WO2018117152A1 - 7H-ピロロ[2,3-d]ピリミジン誘導体の製造方法及びその合成中間体 - Google Patents
7H-ピロロ[2,3-d]ピリミジン誘導体の製造方法及びその合成中間体 Download PDFInfo
- Publication number
- WO2018117152A1 WO2018117152A1 PCT/JP2017/045729 JP2017045729W WO2018117152A1 WO 2018117152 A1 WO2018117152 A1 WO 2018117152A1 JP 2017045729 W JP2017045729 W JP 2017045729W WO 2018117152 A1 WO2018117152 A1 WO 2018117152A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- salt
- acid
- added
- Prior art date
Links
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 144
- JJTNLWSCFYERCK-UHFFFAOYSA-N 7h-pyrrolo[2,3-d]pyrimidine Chemical class N1=CN=C2NC=CC2=C1 JJTNLWSCFYERCK-UHFFFAOYSA-N 0.000 title abstract description 4
- 150000003839 salts Chemical class 0.000 claims abstract description 207
- 150000007524 organic acids Chemical class 0.000 claims abstract description 77
- 150000001875 compounds Chemical class 0.000 claims description 655
- 238000000034 method Methods 0.000 claims description 232
- 239000013078 crystal Substances 0.000 claims description 69
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 claims description 60
- 230000008569 process Effects 0.000 claims description 40
- 239000002253 acid Substances 0.000 claims description 37
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 30
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 29
- NAWXUBYGYWOOIX-SFHVURJKSA-N (2s)-2-[[4-[2-(2,4-diaminoquinazolin-6-yl)ethyl]benzoyl]amino]-4-methylidenepentanedioic acid Chemical compound C1=CC2=NC(N)=NC(N)=C2C=C1CCC1=CC=C(C(=O)N[C@@H](CC(=C)C(O)=O)C(O)=O)C=C1 NAWXUBYGYWOOIX-SFHVURJKSA-N 0.000 claims description 28
- 230000005855 radiation Effects 0.000 claims description 23
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 claims description 22
- -1 lithium aluminum hydride Chemical compound 0.000 claims description 22
- 125000000217 alkyl group Chemical group 0.000 claims description 20
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical group [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 claims description 17
- 239000012320 chlorinating reagent Substances 0.000 claims description 15
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 15
- 239000012280 lithium aluminium hydride Substances 0.000 claims description 13
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 claims description 11
- 239000011734 sodium Substances 0.000 claims description 11
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 10
- FYRHIOVKTDQVFC-UHFFFAOYSA-M potassium phthalimide Chemical compound [K+].C1=CC=C2C(=O)[N-]C(=O)C2=C1 FYRHIOVKTDQVFC-UHFFFAOYSA-M 0.000 claims description 9
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 claims description 8
- 229910052708 sodium Inorganic materials 0.000 claims description 8
- 125000000612 phthaloyl group Chemical group C(C=1C(C(=O)*)=CC=CC1)(=O)* 0.000 claims description 6
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 claims description 5
- 230000009467 reduction Effects 0.000 claims description 4
- IRAIXRXGRCCERF-JSGCOSHPSA-N (3S,4R)-1-benzyl-3-methyl-1,7-diazaspiro[3.4]octane Chemical compound C(C1=CC=CC=C1)N1C[C@@H]([C@]11CNCC1)C IRAIXRXGRCCERF-JSGCOSHPSA-N 0.000 abstract description 9
- 108010024121 Janus Kinases Proteins 0.000 abstract description 5
- 102000015617 Janus Kinases Human genes 0.000 abstract description 5
- LOWWYYZBZNSPDT-ZBEGNZNMSA-N delgocitinib Chemical compound C[C@H]1CN(C(=O)CC#N)[C@@]11CN(C=2C=3C=CNC=3N=CN=2)CC1 LOWWYYZBZNSPDT-ZBEGNZNMSA-N 0.000 abstract description 5
- 239000003795 chemical substances by application Substances 0.000 abstract description 4
- 230000002401 inhibitory effect Effects 0.000 abstract 1
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 440
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 190
- 239000000243 solution Substances 0.000 description 171
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 168
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 142
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 141
- 239000000203 mixture Substances 0.000 description 134
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 116
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 114
- 239000011541 reaction mixture Substances 0.000 description 107
- 239000002904 solvent Substances 0.000 description 99
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 79
- 239000002585 base Substances 0.000 description 69
- 229940126062 Compound A Drugs 0.000 description 65
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 65
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 65
- 239000007787 solid Substances 0.000 description 65
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 61
- 238000005481 NMR spectroscopy Methods 0.000 description 61
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 59
- 239000012044 organic layer Substances 0.000 description 58
- 238000006243 chemical reaction Methods 0.000 description 57
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 44
- 238000004128 high performance liquid chromatography Methods 0.000 description 43
- 239000012299 nitrogen atmosphere Substances 0.000 description 43
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 42
- 238000005160 1H NMR spectroscopy Methods 0.000 description 39
- 238000001914 filtration Methods 0.000 description 37
- 239000000047 product Substances 0.000 description 37
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 37
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 36
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 36
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 35
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 34
- 239000012046 mixed solvent Substances 0.000 description 34
- 238000003756 stirring Methods 0.000 description 33
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropyl acetate Chemical compound CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 32
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 30
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 30
- 230000035484 reaction time Effects 0.000 description 30
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 29
- 229910052739 hydrogen Inorganic materials 0.000 description 29
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 29
- 239000012453 solvate Substances 0.000 description 27
- 239000000706 filtrate Substances 0.000 description 26
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 26
- 238000005406 washing Methods 0.000 description 26
- 239000002002 slurry Substances 0.000 description 25
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 24
- 229910052757 nitrogen Inorganic materials 0.000 description 24
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 23
- 230000015572 biosynthetic process Effects 0.000 description 22
- 239000012267 brine Substances 0.000 description 22
- 238000000746 purification Methods 0.000 description 22
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 21
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 21
- 238000002425 crystallisation Methods 0.000 description 21
- 230000008025 crystallization Effects 0.000 description 21
- BPTCCCTWWAUJRK-UHFFFAOYSA-N 4-chloro-7h-pyrrolo[2,3-d]pyrimidine Chemical compound ClC1=NC=NC2=C1C=CN2 BPTCCCTWWAUJRK-UHFFFAOYSA-N 0.000 description 20
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 20
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 19
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 19
- MSXVEPNJUHWQHW-UHFFFAOYSA-N 2-methylbutan-2-ol Chemical compound CCC(C)(C)O MSXVEPNJUHWQHW-UHFFFAOYSA-N 0.000 description 18
- FFBHFFJDDLITSX-UHFFFAOYSA-N benzyl N-[2-hydroxy-4-(3-oxomorpholin-4-yl)phenyl]carbamate Chemical compound OC1=C(NC(=O)OCC2=CC=CC=C2)C=CC(=C1)N1CCOCC1=O FFBHFFJDDLITSX-UHFFFAOYSA-N 0.000 description 18
- 238000000921 elemental analysis Methods 0.000 description 18
- 150000007522 mineralic acids Chemical class 0.000 description 18
- SDXAWLJRERMRKF-UHFFFAOYSA-N 3,5-dimethyl-1h-pyrazole Chemical compound CC=1C=C(C)NN=1 SDXAWLJRERMRKF-UHFFFAOYSA-N 0.000 description 17
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 17
- 235000017557 sodium bicarbonate Nutrition 0.000 description 17
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 16
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 16
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 16
- 239000010410 layer Substances 0.000 description 16
- 238000005259 measurement Methods 0.000 description 16
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 16
- 235000019798 tripotassium phosphate Nutrition 0.000 description 16
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 15
- 239000003054 catalyst Substances 0.000 description 15
- 235000006408 oxalic acid Nutrition 0.000 description 15
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 14
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 14
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 14
- 239000003638 chemical reducing agent Substances 0.000 description 13
- 238000001816 cooling Methods 0.000 description 13
- 229940093915 gynecological organic acid Drugs 0.000 description 13
- SKTCDJAMAYNROS-UHFFFAOYSA-N methoxycyclopentane Chemical compound COC1CCCC1 SKTCDJAMAYNROS-UHFFFAOYSA-N 0.000 description 13
- 235000005985 organic acids Nutrition 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 12
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 12
- 229960000583 acetic acid Drugs 0.000 description 12
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 12
- 229910000024 caesium carbonate Inorganic materials 0.000 description 12
- 230000014759 maintenance of location Effects 0.000 description 12
- 0 *C1(CC1)C(N)=O Chemical compound *C1(CC1)C(N)=O 0.000 description 11
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 11
- 102100028999 High mobility group protein HMGI-C Human genes 0.000 description 11
- 101000986379 Homo sapiens High mobility group protein HMGI-C Proteins 0.000 description 11
- 235000011054 acetic acid Nutrition 0.000 description 11
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 11
- 229910052794 bromium Inorganic materials 0.000 description 11
- 239000000460 chlorine Substances 0.000 description 11
- 230000032050 esterification Effects 0.000 description 11
- 238000005886 esterification reaction Methods 0.000 description 11
- 239000001257 hydrogen Substances 0.000 description 11
- 239000000543 intermediate Substances 0.000 description 11
- 239000011259 mixed solution Substances 0.000 description 11
- 239000000725 suspension Substances 0.000 description 11
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 10
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 10
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 10
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 10
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 10
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 10
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 10
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 10
- KKEYFWRCBNTPAC-UHFFFAOYSA-N Terephthalic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-N 0.000 description 10
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 10
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 10
- 238000004458 analytical method Methods 0.000 description 10
- 229910052801 chlorine Inorganic materials 0.000 description 10
- 239000012043 crude product Substances 0.000 description 10
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 10
- 238000002844 melting Methods 0.000 description 10
- 230000008018 melting Effects 0.000 description 10
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 10
- 238000001953 recrystallisation Methods 0.000 description 10
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 10
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 10
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 9
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 9
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 9
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 9
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 9
- 238000000113 differential scanning calorimetry Methods 0.000 description 9
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 8
- 239000007864 aqueous solution Substances 0.000 description 8
- 239000012065 filter cake Substances 0.000 description 8
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 8
- 235000019341 magnesium sulphate Nutrition 0.000 description 8
- 239000012074 organic phase Substances 0.000 description 8
- 239000012071 phase Substances 0.000 description 8
- 229920006395 saturated elastomer Polymers 0.000 description 8
- 238000001228 spectrum Methods 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- VVNYDCGZZSTUBC-LURJTMIESA-N (2s)-5-amino-2-[(2-methylpropan-2-yl)oxycarbonylamino]-5-oxopentanoic acid Chemical compound CC(C)(C)OC(=O)N[C@H](C(O)=O)CCC(N)=O VVNYDCGZZSTUBC-LURJTMIESA-N 0.000 description 7
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 7
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 7
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 7
- 238000007792 addition Methods 0.000 description 7
- 229910000027 potassium carbonate Inorganic materials 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- DDWZYWSLHBDVGR-UHFFFAOYSA-N 3-(3,5-dimethylpyrazol-1-yl)-3-oxopropanenitrile Chemical compound CC=1C=C(C)N(C(=O)CC#N)N=1 DDWZYWSLHBDVGR-UHFFFAOYSA-N 0.000 description 6
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 6
- 238000002835 absorbance Methods 0.000 description 6
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 6
- 239000008346 aqueous phase Substances 0.000 description 6
- 238000001938 differential scanning calorimetry curve Methods 0.000 description 6
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 6
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 6
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 6
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 6
- 239000000523 sample Substances 0.000 description 6
- GAWAYYRQGQZKCR-UWTATZPHSA-N (2r)-2-chloropropanoic acid Chemical compound C[C@@H](Cl)C(O)=O GAWAYYRQGQZKCR-UWTATZPHSA-N 0.000 description 5
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 5
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 5
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 5
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 5
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 5
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 5
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 5
- GJWAPAVRQYYSTK-UHFFFAOYSA-N [(dimethyl-$l^{3}-silanyl)amino]-dimethylsilicon Chemical compound C[Si](C)N[Si](C)C GJWAPAVRQYYSTK-UHFFFAOYSA-N 0.000 description 5
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 5
- 235000010323 ascorbic acid Nutrition 0.000 description 5
- 239000011668 ascorbic acid Substances 0.000 description 5
- 229960005070 ascorbic acid Drugs 0.000 description 5
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 5
- 229940092714 benzenesulfonic acid Drugs 0.000 description 5
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 5
- 238000004296 chiral HPLC Methods 0.000 description 5
- 239000001530 fumaric acid Substances 0.000 description 5
- 239000000174 gluconic acid Substances 0.000 description 5
- 235000012208 gluconic acid Nutrition 0.000 description 5
- 229910052736 halogen Inorganic materials 0.000 description 5
- 150000002367 halogens Chemical class 0.000 description 5
- 239000002198 insoluble material Substances 0.000 description 5
- 238000002955 isolation Methods 0.000 description 5
- 239000004310 lactic acid Substances 0.000 description 5
- 235000014655 lactic acid Nutrition 0.000 description 5
- 229960000448 lactic acid Drugs 0.000 description 5
- 229910052744 lithium Inorganic materials 0.000 description 5
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 5
- 239000011976 maleic acid Substances 0.000 description 5
- 239000001630 malic acid Substances 0.000 description 5
- 235000011090 malic acid Nutrition 0.000 description 5
- 229940099690 malic acid Drugs 0.000 description 5
- 229940098779 methanesulfonic acid Drugs 0.000 description 5
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 5
- 229960005235 piperonyl butoxide Drugs 0.000 description 5
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 5
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 5
- 229910000160 potassium phosphate Inorganic materials 0.000 description 5
- 235000011009 potassium phosphates Nutrition 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- 239000012279 sodium borohydride Substances 0.000 description 5
- 229910000033 sodium borohydride Inorganic materials 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 5
- 239000001384 succinic acid Substances 0.000 description 5
- 239000011975 tartaric acid Substances 0.000 description 5
- 229960001367 tartaric acid Drugs 0.000 description 5
- 235000002906 tartaric acid Nutrition 0.000 description 5
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 4
- GYAWQIHUDRAWSZ-UHFFFAOYSA-N 3-(benzylamino)oxolan-2-one Chemical compound O=C1OCCC1NCC1=CC=CC=C1 GYAWQIHUDRAWSZ-UHFFFAOYSA-N 0.000 description 4
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- OVXYQTBCJONCEU-AVRDEDQJSA-N C[C@@H](C1)[C@]2(CN(Cc3ccccc3)CC2)N1C(OCc1ccccc1)=O Chemical compound C[C@@H](C1)[C@]2(CN(Cc3ccccc3)CC2)N1C(OCc1ccccc1)=O OVXYQTBCJONCEU-AVRDEDQJSA-N 0.000 description 4
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 4
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- 230000002378 acidificating effect Effects 0.000 description 4
- 239000000654 additive Substances 0.000 description 4
- 230000000996 additive effect Effects 0.000 description 4
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 4
- 229960001270 d- tartaric acid Drugs 0.000 description 4
- 238000004090 dissolution Methods 0.000 description 4
- VMDTXBZDEOAFQF-UHFFFAOYSA-N formaldehyde;ruthenium Chemical compound [Ru].O=C VMDTXBZDEOAFQF-UHFFFAOYSA-N 0.000 description 4
- 229950006191 gluconic acid Drugs 0.000 description 4
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 4
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 4
- FEWJPZIEWOKRBE-LWMBPPNESA-N levotartaric acid Chemical compound OC(=O)[C@@H](O)[C@H](O)C(O)=O FEWJPZIEWOKRBE-LWMBPPNESA-N 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 229910017604 nitric acid Inorganic materials 0.000 description 4
- 230000003287 optical effect Effects 0.000 description 4
- 239000007800 oxidant agent Substances 0.000 description 4
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical compound [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 4
- 239000002243 precursor Substances 0.000 description 4
- 239000008213 purified water Substances 0.000 description 4
- 238000000926 separation method Methods 0.000 description 4
- 235000011006 sodium potassium tartrate Nutrition 0.000 description 4
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 3
- AKONGNHJBXKFDP-YWZLYKJASA-N 4-[(3S,4R)-1-benzyl-3-methyl-1,7-diazaspiro[3.4]octan-7-yl]-7H-pyrrolo[2,3-d]pyrimidine Chemical compound C12=C(C(=NC=N2)N2CC[C@@]3([C@H](CN3CC3=CC=CC=C3)C)C2)C=CN1 AKONGNHJBXKFDP-YWZLYKJASA-N 0.000 description 3
- KDVYCTOWXSLNNI-UHFFFAOYSA-N 4-t-Butylbenzoic acid Chemical compound CC(C)(C)C1=CC=C(C(O)=O)C=C1 KDVYCTOWXSLNNI-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- RPNUMPOLZDHAAY-UHFFFAOYSA-N Diethylenetriamine Chemical compound NCCNCCN RPNUMPOLZDHAAY-UHFFFAOYSA-N 0.000 description 3
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 3
- QZUPHAGRBBOLTB-UHFFFAOYSA-N NSC 244302 Chemical compound C=1C=CC=CC=1P(C(C)(C)C)C1=CC=CC=C1 QZUPHAGRBBOLTB-UHFFFAOYSA-N 0.000 description 3
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 3
- 239000005708 Sodium hypochlorite Substances 0.000 description 3
- 238000013019 agitation Methods 0.000 description 3
- 229910052782 aluminium Inorganic materials 0.000 description 3
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 3
- 235000001014 amino acid Nutrition 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- 238000001514 detection method Methods 0.000 description 3
- 229910001873 dinitrogen Inorganic materials 0.000 description 3
- 150000002431 hydrogen Chemical class 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 150000007529 inorganic bases Chemical class 0.000 description 3
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 3
- 150000007530 organic bases Chemical class 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 description 3
- 229940074439 potassium sodium tartrate Drugs 0.000 description 3
- 238000001556 precipitation Methods 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- 238000010898 silica gel chromatography Methods 0.000 description 3
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 3
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- UQOMPPZUXPLVMD-RWANSRKNSA-N (2R)-N-benzyl-2-chloro-N-(2-oxooxolan-3-yl)propanamide Chemical compound C[C@@H](Cl)C(=O)N(Cc1ccccc1)C1CCOC1=O UQOMPPZUXPLVMD-RWANSRKNSA-N 0.000 description 2
- PJXWCRXOPLGFLX-VIFPVBQESA-N (2s)-2-(benzylamino)propan-1-ol Chemical compound OC[C@H](C)NCC1=CC=CC=C1 PJXWCRXOPLGFLX-VIFPVBQESA-N 0.000 description 2
- XNBXEJRSGBEPIM-IINYFYTJSA-N (3R,4R)-1-benzyl-3-methyl-1,7-diazaspiro[3.4]octane-2,8-dione Chemical compound C[C@H]1C(=O)N(Cc2ccccc2)[C@]11CCNC1=O XNBXEJRSGBEPIM-IINYFYTJSA-N 0.000 description 2
- FMOWBIUSRIMEHW-LEUCUCNGSA-N (3S,4R)-3-methyl-1,7-diazaspiro[3.4]octane oxalic acid Chemical compound C(C(=O)O)(=O)O.C[C@H]1CN[C@@]12CNCC2 FMOWBIUSRIMEHW-LEUCUCNGSA-N 0.000 description 2
- MIOPJNTWMNEORI-XVKPBYJWSA-N (R)-camphorsulfonic acid Chemical compound C1C[C@]2(CS(O)(=O)=O)C(=O)C[C@H]1C2(C)C MIOPJNTWMNEORI-XVKPBYJWSA-N 0.000 description 2
- QWUWMCYKGHVNAV-UHFFFAOYSA-N 1,2-dihydrostilbene Chemical group C=1C=CC=CC=1CCC1=CC=CC=C1 QWUWMCYKGHVNAV-UHFFFAOYSA-N 0.000 description 2
- MEKOFIRRDATTAG-UHFFFAOYSA-N 2,2,5,8-tetramethyl-3,4-dihydrochromen-6-ol Chemical compound C1CC(C)(C)OC2=C1C(C)=C(O)C=C2C MEKOFIRRDATTAG-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- JVKRKMWZYMKVTQ-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]pyrazol-1-yl]-N-(2-oxo-3H-1,3-benzoxazol-6-yl)acetamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C=1C=NN(C=1)CC(=O)NC1=CC2=C(NC(O2)=O)C=C1 JVKRKMWZYMKVTQ-UHFFFAOYSA-N 0.000 description 2
- WADSJYLPJPTMLN-UHFFFAOYSA-N 3-(cycloundecen-1-yl)-1,2-diazacycloundec-2-ene Chemical compound C1CCCCCCCCC=C1C1=NNCCCCCCCC1 WADSJYLPJPTMLN-UHFFFAOYSA-N 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- YOZQNFQINJKCQC-ZANVPECISA-N 4-[(3S,4R)-3-methyl-1,7-diazaspiro[3.4]octan-7-yl]-7H-pyrrolo[2,3-d]pyrimidine Chemical compound C[C@H]1CN[C@@]11CN(CC1)C=1C2=C(N=CN=1)NC=C2 YOZQNFQINJKCQC-ZANVPECISA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 2
- UTGJDFJVSAEXKQ-HNNXBMFYSA-N CC(C)C(N(Cc1ccccc1)[C@@H](CCNC(OC)=O)C(OC)=O)=O Chemical compound CC(C)C(N(Cc1ccccc1)[C@@H](CCNC(OC)=O)C(OC)=O)=O UTGJDFJVSAEXKQ-HNNXBMFYSA-N 0.000 description 2
- YZSLNFMZNFBJJG-WCCKRBBISA-N CCl.N[C@H](C(=O)O)CCNC(=O)OC Chemical compound CCl.N[C@H](C(=O)O)CCNC(=O)OC YZSLNFMZNFBJJG-WCCKRBBISA-N 0.000 description 2
- MVIQHRGZWMHDGP-BQBZGAKWSA-N C[C@@H]1[C@]2(CNCC2)NC1 Chemical compound C[C@@H]1[C@]2(CNCC2)NC1 MVIQHRGZWMHDGP-BQBZGAKWSA-N 0.000 description 2
- JOVBPMMTLKGUFN-RWANSRKNSA-N C[C@H](C(N(Cc1ccccc1)SC(CCO1)C1=O)=O)Cl Chemical compound C[C@H](C(N(Cc1ccccc1)SC(CCO1)C1=O)=O)Cl JOVBPMMTLKGUFN-RWANSRKNSA-N 0.000 description 2
- GSNUFIFRDBKVIE-UHFFFAOYSA-N DMF Natural products CC1=CC=C(C)O1 GSNUFIFRDBKVIE-UHFFFAOYSA-N 0.000 description 2
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 2
- XLYOFNOQVPJJNP-ZSJDYOACSA-N Heavy water Chemical compound [2H]O[2H] XLYOFNOQVPJJNP-ZSJDYOACSA-N 0.000 description 2
- 229940122245 Janus kinase inhibitor Drugs 0.000 description 2
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 238000002441 X-ray diffraction Methods 0.000 description 2
- ZVQOOHYFBIDMTQ-UHFFFAOYSA-N [methyl(oxido){1-[6-(trifluoromethyl)pyridin-3-yl]ethyl}-lambda(6)-sulfanylidene]cyanamide Chemical compound N#CN=S(C)(=O)C(C)C1=CC=C(C(F)(F)F)N=C1 ZVQOOHYFBIDMTQ-UHFFFAOYSA-N 0.000 description 2
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 2
- 239000012346 acetyl chloride Substances 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 239000008186 active pharmaceutical agent Substances 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 239000012300 argon atmosphere Substances 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 2
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 2
- SXDBWCPKPHAZSM-UHFFFAOYSA-N bromic acid Chemical compound OBr(=O)=O SXDBWCPKPHAZSM-UHFFFAOYSA-N 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 238000005356 chiral GC Methods 0.000 description 2
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- HHFAWKCIHAUFRX-UHFFFAOYSA-N ethoxide Chemical compound CC[O-] HHFAWKCIHAUFRX-UHFFFAOYSA-N 0.000 description 2
- 238000013213 extrapolation Methods 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- 238000004255 ion exchange chromatography Methods 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 2
- ZRSNZINYAWTAHE-UHFFFAOYSA-N p-methoxybenzaldehyde Chemical compound COC1=CC=C(C=O)C=C1 ZRSNZINYAWTAHE-UHFFFAOYSA-N 0.000 description 2
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 2
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- XHFLOLLMZOTPSM-UHFFFAOYSA-M sodium;hydrogen carbonate;hydrate Chemical compound [OH-].[Na+].OC(O)=O XHFLOLLMZOTPSM-UHFFFAOYSA-M 0.000 description 2
- QJXDSDLNUKLDBP-UHFFFAOYSA-M sodium;n-formylmethanimidate Chemical compound [Na+].O=C[N-]C=O QJXDSDLNUKLDBP-UHFFFAOYSA-M 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 230000000707 stereoselective effect Effects 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 2
- AZNQJNJZVBFRAA-RCDICMHDSA-N (2R,3R)-1-benzyl-2-[2-(1,3-dioxoisoindol-2-yl)ethyl]-3-methyl-4-oxoazetidine-2-carboxylic acid Chemical compound C[C@H]1C(=O)N(Cc2ccccc2)[C@@]1(CCN1C(=O)c2ccccc2C1=O)C(O)=O AZNQJNJZVBFRAA-RCDICMHDSA-N 0.000 description 1
- FLXSHEDAVSGQKN-ZUZCIYMTSA-N (2R,3S)-2-(carboxymethyl)-3-methyl-1-phenylmethoxycarbonylazetidine-2-carboxylic acid Chemical compound C(C1=CC=CC=C1)OC(=O)N1[C@]([C@H](C1)C)(C(=O)O)CC(=O)O FLXSHEDAVSGQKN-ZUZCIYMTSA-N 0.000 description 1
- JEQDSBVHLKBEIZ-UWTATZPHSA-N (2r)-2-chloropropanoyl chloride Chemical compound C[C@@H](Cl)C(Cl)=O JEQDSBVHLKBEIZ-UWTATZPHSA-N 0.000 description 1
- BJFPYGGTDAYECS-UHFFFAOYSA-N (3-chlorophenyl)methanamine Chemical compound NCC1=CC=CC(Cl)=C1 BJFPYGGTDAYECS-UHFFFAOYSA-N 0.000 description 1
- GRRIMVWABNHKBX-UHFFFAOYSA-N (3-methoxyphenyl)methanamine Chemical compound COC1=CC=CC(CN)=C1 GRRIMVWABNHKBX-UHFFFAOYSA-N 0.000 description 1
- RGXUCUWVGKLACF-UHFFFAOYSA-N (3-methylphenyl)methanamine Chemical compound CC1=CC=CC(CN)=C1 RGXUCUWVGKLACF-UHFFFAOYSA-N 0.000 description 1
- BJSMFMJXITVAJZ-IINYFYTJSA-N (3R,4R)-1-benzyl-3-methyl-7-oxa-1-azaspiro[3.4]octane-2,8-dione Chemical compound C[C@H]1C(=O)N(Cc2ccccc2)[C@]11CCOC1=O BJSMFMJXITVAJZ-IINYFYTJSA-N 0.000 description 1
- FTMDJYXSWXRYTJ-UOQJWNSWSA-N (3S)-2-tert-butyl-3-methylazetidine-1,2-dicarboxylic acid Chemical compound C(C)(C)(C)C1(N(C[C@@H]1C)C(=O)O)C(=O)O FTMDJYXSWXRYTJ-UOQJWNSWSA-N 0.000 description 1
- XZMIEUIZQGCIRX-KYSPHBLOSA-N (3S,4R)-1-benzyl-3-methyl-1,7-diazaspiro[3.4]octane oxalic acid Chemical compound C(C(=O)O)(=O)O.C(C1=CC=CC=C1)N1C[C@@H]([C@]12CNCC2)C XZMIEUIZQGCIRX-KYSPHBLOSA-N 0.000 description 1
- YMVFJGSXZNNUDW-UHFFFAOYSA-N (4-chlorophenyl)methanamine Chemical compound NCC1=CC=C(Cl)C=C1 YMVFJGSXZNNUDW-UHFFFAOYSA-N 0.000 description 1
- HMTSWYPNXFHGEP-UHFFFAOYSA-N (4-methylphenyl)methanamine Chemical compound CC1=CC=C(CN)C=C1 HMTSWYPNXFHGEP-UHFFFAOYSA-N 0.000 description 1
- KWEKXPWNFQBJAY-UHFFFAOYSA-N (dimethyl-$l^{3}-silanyl)oxy-dimethylsilicon Chemical compound C[Si](C)O[Si](C)C KWEKXPWNFQBJAY-UHFFFAOYSA-N 0.000 description 1
- UKAUYVFTDYCKQA-UHFFFAOYSA-N -2-Amino-4-hydroxybutanoic acid Natural products OC(=O)C(N)CCO UKAUYVFTDYCKQA-UHFFFAOYSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- IDPURXSQCKYKIJ-UHFFFAOYSA-N 1-(4-methoxyphenyl)methanamine Chemical compound COC1=CC=C(CN)C=C1 IDPURXSQCKYKIJ-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 1
- KKZUMAMOMRDVKA-UHFFFAOYSA-N 2-chloropropane Chemical group [CH2]C(C)Cl KKZUMAMOMRDVKA-UHFFFAOYSA-N 0.000 description 1
- BSKHPKMHTQYZBB-UHFFFAOYSA-N 2-methylpyridine Chemical compound CC1=CC=CC=N1 BSKHPKMHTQYZBB-UHFFFAOYSA-N 0.000 description 1
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 1
- CCZZNDRXPAYZOX-UHFFFAOYSA-N 3-(benzylamino)oxolan-2-one;hydrochloride Chemical compound Cl.O=C1OCCC1NCC1=CC=CC=C1 CCZZNDRXPAYZOX-UHFFFAOYSA-N 0.000 description 1
- LFJJGHGXHXXDFT-UHFFFAOYSA-N 3-bromooxolan-2-one Chemical compound BrC1CCOC1=O LFJJGHGXHXXDFT-UHFFFAOYSA-N 0.000 description 1
- OARNHESMASZJCO-UHFFFAOYSA-N 3-chlorooxolan-2-one Chemical compound ClC1CCOC1=O OARNHESMASZJCO-UHFFFAOYSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- DHMQDGOQFOQNFH-UHFFFAOYSA-M Aminoacetate Chemical compound NCC([O-])=O DHMQDGOQFOQNFH-UHFFFAOYSA-M 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 1
- ODGBFVQDIGIWCF-UWJYYQICSA-N C#C[C@@H]1[C@](CC2)(CN2c2c(cc[nH]3)c3ncn2)N(Cc2ccccc2)C1 Chemical compound C#C[C@@H]1[C@](CC2)(CN2c2c(cc[nH]3)c3ncn2)N(Cc2ccccc2)C1 ODGBFVQDIGIWCF-UWJYYQICSA-N 0.000 description 1
- RJLBGJDNJNDYJB-NXCSSKFKSA-N C(C(=O)O)(=O)OCC1=CC=CC=C1.C(C1=CC=CC=C1)N1C[C@]2([C@H](CN2C(=O)O)C)CC1 Chemical compound C(C(=O)O)(=O)OCC1=CC=CC=C1.C(C1=CC=CC=C1)N1C[C@]2([C@H](CN2C(=O)O)C)CC1 RJLBGJDNJNDYJB-NXCSSKFKSA-N 0.000 description 1
- KSJSSRRJPZJCGB-QMOSYVFLSA-N C(C)(C)(C)Cl.C(C1=CC=CC=C1)N1C([C@H](C1)C)C(=O)O Chemical compound C(C)(C)(C)Cl.C(C1=CC=CC=C1)N1C([C@H](C1)C)C(=O)O KSJSSRRJPZJCGB-QMOSYVFLSA-N 0.000 description 1
- DFZYUBYIKFANPP-ZRCOPKFRSA-N C(C)(C)(C)Cl.C[C@@H]1C(NC1)C(=O)O Chemical compound C(C)(C)(C)Cl.C[C@@H]1C(NC1)C(=O)O DFZYUBYIKFANPP-ZRCOPKFRSA-N 0.000 description 1
- MEYAYRIHVXTCSQ-FORAGAHYSA-N C(CCC(=O)O)(=O)O.C(CCC(=O)O)(=O)O.C(C1=CC=CC=C1)N1C[C@@H]([C@]12CNCC2)C Chemical compound C(CCC(=O)O)(=O)O.C(CCC(=O)O)(=O)O.C(C1=CC=CC=C1)N1C[C@@H]([C@]12CNCC2)C MEYAYRIHVXTCSQ-FORAGAHYSA-N 0.000 description 1
- XVXQIAXXWIOFAB-ZETCQYMHSA-N CC(C)(C)OC(N[C@@H](CCNC(OC)=O)C(O)=O)=O Chemical compound CC(C)(C)OC(N[C@@H](CCNC(OC)=O)C(O)=O)=O XVXQIAXXWIOFAB-ZETCQYMHSA-N 0.000 description 1
- STICJRYAHUGVBG-MERQFXBCSA-N CCl.C(C1=CC=CC=C1)N[C@H](C(=O)O)CCNC(=O)OC Chemical compound CCl.C(C1=CC=CC=C1)N[C@H](C(=O)O)CCNC(=O)OC STICJRYAHUGVBG-MERQFXBCSA-N 0.000 description 1
- LYPDRGQKMFEPKB-SIKLNZKXSA-N C[C@@H](C1)[C@@H](CCN(C)Cc2ccccc2)N1C(OCc1ccc(C)cc1)=O Chemical compound C[C@@H](C1)[C@@H](CCN(C)Cc2ccccc2)N1C(OCc1ccc(C)cc1)=O LYPDRGQKMFEPKB-SIKLNZKXSA-N 0.000 description 1
- KOGBEKQWUYLWAD-ZDUSSCGKSA-N C[C@@H](CO)N(CC1=[O](C)C(C)(C)O1)Cc1ccccc1 Chemical compound C[C@@H](CO)N(CC1=[O](C)C(C)(C)O1)Cc1ccccc1 KOGBEKQWUYLWAD-ZDUSSCGKSA-N 0.000 description 1
- WJPRKLSYKIIRHB-MAUKXSAKSA-N C[C@@H]1[C@@H](CCN(C)c2c(cc[nH]3)c3ncn2)N(Cc2ccccc2)C1 Chemical compound C[C@@H]1[C@@H](CCN(C)c2c(cc[nH]3)c3ncn2)N(Cc2ccccc2)C1 WJPRKLSYKIIRHB-MAUKXSAKSA-N 0.000 description 1
- BAVFROATQUIVKW-RCYCPFCNSA-N C[C@@H]1[C@](CCN2)(C2=O)N(Cc2ccccc2)C1O Chemical compound C[C@@H]1[C@](CCN2)(C2=O)N(Cc2ccccc2)C1O BAVFROATQUIVKW-RCYCPFCNSA-N 0.000 description 1
- LRRNLTTYGRNLMB-DGCLKSJQSA-N C[C@H]([C@@H](CCOC)N1Cc2ccccc2)C1=O Chemical compound C[C@H]([C@@H](CCOC)N1Cc2ccccc2)C1=O LRRNLTTYGRNLMB-DGCLKSJQSA-N 0.000 description 1
- FPEWGNIVGKRSDC-SWLSCSKDSA-N C[C@H]([C@@](C)(CCOC=O)N1Cc2ccccc2)C1=O Chemical compound C[C@H]([C@@](C)(CCOC=O)N1Cc2ccccc2)C1=O FPEWGNIVGKRSDC-SWLSCSKDSA-N 0.000 description 1
- SCMFTQVLCXQDHI-XHDPSFHLSA-N C[C@H]([C@](CCN1)(C1=O)N1Cc2ccccc2)C1=[O]=C Chemical compound C[C@H]([C@](CCN1)(C1=O)N1Cc2ccccc2)C1=[O]=C SCMFTQVLCXQDHI-XHDPSFHLSA-N 0.000 description 1
- JTHGUOBXHVLODU-NHYWBVRUSA-N C[C@H]([C@](CCN1)(CC1=O)N1Cc2ccccc2)C1=O Chemical compound C[C@H]([C@](CCN1)(CC1=O)N1Cc2ccccc2)C1=O JTHGUOBXHVLODU-NHYWBVRUSA-N 0.000 description 1
- YASYEJJMZJALEJ-UHFFFAOYSA-N Citric acid monohydrate Chemical compound O.OC(=O)CC(O)(C(O)=O)CC(O)=O YASYEJJMZJALEJ-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 101000844245 Homo sapiens Non-receptor tyrosine-protein kinase TYK2 Proteins 0.000 description 1
- 101000997835 Homo sapiens Tyrosine-protein kinase JAK1 Proteins 0.000 description 1
- 101000997832 Homo sapiens Tyrosine-protein kinase JAK2 Proteins 0.000 description 1
- 101000934996 Homo sapiens Tyrosine-protein kinase JAK3 Proteins 0.000 description 1
- 238000003109 Karl Fischer titration Methods 0.000 description 1
- FFEARJCKVFRZRR-UHFFFAOYSA-N L-Methionine Natural products CSCCC(N)C(O)=O FFEARJCKVFRZRR-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- 229930195722 L-methionine Natural products 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 241001024304 Mino Species 0.000 description 1
- QMBDPPLSBOIXKA-UHFFFAOYSA-N N,N-dimethylmethanamine phenylmethanamine Chemical compound C1(=CC=CC=C1)CN.CN(C)C QMBDPPLSBOIXKA-UHFFFAOYSA-N 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- QJPWUUJVYOJNMH-VKHMYHEASA-N N[C@@H](CCO1)C1=O Chemical compound N[C@@H](CCO1)C1=O QJPWUUJVYOJNMH-VKHMYHEASA-N 0.000 description 1
- 102100032028 Non-receptor tyrosine-protein kinase TYK2 Human genes 0.000 description 1
- OFILLAZUEIXLAX-UHFFFAOYSA-L O.O.[Na+].[Na+].C(C1=CC=CC=C1)OC(=O)N1C(C(C1)C)(C(=O)[O-])CC(=O)[O-] Chemical compound O.O.[Na+].[Na+].C(C1=CC=CC=C1)OC(=O)N1C(C(C1)C)(C(=O)[O-])CC(=O)[O-] OFILLAZUEIXLAX-UHFFFAOYSA-L 0.000 description 1
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- 102100033438 Tyrosine-protein kinase JAK1 Human genes 0.000 description 1
- 102100033444 Tyrosine-protein kinase JAK2 Human genes 0.000 description 1
- 102100025387 Tyrosine-protein kinase JAK3 Human genes 0.000 description 1
- ZBIKORITPGTTGI-UHFFFAOYSA-N [acetyloxy(phenyl)-$l^{3}-iodanyl] acetate Chemical compound CC(=O)OI(OC(C)=O)C1=CC=CC=C1 ZBIKORITPGTTGI-UHFFFAOYSA-N 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 1
- 150000008041 alkali metal carbonates Chemical class 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 229910000318 alkali metal phosphate Inorganic materials 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 238000000218 anomalous X-ray scattering Methods 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 235000009697 arginine Nutrition 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 125000003460 beta-lactamyl group Chemical group 0.000 description 1
- 238000012925 biological evaluation Methods 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- SXDBWCPKPHAZSM-UHFFFAOYSA-M bromate Inorganic materials [O-]Br(=O)=O SXDBWCPKPHAZSM-UHFFFAOYSA-M 0.000 description 1
- UZZBCNWFINIKRG-JFYKYWLVSA-N butanedioic acid (3S,4R)-3-methyl-1,7-diazaspiro[3.4]octane Chemical compound C(CCC(=O)O)(=O)O.C(CCC(=O)O)(=O)O.C[C@H]1CN[C@@]12CNCC2 UZZBCNWFINIKRG-JFYKYWLVSA-N 0.000 description 1
- PIJXRNNCIJAUOX-UHFFFAOYSA-N butanoic acid;hydrochloride Chemical compound Cl.CCCC(O)=O PIJXRNNCIJAUOX-UHFFFAOYSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical class [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- YOIWTVRIVUMRLF-UHFFFAOYSA-N chloroform hydrate Chemical compound O.ClC(Cl)Cl.ClC(Cl)Cl YOIWTVRIVUMRLF-UHFFFAOYSA-N 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- KMPWYEUPVWOPIM-UHFFFAOYSA-N cinchonidine Natural products C1=CC=C2C(C(C3N4CCC(C(C4)C=C)C3)O)=CC=NC2=C1 KMPWYEUPVWOPIM-UHFFFAOYSA-N 0.000 description 1
- KMPWYEUPVWOPIM-LSOMNZGLSA-N cinchonine Chemical compound C1=CC=C2C([C@@H]([C@H]3N4CC[C@H]([C@H](C4)C=C)C3)O)=CC=NC2=C1 KMPWYEUPVWOPIM-LSOMNZGLSA-N 0.000 description 1
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 238000005443 coulometric titration Methods 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- IEJIGPNLZYLLBP-UHFFFAOYSA-N dimethyl carbonate Chemical compound COC(=O)OC IEJIGPNLZYLLBP-UHFFFAOYSA-N 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- MGHPNCMVUAKAIE-UHFFFAOYSA-N diphenylmethanamine Chemical compound C=1C=CC=CC=1C(N)C1=CC=CC=C1 MGHPNCMVUAKAIE-UHFFFAOYSA-N 0.000 description 1
- GUVUOGQBMYCBQP-UHFFFAOYSA-N dmpu Chemical compound CN1CCCN(C)C1=O GUVUOGQBMYCBQP-UHFFFAOYSA-N 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 239000011982 enantioselective catalyst Substances 0.000 description 1
- DHGXNLZYPSXUSC-UPCLLVRISA-N ethyl (2R,3R)-1-benzyl-2-[2-(1,3-dioxoisoindol-2-yl)ethyl]-3-methyl-4-oxoazetidine-2-carboxylate Chemical compound CCOC(=O)[C@@]1(CCN2C(=O)c3ccccc3C2=O)[C@@H](C)C(=O)N1Cc1ccccc1 DHGXNLZYPSXUSC-UPCLLVRISA-N 0.000 description 1
- MVEAAGBEUOMFRX-UHFFFAOYSA-N ethyl acetate;hydrochloride Chemical compound Cl.CCOC(C)=O MVEAAGBEUOMFRX-UHFFFAOYSA-N 0.000 description 1
- 239000012847 fine chemical Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- FUKUFMFMCZIRNT-UHFFFAOYSA-N hydron;methanol;chloride Chemical compound Cl.OC FUKUFMFMCZIRNT-UHFFFAOYSA-N 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 150000002596 lactones Chemical group 0.000 description 1
- 235000018977 lysine Nutrition 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 229960004452 methionine Drugs 0.000 description 1
- ACTNHJDHMQSOGL-UHFFFAOYSA-N n',n'-dibenzylethane-1,2-diamine Chemical compound C=1C=CC=CC=1CN(CCN)CC1=CC=CC=C1 ACTNHJDHMQSOGL-UHFFFAOYSA-N 0.000 description 1
- DMQSHEKGGUOYJS-UHFFFAOYSA-N n,n,n',n'-tetramethylpropane-1,3-diamine Chemical compound CN(C)CCCN(C)C DMQSHEKGGUOYJS-UHFFFAOYSA-N 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 210000002741 palatine tonsil Anatomy 0.000 description 1
- 238000005191 phase separation Methods 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 239000004810 polytetrafluoroethylene Substances 0.000 description 1
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- 230000018612 quorum sensing Effects 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 239000012488 sample solution Substances 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- PNGLEYLFMHGIQO-UHFFFAOYSA-M sodium;3-(n-ethyl-3-methoxyanilino)-2-hydroxypropane-1-sulfonate;dihydrate Chemical compound O.O.[Na+].[O-]S(=O)(=O)CC(O)CN(CC)C1=CC=CC(OC)=C1 PNGLEYLFMHGIQO-UHFFFAOYSA-M 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 238000012916 structural analysis Methods 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- AAHDLRDOCKVUDL-NBFOIZRFSA-N tert-butyl (3S)-1-benzyl-3-methylazetidine-2-carboxylate Chemical compound C(C1=CC=CC=C1)N1C([C@H](C1)C)C(=O)OC(C)(C)C AAHDLRDOCKVUDL-NBFOIZRFSA-N 0.000 description 1
- BNWCETAHAJSBFG-UHFFFAOYSA-N tert-butyl 2-bromoacetate Chemical compound CC(C)(C)OC(=O)CBr BNWCETAHAJSBFG-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- 239000001226 triphosphate Substances 0.000 description 1
- 235000011178 triphosphate Nutrition 0.000 description 1
- UNXRWKVEANCORM-UHFFFAOYSA-N triphosphoric acid Chemical compound OP(O)(=O)OP(O)(=O)OP(O)(O)=O UNXRWKVEANCORM-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 239000011592 zinc chloride Substances 0.000 description 1
- 235000005074 zinc chloride Nutrition 0.000 description 1
- CITILBVTAYEWKR-UHFFFAOYSA-L zinc trifluoromethanesulfonate Substances [Zn+2].[O-]S(=O)(=O)C(F)(F)F.[O-]S(=O)(=O)C(F)(F)F CITILBVTAYEWKR-UHFFFAOYSA-L 0.000 description 1
- ZMLPZCGHASSGEA-UHFFFAOYSA-M zinc trifluoromethanesulfonate Chemical compound [Zn+2].[O-]S(=O)(=O)C(F)(F)F ZMLPZCGHASSGEA-UHFFFAOYSA-M 0.000 description 1
Images















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/22—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C269/00—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C269/00—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C269/06—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups by reactions not involving the formation of carbamate groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D205/00—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom
- C07D205/02—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D205/06—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D205/08—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with one oxygen atom directly attached in position 2, e.g. beta-lactams
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/26—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D307/30—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D307/32—Oxygen atoms
- C07D307/33—Oxygen atoms in position 2, the oxygen atom being in its keto or unsubstituted enol form
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Abstract
Description
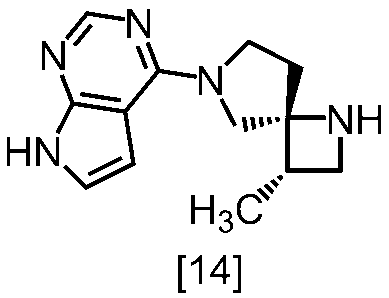
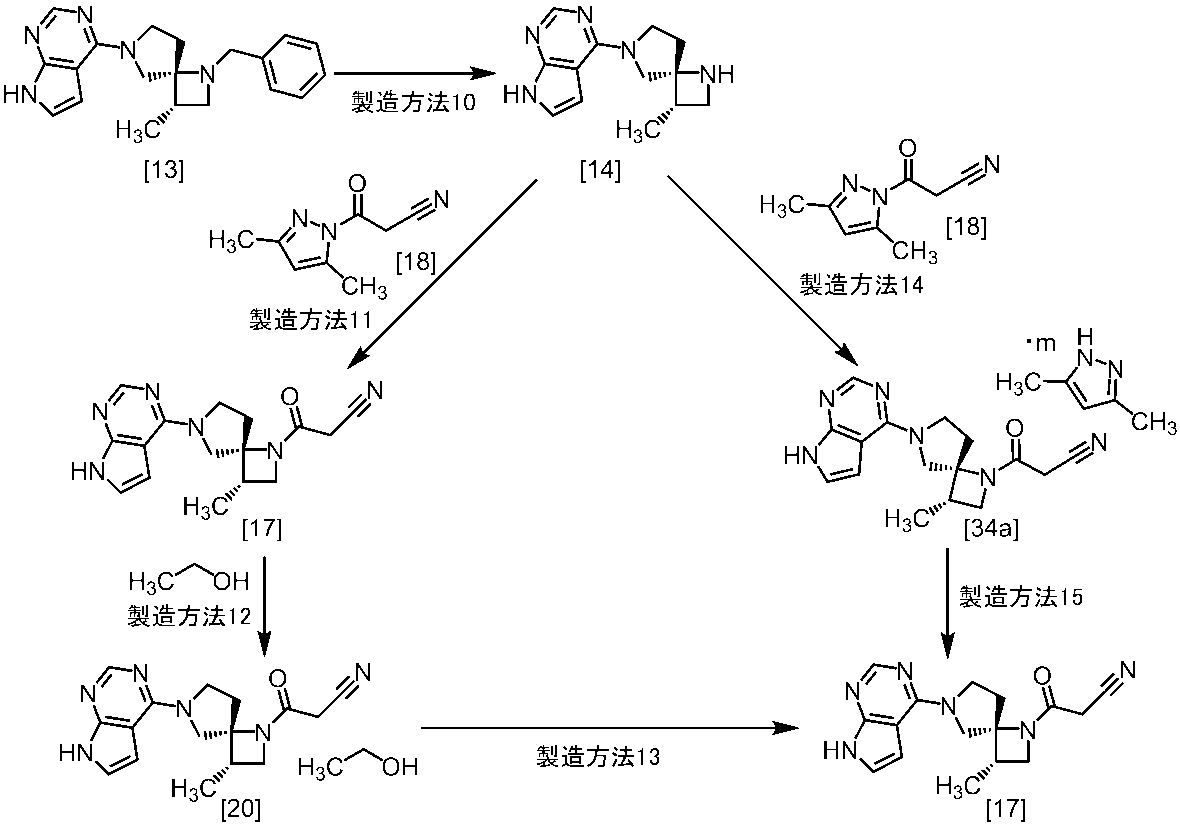
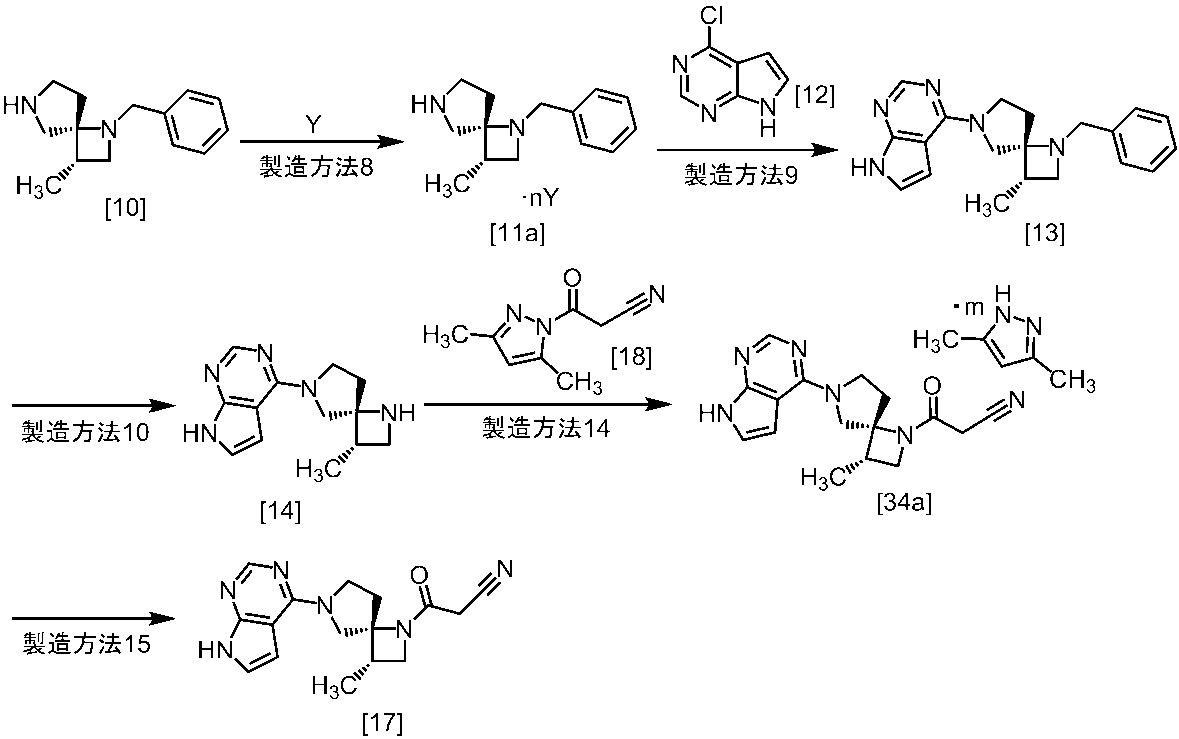
式[13]
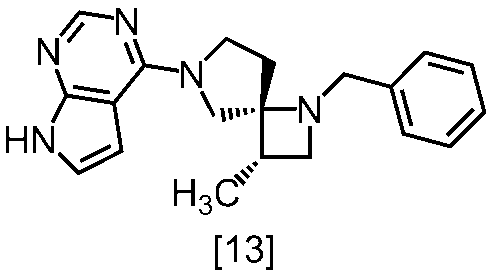
式[17]

(1)式[13]の化合物又はその塩からベンジルを除去することにより式[14]
(2)式[14]の化合物又はその塩を式[18]

無機酸としては、例えば塩酸、硝酸、硫酸、リン酸、臭化水素酸等が挙げられる。好ましい無機酸は塩酸である。
有機酸としては、例えばシュウ酸、マロン酸、マレイン酸、クエン酸、フマル酸、テレフタル酸、乳酸、リンゴ酸、コハク酸、酒石酸、酢酸、トリフルオロ酢酸、グルコン酸、アスコルビン酸、メタンスルホン酸、ベンゼンスルホン酸、p-トルエンスルホン酸、10-カンファースルホン酸等が挙げられる。好ましい有機酸は、シュウ酸、L-酒石酸、D-酒石酸、コハク酸、(+)-10-カンファースルホン酸及び(-)-10-カンファースルホン酸である。より好ましい有機酸は、シュウ酸、コハク酸、L-酒石酸、D-酒石酸及び(+)-10-カンファースルホン酸である。
無機塩基との塩としては、例えばナトリウム塩、カリウム塩、カルシウム塩、マグネシウム塩、アンモニウム塩等が挙げられる。
有機塩基としては、例えばメチルアミン、ジエチルアミン、トリメチルアミン、トリエチルアミン、エタノールアミン、ジエタノールアミン、トリエタノールアミン、エチレンジアミン、トリス(ヒドロキシメチル)メチルアミン、ジシクロヘキシルアミン、N,N-ジベンジルエチレンジアミン、グアニジン、ピリジン、ピコリン、コリン、シンコニン、メグルミン等が挙げられる。
アミノ酸としては、例えばリジン、アルギニン、アスパラギン酸、グルタミン酸等が挙げられる。
溶媒和物とは、本発明に係る化合物又はその塩に、溶媒の分子が配位したものであり、水和物も包含される。溶媒和物は、製薬上許容される溶媒和物が好ましく、例えば本発明に係る化合物又はその塩の水和物、エタノール和物、DMSO和物、1-プロパノール和物、2-プロパノール和物、クロロホルム和物、ジオキサン和物、アニソール和物、アセトン和物、エチレングリコール和物、ジメチルアセトアミド和物等が挙げられる。
本発明に係る化合物は、炭素-炭素二重結合を有する場合がある。その場合、本発明に係る化合物は、E体、Z体、又はE体とZ体の混合物として存在し得る。
本発明に係る化合物は、シス/トランス異性体として認識すべき立体異性体として存在する場合がある。その場合、本発明に係る化合物は、シス体、トランス体、又はシス体とトランス体の混合物として存在し得る。
本発明に係る化合物は、1又はそれ以上の不斉炭素原子を有する場合がある。その場合、本発明に係る化合物は、単一のエナンチオマー、単一のジアステレオマー、エナンチオマーの混合物又はジアステレオマーの混合物として存在する場合がある。
本発明に係る化合物は、アトロプ異性体として存在する場合がある。その場合、本発明に係る化合物は、個々のアトロプ異性体又は異なるアトロプ異性体の混合物として存在し得る。
本発明に係る化合物は、上記の異性体を生じさせる構造上の特徴を同時に複数含み得る。また、本発明に係る化合物は、上記の異性体をあらゆる比率で含み得る。


例えば、エナンチオマー混合物と、実質的に純粋なエナンチオマーであってキラル補助剤(chiral auxiliary)として知られている化合物とを反応させて形成させたジアステレオマー混合物から、分別結晶化やクロマトグラフィーのような標準的な方法により、異性体比率を高めた又は実質的に純粋な単一のジアステレオマーを分離することができる。この分離されたジアステレオマーを、付加されたキラル補助剤を開裂反応にて除去することにより、目的のエナンチオマーに変換することができる。
また、当分野でよく知られた、キラル固定相を使用するクロマトグラフィー法によって、エナンチオマー混合物を直接分離して目的のエナンチオマーを得ることもできる。
あるいは、目的のエナンチオマーを、実質的に純粋な光学活性出発原料を用いることにより、又は、プロキラル(prochiral)な合成中間体に対しキラル補助剤や不斉触媒を用いた立体選択的合成(すなわち、不斉誘導)を行うことによっても得ることができる。
各工程において、反応は溶媒中で行ってもよい。
各工程で得られる化合物は、必要に応じて、蒸留、再結晶、カラムクロマトグラフィー等の公知の方法で単離及び精製することができるが、場合によっては、単離又は精製せず次の工程に進むことができる。
本明細書において、室温とは温度を制御していない状態を示し、一つの態様として1℃から40℃を意味する。反応温度は、記載された温度±5℃、好ましくは±2℃を含むことができる。
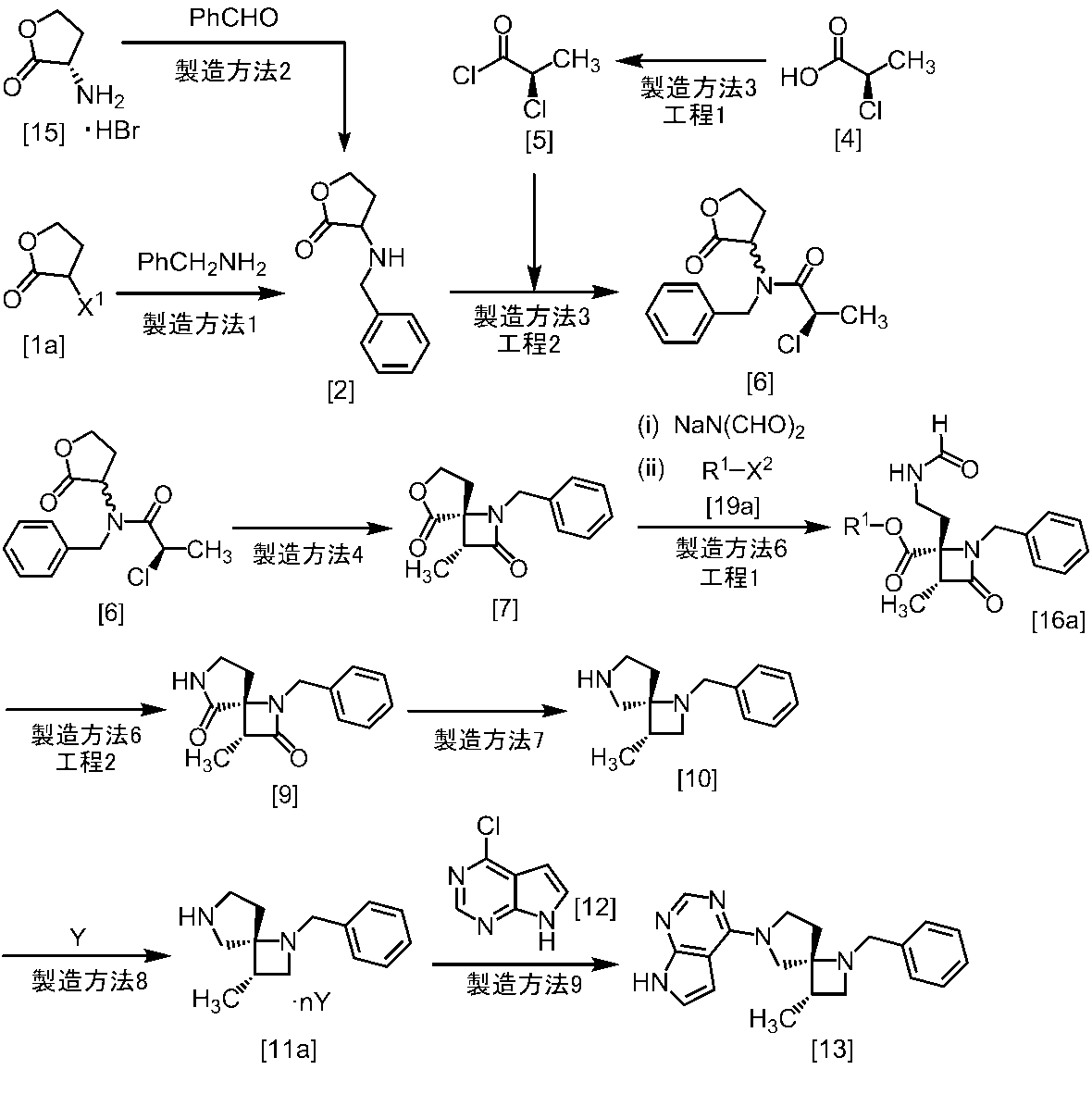
[スキーム中、X1は塩素または臭素である。R1はC1-4アルキル又はベンジルである。X2はハロゲンである。Yは酸である。nは0.5から2の任意の数字である。mは0.4から0.5の任意の数字である。]
スキーム1


[スキーム中、X1は塩素または臭素である。R1はC1-4アルキル又はベンジルである。X2はハロゲンである。Yは酸である。nは0.5から2の任意の数字である。mは0.4から0.5の任意の数字である。]
スキーム2
[スキーム中、R2はメチル、エチル又はベンジルである。R3はメチル、エチル又はベンジルである。Yは酸である。nは0.5から2の任意の数字である。]
スキーム3-1
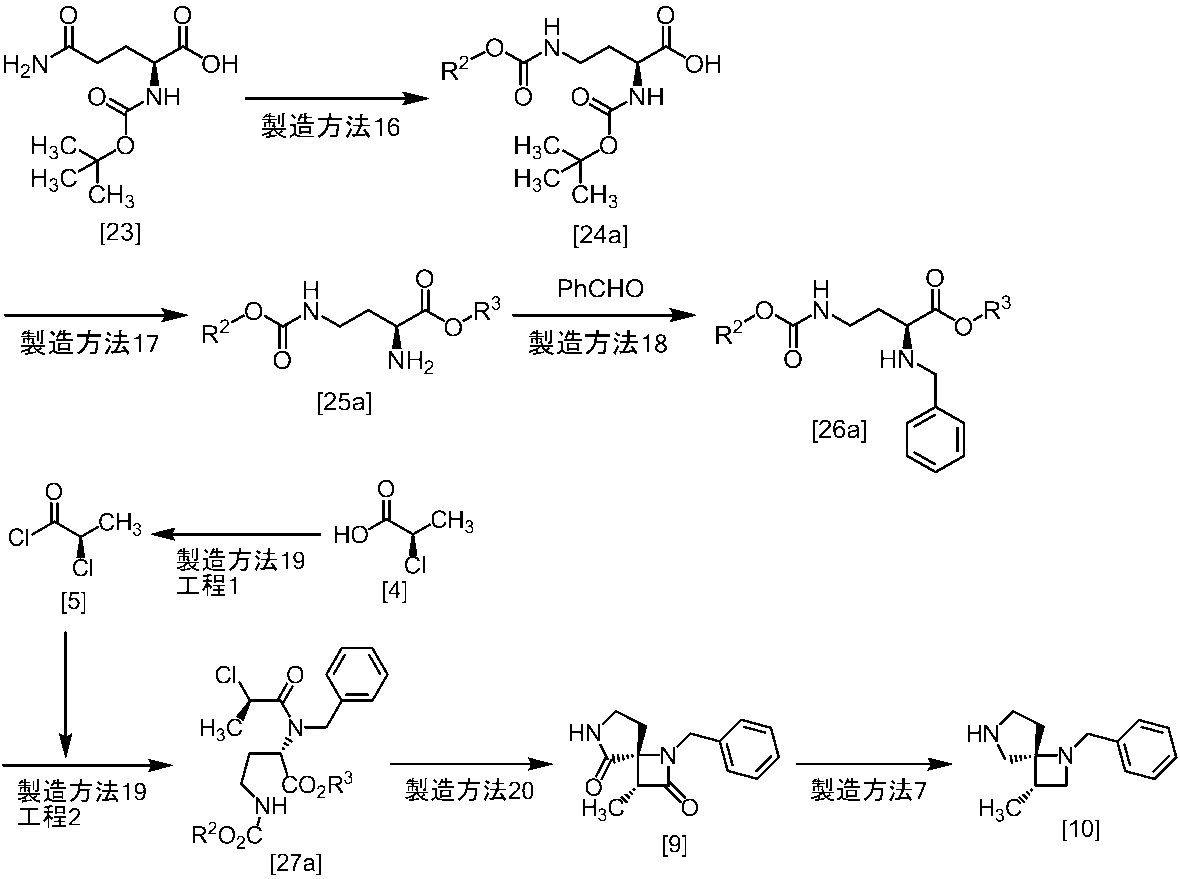


[スキーム中、Yは酸である。nは0.5から2の任意の数字である。]
スキーム4-1





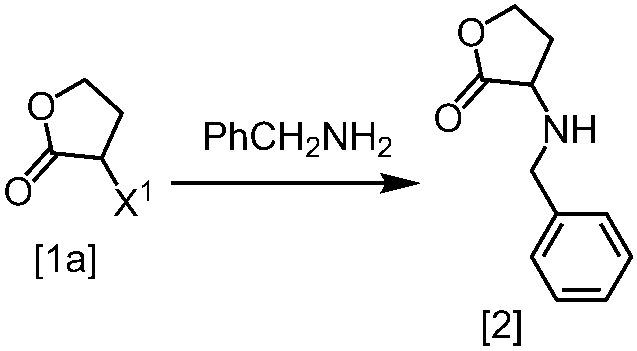
式[2]の化合物は、式[1a]の化合物とベンジルアミンを、塩基存在下、反応させることにより製造することができる。ここで、ベンジルアミンに換えて、4-クロロベンジルアミン、3-クロロベンジルアミン、4-メトキシベンジルアミン、3-メトキシベンジルアミン、4-メチルベンジルアミン、3-メチルベンジルアミン、ベンズヒドリルアミン、トリフェニルメチルアミン等を用いることができる。
式[1a]の化合物としては、BBL、3-クロロジヒドロフラン-2-オンが例示される。好ましい式[1a]の化合物はBBLである。
溶媒としては、例えばTHF、アセトニトリル、DMF、ジメチルアセトアミド、N-メチルピロリドン、DMSOが例示される。好ましい溶媒はアセトニトリルである。
塩基としては、例えばリン酸三カリウム、炭酸カリウム、炭酸セシウムが例示される。好ましい塩基はリン酸三カリウムである。塩基は、例えば式[1a]の化合物に対して2.0当量から5.0当量の量にて用いることができ、好ましくは3.0当量±0.5当量である。
反応温度は、例えば室温から60℃であり、好ましくは45℃±5℃である。別の好ましい態様は、50℃±5℃である。
反応時間は、例えば5時間から48時間であり、好ましくは5時間から30時間である。
酸としては、例えば有機酸又は無機酸が含まれる。
有機酸としては、例えばシュウ酸、マロン酸、マレイン酸、クエン酸、フマル酸、テレフタル酸、乳酸、リンゴ酸、コハク酸、酒石酸、酢酸、トリフルオロ酢酸、グルコン酸、アスコルビン酸、メタンスルホン酸、ベンゼンスルホン酸、p-トルエンスルホン酸等が含まれる。
無機酸としては、例えば塩酸、硝酸、硫酸、リン酸、臭化水素酸等が例示される。好ましい無機酸は、塩酸である。
式[2]の化合物の塩としては、好ましくは、一塩酸塩である。
式[2]の化合物は無機酸と塩を形成させることにより結晶として得ることができる。
式[2]の化合物の塩は、例えば一塩酸塩であって、CuKα放射を使用して測定した回折角(2θ)が8.5°±0.2°、18.9°±0.2°、21.0°±0.2°、21.4°±0.2°又は24.4°±0.2°に少なくとも一個(例えば、少なくとも1、2、3、4、または5個)のピークを有する粉末X線回折パターンを示す結晶である。
好ましくは、式[2]の化合物の塩は、一塩酸塩であって、CuKα放射を使用して測定した回折角(2θ)が8.5°±0.1°、18.9°±0.1°、21.0°±0.1°、21.4°±0.1°又は24.4°±0.1°に少なくとも一個(例えば、少なくとも1、2、3、4、または5個)のピークを有する粉末X線回折パターンを示す結晶である。
より好ましくは、式[2]の化合物の塩は、一塩酸塩であって、CuKα放射を使用して測定した回折角(2θ)が8.5°±0.06°、18.9°±0.06°、21.0°±0.06°、21.4°±0.06°又は24.4°±0.06°に少なくとも一個(例えば、少なくとも1、2、3、4、または5個)のピークを有する粉末X線回折パターンを示す結晶である。

溶媒としては、例えばDMSO、DMF、ジメチルアセトアミド、N-メチルピロリドン、クロロホルム、THFが例示される。好ましい溶媒はDMFである。
酸の例としては、酢酸が挙げられる。
還元剤としては、例えば水素化トリアセトキシホウ素ナトリウム、シアノ水素化ホウ素ナトリウムが例示される。好ましい還元剤は水素化トリアセトキシホウ素ナトリウムである。還元剤は、例えば式[15]の化合物に対して1.0当量から3.0当量の量にて用いることができ、好ましくは1.2当量±0.2当量である。
反応温度は、例えば0℃から60℃であり、好ましくは室温である。
反応時間は、例えば0.5時間から24時間であり、好ましくは1時間から5時間である。

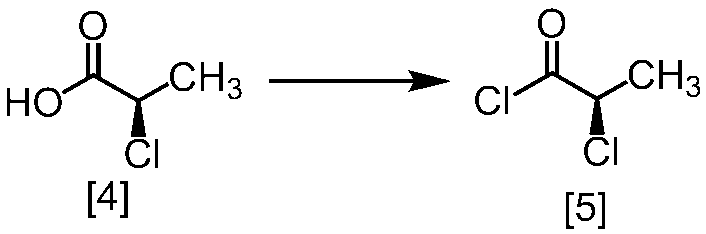
溶媒としては、例えばトルエン、THF、DMF、アセトニトリル、アセトニトリルとDMFとの混合溶媒、及びトルエンとDMFとの混合溶媒が例示される。好ましい溶媒はトルエン、アセトニトリル、アセトニトリルとDMFとの混合溶媒、又はトルエンとDMFとの混合溶媒である。式[5]の化合物は、無溶媒で製造してもよい。
クロロ化剤としては、例えば塩化チオニル、塩化オキサリル、塩化ホスホリルが例示される。好ましいクロロ化剤は塩化チオニルである。クロロ化剤は、例えば式[4]の化合物に対して0.9当量から1.5当量の量にて用いることができ、好ましくは0.95当量から1.15当量である。
反応温度は、通常の知識に基づき適宜調節しうる。クロロ化剤として塩化オキサリルを用いた場合、反応温度は例えば-20℃から10℃であり、好ましくは-10℃から0℃である。クロロ化剤として塩化チオニルを用いた場合、反応温度は45℃から75℃であり、好ましくは65℃±5℃である。クロロ化剤として塩化チオニルを用いた場合の別の好ましい反応温度は、-20℃から10℃であり、より好ましくは-20℃から0℃である。
反応時間は、例えば0.5時間から5時間であり、好ましくは0.5時間から3時間であり、より好ましくは1時間から2時間である。
式[5]の化合物は、例えば減圧下又は常圧下における蒸留により精製し得、好ましくは常圧下である。
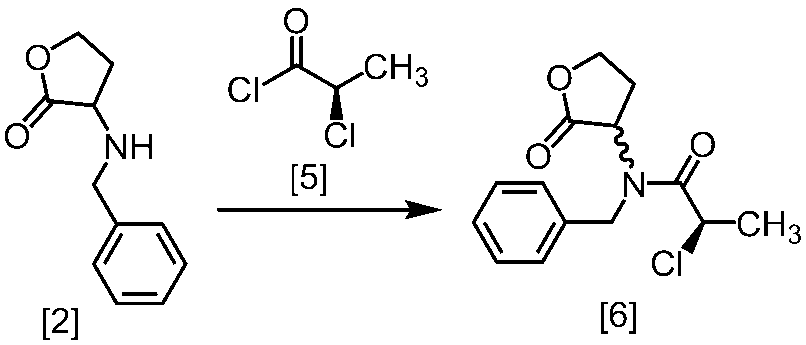
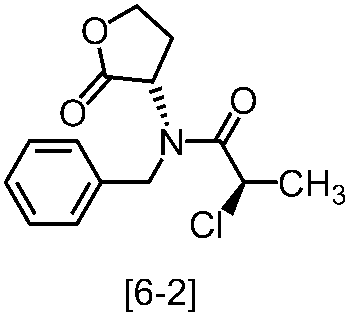
溶媒としては、例えばトルエン、酢酸エチル、THF、これらの混合溶媒が例示される。好ましい溶媒はトルエンと酢酸エチルとの混合溶媒である。
塩基としては、例えば2,6-ルチジン、N,N-ジイソプロピルエチルアミンが例示される。好ましい塩基は2,6-ルチジンである。塩基は、例えば式[2]の化合物に対して1.0当量から5.0当量の量にて用いることができ、好ましくは3.0当量±0.5当量である。また、例えば式[2]の化合物の塩に対して2.0当量から5.0当量の量にて用いることができ、好ましくは4.0当量±0.5当量である。
反応温度は、例えば-20℃から20℃であり、好ましくは-10℃から10℃である。
反応時間は、例えば1時間から5時間であり、好ましくは2時間から3時間である。
式[6]の化合物は結晶として得ることができる。結晶化に用いる溶媒としては、トルエン;2-プロパノール;CPME;酢酸エチル;トルエン、2-プロパノール、CPME及び酢酸エチルから選択される二以上の溶媒の組合せ;2-プロパノールと水の混合溶媒;トルエン、2-プロパノール、CPME又は酢酸エチルとヘプタンとの混合溶媒が例示される。好ましい溶媒は、トルエンとヘプタンの混合溶媒、2-プロパノールとヘプタンの混合溶媒である。
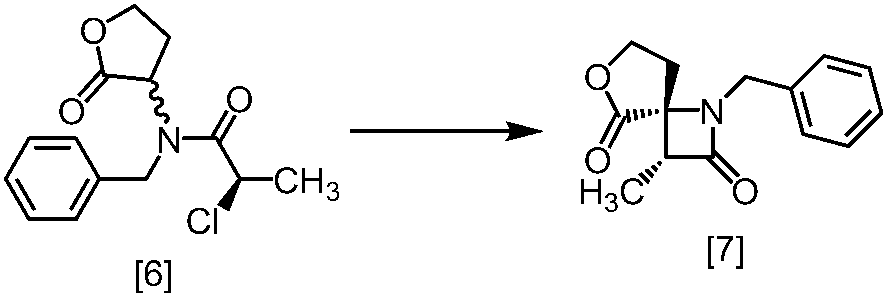
溶媒としては、例えばTHF、アセトニトリル、トルエン、DMSO、DMF、ジメチルアセトアミド、N-メチルピロリドン、これらの混合溶媒が例示される。好ましい溶媒はDMSO、THF又はトルエンとTHFの混合溶媒である。
塩基としては、例えば1,8-ジアザビシクロ[5.4.0]-7-ウンデセン、リチウムヘキサメチルジシラジド、ナトリウムヘキサメチルジシラジド、カリウムヘキサメチルジシラジド、リチウムジイソプロピルアミド、リン酸三カリウム、炭酸セシウム、tert-ブチルイミノ-トリ(ピロリジノ)ホスホランが例示される。好ましい塩基は、リチウムヘキサメチルジシラジド、炭酸セシウム又はリン酸三カリウムである。より好ましい塩基は、リチウムヘキサメチルジシラジドである。
塩基としてリチウムヘキサメチルジシラジドを使用する方法においては、塩基は、例えば式[6]の化合物に対して0.9当量から1.2当量の量にて用いることができ、好ましくは1.0当量±0.05当量である。反応温度は、例えば-20℃から5℃であり、好ましくは-15℃から0℃である。反応時間は、例えば0.5時間から5時間であり、好ましくは1時間から3時間である。
塩基として炭酸セシウム又はリン酸三カリウムを使用する方法においては、塩基は、例えば式[6]の化合物に対して2.0当量から5.0当量の量にて用いることができ、好ましくは3.0当量±0.5当量である。反応温度は、例えば室温から60℃であり、好ましくは室温である。反応時間は、例えば10時間から30時間であり、好ましくは10時間から24時間である。

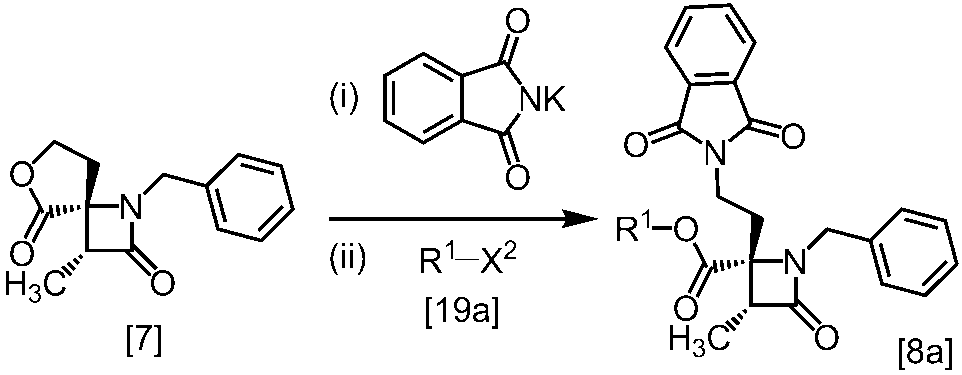
式[8a]の化合物は、式[7]の化合物とフタルイミドカリウムを反応させた後、式[19a]の化合物でエステル化することにより製造することができる。フタルイミドカリウムは、例えば式[7]の化合物に対して1.0当量から2.0当量の量にて用いることができ、好ましくは1.1当量±0.05当量である。
式[19a]の化合物としては、例えばヨウ化メチル、ヨウ化エチル、臭化ベンジルが例示される。好ましい式[19a]の化合物はヨウ化エチルである。式[19a]の化合物は、例えば式[7]の化合物に対して1.0当量から2.0当量の量にて用いることができ、好ましくは1.3当量±0.05当量である。
溶媒としては、例えばDMF、ジメチルアセトアミド、DMSO、N-メチルピロリドン、トルエン、これらの混合溶媒が例示される。好ましい溶媒はDMF、DMSO又はDMSOとトルエンの混合溶媒である。
反応温度は、フタルイミドカリウムとの反応においては、例えば80℃から150℃であり、好ましくは90℃から115℃である。エステル化においては、例えば室温から80℃であり、好ましくは室温から60℃である。
反応時間は、フタルイミドカリウムとの反応においては、例えば10時間から30時間であり、好ましくは10時間から24時間である。エステル化においては、例えば1時間から6時間であり、好ましくは4時間から5時間である。エステル化における別の好ましい反応時間は、1時間から2時間である。
式[8a]の化合物は、式[22]
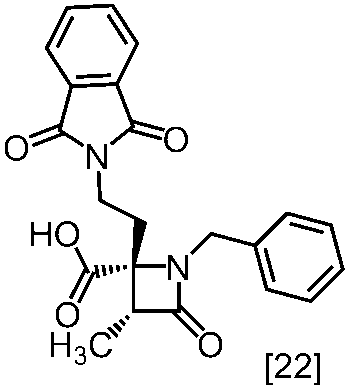
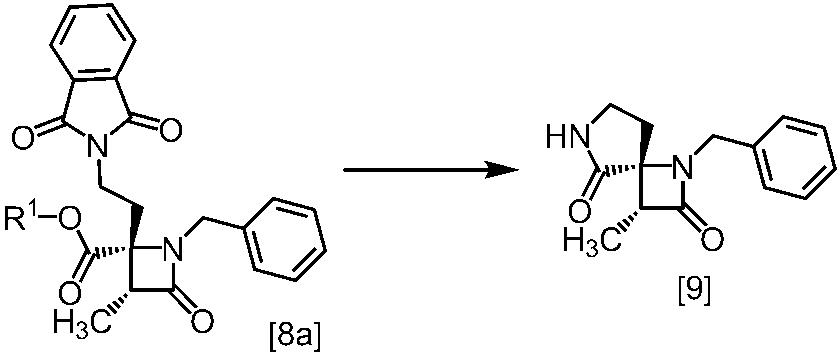
式[9]の化合物は、式[8a]の化合物のフタロイルを除去することにより製造することができる。フタロイルを除去する方法は公知の方法を用いればよく、例えば、式[8a]の化合物とエチレンジアミン又はジエチレントリアミンを反応させることにより、式[9]の化合物を製造することができる。
エチレンジアミン及びジエチレントリアミンは、例えば式[8a]の化合物に対して1.0当量から10当量の量にて用いることができ、好ましくは5.0当量±0.5当量である。
溶媒としては、例えばメタノール、エタノール、1-プロパノール、2-プロパノール、1-ブタノール、2-ブタノール、2-メチル-2-プロパノールが例示される。好ましい溶媒は2-ブタノールである。
反応温度は、例えば60℃から105℃であり、好ましくは80℃から95℃である。
反応時間は、例えば1時間から6時間であり、好ましくは2時間から5時間である。
化合物[9]を再結晶溶媒に溶解させる温度は、例えば40℃から80℃である。好ましい温度は、CPMEを用いる場合は50℃から60℃であり、トルエンを用いる場合は65℃から75℃である。
再結晶化時間は、例えば3時間から10時間であり、好ましくは3時間から5時間である。
式[9]の化合物は、例えばCuKα放射を使用して測定した回折角(2θ)が10.6°±0.2°、16.0°±0.2°、17.5°±0.2°、18.3°±0.2°又は19.2°±0.2°に少なくとも一個(例えば、少なくとも1、2、3、4、または5個)のピークを有する粉末X線回折パターンを示す結晶である。
好ましくは、式[9]の化合物は、CuKα放射を使用して測定した回折角(2θ)が10.6°±0.1°、16.0°±0.1°、17.5°±0.1°、18.3°±0.1°又は19.2°±0.1°に少なくとも一個(例えば、少なくとも1、2、3、4、または5個)のピークを有する粉末X線回折パターンを示す結晶である。
より好ましくは、式[9]の化合物は、CuKα放射を使用して測定した回折角(2θ)が10.6°±0.06°、16.0°±0.06°、17.5°±0.06°、18.3°±0.06°又は19.2°±0.06°に少なくとも一個(例えば、少なくとも1、2、3、4、または5個)のピークを有する粉末X線回折パターンを示す結晶である。


式[16a]の化合物は、式[7]の化合物とジホルミルアミドナトリウムを反応させた後、式[19a]の化合物でエステル化することにより製造することができる。ジホルミルアミドナトリウムは、例えば式[7]の化合物に対して2.0当量から5.0当量の量にて用いることができ、好ましくは3.0当量±0.5当量である。
式[19a]の化合物としては、例えばヨウ化メチル、ヨウ化エチル、臭化ベンジルが例示される。好ましい式[19a]の化合物はヨウ化エチルである。式[19a]の化合物は、例えば式[7]の化合物に対して2.0当量から5.0当量の量にて用いることができ、好ましくは3.0当量±0.5当量である。
溶媒としては、例えばDMSO、ジメチルアセトアミド、N-メチルピロリドンが例示される。好ましい溶媒はDMSOである。
反応温度は、ジホルミルアミドナトリウムとの反応においては、例えば80℃から150℃であり、好ましくは100℃±5℃である。エステル化においては、例えば室温から80℃であり、好ましくは室温から50℃である。
反応時間は、ジホルミルアミドナトリウムとの反応においては、例えば10時間から30時間であり、好ましくは10時間から24時間である。エステル化においては、例えば3時間から6時間であり、好ましくは4時間から5時間である。

式[9]の化合物は、塩基存在下、式[16a]の化合物のホルミルを除去することにより製造することができる。
溶媒としては、例えばアセトニトリル、THF、DMF、DMSO、ジメチルアセトアミド、N-メチルピロリドンが例示される。好ましい溶媒はアセトニトリルである。
塩基としては、例えばリチウムヘキサメチルジシラジド、ナトリウムヘキサメチルジシラジド、カリウムヘキサメチルジシラジド、リチウムジイソプロピルアミド、炭酸セシウム、リン酸三カリウム、tert-ブチルイミノ-トリ(ピロリジノ)ホスホランが例示される。好ましい塩基は炭酸セシウムである。塩基は、例えば式[16a]の化合物に対して1.0当量から2.0当量の量にて用いることができ、好ましくは1.5当量±0.05当量である。
反応温度は、例えば0℃から50℃であり、好ましくは室温である。
反応時間は、例えば3時間から6時間であり、好ましくは4時間から5時間である。

溶媒としては、例えばトルエン、CPME、THF及び2-メチルテトラヒドロフランが例示される。好ましい溶媒はトルエン、THF又はこれらの混合溶媒である。
還元剤としては、例えば、ドデカカルボニル3ルテニウム触媒又はトリフルオロメタンスルホン酸亜鉛触媒の存在下使用されるTMDS、及び酸の存在下使用される水素化アルミニウムリチウムが例示される。酸としては、硫酸、塩化アルミニウム、塩化亜鉛、クロロトリメチルシランが例示される。好ましい還元剤はドデカカルボニル3ルテニウム触媒存在下使用されるTMDS、又はクロロトリメチルシランもしくは塩化アルミニウムの存在下使用される水素化アルミニウムリチウムである。
還元剤は、例えば式[9]の化合物に対して3.0当量から15.0当量の量にて用いることができ、好ましくは10.0当量±0.5当量である。
触媒は、例えば式[9]の化合物に対して0.05当量から0.5当量の量にて用いることができ、好ましくは0.1当量から0.3当量である。
ドデカカルボニル3ルテニウム触媒を使用する場合、さらに添加剤を加えてもよい。添加剤としては、例えばTMEDA、N,N,N’,N’-テトラメチル-1,3-ジアミノプロパンが例示される。好ましい添加剤はTMEDAである。添加剤は、例えば式[9]の化合物に対して0.05当量から0.5当量の量にて用いることができ、好ましくは0.1当量から0.3当量である。
反応温度は、例えば40℃から100℃であり、好ましくは60℃から80℃である。
反応時間は、例えば10時間から50時間であり、好ましくは35時間から45時間である。
還元剤は一度に添加してもよく、又は、2回以上に分けて添加してもよい。好ましい方法は、2回以上に分けて添加する方法である。
(1)一度に添加する方法においては、還元剤は、酸存在下、例えば式[9]の化合物に対して1.0当量から5.0当量の量にて用いることができ、好ましくは3.0当量±0.5当量である。この場合の好ましい酸は、塩化アルミニウム又はクロロトリメチルシランである。塩化アルミニウム又はクロロトリメチルシランは、例えば式[9]の化合物に対して1.0当量から5.0当量の量にて用いることができ、好ましくは3.0当量±0.5当量である。
反応温度は、例えば室温から60℃であり、好ましくは40℃から50℃である。反応時間は、例えば10時間から30時間であり、好ましくは15時間から24時間である。
(2)2回以上に分けて添加する方法において、1段階目の還元剤は、酸存在下、例えば式[9]の化合物に対して1.0当量から5.0当量の量にて用いることができ、好ましくは2.5当量±0.5当量である。この場合の好ましい酸は、クロロトリメチルシランである。クロロトリメチルシランは、例えば式[9]の化合物に対して1.0当量から5.0当量の量にて用いることができ、好ましくは2.5当量±0.5当量である。2段階目の還元剤は、例えば式[9]の化合物に対して0.3当量から3.0当量の量にて用いることができ、好ましくは0.5当量±0.1当量である。
反応温度は、1段階目は、例えば-20℃から10℃であり、好ましくは-20℃から5℃である。2段階目は、例えば室温から60℃であり、好ましくは40℃から50℃である。2段階目の別の好ましい反応温度は、45℃から55℃である。
反応時間は、1段階目は、例えば0.5時間から3時間であり、好ましくは1時間から2時間である。2段階目は、例えば5時間から30時間であり、好ましくは5時間から24時間である。
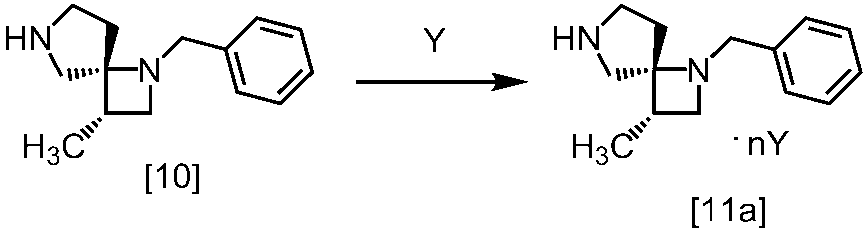
式[11a]の化合物は、式[10]の化合物を酸を用いて塩を形成させることにより製造することができる。
溶媒としては、例えば水、メタノール、エタノール、1-プロパノール、2-プロパノール、THF、これらの混合溶媒が例示される。好ましい溶媒は2-プロパノールである。
酸としては、例えば有機酸又は無機酸が含まれる。
有機酸としては、例えばシュウ酸、マロン酸、マレイン酸、クエン酸、フマル酸、テレフタル酸、乳酸、リンゴ酸、コハク酸、酒石酸、酢酸、トリフルオロ酢酸、グルコン酸、アスコルビン酸、メタンスルホン酸、ベンゼンスルホン酸、p-トルエンスルホン酸、10-カンファースルホン酸等が含まれる。好ましい有機酸は、シュウ酸、L-酒石酸、D-酒石酸、コハク酸、(+)-10-カンファースルホン酸又は(-)-10-カンファースルホン酸である。より好ましい有機酸は、シュウ酸、コハク酸、L-酒石酸、D-酒石酸又は(+)-10-カンファースルホン酸である。
無機酸としては、例えば塩酸、硝酸、硫酸、リン酸、臭化水素酸等が例示される。
反応温度は、例えば室温から80℃であり、好ましくは室温から70℃である。
反応時間は、例えば6時間から15時間であり、好ましくは8時間から12時間である。
式[11a]の化合物の例としては、好ましくは式[10]の化合物の二コハク酸塩である。
式[11a]の化合物は、例えば式[10]の化合物の二コハク酸塩であって、CuKα放射を使用して測定した回折角(2θ)が4.8°±0.2°、11.2°±0.2°、16.2°±0.2°、18.1°±0.2°又は20.1°±0.2°に少なくとも一個(例えば、少なくとも1、2、3、4、または5個)のピークを有する粉末X線回折パターンを示す結晶である。
好ましくは、式[11a]の化合物は、式[10]の化合物の二コハク酸塩であって、CuKα放射を使用して測定した回折角(2θ)が4.8°±0.1°、11.2°±0.1°、16.2°±0.1°、18.1°±0.1°又は20.1°±0.1°に少なくとも一個(例えば、少なくとも1、2、3、4、または5個)のピークを有する粉末X線回折パターンを示す結晶である。
より好ましくは、式[11a]の化合物は、式[10]の化合物の二コハク酸塩であって、CuKα放射を使用して測定した回折角(2θ)が4.8°±0.06°、11.2°±0.06°、16.2°±0.06°、18.1°±0.06°又は20.1°±0.06°に少なくとも一個(例えば、少なくとも1、2、3、4、または5個)のピークを有する粉末X線回折パターンを示す結晶である。
式[11a]の化合物の別の例は、好ましくは式[10]の化合物のヘミシュウ酸塩である。
式[11a]の化合物は、再結晶又は溶媒に式[11a]の化合物を懸濁させた混合液を撹拌すること(以下、「スラリー撹拌」という。)で精製することができる。あるいは、式[11a]の化合物は再結晶及びスラリー撹拌の両方により精製してもよく、その順番は問わない。好ましい精製方法は、スラリー撹拌である。
再結晶及びスラリー撹拌に用いる溶媒としては、例えばメタノール、エタノール、1-プロパノール、2-プロパノール、THF、トルエン、これらの混合溶媒が例示される。再結晶に好ましい溶媒は、メタノールとトルエンの混合溶媒である。スラリー撹拌に好ましい溶媒は、2-プロパノールである。
スラリー撹拌の温度は、例えば0℃から60℃であり、好ましくは30℃から35℃である。別の好ましいスラリー撹拌の温度は、35℃から40℃である。
スラリー撹拌の時間は、例えば1時間から15時間であり、好ましくは2時間から12時間である。

溶媒としては、例えばtert-ブタノール、水、エタノール、メタノール、2-プロパノール、及びこれらの混合溶媒が例示される。好ましい溶媒はtert-ブタノール又は2-プロパノールと水との混合溶媒である。
塩基としては、例えばリン酸三カリウムなどのアルカリ金属リン酸塩、炭酸カリウムなどのアルカリ金属炭酸塩、水酸化カリウムなどのアルカリ金属水酸化物又はこれらの混合物が例示される。好ましい塩基はリン酸三カリウム、又はリン酸三カリウムと水酸化カリウムとの混合物である。塩基は、例えば式[11a]の化合物に対して4.0当量から10.0当量の量にて用いることができ、好ましくは5.0当量から8.0当量である。
CPPY[12]は、例えば式[11a]の化合物に対して0.95当量から1.10当量の量にて用いることができ、好ましくは1.02当量±0.02当量である。
反応温度は、例えば室温から85℃であり、好ましくは80℃±5℃である。別の好ましい反応温度は、75℃±5℃である。
反応時間は、例えば1時間から10時間であり、好ましくは2時間から8時間である。
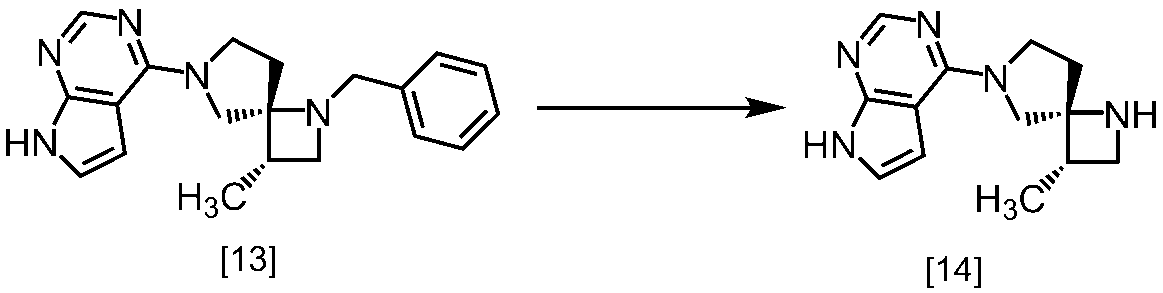
溶媒としては、例えばtert-ブタノール、水、エタノール、2-プロパノール、及びこれらの混合溶媒が例示される。好ましい溶媒はtert-ブタノールと水との混合溶媒、又は2-プロパノールと水との混合溶媒である。
触媒としては、例えば5%パラジウム炭素(50%含水品)、10%パラジウム炭素(50%含水品)、パラジウム炭素、水酸化パラジウム炭素、パラジウム黒又はパラジウムシリカゲルが例示される。好ましい触媒は、5%パラジウム炭素(50%含水品)又は10%パラジウム炭素(50%含水品)である。触媒は、例えば式[13]の化合物の重量に対して0.01倍量から0.5倍量用いることができ、好ましくは0.05倍量から0.2倍量である。
酸の例としては、酢酸が挙げられる。
水素ガス圧は、好ましくは常圧である。0.1MPa程度加圧してもよい。
反応温度は、例えば室温から80℃であり、好ましくは55℃±5℃である。別の好ましい態様は、50℃±5℃である。
反応時間は、例えば2時間から10時間であり、好ましくは3時間から8時間である。

溶媒としては、例えばアセトニトリル、THFが例示される。好ましい溶媒はアセトニトリルである。
塩基としては、例えばトリエチルアミン、N,N-ジイソプロピルエチルアミンが例示される。好ましい塩基はトリエチルアミンである。
DPCN[18]は、例えば式[17]の化合物に対して0.95当量から1.2当量用いることができ、好ましくは1.05当量±0.10当量である。
反応温度は、例えば室温から80℃であり、好ましくは40℃から50℃である。
反応時間は、例えば1時間から12時間であり、好ましくは2時間から6時間である。
溶媒としては、例えばアセトニトリル、テトラヒドロフランが例示される。好ましい溶媒はアセトニトリルである。
DPCNは、例えば式[14]の化合物に対して0.95当量から1.2当量用いることができ、好ましくは1.05当量±0.05当量である。
反応温度は、例えば室温から80℃であり、好ましくは70℃から80℃である。
反応時間は、例えば0.5時間から12時間であり、好ましくは0.5時間から6時間である。

の化合物又はその塩と式[14]の化合物を反応させることにより製造することができる。式[14]の化合物及び式[17]の化合物は、それらの塩であってもよく、フリー体から塩、塩からフリー体の形成は、公知の方法に従って行えばよい。
R4が水素の場合、式[17]の化合物は、式[35a]の化合物又はその塩と式[14]の化合物を、塩基及び縮合剤存在下、反応させることにより製造することができる。
縮合剤としては、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(WSC・HCl)と1-ヒドロキシベンゾトリアゾール(HOBt)の組み合わせ、ヘキサフルオロリン酸(ベンゾトリアゾール-1-イルオキシ)トリスピロリジノホスホニウム(PyBOP)等が例示される。好ましい縮合剤はPyBOPである。
R4がメチル又はエチルの場合、式[17]の化合物は、式[35a]の化合物と式[14]の化合物を縮合させることにより製造することができる。本反応は、3,5-ジメチル-1H-ピラゾールと触媒量のジアザビシクロウンデセン(DBU)の存在下、行うことが好ましい。

溶媒としては、例えばエタノール、1-プロパノール、2-プロパノール、クロロホルム、ジオキサン、アニソール、アセトン、エチレングリコール、ジメチルアセトアミド、水が例示される。好ましい溶媒はエタノールである。
本工程は、化合物[17]の製造に必ずしも必要ではないが、化合物[17]の純度を向上させるために、実施してもよい。
式[20]の化合物は、例えばCuKα放射を使用して測定した回折角(2θ)が8.3°±0.2°、12.7°±0.2°、13.0°±0.2°、20.0°±0.2°又は24.1°±0.2°に少なくとも一個(例えば、少なくとも1、2、3、4、または5個)のピークを有する粉末X線回折パターンを示す結晶である。
好ましくは、式[20]の化合物は、CuKα放射を使用して測定した回折角(2θ)が8.3°±0.1°、12.7°±0.1°、13.0°±0.1°、20.0°±0.1°又は24.1°±0.1°に少なくとも一個(例えば、少なくとも1、2、3、4、または5個)のピークを有する粉末X線回折パターンを示す結晶である。
より好ましくは、式[20]の化合物は、CuKα放射を使用して測定した回折角(2θ)が8.3°±0.06°、12.7°±0.06°、13.0°±0.06°、20.0°±0.06°又は24.1°±0.06°に少なくとも一個(例えば、少なくとも1、2、3、4、または5個)のピークを有する粉末X線回折パターンを示す結晶である。

結晶化に用いる溶媒としては、例えば1-ブタノール、1-プロパノール又は2-メチル-2-ブタノールが例示される。好ましい溶媒は1-ブタノールである。溶媒は、例えば式[20]の化合物の重量に対して8.0倍量から20倍量用いることができ、好ましくは8.5倍量±0.5倍量である。
化合物[20]を結晶化溶媒に溶解させる温度は、例えば100℃から117℃であり、好ましくは110℃±5℃である。
結晶化時間は、例えば15時間から48時間であり、好ましくは18時間から24時間である。
溶媒としては、例えば1-ブタノール、1-プロパノール又は2-メチル-2-ブタノールが例示される。好ましい溶媒は1-ブタノールである。溶媒は、例えば式[17]の化合物の重量に対して18倍量から22倍量用いることができ、好ましくは20倍量±0.5倍量である。
結晶溶解温度は、例えば85℃から100℃であり、好ましくは90℃から100℃である。
再結晶化時間は、例えば10時間から48時間であり、好ましくは10時間から24時間である。

式[34a]の化合物は、式[14]の化合物と1-シアノアセチル-3,5-ジメチル-1H-ピラゾール(DPCN)[18]を縮合させることにより製造することができる。式[14]の化合物は、それらの塩であってもよく、フリー体から塩、塩からフリー体の形成は、公知の方法に従って行えばよい。
好ましい溶媒はアセトニトリルである。
DPCN[18]は、例えば式[14]の化合物に対して0.95当量から1.2当量用いることができ、好ましくは1.1当量±0.05当量である。別の好ましい態様は、1.05当量±0.05当量である。
反応温度は、例えば室温から80℃であり、好ましくは70℃から80℃である。
反応時間は、例えば0.5時間から12時間であり、好ましくは0.5時間から6時間である。
本工程は、化合物[17]の製造に必ずしも必要ではないが、化合物[17]の純度を向上させるために、実施してもよい。
式[34a]の化合物は、例えばCuKα放射を使用して測定した回折角(2θ)が4.6°±0.2°、18.6°±0.2°又は20.9°±0.2°に少なくとも一個(例えば、少なくとも1、2または3個)のピークを有する粉末X線回折パターンを示す結晶である。
好ましくは、式[34a]の化合物は、CuKα放射を使用して測定した回折角(2θ)が4.6°±0.1°、18.6°±0.1°又は20.9°±0.1°に少なくとも一個(例えば、少なくとも1、2または3個)のピークを有する粉末X線回折パターンを示す結晶である。
より好ましくは、式[34a]の化合物は、CuKα放射を使用して測定した回折角(2θ)が4.6°±0.06°、18.6°±0.06°又は20.9°±0.06°に少なくとも一個(例えば、少なくとも1、2または3個)のピークを有する粉末X線回折パターンを示す結晶である。
さらに、式[34a]の化合物はまた、例えばCuKα放射を使用して測定した回折角(2θ)が4.6°±0.2°、12.6°±0.2°、16.1°±0.2°、18.6°±0.2°又は20.9°±0.2°に少なくとも一個(例えば、少なくとも1、2、3、4または5個)のピークを有する粉末X線回折パターンを示す結晶であってもよい。
好ましくは、式[34a]の化合物はまた、CuKα放射を使用して測定した回折角(2θ)が4.6°±0.1°、12.6°±0.1°、16.1°±0.1°、18.6°±0.1°又は20.9°±0.1°に少なくとも一個(例えば、少なくとも1、2、3、4または5個)のピークを有する粉末X線回折パターンを示す結晶であり得る。
より好ましくは、式[34a]の化合物はまた、CuKα放射を使用して測定した回折角(2θ)が4.6°±0.06°、12.6°±0.06°、16.1°±0.06°、18.6°±0.06°又は20.9°±0.06°に少なくとも一個(例えば、少なくとも1、2、3、4または5個)のピークを有する粉末X線回折パターンを示す結晶であり得る。

式[17]の化合物は、式[34a]の化合物を溶解後、結晶化させることにより精製することができる。結晶化において2,6-ジ-tert-ブチル-4-メチルフェノール(BHT)を加えて、精製を実施してもよい。
結晶化に用いる溶媒としては、例えば1-ブタノール、1-プロパノール又は2-メチル-2-ブタノールが例示される。好ましい溶媒は1-ブタノールである。溶媒は、例えば式[34a]の化合物の重量に対して8.0倍量から20倍量用いることができ、好ましくは8.5倍量±0.5倍量である。
化合物[34a]を結晶化溶媒に溶解させる温度は、例えば100℃から117℃であり、好ましくは110℃±5℃である。
結晶化時間は、例えば15時間から48時間であり、好ましくは18時間から24時間である。

式[24a]の化合物は、式[23]の化合物をメタノール、エタノール又はベンジルアルコール及び酸化剤と反応させることにより製造することができる。式[23]の化合物及び式[24a]の化合物は、それらの塩であってもよく、フリー体から塩、塩からフリー体の形成は、公知の方法に従って行えばよい。
溶媒としては、例えばメタノール、エタノール又はベンジルアルコール、及びメタノールと水、THF又はトルエンとの混合溶媒が例示される。好ましい溶媒はメタノールである。
酸化剤としては、例えば臭素、次亜塩素酸ナトリウム、オキソン及び(ジアセトキシヨード)ベンゼンが例示される。好ましい酸化剤は次亜塩素酸ナトリウム又は臭素である。酸化剤は、例えば式[23]の化合物に対して0.9から2.0当量、好ましくは1.1±0.05当量用いることができる。
反応温度は、例えば0℃から60℃、好ましくは40℃から50℃である。
反応時間は、例えば1時間から5時間、好ましくは2時間である。

式[25a]の化合物又はその塩は、式[24a]の化合物を、メタノール、エタノール又はベンジルアルコール及び酸と反応させることにより製造することができる。式[24a]の化合物は、それらの塩であってもよく、フリー体から塩、塩からフリー体の形成は、公知の方法に従って行えばよい。
溶媒としては、例えばメタノール、エタノール又はベンジルアルコール、及びメタノールと水、THF又はトルエンとの混合溶媒が例示される。好ましい溶媒はメタノールである。
酸又は酸前駆体としては、例えば塩酸、塩化アセチル、塩化チオニル、塩化ホスホリル、塩化オキサリルが例示される。好ましい酸又は酸前駆体は、それぞれ塩酸又は塩化チオニルである。より好ましい酸又は酸前駆体は塩化チオニルである。酸又は酸前駆体は、例えば式[24a]の化合物に対して1.0から20.0当量、好ましくは2.0当量用いることができる。
反応温度は、0℃から50℃、好ましくは15℃から25℃である。
反応時間は、例えば2時間から21時間、好ましくは2時間である。
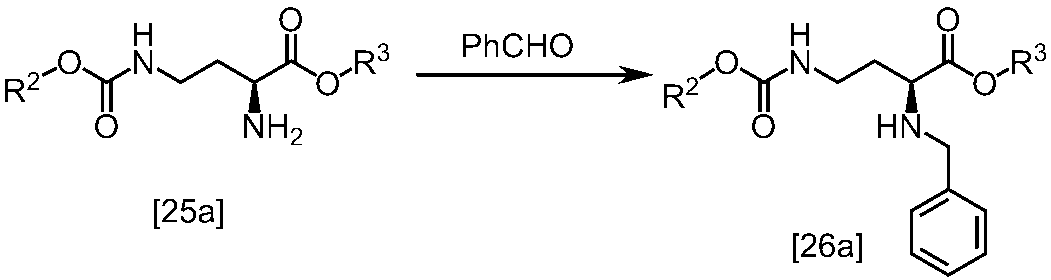
式[26a]の化合物又はその塩は、式[25a]の化合物又はその塩をベンズアルデヒド及び塩基と反応させた後、得られた化合物を還元することにより製造することができる。式[25a]の化合物及び式[26a]の化合物は、それらの塩で用いても製造してもよく、フリー体から塩、塩からフリー体の形成は、公知の方法に従って行えばよい。
溶媒としては、例えばメタノール、エタノール、2-プロパノール、アセトニトリル及び1,2-ジクロロエタンが例示される。好ましい溶媒はメタノールである。
塩基としては、例えばトリエチルアミン及びN,N-ジイソプロピルエチルアミンが例示される。好ましい塩基はトリエチルアミンである。
還元剤としては、例えば水素化ホウ素ナトリウム、トリアセトキシ水素化ホウ素ナトリウム、シアノ水素化ホウ素ナトリウム及び水素ガスが例示される。好ましい還元剤は水素化ホウ素ナトリウムである。還元剤は、例えば式[25a]の化合物に対して0.95から1.2当量、好ましくは1.1±0.05当量用いることができる。
反応物であるベンズアルデヒドは、例えば式[25a]の化合物に対して0.95から2.0当量、好ましくは1.1±0.05当量用いることができる。
水素化ホウ素ナトリウムを添加する間の反応温度は、例えば-30から-5℃、好ましくは-20℃から-15℃である。水素化ホウ素ナトリウムの添加後、反応温度は0℃から25℃、好ましくは20℃から25℃である。
反応時間は、例えば2時間から21時間、好ましくは3時間から6時間である。
塩酸塩を形成させるための溶媒としては、例えばメチルtert-ブチルエーテル、2-プロパノール、酢酸エチル及び酢酸2-プロピルが例示される。好ましい溶媒は酢酸エチル又は酢酸2-プロピルである。より好ましい溶媒は酢酸2-プロピルである。
塩酸塩の形成に用いる酸、塩酸は、例えば式[26a]の化合物に対して0.95から5.0当量、好ましくは1.5から2.5当量用いることができる。
反応温度は、例えば-10℃から50℃、好ましくは0℃から10℃である。
反応時間は、例えば30分から3時間、好ましくは1時間から2時間である。
精製用溶媒としては、例えばメタノール、エタノール、2-プロパノール、1-ブタノール及び酢酸2-プロピルが例示される。好ましい溶媒は酢酸2-プロピルである。
精製温度は、例えば室温から60℃、好ましくは40℃から50℃である。
精製時間は、例えば2時間から12時間、好ましくは3時間から6時間である。
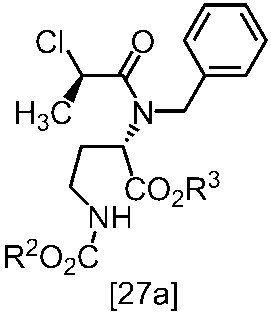
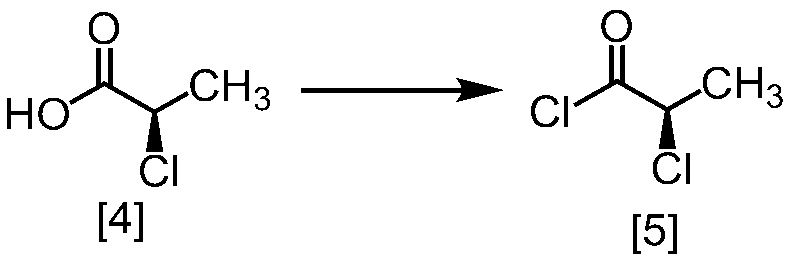
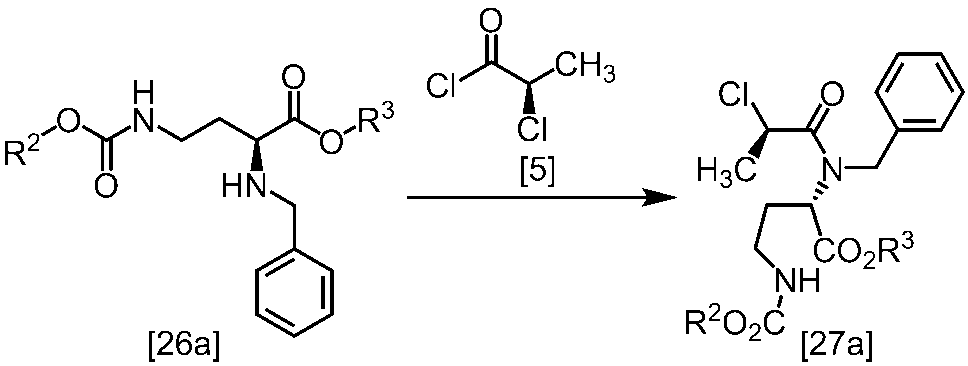
式[27a]の化合物は、式[26a]の化合物又はその塩を、塩基存在下、式[5]の化合物と反応させることにより製造することができる。本工程において、式[26a]の化合物又はその塩に換えて式[26b]の化合物

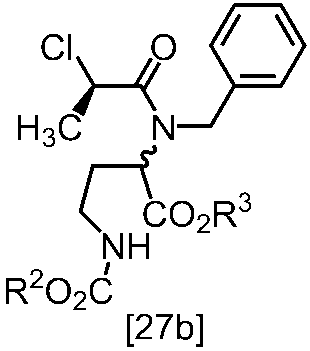
溶媒としては、例えばトルエン、酢酸エチル、THF、メチルtert-ブチルエーテル、アセトニトリル、これらの混合溶媒及びトルエンと水との混合溶媒が例示される。好ましい溶媒はアセトニトリル、トルエン、又はトルエンと水との混合溶媒である。
塩基としては、例えば2,6-ルチジン、N,N-ジイソプロピルエチルアミン、トリエチルアミン、ピリジン、リン酸三カリウム及び炭酸カリウムが例示される。好ましい塩基は、炭酸カリウム、又は2,6-ルチジンとN,N-ジイソプロピルエチルアミンとの組合せである。塩基は、例えば式[26a]の化合物に対して1.0から5.0当量、好ましくは3.0から4.0当量用いることができる。
反応温度は、-20℃から20℃、好ましくは-10℃から5℃である。
反応時間は、例えば1時間から5時間、好ましくは1時間から3時間である。
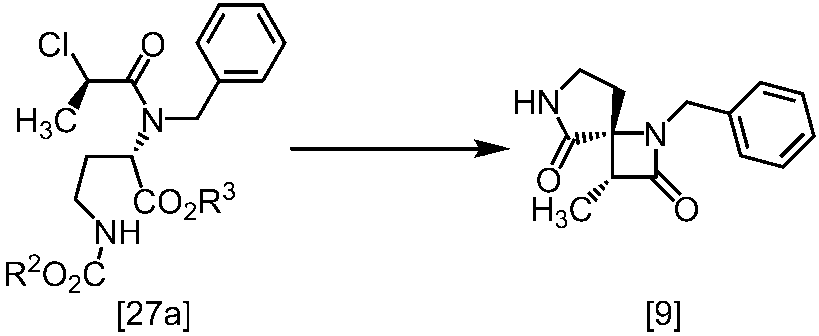
式[9]の化合物は、式[27a]の化合物を塩基存在下、二重環化反応させることにより製造することができる。あるいは、式[27a]の化合物の立体異性体混合物から、式[9]の化合物を含む鏡像異性体混合物を得、次いでキラル技術により分離することで、式[9]の化合物を製造することもできる。
溶媒としては、例えばTHF、アセトニトリル、トルエン、DMSO、DMF、ジメチルアセトアミド、N-メチルピロリドン、炭酸ジメチル及びこれらの任意の混合溶媒が例示される。好ましい溶媒は、アセトニトリル又はDMSOである。
塩基としては、例えばリチウムヘキサメチルジシラジド、ナトリウムヘキサメチルジシラジド、カリウムヘキサメチルジシラジド、リチウムジイソプロピルアミド、リン酸三カリウム、炭酸セシウム、tert-ブチルイミノ-トリ(ピロリジノ)ホスホラン、カリウムtert-ブトキシド及びリチウム2-メチル-2-ブトキシドが例示される。好ましい塩基は、リチウムヘキサメチルジシラジド、炭酸セシウム又はリチウム2-メチル-2-ブトキシドである。より好ましい塩基は、リチウム2-メチル-2-ブトキシドである。
リチウム2-メチル-2-ブトキシドを塩基として用いる場合、塩基は、例えば式[27a]の化合物に対し、1.0から3.0当量、好ましくは3.0当量用いることができる。この場合、反応温度は、例えば-20℃から5℃、好ましくは-10℃から0℃であり、反応時間は、例えば1時間から5時間である。
炭酸セシウムを塩基として用いる場合、塩基は、例えば式[27a]の化合物に対し、2.0から5.0当量、好ましくは2.5当量用いることができる。この場合、反応温度は、例えば15℃から50℃、好ましくは20℃から25℃であり、反応時間は、例えば10時間から30時間、好ましくは15時間から20時間である。

式[29a]の化合物は、酸を用いて式[28]の化合物の塩を形成させることにより製造することができる。
溶媒としては、限定されるものではないが、水、メタノール、エタノール、1-プロパノール、イソプロパノール、アセトニトリル、アセトン、トルエン、メチルtert-ブチルエーテル、テトラヒドロフラン及びこれらの任意の混合溶媒が例示される。好ましい溶媒は、テトラヒドロフランとトルエンとの混合溶媒である。
酸としては、例えば有機酸又は無機酸が例示される。
有機酸としては、例えばシュウ酸、マロン酸、マレイン酸、クエン酸、フマル酸、テレフタル酸、乳酸、リンゴ酸、コハク酸、酒石酸、酢酸、トリフルオロ酢酸、グルコン酸、アスコルビン酸、メタンスルホン酸、ベンゼンスルホン酸、p-トルエンスルホン酸、10-カンファースルホン酸等が例示される。好ましい有機酸はシュウ酸である。
無機酸としては、例えば塩酸、硝酸、硫酸、リン酸、臭化水素酸等が例示される。
反応温度は、例えば0℃から80℃、好ましくは30℃から60℃である。反応時間は、例えば30分から24時間、好ましくは1時間から3時間である。
式[29a]の化合物の例としては、好ましくは式[28]の化合物のシュウ酸塩である。
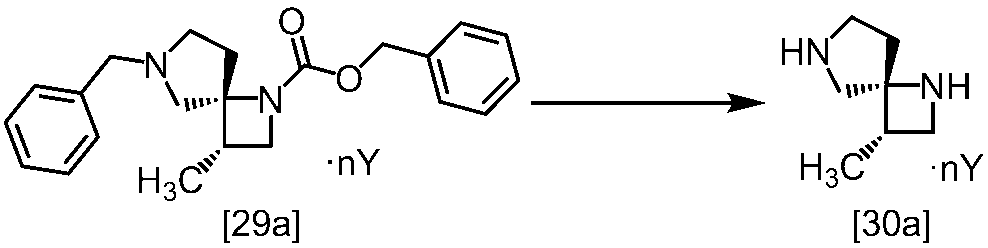
式[30a]の化合物は、式[29a]の化合物から保護基(すなわち、ベンジルオキシカルボニル及びベンジル)を除去することにより製造することができる。脱保護は公知の方法を用いてもよく、例えば式[30a]の化合物は、中性、塩基性又は酸性条件下、触媒存在下にて式[29a]の化合物に水素ガスを添加することにより製造することができる。式[29a]の化合物及び式[30a]の化合物は、それらのフリー体で用いても製造してもよく、フリー体から塩、塩からフリー体の形成は、公知の方法に従って行えばよい。
溶媒としては、例えばtert-ブタノール、水、イソプロパノール、1-プロパノール、エタノール及びこれらの任意の混合溶媒が例示される。
触媒としては、例えば5%パラジウム炭素(50%含水品)、10%パラジウム炭素(50%含水品)、パラジウム炭素、水酸化パラジウム炭素又はパラジウム黒が例示される。好ましい触媒は、5%パラジウム炭素(50%含水品)又は10%パラジウム炭素(50%含水品)である。触媒は、例えば式[29a]の化合物の重量に対して0.01倍量から0.5倍量用いることができ、好ましくは0.05倍量から0.2倍量である。
水素ガス圧は、1から5barである。好ましい水素ガス圧は2から4barである。
反応温度は、例えば室温から80℃、好ましくは40℃±20℃である。
反応時間は、例えば2時間から24時間、好ましくは12時間から24時間である。
塩形成のための有機酸としては、シュウ酸、マロン酸、マレイン酸、クエン酸、フマル酸、テレフタル酸、乳酸、リンゴ酸、コハク酸、酒石酸、酢酸、トリフルオロ酢酸、グルコン酸、アスコルビン酸、メタンスルホン酸、ベンゼンスルホン酸、p-トルエンスルホン酸、10-カンファースルホン酸等が例示される。好ましい有機酸は、シュウ酸及びコハク酸である。
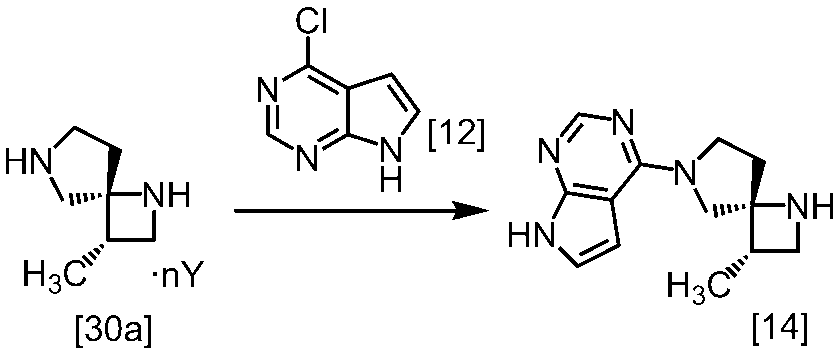
式[14]の化合物は、塩基存在下、式[30a]の化合物を4-クロロ-7H-ピロロ[2,3-d]ピリミジン(CPPY)[12]又はその塩と縮合させることにより製造することができる。式[30a]の化合物はまた、そのフリー体で用いてもよい。式[14]の化合物は、その塩として製造してもよく、フリー体から塩、塩からフリー体の形成は、公知の方法に従って行えばよい。
溶媒としては、例えばエタノール、2-プロパノール、1-プロパノール、2-プロパノール、tert-ブタノール、アセトニトリル、THF及びこれらと水との混合溶媒が例示される。好ましい溶媒は、tert-ブタノールと水との混合溶媒である。
塩基としては、例えばリン酸カリウム、炭酸カリウム、水酸化カリウム、トリエチルアミン及びN,N-ジイソプロピルエチルアミンが例示される。好ましい塩基は、リン酸カリウムのみ又はこれと水酸化カリウムとの組合せである。
反応物であるCPPY[12]は、例えば式[30a]の化合物に対して0.95から1.05当量、好ましくは1.0±0.02当量用いてもよい。
反応温度は、例えば室温から80℃、好ましくは40℃から50℃である。
反応時間は、例えば2時間から48時間、好ましくは12時間から24時間である。

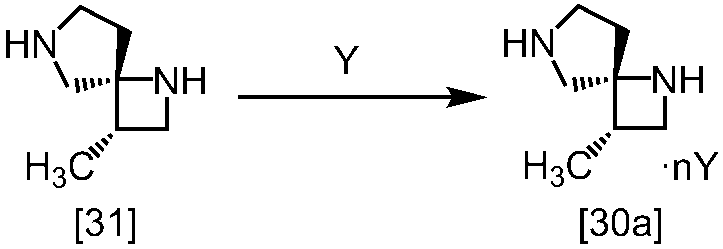
式[30a]の化合物は、製造方法21と同様に製造することができる。
本方法で用いる好ましい有機溶媒はコハク酸である。
(1)少ない工程数で化合物A(化合物[17])を製造することが可能な方法であること。
(2)大量合成には不向きな超低温反応及びオゾン酸化反応を使用しない製造方法であること。
(3)製造方法1において、ラクトン環の開裂が抑えられることによりBABL[2]が主生成物として得られ、化合物A(化合物[17])の収率向上につながること。
(4)BABL[2]の塩を利用した単離工程でビスベンジル付加体及び未反応のベンジルアミン等を除去することにより、製造方法3の工程2における副反応を低減することが可能であり、化合物A(化合物[17])の収率向上につながること。
(5)RR-AOBL[7]を経由することにより、特殊な設備や試薬等を用いることなく、立体選択的にβ-ラクタム環を構築することが可能な製造方法であること。
(6)RR-MDDO[9]の製造において、窒素源としてフタルイミドカリウム又はジホルミルアミドナトリウムを用いることで、RR-AOBL[7]の7位で環が開裂するため、RR-AOPE[8a]、化合物[16a]をそれぞれ得ることが可能であり、工程数の削減及び化合物A(化合物[17])の収率向上につながること。
(7)RR-MDDO[9]を利用した単離工程を経由することにより、化学純度及び光学純度の高い化合物A(化合物[17])の製造が可能であること。
(8)SR-MDBN[10]の塩を利用した単離工程を経由することにより、光学純度の高い化合物A(化合物[17])の製造が可能であること。
(9)反応混合液から直接単離することのできる、安定性の高い化合物[34a]を利用することにより、抽出及びシリカゲルカラムクロマトグラフィーによる単離精製工程を省くことが可能であり、さらに、化学純度の高い化合物A(化合物[17])の製造が可能であること。
項1: 式[13]

式[17]

(1)式[13]の化合物又はその塩からベンジルを除去することにより式[14]
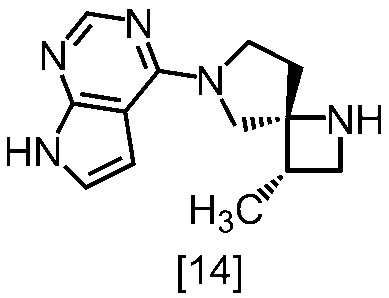
(2)式[14]の化合物又はその塩をシアノアセチル化することにより式[17]の化合物又はその塩を得る工程。


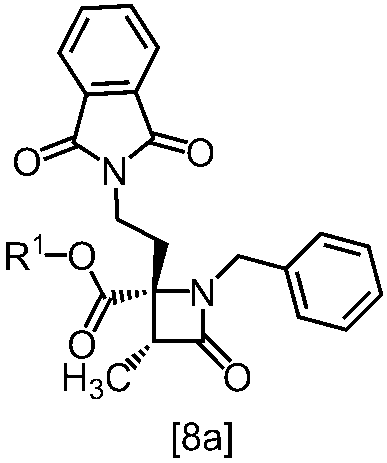
の化合物からフタロイルを除去することにより式[9]の化合物を得る工程をさらに含む、項6又は7に記載の方法。

の化合物からホルミルを除去することにより式[9]の化合物を得る工程をさらに含む、項6又は7に記載の方法。
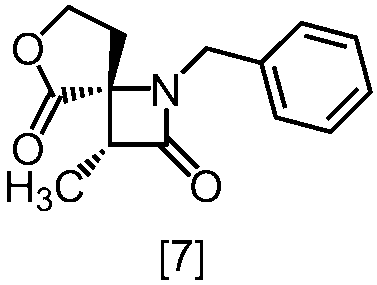
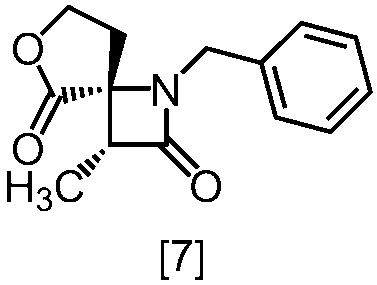



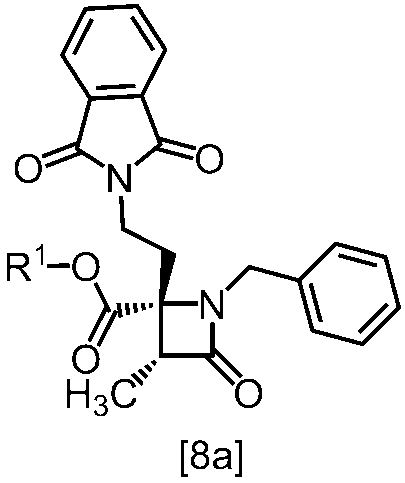
の化合物からフタロイルを除去することにより式[9]の化合物を得る工程をさらに含む、項17又は18に記載の方法。
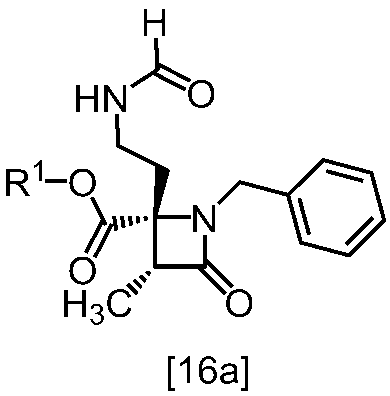
の化合物からホルミルを除去することにより式[9]の化合物を得る工程をさらに含む、項17又は18に記載の方法。



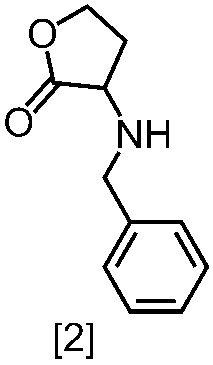
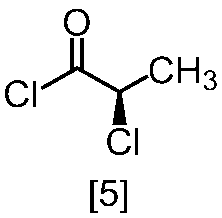


の化合物とベンジルアミンを塩基の存在下で反応させることにより式[2]の化合物又はその塩を得る工程をさらに含む、項28に記載の方法。
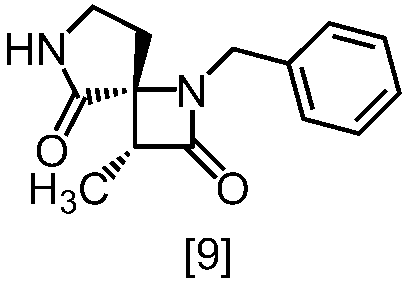

の化合物からフタロイルを除去することにより式[9]の化合物を得る工程を含む方法。

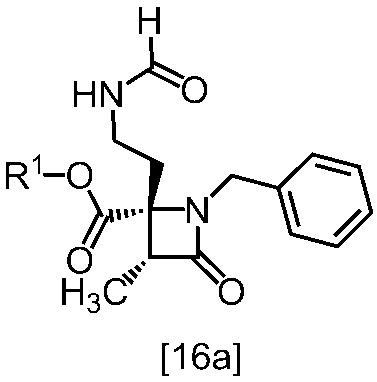
の化合物からホルミルを除去することにより式[9]の化合物を得る工程を含む方法。




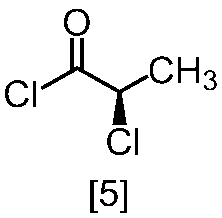


の化合物とベンジルアミンを塩基の存在下で反応させることにより式[2]の化合物又はその塩を得る工程をさらに含む、項40に記載の方法。
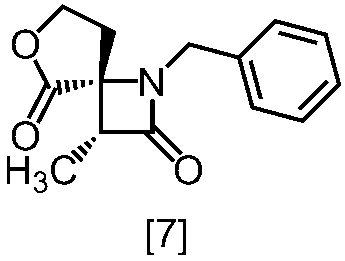




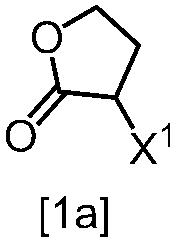
の化合物とベンジルアミンを塩基の存在下で反応させることにより式[2]の化合物又はその塩を得る工程をさらに含む、項48に記載の方法。

の化合物を環化させることにより式[9]の化合物を得る工程をさらに含む、項6又は17に記載の方法。

の化合物又はその塩と式[5]
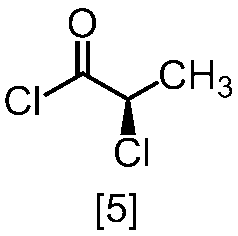
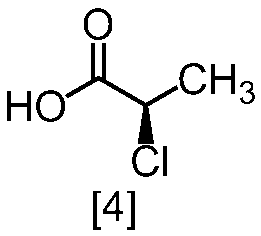

の化合物又はその塩とベンズアルデヒドを反応させることにより式[26a]の化合物又はその塩を得る工程をさらに含む、項52又は53に記載の方法。
の化合物又はその塩をエステル化することにより式[25a]の化合物又はその塩を得る工程をさらに含む、項54に記載の方法。



の化合物を環化させることにより式[9]の化合物を得る工程を含む方法。

の化合物又はその塩と式[5]
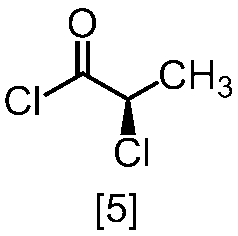
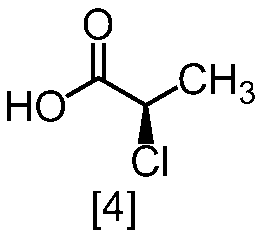
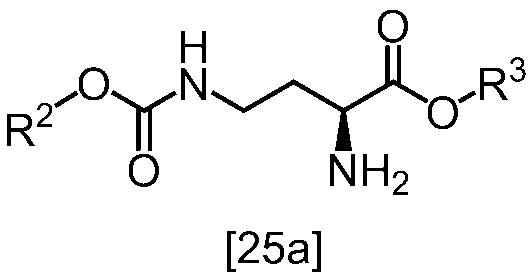
の化合物又はその塩とベンズアルデヒドを反応させることにより式[26a]の化合物又はその塩を得る工程をさらに含む、項59に記載の方法。
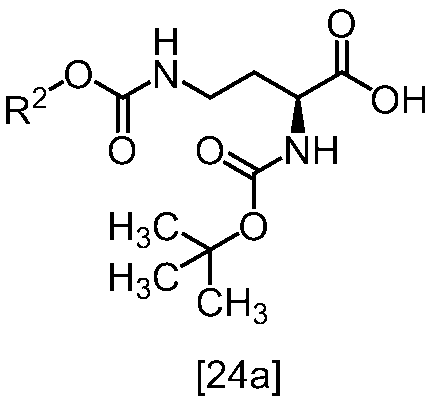
の化合物又はその塩をエステル化することにより式[25a]の化合物又はその塩を得る工程をさらに含む、項60に記載の方法。
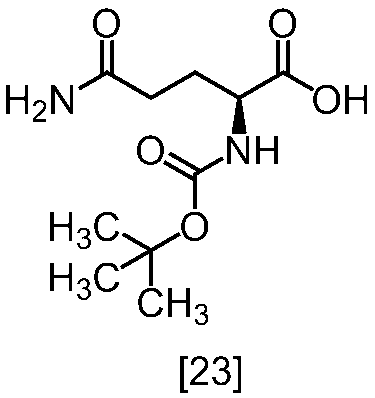
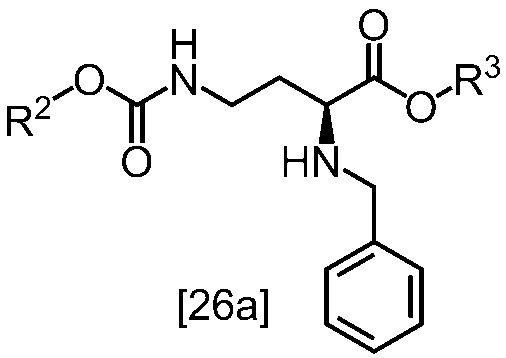
の化合物又はその塩を製造する方法であって、式[25a]

の化合物又はその塩とベンズアルデヒドを反応させることにより式[26a]の化合物又はその塩を得る工程を含む方法。

の化合物又はその塩をエステル化することにより式[25a]の化合物又はその塩を得る工程をさらに含む、項63に記載の方法。
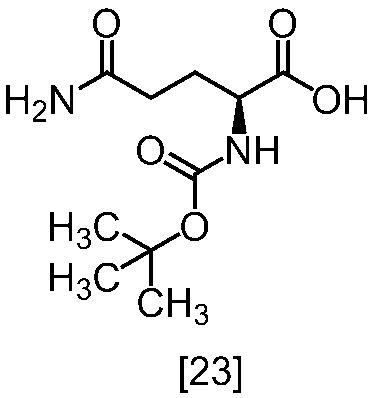


(1)式[31]の化合物又はその有機酸との塩と式[12]

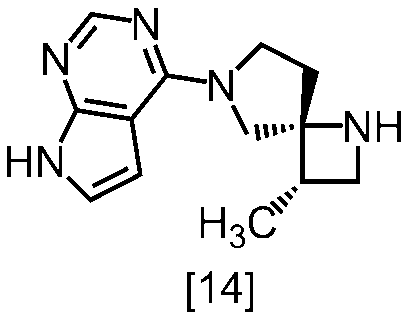
(2)式[14]の化合物又はその塩をシアノアセチル化することにより式[17]の化合物又はその塩を得る工程。
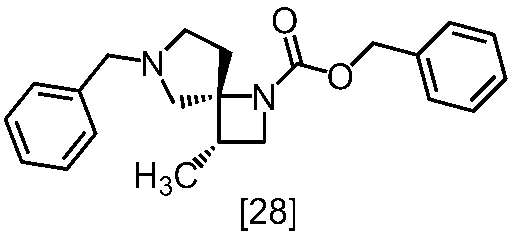
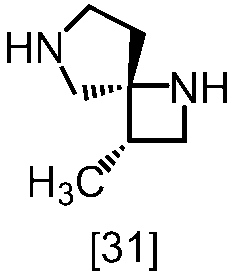




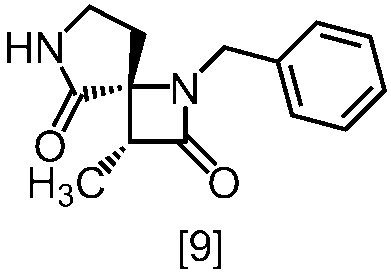
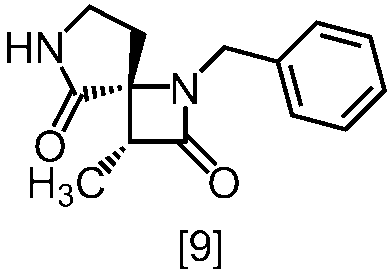
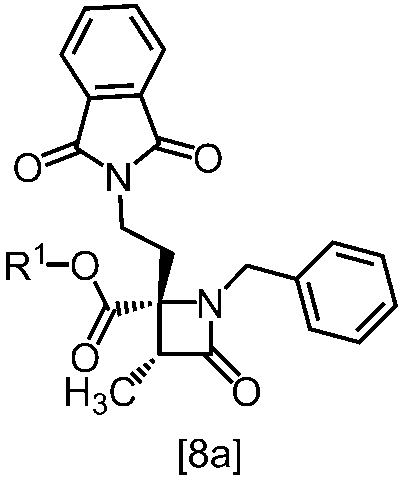
の化合物。
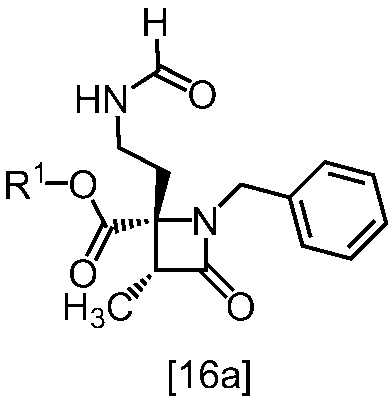
の化合物。
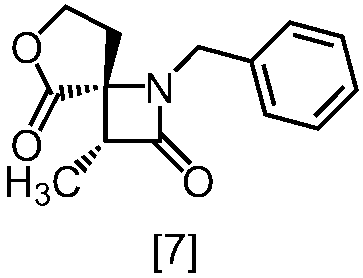
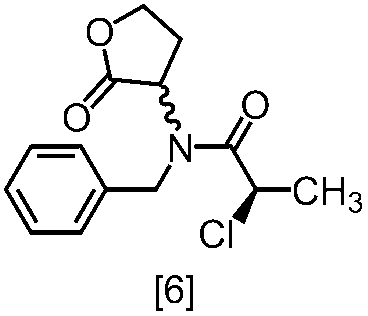
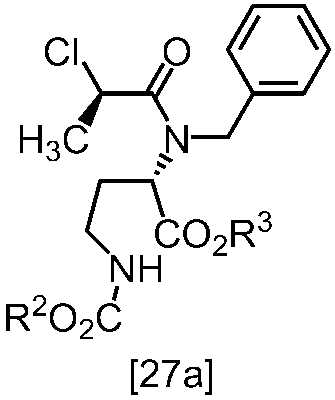
の化合物。

の化合物又はその塩。

の化合物又はその塩。
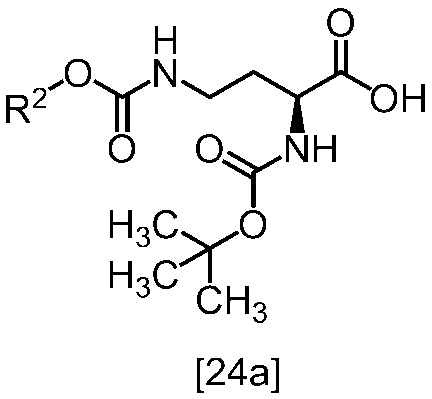
の化合物又はその塩。
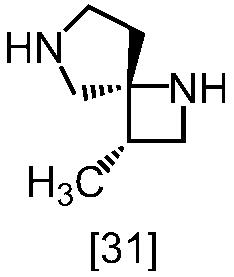

化合物[9]の製造(実施例4及び30)、化合物[11-3]の製造(実施例33)、化合物[20]の製造(実施例13)、化合物[29]の製造(実施例36)、化合物[30-2]の製造(実施例39)及び化合物A(化合物[17])の製造(実施例15及び41)並びに化合物A(化合物[17])の精製(実施例14、16及び20)の結晶化過程において、結晶化促進のために、種晶を用いた。この化合物の結晶は、種晶を用いなくても、実施例に記載の方法に準じた方法で得ることができる。
BBL:3-ブロモジヒドロフラン-2-オン
BABL:3-ベンジルアミノジヒドロフラン-2-オン
BABL-HC:3-ベンジルアミノジヒドロフラン-2-オン一塩酸塩
BHT:2,6-ジ-tert-ブチル-4-メチルフェノール
R-CPRA:(R)-2-クロロプロピオン酸
R-CPRC:(R)-2-クロロプロピオン酸クロリド
R-CPBL:(R)-N-ベンジル-2-クロロ-N-(2-オキソテトラヒドロフラン-3-イル)-プロピオン酸アミド)
RR-AOBL:(3R,4R)-1-ベンジル-3-メチル-6-オキサ-1-アザスピロ[3.4]オクタン-2,5-ジオン
RR-AOPE:(2R,3R)-1-ベンジル-2-[2-(1,3-ジオキソ-1,3-ジヒドロイソインドール-2-イル)-エチル]-3-メチル-4-オキソアゼチジン-2-カルボン酸エチルエステル
RR-AOPA:(2R,3R)-1-ベンジル-2-[2-(1,3-ジオキソ-1,3-ジヒドロイソインドール-2-イル)-エチル]-3-メチル-4-オキソアゼチジン-2-カルボン酸
RR-MDDO:(3R,4R)-1-ベンジル-3-メチル-1,6-ジアザスピロ[3.4]オクタン-2,5-ジオン)
SR-MDBN:(3S,4R)-1-ベンジル-3-メチル-1,6-ジアザスピロ[3.4]オクタン
SR-MDBN-DSU:(3S,4R)-1-ベンジル-3-メチル-1,6-ジアザスピロ[3.4]オクタン二コハク酸塩
SR-MDBP:4-[(3S,4R)-1-ベンジル-3-メチル-1,6-ジアザスピロ[3.4]オクト-6-イル]-7H-ピロロ[2,3-d]ピリミジン
SR-MDOP:4-[(3S,4R)-3-メチル-1,6-ジアザスピロ[3.4]-オクタン-6-イル]-7H-ピロロ[2,3-d]ピリミジン
化合物A(化合物[17]):3-[(3S,4R)-3-メチル-6-(7H-ピロロ[2,3-d]ピリミジン-4-イル)-1,6-ジアザスピロ[3.4]オクタン-1-イル]-3-オキソプロパンニトリル
CPPY:4-クロロ-7H-ピロロ[2,3-d]ピリミジン
DPCN:1-シアノアセチル-3,5-ジメチル-1H-ピラゾール
THF:テトラヒドロフラン
CPME:シクロペンチルメチルエーテル
DMF:ジメチルホルムアミド
DMSO:ジメチルスルホキシド
TMDS:1,1,3,3-テトラメチルジシロキサン
TMEDA:N,N,N’,N’-テトラメチルエチレンジアミン
TMSCl:クロロトリメチルシラン
LHMDS:リチウムヘキサメチルジシラジド
TBBA:ブロモ酢酸tert-ブチルエステル
Boc-Gln-OH:(tert-ブトキシカルボニル)-L-グルタミン
Boc-Dab(MeOCO)-OH:(S)-2-((tert-ブトキシカルボニル)アミノ)-4-((メトキシカルボニル)アミノ)ブタン酸
SR-ZMDB:(3S,4R)-6-ベンジル-3-メチル-1,6-ジアザスピロ[3.4]オクタン-1-カルボン酸ベンジル
SR-ZMDB-OX:(3S,4R)-6-ベンジル-3-メチル-1,6-ジアザスピロ[3.4]オクタン-1-カルボン酸ベンジルシュウ酸塩
S-BAPO:(S)-2-(ベンジルアミノ)プロパン-1-オール
S-BBMO:(S)-N-ベンジル-N-(1-ヒドロキシプロパン-2-イル)グリシン酸tert-ブチル
R-BCAB:(R)-N-ベンジル-N-(2-クロロプロピル)グリシン酸tert-ブチル
S-MABB:(3S)-1-ベンジル-3-メチルアゼチジン-2-カルボン酸tert-ブチル
S-MABB-HC:(3S)-1-ベンジル-3-メチルアゼチジン-2-カルボン酸tert-ブチル塩酸塩
S-MACB-HC:(3S)-3-メチルアゼチジン-2-カルボン酸tert-ブチル塩酸塩
S-ZMAB:2-(tert-ブチル)(3S)-3-メチルアゼチジン-1,2-ジカルボン酸1-ベンジル
RS-ZMBB:2-(tert-ブチル)(2R,3S)-2-(2-(tert-ブトキシ)-2-オキソエチル)-3-メチルアゼチジン-1,2-ジカルボン酸1-ベンジル
RS-ZMAA:(2R,3S)-1-((ベンジルオキシ)カルボニル)-2-(カルボキシメチル)-3-メチルアゼチジン-2-カルボン酸
RS-ZMAA-DN・2H2O:(2R,3S)-1-((ベンジルオキシ)カルボニル)-2-(カルボキシメチル)-3-メチルアゼチジン-2-カルボン酸二ナトリウム塩二水和物
RS-ZMOO:(2R,3S)-2-(2-ヒドロキシエチル)-2-(ヒドロキシメチル)-3-メチルアゼチジン-1-カルボン酸ベンジル
RS-ZMSS:(2R,3S)-3-メチル-2-(2-((メチルスルホニル)オキシ)エチル)-2-(((メチルスルホニル)オキシ)メチル)アゼチジン-1-カルボン酸ベンジル
実施例中の記号は次のような意味である。
s:シングレット(singlet)
d:ダブレット(doublet)
t:トリプレット(triplet)
q:カルテット(quartet)
dd:ダブルダブレット(double doublet)
dq:ダブルカルテット(double quartet)
ddd:ダブルダブルダブレット(double double doublet)
brs:ブロードシングレット(broad singlet)
m:マルチプレット(multiplet)
J:カップリング定数(coupling constant)
測定機器:X’Pert Pro(スペクトリス社)
測定条件:対陰極 :銅
X線管球の管電流と管電圧 :45kV、40mA
試料の回転速度 :毎回1秒
入射側のソーラースリット :0.02rad
入射側の縦発散スリット :15mm
入射側の発散スリット :自動、照射幅15mm
入射側の散乱スリット :1°
受光側のフィルタ :ニッケルフィルタ
受光側のソーラースリット :0.02rad
受光側の発散スリット :自動、照射幅15mm
検出器 :X’Celerator
検出器のモード :スキャニング
検出器の有効幅 :2.122°
走査軸 :ゴニオ
走査モード :連続
走査範囲 :3°から60°
単位ステップあたりの時間 :10秒
測定機器:イオンクロマトグラフLC-20系(島津製作所)
測定条件:電気伝導度検出器SHIMADZU CDD-10A VP
アニオン分析用カラムSHIMADZU SHIM-PAC IC-A3
カチオン分析用カラムSHIMADZU SHIM-PAC IC-C1
測定機器:水分含量測定用電量滴定装置CA-06(三菱化学株式会社)
測定条件:サンプル量:約20mg
試薬:陽極液Aquamicron AX(APIコーポレーション)
陰極液Aquamicron CXU(APIコーポレーション)
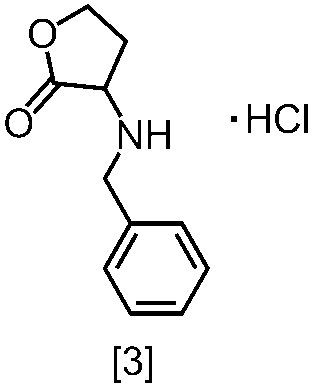

窒素雰囲気下、リン酸三カリウム(1466.7g,6.9mol)、アセトニトリル(3.8L)、ベンジルアミン(246.8g,2.30mol)及びBBL[1](380g,2.30mol)を室温で順次添加した。この反応混合液を40℃から45℃にて21時間撹拌後、室温に冷却した。反応混合液中の不溶物をろ去し、アセトニトリル(760mL)で洗浄した。ろ液と洗液を合わせて減圧下にて濃縮した。この濃縮残渣にトルエン(3.8L)、20%食塩水(1.14L)、酢酸(20.75g)を加え撹拌した後、有機層を分取した。得られた有機層を20%食塩水(760mL)と5%重曹水(380mL)の混合液で洗浄後、減圧下にて有機層の溶媒を留去した。濃縮残渣にトルエン(380mL)を加えて、不溶物をろ去し、トルエン(380mL)で洗浄した。ろ液と洗液を合わせて減圧下にて濃縮した。濃縮残渣に酢酸エチル(1.52L)を加え濃縮する操作を2回繰り返した後、酢酸エチル(760mL)を加え、BABL[2]の酢酸エチル溶液(440g,2.30mol相当)を得た。得られたBABL[2]の酢酸エチル溶液を収率100%として次工程に用いた。
同じ方法で合成した粗BABL[2]を濃縮乾固し、MSを測定した。
MS: m/z = 192 [M+H]+
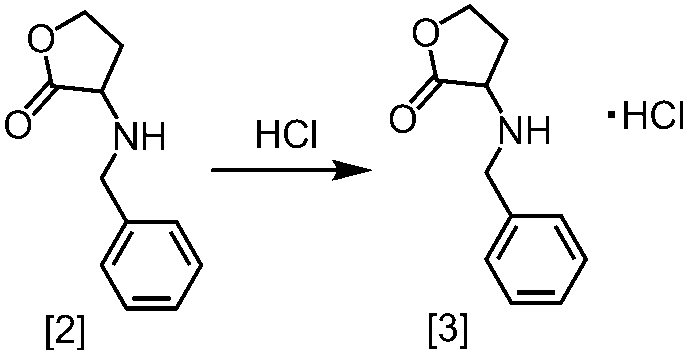
窒素雰囲気下、BABL[2]の酢酸エチル溶液(440g,2.30mol相当)に室温にてメタノール(380mL)を添加した。この混合液に4N塩酸-酢酸エチル溶液(575mL,2.30mol)を0℃にて加え、反応混合液を0℃にて1.5時間撹拌した。析出した固体をろ取し、得られた固体を酢酸エチル(760mL)で2回洗浄した。得られた湿固体を減圧下にて乾燥することでBABL-HC[3](385.1g,1.69mol)を収率73.4%で得た。
同じ方法で合成したBABL-HC[3]について、NMR及び融点を測定し、元素分析を行った。
1H-NMR (DMSO-d6) δ: 10.11 (1H, brs), 7.55-7.41 (5H, m), 4.47 (1H, t, J = 8.8 Hz), 4.36-4.22 (4H, m), 3.31 (1H, brs), 2.61-2.54 (1H, m), 2.49-2.41 (1H, m).
融点:206℃から208℃
元素分析:C 58.1wt%, H 6.2wt%, N 6.1wt% (理論値 C 58.0wt%, H 6.2wt%, N 6.2wt%)
同じ方法で合成したBABL-HC[3]について、粉末X線回折法で回折角2θと回折強度を測定した。得られたスペクトルを図1に示す。
図1の各ピークは以下の表のとおりである。

工程1

非特許文献1に記載された方法に従い合成したアミノラクトン臭素酸塩[15](1.0g,5.49mmol)、DMF(15mL)、酢酸(0.1mL)及びベンズアルデヒド(0.62mL,6.04mmol)を順次添加し、0℃に冷却した。この反応混合液に水素化トリアセトキシホウ素ナトリウム(1.39g,6.59mmol)を加え、室温で1時間撹拌した。反応混合液に1M塩酸を加え、トルエンで洗浄した。得られた水層に飽和重曹水を加え、生成物を酢酸エチルで3回抽出した。有機層を合わせて、飽和食塩水で洗浄後、減圧下にて濃縮し、BABL[2-2](976mg,5.11mmol相当)を得た。
同じ方法で合成した粗BABL[2-2]を濃縮乾固し、MSを測定した。
MS: m/z = 192 [M+H]+
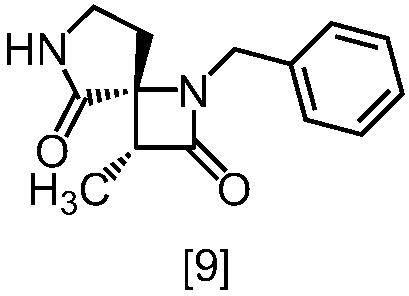

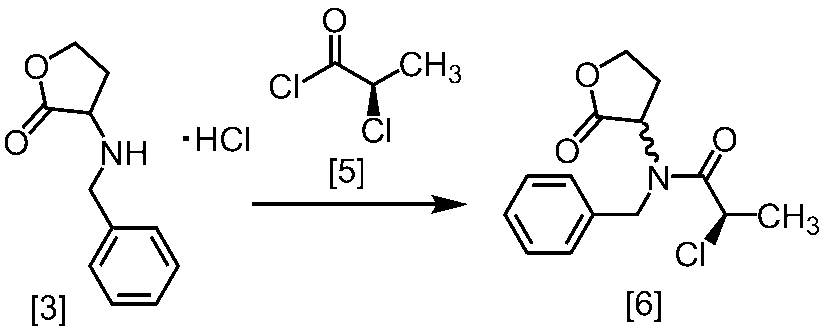
窒素雰囲気下、DMF(260mL)に0℃にて塩化チオニル(107.8mL,1.48mol)を加え、同温度で30分間撹拌した。この溶液に0℃にてR-CPRA[4](148.7g,1.37mol)のトルエン(260mL)溶液を滴下し、同温度で1時間撹拌し、R-CPRC[5](1.37mol相当)のトルエン溶液を得た。
窒素雰囲気下、BABL-HC[3](260g,1.14mol)、酢酸エチル(2L)及び2,6-ルチジン(489.4g,4.57mol)を順次添加し、室温で20分間撹拌した。この混合液を0℃に冷却し、先に得たR-CPRC[5](1.37mol相当)のトルエン溶液を5℃以下にて滴下し、同温度にて2時間撹拌した。この反応混合液に1M塩酸(1.3L)を加えて撹拌した後、有機層を分取し、次いで5%重曹水(1.3L)で2回、水(1.3L)で順次洗浄した。得られた有機層を減圧下で濃縮し、濃縮残渣にトルエン(780mL)を加え再度減圧下濃縮した。この操作を再度行い、濃縮残渣にDMSO(750mL)を加え、粗R-CPBL[6]のDMSO溶液(1187.94g,1.14mol相当)を得た。得られたR-CPBL[6]を収率100%として次工程に用いた。
同じ方法で合成した粗R-CPBL[6]のトルエン溶液の一部を濃縮乾固し、NMR、MS及び融点を測定し、元素分析を行った。
1H-NMR (DMSO-d6) δ: (3:2のジアステレオマー混合物)5.03と4.99 (1H, q, J = 6.5 Hz, 塩素の付け根のプロトン), 1.51と1.47 (3H, d, J = 6.5 Hz, メチルのプロトン).
MS: m/z = 282 [M+H]+
融点:101℃から104℃
元素分析:C 59.8wt%, H 5.7wt%, N 4.9wt% (理論値 C 59.7wt%, H 5.7wt%, N 5.0wt%)
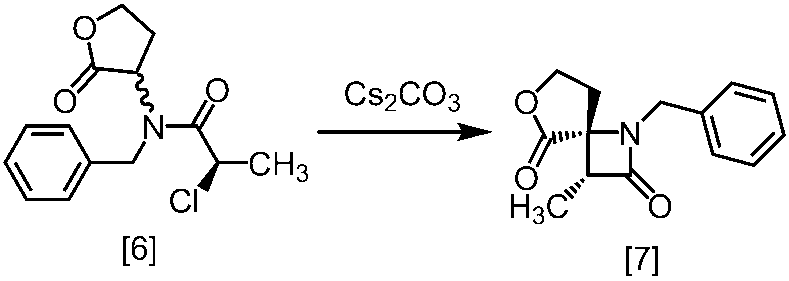
窒素雰囲気下、先に得た粗R-CPBL[6]のDMSO溶液(1161.8g,1.12mol相当)に、室温にてDMSO(250mL)及び炭酸セシウム(728.1g,2.23mol)を順次添加し、室温で一晩撹拌した。20℃以下にした2M塩酸(1.78L)にこの反応混合液を滴下し、酢酸エチル(2.5L)で生成物を抽出した。得られた有機層を5%重曹水(1.3L)で洗浄し、次いで20%食塩水(1.3L)で2回洗浄した後、減圧下で濃縮し、粗RR-AOBL[7](353.9g,1.12mol相当、ジアステレオマー比97:3)を得た。得られた粗RR-AOBL[7]を収率100%として次工程に用いた。
同じ方法で合成した粗RR-AOBL[7]を濃縮乾固し、NMRとMSを測定した。
1H-NMR (CDCl3) δ: 7.37-7.27 (5H, m), 4.86 (1H, d, J = 15.3 Hz), 4.21 (1H, ddd, J = 9.9, 5.2, 4.0 Hz), 4.13-4.06 (1H, m), 4.02 (1H, d, J = 15.3 Hz), 3.36 (1H, q, J = 7.5 Hz), 2.13-2.10 (2H, m), 1.31 (3H, d, J = 7.3 Hz).
MS: m/z = 246 [M+H]+
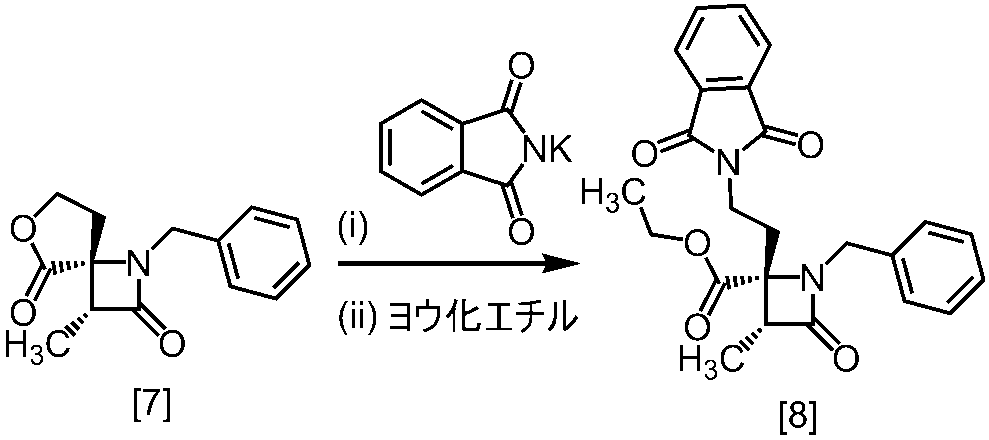
窒素雰囲気下、室温にて粗RR-AOBL[7](220.15g,0.97mol相当)、DMF(1.5L)及びフタルイミドカリウム(232.81g,1.26mol)を順次添加し、80℃から100℃で一晩撹拌した。この反応混合液を50℃付近まで冷却した後、ヨウ化エチル(226.21g,1.45mol)を滴下し、40℃から50℃で4時間撹拌した。反応混合液を0℃付近まで冷却した後、20%食塩水(1.1L)を加え、トルエン(1.1L)で生成物を抽出した。得られた有機層を20%食塩水(1.1L)、水(1.1L)で順次洗浄後、減圧下で濃縮した。濃縮残渣に2-ブタノール(1.1L)を加え、減圧下濃縮した。この操作を再度繰り返し、粗RR-AOPE[8]の2-ブタノール溶液(809.67g,0.97mol相当)を得た。得られたRR-AOPE[8]の2-ブタノール溶液を収率100%として次工程に用いた。
同じ方法で合成した粗RR-AOPE[8]を濃縮乾固し、NMRとMSを測定した。
1H-NMR (CDCl3) δ: 7.83-7.80 (2H, m), 7.73-7.69 (2H, m), 7.37-7.32 (4H, m), 7.29-7.25 (1H, m), 4.79 (1H, d, J = 15.7 Hz), 4.40 (1H, d, J = 15.7 Hz), 4.15-4.06 (2H, m), 3.68-3.61 (1H, m), 3.52-3.44 (1H, m), 3.37 (1H, q, J = 7.5 Hz), 2.27-2.12 (2H, m), 1.26-1.22 (6H, m).
MS: m/z = 421 [M+H]+
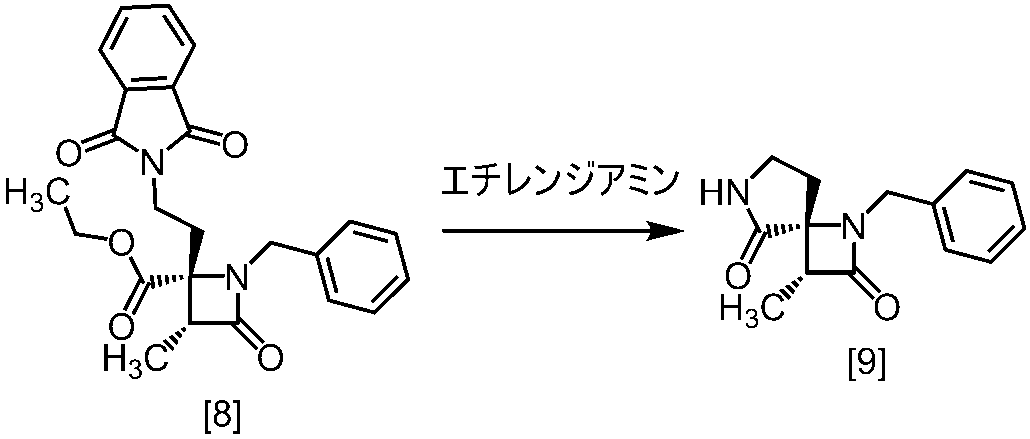
窒素雰囲気下、室温にてRR-AOPE[8]の2-ブタノール溶液(36.78g,43.9mmol相当)にエチレンジアミン(10.56g,175.6mmol)を添加し、80℃から90℃で4時間撹拌した。この反応混合液を50℃付近まで冷却した後、THF(120mL)を加え、40℃から50℃で1時間、室温で一晩撹拌した。不溶物をろ去した後、ろ液に25%硫酸水素カリウム水溶液(170mL)を加え、分液した。得られた有機層を7.5%重曹水と飽和食塩水の混合液(1/4,50mL)で洗浄し、減圧下濃縮した。濃縮残渣に酢酸エチル(200mL)及びカルボラフィン20(0.5g,日本エンバイロケミカルズ製)を加え、室温で30分間撹拌した。この混合液中の不溶物をろ去後、ろ液を濃縮し、濃縮残渣にCPME(50mL)を加えて濃縮した(粗RR-MDDO[9])。得られた濃縮残渣にCPME(40mL)を加え、50℃から60℃に加熱した。この溶液にジイソプロピルエーテル(40mL)を滴下し、同温度で1時間、室温に冷却して2時間撹拌した。析出した固体をろ取し、得られた固体をCPME/ジイソプロピルエーテルの混液(1:1,20mL)で洗浄後、減圧下50℃にて乾燥することで、RR-MDDO[9](6.70g,27.4mmol)をBABL-HC[3]より収率62.5%で得た。
同じ方法で合成したRR-MDDO[9]について、NMR、MS及び融点を測定し、元素分析を行った。
1H-NMR (CDCl3) δ: 7.33-7.26 (5H, m), 5.92 (1H, brs), 4.85 (1H, d, J = 15.5 Hz), 3.99 (1H, d, J = 15.5 Hz), 3.27-3.18 (2H,m), 3.16-3.10 (1H, m), 2.07-1.99 (2H, m), 1.28 (3H, d, J = 7.6 Hz).
MS: m/z = 245 [M+H]+
融点:125℃から127℃
元素分析:C 68.9wt%, H 6.6wt%, N 11.4wt% (理論値 C 68.8wt%, H 6.6wt%, N 11.5wt%)
同じ方法で合成したRR-MDDO[9]について、粉末X線回折法で回折角2θと回折強度を測定した。得られたスペクトルを図2に示す。
図2の各ピークは以下の表のとおりである。
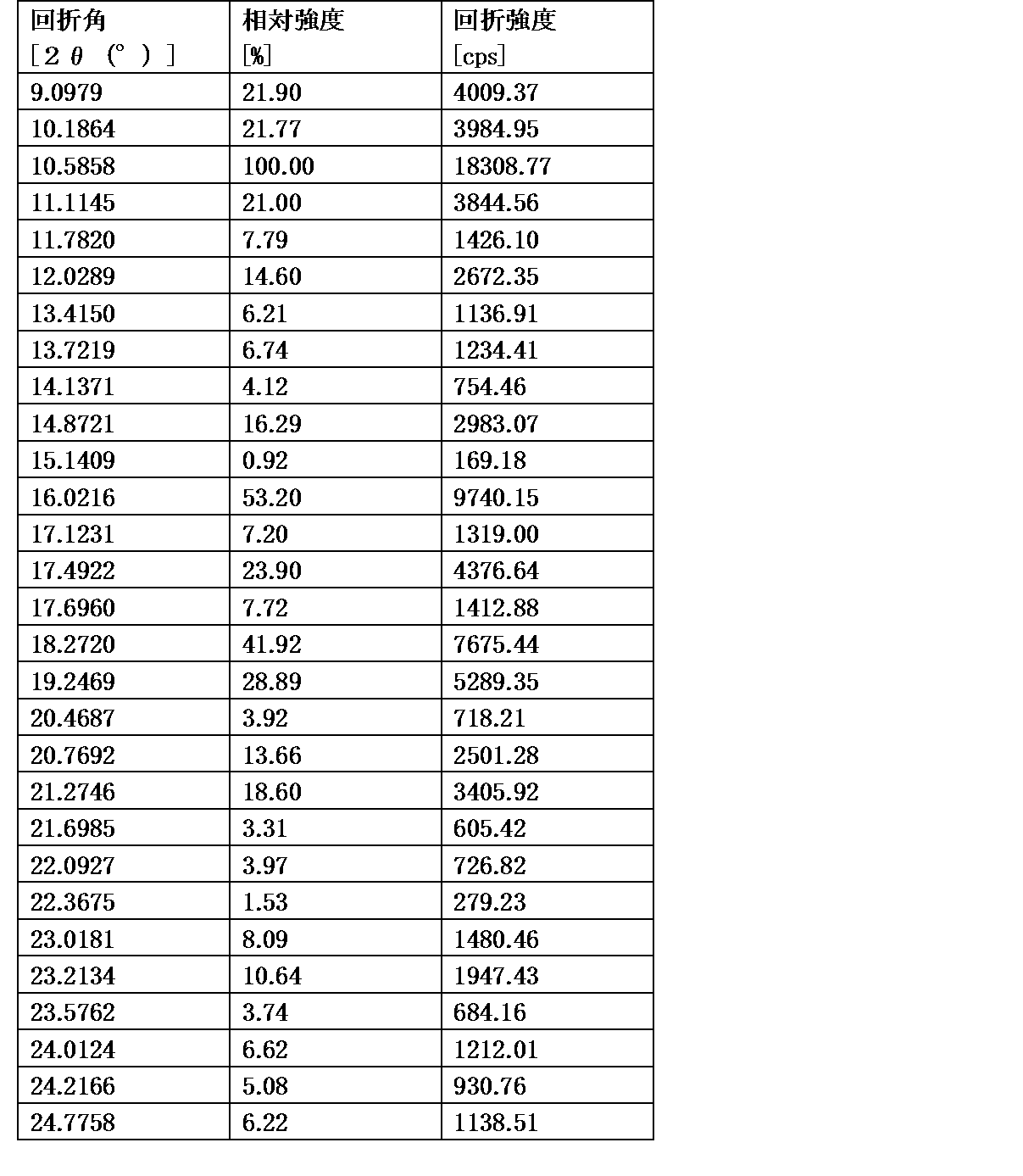
HPLCの測定機器及び条件を以下に示す。
測定機器:Nexeraシステム(島津製作所)
測定条件:
カラム:CHIRAL PAK IF-3:3um,250mm×4.6mm(ダイセル)
カラム温度:40℃
流速:1.0mL/min.
分析時間:35min.
検出波長:UV(220nm)
移動相:ヘキサン/2-プロパノール=80/20
上記HPLC測定条件におけるRR-MDDO[9]の保持時間は、は約14.6分であった。立体異性体の保持時間は、SS体が約10.9分、RS体が約16.5分、SR体が約18.6分であった。

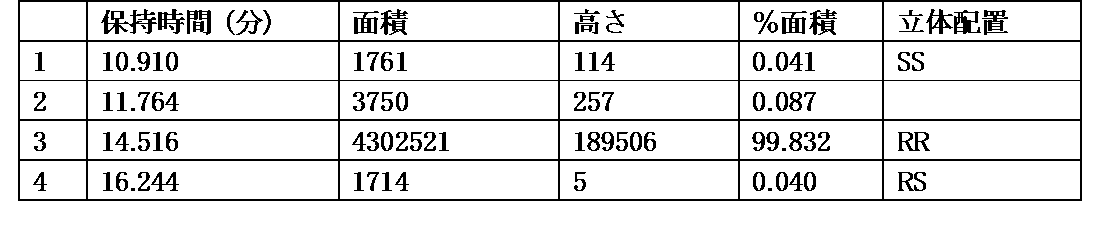
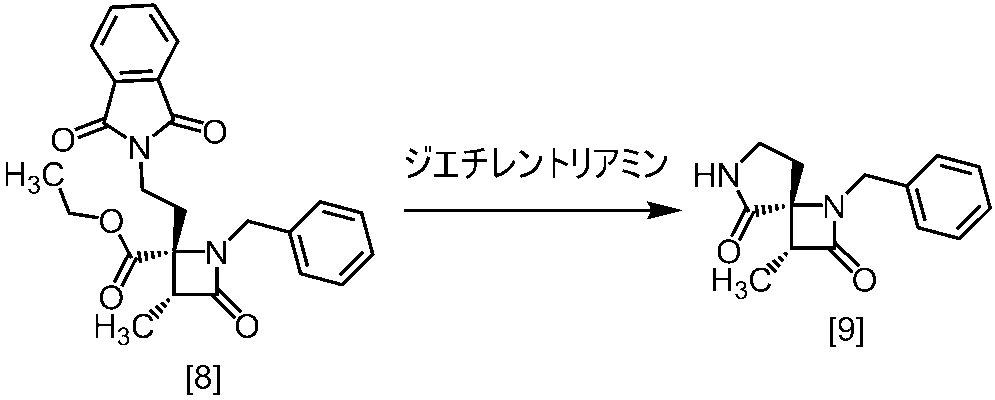
得られたRR-MDDO[9]について、NMRとMSを測定した。
1H-NMR (DMSO-D6) δ: 8.10 (1H, brs), 7.35-7.24 (5H, m), 4.55 (1H, d, J = 16.0 Hz), 3.95 (1H, d, J = 16.0 Hz), 3.35 (1H, q, J = 7.6 Hz), 3.15-3.05 (2H, m), 2.17-2.12 (1H, m), 2.07-1.99 (1H, m), 1.07 (3H, d, J = 7.4 Hz).
MS: m/z = 245 [M+H]+
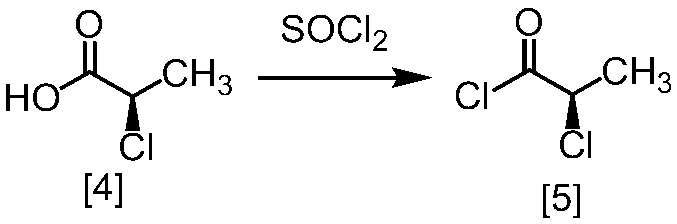

窒素雰囲気下、BABL[2](1.16g,6.06mmol)、酢酸エチル(9mL)及び2,6-ルチジン(1.95g,18.18mmol)を順次添加し、室温で20分間撹拌した。この混合液を0℃に冷却し、先に得たR-CPRC[5](923mg,7.88mmol相当)のトルエン溶液を5℃以下にて滴下し、同温度にて2時間撹拌した。この反応混合液に1M塩酸(5mL)を加えて撹拌した後、有機層を分取し、次いで7.5%重曹水(6mL)で2回、水(6mL)で順次洗浄した。得られた有機層にカルボラフィン20(0.2g,日本エンバイロケミカルズ製)を加え、室温にて一晩撹拌した。不溶物をろ去し、酢酸エチルで洗浄後、ろ液と洗液を合わせて減圧下で濃縮した。得られた濃縮残渣をトルエン(6mL)及びヘプタン(6mL)で結晶化することにより、R-CPBL[6](1.19g,4.22mmol)を収率98.2%で得た。
同じ方法で合成したR-CPBL[6]について、NMR、MS及び融点を測定し、元素分析を行った。
1H-NMR (DMSO-d6) δ: (3:2のジアステレオマー混合物)5.03と4.99 (1H, q, J = 6.5 Hz, 塩素の付け根のプロトン), 1.51と1.47 (3H, d, J = 6.5 Hz, メチルのプロトン).
MS: m/z = 282 [M+H]+
融点:101℃から104℃
元素分析:C 59.8wt%, H 5.7wt%, N 4.9wt% (理論値 C 59.7wt%, H 5.7wt%, N 5.0wt%)

窒素雰囲気下、室温にてR-CPBL[6](5.58g,24.5mmol相当)、DMSO(22mL)及びリン酸三カリウム(15.6g,73.5mmol)を順次添加し、30℃から40℃で24時間撹拌した。3M塩酸(33.5mL)に、室温まで冷却した反応混合液を滴下し、酢酸エチルで生成物を抽出した。得られた有機層を7.5%重曹水で1回、20%食塩水で2回、順次洗浄した後、減圧下で濃縮した。濃縮残渣にトルエン(400mL)を加え濃縮し、RR-AOBL[7]のトルエン溶液(6.44g,24.5mmol相当)を得た。得られたRR-AOBL[7]のトルエン溶液を収率100%として次工程に用いた。
同じ方法で合成した粗RR-AOBL[7]を濃縮乾固し、NMRとMSを測定した。
1H-NMR (CDCl3) δ: 7.37-7.27 (5H, m), 4.86 (1H, d, J = 15.3 Hz), 4.21 (1H, ddd, J = 9.9, 5.2, 4.0 Hz), 4.13-4.06 (1H, m), 4.02 (1H, d, J = 15.3 Hz), 3.36 (1H, q, J = 7.5 Hz), 2.13-2.10 (2H, m), 1.31 (3H, d, J = 7.3 Hz).
MS: m/z = 246 [M+H]+
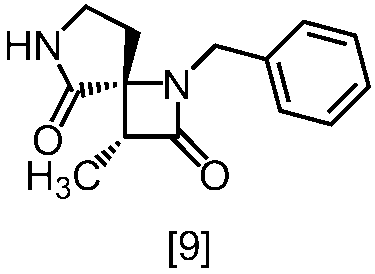
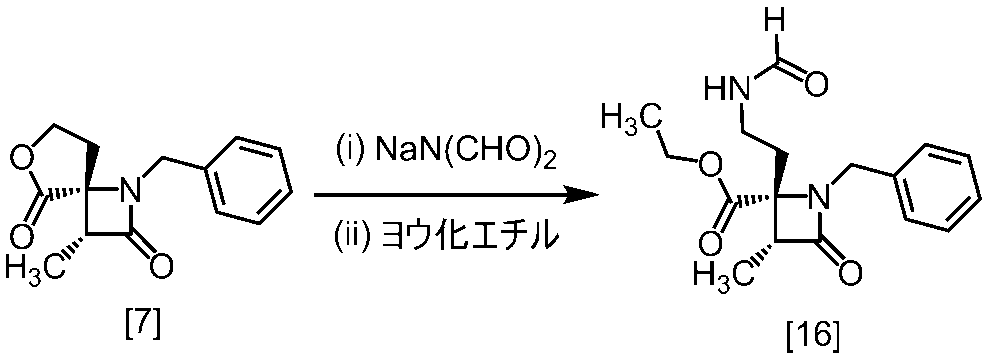
窒素雰囲気下、室温にてRR-AOBL[7](3.0g,12.2mmol)、DMSO(20mL)、及びジホルミルアミドナトリウム(3.48g,36.6mmol)を順次添加し、100℃で18時間撹拌した。この反応混合液を45℃付近まで冷却した後、ヨウ化エチル(3.0mL,37.8mmol)を滴下し、45℃で5時間撹拌した。反応混合液を室温まで冷却した後、5%炭酸カリウム水溶液を加え、生成物をトルエンで抽出した。得られた有機層を5%炭酸カリウム水溶液、20%食塩水で順次洗浄後、減圧下で濃縮した。得られた粗化合物[16]を収率100%として次工程に用いた。
同じ方法で合成した粗化合物[16]を濃縮乾固し、NMRとMSを測定した。
1H-NMR (CDCl3) δ: 8.82 (1H, s), 7.98 (1H, s), 7.37-7.14 (5H, m), 4.85 (1H, d, J = 15.7 Hz), 4.23 (2H, q, J = 7.1 Hz), 4.20 (1H, d, J =15.7 Hz), 3.71 (1H, q, J = 7.1 Hz), 3.27-3.17 (1H, m), 3.11-3.02 (1H, m), 2.06-1.98 (1H, m), 1.95-1.85 (1H, m), 1.30 (3H, t, J = 7.1 Hz), 1.21 (3H, d, J = 7.1 Hz).
MS: m/z = 319 [M+H]+
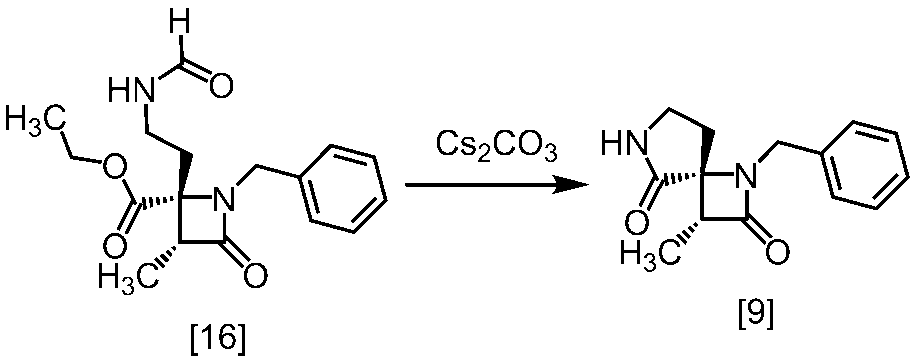
窒素雰囲気下、室温にて粗化合物[16](3.05mmol相当)、アセトニトリル(5mL)、炭酸セシウム(1.49g,4.58mmol)を添加し、室温で4時間撹拌した。この反応混合液に25%硫酸水素カリウム水溶液(5mL)を加え、生成物をクロロホルム(5mL)で3回抽出した。得られた有機層を合わせて、飽和重曹水(5mL)で洗浄後、減圧下濃縮した。濃縮残渣を薄層シリカゲルクロマトグラフィー(展開溶媒:酢酸エチル)にて精製し、RR-MDDO[9](638mg,2.61mmol)を収率85.6%で得た。
同じ方法で合成したRR-MDDO[9]について、NMRとMSを測定した。
1H-NMR (CDCl3) δ: 7.33-7.26 (5H, m), 5.92 (1H, brs), 4.85 (1H, d, J = 15.5 Hz), 3.99 (1H, d, J = 15.5 Hz), 3.27-3.18 (2H,m), 3.16-3.10 (1H, m), 2.07-1.99 (2H, m), 1.28 (3H, d, J = 7.6 Hz).
MS: m/z = 245 [M+H]+
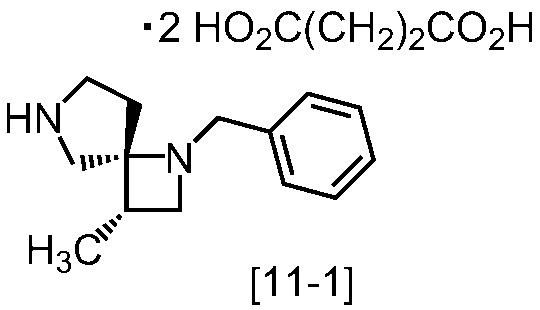
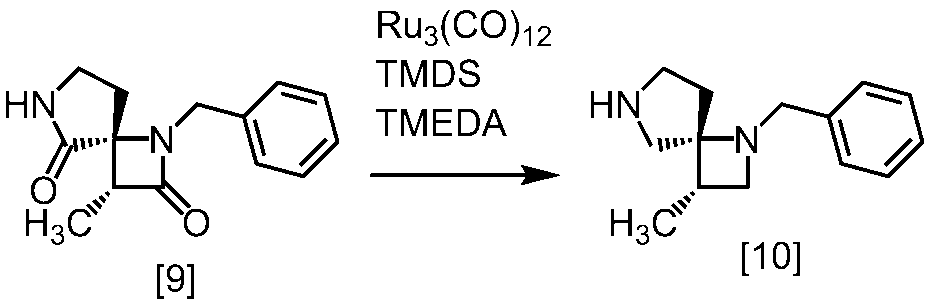
窒素雰囲気下、RR-MDDO[9](1.0g,4.09mmol)、トルエン(10mL)、ドデカカルボニル3ルテニウム(261mg,0.41mmol)、TMDS(5.49g,40.9mmol)及びTMEDA(0.061mL,0.41mmol)を順次加え、70℃で40時間撹拌した。この反応混合液を室温まで冷却し減圧下濃縮した後、得られた濃縮残渣に2M塩酸(10mL)及びTHF(10mL)を加え分液した。得られた水層にCPME(10mL)及び25%水酸化ナトリウム水溶液(5mL)を加え、分液した。得られた有機層を飽和食塩水(5mL)で洗浄し、減圧下濃縮した。この濃縮残渣に2-プロパノール(10mL)を加え、粗SR-MDBN[10]の2-プロパノール溶液(4.09mmol相当)を得た。得られたSR-MDBN[10]の2-プロパノール溶液を収率100%として次工程に用いた。
同じ方法で合成した粗SR-MDBN[10]を濃縮乾固し、NMRとMSを測定した。
1H-NMR (CDCl3) δ: 7.34-7.20 (5H, m), 3.62 (1H, d, J = 12.9 Hz), 3.59 (1H, d, J = 12.9 Hz), 3.21 (1H, dd, J = 7.5, 6.6 Hz), 2.99 (1H, d, J = 12.1 Hz), 2.95 (1H, d, J = 12.1 Hz), 2.84 (2H, t, J = 7.3 Hz), 2.68 (1H, t, J = 5.8 Hz), 2.43-2.35 (1H, m), 2.22-2.15 (1H, m), 1.81-1.74 (2H, m), 1.13 (3H, d, J = 6.9 Hz).
MS: m/z = 217 [M+H]+

窒素雰囲気下、コハク酸(966mg,8.18mmol)及び2-プロパノール(5mL)を添加し、70℃に加熱した。この懸濁液に粗SR-MDBN[10]の2-プロパノール溶液(4.09mmol相当)を70℃にて滴下した後、室温まで冷却し、8時間撹拌した。析出した固体をろ取し、得られた固体を2-プロパノール(3mL)で2回洗浄後、減圧下40℃にて乾燥することで、SR-MDBN-DSU[11-1](1.25g,2.77mmol)を収率67.7%で得た。
同じ方法で合成したSR-MDBN-DSU[11-1]について、NMR及び融点を測定し、元素分析を行った。
1H-NMR (D2O) δ: 7.43-7.38 (5H, m), 4.30 (1H, d, J = 12.1 Hz), 4.24 (1H, d, J = 12.1 Hz), 3.96 (1H, dd, J = 10.1, 8.9 Hz), 3.85 (1H, d, J = 14.5 Hz), 3.77 (1H, d, J = 14.5 Hz), 3.45-3.33 (3H, m), 2.99-2.91 (1H, m), 2.89-2.81 (1H, m), 2.53-2.47 (1H, m), 1.17 (3H, d, J = 7.3 Hz).
融点:126℃から128℃
元素分析:C 58.4wt%, H 7.1wt%, N 6.1wt% (理論値 C 58.4wt%, H 7.1wt%, N 6.2wt%)
同じ方法で合成したSR-MDBN-DSU[11-1]について、粉末X線回折法で回折角2θと回折強度を測定した。得られたスペクトルを図5に示す。
図5の各ピークは以下の表のとおりである。

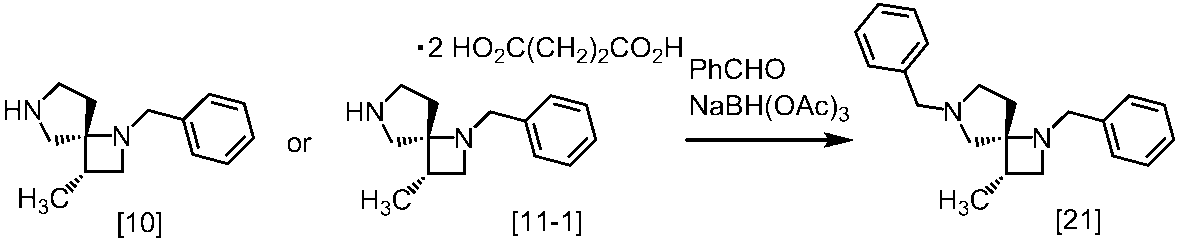
測定機器:Nexeraシステム(島津製作所)
測定条件:
カラム:CHIRALCEL OJ-RH:3um,4.6mm×150mm(ダイセル)
カラム温度:25℃
流速:0.8mL/min.
分析時間:15min.
検出波長:UV(210nm)
移動相:メタノール/ジエチルアミン=100/0.1(v/v)
上記HPLC測定条件における化合物[21]の保持時間は、約5.2分であった。立体異性体の保持時間は、RR体が約4.4分、SS体が約7.2分、RS体が8.6分であった。


SR-MDBN(446mg,2.06mmol)の2-プロパノール(2.9mL)溶液に、室温にてコハク酸(243mg,2.06mmol)を加えた。室温にて撹拌した後、超音波下で結晶を析出させた。析出した結晶をろ取して2-プロパノール(2mL)で洗浄した後、室温にて減圧乾燥した。SR-MDBNの一コハク酸塩(467mg,1.39mmol)を収率67.8%で得た。
得られたSR-MDBNの一コハク酸塩について、示差走査熱量を測定し、元素分析を実施した。
示差走査熱量測定:
示差走査熱量測定装置DSC-60A(島津製作所社製)を用いて、昇温速度5℃/分(アルミニウム製密閉パン)で測定した。測定により得られたDSC曲線を図8に示した。DSC曲線上の吸熱ピークのエンタルピーは、97.56J/gであり、吸熱温度は、118.53℃であり、補外開始温度は、117.25℃であった。
元素分析:C 64.69 wt%, H 7.78 wt%, N 8.34 wt% (理論値 C 64.65 wt%, H 7.84 wt%, N 8.38 wt%)

得られたSR-MDBN-DSU[11-1]について、NMRを測定した。
1H-NMR (DMSO-D6) δ: 11.99 (4H, brs), 7.33-7.21 (5H, m), 3.69 (1H, d, J = 12.9 Hz), 3.48 (1H, d, J = 12.9 Hz), 3.24 (1H, d, J = 12.7 Hz), 3.23-3.17 (1H, m), 3.11-3.05 (2H, m), 2.93 (1H, d, J = 12.7 Hz), 2.64 (1H, dd, J = 6.9, 3.7 Hz), 2.33 (8H, s), 2.31-2.23 (2H, m), 2.02-1.95 (1H, m), 1.15 (3H, d, J = 6.9 Hz).
HPLCの測定機器及び条件を以下に示す。
測定機器:LC-10システム(島津製作所)
測定条件:
カラム:CHIRALPAK IE-3:3um,4.6mm×250mm(ダイセル)
カラム温度:40℃
流速:1.0mL/min.
分析時間:30min.
検出波長:UV(220nm)
移動相:n-ヘキサン/エタノール/イソプロピルアミン=95/5/0.1(体積比)
上記HPLC測定条件における化合物[11-1]の保持時間は、約16.4分であった。エナンチオマー体であるRS-MDBN-DSUの保持時間は、約21.9分であった。
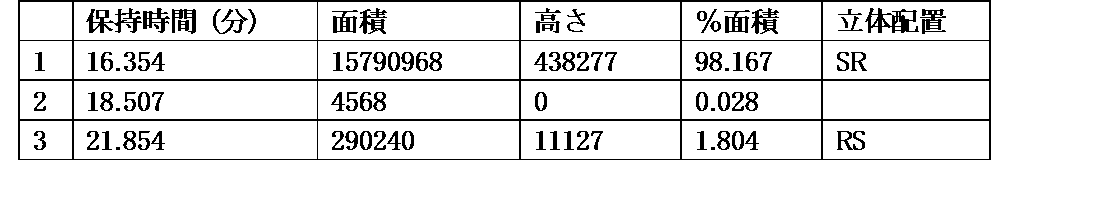


同じ方法で合成した粗SR-MDBN[10]を濃縮乾固し、NMRとMSを測定した。
1H-NMR (CDCl3) δ: 7.34-7.20 (5H, m), 3.62 (1H, d, J = 12.9 Hz), 3.59 (1H, d, J = 12.9 Hz), 3.21 (1H, dd, J = 7.5, 6.6 Hz), 2.99 (1H, d, J = 12.1 Hz), 2.95 (1H, d, J = 12.1 Hz), 2.84 (2H, t, J = 7.3 Hz), 2.68 (1H, t, J = 5.8 Hz), 2.43-2.35 (1H, m), 2.22-2.15 (1H, m), 1.81-1.74 (2H, m), 1.13 (3H, d, J = 6.9 Hz).
MS: m/z = 217 [M+H]+
(A)SR-MDBN(化合物[10])の製造
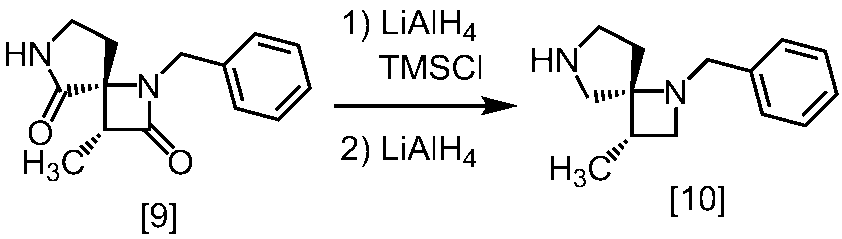
同じ方法で合成した粗SR-MDBN[10]の2-プロパノール溶液の一部を濃縮乾固し、NMRとMSを測定した。
1H-NMR (DMSO-D6) δ: 7.28-7.17 (5H, m), 3.56 (1H, d, J = 13.9 Hz), 3.52 (1H, d, J = 13.2 Hz), 3.20 (1H, brs), 3.09-3.05 (1H, m), 2.80 (1H, d, J = 11.3 Hz), 2.73-2.67 (3H, m), 2.56-2.53 (1H, m), 2.28-2.20 (1H, m), 2.09-2.02 (1H, m), 1.69-1.63 (1H, m), 1.06 (3H, d, J = 6.9 Hz).
MS: m/z = 217 [M+H]+
(B)SR-MDBN(化合物[10])の製造
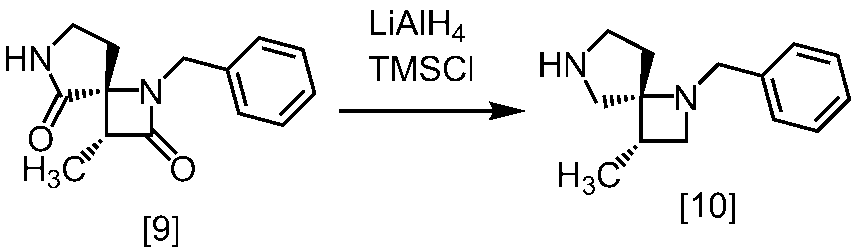
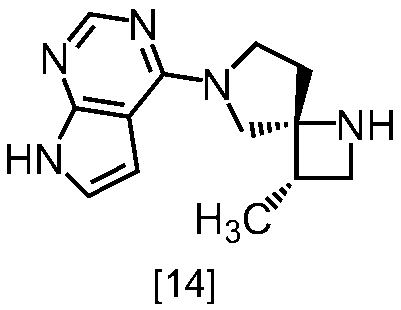
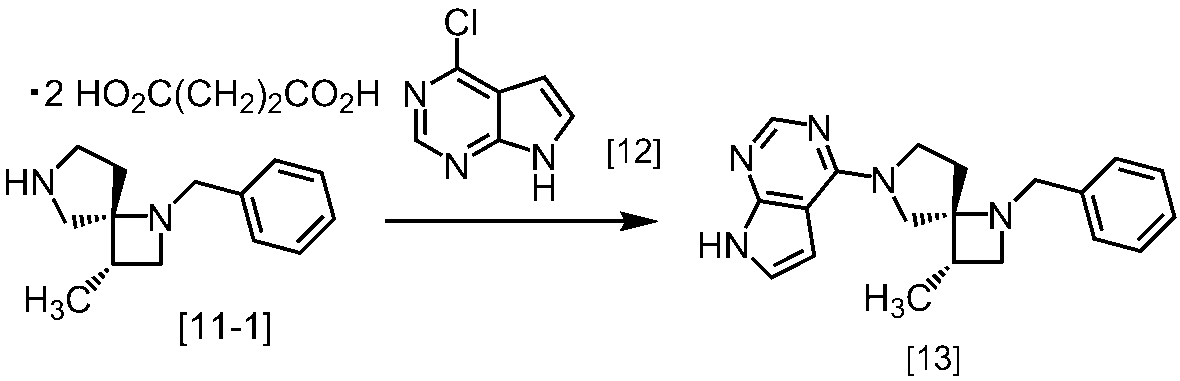
窒素雰囲気下、リン酸三カリウム(14.1g,66.3mmol)に精製水(30mL)を添加した。この溶液に30℃から40℃にてSR-MDBN-DSU[11-1](5.0g,11.0mmol)、CPPY[12](1.73g,11.3mmol)及びにtert-ブタノール(15mL)を順次添加した。反応混合液を75℃から85℃にて2.5時間撹拌した後、室温まで冷却した。この反応混合液を分液することで、粗SR-MDBP[13]の含水tert-ブタノール溶液(43.16g,11.0mmol相当)を得た。得られた粗SR-MDBP[13]の含水tert-ブタノール溶液を収率100%として次工程に用いた。
同じ方法で合成した粗SR-MDBP[13]を濃縮乾固し、NMRとMSを測定した。
1H-NMR (DMSO-d6) δ: 11.57 (1H, s), 8.09 (1H, s), 7.29-7.15 (5H, m), 7.10 (1H, t, J = 2.8 Hz), 6.57 (1H, brs), 3.75-3.53 (6H, m), 3.23 (1H, dd, J = 7.4, 6.5 Hz), 2.70 (1H, t, J = 5.8 Hz), 2.36 (1H, dt, J = 19.5, 7.2 Hz), 2.29-2.22 (1H, m), 2.14-2.07 (1H, m), 1.07 (3H, d, J = 7.2 Hz).
MS: m/z = 344 [M+H]+
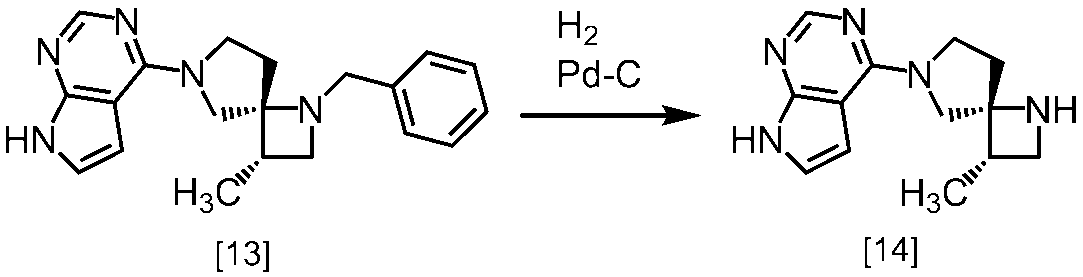
粗SR-MDBP[13]の含水tert-ブタノール溶液(43.16g,11.0mmol相当)に精製水(3.7mL)、酢酸(1.32g,22.0mmol)及び10%パラジウム炭素(川研ファインケミカル社製 Mタイプ,52.6%含水,370mg)を順次添加した。この混合液を水素で置換し、常圧水素存在下で55℃にて7時間撹拌した。反応液を室温まで冷却後、窒素で置換し、トルエン(17mL)及び8M水酸化ナトリウム水溶液(15.5mL,44.0mmol)を加え、45℃で6時間撹拌した。反応混合液を室温まで冷却した後、10%パラジウム炭素を濾過で除去した。反応容器と10%パラジウム炭素をtert-ブタノールとトルエン混合液(1:1,7mL)で洗浄し、ろ液と洗浄液を合わせた後、分液した。得られた有機層を10%食塩水(7mL)で洗浄し、減圧下にて濃縮した。濃縮残渣にトルエン(17mL)を添加し濃縮する操作を3回繰り返した後、濃縮残渣に再度トルエン(20mL)を加え、15℃から30℃にて1時間、次いで0℃から10℃にて1時間撹拌した。析出した固体を濾取し、得られた固体をトルエン(5mL)で洗浄した。得られた湿固体を減圧下にて乾燥することで、SR-MDOP[14](2.45g,10.07mmol)を収率91.5%で得た。
同じ方法で合成したSR-MDOP[14]について、NMR、MSを測定した。
1H-NMR (DMSO-d6) δ: 11.57 (brs, 1H), 8.07 (s, 1H), 7.10 (d, 1H, J = 3.2 Hz), 6.58 (d, 1H, J = 3.2 Hz), 3.92-3.59 (m, 4H), 3.49 (dd, 1H, J = 8.3, 7.2 Hz), 2.93 (dd, 1H, J = 7.2, 6.1 Hz), 2.61-2.53 (m, 2H), 2.12-2.01 (m, 2H), 1.10 (d, 3H, J = 6.9 Hz).
MS: m/z = 244 [M+H]+

窒素雰囲気下、SR-MDOP[14](5.00g,20.5mmol)にアセトニトリル(60mL)及びトリエチルアミン(416mg,4.11mmol)を添加した後、DPCN[18](3.69g,22.6mmol)のアセトニトリル(35mL)溶液を45℃にて滴下し、使用した滴下ロートをアセトニトリル(5.0mL)で洗浄し、洗浄液を反応混合液に添加した。反応混合液を45℃にて3時間撹拌した後、室温まで冷却した。反応後混合液に5%重曹水(25mL)、10%食塩水(25mL)及び酢酸エチル(50mL)を加えて撹拌した後、有機層を分取した。減圧下にて有機層の溶媒を留去した。濃縮残渣にTHF(50mL)を加えて濃縮する操作を4回繰り返した。濃縮残渣にTHF(50mL)を加え、溶液の水分含量が5.5重量%になるように水を添加し、析出した不溶物を濾過で除去した。反応容器と濾過残渣をTHF(15mL)で洗浄し、洗浄液を濾液に添加した後、減圧下にて濾液の溶媒を留去した。濃縮残渣にエタノール(50mL)及び下記実施例11の方法で予め調整した化合物A(化合物[17])の結晶(5.1mg)を添加し、室温にて1時間撹拌した後、減圧下にて溶媒を留去し、エタノール(50mL)を加え再度濃縮した。濃縮残渣にエタノール(15mL)を加え、室温にて1時間撹拌した。析出した固体を濾取し、得られた固体をエタノール(20mL)で洗浄した。得られた湿固体を減圧下にて乾燥することで、化合物A(化合物[17])の1-エタノール和物[20](6.26g,17.6mmol)を収率85.5%で得た。
同じ方法で合成した化合物A(化合物[17])の1-エタノール和物のNMRとMSを測定した。
1H-NMR (DMSO-d6) δ: 11.59 (brs, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.3 Hz), 6.58 (dd, 1H, J = 3.5, 1.8 Hz), 4.34 (t, 1H, J = 5.1 Hz), 4.16 (t, 1H, J = 8.3 Hz), 4.09-3.92 (m, 3H), 3.84-3.73 (m, 1H), 3.71 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 19.0 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 3.44 (dq, 2H, J = 6.7, 5.1 Hz), 2.69-2.60 (m, 2H), 2.23-2.13 (m, 1H), 1.12 (d, 3H, J = 7.1 Hz), 1.06 (t, 3H, J = 6.7 Hz).
MS: m/z = 311 [M+H]+
図11の各ピークは以下の表のとおりである。


窒素雰囲気下、化合物A(化合物[17])の1-エタノール和物[20](4.00g,11.2mmol)及び1-ブタノール(32mL)を混合し、110℃にて溶解させた。85℃に冷却後、本法と同じ方法で予め調製した化合物A(化合物[17])の結晶(4.0mg)を添加し、85℃にて2時間、75℃にて1時間、室温にて16時間撹拌した。析出した固体を濾取し、得られた固体を1-ブタノール(8.0mL)及び酢酸エチル(8.0mL)で順次洗浄した。得られた湿固体を減圧下にて乾燥することで、化合物A(化合物[17])(3.18g,10.2mmol)を収率91.3%で得た。
同じ方法で合成した化合物A(化合物[17])のNMRとMSを測定した。
1H-NMR (DMSO-d6) δ: 11.59 (brs, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.5 Hz), 6.58 (dd, 1H, J = 3.5, 1.8 Hz), 4.16 (t, 1H, J = 8.3 Hz), 4.09-3.93 (m, 3H), 3.84-3.73 (m, 1H), 3.71 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 19.0 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 2.69-2.59 (m, 2H), 2.23-2.13 (m, 1H), 1.12 (d, 3H, J = 7.2 Hz).
MS: m/z = 311 [M+H]+
(1)単結晶作製方法
LaPhaロボバイアル2.0mL広口バイアルに、10mgの化合物A(化合物[17])を入れ、クロロホルム0.5mLを加えて蓋をし、化合物A(化合物[17])を完溶させた。溶媒をゆっくりと蒸発させるため、テルモシリンジ針で、蓋に取り付けられたセプタムに穴を開け、バイアルを室温静置した。得られた単結晶を構造解析に使用した。
(2)測定器
ビームライン:SPring-8 BL32B2
検出器:Rigaku R-AXIS V diffractometer
(3)測定方法
0.71068Åの放射光を単結晶に当て、X線回折データを測定した。
(4)分析方法
得られた化合物A(化合物[17])のクロロホルム和物が有する塩素原子のX線異常散乱効果を利用する方法により、化合物A(化合物[17])の絶対立体配置を(3S,4R)と決定した。化合物A(化合物[17])の絶対立体配置より化合物A(化合物[17])の各製造中間体の立体構造を特定した。
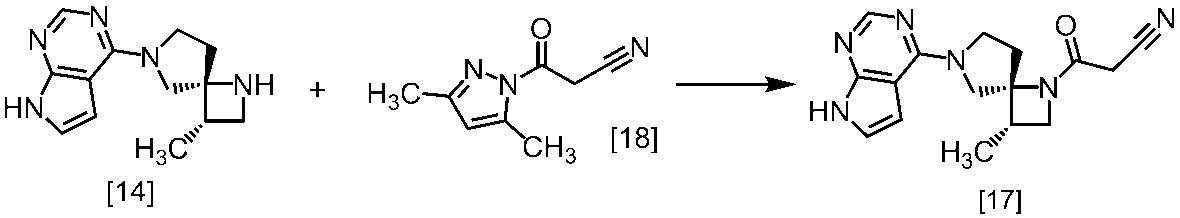
同じ方法で合成した化合物AのNMRとMSを測定した。
1H-NMR (DMSO-d6) δ: 11.60 (s, 1H), 8.09 (s, 1H), 7.12 (dd, 1H, J = 3.0, 2.7 Hz), 6.58 (brs, 1H), 4.16 (t, 1H, J = 8.4 Hz), 4.11-3.91 (m, 3H), 3.88-3.72 (m, 1H), 3.68 (d, 2H, J = 2.1 Hz), 3.57 (dd, 1H, J = 8.4, 6.0 Hz), 2.70-2.56 (m, 2H), 2.24-2.10 (m, 1H), 1.12 (d, 3H, J = 7.2 Hz).
MS: m/z = 311 [M+H]+
窒素雰囲気下、上記実施例15で製造した化合物A(化合物[17])(100g,322mmol)及び1-ブタノール(1.8L)を混合し、90℃から100℃にて溶解させた。この溶液を85℃から100℃にて濾過し、溶解容器と濾過残渣を1-ブタノール(200mL)で洗浄し、洗浄液を濾液に添加した。濾液を60℃から70℃に冷却した後、本法と同じ方法で予め調製した化合物Aの結晶(100mg)を添加した。この混合液を60℃から70℃にて2時間撹拌した後、30℃まで3時間かけて冷却した。20℃から30℃にて1時間撹拌した後、0℃から5℃にて4時間撹拌した。析出した固体を濾取し、得られた固体を1-ブタノール(200mL)及び酢酸エチル(200mL)で順次洗浄した。得られた湿固体を減圧下にて乾燥することで、化合物A(化合物[17])(91.7g,295mmol)を収率91.7%で得た。得られた化合物は粉末X線回折等で分析し、化合物Aであることを確認した。
同じ方法で合成した化合物AのNMRとMSを測定した。
1H-NMR (DMSO-d6) δ: 11.60 (s, 1H), 8.09 (s, 1H), 7.12 (dd, 1H, J = 2.7, 2.4 Hz), 6.59 (brs, 1H), 4.16 (t, 1H, J = 8.2 Hz), 4.11-3.91 (m, 3H), 3.86-3.72 (m, 1H), 3.68 (d, 2H, J = 2.1 Hz), 3.58 (dd, 1H, J = 8.1, 6.0 Hz), 2.71-2.56 (m, 2H), 2.27-2.09 (m, 1H), 1.12 (d, 3H, J = 6.9 Hz).
MS: m/z = 311 [M+H]+

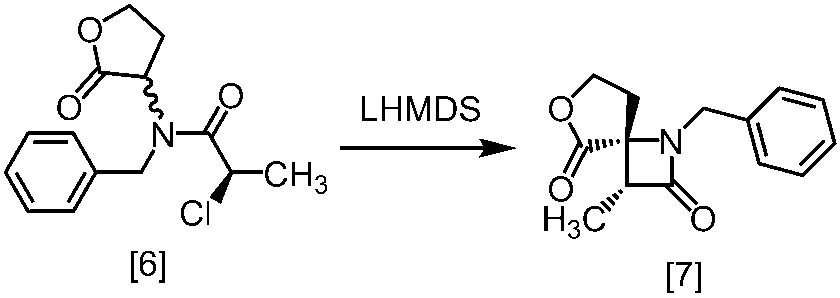
同じ方法で合成した粗RR-AOBL[7]のトルエン溶液の一部を濃縮乾固し、NMRとMSを測定した。
1H-NMR (DMSO-D6) δ: 7.36-7.25 (5H, m), 4.54 (1H, d, J = 15.7 Hz), 4.35-4.30 (1H, m), 4.24-4.18 (1H, m), 4.13 (1H, d, J = 15.7 Hz), 3.60 (1H, q, J = 7.4 Hz), 2.46-2.35 (2H, m), 1.10 (3H, d, J = 7.4 Hz).
MS: m/z = 246 [M+H]+
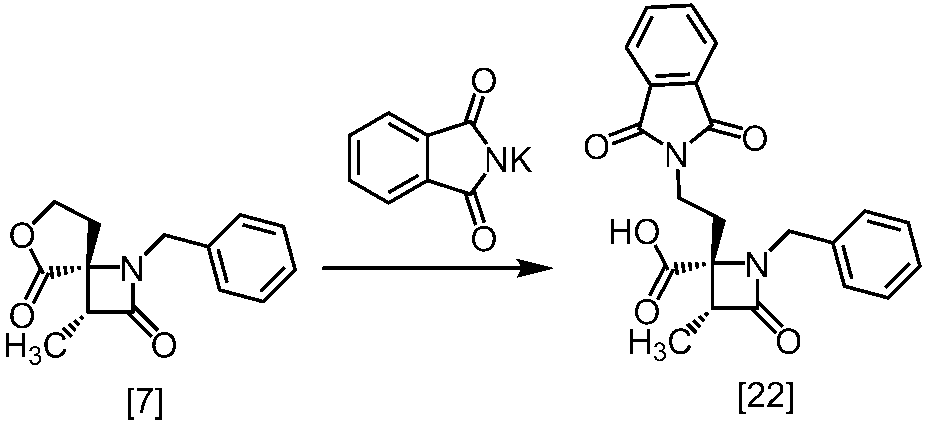
得られたRR-AOPA[22]について、NMRとMSを測定した。
1H-NMR (DMSO-D6) δ: 13.45 (1H, brs), 7.84-7.81 (4H, m), 7.39-7.31 (4H, m), 7.24 (1H, t, J = 7.2 Hz), 4.63 (1H, d, J = 16.1 Hz), 4.33 (1H, d, J = 16.1 Hz), 3.64-3.57 (1H, m), 3.52-3.44 (1H, m), 3.36 (1H, q, J = 7.5 Hz), 2.30-2.15 (2H, m), 1.09 (3H, d, J = 7.5 Hz).
MS: m/z = 393 [M+H]+

合成した化合物A(化合物[17])の3,5-ジメチルピラゾールとの共結晶(モル比2:1)のNMR、元素分析と示差走査熱量を測定した。
1H-NMR (DMSO-d6) δ: 11.98 (br s, 0.5H), 11.59 (br s, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.2 Hz), 6.58 (dd, 1H, J = 3.5, 1.4 Hz), 5.73 (s, 0.5H), 4.16 (t, 1H, J = 8.3 Hz), 4.09-3.93 (m, 3H), 3.84-3.74 (m, 1H), 3.70 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 19.0 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 2.70-2.58 (m, 2H), 2.22-2.12 (m, 1H), 2.12 (s, 3H), 1.12 (d, 3H, J = 7.2 Hz).
元素分析:C 61.9wt%, H 6.1wt%, N 27.2wt% (理論値 C 62.0wt%, H 6.2wt%, N 27.4wt%)
示差走査熱量測定:
示差走査熱量測定装置DSC-60A(島津製作所社製)を用いて、昇温速度5℃/分(アルミニウム製密閉パン)で測定した。測定により得られたDSC曲線を図12に示した。DSC曲線上の吸熱ピークのエンタルピーは、100.26J/gであり、吸熱温度は、173.66℃であり、補外開始温度は、172.36℃であった。得られたスペクトルを図12に示す。
図13の各ピークは以下の表のとおりである。

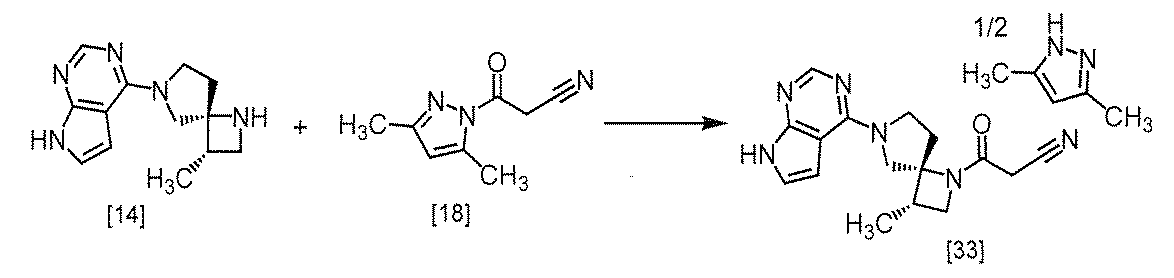
合成した化合物A(化合物[17])の3,5-ジメチルピラゾールとの共結晶(モル比2:1)のNMR、元素分析と示差走査熱量を測定した。
1H-NMR (DMSO-d6) δ: 11.99 (br s, 0.5H), 11.59 (br s, 1H), 8.11 (s, 1H), 7.11 (s, 1H), 6.58 (d, 1H, J = 3.0 Hz), 5.73 (s, 0.5H), 4.16 (t, 1H, J = 8.4 Hz), 4.10-3.92 (m, 3H), 3.85-3.74 (m, 1H), 3.70 (d, 1H, J = 19.1 Hz), 3.65 (d, 1H, J = 19.1 Hz), 3.57 (dd, 1H, J = 7.9, 6.1 Hz), 2.70-2.58 (m, 2H), 2.22-2.14 (m, 1H), 2.12 (s, 3H), 1.12 (d, 3H, J = 6.9 Hz).
元素分析:C 62.0wt%, H 6.2wt%, N 27.2wt% (理論値 C 62.0wt%, H 6.2wt%, N 27.4wt%)
示差走査熱量測定:
示差走査熱量測定装置DSC-60A(島津製作所社製)を用いて、昇温速度5℃/分(アルミニウム製密閉パン)で測定した。測定により得られたDSC曲線を図14に示した。DSC曲線上の吸熱ピークのエンタルピーは、78.02J/gであり、吸熱温度は、173.81℃であり、補外開始温度は、172.02℃であった。得られたスペクトルを図14に示す。
図15の各ピークは以下の表のとおりである。

(A)化合物A(化合物[17])の製造(精製)
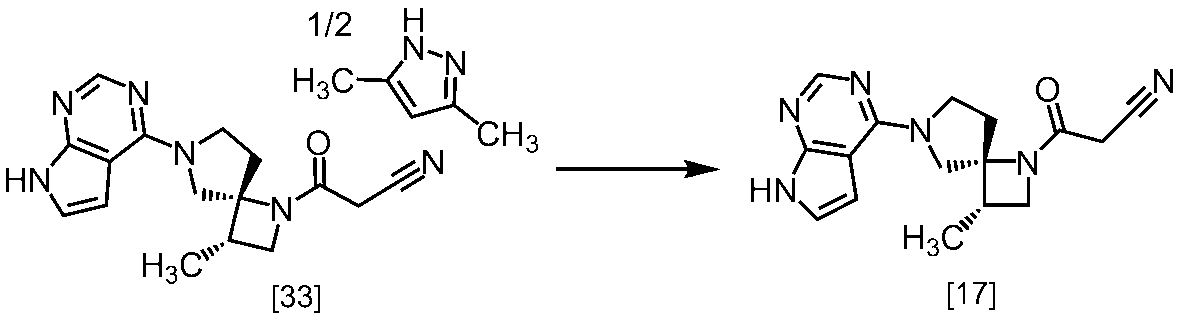
同じ方法で合成した化合物A(化合物[17])のNMRとMSを測定した。
1H-NMR (DMSO-d6) δ: 11.58 (br s, 1H), 8.08 (s, 1H), 7.11 (dd, 1H, J = 3.5, 2.3 Hz), 6.58 (dd, 1H, J = 3.5, 1.6 Hz), 4.16 (t, 1H, J = 8.4 Hz), 4.10-3.94 (m, 3H), 3.84-3.74 (m, 1H), 3.70 (d, 1H, J = 19.0 Hz), 3.65 (d, 1H, J = 18.7 Hz), 3.58 (dd, 1H, J = 8.2, 5.9 Hz), 2.70-2.59 (m, 2H), 2.23-2.12 (m, 1H), 1.12 (d, 3H, J = 7.2 Hz).
MS: m/z = 311 [M+H]+
(B)化合物A(化合物[17])の製造(精製)

窒素雰囲気下、式[33]の化合物(10.0g,27.9mmol)を2-メチル-2-ブタノール(150mL)に110℃で溶解した。75℃にて化合物[17]の種晶を溶液に加えた。溶液を0から5℃に冷却した。固体を2-メチル-2-ブタノール(20mL)、次いで酢酸エチル(20mL)で洗浄した。析出した固体をフィルターで集め、得られた固体を2-メチル-2-ブタノール(20mL)、次いで酢酸エチル(20mL)で洗浄した。得られた湿固体を減圧乾燥し、化合物[17](7.6g,24mmol)を得た。
溶媒を留去して固体を得、これを水(250mL)に溶解した後、酢酸エチル(315mL)を添加した。混合物を激しく撹拌し、水相のpHが~2となるように塩酸水溶液(2M,290mL)を滴下した。相を分離し、水相を酢酸エチル(315mL)で抽出した。集めた有機相を濃縮し、~95%純度の粗生成物[24](71.31g)を得た。
化合物はさらなる精製なしで次の工程に用いた。

反応混合物を20℃から25℃に冷却し、亜硫酸ナトリウム(61.4g,0.49mol)の水溶液(300mL)を添加した。混合物を10分間撹拌した後、濃縮した。酢酸2-プロピル(1.8L)を少しずつ濃縮残渣に加え、混合物を濃縮乾固した。濃縮残渣に水(300mL)及び酢酸2-プロピル(3L)を加えた。20℃から25℃にて塩酸水溶液(2M,2.75L;pH2.56)でpHを2から3に調節した。相を分離し、水相を酢酸2-プロピル(2×1.5L)で抽出した。有機相を集め、水(1.2L)で洗浄した。相を分離し、有機相を濃縮してBoc-Dab(MeOCO)-OH[24](669.4g,2.42mol)を96.3面積%のHPLC純度で得た。
化合物はさらなる精製なしで次の工程に用いた。
1H NMR (600 MHz, DMSO-d6) δ 12.47 (brs, 1H), 7.15-7.05 (m, 2H), 3.88 (td, J = 9.1, 4.7 Hz, 1H), 3.50 (s, 3H), 3.06-2.96 (m, 2H), 1.87-1.78 (m, 1H), 1.70-1.60 (m, 1H), 1.38 (s, 9H). 化合物は少量の回転異性体を示す:6.81-6.68 (m), 3.82-3.75 (m), 1.34 (s).
LC-MS: m/z = 275 [M-H]-
窒素ガスを反応混合物に30分間バブリングし、塩化水素を除去した。反応混合物を濃縮して乾燥した。メタノール(1L)を残渣に添加し、濃縮乾固し、化合物[25](555.9g,2.42mol)を89.7面積%のHPLC純度で得た。
反応混合物を濃縮した。トルエン(200mL)を添加し、溶液を濃縮した。これをさらなるトルエン(200mL)で繰り返し、化合物[25]を得た。化合物をさらなる精製なしで次の工程に用いた。

1H NMR (600 MHz, DMSO-d6) δ 8.62 (brs, 3H), 7.26 (t, J = 5.8 Hz, 1H), 4.02 (t, J = 6.6 Hz, 1H), 3.74 (s, 3H), 3.52 (s, 3H), 3.17-3.08 (m, 2H), 2.01-1.88 (m, 2H).
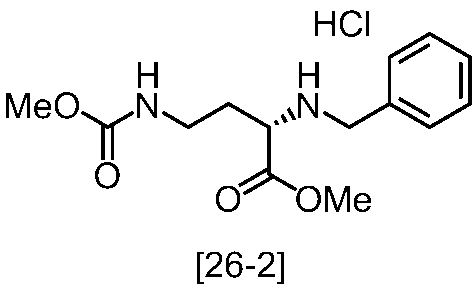
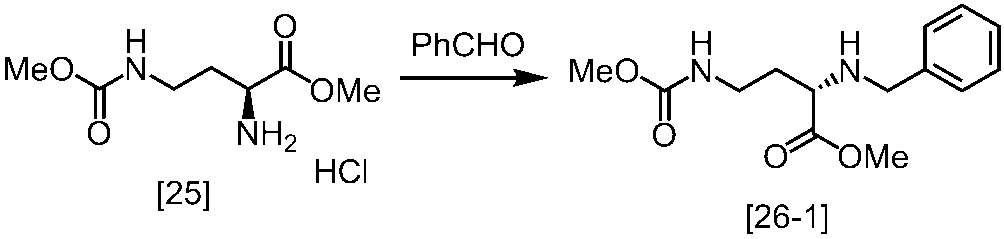
反応混合物を水(1.85L)でクエンチし、15分間撹拌した。メタノールを40℃にて留去し、酢酸2-プロピル(1.85L)を残渣に添加した。相を分離した後、水性乳濁層を酢酸2-プロピル(1.85L)で抽出した。乳濁液をろ過した。集めた有機相を炭酸水素ナトリウム水溶液(20%,1.85L)及びブライン(1.85L)で洗浄した。有機溶液を濃縮し、粗アミン[26-1]((S)-2-(ベンジルアミノ)-4-((メトキシカルボニル)アミノ)ブタン酸メチル)(197.3g,703.8mmol)を92.7面積%の純度で得た。
1H NMR (600 MHz, CDCl3) δ 7.35-7.29 (m, 4H), 7.28-7.25 (m, 1H), 5.49 (brs, 1H), 3.82 (d, J = 12.9 Hz, 1H), 3.73 (s, 3H), 3.65 (s, 3H), 3.61 (d, J = 12.9 Hz, 1H), 3.43-3.36 (m, 1H), 3.30 (dd, J = 9.0, 4.5 Hz, 1H), 3.28-3.23 (m, 1H), 1.95-1.87 (m, 1H), 1.75-1.65 (m, 1H).
13C NMR (151 MHz, CDCl3) δ 175.4, 157.1, 139.6, 128.6, 128.5, 127.4, 59.4, 52.4, 52.1 (2C), 39.1, 32.8.

懸濁液をろ過し、ろ過ケーキを冷却した酢酸2-プロピル(2×500mL)で洗浄し、45℃から50℃、減圧下にて乾燥し、粗化合物[26-2](520.6g,1.64mol)を97.5面積%の純度で得た。
1H NMR (600 MHz, DMSO-d6) δ 10.37 (brs, 1H), 9.85 (brs, 1H), 7.63-7.53 (m, 2H), 7.46-7.37 (m, 3H), 7.26 (t, J = 5.9 Hz, 1H), 4.20 (d, J = 13.0 Hz, 1H), 4.13 (d, J = 13.1 Hz, 1H), 4.06-4.00 (m, 1H), 3.72 (s, 3H), 3.52 (s, 3H), 3.16 (dq, J = 13.6, 5.9 Hz, 1H), 3.07 (dq, J = 13.7, 6.9 Hz, 1H), 2.18 (dq, J = 13.5, 7.3 Hz, 1H), 2.05 (dq, J = 14.0, 7.8 Hz, 1H).
13C NMR (151 MHz, DMSO-d6) δ 168.9, 156.7, 131.5, 130.5 (2C), 129.1, 128.6 (2C), 56.4, 52.9, 51.4, 49.1, 36.5, 29.0.
LC-MS: m/z = 281 [M+H]+

固体をろ去し、冷却した酢酸2-プロピル(2×500mL)で洗浄した後、45℃から50℃、減圧下、乾燥し、化合物[26-2](1.063kg)を99.5面積%のHPLC純度で得た。


反応混合物を常圧にて窒素ガス気流下、蒸留した。主なフラクションを105℃から110℃にて集め、化合物[5](118.24g,931.3mmol)を液体として、99面積%のHPLC純度及びキラルHPLCによる96.5面積%のエナンチオマー純度で得た。
1H NMR (600 MHz, CDCl3) δ 4.66 (q, J = 7.0 Hz, 1H), 1.82 (d, J = 7.1 Hz, 3H).

水(1.25L)を反応混合物に添加した。15分撹拌後、相を分離し、水相をトルエン(2×500mL)で抽出した。集めた有機相をろ過し、溶媒を減圧留去し、化合物[27](312.79g,843.46mmol)を97.90面積%のHPLC純度で得た。
1H NMR (600 MHz, CDCl3) δ 7.59-7.13 (m, 5H), 4.92-4.26* [m, 5H; 4.86 (d, J = 17.0 Hz), 4.80-4.62 (m), 4.58-4.53 (d, J = 16.9 Hz), 4.52 (q, J = 6.6 Hz), 4.49-4.44 (m), 4.30 (t, J = 6.8 Hz)], 3.70* (s, 2.3H), 3.64 (s, 3H), 3.55* (s, 0.7H), 3.28-3.01 (m, 2H), 2.28 (dq, J = 13.9, 6.8 Hz, 1H), 2.01-1.84 (m, 1H), 1.76* (d, J = 6.4 Hz, 0.7H), 1.64* (d, J = 6.5 Hz, 2.3H). *は回転異性体ピークを示す。
LC-MS: m/z = 371 [M+H]+

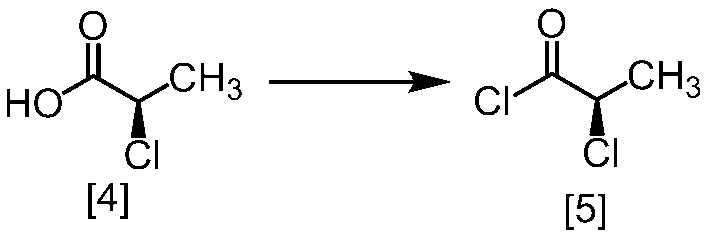

反応混合物を塩酸水溶液(2M,200mL)を滴下してクエンチした後、トルエン(165mL)及び酢酸エチル(165mL)を添加した。二相混合物を10分間激しく撹拌した。有機相を酸性水相から分離し、硫酸ナトリウムで乾燥し、ろ過濃縮して粗生成物(39.5g)を得た。アセトニトリル(100mL)を添加し、混合物を濃縮した。HPLC分析により生成物[27]が97:3のジアステレオ異性体比で形成されたことが示された。
懸濁液を半分の容積まで濃縮した。セシウム塩をろ去し、トルエン(3×40mL)で洗浄し、ろ液を~25mLに濃縮した。Tonsil(登録商標)(3g)フィルターエイドを添加して溶液を透明化した後、ろ去した。得られた溶液を2℃から6℃で終夜結晶化させた。析出物をろ去し、トルエン(3×4mL)で洗浄した。生成物を30℃にて減圧乾燥し、化合物[9](9.31g,38.1mmol)を98.85面積%のHPLC純度及びキラルHPLCによる99.55面積%のエナンチオマー純度で得た。

混合物を0℃以下に冷却し、塩化ナトリウム水溶液(飽和,330mL)及び酢酸2-プロピル(790mL)を混合物に添加した。次いで、クエン酸水溶液(50重量%,159g)を添加してpH5から6に調節した。20分撹拌後、相を分離し、水相を酢酸2-プロピル(2×263mL)で抽出した。集めた有機相を減圧下、半分の容積まで濃縮し、塩化ナトリウム水溶液(飽和,263mL)で洗浄した。有機相を濃縮し、トルエン(800mL)を添加した。溶液を、100%の理論収率に基づき、生成物グラムあたり6mLまで濃縮した。
得られた溶液を60℃に加熱した後、冷却した。40℃にて種晶を添加し、20℃から25℃に冷却した。次いで、ヘプタン(304mL,3mL/グラム)を撹拌混合物に滴下した。添加完了後、混合物を20℃にて1時間撹拌した。析出した固体をろ取した。固体をトルエン及びヘプタン(1:2)混合液(202mL)で洗浄し、40℃にて減圧乾燥し、化合物[9](70.39g,288.1mmol)を98.53面積%のHPLC純度及びキラルHPLCによる97.93面積%のエナンチオマー純度で得た。
生成物をトルエン(1.5mL/生成物グラム)中、50℃にて1時間撹拌した。固体を20℃にてろ取し、トルエン(2×15mL)ですすぎ、40℃にて減圧下乾燥した。生成物[9](64.79g,265.2mmol)を99.78面積%のHPLC純度及びキラルHPLCによる99.56面積%のエナンチオマー純度で得た。
1H NMR (600 MHz, CDCl3) δ 7.34-7.26 (m, 5H), 6.51 (brs, 1H), 4.86 (d, J = 15.5 Hz, 1H), 3.99 (d, J = 15.5 Hz, 1H), 3.27 (q, J = 7.5 Hz, 1H), 3.24-3.20 (m, 1H), 3.15 (ddt, J = 10.0, 8.1, 2.3 Hz, 1H), 2.05 (ddd, J = 13.3, 6.8, 2.5 Hz, 1H), 2.01 (dt, J = 13.3, 8.6 Hz, 1H), 1.29 (d, J = 7.5 Hz, 3H).
13C NMR (151 MHz, CDCl3) δ 174.2, 169.8, 136.4, 128.8 (2C), 128.3 (2C), 127.8, 65.8, 55.8, 44.5, 38.3, 30.7, 9.8.
LC-MS: m/z = 245 [M+H]+

反応混合物を0℃に冷却した後、水(46.5mL)を滴下した。テトラヒドロフラン(500mL)を得られたスラリーに添加した。次いで、水酸化ナトリウム水溶液(15重量%,46.5mL,6.98g,174mmol)を添加し、さらに水(139.5mL)を懸濁液に添加した。混合物を室温とした後、セライト(登録商標)フィルターエイド及び硫酸ナトリウム(60g)を添加した。撹拌を15分間継続した。固体をセライト(登録商標)フィルターエイドのベッドを通してろ去した。ろ過ケーキをテトラヒドロフラン(4×250mL)で洗浄し、ろ液を減圧濃縮し、生成物[10](64.71g,299.1mmol)を、92.37面積%のHPLC純度及びキラルGCによる99.45面積%のエナンチオマー純度で得た。
1H NMR (600 MHz, CDCl3) δ 7.33-7.27 (m, 4H), 7.23-7.19 (m, 1H), 3.62 (d, J = 13.1 Hz, 1H), 3.58 (d, J = 13.0 Hz, 1H), 3.20 (dd, J = 7.5, 6.5 Hz, 1H), 2.95 (s, 2H), 2.82 (dd, J = 7.7, 6.7 Hz, 2H), 2.66 (dd, J = 6.5, 5.5 Hz, 1H), 2.39 (pd, J = 7.1, 5.4 Hz, 1H), 2.17 (dt, J = 13.1, 7.5 Hz, 1H), 1.76 (dt, J = 13.4, 6.8 Hz, 1H), 1.12 (d, J = 7.0 Hz, 3H), 1.68 (brs, 1H).
13C NMR (151 MHz, CDCl3) δ 139.2, 128.7, 128.4, 126.9, 75.7, 57.8, 55.7, 48.9, 45.6, 35.9, 35.8, 15.6.
LC-MS: m/z = 217 [M+H]+
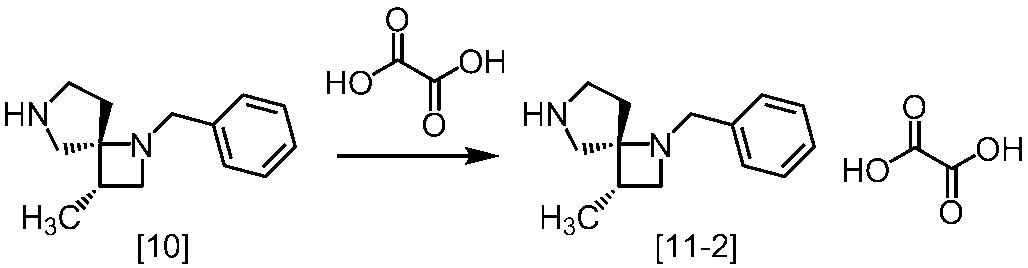
形成された析出物をろ取し、減圧乾燥して化合物[11-2](0.204g)を得た。
1H NMR (600 MHz, DMSO-d6) δ 7.33-7.27 (m, 4H), 7.26-7.21 (m, 1H), 3.74 (d, J = 13.0 Hz, 1H), 3.52 (d, J = 13.0 Hz, 1H), 3.29 (d, J = 12.7 Hz, 1H), 3.24 (ddd, J = 11.4, 7.8, 5.5 Hz, 1H), 3.16-3.09 (m, 2H), 3.02 (d, J = 12.7 Hz, 1H), 2.68 (dd, J = 6.7, 3.8 Hz, 1H), 2.36-2.27 (m, 1H), 2.03 (ddd, J = 12.9, 7.2, 5.5 Hz, 1H), 1.16 (d, J = 7.1 Hz, 3H).
13C NMR (151 MHz, DMSO-d6) δ 164.2, 137.8, 128.4, 128.2, 126.9, 73.6, 56.2, 53.7, 46.1, 43.5, 34.6, 30.2, 15.9.

化合物[10](52.47g,218.3mmol)を2-プロパノール(367mL)に溶解し、混合物を60℃に加熱した。温度を60℃に維持しながら、シュウ酸(9.83g,109.1mmol)の2-プロパノール(157mL)溶液を2時間かけて滴下した。滴下終了後、混合物を60℃にて25分間撹拌した後、化合物[11-3]の結晶を種晶として添加した。
60℃にてさらに35分間撹拌した後、形成された懸濁液を同温にてろ過し、固体を20℃から25℃にて2-プロパノール(2×25mL)で洗浄した。生成物を減圧下、40℃にて乾燥し、化合物[11-3](48.65g,186.14mmol)を99.12面積%のHPLC純度及びキラルHPLCによる99.6面積%のエナンチオマー純度で得た。
1H NMR (600 MHz, D2O) δ 7.45-7.36 (m, 5H), 3.76 (d, J = 12.2 Hz, 1H), 3.71 (d, J = 12.2 Hz, 1H), 3.52 (d, J = 13.2 Hz, 1H), 3.42 (t, J = 7.9 Hz, 1H), 3.33 (ddd, J = 12.5, 7.8, 5.0 Hz, 1H), 3.27-3.20 (m, 2H), 2.86 (dd, J = 7.7, 4.9 Hz, 1H), 2.48 (ddt, J = 11.7, 9.4, 7.9 Hz, 2H), 2.24 (ddd, J = 13.5, 6.9, 5.0 Hz, 1H), 1.17 (d, J = 7.2 Hz, 3H).
13C NMR (151 MHz, D2O) δ 176.25, 138.73, 132.28 (2C), 131.62 (2C), 130.73, 76.91, 58.99, 57.34, 48.48, 46.69, 37.54, 34.33, 17.38.
(3S,4R)-1-ベンジル-3-メチル-1,6-ジアザスピロ[3.4]オクタンヘミシュウ酸塩(化合物[11-3])のスラリー撹拌
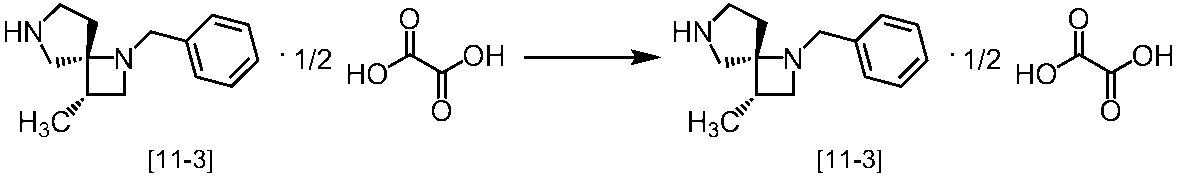
懸濁液を0℃にてろ過し、冷却した2-プロパノール(2×8mL)で洗浄した。固体を40℃にて減圧乾燥し、生成物[11-3](45.95g,175.8mmol)を99.50面積%のHPLC純度及びキラルGCによる99.93面積%のエナンチオマー純度で得た。
工程2B
(3S,4R)-1-ベンジル-3-メチル-1,6-ジアザスピロ[3.4]オクタンヘミシュウ酸塩(化合物[11-3])の再結晶
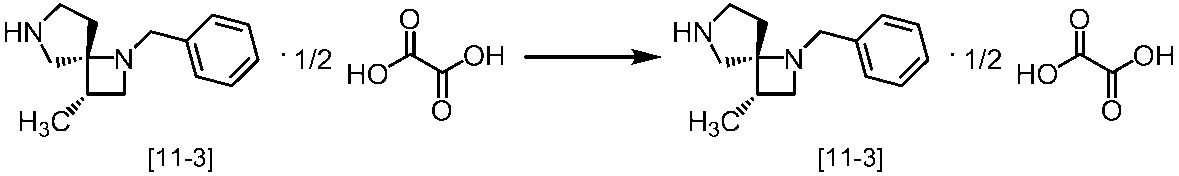
(A)SR-MDBP(化合物[13])の製造

水層を除去し、得られた粗SR-MDBP[13]の2-プロパノール水溶液を100%収率として次の工程に用いた。
(B)SR-MDBP(化合物[13])の製造
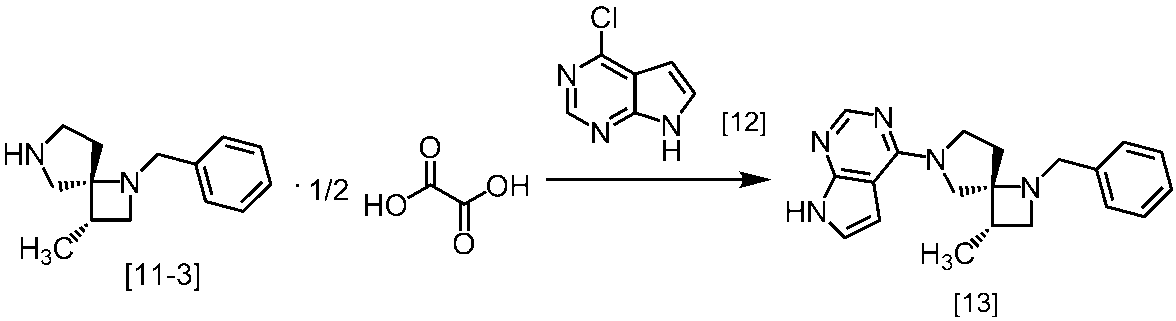
水層を除去し、得られた粗製SR-MDBP[13]の水性2-プロパノール溶液を収率100%と仮定して次の工程に用いた。
(A)SR-MDOP(化合物[14])の製造
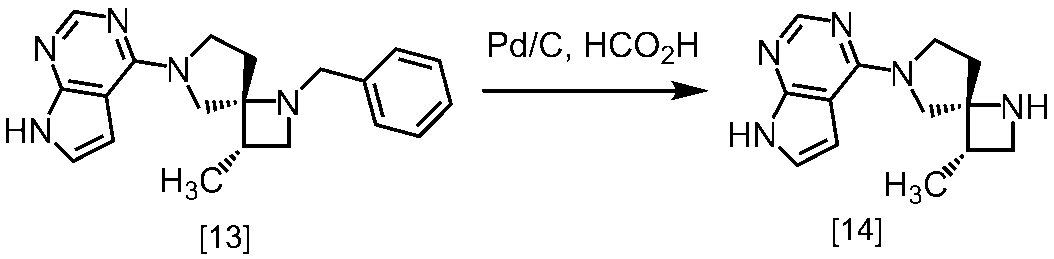
反応混合物を室温に冷却し、パラジウム炭素をろ去し、2-プロパノール及び水の混合液(9:1,12mL)ですすいだ。集めたろ液に、水酸化ナトリウム水溶液(8M,15mL),塩化ナトリウム(7.5g)、2-プロパノール(15mL)及びトルエン(55mL)を添加した。層を分離し、有機層をブライン(20重量%,10mL)で洗浄して濃縮した。残渣(25g)をトルエン及び2-プロパノールの混合液(1:1,35mL)で希釈し、ろ過した。得られた溶液を50℃にて濃縮し、残渣(~25g)を得た。トルエン(35mL)を添加後50℃にて濃縮することで残渣(~25g)を得る操作を2回繰り返し、固体が形成された。
トルエンを濃縮スラリー(~25g)に添加して、スラリーの総重量を48gとした。スラリーを4℃にて16時間維持した後、0℃にて30分間撹拌した。スラリーをろ過し、ろ過ケーキをトルエン(14mL)で洗浄した。固体を50℃にて減圧乾燥し、SR-MDOP[14](4.21g,17.3mmol)を得た。
(B)SR-MDOP(化合物[14])の製造

反応混合物を室温まで冷却し、パラジウムシリカゲルをろ過し、2-プロパノール及び水の混合液(9:1,60mL)ですすいだ。集めたろ液に水性水酸化ナトリウム(8M,57mL)、塩化ナトリウム(29.1g)及びトルエン(199mL)を加えた。層を分離し、有機層を飽和食塩水(20重量%,38mL)で洗浄し、濃縮した。残渣(70g)をトルエンと2-プロパノールの混合液(1:1,140mL)で希釈し、濃縮した。残渣(100g)をトルエンと2-プロパノールの混合液(1:1,140mL)で希釈し、ろ過した。得られた溶液を濃縮した。残渣(80g)をトルエン(140mL)で希釈し、析出が見られるまで濃縮した。混合物を50℃にて1時間撹拌し、残渣120gまで濃縮した。残渣をトルエン(140mL)で希釈し、残渣120gまで濃縮し、トルエン(92mL)で希釈した。スラリーを4℃にて4日間保持した後、0℃にて90分間撹拌した。スラリーをろ過し、ろ過ケーキをトルエン(50mL)で洗浄した。固体を減圧下50℃にて乾燥し、SR-MDOP[14](17.3g,71.1mmol)を得た。
結晶化混合物を0℃にて1時間撹拌し、析出した固体をろ取した。得られた固体を冷却したテトラヒドロフラン(40mL)で洗浄した。固体を30℃にて減圧乾燥し、化合物[29](11.1g,25.2mmol)を得た。
1H-NMR (D2O, 内部標準d4-TMSP) δ 7.71-7.05 (m, 10H), 5.30-4.98 (m, 2H), 4.56-1.98 (m, 11H), 1.28-0.98 (m, 3H).
13C NMR (151 MHz, DMSO-d6) δ 164.2, 154.2, 153.9, 136.6, 136.5, 133.2, 132.7, 130.0, 129.8, 128.8, 128.7, 128.5, 128.4, 128.2, 127.9, 127.6, 73.6, 73.4, 66.1, 65.6, 57.6, 55.5, 55.1, 52.6, 52.4, 51.7, 51.2, 37.3, 36.5, 35.5, 34.6, 15.2, 15.0.
元素分析:C 65.5重量%, H 6.3重量%及びN 6.3重量%
理論値:C 65.4重量%, H 6.4重量%及びN 6.4重量%
水素雰囲気をアルゴン雰囲気に置換し、混合物を水(0.5mL)を用いてセライト(登録商標)ろ過した。集めたろ液を濃縮し、減圧乾燥して化合物[30-1](24mg,0.11mmol)を得た。
1H NMR (600 MHz, D2O, 内部標準d4-TMSP) δ 4.17 (dd, J = 10.7, 8.8 Hz, 1H), 3.99 (dd, J = 14.2, 1.4 Hz, 1H), 3.73 (d, J = 14.2 Hz, 1H), 3.65 (dd, J = 10.7, 6.9 Hz, 1H), 3.58 (ddd, J = 12.7, 8.7, 4.2 Hz, 1H), 3.48 (ddd, J = 12.2, 9.8, 7.1 Hz, 1H), 3.17 (dqd, J = 8.8, 7.3 Hz, 6.9 Hz, 1H), 2.84 (dddd, J = 15.1, 7.2, 4.2, 1.4 Hz, 1H), 2.50 (dt, J = 15.0, 9.3 Hz, 1H), 1.29 (d, J = 7.3 Hz, 3H).
13C NMR (151 MHz, D2O) δ 175.9, 78.0, 51.6, 51.1, 46.7, 37.8, 37.4, 16.6.

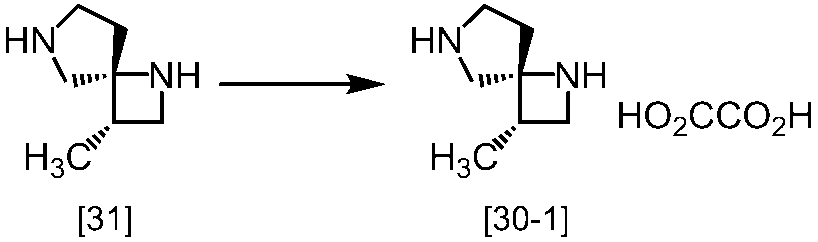
混合物をろ過し、集めた固体をエタノール(0.30mL)で洗浄し、50℃にて減圧乾燥して化合物[30-1](205mg,0.947mmol)を得た。
1H NMR (600 MHz, D2O, 内部標準d4-TMSP) δ 4.17 (dd, J = 10.7, 8.8 Hz, 1H), 3.99 (dd, J = 14.2, 1.4 Hz, 1H), 3.73 (d, J = 14.2 Hz, 1H), 3.65 (dd, J = 10.7, 6.9 Hz, 1H), 3.58 (ddd, J = 12.7, 8.7, 4.2 Hz, 1H), 3.48 (ddd, J = 12.2, 9.8, 7.1 Hz, 1H), 3.17 (dqd, J = 8.8, 7.3 Hz, 6.9 Hz, 1H), 2.84 (dddd, J = 15.1, 7.2, 4.2, 1.4 Hz, 1H), 2.50 (dt, J = 15.0, 9.3 Hz, 1H), 1.29 (d, J = 7.3 Hz, 3H).
13C NMR (151 MHz, D2O) δ 175.9, 78.0, 51.6, 51.1, 46.7, 37.8, 37.4, 16.6.


圧を開放し、反応容器を窒素でパージした。触媒をエタノールで飽和させたセライト(登録商標)フィルターエイドでろ別した。ろ過ケーキをエタノール(10kg)で洗浄した。ろ液を30Lから35Lまで減圧濃縮し、粗生成物[31]を得た。

生成物をろ取し、冷却したエタノール(2×13.7kg)で洗浄した。生成物を減圧乾燥して化合物[30-2](5.143kg,14.19mol)を99.3面積%のHPLC純度で得た。
1H NMR (600 MHz, D2O, 内部標準d4-TMSP) δ 4.18 (dd, J = 10.7, 8.9 Hz, 1H), 3.97 (dd, J = 14.3, 1.4 Hz, 1H), 3.74 (d, J = 14.3 Hz, 1H), 3.64 (dd, J = 10.8, 6.8 Hz, 1H), 3.58 (ddd, J = 12.6, 8.6, 4.2 Hz, 1H), 3.47 (ddd, J = 12.3, 9.9, 7.1 Hz, 1H), 3.15 (ddd, J =8.9, 7.3, 6.8 Hz, 1H), 2.84 (dddd, J = 15.1, 7.1, 4.3, 1.4 Hz, 1H), 2.56 - 2.47 (m, 9H), 1.28 (d, J = 7.3 Hz, 3H).
13C NMR (151 MHz, D2O) δ 182.8, 78.0, 51.6, 51.1, 46.8, 37.8, 37.6, 34.3, 16.6.

トルエン(16L)を添加し、反応混合物の温度を20℃から30℃に下げた。有機層をブライン(飽和,12.2kg)及び水(4.1kg)の混合物で洗浄した。有機層を16L以下の容積に減圧濃縮した。2-メチルプロパン-2-オール(7.42kg)及びトルエン(8L)を反応容器に添加し、混合物を16L以下の容積に減圧濃縮した。2-メチルプロパン-2-オール(9.04kg)及びトルエン(10L)を残渣に添加した。混合物を35℃から40℃にてろ過し、溶液を16L以下の容積に減圧濃縮した。残渣を45℃から50℃に加熱し、トルエン(16L)を添加した。混合物を45℃から50℃にて1時間撹拌した後、16L以下の容積に減圧濃縮した。トルエン(16L)をスラリーに添加し、0℃から5℃にて1時間以上撹拌した。
形成された析出物をろ取し、ろ過ケーキをトルエン(8L)及びヘプタン(8L)で洗浄した。固体を50℃にて20時間減圧乾燥し、SR-MDOP[14](2.445kg,10.05mol)を95.25%のHPLC純度で得た。
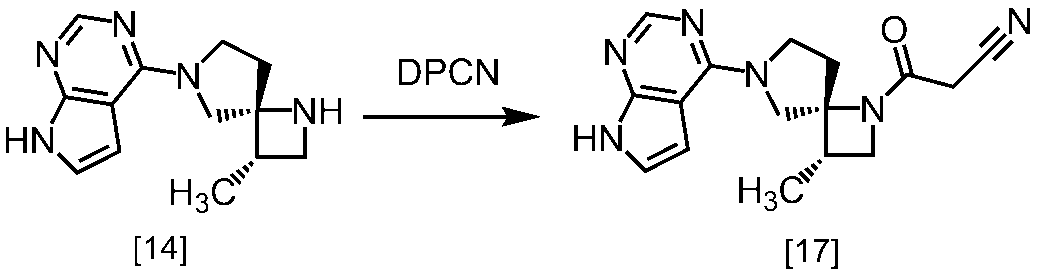
エタノール(2L)を反応混合物に充填し、24L以下の容積に減圧濃縮した。エタノール(24L)を残渣に添加し、溶液を35℃から40℃に加熱した。得られた溶液をエタノール(2L)を用いて細かいフィルターに通して除塵ろ過した。得られた透明溶液を24L以下の容積に減圧濃縮した。エタノール(24L)を得られたスラリーに添加した。スラリーを24L以下の容積に減圧濃縮した。エタノール(4L)をスラリーに添加した。スラリーを19Lの容積に減圧濃縮した。結晶化混合物を0℃から10℃にて2時間撹拌した。
形成された析出物をろ取し、ろ過ケーキをエタノール(10L)で洗浄した。生成物を20時間減圧乾燥し、化合物[17](3.11kg,8.73mol)のエタノール和物を99.85%のHPLC純度で得た。
ブタン-1-オール(43.7kg)を化合物[17](3.10kg)のエタノール和物に添加した。混合物を90℃から95℃に昇温し、2時間撹拌撹拌した。溶液を70℃から72℃に1時間で冷却し、化合物[17](14.6g,47.0mmol)を種晶として添加し、12時間かけて8℃に冷却した。
形成された析出物をろ取し、ろ過ケーキをブタン-1-オール(5.2kg)及び酢酸エチル(6L)で洗浄した。固体を55℃から65℃にて22時間減圧乾燥し、化合物[17](2.46kg,7.93mol)を99.91面積%のHPLC純度で得た。


同じ方法で合成したS-BBMO[38]粗生成物を濃縮乾固し、NMRとMSを測定した。
1H-NMR (DMSO-d6) δ: 7.36-7.13 (5H, m), 4.26 (1H, dd, J = 6.8, 3.9 Hz), 3.72 (2H, dd, J = 14.2, 6.8 Hz), 3.47-3.38 (1H, m), 3.30-3.08 (3H, m), 2.79 (1H, sext, J = 6.8 Hz), 1.35 (9H, s), 0.96 (3H, d, J = 6.8 Hz).
MS: m/z = 280 [M+H]+

同じ方法で合成したR-BCAB粗生成物を濃縮乾固し、NMRとMSを測定した。
1H-NMR (DMSO-d6) δ: 7.28-7.11 (5H, m), 4.24-4.11 (1H, m), 3.80 (2H, d, J = 3.6 Hz), 3.24 (2H, d, J = 3.6 Hz), 2.98-2.78 (2H, m), 1.46-1.37 (12H, m).
MS: m/z = 298 [M+H]+
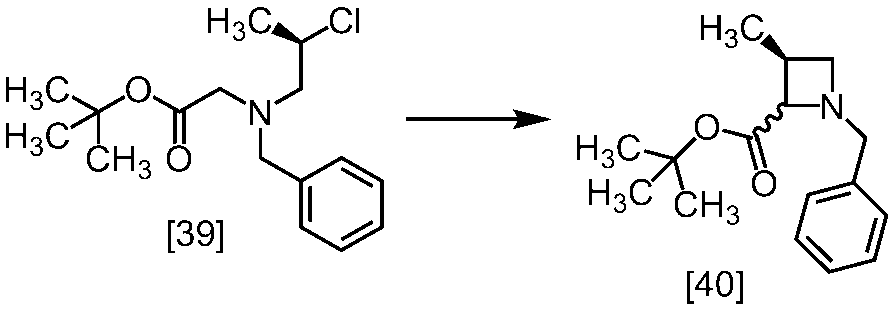
同じ方法で合成したS-MABB[40]粗生成物を濃縮乾固し、NMRとMSを測定した。
1H-NMR (DMSO-d6) δ: 7.28-7.25 (10H, m), 3.75 (1H, d, J = 12.7 Hz), 3.68 (1H, d, J = 1.4 Hz), 3.66 (1H, d, J = 6.7 Hz), 3.46 (2H, d, J = 12.7 Hz), 3.30-3.17 (2H, m), 2.95 (1H, dd, J = 6.2, 1.2 Hz), 2.77 (1H, dd, J = 6.1, 2.2 Hz), 2.65-2.55 (1H, m), 2.48-2.40 (2H, m), 1.35 (9H, s), 1.35 (9H, s), 1.12 (3H, d, J = 7.2 Hz), 1.09 (3H, d, J = 6.2 Hz).
MS: m/z = 262 [M+H]+
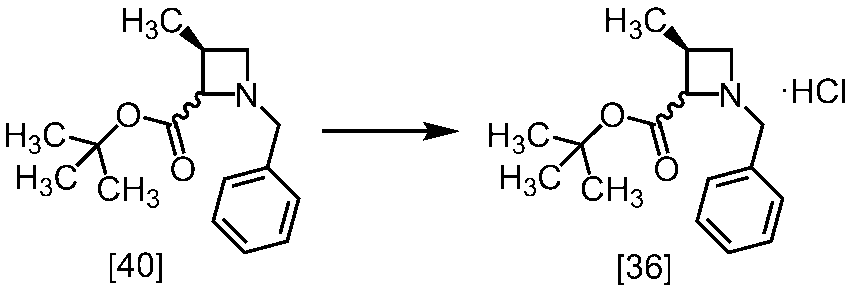
同じ方法で合成したS-MABB-HC[36]のNMR、MS及びCl含量を測定した。
1H-NMR (DMSO-d6) δ: 11.08 (1H, br s), 10.94 (1H, br s), 7.52-7.42 (10H, m), 5.34 (1H, t, J = 8.4 Hz), 4.90 (1H, br s), 4.45-4.10 (5H, m), 3.92-3.49 (3H, br m), 3.10-2.73 (2H, br m), 1.35 (9H, s), 1.29 (9H, s), 1.24 (3H, d, J = 6.7 Hz), 1.17 (3H, d, J = 7.4 Hz).
MS: m/z = 262 [M+H-HCl]+
Cl含量(イオンクロマトグラフィー):11.9%(理論値:11.9%)
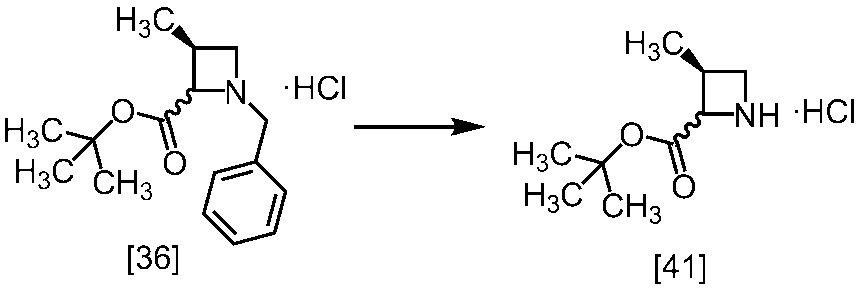
同じ方法で合成したS-MACB-HC[41]粗生成物を濃縮乾固し、NMRとMSを測定した。
1H-NMR (DMSO-d6) δ: 9.60 (br s, 1H), 4.97 (d, 1H, J = 9.2 Hz), 4.61 (d, 1H, J = 8.4 Hz), 4.01 (dd, 1H, J = 10.0, 8.4 Hz), 3.78-3.74 (m, 1H), 3.54 (dd, 1H, J = 9.6, 8.4 Hz), 3.35 (dd, 1H, J = 10.0, 6.0 Hz), 3.15-3.03 (m, 1H), 3.00-2.88 (m, 1H), 1.49 (s, 9H), 1.47 (s, 9H), 1.22 (d, 3H, J = 6.8 Hz), 1.14 (d, 3H, J = 7.2 Hz).
MS: m/z = 172 [M+H]+ (フリー体)

同じ方法で合成したS-ZMAB[42]粗生成物を濃縮乾固し、NMRとMSを測定した。
1H-NMR (CDCl3) δ: 7.38-7.28 (m, 10H), 5.16-5.04 (m, 4H), 4.60 (d, 1H, J = 9.2 Hz), 4.18-4.12 (m, 2H), 4.04 (t, 1H, J = 8.6 Hz), 3.66 (dd, 1H, J = 7.6, 7.2 Hz), 3.50 (dd, 1H, J = 8.0, 5.2 Hz), 3.05-2.94 (m, 1H), 2.60-2.50 (m, 1H), 1.43 (br s, 18H), 1.33 (d, 3H, J = 6.5 Hz), 1.15 (d, 3H, J = 7.2 Hz).
MS: m/z = 328 [M+Na]+
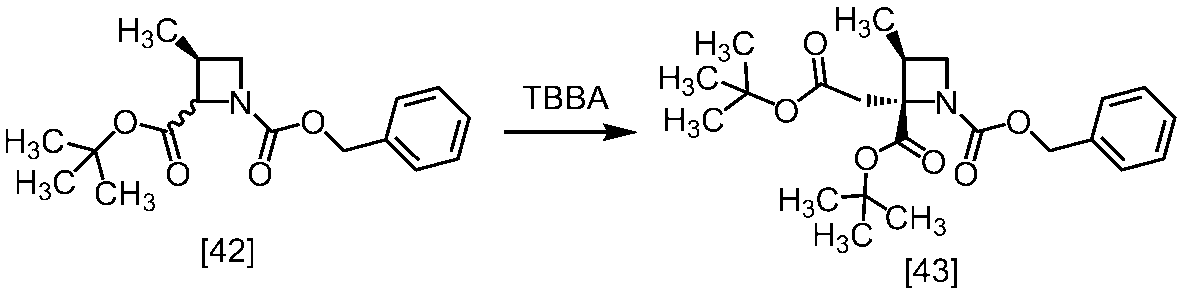
同じ方法で合成したRS-ZMBB[43]粗生成物を濃縮乾固し、NMRとMSを測定した。
1H-NMR (DMSO-d6) δ: 7.38-7.29 (m, 5H), 5.09-4.96 (m, 2H), 3.91 (t, 0.4H, J = 8.0 Hz), 3.79 (t, 0.6H, J = 8.0 Hz), 3.55 (t, 0.4H, J = 7.2 Hz), 3.46 (t, 0.6H, J = 7.5 Hz), 3.14-3.04 (m, 1H), 2.83-2.72 (m, 2H), 1.38 (br s, 9H), 1.37 (br s, 3.6H), 1.34 (br s, 5.4H), 1.12-1.09 (m, 3H).
MS: m/z = 420 [M+H]+
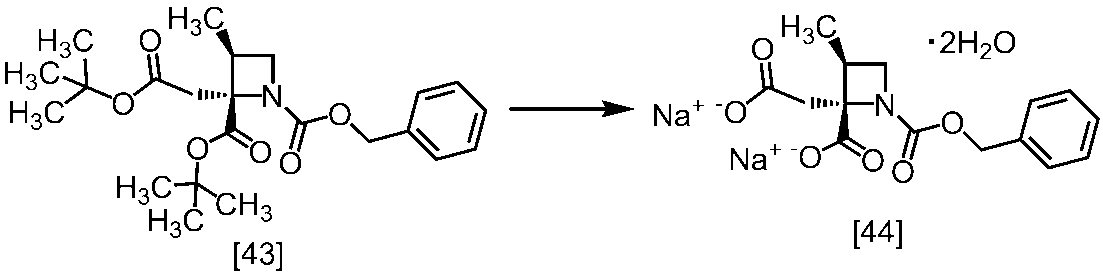
同じ方法で合成したRS-ZMAA-DN・2H2O[44]のNMR、MS、Na含量及び水分含量を測定した。
1H-NMR (DMSO-d6) δ: 7.32-7.22 (m, 5H), 4.97 (d, 1H, J = 12.7 Hz), 4.84 (d, 1H, J = 12.7 Hz), 3.79 (t, 1H, J = 8.0 Hz), 3.29 (d, 1H, J = 14.8 Hz), 3.16-3.12 (m, 1H), 2.17-2.09 (m, 2H), 1.07 (d, 3H, J = 6.9 Hz).
MS: m/z = 352 [M+H]+ (無水物)
Na含量(イオンクロマトグラフィー):13.3%(水分含量補正後)(理論値;13.1%)
水分含量(カール・フィッシャー法):9.8%(理論値;9.3%)
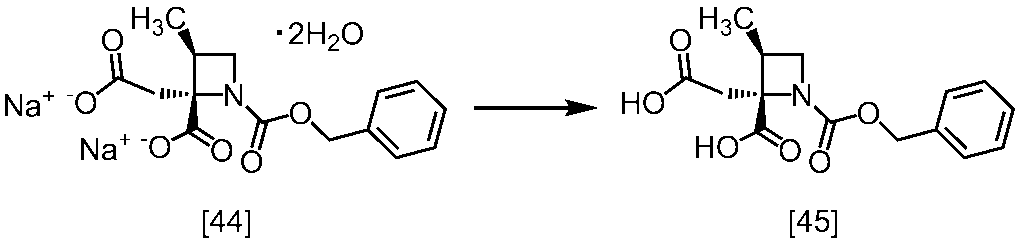
同じ方法で合成したRS-ZMAA[45]を濃縮し、NMRとMSを測定した。
1H-NMR (DMSO-D6) δ: 7.35-7.28 (m, 5H), 5.06-4.94 (m, 2H), 3.86 (dt, 1H, J = 48.4, 7.9 Hz), 3.50 (dt, 1H, J = 37.9, 7.4 Hz), 3.16-3.02 (br m, 1H), 2.91-2.77 (br m, 2H), 1.08 (d, 3H, J = 6.9 Hz)
MS: m/z = 308 [M+H]+

同じ方法で合成したRS-ZMOO[46]を濃縮し、NMRとMSを測定した。
1H-NMR (CDCl3) δ: 7.39-7.30 (m, 5H), 5.10 (s, 2H), 4.15-4.01 (br m, 2H), 3.83-3.73 (br m, 3H), 3.48 (dd, 1H, J = 8.3, 6.4 Hz), 2.59-2.50 (br m, 1H), 2.46-2.40 (br m, 1H), 2.07-1.99 (m, 1H), 1.14 (d, 3H, J = 7.2 Hz)
MS: m/z = 280 [M+H]+
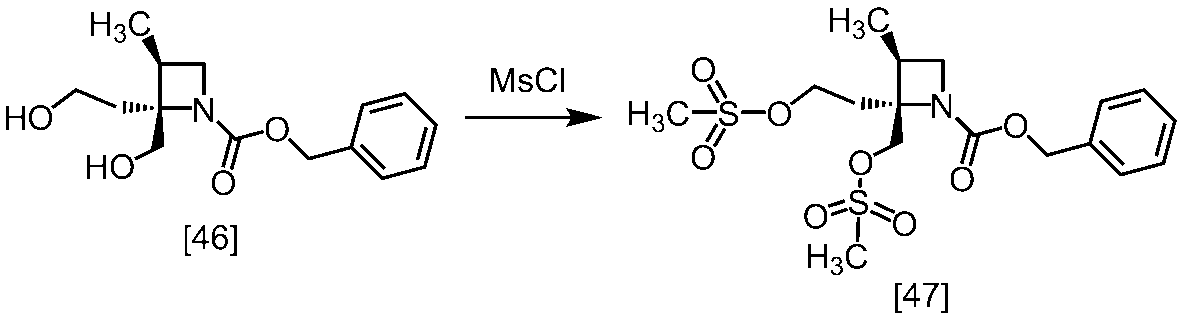
同じ方法で合成したRS-ZMSS[47]を濃縮乾固し、NMRとMSを測定した。
1H-NMR (DMSO-D6) δ: 7.37-7.27 (br m, 5H), 5.10-4.98 (m, 2H), 4.58-4.22 (br m, 4H), 3.84 (dt, 1H, J = 45.6, 8.1 Hz), 3.48-3.33 (br m, 1H), 3.17-3.10 (m, 6H), 2.81-2.74 (br m, 1H), 2.22-2.12 (m, 2H)
MS: m/z = 436 [M+H]+
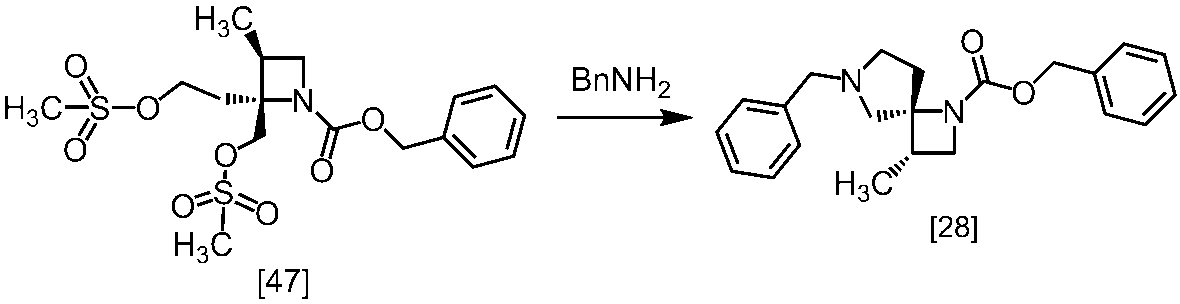
1H-NMR (CDCl3) δ: 7.35-7.20 (m, 10H), 5.08 (d, 2H, J = 23.6 Hz), 3.94 (q, 1H, J = 7.9 Hz), 3.73-3.42 (br m, 2H), 3.30-3.23 (m, 1H), 3.05 (dd, 1H, J = 19.7, 9.5 Hz), 2.79 (dt, 1H, J = 69.6, 6.1 Hz), 2.57-2.32 (br m, 4H), 1.96-1.89 (m, 1H), 1.09 (d, 3H, J = 6.9 Hz)
MS: m/z = 351 [M+H]+
Claims (43)
- 式[13]
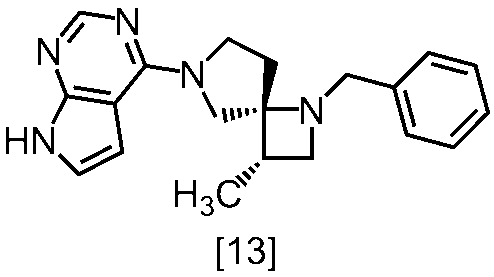
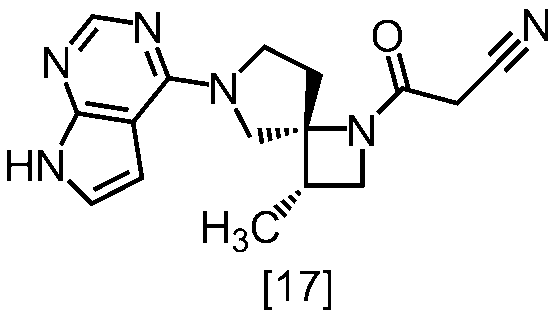
(1)式[13]の化合物又はその塩からベンジルを除去することにより式[14]
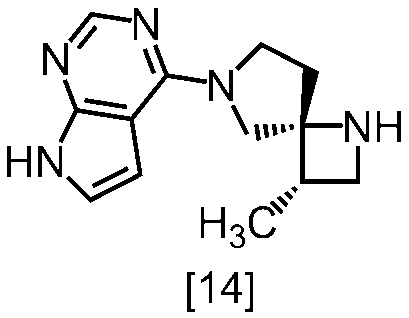
(2)式[14]の化合物又はその塩をシアノアセチル化することにより式[17]の化合物又はその塩を得る工程。 - 式[10]
- 式[10]の化合物に有機酸を添加することにより式[10]の化合物の有機酸との塩を得る工程をさらに含む、請求項2に記載の方法。
- 有機酸との塩が二コハク酸塩、シュウ酸塩又はヘミシュウ酸塩である、請求項2又は3に記載の方法。
- 有機酸との塩がヘミシュウ酸塩である、請求項2又は3に記載の方法。
- 式[9]
- 還元が酸及び水素化アルミニウムリチウムの存在下で行われる、請求項6に記載の方法。
- 式[8a]

の化合物からフタロイルを除去することにより式[9]の化合物を得る工程をさらに含む、請求項6又は7に記載の方法。 - 式[16a]

の化合物からホルミルを除去することにより式[9]の化合物を得る工程をさらに含む、請求項6又は7に記載の方法。 - 式[7]
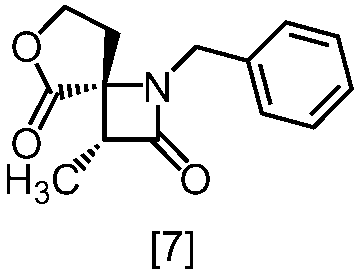
- 式[7]

- 式[6]

- 塩基がリチウムヘキサメチルジシラジドである、請求項12に記載の方法。
- 式[27a]
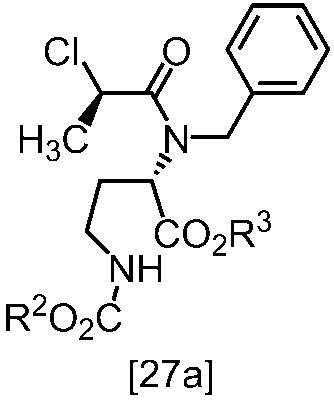
の化合物を環化させることにより式[9]の化合物を得る工程をさらに含む、請求項6に記載の方法。 - 式[26a]
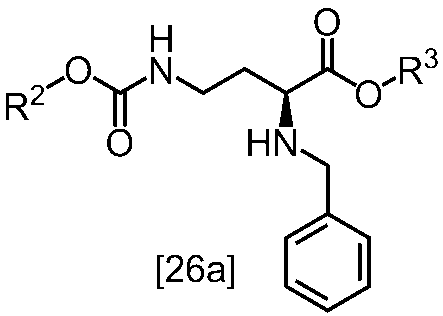
の化合物又はその塩と式[5]
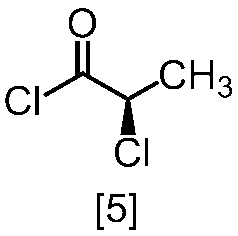
- 式[4]

- 式[25a]
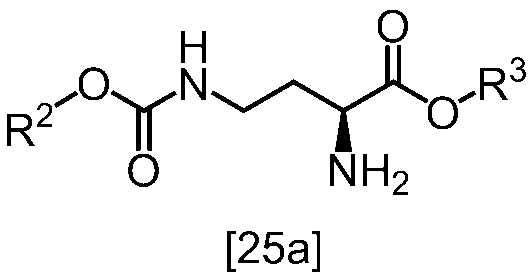
の化合物又はその塩とベンズアルデヒドを反応させることにより式[26a]の化合物又はその塩を得る工程をさらに含む、請求項15又は16に記載の方法。 - 式[24a]

の化合物又はその塩をエステル化することにより式[25a]の化合物又はその塩を得る工程をさらに含む、請求項17に記載の方法。 - 式[31]
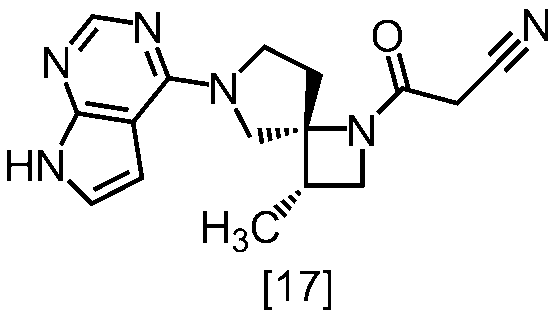
(1)式[31]の化合物又はその有機酸との塩と式[12]


(2)式[14]の化合物又はその塩をシアノアセチル化することにより式[17]の化合物又はその塩を得る工程。 - 式[31]の化合物に有機酸を添加することにより式[31]の化合物の有機酸との塩を得る工程をさらに含む、請求項19に記載の方法。
- 有機酸との塩が二コハク酸塩又はシュウ酸塩である、請求項19又は20に記載の方法。
- 式[28]

- 式[28]の化合物に有機酸を添加することにより式[28]の化合物の有機酸との塩を得る工程をさらに含む、請求項22に記載の方法。
- 式[28]の化合物の有機酸との塩がシュウ酸塩である、請求項22又は23に記載の方法。
- 式[13]

- 式[10]
- 有機酸との塩が二コハク酸塩、シュウ酸塩又はヘミシュウ酸塩である、請求項26に記載の塩。
- 有機酸との塩がヘミシュウ酸塩である、請求項26に記載の塩。
- 式[10]
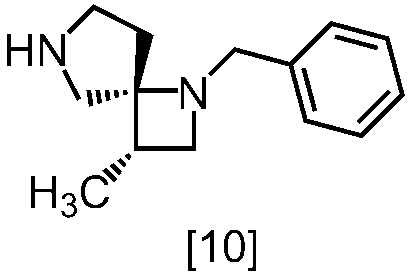
- 式[9]

- 式[9]

- 式[8a]
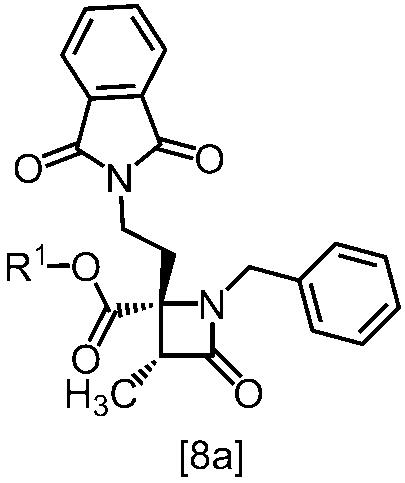
の化合物。 - 式[16a]
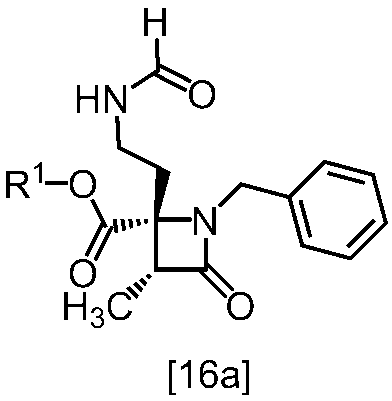
の化合物。 - 式[7]
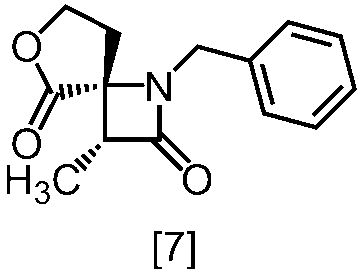
- 式[6]
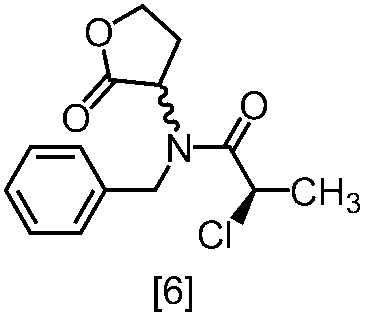
- 式[27a]
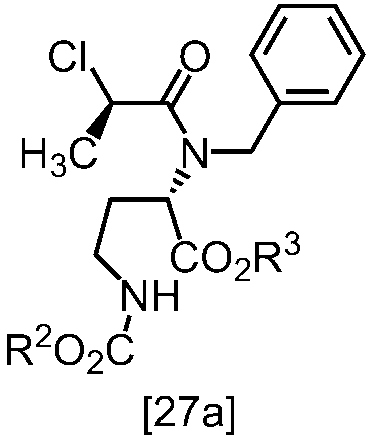
の化合物。 - 式[26a]

の化合物又はその塩。 - 式[25a]

の化合物又はその塩。 - 式[24a]
の化合物又はその塩。 - 式[31]
- 有機酸との塩が二コハク酸塩又はシュウ酸塩である、請求項40に記載の塩。
- 式[28]

- 有機酸との塩がシュウ酸塩である、請求項42に記載の塩。
Priority Applications (15)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BR112019012688-0A BR112019012688A2 (pt) | 2016-12-21 | 2017-12-20 | método para produção de derivado de 7h-pirrol[2,3-d]pirimidina e intermediário sintético do mesmo |
| AU2017384317A AU2017384317C1 (en) | 2016-12-21 | 2017-12-20 | PROCESS FOR PREPARING 7H-PYRROLO[2,3-d]PYRIMIDINE DERIVATIVES AND SYNTHETIC INTERMEDIATES THEREOF |
| EP17885424.6A EP3560932A4 (en) | 2016-12-21 | 2017-12-20 | PROCESS FOR THE PREPARATION OF 7H-PYRROLO [2,3-D] PYRIMIDINE DERIVATIVES AND SYNTHETIC INTERMEDIATE THEREOF |
| RU2019122691A RU2776802C2 (ru) | 2016-12-21 | 2017-12-20 | Способ получения производных 7h-пирроло[2,3-d]пиримидина и промежуточных продуктов для их синтеза |
| CN201780086625.6A CN110300756B (zh) | 2016-12-21 | 2017-12-20 | 7H-吡咯并[2,3-d]嘧啶衍生物的制备方法及其合成中间体 |
| CN202410021834.4A CN117865992A (zh) | 2016-12-21 | 2017-12-20 | 7H-吡咯并[2,3-d]嘧啶衍生物的制备方法及其合成中间体 |
| MX2019007542A MX2019007542A (es) | 2016-12-21 | 2017-12-20 | Proceso para la preparación de derivados de 7h-pirrolo [2, 3-d] pirimidina y sus intermedios sintéticos. |
| CA3047891A CA3047891A1 (en) | 2016-12-21 | 2017-12-20 | Process for preparing 7h-pyrrolo[2,3-d]pyrimidine derivatives and synthetic intermediates thereof |
| JP2018518551A JP6434678B2 (ja) | 2016-12-21 | 2017-12-20 | 7H−ピロロ[2,3−d]ピリミジン誘導体の製造方法及びその合成中間体 |
| US16/470,888 US11673900B2 (en) | 2016-12-21 | 2017-12-20 | Process for preparing 7H-pyrrolo[2,3-d]pyrimidine derivatives and synthetic intermediates thereof |
| KR1020197020824A KR102546513B1 (ko) | 2016-12-21 | 2017-12-20 | 7H-피롤로[2,3-d]피리미딘 유도체의 제조 방법 및 그의 합성 중간체 |
| IL288080A IL288080B2 (en) | 2016-12-21 | 2017-12-20 | A process for the preparation of H7-pyrrolo[3,2-d]pyrimidine derivatives and their artificial intermediates |
| IL267385A IL267385B2 (en) | 2016-12-21 | 2019-06-16 | A process for the preparation of h7-pyrrolo[3,2-d]pyrimidine derivatives and their artificial intermediates |
| AU2022203506A AU2022203506B2 (en) | 2016-12-21 | 2022-05-24 | PROCESS FOR PREPARING 7H-PYRROLO[2,3-d]PYRIMIDINE DERIVATIVES AND SYNTHETIC INTERMEDIATES THEREOF |
| US18/136,661 US20240140967A1 (en) | 2016-12-21 | 2023-04-19 | PROCESS FOR PREPARING 7H-PYRROLO[2,3-d]PYRIMIDINE DERIVATIVES AND SYNTHETIC INTERMEDIATES THEREOF |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016247607 | 2016-12-21 | ||
| JP2016-247607 | 2016-12-21 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US16/470,888 A-371-Of-International US11673900B2 (en) | 2016-12-21 | 2017-12-20 | Process for preparing 7H-pyrrolo[2,3-d]pyrimidine derivatives and synthetic intermediates thereof |
| US18/136,661 Continuation US20240140967A1 (en) | 2016-12-21 | 2023-04-19 | PROCESS FOR PREPARING 7H-PYRROLO[2,3-d]PYRIMIDINE DERIVATIVES AND SYNTHETIC INTERMEDIATES THEREOF |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018117152A1 true WO2018117152A1 (ja) | 2018-06-28 |
Family
ID=62627423
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/045729 WO2018117152A1 (ja) | 2016-12-21 | 2017-12-20 | 7H-ピロロ[2,3-d]ピリミジン誘導体の製造方法及びその合成中間体 |
Country Status (12)
| Country | Link |
|---|---|
| US (2) | US11673900B2 (ja) |
| EP (1) | EP3560932A4 (ja) |
| JP (2) | JP6434678B2 (ja) |
| KR (1) | KR102546513B1 (ja) |
| CN (2) | CN110300756B (ja) |
| AU (2) | AU2017384317C1 (ja) |
| BR (1) | BR112019012688A2 (ja) |
| CA (1) | CA3047891A1 (ja) |
| IL (2) | IL288080B2 (ja) |
| MX (1) | MX2019007542A (ja) |
| TW (2) | TWI816651B (ja) |
| WO (1) | WO2018117152A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111560021A (zh) * | 2020-06-30 | 2020-08-21 | 苏州明锐医药科技有限公司 | 一种德高替尼中间体及其制备方法 |
| US10822354B2 (en) | 2015-07-07 | 2020-11-03 | Japan Tobacco Inc. | Method for producing 7h-pyrrolo[2, 3-d]pyrimidine derivative and intermediate thereof |
| US11673900B2 (en) | 2016-12-21 | 2023-06-13 | Japan Tobacco Inc. | Process for preparing 7H-pyrrolo[2,3-d]pyrimidine derivatives and synthetic intermediates thereof |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111574540B (zh) * | 2020-06-30 | 2023-08-29 | 云南华派医药科技有限公司 | 一种德高替尼的制备方法 |
| CN111606929B (zh) * | 2020-06-30 | 2023-07-07 | 中瀚(齐河县)生物医药科技有限公司 | 德高替尼的制备方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010504351A (ja) * | 2006-09-22 | 2010-02-12 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | バソプレッシン拮抗薬として使用されるスピロベンズアゼピン類 |
| WO2011013785A1 (ja) | 2009-07-31 | 2011-02-03 | 日本たばこ産業株式会社 | 含窒素スピロ環化合物及びその医薬用途 |
| JP2016512195A (ja) * | 2013-03-05 | 2016-04-25 | セラヴァンス バイオファーマ アール&ディー アイピー, エルエルシー | ネプリライシン阻害剤 |
| WO2017006968A1 (ja) * | 2015-07-07 | 2017-01-12 | 日本たばこ産業株式会社 | 7H-ピロロ[2,3-d]ピリミジン誘導体の製造方法及びその中間体 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7662960B2 (en) | 2001-04-26 | 2010-02-16 | Choongwae Pharma Corporation | Beta-strand mimetics and method relating thereto |
| WO2004108731A1 (en) * | 2003-05-30 | 2004-12-16 | Choongwae Pharma Corporation | Beta-strand mimetics and method relating thereto |
| US7230119B2 (en) | 2004-08-25 | 2007-06-12 | Biocryst Pharmaceuticals, Inc. | Process for the preparation of substituted pyrrolidine derivatives and intermediates |
| MX2013008257A (es) | 2011-01-19 | 2013-08-12 | Galapagos Nv | Derivados de azetidina utiles para el tratamiento de enfermedades metabolicas e inflamatorias. |
| CN103570601B (zh) | 2012-07-20 | 2016-03-30 | 重庆博腾制药科技股份有限公司 | 一种光学活性药物中间体的制备方法 |
| JP6500634B2 (ja) * | 2015-06-24 | 2019-04-17 | 株式会社Ihi | 溶接装置及び溶接方法 |
| TWI816651B (zh) | 2016-12-21 | 2023-10-01 | 日商日本煙草產業股份有限公司 | 7H-吡咯并[2,3-d]嘧啶衍生物的製造方法及其合成中間體 |
| US11339181B2 (en) * | 2016-12-21 | 2022-05-24 | Japan Tobacco Inc. | Crystalline forms of a Janus kinase inhibitor |
-
2017
- 2017-12-20 TW TW106144830A patent/TWI816651B/zh active
- 2017-12-20 US US16/470,888 patent/US11673900B2/en active Active
- 2017-12-20 MX MX2019007542A patent/MX2019007542A/es unknown
- 2017-12-20 AU AU2017384317A patent/AU2017384317C1/en active Active
- 2017-12-20 KR KR1020197020824A patent/KR102546513B1/ko active IP Right Grant
- 2017-12-20 EP EP17885424.6A patent/EP3560932A4/en active Pending
- 2017-12-20 CA CA3047891A patent/CA3047891A1/en active Pending
- 2017-12-20 JP JP2018518551A patent/JP6434678B2/ja active Active
- 2017-12-20 CN CN201780086625.6A patent/CN110300756B/zh active Active
- 2017-12-20 WO PCT/JP2017/045729 patent/WO2018117152A1/ja unknown
- 2017-12-20 TW TW112133483A patent/TW202400613A/zh unknown
- 2017-12-20 BR BR112019012688-0A patent/BR112019012688A2/pt active Search and Examination
- 2017-12-20 IL IL288080A patent/IL288080B2/en unknown
- 2017-12-20 CN CN202410021834.4A patent/CN117865992A/zh active Pending
-
2018
- 2018-09-28 JP JP2018185491A patent/JP7098496B2/ja active Active
-
2019
- 2019-06-16 IL IL267385A patent/IL267385B2/en unknown
-
2022
- 2022-05-24 AU AU2022203506A patent/AU2022203506B2/en active Active
-
2023
- 2023-04-19 US US18/136,661 patent/US20240140967A1/en active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010504351A (ja) * | 2006-09-22 | 2010-02-12 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | バソプレッシン拮抗薬として使用されるスピロベンズアゼピン類 |
| WO2011013785A1 (ja) | 2009-07-31 | 2011-02-03 | 日本たばこ産業株式会社 | 含窒素スピロ環化合物及びその医薬用途 |
| JP2016512195A (ja) * | 2013-03-05 | 2016-04-25 | セラヴァンス バイオファーマ アール&ディー アイピー, エルエルシー | ネプリライシン阻害剤 |
| WO2017006968A1 (ja) * | 2015-07-07 | 2017-01-12 | 日本たばこ産業株式会社 | 7H-ピロロ[2,3-d]ピリミジン誘導体の製造方法及びその中間体 |
Non-Patent Citations (8)
| Title |
|---|
| BANKOWSKI, KRZYSZTOF ET AL.: "Synthesis, biological activity and resistance to proteolytic digestion of new cyclic dermorphin/deltorphin analogues", EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, vol. 63, 19 February 2013 (2013-02-19), pages 457 - 467, XP029233981, ISSN: 0223-5234, DOI: 10.1016/j.ejmech.2013.02.019 * |
| DROUILLAT, BRUNO ET AL.: "Insight into the Regioselectivity of Nucleophilic Ring-Opening of Azetidinium Ions Containing Quaternary Carbon Atoms", EUROPEAN JOURNAL OF ORGANIC CHEMISTRY, vol. 2012, no. 30, 7 September 2012 (2012-09-07), pages 6005 - 6012, XP055605878, ISSN: 1099-0690, DOI: 10.1002/ejoc.201200890 * |
| KUNZ, WALTER ET AL.: "91. Bildung von 4-, 5-und 6gliedrigen Heterocyclen durch ambidoselektive Ringschlusse von Enolat-Ionen", HELVETICA CHIMICA ACTA, vol. 62, no. 3, 1 January 1979 (1979-01-01), pages 872 - 881, XP055613274 * |
| LOH, T. -P. ET AL.: "A remote substituent as a control element in indium-mediated allylation reactions in aqueous media: highly diastereoselective synthesis of 1, 3-amino alcohols", TETRAHEDRON LETTERS, vol. 41, no. 33, 2000, pages 6511 - 6515, XP004215096, ISSN: 0040-4039, DOI: 10.1016/S0040-4039(00)01040-6 * |
| MAJER, ZSUZSANNA ET AL.: "Spirobis [2-pyrrolidinones] . II. Synthesis and absolute configuration of 1, 7-diazaspiro[4,4]nonane-2, 6-dione", COLLECTION OF CZECHOSLOVAK CHEMICAL COMMUNICATIONS, vol. 47, no. 3, 1982, pages 950 - 960, XP055605862, ISSN: 0366-547X, DOI: 10.1135/cccc19820950 * |
| NUNEZ-VILLANUEVA, DIEGO ET AL.: "Quaternary a, alpha-2-Oxoazepane a-Amino Acids: Synthesis from Ornithine-Derived beta-Lactams and Incorporation into Model Dipeptides", JOURNAL OF ORGANIC CHEMISTRY- JOC, vol. 76, no. 16, 30 June 2011 (2011-06-30), pages 6592 - 6603, XP055605874, ISSN: 0022-3263, DOI: 10.1021/jo200894d·Source: * |
| STACY, DM ET AL.: "Synthesis and biological evaluation of triazole-containing N-acyl homoserine lactones as quorum sensing modulators", ORG BIOMOL CHEM., vol. 11, no. 6, 14 February 2013 (2013-02-14), pages 938 - 954, XP055094516, DOI: 10.1039/C2OB27155A |
| VAN NISPEN, J. W. ET AL.: "Synthesis and biological activities of two ACTH-analogues containing L-norarginine in position 8", INTERNATIONAL JOURNAL OF PEPTIDE & PROTEIN RESEARCH, vol. 9, no. 3, March 1977 (1977-03-01), pages 193 - 202, XP055605866, ISSN: 0367-8377 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10822354B2 (en) | 2015-07-07 | 2020-11-03 | Japan Tobacco Inc. | Method for producing 7h-pyrrolo[2, 3-d]pyrimidine derivative and intermediate thereof |
| US11673900B2 (en) | 2016-12-21 | 2023-06-13 | Japan Tobacco Inc. | Process for preparing 7H-pyrrolo[2,3-d]pyrimidine derivatives and synthetic intermediates thereof |
| CN111560021A (zh) * | 2020-06-30 | 2020-08-21 | 苏州明锐医药科技有限公司 | 一种德高替尼中间体及其制备方法 |
| CN111560021B (zh) * | 2020-06-30 | 2023-05-26 | 上海鲲博玖瑞医药科技发展有限公司 | 一种德高替尼中间体及其制备方法 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6434678B2 (ja) | 7H−ピロロ[2,3−d]ピリミジン誘導体の製造方法及びその合成中間体 | |
| JP6871856B2 (ja) | 7H−ピロロ[2,3−d]ピリミジン誘導体の製造方法及びその中間体 | |
| JP6359791B1 (ja) | 7H−ピロロ[2,3−d]ピリミジン誘導体の製造方法及びその共結晶 | |
| AU2015414743B2 (en) | Process for the preparation of kinase inhibitors and intermediates thereof | |
| RU2776802C2 (ru) | Способ получения производных 7h-пирроло[2,3-d]пиримидина и промежуточных продуктов для их синтеза |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2018518551 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17885424 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3047891 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112019012688 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2017384317 Country of ref document: AU Date of ref document: 20171220 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20197020824 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2017885424 Country of ref document: EP Effective date: 20190722 |
|
| ENP | Entry into the national phase |
Ref document number: 112019012688 Country of ref document: BR Kind code of ref document: A2 Effective date: 20190619 |