WO2016208576A1 - (6S,9aS)-N-ベンジル-6-[(4-ヒドロキシフェニル)メチル]-4,7-ジオキソ-8-({6-[3-(ピペラジン-1-イル)アゼチジン-1-イル]ピリジン-2-イル}メチル)-2-(プロプ-2-エン-1-イル)-オクタヒドロ-1H-ピラジノ[2,1-c][1,2,4]トリアジン-1-カルボキサミド化合物の結晶 - Google Patents
(6S,9aS)-N-ベンジル-6-[(4-ヒドロキシフェニル)メチル]-4,7-ジオキソ-8-({6-[3-(ピペラジン-1-イル)アゼチジン-1-イル]ピリジン-2-イル}メチル)-2-(プロプ-2-エン-1-イル)-オクタヒドロ-1H-ピラジノ[2,1-c][1,2,4]トリアジン-1-カルボキサミド化合物の結晶 Download PDFInfo
- Publication number
- WO2016208576A1 WO2016208576A1 PCT/JP2016/068381 JP2016068381W WO2016208576A1 WO 2016208576 A1 WO2016208576 A1 WO 2016208576A1 JP 2016068381 W JP2016068381 W JP 2016068381W WO 2016208576 A1 WO2016208576 A1 WO 2016208576A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ppm
- methyl
- diffraction
- crystals
- crystal
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- XOGYEQGEEISWDY-UHFFFAOYSA-N CCOC(CNCc1cccc(F)n1)OCC Chemical compound CCOC(CNCc1cccc(F)n1)OCC XOGYEQGEEISWDY-UHFFFAOYSA-N 0.000 description 1
- MAZRAHBUWVAKAD-SFHVURJKSA-N CC[C@@H](C(OC)=O)NC(OCC1c2ccccc2-c2c1cccc2)=O Chemical compound CC[C@@H](C(OC)=O)NC(OCC1c2ccccc2-c2c1cccc2)=O MAZRAHBUWVAKAD-SFHVURJKSA-N 0.000 description 1
- QQQVLIVWQMWACQ-KRWDZBQOSA-N COC([C@H](CO)NC(OCC1c2ccccc2-c2ccccc12)=O)=O Chemical compound COC([C@H](CO)NC(OCC1c2ccccc2-c2ccccc12)=O)=O QQQVLIVWQMWACQ-KRWDZBQOSA-N 0.000 description 1
- BYTJTXKQBSNGCL-UHFFFAOYSA-N Fc1cc(OCc2ccccc2)ccc1Br Chemical compound Fc1cc(OCc2ccccc2)ccc1Br BYTJTXKQBSNGCL-UHFFFAOYSA-N 0.000 description 1
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- the present invention relates to (6S, 9aS) -N-benzyl-6-[(4-hydroxyphenyl) methyl] -4,7-dioxo-8-( ⁇ 6- [3- (piperazin-1-yl) azetidine- 1-yl] pyridin-2-yl ⁇ methyl) -2- (prop-2-en-1-yl) -octahydro-1H-pyrazino [2,1-c] [1,2,4] triazine-1- It relates to crystals of carboxamide compounds.
- Wnt signaling pathway is conserved regardless of differences between species, and has been clarified as an important pathway related to the development, differentiation and maintenance of living organisms.
- Wnt signaling pathway is constantly activated by adenomato polyposi coli (APC) and ⁇ -catenin inhibitory mutation and activation mutation, respectively.
- APC adenomato polyposi coli
- ⁇ -catenin inhibitory mutation and activation mutation respectively. It is known that In pancreatic cancer, blood cancer, liver cancer and the like, it is known that Wnt signaling pathway is activated after treatment with an existing antitumor agent.
- Non-Patent Documents 1 and 2 describe that when Wnt signaling pathway is inhibited, excellent antitumor activity is described, and Non-Patent Documents 12, 13 and 14 describe fibers by inhibiting Wnt signaling pathway. It is described that it exhibits an excellent effect on the disease. As a new target for tumor treatment and fibrosis treatment, Wnt signaling pathway is getting attention.
- Non-Patent Documents 3, 4, 5, 6, 7, 8, 9, 10 and 11 describe compounds or antibodies that inhibit Wnt signaling pathway, which are Tankyase, Traf2- and Nck-interacting kinase ( TNIK), Porcupine, Frizzled Receptor, etc. have been reported.
- (6S, 9aS) -N-benzyl-8-( ⁇ 6- [3- (4-ethylpiperazin-1-yl) azetidin-1-yl] pyridin-2-yl] is a compound represented by the following formula: ⁇ Methyl) -6- (2-fluoro-4-hydroxybenzyl) -4,7-dioxo-2- (prop-2-en-1-yl) hexahydro-2H-pyrazino [2,1-c] [1 , 2,4] triazine-1 (6H) -carboxamide (hereinafter referred to as “compound 1”) has a Wnt Pathway modulating action.
- the subject of this invention is providing the crystal
- the present invention provides the following [1] to [12].
- Powder X-ray diffraction further has diffraction peaks at diffraction angles (2 ⁇ ⁇ 0.2 °) of 14.2 °, 16.0 °, 18.9 °, 19.7 ° and 23.1 °.
- the crystals of Compound 1 according to the present invention have the properties as shown in the examples and can be used as drug substances for pharmaceuticals.
- FIG. 1 is a graph showing the results of Test Example 2.
- the vertical axis represents the number of polyps per mouse.
- FIG. 2 is a graph showing the results of Test Example 3.
- the horizontal axis indicates the number of days elapsed, and the vertical axis indicates the relative tumor volume (RTV) with respect to the tumor volume on day 0.
- FIG. 3 is a graph showing the results of Test Example 3.
- the horizontal axis represents the number of days elapsed, and the vertical axis represents the relative weight (RBW) with respect to the weight on the 0th day.
- 4 is a powder X-ray diffraction pattern of the crystal of Compound 1 in Test Example 4.
- FIG. The horizontal axis indicates the diffraction angle (2 ⁇ ), and the vertical axis indicates the peak intensity.
- FIG. 1 is a graph showing the results of Test Example 2.
- the vertical axis represents the number of polyps per mouse.
- FIG. 2 is a graph showing the results of Test Example 3.
- FIG. 5 is a graph showing the results of Test Example 5.
- the horizontal axis represents temperature
- the left vertical axis represents the amount of change in thermogravimetric analysis (TG)
- the right vertical axis represents the amount of change in differential thermal analysis (DTA).
- 6 is a 13 C solid state NMR spectrum of the crystal of Compound 1 in Test Example 6.
- FIG. The horizontal axis represents chemical shift ( ⁇ ), and the vertical axis represents peak intensity.
- the diffraction peaks in the powder X-ray diffraction described above are unique to the crystal of Compound 1, and are characteristic diffraction peaks of the crystal.
- the above diffraction angle value includes a numerical value within a range of about ⁇ 0.2 °. Need to be understood. Accordingly, not only crystals whose diffraction angles of peaks in powder X-ray diffraction completely match but also crystals whose diffraction angles of peaks match with an error of about ⁇ 0.2 ° are the same and are included in the present invention.
- the peak intensity or half-value width of the diffraction angle (2 ⁇ ) in powder X-ray diffraction is the difference in measurement conditions and the size and shape of each particle of a powder crystal used as a measurement sample even if the crystal form is the same. Due to variations, it varies from measurement to measurement, and a constant peak intensity or full width at half maximum is not always indicated. Therefore, in the comparison of powder X-ray diffraction patterns, even if there is a difference in peak intensity or half-value width at the same diffraction angle (2 ⁇ ), the difference does not mean that the measured crystal forms are different from each other. Absent.
- the crystal of the compound showing a powder X-ray diffraction pattern having such a difference with respect to the diffraction peak characteristic of the specific crystal of the present invention is the same crystal form as the crystal of the compound of the present invention.
- “having a powder X-ray diffraction pattern substantially the same as the powder X-ray diffraction pattern shown in FIG. 4” means that the powder X-ray diffraction pattern having a certain characteristic diffraction peak is In addition to the case where the powder X-ray diffraction pattern shown in FIG.
- the peak intensity or the half-value width is different or the diffraction angle of the characteristic diffraction peak coincides with an error range of ⁇ 0.2 °.
- the powder X-ray diffraction pattern is the same as the powder X-ray diffraction pattern shown in FIG. Therefore, it means that all crystals having such a powder X-ray diffraction pattern are the same crystals as the crystal of the present invention.
- the chemical shift ( ⁇ ) in the 13 C solid-state NMR spectrum can cause an error within a range of ⁇ 0.5 ppm.
- the value of should be understood as including values in the range of about ⁇ 0.5 ppm. Therefore, the present invention includes not only crystals in which the chemical shift in the 13 C solid state NMR spectrum is completely matched but also crystals in which the chemical shift is matched with an error of about ⁇ 0.5 ppm. Therefore, in this specification, for example, “having a peak at 12.6 ppm of chemical shift ( ⁇ ⁇ 0.5 ppm)” means “having a peak at 12.1 ppm to 13.1 ppm of chemical shift ( ⁇ )”.
- Compound 1 crystals can be produced by the above-described method for producing Compound 1, or by dissolving Compound 1 with heating in a solvent, cooling with stirring and crystallization. Can also be manufactured.
- Compound 1 used for crystallization may be in any form, may be a solvate, hydrate, or anhydride, and may be amorphous or crystalline (including those composed of a plurality of crystal polymorphs). It may be a mixture of these, but is preferably an anhydride.
- Solvents used for crystallization are, for example, alcohol solvents such as methanol, ethanol, 1-propanol and 2-propanol; acetonitrile; amide solvents such as N, N-dimethylformamide; ester systems such as ethyl acetate and isopropyl acetate. Solvents; saturated hydrocarbon solvents such as hexane and heptane; ketone solvents such as acetone and 2-butanone; ether solvents such as t-butyl methyl ether or water. These solvents may be used alone or in combination of two or more. When crystallization is performed by mixing two or more solvents, for example, it is preferable to use heptane and 1-propanol in combination.
- the amount of the solvent used can be appropriately selected with the lower limit being the amount in which compound 1 is dissolved by heating or the amount by which the suspension can be stirred, and the upper limit being the amount that does not significantly reduce the yield of crystals.
- the crystals obtained by the above method are in a single crystal form.
- This crystal form is stable, does not easily transition to another crystal form or amorphous, has good physical properties, and is suitable for formulation.
- seed crystals (such as crystals of desired compound 1) may or may not be added.
- the temperature at which the seed crystal is added is not particularly limited, but is preferably 0 to 100 ° C.
- the temperature at which compound 1 is dissolved by heating may be appropriately selected at a temperature at which compound 1 is dissolved depending on the solvent, but is preferably in the range of the temperature at which the recrystallization solvent starts to reflux from 50 ° C. More preferably, it is 60 to 100 ° C.
- Cooling during crystallization can give crystals containing different forms of crystals (polymorphs) when rapidly cooled, so it is desirable to adjust the cooling rate appropriately in consideration of the effects on crystal quality and particle size.
- cooling is performed at a rate of 5 to 40 ° C./hour, for example. More preferable is cooling at a rate of 5 to 25 ° C./hour, for example.
- the final crystallization temperature can be appropriately selected from the yield and quality of crystals, but is preferably -25 to 30 ° C.
- the crystallized crystal is separated by a normal filtration operation. If necessary, the crystal separated by filtration is washed with a solvent, and further dried to obtain a target crystal.
- a solvent used for washing the crystals, the same crystallization solvent can be used.
- Preferred examples of such a solvent include ethanol, acetone, 2-butanone, ethyl acetate, diethyl ether, t-butyl methyl ether, hexane, heptane and the like. These solvents may be used alone or in combination of two or more.
- the crystals separated by the filtration operation can be appropriately dried by being left in the air or a nitrogen stream, or by heating.
- the drying time may be appropriately selected according to the production amount, drying apparatus, drying temperature, etc. until the residual solvent falls below a predetermined amount. Moreover, drying can be performed under ventilation or under reduced pressure. The degree of vacuum may be appropriately selected according to the production amount, the drying device, the drying temperature, and the like. The obtained crystals can be left in the air after drying, if necessary.
- a pharmaceutical composition can be produced by adding a pharmaceutically acceptable additive to the crystal of Compound 1 as necessary.
- the dosage form of the pharmaceutical composition include oral preparations (tablets, granules, powders, capsules, syrups, etc.), injections (for intravenous administration, intramuscular administration, subcutaneous administration, and intraperitoneal administration). Etc.), external preparations (transdermal absorption preparations (ointments, patches, etc.), eye drops, nasal drops, suppositories, etc.).
- These solid preparations such as tablets, capsules, granules and powders can usually contain 0.001 to 99.5% by mass, preferably 0.001 to 90% by mass of Compound 1 crystals.
- excipients When producing an oral solid preparation, excipients, binders, disintegrants, lubricants, coloring agents, etc. are added to the crystals of compound 1 as necessary, and tablets, granules are prepared by conventional methods. , Powders and capsules. In addition, tablets, granules, powders, capsules and the like may be coated as necessary.
- the dose of the pharmaceutical agent according to the present invention usually varies depending on symptoms, age, sex, weight, etc., and may be an amount sufficient to produce a desired effect. For example, in the case of an adult, about 0.1 to 5000 mg (preferably 0.5 to 1000 mg) per day is used once a day or a plurality of days or divided into 2 to 6 times a day. .
- the crystals of Compound 1 according to the present invention can be produced, for example, by the methods described in the following production examples and examples. However, these are illustrative, and the crystal of the compound according to the present invention is not limited to the following specific examples in any case.
- YMC GEL SILICA (YMC Co., Ltd., catalog code: SL06I52W) is used as a silica gel for purification used in silica gel column chromatography, and an NH silica gel column is used.
- the silica gel for purification used in the chromatography is NH Siliga Gel (Fuji Silysia Chemical LTD., Catalog code: DM2035), and the silica gel for purification used in the ODS silica gel column chromatography is YAMAZEN GEL ODS-SM. (YAMAZEN Corporation, catalog codes: W113, W116, etc.) were used.
- the TLC plate for purification used in silica gel thin layer chromatography is TLC Silica gel 60F 254 (Merck KGaA, catalog code: 1.0571.0001), and is used in NH silica gel thin layer chromatography.
- TLC plate for purification NH SILICA GEL TLC plate (Fuji Silysia Chemical LTD., Catalog code: T050908) was used.
- NMP N-methylpyrrolidinone
- THF Tetrahydrofuran
- HATU O- (7-azabenzotriazol-1-yl) -N, N, N ′, N′-tetramethyluronium hexafluorophosphate
- HBTU O- (benzotriazole -1-yl) -N, N, N ′, N′-tetramethyluronium hexafluorophosphate
- TFA trifluoroacetic acid
- DMF N, N-dimethylformamide
- DMSO dimethyl sulfoxide
- the reaction mixture was quenched with sodium bicarbonate and water and extracted with ethyl acetate.
- the organic layer was dried over anhydrous magnesium sulfate and filtered.
- the title compound (1.14 g, yield: 74%) was obtained.
- the reaction mixture was filtered and the filtrate was concentrated under reduced pressure. Water was added to the residue and extracted with ethyl acetate. The organic layer was washed with water, saturated aqueous sodium thiosulfate solution, and saturated brine. The organic layer was dried over anhydrous magnesium sulfate and filtered. The solvent was evaporated under reduced pressure, diethyl ether and heptane were added to the resulting residue, and the precipitated solid was collected by filtration to obtain the title compound (3.82 g, yield: 68%).
- Test Example 1 Detection of Wnt signal pcDNA3.1 (+) (invitrogen) was cleaved with restriction enzymes BglII and NotI, and adapters BEHKS (restriction enzyme sites BglII, EcoRI, HindIII, KpnI, Plasmid pNeo-HKS was constructed by inserting SacI and NotI).
- BEHKS-R 5′-ggccgcgagctctctagagagtaccctcgagaagctttgaattca-3 ′ SEQ ID NO: 2
- an approximately 2700 bp fragment (including Wnt responsive sequence and luciferase gene) cleaved from the TOPglow plasmid contained in TOPglow / FOPglow TCF Reporter Kit (upstate Catalog # 17-366) using restriction enzymes HindIII and KpnI
- the plasmid pNeo-TOP was constructed by inserting between pNeo-HKS between HindIII and KpnI.
- Plasmid pNeo-TOP was introduced into human fetal kidney cell line HEK293, and compounds were selected using G418, and then a cell clone was established by the limiting dilution method. This cell clone was subjected to a Wnt signal detection test. This cell clone was subcultured in D-MEM high glucose medium (manufactured by Wako Pure Chemical Industries, Ltd.) containing 10% FBS, and cells in the growth phase were used for the test. After recovering the cells using trypsin-EDTA, the number of cells was counted and suspended in D-MEM high glucose medium containing 10% FBS so as to be 2 ⁇ 10 5 cells / mL.
- D-MEM high glucose medium manufactured by Wako Pure Chemical Industries, Ltd.
- the cell suspension was added to a 96-well plate for cell culture (manufactured by Greiner, product number 655083) at 0.1 mL / well and cultured overnight in a 5% CO 2 incubator (37 ° C.). After culturing, the test substance dissolved in DMSO was diluted with D-MEM high glucose medium containing 10% FBS and 80 mM LiCl to obtain a test solution. 0.1 mL of the test solution was added to each well and cultured in a 5% CO 2 incubator (37 ° C.). Six hours after the addition of the test solution, the supernatant of each well was removed, and 50 ⁇ L of Bright-Glo TM Luciferase substrate (Promega, product number E2620) was added.
- Bright-Glo TM Luciferase substrate Promega, product number E2620
- the plate was put on a plate mixer for several seconds, and then the luminescence of each well was measured using an EnVision TM Multilabel plate reader (Perkin Elmer). Luminescence of wells to which no test solution was added and LiCl was added was defined as 100% Wnt signal activity, and luminescence of wells to which neither a test substance nor LiCl was added was defined as 0% Wnt signal activity. As a result, the Wnt signal activity rate (%) of each well was determined, and the concentration (IC 50 ) required to inhibit the Wnt signal activity of the test substance by 50% was calculated. The IC 50 of the compound of Example 1 was 0.06 ⁇ M.
- Test Example 2 Intestinal polyp inhibitory action of APC Min / + mice
- the APC gene (Adenomatous polyposis coli gene), which is a Wnt signal degradation regulator, is called a colon cancer suppressor gene and is also a causative gene of human familial colorectal adenomatosis It has become.
- the colonic mucosal cells start disordered abnormal growth and form a colonic polyp that can be called a precancerous lesion. Thus, it is known to play an important role in the early stage of the colorectal cancer development process.
- This APC gene mutant mouse develops many polyps in the intestinal tract in the same way as patients with familial colorectal adenomatosis, and elucidates the mechanisms of colon cancer carcinogenesis and invasion based on WNT signals. It is useful and has become the standard model used for research on prevention, diagnosis and treatment of colorectal cancer.
- the grouping of APC Min / + mice (C57BL / 6J-APC ⁇ Min> / J Hetero, female, produced by Sampranet Co., Ltd.) was made so that the average values of the body weights of the mice on the first day of administration were almost equal. ,went.
- test substance (compound of Example 1) was dissolved in 0.1N HCl so as to have a dose concentration, and stored in a refrigerator at 4 ° C. to obtain an evaluation sample.
- control (vehicle) group only the administration solvent was used as an evaluation sample.
- the test specimen was orally administered twice a day for 4 consecutive days at doses of 50 mg / kg and 75 mg / kg, followed by a 3-day drug holiday for one cycle, for a total of 16 days (4 days ⁇ 4 Cycle). The experiment was conducted using 6-7 mice per group. For each of the control group and the test substance-administered group, the value of the weight of the last day relative to the weight of the first day (relative body weight: RBW) was calculated.
- RBW relative body weight
- the test substance administration group in which the RBW of the test substance administration group / RBW of the control group was 0.8 or more was determined to be a safe administration group.
- the actual number and standard error of the number of polyps after administration of the test substance relative to the number of control polyps on the last day (from the first day of administration to the 25th day) are shown in FIG.
- the polyps counted in the small intestine and colon were counted.
- Statistical analysis (Dunnett's test) of the test substance administration group with respect to the control group was performed, and the P value was described.
- Test Example 3 Antitumor effect in human K562 subcutaneous transplantation model Human chronic myeloid leukemia cell line K562 cultured in RPMI-1640 culture medium containing 10% FBS and penicillin / streptomycin was added to PBS (Wako Pure Chemical Industries, Ltd. Cat # 045). -29795) and MATRIGEL (BD Bioscience Cat # 354234) prepared at a concentration of 2 ⁇ 10 8 cells / mL are mixed 1: 1, and a cell suspension of 1 ⁇ 10 8 cells / mL is mixed. Prepared.
- the obtained cell suspension was transplanted at a dose of 100 ⁇ L into the right flank subcutaneous part of a 6-week-old nude mouse (CAnN.Cg-Foxn1nu / CrlCrlj, female, Charles River Japan Co., Ltd.). Seven days after transplantation, the short axis and long axis of the tumor were measured using an electronic digital caliper (Digimatic TM caliper, Mitutoyo Corporation), and the tumor volume was calculated by the following formula.
- Tumor volume (mm 3 ) major axis (mm) ⁇ minor axis (mm) ⁇ minor axis (mm) / 2
- Nude mice were grouped in such a manner that the average values of the tumor volumes were substantially equal based on the tumor volume on the first day of administration.
- a test substance compound of Example 1 was dissolved in 0.1N HCl and prepared to give a dose of 10 mL / kg to obtain an evaluation sample.
- Dasatinib, Free Base LC Laboratories, Cat. No.
- Tumor volume over time from the first day to the 28th day for each of the control group, the test substance single administration group, the Dasatinib single administration group, and the test substance and Dasatinib combination administration group (hereinafter also referred to as “combination administration group”). And the body weight was measured.
- the control group and the test substance single administration group were measured from the first day of administration to the 11th day.
- a tumor volume value (relative tumor volume: RTV) and a body weight value (relative body weight: RBW) with respect to the tumor volume on day 0 were calculated. Each is shown in FIG.
- Test Example 4 Powder X-ray Diffraction Crystal of Compound 1 obtained in Example 2 was placed on a sample stage of a powder X-ray diffractometer and analyzed under the following conditions. The result is shown in FIG. (Measurement condition)
- Target Copper detector: Scintillation counter tube voltage: 50 kV Tube current: 300mA
- Slit Divergence slit 0.5 mm, scattering slit open, light receiving slit open
- Scan speed 5 ° / min
- Sampling interval 0.02 ° Scan range: 5 ° to 35 °
- Sample holder Aluminum holder
- Test Example 5 Thermal Analysis The crystals obtained in Example 2 were precisely weighed in an aluminum sample pan, and thermogravimetric analysis (TG) and differential thermal analysis (DTA) were performed under the following conditions. The result is shown in FIG. (Measurement condition) Atmosphere: 40 mL / min under nitrogen gas flow Control: Empty aluminum sample pan heating rate: 10 ° C./min Sampling interval: 1 sec Measurement temperature range: 40 to 300 ° C
- Test Example 6 13 C solid state NMR spectrum
- the 13 C solid state NMR spectrum was measured under the following conditions. The results are shown in FIG. (Measurement condition) Device used: Avance 400 MHz (manufactured by BRUKER) 7 mm-CPMAS probe (manufactured by BRUKER) Measurement nucleus: 13 C (resonance frequency 100.6248425 MHz) Measurement temperature: Room temperature (22 ° C) Pulse mode: CPTOSS measurement rotation speed: 5000Hz Pulse repetition time: 4 sec Contact time: 1msec Integration number: 8000 times Reference substance: Glycine (External standard: 176.03 ppm)
- 13 C solid state NMR spectrum was obtained by CPTOSS method (spinning sideband elimination method) of carbon nucleus (resonance frequency 100.6 MHz) using Avance 400 MHz (manufactured by BRUKER) equipped with 7 mm-CPMAS probe manufactured by BRUKER. .
- a sample tube containing approximately 300 mg of a solid sample was rotated at 5 kHz, and the measurement was performed at a contact time of 1 msec, a pulse waiting time of 4 sec, an integration number of 8000 times, and room temperature (22 ° C.).
- the chemical shift was corrected by an external standard method in which the carbonyl carbon of glycine was 176.03 ppm.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description

一般に、医薬品として用いられる化合物およびその塩ならびにそれらの結晶の物性は、薬物のバイオアベイラビリティー、原薬の純度、製剤の処方などに大きな影響を与える。したがって、本発明の課題は、医薬品の原薬としての利用可能性を有する化合物1の結晶を提供することにある。
すなわち、本発明は、以下の[1]~[12]を提供する。
[1](6S,9aS)-N-ベンジル-8-({6-[3-(4-エチルピペラジン-1-イル)アゼチジン-1-イル]ピリジン-2-イル}メチル)-6-(2-フルオロ-4-ヒドロキシベンジル)-4,7-ジオキソ-2-(プロプ-2-エン-1-イル)ヘキサヒドロ-2H-ピラジノ[2,1-c][1,2,4]トリアジン-1(6H)-カルボキサミドの結晶。

[2]粉末X線回折において、回折角度(2θ±0.2°)5.8°に回折ピークを有する、[1]に記載の結晶。
[3]粉末X線回折において、回折角度(2θ±0.2°)5.8°、6.4°および10.1°に回折ピークを有する、[1]に記載の結晶。
[4]粉末X線回折において、さらに回折角度(2θ±0.2°)8.0°および12.8°に回折ピークを有する、[3]に記載の結晶。
[5]粉末X線回折において、さらに回折角度(2θ±0.2°)14.2°、16.0°、18.9°、19.7°および23.1°に回折ピークを有する、[4]に記載の結晶。
[6]13C固体NMRスペクトルにおいて、化学シフト(δ±0.5ppm)154.7ppmにピークを有する、(6S,9aS)-N-ベンジル-8-({6-[3-(4-エチルピペラジン-1-イル)アゼチジン-1-イル]ピリジン-2-イル}メチル)-6-(2-フルオロ-4-ヒドロキシベンジル)-4,7-ジオキソ-2-(プロプ-2-エン-1-イル)ヘキサヒドロ-2H-ピラジノ[2,1-c][1,2,4]トリアジン-1(6H)-カルボキサミドの結晶。
[7]13C固体NMRスペクトルにおいて、さらに化学シフト(δ±0.5ppm)141.1ppmおよび158.1ppmにピークを有する、[6]の結晶。
[8]13C固体NMRスペクトルにおいて、さらに化学シフト(δ±0.5ppm)134.0ppmおよび165.1ppmにピークを有する、[7]記載の結晶。
[9]13C固体NMRスペクトルにおいて、さらに化学シフト(δ±0.5ppm)12.6ppm、55.5ppmおよび118.5ppmにピークを有する、[8]記載の結晶。
[10]図4で示される粉末X線パターンと実質的に同一の粉末X線回折パターンを有する、(6S,9aS)-N-ベンジル-8-({6-[3-(4-エチルピペラジン-1-イル)アゼチジン-1-イル]ピリジン-2-イル}メチル)-6-(2-フルオロ-4-ヒドロキシベンジル)-4,7-ジオキソ-2-(プロプ-2-エン-1-イル)ヘキサヒドロ-2H-ピラジノ[2,1-c][1,2,4]トリアジン-1(6H)-カルボキサミドの結晶。
[11]図6で示される13C固体NMRスペクトルと実質的に同一の13C固体NMRスペクトルを有する、(6S,9aS)-N-ベンジル-8-({6-[3-(4-エチルピペラジン-1-イル)アゼチジン-1-イル]ピリジン-2-イル}メチル)-6-(2-フルオロ-4-ヒドロキシベンジル)-4,7-ジオキソ-2-(プロプ-2-エン-1-イル)ヘキサヒドロ-2H-ピラジノ[2,1-c][1,2,4]トリアジン-1(6H)-カルボキサミドの結晶。
[12][1]~[11]のいずれか一に記載の結晶を含む、医薬組成物。
回折角度(2θ±0.2°)5.8°に回折ピークを有する、化合物1の結晶;
回折角度(2θ±0.2°)5.8°、6.4°および10.1°に回折ピークを有する、化合物1の結晶;
回折角度(2θ±0.2°)5.8°、6.4°、8.0°、10.1°および12.8°に回折ピークを有する、化合物1の結晶;
回折角度(2θ±0.2°)5.8°、6.4°、8.0°、10.1°、12.8°、14.2°、16.0°、18.9°、19.7°および23.1°に回折ピークを有する、化合物1の結晶などを挙げることができる。
化学シフト(δ±0.5ppm)154.7ppmにピークを有する、化合物1の結晶;
化学シフト(δ±0.5ppm)141.1ppm,154.7ppmおよび158.1ppmにピークを有する、化合物1の結晶;
化学シフト(δ±0.5ppm)134.0ppm,141.1ppm,154.7ppm,158.1ppm,および165.1ppmにピークを有する、化合物1の結晶;
化学シフト(δ±0.5ppm)12.6ppm,55.5ppm,118.5ppm,134.0ppm,141.1ppm,154.7ppm,158.1ppm,および165.1ppmにピークを有する、化合物1の結晶などを挙げることができる。
化合物1は、後述する実施例、製造例に記載の方法で合成することができる。
化合物1の結晶は、上述の化合物1の製造方法により製造することができ、または、化合物1を、溶媒中で加熱溶解し、攪拌下冷却して晶析することにより、製造することもできる。
NMP:N-メチルピロリジノン
THF:テトラヒドロフラン
HATU:O-(7-アザベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウム ヘキサフルオロフォスフェート
HBTU:O-(ベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウム ヘキサフルオロフォスフェート
TFA:トリフルオロ酢酸
DMF:N,N-ジメチルホルムアミド
DMSO:ジメチルスルホキシド
(6S,9aS)-N-ベンジル-8-({6-[3-(4-エチルピペラジン-1-イル)アゼチジン-1-イル]ピリジン-2-イル}メチル)-6-(2-フルオロ-4-ヒドロキシベンジル)-4,7-ジオキソ-2-(プロプ-2-エン-1-イル)ヘキサヒドロ-2H-ピラジノ[2,1-c][1,2,4]トリアジン-1(6H)-カルボキサミド
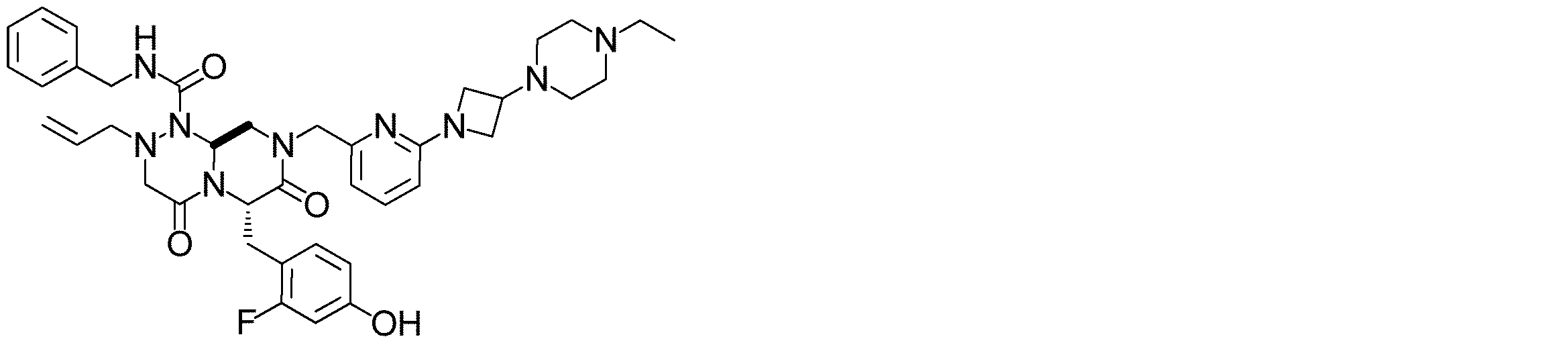
製造例1-1-6に記載の(6S,9aS)-N-ベンジル-6-((2-フルオロ-4-ヒドロキシフェニル)メチル)-8-((6-フルオロピリジン-2-イル)メチル)-4,7-ジオキソ-2-(プロプ-2-エン-1-イル)-オクタヒドロ-1H-ピラジノ[2,1-c][1,2,4]トリアジン-1-カルボキサミド(397mg、0.689mmol)とNMP(10mL)の混合溶液に、室温で製造例1-3-2に記載の1-(アゼチジン-3-イル)-4-エチルピペラジンとベンジルベンゼンの混合物(1.16g)を加え、140℃にて12時間マイクロウェーブ照射した。室温まで冷却した後、反応混合物に水を加え、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥し、ろ過した。溶媒を減圧下留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(メタノール)で精製した後、更にNHシリカゲルカラムクロマトグラフィー(酢酸エチル:メタノール=20:1)で精製し、標記化合物(402mg、収率:80%)を得た。
1H-NMR Spectrum (400 MHz, CDCl3)δ (ppm): 1.00(3H, t, J=6.8 Hz), 2.10-2.65(10H, m), 3.10-3.21(2H, m),3.41-3.74(8H, m), 3.84-3.89(1H, m), 3.95-4.05(2H, m), 4.17-4.23(2H, m),4.51(1H, dd, J=6.8 Hz, 15.6 Hz), 4.95(1H, d, J=13.6 Hz), 5.20-5.30(3H, m),5.50-5.60(1H, m), 5.70-5.80(1H, m), 5.82-5.87(1H, m), 6.24(1H, d, J=8.0 Hz),6.41(1H, dd, J=2.0 Hz, 11.2 Hz), 6.47(1H, dd, J=8.8 Hz, 8.8 Hz), 6.69(1H, d,J=7.2 Hz), 6.80-6.86(1H, m), 7.20-7.31(3H, m), 7.35-7.46(3H, m).
ESI-MS(m/z): 726.57 [M+H]+.
市販の2,2-ジエトキシエタン-1-アミン(926μL、6.39mmol)とTHF(10.0mL)、酢酸(1.00mL)の混合溶液に、室温で市販の6-フルオロピリジン-2-カルバルデヒド(800mg、6.39mmol)を加え、室温で25分撹拌した。続いて、トリアセトキシ水素化ホウ素ナトリウム(2.71g、12.8mmol)を室温で加え、1時間10分撹拌した。反応混合物に炭酸水素ナトリウムと水を加えて反応を止め、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥し、ろ過した。溶媒を減圧下留去し、得られた残渣をNHシリカゲルカラムクロマトグラフィー(ヘプタン:酢酸エチル=1:1)で精製した後、更にシリカゲルカラムクロマトグラフィー(酢酸エチル:メタノール=20:1)で精製し、標記化合物(1.14g、収率:74%)を得た。
1H-NMR Spectrum (400 MHz, CDCl3)δ (ppm): 1.22(6H, t, J=7.2 Hz), 2.76(2H, d, J=5.5 Hz), 3.50-3.61(2H, m),3.65-3.76(2H, m), 3.89(2H, s), 4.64(1H, t, J=5.5 Hz), 6.80(1H, dd, J=2.8 Hz,8.2 Hz), 7.22(1H, dd, J=2.4 Hz, 7.3 Hz), 7.74(1H, q, J=7.9 Hz).
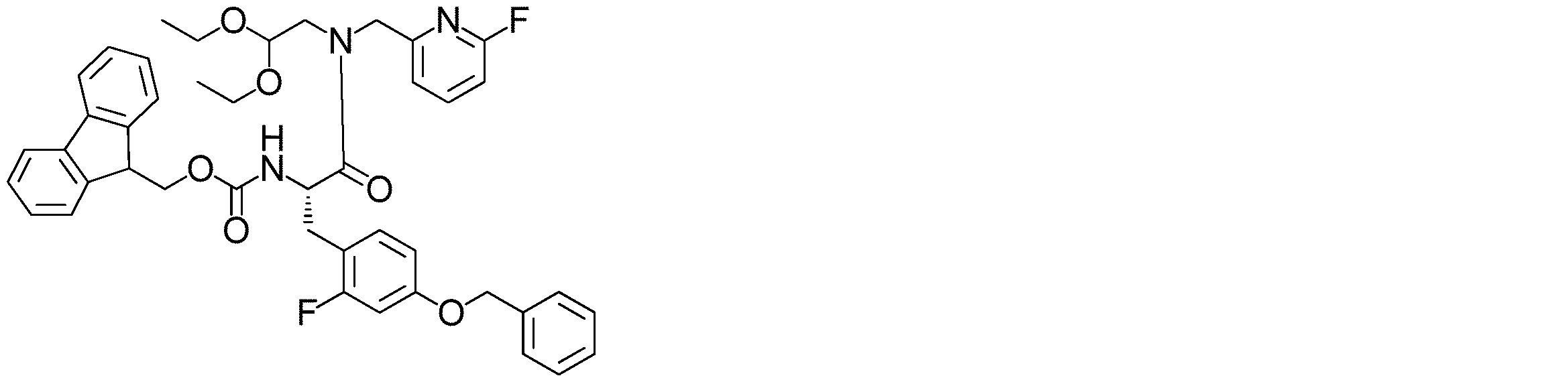
製造例1-1-1に記載の(2,2-ジエトキシエチル)((6-フルオロピリジン-2-イル)メチル)アミン(3.50g、14.4mmol)とジクロロメタン(25mL)の混合溶液に、室温で製造例1-2-7に記載の(2S)-3-(4-(ベンジルオキシ)-2-フルオロフェニル)-2-(((9H-フルオレン-9-イルメトキシ)カルボニル)アミノ)プロパノイック アシッド(7.76g、15.1mmol)、N-メチルモルホリン(2.06mL、18.7mmol)、HATU(6.04g、15.8mmol)を加え、室温で13時間撹拌した。反応混合物に炭酸水素ナトリウムと水を加え、ジクロロメタンで抽出した。有機層を無水硫酸マグネシウムで乾燥し、ろ過した。溶媒を減圧下留去し、標記化合物の粗体(14.4g)を得た。これ以上精製することなく、次の反応に用いた。
ESI-MS(m/z): 758.50 [M+Na]+.

製造例1-1-2に記載の9H-フルオレン-9-イルメチル N-((1S)-2-(4-(ベンジルオキシ)-2-フルオロフェニル)-1-((2,2-ジエトキシエチル)((6-フルオロピリジン-2-イル)メチル)カルバモイル)エチル)カーバメート(14.4g)とTHF(30mL)の混合溶液に、室温でジエチルアミン(5.27mL、50.4mmol)を加え、室温で2時間撹拌した。反応混合物を減圧下濃縮し、残渣にメタノール、水、ヘプタンを加え分配した。水層をヘプタンで洗浄した後、減圧下濃縮した。残渣に水を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥し、ろ過した。溶媒を減圧下留去し、得られた残渣をNHシリカゲルカラムクロマトグラフィー(ヘプタン:酢酸エチル=1:1ついで酢酸エチル)で精製し、標記化合物(6.87g、収率:93%)を得た。
ESI-MS(m/z): 514.32 [M+H]+.
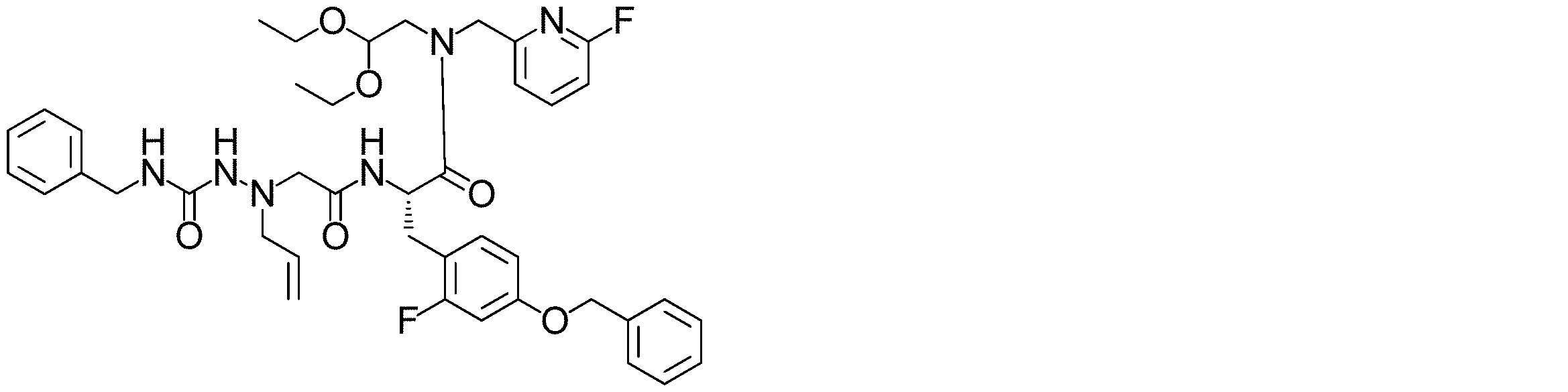
製造例1-1-3に記載の(2S)-2-アミノ-3-(4-(ベンジルオキシ)-2-フルオロフェニル)-N-(2,2-ジエトキシエチル)-N-((6-フルオロピリジン-2-イル)メチル)プロパンアミド(4.87g、9.48mmol)とジクロロメタン(100mL)の混合溶液に、WO2009/148192に記載の2-(((ベンジルカルバモイル)アミノ)(プロプ-2-エン-1-イル)アミノ)アセティック アシッド(2.62g、9.95mmol)、トリエチルアミン(2.64mL、19.0mmol)及びHBTU(3.96g、10.4mmol)を室温で加え、室温で1時間撹拌した。反応混合物を減圧下濃縮し、得られた残渣をNHシリカゲルカラムクロマトグラフィー(酢酸エチルついで酢酸エチル:メタノール=30:1)で精製し、標記化合物(7.28g、収率:定量的)を得た。
ESI-MS(m/z): 759.43 [M+H]+.

製造例1-1-4に記載の(2S)-2-(2-(((ベンジルカルバモイル)アミノ)(プロプ-2-エン-1-イル)アミノ)アセトアミド)-3-(4-(ベンジルオキシ)-2-フルオロフェニル)-N-(2,2-ジエトキシエチル)-N-((6-フルオロピリジン-2-イル)メチル)プロパンアミド(7.28g、9.48mmol)とギ酸(50mL)の混合溶液を室温で15時間15分撹拌した。反応混合物を減圧下濃縮し、残渣にアンモニア水溶液を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥し、ろ過した。溶媒を減圧下留去し、得られた残渣をNHシリカゲルカラムクロマトグラフィー(ヘプタン:酢酸エチル=1:1)で精製し、標記化合物(5.04g、収率:80%)を得た。
ESI-MS(m/z): 667.39 [M+H]+.

製造例1-1-5に記載の(6S,9aS)-N-ベンジル-6-((4-(ベンジルオキシ)-2-フルオロフェニル)メチル)-8-((6-フルオロピリジン-2-イル)メチル)-4,7-ジオキソ-2-(プロプ-2-エン-1-イル)-オクタヒドロ-1H-ピラジノ[2,1-c][1,2,4]トリアジン-1-カルボキサミド(5.04g、7.56mmol)とTFA(20mL)の混合溶液に、室温でチオアニソール(3.55mL、30.2mmol)を加え、室温で13時間50分撹拌した。反応混合物を減圧下濃縮し、残渣に炭酸水素ナトリウムと水を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥し、ろ過した。溶媒を減圧下留去し、得られた残渣をNHシリカゲルカラムクロマトグラフィー(酢酸エチル:メタノール=20:1)で精製し、標記化合物(4.34g、収率:定量的)を得た。
ESI-MS(m/z): 577.31 [M+H]+.

市販のL-セリン メチルエステル 塩酸塩(10.0g、64.3mmol)と1,4-ジオキサン(15mL)、水(90mL)の混合溶液に、室温で炭酸水素ナトリウム(10.8g、129mmol)を加え、室温で15分撹拌した。続いて、室温で2,5-ジオキソピロリジン-1-イル 9H-フルオレン-9-イルメチル カルボネート(21.7g、64.3mmol)の1,4-ジオキサン(60mL)溶液を加え、室温で14時間撹拌した。反応混合物に水を加え、酢酸エチルで3回抽出し、合わせた有機層を水及び飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥し、ろ過した。溶媒を減圧下留去し、得られた残渣にジエチルエーテル及びヘプタンを加え、析出した固体をろ取し、標記化合物(22.3g、収率:定量的)を得た。
1H-NMR Spectrum (400 MHz, CDCl3)δ (ppm): 2.00-2.15(1H, m), 3.81(3H, s), 3.89-4.07(2H, m), 4.20-4.28(1H, m),4.39-4.53(3H, m), 5.63-5.74(1H, m), 7.29-7.37(2H, m), 7.38-7.46(2H, m),7.55-7.65(2H, m), 7.74-7.82(2H, m).

製造例1-2-1に記載のメチル (2S)-2-(((9H-フルオレン-9-イルメトキシ)カルボニル)アミノ)-3-ヒドロキシプロパノエート(5.00g、14.6mmol)とピリジン(25mL)の混合溶液に、0℃で4-ジメチルアミノピリジン(18.0mg、0.146mmol)、p-トルエンスルホニル クロリド(5.58g、29.3mmol)を加え、0℃で7時間撹拌した。反応混合物に水を加え、酢酸エチルで2回抽出した。合わせた有機層を1N塩酸で3回、飽和炭酸水素ナトリウム水溶液、および飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥し、ろ過した。溶媒を減圧下留去し、得られた残渣に酢酸エチル、ジエチルエーテルおよびヘプタンを加え、析出した固体をろ取し、標記化合物(6.20g、収率:85%)を得た。
1H-NMR Spectrum (400 MHz, CDCl3)δ (ppm): 2.37(3H, s), 3.74(3H, s), 4.16-4.23(1H, m), 4.23-4.31(1H, m),4.32-4.40(2H, m), 4.41-4.48(1H, m), 4.54-4.62(1H, m), 5.63-5.66(1H, m),7.26-7.37(4H, m), 7.38-7.45(2H, m), 7.56-7.64(2H, m), 7.72-7.81(4H, m).

製造例1-2-2に記載のメチル (2S)-2-(((9H-フルオレン-9-イルメトキシ)カルボニル)アミノ)-3-(((4-メチルベンゼン)スルホニル)オキシ)プロパノエート(6.20g、12.5mmol)とアセトン(50mL)の混合溶液に、室温でヨウ化ナトリウム(9.38g、62.6mmol)を加え、室温で90時間50分撹拌した。反応混合物をろ過し、ろ液を減圧下濃縮した。残渣に水を加え、酢酸エチルで抽出した。有機層を水、飽和チオ硫酸ナトリウム水溶液、および飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥し、ろ過した。溶媒を減圧下留去し、得られた残渣にジエチルエーテルおよびヘプタンを加え、析出した固体をろ取し、標記化合物(3.82g、収率:68%)を得た。
1H-NMR Spectrum (400 MHz, CDCl3)δ (ppm): 3.55-3.66(2H, m), 3.84(3H, s), 4.20-4.30(1H, m), 4.35-4.48(2H, m),4.56-4.62(1H, m), 5.63-5.72(1H, m), 7.30-7.37(2H, m), 7.38-7.45(2H, m), 7.62(2H,d, J=7.2 Hz), 7.78(2H, d, J=7.5 Hz).

市販の4-ブロモ-3-フルオロフェノール(15.0g、78.5mmol)とDMF(30mL)の混合溶液に、室温で炭酸カリウム(21.7g、157mmol)、ベンジルブロミド(10.2mL、86.4mmol)を加え、室温で20分撹拌した後に、70℃で40分撹拌した。反応混合物を室温に冷却した後、水を加え、酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥し、ろ過した。溶媒を減圧下留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(ヘプタン:酢酸エチル=5:1)で精製し、標記化合物(22.7g、収率:定量的)を得た。
1H-NMR Spectrum (400 MHz, CDCl3)δ (ppm): 5.04(2H, s), 6.65-6.72(1H, m), 6.75-6.80(1H, m), 7.30-7.45(6H, m).

製造例1-2-4に記載の4-(ベンジルオキシ)-1-ブロモ-2-フルオロベンゼン(187g、665mmol)と1,4-ジオキサン(300mL)の混合溶液に、室温でヨウ化銅(I)(12.6g、66.1mmol)、ヨウ化ナトリウム(200g、1.33mol)、N,N’-ジメチルエチレンジアミン(14.0mL、132mmol)を加え、窒素雰囲気下、110~115℃にて19時間撹拌した。反応混合物を室温に冷却した後、水と酢酸エチルを加え、セライトを用いてろ過し、ろ液を分配した。得られた水層を酢酸エチルで抽出した。得られた有機層を合わせ、シリカゲルを敷いたグラスフィルターでろ過した。シリカゲルを酢酸エチルで洗浄し、得られた有機層を合わせ、溶媒を減圧下留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘプタン:酢酸エチル=7:1ついで4:1)で精製し、標記化合物(195g、収率:89%)を得た。
1H-NMR Spectrum (400 MHz, CDCl3)δ (ppm): 5.04(2H, s), 6.57-6.62(1H, m), 6.73(1H, dd, J=2.8Hz, 10.0 Hz),7.31-7.43(5H, m), 7.55-7.60(1H, m).

亜鉛粉末(51.6g、789mmol)を1N塩酸(100mL)に加え、超音波振動にかけた後、静止させ上澄み液を除いた。この操作を2回行った。得られた亜鉛の残渣に水(300mL)を加え、攪拌した後、静止させ上澄み液を除く操作を3回行った。更にアセトン(300mL)を加え、攪拌した後、静止させ上澄み液を除き、続いてジエチルエーテル(300mL)を加え、攪拌した後、静止させ上澄み液を除き、残渣を減圧下乾燥させ、活性化亜鉛を得た。この活性化亜鉛に、窒素雰囲気下、室温でDMF(120mL)、ヨウ素(3.36g、13.2mmol)を加え、室温で45分撹拌した。反応混合物に、窒素雰囲気下、室温で製造例1-2-3に記載のメチル (2R)-2-(((9H-フルオレン-9-イルメトキシ)カルボニル)アミノ)-3-ヨードプロパノエート(120g、266mmol)のDMF(500mL)溶液を30分かけて加え、室温で40分間撹拌した。窒素雰囲気下、室温で反応混合物に、製造例1-2-5に記載の4-(ベンジルオキシ)-2-フルオロ-1-ヨードベンゼン(104g、318mmol)、トリス(ジベンジリデンアセトン)パラジウム(0)(6.00g、6.55mmol)、2-ジシクロヘキシルホスフィノ-2’,6’-ジメトキシビフェニル(5.40g、13.2mmol)を加え、室温で20時間40分撹拌した。反応混合物に水と酢酸エチルを加え、セライトを用いてろ過した。ろ液を分配し、更に水層を酢酸エチルで3回抽出した。合わせた有機層を水および飽和食塩水で洗浄した。有機層を無水硫酸マグネシウムで乾燥し、ろ過した。溶媒を減圧下留去し、得られた残渣にジエチルエーテル(1.00L)、ヘプタン(1.00L)を加え、析出した固体をろ取した。ろ取した固体にジエチルエーテル(500mL)、ヘプタン(500mL)を加え、析出した固体をろ取し、標記化合物(107g、収率:77%)を得た。
1H-NMR Spectrum (400 MHz, CDCl3)δ (ppm): 3.03-3.20(2H, m), 3.75(3H, s), 4.20(1H, t, J=6.6 Hz), 4.25-4.38(1H,m), 4.43(1H, dd, J=7.1 Hz, 10.4 Hz), 4.58-4.70(1H, m), 4.99(2H, s), 5.33(1H, d,J=8.4 Hz), 6.63-6.72(2H, m), 6.94-7.03(1H, m), 7.26-7.48(9H, m), 7.52-7.62(2H,m), 7.77(2H, d, J=7.7 Hz).

製造例1-2-6に記載のメチル (2S)-3-(4-(ベンジルオキシ)-2-フルオロフェニル)-2-(((9H-フルオレン-9-イルメトキシ)カルボニル)アミノ)プロパノエート(60.0g、114mmol)と酢酸エチル(1331mL)の混合溶液に、室温でヨウ化リチウム(92.0g、685mmol)を加え、加熱還流下23時間45分撹拌した。反応混合物を0℃に冷却し、析出した固体をろ取した。得られた固体に1N塩酸(228mL)を加え、酢酸エチルで抽出した。有機層を無水硫酸マグネシウムで乾燥し、ろ過した。溶媒を減圧下留去し、標記化合物(42.2g、収率:72%)を得た。
1H-NMR Spectrum (400 MHz, CDCl3)δ (ppm): 3.05-3.15(1H, m), 3.18-3.30(1H, m), 4.15-4.23(1H, m), 4.25-4.50(2H, m),4.60-4.70(1H, m), 4.99(2H, m), 5.29(1H, d, J=7.6 Hz), 6.64-6.73(2H, m),7.06(1H, dd, J=8.0, 9.6 Hz), 7.24-7.44(9H, m), 7.55(2H, dd, J=6.4, 6.4 Hz),7.76(2H, d, J=7.6 Hz).
ESI-MS(m/z): 512.30 [M+H]+.

市販の1-(ジフェニルメチル)アゼチジン-3-オン(10.1g、42.6mmol)とTHF(100mL)、酢酸(5.00mL)の混合溶液に、室温でエチルピペラジン(6.48mL、51.1mmol)を加え、室温で45分間撹拌した。反応混合物に、室温でトリアセトキシ水素化ホウ素ナトリウム(18.1g、85.1mmol)を加え、室温で15時間撹拌した。反応混合物に炭酸水素ナトリウム、水を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、ろ過した。溶媒を減圧下留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル~メタノール)で精製した後、NHシリカゲルカラムクロマトグラフィー(ヘプタン:酢酸エチル=2:1ついで1:1)で精製し、標記化合物(12.7g、収率:89%)を得た。
1H-NMR Spectrum (400 MHz, CDCl3)δ (ppm): 1.07(3H, t, J=7.6 Hz), 2.20-2.65(10H, m), 2.85-2.93(2H, m),2.95-3.05(1H, m), 3.35-3.45(2H, m), 4.41(1H, s), 7.15-7.20(2H, m),7.23-7.29(4H, m), 7.37-7.42(4H, m).

製造例1-3-1に記載の1-(1-(ジフェニルメチル)アゼチジン-3-イル)-4-エチルピペラジン(12.7g、37.9mmol)とメタノール(50mL)の混合溶液に、室温で水酸化パラジウム-炭素(5.00g)を加え、水素雰囲気下、室温、0.35MPa~0.40MPaで10時間撹拌した。反応混合物を窒素雰囲気に置換した後、セライトを用いてろ過した。ろ液を減圧下濃縮し、標記化合物をベンジルベンゼンとの混合物(12.4g)として得た。これ以上精製することなく、次の反応に用いた。
1H-NMR Spectrum (400 MHz, CDCl3)δ (ppm): 1.09(3H, t, J=7.2 Hz), 2.10-2.80(10H, m), 3.20-3.30(1H, m),3.53-3.60(2H, m), 3.60-3.70(2H, m).
(6S,9aS)-N-ベンジル-8-({6-[3-(4-エチルピペラジン-1-イル)アゼチジン-1-イル]ピリジン-2-イル}メチル)-6-(2-フルオロ-4-ヒドロキシベンジル)-4,7-ジオキソ-2-(プロプ-2-エン-1-イル)ヘキサヒドロ-2H-ピラジノ[2,1-c][1,2,4]トリアジン-1(6H)-カルボキサミドの結晶
実施例1に記載の(6S,9aS)-N-ベンジル-8-({6-[3-(4-エチルピペラジン-1-イル)アゼチジン-1-イル]ピリジン-2-イル}メチル)-6-(2-フルオロ-4-ヒドロキシベンジル)-4,7-ジオキソ-2-(プロプ-2-エン-1-イル)ヘキサヒドロ-2H-ピラジノ[2,1-c][1,2,4]トリアジン-1(6H)-カルボキサミド(150mg)に酢酸イソプロピル(1.5mL)を加え85℃で1時間、さらに室温で終夜攪拌した。得られた懸濁物を濾取し、4℃に冷却した酢酸イソプロピルで洗浄した後、45℃で減圧乾燥し、白色固体を141.2mg得た。得られた固体のうち100mgに1-プロパノール(0.4mL)を加え95℃で加熱溶解した。1-プロパノール(0.1mL)を追加し、室温で攪拌した。得られた懸濁物にヘプタン(1.5mL)を室温で加えた。懸濁物を濾取し、4℃に冷却したヘプタンで洗浄した後、45℃で減圧乾燥し、標記化合物を92.1mg得た。
1H-NMR(600 MHz, CD3OD) δ (ppm): 1.15 (t, J=7 Hz, 3 H), 2.0-3.1 (br, 8 H),3.13 (dd, J=14, 7 Hz, 1 H), 3.32 (m, 1 H), 3.33 (m, 1 H), 3.44 (dd, J=14, 5 Hz,1 H), 3.46 (d, J=17 Hz, 1 H), 3.54 (dd, J=12, 4 Hz, 1 H), 3.62 (dd, J=13, 6 Hz,1 H), 3.69 (dd, J=13, 7 Hz, 1 H), 3.79 (dd, J=8, 5 Hz, 1 H), 3.86 (dd, J=8, 6Hz, 1 H), 3.89 (t, J=11 Hz, 1 H), 4.03 (t, J=8 Hz, 1 H), 4.06 (t, J=8 Hz, 1 H),4.25 (d, J=15 Hz, 1 H), 4.33 (d, J=16 Hz, 1 H), 4.41 (d, J=16 Hz, 1 H), 4.77(d, J=16Hz, 1 H), 5.12 - 5.18 (m, 2H), 5.27 (dd, J=7, 5 Hz, 1 H), 5.84 (m, 2H), 6.30 (d, J=8 Hz, 1 H), 6.38 (m, 1 H), 6.40 (m,1 H), 6.51 (d, J=8 Hz, 1 H),6.88 (dd, J=9, 8 Hz, 1 H), 7.25 (dd, J=8, 7 Hz, 1 H), 7.28 (d, J=8 Hz, 2 H),7.34 (dd, J=8, 7 Hz, 2 H), 7.47 (dd, J=8, 8 Hz, 1 H)
pcDNA3.1(+)(invitrogen)を、制限酵素BglIIおよびNotIを用いて切断し、以下に示す配列を有するアダプターBEHKS(制限酵素サイトBglII、EcoRI、HindIII、KpnI、SacIおよびNotIを含む)を挿入することにより、プラスミドpNeo-HKSを作製した。
BEHKS-F 5’-gatctgaattcaagcttctcgagggtacctctagagagctcgc-3’ (配列番号1)
BEHKS-R 5’-ggccgcgagctctctagaggtaccctcgagaagcttgaattca-3’ (配列番号2)
次に、TOPglow/FOPglow TCF Reporter Kit(upstate Catalog#17-366)に含まれるTOPglowプラスミドから、制限酵素HindIIIおよびKpnIを用いて切断した約2700bpフラグメント(Wnt応答性配列およびルシフェラーゼ遺伝子を含む)を、pNeo-HKSのHindIII-KpnI間へ挿入してプラスミドpNeo-TOPを作製した。ヒト胎児由来腎臓細胞株HEK293にプラスミドpNeo-TOPを導入し、G418を用いて化合物を選択した後、限界希釈法により細胞クローン株を樹立した。本細胞クローン株をWntシグナル検出試験に供した。
本細胞クローン株は、10%FBSを含むD-MEM 高グルコース培地(和光純薬工業(株)製)で継代培養し、試験には増殖期の細胞を使用した。トリプシン-EDTAを用いて細胞を回収後、細胞数を計測して2×105個/mLになるように10%FBSを含むD-MEM 高グルコース培地に懸濁した。その細胞懸濁液を細胞培養用96ウェルプレート(グライナー社製、製品番号655083)に0.1mL/well加え、5%CO2インキュベーター中(37℃)で一晩培養した。培養後、DMSOで溶解した被検物質を10%FBSおよび80mM LiClを含むD-MEM 高グルコース培地で希釈して被検溶液を得た。上記被検溶液0.1mLを各ウェルに添加し、5%CO2インキュベーター中(37℃)で培養した。被検溶液添加してから6時間後に各ウェルの上清を取り除き、50μLのBright-GloTM Luciferase基質(Promega社製、製品番号E2620)を添加した。そのプレートをプレートミキサーに数秒かけた後に、EnVisionTM Multilabelプレートリーダー(パーキンエルマー社製)を用いて各ウェルの発光を測定した。被検溶液を添加せず、LiClを添加したウェルの発光度を100%のWntシグナル活性として、また、被検物質およびLiClのどちらも添加していないウェルの発光度を0%のWntシグナル活性として、各ウェルのWntシグナル活性率(%)を求め、被検物質のWntシグナル活性を50%阻害するのに必要な濃度(IC50)を算出した。実施例1化合物のIC50は、0.06μMであった。
Wntシグナルの分解制御因子であるAPC遺伝子(Adenomatous polyposis coli gene)は大腸癌抑制遺伝子と呼ばれ、ヒト家族性大腸腺腫症の原因遺伝子にもなっている。APC遺伝子に変異が生じると、大腸粘膜細胞は無秩序な異常増殖を開始し、前癌病変とも言うべき大腸ポリープを形成する。このように大腸癌発生プロセスの初期段階において重要な役割を担っていることが知られている。
このAPC遺伝子の突然変異マウス(APCMin/+マウス)は、家族性大腸腺腫症患者と同様に腸管に多くのポリープを発症し、WNTシグナルに基づく大腸がんの発癌や浸潤のメカニズムの解明に有用であり、大腸癌の予防、診断、治療の研究に使われているスタンダードモデルとなっている。APCMin/+マウス(C57BL/6J-APC<Min>/J Hetero、雌、株式会社サンプラネットにて生産)の群分けは、投与初日の当該マウスの体重の平均値がほぼ等しくなるようにして、行った。被験物質(実施例1化合物)を0.1N HClで投与濃度となるように溶解し、4℃の冷蔵庫内で保管して、評価検体を得た。コントロール(Vehicle)群には、投与用溶媒のみを評価検体として用いた。評価検体は50mg/kgおよび75mg/kgの投与容量で1日2回4日連続して経口投与した後、3日間の休薬期間を設けることを1サイクルとし、計16日間(4日間×4サイクル)投与した。なお、実験は1群6-7匹のマウスを用いて行った。コントロール群、被験物質投与群それぞれに対し、初日の体重に対する最終日の体重の値(relative body weight: RBW)を算出した。被験物質投与群のRBW/コントロール群のRBWが0.8以上の被験物質投与群を安全に投与可能な群と判定した。これに該当する被験物質投与群について、最終日(投与初日から25日目)におけるコントロールのポリープ数に対する被験物質投与後のポリープ数の実数および標準誤差を図1に示した。なお、ポリープは小腸と結腸に形成したものをカウントした。コントロール群に対する被験物質投与群の統計解析(Dunnett’s test)を行い、P値を記載した。
10%FBS、ペニシリン/ストレプトマイシンを含むRPMI-1640培養液で培養したヒト慢性骨髄性白血病細胞株K562をPBS(和光純薬工業株式会社 Cat#045-29795)にて2×108cells/mL濃度に調製したものと、MATRIGEL(BDバイオサイエンス Cat# 354234)とを1:1で混合し、1×108cells/mLの細胞懸濁液を調製した。得られた細胞懸濁液を、6週齢のヌードマウス(CAnN.Cg-Foxn1nu/CrlCrlj、雌、日本チャールス・リバー株式会社)の右脇腹皮下部に100μLの用量で移植した。移植から7日後に電子デジタルノギス(デジマチックTMキャリパ、株式会社ミツトヨ)を用いて腫瘍の短径、長径を計測し、以下の計算式で腫瘍体積を算出した。
腫瘍体積(mm3)=長径(mm)×短径(mm)×短径(mm)/2
ヌードマウスの群分けは、投与初日の腫瘍体積をもとに腫瘍体積の平均値がほぼ等しくなるようにして、行った。被験物質(実施例1化合物)を0.1N HClに溶解し、10mL/kgの投与用量となるように調製し、評価検体を得た。また、Dasatinib,Free Base(LC Laboratories,Cat.No.D-3307)を、10mL/kgの投与用量となるように、大塚蒸留水(大塚製薬株式会社、 Cat#1324)とPROPYLENE GLYCOL(和光純薬工業株式会社、Cat#164-04996)の1:1溶液に溶解し、Dasatinib投与液を調製した。評価検体は1日2回(bid)で投与初日より5日間連続して経口投与を行った。Dasatinib投与液は1日1回(qd)5日間連続して経口投与した後、2日間の休薬期間を設けることを1サイクルとし、合計2サイクル投与した。コントロール群は無投与とした。なお、実験は1群9-10匹のマウスを用いて行った。コントロール群、被験物質単独投与群、Dasatinib単独投与群、ならびに、被験物質およびDasatinib併用投与群(以下、「併用投与群」ともいう。)それぞれに対し、初日から28日目まで経時的に腫瘍体積および、体重を測定した。コントロール群、被験物質単独投与群は、投与初日から11日目まで測定した。測定毎に0日目の腫瘍体積に対する腫瘍体積の値(relative tumor volume: RTV)および、体重の値(relative body weight: RBW)を算出し、投与初日から28日目までのグラフを図2,図3にそれぞれ示した。また、28日目のRTV値を使用し、Dasatinib単独投与群に対する併用投与群の統計解析(Dunnett’s test)を行い、P値が0.05以下であった群にアスタリスク(*)を付けた。さらに、28日目に腫瘍が目視および触知できない(non-palpable tumor)個体数を表1に示した。その時、Dasatinib単独投与群に対する被験物質Dasatinib併用投与群の統計解析(Fisher’s test)を行い、P値が0.05以下に関して*を、P値0.01以下の群には***を付けた。

実施例2で得られた化合物1の結晶を、粉末X線回折装置の試料台に載せ、下記の条件で分析した。その結果を図4に示す。
(測定条件)
ターゲット:銅
検出器:シンチレーションカウンター
管電圧:50kV
管電流:300mA
スリット:発散スリット0.5mm、散乱スリット開放、受光スリット開放
スキャン速度:5°/min
サンプリング間隔:0.02°
スキャン範囲:5°~35°
サンプルホルダー:アルミニウム製ホルダー
実施例2で得られた結晶をアルミニウム製試料パンに精密に秤取し、以下の条件で熱重量分析(TG)及び示差熱分析(DTA)を行った。その結果を図5に示す。
(測定条件)
雰囲気:40mL/min窒素ガス気流下
対照:空のアルミニウム製試料パン
昇温速度:10℃/min
サンプリング間隔:1sec
測定温度範囲:40~300℃
13C固体NMRスペクトルは以下の条件で測定した。結果を図6に示す。
(測定条件)
使用装置:Avance400MHz(BRUKER社製)7mm-CPMASプローブ(BRUKER社製)
測定核:13C(共鳴周波数 100.6248425MHz)
測定温度:室温(22℃)
パルスモード:CPTOSS測定
回転数:5000Hz
パルス繰り返し時間:4sec
コンタクトタイム:1msec
積算回数:8000回
基準物質:グリシン(外部基準:176.03ppm)
Claims (12)
- (6S,9aS)-N-ベンジル-8-({6-[3-(4-エチルピペラジン-1-イル)アゼチジン-1-イル]ピリジン-2-イル}メチル)-6-(2-フルオロ-4-ヒドロキシベンジル)-4,7-ジオキソ-2-(プロプ-2-エン-1-イル)ヘキサヒドロ-2H-ピラジノ[2,1-c][1,2,4]トリアジン-1(6H)-カルボキサミドの結晶。

- 粉末X線回折において、回折角度(2θ±0.2°)5.8°に回折ピークを有する、請求項1に記載の結晶。
- 粉末X線回折において、回折角度(2θ±0.2°)5.8°、6.4°および10.1°に回折ピークを有する、請求項1に記載の結晶。
- 粉末X線回折において、さらに回折角度(2θ±0.2°)8.0°および12.8°に回折ピークを有する、請求項3記載の結晶。
- 粉末X線回折において、さらに回折角度(2θ±0.2°)14.2°、16.0°、18.9°、19.7°および23.1°に回折ピークを有する、請求項4に記載の結晶。
- 13C固体NMRスペクトルにおいて、化学シフト(δ±0.5ppm)154.7ppmにピークを有する、(6S,9aS)-N-ベンジル-8-({6-[3-(4-エチルピペラジン-1-イル)アゼチジン-1-イル]ピリジン-2-イル}メチル)-6-(2-フルオロ-4-ヒドロキシベンジル)-4,7-ジオキソ-2-(プロプ-2-エン-1-イル)ヘキサヒドロ-2H-ピラジノ[2,1-c][1,2,4]トリアジン-1(6H)-カルボキサミドの結晶。
- 13C固体NMRスペクトルにおいて、さらに化学シフト(δ±0.5ppm)141.1ppmおよび158.1ppmにピークを有する、請求項6記載の結晶。
- 13C固体NMRスペクトルにおいて、さらに化学シフト(δ±0.5ppm)134.0ppmおよび165.1ppmにピークを有する、請求項7記載の結晶。
- 13C固体NMRスペクトルにおいて、さらに化学シフト(δ±0.5ppm)12.6ppm、55.5ppmおよび118.5ppmにピークを有する、請求項8記載の結晶。
- 図4で示される粉末X線パターンと実質的に同一の粉末X線回折パターンを有する、(6S,9aS)-N-ベンジル-8-({6-[3-(4-エチルピペラジン-1-イル)アゼチジン-1-イル]ピリジン-2-イル}メチル)-6-(2-フルオロ-4-ヒドロキシベンジル)-4,7-ジオキソ-2-(プロプ-2-エン-1-イル)ヘキサヒドロ-2H-ピラジノ[2,1-c][1,2,4]トリアジン-1(6H)-カルボキサミドの結晶。
- 図6で示される13C固体NMRスペクトルと実質的に同一の13C固体NMRスペクトルを有する、(6S,9aS)-N-ベンジル-8-({6-[3-(4-エチルピペラジン-1-イル)アゼチジン-1-イル]ピリジン-2-イル}メチル)-6-(2-フルオロ-4-ヒドロキシベンジル)-4,7-ジオキソ-2-(プロプ-2-エン-1-イル)ヘキサヒドロ-2H-ピラジノ[2,1-c][1,2,4]トリアジン-1(6H)-カルボキサミドの結晶。
- 請求項1~11のいずれか一項に記載の結晶を含む、医薬組成物。
Priority Applications (13)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016572771A JP6126319B1 (ja) | 2015-06-23 | 2016-06-21 | (6S,9aS)−N−ベンジル−6−[(4−ヒドロキシフェニル)メチル]−4,7−ジオキソ−8−({6−[3−(ピペラジン−1−イル)アゼチジン−1−イル]ピリジン−2−イル}メチル)−2−(プロプ−2−エン−1−イル)−オクタヒドロ−1H−ピラジノ[2,1−c][1,2,4]トリアジン−1−カルボキサミド化合物の結晶 |
| KR1020177033876A KR102559211B1 (ko) | 2015-06-23 | 2016-06-21 | (6S,9aS)-N-벤질-8-({6-[3-(4-에틸피페라진-1-일)아제티딘-1-일]피리딘-2-일}메틸)-6-(2-플루오로-4-하이드록시벤질)-4,7-디옥소-2-(프로프-2-엔-1-일)헥사하이드로-2H-피라지노[2,1-c][1,2,4]트리아진-1(6H)-카복사마이드의 결정 |
| US15/577,858 US10259817B2 (en) | 2015-06-23 | 2016-06-21 | Crystal (6S,9aS)-N-benzyl-8-({6-[3-(4-ethylpiperazin-1-yl)azetidin-1-yl]pyridin-2-yl}methyl)-6-(2-fluoro-4-hydroxybenzyl)-4,7-dioxo-2-(prop-2-en-1-yl)hexahydro-2H-pyrazino[2,1-c][1,2,4]triazine-1(6H)-carboxamide |
| CA2986999A CA2986999C (en) | 2015-06-23 | 2016-06-21 | Crystal of (6s,9as)-n-benzyl-8-({6-[3-(4-ethylpiperazin-1-yl)azetidin-1-yl]pyridin-2-yl}methyl)-6-(2-fluoro-4-hydroxybenzyl)-4,7-dioxo-2-(prop-2-en-1-yl)hexahydro-2h-pyrazino[2,1-c][1,2,4]triazine-1(6h)-carboxamide |
| EP16814346.9A EP3315499B1 (en) | 2015-06-23 | 2016-06-21 | Crystal of (6S,9aS)-N-benzyl-8-({6-[3-(4-ethylpiperazin-1-yl)azetidin-1-yl]pyridin-2-yl}methyl)-6-(2-fluoro-4-hydroxybenzyl)-4,7-dioxo-2-(prop-2-en-1-yl)hexahydro-2H-pyrazino[2,1-c][1,2,4]triazine-1(6H)-carboxamide |
| AU2016284383A AU2016284383B2 (en) | 2015-06-23 | 2016-06-21 | Crystal of (6S,9aS)-N-benzyl-8-({6-[3-(4-ethylpiperazin-1- yl)azetidin-1-yl]pyridin-2-yl}methyl)-6-(2-fluoro-4- hydroxybenzyl)-4,7-dioxo-2-(prop-2-en-1-yl)hexahydro-2Hpyrazino[ 2,1-c][1,2,4]triazine-1-(6H)-carboxamide |
| BR112017025519-7A BR112017025519B1 (pt) | 2015-06-23 | 2016-06-21 | Cristal do composto (6s,9as)-n-benzil-6-[(4-hidroxifenil)metil]-4,7-dioxo-8-({6-[3-(piperazina-1-il) azetidin-1-il]piridin-2-il}metil)-2-(prop-2-en-1-il)-octaidro-1hpirazino[2,1-c] [1,2,4]triazina-1-carboxamida |
| ES16814346T ES2870013T3 (es) | 2015-06-23 | 2016-06-21 | Cristal de (6S,9aS)-N-bencil-8-({6-[3-(4-etilpiperazin-1-il)azetidin-1-il]piridin-2-il}metil)-6-(2-fluoro-4-hidroxibencil)-4,7-dioxo-2-(prop-2-en-1-il)hexahidro-2H-pirazino[2,1-c][1,2,4]triazin-1(6H)-carboxamida |
| CN201680030060.5A CN107614504B (zh) | 2015-06-23 | 2016-06-21 | 甲酰胺化合物的晶体 |
| MX2017015257A MX2017015257A (es) | 2015-06-23 | 2016-06-21 | Cristal de (6s, 9as)-n-bencil-8-({6-[3-(4-etilpiperazin-1-il)azeti din-1-il]piridin-2-il}metil)-6-(2-(fluoruro-4-hidroxibencil)-4,7- dioxo-2-prop-2-en-1-il)hexahidro-2h-pirazino[2,1-c] [1,2,4]triazon-1-(6h)-carboxamida. |
| SG11201709593VA SG11201709593VA (en) | 2015-06-23 | 2016-06-21 | CRYSTAL OF (6S,9aS)-N-BENZYL-6-[(4-HYDROXYPHENYL)METHYL]- 4,7-DIOXO-8-({6-[3-(PIPERAZIN-1-YL)AZETIDIN-1-YL]PYRIDIN- 2-YL}METHYL)-2-(PROP-2-EN-1-YL)-OCTAHYDRO-1H-PYRAZINO[2,1-c][1,2,4]TRIAZINE-1-CARBOXAMIDE COMPOUND |
| RU2017141763A RU2723551C2 (ru) | 2015-06-23 | 2016-06-21 | Кристалл (6S,9aS)-N-бензил-8-({ 6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил} метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамида |
| IL255901A IL255901B (en) | 2015-06-23 | 2017-11-26 | Crystal of (6s,9as)–n–benzyl–8–({6–[3–(4–ethylpiperazin–1–yl)azetidin–1–yl]pyridin–2–yl}methyl)–6–(2– fluoro-4-hyfroxybenzyl)-4,7-dioxo-2-(prop-2-en-1-yl)hexahydro-2h-pyrazino[2,1-c][1,2,4]triazine-1(6h )-carboxamide |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562183577P | 2015-06-23 | 2015-06-23 | |
| US62/183577 | 2015-06-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016208576A1 true WO2016208576A1 (ja) | 2016-12-29 |
Family
ID=57586245
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/068381 Ceased WO2016208576A1 (ja) | 2015-06-23 | 2016-06-21 | (6S,9aS)-N-ベンジル-6-[(4-ヒドロキシフェニル)メチル]-4,7-ジオキソ-8-({6-[3-(ピペラジン-1-イル)アゼチジン-1-イル]ピリジン-2-イル}メチル)-2-(プロプ-2-エン-1-イル)-オクタヒドロ-1H-ピラジノ[2,1-c][1,2,4]トリアジン-1-カルボキサミド化合物の結晶 |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US10259817B2 (ja) |
| EP (1) | EP3315499B1 (ja) |
| JP (1) | JP6126319B1 (ja) |
| KR (1) | KR102559211B1 (ja) |
| CN (1) | CN107614504B (ja) |
| AU (1) | AU2016284383B2 (ja) |
| CA (1) | CA2986999C (ja) |
| ES (1) | ES2870013T3 (ja) |
| IL (1) | IL255901B (ja) |
| MX (1) | MX2017015257A (ja) |
| RU (1) | RU2723551C2 (ja) |
| SG (1) | SG11201709593VA (ja) |
| WO (1) | WO2016208576A1 (ja) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018147275A1 (ja) * | 2017-02-08 | 2018-08-16 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | 腫瘍治療用医薬組成物 |
| US10822307B2 (en) | 2014-08-28 | 2020-11-03 | Eisai R&D Management Co., Ltd. | High-purity quinoline derivative and method for manufacturing same |
| US11090386B2 (en) | 2015-02-25 | 2021-08-17 | Eisai R&D Management Co., Ltd. | Method for suppressing bitterness of quinoline derivative |
| US11369623B2 (en) | 2015-06-16 | 2022-06-28 | Prism Pharma Co., Ltd. | Anticancer combination of a CBP/catenin inhibitor and an immune checkpoint inhibitor |
| US11547705B2 (en) | 2015-03-04 | 2023-01-10 | Merck Sharp & Dohme Llc | Combination of a PD-1 antagonist and a VEGF-R/FGFR/RET tyrosine kinase inhibitor for treating cancer |
| US11598776B2 (en) | 2011-06-03 | 2023-03-07 | Eisai R&D Management Co., Ltd. | Biomarkers for predicting and assessing responsiveness of thyroid and kidney cancer subjects to lenvatinib compounds |
| WO2023038030A1 (ja) | 2021-09-08 | 2023-03-16 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | 固形腫瘍治療用医薬組成物 |
| WO2023190748A1 (ja) | 2022-03-31 | 2023-10-05 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | 腫瘍治療用医薬組成物 |
| WO2024209717A1 (ja) * | 2023-04-06 | 2024-10-10 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | 腫瘍治療用医薬組成物 |
| US12220398B2 (en) | 2015-08-20 | 2025-02-11 | Eisai R&D Management Co., Ltd. | Tumor therapeutic agent |
| US12226409B2 (en) | 2017-05-16 | 2025-02-18 | Eisai R&D Management Co., Ltd. | Treatment of hepatocellular carcinoma |
| US12508313B2 (en) | 2009-08-19 | 2025-12-30 | Eisai R&D Management Co., Ltd. | Quinoline derivative-containing pharmaceutical composition |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2020374883A1 (en) | 2019-10-29 | 2022-05-26 | Eisai R&D Management Co., Ltd. | Combination of a PD-1 antagonist, a VEGFR/FGFR/RET tyrosine kinase inhibitor and a CBP/beta-catenin inhibitor for treating cancer |
| AU2021236240B2 (en) | 2020-03-12 | 2024-07-25 | 3+2 Pharma LLC | CBP/catenin signaling pathway inhibitors and uses thereof |
| CN115784934A (zh) * | 2022-11-22 | 2023-03-14 | 上海吉奉生物科技有限公司 | 一种酪氨酸衍生物的合成方法 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006123517A1 (ja) * | 2005-04-26 | 2006-11-23 | Kissei Pharmaceutical Co., Ltd. | ヒドロキシノルエフェドリン誘導体塩酸塩の結晶多形 |
| JP2011500666A (ja) * | 2007-10-15 | 2011-01-06 | チュンウェ ファーマ コーポレーション | リバースターン類似体の新規化合物およびその用途(3) |
| JP2011522037A (ja) * | 2008-06-06 | 2011-07-28 | PRISM BioLab株式会社 | アルファへリックスミメティック及び関連の方法 |
| JP2013540774A (ja) * | 2010-10-14 | 2013-11-07 | ジェイダブリュ ファーマセウティカル コーポレーション | リバースターン類似体の新規な化合物およびその製造方法と用途 |
| WO2015098855A1 (ja) * | 2013-12-25 | 2015-07-02 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | ピラジノ[2,1-c][1,2,4]トリアジン化合物の結晶(2) |
| WO2015098853A1 (ja) * | 2013-12-25 | 2015-07-02 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | (6S,9aS)-N-ベンジル-6-[(4-ヒドロキシフェニル)メチル]-4,7-ジオキソ-8-({6-[3-(ピペラジン-1-イル)アゼチジン-1-イル]ピリジン-2-イル}メチル)-2-(プロプ-2-エン-1-イル)-オクタヒドロ-1H-ピラジノ[2,1-c][1,2,4]トリアジン-1-カルボキサミド化合物 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9714129D0 (en) | 1997-07-04 | 1997-09-10 | Pfizer Ltd | Azetidines |
| ES2245955T3 (es) | 1999-12-21 | 2006-02-01 | Sugen, Inc. | 7-aza-indolin-2-onas 4-sustituidas y su utilizacion como inhibidores de proteina-quinasa. |
| WO2003031448A1 (en) | 2001-10-12 | 2003-04-17 | Choongwae Pharma Corporation | Reverse-turn mimetics and method relating thereto |
| US8080657B2 (en) | 2001-10-12 | 2011-12-20 | Choongwae Pharma Corporation | Compounds of reverse turn mimetics and the use thereof |
| UA103918C2 (en) | 2009-03-02 | 2013-12-10 | Айерем Элелси | N-(hetero)aryl, 2-(hetero)aryl-substituted acetamides for use as wnt signaling modulators |
| MX340424B (es) | 2009-04-15 | 2016-07-08 | Jw Pharmaceutical Corp | Compuestos novedosos de mimeticos de cambio inverso; metodo para fabricarlos y su uso. |
| WO2012115286A1 (en) | 2011-02-25 | 2012-08-30 | Prism Biolab Corporation | Alpha helix mimetics and methods relating thereto |
| PH12017500997A1 (en) | 2012-04-04 | 2018-02-19 | Samumed Llc | Indazole inhibitors of the wnt signal pathway and therapeutic uses thereof |
| EP3456325A3 (en) | 2013-02-22 | 2019-05-22 | Samumed, LLC | Gamma-diketones as wnt/beta-catenin signalling pathway activators |
-
2016
- 2016-06-21 SG SG11201709593VA patent/SG11201709593VA/en unknown
- 2016-06-21 CN CN201680030060.5A patent/CN107614504B/zh active Active
- 2016-06-21 JP JP2016572771A patent/JP6126319B1/ja active Active
- 2016-06-21 KR KR1020177033876A patent/KR102559211B1/ko active Active
- 2016-06-21 CA CA2986999A patent/CA2986999C/en active Active
- 2016-06-21 AU AU2016284383A patent/AU2016284383B2/en active Active
- 2016-06-21 WO PCT/JP2016/068381 patent/WO2016208576A1/ja not_active Ceased
- 2016-06-21 MX MX2017015257A patent/MX2017015257A/es unknown
- 2016-06-21 RU RU2017141763A patent/RU2723551C2/ru active
- 2016-06-21 EP EP16814346.9A patent/EP3315499B1/en active Active
- 2016-06-21 ES ES16814346T patent/ES2870013T3/es active Active
- 2016-06-21 US US15/577,858 patent/US10259817B2/en active Active
-
2017
- 2017-11-26 IL IL255901A patent/IL255901B/en active IP Right Grant
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006123517A1 (ja) * | 2005-04-26 | 2006-11-23 | Kissei Pharmaceutical Co., Ltd. | ヒドロキシノルエフェドリン誘導体塩酸塩の結晶多形 |
| JP2011500666A (ja) * | 2007-10-15 | 2011-01-06 | チュンウェ ファーマ コーポレーション | リバースターン類似体の新規化合物およびその用途(3) |
| JP2011522037A (ja) * | 2008-06-06 | 2011-07-28 | PRISM BioLab株式会社 | アルファへリックスミメティック及び関連の方法 |
| JP2013540774A (ja) * | 2010-10-14 | 2013-11-07 | ジェイダブリュ ファーマセウティカル コーポレーション | リバースターン類似体の新規な化合物およびその製造方法と用途 |
| WO2015098855A1 (ja) * | 2013-12-25 | 2015-07-02 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | ピラジノ[2,1-c][1,2,4]トリアジン化合物の結晶(2) |
| WO2015098853A1 (ja) * | 2013-12-25 | 2015-07-02 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | (6S,9aS)-N-ベンジル-6-[(4-ヒドロキシフェニル)メチル]-4,7-ジオキソ-8-({6-[3-(ピペラジン-1-イル)アゼチジン-1-イル]ピリジン-2-イル}メチル)-2-(プロプ-2-エン-1-イル)-オクタヒドロ-1H-ピラジノ[2,1-c][1,2,4]トリアジン-1-カルボキサミド化合物 |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12508313B2 (en) | 2009-08-19 | 2025-12-30 | Eisai R&D Management Co., Ltd. | Quinoline derivative-containing pharmaceutical composition |
| US11598776B2 (en) | 2011-06-03 | 2023-03-07 | Eisai R&D Management Co., Ltd. | Biomarkers for predicting and assessing responsiveness of thyroid and kidney cancer subjects to lenvatinib compounds |
| US11186547B2 (en) | 2014-08-28 | 2021-11-30 | Eisai R&D Management Co., Ltd. | High-purity quinoline derivative and method for manufacturing same |
| US10822307B2 (en) | 2014-08-28 | 2020-11-03 | Eisai R&D Management Co., Ltd. | High-purity quinoline derivative and method for manufacturing same |
| US11090386B2 (en) | 2015-02-25 | 2021-08-17 | Eisai R&D Management Co., Ltd. | Method for suppressing bitterness of quinoline derivative |
| US12083112B2 (en) | 2015-03-04 | 2024-09-10 | Eisai R&D Management Co., Ltd. | Combination of a PD-1 antagonist and a VEGFR/FGFR/RET tyrosine kinase inhibitor for treating cancer |
| US11547705B2 (en) | 2015-03-04 | 2023-01-10 | Merck Sharp & Dohme Llc | Combination of a PD-1 antagonist and a VEGF-R/FGFR/RET tyrosine kinase inhibitor for treating cancer |
| US11369623B2 (en) | 2015-06-16 | 2022-06-28 | Prism Pharma Co., Ltd. | Anticancer combination of a CBP/catenin inhibitor and an immune checkpoint inhibitor |
| US12220398B2 (en) | 2015-08-20 | 2025-02-11 | Eisai R&D Management Co., Ltd. | Tumor therapeutic agent |
| RU2750539C2 (ru) * | 2017-02-08 | 2021-06-29 | Эйсай Ар Энд Ди Менеджмент Ко., Лтд. | Фармацевтическая композиция для лечения опухоли |
| JPWO2018147275A1 (ja) * | 2017-02-08 | 2019-11-07 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | 腫瘍治療用医薬組成物 |
| EP3581183A4 (en) * | 2017-02-08 | 2020-12-09 | Eisai R&D Management Co., Ltd. | PHARMACEUTICAL COMPOSITION FOR ONCOTHERAPY |
| KR20190110525A (ko) * | 2017-02-08 | 2019-09-30 | 에자이 알앤드디 매니지먼트 가부시키가이샤 | 종양-치료용 약제학적 조성물 |
| CN110072528B (zh) * | 2017-02-08 | 2022-04-26 | 卫材R&D管理有限公司 | 治疗肿瘤的药物组合物 |
| KR102539920B1 (ko) | 2017-02-08 | 2023-06-05 | 에자이 알앤드디 매니지먼트 가부시키가이샤 | 종양-치료용 약제학적 조성물 |
| CN110072528A (zh) * | 2017-02-08 | 2019-07-30 | 卫材R&D管理有限公司 | 治疗肿瘤的药物组合物 |
| EP3581183B1 (en) | 2017-02-08 | 2023-11-29 | Eisai R&D Management Co., Ltd. | Tumor-treating pharmaceutical composition |
| US12303505B2 (en) | 2017-02-08 | 2025-05-20 | Eisai R&D Management Co., Ltd. | Tumor-treating pharmaceutical composition |
| WO2018147275A1 (ja) * | 2017-02-08 | 2018-08-16 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | 腫瘍治療用医薬組成物 |
| US12226409B2 (en) | 2017-05-16 | 2025-02-18 | Eisai R&D Management Co., Ltd. | Treatment of hepatocellular carcinoma |
| WO2023038030A1 (ja) | 2021-09-08 | 2023-03-16 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | 固形腫瘍治療用医薬組成物 |
| KR20240056487A (ko) | 2021-09-08 | 2024-04-30 | 에자이 알앤드디 매니지먼트 가부시키가이샤 | 고형 종양을 치료하기 위한 약제학적 조성물 |
| KR20240167633A (ko) | 2022-03-31 | 2024-11-27 | 에자이 알앤드디 매니지먼트 가부시키가이샤 | 종양 치료용 약학 조성물 |
| WO2023190748A1 (ja) | 2022-03-31 | 2023-10-05 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | 腫瘍治療用医薬組成物 |
| WO2024209717A1 (ja) * | 2023-04-06 | 2024-10-10 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | 腫瘍治療用医薬組成物 |
| EP4656193A1 (en) | 2023-04-06 | 2025-12-03 | Eisai R&D Management Co., Ltd. | Pharmaceutical composition for treating tumors |
Also Published As
| Publication number | Publication date |
|---|---|
| BR112017025519A2 (ja) | 2018-08-07 |
| KR20180019083A (ko) | 2018-02-23 |
| MX2017015257A (es) | 2018-02-19 |
| ES2870013T3 (es) | 2021-10-26 |
| RU2017141763A3 (ja) | 2019-08-23 |
| JP6126319B1 (ja) | 2017-05-10 |
| US10259817B2 (en) | 2019-04-16 |
| AU2016284383A1 (en) | 2017-12-14 |
| EP3315499B1 (en) | 2021-04-07 |
| CN107614504A (zh) | 2018-01-19 |
| CA2986999A1 (en) | 2016-12-29 |
| JPWO2016208576A1 (ja) | 2017-06-29 |
| US20180141950A1 (en) | 2018-05-24 |
| CN107614504B (zh) | 2019-12-24 |
| KR102559211B1 (ko) | 2023-07-25 |
| RU2017141763A (ru) | 2019-07-23 |
| RU2723551C2 (ru) | 2020-06-16 |
| EP3315499A1 (en) | 2018-05-02 |
| IL255901A (en) | 2018-01-31 |
| CA2986999C (en) | 2023-08-08 |
| EP3315499A4 (en) | 2018-12-19 |
| AU2016284383B2 (en) | 2020-08-20 |
| IL255901B (en) | 2020-07-30 |
| SG11201709593VA (en) | 2017-12-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6126319B1 (ja) | (6S,9aS)−N−ベンジル−6−[(4−ヒドロキシフェニル)メチル]−4,7−ジオキソ−8−({6−[3−(ピペラジン−1−イル)アゼチジン−1−イル]ピリジン−2−イル}メチル)−2−(プロプ−2−エン−1−イル)−オクタヒドロ−1H−ピラジノ[2,1−c][1,2,4]トリアジン−1−カルボキサミド化合物の結晶 | |
| ES2728353T3 (es) | Compuesto de (6S,9aS)-N-bencil-6-[(4-hidroxifenil)metil]-4,7-dioxo-8-({6-[3-(piperazin-1-il)azetidin-1-il]piridin-2-il}metil)-2-(prop-2-en-1-il)-octahidro-1H-pirazino[2,1-c][1,2,4]triazin-1-carboxamida | |
| TWI573790B (zh) | 作為活化激酶抑制劑之經取代吲唑衍生物 | |
| KR20190005838A (ko) | 이중 lsd1/hdac 억제제로서 사이클로프로필-아마이드 화합물 | |
| AU2022405082B2 (en) | Lpa receptor antagonists and uses thereof | |
| JP7481435B2 (ja) | Crac阻害剤としての2h-ベンゾピラン誘導体 | |
| CN109641909A (zh) | 雷帕霉素信号通路抑制剂的机理靶标及其治疗应用 | |
| US20200207782A1 (en) | Chemically activated water-soluble prodrug | |
| BR112017025519B1 (pt) | Cristal do composto (6s,9as)-n-benzil-6-[(4-hidroxifenil)metil]-4,7-dioxo-8-({6-[3-(piperazina-1-il) azetidin-1-il]piridin-2-il}metil)-2-(prop-2-en-1-il)-octaidro-1hpirazino[2,1-c] [1,2,4]triazina-1-carboxamida | |
| HK1222656B (en) | (6s,9as)-n-benzyl-6-[(4-hydroxyphenyl)methyl]-4,7-dioxo-8-({6-[3-(piperazine-1-yl)azetidine-1-yl]pyridine-2-yl}methyl)-2-(prop-2-en-1-yl)-octahydro-1h-pyrazino[2,1-c][1,2,4]triazine-1-carboxamide compound | |
| NZ720718B2 (en) | (6S,9aS)-N-BENZYL-6-[(4-HYDROXYPHENYL)METHYL]-4,7-DIOXO-8-({6-[3-(PIPERAZINE-1-YL)AZETIDINE-1-YL]PYRIDINE-2-YL}METHYL)-2-(PROP-2-EN-1-YL)-OCTAHYDRO-1H-PYRAZINO[2,1-c][1,2,4]TRIAZINE-1-CARBOXAMIDE COMPOUND | |
| NZ713233B2 (en) | (6S,9aS)-N-BENZYL-6-[(4-HYDROXYPHENYL)METHYL]-4,7-DIOXO-8-({6-[3-(PIPERAZINE-1-YL)AZETIDINE-1-YL]PYRIDINE-2-YL}METHYL)-2-(PROP-2-EN-1-YL)-OCTAHYDRO-1H-PYRAZINO[2,1-c][1,2,4]TRIAZINE-1-CARBOXAMIDE COMPOUND |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2016572771 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16814346 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11201709593V Country of ref document: SG |
|
| ENP | Entry into the national phase |
Ref document number: 2986999 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 20177033876 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 255901 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2017/015257 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15577858 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2016284383 Country of ref document: AU Date of ref document: 20160621 Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2017141763 Country of ref document: RU |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112017025519 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112017025519 Country of ref document: BR Kind code of ref document: A2 Effective date: 20171128 |