WO2018196747A1 - 吲哚胺2,3-双加氧酶抑制剂与应用 - Google Patents
吲哚胺2,3-双加氧酶抑制剂与应用 Download PDFInfo
- Publication number
- WO2018196747A1 WO2018196747A1 PCT/CN2018/084267 CN2018084267W WO2018196747A1 WO 2018196747 A1 WO2018196747 A1 WO 2018196747A1 CN 2018084267 W CN2018084267 W CN 2018084267W WO 2018196747 A1 WO2018196747 A1 WO 2018196747A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- membered
- hydrogen
- group
- halo
- Prior art date
Links
- 0 *N=C([C@]1NON=C1NC1CCNCC1)Nc(cc1Br)ccc1F Chemical compound *N=C([C@]1NON=C1NC1CCNCC1)Nc(cc1Br)ccc1F 0.000 description 6
- HZXPDQRNVWNKOY-UHFFFAOYSA-N CC(CC1)CCS1(=O)=O Chemical compound CC(CC1)CCS1(=O)=O HZXPDQRNVWNKOY-UHFFFAOYSA-N 0.000 description 2
- BWAKVVQJIWKENI-JTQLQIEISA-N Bc1cc(N/C(/c2n[o]nc2N[C@@H](CC2)CS2(=O)=O)=N\CCO)ccc1F Chemical compound Bc1cc(N/C(/c2n[o]nc2N[C@@H](CC2)CS2(=O)=O)=N\CCO)ccc1F BWAKVVQJIWKENI-JTQLQIEISA-N 0.000 description 1
- AUPXWLROVDJWFB-UHFFFAOYSA-N C=[N-](C(CC1)CCS1(=O)=O)c1n[o]nc1C(Nc(cc1Br)ccc1F)=[Ne] Chemical compound C=[N-](C(CC1)CCS1(=O)=O)c1n[o]nc1C(Nc(cc1Br)ccc1F)=[Ne] AUPXWLROVDJWFB-UHFFFAOYSA-N 0.000 description 1
- CWOCGQMZNQQHFT-UHFFFAOYSA-N CC(C)(C)OC(NS(N(CC1)CCC1Nc1n[o]nc1C(N1c(cc2Br)ccc2F)=NOC1=O)(=O)=O)=O Chemical compound CC(C)(C)OC(NS(N(CC1)CCC1Nc1n[o]nc1C(N1c(cc2Br)ccc2F)=NOC1=O)(=O)=O)=O CWOCGQMZNQQHFT-UHFFFAOYSA-N 0.000 description 1
- IAAWASZQYJLKFI-UHFFFAOYSA-N CC(C1)CC(C2)C1CS2(=O)=O Chemical compound CC(C1)CC(C2)C1CS2(=O)=O IAAWASZQYJLKFI-UHFFFAOYSA-N 0.000 description 1
- CMJLMPKFQPJDKP-UHFFFAOYSA-N CC(CC1)CS1(=O)=O Chemical compound CC(CC1)CS1(=O)=O CMJLMPKFQPJDKP-UHFFFAOYSA-N 0.000 description 1
- APUAFDHBPUABDQ-UHFFFAOYSA-N CS(N(CC1C2)C2CC1Nc1n[o]nc1/C(/Nc(cc1)cc(Br)c1F)=N/O)(=O)=O Chemical compound CS(N(CC1C2)C2CC1Nc1n[o]nc1/C(/Nc(cc1)cc(Br)c1F)=N/O)(=O)=O APUAFDHBPUABDQ-UHFFFAOYSA-N 0.000 description 1
- XCBOUPGRTHCXBN-UHFFFAOYSA-N NS(N(CC1)CCC1Nc1n[o]nc1/C(/Nc(cc1Br)ccc1F)=N/O)(=O)=O Chemical compound NS(N(CC1)CCC1Nc1n[o]nc1/C(/Nc(cc1Br)ccc1F)=N/O)(=O)=O XCBOUPGRTHCXBN-UHFFFAOYSA-N 0.000 description 1
- AUGHTHCFGCFSTK-UHFFFAOYSA-N O=C1ON=C(c2n[o]nc2NC2CCNCC2)N1c(cc1)cc(Br)c1F Chemical compound O=C1ON=C(c2n[o]nc2NC2CCNCC2)N1c(cc1)cc(Br)c1F AUGHTHCFGCFSTK-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/08—1,2,5-Oxadiazoles; Hydrogenated 1,2,5-oxadiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
Definitions
- the invention belongs to the technical field of medicine, and relates to a guanamine 2,3-dioxygenase (IDO) inhibitor compound, a pharmaceutically acceptable salt thereof and a stereoisomer thereof, and a pharmaceutical preparation and a pharmaceutical composition thereof And its use in the manufacture of a medicament for the treatment of a related disorder mediated by amidoxime 2,3-dioxygenase (IDO) abnormalities.
- IDO amidoxime 2,3-dioxygenase
- Tryptophan is an essential amino acid in the body that is used in vivo for the biosynthesis of proteins, niacin and the neurotransmitter serotonin (serotonin).
- Indoleamine 2,3-dioxygenase is a monomeric enzyme containing intracellular heme, which is widely distributed in many tissues and cells of humans and animals. IDO catalyzes the first step of the guanidine oxidative cleavage of L-tryptophan to form kynurenine, which is also a critical rate-limiting reaction. IDO is expressed at a low level in the normal state of the body, and abnormally high expression can be detected under the disease state.
- IDO plays a crucial role in the pathogenesis of neurological diseases, especially Alzheimer's disease and depression. IDO also has an immune tolerance function, and IDO on tumor cells and antigen presenting cells can induce immune tolerance of T cells to tumor antigens. In addition, IDO is also involved in the pathogenesis of chronic infection, HIV-infection, AIDS, and autoimmune diseases. Therefore, IDO has proven to be an important drug discovery target, and the development of IDO inhibitors is urgently needed.
- IDO mainly affects the function of the brain through two mechanisms, which leads to the occurrence of nervous system diseases: (1) in the inflammatory reaction, by reducing the circulating tryptophan concentration by metabolizing tryptophan, thereby making 5-hydroxytryptamine Decreased levels, leading to depression; (2) catalyzing the metabolism of tryptophan by kynurenine pathway to accumulate kynurenine and neurotoxic quinolinic acid, leading to the occurrence of neurological diseases. Therefore, IDO inhibitors have broad prospects in the treatment of neurological diseases such as Alzheimer's disease and depression.
- IDO has also become a very important small molecule regulatory target for anti-tumor immunotherapy. Its regulation of the immune system mainly includes the following aspects: (1) high expression of IDO can lead to local tryptophan depletion, because T cell color The depletion of amino acids is particularly sensitive, so when the tryptophan concentration is lowered, the proliferation of T cells will be arrested in the G1 phase. (2) IDO-dependent tryptophan degradation leads to an increase in kynurenine levels and induces oxygen free radical-mediated T cell apoptosis. (3) Up-regulation of dendritic cell IDO expression enhances local regulatory T cell (Treg)-mediated immunosuppression by degrading local tryptophan, promoting the body's peripheral immune tolerance to tumor-specific antigens.
- Treg local regulatory T cell
- IDO small molecule inhibitors have been developed at home and abroad to treat or prevent IDO-related diseases.
- patent WO99/29310 reports the use of IDO inhibitors 1-methyl-DL-tryptophan, 4-(3-benzofuran)-DL-alanine, 4-(3-benzothiophene)-DL. - Alanine and 6-nitro-L-tryptophan, altering T-cell mediated immunity, including altering the concentration of local extracellular tryptophan and tryptophan metabolites.
- Patent WO 2004/094409 also reports some compounds which specifically have IDO inhibition.
- US 2004/0234623 the use of an IDO inhibitor to treat cancer and infection is reported.
- IDO indoleamine 2,3-dioxygenase
- Epacadostat II, Under Active Development
- NLG-919 I, Under Active Development
- the invention aims to study a series of novel and highly effective IDO inhibitors, which have good drug-forming properties and can be used for preventing and/or treating Alzheimer's disease, cataract, cellular immune activation-related infections, autoimmune diseases, AIDS, cancer. , drugs with depression or abnormal metabolism of tryptophan.
- the technical problem to be solved by the present invention is to provide a novel class of IDO inhibitors which have excellent inhibitory activity against IDO and can be used for the treatment of diseases mediated by IDO abnormalities.
- X and Y are each independently selected from CH or N;
- Z is selected from O or S
- R 1 is selected from cycloalkyl, heterocyclic, heteroaryl, optionally substituted by R 1a , and R 1a is selected from hydrogen, halogen, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 Alkynyl, halo C 1-6 alkyl, -CN, -NO 2 , -OR b , -C(O)R c , -C(O)OR b , -OC(O)R c , -C( O) NR e R f , -OC(O)NR e R f , -NR e R f , -NR d C(O)R c , -NR d C(O)NR e R f , -NR d C( O) OR b , -SR b , -S(O)R c , -S(O)NR e R f , -S(O) 2 R c
- R 2 is selected from cycloalkyl, heterocyclic, heteroaryl or aryl, optionally substituted by R 2a , and R 2a is selected from hydrogen, halogen, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, halo C 1-6 alkyl, -CN, -NO 2 , -OR b , -C(O)R c , -C(O)OR b , -OC(O)R c , -C(O)NR e R f , -OC(O)NR e R f , -NR e R f , -NR d C(O)R c , -NR d C(O)NR e R f , -NR d C(O)OR b , -SR b , -S(O)R c , -S(O)NR e R f , -S(O) 2 R
- R 3 is selected from hydrogen, C 1-6 alkyl or cycloalkyl
- R b is selected from hydrogen, C 1-6 alkyl, halo C 1-6 alkyl, C 1-6 alkoxy C 1-6 alkyl, cycloalkyl, heterocyclyl, heteroaryl or aryl ;
- R c is selected from the group consisting of hydrogen, C 1-6 alkyl, halo C 1-6 alkyl, C 1-6 alkoxy, cycloalkyl, heterocyclyl, heteroaryl or aryl;
- R d , R e , R f are each independently selected from hydrogen, C 1-6 alkyl, halo C 1-6 alkyl, cycloalkyl, heterocyclyl, heteroaryl or aryl;
- n 0, 1, 2, 3, 4 or 5;
- n 0, 1, 2, 3, 4 or 5.
- X and Y are each independently selected from CH or N;
- Z is selected from O or S
- R 1 is selected from a 3-14 membered cycloalkyl group, a 3-14 membered heterocyclic group, a 5-14 membered heteroaryl group, optionally substituted by R 1a , and R 1a is selected from the group consisting of hydrogen, halogen, and C 1-6 alkyl.
- R 2 is selected from a 3-8 membered cycloalkyl group, a 3-14 membered heterocyclic group, a 5-14 membered heteroaryl group or a 6-14 membered aryl group, which may be optionally substituted by R 2a , and R 2a is selected from the group consisting of hydrogen and halogen.
- R 3 is selected from hydrogen, C 1-6 alkyl or 3-8 membered cycloalkyl
- R b is selected from the group consisting of hydrogen, C 1-6 alkyl, halo C 1-6 alkyl, C 1-6 alkoxy C 1-6 alkyl, 3-8 membered cycloalkyl, 3-8 membered heterocyclic ring a 5-8 membered heteroaryl group or a 6-14 membered aryl group;
- R c is selected from the group consisting of hydrogen, C 1-6 alkyl, halogenated C 1-6 alkyl, C 1-6 alkoxy, 3-8 membered cycloalkyl, 3-8 membered heterocyclic, 5-8 member a heteroaryl group or a 6-14 membered aryl group;
- R d , R e , and R f are each independently selected from the group consisting of hydrogen, C 1-6 alkyl, halo C 1-6 alkyl, 3-8 membered cycloalkyl, 3-8 membered heterocyclic, 5-8 a heteroaryl group or a 6-14 membered aryl group;
- n 0, 1, 2, 3, 4 or 5;
- n 0, 1, 2, 3, 4 or 5.
- R 1 is selected from a 3-12 membered heterocyclic group, a 5-12 membered heteroaryl group, which may be optionally substituted by R 1a , and R 1a is selected from the group consisting of hydrogen, halogen, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, halo C 1-6 alkyl, -CN, -NO 2 , -OR b , -C(O)R c , -C(O)OR b , -OC(O)R c , -C(O)NR e R f , -OC(O)NR e R f , -NR e R f , -NR d C(O)R c , -NR d C(O)NR e R f ,- NR d C(O)OR b , -SR b , -S(O)R c , -S(O)NR e
- R 2 is selected from 6-14 membered aryl or 5-12 membered heteroaryl, optionally substituted by R 2a , and R 2a is selected from the group consisting of hydrogen, halogen, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, halo C 1-6 alkyl, -CN, -NO 2 , -OR b , -C(O)R c , -C(O)OR b , -OC(O)R c , -C(O)NR e R f , -OC(O)NR e R f , -NR e R f , -NR d C(O)R c , -NR d C(O)NR e R f , -NR d C(O)OR b , -SR b , -S(O)R c , -S(O)NR e R f , -S(
- R 3 is selected from hydrogen or C 1-6 alkyl
- R b is selected from hydrogen, C 1-6 alkyl, halo C 1-6 alkyl or C 1-6 alkoxy C 1-6 alkyl;
- R c is selected from hydrogen, C 1-6 alkyl, halo C 1-6 alkyl or C 1-6 alkoxy;
- R d , R e , R f are each independently selected from hydrogen, C 1-6 alkyl or halogenated C 1-6 alkyl;
- n 0, 1, 2 or 3;
- n 0, 1, 2 or 3.
- R 1 is further preferably a 3-8 membered heterocyclic group, a 5-6 membered heteroaryl group, a 6-12 membered heterocyclic group, a 6-12 membered bridged heterocyclic group or a 6-12 membered spiroheterocyclic group. It is optionally substituted by R 1a and R 1a is as defined above.
- R 1 is further preferably a 4-6 membered saturated heterocyclic group, an 8-membered saturated heterocyclic group, a 7-8 membered saturated bridged heterocyclic group or a 7-membered saturated spiroheterocyclic group, which may be optionally substituted by R 1a , R 1a Selected from hydrogen, halogen, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, halo C 1-6 alkyl, -CN, -NO 2 , -OR b , -C( O) R c , -C(O)OR b , -OC(O)R c , -C(O)NR e R f , -OC(O)NR e R f , -NR e R f , -NR d C(O)R c , -NR d C(O)NR e R f , -NR d C(O)OR
- R b is selected from hydrogen, C 1-6 alkyl, halo C 1-6 alkyl or C 1-6 alkoxy C 1-6 alkyl;
- R c is selected from hydrogen, C 1-6 alkyl, halo C 1-6 alkyl or C 1-6 alkoxy;
- R d , R e , and R f are each independently selected from hydrogen, C 1-6 alkyl or halogenated C 1-6 alkyl.
- R 1 is Ring A is a 3-12 membered mono, snail, bridged heterocyclic group
- R 2 is selected from a 6-14 membered aryl group.
- R 1 is Ring A is a 3-8 membered monoheterocyclic group, a 6-12 membered heterocyclic group, a 6-12 membered bridged heterocyclic group or a 6-12 membered spiroheterocyclic group;
- R 2 is a phenyl group.
- R 1 is Ring A is a 4-6 membered saturated heterocyclic group, an 8 membered saturated heterocyclic group, a 7-8 membered saturated bridged heterocyclic group or a 7 membered saturated spiroheterocyclic group;
- R 2 is a phenyl group.
- R 1 is T 1 and T 2 are each independently selected from CR 4 R 4 ', NR 4 or O, and t 1 and t 2 are each independently 0, 1 , 2 or 3, and t 1 and t 2 are not 0 at each time; 1 may be optionally substituted by R 1a , and R 1a is selected from the group consisting of hydrogen, halogen, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, halo C 1-6 alkyl, -CN, -NO 2 , -OR b , -C(O)R c , -C(O)OR b , -OC(O)R c , -C(O)NR e R f , -OC(O)NR e R f , -NR e R f , -NR d C(O)R c , -NR d C(O)NR e R f , -NR d C(O)
- R 2 is selected from phenyl, optionally substituted by R 2a , and R 2a is selected from hydrogen, halogen or C 1-6 alkyl;
- R b is selected from hydrogen, C 1-6 alkyl, halo C 1-6 alkyl or C 1-6 alkoxy C 1-6 alkyl;
- R c is selected from hydrogen, C 1-6 alkyl, halo C 1-6 alkyl or C 1-6 alkoxy;
- R d , R e , and R f are each independently selected from hydrogen, C 1-6 alkyl or halogenated C 1-6 alkyl.
- R 1 is t 1 and t 2 are each independently 0, 1 , 2 or 3, and t 1 and t 2 are not simultaneously 0; R 1 may be optionally substituted by R 1a , and R 1a is selected from hydrogen, halogen, C 1-6 Alkyl, C 2-6 alkenyl, C 2-6 alkynyl, halo C 1-6 alkyl, -CN, -NO 2 , -OR b , -C(O)R c , -C(O) OR b , -OC(O)R c , -C(O)NR e R f , -OC(O)NR e R f , -NR e R f , -NR d C(O)R c , -NR d C(O)NR e R f , -NR d C(O)OR b , -SR b , -S(O)R c , -S(O)NR
- R b is selected from hydrogen, C 1-6 alkyl, halo C 1-6 alkyl or C 1-6 alkoxy C 1-6 alkyl;
- R c is selected from hydrogen, C 1-6 alkyl, halo C 1-6 alkyl or C 1-6 alkoxy;
- R d , R e , and R f are each independently selected from hydrogen, C 1-6 alkyl or halogenated C 1-6 alkyl.
- R 1 is t 1 and t 2 are independently 0, 1, 2, 3, respectively, and t 1 and t 2 are not simultaneously 0, R 1 may be optionally substituted by R 1a , and R 1a is selected from hydrogen, halogen or C 1-6 alkyl;
- R 2 is selected from phenyl, optionally substituted by R 2a , and R 2a is selected from hydrogen, halogen or C 1-6 alkyl;
- R 3 is selected from hydrogen or C 1-6 alkyl
- n 0, 1 or 2;
- n 0, 1, 2, 3, 4 or 5.
- R 1 is Ring A is 6-12 membered heterocyclyl, 6-12 membered bridged heterocyclic or 6-12 membered spiroheterocyclyl, optionally substituted by R 1a , and R 1a is selected from hydrogen, halogen, C 1-6 Alkyl, C 2-6 alkenyl, C 2-6 alkynyl, halo C 1-6 alkyl, -CN, -NO 2 , -OR b , -C(O)R c , -C(O) OR b , -OC(O)R c , -C(O)NR e R f , -OC(O)NR e R f , -NR e R f , -NR d C(O)R c , -NR d C(O)NR e R f , -NR d C(O)OR b , -SR b , -S(O)R c , -S
- R 2 is selected from phenyl, optionally substituted by R 2a , and R 2a is selected from hydrogen, halogen or C 1-6 alkyl;
- R b is selected from hydrogen, C 1-6 alkyl, halo C 1-6 alkyl or C 1-6 alkoxy C 1-6 alkyl;
- R c is selected from hydrogen, C 1-6 alkyl, halo C 1-6 alkyl or C 1-6 alkoxy;
- R d , R e , and R f are each independently selected from hydrogen, C 1-6 alkyl or halogenated C 1-6 alkyl.
- R 1 is Ring A is a saturated 6-12 membered fused heterocyclyl, 6-12 membered saturated heterocyclic group bridged or spiro saturated 6-12 membered heterocyclyl may optionally be substituted with R 1a, R 1a is selected from hydrogen, halo, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, halo C 1-6 alkyl, -CN, -NO 2 , -OR b , -C(O)R c , -C (O)OR b , -OC(O)R c , -C(O)NR e R f , -OC(O)NR e R f , -NR e R f , -NR d C(O)R c , -NR d C(O)NR e R f , -NR d C(O)OR b , -SR b , -S(O)R c
- R b is selected from hydrogen, C 1-6 alkyl, halo C 1-6 alkyl or C 1-6 alkoxy C 1-6 alkyl;
- R c is selected from hydrogen, C 1-6 alkyl, halo C 1-6 alkyl or C 1-6 alkoxy;
- R d , R e , and R f are each independently selected from hydrogen, C 1-6 alkyl or halogenated C 1-6 alkyl.
- R 1 is selected from a 5-6 membered heteroaryl group, optionally substituted by R 1a , and R 1a is selected from the group consisting of hydrogen, halogen, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, halo Generation C 1-6 alkyl, -CN, -NO 2 , -OR b , -C(O)R c , -C(O)OR b , -OC(O)R c , -C(O)NR e R f , -OC(O)NR e R f , -NR e R f , -NR d C(O)R c , -NR d C(O)NR e R f , -NR d C(O)OR b , -SR b , -S(O)R c , -S(O)NR e R f , -S(O) 2 R
- R 2 is selected from phenyl, optionally substituted by R 2a , and R 2a is selected from hydrogen, halogen or C 1-6 alkyl;
- R b is selected from hydrogen, C 1-6 alkyl, halo C 1-6 alkyl or C 1-6 alkoxy C 1-6 alkyl;
- R c is selected from hydrogen, C 1-6 alkyl, halo C 1-6 alkyl or C 1-6 alkoxy;
- R d , R e , and R f are each independently selected from hydrogen, C 1-6 alkyl or halogenated C 1-6 alkyl.
- R 1 is selected from These groups may optionally be substituted with R 1a, R 1a is selected from hydrogen, halo, C 1-4 alkyl, halo C 1-4 alkyl, -CN, -NO 2, -OR b , -C (O R c , -C(O)OR b , -OC(O)R c , -C(O)NR e R f , -OC(O)NR e R f , -NR e R f , -NR d C (O) R c , -NR d C(O)NR e R f , -NR d C(O)OR b , -S(O) 2 R c or -S(O) 2 NR e R f ;
- R 2 is selected from phenyl, optionally substituted by R 2a , and R 2a is selected from hydrogen, halogen or C 1-4 alkyl;
- R 3 is selected from hydrogen or C 1-4 alkyl
- R b , R c , R d , R e , R f are each independently selected from hydrogen or C 1-4 alkyl;
- n 0, 1, 2 or 3;
- n 0, 1, 2 or 3.
- R 1 is preferably: These groups may optionally be substituted with R 1a, R 1a is selected from hydrogen, halo, C 1-4 alkyl, halo C 1-4 alkyl, -CN, - NO 2, -OR b, -C ( O) R c , -C(O)OR b , -OC(O)R c , -C(O)NR e R f , -NR e R f , -NR d C(O)R c , -S( O) 2 R c or -S(O) 2 NR e R f ,
- R b , R c , R d , R e , and R f are each independently selected from hydrogen or C 1-4 alkyl.
- R 1 is selected from These groups may optionally be substituted with R 1a, R 1a is selected from hydrogen, halo, C 1-4 alkyl.
- R 1 is selected from These groups may optionally be substituted with R 1a, R 1a is selected from hydrogen, halo, C 1-4 alkyl.
- the present invention provides the following compounds, pharmaceutically acceptable salts thereof or stereoisomers thereof:
- compositions comprising a compound of Formula I and Formula II, a pharmaceutically acceptable salt thereof, and a stereoisomer thereof, which may optionally contain one or more pharmaceutically acceptable carriers.
- the pharmaceutical composition can be administered to a patient or subject in need of such treatment by any suitable mode of administration, such as oral, parenteral, rectal or pulmonary administration.
- the pharmaceutical composition can be prepared into a conventional solid preparation such as a tablet, a capsule, a pill, a granule, etc.; or an oral liquid preparation such as an oral solution or an oral suspension. , syrup, and the like.
- the pharmaceutical composition can be prepared as an injection, including an injection, a sterile powder for injection, and a concentrated solution for injection.
- the injection can be produced by a conventional method in the prior art, and when the injection is placed, an additional agent may be added, or a suitable additive may be added depending on the nature of the drug.
- the pharmaceutical composition can be formulated as a suppository or the like.
- the pharmaceutical composition can be formulated as an inhalant or a spray.
- Another aspect of the present invention provides the use of a compound of Formula I and Formula II, a pharmaceutically acceptable salt thereof, and a stereoisomer thereof for the preparation of a medicament for treating an abnormality mediated by IDO, particularly treatment and Cancer-associated tumor-specific immunosuppression.
- the disease in which the IDO abnormality is mediated is an infectious disease, a nervous system disease, a cancer, or a non-cancerous proliferative disease.
- infectious diseases include influenza virus, hepatitis C virus (HCV), human papillomavirus (HPV), cytomegalovirus (CMV), EB virus (EBV), poliovirus, varicella zoster virus, Disease caused by Coxsackie virus, human immunodeficiency virus (HIV) infection;
- the nervous system diseases include: Alzheimer's disease, depression;
- the cancer includes lung cancer, squamous cell carcinoma, bladder cancer, Gastric cancer, ovarian cancer, peritoneal cancer, breast cancer, ductal carcinoma of the breast, head and neck cancer, endometrial cancer, uterine body cancer, rectal cancer, liver cancer, kidney cancer, renal pelvic cancer, esophageal cancer, esophageal adenocarcinoma, glioma, Prostate cancer, thyroid cancer,
- halogen as used in the present invention means fluorine, chlorine, bromine, iodine or the like, preferably a fluorine atom or a chlorine atom.
- halo means that any carbon atom in the substituent may be substituted by one or more of the same or different halogens.
- Halogen is as defined above.
- C 1-6 alkyl group as used in the present invention means a straight or branched alkyl group derived from a hydrocarbon having 1 to 6 carbon atoms and removed by a hydrogen atom, such as a methyl group, an ethyl group, a n-propyl group, Isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, isopentyl, 2-methylbutyl, neopentyl, 1-ethylpropyl, n-hexyl, iso-hexyl Base, 4-methylpentyl, 3-methylpentyl, 2-methylpentyl, 1-methylpentyl, 3,3-dimethylbutyl, 2,2-dimethylbutyl, 1,1-dimethylbutyl, 1,2-dimethylbutyl, 1,3-dimethylbutyl, 2,3-dimethylbutyl, 2-eth
- C 2-6 alkenyl group as used in the present invention means an olefin moiety having 2 to 6 carbon atoms containing a carbon-carbon double bond, and a linear or branched olefin group derived by removing one hydrogen atom, such as a vinyl group, 1- Propylene, 2-propenyl, 1-butenyl, 2-butenyl, 1,3-butadienyl, 1-pentenyl, 2-pentenyl, 3-pentenyl, 1,3 - pentadienyl, 1,4-pentadienyl, 1-hexenyl, 1,4-hexadienyl, and the like.
- C 2-6 alkynyl group as used in the present invention means an alkyne moiety having 2 to 6 carbon atoms containing a carbon-carbon oxime bond, and a linear or branched alkyne group derived by removing a hydrogen atom, such as ethynyl group, C. Alkynyl, 2-butynyl, 2-pentynyl, 3-pentynyl, 4-methyl-2-pentynyl, 2-hexynyl, 3-hexynyl, and the like.
- C 1-6 alkoxy refers to the present invention as hereinbefore defined “C 1-6 alkyl” through an oxygen atom to the parent molecular moiety attached, i.e., "C 1-6 alkyl -O a "group such as methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, tert-butoxy, n-pentyloxy, neopentyloxy and n-hexyloxy and the like.
- the "C 1-4 alkoxy group” refers to the above-mentioned example having 1 to 4 carbon atoms, that is, a "C 1-4 alkyl-O-” group.
- the "fused ring” as used in the present invention means a polycyclic ring structure formed by joining two, two or more cyclic structures in a snail, a snail, or a bridge.
- the parallel ring refers to a fused ring structure formed by two or more ring structures sharing two adjacent ring atoms with each other (ie, sharing one bond).
- the bridged ring refers to a fused ring structure formed by two or more ring-shaped structures sharing two non-adjacent ring atoms with each other.
- the spiro ring refers to a fused ring structure formed by two or more ring structures sharing one ring atom with each other.
- the "cycloalkyl group” of the present invention may be a 3-14 membered cycloalkyl group, including a monocyclic cycloalkyl group or a fused ring cycloalkyl group, which may be saturated, partially saturated or unsaturated, but not aromatic. of.
- the monocyclic cycloalkyl group may be a 3-8 membered cycloalkyl group or a 5-7 membered cycloalkyl group, and examples thereof include, but are not limited to, a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexane group, and a ring.
- the fused ring cycloalkyl group includes a cyclocycloalkyl group, a bridged cycloalkyl group, a spirocycloalkyl group, and may be saturated, partially saturated or unsaturated, but not aromatic.
- the cyclocyclocycloalkyl group may be a 6-12 membered cyclocycloalkyl group, a 7-10 membered cyclocycloalkyl group, and examples thereof include, but are not limited to, bicyclo [3.1.1] heptyl, bicyclo [2.2 .1] heptyl, bicyclo[2.2.2]octyl, bicyclo[3.2.2]nonanyl, bicyclo[3.3.1]nonanyl and bicyclo[4.2.1]nonanyl.
- the spiro group may be a 6-12 membered spiro group, a 7-11 membered spiro group, examples of which include, but are not limited to:
- the bridged ring group may be a 6-12 membered bridged ring group and a 7-11 membered bridged ring group, and examples thereof include, but are not limited to:
- heterocyclic group as used in the present invention means a non-aromatic cyclic group in which at least one ring carbon atom is replaced by a hetero atom selected from O, S, N, preferably 1-3 hetero atoms, including carbon.
- a hetero atom selected from O, S, N, preferably 1-3 hetero atoms, including carbon.
- an atom or a sulfur atom is substituted by oxo, for example, a carbon atom is replaced by C(O), S(O), or S(O)2.
- the "heterocyclic group” may be a 3-14 membered heterocyclic group or a 3-12 membered heterocyclic group, and includes a monoheterocyclic group or a fused heterocyclic group.
- the monoheterocyclic group may be a 3-8 membered heterocyclic group, a 3-8 membered saturated heterocyclic group, a 3-6 membered heterocyclic group, a 4-6 membered heterocyclic group, a 5-7 membered heterocyclic group, 5-6.
- a heterocyclic group a 5-6 membered oxygen-containing heterocyclic group, a 5-6 membered nitrogen-containing heterocyclic group, a 5-6 membered saturated heterocyclic group, a 5-7 membered saturated heterocyclic group or the like.
- Examples thereof include, but are not limited to, aziridine, 2H-azepine, diaziryl, 3H-diazacyclopropenyl, azetidinyl, 1,4-dioxo Heterocyclohexane, 1,3-dioxanyl, 1,3-dioxolyl, 1,4-dioxadienyl, tetrahydrofuranyl, dihydropyrrolyl , pyrrolidinyl, imidazolidinyl, 4,5-dihydroimidazolyl, pyrazolidinyl, 4,5-dihydropyrazolyl, 2,5-dihydrothienyl, tetrahydrothiophenyl, 4,5 -dihydrothiazolyl, piperidinyl, piperazinyl, morpholinyl, hexahydropyrimidinyl, hexahydropyridazinyl, 4,5-d
- the fused heterocyclic ring includes a heterocyclic group, a spiroheterocyclic group, a bridged heterocyclic group, and may be saturated, partially saturated or unsaturated, but not aromatic.
- the heterocyclic group may be 6-12 members and a heterocyclic group, a 7-10 membered heterocyclic group, a 6-12 membered saturated heterocyclic group, a 7-8 membered saturated heterocyclic group, and an 8-membered saturated group.
- heterocyclic group examples thereof include, but are not limited to, 3-azabicyclo[3.10.]hexane, 3,6-diazabicyclo[3.2.0]heptane, 3,8-diazabicyclo[ 4.2.0] Octyl, 3,7-diazabicyclo[4.2.0]octyl, octahydropyrrolo[3,4-c]pyrrole, octahydropyrrolo[3,4-b]pyrrole , octahydropyrrolo[3,4-b][1,4]oxazinyl, octahydro-1H-pyrrolo[3,4-c]pyridine, 2,3-dihydrobenzofuran-2-yl , 2,3-dihydrobenzofuranyl-3-yl, indan-1-yl, indan-2-yl, indan-3-yl, 2,3 dihydrobenzothiophene -2 base, octahydro-1
- the spiroheterocyclyl group may be a 6-12 membered spiroheterocyclyl group, a 7-11 membered spiroheterocyclyl group, a 6-12 membered saturated spiroheterocyclyl group, a 7-membered saturated spiroheterocyclyl group, examples of which include but not Limited to:
- the bridged heterocyclic group may be a 6-12 membered bridged heterocyclic group, a 7-11 membered bridged heterocyclic group, and a 6-12 membered saturated bridged ring group.
- Examples of the 7-8 membered saturated bridged ring group include, but are not limited to:
- the aryl group means an aromatic carbocyclic group including a 6-8 membered monocyclic aryl group and an 8-14 membered fused ring aryl group.
- a 6-8 membered monocyclic aryl group such as phenyl, cyclooctyltetraenyl or the like.
- 8-14 membered fused ring aryl such as naphthalene, phenanthrene, and the like.
- heteroaryl as used in the present invention may be a 5-14 membered heteroaryl group, and means an aromatic cyclic group in which at least one ring carbon atom is replaced by a hetero atom selected from O, S, N, preferably 1 - 3 heteroatoms including both carbon atoms and sulfur atoms by oxo, for example, carbon atoms are replaced by C(O), S(O), S(O)2.
- Heteroaryl groups include monoheteroaryl and fused heteroaryl.
- the monoheteroaryl group may be a 5-7 membered heteroaryl group, a 5-6 membered heteroaryl group, and examples thereof include, but are not limited to, furyl, imidazolyl, isoxazolyl, thiazolyl, isothiazolyl, oxadiazolyl , oxazolyl, isoxazolyl, pyridyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl, pyrazolyl, pyrrolyl, tetrazolyl, thiadiazolyl, thiazolyl, thienyl, Triazolyl and triazinyl.
- the fused heteroaryl group may be a 8-12 membered heteroaryl group, a 9-10 membered heteroaryl group, and examples thereof include, but are not limited to, benzimidazolyl, benzofuranyl, benzothienyl, benzothienyl, Benzooxadiazolyl, benzothiazolyl, porphyrinyl, oxazolyl, indolyl, isoquinolyl, naphthyridinyl, indolyl, quinolyl.
- a “pharmaceutically acceptable salt” as used herein refers to a pharmaceutically acceptable acid and base addition salt of a compound of Formula I or Formula II.
- a salt formed by an acidic functional group such as -COOH, -OH, -SO 3 H, etc.
- a suitable inorganic or organic cation (base) in the compound including a salt, an ammonium salt formed with an alkali metal or an alkaline earth metal, and a salt formed by a nitrogen-containing organic base
- a salt formed by a basic functional group such as -NH2, etc.
- a suitable inorganic or organic anion (acid) in the compound including a mineral acid salt and an organic acid salt.
- Such “pharmaceutically acceptable salts” include, but are not limited to, acid salts: hydrochloric acid, phosphoric acid, hydrobromic acid, sulfuric acid, sulfurous acid, formic acid, toluenesulfonic acid, methanesulfonic acid, nitric acid, benzoic acid, citric acid, Tartaric acid, maleic acid, hydroiodic acid, alkanoic acid (such as acetic acid, HOOC-(CH 2 ) n-COOH (where n is 0 to 4)), and the like; salts of the base: sodium, potassium, calcium, ammonium, and the like.
- stereoisomer as used in the present invention means that when an asymmetric carbon atom is present in the compound of formula I or formula II, an enantiomer is produced; when the compound has a carbon-carbon double bond or a cyclic structure, it is produced.
- a cis-trans isomer when a compound is present in a ketone or a oxime, a tautomer is produced, and all enantiomers, diastereomers, racemic isomers, cis of the compound of Formula I or Formula II are produced.
- Trans isomers, tautomers, geometric isomers, epimers, and mixtures thereof are included within the scope of the invention.
- PE petroleum ether
- the raw material malononitrile (50.0g, 0.76mol, 1.0eq) was added to a four-necked bottle, dissolved by heating (about 50 ° C), water (0.5 L) was added, and the ice water bath was about 10 ° C, and sodium nitrite was added in portions (57.72). g, 0.83 mol, 1.1 eq), after addition, hydrochloric acid (6N, 8.5 mL) was added dropwise at 10 ° C or lower, and the mixture was stirred for 0.5 h until the temperature was constant. The above reaction solution was recorded as A.
- the mixture was stirred for 0.5 h in an ice water bath until the temperature was constant, the ice water bath was removed, and the mixture was heated to reflux for 12 hours.
- hydrochloric acid (6N, 120 mL) is added dropwise in an ice water bath (0 ° C), the pH is adjusted to 7, the stirring is continued for 40 min, the reaction solution is suction filtered, the filter cake is washed with water, and the filter cake is drained to obtain the target compound ( 101 g, yield: 93.5%).
- Step 3 Synthesis of 4-amino-N-(3-bromo-4-fluorophenyl)-N'-hydroxy-1,2,5-oxadiazole-3-carboxamidine
- Step 1 4-(3-Bromo-4-fluorophenyl)-3-(4-((1,1-dioxotetrahydro-2H-thiopyran-4-yl)amino)-1,2, Synthesis of 5-oxadiazol-3-yl)-1,2,4-oxadiazol-5(4H)one
- Step 2 N-(3-Bromo-4-fluorophenyl)-4-((1,1-dioxotetrahydro-2H-thiopyran-4-yl)amino)-N'-hydroxy-1, Synthesis of 2,5-oxadiazole-3-carboxamidine
- Step 1 4-(3-Bromo-4-fluorophenyl)-3-(4-((1-(methylsulfonyl)piperidin-4-yl)amino)-1,2,5-oxa 2 Synthesis of oxazol-3-yl)-1,2,4-oxadiazol-5(4H)-one
- Step 1 4–((4-(4-(3-Bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-1,2,4-oxadiazol-3-yl) Synthesis of tert-butyl ester of -1,2,5-oxadiazol-3-yl)amino)piperidine-1-carboxylate
- Step 2 4-(3-Bromo-4-fluorophenyl)-3-(4-(piperidin-4-ylamino)-1,2,5-oxadiazol-3-yl)-1,2 Synthesis of 4-oxadiazol-5(4H)-one
- Step 3 ((4-(4-(4-(4-(3-Bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-1,2,4-oxadiazol-3- Synthesis of tert-butyl ester of 1,1,2,5-oxadiazol-3-yl)amino)piperidin-1-yl)sulfonyl)carbamate
- tert-Butyl alcohol (34.1 mg, 0.46 mmol, 1.2 eq) was dissolved in DCM (3 mL), cooled to 0 ° C, chlorosulfonyl isocyanate (65.1 mg, 0.46 mmol) was added dropwise, and reacted at 0 ° C for 1.5 hours. Add dropwise to 4-(3-bromo-4-fluorophenyl)-3-(4-(piperidin-4-ylamino)-1,2,5-oxadiazole-3 previously cooled to below 0 °C.
- Step 4 4-((4-(4-(3-Bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-1,2,4-oxadiazol-3-yl) Synthesis of -1,2,5-oxadiazol-3-yl)amino)piperidine-1-sulfonamide
- Step 5 N-(3-Bromo-4-fluorophenyl)-N'-hydroxy-4-((1-sulfamoylpyridin-4-yl)amino)-1,2,5-oxadiazole- Synthesis of 3-mercapto
- Step 1 4-((4-(N-(3-Bromo-4-fluorophenyl)-N'-hydroxymethylindolyl)-1,2,5-oxadiazol-3-yl)amino)piperidin Synthesis of tert-butyl pyridine-1-carboxylate
- Step 2 Synthesis of N-(3-bromo-4-fluorophenyl)-N'-hydroxy-4-(piperidin-4-ylamino)-1,2,5-oxadiazole-3-carboxamidine
- Step 1 5-((4-(4-(3-Bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-1,2,4-oxadiazol-3-yl) Synthesis of -1,2,5-oxadiazol-3-yl)amino)hexahydrocyclopenta[c]pyrrole-2(1H)-carboxylic acid tert-butyl ester
- Step 2 4-(3-Bromo-4-fluorophenyl)-3-(4-((octahydrocyclopenta[c]pyrrole-5-yl)amino)-1,2,5-acean Synthesis of oxazol-3-yl)-1,2,4-oxadiazol-5(4H)-one
- Step 3 4-(3-Bromo-4-fluorophenyl)-3-(4-((2-(methylsulfonyl)octahydrocyclopenta[c]pyrrole-5-yl)amino) Synthesis of -1,2,5-oxadiazol-3-yl)-1,2,4-oxadiazol-5(4H)-one
- Step 4 N-(3-Bromo-4-fluorophenyl)-N'-hydroxy-4-((2-(methylsulfonyl)octahydrocyclopenta[c]pyrrole-5-yl) Synthesis of Amino)-1,2,5-oxadiazole-3-carboxamidine
- Step 1 ((5-((4-(4-(3-Bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-1,2,4-oxadiazol-3- Synthesis of tert-butyl-1,2,5-oxadiazol-3-yl)amino)hexahydrocyclopenta[c]pyrrole-2(1H)-yl)sulfonyl)carbamate
- Step 2 5-((4-(4-(3-Bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-1,2,4-oxadiazol-3-yl) Synthesis of -1,2,5-oxadiazol-3-yl)amino)hexahydrocyclopenta[c]pyrrole-2(1H)-sulfonamide
- Step 3 N-(3-Bromo-4-fluorophenyl)-N'-hydroxy-4-((2-sulfinyl octahydrocyclopenta[c]pyrrole-5-yl)amino)- Synthesis of 1,2,5-oxadiazole-3-carboxamidine
- Step 1 3-(4-((2-oxaspiro[3.3]heptan-6-yl)amino)-1,2,5-oxadiazol-3-yl)-4-(3-bromo- Synthesis of 4-fluorophenyl)-1,2,4-oxadiazol-5(4H)-one
- Step 2 4-((2-oxaspiro[3.3]heptan-6-yl)amino)-N-(3-bromo-4-fluorophenyl)-N'-hydroxy-1,2,5- Synthesis of Oxadiazole-3-carboxamidine
- Step 1 4-(3-Bromo-4-fluorophenyl)-3-(4-((2,2-dioxo-2-thiaspiro[3.3]heptan-6-yl)amino)- Synthesis of 1,2,5-oxadiazol-3-yl)-1,2,4-oxadiazol-5(4H)-one
- Step 2 N-(3-Bromo-4-fluorophenyl)-4-((2,2-dioxo-2-thiaspiro[3.3]heptan-6-yl)amino)-N'- Synthesis of Hydroxy-1,2,5-oxadiazole-3-carboxamidine
- Step 1 4-(3-Bromo-4-fluorophenyl)-3-(4-((tetrahydrofuran-3-yl)amino)-1,2,5-oxadiazol-3-yl)-1, Synthesis of 2,4-oxadiazol-5-(4H)-one
- Step 2 N-(3-Bromo-4-fluorophenyl)-N'-hydroxy-4-((tetrahydrofuran-3-yl)amino)-1,2,5-oxadiazole-3-carboxamidine synthesis
- Step 1 4-(3-Bromo-4-fluorophenyl)-3-(4-((1,1-dioxotetrahydrothiophen-3-yl)amino)-1,2,5-oxa 2 Synthesis of oxazol-3-yl)-1,2,4-oxadiazol-5(4H)-one
- Step 1 6-((4-(4-(3-Bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-1,2,4-oxadiazol-3-yl) Synthesis of tert-butyl ester of -1,2,5-oxadiazol-3-yl)amino)-2-azaspiro[3.3]heptane-2-carboxylate
- Step 2 3-(4-((2-Azaspiro[3.3]heptan-6-yl)amino)-1,2,5-oxadiazol-3-yl)-4-(3-bromo- Synthesis of 4-fluorophenyl)-1,2,4-oxadiazol-5(4H)-one
- Step 3 4-(3-Bromo-4-fluorophenyl)-3-(4-((2-(methylsulfonyl)-2-azaspiro[3.3]heptan-6-yl)amino) Synthesis of -1,2,5-oxadiazol-3-yl)-1,2,4-oxadiazol-5(4H)-one
- Step 4 N-(3-Bromo-4-fluorophenyl)-N'-hydroxy-4-((2-(methylsulfonyl)-2-azaspiro[3.3]heptane-6-yl) Synthesis of Amino)-1,2,5-oxadiazole-3-carboxamidine
- Step 1 ((6-((4-(4-(3-Bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-1,2,4-oxadiazole-3- Synthesis of tert-butyl ester of 1,1,2,5-oxadiazol-3-yl)amino)-2-azaspiro[3.3]heptan-2-yl)sulfonyl)carbamate
- Step 2 6-((4-(4-(3-Bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-1,2,4-oxadiazol-3-yl) Synthesis of -1,2,5-oxadiazol-3-yl)amino)-2-azaspiro[3.3]heptane-2-sulfonamide
- Step 3 N-(3-Bromo-4-fluorophenyl)-N'-hydroxy-4-((2-sulfamoyl-2-azaspiro[3.3]heptan-6-yl)amino)- Synthesis of 1,2,5-oxadiazole-3-carboxamidine
- Step 1 5-((4-(4-(3-Bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-1,2,4-oxadiazol-3-yl) Synthesis of tert-butyl ester of -1,2,5-oxadiazol-3-yl)amino)-2-azabicyclo[2.2.1]heptane-2-carboxylate
- Step 2 3-(4-((2-Azabicyclo[2.2.1]heptan-5-yl)amino)-1,2,5-oxadiazol-3-yl)-4-(3- Synthesis of bromo-4-fluorophenyl)-1,2,4-oxadiazol-5(4H)-one
- Step 3 ((5-((4-(4-(3-Bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-1,2,4-oxadiazole-3- Synthesis of tert-butyl ester of 1,1,2,5-oxadiazol-3-yl)amino)-2-azabicyclo[2.2.1]heptan-2-yl)sulfonyl)carbamate
- tert-Butyl alcohol (1.02 g, 118.0 mmol, 1.2 eq) was dissolved in DCM (10 mL), cooled to 0 ° C, chlorosulfonyl isocyanate (1.87 g) was added dropwise, and reacted for 1.5 hours, and the reaction solution (1.15 mL) was taken up.
- Step 4 5-((4-(4-(3-Bromo-4-fluorophenyl)-5-oxo-4,5-dihydro-1,2,4-oxadiazol-3-yl) Synthesis of -1,2,5-oxadiazol-3-yl)amino)-2-azabicyclo[2.2.1]heptane-2-sulfonamide
- Step 5 N-(3-Bromo-4-fluorophenyl)-N'-hydroxy-4-((2-sulfamoyl-2-azabicyclo[2.2.1]heptane-5-yl)amino Synthesis of-1,2,5-oxadiazole-3-carboxamidine
- Step 1 4-(3-Bromo-4-fluorophenyl)-3-(4-((2-(methylsulfonyl)-2-azabicyclo[2.2.1]heptane-5-yl)) Synthesis of Amino)-1,2,5-oxadiazolidine-3-yl)-1,2,4-oxadiazol-5(4H)-one
- EtOAc EtOAc
- EtOAcjjjjjjjjjjjj (3-Bromo-4-fluorophenyl)-3-(4-((2-(methylsulfonyl)-2-azabicyclo[2.2.1]heptane-5-yl)amino)-1, 2,5-oxadiazolidine-3-yl)-1,2,4-oxadiazol-5(4H)-one (267 mg, yield: 78.1%).
- Step 2 N-(3-Bromo-4-fluorophenyl)-N'-hydroxy-4-((2-(methylsulfonyl)-2-azabicyclo[2.2.1]heptane-5- Synthesis of yl)amino)-1,2,5-oxadiazole-3-carboxamidine
- Step 1 3-(4-((8-oxabicyclo[3.2.1]octane-3-yl)amino)-1,2,5-oxadiazol-3-yl)-4-(3- Synthesis of bromo-4-fluorophenyl)-1,2,4-oxadiazol-5(4H)-one
- Step 2 4-((8-oxabicyclo[3.2.1]octane-3-yl)amino)-N-(3-bromo-4-fluorophenyl)-N'-hydroxy-1,2, Synthesis of 5-oxadiazole-3-carboxamidine
- Compound 26 was prepared by the preparation method of Example 2.
- Test substance A compound of the invention, prepared by the corresponding examples of the invention.
- Hela cell line (purchased from ATCC, Cat. No. CCL-2, 4965442)
- rhIFN- ⁇ Recombinant human IFN- ⁇ (rhIFN- ⁇ , purchased from Peprotech, Cat. No. 300-02)
- MEM cell culture medium (purchased from Invitrogen, Cat. No. 11095098)
- Fetal bovine serum purchased from Invitrogen, Cat. No. 10099-141, Lot. No. 8153379)
- Hela cell suspension was prepared from fresh MEM cell culture medium, added to a 96-well cell culture plate at 5000 cells/well, and cultured at 5% carbon dioxide at 37 ° C overnight.
- Preparation of compound The compound was formulated into 2 mM in DMSO, and the compound was diluted 5-fold in DMSO to obtain a compound dilution mother solution (200 ⁇ ) of 6 to 9 concentration gradients, and a 10 ⁇ dilution solution was prepared in a ratio of cell culture medium and liquid. .
- Dosing incubation After the cells were plated overnight, 10 ⁇ L of the corresponding compound dilution mother solution (10 ⁇ ) and 10 ⁇ L of 500 ng/ml rhIFN- ⁇ were added to each well, and the final volume of each well was 100 ⁇ L.
- the negative control wells contained 100 ⁇ L of 0.5% DMSO cell culture medium and Hela cells.
- the positive control wells were filled with negative concentration of 50 ng/ml of rhIFN- ⁇ , and the background control wells contained 100 ⁇ L of cell culture medium. Incubate for 48 hours in a 37 ° C incubator and observe the cell morphology under an inverted microscope.
- Detection 80 ⁇ L of the supernatant was added to a Corning 3894 plate, 10 ⁇ L of 6.1 N trichloroacetic acid was added to each well, shaken for 2 minutes, placed in a 50-degree incubator for 30 minutes, centrifuged, and 2500 rpm for 10 minutes. 70 ⁇ L of the supernatant was transferred to a Corning 3635 UV plate, 70 ⁇ L of the reaction solution was added, and the mixture was shaken for 2 minutes to make the reaction uniform, and the OD value data at 480 nm was detected using EnSpire (PE).
- PE EnSpire
- the compounds of the present invention have excellent Hela cell activity.
- Test substance A compound of the invention, prepared by the corresponding examples of the invention.
- IDO-1 (provided by CP, batch number: 20160706)
- Test object IDO1(nM) Compound 13 220 Compound 15 220 Compound 15-P1 200 Compound 15-P2 240 Compound 16 170
- the compound of the present invention has excellent IDO enzymatic activity.
- Test substance A compound of the invention, prepared by the corresponding examples of the invention.
- Animal administration and sample collection intragastric administration: Compounds 2 and 13 were prepared by dissolving 10% DMA + 10% (30% Solutol) + 80% physiological saline. Compound 15 was treated with 10% DMA + 10% PEG400 + 80. The solution was prepared by dissolving % physiological saline, and the SD rats were intragastrically administered at a dose of 5.0 mg/kg. The time of blood collection was: 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h, 24 h.
- Intravenous administration Compounds 2 and 13 were dissolved in 10% DMA + 10% (30%solutol) + 80% physiological saline to prepare a solution, and compound 15 was dissolved in 10% DMA + 10% PEG 400 + 80% physiological saline to prepare a solution.
- SD rats were administered statically at a dose of 1 mg/kg. The time points of blood collection were: 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h, 24 h.
- the animals were fixed, and the tail was heated in a water bath 10 min before each time point, and about 100 ⁇ L of blood was collected through the tail vein, and the blood was collected and placed in an anti-coagulation tube containing EDTA-K2. Blood samples were centrifuged at 8000 rpm for 6 min at 4 ° C to obtain plasma samples, which must be prepared within 30 min after blood collection. Store in a -80 ° C freezer before plasma testing.
- the concentration of the test substance was output using AB's Analyst 1.6.3.
- the compounds of the present invention have excellent pharmacokinetic properties.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Virology (AREA)
- Engineering & Computer Science (AREA)
- Oncology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Communicable Diseases (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Psychiatry (AREA)
- Biotechnology (AREA)
- Pain & Pain Management (AREA)
- Pulmonology (AREA)
- AIDS & HIV (AREA)
- Tropical Medicine & Parasitology (AREA)
- Hospice & Palliative Care (AREA)
- Hematology (AREA)
- Transplantation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
本发明属于医药技术领域,具体涉及式I所示的化合物、其药学上可接受的盐及其立体异构体,X、Y、Z、R 1、R 2、R 3、R 1a、R 2a、m、n如说明书中所定义;本发明还涉及这些化合物的药物制剂、药物组合物,及其在制备治疗由吲哚胺2,3-双加氧酶(IDO)异常介导的相关疾病的药物中的用途。
Description
本发明属于医药技术领域,涉及吲哚胺2,3-双加氧酶(IDO)抑制剂化合物、其药学上可接受的盐及其立体异构体,及这些化合物的药物制剂、药物组合物,及其在制备治疗由吲哚胺2,3-双加氧酶(IDO)异常介导的相关疾病的药物中的用途。
色氨酸(Trp)是体内的必需氨基酸,在体内用于生物合成蛋白质,烟酸和神经递质5-羟色胺(血清素)。吲哚胺2,3-双加氧酶(IDO)是一种细胞内含亚铁血红素的单体酶,广泛分布于人和动物的许多组织和细胞中。IDO可催化L-色氨酸的吲哚环氧化裂解生成犬尿氨酸(kynurenine)的第一步反应,该步骤也是关键限速反应。IDO在机体正常状态下呈低水平表达,在疾病状态下,可以检测到其异常高表达。IDO在神经系统疾病(尤其是阿尔茨海默症和抑郁症)的发病机制中发挥着至关重要的作用。IDO还具有免疫耐受功能,肿瘤细胞、抗原递呈细胞上的IDO均可诱导T细胞对肿瘤抗原的免疫耐受。除此之外,IDO也参与慢性感染、HIV-感染、AIDS、自身免疫疾病等发病过程。因此,IDO已被证实是一个重要的药物发现靶标,IDO抑制剂的开发非常急需。
IDO主要通过两种机制影响脑部的功能,从而导致神经系统疾病的发生:(1)在炎症反应时,通过代谢色氨酸,降低了循环的色氨酸浓度,从而使5-羟基色胺水平降低,导致抑郁;(2)催化色氨酸循犬尿氨酸途径代谢使犬尿氨酸和神经毒性喹啉酸累积,从而导致神经系统疾病的发生。因此,IDO抑制剂在治疗阿尔茨海默病、抑郁症等神经系统疾病方面具有广阔的前景。
IDO也已经成为抗肿瘤免疫疗法非常重要的小分子调控靶点,其对于免疫系统的调控主要包括以下几方面:(1)IDO高表达可导致细胞局部的色氨酸耗竭,因为T细胞对色氨酸的耗竭特别敏感,因此在色氨酸浓度降低时,T细胞的增殖就会停滞于G1期。(2)IDO依赖性的色氨酸降解导致犬尿氨酸水平的提高,诱导氧自由基介导的T细胞凋亡。(3)上调树突状细胞IDO的表达通过降解局部色氨酸而加强局部调节性T细胞(Treg)介导的免疫抑制,促使机体对肿瘤特异性抗原的外周免疫耐受。
目前,国内外也已经开展了一系列IDO小分子抑制剂的开发工作,用于治疗或者预防IDO相关的疾病。比如在专利WO99/29310报道了利用IDO抑制剂1-甲基-DL-色氨酸、4-(3-苯并呋喃)-DL-丙氨酸、4-(3-苯并噻吩)-DL-丙氨酸和6-硝基-L-色氨酸,改变T-细胞介导的免疫力,包括改变局部细胞外的色氨酸和色氨酸代谢物的浓度。
专利WO2004/094409也报道了一些具体有IDO抑制作用的化合物。在专利US2004/0234623中,报道了利用一种IDO抑制剂治疗癌症和感染。
但是现有的吲哚胺2,3-双加氧酶(IDO)抑制剂普遍抑制效力低下,目前尚无吲哚胺2,3-双加氧酶(IDO)抑制剂药物问世。在研药物中,较为有代表性的IDO抑制剂是Epacadostat(II期,Under Active Development)和NLG-919(I期,Under Active Development)。其结构式如下所示:

本发明致力于研究一系列新型高效的IDO抑制剂,具有良好的成药性,可用于预防和/或治疗阿尔茨海默病、白内障、细胞免疫激活相关的感染、自身免疫性疾病、艾滋病、癌症、抑郁症或色氨酸代谢异常的药物。
发明内容
本发明所要解决的技术问题是提供一类新型的IDO抑制剂,此类化合物对IDO具有很好的抑制活性,可用于治疗由IDO异常介导的疾病。
本发明提供如下技术方案:
1.式I所示的化合物、其药学上可接受的盐及其立体异构体:

其中,
 代表顺式异构体、反式异构体或顺反异构体的混合物;
代表顺式异构体、反式异构体或顺反异构体的混合物;
X和Y分别独立的选自CH或N;
Z选自O或S;
R
1选自环烷基、杂环基、杂芳基,可任选被R
1a取代,R
1a选自氢、卤素、C
1-6烷基、C
2-6烯基、C
2-6炔基、卤代C
1-6烷基、-CN、-NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-SR
b、-S(O)R
c、-S(O)NR
eR
f、-S(O)
2R
c、-S(O)
2NR
eR
f、环烷基、杂环基、杂芳基或芳基;
R
2选自环烷基、杂环基、杂芳基或芳基,可任选被R
2a取代, R
2a选自氢、卤素、C
1-6烷基、C
2-6烯基、C
2-6炔基、卤代C
1-6烷基、-CN、-NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-SR
b、-S(O)R
c、-S(O)NR
eR
f、-S(O)
2R
c、-S(O)
2NR
eR
f、环烷基、杂环基、杂芳基或芳基;
R
3选自氢、C
1-6烷基或环烷基;
R
b选自氢、C
1-6烷基、卤代C
1-6烷基、C
1-6烷氧基C
1-6烷基、环烷基、杂环基、杂芳基或芳基;
R
c选自氢、C
1-6烷基、卤代C
1-6烷基、C
1-6烷氧基、环烷基、杂环基、杂芳基或芳基;
R
d、R
e、R
f分别独立的选自氢、C
1-6烷基、卤代C
1-6烷基、环烷基、杂环基、杂芳基或芳基;
m为0、1、2、3、4或5;
n为0、1、2、3、4或5。
2.如方案1所述的化合物、其药学上可接受的盐及其立体异构体:
其中,
 代表顺式异构体、反式异构体或顺反异构体的混合物;
代表顺式异构体、反式异构体或顺反异构体的混合物;
X和Y分别独立的选自CH或N;
Z选自O或S;
R
1选自3-14元环烷基、3-14元杂环基、5-14元杂芳基,可任选被R
1a取代,R
1a选自氢、卤素、C
1-6烷基、C
2-6烯基、C
2-6炔基、卤代C
1-6烷基、-CN、-NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-SR
b、-S(O)R
c、-S(O)NR
eR
f、-S(O)
2R
c、-S(O)
2NR
eR
f、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;
R
2选自3-8元环烷基、3-14元杂环基、5-14元杂芳基或6-14元芳基,可任选被R
2a取代,R
2a选自氢、卤素、C
1-6烷基、C
2-6烯基、C
2-6炔基、卤代C
1-6烷基、-CN、-NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-SR
b、-S(O)R
c、-S(O)NR
eR
f、-S(O)
2R
c、-S(O)
2NR
eR
f、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;
R
3选自氢、C
1-6烷基或3-8元环烷基;
R
b选自氢、C
1-6烷基、卤代C
1-6烷基、C
1-6烷氧基C
1-6烷基、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;
R
c选自氢、C
1-6烷基、卤代C
1-6烷基、C
1-6烷氧基、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;
R
d、R
e、R
f分别独立的选自氢、C
1-6烷基、卤代C
1-6烷基、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;
m为0、1、2、3、4或5;
n为0、1、2、3、4或5。
3.如方案2所述的化合物、其药学上可接受的盐及其立体异构体,具有如下式II所示的结构:

其中,
 代表顺式异构体、反式异构体或顺反异构体的混合物;
代表顺式异构体、反式异构体或顺反异构体的混合物;
R
1选自3-12元杂环基、5-12元杂芳基,可任选被R
1a取代, R
1a选自氢、卤素、C
1-6烷基、C
2-6烯基、C
2-6炔基、卤代C
1-6烷基、-CN、-NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-SR
b、-S(O)R
c、-S(O)NR
eR
f、-S(O)
2R
c、-S(O)
2NR
eR
f、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;
R
2选自6-14元芳基或5-12元杂芳基,可任选被R
2a取代,R
2a选自氢、卤素、C
1-6烷基、C
2-6烯基、C
2-6炔基、卤代C
1-6烷基、-CN、-NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-SR
b、-S(O)R
c、-S(O)NR
eR
f、-S(O)
2R
c、-S(O)
2NR
eR
f、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;
R
3选自氢或C
1-6烷基;
R
b选自氢、C
1-6烷基、卤代C
1-6烷基或C
1-6烷氧基C
1-6烷基;
R
c选自氢、C
1-6烷基、卤代C
1-6烷基或C
1-6烷氧基;
R
d、R
e、R
f分别独立的选自氢、C
1-6烷基或卤代C
1-6烷基;
m为0、1、2或3;
n为0、1、2或3。
R
1进一步优选为3-8元杂环基、5-6元杂芳基、6-12元并杂环基、6-12元桥杂环基或6-12元螺杂环基,可任选被R
1a取代,R
1a如上文所定义。
R
1进一步优选为4-6元饱和杂环基、8元饱和并杂环基、7-8元饱和桥杂环基或7元饱和螺杂环基,可任选被R
1a取代,R
1a选自氢、卤素、C
1-6烷基、C
2-6烯基、C
2-6炔基、卤代C
1-6烷基、-CN、-NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-SR
b、-S(O)R
c、-S(O)NR
eR
f、-S(O)
2R
c或-S(O)
2NR
eR
f;
R
b选自氢、C
1-6烷基、卤代C
1-6烷基或C
1-6烷氧基C
1-6烷基;
R
c选自氢、C
1-6烷基、卤代C
1-6烷基或C
1-6烷氧基;
R
d、R
e、R
f分别独立的选自氢、C
1-6烷基或卤代C
1-6烷基。
优选地,R
1为
 环A为3-12元单、并、螺、桥杂环基;
环A为3-12元单、并、螺、桥杂环基;
R
2选自6-14元芳基。
优选地,R
1为
 环A为3-8元单杂环基、6-12元并杂环基、6-12元桥杂环基或6-12元螺杂环基;
环A为3-8元单杂环基、6-12元并杂环基、6-12元桥杂环基或6-12元螺杂环基;
R
2为苯基。
优选地,R
1为
 环A为4-6元饱和杂环基、8元饱和并杂环基、7-8元饱和桥杂环基或7元饱和螺杂环基;
环A为4-6元饱和杂环基、8元饱和并杂环基、7-8元饱和桥杂环基或7元饱和螺杂环基;
R
2为苯基。
4.如方案3所述的化合物、其药学上可接受的盐及其立体异构体:
其中,
R
1为
 T
1和T
2分别独立地选自CR
4R
4’、NR
4或O,t
1和t
2分别独立地为0、1、2或3,且t
1和t
2不同时为0;R
1可任选被R
1a取代,R
1a选自氢、卤素、C
1-6烷基、C
2-6烯基、C
2-6炔基、卤代C
1-6烷基、-CN、-NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-SR
b、-S(O)R
c、-S(O)NR
eR
f、-S(O)
2R
c、-S(O)
2NR
eR
f、5-7元环烷基、5-7元杂环基、5-7元杂芳基或苯基;R
4和R
4’分别独立地选自氢或C
1-6烷基;
T
1和T
2分别独立地选自CR
4R
4’、NR
4或O,t
1和t
2分别独立地为0、1、2或3,且t
1和t
2不同时为0;R
1可任选被R
1a取代,R
1a选自氢、卤素、C
1-6烷基、C
2-6烯基、C
2-6炔基、卤代C
1-6烷基、-CN、-NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-SR
b、-S(O)R
c、-S(O)NR
eR
f、-S(O)
2R
c、-S(O)
2NR
eR
f、5-7元环烷基、5-7元杂环基、5-7元杂芳基或苯基;R
4和R
4’分别独立地选自氢或C
1-6烷基;
R
2选自苯基,可任选被R
2a取代,R
2a选自氢、卤素或C
1-6烷基;
R
b选自氢、C
1-6烷基、卤代C
1-6烷基或C
1-6烷氧基C
1-6烷基;
R
c选自氢、C
1-6烷基、卤代C
1-6烷基或C
1-6烷氧基;
R
d、R
e、R
f分别独立的选自氢、C
1-6烷基或卤代C
1-6烷基。
5.如方案4所述的化合物、其药学上可接受的盐及其立体异构体:
其中,
R
1为
 t
1和t
2分别独立地为0、1、2或3,且t
1和t
2不同时为0;R
1可任选被R
1a取代,R
1a选自氢、卤素、C
1-6烷基、C
2-6烯基、C
2-6炔基、卤代C
1-6烷基、-CN、-NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-SR
b、-S(O)R
c、-S(O)NR
eR
f、-S(O)
2R
c或-S(O)
2NR
eR
f;R
4和R
4’分别独立地选自氢或C
1-6烷基;
t
1和t
2分别独立地为0、1、2或3,且t
1和t
2不同时为0;R
1可任选被R
1a取代,R
1a选自氢、卤素、C
1-6烷基、C
2-6烯基、C
2-6炔基、卤代C
1-6烷基、-CN、-NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-SR
b、-S(O)R
c、-S(O)NR
eR
f、-S(O)
2R
c或-S(O)
2NR
eR
f;R
4和R
4’分别独立地选自氢或C
1-6烷基;
R
b选自氢、C
1-6烷基、卤代C
1-6烷基或C
1-6烷氧基C
1-6烷基;
R
c选自氢、C
1-6烷基、卤代C
1-6烷基或C
1-6烷氧基;
R
d、R
e、R
f分别独立的选自氢、C
1-6烷基或卤代C
1-6烷基。
优选地,R
1为
 t
1和t
2分别独立地为0、1、2、3,且t
1和t
2不同时为0,R
1可任选被R
1a取代,R
1a选自氢、卤素或C
1-6烷基;
t
1和t
2分别独立地为0、1、2、3,且t
1和t
2不同时为0,R
1可任选被R
1a取代,R
1a选自氢、卤素或C
1-6烷基;
R
2选自苯基,可任选被R
2a取代,R
2a选自氢、卤素或C
1-6烷基;
R
3选自氢或C
1-6烷基;
m为0、1或2;
n为0、1、2、3、4或5。
6.如方案3所述的化合物、其药学上可接受的盐及其立体异构体:
其中,
R
1为
 环A为6-12元并杂环基、6-12元桥杂环基或6-12元螺杂环基,可任选被R
1a取代,R
1a选自氢、卤素、C
1-6烷基、C
2-6烯基、C
2-6炔基、卤代C
1-6烷基、-CN、-NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-SR
b、-S(O)R
c、-S(O)NR
eR
f、-S(O)
2R
c、-S(O)
2NR
eR
f、5-7元环烷基、5-7元杂环基、5-7元杂芳基或苯基;
环A为6-12元并杂环基、6-12元桥杂环基或6-12元螺杂环基,可任选被R
1a取代,R
1a选自氢、卤素、C
1-6烷基、C
2-6烯基、C
2-6炔基、卤代C
1-6烷基、-CN、-NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-SR
b、-S(O)R
c、-S(O)NR
eR
f、-S(O)
2R
c、-S(O)
2NR
eR
f、5-7元环烷基、5-7元杂环基、5-7元杂芳基或苯基;
R
2选自苯基,可任选被R
2a取代,R
2a选自氢、卤素或C
1-6烷基;
R
b选自氢、C
1-6烷基、卤代C
1-6烷基或C
1-6烷氧基C
1-6烷基;
R
c选自氢、C
1-6烷基、卤代C
1-6烷基或C
1-6烷氧基;
R
d、R
e、R
f分别独立的选自氢、C
1-6烷基或卤代C
1-6烷基。
7.如方案6所述的化合物、其药学上可接受的盐及其立体异构体:
R
1为
 环A为6-12元饱和并杂环基、6-12元饱和桥杂环基或6-12元饱和螺杂环基,可任选被R
1a取代,R
1a选自氢、卤素、C
1-6烷基、C
2-6烯基、C
2-6炔基、卤代C
1-6烷基、-CN、-NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-SR
b、-S(O)R
c、-S(O)NR
eR
f、-S(O)
2R
c或-S(O)
2NR
eR
f;
环A为6-12元饱和并杂环基、6-12元饱和桥杂环基或6-12元饱和螺杂环基,可任选被R
1a取代,R
1a选自氢、卤素、C
1-6烷基、C
2-6烯基、C
2-6炔基、卤代C
1-6烷基、-CN、-NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-SR
b、-S(O)R
c、-S(O)NR
eR
f、-S(O)
2R
c或-S(O)
2NR
eR
f;
R
b选自氢、C
1-6烷基、卤代C
1-6烷基或C
1-6烷氧基C
1-6烷基;
R
c选自氢、C
1-6烷基、卤代C
1-6烷基或C
1-6烷氧基;
R
d、R
e、R
f分别独立的选自氢、C
1-6烷基或卤代C
1-6烷基。
8.如方案3所述的化合物、其药学上可接受的盐及其立体异构体:
R
1选自5-6元杂芳基,可任选被R
1a取代,R
1a选自氢、卤素、C
1-6烷基、C
2-6烯基、C
2-6炔基、卤代C
1-6烷基、-CN、-NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-SR
b、-S(O)R
c、-S(O)NR
eR
f、-S(O)
2R
c、-S(O)
2NR
eR
f、5-7元环烷基、5-7元杂环基、5-7元杂芳基或苯基;
R
2选自苯基,可任选被R
2a取代,R
2a选自氢、卤素或C
1-6烷基;
R
b选自氢、C
1-6烷基、卤代C
1-6烷基或C
1-6烷氧基C
1-6烷基;
R
c选自氢、C
1-6烷基、卤代C
1-6烷基或C
1-6烷氧基;
R
d、R
e、R
f分别独立的选自氢、C
1-6烷基或卤代C
1-6烷基。
9.如方案3所述的化合物、其药学上可接受的盐及其立体异构体:
R
1选自


 这些基团可任选被R
1a取代,R
1a选自氢、卤素、C
1-4烷基、卤代C
1-4烷基、-CN、-NO
2、-OR
b、 -C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-S(O)
2R
c或-S(O)
2NR
eR
f;
这些基团可任选被R
1a取代,R
1a选自氢、卤素、C
1-4烷基、卤代C
1-4烷基、-CN、-NO
2、-OR
b、 -C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-OC(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-NR
dC(O)NR
eR
f、-NR
dC(O)OR
b、-S(O)
2R
c或-S(O)
2NR
eR
f;
R
2选自苯基,可任选被R
2a取代,R
2a选自氢、卤素或C
1-4烷基;
R
3选自氢或C
1-4烷基;
R
b、R
c、R
d、R
e、R
f分别独立的选自氢或C
1-4烷基;
m为0、1、2或3;
n为0、1、2或3。
R
1优选为:


 这些基团可任选被R
1a取代,R
1a选自氢、卤素、C
1-4烷基、卤代C
1-4烷基、-CN、--NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-S(O)
2R
c或-S(O)
2NR
eR
f,
这些基团可任选被R
1a取代,R
1a选自氢、卤素、C
1-4烷基、卤代C
1-4烷基、-CN、--NO
2、-OR
b、-C(O)R
c、-C(O)OR
b、-OC(O)R
c、-C(O)NR
eR
f、-NR
eR
f、-NR
dC(O)R
c、-S(O)
2R
c或-S(O)
2NR
eR
f,
R
b、R
c、R
d、R
e、R
f分别独立的选自氢或C
1-4烷基。
优选地,R
1选自
 这些基团可任选被R
1a取代,R
1a选自氢、卤素、C
1-4烷基。
这些基团可任选被R
1a取代,R
1a选自氢、卤素、C
1-4烷基。
优选地,R
1选自

 这些基团可任选被R
1a取代,R
1a选自氢、卤素、C
1-4烷基。
这些基团可任选被R
1a取代,R
1a选自氢、卤素、C
1-4烷基。
本发明提供如下化合物、其药学上可接受的盐或其立体异构体:




本发明的另一技术方案是提供含有式I和式II所示化合物、其药学上可接受的盐及其立体异构体的药物组合物,可以任选含有一种或多种药用载体,制成药学上可接受的任一剂型。所述的药物组合物可以以任何合适的给药方式,例如口服、肠胃外、直肠或经肺给药等方式施用于需要这种治疗的患者或受试者。用于口服给药时,所述药物组合物可制成常规的固体制剂,如片剂、胶囊剂、丸剂、颗粒剂等;也可制成口服液体制剂,如口服溶液剂、口服混悬剂、糖浆剂等。制成口服制剂时,可以加入适宜的填充剂、粘合剂、崩解剂、润滑剂等。用于肠胃外给药时,所述药物组合物可制成注射剂、包括注射液、注射用无菌粉末与注射用浓溶液。制成注射剂时,可采用现有制药领域中的常规方法生产,配置注射剂时,可以不加入附加剂,也可以根据药物的性质加入适宜的附加剂。用于直肠给药时,所述药物组合物可制成栓剂等。用于经肺给药时,所述药物组合物可制成吸入剂或喷雾剂等。
本发明的另一技术方案是提供式I和式II所示化合物、其药学上可接受的盐及其立体异构体在制备治疗IDO异常介导的疾病的药物中的应用,尤其是治疗与癌症有关的肿瘤特异性免疫抑制。
所述的IDO异常介导的疾病为传染性疾病、神经系统疾病、癌症或非癌性增殖性疾病。所述传染性疾病包括由流感病毒、丙型肝炎病毒(HCV)、人乳头瘤病毒(HPV)、巨细胞病毒(CMV)、E-B病 毒(EBV)、脊髓灰质炎病毒、水痘带状疱疹病毒、柯萨奇病毒、人免疫缺陷病毒(HIV)感染所导致的疾病;所述神经系统疾病包括:阿尔茨海默症、抑郁症;所述的癌症包括肺癌、鳞状上皮细胞癌、膀胱癌、胃癌、卵巢癌、腹膜癌、乳腺癌、乳腺导管癌、头颈癌、子宫内膜癌、宫体癌、直肠癌、肝癌、肾癌、肾盂癌、食管癌、食管腺癌、神经胶质瘤、前列腺癌、甲状腺癌、女性生殖系统癌症、原位癌、淋巴瘤、神经纤维瘤病、骨癌、皮肤癌、脑癌、结肠癌、睾丸癌、胃肠道间质瘤、口腔癌、咽癌、多发性骨髓瘤、白血病、非霍奇金淋巴瘤、大肠绒毛腺瘤、黑色素瘤、细胞瘤、和肉瘤;所述的非癌性增值性疾病,是指因为免疫器官、免疫组织或免疫细胞异常增生(包括良性或恶性)所致的一组疾病,表现出免疫功能异常或免疫球蛋白水平增高,例如骨髓增生性疾病、和淋巴增值性疾病。
发明详述
本发明所述的“卤素”是指氟、氯、溴、碘等,优选氟原子,氯原子。
本发明所述的“卤代”是指取代基中的任一碳原子可被一个或多个相同或不同的卤素取代。“卤素”如前文所定义。
本发明所述的“C
1-6烷基”指含有1-6个碳原子的烃部分去除一个氢原子衍生的直链或支链的烷基,如甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、2-甲基丁基、新戊基、1-乙基丙基、正己基、异己基、4-甲基戊基、3-甲基戊基、2-甲基戊基、1-甲基戊基、3,3-二甲基丁基、2,2-二甲基丁基、1,1-二甲基丁基、1,2-二甲基丁基、1,3-二甲基丁基、2,3-二甲基丁基、2-乙基丁基和1-甲基-2-甲基丙基等。所述“C
1-4烷基”指含有1-4个碳原子的上述实例。
本发明所述的“C
2-6烯基”指含有碳碳双键的2-6个碳原子的烯烃部分去除一个氢原子衍生的直链或支链的烯烃基,如乙烯基、1-丙烯基、2-丙烯基、1-丁烯基、2-丁烯基、1,3-丁二烯基、1-戊烯基、2-戊烯基、3-戊烯基、1,3-戊二烯基、1,4-戊二烯基、1-己烯基、1,4-己二烯基、等。
本发明所述的“C
2-6炔基”指含有碳碳叁键的2-6个碳原子的炔烃部分去除一个氢原子衍生的直链或支链的炔烃基,如乙炔基、丙炔基、2-丁炔基、2-戊炔基、3-戊炔基、4-甲基-2-戊炔基、2-己炔基、3-己炔基等。
本发明所述的“C
1-6烷氧基”是指前文所定义的“C
1-6烷基”通过氧原子与母体分子部分连接的基团,即“C
1-6烷基-O-”基团,如甲氧基、乙氧基、丙氧基、异丙氧基、正丁氧基、叔丁氧基、正戊氧基、新戊氧基和正己氧基等。所述的“C
1-4烷氧基”指含有1-4个碳原子的上述实例,即“C
1-4烷基-O-”基团。
本发明所述的“稠环”是指由两个或两个以上环状结构以并、螺、桥的连接方式所形成的多环系结构。所述的并环是指由两个或两个以上环状结构彼此公用两个相邻的环原子(即共用一个键)所形成的稠环结构。所述的桥环是指有两个或两个以上环装结构彼此共用两个非相邻的环原子所形成的稠环结构。所述的螺环是指由两个或两个以上环状结构彼此共用一个环原子所形成的稠环结构。
本发明所述的“环烷基”,可以是3-14元环烷基,包括单环环烷基或者稠环环烷基,可以是饱和的、部分饱和的或不饱和,但不是芳香性的。单环环烷基可以为3-8元环烷基、5-7元环烷基,其实例包括但不限于:环丙烷基、环丁烷基、环戊烷基、环己烷基、环庚烷基、环辛烷基、环丁烯基、环戊烯基、环己烯基、1,4- 环己二烯基、环庚烯基、1,4-环庚二烯基、环辛烯基、1,5-环辛二烯基等。稠环环烷基包括并环环烷基、桥环烷基、螺环烷基,可以是饱和的、部分饱和的或不饱和的,但不是芳香性的。
所述的并环环烷基可以为6-12元并环环烷基、7-10元并环环烷基,其实例包括但不限于:双环[3.1.1]庚烷基、双环[2.2.1]庚烷基、双环[2.2.2]辛烷基、双环[3.2.2]壬烷基、双环[3.3.1]壬烷基和双环[4.2.1]壬烷基。
所述的螺环基可以为6-12元螺环基、7-11元螺环基,其实例包括但不限于:


所述的桥环基可以为6-12元桥环基、7-11元桥环基,其实例包括但不限于:

本发明所述的“杂环基”是指至少一个环碳原子被选自O、S、N的杂原子替代的非芳香性的环状基团,优选1-3个杂原子,同时包括碳原子、硫原子被氧代的情况,例如碳原子被C(O)、S(O)、S(O)2替代。
“杂环基”,可以为3-14元杂环基、3-12元杂环基,包括单杂环基或者稠杂环基。单杂环基可以为3-8元杂环基、3-8元饱和杂环基、3-6元杂环基、4-6元杂环基、5-7元杂环基、5-6元杂环基、5-6元含氧杂环基、5-6元含氮杂环基、5-6元饱和杂环基、5-7元饱和杂环基等。其实例包括但不限于:氮杂环丙烷基、2H-氮杂环丙烷基、二氮杂环丙烷基、3H-二氮杂环丙烯基、氮杂环丁烷基、1,4-二氧杂环己烷基、1,3-二氧杂环己烷基、1,3-二氧杂环戊烷基、1,4-二氧杂环己二烯基、四氢呋喃基、二氢吡咯基、吡咯烷基、咪唑烷基、4,5-二氢咪唑基、吡唑烷基、4,5-二氢吡 唑基、2,5-二氢噻吩基、四氢噻吩基、4,5-二氢噻唑基、哌啶基、哌嗪基、吗啉基、六氢嘧啶基、六氢哒嗪基、4,5-二氢噁唑基、4,5-二氢异噁唑基、2,3-二氢异噁唑基、2H-1,2-噁嗪基、6H-1,3-噁嗪基、4H-1,3-噻嗪基、6H-1,3-噻嗪基、2H-吡喃基、2H-吡喃-2-酮基、3,4-二氢-2H-吡喃基、1,1-二氧代四氢噻喃、1,1-二氧代四氢噻吩、1,1-二氧化硫杂环丁烷等。
稠杂环包括并杂环基、螺杂环基、桥杂环基,可以是饱和的、部分饱和的或不饱和的,但不是芳香性的。
所述的并杂环基可以为6-12元并杂环基、7-10元并杂环基、6-12元饱和并杂环基、7-8元饱和并杂环基、8元饱和并杂环基,其实例包括但不限于:3-氮杂双环[3.10.]己烷基、3,6-二氮杂双环[3.2.0]庚烷、3,8-二氮杂双环[4.2.0]辛烷基、3,7-二氮杂双环[4.2.0]辛烷基、八氢吡咯并[3,4-c]吡咯、八氢吡咯并[3,4-b]吡咯、八氢吡咯并[3,4-b][1,4]噁嗪基、八氢-1H-吡咯并[3,4-c]吡啶、2,3-二氢苯并呋喃-2-基、2,3-二氢苯并呋喃基-3-基、二氢吲哚-1-基、二氢吲哚-2-基、二氢吲哚3-基、2,3二氢苯并噻吩-2基、八氢-1H-吲哚基、八氢苯并呋喃基、八氢环戊二烯并[c]吡咯、六氢环戊二烯并[c]呋喃、2,2-二氧代六氢环戊二烯并[c]噻吩。
所述的螺杂环基可以为6-12元螺杂环基、7-11元螺杂环基、6-12元饱和螺杂环基、7元饱和螺杂环基,其实例包括但不限于:

所述的桥杂环基可以为6-12元桥杂环基、7-11元桥杂环基、6-12元饱和桥环基,7-8元饱和桥环基实例包括但不限于:


芳基是指芳香性的碳环基团,包括6-8元单环芳基和8-14元稠环芳基。6-8元单环芳基例如苯基、环辛四烯基等。8-14元稠环芳基例如萘、菲等。
本发明所述的“杂芳基”可以是5-14元杂芳基,是指至少一个环碳原子被选自O、S、N的杂原子替代的芳香性的环状基团,优选1-3个杂原子,同时包括碳原子、硫原子被氧代的情况,例如碳原子被C(O)、S(O)、S(O)2替代。杂芳基包括单杂芳基和稠杂芳基。单杂芳基可以为5-7元杂芳基、5-6元杂芳基,其实例包括但不仅限于呋喃基、咪唑基、异噁唑基、噻唑基、异噻唑基、噁二唑基、噁唑基、异噁唑基、吡啶基、吡啶酮基、哒嗪基、嘧啶基、吡嗪基、吡唑基、吡咯基、四唑基、噻二唑基、噻唑基、噻吩基、三唑基和三嗪基。稠杂芳基可以为8-12元并杂芳基、9-10元并杂芳基,其实例包括但不限于苯并咪唑基、苯并呋喃基、苯并噻吩基、苯并噻吩基、苯并噁二唑基、苯并噻唑基、噌啉基、吲唑基、吲哚基、异喹啉基、萘啶基、嘌呤基、喹啉基。
本发明所述的“药学上可接受的盐”是指式I或式II化合物的可药用的酸和碱的加成盐。当化合物中存在酸性官能团(如-COOH、-OH、-SO
3H等)与适当的无机或有机阳离子(碱)形成的盐,包括与碱金属或碱土金属形成的盐、铵盐,以及与含氮有机碱形成的盐;当化合物中存在碱性官能团(如-NH2等)与适当的无机或 者有机阴离子(酸)形成的盐,包括与无机酸盐、有机酸盐。这样的“药学上可接受的盐”包括但不限于,酸的盐:盐酸、磷酸、氢溴酸、硫酸、亚硫酸、甲酸、甲苯磺酸、甲磺酸、硝酸、苯甲酸、柠檬酸、酒石酸、马来酸、氢碘酸、链烷酸(如乙酸、HOOC-(CH
2)n-COOH(其中n是0~4))等;碱的盐:钠、钾、钙、铵等。
本发明所述的“立体异构体”是指当式I或式II化合物存在不对称碳原子时,会产生对映异构体;当化合物存在碳碳双键或环状结构时,会产生顺反异构体;当化合物存在酮或肟时,会产生互变异构体,所有式I或式II化合物的对映异构体、非对映异构体、消旋异构体、顺反异构体、互变异构体、几何异构体、差向异构体及其混合物,均包括在本发明范围中。
例如:
当
 存在时,可能产生
存在时,可能产生

当
 存在时,可能产生
存在时,可能产生

EA:乙酸乙酯
PE:石油醚
DMA:N,N-二甲基乙酰胺
THF:四氢呋喃
DCM:二氯甲烷
TEA:三乙醇胺
TFA:三氟乙酸
制备例1 4-氨基-N-(3-溴-4-氟苯基)-N'-羟基-1,2,5-噁二唑 -3-甲脒的合成

步骤1:4-氨基-N'-羟基-1,2,5-噁二唑-3-甲脒的合成

将原料丙二腈(50.0g,0.76mol,1.0eq)加入四口瓶中,加热溶解(约50℃),加入水(0.5L),冰水浴10℃左右,分批加入亚硝酸钠(57.72g,0.83mol,1.1eq),加毕,10℃以下滴加盐酸(6N,8.5mL),滴毕,冰水浴下搅拌0.5h至温度恒定不变,将上述反应液记为A。将H
2NOH·HCl(158.4g,2.28mol,3.0eq)溶解在水(255mL)中,向其中加入氢氧化钾(127.9g,2.28mol,3.0eq)的水(255mL)溶液,加毕,室温(25℃)搅拌10min,将上述反应液记为B。将A反应液用冰水浴降温至0℃-10℃,将B反应液滴加到A反应液中,滴毕,冰水浴下搅拌0.5h至温度恒定不变,撤去冰水浴,加热回流12h。待反应完毕,冰水浴(0℃)下,滴加入盐酸(6N,120mL),调节pH=7,继续搅拌40min,反应液抽滤,滤饼用水打洗,滤饼抽干,得到目标化合物(101g,收率:93.5%)。
步骤2:4-氨基-N-羟基-1,2,5-噁二唑-3-甲亚胺酰氯的合成

将4-氨基-N'-羟基-1,2,5-噁二唑-3-甲脒(101g,0.71mol,1.0eq)加入反应瓶中,加入水(1.4L)搅拌,搅拌下加入HCl(6N,350mL),AcOH(710mL),加热(42℃-45℃)搅拌至溶液澄清,加入NaCl(124.5g,2.13mol,3.0eq),于冰水浴下搅拌冰水浴下滴加NaNO
2(48.3g,0.70mol,0.98eq)的水(168mL)溶液,3.5h滴加完毕,加毕,冰水浴下搅拌1.5h,缓慢升温至室温(25℃)搅拌,约1h,反应液抽滤,滤饼用水洗涤,滤饼抽干,得到4-氨基-N-羟基-1,2,5-噁二唑-3-甲亚胺酰氯(粗品36.78g,收率:32%)。
步骤3:4-氨基-N-(3-溴-4-氟苯基)-N'-羟基-1,2,5-噁二唑-3-甲脒的合成

将4-氨基-N-羟基-1,2,5-噁二唑-3-甲亚胺酰氯(36.78g,0.23mol,1.0eq)溶于水(318ml)中,升温搅拌至60℃,加入3-溴-4-氟苯胺(47.50g,0.25mol,1.1eq),于60℃下搅拌约10min,滴加NaHCO
3(29.40g,0.35mmol,1.5eq)的水(318mL)溶液,20min滴加完毕,滴毕于60℃下搅拌30min,冷却至室温,反应液抽滤,滤饼用水洗涤,滤饼抽干,用水打浆过夜,次日抽滤,得灰色固体,烘干,得目标产物(61.21g,收率:84.2%)。
1HNMR(400MHz,d
6-DMSO)δ(ppm):11.44(s,1H),8.88(s,1H),7.17-7.21(t,1H),7.09-7.12(d,1H),6.75-6.79(m,1H),6.26(s,2H).
分子式:C
9H
7BrFN
5O
2,分子量:316.09,LC-MS(Neg,m/z)=314.0[M-H
+].
制备例2 4-(3-溴-4-氟苯基)-3-(4-硝基-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮的合成

步骤1:3-(4-氨基-1,2,5-噁二唑-3-基)-4-(3-溴 4-氟苯基)-1,2,4-噁二唑-5(4H)-酮的合成

将4-氨基-N-(3-溴-4-氟苯基)-N'-羟基-1,2,5-噁二唑-3-甲脒(5.0g,15.8mmol,1.0eq)溶解在乙酸乙酯(65mL)中,加入羰基二咪唑(3.85g,23.7mmol,1.5eq),60℃加热反应0.5h,TLC监测反应完全,反应液冷却至室温,用1mol/L盐酸洗涤(65mL×2),合并有机相,分出有机相,浓缩干,粗品经过甲基叔丁基醚打浆抽滤抽干得3-(4-氨基-1,2,5-噁二唑-3-基)-4-(3-溴-4-氟苯基)-1,2,4-噁二唑-5(4H)-酮(3.53g,收率:65%)。
步骤2:4-(3-溴-4-氟苯基)-3-(4-硝基-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮的合成

将3-(4-氨基-1,2,5-噁二唑-3-基)-4-(3-溴-4-氟苯基)-1,2,4-噁二唑-5(4H)-酮(264mg,0.8mmol,1.0eq)溶于三 氟乙酸(5mL)中,加入双氧水(3mL,30%),45℃反应18h,TLC监测反应完全,反应液冷却至室温,冰浴下加入饱和碳酸氢钠溶液直到溶液不冒气泡为止,加入EA萃取(3×30mL),无水硫酸钠干燥,抽滤,滤液浓缩,粗品经硅胶柱层析(PE:EA=10:1-2:1),得到4-(3-溴-4-氟苯基)-3-(4-硝基-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)酮(189.9mg,收率:66%)。
1HNMR(400MHz,DMSO-d
6):8.06-8.04(m,1H),7.69-7.65(m,1H),7.60-7.55(m,1H).
本制备例所制备的4-(3-溴-4-氟苯基)-3-(4-硝基-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮可用于下述实施例。
实施例1 N-(3-溴-4-氟苯基)-N'-羟基-4-((四氢-2H-吡喃-4-基)氨基)-1,2,5-噁二唑-3-甲脒(化合物1)的合成

将4-(3-溴-4-氟苯基)-3-(4-硝基-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(189.9mg,0.51mmol,1.0eq)溶于THF(15mL)中,加入四氢-2H-吡喃-4-氨(103.2mg,1.02mmol,2.0eq)和2mol/L(1.5mL)氢氧化钠溶液,搅拌反应30min,TLC监测反应完全,用1mol/L盐酸溶液调节溶液pH=2-3,EA萃取(3×20mL),合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品经 硅胶柱层析(PE:EA=10:1-1:1),得到N-(3-溴-4-氟苯基)-N'-羟基-4-((四氢-2H-吡喃-4-基)氨基)-1,2,5-噁二唑-3-甲脒(61.3mg,收率:30%)。
1HNMR(400MHz,DMSO-d
6)δ(ppm):11.47(s,1H),
8.89(s,1H),7.11-7.19(m,2H),6.79(s,1H),6.15-6.16(m,1H),3.42(m,1H),3.32-3.39(m,2H),1.91-19.4(m,1H),1.46-1.49(m,2H).
分子式:C
14H
15BrFN
5O
3分子量:400.21LC-MS(Neg,m/z)=400.0[M-H
+].
实施例2:N-(3-溴-4-氟苯基)-4-((1,1-二氧代四氢-2H-噻喃-4-基)氨基)-N'-羟基-1,2,5-噁二唑-3-甲脒(化合物2)的合成

步骤1:4-(3-溴-4-氟苯基)-3-(4-((1,1-二氧代四氢-2H-噻喃-4-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)酮的合成

将4-(3-溴-4-氟苯基)-3-(4-硝基-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)酮(250.4mg,0.67mmol,1.0eq)溶于DMA(4mL)中,冰浴下加入4-氨基四氢-2H-噻喃1,1-二氧化物(200.8mg,1.35mmol,2.0eq),冰浴下搅拌反应1h,TLC监测反应完全,加入水(30mL),EA萃取(3×30mL),合并有机相, 无水硫酸镁干燥,过滤,浓缩,粗品经柱层析(PE:EA=10:1~1:1),得到4-(3-溴-4-氟苯基)-3-(4-((1,1-二氧代四氢-2H-噻喃-4-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)酮(78.9mg,收率:25%)。
步骤2:N-(3-溴-4-氟苯基)-4-((1,1-二氧代四氢-2H-噻喃-4-基)氨基)-N'-羟基-1,2,5-噁二唑-3-甲脒的合成

将4-(3-溴-4-氟苯基)-3-(4-((1,1-二氧代四氢-2H-噻喃-4-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)酮(78.9mg,0.17mmol,1.0eq)溶于THF(10mL)中,加入2mol/L(2mL)氢氧化钠溶液,搅拌反应30min,TLC监测反应完全,用1mol/L盐酸溶液调节溶液pH=2~3,EA萃取(3×20mL),合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品经硅胶柱层析(PE:EA=10:1~1:1),得到N-(3-溴-4-氟苯基)-4-((1,1-二氧代四氢-2H-噻喃-4-基)氨基)-N'-羟基-1,2,5-噁二唑-3-甲脒(26mg,收率:35%)。
1HNMR(400MHz,DMSO-d
6)δ(ppm):11.42(s,1H),8.91(s,1H),7.18(m,1H),7.09-7.10(m,1H),6.77-6.79(m,1H),6.37-6.39(m,1H),3.70-3.72(m,1H),3.07-3.07(m,2H),2.20(3,3H),2.02-2.05(m,3H).
分子式:C
14H
15BrFN
5O
4S分子量:448.27 LC-MS(m/z)=448.0[M-H
+].
实施例3:N-(3-溴-4-氟苯基)-N'-羟基-4-((1-(甲基磺酰基)哌啶-4-基)氨基)-1,2,5-噁二唑-3-甲脒(化合物3)的合成

步骤1:4-(3-溴-4-氟苯基)-3-(4-((1-(甲基磺酰基)哌啶-4-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮的合成

将4-(3-溴-4-氟苯基)-3-(4-(哌啶-4-基氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(161.9mg,0.382mmol)(化合物4步骤2制备所得)溶于DCM(3mL)中,加入三乙胺(116.0mg,1.146mmol,3.0eq),滴加甲基磺酰氯(62.3mg,0.573mmol,1.5eq),反应1h,TLC监测反应完全。加入水(10mL),EA萃取(3×15mL),合并有机相,无水硫酸镁干燥,过滤,在旋转蒸发仪上浓缩得粗品4-(3-溴-4-氟苯基)-3-(4-((1-(甲基磺酰基)哌啶-4-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(170.0mg,收率:88.5%)。
步骤2:N-(3-溴-4-氟苯基)-N'-羟基-4-((1-(甲基磺酰基)哌啶-4-基)氨基)-1,2,5-噁二唑-3-甲脒的合成

将4-(3-溴-4-氟苯基)-3-(4-((1-(甲基磺酰基)哌啶-4-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(160.0mg,0.32mmol,1.0eq)溶于THF(2mL)中,加入氢氧化钠水溶液(10%)(3mL),室温搅拌反应30min,TLC监测反应完全,用饱和氯化铵水溶液中和,EA萃取(20mL×3),合并有机相,无水硫酸钠干燥,过滤,滤液浓缩,粗品经硅胶柱层析分离(洗脱剂DCM:MeOH=80:1,60:1),得到N-(3-溴-4-氟苯基)-N'-羟基-4-((1-(甲基磺酰基)哌啶-4-基)氨基)-1,2,5-噁二唑-3-甲脒(35.0mg,收率:23%)。
1HNMR(400MHz,DMSO-d
6)δ(ppm):11.47(s,1H),8.90(s,1H),7.16-7.21(t,2H),6.76-6.78(d,1H),6.19-6.21(d,1H),5.32(s,1H),3.49-3.52(d,3H),3.44(s,2H),3.07(s,3H),2.87(s,6H),1.98-2.05(m,4H),1.52-1.58(t,3H).
分子式:C
15H
18BrFN
6O
4S,分子量:477.31,LC-MS(Pos,m/z)=477.1[M+H
+].
实施例4:N-(3-溴-4-氟苯基)-N'-羟基-4-((1-氨磺酰基吡啶-4-基)氨基)-1,2,5-噁二唑-3-甲脒(化合物4)的合成

步骤1:4–((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)哌啶-1-甲酸叔丁酯的合成

将4-(3-溴-4-氟苯基)-3-(4-硝基-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(500.0mg,1.34mmol,1.0eq)溶于THF(5mL)中,加入4-氨基哌啶-1-羧酸叔丁酯(536.4mg,2.68mmol,2.0eq)和三乙胺(738.7mg,7.3mmol,5.4eq),加热到75℃搅拌反应过夜,TLC监测反应完全。加入水(15mL),EA萃取(3×20mL),合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品经硅胶柱层析(PE:EA=10:1~1:1),得到4–((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5噁二唑-3-基)氨基)哌啶-1-甲酸叔丁酯(400.0mg,收率:56.9%)。
步骤2:4-(3-溴-4-氟苯基)-3-(4-(哌啶-4-基氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮的合成

将4-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5噁二唑-3-基)氨基)哌啶-1-甲酸叔丁酯(400 mg,0.76mmol,1.0eq)溶于DCM(6mL)中,冷却至0℃,加入三氟乙酸(3mL),升至室温反应2h,TLC监测反应完全,反应液减压浓缩至,得到4-(3-溴-4-氟苯基)-3-(4-(哌啶-4-基氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(380mg,收率:以100%计)直接投下步反应。
步骤3:((4-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)哌啶-1-基)磺酰基)氨基甲酸叔丁酯的合成

将叔丁醇(34.1mg,0.46mmol,1.2eq)溶于DCM(3mL)中冷却至0℃,滴加氯磺酰基异氰酸酯(65.1mg,0.46mmol),0℃下反应1.5小时,反应液缓慢滴加到预先冷却至0℃以下的4-(3-溴-4-氟苯基)-3-(4-(哌啶-4-基氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(161.9mg,0.382mmol,1.0eq),TEA(116mg,3eq)和DCM(4mL)的混合液中,0℃以下搅拌反应2小时,TLC监测反应完全,加入水20mL搅拌,乙酸乙酯萃取(20mL×3),分液,合并有机相,无水硫酸钠干燥,过滤,滤液浓缩,粗品,得到((4-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)哌啶-1-基)磺酰基)氨基甲酸叔丁酯(240mg,收率:以100%计)。
步骤4:4-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)哌啶-1-磺酰胺的合成

将((4-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)哌啶-1-基)磺酰基)氨基甲酸叔丁酯(230.3mg)溶于DCM(4mL)中,冰浴至0℃加入TFA(2mL),搅拌升至室温反应2h,TLC监测反应完全,减压浓缩至干,得到4-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)哌啶-1-磺酰胺(200mg粗品,收率:以100%计),不经纯化直接投下步反应。
步骤5:N-(3-溴-4-氟苯基)-N'-羟基-4-((1-氨磺酰基吡啶-4-基)氨基)-1,2,5-噁二唑-3-甲脒的合成

将4-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)哌啶-1-磺酰胺(192.15mg粗品)溶于THF(3mL)中,加入氢氧化钠水溶液(10%)(6mL),室温搅拌反应1h,TLC监测反应完全,用饱和氯化铵水溶液中和,EA萃取(20mL×3),分液,合并有机相,无水硫酸钠干燥,过滤,滤液浓缩,粗品经硅胶柱层析分离(洗脱剂DCM:MeOH=100:1-80:1),得到N-(3-溴-4-氟苯基)-N'-羟基 -4-((1-氨磺酰基吡啶-4-基)氨基)-1,2,5-噁二唑-3-甲脒(30.0mg,收率:16.5%)。
1HNMR(400MHz,DMSO-d
6)δ(ppm):11.48(s,1H),8.89(s,1H),7.11-7.21(m,2H),6.74-6.81(m,3H),3.37-3.45(m,3H),2.66-2.73(m,2H),2.04-2.06(d,2H),1.57-1.60(t,2H).分子式:C
14H
17BrFN
7O
4S,分子量:478.30,LC-MS(Pos,m/z)=478.0[M+H
+].
实施例5:N-(3-溴-4-氟苯基)-N'-羟基-4-(哌啶-4-基氨基)-1,2,5-噁二唑-3-甲脒(化合物5)的合成

步骤1:4-((4-(N-(3-溴-4-氟苯基)-N'-羟基甲脒基)-1,2,5-噁二唑-3-基)氨基)哌啶-1-甲酸叔丁酯的合成

将4-(3-溴-4-氟苯基)-3-(4-硝基-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(500.0mg,1.34mmol,1.0eq)溶于THF(5mL)中,加入4-氨基哌啶-1-羧酸叔丁酯(536.4mg,2.68mmol,2.0eq)和三乙胺(738.7mg,7.3mmol,5.4eq)加热到75℃搅拌反应过夜,TLC监测反应完全。加入水(15mL),EA萃取(3×20mL),合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品经硅胶柱层析(PE:EA=10:1~1:1),得到4-((4-(N-(3-溴-4-氟 苯基)-N'-羟基甲脒基)-1,2,5-噁二唑-3-基)氨基)哌啶-1-甲酸叔丁酯(226.0mg,收率:33.9%)。
步骤2:N-(3-溴-4-氟苯基)-N'-羟基-4-(哌啶-4-基氨基)-1,2,5-噁二唑-3-甲脒的合成

将4-((4-(N-(3-溴-4-氟苯基)-N'-羟基甲脒基)-1,2,5-噁二唑-3-基)氨基)哌啶-1-甲酸叔丁酯(100.0mg,0.2mmol,1.0eq)溶于DCM(2mL)中,冷却至0℃,加入三氟乙酸(1mL),升至室温反应2h,TLC监测反应完全,加入饱和碳酸氢钠溶液,调节PH~7,EA(3×10mL)合并有机相,无水硫酸钠干燥,粗品经硅胶柱层析(EA:MeOH=30:1,20:1,10:1)得到N-(3-溴-4-氟苯基)-N'-羟基-4-(哌啶-4-基氨基)-1,2,5-噁二唑-3-甲脒(20.0mg,收率:25.0%)。
1HNMR(400MHz,DMSO-d
6)δ(ppm):11.49(s,1H),8.91(s,1H),8.64(s,1H),8.44(s,1H),7.16-7.20(t,1H),7.09(s,1H),6.79(s,1H),6.13-6.33(d,1H),5.32(s,1H),4.01-4.03(d,1H),2.08-2.11(d,3H),1.98(s,3H),1.63-1.65(d,3H).分子式:C
14H
16BrFN
6O
2,分子量:399.22,LC-MS(Pos,m/z)=399.0[M+H
+].
实施例6:N-(3-溴-4-氟苯基)-N'-羟基-4-((2-(甲基磺酰基)八氢环戊二烯并[c]吡咯-5-基)氨基)-1,2,5-噁二唑-3-甲脒(化合物8)合成

步骤1:5-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)六氢环戊二烯并[c]吡咯-2(1H)-羧酸叔丁酯的合成

将4-(3-溴-4-氟苯基)-3-(4-硝基-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(543.0mg,1.5mmol,1.0eq)溶于THF(6.0mL)中,加入5-氨基六氢环戊二烯并[c]吡咯-2(1H)-羧酸叔丁酯(575.3mg,2.2mmol,1.5eq)和三乙胺(819.6mg,8.1mmol,5.4eq),搅拌反应1.0h,TLC监测反应完全,向反应液中加入15.0mL水和6.0mL乙酸乙酯分液,水相乙酸乙酯萃取(2×6.0mL),合并有机相,饱和食盐水洗涤(15.0mL)无水硫酸钠干燥,过滤,浓缩,得到5-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)六氢环戊二烯并[c]吡咯-2(1H)-羧酸叔丁酯(810.6mg,收率:100%)。
步骤2:4-(3-溴-4-氟苯基)-3-(4–((八氢环戊二烯并[c]吡咯-5-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮的合成

将5-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)六氢环戊二烯并[c]吡咯-2(1H)-羧酸叔丁酯(581.0mg,1.1mmol,1.0eq)溶于DCM(6.0mL)中,加入三氟乙酸(3.0mL),室温搅拌反应2.0h,TLC监测反应完全。将反应液浓缩干,加入饱和碳酸氢钠(10.0mL)调节PH至中性,加入乙酸乙酯萃取(3×5.0mL),合并有机相,有机相饱和食盐水洗涤,无水硫酸钠干燥,过滤,浓缩得4-(3-溴-4-氟苯基)-3-(4–((八氢环戊二烯并[c]吡咯-5-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(278.9mg,收率:56.2%)。
步骤3:4-(3-溴-4-氟苯基)-3-(4–((2-(甲基磺酰基)八氢环戊二烯并[c]吡咯-5-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮的合成

将4-(3-溴-4-氟苯基)-3-(4–((八氢环戊二烯并[c]吡咯-5-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(230.1mg,0.51mmol,1.0eq)溶于二氯甲烷(4.0mL)中,加入三乙胺(154.8mg,1.53mmol,3.0eq),缓慢滴加甲磺酰氯(87.2mg, 0.76mmol,1.5eq),室温搅拌反应2.0h,TLC监测反应完全,向反应液中加入10.0mL水,乙酸乙酯萃取(5.0mL×3),有机相饱和食盐水(15.0mL)洗涤,无水硫酸钠干燥,过滤,得4-(3-溴-4-氟苯基)-3-(4–((2-(甲基磺酰基)八氢环戊二烯并[c]吡咯-5-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(250.7mg,收率:92.9%)。
步骤4:N-(3-溴-4-氟苯基)-N'-羟基-4-((2-(甲基磺酰基)八氢环戊二烯并[c]吡咯-5-基)氨基)-1,2,5-噁二唑-3-甲脒的合成

将4-(3-溴-4-氟苯基)-3-(4–((2-(甲基磺酰基)八氢环戊二烯并[c]吡咯-5-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(250.7mg,0.47mmol,1.0eq)溶于THF(6.0mL)中,加入氢氧化钠溶液(10%,3.0mL)室温搅拌反应半小时,TLC监测反应完全,向反应液中加入饱和氯化铵溶液15.0mL,加入乙酸乙酯萃取(8.0mL×3),合并有机相,有机相饱和食盐水(15.0mL)洗涤,无水硫酸钠干燥,过滤,浓缩,制砂,粗品硅胶柱层析(DCM:MeOH=200:1~100:1)得N-(3-溴-4-氟苯基)-N'-羟基-4-((2-(甲基磺酰基)八氢环戊二烯并[c]吡咯-5-基)氨基)-1,2,5-噁二唑-3-甲脒(65.7mg,收率:27.8%)。
1HNMR(400MHz,DMSO-d
6)δ(ppm):11.44(s,1H),8.87(s,1H),7.11-7.21(m,2H),6.76-6.80(m,1H),6.27(d,1H), 3.73-3.76(m,1H),3.20-3.22(m,2H),3.07-3.10(m,2H),2.80(s,3H),2.68(m,2H),2.33-2.37(m,2H),1.28-1.35(m,2H).
分子式:C
17H
20BrFN
6O
4S分子量:503.35 LC-MS(Neg,m/z)=501.55[M+H
+].
实施例7:N-(3-溴-4-氟苯基)-N'-羟基-4-((2-氨基磺酰基八氢环戊二烯并[c]吡咯-5-基)氨基)-1,2,5-噁二唑-3-甲脒(化合物9)的合成

步骤1:((5-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)六氢环戊二烯并[c]吡咯-2(1H)-基)磺酰基)氨基甲酸叔丁酯的合成

将磺酰氯异氰酸酯(104.7mg,0.74mmol,1.2eq)和叔丁醇(54.8mg,0.74mmol,1.2eq)溶于二氯甲烷(8.0mL)中,冰浴降温至0℃,加入三乙胺(154.8mg,1.53mmol,3.0eq),该温度下搅拌反应1.0h,向反应液中缓慢滴加2.0mL二氯甲烷溶解的4-(3-溴-4-氟苯基)-3-(4-((八氢环戊二烯并[c]吡咯-5-基) 氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(278.9mg,0.62mmol,1.0eq),室温搅拌反应1.5h,TLC监测反应完全,向反应液中加入饱和氯化铵溶液(15.0mL),乙酸乙酯萃取(8.0mL×3),有机相饱和食盐水(15.0mL)洗涤,无水硫酸钠干燥,过滤,浓缩得((5-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-二唑-3-基)氨基)六氢环戊二烯并[c]吡咯-2(1H)-基)磺酰基)氨基甲酸叔丁酯(234.2mg,收率:59.9%)。
步骤2:5-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)六氢环戊二烯并[c]吡咯-2(1H)-磺酰胺的合成

将((5-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)六氢环戊二烯并[c]吡咯-2(1H)-基)磺酰基)氨基甲酸叔丁酯(234.2mg,0.37mmol,1.0eq)溶于DCM(4.0mL)中,加入三氟乙酸(3.0mL),室温搅拌反应2.0h,TLC监测反应完全。将反应液浓缩,得到250.0mg粗品,收率:100%。
步骤3:N-(3-溴-4-氟苯基)-N'-羟基-4–((2-亚磺酰基八氢环戊二烯并[c]吡咯-5-基)氨基)-1,2,5-噁二唑-3-甲脒的合成

将5-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)六氢环戊二烯并[c]吡咯-2(1H)-磺酰胺(250.0mg,0.47mmol,1.0eq)溶于THF(10.0mL)中,加入氢氧化钠溶液(10%,5.0mL)室温搅拌反应40min,TLC监测反应完全,向反应液中加入饱和氯化铵溶液(20.0mL),加入乙酸乙酯萃取(8.0mL×3),合并有机相,有机相饱和食盐水(15.0mL)洗涤,无水硫酸钠干燥,过滤,浓缩,粗品硅胶柱层析(DCM:MeOH=200:1~100:1)得N-(3-溴-4-氟苯基)-N'-羟基-4-((2-氨基磺酰基八氢环戊二烯并[c]吡咯-5-基)氨基)-1,2,5-噁二唑-3-甲脒(30.2mg,收率:12.7%)。
1HNMR(400MHz,DMSO-d
6)δ(ppm):11.44(s,1H),8.87(s,1H),7.11-7.21(m,2H),6.76-6.80(m,1H),6.27(d,1H),3.73-3.76(m,1H),3.20-3.22(m,2H),2.67-2.69(m,2H),2.33-2.37(m,2H),1.24-1.30(m,2H).
分子式:C
16H
19BrFN
7O
4S分子量:504.34 LC-MS(Neg,m/z)=502.67[M+H
+].
实施例8:4-((2-氧杂螺[3.3]庚烷-6-基)氨基)-N-(3-溴-4-氟苯基)-N'-羟基-1,2,5-噁二唑-3-甲脒(化合物12)的合成

步骤1:3-(4-((2-氧杂螺[3.3]庚烷-6-基)氨基)-1,2,5-噁二唑-3-基)-4-(3-溴-4-氟苯基)-1,2,4-噁二唑-5(4H)-酮的合成

将4-(3-溴-4-氟苯基)-3-(4-硝基-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(329.9mg,0.88mmol,1.0eq)溶解在四氢呋喃(8mL)中,室温下向体系中加入三乙胺(538.3mg,5.32mmol,6.0eq)和2-氧杂螺[3.3]庚烷-6-胺盐酸盐(265.3mg,1.77mmol,2.0eq),加完升至75℃回流搅拌过夜。TLC监测反应完全,反应液冷却后加入用饱和氯化铵水溶液(50mL),用二氯甲烷(150mL×3)萃取,有机相用无水硫酸钠干燥,过滤,浓缩,粗品经硅胶柱层析分离(200-300目硅胶;石油醚和乙酸乙酯=10:1~8:1~6:1)得到3-(4-((2-氧杂螺[3.3]庚烷-6-基)氨基)-1,2,5-噁二唑-3-基)-4-(3-溴-4-氟苯基)-1,2,4-噁二唑-5(4H)-酮(170.4mg,收率:44.1%)。
步骤2:4-((2-氧杂螺[3.3]庚烷-6-基)氨基)-N-(3-溴-4-氟苯基)-N'-羟基-1,2,5-噁二唑-3-甲脒的合成

将3-(4-((2-氧杂螺[3.3]庚烷-6-基)氨基)-1,2,5-噁二唑-3-基)-4-(3-溴-4-氟苯基)-1,2,4-噁二唑-5(4H)-酮(169.4mg,0.38mmol,1.0eq)溶于四氢呋喃(4mL)中,室温搅拌下滴加10% 氢氧化钠溶液(2.5mL),滴毕,室温下搅拌1小时。TLC显示反应完全,向体系中加入水(40mL),用二氯甲烷(100mL×3)萃取,有机相无水硫酸钠干燥,粗品经硅胶柱层析分离(200-300目硅胶,石油醚和乙酸乙酯=10:1~1:1)得到产物4-((2-氧杂螺[3.3]庚烷-6-基)氨基)-N-(3-溴-4-氟苯基)-N'-羟基-1,2,5-噁二唑-3-甲脒(80.3mg,收率:51.2%)。
1HNMR(400MHz,DMSO-d
6)δ(ppm):11.40(s,1H),8.80(s,1H),7.11-7.19(m,2H),6.74(s,1H),6.35(s,1H),4.48-4.59(m,4H),3.76-3.78(s,1H),2.49-2.60(m,2H),2.11(m,2H).分子式:C
15H
15BrFN
5O
3分子量:412.22 LC-MS(Neg,m/z)=412.0[M+H
+].
实施例9:N-(3-溴-4-氟苯基)-4-((2,2-二氧代-2-硫杂螺[3.3]庚烷-6-基)氨基)-N'-羟基1,2,5-噁二唑-3-甲脒(化合物13)的合成

步骤1:4-(3-溴-4-氟苯基)-3-(4-((2,2-二氧代-2-硫杂螺[3.3]庚烷-6-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮的合成

将4-(3-溴-4-氟苯基)-3-(4-硝基-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(200.0mg,0.54mmol,1.0eq)溶于THF(2mL)中,加入6-氨基-2-硫杂螺[3.3]庚烷-2,2-二氧化物(174.1mg,1.08mmol,2.0eq)和二异丙基乙胺(209.4mg,1.62mmol,3.0eq),0℃搅拌2h,TLC监测反应完全。加入水(15mL),EA萃取(3×20mL),合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品经硅胶柱层析(DCM:MeOH=200:1~100:1),得到4-(3-溴-4-氟苯基)-3-(4-((2,2-二氧代-2-硫杂螺[3.3]庚烷-6-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(240.0mg,收率:92.3%)。
步骤2:N-(3-溴-4-氟苯基)-4-((2,2-二氧代-2-硫杂螺[3.3]庚烷-6-基)氨基)-N'-羟基-1,2,5-噁二唑-3-甲脒的合成

将4-(3-溴-4-氟苯基)-3-(4-((2,2-二氧代-2-硫杂螺[3.3]庚烷-6-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(230.0mg,0.47mmol)溶于THF(2mL)中,加入氢氧化钠水溶液(10%)(3mL),室温搅拌反应40min,TLC监测反应完全,加入H
2O,EA萃取(20mL×3),分液,合并有机相,无水硫酸钠干燥,过滤,滤液浓缩,粗品经硅胶柱层析分离(洗脱剂DCM:MeOH=200:1,100:1,80:1),得到N-(3-溴-4-氟苯基)-4-((2,2-二氧代-2-硫杂螺[3.3]庚烷-6-基)氨基)-N'-羟基-1,2,5-噁二唑-3-甲脒(110.0mg,收率:50.8%)。
1HNMR(400MHz,DMSO-d
6)δ(ppm):11.42(s,1H),8.90(s,1H),7.12-7.21(t,1H),7.10-7.12(t,1H),6.74-6.77(m,1H),6.47-6.49(d,1H),4.01-4.29(m,4H),3.91-3.97(m,1H),2.65-2.69(m,2H),2.30-2.50(m,2H).
分子式:C
15H
15BrFN
5O
4S,分子量:460.28,LC-MS(Pos,m/z)=460.0[M+H
+].
实施例10:N-(3-溴-4-氟苯基)-N'-羟基-4-((四氢呋喃-3-基)氨基)-1,2,5-噁二唑-3-甲脒(化合物14)的合成

步骤1:4-(3-溴-4-氟苯基)-3-(4-((四氢呋喃-3-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5-(4H)-酮的合成

将4-(3-溴-4-氟苯基)-3-(4-硝基-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(213.9mg,0.58mmol,1.0eq)溶于THF(2.0mL)中,加入四氢呋喃-3-胺(100.0mg,1.15mmol,2.0eq)和三乙胺(314.2mg,3.1mmol,5.4eq),75℃温度下回流反应过夜,TLC监测反应完全,向反应液中加入水(8mL)和乙酸乙酯(4mL)分液,水相乙酸乙酯萃取(2×4.0mL),合并有机相,饱和食盐水洗涤(10.0mL),无水硫酸钠干燥,过滤,浓缩,粗品经硅胶柱层析(PE:EA=10:1)得到4-(3-溴-4-氟苯基)-3-(4-((四氢呋喃-3-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5-(4H)-酮(102.0mg,收率:42.7%)。
步骤2:N-(3-溴-4-氟苯基)-N'-羟基-4–((四氢呋喃-3-基)氨基)-1,2,5-噁二唑-3-甲脒的合成

将4-(3-溴-4-氟苯基)-3-(4-((四氢呋喃-3-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5-(4H)-酮(102.0mg,0.24mmol,1.0eq)溶于THF(3.0mL)中,加入氢氧化钠溶液(10%,1.5mL),室温搅拌反应半小时,TLC监测反应完全,向反应液中加入饱和氯化铵溶液10.0mL,加入乙酸乙酯萃取(4mL×3),合并有机相,饱和食盐水(10.0mL)洗涤,无水硫酸钠干燥,过滤,浓缩,粗品硅胶柱层析(DCM:MeOH=200:1~100:1)得N-(3-溴-4-氟苯基)-N'-羟基-4–((四氢呋喃-3-基)氨基)-1,2,5-噁二唑-3-甲脒(29.6mg,收率:32.0%)。
1HNMR(400MHz,DMSO-d
6)δ(ppm):11.48(s,1H),8.88(s,1H),6.80-7.18(m,2H),6.77(m,1H),6.30(d,1H),4.04-4.10(m,1H),3.80(m,2H),3.73(m,1H),3.60(m,1H),2.22(m,1H),1.87(m,1H).
分子式:C
13H
13BrFN
5O
3分子量:386.18 LC-MS(Neg,m/z)=384.51[M+H
+].
实施例11:N-(3-溴-4-氟苯基)-4-((1,1-二氧代四氢噻吩-3-基)氨基)-N'-羟基-1,2,5-噁二唑-3-甲脒(化合物15)的合成

步骤1:4-(3-溴-4-氟苯基)-3-(4-((1,1-二氧代四氢噻吩 -3-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮合成

将4-(3-溴-4-氟苯基)-3-(4-硝基-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(300.0mg,0.81mmol,1.0eq)溶于DMA(4mL)中,冰浴下加入3-氨基四氢噻吩1,1-二氧化物盐酸盐(276.7mg,1.61mmol,2.0eq),冰浴下搅拌反应1h,TLC监测反应完全,加入水(30mL),EA萃取(3×30mL),合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品经硅胶柱层析(PE:EA=10:1~1:1)得到4-(3-溴-4-氟苯基)-3-(4-((1,1-二氧代四氢噻吩-3-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(165.1mg,收率:44.6%)。
步骤2:N-(3-溴-4-氟苯基)-4-((1,1-二氧代四氢噻吩-3-基)氨基)-N'-羟基-1,2,5-噁二唑-3-甲脒的合成

将4-(3-溴-4-氟苯基)-3-(4-((1,1-二氧代四氢噻吩-3-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(165.1mg,0.36mmol,1.0eq)溶于THF(15mL)中,加入氢氧化钠溶液(2mol/L,2mL),搅拌反应30min,TLC监测反应完全,用1mol/L盐酸溶液调节溶液pH=2~3,EA萃取(3×20mL),合并有机相,无水硫酸镁干燥,过滤,浓缩,粗品经硅胶柱层析(PE:EA=10:1~1:1),得到N-(3-溴-4-氟苯基)-4–((1,1-二氧代四氢噻吩-3-基) 氨基)-N'-羟基-1,2,5-噁二唑-3-甲脒(48.5mg,收率:31%)。
1HNMR(400MHz,DMSO-d
6)δ(ppm):11.48(s,1H),8.93(s,1H),7.11-7.20(m,2H),6.75-6.78(m,1H),6.63-6.65(m,1H),4.29-4.34(m,1H),3.51-3.53(m,1H),3.48-3.49(m,1H),2.20(3,3H),3.21-3.31-2.05(m,1H),3.07-3.19(m,1H),2.45-2.50(m,1H),2.17-2.19(m,1H).
分子式:C
13H
13BrFN
5O
4S分子量:434.24 LC-MS(m/z)=434.0[M-H
+].
取化合物15:N-(3-溴-4-氟苯基)-4-((1,1-二氧代四氢噻吩-3-基)氨基)-N'-羟基-1,2,5-噁二唑-3-甲脒(30mg),经过手性柱拆分,收集第一个峰,经冻干后得到白色固体(8.3mg,ee值100%),以下称为15-P1,收集第二个峰,经冻干后得白色固体(9.66mg,ee值99.7%),以下称之为15-P2,15-P1和15-P2的结构独立地对应化合物24和25中的每一个,


实施例12:N-(3-溴-4-氟苯基)-N'-羟基-4-((2-(甲基磺酰基)-2-氮杂螺[3.3]庚烷-6-基)氨基)-1,2,5-噁二唑-3-甲脒(化合物16)的合成

步骤1:6-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢 -1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)-2-氮杂螺[3.3]庚烷-2-羧酸叔丁酯的合成

将4-(3-溴-4-氟苯基)-3-(4-硝基-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(372mg,1mmol,1.0eq)和6-氨基-2-氮杂螺[3.3]庚烷-2-羧酸叔丁酯(448mg,2.11mmol,2.0eq)溶于THF(20ml),加入三乙胺(303mg,3mmol.3.0eq),升温至75℃反应2h。TLC(PE:EA=2:1)监测反应完全,加水(50ml)淬灭反应,EA(50mL×2)萃取,合并有机相,无水硫酸钠干燥,过滤,浓缩,粗品经硅胶柱层析(PE:EA=10:1~2:1)得目标化合物(500mg,收率:93%)。
步骤2:3-(4-((2-氮杂螺[3.3]庚烷-6-基)氨基)-1,2,5-噁二唑-3-基)-4-(3-溴-4-氟苯基)-1,2,4-噁二唑-5(4H)-酮的合成

将6-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)-2-氮杂螺[3.3]庚烷-2-羧酸叔丁酯(500mg,0.93mmol)溶于DCM(10mL),加入TFA(2mL),反应室温搅拌2h。TLC(DCM:MeOH=10:1)检测反应完全,反应液直接浓缩,加入DCM(50mL)溶解后真空浓缩,重复两次得目标化合物(790mg粗品,收率100%)。
步骤3:4-(3-溴-4-氟苯基)-3-(4-((2-(甲基磺酰基)-2-氮杂螺[3.3]庚烷-6-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑 -5(4H)-酮的合成

将3-(4-((2-氮杂螺[3.3]庚烷-6-基)氨基)-1,2,5-噁二唑-3-基)-4-(3-溴-4-氟苯基)-1,2,4-噁二唑-5(4H)-酮(200mg,粗品)溶于DCM(5mL),加入三乙胺(162mg,0.16mmol,),搅拌下缓慢加入甲磺酰氯(71mg,0.532mmol),室温搅拌两小时。TLC(DCM:MeOH=10:1)检测原料反应完全。浓缩,粗品经硅胶柱层析(DCM:MeOH=10:1)得产物(90mg,收率:74.2%)。
步骤4:N-(3-溴-4-氟苯基)-N'-羟基-4-((2-(甲基磺酰基)-2-氮杂螺[3.3]庚烷-6-基)氨基)-1,2,5-噁二唑-3-甲脒的合成

将4-(3-溴-4-氟苯基)-3-(4-((2-(甲基磺酰基)-2-氮杂螺[3.3]庚烷-6-基)氨基)-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(90mg,0.175mmol,1.0eq)溶THF(2mL),加入NaOH水溶液(2mol/L,0.5mL),室温反应两个小时。TLC(DCM:MeOH=10:1)检测原料完全,反应液倒入水(10mL)中,DCM(30mL×3)萃取,合并有机相,无水硫酸钠干燥,过滤,浓缩得粗品(80mg),粗品加入DCM(3mL)浆洗1小时,过滤,干燥得N-(3-溴-4-氟苯基)-N'-羟基-4-((2-(甲基磺酰基)-2-氮杂螺[3.3]庚烷-6-基)氨基)-1,2,5-噁二唑-3-甲脒(53mg,收率:62%)。
1HNMR(400MHz,DMSO-d
6)δ(ppm):11.42(s,1H),8.89(s,1H),7.10-7.21(m,1H),6.74-6.78(m,2H),6.39-6.41(m,1H),3.92(s,2H),3.83-3.89(m,1H),3.81(s,2H),2.96(s,3H),2.16-2.56(m,2H),2.13-2.15(m,2H).
分子式:C
16H
18BrFN
6O
4S分子量:488.03LC-MS(Pos,m/z)=489.0[M+H
+].
实施例13:N-(3-溴-4-氟苯基)-N'-羟基-4-((2-氨磺酰基-2-氮杂螺[3.3]庚烷-6-基)氨基)-1,2,5-噁二唑-3-甲脒(化合物17)的合成

步骤1:((6-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)-2-氮杂螺[3.3]庚烷-2-基)磺酰基)氨基甲酸叔丁酯的合成

将叔丁醇(45mg,0.6mmol,1.2eq)溶于DCM(10mL)中冷却至0℃,滴加氯磺酰基异氰酸(77.8mg,0.55mmol,1.1eq),反应1.5小时,吸取该反应液(1.15mL),缓慢滴加到预先冷却至0℃以下的3-(4-((2-氮杂螺[3.3]庚烷-6-基)氨基)-1,2,5-噁二唑-3-基)-4-(3-溴-4-氟苯基)-1,2,4-噁二唑-5(4H)-酮(化合物16步骤2制备所得)(218mg,0.50mmol,1.0eq)、TEA(125mg,1.5mmol,3.0eq)和DCM(5mL)的混合液中,0℃以下搅拌反应2 小时,TLC监测反应完全,加入水搅拌,DCM萃取(20mL×3),分液,合并有机相,无水硫酸钠干燥,过滤,滤液浓缩,粗品经硅胶柱层析分离(洗脱剂DCM:MeOH=10:1)得到((6-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)-2-氮杂螺[3.3]庚烷-2-基)磺酰基)氨基甲酸叔丁酯(120mg,收率38.9%)
步骤2:6-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)-2-氮杂螺[3.3]庚烷-2-磺酰胺的合成

将((6-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)-2-氮杂螺[3.3]庚烷-2-基)磺酰基)氨基甲酸叔丁酯(100mg)溶于DCM(2mL)中,加入TFA(0.5mL),室温反应30min,TLC监测反应完全,减压浓缩至干,得到6-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)-2-氮杂螺[3.3]庚烷-2-磺酰胺(120mg粗品,收率100%),不经纯化直接投下步反应。
步骤3:N-(3-溴-4-氟苯基)-N'-羟基-4-((2-氨磺酰基-2-氮杂螺[3.3]庚烷-6-基)氨基)-1,2,5-噁二唑-3-甲脒的合成

将6-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁 二唑-3-基)-1,2,5-噁二唑-3-基)氨基)-2-氮杂螺[3.3]庚烷-2-磺酰胺(120mg粗品)溶THF(2mL),加入NaOH水溶液(2mmol/L,0.5mL),室温反应2小时。TLC(DCM:MeOH=10:1)检测原料完全,将反应液倒入水(10mL)中,DCM萃取(30mL×3),合并有机相,无水硫酸钠干燥,过滤,浓缩,粗品经硅胶柱层析(DCM:MeOH=10:1)纯化得N-(3-溴-4-氟苯基)-N'-羟基-4-((2-氨磺酰基-2-氮杂螺[3.3]庚烷-6-基)氨基)-1,2,5-噁二唑-3-甲脒(25mg,三步收率:31.4%)。
1HNMR(400MHz,DMSO-d
6)δ(ppm):7.79(s,1H),7.24-7.60(m,2H),6.80-6.83(m,1H),6.58(s,2H),3.81-4.13(m,4H),3.06(s,3H),2.33-2.36(m,2H),1.87-1.89(dd,2H).
分子式:C
15H
17BrFN
7O
4S分子量:489.02LC-MS(Pos,m/z)=489.9[M+H
+].
实施例14:N-(3-溴-4-氟苯基)-N'-羟基-4-((2-氨磺酰基-2-氮杂双环[2.2.1]庚烷-5-基)氨基)-1,2,5-噁二唑-3-甲脒(化合物18)的合成

步骤1:5-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)-2-氮杂双环[2.2.1]庚烷-2-甲酸叔丁酯的合成

将4-(3-溴-4-氟苯基)-3-(4-硝基-1,2,5-噁二唑-3-基)-1,2,4-噁二唑-5(4H)-酮(701mg,1.884mmol,1.0eq)溶解在THF(45mL)中,加入5-氨基-2-氮杂双环[2.2.1]庚烷-2-羧酸叔丁酯(780mg,3.67mmol,2eq),加入TEA(1029mg,10.174mmol,5.4eq),70℃加热反应2h,TLC监测反应完全,反应液冷却至室温,用1mol/L盐酸调节反应液pH=2,EA萃取(20mL×3),分液,合并有机相,无水硫酸钠干燥,抽滤,滤液浓缩,柱层析分离(洗脱剂DCM:MeOH=100:1)得到5-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)-2-氮杂双环[2.2.1]庚烷-2-甲酸叔丁酯(粗品1072mg)。
步骤2:3-(4-((2-氮杂双环[2.2.1]庚烷-5-基)氨基)-1,2,5-噁二唑-3-基)-4-(3-溴-4-氟苯基)-1,2,4-噁二唑-5(4H)-酮的合成

将5-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)-2-氮杂双环[2.2.1]庚烷-2-甲酸叔丁酯(粗品1072mg)溶于THF(10mL)中,冷却至0℃,加入三氟乙酸(3.5mL),升至室温反应30min,TLC监测反应完全,反应液减压浓缩至干,得到3-(4-((2-氮杂双环[2.2.1]庚烷-5-基)氨基)-1,2,5-噁二唑-3-基)-4-(3-溴-4-氟苯基)-1,2,4-噁二唑-5(4H)-酮(872mg)直接投下步反应。
步骤3:((5-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)-2-氮杂双环[2.2.1]庚烷-2-基)磺酰基)氨基甲酸叔丁酯的合成

将叔丁醇(1.02g,118.0mmol,1.2eq)溶于DCM(10mL)中冷却至0℃,滴加氯磺酰基异氰酸酯(1.87g),反应1.5小时,吸取该反应液(1.15mL),缓慢滴加到预先冷却至0℃以下的中间体3-(4-((2-氮杂双环[2.2.1]庚烷-5-基)氨基)-1,2,5-噁二唑-3-基)-4-(3-溴-4-氟苯基)-1,2,4-噁二唑-5(4H)-酮(580mg,1.327mmol,1.0eq)、TEA(671.4mg,5eq)和DCM(5mL)的混合液中,0℃以下搅拌反应1小时,TLC监测反应完全,加入水20mL搅拌,DCM萃取(20mL×3),分液,合并有机相,无水硫酸钠干燥,过滤,滤液浓缩,粗品经硅胶柱层析分离(洗脱剂DCM:MeOH=100:1),得到((5-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)-2-氮杂双环[2.2.1]庚-2-基)磺酰基)氨基甲酸叔丁酯(621mg,收率:75.9%)。
步骤4:5-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)-2-氮杂双环[2.2.1]庚烷-2-磺酰胺的合成

将((5-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)-2-氮杂双环[2.2.1]庚烷-2-基)磺酰基)氨基甲酸叔丁酯(621mg)溶于DCM(4mL)中,加 入TFA(2mL),搅拌升至室温反应30min,TLC监测反应完全,减压浓缩得到5-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)-2-氮杂双环[2.2.1]庚烷-2-磺酰胺(600mg)直接投下步反应。
步骤5:N-(3-溴-4-氟苯基)-N'-羟基-4-((2-氨磺酰基-2-氮杂双环[2.2.1]庚烷-5-基)氨基)-1,2,5-噁二唑-3-甲脒的合成

将5-((4-(4-(3-溴-4-氟苯基)-5-氧代-4,5-二氢-1,2,4-噁二唑-3-基)-1,2,5-噁二唑-3-基)氨基)-2-氮杂双环[2.2.1]庚烷-2-磺酰胺(400mg粗品)溶于THF(4mL)中,加入2N氢氧化钠水溶液(4mL),室温搅拌反应30min,TLC监测反应完全,用饱和氯化铵水溶液中和,DCM萃取(20mL×3),分液,合并有机相,无水硫酸钠干燥,过滤,滤液浓缩,粗品经硅胶柱层析分离(洗脱剂DCM:MeOH=150:1),得到N-(3-溴-4-氟苯基)-N'-羟基-4-((2-氨磺酰基-2-氮杂双环[2.2.1]庚烷-5-基)氨基)-1,2,5-噁二唑-3-甲脒(49mg,三步收率:15%)。
1HNMR(400MHz,DMSO-d
6)δ(ppm):11.59(s,1H),8.88(s,1H),7.16-7.21(m,2H),6.85(s,1H),6.75-6.85(d,2H),6.35-6.36(d,1H),3.94(s,1H),3.89(s,1H),3.13-3.16(d,2H),2.88(s,1H),2.20-2.25(t,1H),1.82-1.84(d,1H),1.53-1.55(d,1H),1.40-1.43(d,1H).
分子式:C
15H
17BrFN
7O
4S,分子量:489.02,LC-MS(Neg,m/z)=488.0[M-H
+].
实施例15:N-(3-溴-4-氟苯基)-N'-羟基-4-((2-(甲基磺酰基)-2-氮杂双环[2.2.1]庚烷-5-基)氨基)-1,2,5-噁二唑-3-甲脒(化合物19)的合成

步骤1:4-(3-溴-4-氟苯基)-3-(4-((2-(甲基磺酰基)-2-氮杂双环[2.2.1]庚烷-5-基)氨基)-1,2,5-噁二唑吡啶-3-基)-1,2,4-噁二唑-5(4H)-酮的合成

将3-(4-((2-氮杂双环[2.2.1]庚烷-5-基)氨基)-1,2,5-噁二唑-3-基)-4-(3-溴-4-氟苯基)-1,2,4-噁二唑-5(4H)-酮(化合物18步骤2制备所得)(290mg,0.663mmol,1.0eq)溶于二氯甲烷(2mL)中,加入TEA(268mg,2.653mmol,4eq),加入甲磺酰氯(75.9mg,0.663mmol,1.0eq)。加入水搅拌,DCM萃取(20mL×3),分液,合并有机相,无水硫酸钠干燥,过滤,滤液浓缩,粗品经制备薄层色谱纯化(DCM:MeOH=15:1),得到4-(3-溴-4-氟苯基)-3-(4-((2-(甲基磺酰基)-2-氮杂双环[2.2.1]庚烷-5-基)氨基)-1,2,5-噁二唑吡啶-3-基)-1,2,4-噁二唑-5(4H)-酮(267mg,收率:78.1%)。
步骤2:N-(3-溴-4-氟苯基)-N'-羟基-4-((2-(甲基磺酰基)-2-氮杂双环[2.2.1]庚烷-5-基)氨基)-1,2,5-噁二唑-3-甲脒的合成

将4-(3-溴-4-氟苯基)-3-(4-((2-(甲基磺酰基)-2-氮杂双环[2.2.1]庚烷-5-基)氨基)-1,2,5-噁二唑吡啶-3-基)-1,2,4-二唑-5(4H)-酮(257mg)溶于THF(2mL)中,加入2N氢氧化钠水溶液(2mL),室温搅拌反应30min,TLC监测反应完全,用饱和氯化铵水溶液中和,DCM萃取(20mL×3),分液,合并有机相,无水硫酸钠干燥,过滤,滤液浓缩,粗品经硅胶制备版(DCM:MeOH=20:1)分离,得到N-(3-溴-4-氟苯基)-N'-羟基-4-((2-(甲基磺酰基)-2-氮杂双环[2.2.1]庚烷-5-基)氨基)-1,2,5-噁二唑-3-甲脒(110mg,收率:45%)。
1HNMR(400MHz,DMSO-d
6)δ(ppm):11.57(s,1H),8.89(s,1H),7.14-7.21(m,2H),6.82-6.89(m,1H),6.42-6.43(d,1H),4.04(s,1H),3.87-3.91(m,1H),3.15-3.23(m,2H),2.95(s,1H),2.91(s,3H),2.23-2.30(t,1H),1.62-1.74(t,2H),1.39-1.43(d,1H).
分子式:C
16H
18BrFN
6O
4S,分子量:488.03,LC-MS(Neg,m/z)=487.0[M-H
+].
实施例16:4-((8-氧杂双环[3.2.1]辛烷-3-基)氨基)-N-(3-溴-4-氟苯基)-N'-羟基-1,2,5-噁二唑-3-甲脒(化合物22)的合成

步骤1:3-(4-((8-氧杂双环[3.2.1]辛烷-3-基)氨基)-1,2,5- 噁二唑-3-基)-4-(3-溴-4-氟苯基)-1,2,4-噁二唑-5(4H)-酮的合成

将4-(3-溴-4-氟苯基)-3-(4-硝基-1,2,5-噁二唑-3-基)-1,2,4-二唑-5(4H)-酮(200mg,0.538mmol,1.0eq)溶解于THF(15ml)中,加入8-氧杂双环[3.2.1]辛烷-3-胺盐酸盐(175.9mg,1.075mmol,2.0eq)和三乙胺(293.7mg,2.903mmol,5.4eq),升温至70℃,反应2h后,TLC监测显示反应完成。用1mol/L的HCl调pH值至2左右,加水(10mL)和EA(20mL),搅拌,分液,有机相无水硫酸钠干燥,浓缩,粗品经硅胶柱层析(PE:EA=5:1)得(8-氧杂双环[3.2.1]辛烷-3-基)氨基)-1,2,5-噁二唑-3-基)-4-(3-溴-4-氟苯基)-1,2,4-噁二唑-5(4H)-酮(120mg,收率:49%)。
步骤2:4-((8-氧杂双环[3.2.1]辛烷-3-基)氨基)-N-(3-溴-4-氟苯基)-N'-羟基-1,2,5-噁二唑-3-甲脒的合成

将3-(4-((8-氧杂双环[3.2.1]辛烷-3-基)氨基)-1,2,5-噁二唑-3-基)-4-(3-溴-4-氟苯基)-1,2,4-噁二唑-5(4H)-酮(120mg,0.265mmol,1.0eq)溶于THF(1mL)中,加入2mol/L的NaOH溶液,搅拌2h后,TLC监测显示反应完全,加入饱和NH
4Cl溶液 (4mL),用DCM萃取(20mL),TLC显示水相无产品,合并有机相,干燥,浓缩,通过制备薄层色谱纯化(DCM:MeOH=20:1)得4-((8-氧杂双环[3.2.1]辛烷-3-基)氨基)-N-(3-溴-4-氟苯基)-N'-羟基-1,2,5-噁二唑-3-甲脒(80mg,收率:71%)。
1HNMR(400MHz,DMSO-d
6)δ(ppm):11.56(s,1H),11.46(s,1H),8.87(s,2H),7.14-7.19(m,3H),6.80(m,2H),6.45(m,1H),6.00(m,1H),4.30-4.35(m,3H),3.70(m,1H),1.75-2.10(m,12H),1.48-1.54(m,2H).
分子式:C
16H
17BrFN
5O
3分子量:426.25 LC-MS(Neg,m/z)=426.0[M-H
+].
实施例17 N-(3-溴-4-氟苯基)-4-((1,1-二氧化硫杂环丁烷-3-基)氨基)-N'-羟基-1,2,5-恶二唑-3-甲脒(化合物26)的合成

参照实施例2的制备方法制备得到化合物26。
1HNMR(400MHz,CDCl
3)δ(ppm):7.56-7.54(d,1H),7.07-7.02(t,1H),7.00-6.91(m,1H),6.47-6.42(t,1H),4.65-4.59(m,2H),4.55-4.50(m,1H),4.17-4.13(m,2H).
分子式:C
12H
11BrFN
5O
4S分子量:420.21 LC-MS(Pos,m/z)=420.0[M+H
+]
生物实验例
实验例1:Hela细胞评价方法
测试物:本发明化合物,由本发明相应的实施例所制备。
一、实验材料及仪器
Hela细胞株(购自ATCC,Cat.No.CCL-2,4965442)
重组人IFN-γ(rhIFN-γ,购自Peprotech,Cat.No.300-02)
MEM细胞培养基(购自Invitrogen,Cat.No.11095098)
胎牛血清(购自Invitrogen,Cat.No.10099-141,Lot.No.8153379)
6.1 N 三氯乙酸(购自Sigma,Cat.No.T0699)
二、试验步骤
细胞铺板:用新鲜MEM细胞培养基制取Hela细胞悬液,以5000个细胞/孔加入96孔细胞培养板中,5%二氧化碳37℃过夜培养。
准备化合物:用DMSO将化合物配制成2mM,以DMSO将化合物5倍稀释,得到6~9个浓度梯度的化合物稀释母液(200×),按比例用细胞培养基、液配成10×稀释液。。
加药孵育:细胞种板过夜后,每孔中加入10μL相应化合物稀释母液(10×)和10μL 500ng/ml rhIFN-γ,各孔终体积为100μL。阴性对照孔中含有100μL 0.5%DMSO细胞培养液培养及Hela细胞,阳性对照孔为阴性对照孔中添加终浓度为50ng/ml的rhIFN-γ,背景对照孔中含有100μL细胞培养液。在37℃培养箱中孵育48小时,在倒置显微镜下观察细胞形态。
检测:取80μL上清加到Corning3894板中,每孔加10μL 6.1N 三氯乙酸,振荡2分钟,放入50度恒温箱中反应30分钟,离心,2500转10分钟。取上清70μL转到Corning3635 UV板中,加入70μL反应液,振荡2分钟,使其反应均匀,使用EnSpire(PE)检测480nm时的OD值数据。
三、试验结果
表1本发明化合物的细胞学测试
| 测试物 | Hela(nM) |
| 化合物1 | 109 |
| 化合物2 | 39.73 |
| 化合物3 | 70.97 |
| 化合物4 | 78.78 |
| 化合物16 | 78.27 |
| 化合物12 | 104.63 |
| 化合物13 | 68.63 |
| 化合物15 | 51.61 |
| 化合物15-P1 | 19.4 |
| 化合物15-P2 | 119 |
| 化合物26 | 76 |
由上述结果可以看出,本发明化合物具有优异的Hela细胞活性。
实验例2:IDO1酶学评价方法
测试物:本发明化合物,由本发明相应的实施例所制备。
一、实验材料及仪器
IDO-1(CP提供,批号:20160706)
过氧化氢酶(购自Sigma,Cat.No.C9322-5G)
L-色氨酸(购自Sigma,Cat.No.93659-10G)
抗坏血酸盐(购自Sigma,Cat.No.11140-250G)
亚甲蓝(购自Sigma,Cat.No.M9140-100G)
DMSO(购自Sigma,Cat.No.D2650)
96孔板(购自Sigma,Cat.No.3635)
二、试验步骤
准备化合物:用DMSO将化合物配制成1mM化合物母液,以DMSO将化合物3倍稀释,得到9个浓度梯度的化合物稀释母液(100×)。
加药孵育:取2μL 100×的化合物稀释母液加入96孔板中,加入100μL酶液(IDO-1),室温孵育10min。以100%DMSO作为阴性对照,以NLG-919作为阳性对照。加入100μL底物混合液(L-色氨酸,抗坏血酸盐,亚甲蓝,过氧化氢酶)孵育。
检测:使用Spectramax检测321 nm时动态OD值数据。
计算:抑制率按照如下公式计算,使用Spectramax程序拟合曲线斜率,使用GraphPad Prism5.0拟合IC50:

三、试验结果
表2本发明化合物的酶学测试
| 测试物 | IDO1(nM) |
| 化合物13 | 220 |
| 化合物15 | 220 |
| 化合物15-P1 | 200 |
| 化合物15-P2 | 240 |
| 化合物16 | 170 |
由上述结果可以看出,本发明化合物具有优异的IDO酶学活性。
实施例3:化合物的大鼠PK评价
Solutol:15-羟基硬脂酸聚乙二醇酯
DMA:N,N-二甲基乙酰胺
测试物:本发明化合物,由本发明相应的实施例所制备。
动物给药及样品采集:灌胃给药:化合物2和13用10%DMA+10%(30%Solutol)+80%生理盐水溶解制备溶液剂,化合物15用10%DMA+10%PEG400+80%生理盐水溶解制备溶液剂,SD大鼠以5.0mg/kg的剂量灌胃给药,采血时间点为:15min,30min,1h,2h,4h,6h,8h,24h。
静脉给药:化合物2和13用10%DMA+10%(30%solutol)+80%生理盐水溶解制备溶液剂,化合物15用10%DMA+10%PEG400+80%生理盐水溶解制备溶液剂,SD大鼠以1mg/kg的剂量静推给药,采血时间点为:5min,15min,30min,1h,2h,4h,6h,8h,24h。
固定动物,每个时间点前10min用水浴锅加热尾部,通过尾静脉采集100μL左右的血液,血液采集后放置到含有EDTA-K2抗凝管中。血液样品在4℃条件下8000rpm离心6min得到血浆样品,血浆必需在血液采集后的30min内制备。血浆测试前存放在-80℃冰箱内。
样品分析方法:
从冰箱中取出待测样品(-80℃),室温自然融化后涡旋5min,精密吸取20μL血浆样品至1.5mL离心管中;加入200μL浓度为50ng/mL的内标工作溶液,混匀;涡旋5min后,离心5min(12000rpm);精密吸取50μL上清液至预先加有150μL/每孔水的96-孔板中;涡旋混匀5min,进样体积为20μL,进行LC-MS/MS测定。
数据处理方法:
受试物浓度使用AB公司的Analyst 1.6.3输出结果。
Microsoft Excel计算均值、标准差、变异系数等参数(Analyst 1.6.3直接输出的不用计算),PK参数采用Pharsight Phoenix 6.1 软件NCA计算(Tmax为中位数)。
结果:
表3本发明化合物的大鼠体内PK测试
(IV:1mg/kg,PO:5mg/kg,n=3)
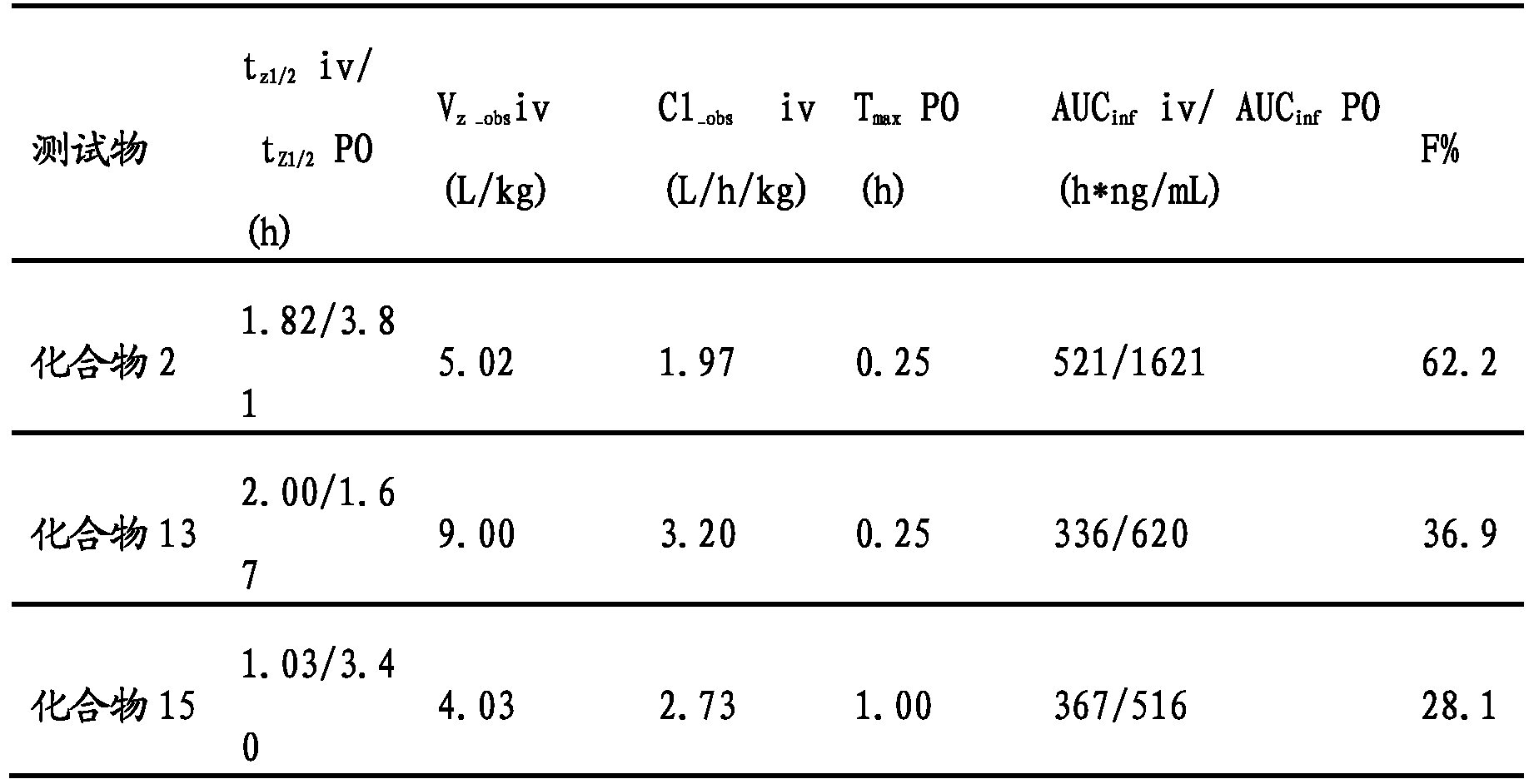
由上述结果可以看出,本发明化合物具有优异的药代动力学性质。
Claims (12)
- 式I所示的化合物、其药学上可接受的盐及其立体异构体:
 其中,代表顺式异构体、反式异构体或顺反异构体的混合物;
其中,代表顺式异构体、反式异构体或顺反异构体的混合物; X和Y分别独立的选自CH或N;Z选自O或S;R 1选自环烷基、杂环基、杂芳基,可任选被R 1a取代,R 1a选自氢、卤素、C 1-6烷基、C 2-6烯基、C 2-6炔基、卤代C 1-6烷基、-CN、-NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-SR b、-S(O)R c、-S(O)NR eR f、-S(O) 2R c、-S(O) 2NR eR f、环烷基、杂环基、杂芳基或芳基;R 2选自环烷基、杂环基、杂芳基或芳基,可任选被R 2a取代,R 2a选自氢、卤素、C 1-6烷基、C 2-6烯基、C 2-6炔基、卤代C 1-6烷基、-CN、-NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-SR b、-S(O)R c、-S(O)NR eR f、-S(O) 2R c、-S(O) 2NR eR f、环烷基、杂环基、杂芳基或芳基;R 3选自氢、C 1-6烷基或环烷基;R b选自氢、C 1-6烷基、卤代C 1-6烷基、C 1-6烷氧基C 1-6烷基、环 烷基、杂环基、杂芳基或芳基;R c选自氢、C 1-6烷基、卤代C 1-6烷基、C 1-6烷氧基、环烷基、杂环基、杂芳基或芳基;R d、R e、R f分别独立的选自氢、C 1-6烷基、卤代C 1-6烷基、环烷基、杂环基、杂芳基或芳基;m为0、1、2、3、4或5;n为0、1、2、3、4或5。
X和Y分别独立的选自CH或N;Z选自O或S;R 1选自环烷基、杂环基、杂芳基,可任选被R 1a取代,R 1a选自氢、卤素、C 1-6烷基、C 2-6烯基、C 2-6炔基、卤代C 1-6烷基、-CN、-NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-SR b、-S(O)R c、-S(O)NR eR f、-S(O) 2R c、-S(O) 2NR eR f、环烷基、杂环基、杂芳基或芳基;R 2选自环烷基、杂环基、杂芳基或芳基,可任选被R 2a取代,R 2a选自氢、卤素、C 1-6烷基、C 2-6烯基、C 2-6炔基、卤代C 1-6烷基、-CN、-NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-SR b、-S(O)R c、-S(O)NR eR f、-S(O) 2R c、-S(O) 2NR eR f、环烷基、杂环基、杂芳基或芳基;R 3选自氢、C 1-6烷基或环烷基;R b选自氢、C 1-6烷基、卤代C 1-6烷基、C 1-6烷氧基C 1-6烷基、环 烷基、杂环基、杂芳基或芳基;R c选自氢、C 1-6烷基、卤代C 1-6烷基、C 1-6烷氧基、环烷基、杂环基、杂芳基或芳基;R d、R e、R f分别独立的选自氢、C 1-6烷基、卤代C 1-6烷基、环烷基、杂环基、杂芳基或芳基;m为0、1、2、3、4或5;n为0、1、2、3、4或5。 - 如权利要求1所述的化合物、其药学上可接受的盐及其立体异构体:其中,代表顺式异构体、反式异构体或顺反异构体的混合物;
 X和Y分别独立的选自CH或N;Z选自O或S;R 1选自3-14元环烷基、3-14元杂环基、5-14元杂芳基,可任选被R 1a取代,R 1a选自氢、卤素、C 1-6烷基、C 2-6烯基、C 2-6炔基、卤代C 1-6烷基、-CN、-NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-SR b、-S(O)R c、-S(O)NR eR f、-S(O) 2R c、-S(O) 2NR eR f、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;R 2选自3-8元环烷基、3-14元杂环基、5-14元杂芳基或6-14元芳基,可任选被R 2a取代,R 2a选自氢、卤素、C 1-6烷基、C 2-6烯基、C 2-6炔基、卤代C 1-6烷基、-CN、-NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-SR b、-S(O)R c、-S(O)NR eR f、-S(O) 2R c、-S(O) 2NR eR f、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;R 3选自氢、C 1-6烷基或3-8元环烷基;R b选自氢、C 1-6烷基、卤代C 1-6烷基、C 1-6烷氧基C 1-6烷基、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;R c选自氢、C 1-6烷基、卤代C 1-6烷基、C 1-6烷氧基、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;R d、R e、R f分别独立的选自氢、C 1-6烷基、卤代C 1-6烷基、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;m为0、1、2、3、4或5;n为0、1、2、3、4或5。
X和Y分别独立的选自CH或N;Z选自O或S;R 1选自3-14元环烷基、3-14元杂环基、5-14元杂芳基,可任选被R 1a取代,R 1a选自氢、卤素、C 1-6烷基、C 2-6烯基、C 2-6炔基、卤代C 1-6烷基、-CN、-NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-SR b、-S(O)R c、-S(O)NR eR f、-S(O) 2R c、-S(O) 2NR eR f、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;R 2选自3-8元环烷基、3-14元杂环基、5-14元杂芳基或6-14元芳基,可任选被R 2a取代,R 2a选自氢、卤素、C 1-6烷基、C 2-6烯基、C 2-6炔基、卤代C 1-6烷基、-CN、-NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-SR b、-S(O)R c、-S(O)NR eR f、-S(O) 2R c、-S(O) 2NR eR f、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;R 3选自氢、C 1-6烷基或3-8元环烷基;R b选自氢、C 1-6烷基、卤代C 1-6烷基、C 1-6烷氧基C 1-6烷基、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;R c选自氢、C 1-6烷基、卤代C 1-6烷基、C 1-6烷氧基、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;R d、R e、R f分别独立的选自氢、C 1-6烷基、卤代C 1-6烷基、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;m为0、1、2、3、4或5;n为0、1、2、3、4或5。 - 如权利要求1或2所述的化合物、其药学上可接受的盐及其立体异构体,具有如下式II所示的结构:
 其中,代表顺式异构体、反式异构体或顺反异构体的混合物;
其中,代表顺式异构体、反式异构体或顺反异构体的混合物; R 1选自3-12元杂环基、5-12元杂芳基,可任选被R 1a取代,R 1a选自氢、卤素、C 1-6烷基、C 2-6烯基、C 2-6炔基、卤代C 1-6烷基、-CN、-NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-SR b、-S(O)R c、-S(O)NR eR f、-S(O) 2R c、-S(O) 2NR eR f、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;R 2选自6-14元芳基或5-12元杂芳基,可任选被R 2a取代,R 2a选自氢、卤素、C 1-6烷基、C 2-6烯基、C 2-6炔基、卤代C 1-6烷基、-CN、 -NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-SR b、-S(O)R c、-S(O)NR eR f、-S(O) 2R c、-S(O) 2NR eR f、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;R 3选自氢或C 1-6烷基;R b选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基C 1-6烷基;R c选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基;R d、R e、R f分别独立的选自氢、C 1-6烷基或卤代C 1-6烷基;m为0、1、2或3;n为0、1、2或3;优选地,R 1为环A为3-12元单、并、螺、桥杂环基;
R 1选自3-12元杂环基、5-12元杂芳基,可任选被R 1a取代,R 1a选自氢、卤素、C 1-6烷基、C 2-6烯基、C 2-6炔基、卤代C 1-6烷基、-CN、-NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-SR b、-S(O)R c、-S(O)NR eR f、-S(O) 2R c、-S(O) 2NR eR f、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;R 2选自6-14元芳基或5-12元杂芳基,可任选被R 2a取代,R 2a选自氢、卤素、C 1-6烷基、C 2-6烯基、C 2-6炔基、卤代C 1-6烷基、-CN、 -NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-SR b、-S(O)R c、-S(O)NR eR f、-S(O) 2R c、-S(O) 2NR eR f、3-8元环烷基、3-8元杂环基、5-8元杂芳基或6-14元芳基;R 3选自氢或C 1-6烷基;R b选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基C 1-6烷基;R c选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基;R d、R e、R f分别独立的选自氢、C 1-6烷基或卤代C 1-6烷基;m为0、1、2或3;n为0、1、2或3;优选地,R 1为环A为3-12元单、并、螺、桥杂环基; R 2选自6-14元芳基;优选地,R 1为环A为3-8元单杂环基、6-12元并杂环基、6-12元桥杂环基或6-12元螺杂环基;
R 2选自6-14元芳基;优选地,R 1为环A为3-8元单杂环基、6-12元并杂环基、6-12元桥杂环基或6-12元螺杂环基; R 2为苯基。
R 2为苯基。 - 如权利要求3所述的化合物、其药学上可接受的盐及其立体异构体:其中,R 1为T 1和T 2分别独立地选自CR 4R 4’、NR 4或O,t 1和t 2分别独立地为0、1、2或3,且t 1和t 2不同时为0;R 1可任选被R 1a取代,R 1a选自氢、卤素、C 1-6烷基、C 2-6烯基、C 2-6炔基、 卤代C 1-6烷基、-CN、-NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-SR b、-S(O)R c、-S(O)NR eR f、-S(O) 2R c、-S(O) 2NR eR f、5-7元环烷基、5-7元杂环基、5-7元杂芳基或苯基;R 4和R 4’分别独立地选自氢或C 1-6烷基;
 R 2选自苯基,可任选被R 2a取代,R 2a选自氢、卤素或C 1-6烷基;R b选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基C 1-6烷基;R c选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基;R d、R e、R f分别独立的选自氢、C 1-6烷基或卤代C 1-6烷基。
R 2选自苯基,可任选被R 2a取代,R 2a选自氢、卤素或C 1-6烷基;R b选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基C 1-6烷基;R c选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基;R d、R e、R f分别独立的选自氢、C 1-6烷基或卤代C 1-6烷基。 - 如权利要求4所述的化合物、其药学上可接受的盐及其立体异构体:其中,R 1为t 1和t 2分别独立地为0、1、2或3,且t 1和t 2不同时为0;R 1可任选被R 1a取代,R 1a选自氢、卤素、C 1-6烷基、C 2-6烯基、C 2-6炔基、卤代C 1-6烷基、-CN、-NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-SR b、-S(O)R c、-S(O)NR eR f、-S(O) 2R c或-S(O) 2NR eR f;R 4和R 4’分别独立地选自氢或C 1-6烷基;
 R b选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基C 1-6烷基;R c选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基;R d、R e、R f分别独立的选自氢、C 1-6烷基或卤代C 1-6烷基。优选地,R 1为t 1和t 2分别独立地为0、1、2、3,且t 1和t 2不同时为0,R 1可任选被R 1a取代,R 1a选自氢、卤素或C 1-6烷基;
R b选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基C 1-6烷基;R c选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基;R d、R e、R f分别独立的选自氢、C 1-6烷基或卤代C 1-6烷基。优选地,R 1为t 1和t 2分别独立地为0、1、2、3,且t 1和t 2不同时为0,R 1可任选被R 1a取代,R 1a选自氢、卤素或C 1-6烷基; R 2选自苯基,可任选被R 2a取代,R 2a选自氢、卤素或C 1-6烷基;R 3选自氢或C 1-6烷基;m为0、1或2;n为0、1、2、3、4或5。
R 2选自苯基,可任选被R 2a取代,R 2a选自氢、卤素或C 1-6烷基;R 3选自氢或C 1-6烷基;m为0、1或2;n为0、1、2、3、4或5。 - 如权利要求3所述的化合物、其药学上可接受的盐及其立体异构体:其中,R 1为环A为6-12元并杂环基、6-12元桥杂环基或6-12元螺杂环基,可任选被R 1a取代,R 1a选自氢、卤素、C 1-6烷基、C 2-6烯基、C 2-6炔基、卤代C 1-6烷基、-CN、-NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-SR b、-S(O)R c、-S(O)NR eR f、-S(O) 2R c、-S(O) 2NR eR f、5-7元环烷基、5-7元杂环基、5-7元杂芳基或苯基;
 R 2选自苯基,可任选被R 2a取代,R 2a选自氢、卤素或C 1-6烷基;R b选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基C 1-6烷基;R c选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基;R d、R e、R f分别独立的选自氢、C 1-6烷基或卤代C 1-6烷基。
R 2选自苯基,可任选被R 2a取代,R 2a选自氢、卤素或C 1-6烷基;R b选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基C 1-6烷基;R c选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基;R d、R e、R f分别独立的选自氢、C 1-6烷基或卤代C 1-6烷基。 - 如权利要求6所述的化合物、其药学上可接受的盐及其立体异构体:R 1为环A为6-12元饱和并杂环基、6-12元饱和桥杂环基或6-12元饱和螺杂环基,可任选被R 1a取代,R 1a选自氢、 卤素、C 1-6烷基、C 2-6烯基、C 2-6炔基、卤代C 1-6烷基、-CN,-NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-SR b、-S(O)R c、-S(O)NR eR f、-S(O) 2R c或-S(O) 2NR eR f;
 R b选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基C 1-6烷基;R c选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基;R d、R e、R f分别独立的选自氢、C 1-6烷基或卤代C 1-6烷基。
R b选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基C 1-6烷基;R c选自氢、C 1-6烷基、卤代C 1-6烷基或C 1-6烷氧基;R d、R e、R f分别独立的选自氢、C 1-6烷基或卤代C 1-6烷基。 - 如权利要求3所述的化合物、其药学上可接受的盐及其立体异构体:R 1选自

 这些基团可任选被R 1a取代,R 1a选自氢、卤素、C 1-4烷基、卤代C 1-4烷基、-CN、-NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-S(O) 2R c或-S(O) 2NR eR f;
这些基团可任选被R 1a取代,R 1a选自氢、卤素、C 1-4烷基、卤代C 1-4烷基、-CN、-NO 2、-OR b、-C(O)R c、-C(O)OR b、-OC(O)R c、-C(O)NR eR f、-OC(O)NR eR f、-NR eR f、-NR dC(O)R c、-NR dC(O)NR eR f、-NR dC(O)OR b、-S(O) 2R c或-S(O) 2NR eR f; R 2选自苯基,可任选被R 2a取代,R 2a选自氢、卤素或C 1-4烷基;R 3选自氢或C 1-4烷基;R b、R c、R d、R e、R f分别独立的选自氢或C 1-4烷基;m为0、1、2或3;n为0、1、2或3,优选地,R 1选自
R 2选自苯基,可任选被R 2a取代,R 2a选自氢、卤素或C 1-4烷基;R 3选自氢或C 1-4烷基;R b、R c、R d、R e、R f分别独立的选自氢或C 1-4烷基;m为0、1、2或3;n为0、1、2或3,优选地,R 1选自 这些基团可任选被R 1a取代,R 1a选自氢、卤素、C 1-4烷基,
这些基团可任选被R 1a取代,R 1a选自氢、卤素、C 1-4烷基, 优选地,R 1选自
优选地,R 1选自 这些基团可任选被R 1a取代,R 1a选自氢、卤素、C 1-4烷基。
这些基团可任选被R 1a取代,R 1a选自氢、卤素、C 1-4烷基。
- 如权利要求1所述的化合物、其药学上可接受的盐及其立体异构体,所述的化合物选自:



- 含有权利要求1-9任一项所述的化合物、其药学上可接受 的盐及其立体异构体的药物组合物,其特征在于包含一种或多种药用载体。
- 权利要求1-9任一项所述的化合物、其药学上可接受的盐及其立体异构体在制备治疗IDO异常介导的疾病的药物中的应用。
- 权利要求11所述的应用,其中IDO异常介导的疾病为传染性疾病、神经系统疾病、癌症或非癌性增殖性疾病,所述传染性疾病包括由流感病毒、丙型肝炎病毒、人乳头瘤病毒、巨细胞病毒、E-B病毒、脊髓灰质炎病毒、水痘带状疱疹病毒、柯萨奇病毒、或人免疫缺陷病毒感染所导致的疾病;所述神经系统疾病包括:阿尔茨海默症、和抑郁症;所述的癌症包括肺癌、鳞状上皮细胞癌、膀胱癌、胃癌、卵巢癌、腹膜癌、乳腺癌、乳腺导管癌、头颈癌、子宫内膜癌、宫体癌、直肠癌、肝癌、肾癌、肾盂癌、食管癌、食管腺癌、神经胶质瘤、前列腺癌、甲状腺癌、女性生殖系统癌症、原位癌、淋巴瘤、神经纤维瘤病、骨癌、皮肤癌、脑癌、结肠癌、睾丸癌、胃肠道间质瘤、口腔癌、咽癌、多发性骨髓瘤、白血病、非霍奇金淋巴瘤、大肠绒毛腺瘤、黑色素瘤、细胞瘤、和肉瘤;所述的非癌性增值性疾病,包括骨髓增生性疾病、和淋巴增值性疾病。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US16/607,827 US11046679B2 (en) | 2017-04-24 | 2018-04-24 | Indoleamine 2,3-dioxygenase inhibitor and application |
| EP18792247.1A EP3617204A1 (en) | 2017-04-24 | 2018-04-24 | Indoleamine 2,3-dioxygenase inhibitor and application |
| JP2019557849A JP6860237B2 (ja) | 2017-04-24 | 2018-04-24 | インドールアミン2,3−ジオキシゲナーゼ阻害剤及び適用 |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201710270704 | 2017-04-24 | ||
| CN201710270704.4 | 2017-04-24 | ||
| CN201710543401.5 | 2017-07-05 | ||
| CN201710543401 | 2017-07-05 | ||
| CN201711478213 | 2017-12-29 | ||
| CN201711478213.5 | 2017-12-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018196747A1 true WO2018196747A1 (zh) | 2018-11-01 |
Family
ID=63919479
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2018/084267 WO2018196747A1 (zh) | 2017-04-24 | 2018-04-24 | 吲哚胺2,3-双加氧酶抑制剂与应用 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US11046679B2 (zh) |
| EP (1) | EP3617204A1 (zh) |
| JP (1) | JP6860237B2 (zh) |
| CN (1) | CN108727361A (zh) |
| WO (1) | WO2018196747A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20210198247A1 (en) * | 2017-12-20 | 2021-07-01 | Hinova Pharmaceuticals Inc. | Indoleamine-2,3-dioxygenase inhibitor, the preparative method and the use thereof |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20200361919A1 (en) * | 2017-09-01 | 2020-11-19 | Nanjing Transthera Biosciences Co. Ltd. | Deuterated indoleamine 2,3-dioxygenase inhibitor and application thereof |
| CN110343098B (zh) * | 2018-04-04 | 2023-05-26 | 中国科学院上海药物研究所 | 一类噁二唑类化合物及其制备方法、药物组合物和用途 |
| CN111689952A (zh) * | 2019-03-14 | 2020-09-22 | 复旦大学 | 含硫代四元环结构的2,3-双加氧酶抑制剂及其制备方法和用途 |
| CN115869305A (zh) * | 2021-09-30 | 2023-03-31 | 山东新时代药业有限公司 | 一种治疗老年痴呆的药物组合物 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999029310A2 (en) | 1997-12-05 | 1999-06-17 | Medical College Of Georgia Research Institute, Inc. | Regulation of t cell-mediated immunity by tryptophan and its analogs |
| WO2004094409A1 (en) | 2003-03-27 | 2004-11-04 | Lankenau Institute For Medical Research | Novel ido inhibitors and methods of use |
| US20040234623A1 (en) | 2003-04-01 | 2004-11-25 | Medical College Of Georgia Research Institute, Inc. | Use of inhibitors of indoleamine-2,3-dioxygenase in combination with other therapeutic modalities |
| CN101212967A (zh) * | 2005-05-10 | 2008-07-02 | 因塞特公司 | 吲哚胺2,3-双加氧酶调节剂及其用法 |
| CN105481789A (zh) * | 2014-09-15 | 2016-04-13 | 中国科学院上海有机化学研究所 | 一种吲哚胺-2,3-双加氧酶抑制剂及其制备方法 |
| CN105646389A (zh) * | 2016-01-28 | 2016-06-08 | 中国科学院上海有机化学研究所 | 一种作为吲哚胺-2,3-双加氧酶抑制剂的氨基磺酸脂及其制备方法和用途 |
| CN106883194A (zh) * | 2015-12-16 | 2017-06-23 | 江苏恒瑞医药股份有限公司 | 噁二唑类衍生物、其制备方法及其在医药上的应用 |
| CN107304191A (zh) * | 2016-04-20 | 2017-10-31 | 上海翰森生物医药科技有限公司 | 吲哚胺2,3‑双加氧酶抑制剂及其制备方法与应用 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20080182882A1 (en) * | 2006-11-08 | 2008-07-31 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| US10988487B2 (en) * | 2016-08-29 | 2021-04-27 | Merck Sharp & Dohme Corp. | Substituted n′-hydroxycarbamimidoyl-1,2,5-oxadiazole compounds as indoleamine 2,3-dioxygenase (IDO) inhibitors |
| CN106967005B (zh) | 2017-04-07 | 2019-07-16 | 上海肇钰医药科技有限公司 | 一种能抑制ido的化合物、其制备方法及其用途 |
-
2018
- 2018-04-24 US US16/607,827 patent/US11046679B2/en active Active
- 2018-04-24 WO PCT/CN2018/084267 patent/WO2018196747A1/zh active Search and Examination
- 2018-04-24 JP JP2019557849A patent/JP6860237B2/ja active Active
- 2018-04-24 EP EP18792247.1A patent/EP3617204A1/en not_active Withdrawn
- 2018-04-24 CN CN201810372792.3A patent/CN108727361A/zh active Pending
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999029310A2 (en) | 1997-12-05 | 1999-06-17 | Medical College Of Georgia Research Institute, Inc. | Regulation of t cell-mediated immunity by tryptophan and its analogs |
| WO2004094409A1 (en) | 2003-03-27 | 2004-11-04 | Lankenau Institute For Medical Research | Novel ido inhibitors and methods of use |
| US20040234623A1 (en) | 2003-04-01 | 2004-11-25 | Medical College Of Georgia Research Institute, Inc. | Use of inhibitors of indoleamine-2,3-dioxygenase in combination with other therapeutic modalities |
| CN101212967A (zh) * | 2005-05-10 | 2008-07-02 | 因塞特公司 | 吲哚胺2,3-双加氧酶调节剂及其用法 |
| CN105481789A (zh) * | 2014-09-15 | 2016-04-13 | 中国科学院上海有机化学研究所 | 一种吲哚胺-2,3-双加氧酶抑制剂及其制备方法 |
| CN106883194A (zh) * | 2015-12-16 | 2017-06-23 | 江苏恒瑞医药股份有限公司 | 噁二唑类衍生物、其制备方法及其在医药上的应用 |
| CN105646389A (zh) * | 2016-01-28 | 2016-06-08 | 中国科学院上海有机化学研究所 | 一种作为吲哚胺-2,3-双加氧酶抑制剂的氨基磺酸脂及其制备方法和用途 |
| CN107304191A (zh) * | 2016-04-20 | 2017-10-31 | 上海翰森生物医药科技有限公司 | 吲哚胺2,3‑双加氧酶抑制剂及其制备方法与应用 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3617204A4 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20210198247A1 (en) * | 2017-12-20 | 2021-07-01 | Hinova Pharmaceuticals Inc. | Indoleamine-2,3-dioxygenase inhibitor, the preparative method and the use thereof |
| US11447477B2 (en) * | 2017-12-20 | 2022-09-20 | Hinova Pharmaceuticals Inc. | Indoleamine-2,3-dioxygenase inhibitor, the preparative method and the use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6860237B2 (ja) | 2021-04-21 |
| US20200181131A1 (en) | 2020-06-11 |
| JP2020517690A (ja) | 2020-06-18 |
| EP3617204A4 (en) | 2020-03-04 |
| EP3617204A1 (en) | 2020-03-04 |
| CN108727361A (zh) | 2018-11-02 |
| US11046679B2 (en) | 2021-06-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2019267824B2 (en) | Tetrahydro-imidazo(4,5-C)pyridine derivatives as PD-L1 immunomodulators | |
| JP7559059B2 (ja) | 免疫調節剤としてのピリド[3,2-d]ピリミジン化合物 | |
| WO2022184178A1 (en) | Kras g12d inhibitors | |
| CN103547579B (zh) | 用作ido抑制剂的稠合咪唑衍生物 | |
| WO2018196747A1 (zh) | 吲哚胺2,3-双加氧酶抑制剂与应用 | |
| CA2883894C (en) | Compounds and methods for kinase modulation, and indications therefor | |
| RU2689777C1 (ru) | Конденсированные трициклические производные бензимидазолов в качестве модуляторов активности tnf | |
| CA3095758A1 (en) | Heterocyclic compounds as immunomodulators | |
| RU2707073C2 (ru) | Трициклическое спиро-соединение | |
| KR20220042204A (ko) | Rip1 억제 화합물 및 그를 제조 및 사용하는 방법 | |
| US20150328228A1 (en) | Novel 4-(indol-3-yl)-pyrazole derivatives, pharmaceutical compositions and methods for use | |
| TW201311685A (zh) | 咪唑並吡啶化合物 | |
| JPWO2020045334A1 (ja) | 光学活性なアザビシクロ環誘導体 | |
| JP6972384B2 (ja) | 複素環化合物 | |
| JP2024522494A (ja) | トリアジン誘導体およびがんの処置におけるその使用 | |
| TW202134229A (zh) | 環烷基尿素衍生物 | |
| CA2863517A1 (en) | Substituted pyrimidine compounds and their use as syk inhibitors | |
| WO2018028664A1 (zh) | Fgfr4抑制剂及其制备方法和应用 | |
| WO2019158070A1 (zh) | A2a和/或a2b受体拮抗剂 | |
| KR20190120786A (ko) | 디히드로오로테이트 옥시게나제 억제제로서의 1,4,6-삼치환된-2-알킬-1H-벤조[d]이미다졸 유도체 | |
| US11939328B2 (en) | Quinoline compounds as inhibitors of KRAS | |
| WO2022089389A1 (zh) | 杂环化合物及其制备方法、药物组合物和应用 | |
| JP2018087189A (ja) | 医薬用途 | |
| JP2021134218A (ja) | 光学活性なアザビシクロ環誘導体からなる医薬 | |
| WO2021088845A1 (zh) | 咪唑烷酮类化合物及其制备方法与应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18792247 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2019557849 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2018792247 Country of ref document: EP Effective date: 20191125 |