4時間~5日間である。
・工程(2)
製造方法(1)の工程(2)において、ポリマー(P2)を中性条件下又は弱酸性条件下で加水分解し、ポリマー(P2)の繰り返し単位(I’)のアセタール構造をアルデヒド、又はケタール構造をケトンに変換する。
加水分解は、ポリマー(P2)の繰り返し単位(I’)のアセタール構造をアルデヒド、又はケタール構造をケトンに変換できる条件であれば特に限定されない。例えば、(i)0.1N塩酸で30分程度処理する方法、(ii)アセトン及びインジウム(III)トリフルオロメタンスルホネート(触媒)の存在下で処理する方法、(iii)30℃の水中で触媒量のテトラキス(3,5-トリフルオロメチルフェニル)ホウ酸ナトリウムを用いる方法、(iv)室温でウェットニトロメタン中、1~5モル%のEr(OTf)3を用いる方法、(v)ほぼ中性のpH条件下、室温でウェットニトロメタン中、触媒量のセリウム(III)トリフレートを用いる方法等、公知の方法が挙げられる。
製造方法(1)の工程(2)において、ポリマー(P2)を中性条件下又は弱酸性条件下で加水分解し、ポリマー(P2)の繰り返し単位(I’)のアセタール構造をアルデヒド、又はケタール構造をケトンに変換する。
加水分解は、ポリマー(P2)の繰り返し単位(I’)のアセタール構造をアルデヒド、又はケタール構造をケトンに変換できる条件であれば特に限定されない。例えば、(i)0.1N塩酸で30分程度処理する方法、(ii)アセトン及びインジウム(III)トリフルオロメタンスルホネート(触媒)の存在下で処理する方法、(iii)30℃の水中で触媒量のテトラキス(3,5-トリフルオロメチルフェニル)ホウ酸ナトリウムを用いる方法、(iv)室温でウェットニトロメタン中、1~5モル%のEr(OTf)3を用いる方法、(v)ほぼ中性のpH条件下、室温でウェットニトロメタン中、触媒量のセリウム(III)トリフレートを用いる方法等、公知の方法が挙げられる。
〔ポリマー(P)の製造方法(2)〕
ポリマー(P)の製造方法(以下、「製造方法(2)」という場合がある)は、下記一般式(II’)で表される繰り返し単位(II’)を有するポリマー(P1)と下記一般式(1a)で表される化合物(1a)とを反応させて、下記一般式(I’)で表される繰り返し単位及び前記繰り返し単位(II’)を有するポリマー(P2)を得る工程(1)と、
前記ポリマー(P2)をアルカリ条件下の加水分解、エステル交換反応、アミノリシス、並びにアルカリ条件下の加水分解及びアミドカップリングからなる群より選ばれる少なくとも1種の処理に付し、下記一般式(I’)で表される繰り返し単位(I’)及び下記一般式(II’)で表される繰り返し単位(II’)を有するポリマー(P3)を得る工程(2a)と、
前記ポリマー(P3)を中性条件下又は弱酸性条件下で加水分解して、下記一般式(I)で表される繰り返し単位(I)及び下記一般式(II)で表される繰り返し単位(II)を有するポリマー(P)を得る工程(2b)と、
を含む。
ポリマー(P)の製造方法(以下、「製造方法(2)」という場合がある)は、下記一般式(II’)で表される繰り返し単位(II’)を有するポリマー(P1)と下記一般式(1a)で表される化合物(1a)とを反応させて、下記一般式(I’)で表される繰り返し単位及び前記繰り返し単位(II’)を有するポリマー(P2)を得る工程(1)と、
前記ポリマー(P2)をアルカリ条件下の加水分解、エステル交換反応、アミノリシス、並びにアルカリ条件下の加水分解及びアミドカップリングからなる群より選ばれる少なくとも1種の処理に付し、下記一般式(I’)で表される繰り返し単位(I’)及び下記一般式(II’)で表される繰り返し単位(II’)を有するポリマー(P3)を得る工程(2a)と、
前記ポリマー(P3)を中性条件下又は弱酸性条件下で加水分解して、下記一般式(I)で表される繰り返し単位(I)及び下記一般式(II)で表される繰り返し単位(II)を有するポリマー(P)を得る工程(2b)と、
を含む。

前記一般式(I’)、(II’)、(I)及び(II)中、m、L,X、Rx、Rx1及びRx2は、前記一般式(I)及び(II)中のm、L,X、Rx、Rx1及びRx2と同様である。
前記一般式(1a)及び(I’)中、R1、Ra11、Ra12は前記と同様である。
前記一般式(1a)及び(I’)中、R1、Ra11、Ra12は前記と同様である。
・工程(1)
製造方法(2)の工程(1)は、製造方法(1)の工程(1)と同様である。
製造方法(2)の工程(1)は、製造方法(1)の工程(1)と同様である。
・工程(2a)
製造方法(2)の工程(2a)では、ポリマー(P2)を所定の処理に付すことにより、繰り返し単位(I’)がアセタール構造で保護された状態で、繰り返し単位(II’)の側鎖に所望の官能基を導入することができる。
アルカリ条件下の加水分解は、例えば、0.5NのNaOH溶液とDMSOとの混合物(体積比:50/50)中、室温で30分処理する方法、DMSO中トリエチルアミンで室温にて1時間処理する方法、DMSO中ジイソプロピルエチルアミンで室温にて1時間処理する方法等が挙げられる。アルカリ条件下の加水分解により得られるカルボン酸残基は、後述するミセルのコア中のプロトンを引き寄せ、ヒドラゾン結合の加水分解を容易にし、低pH条件下において生体材料の放出を可能とする。
アミノリシスは、例えば、エチレンジアミン又はジアミノプロパンによりエステルを開裂してアミノ官能基を導入することができる。アミノ基導入により、蛍光色素と結合させることができる。また、他のカルボン酸基を有する画像診断剤と、公知のアミノカップリングに付すこともできる。アセタール構造及びケタール構造は、このようなアミノ基導入条件下では安定なので、ポリマーの多官能ナノキャリアデザインに供することができる。
アルカリ条件下の加水分解及びアミドカップリングは、例えば、アルカリ条件下の加水分解によりエステル残基を処理後、生成したカルボン酸をエステル交換反応もしくは公知のカップリング剤を用いたアミドカップリングに付すことができる。ヒドロキシ/アミン官能基による適切な構造モチーフにより、ポリマーの親水性/疎水性のバランスを所望のものとすることができ、極性又は非極性溶媒中での自己組織化に寄与する。
製造方法(2)の工程(2a)では、ポリマー(P2)を所定の処理に付すことにより、繰り返し単位(I’)がアセタール構造で保護された状態で、繰り返し単位(II’)の側鎖に所望の官能基を導入することができる。
アルカリ条件下の加水分解は、例えば、0.5NのNaOH溶液とDMSOとの混合物(体積比:50/50)中、室温で30分処理する方法、DMSO中トリエチルアミンで室温にて1時間処理する方法、DMSO中ジイソプロピルエチルアミンで室温にて1時間処理する方法等が挙げられる。アルカリ条件下の加水分解により得られるカルボン酸残基は、後述するミセルのコア中のプロトンを引き寄せ、ヒドラゾン結合の加水分解を容易にし、低pH条件下において生体材料の放出を可能とする。
アミノリシスは、例えば、エチレンジアミン又はジアミノプロパンによりエステルを開裂してアミノ官能基を導入することができる。アミノ基導入により、蛍光色素と結合させることができる。また、他のカルボン酸基を有する画像診断剤と、公知のアミノカップリングに付すこともできる。アセタール構造及びケタール構造は、このようなアミノ基導入条件下では安定なので、ポリマーの多官能ナノキャリアデザインに供することができる。
アルカリ条件下の加水分解及びアミドカップリングは、例えば、アルカリ条件下の加水分解によりエステル残基を処理後、生成したカルボン酸をエステル交換反応もしくは公知のカップリング剤を用いたアミドカップリングに付すことができる。ヒドロキシ/アミン官能基による適切な構造モチーフにより、ポリマーの親水性/疎水性のバランスを所望のものとすることができ、極性又は非極性溶媒中での自己組織化に寄与する。
・工程(2b)
製造方法(2)の工程(2b)において、ポリマー(P3)を弱酸性条件下で加水分解し、ポリマー(P3)の繰り返し単位(I’)のアセタール構造をアルデヒドに変換する。加水分解の条件は、製造方法(1)の工程(2)と同様である。
製造方法(2)の工程(2b)において、ポリマー(P3)を弱酸性条件下で加水分解し、ポリマー(P3)の繰り返し単位(I’)のアセタール構造をアルデヒドに変換する。加水分解の条件は、製造方法(1)の工程(2)と同様である。
≪ポリマー(P)と化合物(S)との薬物複合体≫
ポリマー(P)と化合物(S)との結合は、化合物(S)にアルデヒド基又はケトン基とシッフ塩基を形成し得る窒素原子含有基(以下、「シッフ塩基形成基」という場合がある)がある場合には、当該シッフ塩基形成基と、ポリマー(P)の繰り返し単位(I)に含まれるアルデヒド基又はケトン基とを反応させることにより行うことができる。そのようなシッフ塩基としては、例えば、アミノ基、イミノ基、ヒドラジド基等が挙げられる。
また、化合物(S)がシッフ塩基形成基を有しない場合には、シッフ塩基形成基を化合物(S)に導入すればよい。シッフ塩基形成基の導入は、公知の方法により行うことができる。
ポリマー(P)と化合物(S)との結合は、化合物(S)にアルデヒド基又はケトン基とシッフ塩基を形成し得る窒素原子含有基(以下、「シッフ塩基形成基」という場合がある)がある場合には、当該シッフ塩基形成基と、ポリマー(P)の繰り返し単位(I)に含まれるアルデヒド基又はケトン基とを反応させることにより行うことができる。そのようなシッフ塩基としては、例えば、アミノ基、イミノ基、ヒドラジド基等が挙げられる。
また、化合物(S)がシッフ塩基形成基を有しない場合には、シッフ塩基形成基を化合物(S)に導入すればよい。シッフ塩基形成基の導入は、公知の方法により行うことができる。
例えば、K252aにはシッフ塩基形成基が存在しないため、後述する実施例で示すように、ヒドラジド基を導入してK252a-Hとすることにより、ポリマー(P)に結合させることができる。ポリマー(P)とK252aとの薬物複合体としては、後述する[第2の態様]に記載の繰り返し単位(ka-1)及び繰り返し単位(II)を有するポリマーが挙げられる。
ポリマー(P)と化合物(S)との薬物複合体は、下記一般式(Ia)で表される繰り返し単位(Ia)、及び下記一般式(II)で表される繰り返し単位(II)を有するポリマーであることが好ましい。

前記一般式(Ia)及び(II)中、m、L、R1、X、Rx、Rx1及びRx2は、前記一般式(I)及び(II)中のm、L、R1、X、Rx、Rx1及びRx2と同様である。
前記一般式(Ia)中、BMは化合物(S)から誘導される基を表す。化合物(S)から誘導される基としては、K252a-Hから誘導される基が好適に例示される。
前記一般式(Ia)中、BMは化合物(S)から誘導される基を表す。化合物(S)から誘導される基としては、K252a-Hから誘導される基が好適に例示される。
ポリマー(P)では、導入するアルデヒド基又はケトン基の量(繰り返し単位(I)の量)を制御することができ、アルデヒド基又はケトン基に結合させる薬物の量も制御することができる。そのため、化合物(S)の投与量を適切に制御することができる。
また、ポリマー(P)においては、繰り返し単位(I)に導入するアルデヒド基又はケトン基(上記一般式(I)中の-L-C(=O)-R1で表される基)を、芳香族アルデヒド基(一般式(I)中のLが2価の芳香族炭化水素基、R1が水素原子)、脂肪族アルデヒド基(一般式(I)中のLが2価の脂肪族炭化水素基、R1が水素原子)、芳香族ケトン基(一般式(I)中のLが2価の芳香族炭化水素基、R1が脂肪族炭化水素基又は芳香族炭化水素基、;R1は好ましくは脂肪族炭化水素基、より好ましくはメチル基)、脂肪族ケトン基(一般式(I)中のLが2価の脂肪族炭化水素基、R1が脂肪族炭化水素基又は芳香族炭化水素基;R1は好ましくは脂肪族炭化水素基、より好ましくはメチル基)から選択することができる。
芳香族アルデヒド基又は芳香族ケトン基とシッフ塩基との結合は、脂肪族アルデヒド基又は脂肪族ケトン基とシッフ塩基との結合よりも安定であるため、ポリマー(P)の繰り返し単位(I)に芳香族アルデヒド基を導入した場合には、ポリマー(P)の繰り返し単位(I)に化合物(S)がより安定に保持される。そのため、疾患の状態や薬物の種類により、ポリマー(P)の繰り返し単位(I)に導入するアルデヒド基又はケトン基の種類を選択することにより、薬物の徐放性を制御することができる。さらに、ポリマー(P)と化合物(S)との薬物複合体では、化合物(S)がポリマー(P)に保持されている間、化合物(S)は安定に維持され、毒性も緩和されるため、副作用を軽減して治療効果を高めることができる。
ポリマー(P)と化合物(S)との薬物複合体においては、生体内に投与したときの抗腫瘍効果の観点から、ポリマー(P)の繰り返し単位(I)に脂肪族ケトン基を導入することが好ましい。すなわち、上記一般式(Ia)中、Lは2価の脂肪族炭化水素基(好ましくはメチレン基又はエチレン基)であることが好ましく、R1は脂肪族炭化水素基(好ましくはメチル基)であることが好ましい。
また、ポリマー(P)においては、繰り返し単位(I)に導入するアルデヒド基又はケトン基(上記一般式(I)中の-L-C(=O)-R1で表される基)を、芳香族アルデヒド基(一般式(I)中のLが2価の芳香族炭化水素基、R1が水素原子)、脂肪族アルデヒド基(一般式(I)中のLが2価の脂肪族炭化水素基、R1が水素原子)、芳香族ケトン基(一般式(I)中のLが2価の芳香族炭化水素基、R1が脂肪族炭化水素基又は芳香族炭化水素基、;R1は好ましくは脂肪族炭化水素基、より好ましくはメチル基)、脂肪族ケトン基(一般式(I)中のLが2価の脂肪族炭化水素基、R1が脂肪族炭化水素基又は芳香族炭化水素基;R1は好ましくは脂肪族炭化水素基、より好ましくはメチル基)から選択することができる。
芳香族アルデヒド基又は芳香族ケトン基とシッフ塩基との結合は、脂肪族アルデヒド基又は脂肪族ケトン基とシッフ塩基との結合よりも安定であるため、ポリマー(P)の繰り返し単位(I)に芳香族アルデヒド基を導入した場合には、ポリマー(P)の繰り返し単位(I)に化合物(S)がより安定に保持される。そのため、疾患の状態や薬物の種類により、ポリマー(P)の繰り返し単位(I)に導入するアルデヒド基又はケトン基の種類を選択することにより、薬物の徐放性を制御することができる。さらに、ポリマー(P)と化合物(S)との薬物複合体では、化合物(S)がポリマー(P)に保持されている間、化合物(S)は安定に維持され、毒性も緩和されるため、副作用を軽減して治療効果を高めることができる。
ポリマー(P)と化合物(S)との薬物複合体においては、生体内に投与したときの抗腫瘍効果の観点から、ポリマー(P)の繰り返し単位(I)に脂肪族ケトン基を導入することが好ましい。すなわち、上記一般式(Ia)中、Lは2価の脂肪族炭化水素基(好ましくはメチレン基又はエチレン基)であることが好ましく、R1は脂肪族炭化水素基(好ましくはメチル基)であることが好ましい。
また、公知のリンカー試薬を用いて、化合物(S)とポリマー(P)とを結合させてもよい。リンカー試薬がシッフ塩基形成基を含む場合には、当該シッフ塩基形成基を用いて、ポリマー(P)に化合物(S)を結合させることができる。
一方、リンカー試薬が、シッフ塩基形成基を有しない場合には、化合物(S)にリンカーを結合させた化合物に、シッフ塩基形成基を導入してもよい。利用可能なリンカー試薬は、特に限定されないが、例えば、下記式(L1)で表されるグルタチオン(GSH)/グルタチオン-S-トランスフェラーゼ(GST)感応性リンカー試薬(リンカー試薬(L1);国際公開第2012/039499号、Huang CH et al., ChemMedChem. 2017 Jan 5;12(1):19-22.; Zhang J et al., J Am Chem Soc. 2011 Sep 7;133(35):14109-19.)が挙げられる。

例えば、下記一般式(SL1)で表される化合物(SL1)は、化合物(S-2-1)に前記リンカー試薬(L1)を反応させて得た化合物である。下記化合物(SL1)を、無水ヒドラジン又はヒドラジン水和物の存在下、無溶媒で還流することにより、シッフ塩基形成基を含む下記化合物(SL1-H)を得ることができる。あるいは、化合物(SL1-H)は、例えば、化合物(SL1)をメタノール、エタノール、プロパノール、イソプロパノール、ブタノール、tert-ブタノール、N,N-ジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、テトラヒドロフラン(THF)、ジオキサン、アセトニトリル、トルエン等の溶媒に溶解し、無水ヒドラジンに添加して反応させることによっても製造することができる。反応温度及び反応時間は、特に限定されないが、例えば室温~50℃で30分~15時間が例示される。

上記化合物(SL1-H)をポリマー(P)に結合させた薬物複合体は、下記一般式(Ia-S)で表される繰り返し単位(Ia-S)、及び下記一般式(II)で表される繰り返し単位(II)を有する。

上記繰り返し単位(Ia-S)及び繰り返し単位(II)を有する薬物複合体は、pH感応性の結合(I)及びGSH/GST感応性の結合(II)を含む。そのため、上記薬物複合体を生体内に投与すると、まず、低pH環境の腫瘍組織周辺で結合(I)が切断し、化合物(SL1-H)が放出される。さらに、化合物(SL1-H)が腫瘍細胞に取り込まれ、GSH/GSTの作用により、結合(II)が切断し、化合物(S-2-1)が放出される。そのため、生体内においけるポリマー(P)からの化合物(S)の放出を、2段階で制御することができる。
≪他のポリマーとの薬物複合体≫
化合物(S)は、上記ポリマー(P)以外のポリマーとの薬物複合体としてもよい。その場合、化合物(S)とポリマーとの結合は、化合物(S)及びポリマーが含む官能基に応じて、公知の方法を用いて行うことができる。例えば、化合物(S)をポリマーに直接結合させてもよく、化合物(S)にポリマーに結合可能な官能基を導入し、前記官能基を介してポリマーに結合させてもよい。また、公知のリンカー試薬を用いてもよい。リンカー試薬は、特に限定されないが、例えば、上記リンカー試薬(L1)が挙げられる。また、化合物(S)にリンカーを導入した後、活性エステル化剤により活性エステルを導入し、ヒドロキシ基又はアミノ基を有するポリマーに結合させてもよい。また、逆に、ポリマーに活性エステルを導入してもよい。
化合物(S)は、上記ポリマー(P)以外のポリマーとの薬物複合体としてもよい。その場合、化合物(S)とポリマーとの結合は、化合物(S)及びポリマーが含む官能基に応じて、公知の方法を用いて行うことができる。例えば、化合物(S)をポリマーに直接結合させてもよく、化合物(S)にポリマーに結合可能な官能基を導入し、前記官能基を介してポリマーに結合させてもよい。また、公知のリンカー試薬を用いてもよい。リンカー試薬は、特に限定されないが、例えば、上記リンカー試薬(L1)が挙げられる。また、化合物(S)にリンカーを導入した後、活性エステル化剤により活性エステルを導入し、ヒドロキシ基又はアミノ基を有するポリマーに結合させてもよい。また、逆に、ポリマーに活性エステルを導入してもよい。
例えば、化合物(S)にリンカー試薬(L1)を結合させて、上記化合物(SL1)を得た後、化合物(SL1)を脱メチル化し、活性エステル化剤(N-ヒドロキシコハク酸イミド(NHS)、1-ヒドロキシベンゾトリアゾール(HOBt)、1-ヒドロキシ-7-アザベンゾトリアゾール(HOAt)、ペンタフルオロフェノール、p-ニトロフェノール等)を反応させて、下記式(SL2)で表される活性エステルを含む化合物(SL2)を得てもよい。

上記活性エステルを含む化合物(SL2)を、アミノ基又はヒドロキシ基を有する任意のポリマーと反応させることにより、化合物(S-2-1)と当該ポリマーとの薬物複合体を得ることができる。

≪薬物複合体のミセル≫
本実施形態の医薬組成物に含有される化合物(S)とポリマーとの薬物複合体は、ミセルを形成していてもよい。薬物複合体がミセルを形成する場合、薬物複合体が含むポリマーは、親水性ポリマーセグメントと疎水性ポリマーセグメントとを有することが好ましい。化合物(S)とポリマーとの薬物複合体のミセルは、公知の手法により調製することができる。例えば、薬物複合体を親油性又は親水性の溶媒に溶解又は懸濁し、当該溶解液又は懸濁液を親水性又は親油性の溶媒に滴下して撹拌することにより、薬物複合体のミセルを調製することができる。
本実施形態の医薬組成物に含有される化合物(S)とポリマーとの薬物複合体は、ミセルを形成していてもよい。薬物複合体がミセルを形成する場合、薬物複合体が含むポリマーは、親水性ポリマーセグメントと疎水性ポリマーセグメントとを有することが好ましい。化合物(S)とポリマーとの薬物複合体のミセルは、公知の手法により調製することができる。例えば、薬物複合体を親油性又は親水性の溶媒に溶解又は懸濁し、当該溶解液又は懸濁液を親水性又は親油性の溶媒に滴下して撹拌することにより、薬物複合体のミセルを調製することができる。
(任意成分)
本実施形態の医薬組成物は、化合物(S)に加えて、他の成分を含んでいてもよい。他の成分としては、例えば、薬学的に許容される担体が挙げられる。「薬学的に許容される担体」とは、有効成分の生理活性を阻害せず、且つ、その投与対象に対して実質的な毒性を示さない担体を意味する。「実質的な毒性を示さない」とは、その成分が通常使用される投与量において、投与対象に対して毒性を示さないことを意味する。薬学的に許容される担体としては、医薬分野において常用されるものを特に制限なく使用することができ、例えば、水、生理食塩水、リン酸緩衝液、DMSO、ジメチルアセトアミド、エタノール、グリセロール、ミネラルオイル等を挙げることができる。
また、他の成分としては、溶剤、溶解補助剤、懸濁化剤、等張化剤、緩衝剤、pH調整剤、賦形剤、安定剤、抗酸化剤、浸透圧調整剤、防腐剤、着色剤、香料等が挙げられる。
また、本実施形態の医薬組成物は、化合物(S)以外の抗がん剤、又は抗がん剤以外の活性成分を含んでいてもよい。抗がん剤以外の活性成分としては、抗炎症剤、鎮痛剤、解熱剤、消炎剤、血行促進剤、刺激緩和剤、抗生物質、生薬等が挙げられるが、これらに限定されない。
本実施形態の医薬組成物は、化合物(S)に加えて、他の成分を含んでいてもよい。他の成分としては、例えば、薬学的に許容される担体が挙げられる。「薬学的に許容される担体」とは、有効成分の生理活性を阻害せず、且つ、その投与対象に対して実質的な毒性を示さない担体を意味する。「実質的な毒性を示さない」とは、その成分が通常使用される投与量において、投与対象に対して毒性を示さないことを意味する。薬学的に許容される担体としては、医薬分野において常用されるものを特に制限なく使用することができ、例えば、水、生理食塩水、リン酸緩衝液、DMSO、ジメチルアセトアミド、エタノール、グリセロール、ミネラルオイル等を挙げることができる。
また、他の成分としては、溶剤、溶解補助剤、懸濁化剤、等張化剤、緩衝剤、pH調整剤、賦形剤、安定剤、抗酸化剤、浸透圧調整剤、防腐剤、着色剤、香料等が挙げられる。
また、本実施形態の医薬組成物は、化合物(S)以外の抗がん剤、又は抗がん剤以外の活性成分を含んでいてもよい。抗がん剤以外の活性成分としては、抗炎症剤、鎮痛剤、解熱剤、消炎剤、血行促進剤、刺激緩和剤、抗生物質、生薬等が挙げられるが、これらに限定されない。
化合物(S)は、リポソームやミセル等の公知のドラッグデリバリー用粒子に封入又は担持されたのもであってもよい。ドラッグデリバリー用粒子への封入や担持は、公知の方法により行うことができる。本実施形態の医薬組成物は、そのようなドラッグデリバリー用粒子もまた任意成分として含み得る。
(抗がん剤耐性のがん)
本実施形態の医薬組成物は、抗がん剤耐性のがんを治療又は予防するために用いることができる。「抗がん剤耐性のがん」とは、1種以上の抗がん剤に対して耐性を示すがんを意味する。「抗がん剤」とは、がんの治療又は予防のために用いられる化合物であって、少なくとも1種のがん細胞の増殖を抑制する作用を有する化合物を意味する。がんが耐性を有する抗がん剤は、好ましくは、当該がんの治療用として開発された抗がん剤である。より好ましくは、抗がん剤は、当該がんの治療用として承認された抗がん剤である。例えば、当該がんの標準治療に用いられている抗がん剤である。
そのような標準治療に用いられる抗がん剤に対する耐性を獲得したがんは、有効な薬物療法を行なうことができず、難治性である。しかしながら、後述する実施例に示すように、化合物(S)は、抗がん剤耐性のがんに対し、高い抗腫瘍活性を示す。そのため、本実施形態の医薬組成物は、抗がん剤耐性のがんを治療又は予防するために有効に用いることができる。例えば、がん患者に対して既存の抗がん剤で治療を行い、当該抗がん剤の効果が得られない場合、又は当該抗がん剤の効果が得られなくなった場合に、本実施形態の医薬組成物を好適に用いることができる。本実施形態の医薬組成物は、セカンドライン以降のがん治療に用いる抗がん剤として好適に利用することができる。本実施形態の医薬組成物は、ファーストラインのがん治療に用いてもよい。
本実施形態の医薬組成物は、抗がん剤耐性のがんを治療又は予防するために用いることができる。「抗がん剤耐性のがん」とは、1種以上の抗がん剤に対して耐性を示すがんを意味する。「抗がん剤」とは、がんの治療又は予防のために用いられる化合物であって、少なくとも1種のがん細胞の増殖を抑制する作用を有する化合物を意味する。がんが耐性を有する抗がん剤は、好ましくは、当該がんの治療用として開発された抗がん剤である。より好ましくは、抗がん剤は、当該がんの治療用として承認された抗がん剤である。例えば、当該がんの標準治療に用いられている抗がん剤である。
そのような標準治療に用いられる抗がん剤に対する耐性を獲得したがんは、有効な薬物療法を行なうことができず、難治性である。しかしながら、後述する実施例に示すように、化合物(S)は、抗がん剤耐性のがんに対し、高い抗腫瘍活性を示す。そのため、本実施形態の医薬組成物は、抗がん剤耐性のがんを治療又は予防するために有効に用いることができる。例えば、がん患者に対して既存の抗がん剤で治療を行い、当該抗がん剤の効果が得られない場合、又は当該抗がん剤の効果が得られなくなった場合に、本実施形態の医薬組成物を好適に用いることができる。本実施形態の医薬組成物は、セカンドライン以降のがん治療に用いる抗がん剤として好適に利用することができる。本実施形態の医薬組成物は、ファーストラインのがん治療に用いてもよい。
がんが抗がん剤耐性であるか否かは、例えば、抗がん剤非存在下でのがん細胞の増殖と比較して、抗がん剤存在下におけるがん細胞の増殖が抑制されるか否かにより判定し得る。抗がん剤非存在下のがん細胞増殖率と比較して、抗がん剤存在下のがん細胞の増殖抑制率が、例えば50%以下、好ましくは30%以下、より好ましくは20%以下、さらに好ましくは10%以下である場合に、当該がん細胞は当該抗がん剤に対して耐性であると判定し得る。あるいは、抗がん剤感受性であるがん細胞の増殖率と比較して、例えば増殖率が2倍以上、好ましくは5倍以上、より好ましくは10倍以上である場合に、当該がん細胞は当該抗がん剤に対して耐性であると判定し得る。
あるいは、がんが抗がん剤耐性であるか否かは、例えば、抗がん剤のIC50により評価してもよい。がん細胞に対する抗がん剤のIC50が、例えば、10nM以上、好ましくは20nM以上、より好ましくは30nM以上、さらに好ましくは50nM以上である場合に、当該がん細胞は当該抗がん剤に対して耐性であると判定し得る。あるいは、がん細胞に対する抗がん剤のIC50が、例えば、1μM以上、好ましくは3μM以上、より好ましくは5μM以上、さらに好ましくは10μM以上である場合に、当該がん細胞は当該抗がん剤に対して耐性であると判定し得る。
また、抗がん剤耐性のがんは、抗がん剤不応のがんであるともいえる。すなわち、がん患者に抗がん剤を投与して、当該がん患者のがんが縮小しない場合、がんが増殖する場合、又はがんの増殖抑制率が低い場合に、当該がんは当該抗がん剤にして耐性(不応)であると判定し得る。
あるいは、がんが抗がん剤耐性であるか否かは、例えば、抗がん剤のIC50により評価してもよい。がん細胞に対する抗がん剤のIC50が、例えば、10nM以上、好ましくは20nM以上、より好ましくは30nM以上、さらに好ましくは50nM以上である場合に、当該がん細胞は当該抗がん剤に対して耐性であると判定し得る。あるいは、がん細胞に対する抗がん剤のIC50が、例えば、1μM以上、好ましくは3μM以上、より好ましくは5μM以上、さらに好ましくは10μM以上である場合に、当該がん細胞は当該抗がん剤に対して耐性であると判定し得る。
また、抗がん剤耐性のがんは、抗がん剤不応のがんであるともいえる。すなわち、がん患者に抗がん剤を投与して、当該がん患者のがんが縮小しない場合、がんが増殖する場合、又はがんの増殖抑制率が低い場合に、当該がんは当該抗がん剤にして耐性(不応)であると判定し得る。
本実施形態の医薬組成物を適用するがんは、抗がん剤耐性のがんであれば、がんの種類は特に限定されない。がんとしては、例えば、肺がん、膵臓がん、頭頸部がん、中皮腫、神経芽腫、肝臓がん、悪性黒色腫、子宮がん、膀胱がん、胆道がん、食道がん、骨肉腫、精巣腫瘍、甲状腺がん、急性骨髄性白血病、脳腫瘍、前立腺がん、頭頸部扁平上皮がん、大腸がん、腎がん、卵巣がん、及び乳がん等が挙げられる。本実施形態の医薬組成物を適用するがんとしては、例えば、肺がん、膵臓がん、頭頸部がん、中皮腫を好ましく例示できる。
本実施形態の医薬組成物を適用し得るがんが耐性を示す抗がん剤の具体例としては、ゲフィチニブ、アファチニブ、オシメルチニブ、ダサチニブ、エルロチニブ、ゲムシタビン、シスプラチン、ペメトレキセド、及びミドスタウリン等が挙げられる。
本実施形態の医薬組成物を適用し得る抗がん剤耐性のがんの具体例を以下に例示する。
(1)ゲフィチニブ、アファチニブ、オシメルチニブ、ダサチニブ、エルロチニブ、ゲムシタビン、シスプラチン、ペメトレキセド、及びミドスタウリンからなる群より選択される少なくとも1種の抗がん剤に耐性を示すがん。
(2)ゲフィチニブ、アフィチニブ、オシメルチニブ、及びダサチニブからなる群より選択される少なくとも1種の抗がん剤に耐性を示す肺がん(特に非小細胞肺がん)。中でも、オシメルチニブに耐性を示す肺がん(特に非小細胞肺がん)。より好ましくは、ゲフィチニブ、アフィチニブ、及びオシメルチニブに耐性を示す肺がん。
(3)ゲムシタビン、及びエルロチニブからなる群より選択される少なくとも1種の抗がん剤に耐性を示す膵臓がん。より好ましくは、ゲムシタビン、及びエルロチニブに耐性を示す膵臓がん。
(4)シスプラチン、エルロチニブ、ゲフィチニブ、及びミドスタウリンからなる群より選択される少なくとも1種の抗がん剤に耐性を示す頭頸部がん。より好ましくは、シスプラチン及びエルロチニブに耐性を示す頭頚部がん、またはエルロチニブ、ゲフィチニブ、及びミドスタウリンに耐性を示す頭頚部がん。
(5)シスプラチン、ペメトレキセド、及びミドスタウリンからなる群より選択される少なくとも1種の抗がん剤に耐性を示す悪性中皮腫。より好ましくは、シスプラチン、ペメトレキセド、及びミドスタウリンに耐性を示す悪性中皮腫。
(6)キナーゼを標的とする分子標的薬に耐性を示すがん。前記分子標的薬としては、ゲフィチニブ、アファチニブ、オシメルチニブ、ダサチニブ、エルロチニブ、及びミドスタウリン等が例示される。
(7)EGFR、ABL1、ALK1、HER2、c-Kit、FGFR1、FGFR2、FGFR3、c-Src、PDGFRa、RET、DDR2、TRKA、及びFlt-3からなる群より選択される少なくとも1種のキナーゼを標的とする分子標的薬に耐性を示すがん。
(8)ゲートキーパー変異を有するキナーゼを発現するがん。「ゲートキーパー変異」とは、ゲートキーパー部位の変異をいう。「ゲートキーパー部位」とは、キナーゼのATP結合ポケットの一番奥に位置するアミノ酸残基の部位をいう。
(9)d746-750、T790M、C797S、及びL858Rからなる群より選択される少なくとも1種の変異を有するEGFRを発現するがん。中でも、d746-750及びT790Mの変異を有するEGFR;d746-750、T790M、及びC797Sの変異を有するEGFR;T790M及びL858Rの変異を有するEGFR;又は、T790M、C797S、及びL858Rの変異を有するEGFRを発現するがん。前記において、「d」はアミノ酸残基の欠失を意味し、例えば、「d746-750」は、746~750位のアミノ酸残基の欠失を意味する。本明細書において、アミノ酸残基の欠失は、前記と同様に表記される。前記において、数字で示されるアミノ酸の位置番号の左右にアミノ酸の1文字表記が記載された表記は、数字で示される位置番号のアミノ酸残基の変異を示し、位置番号の左側が元のアミノ酸残基を示し、位置番号の右側が変異後のアミノ酸残基を示す。例えば、「T790M」は、790位のスレオニン(T)残基がメチオニン(M)残基に変異していることを示す。本明細書において、アミノ酸残基の変異は、前記と同様に表記される。
(10)T315I及びG1269Aからなる群より選択される少なくとも1種の変異を有するABL1を発現するがん。
(11)L1196M及びG1269Aからなる群より選択される少なくとも1種の変異を有するALK1を発現するがん。
(12)P780_Y781insGSPの変異を有するHer2を発現するがん。
(13)D816E、D816F、D816H、D816I、D816V、D816Y、T670I、及びV559Dからなる群より選択される少なくとも1種のc-Kitを発現するがん。中でも、T670I及びV559Dからなる群より選択される少なくとも1種のc-Kitを発現するがん。
(14)V561Mの変異を有するFGFR1を発現するがん。
(15)V564Fの変異を有するFGFR2を発現するがん。
(16)V565Mの変異を有するFGFR3を発現するがん。
(17)T341Mの変異を有するc-Srcを発現するがん。
(18)T674Iの変異を有するPDGFRaを発現するがん。
(19)V804Lの変異を有するRETを発現するがん。
(20)T654Mの変異を有するDDR2を発現するがん。
(21)G667Cの変異を有するTRKAを発現するがん。
(22)D835Yの変異を有するFlt-3を発現するがん。
(23)図24に記載の少なくとも1種の変異を有するキナーゼを発現するがん。
(24)後述の表3に記載のキナーゼの少なくとも1種を発現するがん。中でも、AuroraA、AuroraB、CAMK2a、CAMK2d、CyclinC、CyclinA、CyclinA1、CHK1、DDR1、DYRK3、AK3、MEK3、MEK5、PDK1、PIM3、PKCmu、PKC nu、TRKB、及びTRKCからなる群より選択される少なくとも1種のキナーゼを発現するがん。
(25)図35に記載の少なくとも1種のキナーゼを発現するがん。
(1)ゲフィチニブ、アファチニブ、オシメルチニブ、ダサチニブ、エルロチニブ、ゲムシタビン、シスプラチン、ペメトレキセド、及びミドスタウリンからなる群より選択される少なくとも1種の抗がん剤に耐性を示すがん。
(2)ゲフィチニブ、アフィチニブ、オシメルチニブ、及びダサチニブからなる群より選択される少なくとも1種の抗がん剤に耐性を示す肺がん(特に非小細胞肺がん)。中でも、オシメルチニブに耐性を示す肺がん(特に非小細胞肺がん)。より好ましくは、ゲフィチニブ、アフィチニブ、及びオシメルチニブに耐性を示す肺がん。
(3)ゲムシタビン、及びエルロチニブからなる群より選択される少なくとも1種の抗がん剤に耐性を示す膵臓がん。より好ましくは、ゲムシタビン、及びエルロチニブに耐性を示す膵臓がん。
(4)シスプラチン、エルロチニブ、ゲフィチニブ、及びミドスタウリンからなる群より選択される少なくとも1種の抗がん剤に耐性を示す頭頸部がん。より好ましくは、シスプラチン及びエルロチニブに耐性を示す頭頚部がん、またはエルロチニブ、ゲフィチニブ、及びミドスタウリンに耐性を示す頭頚部がん。
(5)シスプラチン、ペメトレキセド、及びミドスタウリンからなる群より選択される少なくとも1種の抗がん剤に耐性を示す悪性中皮腫。より好ましくは、シスプラチン、ペメトレキセド、及びミドスタウリンに耐性を示す悪性中皮腫。
(6)キナーゼを標的とする分子標的薬に耐性を示すがん。前記分子標的薬としては、ゲフィチニブ、アファチニブ、オシメルチニブ、ダサチニブ、エルロチニブ、及びミドスタウリン等が例示される。
(7)EGFR、ABL1、ALK1、HER2、c-Kit、FGFR1、FGFR2、FGFR3、c-Src、PDGFRa、RET、DDR2、TRKA、及びFlt-3からなる群より選択される少なくとも1種のキナーゼを標的とする分子標的薬に耐性を示すがん。
(8)ゲートキーパー変異を有するキナーゼを発現するがん。「ゲートキーパー変異」とは、ゲートキーパー部位の変異をいう。「ゲートキーパー部位」とは、キナーゼのATP結合ポケットの一番奥に位置するアミノ酸残基の部位をいう。
(9)d746-750、T790M、C797S、及びL858Rからなる群より選択される少なくとも1種の変異を有するEGFRを発現するがん。中でも、d746-750及びT790Mの変異を有するEGFR;d746-750、T790M、及びC797Sの変異を有するEGFR;T790M及びL858Rの変異を有するEGFR;又は、T790M、C797S、及びL858Rの変異を有するEGFRを発現するがん。前記において、「d」はアミノ酸残基の欠失を意味し、例えば、「d746-750」は、746~750位のアミノ酸残基の欠失を意味する。本明細書において、アミノ酸残基の欠失は、前記と同様に表記される。前記において、数字で示されるアミノ酸の位置番号の左右にアミノ酸の1文字表記が記載された表記は、数字で示される位置番号のアミノ酸残基の変異を示し、位置番号の左側が元のアミノ酸残基を示し、位置番号の右側が変異後のアミノ酸残基を示す。例えば、「T790M」は、790位のスレオニン(T)残基がメチオニン(M)残基に変異していることを示す。本明細書において、アミノ酸残基の変異は、前記と同様に表記される。
(10)T315I及びG1269Aからなる群より選択される少なくとも1種の変異を有するABL1を発現するがん。
(11)L1196M及びG1269Aからなる群より選択される少なくとも1種の変異を有するALK1を発現するがん。
(12)P780_Y781insGSPの変異を有するHer2を発現するがん。
(13)D816E、D816F、D816H、D816I、D816V、D816Y、T670I、及びV559Dからなる群より選択される少なくとも1種のc-Kitを発現するがん。中でも、T670I及びV559Dからなる群より選択される少なくとも1種のc-Kitを発現するがん。
(14)V561Mの変異を有するFGFR1を発現するがん。
(15)V564Fの変異を有するFGFR2を発現するがん。
(16)V565Mの変異を有するFGFR3を発現するがん。
(17)T341Mの変異を有するc-Srcを発現するがん。
(18)T674Iの変異を有するPDGFRaを発現するがん。
(19)V804Lの変異を有するRETを発現するがん。
(20)T654Mの変異を有するDDR2を発現するがん。
(21)G667Cの変異を有するTRKAを発現するがん。
(22)D835Yの変異を有するFlt-3を発現するがん。
(23)図24に記載の少なくとも1種の変異を有するキナーゼを発現するがん。
(24)後述の表3に記載のキナーゼの少なくとも1種を発現するがん。中でも、AuroraA、AuroraB、CAMK2a、CAMK2d、CyclinC、CyclinA、CyclinA1、CHK1、DDR1、DYRK3、AK3、MEK3、MEK5、PDK1、PIM3、PKCmu、PKC nu、TRKB、及びTRKCからなる群より選択される少なくとも1種のキナーゼを発現するがん。
(25)図35に記載の少なくとも1種のキナーゼを発現するがん。
抗がん剤耐性のがんは、上記(1)~(25)からなる群より選択される少なくとも1つの特徴を有していることが好ましく、上記(1)~(25)からなる群より選択される複数の特徴を有していることがより好ましい。
本実施形態の医薬組成物の剤型は、特に限定されず、公知の製剤方法を適用することができる。本実施形態の医薬組成物の剤型としては、例えば、乳剤、エマルション剤、液剤、ゲル状剤、カプセル剤、軟膏剤、貼付剤、バップ剤、顆粒剤、錠剤等を例示できるが、これらに限定されない。
本実施形態の医薬組成物の投与経路は、特に限定されず、経口又は非経口経路で投与することができる。非経口経路は、経口以外の全ての投与経路、例えば、静脈内、筋肉内、皮下、鼻腔内、皮内、点眼、脳内、直腸内、腟内及び腹腔内等への投与を包含する。また、投与は、局所投与であってもよく、全身投与であってもよい。
本実施形態の医薬組成物は、単回投与又は複数回投与を行うことが可能であり、その投与期間及び間隔は、疾患の種類及び状態等、投与経路、投与対象の年齢、体重及び性別等によって、適宜選択することができる。投与間隔としては、例えば、1日1~3回、2~3日に1回、週1~3回、10日1回等が例示できるが、これらに限定されない。
本実施形態の医薬組成物の投与量、その投与期間及び間隔は、薬物の種類、疾患の種類及び状態等、投与経路、投与対象の年齢、体重及び性別等によって、適宜選択することができる。本実施形態の医薬組成物の投与量は、化合物(S)の治療的有効量とすることができる。「治療的有効量」とは、抗がん剤耐性がんの治療又は予防のために有効な化合物(S)の量を意味する。例えば、投与1回につき、化合物(S)の投与量として、体重1kgあたり0.01~1000mg程度、0.1~100mg程度、0.5~50mg程度、1~10mg程度とすることができる。
本実施形態の医薬組成物は、単回投与又は複数回投与を行うことが可能であり、その投与期間及び間隔は、疾患の種類及び状態等、投与経路、投与対象の年齢、体重及び性別等によって、適宜選択することができる。投与間隔としては、例えば、1日1~3回、2~3日に1回、週1~3回、10日1回等が例示できるが、これらに限定されない。
本実施形態の医薬組成物の投与量、その投与期間及び間隔は、薬物の種類、疾患の種類及び状態等、投与経路、投与対象の年齢、体重及び性別等によって、適宜選択することができる。本実施形態の医薬組成物の投与量は、化合物(S)の治療的有効量とすることができる。「治療的有効量」とは、抗がん剤耐性がんの治療又は予防のために有効な化合物(S)の量を意味する。例えば、投与1回につき、化合物(S)の投与量として、体重1kgあたり0.01~1000mg程度、0.1~100mg程度、0.5~50mg程度、1~10mg程度とすることができる。
[第2の態様]
<化合物>
他の態様において、本発明は、下記一般式(k1)で表される化合物を提供する。
<化合物>
他の態様において、本発明は、下記一般式(k1)で表される化合物を提供する。

(b)-CO(CH2)jR4[ここで、jは1から6であり、R4は、
(i)水素、ハロゲン、-N3、
(ii)-NR5R6(ここで、R5およびR6は上記に定義されたとおりである)、
(iii)-SR7(ここで、R7は、水素、置換もしくは非置換の低級アルキル、置換もしくは非置換の低級アルケニル、置換もしくは非置換の低級アルキニル、置換もしくは非置換のアリール、置換もしくは非置換のヘテロアリール、置換もしくは非置換のアラルキル、-(CH2)aCO2R10(ここで、aは、1または2であり、R10は、水素および置換もしくは非置換の低級アルキルからなる群から選択される)および-(CH2)aCO2NR5R6、
(iv)-OR8、-OCOR8(ここで、R8は、水素、置換もしくは非置換の低級アルキル、置換もしくは非置換の低級アルケニル、置換もしくは非置換の低級アルキニル、置換もしくは非置換のアリール、置換もしくは非置換のヘテロアリールから選択される)
からなる群から選択される]、
(c)-CH(OH)(CH2)jR4(ここで、jおよびR4は、上記に定義したとおりである);
(d)-(CH2)dCHR11CO2R12または-(CH2)dCHR11CONR5R6(ここで、dは、0から5であり、R11は、水素、-CONR5R6、または-CO2R13であり、R13は、水素または置換もしくは非置換の低級アルキルであり、R12は、水素または置換もしくは非置換の低級アルキルである);
(e)-(CH2)kR14[ここで、kは2から6であり、R14は、ハロゲン、置換もしくは非置換のアリール、置換もしくは非置換のヘテロアリール、-COOR15、-OR15(ここで、R15は、水素、置換もしくは非置換の低級アルキル、置換もしくは非置換の低級アルケニル、置換もしくは非置換の低級アルキニル、置換もしくは非置換のアリール、置換もしくは非置換のヘテロアリールまたはアシルである)、-SR7(ここで、R7は、上記に定義したとおりである)、-CONR5R6、-NR5R6(ここで、R5およびR6は、上記に定義したとおりである)または-N3である];
(f)-CH=CH(CH2)m1R16[ここで、m1は、0から4であり、R16は、水素、置換もしくは非置換の低級アルキル、置換もしくは非置換の低級アルケニル、置換もしくは非置換の低級アルキニル、置換もしくは非置換のアリール、置換もしくは非置換のヘテロアリール、-COOR15、-OR15(ここで、R15は上記に定義したとおりである)、-CONR5R6または-NR5R6(ここで、R5およびR6は上記に定義したとおりである)];
(g)-CH=C(CO2R12)2(ここで、R12は、上記に定義したとおりである);
(h)-C≡C(CH2)nR16(ここで、nは0から4であり、R16は、上記に定義したとおりである);
(i)-CH2OR22(ここで、R22は、3個の低級アルキル基が同じかもしくは異なっているトリ-低級アルキルシリルであるか、またはR22は、R8と同じ意味を有する);
(j)-CH(SR23)2および-CH2-SR7(ここで、R23は、低級アルキル、低級アルケニルまたは低級アルキニルであり、R7は、上記に定義したとおりである)
からなる群から、それぞれ独立して、選択され;
Rk3は、水素、ハロゲン、アシル、カルバモイル、置換もしくは非置換の低級アルキル、置換もしくは非置換のアルケニル、置換もしくは非置換の低級アルキニルまたはアミノであり;
Wk1およびWk2は、独立して、水素、ヒドロキシであるか、またはWk1およびWk2は一緒に酸素を表す。]
用語「低級アルキル」は、単独で用いられるかまたは他の基と合わせて用いられる場合、1~6個の炭素原子、好ましくは1~5個、より好ましくは1~4個、特に好ましくは1~3個または1~2個の炭素原子を含む直鎖または分岐の低級アルキル基を意味する。
これらの基には、特に、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、アミル、イソアミル、ネオペンチル、1-エチルプロピル、ヘキシルなどがある。「低級アルコキシ」、「低級アルコキシカルボニル」、「低級アルキルアミノカルボニル」、「低級ヒドロキシアルキル」および「トリ-低級アルキルシリル」の低級アルキル部分は、上記に定義した「低級アルキル」と同じ意味を有する。
これらの基には、特に、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、アミル、イソアミル、ネオペンチル、1-エチルプロピル、ヘキシルなどがある。「低級アルコキシ」、「低級アルコキシカルボニル」、「低級アルキルアミノカルボニル」、「低級ヒドロキシアルキル」および「トリ-低級アルキルシリル」の低級アルキル部分は、上記に定義した「低級アルキル」と同じ意味を有する。
「低級アルケニル」基は、直鎖または分岐であってもよく、Z型またはE型であってもよいC2~C6アルケニル基として定義する。このような基には、ビニル、プロペニル、1-ブテニル、イソブテニル、2-ブテニル、1-ペンテニル、(Z)-2-ペンテニル、(E)-2-ペンテニル、(Z)-4-メチル-2-ペンテニル、(E)-4-メチル-2-ペンテニル、ペンタジエニル、例えば、1,3または2,4-ペンタジエニルなどがある。より好ましいC2~C6-アルケニル基は、C2~C5-、C2~C4-アルケニル基、さらにより好ましくはC2~C3-アルケニル基である。
用語「低級アルキニル」基は、直鎖または分岐であってもよいC2~C6-アルキニル基を意味し、エチニル、プロピニル、1-ブチニル、2-ブチニル、1-ペンチニル、2-ペンチニル、3-メチル-1-ペンチニル、3-ペンチニル、1-ヘキシニル、2-ヘキシニル、3-ヘキシニルなどがある。より好ましいC2~C6-アルキニル基は、C2~C5-、C2~C4-アルキニル基、さらにより好ましくはC2~C3-アルキニル基である。
用語「アリール」基は、6から14個までの環状炭素原子を含むC6~C14-アリール基を意味する。これらの基は、単環式、二環式、または三環式であってもよく、縮合環である。好ましいアリール基には、フェニル、ビフェニル、ナフチル、アントラセニル、フェナンスレニルなどがある。「アリールカルボニル」基および「アリールアミノカルボニル」基のアリール部分は、上記定義と同じ意味を有する。
用語「ヘテロアリール」基は、窒素、硫黄または酸素から独立して選択される1から3個のヘテロ原子を含み得、C3~C13-ヘテロアリール基をいう。これらの基は、単環式、二環式または三環式であってもよい。本発明のC3~C13ヘテロアリール基には、ヘテロ芳香族、ならびに飽和および部分飽和ヘテロ環基などがある。これらのヘテロ環は、単環式、二環式、三環式であってもよい。好ましい5または6員ヘテロ環基は、チエニル、フリル、ピロリル、ピリジル、ピラニル、モノホリニル、ピラジニル、メチルピロリル、およびピリダジニルである。C3~C13-ヘテロアリールは、二環式ヘテロ環基であってもよい。好ましい二環式ヘテロ環基は、ベンゾフリル、ベンゾチエニル、インドリル、イミダゾリル、およびピリミジニルである。最も好ましいC3~C13-ヘテロアリールは、フリルおよびピリジルである。
用語「低級アルコキシ」には、1から6個の炭素原子、好ましくは1から5個、より好ましくは1から4個、特に好ましくは1から3個または1から2個の炭素原子を含むアルコキシ基が含まれ、直鎖または分岐であってもよい。これらの基には、メトキシ、エトキシ、プロポキシ、ブトキシ、イソプロポキシ、tert-ブトキシ、ペントキシ、ヘキソキシなどがある。
用語「アシル」には、1から6個の炭素原子、好ましくは1から5個、1から4個、1から3個または1から2個の炭素原子を含む低級アルカノイルが含まれ、直鎖または分岐であってもよい。これらの基には、好ましくは、ホルミル、アセチル、プロピオニル、ブチリル、イソブチリル、tert-ブチリル、ペンタノイルおよびヘキサノイルなどがある。「アシルオキシ」基のアシル部分は、上記定義と同じ意味を有する。
用語「ハロゲン」には、フルオロ、クロロ、ブロモ、ヨードなどがある。
用語「アラルキル」基は、アルキル基がアリールによって置換されているC7~C15-アラルキルをいう。該アルキル基およびアリールは、上記に定義したC1~C6-アルキル基およびC6~C14-アリール基から選択することができ、ここで、炭素原子の総数は7から15個である。好ましいC7~C15-アラルキル基は、ベンジル、フェニルエチル、フェニルプロピル、フェニルイソプロピル、フェニルブチル、ジフェニルメチル、1,1-ジフェニルエチル、1,2-ジフェニルエチルである。「アラルキルオキシ」基のアラルキル部分は、上記定義と同じ意味を有する。
置換低級アルキル基、置換低級アルケニル基、および置換低級アルキニル基は、低級アルキル、ヒドロキシ、低級アルコキシ、カルボキシル、低級アルコキシカルボニル、ニトロ、ハロゲン、アミノ、モノまたはジ低級アルキルアミノ、ジオキソラン、ジオキサン、ジチオラン、およびジチオンなどの独立して選択される1から3個の置換基を有する。置換低級アルキル基、置換低級アルケニル基および置換低級アルキニル基の低級アルキル置換部分、ならびに置換低級アルキル基、置換低級アルケニル基および置換低級アルキニル基の低級アルコキシ置換基、低級アルコキシカルボニル置換基、モノまたはジ低級アルキルアミノ置換基の低級アルキル部分は、上記に定義した「低級アルキル」と同じ意味を有する。
置換アリール基、置換ヘテロアリール基および置換アラルキル基はそれぞれ、低級アルキル、ヒドロキシ、低級アルコキシ、カルボキシ、低級アルコキシカルボニル、ニトロ、アミノ、モノまたはジ低級アルキルアミノ、およびハロゲンなどの独立して選択される1から3個の置換基を有する。
窒素原子と結合したR5およびR6によって形成されるヘテロ環基には、ピロリジニル、ピペリジニル、ピペリジノ、モルホリニル、モルホリノ、チオモルホリノ、N-メチルピペラジニル、インドリル、およびイソインドリルなどがある。
α-アミノ酸基には、グリシン、アラニン、プロリン、グルタミン酸およびリジンなどがあり、L-型、D-型またはラセミ体の型であってもよい。
好ましくは、Rk1およびRk2は、水素、ハロゲン、ニトロ、-CH2OH、-(CH2)kR14、-CH=CH(CH2)mR16、-C≡C(CH2)nR15、-CO(CH2)jR4(ここで、R4は-SR7である)、CH2O-(置換または非置換の)低級アルキル(ここで、置換低級アルキルは、好ましくは、メトキシメチル、メトキシエチルまたはエトキシメチルである)、-NR5R6からなる群から独立して選択される。より好ましくは、Rk1およびRk2は、水素である。
Rk1およびRk2の上記好ましい意味において、残基R14は、好ましくは、フェニル、ピリジル、イミダゾリル、チアゾリル、テトラゾリル、-COOR15、-OR15(ここで、R15は、好ましくは、水素、メチル、エチル、フェニルまたはアシルから選択される)、-SR7(ここで、R7は、好ましくは、置換または非置換の低級アルキル、2-チアゾリンおよびピリジルから選択される)および-NR5R6(ここで、R5およびR6は、好ましくは、水素、メチル、エチル、フェニル、カルバモイルおよび低級アルキルアミノカルボニルから選択される)から選択される。さらに、残基R16は、好ましくは、水素、メチル、エチル、フェニル、イミダゾール、チアゾール、テトラゾール、-COOR15、-OR15および-NR5R6(ここで、残基R15、R5およびR6は、上記した好ましい意味を有する)から選択される。Rk1およびRk2の上記好ましい意味において、残基R7は、好ましくは、置換または非置換の低級アルキル、置換または非置換のフェニル、ピリジル、ピリミジニル、チアゾールおよびテトラゾールからなる群から選択される。さらに、kは、好ましくは2、3または4であり、jは、好ましくは1または2であり、m1およびnは、独立して、好ましくは0または1である。
好ましくは、Rk3は、水素またはアセチル、最も好ましくは水素である。
好ましくは、それぞれのWk1およびWk2は水素である。
前記化合物(K)は、下記式(k1-1)で表されることが好ましい。

<化合物(K)の製造方法>
化合物(K)は、例えば、下記一般式(k0)で表される化合物を、無水ヒドラジン又はヒドラジン水和物の存在下、無溶媒で還流して製造することができる。また、例えば、下記一般式(k0)で表される化合物をメタノール、エタノール、プロパノール、イソプロパノール、ブタノール、tert-ブタノール、N,N-ジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、テトラヒドロフラン(THF)、ジオキサン、アセトニトリル、トルエン等の溶媒に溶解し、無水ヒドラジンに添加して反応させることにより製造することができる。反応温度及び反応時間は特に限定されないが、例えば室温~50℃で30分~15時間反応させることができる。
化合物(K)は、例えば、下記一般式(k0)で表される化合物を、無水ヒドラジン又はヒドラジン水和物の存在下、無溶媒で還流して製造することができる。また、例えば、下記一般式(k0)で表される化合物をメタノール、エタノール、プロパノール、イソプロパノール、ブタノール、tert-ブタノール、N,N-ジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、テトラヒドロフラン(THF)、ジオキサン、アセトニトリル、トルエン等の溶媒に溶解し、無水ヒドラジンに添加して反応させることにより製造することができる。反応温度及び反応時間は特に限定されないが、例えば室温~50℃で30分~15時間反応させることができる。

<薬物複合体>
本発明の第二の態様は、下記一般式(ka)で表される繰り返し単位(ka)、及び下記一般式(II)で表される繰り返し単位(II)を有するポリマーを含有する薬物複合体(以下、「薬物複合体(K)」という場合がある。)である。
本発明の第二の態様は、下記一般式(ka)で表される繰り返し単位(ka)、及び下記一般式(II)で表される繰り返し単位(II)を有するポリマーを含有する薬物複合体(以下、「薬物複合体(K)」という場合がある。)である。

前記一般式(ka)中、mは1が好ましい。
前記一般式(ka)中、Lはメチレン基、エチレン基、プロピレン基、ベンジレン基が好ましく、メチレン基、エチレン基又はベンジレン基がより好ましい。
前記一般式(ka)中、R1は水素原子又は脂肪族炭化水素基が好ましく、水素原子又はメチル基がより好ましい。
前記一般式(ka)中、Rk1およびRk2はそれぞれ独立に、水素、ハロゲン、ニトロ、-CH2OH、-(CH2)kR14、-CH=CH(CH2)mR16、-C≡C(CH2)nR15、-CO(CH2)jR4(ここで、R4は-SR7である)、CH2O-(置換または非置換の)低級アルキル(ここで、置換低級アルキルは、好ましくは、メトキシメチル、メトキシエチルまたはエトキシメチルである)又は-NR5R6であることが好ましく、水素であることがより好ましい。
前記一般式(ka)中、Rk3は、水素またはアセチルであることが好ましく、水素であることがより好ましい。
前記一般式(ka)中、Wk1およびWk2は水素であることが好ましい。
前記一般式(II)中、mは1が好ましい。
前記一般式(II)中、XはORxが好ましく、OH(ヒドロキシ基)がより好ましい。
前記一般式(ka)中、Lはメチレン基、エチレン基、プロピレン基、ベンジレン基が好ましく、メチレン基、エチレン基又はベンジレン基がより好ましい。
前記一般式(ka)中、R1は水素原子又は脂肪族炭化水素基が好ましく、水素原子又はメチル基がより好ましい。
前記一般式(ka)中、Rk1およびRk2はそれぞれ独立に、水素、ハロゲン、ニトロ、-CH2OH、-(CH2)kR14、-CH=CH(CH2)mR16、-C≡C(CH2)nR15、-CO(CH2)jR4(ここで、R4は-SR7である)、CH2O-(置換または非置換の)低級アルキル(ここで、置換低級アルキルは、好ましくは、メトキシメチル、メトキシエチルまたはエトキシメチルである)又は-NR5R6であることが好ましく、水素であることがより好ましい。
前記一般式(ka)中、Rk3は、水素またはアセチルであることが好ましく、水素であることがより好ましい。
前記一般式(ka)中、Wk1およびWk2は水素であることが好ましい。
前記一般式(II)中、mは1が好ましい。
前記一般式(II)中、XはORxが好ましく、OH(ヒドロキシ基)がより好ましい。
前記薬物複合体(K)は、下記一般式(ka-1)で表される繰り返し単位(ka-1)、及び下記一般式(II)で表される繰り返し単位(II)を有するポリマーを含有することが好ましい。

前記一般式(ka-1)中、mは1が好ましい。
前記一般式(ka-1)中、Lはメチレン基、エチレン基、プロピレン基、ベンジレン基が好ましく、メチレン基、エチレン基又はベンジレン基がより好ましい。
前記一般式(ka-1)中、R1は水素原子又は脂肪族炭化水素基が好ましく、水素原子又はメチル基がより好ましい。
前記一般式(ka-1)中、Lはメチレン基、エチレン基、プロピレン基、ベンジレン基が好ましく、メチレン基、エチレン基又はベンジレン基がより好ましい。
前記一般式(ka-1)中、R1は水素原子又は脂肪族炭化水素基が好ましく、水素原子又はメチル基がより好ましい。
前記一般式(II)中、XはORxが好ましく、OH(ヒドロキシ基)がより好ましい。
化合物(K)又は薬物複合体(K)は、既存のがん治療薬が有効性を示さない悪性がんや転移がんに対して、有効性を示す。そのため、化合物(K)及び薬物複合体(K)は、既存のがん治療薬に対して耐性であるがんの治療薬として有用である。
化合物(K)は、薬物複合体(K)とすることにより、第1の態様の薬物複合体と同様に、化合物(K)の徐放性を制御することができる。化合物(K)がポリマーに保持されている間、化合物(K)は安定に維持され、毒性も緩和されるため、副作用を軽減して治療効果を高めることができる。
また、薬物複合体(K)を含有するミセルを調製することにより、第1の態様のミセルと同様に、生体内のpH環境に依存して、薬物複合体(K)からの化合物(K)のリリースを制御することができる。薬物複合体(K)を含有するミセルの調製は、第1の態様の薬物複合体のミセルと同様に、公知の方法により調製することができる。例えば、薬物複合体(K)を親油性又は親水性の溶媒に溶解又は懸濁し、当該溶解液又は懸濁液を親水性又は親油性の溶媒に滴下して撹拌することにより、薬物複合体(K)を含有するミセルを調製することができる。
なお、薬物複合体(K)は、第1の態様の薬物複合体において、薬物が化合物(K)である態様であり、第1の態様の薬物複合体に包含される。また、薬物複合体(K)は、第1の態様における一般式(Ia)において、BMが化合物(K)から誘導される基である態様であるということもできる。
また、薬物複合体(K)を含有するミセルを調製することにより、第1の態様のミセルと同様に、生体内のpH環境に依存して、薬物複合体(K)からの化合物(K)のリリースを制御することができる。薬物複合体(K)を含有するミセルの調製は、第1の態様の薬物複合体のミセルと同様に、公知の方法により調製することができる。例えば、薬物複合体(K)を親油性又は親水性の溶媒に溶解又は懸濁し、当該溶解液又は懸濁液を親水性又は親油性の溶媒に滴下して撹拌することにより、薬物複合体(K)を含有するミセルを調製することができる。
なお、薬物複合体(K)は、第1の態様の薬物複合体において、薬物が化合物(K)である態様であり、第1の態様の薬物複合体に包含される。また、薬物複合体(K)は、第1の態様における一般式(Ia)において、BMが化合物(K)から誘導される基である態様であるということもできる。
化合物(K)及び薬物複合体(K)は、それぞれ、上記第1の態様の医薬組成物における化合物(S)及びその薬物複合体としてそれぞれ用いることができる。
[他の態様]
他の態様において、本発明は、抗がん剤耐性のがんを治療又は予防するための医薬組成物の製造における、化合物(S)若しくはその薬学的に許容される塩、又は薬学的に許容されるポリマーに前記化合物(S)若しくはその薬学的に許容される塩が結合した薬物複合体の使用、を提供する。
他の態様において、本発明は、化合物(S)若しくはその薬学的に許容される塩、又は薬学的に許容されるポリマーに前記化合物(S)若しくはその薬学的に許容される塩が結合した薬物複合体を対象(例えば、抗がん剤耐性のがんに罹患した患者)に投与することを含む、抗がん剤耐性のがんを治療又は予防する方法、を提供する。
他の態様において、本発明は、抗がん剤耐性のがんの治療又は予防に使用するための化合物(S)若しくはその薬学的に許容される塩、又は薬学的に許容されるポリマーに前記化合物(S)若しくはその薬学的に許容される塩が結合した薬物複合体、を提供する。
他の態様において、本発明は、抗がん剤耐性のがんを治療又は予防するための化合物(S)若しくはその薬学的に許容される塩、又は薬学的に許容されるポリマーに前記化合物(S)若しくはその薬学的に許容される塩が結合した薬物複合体の使用、を提供する。
他の態様において、本発明は、抗がん剤耐性のがんを治療又は予防するための医薬組成物の製造における、化合物(S)若しくはその薬学的に許容される塩、又は薬学的に許容されるポリマーに前記化合物(S)若しくはその薬学的に許容される塩が結合した薬物複合体の使用、を提供する。
他の態様において、本発明は、化合物(S)若しくはその薬学的に許容される塩、又は薬学的に許容されるポリマーに前記化合物(S)若しくはその薬学的に許容される塩が結合した薬物複合体を対象(例えば、抗がん剤耐性のがんに罹患した患者)に投与することを含む、抗がん剤耐性のがんを治療又は予防する方法、を提供する。
他の態様において、本発明は、抗がん剤耐性のがんの治療又は予防に使用するための化合物(S)若しくはその薬学的に許容される塩、又は薬学的に許容されるポリマーに前記化合物(S)若しくはその薬学的に許容される塩が結合した薬物複合体、を提供する。
他の態様において、本発明は、抗がん剤耐性のがんを治療又は予防するための化合物(S)若しくはその薬学的に許容される塩、又は薬学的に許容されるポリマーに前記化合物(S)若しくはその薬学的に許容される塩が結合した薬物複合体の使用、を提供する。
本発明を実施例に基づいて説明する。ただし、本発明の実施態様は、これら実施例の記載に限定されるものではない。
<ポリマーの合成>
[ポリマー合成例1]メトキシ-ポリ(エチレングリコール)-b-ポリ(β-ベンジル-アスパルタミド)の合成
α-メトキシ-ω-アミノ ポリ(エチレングリコール)の末端第一級アミノ基によって開始される、β-ベンジルアスパラギン酸 N-カルボキシアルデヒドの開環重合により、メトキシ-ポリ(エチレングリコール)-b-ポリ(β-ベンジル-アスパルタミド)(MeO-PEG-PBLA;PEG分子量=12kDa;PBLAの重合度=40)コポリマーを合成した。ω-アミン基は、アセチル化によりブロックした。
[ポリマー合成例1]メトキシ-ポリ(エチレングリコール)-b-ポリ(β-ベンジル-アスパルタミド)の合成
α-メトキシ-ω-アミノ ポリ(エチレングリコール)の末端第一級アミノ基によって開始される、β-ベンジルアスパラギン酸 N-カルボキシアルデヒドの開環重合により、メトキシ-ポリ(エチレングリコール)-b-ポリ(β-ベンジル-アスパルタミド)(MeO-PEG-PBLA;PEG分子量=12kDa;PBLAの重合度=40)コポリマーを合成した。ω-アミン基は、アセチル化によりブロックした。
[ポリマー合成例2]芳香族アセタール基導入ポリマーの合成
PEG-PBLAポリマー(220mg、0.011mmol)をDMF(2mL)で溶解し、{〔4-(ジメトキシメチル)フェニル〕メタンアミン}(100μL、0.58mmol)を添加して、徐々に温度を上げて40℃で4日間撹拌した。特性分析のために、反応混合物の一部をエーテル沈殿し、芳香族アセタール基導入ポリマーを回収した。
反応スキームを図1に示す。また、得られた芳香族アセタール基導入ポリマーの1H-NMR解析結果を図2に示す。
1H-NMR解析により25個のベンジルエステルユニットのアミドへの置換が、確認された。残りのベンジルエステル(40-25=15)は、おそらくエーテル沈殿操作中に、加水分解された。
PEG-PBLAポリマー(220mg、0.011mmol)をDMF(2mL)で溶解し、{〔4-(ジメトキシメチル)フェニル〕メタンアミン}(100μL、0.58mmol)を添加して、徐々に温度を上げて40℃で4日間撹拌した。特性分析のために、反応混合物の一部をエーテル沈殿し、芳香族アセタール基導入ポリマーを回収した。
反応スキームを図1に示す。また、得られた芳香族アセタール基導入ポリマーの1H-NMR解析結果を図2に示す。
1H-NMR解析により25個のベンジルエステルユニットのアミドへの置換が、確認された。残りのベンジルエステル(40-25=15)は、おそらくエーテル沈殿操作中に、加水分解された。
[ポリマー合成例3]芳香族アルデヒド基含有ポリマーの合成
アミノリシス反応後、反応混合物を酸性化し(0.1N HCl、100μL、30分)、その後、水で透析し、ポリマーを凍結乾燥することにより、透析袋からアルデヒド基導入ポリマーを回収した。反応スキームを図1に示す。また、得られた芳香族アルデヒド基含有ポリマーの1H-NMR解析結果を図3に示す。27個のアルデヒドユニットが1H-NMRスペクトラムで確認された。
アミノリシス反応後、反応混合物を酸性化し(0.1N HCl、100μL、30分)、その後、水で透析し、ポリマーを凍結乾燥することにより、透析袋からアルデヒド基導入ポリマーを回収した。反応スキームを図1に示す。また、得られた芳香族アルデヒド基含有ポリマーの1H-NMR解析結果を図3に示す。27個のアルデヒドユニットが1H-NMRスペクトラムで確認された。
[ポリマー合成例4]脂肪族アルデヒド基含有ポリマーの合成
脂肪族アルデヒド基含有ポリマーの合成は、{〔4-(ジメトキシメチル)フェニル〕メタンアミン}に替えて、1-アミノ-3,3-ジエトキシプロパンを使用した以外は、芳香族アルデヒド基含有ポリマーの合成と同様の方法で行った。反応スキームを図1に示す。また、中間体である脂肪族アセタール基含有ポリマーの1H-NMR解析結果を図4に示す。また、得られた脂肪族アルデヒド基含有ポリマーの1H-NMR解析結果を図5に示す。23個のアルデヒドユニットが1H-NMRスペクトラムで確認された。
脂肪族アルデヒド基含有ポリマーの合成は、{〔4-(ジメトキシメチル)フェニル〕メタンアミン}に替えて、1-アミノ-3,3-ジエトキシプロパンを使用した以外は、芳香族アルデヒド基含有ポリマーの合成と同様の方法で行った。反応スキームを図1に示す。また、中間体である脂肪族アセタール基含有ポリマーの1H-NMR解析結果を図4に示す。また、得られた脂肪族アルデヒド基含有ポリマーの1H-NMR解析結果を図5に示す。23個のアルデヒドユニットが1H-NMRスペクトラムで確認された。
[ポリマー合成例5]脂肪族ケトン基含有ポリマーの合成
PEG-PBLAポリマー(318mg、0.016mmol)をDMF(3mL)で溶解し、得られた溶液に3,3-ジメトキシブタン(200μL)を添加した。反応液を40℃で72時間撹拌し、HCl溶液(100μL、0.1N)添加して1時間撹拌した。水で透析し、ポリマーを凍結乾燥することにより、側鎖に脂肪族ケトンが導入されたポリマーを回収した。
反応スキームを図6に示す。また、得られた脂肪族ケトン基含有ポリマーの1H-NMR解析結果を図7に示す。35個の脂肪族ケトンユニットがポリマーに導入されていることが確認された。
PEG-PBLAポリマー(318mg、0.016mmol)をDMF(3mL)で溶解し、得られた溶液に3,3-ジメトキシブタン(200μL)を添加した。反応液を40℃で72時間撹拌し、HCl溶液(100μL、0.1N)添加して1時間撹拌した。水で透析し、ポリマーを凍結乾燥することにより、側鎖に脂肪族ケトンが導入されたポリマーを回収した。
反応スキームを図6に示す。また、得られた脂肪族ケトン基含有ポリマーの1H-NMR解析結果を図7に示す。35個の脂肪族ケトンユニットがポリマーに導入されていることが確認された。
[実施例1]K252a-ヒドラジド(K252a-H)の合成
K252aは、BOC Science(US)から購入した。K252a(20mg)を無水メタノール(200μL)に溶解し、無水ヒドラジン(300μL)に添加した。反応混合物を40℃で15時間撹拌した。反応混合物をエバポレートして乾燥体を得た。エバポレーションでは、トルエンを共エバポレーション溶媒として用いた。得られた生成物を、さらなる精製を行うことなく、薬物複合体とミセルの調製に使用した。得られた生成物の1H-NMR解析結果を図8に示す。得られた生成物では、K252aのメチル基が消失し、ヒドラジド基プロトンのピークが確認されたことから、K252a-Hが得られたことが確認できた。また、K252a-HをHPLCで分析した。分析結果を図9に示す。K252a-HとK252aとは、リテンションタイムが異なっていることから、両者は異なる構造であることが確認された。なお、HPLC解析の条件は以下の通りである。
TSK-GEL ODS-100Vカラム 4.6×150mm、粒子径5μm(東ソー株式会社)
カラム圧:10.7MPa
カラム温度:40℃付近の一定の温度
移動相:メタノール/ギ酸緩衝液(pH3.0)混液(3:2)流速:1.2mL/分、20~30分
UV検出:波長290nm
K252aは、BOC Science(US)から購入した。K252a(20mg)を無水メタノール(200μL)に溶解し、無水ヒドラジン(300μL)に添加した。反応混合物を40℃で15時間撹拌した。反応混合物をエバポレートして乾燥体を得た。エバポレーションでは、トルエンを共エバポレーション溶媒として用いた。得られた生成物を、さらなる精製を行うことなく、薬物複合体とミセルの調製に使用した。得られた生成物の1H-NMR解析結果を図8に示す。得られた生成物では、K252aのメチル基が消失し、ヒドラジド基プロトンのピークが確認されたことから、K252a-Hが得られたことが確認できた。また、K252a-HをHPLCで分析した。分析結果を図9に示す。K252a-HとK252aとは、リテンションタイムが異なっていることから、両者は異なる構造であることが確認された。なお、HPLC解析の条件は以下の通りである。
TSK-GEL ODS-100Vカラム 4.6×150mm、粒子径5μm(東ソー株式会社)
カラム圧:10.7MPa
カラム温度:40℃付近の一定の温度
移動相:メタノール/ギ酸緩衝液(pH3.0)混液(3:2)流速:1.2mL/分、20~30分
UV検出:波長290nm
K252aからのK252a-Hの合成スキームを以下に示す。

[実施例2]K252a-Hとポリマーとの薬物複合体の調製
芳香族アルデヒド基含有ポリマー又は脂肪族ケトン基含有ポリマー、及びK252a-Hを、質量比2:1で混合し、DMSOに溶解した(1mLのDMSOあたり、15mgの薬物/ポリマー混合物を溶解)。芳香族アルデヒド基含有ポリマーとK252a-Hとの結合反応は、反応混合物を、40℃で一晩攪拌することにより行った。脂肪族ケトン基含有ポリマーとK252a-Hとの結合反応は、反応混合物を、96時間攪拌することにより行った。反応を1H-NMRでモニターし、K252a-Hがポリマーに完全に結合したことを確認した。脂肪族ケトン基含有ポリマーとK252a-Hとの結合を確認した1H-NMRの解析結果を図10に示す。脂肪族アルデヒド基含有ポリマーとK252a-Hとの結合を確認した1H-NMRの解析結果を図11に示す。
芳香族アルデヒド基含有ポリマー又は脂肪族ケトン基含有ポリマー、及びK252a-Hを、質量比2:1で混合し、DMSOに溶解した(1mLのDMSOあたり、15mgの薬物/ポリマー混合物を溶解)。芳香族アルデヒド基含有ポリマーとK252a-Hとの結合反応は、反応混合物を、40℃で一晩攪拌することにより行った。脂肪族ケトン基含有ポリマーとK252a-Hとの結合反応は、反応混合物を、96時間攪拌することにより行った。反応を1H-NMRでモニターし、K252a-Hがポリマーに完全に結合したことを確認した。脂肪族ケトン基含有ポリマーとK252a-Hとの結合を確認した1H-NMRの解析結果を図10に示す。脂肪族アルデヒド基含有ポリマーとK252a-Hとの結合を確認した1H-NMRの解析結果を図11に示す。
[実施例3]K252a-H結合ポリマーミセルの調製
(ミセルの調製)
DMSO中の反応混合物をジメチルアセトアミド(DMAc)で透析し、DMSOからDMAcへの溶媒交換を行った(4時間透析、透析溶媒は1度交換)。また、透析により、遊離のK252a-Hを除去した。上記のように得られたポリマーとK252a-Hとの薬物複合体のDMAc溶液をミセルの調製に使用した。前記DMAc溶液を、容量比で水10に対して1の割合で、水に滴下してボルテックスし、ミセルを調製した。この溶液を、分画分子量(MWCO)3500Daの透析バッグで24時間、水で透析した。透析媒体は、透析中に5回交換した。透析バッグ中の溶液をフィルター(0.22μm)ろ過し、100kDa MWCOのフィルターメンブラン(Amicon)を用いた限外濾過により濃縮した。なお、K252a-Hと上記ポリマーとの薬物複合体のミセルを、「K252a-H結合ポリマーミセル」ということがある。
(ミセルの調製)
DMSO中の反応混合物をジメチルアセトアミド(DMAc)で透析し、DMSOからDMAcへの溶媒交換を行った(4時間透析、透析溶媒は1度交換)。また、透析により、遊離のK252a-Hを除去した。上記のように得られたポリマーとK252a-Hとの薬物複合体のDMAc溶液をミセルの調製に使用した。前記DMAc溶液を、容量比で水10に対して1の割合で、水に滴下してボルテックスし、ミセルを調製した。この溶液を、分画分子量(MWCO)3500Daの透析バッグで24時間、水で透析した。透析媒体は、透析中に5回交換した。透析バッグ中の溶液をフィルター(0.22μm)ろ過し、100kDa MWCOのフィルターメンブラン(Amicon)を用いた限外濾過により濃縮した。なお、K252a-Hと上記ポリマーとの薬物複合体のミセルを、「K252a-H結合ポリマーミセル」ということがある。
(ミセルサイズとPDIの測定)
ミセルのサイズと多分散指数(PDI)を、動的光散乱(DLS)手法により求めた。
測定は、入射ビームとして緑色レーザー(532nm)を用い、173°の検出角度で、Zetasizer nano ZS(Malvern instruments,UK)を使用して、25℃の温度条件で行った。結果を図12A及び図12Bに示す。図12Aは、芳香族アルデヒド基含有ポリマーとK252a-Hとの薬物複合体のミセル(以下、「K252a-H結合芳香族ポリマーミセル」という。)であり、図12Bは、脂肪族ケトン基含有ポリマーとK252a-Hとの薬物複合体のミセル(以下、「K252a-H結合脂肪族ポリマーミセル」という。)である。K252a-H結合脂肪族ポリマーミセルは、K252a-H結合芳香族ポリマーミセルと比較して、ミセルサイズが小さいことが確認された。そのため、K252a-H結合脂肪族ポリマーミセルは、K252a-H結合芳香族ポリマーミセルよりも、がんへの集積性が高いと予想される。
ミセルのサイズと多分散指数(PDI)を、動的光散乱(DLS)手法により求めた。
測定は、入射ビームとして緑色レーザー(532nm)を用い、173°の検出角度で、Zetasizer nano ZS(Malvern instruments,UK)を使用して、25℃の温度条件で行った。結果を図12A及び図12Bに示す。図12Aは、芳香族アルデヒド基含有ポリマーとK252a-Hとの薬物複合体のミセル(以下、「K252a-H結合芳香族ポリマーミセル」という。)であり、図12Bは、脂肪族ケトン基含有ポリマーとK252a-Hとの薬物複合体のミセル(以下、「K252a-H結合脂肪族ポリマーミセル」という。)である。K252a-H結合脂肪族ポリマーミセルは、K252a-H結合芳香族ポリマーミセルと比較して、ミセルサイズが小さいことが確認された。そのため、K252a-H結合脂肪族ポリマーミセルは、K252a-H結合芳香族ポリマーミセルよりも、がんへの集積性が高いと予想される。
[実施例4]K252a-Hと薬物複合体ミセルの水に対する可溶化試験
K252a-H粉末を、蒸留水又はメタノールに1mg/mLで溶解し、よく混合した。その後、孔径22μmのフィルターでろ過した。前記のように調製したろ液について、分光光度計(ジャスコ V670)を用いて紫外吸収スペクトルを測定した。結果を図13に示す。図13の結果から、K252a-Hは、水には1μg/mL以下でしか溶解しないことが確認された。
一方、K252a-H結合芳香族ポリマーミセル及びK252a-H結合脂肪族ポリマーミセルを、水に溶解し、HPLCでK252a-Hの濃度測定を行ったところ、水に対して2mg/mL以上の溶解性を示すことが確認された(図示せず)。以上の結果より、K252a-Hのような水に難溶の化合物であっても、ミセル化することによって、水に対する溶解性を増大できることが示された。
K252a-H粉末を、蒸留水又はメタノールに1mg/mLで溶解し、よく混合した。その後、孔径22μmのフィルターでろ過した。前記のように調製したろ液について、分光光度計(ジャスコ V670)を用いて紫外吸収スペクトルを測定した。結果を図13に示す。図13の結果から、K252a-Hは、水には1μg/mL以下でしか溶解しないことが確認された。
一方、K252a-H結合芳香族ポリマーミセル及びK252a-H結合脂肪族ポリマーミセルを、水に溶解し、HPLCでK252a-Hの濃度測定を行ったところ、水に対して2mg/mL以上の溶解性を示すことが確認された(図示せず)。以上の結果より、K252a-Hのような水に難溶の化合物であっても、ミセル化することによって、水に対する溶解性を増大できることが示された。
[実施例5]K252a-H結合ポリマーミセルからのK252a-Hの放出
K252a-H結合脂肪族ポリマーミセルを、pH1の水性緩衝液(メタノール:ギ酸緩衝液(pH3)=3:2)中で、37℃で1時間インキュベートした。その後、HPLC解析を行った。HPLC条件は、実施例1と同様である。結果を図14に示す。pH1水性緩衝液処理後のサンプルで、K252a-Hと同一のピークが確認されたことから、酸性条件下で、K252a-H結合脂肪族ポリマーミセルからK252a-Hが放出されることが示された。
K252a-H結合脂肪族ポリマーミセルを、pH1の水性緩衝液(メタノール:ギ酸緩衝液(pH3)=3:2)中で、37℃で1時間インキュベートした。その後、HPLC解析を行った。HPLC条件は、実施例1と同様である。結果を図14に示す。pH1水性緩衝液処理後のサンプルで、K252a-Hと同一のピークが確認されたことから、酸性条件下で、K252a-H結合脂肪族ポリマーミセルからK252a-Hが放出されることが示された。
[実施例6]K252a類の肺がん細胞に対する細胞毒性試験
表1に示す肺がん細胞株を用いて、K252a、K252a-H、K252a-H結合芳香族ポリマーミセル及びK252a-H結合脂肪族ポリマーミセル(以下、まとめて「K252a類」ということがある。)の細胞毒性試験を行った。
以下の実施例において、使用した細胞株はアメリカン・タイプ・.カルチャー・コレクション(American Type Culture Collection,ATCC)またはJCRB細胞バンク(国立研究開発法人医薬基盤・健康・栄養研究所)より購入した。PC14 PE6は、法正先生(鳥取大学)より分与してもらった。培地は、特に記載していない限り、DMEM、RPMI又はE-MEMを使用した。Gefetinib、Afatinib、Erlotinib、Cisplatin、Pemetrexed、Gemicitabineは、フナコシ株式会社より購入した。Osimertinibは、Selleck Chemicals社より購入した。
表1に示す肺がん細胞株を用いて、K252a、K252a-H、K252a-H結合芳香族ポリマーミセル及びK252a-H結合脂肪族ポリマーミセル(以下、まとめて「K252a類」ということがある。)の細胞毒性試験を行った。
以下の実施例において、使用した細胞株はアメリカン・タイプ・.カルチャー・コレクション(American Type Culture Collection,ATCC)またはJCRB細胞バンク(国立研究開発法人医薬基盤・健康・栄養研究所)より購入した。PC14 PE6は、法正先生(鳥取大学)より分与してもらった。培地は、特に記載していない限り、DMEM、RPMI又はE-MEMを使用した。Gefetinib、Afatinib、Erlotinib、Cisplatin、Pemetrexed、Gemicitabineは、フナコシ株式会社より購入した。Osimertinibは、Selleck Chemicals社より購入した。

2000細胞/50μLの細胞溶液を適量調整し、96ウェルプレートの列2から2まで50μLずつ播種した。列1に培地(10% FBSを含むDMEM培地)のみを加えた。播種後、24時間インキュベートした。
24ウェルプレートの10個の各ウェルに、300μLの培地を加えた。薬物溶液100μLを24ウェルプレートのウェル1Aに加え、ピペッティング(10回程度)で撹拌した後、そこから100μLをウェル2Bへ移し、同様に撹拌した。同様の操作を10回繰り返し、10種類の濃度の薬物溶液を調整した。
上記で調整した薬物溶液を、がん細胞を播種した96ウェルプレートの列3から列12までミセル濃度の高い順に50μLずつ加えた。列1と2には、培地のみを50μL加えた。その後、48時間インキュベートした。
48時間後に、cell-counting kit-8溶液(DOJINDO)を各ウェルに10μLずつ添加した。再びインキュベーターし、30分後、1時間後、及び2時間後に、450nmの吸光度を測定した。
24ウェルプレートの10個の各ウェルに、300μLの培地を加えた。薬物溶液100μLを24ウェルプレートのウェル1Aに加え、ピペッティング(10回程度)で撹拌した後、そこから100μLをウェル2Bへ移し、同様に撹拌した。同様の操作を10回繰り返し、10種類の濃度の薬物溶液を調整した。
上記で調整した薬物溶液を、がん細胞を播種した96ウェルプレートの列3から列12までミセル濃度の高い順に50μLずつ加えた。列1と2には、培地のみを50μL加えた。その後、48時間インキュベートした。
48時間後に、cell-counting kit-8溶液(DOJINDO)を各ウェルに10μLずつ添加した。再びインキュベーターし、30分後、1時間後、及び2時間後に、450nmの吸光度を測定した。
結果を図15に示す。Gefenitib、Afatinib、及びOsimetinibは、現在FDAに認可されている肺がんの分子標的治療薬である。図15の結果から、K252a類(特に、K252a、K252a-H、及びK252a-H結合脂肪族ポリマーミセル)は、既存の肺がん治療薬耐性の肺がんに対して、高い細胞傷害活性を示すことが示された。
図15のPC14株とPC14・PE6株とを抽出したものを図16に示す。PC14・PE6株は、PC14株をマウスの尾静脈に投与し、胸腔内に転移したがん細胞を樹立する作業を6回行ったがん細胞株である(Yano et al., Oncology Research 9:573-579 (1997))。そのため、PC14・PE6株は、極めて転移能が高く、悪性化したがん細胞株である。図15及び図16に示すように、PC14株に対しては、Gefenitib、Afatinib、及びOsimetinibは高い細胞傷害活性を示した。しかし、悪性化したPC14・PE6株に対しては、これらの既存の分子標的薬は、細胞傷害活性が著しく低下した。一方、K252a類(特に、K252a-H及びK252a-H結合脂肪族ポリマーミセル)では、PC14・PE6株に対する細胞傷害活性の方が高くなった。これらの結果は、K252a類は、悪性化した転移がんに対して、より有効であることが示している。なお、K252a-H結合芳香族ポリマーミセルとK252a-H結合脂肪族ポリマーミセルとでは、K252a-H結合脂肪族ポリマーミセルの方ががん細胞に対する細胞傷害活性が高い傾向にあった。
[実施例7]K252a類の膵がん細胞に対する細胞毒性試験
各種膵がん細胞株を用いて、上記と同様に、K252a類の細胞毒性試験を行った。結果を図17に示す。BXPC3-F3株は、マウスへのBXPC3株の同所移植後、肝転移したがん細胞株を樹立することを3回繰り返して悪性化させた転移株である。また、KP-3Lは、ヒト肝転移膵臓がんをヌードマウスの脾臓に移植後、肝転移したがん細胞株を樹立した転移株である(国立研究開発法人 医薬基盤・健康・栄養研究所 JCRB細胞バンクより入手)。また、Gemcitabine及びErlotinibは、現在認可されている膵がん治療薬である。
図17に示すように、K252a類は、Gemcitabine及びErlotinibの効果が低いK-ras変異株及び悪性化転移株に対して、有効であることが確認された。なお、K252a-H結合芳香族ポリマーミセルとK252a-H結合脂肪族ポリマーミセルとでは、K252a-H結合脂肪族ポリマーミセルの方ががん細胞に対する細胞傷害活性が高い傾向にあった。
各種膵がん細胞株を用いて、上記と同様に、K252a類の細胞毒性試験を行った。結果を図17に示す。BXPC3-F3株は、マウスへのBXPC3株の同所移植後、肝転移したがん細胞株を樹立することを3回繰り返して悪性化させた転移株である。また、KP-3Lは、ヒト肝転移膵臓がんをヌードマウスの脾臓に移植後、肝転移したがん細胞株を樹立した転移株である(国立研究開発法人 医薬基盤・健康・栄養研究所 JCRB細胞バンクより入手)。また、Gemcitabine及びErlotinibは、現在認可されている膵がん治療薬である。
図17に示すように、K252a類は、Gemcitabine及びErlotinibの効果が低いK-ras変異株及び悪性化転移株に対して、有効であることが確認された。なお、K252a-H結合芳香族ポリマーミセルとK252a-H結合脂肪族ポリマーミセルとでは、K252a-H結合脂肪族ポリマーミセルの方ががん細胞に対する細胞傷害活性が高い傾向にあった。
[実施例8]K252a類の頭頸部がん細胞に対する細胞毒性試験
各種頭頚部がん細胞株を用いて、上記と同様に、K252a類の細胞毒性試験を行った。結果を図18に示す。なお、CDDPは、頭頸部がんの標準治療薬であるシスプラチンを示す。SAS株、FaDu株、HSC2株はいずれもシスプラチン耐性の細胞株である。
図18に示すように、K252a類は、いずれの頭頸部がん細胞株に対しても、シスプラチン及びErlotinibよりも高い細胞傷害活性を示した。特に、FaDu株では、K252a-H結合脂肪族ポリマーミセルの細胞傷害活性が高かった。この結果は、K252a類が、シスプラチン耐性株に対して、有効性があることを示す。
各種頭頚部がん細胞株を用いて、上記と同様に、K252a類の細胞毒性試験を行った。結果を図18に示す。なお、CDDPは、頭頸部がんの標準治療薬であるシスプラチンを示す。SAS株、FaDu株、HSC2株はいずれもシスプラチン耐性の細胞株である。
図18に示すように、K252a類は、いずれの頭頸部がん細胞株に対しても、シスプラチン及びErlotinibよりも高い細胞傷害活性を示した。特に、FaDu株では、K252a-H結合脂肪族ポリマーミセルの細胞傷害活性が高かった。この結果は、K252a類が、シスプラチン耐性株に対して、有効性があることを示す。
[実施例9]K252a類の悪性中皮腫細胞に対する細胞毒性試験
悪性中皮腫細胞株MSTO-211Hを用いて、上記と同様に、K252a類の細胞毒性試験を行った。結果を図19に示す。なお、CDDPはシスプラチンを示し、悪性中皮腫の標準治療薬である。Pemetrexedも悪性中皮腫の標準治療薬である。
図19に示すように、K252a類は、シスプラチン、Pemetrexed及びMidostaurinよりも高い細胞傷害活性を示した。
悪性中皮腫細胞株MSTO-211Hを用いて、上記と同様に、K252a類の細胞毒性試験を行った。結果を図19に示す。なお、CDDPはシスプラチンを示し、悪性中皮腫の標準治療薬である。Pemetrexedも悪性中皮腫の標準治療薬である。
図19に示すように、K252a類は、シスプラチン、Pemetrexed及びMidostaurinよりも高い細胞傷害活性を示した。
[実施例10]K252a-Hのホットスポットキナーゼプロファイリング
Rection Biology社に委託して、Monthly 202 mutant kinase panelを用いて、K252a-Hのキナーゼプロファイリングを行った。キナーゼアッセイの条件は、以下の通りである。K252aは、DMSOに溶解し、100nMでアッセイを行った。
バッファー条件:20mM HEPES、pH7.5、10mM MgCl2、1mM EGTA、(適用可能であれば、2mM MnCl2)
ATP濃度:10μM
反応時間:2時間
Rection Biology社に委託して、Monthly 202 mutant kinase panelを用いて、K252a-Hのキナーゼプロファイリングを行った。キナーゼアッセイの条件は、以下の通りである。K252aは、DMSOに溶解し、100nMでアッセイを行った。
バッファー条件:20mM HEPES、pH7.5、10mM MgCl2、1mM EGTA、(適用可能であれば、2mM MnCl2)
ATP濃度:10μM
反応時間:2時間
参考として、第1世代、第2世代、及び第3世代のチロシンキナーゼ阻害剤(tyrosine kinase inhibitor:TKI)と、その耐性変異を図20及び図21に示す。図20及び図21に示されるように、第1~第3世代TKIは、C797S変異に有効ではないため、C797S変異に有効なTKIが求められている。
EGFR変異に対するK252a-Hのキナーゼアッセイ結果を図22に示す。図22に示すように、K252a-Hは、多くのTKIが有効性を示さないゲートキーパー変異(d746-750/T790M、d746-750/T790M/C797S、L858R,T790Mなど)を有するキナーゼに対して、特異的に高い阻害活性を示した。
図23A~Cに、K252a-Hのキナーゼプロファイリングの結果を示す。図24に、K252a-Hに対するキナーゼプロファイリングの結果、K252a-Hによりキナーゼ活性が30%以下に抑制された突然変異キナーゼの一覧を示す。K252a-Hは、がん遺伝子であるc-kit/Flt3/RETの強力な阻害剤であることが確認された。
図23A~Cに、K252a-Hのキナーゼプロファイリングの結果を示す。図24に、K252a-Hに対するキナーゼプロファイリングの結果、K252a-Hによりキナーゼ活性が30%以下に抑制された突然変異キナーゼの一覧を示す。K252a-Hは、がん遺伝子であるc-kit/Flt3/RETの強力な阻害剤であることが確認された。
[実施例11]K252a-H及びスタウロスポリンのキナーゼプロファイリング
Rection Biology社に委託して、K252a-H及びスタウロスポリンのキナーゼプロファイリングを行った。キナーゼアッセイにおけるK252a又はスタウロスポリンの濃度は、指定した各濃度で行った。キナーゼパネルには、Monthly wild type kinase pannel又はMonthly Mutant kinase pannelを用いた。アッセイは、各濃度につきduplicateで行った。試験した各薬物濃度のキナーゼ活性及びIC50のデータを取得し、duplicateで3回行うことによりN=6で平均値をもとめた。各薬物濃度のキナーゼ活性を活性阻害率(%)に変換し、Prism7により解析してグラフ化した。
Rection Biology社に委託して、K252a-H及びスタウロスポリンのキナーゼプロファイリングを行った。キナーゼアッセイにおけるK252a又はスタウロスポリンの濃度は、指定した各濃度で行った。キナーゼパネルには、Monthly wild type kinase pannel又はMonthly Mutant kinase pannelを用いた。アッセイは、各濃度につきduplicateで行った。試験した各薬物濃度のキナーゼ活性及びIC50のデータを取得し、duplicateで3回行うことによりN=6で平均値をもとめた。各薬物濃度のキナーゼ活性を活性阻害率(%)に変換し、Prism7により解析してグラフ化した。
Monthly Mutant kinase pannelを用いたキナーゼプロファイリング結果を図25~34Dに示す。図25~32中、実線で囲んだキナーゼは、薬剤耐性に関与する変異を有し、試験した薬剤による阻害活性が高いものを示す。これらの変異を表2にまとめた。スタウロスポリン及びK252aは、表2に記載の変異を有するキナーゼの阻害活性が高いことが示された。また、スタウロスポリン及びK252aは、EGFRのオシメルチニブ耐性の3重変異(d746-750/T790M/C797S,L858R/T790M/C797S)に対して、高い阻害効果を有することが示された。また、HER2のゲートキーパー変異(T798I)を有するキナーゼは、アッセイに用いたキナーゼパネルにはなかったが、HER2はEGFRのホモログであるため、EGFRのゲートキーパー変異と同じくスタウロスポリン及びK252aによる阻害効果が高いと予想される。図33A~Dは、500nMのK252a-Hによるキナーゼプロファイリングの結果を示す。図34A~Dは、20nMのスタウロスポリンによるキナーゼプロファイリングの結果を示す。

Monthly wild type kinase pannelを用いたキナーゼプロファイリングの結果から、各wild typeキナーゼに対するK252a-HのIC50を求めた。その結果、K252a-HのIC50が5nM以下であったキナーゼを表3に示す。表3中、下線で示すキナーゼは、薬剤耐性に関与するとされるキナーゼである。

Monthly wild type kinase pannelを用いたキナーゼプロファイリングの結果から、各wild typeキナーゼに対するスタウロスポリンのIC50を求めた。その結果、スタウロスポリンのIC50が0.2nM以下であったキナーゼを図35に示す。図35中、下線で示すキナーゼは、がん(特に薬剤耐性がん)の治療標的として重要とされるキナーゼである。スタウロスポリンは、CaMK2a、CaMK2b、JAK3、ROS1、RSK4、STK22、TRKBに対するIC50が0.1nM以下であり、特に高い阻害効果を示した。これらの結果は、低濃度のスタウロスポリンが、これらのキナーゼを発現するがんに対して、特異性が高いことを示している。
[実施例12]K252a-H及びスタウロスポリンのがん細胞に対する細胞毒性(細胞株)
PC14、A549、H358、H2228、PancI、Mia-Paca2、SAS、FaDU、及びHSC2は、ATCCより購入した。H460、NCI-1650、H1975、H520、BxPC3、KP3L、及びAsPC-1は、JCRB細胞バンクより購入した。
PC14-PE6は、PC14をマウスに尾静脈投与し、胸腔内へ転移した細胞を樹立する作業を6回行った、極めて転移能の高い、悪性化した細胞株(Yano et al , OncologyResearch 9:573-579 (1997))である。PC14-PE6に対して、さらに、Cignal Lenti positive control(Luc)(Qiagen)をtransductionし、クローニングを行い、PC14-PE6-Lucを作製した。
BxPC3-F3は、BxPC3を悪性化させた肝転移株である。BxPC3-F3は、マウスの膵臓にBxPC3を同所移植後、肝転移した細胞を肝臓からトリプシン処理によって単離して、細胞株を樹立し、その作業(細胞株樹立)を3回繰り返すことにより作製した。
PC14、A549、H358、H2228、PancI、Mia-Paca2、SAS、FaDU、及びHSC2は、ATCCより購入した。H460、NCI-1650、H1975、H520、BxPC3、KP3L、及びAsPC-1は、JCRB細胞バンクより購入した。
PC14-PE6は、PC14をマウスに尾静脈投与し、胸腔内へ転移した細胞を樹立する作業を6回行った、極めて転移能の高い、悪性化した細胞株(Yano et al , OncologyResearch 9:573-579 (1997))である。PC14-PE6に対して、さらに、Cignal Lenti positive control(Luc)(Qiagen)をtransductionし、クローニングを行い、PC14-PE6-Lucを作製した。
BxPC3-F3は、BxPC3を悪性化させた肝転移株である。BxPC3-F3は、マウスの膵臓にBxPC3を同所移植後、肝転移した細胞を肝臓からトリプシン処理によって単離して、細胞株を樹立し、その作業(細胞株樹立)を3回繰り返すことにより作製した。
(抗がん剤)
抗がん剤(Gefetinib、Afetinib、Osimertinib、Gemicitabine、Cisplatin、Erlotinib、Pemetrexed、Midostaurin)は、フナコシより購入した。
抗がん剤(Gefetinib、Afetinib、Osimertinib、Gemicitabine、Cisplatin、Erlotinib、Pemetrexed、Midostaurin)は、フナコシより購入した。
(細胞毒性の測定方法)
試験薬剤の細胞毒性は、cell counting kit8(DOJINDO,JAPAN)を用いて、以下の方法で測定した。1~3×103cell/ウェルでがん細胞株を播種した。翌日、培地を交換し、50μLの培地を加えた。さらに、試験薬剤の希釈シリーズを作製し、50μLをウェルに添加して攪拌した。72時間後、10μLのcell counting kit 8を加え、1時間経過後、マイクロプレートリーダー(TECAN)で450nmの吸光度を測定した。測定した吸光度に基づき、下記の式(1)により、試験薬剤毎に各希釈率における細胞生存率を算出した。前記算出された細胞生存率に基づき、試験薬剤のIC50を算出した。
試験薬剤の細胞毒性は、cell counting kit8(DOJINDO,JAPAN)を用いて、以下の方法で測定した。1~3×103cell/ウェルでがん細胞株を播種した。翌日、培地を交換し、50μLの培地を加えた。さらに、試験薬剤の希釈シリーズを作製し、50μLをウェルに添加して攪拌した。72時間後、10μLのcell counting kit 8を加え、1時間経過後、マイクロプレートリーダー(TECAN)で450nmの吸光度を測定した。測定した吸光度に基づき、下記の式(1)により、試験薬剤毎に各希釈率における細胞生存率を算出した。前記算出された細胞生存率に基づき、試験薬剤のIC50を算出した。
Ac:陰性対照(培地、細胞及びCell Counting Kit溶液の入ったウェル(試験薬剤なし))の吸光度
Ab:ブランク(培地及びCell Counting Kit溶液の入ったウェル(細胞なし))の吸光度
(非小細胞肺がん)
非小細胞肺がん細胞株に対する細胞毒性試験の結果を図36に示す。各細胞株の特徴は、上記表1に示すとおりである。公知の肺がん治療薬(Gefetinib、Afatinib、Osimertinib、Dasatinib)は、PC14に対して高い細胞毒性を示したが、Gefetinib、Afatinib、及びOsimertinibは、PC14の悪性転移株であるPC14-PE6に対しては、細胞毒性が低かった。
一方、スタウロスポリン、K252a、及びK252a-Hは、PC14よりもPC14ー-PE6に対して、より高い効果を示した。
PC14は、公知の肺がん治療薬(Gefetinib、Afatinib、Osimertinib、Dasatinib)に対して感受性を示したが、PC14-PE6、A549、H358、NCI1650、及びH520は、公知の肺がん治療薬に耐性であった。一方、スタウロスポリン、K252a、及びK252a-Hは、これらの肺がん治療薬耐性株に対しても高い細胞毒性を示す傾向にあった。
非小細胞肺がん細胞株に対する細胞毒性試験の結果を図36に示す。各細胞株の特徴は、上記表1に示すとおりである。公知の肺がん治療薬(Gefetinib、Afatinib、Osimertinib、Dasatinib)は、PC14に対して高い細胞毒性を示したが、Gefetinib、Afatinib、及びOsimertinibは、PC14の悪性転移株であるPC14-PE6に対しては、細胞毒性が低かった。
一方、スタウロスポリン、K252a、及びK252a-Hは、PC14よりもPC14ー-PE6に対して、より高い効果を示した。
PC14は、公知の肺がん治療薬(Gefetinib、Afatinib、Osimertinib、Dasatinib)に対して感受性を示したが、PC14-PE6、A549、H358、NCI1650、及びH520は、公知の肺がん治療薬に耐性であった。一方、スタウロスポリン、K252a、及びK252a-Hは、これらの肺がん治療薬耐性株に対しても高い細胞毒性を示す傾向にあった。
(膵臓がん)
膵臓がん細胞株に対する細胞毒性試験の結果を図37に示す。図37中、BxPC3-F3は、BxPC3の悪性化肝転移株である。KP3Lは、ヒトの肝転移膵臓がんをヌードマウス脾臓に移植後、マウス肝臓に転移した転移株である。膵臓がんの標準治療薬(Gemicitabine、Erlotinib)は、BxPC3に対して高い細胞毒性を示したが、BxPC3-F3及びKP3Lに対しては、細胞毒性が低かった。
一方、スタウロスポリン、K252a、及びK252a-Hは、これらの膵臓がん治療薬耐性株に対しても高い細胞毒性を示した。
膵臓がん細胞株に対する細胞毒性試験の結果を図37に示す。図37中、BxPC3-F3は、BxPC3の悪性化肝転移株である。KP3Lは、ヒトの肝転移膵臓がんをヌードマウス脾臓に移植後、マウス肝臓に転移した転移株である。膵臓がんの標準治療薬(Gemicitabine、Erlotinib)は、BxPC3に対して高い細胞毒性を示したが、BxPC3-F3及びKP3Lに対しては、細胞毒性が低かった。
一方、スタウロスポリン、K252a、及びK252a-Hは、これらの膵臓がん治療薬耐性株に対しても高い細胞毒性を示した。
(頭頸部がん)
膵臓がん細胞株に対する細胞毒性試験の結果を図38に示す。図38中、「CDDP」は、シスプラチンを表す。いずれの細胞株においても、スタウロスポリン、K252a、及びK252a-Hは、公知の頭頸部がん治療薬(Midostaurin、シスプラチン、Erlotinib、Gefetinib)よりも高い細胞毒性を示した。特に、Midostaurin耐性株であるSASに対しても、スタウロスポリン、K252a、及びK252a-Hは、高い細胞毒性を示した。
膵臓がん細胞株に対する細胞毒性試験の結果を図38に示す。図38中、「CDDP」は、シスプラチンを表す。いずれの細胞株においても、スタウロスポリン、K252a、及びK252a-Hは、公知の頭頸部がん治療薬(Midostaurin、シスプラチン、Erlotinib、Gefetinib)よりも高い細胞毒性を示した。特に、Midostaurin耐性株であるSASに対しても、スタウロスポリン、K252a、及びK252a-Hは、高い細胞毒性を示した。
(悪性中皮腫)
悪性中皮腫細胞株に対する細胞毒性試験の結果を図39に示す。図39中、「CDDP」は、シスプラチンを表す。スタウロスポリン、K252a、及びK252a-Hは、公知の悪性中皮腫治療薬(Midostaurin、シスプラチン、Pemetrexed)よりも高い細胞毒性を示した。
悪性中皮腫細胞株に対する細胞毒性試験の結果を図39に示す。図39中、「CDDP」は、シスプラチンを表す。スタウロスポリン、K252a、及びK252a-Hは、公知の悪性中皮腫治療薬(Midostaurin、シスプラチン、Pemetrexed)よりも高い細胞毒性を示した。
[実施例13]K252a-Hのin vivo抗腫瘍活性(非小細胞肺がん)
Balb/cヌードマウスにイソフルラン麻酔を行い、5x105 cells/20%マトリゲル 50μLの非小細胞肺がん細胞株(PC14-PE6-luc:PC14-PE6にルシフェラーゼ遺伝子を導入した細胞)を、直接、前記マウスの肺に注入した。
注射針の先端から5mmの位置に目印をつけ、肋骨の3~4番目の位置から肺の下葉に向かって、前記目印まで注射針を差し込み、非小細胞肺がん細胞株の20%マトリゲル懸濁液50μLを、直接、肺に注射した。このようにして、ヒト非小細胞肺がん同所移植モデルマウスを作製した。
がん細胞注入の5日後から、ヒト非小細胞肺がん同所移植モデルマウスに、1mg/体重kgの投与量で、週に2回、薬剤を尾静脈注投与した。週2回、イソフルラン麻酔下で、IVISスペクトル(Xwnogen Corporation)を用いて、腫瘍の成長をモニタリングし、薬剤の抗腫瘍効果を評価した。腫瘍の大きさ及び生存日数の統計処理は、Prism7を使用して行った。試験薬剤には、K252a―H、及びK252a-H結合脂肪族ポリマーミセル(実施例3で調製)を用いた。陰性対照としてPBSを用いた。
Balb/cヌードマウスにイソフルラン麻酔を行い、5x105 cells/20%マトリゲル 50μLの非小細胞肺がん細胞株(PC14-PE6-luc:PC14-PE6にルシフェラーゼ遺伝子を導入した細胞)を、直接、前記マウスの肺に注入した。
注射針の先端から5mmの位置に目印をつけ、肋骨の3~4番目の位置から肺の下葉に向かって、前記目印まで注射針を差し込み、非小細胞肺がん細胞株の20%マトリゲル懸濁液50μLを、直接、肺に注射した。このようにして、ヒト非小細胞肺がん同所移植モデルマウスを作製した。
がん細胞注入の5日後から、ヒト非小細胞肺がん同所移植モデルマウスに、1mg/体重kgの投与量で、週に2回、薬剤を尾静脈注投与した。週2回、イソフルラン麻酔下で、IVISスペクトル(Xwnogen Corporation)を用いて、腫瘍の成長をモニタリングし、薬剤の抗腫瘍効果を評価した。腫瘍の大きさ及び生存日数の統計処理は、Prism7を使用して行った。試験薬剤には、K252a―H、及びK252a-H結合脂肪族ポリマーミセル(実施例3で調製)を用いた。陰性対照としてPBSを用いた。
結果を図40及び図41に示す。図40は腫瘍の成長を示し、図41はカプランマイヤー法によるマウスの生存曲線を示す。K252a-H結合脂肪族ポリマーミセルを投与した場合、PBSを投与した場合と比較して、腫瘍の成長が有意に抑制され、生存期間も有意に延長した。K252a-Hを投与した場合、PBSを投与した場合と比較して、生存期間が延長する傾向があった。これらの結果から、K252a類(特に、K252a-H結合脂肪族ポリマーミセル)が、オシメルチニブ耐性肺がんに対して、in vivoでも抗腫瘍効果を示すことが確認された。
(膵臓がん:同所移植モデルマウスを用いた試験)
がん細胞株としてBxPC3-F3-luc(BxPC3-F3にルシフェラーゼ遺伝子を導入した細胞)を用い、がん細胞株を膵臓に調節注入したこと以外は、上記「(非小細胞肺がん)」で記載した方法と同様の方法で、ヒト膵がん同所移植モデルマウスを作製した。
上記のように作製したヒト膵がん同所移植モデルマウスに、薬剤の種類と薬剤の投与量以外は、上記「(非小細胞肺がん)」で記載した方法と同様の方法で、薬剤の投与を行った。
薬剤として、K252a-H、K252a-H結合脂肪族ポリマーミセル、K252a-H結合芳香族ポリマーミセル(実施例3で調製)、及びゲムシタビンを用いた。陰性対照としてPBSを用いた。各薬剤の1回あたりの投与量は、K252a-H、K252a-H結合脂肪族ポリマーミセル、及びK252a-H結合芳香族ポリマーミセルについては、3mg/体重kgとし、ゲムシタビンについては、5mg/体重kgとした。
がん細胞株としてBxPC3-F3-luc(BxPC3-F3にルシフェラーゼ遺伝子を導入した細胞)を用い、がん細胞株を膵臓に調節注入したこと以外は、上記「(非小細胞肺がん)」で記載した方法と同様の方法で、ヒト膵がん同所移植モデルマウスを作製した。
上記のように作製したヒト膵がん同所移植モデルマウスに、薬剤の種類と薬剤の投与量以外は、上記「(非小細胞肺がん)」で記載した方法と同様の方法で、薬剤の投与を行った。
薬剤として、K252a-H、K252a-H結合脂肪族ポリマーミセル、K252a-H結合芳香族ポリマーミセル(実施例3で調製)、及びゲムシタビンを用いた。陰性対照としてPBSを用いた。各薬剤の1回あたりの投与量は、K252a-H、K252a-H結合脂肪族ポリマーミセル、及びK252a-H結合芳香族ポリマーミセルについては、3mg/体重kgとし、ゲムシタビンについては、5mg/体重kgとした。
結果を図42に示す。ゲムシタビンを投与した場合には、PBSを投与した場合と同様に腫瘍が成長した。一方、K252a類(K252a-H、K252a-H結合脂肪族ポリマーミセル、K252a-H結合芳香族ポリマーミセル)を投与した場合には、PBSを投与した場合と比較して、腫瘍の成長が抑制された。特に、K252a-H結合脂肪族ポリマーミセルを投与した場合には、腫瘍の成長が有意に抑制された。これらの結果から、K252a類(特に、K252a-H結合脂肪族ポリマーミセル)が、ゲムシタビン耐性膵臓がんに対して、in vivoでも抗腫瘍効果を示すことが確認された。
(膵臓がん:膵がん自然発症マウスを用いた試験)
膵がんを自然発症するトランスジェニックマウス(Capiler corp)を購入し、膵がん自然発症モデルマウス(EL-1-luc/TAg)を作製した(Proc Natl Acad Sci U S A. 2013 Jul 9; 110(28): 11397-11402.)。膵がんが発生する13~15週令から、薬剤の投与を開始した。薬剤の投与方法は、「(非小細胞肺がん)」で記載した方法と同様とした。薬剤として、K252a-H結合脂肪族ポリマーミセル及びK252a-H結合芳香族ポリマーミセルを用いた。陰性対照としてPBSを用いた。各薬剤の1回あたりの投与量は、1mg/体重kgとした。
膵がんを自然発症するトランスジェニックマウス(Capiler corp)を購入し、膵がん自然発症モデルマウス(EL-1-luc/TAg)を作製した(Proc Natl Acad Sci U S A. 2013 Jul 9; 110(28): 11397-11402.)。膵がんが発生する13~15週令から、薬剤の投与を開始した。薬剤の投与方法は、「(非小細胞肺がん)」で記載した方法と同様とした。薬剤として、K252a-H結合脂肪族ポリマーミセル及びK252a-H結合芳香族ポリマーミセルを用いた。陰性対照としてPBSを用いた。各薬剤の1回あたりの投与量は、1mg/体重kgとした。
図43は、自然発症膵がんEL1-luc/TAg細胞をマウス膵臓より取りだし、細胞株化したものに対するスタウロスポリン及びK252a類のIC50を示す。IC50は、実施例12に記載の方法と同様の方法で算出した。図43に示すように、EL-1/TAgは、ゲムシタビン及びエルロチニブに耐性を有する。一方、EL-1/TAgは、スタウロスポリン及びK252a類に対して感受性であった。
図44及び図45は、膵がん自然発症モデルマウスを用いた抗腫瘍効果評価試験の結果を示す図である。図44は腫瘍の成長を示し、図45はカプランマイヤー法によるマウスの生存曲線を示す。K252a-H結合脂肪族ポリマーミセル及びK252a-H結合脂肪族ポリマーミセルのいずれを投与した場合も、PBSを投与した場合と比較して、腫瘍の成長が有意に抑制された。K252a-H結合脂肪族ポリマーミセルを投与した場合は、PBSを投与した場合と比較して、生存期間が有意に延長した。K252a-H結合芳香族ポリマーミセルを投与した場合も、PBSを投与した場合と比較して、生存期間が延長する傾向があった。これらの結果から、K252a類(特に、K252a-H結合脂肪族ポリマーミセル)が、ゲムシタビン及びエルロチニブ耐性膵がんに対して、in vivoでも抗腫瘍効果を示すことが確認された。
[実施例14]K252a類のリンパ腫に対する細胞毒性
実施例12に記載した方法と同様の方法で、リンパ腫細胞株に対するK252a類のIC50を算出した。
結果を図46に示す。リンパ腫細胞株に対しても、K252a類(特に、K252a、K252a-H、K252a―H結合脂肪族ポリマーミセル)は、高い細胞毒性を示した。
実施例12に記載した方法と同様の方法で、リンパ腫細胞株に対するK252a類のIC50を算出した。
結果を図46に示す。リンパ腫細胞株に対しても、K252a類(特に、K252a、K252a-H、K252a―H結合脂肪族ポリマーミセル)は、高い細胞毒性を示した。
[実施例15]ABCトランスポーター(MDR-1)の薬剤排出阻害効果
ヒト腎がん細胞株である786-0は、MDR-1(ABCB-1,p-gp)を高発現している細胞株である(データは示さず)。そこで、786-0を用いて、MDR-1の薬剤排出活性に対するスタウロスポリン及びK252a類の影響を評価した。
薬剤排出活性の評価は、eFluxx-IDTM Green Multidrug Resistance Assay kit(Enzo Life Science)を用いて行った。786-0細胞をウェルプレートで培養後、トリプシン・EDTA溶液を加えてインキュベートし、細胞をウェルから剥がした。剥がした細胞を回収して培地で洗浄した後、1x106cell/mLの濃度で細胞を培地に懸濁して、細胞懸濁液を得た。細胞懸濁液を37℃で10分間以上予備加温しておき、250μLの細胞懸濁液(2.5x105 cell)に対して、希釈系列の薬剤を添加した。
薬剤として、スタウロスポリン、K252a、K252a-H、及びスニチニブを用いた。陽性対照としてMDR-1阻害剤であるベラパミル(終濃度30μM)を用いた。
薬剤添加後、eFluxx-IDTM Green検出試薬を加えて、37℃で30分間インキュベートし、終濃度0.5μg/mLのDAPIを加えて、フローサイトメーター(BD,Cell Analyzer LSRFortessa X-20)で解析した。フローサイトメーターによる解析結果から、eFluxx-GFPの排出量が半分になる薬剤濃度としてLD50を算出した。
ヒト腎がん細胞株である786-0は、MDR-1(ABCB-1,p-gp)を高発現している細胞株である(データは示さず)。そこで、786-0を用いて、MDR-1の薬剤排出活性に対するスタウロスポリン及びK252a類の影響を評価した。
薬剤排出活性の評価は、eFluxx-IDTM Green Multidrug Resistance Assay kit(Enzo Life Science)を用いて行った。786-0細胞をウェルプレートで培養後、トリプシン・EDTA溶液を加えてインキュベートし、細胞をウェルから剥がした。剥がした細胞を回収して培地で洗浄した後、1x106cell/mLの濃度で細胞を培地に懸濁して、細胞懸濁液を得た。細胞懸濁液を37℃で10分間以上予備加温しておき、250μLの細胞懸濁液(2.5x105 cell)に対して、希釈系列の薬剤を添加した。
薬剤として、スタウロスポリン、K252a、K252a-H、及びスニチニブを用いた。陽性対照としてMDR-1阻害剤であるベラパミル(終濃度30μM)を用いた。
薬剤添加後、eFluxx-IDTM Green検出試薬を加えて、37℃で30分間インキュベートし、終濃度0.5μg/mLのDAPIを加えて、フローサイトメーター(BD,Cell Analyzer LSRFortessa X-20)で解析した。フローサイトメーターによる解析結果から、eFluxx-GFPの排出量が半分になる薬剤濃度としてLD50を算出した。
結果を図47に示す。スタウロスポリン、K252a、及びK252a-Hは、最もよく用いられるMDR-1阻害剤であるベラパミルよりもLD50が低く、MDR-1阻害効果が高いことが確認された。
[実施例16]ヒトα1-AGPの影響
ヒト血清蛋白質であるヒトα1-AGP(acid glycoprotein)の影響を評価するため、ヒトα1-AGP(以下、「hAGP」とも記載する。)の存在下で、K252a-H及びK252a-H結合脂肪族ポリマーミセルの細胞属性を評価した。
細胞毒性の評価は、cell counting kit8(DOJINDO,JAPAN)を用いて行った。1~3×103cell/ウェルで、肺がん細胞株であるH460をウェルに播種し、培養を行った。翌日、培地を交換し、終濃度0.5mg/mLとなるようにhAGPを加えた。対照として、hAGPを含まない培地を50μL加えたものも用意した。次いで、1μMとなるように薬剤(K252a-H又はK252a-H結合脂肪族ポリマーミセル)を添加した。48時間後、10μLのcell counting kit 8を加えた。1時間経過後、マイクロプレートリーダー(TECAN)で450nmの吸光度を測定した。測定した吸光度に基づき、上記式(1)により、細胞生存率を算出した。
ヒト血清蛋白質であるヒトα1-AGP(acid glycoprotein)の影響を評価するため、ヒトα1-AGP(以下、「hAGP」とも記載する。)の存在下で、K252a-H及びK252a-H結合脂肪族ポリマーミセルの細胞属性を評価した。
細胞毒性の評価は、cell counting kit8(DOJINDO,JAPAN)を用いて行った。1~3×103cell/ウェルで、肺がん細胞株であるH460をウェルに播種し、培養を行った。翌日、培地を交換し、終濃度0.5mg/mLとなるようにhAGPを加えた。対照として、hAGPを含まない培地を50μL加えたものも用意した。次いで、1μMとなるように薬剤(K252a-H又はK252a-H結合脂肪族ポリマーミセル)を添加した。48時間後、10μLのcell counting kit 8を加えた。1時間経過後、マイクロプレートリーダー(TECAN)で450nmの吸光度を測定した。測定した吸光度に基づき、上記式(1)により、細胞生存率を算出した。
結果を図48に示す。K252a-H結合脂肪族ポリマーミセルでは、hAGP存在下でも細胞生存率が低く維持されていた。この結果より、K252a-H結合脂肪族ポリマーミセルでは、K252aと比較してhAGPに対する親和性が低くなり、hAGPによる不活性化を回避して、がん細胞に対する細胞毒性を維持することが確認された。
[実施例17]スタウロスポリン内包ミセルの調製
スタウロスポリン内包ミセルを調製するため、スタウロスポリンに下記式(L1)で表されるリンカー試薬(L1)を結合させた。リンカー結合反応のスキームを図49に示す。図49中、「DIC」は、ジイソプロピルカルボジイミドを表す。図49中の活性化エステルとしては、N-ヒドロキシコハク酸イミド(NHS)、1-ヒドロキシベンゾトリアゾール(HOBt)、1-ヒドロキシ-7-アザベンゾトリアゾール(HOAt)、ペンタフルオロフェノール、p-ニトロフェノール等から誘導される活性エステルが挙げられる。
スタウロスポリン内包ミセルを調製するため、スタウロスポリンに下記式(L1)で表されるリンカー試薬(L1)を結合させた。リンカー結合反応のスキームを図49に示す。図49中、「DIC」は、ジイソプロピルカルボジイミドを表す。図49中の活性化エステルとしては、N-ヒドロキシコハク酸イミド(NHS)、1-ヒドロキシベンゾトリアゾール(HOBt)、1-ヒドロキシ-7-アザベンゾトリアゾール(HOAt)、ペンタフルオロフェノール、p-ニトロフェノール等から誘導される活性エステルが挙げられる。

N,N-ジメチルホルムアミド(DMF)中で、上記結合反応を行い、反応終了後、分液により水溶成分を除去した。薄層クロマトグラフィーにより、スタウロスポリンの完全な消費と新フラクションの生成を確認した。有機相成分の1H NMR解析結果を図50に示す。芳香族成分の増加と、エステル成分(~4.0 ppm)の出現により、スタウロスポリンにリンカーが結合していることが示唆された。
活性エステルリンカーを結合したスタウロスポリンは、ポリエチレングリコール(PEG)等のポリマーに結合させて、ポリマーとの薬物複合体を形成させることができる。図51上図は、リンカーを介して、スタウロスポリンに、ポリエチレングリコールを結合させる反応である。図51下図は、リンカーを介して、スタウロスポリンにPEG-ポリアミノ酸ブロックコポリマーを結合させる反応である。(図51下図)。
ポリマー-リンカー-スタウロスポリン複合体は、細胞内で、グルタチオントランスフェラーゼ(GST)によりグルタチオン(GSH)と反応し、スタウロスポリンが放出される。下記に、PEG-リンカー-スタウロスポリン複合体から、グルタチオントランスフェラーゼ(GST)により、スタウロスポリンが放出される反応を示す。

スタウロスポリンに、リンカー試薬(L1)を反応させて得られた化合物(S’L1)は、実施例1と同様に、無水メタノール溶媒下で、無水ヒドラジンと反応させることにより、ヒドラジド基を導入することができる(化合物(S’L1-H))。当該スキームを下記に示す。

さらに、実施例2と同様に、化合物(S’L1-H)を、ポリマー合成例1~5で合成したポリマーに結合させることができる。化合物(S’L1-H)を、芳香族アルデヒド基含有ポリマーに結合させた薬物複合体を下記に例示する。下記式において、結合(I)は、pH感受性の結合を表す。結合(II)は、GST存在下でGSHと反応するGSH感受性の結合を表す。

化合物(S’L1-H)を、脂肪族ケトン基含有ポリマーに結合させた薬物複合体を下記に例示する。

上記結合(I)及び結合(II)を含む薬物複合体を生体に投与すると、まず、腫瘍組織周辺の低pH環境下で、結合(I)が切断され、化合物(S’L1-H)が放出される。次に、化合物(S’L1-H)が腫瘍細胞内に取り込まれ、GSTによりGSHと反応して、結合(II)が切断され、スタウロスポリンが生じる。当該スタウロスポリンにより、腫瘍細胞が殺傷される。
Claims (9)
- 下記一般式(S)で表される化合物(S)、若しくはその薬学的に許容される塩、又は薬学的に許容されるポリマーに前記化合物(S)若しくはその薬学的に許容される塩が結合した薬物複合体を含む、抗がん剤耐性のがんを治療又は予防するための医薬組成物。

X及びYは、それぞれ独立に、H、-OH、Cl、プロポキシ、又はエチルチオメチルであり;
R6は、H、C1-3アルキル、-NH2、ベンジル、


R7及びR8は、それぞれ独立に、H、-OH、又はメトキシであるか、或いは一緒になってO=を形成してもよく;
R9及びR10は、それぞれ独立に、H、メチル、β-D-グルコピラノシル、4-O-メチル-β-D-グルコピラノシル、シアノエチル、又は



R11は、メチルであり;
R12は、Hであり;
R13及びR14は、それぞれ独立に、H、メトキシ、-OH、ヒドロキシメチル、メチルカルボキシレート、メチルアミノ、メチルアミノメチル、プロピルアミノメチル、ジメチルアミノメチル、メトキシカルボニル、


R15及びR16は、それぞれ独立に、H、-OH、又は

R17及びR18は、H、-OH、メチルアミノ、ジメチルアミノ、オキシム、又は下記のいずれかの式で表される基

R19は、メトキシ、ヒドロキシ、-NH-NH2、又は活性エステルを含む基である。] - 前記抗がん剤が、ゲフィチニブ、アファチニブ、オシメルチニブ、ダサチニブ、エルロチニブ、ゲムシタビン、シスプラチン、ペメトレキセド、及びミドスタウリンからなる群より選択される少なくとも1種の抗がん剤である、請求項1に記載の医薬組成物。
- 前記抗がん剤が、キナーゼを標的とする分子標的薬である、請求項1又は2に記載の医薬組成物。
- 前記キナーゼが、EGFR、ABL1、ALK1、HER2、c-Kit、FGFR1、FGFR2、FGFR3、c-Src、PDGFRa、RET、DDR2、TRKA、及びFlt-3からなる群より選択される少なくとも1種のキナーゼである、請求項3に記載の医薬組成物。
- 前記抗がん剤耐性のがんが、ゲートキーパー変異を有するキナーゼを発現するがんである、請求項1~4のいずれか一項に記載の医薬組成物。
- 前記キナーゼが、d746-750、T790M、及びC797Sの変異を有するEGFRである、請求項5に記載の医薬組成物。
- 前記化合物(S)が、スタウロスポリン、7-ヒドロキシスタウトスポリン、KT5926、スタウロスポリン・アグリコン、SF2370、KT5823、4’-N-ベンゾイルスタウロスポリン、Go6976、N,N-ジメチルスタウロスポリン、NA 0359、N-エトキシカルボニル-7-オキソスタウロスポリン、KT-6124、CGP42700、4’-デメチルアミノ-4’,5’-ジヒドロキシスタウロスポリン、7-オキソスタウロスポリン、CEP751、NA0346、NA0359、3’-デメトキシ-3’-ヒドロキシスタウロスポリン、KT 6006、7-O-メチル-UCN 01、TAN 999、NA 0346、NA 0345、NA 0344、CGP 44171A、SCH 47112、N,N-ジメチルスタウロスポリン、TAN 1030A、レスタウルチニブ、4’-デメチルアミノ-4’-ヒドロキシスタウロスポリン、AFN941、エドテカリン、ベカテカリン、K252a、及びK252aヒドラジドからなる群より選択される少なくとも1種の化合物である、請求項1~6のいずれか一項に記載の医薬組成物。
- 前記化合物(S)が、スタウロスポリン、K252a、及びK252aヒドラジドからなる群より選択される少なくとも1種の化合物である、請求項7に記載の医薬組成物。
- 前記薬物複合体がミセルを形成している、請求項1~8のいずれか一項に記載の医薬組成物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018030208A JP7223502B6 (ja) | 2018-02-22 | 2018-02-22 | 医薬組成物 |
| JP2018-030208 | 2018-02-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019163943A1 true WO2019163943A1 (ja) | 2019-08-29 |
Family
ID=67688423
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2019/006780 WO2019163943A1 (ja) | 2018-02-22 | 2019-02-22 | 医薬組成物 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP7223502B6 (ja) |
| WO (1) | WO2019163943A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111303165A (zh) * | 2019-09-19 | 2020-06-19 | 杭州科兴生物化工有限公司 | 一类星孢菌素类衍生物及其制备方法和应用 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10501526A (ja) * | 1994-06-01 | 1998-02-10 | チバ−ガイギー アクチェンゲゼルシャフト | 多薬物耐性に対する作用物質としてのカルバゾール誘導体 |
| JP2009506000A (ja) * | 2005-08-25 | 2009-02-12 | クレアビリス・セラピューティクス・エスピーエー | K‐252aおよびその誘導体のポリマー結合体 |
| WO2016024595A1 (ja) * | 2014-08-11 | 2016-02-18 | 国立大学法人東京大学 | エピルビシン複合化ブロック共重合体と、抗癌剤とを含むミセル、及び当該ミセルを含む癌又は耐性癌、転移癌の治療に適用可能な医薬組成物 |
| WO2018038165A1 (ja) * | 2016-08-23 | 2018-03-01 | 公益財団法人川崎市産業振興財団 | ポリマー、ポリマーの製造方法、及び薬物複合体 |
-
2018
- 2018-02-22 JP JP2018030208A patent/JP7223502B6/ja active Active
-
2019
- 2019-02-22 WO PCT/JP2019/006780 patent/WO2019163943A1/ja active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10501526A (ja) * | 1994-06-01 | 1998-02-10 | チバ−ガイギー アクチェンゲゼルシャフト | 多薬物耐性に対する作用物質としてのカルバゾール誘導体 |
| JP2009506000A (ja) * | 2005-08-25 | 2009-02-12 | クレアビリス・セラピューティクス・エスピーエー | K‐252aおよびその誘導体のポリマー結合体 |
| WO2016024595A1 (ja) * | 2014-08-11 | 2016-02-18 | 国立大学法人東京大学 | エピルビシン複合化ブロック共重合体と、抗癌剤とを含むミセル、及び当該ミセルを含む癌又は耐性癌、転移癌の治療に適用可能な医薬組成物 |
| WO2018038165A1 (ja) * | 2016-08-23 | 2018-03-01 | 公益財団法人川崎市産業振興財団 | ポリマー、ポリマーの製造方法、及び薬物複合体 |
Non-Patent Citations (10)
| Title |
|---|
| CONSEIL, G. ET AL.: "Protein Kinase C Effectors Bind to Mul tidrug ABC Transporters and Inhibit Their Activity", BIOCHEMISTRY, vol. 40, no. 8, 2001, pages 2564 - 2571, XP055633336, DOI: 10.1021/bi002453m * |
| FESTUCCIA, C. ET AL.: "Uncoupling of the epidermal growth factor receptor from downstream signal transduction molecules guides the acquired resistance to gefitinib in prostate cancer cells", ONCOLOGY REPORTS, vol. 18, no. 2, 2007, pages 503 - 511, XP055633345 * |
| KONG, L. ET AL.: "Structural pharmacological studies on EGFR T790M/C797S", BIOCHEM BIOPHYS RES COMMUN., vol. 488, no. 2, 2017, pages 266 - 272, XP085033851, DOI: 10.1016/j.bbrc.2017.04.138 * |
| LAI, P. ET AL.: "Lestaurtinib is Cytotoxic to Oxaliplatin-resistant Transitional Cell Carcinoma Cell Line T24 In Vitro", TZU CHI MEDICAL JOURNAL, vol. 22, no. 3, 2010, pages 125 - 130, XP027457833, DOI: 10.1016/S1016-3190(10)60056-0 * |
| LEE, H. ET AL.: "Noncovalent Wild-type-Sparing Inhibitors of EGFR T790M", CANCER DISCOVERY, vol. 3, no. 2, 2012, pages 168 - 181, XP002725587 * |
| MIYAMOTO, K. ET AL.: "Inhibition of multi drug resistance by a new staurosporine derivative, NA- 382, in vitro and in vivo", CANCER RESEARCH, vol. 53, no. 7, 1993, pages 1555 - 1559, XP055633350 * |
| SHI, Z. ET AL.: "S-Phase arrest by nucleoside analogues and abrogation of survival without cell cycle progression by 7-hydroxystaurosporine", CANCER RESEARCH, vol. 61, no. 3, 2001, pages 1065 - 1072, XP055633340 * |
| SONG, X. ET AL.: "Staurosporine scaffold-based rational discovery of the wild-type sparing reversible inhibitors of EGFR T790M gatekeeper mutant in lung cancer with analog-sensitive kinase technology", JOURNAL OF MOLECULAR RECOGNITION., vol. 30, no. 4, 2017, pages e2590, XP055633338 * |
| UTZ, I. ET AL.: "The protein kinase C inhibitor CGP 41251, a staurosporine derivative with antitumor activity, reverses multidrug resistance", INT J CANCER., vol. 57, no. 1, 1994, pages 104 - 110, XP055406335, DOI: 10.1002/ijc.2910570119 * |
| YAO, J. ET AL.: "Systematic profiling of chemotherapeutic drug response to EGFR gatekeeper mutation in non-small cell lung cancer", COMPUTATIONAL BIOLOGY AND CHEMISTRY, vol. 64, 2016, pages 126 - 133, XP055633341 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111303165A (zh) * | 2019-09-19 | 2020-06-19 | 杭州科兴生物化工有限公司 | 一类星孢菌素类衍生物及其制备方法和应用 |
| CN111333659A (zh) * | 2019-09-19 | 2020-06-26 | 杭州科兴生物化工有限公司 | 一类星孢菌素类衍生物及其制备方法和应用 |
| CN111471050A (zh) * | 2019-09-19 | 2020-07-31 | 杭州科兴生物化工有限公司 | 一类星孢菌素类衍生物及其制备方法和应用 |
| CN111471050B (zh) * | 2019-09-19 | 2021-07-06 | 杭州科兴生物化工有限公司 | 一类星孢菌素类衍生物及其制备方法和应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP7223502B6 (ja) | 2024-02-08 |
| JP2019142823A (ja) | 2019-08-29 |
| JP7223502B2 (ja) | 2023-02-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Sanches et al. | Is prodrug design an approach to increase water solubility? | |
| JP6013424B2 (ja) | Hpma−ドセタキセルコンジュゲートおよびその使用 | |
| WO2018038165A1 (ja) | ポリマー、ポリマーの製造方法、及び薬物複合体 | |
| KR101721865B1 (ko) | 항암제의 전달을 위한 폴리머 시스템 | |
| JP2010526091A (ja) | 癌の処置のための生物学的な標的基の改変 | |
| JP2010526091A5 (ja) | ||
| CN110585441B (zh) | 一种靶向乏氧肿瘤的短肽小分子自组装纳米材料及其制备方法和应用 | |
| Zhao et al. | An iTEP-salinomycin nanoparticle that specifically and effectively inhibits metastases of 4T1 orthotopic breast tumors | |
| Pes et al. | Novel auristatin E-based albumin-binding prodrugs with superior anticancer efficacy in vivo compared to the parent compound | |
| CN110573166A (zh) | 用于癌症治疗的吉西他滨衍生物 | |
| Zhou et al. | A linear polyethylenimine (LPEI) drug conjugate with reversible charge to overcome multidrug resistance in cancer cells | |
| Guo et al. | Supramolecular nanofibers increase the efficacy of 10-hydroxycamptothecin by enhancing nuclear accumulation and depleting cellular ATP | |
| WO2019163943A1 (ja) | 医薬組成物 | |
| Wong et al. | Linear-like polypeptide-based micelle with pH-sensitive detachable PEG to deliver dimeric camptothecin for cancer therapy | |
| WO2017145164A9 (en) | Polymeric conjugates and uses thereof | |
| KR20180118226A (ko) | 증식성 질환을 위한 조합 치료 | |
| Zhang et al. | Modulating tumor-stromal crosstalk via a redox-responsive nanomedicine for combination tumor therapy | |
| JP2022512826A (ja) | MetAP2阻害剤のバイオマーカーとその応用 | |
| CN114980932A (zh) | Sortilin结合缀合化合物、其组合物及其用于治疗癌症的用途 | |
| Feng et al. | A redox-responsive nanosystem to suppress chemoresistant lung cancer through targeting STAT3 | |
| JP7084740B2 (ja) | 薬物複合体、ミセル、及び医薬組成物 | |
| KR20230167033A (ko) | 암 줄기 세포를 표적화하기 위한 방법 및 소르틸린 결합 접합체 화합물 | |
| Liu et al. | Bioresponsive nanocomplex integrating cancer-associated fibroblast deactivation and immunogenic chemotherapy for rebuilding immune-excluded tumors | |
| Wang et al. | Enhancing cancer therapy: The role of drug delivery systems in STAT3 inhibitor efficacy and safety | |
| Fujita et al. | Drug–drug conjugates of MEK and Akt inhibitors for RAS-mutant cancers |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19757059 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 19757059 Country of ref document: EP Kind code of ref document: A1 |