WO2016063872A1 - メタノール製造方法及びメタノール製造装置 - Google Patents
メタノール製造方法及びメタノール製造装置 Download PDFInfo
- Publication number
- WO2016063872A1 WO2016063872A1 PCT/JP2015/079580 JP2015079580W WO2016063872A1 WO 2016063872 A1 WO2016063872 A1 WO 2016063872A1 JP 2015079580 W JP2015079580 W JP 2015079580W WO 2016063872 A1 WO2016063872 A1 WO 2016063872A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- gas
- methanol
- reactor
- synthesis
- final
- Prior art date
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/15—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of oxides of carbon exclusively
- C07C29/151—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of oxides of carbon exclusively with hydrogen or hydrogen-containing gases
- C07C29/152—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of oxides of carbon exclusively with hydrogen or hydrogen-containing gases characterised by the reactor used
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/80—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with zinc, cadmium or mercury
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/03—Precipitation; Co-precipitation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J8/00—Chemical or physical processes in general, conducted in the presence of fluids and solid particles; Apparatus for such processes
- B01J8/02—Chemical or physical processes in general, conducted in the presence of fluids and solid particles; Apparatus for such processes with stationary particles, e.g. in fixed beds
- B01J8/04—Chemical or physical processes in general, conducted in the presence of fluids and solid particles; Apparatus for such processes with stationary particles, e.g. in fixed beds the fluid passing successively through two or more beds
- B01J8/0492—Feeding reactive fluids
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J8/00—Chemical or physical processes in general, conducted in the presence of fluids and solid particles; Apparatus for such processes
- B01J8/02—Chemical or physical processes in general, conducted in the presence of fluids and solid particles; Apparatus for such processes with stationary particles, e.g. in fixed beds
- B01J8/04—Chemical or physical processes in general, conducted in the presence of fluids and solid particles; Apparatus for such processes with stationary particles, e.g. in fixed beds the fluid passing successively through two or more beds
- B01J8/0496—Heating or cooling the reactor
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C31/00—Saturated compounds having hydroxy or O-metal groups bound to acyclic carbon atoms
- C07C31/02—Monohydroxylic acyclic alcohols
- C07C31/04—Methanol
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2208/00—Processes carried out in the presence of solid particles; Reactors therefor
- B01J2208/00008—Controlling the process
- B01J2208/00017—Controlling the temperature
- B01J2208/00106—Controlling the temperature by indirect heat exchange
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2208/00—Processes carried out in the presence of solid particles; Reactors therefor
- B01J2208/00796—Details of the reactor or of the particulate material
- B01J2208/00893—Feeding means for the reactants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/52—Improvements relating to the production of bulk chemicals using catalysts, e.g. selective catalysts
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Catalysts (AREA)
Abstract
Description

特許文献3は、反応器における原料分圧を高くすることで、過反応や高温につながると述べている。そして、この高温によって触媒劣化速度が高くなることにつながる可能性があると指摘している。そこで、特許文献3に記載の技術は、期待される触媒寿命を短くせず、経済的に成り立つ手法で大量の目的物を得る手法として、合成ループ内に反応器を複数配置し、それぞれの反応器後に分離器を配置し、原料ガスを複数の反応器前に供給することも可能な方法で、反応器間で昇圧することを特徴とした技術を提案している。特許文献3は、上記技術により、循環ガス量を低減し、触媒層温度を制御し、それにより、受け入れ可能な触媒寿命を達成した上で、達成すべき所望の生成物の生産を可能にしたと記載している。そして、実施例においては、循環ガス量の23%又は約28%を削減したことが示されている。
加えて、メタノール合成に伴い生じる熱によって触媒のシンタリングが促進されたり、非特許文献1に記載のように、メタノール合成に伴い生成する水が触媒劣化を促進させたりする。そこで、メタノール合成では、触媒を有効に利用するに当たって触媒の負荷を平準化し、効率的に触媒を利用することが求められている。
特許文献3は、循環ガス量を低減し、触媒層温度を制御することで受け入れ可能な触媒寿命を達成した上で、達成すべき所望の生成物の生産を可能にしたと記載している。しかしながら、その実施例では、循環ガス量が既存技術に対して72%又は77%に低減できたことを示しているにすぎず、カーボン収率についての記載もない。カーボン収率について考慮しないのであれば、循環ガス量を低減したとしても、原料ガス量を増加して生産量を維持することが可能であり、技術的な革新性がない。
また、メイクアップガス入口に対して、合成ループにおける最も遠い位置にパージガスの取り出しを設けるのが、カーボン収率の観点から最も都合がよい。一方で、循環機の処理ガス量の観点では、合成ループにおけるパージガスの取り出し位置は循環機の直前がよい。特許文献3に開示されたプロセスは、本来であれば循環機によって昇圧する必要のないガス(合成ループから取り出すパージガス)を昇圧するプロセスとなっている。これでは、循環器の処理ガス量が増大し、エネルギー使用量が増加してしまうので、適切ではない。
加えて、最終の凝縮分離工程ではない凝縮分離工程において、冷却水削減の目的又は機器費削減の目的で、水冷熱交換器を用いずにエアフィンクーラーのみを用いて、冷却器出口ガス温度を55~90℃としてメタノール分離割合を低下させる場合、その凝縮分離の工程に用いる機器の後段に循環機を配置すると、導入される凝縮性気体の総量が増加する。この場合、循環機内で凝縮液滴が発生する確率が高まる。循環機内で凝縮液滴が発生すると機械的な故障およびエネルギー損失の原因となり、この位置に循環機を配置するのは適切ではない。
また、プラントの大型化による経済性向上を追求する場合に行われる並列化については、例えば反応器を並列化する場合、反応器に導入できるガス量が増加するため大型化は可能となる。しかしながら、そのような並列化は、一般的に収率改善や循環比低減にはつながるものではない。
[1]水素と一酸化炭素と二酸化炭素とを含む合成ガスからメタノールを合成する合成工程と、前記合成工程を経て得られた反応混合物から未反応ガスを分離する分離工程と、を有するメタノール製造方法であって、少なくとも2つの前記合成工程と、少なくとも2つの前記分離工程とを有する合成ループを有し、
前記合成ループにおいて、最終合成工程の後の最終分離工程で最終反応混合物から分離した最終未反応ガスからパージガスを取り除いた残りのガスを循環機で昇圧し、水素と一酸化炭素と二酸化炭素とを含むメイクアップガスの10~90モル%と混合して第1混合ガスを得る第1混合工程と、前記第1混合ガスからメタノールを合成する第1合成工程と、前記第1合成工程で得られた第1反応混合物から第1未反応ガスを分離する第1分離工程と、前記第1未反応ガスと前記メイクアップガスの10~90モル%のうちの少なくとも一部とを混合して第2混合ガスを得る第2混合工程と、最終的にメタノールを合成する前記最終合成工程と、前記最終合成工程で得られた前記最終反応混合物から前記最終未反応ガスを分離する前記最終分離工程と、を有し、少なくとも前記最終合成工程において、加圧沸騰水との間接熱交換により触媒層の反応温度を制御する、メタノール製造方法。
[2]合成ループが有する前記少なくとも2つの分離工程のうちの少なくとも1つの分離工程が、ガス状の前記反応混合物を冷却することで生じるメタノールを含む液を気液分離器によって分離する工程である、[1]記載のメタノール製造方法。
[3]前記第1未反応ガスに、前記メイクアップガスのうちの40~70モル%を混合する、[1]又は[2]に記載のメタノール製造方法。
[4]前記第1分離工程において、前記第1反応混合物に含まれるメタノールのうちの35~100モル%と前記第1未反応ガスとを分離する、[1]~[3]のいずれか1つに記載のメタノール製造方法。
[5]前記第1分離工程において、前記第1反応混合物に含まれるメタノールのうちの75~96モル%と前記第1未反応ガスとを分離する、[1]~[4]のいずれか1つに記載のメタノール製造方法。
[6]前記メイクアップガスのモル流量に対する前記最終未反応ガスからパージガスを取り除いた残りのガスのモル流量の比である循環比が、0.6~2.0である、[1]~[5]のいずれか1項に記載のメタノール製造方法。
[7]前記循環比が0.8~1.5である、[6]記載のメタノール製造方法。
[8]前記加圧沸騰水が220~260℃である、[1]~[7]のいずれか1つに記載のメタノール製造方法。
[9]前記最終合成工程は、前記第2混合ガスからメタノールを合成する工程、又は、その工程で得られた第2反応混合物から第2未反応ガスを分離し、その第2未反応ガスとメイクアップガスの一部とを混合して得られる第3混合ガス又は前記第2未反応ガスから、メタノールを合成する工程である、[1]~[8]のいずれか1つに記載のメタノール製造方法。
[10]前記最終合成工程は、前記第2混合ガスからメタノールを合成する工程である、[1]~[9]のいずれか1つに記載のメタノール製造方法。
[11]前記合成工程において用いられる触媒が銅原子及び亜鉛原子を原子比(銅/亜鉛)2.0~3.0で含み、かつアルミニウム原子を含む、[1]~[10]のいずれか1つに記載のメタノール製造方法。
[12]前記合成工程において用いられる触媒が、銅原子及び亜鉛原子を原子比(銅/亜鉛)2.1~3.0で含み、アルミナを3~20質量%含み、かつ銅を含む水溶液と亜鉛を含む水溶液とアルカリ水溶液とを混合して銅及び亜鉛を含む沈殿物を生成する工程と、前記沈殿物と擬ベーマイト構造を有するアルミナ水和物とを混合して混合物を得る工程と、前記混合物を密度が2.0~3.0g/mLになるように成型する工程とを有する製造方法によって調製される、[1]~[11]のいずれか1つに記載のメタノール製造方法。
[13]前記第1未反応ガスに混合する前記メイクアップガスの割合を、前記合成工程における反応器の温度に応じて調整する、[1]~[12]のいずれか一つに記載のメタノール製造方法。
[14]前記合成工程の全てにおいて、加圧沸騰水との間接熱交換により触媒層の反応温度を制御する、[1]~[13]のいずれか1つに記載のメタノール製造方法。
[15]水素と一酸化炭素と二酸化炭素とを含む合成ガスからメタノールを合成する反応器と、前記反応器において得られた反応混合物から未反応ガスを分離する分離装置と、を備えるメタノール製造装置であって、少なくとも2つの前記反応器と、少なくとも2つの前記分離装置とを備える合成ループを有し、前記合成ループにおいて、最終反応器の後の最終分離装置において最終反応混合物から分離した最終未反応ガスからパージガスを取り除いた残りのガスを循環機で昇圧し、水素と一酸化炭素と二酸化炭素とを含むメイクアップガスのうちの10~90モル%混合して第1混合ガスを得る第1混合手段と、前記第1混合ガスからメタノールを合成する第1反応器と、前記第1反応器において得られた第1反応混合物から第1未反応ガスを分離する第1分離装置と、前記第1未反応ガスと前記メイクアップガスのうちの10~90モル%とを混合して第2混合ガスを得る第2混合手段と、最終的にメタノールを合成する前記最終反応器と、前記最終反応器において得られた前記最終反応混合物から前記最終未反応ガスを分離する前記最終分離装置と、を備え、少なくとも前記最終反応器において、加圧沸騰水との間接熱交換により触媒層の反応温度を制御する、メタノール製造装置。
[16]スチームドラムを更に備え、前記第1反応器において、前記加圧沸騰水との間接熱交換により触媒層の反応温度を制御し、前記加圧沸騰水は、前記最終反応器及び前記第1反応器と前記スチームドラムとの間で少なくとも一部が循環する、[15]記載のメタノール製造装置。
[17]少なくとも2つのスチームドラムを更に備え、前記加圧沸騰水は、前記最終反応器と前記少なくとも2つのスチームドラムのうちの1つとの間で少なくとも一部が循環し、かつ、前記第1反応器において、加圧沸騰水との間接熱交換により触媒層の反応温度を制御し、その加圧沸騰水は、前記第1反応器と前記少なくとも2つのスチームドラムのうちの別の1つとの間で少なくとも一部が循環する、[15]記載のメタノール製造装置。
[18]前記最終反応器は、前記第2混合ガスからメタノールを合成する反応器、又は、その反応器において得られた第2反応混合物から第2未反応ガスを分離し、その第2未反応ガスとメイクアップガスとを混合して得られる第3混合ガスから、メタノールを合成する反応器である、[15]~[17]のいずれか1つに記載のメタノール製造装置。
本実施形態においては、少なくとも1つの分離工程が最終合成工程よりも前段に設けられている。また、本実施形態においては、最終分離工程以外の少なくとも1つの分離工程において、その直前の合成工程で得られた反応混合物からメタノールを含む反応生成物と未反応の合成ガス(以下「未反応ガス」という。)とを分離し、この未反応ガスに上記メイクアップガスを混合して得られた混合ガスから、その後段の合成工程においてメタノールを合成する。
本実施形態において、合成ループは、少なくとも1つの合成工程と少なくとも1つの分離工程とを経たガスが最終合成工程と最終分離工程とを経て、最終分離工程において分離された未反応ガスが、循環機を通じて第1合成工程における原料のガスとして用いられることで形成される。合成ループへの物質の出入りのうちの入りとして、メイクアップガスが複数の流れに分割された後に各合成工程前の混合工程から合成ループに導入される。また、物質の出入りのうちの出として、分離工程において反応混合物中の反応生成物が分離されて合成ループ外へ抜き出されると共に、最終合成工程の後の最終分離工程において分離された未反応ガスの一部がパージガスとして合成ループ外に取り出される。なお、本明細書において、「反応混合物」とは、合成工程の出口成分であって、合成工程における反応により生じた成分と未反応成分との混合物であり、通常、メタノールを含むものである。
ここで、合成ループにおけるパージガスの取り出し位置は、循環機の処理ガス量を削減する観点から、合成ループ内での圧力が低くなる箇所が好ましく、循環機の直前がより好ましい。一方で、カーボン収率の観点から、反応混合物中の反応生成物を分離して合成ループ外へ抜き出した後の未反応ガスの一部をパージガスとして分岐することが好ましく、そのパージガスの取り出し位置がメイクアップガスの合流前であるとより好ましい。特許文献3では循環機を複数の反応器の間に存在させる結果、パージガス分を含めて循環機で昇圧することになり、適切な配置ではない。
また、各分離段階での未反応ガスは、次の混合工程、合成工程および分離工程に導かれ、各未反応ガスは直列に全反応器に導入され得る合成ループを形成する。
最終合成工程は、第1合成工程の後に、メタノールを合成する合成工程を経た反応混合物から分離された未反応ガスとメイクアップガスとを混合して得られる混合ガスから、メタノールを合成する工程であれば、特に限定されない。最終合成工程は、上記第2混合ガスからメタノールを合成する工程(第2合成工程)であると好ましい。あるいは、最終合成工程は、第2合成工程で得られた第2反応混合物から第2未反応ガスを分離し、その第2未反応ガスとメイクアップガスとを混合して得られる第3混合ガスから、メタノールを合成する工程(第3合成工程)であっても好ましい。これら第2合成工程及び第3合成工程のうち、最終合成工程は、第2合成工程であることがより好ましい。
また、最終分離工程は、第1分離工程の後に、反応混合物から未反応ガスを分離する工程であれば、特に限定されない。最終分離工程は、第2合成工程で得られた第2反応混合物から第2未反応ガスを分離する第2分離工程であると好ましい。あるいは、最終分離工程は、第3合成工程で得られた第3反応混合物から第3未反応ガスを分離する第3分離工程であると好ましい。これら第2分離工程及び第3分離工程のうち、最終分離工程は、第2分離工程であることがより好ましい。
メイクアップガスは、天然ガスの水蒸気改質ガスや石炭ガス化ガスなどの一酸化炭素(CO)、二酸化炭素(CO2)及び水素(H2)を含む合成原料ガスを圧縮機によって反応圧力まで昇圧したものである。反応圧力は、例えば、4.9~14.7MPa-G(50~150kg/cm2-G)であってもよく、より好ましくは7.8~10.8MPa-G(80~110kg/cm2-G)である。工業的には、メイクアップガスは、例えば天然ガスを原料とした水蒸気改質反応によって得られるものであり、下記式により算出されるCO、CO2及びH2のモル%の関係(M):
M=(H2モル%)/(2×COモル%+3×CO2モル%)
が、1.0より大きく2.0以下であるものが好ましい。さらに好ましいものはMが1.3~1.5である。
本実施形態において、メイクアップガスは、合成ループに導入する前に複数の流れに分割され、合成ループ内に存在する複数の合成工程における原料ガスの一部として、合成ループに導入される。メイクアップガスの分割比率は、各合成工程における合成条件及び各分離工程における分離条件によって好適な範囲が異なる。ただし、最初のメタノール合成工程(第1合成工程)に供給する混合ガス(第1混合ガス)に含まれるメイクアップガスのモル流量は、メイクアップガスの全体量に対して10~90モル%、好ましくは10~70モル%である。次いで第2合成工程に供給する混合ガス(第2混合ガス)に含まれるメイクアップガスのモル流量は、メイクアップガスの全体量に対して10~90モル%、好ましくは10~70モル%である。そして、第3合成工程以降が存在する場合であって、第1合成工程および第2合成工程に供給したメイクアップガスのモル流量の和をメイクアップガスの全体量の100%未満とする場合は、残りのメイクアップガスを第3合成工程以降の各メタノール合成工程に適宜分割する。また、第3合成工程以降が存在する場合であって、第1合成工程および第2合成工程に供給したメイクアップガスのモル流量の和がメイクアップガスの全体量の100%とする場合は、メイクアップガスを第3合成工程以降の各メタノール合成工程には分割供給しない。例えば、1つの実施態様として、分離工程における分離方法として凝縮分離方法を用いて、2つの合成工程と2つの凝縮分離工程とを有する製造方法の場合を説明する。この実施態様では、第1凝縮分離工程の出口ガス温度を20℃~100℃とする場合、最終合成工程(第2合成工程)の直前で合成ループに導入されるメイクアップガスの割合(メイクアップガスの全体量に対する割合。以下同様。)は、カーボン収率及び触媒層の最高温度などの観点から、10~90モル%であると好ましく、より好ましくは30~90モル%、さらに好ましくは40~70モル%である。また、第1凝縮分離工程の出口ガス温度を40℃~80℃とする場合、最終合成工程の直前で合成ループに導入されるメイクアップガスの割合は、上記と同様の観点から、10~90モル%であるとよく、30~90モル%であると好ましく、より好ましくは40~70モル%、さらに好ましくは45~65モル%である。
本実施形態においては、メイクアップガスを合成ループに導入する前に複数の流れに分割し、その分割比率を調整することができる。これにより、合成工程での反応器の温度を容易に制御することが可能となる。
合成工程においては、合成ガスからメタノールを合成する。合成工程において用いられる反応器は、触媒層を有すると共に反応で生じる熱を触媒層から取り除く機構(除熱機構)を有するものであると好ましい。
好ましい触媒の具体例としては、国際公開第2011/048976号の実施例及び比較例、例えば、実施例2及び実施例3に用いられた触媒が挙げられる。また、触媒における銅原子及び亜鉛原子のより好ましい原子比(銅/亜鉛)は、2.1~3.0の範囲である。それに加えて、アルミナを3~20質量%含むメタノール合成触媒がさらに好ましい。かかる触媒は、上述のとおり、例えば、国際公開第2011/048976号に記載の方法により調製することができる。より具体的には、例えば、銅を含む水溶液と亜鉛を含む水溶液とアルカリ水溶液とを混合して銅及び亜鉛を含む沈殿物を生成する工程と、得られた沈殿物と擬ベーマイト構造を有するアルミナ水和物とを混合して混合物を得る工程と、得られた混合物を密度が2.0~3.0g/mLになるように成型する工程とを有する製造方法によって調製される。ここで、成型方法としては、例えば、錠剤化、押出成形及び転動造粒が挙げられる。ただし、本実施形態に用いる触媒は上記の触媒及び上記の調製方法で調製された触媒に限定されるものではなく、同等のメタノール合成活性を有する他の触媒であってもよい。
加圧沸騰水との間接熱交換により触媒層の反応温度を制御するのは、少なくとも最終合成工程においてであればよいが、全ての合成工程において加圧沸騰水との間接熱交換により触媒層の反応温度を制御するのが好ましい。なお、複数の反応器において加圧沸騰水を冷却材に用いる場合、それぞれの反応器における加圧沸騰水の温度は互いに同一であっても異なっていてもよい。
分離工程においては、合成工程を経て得られた反応生成物を含む反応混合物から未反応ガスを分離する。言い換えれば、上記反応混合物に含まれるメタノール又はメタノール及び水と未反応ガスとを分離する。分離方法としては、例えば、合成工程からの出口ガスを冷却し、冷却によって生じる凝縮液を気液分離器によって分離する凝縮分離方法、及び分離膜を用いた膜分離方法が挙げられ、これらの中では凝縮分離方法が好ましい。本実施形態においては、凝縮分離方法を用いた分離工程(凝縮分離工程)が合成ループ内に少なくとも2つ設けられ、それらのうちの1つは、最終合成工程の後の最終凝縮分離工程であることが好ましい。凝縮分離工程において冷却される流体は、当該凝縮分離工程の前の合成工程からの出口ガス(ガス状の反応混合物)であり、合成されたメタノールを含む。メタノールを含む液を凝縮液として得る方法としては、例えば、反応器に供給される合成ガスとの相互熱交換やエアフィンクーラーなどによる空冷、冷却水やブラインなどの冷却材による冷却などが挙げられる。冷却対象となる流体(反応混合物)の冷却前の初期温度と冷却後の目標温度に応じて、凝縮液を得る方法は1種類を単独で又は2種類以上を組み合わせて用いられる。得られた凝縮液は、気液分離器(以下、単に「分離器」ともいう。)を用いて分離することが一般的である。これら冷却器(凝縮器)と分離器との組み合わせとしては、冷却器と分離器とを1つずつ組み合わせたものであってもよく、冷却器と分離器とを複数ずつ組み合わせたものであってもよい。冷却器と分離器とが複数組み合わされた例としては、例えば、特開昭61-257934号公報に記載のものが挙げられる。より具体的には、合成工程を経て得られる反応混合物を冷却し、メタノールを主成分とする反応生成物を凝縮し分離するに際し、凝縮器を2段に分け、前段の凝縮器の伝熱表面温度を反応混合物の露点以下、かつ反応混合物中に含まれるパラフィン類の融点以上の温度に設定し、後段の凝縮器の伝熱表面温度を60℃以下とする手法が挙げられる。
凝縮分離工程においては、冷却によって、メタノール、あるいはメタノール及び水を含む凝縮液が所定量生じるまで反応混合物を冷却する。例えば、メタノール分圧0.69~0.88MPa-G(7.0~9.0kg/cm2-G)の流体(反応混合物)を冷却し凝縮させる場合は、好ましくは、20~100℃、より好ましくは40~80℃に冷却することが好ましい。このとき、メタノール収率向上の観点から、第1凝縮分離工程において、第1合成工程からの出口ガスに含まれるメタノールの分離割合は75モル%より高くすることが好ましい。さらに、その後の第2合成工程での反応制御のために、第1凝縮分離工程における第1合成工程からの出口ガスに含まれるメタノールの分離割合は96モル%より低くすることがより好ましい。冷却水の節約の観点からは、第1凝縮分離工程における冷却は、エアフィンクーラーによる冷却(空冷)のみを用いることが好ましい。この場合、反応混合物の冷却後の目標温度は、同様の観点から55~90℃であることが好ましい。
合成ループにおけるパージガスの取り出し位置は、循環機の処理ガス量を削減する観点から、合成ループ内での圧力が低くなる箇所が好ましく、循環機の直前がより好ましい。加えて、カーボン収率の観点から、反応混合物中の反応生成物を分離して合成ループ外へ抜き出した後の未反応ガスの一部をパージガスとして分岐することが好ましく、そのパージガス取り出し位置がメイクアップガスの合流前であるとより好ましい。さらに、複数の合成工程間の分離工程において、その分離工程の前の合成工程からの出口ガスに含まれるメタノールのうち4~25モル%を分離せずに、その後の合成工程に供給することで、後段の合成工程における反応を制御し、触媒層の過熱を抑制することも可能となる。この場合、その分離工程に用いる分離器の後段であって、続く合成工程に用いる反応器の前段に循環機を配置することは、循環機内で凝縮が発生する恐れがあり、適切ではない。
これらの観点から、循環機は、最終分離工程後の未反応ガスからパージガスを取り除いた残りのガスを第1混合工程において混合する箇所へ循環させるような位置が好適である。
メタノール合成に用いる触媒は、特公昭51-44715号公報の実施例1に記載の方法によって調製された触媒(メタノール合成触媒A)、特開平8-299796号公報の実施例1に記載の方法によって調製された触媒(メタノール合成触媒B)、国際公開第2011/048976号の実施例3に記載の方法によって調製された触媒(メタノール合成触媒C)、又は、特開平8-299796号公報の比較例4に記載の方法によって調製された触媒(メタノール合成触媒D)のいずれかとした。
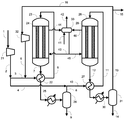
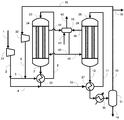
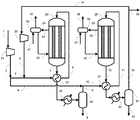
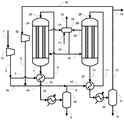
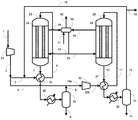
実施例1では図1に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスとして、天然ガスの水蒸気改質反応で生じるガスを利用し、循環比1.0の条件でメタノールの合成を行った。また、反応器23及び28における触媒としてメタノール合成触媒Cを用いた。圧縮機21により9.9MPa-G(101kg/cm2-G)まで昇圧した。昇圧した合成原料ガス(メイクアップガス)のうち40モル%をライン3内に流通させ、反応器23出口のライン7内を流通する反応生成物を含む出口ガス(反応混合物)と熱交換させることで、ライン6における温度が200℃になるよう予熱した。メイクアップガスのうち残りの60モル%をライン4内に流通させた。反応器23としては炭素鋼からなる内管24を有するものを用いた。触媒層における流体の圧力は9.8~9.9MPa-G(100~101kg/cm2-G)、温度は200~262℃の間であった。

比較例1では、図2に示す製造装置を用いた。実施例1との相違点は、第1合成工程の後の第1凝縮分離工程がない点である。具体的には、反応器23を通過し生成したメタノールを含む合成ガスはライン7から予熱器22を通過し、ライン2のメイクアップガスのうちのライン4へ取り出された60%と混合され、ライン10、予熱器27及びライン11から反応器28に供給された。原料ガスの組成及び合計モル流量を実施例1と同様とするとともに、圧縮機21により昇圧した圧力及びライン6とライン11における温度も実施例1と同様にした。また、反応器23の内管24及び反応器28の内管29の材料には炭素鋼を用い、反応器23及び反応器28にはメタノール合成触媒Cを充填した。比較例1は、特許文献1の技術に基づいたものである。

比較例2では、図2に示す製造装置を用いた。比較例1との相違点は、循環比が異なる点であり、循環比を3.0とした。原料ガスの組成及び合計モル流量を実施例1および比較例1と同様とするとともに、圧縮機21により昇圧した圧力及びライン6とライン11における温度も実施例1と同様にした。また、反応器23の内管24及び反応器28の内管29の材料には炭素鋼を用い、反応器23及び反応器28にはメタノール合成触媒Cを充填した。比較例2は、特許文献1の技術に基づいたものである。

このように、本発明によれば、循環比を大きく削減することによって、複数の合成工程間で凝縮分離を行う製造システムであっても、カーボン収率を保ちつつ、エネルギー削減も図れるシステムであることがわかった。特に、好ましい触媒である銅原子及び亜鉛原子を原子比(銅/亜鉛)2.0~3.0で含み、かつアルミニウム原子を含む触媒を用いることにより、カーボン収率を高く維持することとエネルギーを削減することの両立をより良好に達成できることがわかった。
実施例2では図1に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスを実施例1で用いた原料ガスと同様の組成とし、循環比1.1の条件でメタノールの合成を行った。また、メイクアップガスのうち50モル%をライン3内に流通させ、残りの50モル%をライン4内に流通させた。ライン15から系外に抜き出されるパージガス量を、循環比が1.1となるように調整した。圧縮機21により昇圧した圧力及びライン6とライン11における温度は実施例1と同様にした。また、反応器23の内管24及び反応器28の内管29の材料には炭素鋼を用い、反応器23及び反応器28にはメタノール合成触媒Cを充填した。

比較例3では図1に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスの各成分のモル流量及び循環比が実施例2と等しくなる条件とした。この比較例は特許文献2の技術に基づいたものであり、ライン2のメイクアップガスの全量をライン3内に流通させた。圧縮機21により昇圧した圧力及びライン6とライン11における温度は実施例2と同様にした。また、反応器23の内管24及び反応器28の内管29の材料には炭素鋼を用い、反応器23及び反応器28にはメタノール合成触媒Cを充填した。

また、比較例3における循環比は1.1であり、カーボン収率は97.9%であった。
加えて、冷却器25及び冷却器30に導入される合計ガス量を比較すると、実施例2では41880kg-mol/hであるのに対して、比較例3では45825kg-mol/hであり、比較例3では冷却器の負荷が高く、好ましくない。
実施例3では図1に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスを実施例1の原料ガスと同様の組成とし、循環比0.8の条件でメタノールの合成を行った。

実施例4では図1に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスを実施例1の原料ガスと同様の組成とし、循環比0.6の条件でメタノールの合成を行った。

実施例5では図1に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスを実施例1の原料ガスと同様の組成とし、循環比1.6の条件でメタノールの合成を行った。

実施例6では図1に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスを実施例1の原料ガスと同様の組成とし、循環比1.2の条件でメタノールの合成を行った。

実施例7では図1に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスを実施例1の原料ガスと同様の組成とし、循環比1.0の条件でメタノールの合成を行った。

実施例8では図3に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスを実施例1の原料ガスと同様の組成とし、循環比1.0の条件でメタノールの合成を行った。

実施例9では図1に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスを実施例1の原料ガスと同様の組成とし、循環比1.5の条件でメタノールの合成を行った。

実施例10では図1に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスを実施例1の原料ガスと同様の組成とし、循環比1.7の条件でメタノールの合成を行った。

実施例11では図1に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスを実施例1の原料ガスと同様の組成とし、循環比1.2の条件でメタノールの合成を行った。

実施例12では図1に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスを実施例1の原料ガスと同様の組成とし、循環比1.2の条件でメタノールの合成を行った。

実施例13では図1に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスを実施例1の原料ガスと同様の組成とし、循環比1.4の条件でメタノールの合成を行った。

実施例14では図1に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスを実施例1の原料ガスと同様の組成とし、循環比1.4の条件でメタノールの合成を行った。

実施例15では図1に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスを実施例1の原料ガスと同様の組成とし、循環比0.9の条件でメタノールの合成を行った。

比較例4では図4に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスのモル流量を実施例15と同様とし、循環比0.9の条件でメタノールの合成を行った。
このとき循環比は、メイクアップガスのモル流量に対する循環ガスのモル流量で定義しており、循環ガスのモル流量は、ライン16aとライン16bのモル流量の合計である。

なお、比較例4では各合成ループは、1つの分離工程を有するのみであるので、それぞれのループで分離工程は同条件とし、表24中で第2分離工程欄に該温度およびメタノール分離割合を記載した。
比較例5では図5に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスのモル流量を実施例15と同様とし、循環比0.9の条件でメタノールの合成を行った。
このとき循環比は、メイクアップガスのモル流量に対する循環ガスのモル流量で定義しており、循環ガスのモル流量は、ライン5のモル流量である。

実施例16では図6に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスは、天然ガスの水蒸気改質反応で生じるガスを利用し、循環比0.9の条件でメタノールの合成を行った。また、反応器23a、23b及び23cにおける触媒としてメタノール合成触媒Cを用いた。圧縮機21により9.9MPa-G(101kg/cm2-G)まで昇圧した。昇圧した合成原料ガス(メイクアップガス)のうち30モル%をライン3a内に、30モル%をライン3b内に、40モル%をライン3c内にそれぞれ流通させた。ライン3a内を流通したガスとライン5からの循環ガスとを混合した混合ガスを、反応器23a出口のライン7a内を流通する反応生成物を含む出口ガス(反応混合物)と熱交換させることで、200℃に予熱した。反応器23aとしては炭素鋼からなる内管24aを有するものを用いた。触媒層における流体の圧力は9.8MPa-G(100kg/cm2-G)、温度は200~261℃の間となった。

実施例17では図1に示す製造装置を用いた。各条件は以下のとおりとした。すなわち、原料ガスを実施例1の原料ガスと同様の組成とし、循環比1.2の条件でメタノールの合成を行った。

比較例6では、図7に示す製造装置を用いた。実施例13との相違点は、循環機の位置である。すなわち、実施例13においては、第2気液分離器31からライン13に取り出された未反応ガスからライン15に取り出したパージガスを除いたライン16の循環ガスを循環機32にて昇圧するようにした。一方、比較例6においては、第1気液分離器26からライン8に取り出した未反応ガスにライン4に取り出されたメイクアップガスの一部が混合されたライン10aを循環機32にて昇圧するようにした。この循環機32の位置が異なることに起因して、反応器23及び反応器28のそれぞれの入口圧力が実施例と比較例で異なる結果となった。
原料ガスの組成及び合計モル流量を実施例13と同様とするとともに、循環機32の吐出圧力を同様にした。また、反応器23の内管24及び反応器28の内管29の材料にはステンレス鋼を用い、反応器23及び反応器28にはメタノール合成触媒Cを充填した。比較例6は、特許文献3の技術に基づいたものである。




Claims (18)
- 水素と一酸化炭素と二酸化炭素とを含む合成ガスからメタノールを合成する合成工程と、前記合成工程を経て得られた反応混合物から未反応ガスを分離する分離工程と、を有するメタノール製造方法であって、
少なくとも2つの前記合成工程と、少なくとも2つの前記分離工程とを有する合成ループを有し、
前記合成ループにおいて、最終合成工程の後の最終分離工程で最終反応混合物から分離した最終未反応ガスからパージガスを取り除いた残りのガスを循環機で昇圧し、水素と一酸化炭素と二酸化炭素とを含むメイクアップガスの10~90モル%と混合して第1混合ガスを得る第1混合工程と、前記第1混合ガスからメタノールを合成する第1合成工程と、前記第1合成工程で得られた第1反応混合物から第1未反応ガスを分離する第1分離工程と、前記第1未反応ガスと前記メイクアップガスの10~90モル%のうちの少なくとも一部とを混合して第2混合ガスを得る第2混合工程と、最終的にメタノールを合成する前記最終合成工程と、前記最終合成工程で得られた前記最終反応混合物から前記最終未反応ガスを分離する前記最終分離工程と、を有し、少なくとも前記最終合成工程において、加圧沸騰水との間接熱交換により触媒層の反応温度を制御する、メタノール製造方法。 - 合成ループが有する前記少なくとも2つの分離工程のうちの少なくとも1つの分離工程が、ガス状の前記反応混合物を冷却することで生じるメタノールを含む液を気液分離器によって分離する工程である、請求項1記載のメタノール製造方法。
- 前記第1未反応ガスに、前記メイクアップガスのうちの40~70モル%を混合する、請求項1又は2に記載のメタノール製造方法。
- 前記第1分離工程において、前記第1反応混合物に含まれるメタノールのうちの35~100モル%と前記第1未反応ガスとを分離する、請求項1~3のいずれか1項に記載のメタノール製造方法。
- 前記第1分離工程において、前記第1反応混合物に含まれるメタノールのうちの75~96モル%と前記第1未反応ガスとを分離する、請求項1~4のいずれか1項に記載のメタノール製造方法。
- 前記メイクアップガスのモル流量に対する前記最終未反応ガスからパージガスを取り除いた残りのガスのモル流量の比である循環比が、0.6~2.0である、請求項1~5のいずれか1項に記載のメタノール製造方法。
- 前記循環比が0.8~1.5である、請求項6記載のメタノール製造方法。
- 前記加圧沸騰水が220~260℃である、請求項1~7のいずれか1項に記載のメタノール製造方法。
- 前記最終合成工程は、前記第2混合ガスからメタノールを合成する工程、又は、その工程で得られた第2反応混合物から第2未反応ガスを分離し、その第2未反応ガスとメイクアップガスの一部とを混合して得られる第3混合ガス又は前記第2未反応ガスから、メタノールを合成する工程である、請求項1~8のいずれか1項に記載のメタノール製造方法。
- 前記最終合成工程は、前記第2混合ガスからメタノールを合成する工程である、請求項1~9のいずれか1項に記載のメタノール製造方法。
- 前記合成工程において用いられる触媒が銅原子及び亜鉛原子を原子比(銅/亜鉛)2.0~3.0で含み、かつアルミニウム原子を含む、請求項1~10のいずれか1項に記載のメタノール製造方法。
- 前記合成工程において用いられる触媒が、銅原子及び亜鉛原子を原子比(銅/亜鉛)2.1~3.0で含み、アルミナを3~20質量%含み、かつ銅を含む水溶液と亜鉛を含む水溶液とアルカリ水溶液とを混合して銅及び亜鉛を含む沈殿物を生成する工程と、前記沈殿物と擬ベーマイト構造を有するアルミナ水和物とを混合して混合物を得る工程と、前記混合物を密度が2.0~3.0g/mLになるように成型する工程とを有する製造方法によって調製される、請求項1~11のいずれか1項に記載のメタノール製造方法。
- 前記第1未反応ガスに混合する前記メイクアップガスの割合を、前記合成工程における反応器の温度に応じて調整する、請求項1~12のいずれか1項に記載のメタノール製造方法。
- 前記合成工程の全てにおいて、加圧沸騰水との間接熱交換により触媒層の反応温度を制御する、請求項1~13のいずれか1項に記載のメタノール製造方法。
- 水素と一酸化炭素と二酸化炭素とを含む合成ガスからメタノールを合成する反応器と、前記反応器において得られた反応混合物から未反応ガスを分離する分離装置と、を備えるメタノール製造装置であって、
少なくとも2つの前記反応器と、少なくとも2つの前記分離装置とを備える合成ループを有し、
前記合成ループにおいて、最終反応器の後の最終分離装置において最終反応混合物から分離した最終未反応ガスからパージガスを取り除いた残りのガスを循環機で昇圧し、水素と一酸化炭素と二酸化炭素とを含むメイクアップガスのうちの10~90モル%と混合して第1混合ガスを得る第1混合手段と、前記第1混合ガスからメタノールを合成する第1反応器と、前記第1反応器において得られた第1反応混合物から第1未反応ガスを分離する第1分離装置と、前記第1未反応ガスと前記メイクアップガスのうちの10~90モル%とを混合して第2混合ガスを得る第2混合手段と、最終的にメタノールを合成する前記最終反応器と、前記最終反応器において得られた前記最終反応混合物から前記最終未反応ガスを分離する前記最終分離装置と、を備え、
少なくとも前記最終反応器において、加圧沸騰水との間接熱交換により触媒層の反応温度を制御する、メタノール製造装置。 - スチームドラムを更に備え、
前記第1反応器において、前記加圧沸騰水との間接熱交換により触媒層の反応温度を制御し、
前記加圧沸騰水は、前記最終反応器及び前記第1反応器と前記スチームドラムとの間で少なくとも一部が循環する、請求項15記載のメタノール製造装置。 - 少なくとも2つのスチームドラムを更に備え、
前記加圧沸騰水は、前記最終反応器と前記少なくとも2つのスチームドラムのうちの1つとの間で少なくとも一部が循環し、かつ、
前記第1反応器において、加圧沸騰水との間接熱交換により触媒層の反応温度を制御し、その加圧沸騰水は、前記第1反応器と前記少なくとも2つのスチームドラムのうちの別の1つとの間で少なくとも一部が循環する、請求項15記載のメタノール製造装置。 - 前記最終反応器は、前記第2混合ガスからメタノールを合成する反応器、又は、その反応器において得られた第2反応混合物から第2未反応ガスを分離し、その第2未反応ガスとメイクアップガスとを混合して得られる第3混合ガスから、メタノールを合成する反応器である、請求項15~17のいずれか1項に記載のメタノール製造装置。
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2015336514A AU2015336514B2 (en) | 2014-10-20 | 2015-10-20 | Method for producing methanol and apparatus for producing methanol |
| EP15852599.8A EP3210961B1 (en) | 2014-10-20 | 2015-10-20 | Methanol production method and methanol production apparatus |
| CA2964713A CA2964713C (en) | 2014-10-20 | 2015-10-20 | Method for producing methanol and apparatus for producing methanol |
| CN201580057030.9A CN107074702B (zh) | 2014-10-20 | 2015-10-20 | 甲醇制造方法以及甲醇制造装置 |
| DK15852599.8T DK3210961T3 (da) | 2014-10-20 | 2015-10-20 | Fremgangsmåde til fremstilling af methanol og indretning til fremstilling af methanol |
| US15/519,003 US10252963B2 (en) | 2014-10-20 | 2015-10-20 | Method for producing methanol and apparatus for producing methanol |
| NZ731061A NZ731061A (en) | 2014-10-20 | 2015-10-20 | Method for producing methanol and apparatus for producing methanol |
| JP2016555233A JP6666595B2 (ja) | 2014-10-20 | 2015-10-20 | メタノール製造方法及びメタノール製造装置 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014-214081 | 2014-10-20 | ||
| JP2014214081 | 2014-10-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016063872A1 true WO2016063872A1 (ja) | 2016-04-28 |
Family
ID=55760905
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/079580 WO2016063872A1 (ja) | 2014-10-20 | 2015-10-20 | メタノール製造方法及びメタノール製造装置 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US10252963B2 (ja) |
| EP (1) | EP3210961B1 (ja) |
| JP (1) | JP6666595B2 (ja) |
| CN (1) | CN107074702B (ja) |
| AU (1) | AU2015336514B2 (ja) |
| CA (1) | CA2964713C (ja) |
| DK (1) | DK3210961T3 (ja) |
| NZ (1) | NZ731061A (ja) |
| WO (1) | WO2016063872A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017175760A1 (ja) * | 2016-04-07 | 2017-10-12 | 三菱瓦斯化学株式会社 | メタノール製造方法及びメタノール製造装置 |
| WO2021106132A1 (ja) * | 2019-11-28 | 2021-06-03 | 三菱重工エンジニアリング株式会社 | メタノール製造設備及びメタノール製造方法 |
| EP3401299B1 (de) | 2017-05-12 | 2021-11-03 | L'air Liquide, Société Anonyme Pour L'Étude Et L'exploitation Des Procédés Georges Claude | Reaktor zum durchführen exothermer gleichgewichtsreaktionen |
| WO2023182506A1 (ja) * | 2022-03-25 | 2023-09-28 | 三菱瓦斯化学株式会社 | メタノール製造方法及びメタノール製造装置 |
| WO2024004464A1 (ja) * | 2022-06-30 | 2024-01-04 | 三菱瓦斯化学株式会社 | メタノール製造方法及びメタノール製造装置 |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3208258A1 (en) * | 2016-02-16 | 2017-08-23 | Fundació Privada Institut Català d'Investigació Química (ICIQ) | Methanol production process |
| US11130681B2 (en) * | 2017-03-12 | 2021-09-28 | Haldor Topsoe A/S | Co-production of methanol and ammonia |
| CN109799157B (zh) * | 2019-02-01 | 2021-09-21 | 兖州煤业榆林能化有限公司 | 一种甲醇生产过程中产生的副产物成分分析方法 |
| EP3808725A1 (de) * | 2019-10-16 | 2021-04-21 | L'air Liquide, Société Anonyme Pour L'Étude Et L'exploitation Des Procédés Georges Claude | Verfahren zum mehrstufigen herstellen von methanol |
| GB202109905D0 (en) * | 2021-07-09 | 2021-08-25 | Johnson Matthey Plc | Process for synthesising methanol |
| CN114632477A (zh) * | 2022-03-23 | 2022-06-17 | 中国神华煤制油化工有限公司 | 甲醇合成方法及甲醇合成系统 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5110210B2 (ja) * | 1971-11-16 | 1976-04-02 | ||
| JPS5144715B2 (ja) * | 1973-10-24 | 1976-11-30 | ||
| JPH08299796A (ja) * | 1995-05-11 | 1996-11-19 | Mitsubishi Gas Chem Co Inc | メタノール合成触媒の製造法 |
| JP2002515467A (ja) * | 1998-05-20 | 2002-05-28 | インペリアル・ケミカル・インダストリーズ・ピーエルシー | メタノールの合成 |
| WO2006018610A1 (en) * | 2004-08-20 | 2006-02-23 | Davy Process Technology Ltd | Process for use in gas phase reactions |
| WO2011048976A1 (ja) * | 2009-10-23 | 2011-04-28 | 三菱瓦斯化学株式会社 | メタノール合成触媒 |
| WO2014012601A1 (en) * | 2012-07-18 | 2014-01-23 | Haldor Topsøe A/S | Process and reaction system for the preparation of methanol |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2433294A1 (de) | 1974-07-11 | 1976-01-22 | Albert Berner | Verankerungsvorrichtung fuer holz- und maschinenschrauben |
| JP2695663B2 (ja) | 1989-08-07 | 1998-01-14 | 三菱重工業株式会社 | メタノール合成用触媒の製造方法 |
| JPH0635401A (ja) | 1992-07-16 | 1994-02-10 | Nec Home Electron Ltd | 地図表示装置 |
| JPH10272361A (ja) | 1997-03-31 | 1998-10-13 | Agency Of Ind Science & Technol | メタノール合成及び改質触媒 |
| JP2001205089A (ja) | 2000-01-24 | 2001-07-31 | Natl Inst Of Advanced Industrial Science & Technology Meti | メタノール合成用触媒およびその製造方法 |
| GB2393013B (en) * | 2001-02-08 | 2005-05-04 | Kim Jong-Hae | The method of education and scholastic management for cyber education system utilizing internet |
| US7192987B2 (en) * | 2004-03-05 | 2007-03-20 | Exxonmobil Chemical Patents Inc. | Processes for making methanol streams and uses for the streams |
| DE102004028200B3 (de) * | 2004-05-28 | 2005-12-15 | Hippweb E.K. | Verfahren zur Durchführung heterogen katalytischer exothermer Gasphasenreaktionen für die Methanolsynthese |
| GB0710022D0 (en) * | 2007-05-25 | 2007-07-04 | Johnson Matthey Plc | Methonal process |
| CN101386564B (zh) * | 2008-10-29 | 2011-12-28 | 西南化工研究设计院 | 一种氢气和二氧化碳合成甲醇的工艺方法 |
-
2015
- 2015-10-20 WO PCT/JP2015/079580 patent/WO2016063872A1/ja active Application Filing
- 2015-10-20 EP EP15852599.8A patent/EP3210961B1/en active Active
- 2015-10-20 NZ NZ731061A patent/NZ731061A/en unknown
- 2015-10-20 AU AU2015336514A patent/AU2015336514B2/en active Active
- 2015-10-20 US US15/519,003 patent/US10252963B2/en active Active
- 2015-10-20 DK DK15852599.8T patent/DK3210961T3/da active
- 2015-10-20 CN CN201580057030.9A patent/CN107074702B/zh active Active
- 2015-10-20 CA CA2964713A patent/CA2964713C/en active Active
- 2015-10-20 JP JP2016555233A patent/JP6666595B2/ja active Active
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5110210B2 (ja) * | 1971-11-16 | 1976-04-02 | ||
| JPS5144715B2 (ja) * | 1973-10-24 | 1976-11-30 | ||
| JPH08299796A (ja) * | 1995-05-11 | 1996-11-19 | Mitsubishi Gas Chem Co Inc | メタノール合成触媒の製造法 |
| JP2002515467A (ja) * | 1998-05-20 | 2002-05-28 | インペリアル・ケミカル・インダストリーズ・ピーエルシー | メタノールの合成 |
| WO2006018610A1 (en) * | 2004-08-20 | 2006-02-23 | Davy Process Technology Ltd | Process for use in gas phase reactions |
| WO2011048976A1 (ja) * | 2009-10-23 | 2011-04-28 | 三菱瓦斯化学株式会社 | メタノール合成触媒 |
| WO2014012601A1 (en) * | 2012-07-18 | 2014-01-23 | Haldor Topsøe A/S | Process and reaction system for the preparation of methanol |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3210961A4 * |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017175760A1 (ja) * | 2016-04-07 | 2017-10-12 | 三菱瓦斯化学株式会社 | メタノール製造方法及びメタノール製造装置 |
| US10556849B2 (en) | 2016-04-07 | 2020-02-11 | Mitsubishi Gas Chemical Company, Inc. | Method for producing methanol and apparatus for producing methanol |
| EP3401299B1 (de) | 2017-05-12 | 2021-11-03 | L'air Liquide, Société Anonyme Pour L'Étude Et L'exploitation Des Procédés Georges Claude | Reaktor zum durchführen exothermer gleichgewichtsreaktionen |
| WO2021106132A1 (ja) * | 2019-11-28 | 2021-06-03 | 三菱重工エンジニアリング株式会社 | メタノール製造設備及びメタノール製造方法 |
| JPWO2021106132A1 (ja) * | 2019-11-28 | 2021-06-03 | ||
| JP7319493B2 (ja) | 2019-11-28 | 2023-08-02 | 三菱重工業株式会社 | メタノール製造設備及びメタノール製造方法 |
| WO2023182506A1 (ja) * | 2022-03-25 | 2023-09-28 | 三菱瓦斯化学株式会社 | メタノール製造方法及びメタノール製造装置 |
| WO2024004464A1 (ja) * | 2022-06-30 | 2024-01-04 | 三菱瓦斯化学株式会社 | メタノール製造方法及びメタノール製造装置 |
Also Published As
| Publication number | Publication date |
|---|---|
| DK3210961T3 (da) | 2021-05-03 |
| JPWO2016063872A1 (ja) | 2017-08-03 |
| NZ731061A (en) | 2022-10-28 |
| EP3210961A1 (en) | 2017-08-30 |
| CA2964713A1 (en) | 2016-04-28 |
| AU2015336514A1 (en) | 2017-05-04 |
| CN107074702A (zh) | 2017-08-18 |
| AU2015336514B2 (en) | 2020-02-20 |
| EP3210961A4 (en) | 2018-06-27 |
| US20170240492A1 (en) | 2017-08-24 |
| CA2964713C (en) | 2023-02-28 |
| JP6666595B2 (ja) | 2020-03-18 |
| CN107074702B (zh) | 2020-11-03 |
| EP3210961B1 (en) | 2021-03-24 |
| US10252963B2 (en) | 2019-04-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2016063872A1 (ja) | メタノール製造方法及びメタノール製造装置 | |
| WO2017175760A1 (ja) | メタノール製造方法及びメタノール製造装置 | |
| RU2381058C2 (ru) | Способ проведения гетерогенных каталитических экзотермических газофазных реакций | |
| EP3402773B1 (en) | Methanol process | |
| EA034987B1 (ru) | Способ получения метанола | |
| EP2707355B1 (en) | High-yield process for the synthesis of urea | |
| EP2941417B1 (en) | Urea synthesis process and plant | |
| WO2022229847A2 (en) | Integration for processing effluent of oxidative dehydrogenation (odh) reactor | |
| CA3215019A1 (en) | Integration for feed dilution in oxidative dehydrogenation (odh) reactor system | |
| WO2019008317A1 (en) | PROCESS FOR THE SYNTHESIS OF METHANOL | |
| CA2896706C (en) | Urea plant revamping method | |
| EA025632B1 (ru) | Способ получения смеси, содержащей циклогексанол и циклогексанон | |
| BR112020025251A2 (pt) | Processo e usina para a produção de metanol | |
| EP4197993A1 (en) | Integrated plant and process for the production of methanol from carbon dioxide and hydrogen | |
| WO2017121978A1 (en) | Methanol process |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15852599 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2016555233 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2964713 Country of ref document: CA |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015852599 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15519003 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2015336514 Country of ref document: AU Date of ref document: 20151020 Kind code of ref document: A |