WO2017141772A1 - 樹脂用改質剤、樹脂組成物、およびこれを用いたフィルム - Google Patents
樹脂用改質剤、樹脂組成物、およびこれを用いたフィルム Download PDFInfo
- Publication number
- WO2017141772A1 WO2017141772A1 PCT/JP2017/004427 JP2017004427W WO2017141772A1 WO 2017141772 A1 WO2017141772 A1 WO 2017141772A1 JP 2017004427 W JP2017004427 W JP 2017004427W WO 2017141772 A1 WO2017141772 A1 WO 2017141772A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- resin
- modifier
- acid
- mass
- glycol
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/66—Polyesters containing oxygen in the form of ether groups
- C08G63/668—Polyesters containing oxygen in the form of ether groups derived from polycarboxylic acids and polyhydroxy compounds
- C08G63/672—Dicarboxylic acids and dihydroxy compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/18—Manufacture of films or sheets
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L1/00—Compositions of cellulose, modified cellulose or cellulose derivatives
- C08L1/02—Cellulose; Modified cellulose
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L67/00—Compositions of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Compositions of derivatives of such polymers
- C08L67/02—Polyesters derived from dicarboxylic acids and dihydroxy compounds
- C08L67/025—Polyesters derived from dicarboxylic acids and dihydroxy compounds containing polyether sequences
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2301/00—Characterised by the use of cellulose, modified cellulose or cellulose derivatives
- C08J2301/02—Cellulose; Modified cellulose
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2301/00—Characterised by the use of cellulose, modified cellulose or cellulose derivatives
- C08J2301/08—Cellulose derivatives
- C08J2301/10—Esters of organic acids
- C08J2301/12—Cellulose acetate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2467/00—Characterised by the use of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Derivatives of such polymers
- C08J2467/02—Polyesters derived from dicarboxylic acids and dihydroxy compounds
Definitions
- the present invention relates to a resin modifier, a resin composition, and a film using the same, and more specifically, a resin modifier and a resin composition capable of highly balancing flexibility, transparency, and bleed resistance. And a film using the same.
- Cellulosic resins are generally tougher than other synthetic resins, have excellent characteristics such as excellent transparency, gloss and gloss, and a smooth surface and good feel. For this reason, the use of the cellulose-based resin is very diverse, for example, a sheet, a film, a wire coating, a toy, a medical device or a food packaging material.
- plasticizers include, for example, triphenyl phosphate, tricresyl phosphate, diphenyl phosphate, triethyl phosphate, tributyl phosphate, dimethyl phthalate, diethyl phthalate, dimethoxyethyl phthalate, ethyl phthalyl ethyl glycolate, butyl phthalyl butyl glycol. Rate, toluenesulfonamide, triacetin or pentaerythritol tetraacetate have been used.
- plasticizers can satisfy a wide range of performances such as compatibility with cellulosic resins, plasticization efficiency, non-volatility, stability to heat and light, non-migration, non-extractability, and water resistance. There is nothing, which hinders further expansion of the use of the cellulose resin composition.
- Patent Document 1 proposes a cellulose resin composition obtained by adding a polyester plasticizer to a cellulose resin.
- This polyester plasticizer is obtained by polycondensation of a polybasic acid, a polyhydric alcohol, and an ether alcohol, and 10 to 70 mol% of the polybasic acid is composed of an aromatic dicarboxylic acid. It is.
- the polyester plasticizer disclosed in Patent Document 1 prevents deterioration of retention of thermoplastic resins, particularly cellulose resins.
- the retentivity refers to the property of reducing the mass of the film due to precipitation or volatilization of additives such as plasticizers outside the film in a hot and humid environment.
- the polyester-based plasticizer proposed in Patent Document 1 it is difficult to highly balance the flexibility, transparency and bleed resistance of the resin composition.
- an object of the present invention is to provide a resin modifier, a resin composition, and a film using the same, which can highly balance flexibility, transparency and bleed resistance.
- the resin modifier of the present invention comprises a structural unit derived from an aliphatic dibasic acid, a structural unit derived from an alkylene diol, and a structural unit derived from a polyalkylene ether glycol, and the terminal is a hydroxyl group. Or it consists of a random copolymer which is a carboxyl group.
- the polyalkylene ether glycol is preferably polyethylene ether glycol.
- the alkylene diol includes ethylene glycol, 1,2-propylene glycol, 1,3-propanediol, 2-methyl-1,3-propanediol, 1,3- It is preferably at least one selected from the group consisting of butanediol, 1,4-butanediol, neopentyl glycol, 3-methyl-1,5-pentanediol, and hexanediol.
- the mass ratio of the structural unit derived from the aliphatic dibasic acid, the structural unit derived from the alkylene diol, and the structural unit derived from the polyalkylene ether glycol is: Each is preferably 10 to 80% by mass, 5 to 80% by mass, and 1 to 50% by mass.
- the weight average molecular weight is preferably 1000 to 3000.
- the resin composition of the present invention is characterized in that the resin modifier of the present invention is added to a resin.
- the resin is preferably a cellulose resin.
- the resin modifier is preferably 3 to 100 parts by mass with respect to 100 parts by mass of the resin.
- the film of the present invention is characterized by comprising the resin composition of the present invention.
- the present invention it is possible to provide a modifier for resin, a resin composition, and a film using the same that can highly balance flexibility, transparency and bleed resistance.
- the modifier for resin of the present invention (hereinafter also referred to as “modifier”) includes a structural unit derived from an aliphatic dibasic acid, a structural unit derived from an alkylene diol, and a structure derived from a polyalkylene ether glycol. And a random copolymer having a terminal that is a hydroxyl group or a carboxyl group.
- modifyifier includes a structural unit derived from an aliphatic dibasic acid, a structural unit derived from an alkylene diol, and a structure derived from a polyalkylene ether glycol. And a random copolymer having a terminal that is a hydroxyl group or a carboxyl group.
- examples of the aliphatic dibasic acid constituting the structural unit derived from the aliphatic dibasic acid include oxalic acid, malonic acid, succinic acid, glutaric acid, adipic acid, pimelic acid, Saturated aliphatic dibasic acids such as suberic acid, azelaic acid, sebacic acid, undecanedioic acid, dodecanedioic acid, tridecanedioic acid, tetradecanedioic acid, for example, maleic acid, fumaric acid, itaconic acid, citraconic acid, mesaconic acid And unsaturated aliphatic dibasic acids such as Among these, succinic acid, glutaric acid, adipic acid, pimelic acid, suberic acid, azelaic acid, and sebacic acid are more preferable, and adipic acid is particularly preferable.
- the aliphatic dibasic acid constituting the structural unit derived from the
- examples of the alkylene diol constituting the structural unit derived from the alkylene diol include ethylene glycol, 1,2-propylene glycol, 1,3-propanediol, 1,3-butanediol, 1,4-butanediol, pentanediol, 3-methyl-1,5-pentanediol, hexanediol, neopentyl glycol, dimethylol heptane (2-butyl-2-ethyl-1,3-propanediol), nonanediol 2,4-diethyl-1,5-pentanediol, 2-methyl-1,4-butanediol, octanediol, 2-ethylhexanediol, 2-methyl-1,3-propanediol, etc.
- ethylene glycol, 1,2-propylene glycol, 1,3-propanediol, 2-methyl-1,3-propanediol, 1,3-butanediol, 1,4-butanediol, neopentyl glycol, 3-methyl-1,5-pentanediol and hexanediol are more preferable, and ethylene glycol, 1,2-propylene glycol, 1,3-butanediol, and 3-methyl-1,5-pentanediol are particularly preferable.
- the alkylene diol constituting the structural unit derived from the alkylene diol may be one type or two or more types.
- the polyalkylene ether glycol constituting the structural unit derived from polyalkylene ether glycol includes polymethylene ether glycol, polyethylene ether glycol, poly-1,2-propylene ether glycol, polytrimethylene.
- polyethylene ether glycol is more preferable.
- polyalkylene ether glycols those having a weight average molecular weight of 100 to 600 are more preferred, those having 150 to 300 are more preferred, and those having 150 to 200 are particularly preferred.
- the polyalkylene ether glycol constituting the structural unit derived from the polyalkylene ether glycol may be one type or two or more types.
- the modifier of the present invention comprises a structural unit derived from an aliphatic dibasic acid, a structural unit derived from an alkylene diol, and a structural unit derived from a polyalkylene ether glycol, and the terminal is a hydroxyl group or a carboxyl group.
- the weight average molecular weight is preferably 1000 to 3000, and more preferably 1500 to 2500. By setting the weight average molecular weight within this range, the effects of the present invention can be obtained satisfactorily.
- the acid value of the modifier is preferably 1 or less.
- the mass ratio of the structural unit derived from the aliphatic dibasic acid, the structural unit derived from the alkylene diol, and the structural unit derived from the polyalkylene ether glycol is 10 to 80 masses, respectively. %, 5 to 80% by mass and 1 to 50% by mass. More preferably, the aliphatic dibasic acid is 30 to 70% by mass, the alkylene diol is 10 to 60% by mass, and the polyalkylene ether glycol is 5 to 40% by mass.
- the resin modifier of the present invention can be produced by a known method.
- it can be produced using an aliphatic dibasic acid, an alkylene diol, and a polyalkylene ether glycol in the presence of a catalyst such as dioctyl zirconium oxide, dibutyl tin oxide, or tetraalkyl titanate.
- a catalyst such as dioctyl zirconium oxide, dibutyl tin oxide, or tetraalkyl titanate.
- the ratio of each component used in the production can be appropriately designed according to the type of the component used and the properties of the intended modifier, the molecular weight, etc., but preferably an aliphatic dibasic acid is used.
- alkylene diol is used in an amount of 5 to 80% by mass
- polyalkylene ether glycol is used in a ratio of 1 to 50% by mass. More preferably, an aliphatic dibasic acid is used in an amount of 30 to 70% by mass. Is used in a proportion of 10 to 60% by weight and polyalkylene ether glycol in a proportion of 5 to 40% by weight.
- the resin composition of the present invention is obtained by adding the resin modifier of the present invention to a resin.
- the resin is preferably a cellulose-based resin, but is not limited thereto, and may be a thermoplastic resin or a thermosetting resin.
- a lower fatty acid ester of cellulose is preferable.
- the lower fatty acid means a fatty acid having 6 or less carbon atoms.
- Examples of the lower fatty acid ester of cellulose include, for example, cellulose acetate, cellulose propionate, cellulose butyrate and the like, JP-A-10-45804, JP-A-8-231761, and US Pat. No. 2,319,052.
- Examples thereof include mixed fatty acid esters such as cellulose acetate propionate and cellulose acetate butyrate, cellulose triacetate, etc.
- a cellulose ester synthesized using cotton linter, wood pulp, kenaf or the like as a raw material can be used alone or in combination.
- a cellulose ester synthesized from cotton linter alone or in combination it is preferable to use a cellulose ester synthesized from cotton linter alone or in combination.
- thermoplastic resin examples include polyvinyl chloride resin, polyethylene resin, polypropylene resin, polystyrene resin, polyvinyl acetate resin, polyurethane resin, cellulose resin, acrylic resin, AS ( (Acrylonitrile-styrene) resin, ABS (acrylonitrile-butadiene-styrene) resin, fluorine resin, thermoplastic elastomer, polyamide resin, polyacetal resin, polycarbonate resin, modified polyphenylene ether resin, polyethylene terephthalate resin, polybutylene terephthalate resin, polylactic acid Examples thereof include resins, cyclic polyolefin resins, polyphenylene sulfide resins, and the like. These resins may be used alone or in combination of two or more.
- polyvinyl chloride resin examples include homopolymers such as polyvinyl chloride and polyvinylidene chloride, and vinyl compound copolymers such as vinyl chloride or a copolymer of vinylidene chloride and vinyl acetate. These may be used alone or in combination of two or more.
- polyethylene resin examples include low density polyethylene, medium density polyethylene, and high density polyethylene. These may be used alone or in combination of two or more.
- the polymerization method is not particularly limited, and examples thereof include homopolymers, random polymers, block polymers and the like. These may be used alone or in combination of two or more.
- acrylic resin examples include polymethyl acrylate and polymethyl methacrylate. These may be used alone or in combination of two or more.
- polyamide resin examples include aliphatic polyamides such as nylon-6, nylon-66, nylon-10, nylon-12, and nylon-46, and aromatic polyamides produced from aromatic dicarboxylic acids and aliphatic diamines. It is done. These may be used alone or in combination of two or more.
- polyacetal resin examples include polyformaldehyde, polyacetaldehyde, polypropionaldehyde, polybutyraldehyde, and the like. These may be used alone or in combination of two or more.
- the addition amount of the modifier is 3 to 100 parts by mass, preferably 5 to 80 parts by mass with respect to 100 parts by mass of the resin. This is because if the addition amount of the modifier is less than 3 parts by mass, the effect of imparting flexibility may not be sufficiently obtained, and if it is used in excess of 100 parts by mass, bleeding may occur.
- various additives such as phosphorus antioxidants, phenol antioxidants or sulfur antioxidants, ultraviolet absorbers, hindered amine light stabilizers and the like may be further blended. it can.
- phosphorus antioxidants include triphenyl phosphite, tris (2,4-ditert-butylphenyl) phosphite, tris (nonylphenyl) phosphite, tris (dinonylphenyl) phosphite, tris (mono , Dimixed nonylphenyl) phosphite, bis (2-tert-butyl-4,6-dimethylphenyl) ⁇ ethyl phosphite, diphenyl acid phosphite, 2,2′-methylenebis (4,6-ditert-butylphenyl) ) Octyl phosphite, diphenyl decyl phosphite, phenyl diisodecyl phosphite, tributyl phosphite, tris (2-ethylhexyl) phosphite, tridecyl
- phenolic antioxidants examples include 2,6-ditert-butyl-p-cresol, 2,6-diphenyl-4-octadecyloxyphenol, stearyl (3,5-ditert-butyl-4-hydroxy). Phenyl) propionate, distearyl (3,5-ditert-butyl-4-hydroxybenzyl) phosphonate, tridecyl 3,5-ditert-butyl-4-hydroxybenzylthioacetate, thiodiethylenebis [(3,5- Di-tert-butyl-4-hydroxyphenyl) propionate], 4,4′-thiobis (6-tert-butyl-m-cresol), 2-octylthio-4,6-di (3,5-di-tert-butyl- 4-hydroxyphenoxy) -s-triazine, 2,2′-methylenebis (4-methyl-6-tert-butylphenol), bis [3,3-bi (4-Hydroxy-3-ter
- sulfur-based antioxidants include dialkylthiodipropionates such as dilauryl, dimyristyl, myristylstearyl, and distearyl esters of thiodipropionic acid, and polyols such as pentaerythritol tetra ( ⁇ -dodecylmercaptopropionate). and ⁇ -alkyl mercaptopropionic acid esters.
- Examples of the ultraviolet absorber include 2,4-dihydroxybenzophenone, 2-hydroxy-4-methoxybenzophenone, 2-hydroxy-4-octoxybenzophenone, 2-hydroxy-4-tert-butyl-4 ′-(2- 2-hydroxybenzophenones such as methacryloyloxyethoxyethoxy) benzophenone and 5,5′-methylenebis (2-hydroxy-4-methoxybenzophenone); 2- (2-hydroxy-5-methylphenyl) benzotriazole, 2- ( 2-hydroxy-5-tert-octylphenyl) benzotriazole, 2- (2-hydroxy-3,5-ditert-butylphenyl) -5-chlorobenzotriazole, 2- (2-hydroxy-3-tert-butyl) -5-methylphenyl) -5-chlorobenzotriazole, 2- 2-hydroxy-3-dodecyl-5-methylphenyl) benzotriazole, 2- (2-hydroxy-3-tert-butyl-5-C7-9 mixed al
- hindered amine light stabilizer examples include 2,2,6,6-tetramethyl-4-piperidyl stearate, 1,2,2,6,6-pentamethyl-4-piperidyl stearate, 2,2,6. , 6-tetramethyl-4-piperidylbenzoate, bis (2,2,6,6-tetramethyl-4-piperidyl) sebacate, bis (1,2,2,6,6-pentamethyl-4-piperidyl) sebacate, Bis (1-octoxy-2,2,6,6-tetramethyl-4-piperidyl) sebacate, tetrakis (2,2,6,6-tetramethyl-4-piperidyl) -1,2,3,4-butane Tetracarboxylate, tetrakis (1,2,2,6,6-pentamethyl-4-piperidyl) -1,2,3,4-butanetetracarboxylate, bis (2,2, , 6-tetramethyl-4-piperidyl) .bis (tridecyl
- the resin composition of the present invention may further contain other additives such as fillers, colorants, cross-linking agents, antistatic agents, plate-out preventing agents, surface treatment agents, lubricants, flame retardants, and fluorescent agents as necessary.
- additives such as fillers, colorants, cross-linking agents, antistatic agents, plate-out preventing agents, surface treatment agents, lubricants, flame retardants, and fluorescent agents as necessary.
- Antifungal agents, bactericides, metal deactivators, mold release agents, pigments, processing aids, foaming agents and the like can be blended.
- the resin composition of the present invention contains the modifier of the present invention, and there is no particular limitation other than that.
- the blending method and molding method are not particularly limited, and the blending method includes a method of mixing with a normal blender, mixer, etc., a method of melt kneading with an extruder, etc., a method of mixing with a solvent and casting a solution, etc.
- the molding method include extrusion molding, injection molding, stretched film molding, blow molding and the like.
- molding the resin composition of this invention For example, the thing of a sheet form, a film form, a special shape etc. can be mentioned.
- the film of the present invention is obtained from the resin composition of the present invention.
- a method for producing a cellulose ester film obtained from a cellulose ester will be described.
- the cellulose ester film is produced by casting and drying a dope solution in which cellulose ester is dissolved in a solvent.
- Various additives can be mixed in the dope liquid as necessary.
- the concentration of the cellulose ester in the dope is preferably higher because the drying load after casting on the support can be reduced. However, if the concentration of the cellulose ester is too high, the load during filtration increases and the filtration accuracy is poor. Become.
- the concentration for achieving both of these is preferably 1 to 30% by mass, more preferably 2 to 20% by mass.
- the solvent used for preparing the dope solution may be used alone or in combination, but it is preferable in terms of production efficiency to use a good solvent and a poor solvent of cellulose ester, and the preferred range of the mixing ratio of the good solvent and the poor solvent is
- the good solvent is 70 to 98% by mass, and the poor solvent is 30 to 2% by mass.
- the good solvent and the poor solvent used in the present invention those that dissolve the cellulose ester used alone are defined as good solvents, and those that swell or cannot dissolve alone are defined as poor solvents. Therefore, depending on the average degree of acetylation of cellulose, the good solvent and the poor solvent change. For example, acetone becomes a good solvent for cellulose esters having an average degree of acetylation of 55%, and poor for cellulose esters having an average degree of acetylation of 60%. It becomes a solvent.
- the good solvent and the poor solvent are not uniquely determined in all cases, but when cellulose triacetate is used as the cellulose resin, the good solvent includes an organic halogen compound such as methylene chloride, In the case of dioxolanes and cellulose acetate propionate, methylene chloride, acetone, methyl acetate and the like can be mentioned.
- the poor solvent include methanol, ethanol, n-butanol, cyclohexane and cyclohexanone.
- a method for dissolving the cellulose ester when preparing the dope solution a general method can be used, but heating is performed at a temperature not lower than the boiling point at normal pressure of the solvent and in a range where the solvent does not boil under pressure.
- the method of dissolving with stirring is preferred in order to prevent the generation of massive undissolved material called gel or mako.
- a method in which cellulose ester is mixed with a poor solvent and wetted or swollen, and then mixed with a good solvent and dissolved is also preferably used.
- a known cooling dissolution method may be used. When the cooling dissolution method is used, methyl acetate or acetone can be used as a good solvent.
- the pressurization may be performed by a method of injecting an inert gas such as nitrogen gas or by increasing the vapor pressure of the solvent by heating. Heating is preferably performed from the outside. For example, a jacket type is preferable because temperature control is easy.
- the heating temperature after addition of the solvent is preferably in the range where the solvent does not boil at or above the normal pressure of the solvent used from the viewpoint of the solubility of the cellulose ester, but is required if the heating temperature is too high.
- the pressure increases and productivity is reduced.
- the range of the preferred heating temperature is 45 to 120 ° C, more preferably 60 to 110 ° C, and further preferably 70 to 105 ° C. The pressure is adjusted so that the solvent does not boil at the set temperature.
- the cellulose ester solution is filtered using an appropriate filter medium such as filter paper.
- the filter medium it is preferable that the absolute filtration accuracy is small in order to remove unnecessary substances and the like, but if the absolute filtration accuracy is too small, there is a problem that the filter medium is likely to be clogged. Therefore, a filter medium having an absolute filtration accuracy of 0.008 mm or less is preferable, a filter medium in the range of 0.001 to 0.008 mm is more preferable, and a filter medium in the range of 0.003 to 0.006 mm is more preferable.
- the material of the filter medium is not particularly limited, and a normal filter medium can be used. However, a plastic filter medium such as polypropylene and Teflon (registered trademark) and a metal filter medium such as stainless steel are preferable because fibers do not fall off.
- the dope solution can be filtered by a normal method, but the method of filtering while heating at a temperature not lower than the boiling point of the solvent at a normal pressure and in a range where the solvent does not boil is a differential pressure before and after the filter medium.
- An increase in (hereinafter sometimes referred to as filtration pressure) is small and preferable.
- the filtration temperature is preferably 45 to 120 ° C, more preferably 45 to 70 ° C, and further preferably 45 to 55 ° C. A smaller filtration pressure is preferred.
- the filtration pressure is preferably 1.6 ⁇ 10 6 Pa or less, more preferably 1.2 ⁇ 10 6 Pa or less, and further preferably 1.0 ⁇ 10 6 Pa or less.
- the support used in the casting process is preferably a support having a mirror-finished endless belt-like or drum-like stainless steel.
- the temperature of the support in the casting step is preferably from 0 ° C. to less than the boiling point of the solvent. The higher the temperature, the faster the drying speed. However, if the temperature is too high, foaming may occur or the flatness may deteriorate.
- the support temperature is preferably 0 to 50 ° C, more preferably 5 to 30 ° C.
- the method for controlling the temperature of the support is not particularly limited, and there are a method of blowing warm air or cold air, and a method of bringing a hot water bat into contact with the support.
- the amount of residual solvent upon peeling from the support is preferably 10 to 120%, more preferably 20 to 40% or 60 to 120%, particularly preferably. Is 20-30% or 70-115%.
- the means for drying the film is not particularly limited, and is performed with hot air, infrared rays, a heating roll, a microwave, or the like. It is preferable to carry out with hot air from the point of simplicity.
- the drying temperature is preferably increased stepwise in the range of 40 to 150 ° C., and more preferably 50 to 140 ° C. in order to improve dimensional stability.
- the film thickness of the cellulose ester film is not particularly limited, but is preferably 10 to 500 ⁇ m, and more preferably 20 to 100 ⁇ m.
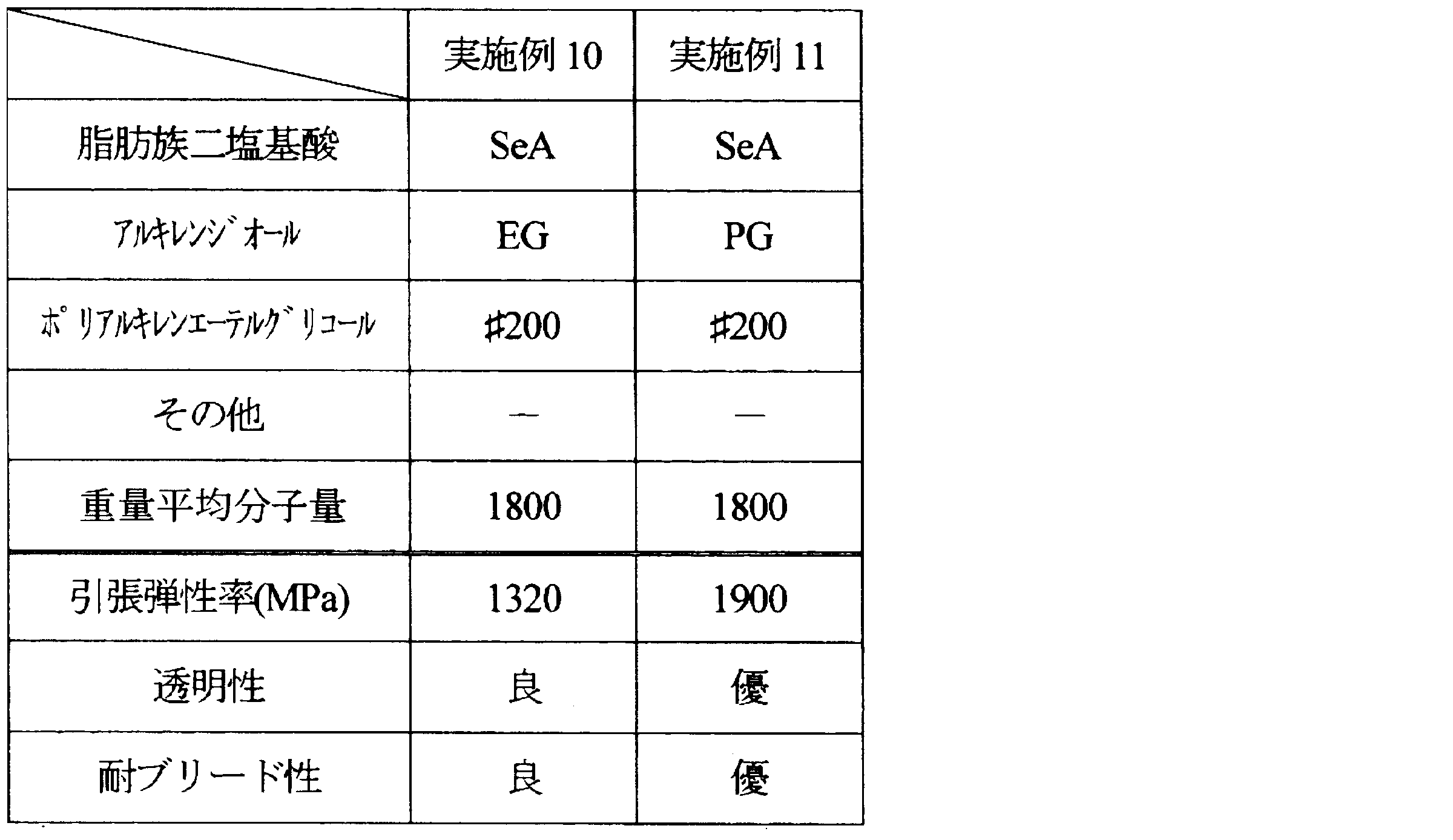
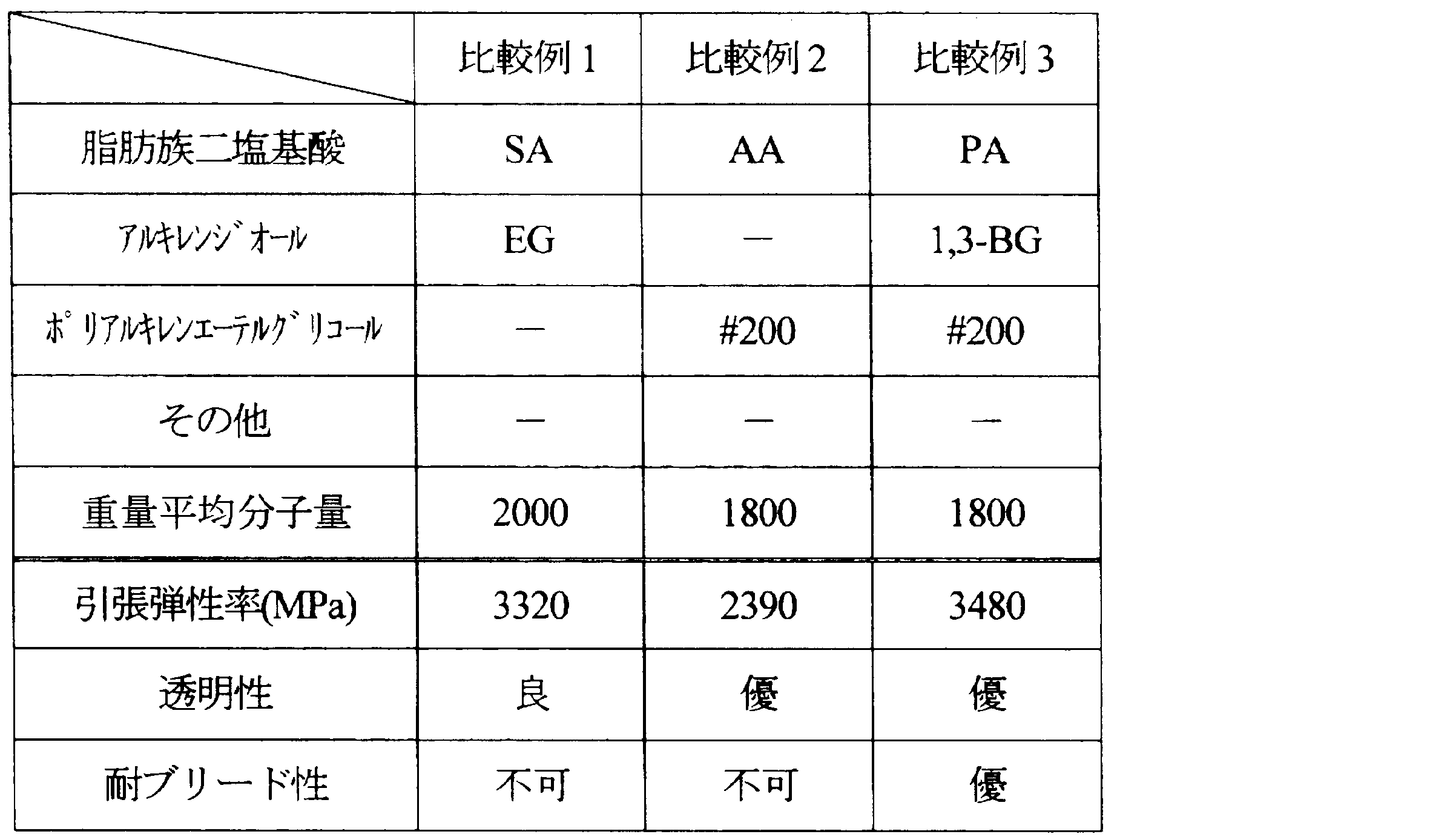
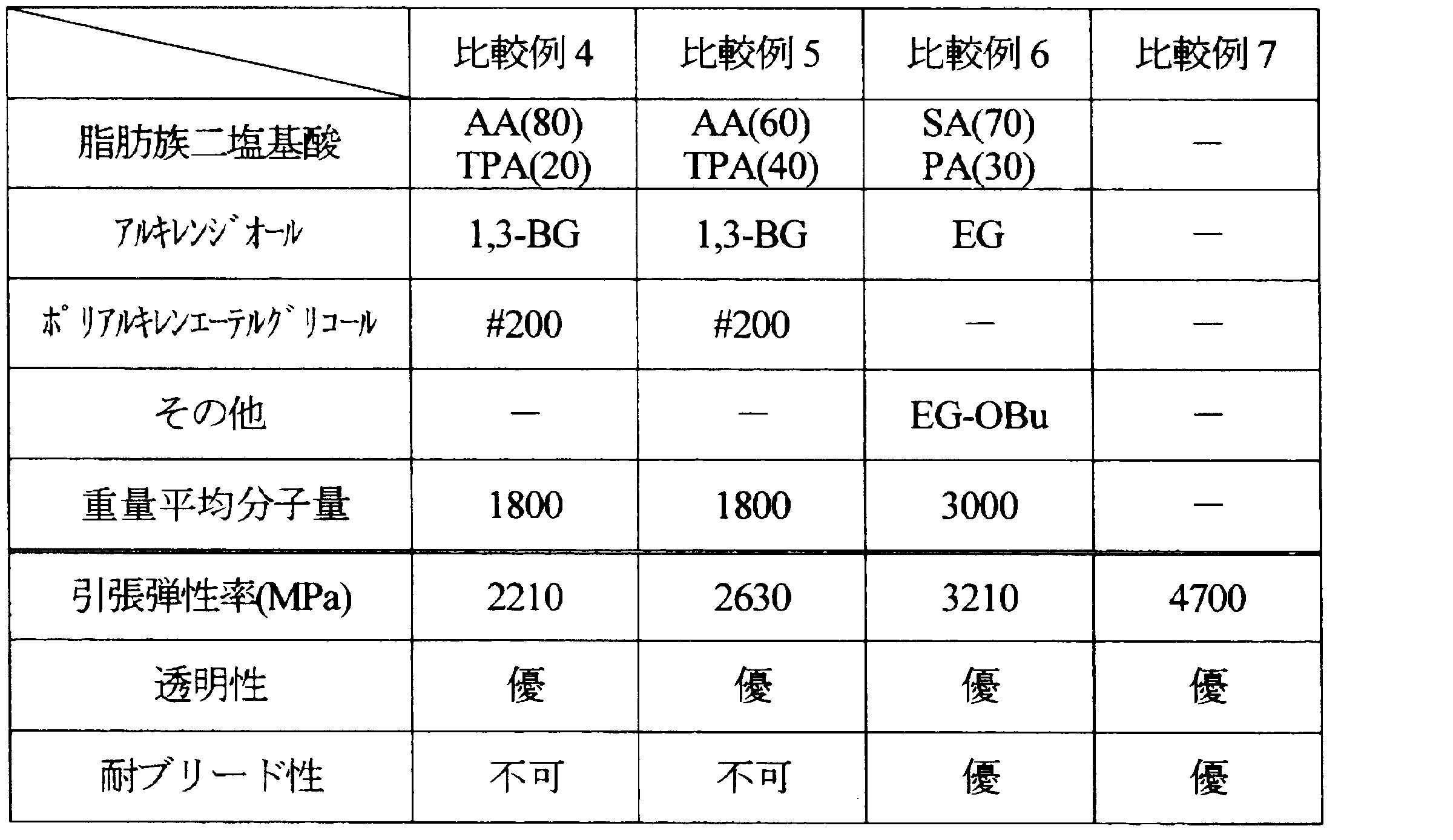
- Examples 1 to 11 and Comparative Examples 1 to 7 Aliphatic dibasic acids, alkylene diols, and polyalkylene ether glycols shown in Tables 1 to 5 below were polycondensed under the following conditions, and the resulting compounds were used as resin modifiers.
- the weight average molecular weight of the obtained compound is as shown in the same table.
- the ratio of each component is as follows.
- Example 1 A 5L 1L flask is equipped with a stirrer, nitrogen blowing tube, thermometer, rectifying tube and ball plug (for sampling), and a water metering receiver and cooling tube are attached to the end of the rectifying tube. Used as a device.
- Example 2 Using the same reaction apparatus as in Example 1, the amount of succinic acid used in Example 1 was changed to 403.9 g (3.42 mol), and ethylene glycol was changed to propylene glycol: 219.3 g (2.88 mol). Except for the above, reaction and purification were carried out in the same manner as in Example 1 to obtain 669 g of the resin modifier of Example 2.
- Example 3 Using the same reactor as in Example 1, the amount of succinic acid used in Example 1 was 378.6 g (3.21 mol), ethylene glycol was 1,3-butanediol: 238.6 g (2.65 mol). The reaction and purification were carried out in the same manner as in Example 1 except that the compound was changed to) to obtain 684 g of the resin modifier of Example 3.
- Example 4 Using the same reaction apparatus as in Example 1, the amount of succinic acid used in Example 1 was 338.8 g (2.87 mol), and ethylene glycol was 3-methyl-1,5-pentanediol: 269.0 g ( Except for changing to 2.28 mol), the reaction and purification were conducted in the same manner as in Example 1 to obtain 671 g of the resin modifier of Example 4.
- Example 5 Using the same reactor as in Example 1, the succinic acid of Example 1 was changed to adipic acid: 449.5 g (3.08 mol), and the amount of ethylene glycol used was changed to 155.4 g (2.50 mol). Except for the above, the reaction and purification were conducted in the same manner as in Example 1 to obtain 679 g of a resin modifier of Example 5.
- Example 6 Using the same reactor as in Example 1, the succinic acid of Example 1 was changed to adipic acid: 424.5 g (2.90 mol), and ethylene glycol was changed to propylene glycol: 176.17 g (2.32 mol). Except for the above, reaction and purification were carried out in the same manner as in Example 1 to obtain 672 g of the modifier for resin of Example 6.
- Example 7 Using the same reactor as in Example 1, the succinic acid of Example 1 was adipic acid: 424.5 g (2.90 mol), and ethylene glycol was 1,3-propanediol: 176.17 g (2.32 mol). The reaction and purification were carried out in the same manner as in Example 1 except for changing to) to obtain 677 g of a resin modifier of Example 7.
- Example 8 Using the same reactor as in Example 1, the succinic acid of Example 1 was adipic acid: 402.9 g (2.76 mol), and ethylene glycol was 1,3-butanediol: 194.0 g (2.15 mol). The reaction and purification were carried out in the same manner as in Example 1 except that) was changed to 668 g of the resin modifier of Example 8.
- Example 9 Using the same reactor as in Example 1, the succinic acid of Example 1 was adipic acid: 367.8 g (2.52 mol), and ethylene glycol was 3-methyl-1,5-pentanediol: 223.16 g ( Except for changing to 1.89 mol), the reaction and purification were conducted in the same manner as in Example 1 to obtain 680 g of the resin modifier of Example 9.
- Example 10 Using the same reactor as in Example 1, the succinic acid in Example 1 was changed to sebacic acid: 469.2 g (2.32 mol), and the amount of ethylene glycol used was changed to 103.78 g (1.67 mol). Except for the above, reaction and purification were conducted in the same manner as in Example 1 to obtain 677 g of the modifier for resin of Example 10.
- Example 11 Using the same reactor as in Example 1, the succinic acid of Example 1 was changed to sebacic acid: 451.5 g (2.23 mol), and ethylene glycol was changed to propylene glycol: 119.9 g (1.58 mol). Except for the above, the reaction and purification were carried out in the same manner as in Example 1 to obtain 668 g of the modifier for resin of Example 11.
- Example creation> In a mixed solvent composed of 90 parts by mass of methylene chloride and 10 parts by mass of methyl alcohol, 4.7 parts by mass of acetylcellulose (acetylation degree 61.5%, polymerization degree 260), Examples 1 to 11 and Comparative Examples 1 to 60 parts by mass of each resin modifier 6 was added to 100 parts by mass of acetylcellulose, and dissolved while stirring. The obtained solution was cast and formed into a film with a thickness of about 80 ⁇ m. The resulting film was evaluated for tensile modulus, transparency and bleed resistance. The evaluation method is as follows. In Comparative Example 7, the tensile modulus, transparency and bleed resistance were evaluated in the same procedure except that no modifier was added.
- ⁇ Tensile modulus> The tensile modulus of the produced film was measured according to the JIS K 6251 standard of the film test method. From the sheet-like molded article, dumbbell No. 3 (total length: 100 mm, parallel part: length 20 mm, width 5 mm) was cut out as a test piece, and the test piece was pulled at a grip interval of 25 mm and a pulling speed of 1 mm / min. The tensile elastic modulus was measured. The obtained results are also shown in Tables 1 to 5. In addition, the tensile elasticity modulus set 3000 MPa or less as the pass.
- SA succinic acid
- EG ethylene glycol
- PG 1,2-propylene glycol
- 1,3-BG 1,3-butanediol
- MPD 3-methyl-1,5-pentanediol
- 200 polyethylene glycol 200 (Aoki Yushi Kogyo Co., Ltd., weight average molecular weight: 200)
- SeA Sebacic acid
- PA phthalic acid
- EG-OBu ethylene glycol monobutyl ether
- TPA terephthalic acid
- the resin composition of the present invention can highly balance flexibility, transparency and bleed resistance.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Manufacturing & Machinery (AREA)
- Materials Engineering (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Polyesters Or Polycarbonates (AREA)
Abstract
Description
本発明の樹脂用改質剤(以下、「改質剤」とも称する)は、脂肪族二塩基酸に由来する構造単位と、アルキレンジオールに由来する構造単位と、ポリアルキレンエーテルグリコールに由来する構造単位と、からなり、末端が水酸基またはカルボキシル基であるランダム共重合体である。かかる樹脂用改質剤を用いれば、樹脂組成物の柔軟性、透明性および耐ブリード性を高度にバランスすることができる。以下、本発明の樹脂用改質剤および樹脂組成物につき、詳細に説明する。
本発明の改質剤においては、脂肪族二塩基酸に由来する構造単位を構成する脂肪族二塩基酸としては、例えば、シュウ酸、マロン酸、コハク酸、グルタル酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸、ウンデカン二酸、ドデカン二酸、トリデカン二酸、テトラデカン二酸等の飽和脂肪族二塩基酸や、例えば、マレイン酸、フマル酸、イタコン酸、シトラコン酸、メサコン酸等の不飽和脂肪族二塩基酸を挙げることができる。これらの中でも、コハク酸、グルタル酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸がより好ましく、アジピン酸が特に好ましい。脂肪族二塩基酸に由来する構造単位を構成する脂肪族二塩基酸は、1種でもよく、2種以上であってもよい。
本発明の改質剤においては、アルキレンジオールに由来する構造単位を構成するアルキレンジオールとしては、例えば、エチレングリコール、1,2-プロピレングリコール、1,3-プロパンジオール、1,3-ブタンジオール、1,4-ブタンジオール、ペンタンジオール、3-メチル-1,5-ペンタンジオール、ヘキサンジオール、ネオペンチルグリコール、ジメチロールへプタン(2-ブチル-2-エチル-1,3-プロパンジオール)、ノナンジオール、2,4-ジエチル-1,5-ペンタンジオール、2-メチル-1,4-ブタンジオール、オクタンジオール、2-エチルヘキサンジオール、2-メチル-1,3-プロパンジオール等を挙げることができる。これらの中でも、エチレングリコール、1,2-プロピレングリコール、1,3-プロパンジオール、2-メチル-1,3-プロパンジオール、1,3-ブタンジオール、1,4-ブタンジオール、ネオペンチルグリコール、3-メチル-1,5-ペンタンジオール、ヘキサンジオールがより好ましく、エチレングリコール、1,2-プロピレングリコール、1,3-ブタンジオール、3-メチル-1,5-ペンタンジオールが特に好ましい。アルキレンジオールに由来する構造単位を構成するアルキレンジオールは、1種でもよく、2種以上であってもよい。
本発明の改質剤においては、ポリアルキレンエーテルグリコールに由来する構造単位を構成するポリアルキレンエーテルグリコールとしては、ポリメチレンエーテルグリコール、ポリエチレンエーテルグリコール、ポリ-1,2-プロピレンエーテルグリコール、ポリトリメチレンエーテルグリコール、ポリテトラメチレンエーテルグリコール、ポリネオペンチレンエーテルグリコール、ポリ(エチレンオキシド・プロピレンオキシド)共重合体、ポリ(エチレンオキシド・テトラヒドロフラン)共重合体、ポリ(エチレンオキシド・プロピレンオキシド・テトラヒドロフラン)共重合体等を挙げることができる。これらの中でも、ポリエチレンエーテルグリコールがより好ましい。また、ポリアルキレンエーテルグリコールの中でも、重量平均分子量が、100~600のものがより好ましく、150~300のものが更に好ましく、150~200のものが特に好ましい。ポリアルキレンエーテルグリコールに由来する構造単位を構成するポリアルキレンエーテルグリコールは、1種でもよく、2種以上であってもよい。
本発明のフィルムは、本発明の樹脂組成物から得られるものであり、以下、一例として、セルロースエステルから得られるセルロースエステルフィルムの製造方法について説明する。
残留溶剤量=〔(加熱処理前のフィルム質量-加熱処理後のフィルム質量)/(加熱処理後のフィルム質量)〕×100(%)
で定義される。なお、残留溶剤量を測定する際の加熱処理とは、フィルムを115℃で1時間加熱することをいう。また、セルロースエステルフィルムの乾燥工程においては、支持体より剥離したフィルムをさらに乾燥し、残留溶剤量を3質量%以下にすることが好ましく、さらに0.5質量%以下が好ましい。フィルム乾燥工程では一般にロール懸垂方式か、テンター方式でフィルムを搬送しながら乾燥する方式が採られる。
下記表1~5に示す脂肪族二塩基酸、アルキレンジオール、およびポリアルキレンエーテルグリコールを以下の条件で重縮合させ、得られた化合物を樹脂用改質剤として用いた。得られた化合物の重量平均分子量は、同表中に示すとおりである。また、各成分の比率は、以下のとおりである。
(実施例1)
五ツ口の1Lフラスコに攪拌機、窒素吹込み管、温度計、精留管および玉栓(サンプリング用)を取付け、さらに精留管の先に水分定量受器、冷却管を取付けたものを反応装置として用いた。上記フラスコに原料のコハク酸:434.1g(3.68モル)、エチレングリコール:196.36g(3.16モル)、ポリエチレンエーテルグリコール(分子量200):240g(1.20モル)およびエステル化触媒としてオクチル酸ジルコニウム:0.0048g(1.22×10-5モル)を加え、常圧下、230℃で生成水を系外に除きながらエステル化反応を行った。生成水の留出がおさまってきた時点で系内を減圧(30~40kPa)に切り替え、反応を継続した。酸価が0.51(目標:1.0未満)となったところでエステル化反応を終了させた。引き続き減圧下(2kPa以下)、180~220℃でエステル交換反応を行った。水酸基価が62.29(目標:60.65~64.12)となった時点で冷却し、120℃でセライトを用いて濾過を行い、677gの実施例1の樹脂用改質剤を得た。
実施例1と同様の反応装置を用いて、実施例1のコハク酸の使用量を403.9g(3.42モル)、エチレングリコールをプロピレングリコール:219.3g(2.88モル)に替えた以外は実施例1と同様に、反応、精製を行い、669gの実施例2の樹脂用改質剤を得た。
実施例1と同様の反応装置を用いて、実施例1のコハク酸の使用量を378.6g(3.21モル)、エチレングリコールを1,3-ブタンジオール:238.6g(2.65モル)に替えた以外は実施例1と同様に、反応、精製を行い、684gの実施例3の樹脂用改質剤を得た。
実施例1と同様の反応装置を用いて、実施例1のコハク酸の使用量を338.8g(2.87モル)、エチレングリコールを3-メチル-1,5-ペンタンジオール:269.0g(2.28モル)に替えた以外は実施例1と同様に、反応、精製を行い、671gの実施例4の樹脂用改質剤を得た。
実施例1と同様の反応装置を用いて、実施例1のコハク酸をアジピン酸:449.5g(3.08モル)、エチレングリコールの使用量を155.4g(2.50モル)に替えた以外は実施例1と同様に、反応、精製を行い、679gの実施例5の樹脂用改質剤を得た。
実施例1と同様の反応装置を用いて、実施例1のコハク酸をアジピン酸:424.5g(2.90モル)、エチレングリコールをプロピレングリコール:176.17g(2.32モル)に替えた以外は実施例1と同様に、反応、精製を行い、672gの実施例6の樹脂用改質剤を得た。
実施例1と同様の反応装置を用いて、実施例1のコハク酸をアジピン酸:424.5g(2.90モル)、エチレングリコールを1,3-プロパンジオール:176.17g(2.32モル)に替えた以外は実施例1と同様に、反応、精製を行い、677gの実施例7の樹脂用改質剤を得た。
実施例1と同様の反応装置を用いて、実施例1のコハク酸をアジピン酸:402.9g(2.76モル)、エチレングリコールを1,3-ブタンジオール:194.0g(2.15モル)に替えた以外は実施例1と同様に、反応、精製を行い、668gの実施例8の樹脂用改質剤を得た。
実施例1と同様の反応装置を用いて、実施例1のコハク酸をアジピン酸:367.8g(2.52モル)、エチレングリコールを3-メチル-1,5-ペンタンジオール:223.16g(1.89モル)に替えた以外は実施例1と同様に、反応、精製を行い、680gの実施例9の樹脂用改質剤を得た。
実施例1と同様の反応装置を用いて、実施例1のコハク酸をセバシン酸:469.2g(2.32モル)、エチレングリコールの使用量を103.78g(1.67モル)に替えた以外は実施例1と同様に、反応、精製を行い、677gの実施例10の樹脂用改質剤を得た。
実施例1と同様の反応装置を用いて、実施例1のコハク酸をセバシン酸:451.5g(2.23モル)、エチレングリコールをプロピレングリコール:119.9g(1.58モル)に替えた以外は実施例1と同様に、反応、精製を行い、668gの実施例11の樹脂用改質剤を得た。
原料としてコハク酸:555.9g(4.71モル)、エチレングリコール:345.3g(5.56モル)を使用した。反応装置は実施例1と同様で、手順についてもエステル交換の目標水酸基価を54.74~57.55とした以外は同様に合成、精製を行い、659gの比較例1の樹脂用改質剤を得た。
原料としてアジピン酸:301.6g(2.06モル)、ポリエチレンエーテルグリコール(分子量200):507.6g(2.54モル)を使用した。実施例1と同様の反応装置および手順にて合成、精製を行い、678gの比較例2の樹脂用改質剤を得た。
原料として無水フタル酸:374.4g(2.53モル)、1,3-ブタンジオール:196.8g(2.18モル)、ポリエチレンエーテルグリコール(分子量200):240.0g(1.20モル)を使用した。実施例1と同様の反応装置および手順にて合成、精製を行い、669gの比較例3の樹脂用改質剤を得た。
原料としてアジピン酸:316.1g(2.16モル)、テレフタル酸:89.8g(0.54モル)、1,3-ブタンジオール:188.8g(2.09モル)、ポリエチレンエーテルグリコール(分子量200):240.0g(1.20モル)を使用した。実施例1と同様の反応装置および手順にて合成、精製を行い、670gの比較例4の樹脂用改質剤を得た。
原料としてアジピン酸:232.5g(1.59モル)、テレフタル酸:176.2g(1.06モル)、1,3-ブタンジオール:183.6g(2.04モル)、ポリエチレンエーテルグリコール(分子量200):240.0g(1.20モル)を使用した。実施例1と同様の反応装置および手順にて合成、精製を行い、662gの比較例5の樹脂用改質剤を得た。
原料としてコハク酸:353.7g(3.00モル)、無水フタル酸:190.12g(1.28モル)、エチレングリコール:300.9g(4.85モル)、エチレングリコールモノブチルエーテル:68.1g(0.58モル)を使用した。反応装置は実施例1と同様で、手順についてもエステル交換の目標水酸基価を20未満とした以外は同様に合成、精製を行い、680gの比較例6の樹脂用改質剤を得た。
メチレンクロライド90質量部とメチルアルコール10質量部とからなる混合溶剤に、アセチルセルロース(酢化度61.5%、重合度260)を4.7質量部、実施例1~11および比較例1~6の樹脂用改質剤を各々アセチルセルロース100質量部に対して60質量部加え、撹拌しながら溶解させた。得られた溶液を流延し、約80μmの厚さで製膜した。得られたフィルムについて、引張弾性率、透明性および耐ブリード性について評価した。評価方法は以下のとおりである。なお、比較例7は、改質剤を添加しなかったこと以外は、同様の手順で引張弾性率、透明性および耐ブリード性について評価した。
フィルムの試験方法のJIS K 6251規格に準じて、作製したフィルムの引張弾性率を測定した。シート状の成形品から、ダンベル3号形(全長:100mm、平行部:長さ20mm、幅5mm)に切り出して試験片とし、つかみ間隔25mm、引張速度1mm/minで試験片を引張り、試験片の引張弾性率を測定した。得られた結果を、表1~5に併記する。なお、引張弾性率は3000MPa以下を合格とした。
得られたサンプルにつき、目視にて透明性を評価した。評価基準は、以下の三段階とした。得られた結果を、表1~5に併記する。
優:非常にクリア
良:濁りがほとんど見られない
可:やや濁りが見られる
作製したフィルムを10cm×10cmのサイズに裁断し、23℃、50%RHで2週間放置した後、サンプル表面のブリードを目視観察した。評価基準は、以下の三段階とした。得られた結果を、表1~5に併記する。
優:ブリードがないもの
良:若干のブリードが認められるもの
不可:ブリードが激しいもの
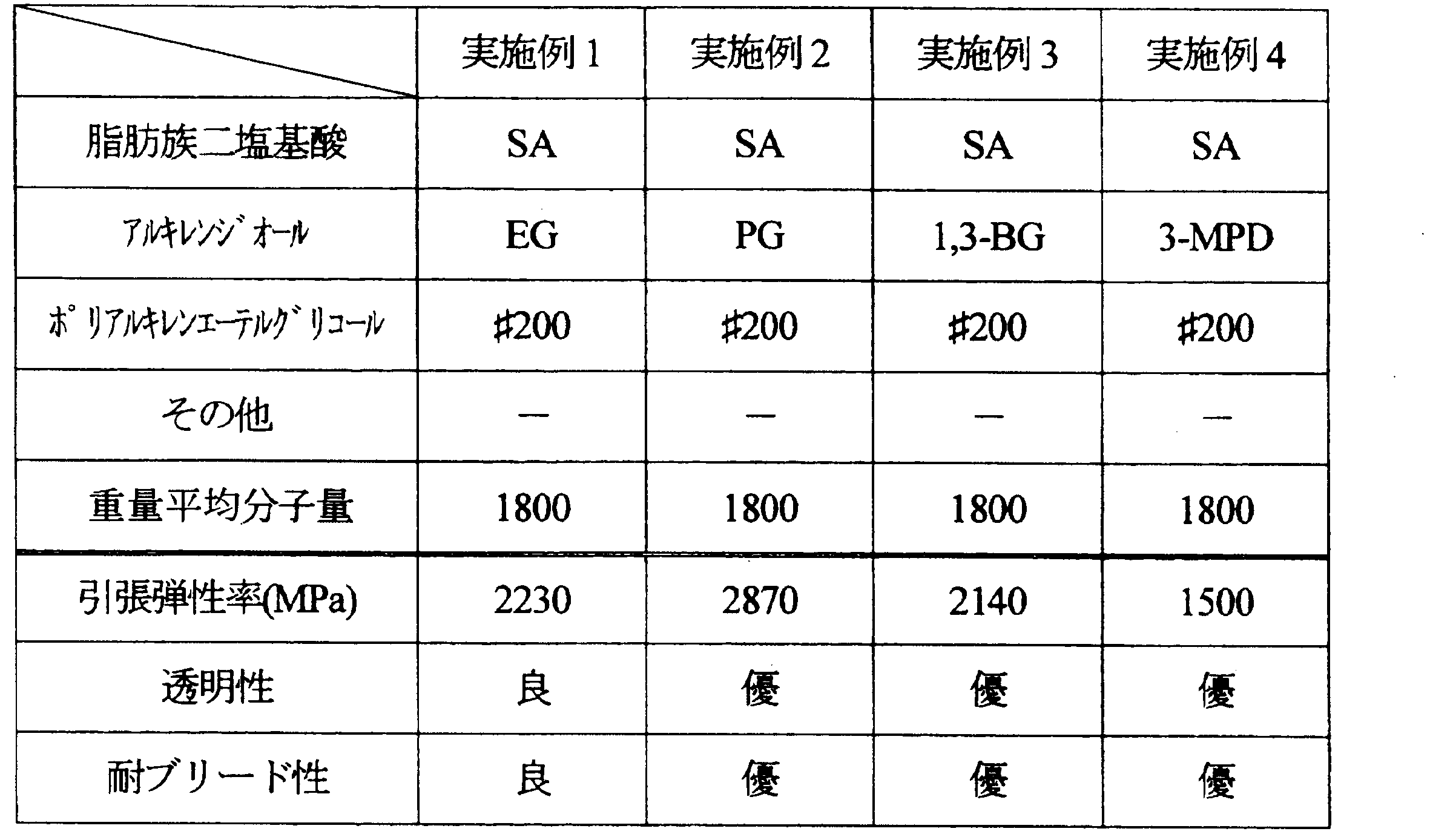
EG:エチレングリコール
PG:1,2-プロピレングリコール
1,3-BG:1,3-ブタンジオール
3-MPD:3-メチル-1,5-ペンタンジオール
#200:ポリエチレングリコール200(青木油脂工業(株),重量平均分子量:200)

1,3-PG:1,3-プロパンジオール
TPA:テレフタル酸
Claims (9)
- 脂肪族二塩基酸に由来する構造単位と、アルキレンジオールに由来する構造単位と、ポリアルキレンエーテルグリコールに由来する構造単位と、からなり、末端が水酸基またはカルボキシル基であるランダム共重合体からなることを特徴とする樹脂用改質剤。
- 前記ポリアルキレンエーテルグリコールが、ポリエチレンエーテルグリコールである請求項1記載の樹脂用改質剤。
- 前記アルキレンジオールが、エチレングリコール、1,2-プロピレングリコール、1,3-プロパンジオール、2-メチル-1,3-プロパンジオール、1,3-ブタンジオール、1,4-ブタンジオール、ネオペンチルグリコール、3-メチル-1,5-ペンタンジオール、ヘキサンジオールからなる群より選ばれる少なくとも1種である請求項1または2記載の樹脂用改質剤。
- 前記脂肪族二塩基酸に由来する構造単位、前記アルキレンジオールに由来する構造単位、および前記ポリアルキレンエーテルグリコールに由来する構造単位の質量比が、各々10~80質量%、5~80質量%および1~50質量%である請求項1~3のうちいずれか一項記載の樹脂用改質剤。
- 重量平均分子量が、1000~3000である請求項1~4のうちいずれか一項記載の樹脂用改質剤。
- 請求項1~5のうちいずれか一項記載の樹脂用改質剤が樹脂に添加されてなることを特徴とする樹脂組成物。
- 前記樹脂が、セルロース系樹脂である請求項6記載の樹脂組成物。
- 前記樹脂100質量部に対して、前記樹脂用改質剤が3~100質量部である請求項6または7記載の樹脂組成物。
- 請求項6~8のうちいずれか一項記載の樹脂組成物からなることを特徴とするフィルム。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17753030.0A EP3418314A4 (en) | 2016-02-15 | 2017-02-07 | MODIFICATOR FOR RESIN, RESIN COMPOSITION AND FILM THEREWITH |
| CN201780011238.6A CN108779235A (zh) | 2016-02-15 | 2017-02-07 | 树脂用改性剂、树脂组合物和使用其的薄膜 |
| KR1020187026416A KR20180110122A (ko) | 2016-02-15 | 2017-02-07 | 수지용 개질제, 수지 조성물 및 이것을 사용한 필름 |
| US16/077,673 US20190048135A1 (en) | 2016-02-15 | 2017-02-07 | Modifier for resin, resin composition, and film in which same is used |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016025843A JP2017145275A (ja) | 2016-02-15 | 2016-02-15 | 樹脂用改質剤、樹脂組成物、およびこれを用いたフィルム |
| JP2016-025843 | 2016-02-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017141772A1 true WO2017141772A1 (ja) | 2017-08-24 |
Family
ID=59625034
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/004427 WO2017141772A1 (ja) | 2016-02-15 | 2017-02-07 | 樹脂用改質剤、樹脂組成物、およびこれを用いたフィルム |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20190048135A1 (ja) |
| EP (1) | EP3418314A4 (ja) |
| JP (1) | JP2017145275A (ja) |
| KR (1) | KR20180110122A (ja) |
| CN (1) | CN108779235A (ja) |
| WO (1) | WO2017141772A1 (ja) |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS497354A (ja) * | 1972-05-11 | 1974-01-23 | ||
| JPH05155809A (ja) * | 1991-12-05 | 1993-06-22 | Satoru Matsumoto | エ−テルエステル末端構造を有するジエステル複合エステル並びにポリエステル |
| JP2006219649A (ja) * | 2005-01-17 | 2006-08-24 | Taoka Chem Co Ltd | 樹脂用可塑剤および樹脂組成物 |
| JP2006241378A (ja) * | 2005-03-04 | 2006-09-14 | Taoka Chem Co Ltd | 樹脂用可塑剤および樹脂組成物 |
| WO2009093503A1 (ja) * | 2008-01-23 | 2009-07-30 | Adeka Corporation | セルロース系樹脂組成物およびセルロース系樹脂フィルム |
| JP2009173741A (ja) * | 2008-01-23 | 2009-08-06 | Adeka Corp | セルロース系樹脂組成物およびセルロース系樹脂フィルム |
| JP2009173740A (ja) * | 2008-01-23 | 2009-08-06 | Adeka Corp | セルロース系樹脂組成物およびセルロース系樹脂フィルム |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2003281227A1 (en) * | 2002-07-03 | 2004-01-23 | Mitsubishi Chemical Corporation | Aliphatic polyester polyether copolymer, process for producing the same and aliphatic polyester composition using the copolymer |
| CN101258195A (zh) * | 2004-11-19 | 2008-09-03 | 伊士曼化工公司 | 稳定的脂族聚酯组合物 |
| CN102504223A (zh) * | 2011-10-27 | 2012-06-20 | 山东东大一诺威聚氨酯有限公司 | 聚醚酯多元醇及其使用方法 |
| JP6600440B2 (ja) * | 2012-06-29 | 2019-10-30 | エフアールエックス ポリマーズ、インク. | ポリエステルコ−ホスホネート |
-
2016
- 2016-02-15 JP JP2016025843A patent/JP2017145275A/ja active Pending
-
2017
- 2017-02-07 US US16/077,673 patent/US20190048135A1/en not_active Abandoned
- 2017-02-07 CN CN201780011238.6A patent/CN108779235A/zh active Pending
- 2017-02-07 EP EP17753030.0A patent/EP3418314A4/en not_active Withdrawn
- 2017-02-07 KR KR1020187026416A patent/KR20180110122A/ko unknown
- 2017-02-07 WO PCT/JP2017/004427 patent/WO2017141772A1/ja active Application Filing
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS497354A (ja) * | 1972-05-11 | 1974-01-23 | ||
| JPH05155809A (ja) * | 1991-12-05 | 1993-06-22 | Satoru Matsumoto | エ−テルエステル末端構造を有するジエステル複合エステル並びにポリエステル |
| JP2006219649A (ja) * | 2005-01-17 | 2006-08-24 | Taoka Chem Co Ltd | 樹脂用可塑剤および樹脂組成物 |
| JP2006241378A (ja) * | 2005-03-04 | 2006-09-14 | Taoka Chem Co Ltd | 樹脂用可塑剤および樹脂組成物 |
| WO2009093503A1 (ja) * | 2008-01-23 | 2009-07-30 | Adeka Corporation | セルロース系樹脂組成物およびセルロース系樹脂フィルム |
| JP2009173741A (ja) * | 2008-01-23 | 2009-08-06 | Adeka Corp | セルロース系樹脂組成物およびセルロース系樹脂フィルム |
| JP2009173740A (ja) * | 2008-01-23 | 2009-08-06 | Adeka Corp | セルロース系樹脂組成物およびセルロース系樹脂フィルム |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3418314A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20180110122A (ko) | 2018-10-08 |
| US20190048135A1 (en) | 2019-02-14 |
| JP2017145275A (ja) | 2017-08-24 |
| CN108779235A (zh) | 2018-11-09 |
| EP3418314A4 (en) | 2019-10-02 |
| EP3418314A1 (en) | 2018-12-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5551618B2 (ja) | セルロース系樹脂組成物およびセルロース系樹脂フィルム | |
| US9334392B2 (en) | Cellulose resin composition | |
| JP5469079B2 (ja) | セルロース系樹脂組成物およびセルロース系樹脂フィルム | |
| JP5312812B2 (ja) | セルロース系樹脂組成物およびセルロース系樹脂フィルム | |
| JP2009173740A (ja) | セルロース系樹脂組成物およびセルロース系樹脂フィルム | |
| JPWO2016158790A1 (ja) | 樹脂用改質剤およびこれを用いた樹脂組成物 | |
| JP6289865B2 (ja) | セルロース系樹脂組成物、およびこれを用いたフィルム | |
| JP2009173741A (ja) | セルロース系樹脂組成物およびセルロース系樹脂フィルム | |
| JP6616563B2 (ja) | リタデーション上昇剤、これを用いたセルロース系樹脂組成物、およびフィルム | |
| JP6092116B2 (ja) | 延伸光学補償フィルム | |
| WO2017141772A1 (ja) | 樹脂用改質剤、樹脂組成物、およびこれを用いたフィルム | |
| JP6802678B2 (ja) | 樹脂用改質剤およびこれを用いた樹脂組成物 | |
| JP6289862B2 (ja) | 防湿剤、セルロース系樹脂組成物、およびこれを用いたフィルム | |
| JP6351945B2 (ja) | セルローストリアセテート樹脂用防湿剤、セルローストリアセテート樹脂組成物、およびフィルム | |
| JP6243189B2 (ja) | セルロース系樹脂組成物、およびこれを用いたフィルム | |
| JP2014224962A (ja) | リタデーション上昇剤 | |
| JP2015083643A (ja) | 防湿剤、セルロース系樹脂組成物、およびこれを用いたフィルム |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17753030 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20187026416 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020187026416 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2017753030 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2017753030 Country of ref document: EP Effective date: 20180917 |