Beschreibung
Herstellung organisch phosphoreszenter Schichten unter Zusatz schwerer Hauptgruppenmetallkomplexe
Die vorliegende Erfindung betrifft ein Verfahren zur Herstellung organisch elektrischer Schichten aufweisend bei Raumtemperatur phosphoreszente organische Emitter, dadurch gekennzeichnet, dass organisch fluoreszente Emitter F zusammen mit Metallkomplexen enthaltend organische Komplexliganden L und mindestens ein schweres Hauptgruppenmetall M ausgewählt aus der Gruppe umfassend In, Tl, Sn, Pb, Sb und Bi , gemeinsam innerhalb einer Schicht abgeschieden werden und das schwere Hauptgruppenmetall M seine Koordinationssphäre unter Aufnahme des organisch fluoreszenten Emitters F ändert.
Prinzipielle Methoden zur Umwandlung von Licht in elektrischen Strom (und umgekehrt) mittels organischer Elektronik sind seit einigen Jahrzehnten bekannt. Technisch durchge- setzt, und derzeit an der Grenze zur Massenmarktreife , haben sich Mehrschichtaufbauten, wie sie zum Beispiel in Figur I für eine organische Licht emittierenden Diode {OLED} oder in Figur II für eine organische Solarzelle schematisch dargestellt sind. Obwohl die Effizienz dieser Komponenten gerade in den letzten Jahren durch den Einsatz optimierter organischer Verbindungsklassen eine deutliche Leistungssteigerung erfuhr, ergeben sich weiterhin vielversprechende Ansatzpunkte noch höhere Quantenausbeuten und damit noch höhere Wirkungsgrade bei gesenkten Materialkosten bereitzustellen.
Einer dieser Ansatzpunkte liegt in der Verwendung
phosphoreszenter Emitter, sogenannter Triplett-Emitter, welche zum Beispiel in PHOLEDs (phosphorescent organic light- emitting devices) Verwendung finden. In Anbetracht der gel- tenden Quantenstatistik sind diese, zumindest theoretisch, in der Lage, einen internen Quanten-Wirkungsgrad von 100% zu erreichen. Dies im Gegensatz zu Dioden mit rein fluoreszenten Emittern, welche aufgrund der Quantenstatistik der injizier-
ten Ladungsträger eine maximale interne Quantenausbeute von nur 25 % aufweisen.
Betrachtet man alleine die interne Quantenausbeute, so sind also Bauteile der organischen Elektronik, welche eine auf
Phosphoreszenz basierende Umwandlung von Strom in Licht {und umgekehrt) nutzen (Triplett-Emitter/Emission) , eher geeignet eine hohe Lumineszenz (cd/m2) bzw. Effizienz (cd/A, lm/ ) bereitzustellen. Im Bereich der Verbindungen, welche zu Trip- lett-Emission befähigt sind, gibt es allerdings mehrere Randbedingungen zu beachten. Zwar tritt Phosphoreszenz auch in Verbindungen der Elemente der vierten und fünften Periode des Periodensystems auf, durchgesetzt haben sich in oben genannten Anwendungen jedoch die Komplexe der Metalle der 6. Perio- de. Je nach Lage der Elemente in dieser Periode ist dabei der Ursprung der Phosphoreszenz innerhalb der Orbitalstruktur der Komplexe unterschiedlich gewichtet.
Bei den Lanthanoiden ist sowohl das HOMO (Highest Occupied Molecular Orbital) als auch das LUMO (Lowest Unoccupied
Molecular Orbital) vorwiegend metallzentriert, d.h. der Anteil der Liganden-Orbitale ist relativ schwach ausgeprägt. Dies führt dazu, dass die Emissionswellenlänge (Farbe) der Emitter fast ausschließlich durch die Bandstruktur des Me- talls festgelegt ist (Beispiele Europium = rot, Terbium = grün) . Aufgrund der starken Abschirmung der f -Elektronen dieser Metalle vermögen an das Metall gekoppelte Liganden die Energien der f"-Konfiguration der Metalle nur um circa 100 cm"1 aufzuspalten, sodass sich die Spektroskopie durch ihr Ligandenfeld der d- Ionen von derjenigen der f-Ionen erheblich unterscheidet. Bei Ionen der Lanthaniden resultiert die Farbe aus Übergängen von f- in unbesetzte s-, p- und d-Orbitale.
Geht man entlang der Periode zu den Elementen Osmium, Iridi- um, Platin und Gold, so spalten Ligandenfeider die Metallorbitale um einen Faktor 10 - lOOmal mehr auf als im Falle der Lanthanoiden. Damit lässt sich durch Variation der Liganden praktisch das gesamte sichtbare Wellenlängenspektrum mit die-
sen Elementen darstellen. Durch die starke Kopplung des Bahndrehimpulses des Metallatoms mit dem Spindrehimpuls der
Elektronen wird in den Emittern Phosphoreszenz erhalten. Das HOMO ist dabei meist metallzentriert, während das LUMO meist ligandzentriert ist. Die strahlenden Übergänge werden daher als Metall-Ligand-Charge-TransferÜbergänge (MLCT) bezeichnet.
Sowohl OLEDs als auch OLEECs (light-emitting organic
electrochemical cell) nutzen derzeit fast ausschließlich Iri- dium-Komplexe als Emitter. Im Falle der OLEDs sind die Emitter-Komplexe ungeladen, im Falle der OLEECs werden ionische d.h. geladene Emitter-Komplexe genutzt. Die Verwendung von Iridium in diesen Bauteilen hat jedoch einen gravierenden Nachteil. Die Jahresproduktion von Iridium liegt weit unter 10 t (3t in 2000) . Dies führt dazu, dass die Material-Kosten einen signifikanten Beitrag zu den Herstellkosten organisch elektrischer Bauteile leisten. Hinzu kommt, dass Iridium- Emitter nicht in der Lage sind, effizient das gesamte Spektrum des sichtbaren Lichts abzubilden. So sind beispielsweise stabile blaue Iridium-Emitter eher selten, welches einer flexiblen Verwendung dieser Materialien in OLED oder OLECC Anwendungen entgegensteht .
In der neueren Literatur finden sich hingegen einige Ansätze, welche ein „Triplett-Harvesting" auch mit nicht auf Iridium basierenden Emittern vorschlagen. So verweisen zum Beispiel Omary et al . in „Enhancement of the Phosphorescence of
Organic Luminophores upon interaction with a Mercury
Trifunctional Lewis Acid" (Mohammad A. Omary, Refaie M. Kas- sab, Mason R. Haneline, O. Elbjeirami, and Francois P.
Gabbai, Inorg. Chem. 2003, 42, 2176-2178) auf die Möglichkeit zur Erreichung einer ausreichenden Phosphoreszenz rein organischer Emitter durch den Einsatz von Quecksilber. Bedingt durch den Schweratome fekt des Quecksilbers in einer Matrix aus organischen Liganden wird ein Singulett-Triplett-
/Triplett-Singulett-Übergang der angeregten Elektronen der organischen Matrix quantenmechanisch ermöglicht (ISC, inter System crossing) , welches in einer deutlichen Verringerung
der Lebensdauer der angeregten elektronischen (Triplett) Zustände resultiert und eine unerwünschte Sättigung der Besetzung dieser Zustände vermeidet. Ursächlich für diesen Mechanismus ist die S in-Bahnkopplung des Quecksilber-Schweratoms mit den angeregten Elektronen der organischen Matrix. Nachteilig hingegen ist, dass der Einsatz von Quecksilber aufgrund von toxischen und umweltpolitischen Aspekten problematisch ist, Eine Möglichkeit eine hinreichende Quantenausbeute auf Basis rein organischer Phosphoreszenz-Emitter zu erhalten wird zum Beispiel durch Bolton et al . in NATURE CHEMISTRY, 2011, 1-6 beschrieben („Activating efficient phosphorescence from purely organic materials by crystal design" , Onas Bolton, Kangwon Lee, Hyong-Jun Kim, Kevin Y. Lin and Jinsang Kim, NATURE CHEMISTRY, 2011, 1-6) . An dieser Stelle wird vorgestellt, dass der Einbau schwerer Halogenide in einen Kristall aus einer organischen Matrix zu hohen Quantenausbeuten durch phosphoreszierende organische Emitter ührt. Nachteilig an dieser Lösung ist hingegen, dass für diesen Effekt anscheinend ein bestimmter Abstand zwischen den schweren Halogeniden und der organischen Matrix und ein kristalliner Aufbau nötig sind. Dies würde einer kostengünstigen industriellen Fertigung von organischen Bauelementen entgegenstehen.
Die WO 2012/016074 AI hingegen beschreibt eine dünne Schicht umfassend eine Verbindung der Formel
wobei Ar
1 und Ar
2 jeweils unabhängig ein C3-30 aromatischer Ring sind; R
1 und R
2 ein Substituent sind; a und b jeweils unabhängig eine ganze Zahl von 0 bis 12 sind, wobei, wenn a 2 oder mehr ist, jeder Rest Rl gegebenenfalls voneinander ver- schieden ist, und zwei Reste R
1 gegebenenfalls miteinander gebunden sind, um eine Ringstruktur zu bilden, und, wenn b 2 oder mehr ist, jeder Rest R
2 gegebenenfalls voneinander verschieden ist und zwei Reste R
2 gegebenenfalls miteinander gebunden sind, um eine Ringstruktur zu bilden; A
1 irgendeine Art einer direkten Bindung, -O-, -S-, -S(=0)-, -S(=0)
2-, -
PR3-, -NR4- und -C(R5)2- ist; R3 ein Wasserstoffatom oder ein Substituent ist; R4 ein Wasserstoffatom oder ein Substituent ist; R5 ein Wasserstoffatom oder ein Substituent ist und zwei Reste Rs gegebenenfalls voneinander verschieden sind; E1 ein einwertiger Rest mit 50 oder weniger Kohlenstoffatomen ist; L1 ein Ligand mit 50 oder weniger Kohlenstoffatomen ist; c eine ganze Zahl von 0 bis 3 ist, wobei, wenn c 2 oder mehr ist, jeder Rest L1 gegebenenfalls voneinander verschieden ist; und jede Kombination einer Kombination von E1 und Ar1 und einer Kombination von E1 und Ar2 gegebenenfalls eine Bindung bildet; und, wenn c 1 bis 3 ist, jede Kombination einer Kombination von L1 und E1, einer Kombination von L1 und Ar1, einer Kombination von L1 und Ar2 und einer Kombination von L1 und L1 gegebenenfalls eine Bindung bildet. Nachteilig hinge- gen ist, dass die beschriebenen Verbindungen nur eine ungenügende Quantenausbeute aufweisen und in Lösung nicht ausreichend stabil sind, sodass sie sich zersetzen.
Die DE 103 60 681 AI offenbart Hauptgruppenmetall - Diketonatokomplexe nach folgender Formel
als phosphoreszente Emitter-Moleküle in organischen Leuchtdioden (OLEDs), worin M Tl(I), Pb(II) und Bi(III) sein können. Desweiteren wird die Verwendung dieser Hauptgruppenmetall- Diketonatokomplexe als Licht-emittierende Schichten in OLEDs, Licht-emittierende Schichten, enthaltend mindestens einen Hauptgruppenmetall-Diketonatokomplex, eine OLED, enthaltend diese Licht-emittierende Schicht, sowie Vorrichtungen, die eine erfindungsgemäßes OLED enthalten offenbart. In Versuchen hingegen konnte gezeigt werden, dass die unter strengen Was- serausschluss synthetisierten, oben genannten Verbindungen keine auf Phosphoreszenz basierende Emission nach elektronischer Anregung zeigen. Hochwahrscheinlich rühren die angeführten phosphoreszenten Emissionen von nicht näher bestimmbaren Oxo-Clustern her, welche sich unkontrolliert, zum Beispiel durch Hydrolyse im Rahmen der Herstellung, gebildet haben. Nachteilig an dieser speziellen Lösung ist, dass das n~ System dieser Acetylacetonat-Liganden, insbesondere der beschrieben vollfluorierten Varianten, wenig ausgeprägt ist und als alleinige Phosphoreszenz-Emitter nur geringe Phosphoreszenz-Ausbeuten erlaubt.
Es ist daher die Aufgabe der vorliegenden Erfindung ein Verfahren bereitzustellen, mit welchem kostengünstig Schichten für organisch elektronische Bauelemente hergestellt werden können, welche durch die Nutzung von Phosphoreszenz eine effektive Umwandlung von Strom in Licht und umgekehrt ermöglichen .
Gelöst wird diese Aufgabe durch die Merkmale des Anspruchs 1. Besondere Ausführungsformen der Erfindung werden in den Unteransprüchen wiedergegeben. Erfindungsgemäß ist ein Verfahren zur Herstellung organisch elektrischer Schichten aufweisend bei Raumtemperatur
phosphoreszente organische Emitter, dadurch gekennzeichnet, dass organisch fluoreszente Emitter F zusammen mit Metallkomplexen enthaltend organische Komplexliganden L und mindestens ein schweres Hauptgruppenmetall M ausgewählt aus der Gruppe umfassend In, Tl, Sn, Pb, Sb und Bi, gemeinsam innerhalb einer Schicht abgeschieden werden und das schwere Hauptgruppenmetall M seine Koordinationssphäre unter Aufnahme des organisch f luoreszenten Emitters F ändert. Überraschenderweise wurde gefunden, dass mittels dieses Verfahrens einfach und kostengünstig, Schichten mit bei Raumtemperatur
phosphoreszenten Emittern erhalten werden, welche eine hohe interne Quanteneffizienz, hohe Leuchtdichten, schnelles Ansprechverhalten und gute Langzeitstabilitäten aufweisen. Auf- grund der Tatsache, dass die Phosphoreszenz nur durch die organischen Emitter hervorgerufen wird, kann durch Modifikation der Liganden, insbesondere deren π-System, die Emissionswellenlänge der Emitter desweiteren durchgestimmt werden. Möglich ist insbesondere der Aufbau heteroleptischer Komplexe oder Anlagerungs -Verbindungen, welche Emissionen über die Orbitale unterschiedlich aufgebauter Liganden/Emitter ermöglicht. Dies erhöht zusätzlich die Vielfalt zur Herstellung rein organischer phosphoreszenter Emitter. Ohne durch die Theorie gebunden zu sein wird innerhalb des erfindungsgemäßen Verfahrens der zur Fluoreszenz befähigte organische Emitter in die Nähe des schweren Hauptgruppenmetalls abgeschieden, wobei sich die Koordinationssphäre des Metalls ändert. Die Änderung der Koordinationssphäre des Metalls kann dabei sowohl eine Erweiterung der Anzahl koordinierter Ligan- den/Emitter, einen Ersatz einer oder mehrerer Liganden durch
Emitter oder sogar in der Reduzierung der Anzahl koordinierter Liganden durch die Addukt- /Anlagerungs -/Kom lexbildung mit dem Emitter bestehen. Dies als Funktion der sterischen
und elektronischen Voraussetzungen der Emitter und Liganden, der Koordinationsstärke der einzelnen Liganden und der
Abscheidegeschwindigkeit und -Temperatur im Rahmen der gewählten Herstellung. Im Falle der Adduktbildung oder Koordi- nation des Emitters an das schwere Hauptgruppenmetallatom muss zwischen dem Schweratom und dem organischen Emittern keine σ-Bindung ausgebildet werden. Es reichen schwache elektrostatische und/oder n-Wechselwirkungen zwischen Metall und Emitter. Durch die Wechselwirkung mit dem Schwermetall- atom kann sich allerdings auch die die Energielage des H0-
MOs/LUMOs des oder der fluoreszenten Emitter ändern. Bedingt durch den Schweratomeffekt kann es zudem zu einer Spin- Bahnkopplung zwischen dem Emitter-Elektronen und dem Metallatomkern kommen, welches dazu führt, dass bisher spin- verbotene elektronische Übergänge quantenmechanisch erlaubt werden. Dadurch sinkt die Lebensdauer der elektronisch angeregten Zustände und somit wird bei Raumtemperatur ein effektiver Phosphoreszenzkanal (mit Triplett-Singulett-Übergängen) eröffnet. Das Metall beteiligt sich nicht direkt an der Emis- sion, es stellt lediglich seinen Bahndrehimpuls zur Verfügung. Dies ist im Gegensatz zu der bei Raumtemperatur rein fluoreszenten Emission der organischen Emitter ohne Schwermetallkoordination . Die erfindungsgemäß erhaltenen Emitterschichten können dabei neutraler oder ionischer Natur sein und somit OLED oder OLEC- typisches Emissionsverhalten zeigen.
Unter einer organisch elektrischen Schicht im Sinne der Er- findung wird eine Schicht umfassend organischen Emitter, Metallkomplexe enthaltend schwere Hauptgruppenmetalle und optional Matrixmaterialien verstanden. Die Schicht kann auch amorph sein, d.h. die einzelnen Bestandteile in dieser
Schicht weisen keine periodische Anordnung über einen größe- ren Bereich (Fernordnung) auf. Insbesondere wird darunter nicht das Vorliegen eines Einkristalls oder kristalliner Bereiche mit einer Größenausdehnung von größer oder gleich 50 nm verstanden. Die in der Schicht vorliegenden Verbindungen
könne aber hingegen eine gewisse Nahordnung (Abstand und Orientierung) zu Ihren nächsten Nachbarn aufweisen. Diese Bereiche sind aber statistisch verteilt. Innerhalb eines
Röntgendiffraktogramms, zum Beispiel erhalten durch eine XRD- Messung (Röntgen- Pulverdiffraktometrie) , zeichnet sich die amorphe Schicht durch einen breiten Halo aus. Überraschenderweise wurde gefunden, dass eine amorphe Anordnung der Bereiche in der Schicht zum Erhalt einer ausreichenden Phosphoreszenz-Ausbeute genügt. Dies im Gegensatz zu experimentellen Ergebnissen, welche eine sehr regelmäßige Anordnung einer
Vielzahl von Metallatomen und Emittern als Bedingung zum Erhalt hoher Phosphoreszenz-Ausbeuten voraussetzen.
Bedingt durch den Einfluss des Schwermetalls wird bei Raumtemperatur ein nennenswerter Phosphoreszen -Beitrag des organischen fluoreszenten Emitters erhalten. Raumtemperatur im Sinne der Erfindung ist der Temperaturbereich von -50°C bis 150°C (der übliche Betriebstemperaturbereich für organische Elektronik) . In diesem Temperaturbereich liefern die
phosphoreszenten Übergänge eines oder mehrerer fluoreszenter Emitter ohne Schwermetallatomeinfluss in der Regel keine nennenswerten Beiträge zur Emission rein organischer Emitter,
Organisch fluoreszente Emitter F sind organische Moleküle, welche entweder partiell oder insgesamt aromatischen Charakter mit delokalisierte π-Elektronen aufweisen können. Desweiteren können diese Moleküle Heteroatome wie N, N-R, 0, S, Se, Si oder Metalle wie Li oder AI, Ga oder Zn aufweisen. R steht in diesen Fall für einen alkyl- oder aromatischen Rest. Diese Moleküle zeigen als Feststoff oder in Lösung nach elektronischer Anregung Fluoreszenz, d.h. elektronische (S1-S0) singulett-singulett Übergänge, Phosphoreszente Übergänge (T- S) sind aufgrund der quantenmechanischen Ausschlussregeln (Spinumkehr) bei Raumtemperatur nicht beobachtbar. Die Le- bensdauer der fluoreszenten Übergänge in den erfindungsgemäß einsetzbaren organischen fluoreszenten Emittern kann ohne Annäherung an das schwere Metallatom in einem Bereich unterhalb von 100 ns liegen.
Bevorzugt kann es sich bei den organisch fluoreszenten Emittern F um C10-C60 Heteroaromaten, desweiteren bevorzugt um C15-C50 Heteroaromaten handeln. In speziellen Anwendungsfäl- len haben sich die Sauerstoff und die Stickstoff enthaltenen Heteroaromaten als besonders günstig erwiesen. Desweiteren können organisch fluoreszente Emitter bevorzugt innerhalb des erfindungsgemäßen Verfahrens eingesetzt werden, deren Trip- lett-Zustand größer oder gleich -5 eV und kleiner oder gleich 5 eV vom SO-Zustand entfernt ist. Mit diesen elektronischen Rahmenbedingungen können sich im Rahmen des erfindungsgemäßen Verfahrens besonders hohe Quantenausbeuten erhalten lassen.
Im Rahmen des erfindungsgemäßen Verfahrens werden Metallkom- plexe enthaltend schwere Hauptgruppenmetalle M aus der Gruppe umfassend In, Tl, Sn, Pb, Sb und Bi eingesetzt. Diese Metallkomplexe können organische Komplexliganden aufweisen und sind besonders aufgrund ihrer Verfügbarkeit, ihres Beschaffungs- preises und ihrer Fähigkeit zur Ausbildung einer ausgeprägten Spin-Bahnkop lung und der Möglichkeit zur Erweiterung der
Koordinationsspähre bevorzugt. Es können auch mehrere unterschiedliche Metalle aus der oben genannten Gruppe im erfindungsgemäß einsetzbaren Metallkomplex enthalten sein. Diese Gruppe ist besonders geeignet, da die in ihr aufgeführten Elemente einen besonders hohen Bahndrehimpuls aufweisen, welcher effektive Phosphoreszenz-Übergänge in den organischen Emittern F ermöglicht. Zudem sind diese Metalle in hoher Reinheit zu relativ niedrigen Preisen verfügbar. In einer besonderen AusführungsVariante kann die Gruppe vorteilhafterweise auch Sn, Pb und Bi umfassen. Diese Metalle besitzen zudem den Vorzug, dass sie sich auch sehr gut aus Lösungen heraus verarbeiten lassen. Bevorzugt kann die Koordination der Metalle mit organischen Liganden erfolgen, welche terminal, bidentat, tridentat oder hetero-bimetallisch mit dem Metallatom verbrückt sind. Vorteilhafte Ausgestaltungen können sich ergeben, wenn die Koor-
dination der Liganden an das Metallatom über zwei Sauerstoff - atome erfolgt. Ohne durch die Theorie gebunden zu sein, können diese Substituenten im Rahmen des Abscheideverfahrens effizient durch die organischen Emitter verdrängt oder die Koordinationssphäre erweitert werden und so zu einer hohen Phosphoreszenzausbeute beitragen. Desweiteren kann mittels dieser Substituenten die Emissionswellenlänge des
Phosphoreszenzlichtes durch Ligand-Ligand-Übergänge abgestimmt werden. Bevorzugt können dieserart koordinierte L gan- den aromatische π-Systeme aufweisen, welche mindestens 10 C- Atome aufweisen. Dies kann zu einer größeren AufWeitung der Emissionswellenlängen bei Ligand-Ligand-Übergängen beitragen.
Das schwere Hauptgruppenmetall M ändert unter Aufnahme des organisch fluoreszenten Emitters F seine Koordinationssphäre. Ohne durch die Theorie gebunden zu sein, wird im erfindungsgemäßen Verfahren dabei der organische Emitter in die Nähe des Hauptgruppenmetalls gebracht. Daraufhin ändert sich die Anordnung der Liganden des Metallkomplexes. Dies bedingt durch van-der-Waals- , Coulomb-, π-σ- oder σ-Wechselwirkungen des organischen Emitters mit dem Metall. Eine o- Wechselwirkungen ist zur Ausbildung der Phosphoreszenz nicht vonnöten, kann aber auch ausgebildet werden. Die Koordinationssphäre des Metalls kann durch die Nähe des organischen Emitters aufgeweitet werden. Es kann auch zur Substitution eines einzelnen oder mehrerer Liganden durch den organischen Emitter kommen. Desweiteren ist es auch möglich, dass die Anzahl der Liganden durch die Änderung der Koordinationssphäre reduziert wird. Dies zum Beispiel durch das Verdrängen einer oder mehrerer Liganden durch die Aufnahme des fluoreszenten organischen Emitters.
Zusätzlich zum Metallkomplex und zum organischen Emitter können im Rahmen des erfindungsgemäßen Verfahrens noch weitere nicht koordinierende Matrixmaterialien innerhalb der Schicht abgeschieden werden. Dieses oder diese Matrixmaterialien können beispielsweise die elektronische Leitfähigkeit der
Schicht beeinflussen oder allgemein Einfluss auf die Beweg-
lichkeit des organischen Emitters oder des Metallkomplexes nehmen. Geeignete Matrixmaterialien können ausgewählt sein aus der Gruppe 2 , 2 ' , 7, 7 ' -tetrakis (carbazol-9-yl) -9, 9- spirobifluorene 2 , 7 -bis (carbazol- 9 -yl) - 9 , 9-ditolylfluorene ; 9 , -bis [ - (carbazol- 9 -yl) -phenyl] fluorene ; 2 , 7-bis (carbazol- 9-yl) -9 , 9 -spirobif luorene; 1 , 4 -bis ( triphenylsilyl) benzene ; 1 , 3 -bis (triphenylsilyl) benzene ; Bis (4 -N, N-diethylamino-2 - methylphenyl) - -methylphenylmethane ; 2, 7-bis (carbazol-9-yl) - 9 , 9~dioctylfluorene ; 4,4' -di (triphenylsilyl) -p-terphenyl ; 4,4· -di (triphenylsilyl) -biphenyl; 9- (4-tert-butylphenyl) -3,6- bis (triphenylsilyl) - 9Hcarbazole ; 9- (4-tert-butylphenyl) -3, 6- ditrityl-9H-carbazole; 9- (4-tert-butylphenyl) -3, 6 -bis (9- (4- methoxyphenyl) -9H-fluoren-9-yl) - H-carbazole ; 2, 6-bis (3- (9H- carbazol - -yl) phenyl) Pyridine; 3 , 5 -bis (3- ( 9H- carbazol- 9- yl) phenyl) yridine, Triphenyl (4 - ( 9-phenyl- 9H- fluoren- 9 - yl) phenyl) silane ; 9 , 9-dimethyl-N, N-diphenyl-7- (4 - (1-phenyl- lHbenzo [d] imidazol-2-yl) phenyl) -9H-fluoren- 2 -amine; 3,5- bis (3- (9H-carbazol-9-yl) phenyl } yridine ; 9, 9-spirobifluoren- 2-yl-diphenyl-phosphine oxide; 9, 9 (5- (triphenylsilyl) -1, 3- phenylene) is (9Hcarbazole) ; 4 , 4 , 8 , 8 , -12 , 12 -hexa-p- tolyl-4H- 8H-12H-12C-Azadibenzo [cd,mn] yrene; 2,2' -bis (4- (carbazol-9- yl) phenyl) -biphenyl ; 2,8- bis (diphenylphosphoryl) dibenzo [b, d] Thiophene; Bis (2- methylphenyl) diphenylsilane; Bis [3 , 5-di (9H-carbazol-9- yl ) henyl] diphenylsilane ; 3 , 6-bis (carbazol- 9-yl) -9- (2-ethyl- hexyl) - 9Hcarbazole ; 3- (diphenylphosphoryl) -9- (4- ( diphenylphosphoryl) henyl) -9H-carbazole ; 3, 6-bis [(3,5- diphenyl) phenyl] -9-phenyl-carbazole; 2, 8-di ( 9H- carbazol- 9- yl) dibenzo [b,d] thiophene; 10- (4 ' - (diphenylamino) biphenyl-4- yl) cridin-9 (10H) -one; 2, 7-bis (diphenylphosphoryl) -9, 9 ' - spirobi [fluorene] ; 1 , 4 -bis (( 9H-carbazol- 9-yl) methyl) benzene ; Bis -4- (N-carbazolyl) henyl) henylphosphineoxide; 2,7- Bis (diphenylphosphoryl) -9- (4 -diphenylamino) henyl- 9 ' -phenyl- fluorene; Di (4 - (6H-indolo [3 , 2-b] quinoxalin-6- yl) phenyl) diphenylsilane; Di (4- (6H-indolo [3 , 2-b] quinoxalin- 6- yl) phenyl) diphenylmethane ; Bis [3 , 5-di ( 9H-carbazol - 9- yl)phenyl] diphenylsilane; 2,6, 14-Tris (carbazol-9- yl ) triptycene ; 2 , 6 , 14 -Tris (diphenylphosphine-
oxide) triptycene ; 2 , 6 , 14 -Tris {diphenyl-amino) triptycene ; 2,7- bis (diphenylphosphoryl) -9-phenyl-9H-carbazole Tris [4- (9- phenylfluoren-9-yl) phenyl] aminebiphenyl-3-amine) ; 2,7- Bis (diphenylphosphoryl) spiro [fluorene-7, 11' -benzofluorene] .
Die vorliegende Erfindung wird in Verbindung mit weiteren Aspekten und Ausführungsformen nachfolgend näher beschrieben. Sie können beliebig miteinander kombiniert werden, sofern sich aus dem Zusammenhang nicht eindeutig das Gegenteil ergibt.
In einer bevorzugten Ausführungsform des Verfahrens kann das schwere Hauptgruppenmetall Bi umfassen. Bismuth hat sich aufgrund seiner ökonomischen und prozesstechnischen Eigenschaf- ten als besonders geeignet erwiesen. Es existiert eine Vielzahl von Komplexverbindungen, welche sich im Rahmen von Nass- oder Gasphasenprozessen besonders effizient mit organischen Fluoreszenz -Emittern verarbeiten lassen. Obwohl Bismut im Periodensystem direkt hinter dem Blei folgt, hat es ganz andere physiologische Eigenschaften. Dadurch, dass es nur schwer über den Magen-Darm-Trakt aufgenommen werden kann, sind Vergiftungen mit Bismut eher selten. Im Gegenteil, Salze von Bismut werden zur Behandlung von Magengeschwüren oder Syphilis in der Medizin eingesetzt. Auch als Kontrastmittel für Röntgenuntersuchungen fand es Verwendung. Von Bismut kommt nur das Isotop mit der Masse 209 natürlich vor. Es ist mit einer Halbwertszeit von 1,9 1019 Jahren ein radioaktiver oc- Strahler. Aus der langen Halbwertszeit ergibt sich für 1 kg eine Aktivität von 0,0033 Bq, Diese ist damit ca. 10 Millio- nen mal geringer als die des in Organismen vorkommenden Kaliums. Ein Kilogramm Kalium enthält von Natur aus 0,012 Prozent also 0,12 Gramm des radioaktiven Isotops 0K mit einer Halbwertszeit ti/ von 1, 248 x 109 Jahren = 39,38xl015 Sekunden, und besitzt eine Atommasse von 39,96. Daraus ergibt sich eine Radioaktivität von 31825 Bq. Damit ist die Radioaktivität des Bismuts für praktische Anwendungen vernachlässigbar klein und wäre von einem Menschen, der den Geigerzähler hält nicht einmal nachweisbar. Bismut besitzt, im Gegensatz zu Iridium
{3/2}und Europium (5/2) , einen Kernspin von (9/2) . Dieser ist in der Lage mit ungepaarten Elektronen zu koppeln, die sich auf Liganden befinden {siehe auch „Synthesen und Eigenschaften neuer Tris (fluorphenyl) antimon- und -bismut- Verbindun- gen. Kristallstruktur von Tkis (2, 6-difluorphenyl) bismut" von T. Lewe et al . Z. anorg. allg. Chem. 622 (1996) 2009-2015), Diese Eigenschaften und die Tatsache, dass die Bismut- im Vergleich zu den Iridiumvorkommen praktisch keiner Beschränkung unterliegen können zu einer dramatisch besseren Edukt- Kostensituation führen.
Bevorzugt können die einsetzbaren Bismuth-Komplexe Bismuth in einer Oxidationszahl von +11, +III oder +V aufweisen. Diese Oxidationszahlen haben sich als Anlagerungspunkt noch weite- rer Liganden, wie zum Beispiel der organischen Emitter F, als besonders geeignet erwiesen. Die Anlagerungskinetik der organischen Emitter scheint mit diesen Oxidationszahlen des
Bismuths gerade auch für eine Gasphasenabscheidung besonders geeignet. Desweiteren lassen sich die Bismuth II/III/V- Komplexe auf Basis ihrer physikalischen Daten, wie zum Beispiel der Verdampfungstemperatur oder der Löslichkeit sehr gut mittels Gasphasenabscheidungs- oder auch Nassprozessen zu Schichten formen. Insbesondere kann die Änderung der Koordinationssphäre des Bi-Metallatoms durch eine Anlagerung von Heteroatomen eines fluoreszenten Emitters erfolgen. Es können sich dabei Anlagerungsverbindungen ergeben, in welchen sich spezielle Metall - Heteroatom-Abstände als besonders vorteilhaft herausgestellt haben. Im Falle von fluoreszenten Emittern, welche über ein
Sauerstoff mit dem Bi-Schwermetallatom wechselwirken können, hat sich eine Herstellung als besonders geeignet erwiesen, im welchen der Bi-0 Abstand größer oder gleich 2,25 Ä und kleiner 2,75 Ä , bevorzugt größer oder gleich 2,3 Ä und kleiner 2,70 Ä und desweiteren bevorzugt größer oder gleich 2,4 Ä und kleiner 2,6 Ä beträgt.
Im Falle einer Wechselwirkung über ein Cl-Heteroatom hat sich eine Herstellung als besonders geeignet erwiesen, im welchen
der Bi-Cl Abstand größer oder gleich 2,3 Ä und kleiner 2,9 Ä, bevorzugt größer oder gleich 2,4 Ä und kleiner 2,80 Ä und desweiteren bevorzugt größer oder gleich 2,45 Ä und kleiner 2,75 Ä beträgt.
Im Falle einer Wechselwirkung über ein N-Heteroatom hat sich eine Herstellung als besonders geeignet erwiesen, im welchen der Bi-N Abstand größer oder gleich 2,3 Ä und kleiner 2,9 Ä, bevorzugt größer oder gleich 2,4 Ä und kleiner 2,80 Ä und desweiteren bevorzugt größer oder gleich 2,45 Ä und kleiner 2, 70 Ä beträgt .
Im Falle einer Wechselwirkung über ein I-Heteroatom hat sich eine Herstellung als besonders geeignet erwiesen, im welchen der Bi-I Abstand größer oder gleich 2,6 Ä und kleiner 3,2 Ä, bevorzugt größer oder gleich 2,7 Ä und kleiner 3,10 Ä und desweiteren bevorzugt größer oder gleich 2,8 Ä und kleiner 3 , 1 Ä beträgt .
Im Falle einer Wechselwirkung über ein Br-Heteroatom hat sich eine Herstellung als besonders geeignet erwiesen, im welchen der Bi-Br Abstand größer oder gleich 2,5 Ä und kleiner 3,1 Ä, bevorzugt größer oder gleich 2,6 Ä und kleiner 3,0 Ä und des- weiteren bevorzugt größer oder gleich 2,7 Ä und kleiner 2,95 Ä beträgt .
Die Bindungslängen können dabei aus Einkristalldaten der betreffenden Verbindungen nach dem Fachmann bekannten Methoden bestimmt werden.
Diese Abstände der Heteroatom-enthaltenden Emitter zu dem Schwermetallatom zeigen einen ausreichenden Schwermetallatomeffekt zur Öffnung des Fluoreszenzkanals im organischen Emitter und ermöglichen zudem eine gute Wechselwirkung des orga- nischen Emitters mit den restlichen Liganden.
In einer weiteren Ausgestaltung des Verfahrens kann der Anteil der durch elektronische inter- und intra-Liganden- Übergänge hervorgerufenen, phosphoreszenten Emission unter rein elektronischer Anregung größer oder gleich 20% und kleiner oder gleich 100% betragen. Die Aufnahme des fluoreszenten Emitters in die Koordinationssphäre des schweren Metallatoms kann einen effektiven „Phosphoreszenz -Kanal" des organischen
Emitters öffnen. Zusätzlich zu einer fluoreszenten Emission können dabei auch zusätzliche Beiträge durch phosphoreszente Strahlung erhalten werden. Dies kann zu einer deutlichen Erhöhung der internen Quantenausbeute der Schicht beitragen. Die Unterscheidung, ob eine Strahlungskomponente
fluoreszenten oder phosphoreszenten Ursprungs ist kann dabei anhand von zeitkorrelierter Einzelphotonenzählung (englisch time correlated Single photon counting, TCSPC-Messungen) bestimmt werden. Mittels TCSPC wird die Laufzeit jedes einzel- nen Photons gemessen und die Verteilung der Laufseiten wird akkumuliert. Komponenten auf einer MikroSekunden Zeitskala können hierbei phosphoreszenten und schnellere Übergänge fluoreszenten Übergängen zugeordnet werden. Betrachtet wird dabei jeweils die mathematische Anpassung an den gemessenen Intensitätsverlauf. Diese Methode ist dem Fachmann bekannt. Beispiele hierzu finden sich im experimentellen Teil.
In einer zusätzlichen Ausgestaltung des Verfahrens können die organisch fluoreszenten Emitter F aus der Gruppe der substi- tuierten oder nicht substituierten C6-C60 Aromaten oder Hete- roaromaten ausgewählt sein. Zum Erhalt eines möglichst großen Phosphoreszenz-Beitrages des organischen fluoreszenten Emitters und einer möglichst stabilen Assoziation des Emitters an das schwere Hauptgruppenmetall können fluoreszente Emitter in diesem Größenbereich besonders vorteilhaft eingesetzt werden. Zudem lassen sich diese Emitter sowohl gut aus der Flüssig- wie auch aus der Gasphase abscheiden. Die einzelnen Moleküle können dabei sowohl vollständig aromatisch durchkonjugiert, wie auch teilweise nicht aromatische Abschnitte aufweisen.
In einer alternativen Ausführungsmöglichkeit des Verfahrens kann die längste Lebensdauer elektronisch angeregter Zustände des organisch fluoreszenten Emitters F nach Aufnahme in die Koordinationssphäre des schweren Hauptgruppenmetalls M bei Raumtemperatur größer oder gleich 0,01 Mikrosekunden und kleiner oder gleich 10000 Mikrosekunden betragen. Durch die Aufnahme des organisch fluoreszenten Emitters F in die Koordinationssphäre des schweren Hauptgruppenmetalls M kann, be-
dingt durch die Spin-Bahnkopplung des Metalls mit den angeregten Elektronen des organischen Emitter, eine Interkombina- tion der Singulett- mit den Triplett-Zuständen ermöglicht werden. Diese kann den Phosphoreszenz-Kanal des Emitters „öffnen", welches zu einer höheren Quantenausbeute und längeren, beobachtbaren Lebensdauern der angeregten elektronischen Zustände des Emitters führen kann. Die Lebensdauern können mit gängigen Verfahren, wie beispielweise in den Beispielen durchgeführt mittels TCSPC bestimmt werden. Die fluoreszenten Übergänge zeichnen sich dabei durch Lebensdauern von 10"9 - KT Sekunden aus, wohingegen die phosphoreszenten Übergänge üblicherweise längere Zeitkonstanten aufweisen. In Abhängigkeit der Zusammensetzung der elektrischen Schicht können auch mehrere Zeitkonstanten oder Lebensdauern vorliegen. Die längste Lebensdauer im Erfindungssinn ist diejenige, welche mit einem Anteil an der Gesamtlebensdauer von größer oder gleich 2,5 % die größte Lebensdauer aufweist.
In einer weiteren Ausgestaltung des Verfahrens können die or- ganisch fluoreszenten Emitter F aus der Gruppe umfassend 4,7- di (9H~carbazol~9-yl) -1, 10 -phenanthrolin, 2, 6-bis (3- {9H- carbazol-9-yl) henyl) yridine, 3 , 5-bis (3- {9H-carbazol-9- yl) phenyl) pyridine, 2, 8-di (9H-carbazol-9- yl) dibenzo [b, d] thiophene, 2,2' ,2" -(1,3 , 5-Benzinetriyl) - tris (1-phenyl-l-H-benzimidazol) , 2- (4-Biphenylyl) -5- (4-tert- butylphenyl) -1, 3 , 4 -oxadiazol , 2, 9-Dimethyl-4 , 7 -diphenyl-1 , 10- phenanthrolin, 8-Hydroxyquinolinolato-lithium, 4- (Naphthalen- 1-yl) -3, 5-diphenyl-4H-l, 2,4-triazol, 1, 3 -Bis [2- (2, 2 ' - bipyridine-6-yl) -1,3,4 -oxadiazo- 5 -yl] benzen, 4 , 7-Diphenyl- 1 , 10 -phenanthrolin, 3 - (4-Biphenylyl) -4 -phenyl-5-tert- butylphenyl-1, 2,4-triazol, Bis (2-methyl-8-quinolinolate) -4- (phenylphenolato) aluminium, 6,6' -Bis [5- (biphenyl-4 -yl) -1,3,4- oxadiazo-2-yl] -2,2' -bipyridyl, 2-phenyl- 9 , 10-di (naphthalen-2 - yl) -anthracen, 2, 7 -Bis [2- (2 , 2 » -bipyridine- 6 -yl) -1,3,4- oxadiazo-5-yl] -9, 9 -dimethylfluoren, 1, 3-Bis [2- (4-tert- butylphenyl) -1,3, 4-oxadiazo-5-yl] benzen, 2- (naphthalen-2-yl) - 4 , 7-diphenyl-l , 10 -phenanthrolin, 2 , 9-Bis (naphthalen-2 -yl) - 4 , 7-diphenyl-l , 10 -phenanthrolin, Tris (2 , 4 , 6-trimethyl-3 -
{ yridin- 3 ~yl) henyl) boran, l-methyl-2 - (4- (naphthalen-2- yl)phenyl) -lH-imidazo [4 , 5-f] [1 ( 10] henanthrolin, 1,3- bi (carbazol-9-yl) benzene , 1 , 3-bis (carbazol-9-yl) pyridine, 1, 3 , 5- tris (carbazol-9-yl) benzene, 9- (3- (9H-carbazol-9- yl) henyl) -3- (4- (1-phenyl-lH-benzo [d] imidazol-2-yl) phenyl) - 9H-carbazole, 2, 6, 14-Tris (carbazol-9-yl) triptycene, 1,3- bis (carbazol-9-yl) benzene, 1,3, 5-tris (carbazol-9-yl) benzene, 3 , 5-di (9H-carbazol-9-yl) bipheny, 9- (3 , 5- bis (diphenylphosphoryl) henyl) -9H-carbazole, Bis [3 , 5-di (9H- carbazol-9-yl)phenyl] diphenylsilane, 2,8- bis (diphenylphosphoryl) dibenzo [b, d] thiophen, poly [3- (carbazol-9-yl) -9- (3 -methyloxetan-3 -ylmethyl) carbazole] , Poly [3- (carbazol- 9 -ylmethyl) -3 -methyloxetane] ausgesucht sein. Diese Verbindungen haben sich zum Einsatz als organisch fluoreszente Emitter F als besonders geeignet, aber nicht einschränkend erwiesen. Sowohl die elektronischen wie auch sterischen Eigenschaften dieser Verbindungen erlauben ausreichende Wechselwirkungen mit den schweren Hauptgruppenmetallen zur „Öffnung" des Phosphoreszenzkanals mit guten internen Quantenausbeuten und langer Standzeit der Schichten. Zudem weisen diese Verbindungen hinreichend große aromatische Bereiche auf, welches zu geeigneten Emissionswellenlängen führen kann. Die gute Wechselwirkung der organisch fluoreszenten Emitter F kann hochwahrscheinlich auch auf deren sterische Gegebenheiten und hier insbesondere auf die geeigneten Koordinationsstellen zum Metallatom zurückgeführt werden. Zudem weisen diese Verbindungen eine gute Verarbeitung in Nass- wie auch Gasphasenabscheidungsprozessen auf . Gemäß einer weiteren Ausgestaltung des erfindungsgemäßen Verfahrens kann der organisch fluoreszente Emitter F, 4,7-di(9H- carbazol-9-yl) -1, 10 -phenanthrolin (BUPHl)sein. BUPH1 zeigt, hochwahrscheinlich bedingt durch seine elektronische
HOMO/LUMO- Struktur , bei Raumtemperatur ohne Koordination an ein schweres Hauptgruppenmetall aus der oben angegebenen
Gruppe nur reine Fluoreszenzemission. Nach Koordination oder Adduktbildung mit dem Hauptgruppenmetall werden mit hohen Quantenausbeuten phosphoreszente Emissionen beobachtbar. Die-
se werden im hohen Maße durch die elektronische Struktur des organischen Emitters und der restlichen Liganden des Komplexes bestimmt. Schichten mit diesem Emitter-Aufbau haben sich dabei als besonders effizient und langlebig erwiesen. Die Langlebigkeit kann dabei wahrscheinlich auf die Größe des organischen Moleküls und dessen geringe Kristallxsationsneigung zurückgeführt werden.
In einer weiteren Ausgestaltung des erfindungsgemäßen Verfah- rens können die Liganden des Metallkomplexes unabhängig voneinander aus der Gruppe umfassend Halogenide und fluorierte oder nicht- fluorierte C2-C20 Alkyl- oder Aryl-Carboxylate, - Alkoholate, -Thiolate, -Cyanate, -Isocyanate, -Thiocyanate , - Acetylacetonate , -Sulfonate ausgewählt sein. Diese Liganden im Metallkomplex können zu einer leichten Verarbeitbarkeit in Nass- wie auch Gasphasenprozessen beitragen und können aufgrund Ihrer Koordinationseigenschaften zum Metallatom zu einer einfachen Änderung der Koordinationssphäre des Metall- atoms beitragen. Innerhalb des Metallkomplexes kann/können dabei nur ein oder aber auch mehrere der oben genannten Liganden vorliegen. Bevorzugt kann der Komplex gemischte Liganden aufweisen. Dies entweder durch Erweiterung der Koordinationssphäre des Metalls oder durch Ersatz eines einzelnen oder mehrerer Liganden. Diese Liganden können zudem zur
Justage der Emissionswellenlänge des organischen Emitters genutzt werden. Dies kann durch elektronische Wechselwirkungen des oder der Liganden mit dem Emitter hervorgerufen werden. Diese Liganden L im Metallkomplex können bevorzugt einen Anteil von größer oder gleich 0 % und kleiner oder gleich 20 % an der gesamten Emissionsausbeute der Schicht ausmachen. Bevorzugt kann dieser Bereich zwischen größer oder gleich 0 % und kleiner oder gleich 10 % und desweiteren bevorzugt zwischen größer oder gleich 0 % und kleiner oder gleich 5 % liegen.
Desweiteren können in einem weiteren Aspekt des Verfahrens die Liganden L des Metallkomplexes unabhängig voneinander aus der Gruppe umfassend C6-C30 Aromaten und Heteroaromaten aus-
gewählt sein. Diese Aromaten oder Heteroaromaten können zu einer leichten Verarbeitbarkeit in Nass- wie auch Gasphasenprozessen beitragen und ermöglichen den organischen Emittern zudem eine einfache Koordination an das Metallatom. Die Wech- selwirkungen der π-Elektronen können zudem die Lage der Phosphoreszenz-Wellenlängen des organischen Emitters beeinflussen und so zu einem veränderten Emissionsspektrum beitragen.
In einer weiteren bevorzugten Ausführungsform kann der Me- tallkomplex ein Bi(III) und mindestens einen Komplexliganden aus der Gruppe der nicht substituierten, partiell fluorierten oder per- fluorierten organischen Carbonsäuren enthalten. Bevorzugt kann der Metallkomplex ein, zwei oder drei dieser organischen Carbonsäuren enthalten. Organische Carbonsäuren können dabei generell aus der Gruppe der aliphatisch, gesättigten Monocarbonsäuren; aliphatisch, ungesättigten Mono- carbonsäuren; aliphatisch, gesättigten Dicarbonsäuren; aliphatisch, gesättigte Tricarbonsäuren; aliphatisch, ungesättigten Dicarbonsäuren; aromatischen Carbonsäuren; heterocyc- lischen Carbonsäuren; aliphatisch, ungesättigten, cyclischen Monocarbonsäuren ausgewählt werden. Besonders bevorzugte partielle oder perfluorierte Liganden L können aus substituierten oder unsubstituierten Verbindungen der Essigsäure, Phe- nylessigsäure und/oder Benzoesäure ausgewählt werden. Beson- ders bevorzugt kann nicht- fluorierte, partiell fluorierte oder perfluorierte Essigsäure eingesetzt werden. In einer weiteren bevorzugten Ausführung form können ein oder mehrere mehrzähnige Liganden L im unverdampften Zustand verbrückend zwischen den Metallatomen des Komplexes angeordnet sein. Die- se Verbindungen lassen sich einfach sowohl aus der Nassphase als auch über ein Gasphasenabscheidungsverfahren verarbeiten und ermöglichen eine gute Anbindung des fluoreszenten Emitters in der Schicht. Sie können derart zu langlebigen Emitter-Bauteilen führen, welche eine sehr gute Quantenausbeute aufweisen.
Besonders bevorzugt können die Bi (III) -Metallkomplexe als Ausgangsstoffe nach unten angegebenen Strukturen dabei entweder ein-kernig
wobei R
1 und R
2 unabhängig voneinander Sauerstoff, Schwefel, Selen, NH oder NR
4 sein können, wobei R
4 ausgewählt ist aus der Gruppe enthaltend Alkyl oder Aryl und mit R
3 verbunden sein kann; und
R3 ausgewählt ist aus der Gruppe enthaltend Alkyl,
langkettiges Alkyl, Alkoxy, langkettiges Alkoxy, Cycloalkyl, Halogenalkyl , Aryl, Arylene, Halogenaryl, Heteroaryl,
Heteroarylene , Heterocycloalkylene, Heterocycloalkyl , Halo- genheteroaryl , Alkenyl, Halogenalkenyl , Alkinyl,
Halogenalkinyl , Ketoaryl, Halogenketoaryl , Ketoheteroaryl , Ketoalkyl, Halogenketoalkyl , Ketoalkenyl, Halogenketoalkenyl, wobei bei geeigneten Resten eine oder mehrere nichtbenachbarte CH2-Gruppen unabhängig voneinander durch -O-, -S- , -NH-, -NR°-, -SiR°R0O~; -CO-, -COO-, -OCO-, -OCO-O-, -S02-, -S-CO-, -CO-S-, -CY1^CY2 oder -C=C- ersetzt sein können und zwar derart, dass O und/oder S Atome nicht direkt miteinander
verbunden sind, ebenfalls optional mit Aryl- oder Heteroaryl bevorzugt enthaltend 1 bis 30 C Atome ersetzt sind (endständige CH3-Gruppen werden wie CH2-Gruppen im Sinne von CH2-H verstanden)
Ohne durch die Theorie gebunden zu sein ergibt sich bei den ein-kernigen Komplexen eine terminale Koordination des Metalls über den/die Liganden, Im Falle eines zwei-kernigen Komplexes ergibt sich eine zwei- oder drei -zähnige Koordination des Metallatoms . Diese Koordinationsgeometrie kann den Zutritt eines fluoreszenten Emitters erleichtern und so zu einer effektiven Komplex oder Adduktbildung beitragen. Des- weiteren zeigen diese Verbindungen in Schichten gute elektrische Eigenschaften und eine geringe Kristallisationsneigung, sodass langlebig hohe Quantenausbeuten erhältlich sein kön- nen.
In einer weiteren Charakteristik des erfindungsgemäßen Verfahrens kann der Metallkomplex eine oder mehrere Verbindungen aus der Gruppe Trisarylbismuth (V) biscarboxylat und
Bi (III) triscarboxylat umfassen. Das Trisarylbismuthcarboxylat ergibt sich nach folgender Formel
wobei Ari , Ar
2 und Ar
3 unabhängig voneinander substituierte oder nicht substituierte, fluorierte order nicht fluorierte Aromaten oder Heteroaromaten bezeichnen. Diese Verbindungen lassen sich besonders leicht mittels Nassphasen- oder Gasphasenabscheidung verarbeiten und ermöglichen eine gute Koordi- nation der organischen fluoreszenten Emitter an das Bi- Zentralatom. Die so erhaltenen Schichten zeichnen sich durch eine hohe Quantenausbeute und eine geringe Kristallisations-
neigung aus. Dieses kann die Lebensdauer der Schichten erhöhen.
Desweiteren kann in einem weiteren Aspekt des Verfahrens der Metallkomplex eine oder mehrere Verbindungen aus der Gruppe ganz oder teilweise fluoriertes Triphenylbismuth (V) bis- ( fluorobenzoat) und Bi (III) pentafluorobenzoat umfassen. Diese Verbindungen lassen sich besonders leicht mittels Nassphasen- oder Gasphasenabscheidung verarbeiten und ermöglichen eine gute Koordination der organischen fluoreszenten Emitter an das Bi- Zentralatom. Die so erhaltenen Schichten zeichnen sich durch eine hohe Quantenausbeute und eine geringe Kristallisationsneigung aus. Dieses kann die Lebensdauer der Schichten erhöhen .
In einer zusätzlichen Charakteristik des Verfahrens kann der Metallkomplex eine oder mehrere Verbindungen aus der Gruppe Bi (III) fluorobenzoat , Bi (III) fluoroalkylbenzoat ,
Bi (III) fluorodialkylbenzoat , Bi (III) fluorotrialkyl-benzoat Bi (III) pentafluorobenzoat und Bi (III) 3 , 5trifluormethyl- benzoat umfassen. Diese Verbindungen mit Bi in der Oxidati- onsstufe III und Benzoat-Sustituenten lassen sich besonders leicht mittels Nassphasen- oder Gasphasenabscheidung verarbeiten und ermöglichen eine gute Koordination der organischen fluoreszenten Emitter an das Bi- Zentralatom . Die so erhaltenen Schichten zeichnen sich durch eine hohe Quantenausbeute und eine geringe Kristallisationsneigung aus. Dieses kann die Lebensdauer der Schichten erhöhen. Die Fluorierung kann dabei sowohl nur ein einzelnes H-Atom bis hin zu einer
Perfluorierung der Verbindung erfassen. Die Alkyl-Gruppen können dabei vorzugsweise Cl-C5-Akyl sein und falls nicht näher spezifiziert, können 1-4 Positionen des Grundkörpers unabhängig voneinander alkyliert sein. In einem weiteren Aspekt des Verfahrens kann der Metallkomplex eine oder mehrere Verbindungen aus der Gruppe der
Trisarylbismuth (V) carboxylate umfassen. Diese Verbindungen mit Bi in der Oxidationsstufe V lassen sich besonders leicht
mittels Nassphasen- oder Gasphasenabscheidung verarbeiten und ermöglichen eine gute Koordination der organischen
fluoreszenten Emitter an das Bi-Zentralatom. Die so erhaltenen Schichten zeichnen sich durch eine hohe Quantenausbeute und eine geringe Kristallisationsneigung aus . Dieses kann die Lebensdauer der Schichten erhöhen. Die Fluorierung kann dabei sowohl nur ein einzelnes H-Atom bis hin zu einer
Perfluorierung der Verbindung erfassen. Eine bevorzugte Verbindung dieser Verbindungslasse ist beispielsweise ganz oder teilweise fluoriertes Triphenylbismuth (V) bis (fluorobenzoat) .
In einer weiteren, bevorzugten Ausführungsform kann der Metallkomplex aus der Gruppe umfassend Bi (III) triscarboxylat , Bi (III) fluoroacetat und Bi (III) trifluoroacetat ausgewählt sein. Gerade die Koordination der Liganden über zwei Sauerstoffatome an das schwere Hauptgruppenmetall kann eine erleichterte Änderung der Koordinationssphäre durch den Eintritt des organischen Fluoreszen -Emitters ermöglichen. Durch diese Konfiguration können besonders stabile und effiziente Schichten mit langen Lebensdauern hergestellt werden.
In einer zusätzlichen Ausgestaltung des Verfahrens kann der Metallkomplex und der organisch fluoreszente Emitter F auf einem Trägersubstrat mittels Ko-Evaporation, Rotations-, Vor- hangbeschichtung, Rakeln oder Drucken abgeschieden werden.
Besonders bevorzugt kann die amorphe Schicht mittels Gasphasenabscheidung oder Nassprozessen hergestellt werden. Mittels dieser Verfahren können der Metallkomplex und der organische Fluorezenz-Emitter zusammen abgeschieden werden und so die amorphe Schicht bilden. Beide Substanzen können im Rahmen eines Ko-Evaporationsverfahrens dabei aus unterschiedlichen Quellen unter Einsatz thermischer Energie sublimiert werden. Mittels dieser Verfahren erhält man besonders homogene und gleichmäßige Schichten. Lösemittelprozesse können bevorzugt so durchgeführt werden, dass die Komponenten aus einem Lösemittel auf ein Substrat abgeschieden werden. Dies kann die Prozessführung vereinfachen und eine günstigere Herstellung ermöglichen. Zusätzlich können noch weitere Materialien wie
zum Beispiel Matrixmaterialien, welche nicht an das Metall - atom koordinieren in dem Lösungsmittel gelöst und/oder innerhalb der Schicht mit abgeschieden werden. Ebenso können diese Matrixmaterialien aus weiteren Quellen zusätzlich mit ver- dampft werden.
In einer weiteren Charakteristik des erfindungsgemäßen Verfahrens kann das molare Verhältnis von Metallkomplex zu organisch fluoreszenten Emitter F größer oder gleich 1:10 und kleiner oder gleich 10:1 betragen. Diese Mengenverhältnisse von Metallkomplex zu organischen Emitter innerhalb der
Schicht haben sich zum Erhalt hoher Leuchtdichten und einer langen Lebensdauer der Schichten als besonders vorteilhaft erwiesen. Höhere Anteile an Metallkomplex können zwar durch die Veränderung der Schichtleitfähigkeit zu einer Erhöhung der Phosphoreszenz-Ausbeute führen, dieser Effekt kann aber auch durch andere Verbindungen bei niedrigeren Materialkosten erreicht werden. Geringere Anteile an Metallkomplex können hingegen zu einer nur ungenügenden Aktivierung des Phospho- reszenz-Pfades führen. Dies kann für die interne Quantenausbeute der Schicht nachteilig sein. Bevorzugt beträgt das molare Verhältnis von Metallkomplex zu organisch fluoreszenten Emitter F größer oder gleich 1:5 bis 5:1 und desweiteren besonders bevorzugt 1:3 bis 3:1.
Zusätzlich kann im erfindungsgemäßen Verfahren die Abschei- dung des Metallkomplex und des organisch fluoreszenten Emitters F mittels eines Ko-Evaporationsverfahrens erfolgen und die Abscheiderate der organisch elektrischen Schicht größer oder gleich 0,1 Ä/s und kleiner oder gleich 200 Ä/s betragen. Die Öffnung des Phosphoreszenz -Kanals des organischen Emitters ist im Wesentlichen mit der Koordinationsänderung des schweren Hauptgruppenmetalls M durch die Aufnahme des oder Adduktbildung mit dem organischen Emitter gekoppelt. Die räumliche Nähe des Emitters zum Metall ermöglicht dabei eine Spin-Bahnkopplung, welche zu einer reduzierten Lebensdauer angeregter Triplett -Zustände des organischen Emitters führt. Überraschenderweise wurde gefunden, dass diese Abstände zwi-
sehen Emitter und Metall auch mittels Ko-Evaporation herbeigeführt werden können. Dies ist deshalb überraschend, da man als Voraussetzung für das Vorliegen hoher Quantenausbeuten einen möglichst definierten Abstand, wie z.B. in
Einkristallen oder bei kristallinen Strukturen gegeben, als Voraussetzung erwarten würde. Dies lässt sich aber bei einer Herstellung mittels Ko-Evaporation nicht erwarten, da die einzelnen Moleküle ungeordnet, amorph, innerhalb einer
Schicht abgeschieden werden. Durch dieses Verfahren lassen sich lösungsmittelfreie Schichten mit langer Lebensdauer erhalten. Die bevorzugte Abscheiderate kann dabei zu einem gleichmäßigen Schichtaufbau beitragen. Kleinere
Abscheideraten sind nicht erfindungsgemäß, da diese die Herstellung aufgrund des Zeitaufwandes deutlich verteuern wür- den. Höhere Raten sind desweiteren nicht erfindungsgemäß, da die Quantenausbeute sich aufgrund einer ungenügenden Abstandseinstellung zwischen Metall und organischen Emitter verringern kann. Bevorzugt kann die Abscheiderate desweiteren größer oder gleich 0,1 Ä/s und kleiner oder gleich 150 Ä/s und weiterhin bevorzugt größer oder gleich 1,0 Ä/s und kleiner oder gleich 100 Ä/s betragen.
Desweiteren erfindungsgemäß ist eine Schicht in einem organisch elektrischen Bauelement hergestellt nach dem erfin- dungsgemäßen Verfahren. Mittels des erfindungsgemäßen Verfahrens lassen sich Schichten in organisch elektrischen Bauelementen herstellen, welche zur Emission und Umwandlung von Licht geeignet sind. Die Schichten können eine Schichtdicke von größer oder gleich 1 nm und kleiner oder gleich 500 m aufweisen und mittels der vorher beschriebenen Verfahren aufgetragen werden. Im Rahmen von Ko-Evaporationsprozessen erhält man die Schicht durch das direkte Aufbringen der Substanzen aus der Gasphase, wohingegen bei Nassprozessen, die Schicht nach Evaporation des oder der Lösemittel erhalten wird.
Zusätzlich kann die erfindungsgemäße Schicht als aktive
Schicht in einem organisch elektrischen Bauelement zur Um-
Wandlung elektrischen Stroms in Licht, von Licht in elektrischen Strom und von Licht in Licht anderer Wellenlänge Verwendung finden. Die erfindungsgemäße Schicht lässt sich demzufolge zur Gewinnung von Strom durch Absorption von Licht - wellen als auch zur Erzeugung von Licht mittels eines elektrischen Stromes nutzen. Desweiteren kann die Schicht auch zur Umwandlung von Lichtwellen in Lichtwellen einer anderen Wellenlänge genutzt werden. Dies beispielsweise durch Aufnahme von Lichtquanten und Abgabe von Lichtquanten anderer Wellen- länge.
Erfindungsgemäß ist weiterhin ein organisches Halbleiterbauelement ausgewählt aus der Gruppe umfassend Photodioden, Solarzellen, organische Leuchtdioden, Licht emittierende elekt- rochemische Zellen enthaltend die erfindungsgemäße Schicht, Das beschriebene Verfahren und die damit herstellbaren
Schichten können entsprechend für absorbierende Bauelemente wie Photodioden oder Solarzellen Verwendung finden. Desweiteren können die Schichten auch für Photo-Konversionsschichten in der Photovoltaik oder Sensorik eingesetzt werden. Das Verfahren ist mit den Standard-Herstellungsschritten dieser Bauelemente kompatibel und derart lassen sich kostengünstig, langlebige und effiziente Bauelemente erhalten. Hinsichtlich weiterer Vorteile und Merkmale der vorbeschriebenen organischen Halbleiterbauelemente wird hiermit explizit auf die Erläuterungen im Zusammenhang mit der erfindungsgemäßen Schicht sowie dem erfindungsgemäßen Verfahren verwiesen. Auch sollen erfindungsgemäße Merkmale und Vorteile des erfin- dungsgemäßen Verfahrens auch für die erfindungsgemäße Schichten und die erfindungsgemäßen organischen Halbleiterbauelemente anwendbar sein und als offenbart gelten und umgekehrt. Unter die Erfindung fallen auch sämtliche Kombinationen aus zumindest zwei von in der Beschreibung und/oder den Ansprü- chen offenbarten Merkmalen.
Die oben beschriebenen Eigenschaften, Merkmale und Vorteile dieser Erfindung sowie die Art und Weise, wie diese erreicht
werden, werden klarer und deutlicher verständlich im Zusammenhang mit der folgenden Beschreibung der Ausführungsbei - spiele, die im Zusammenhang mit den Zeichnungen näher erläutert werden.
Die Eigenschaften der gewählten Edukte und die Eigenschaften der erfindungsgemäßen Schichten werden nachfolgend anhand von Figuren näher erläutert. Die Figuren zeigen Fig. 1 schematisch den Aufbau einer organischen Leuchtdiode 10. Die Leuchtdiode ist aus einer Glas-Schicht 1; Indium-Zinn-Oxid ITO-Schicht 2; Loch- Inj ektor- Schicht 3; Loch-Transportschicht HTL 4; Emitter- Schicht EML 5; Loch-Blocker-Schicht HBL 6; Elektro- nen-Transportschicht ETL 7; Elektronen-
Injektorschicht 8 und einer Kathoden- Schicht 9 aufgebaut ;
Fig. 2 schematisch den Aufbau einer organischen Solarzelle mit PIN-Struktur 20, welche Licht 21 in elektrischen Strom umwandelt. Die Solarzelle besteht aus einer Schicht aus Indium-Zinn-Oxid 22; einer p- dotierten Schicht 23; einer Absorptions-Schicht 24; einer n-dotierten Schicht 25 und einer Metall- Schicht 26;
Fig. 3 die Photolumineszenz -Spektren von festen und in THF gelösten BUPH1 bei Raumtemperatur; Fig. 4 die Photolumineszenz -Spektren von BUPH1 in 2-
Methyl-THF bei 77 K;
Fig . 5 das zeitkorrelierte Einzelphotonenzählungs- (TCSPC) - Spektrum von BUPH1 in THF zur Bestimmung der Fluoreszenz-Lebensdauer des BUPH1;
Fig. 6 das Cyclo-Voltagramm von BUPH1 in Acetonitril;
die UV-Absorptionsspektren von Bi(tfa)3, Bi(pFBz}3 und Bi (3 , StfmBz) 3 zusammen mit einer Taue- Darstellung ; die Auftragung der molaren Extinktionskoeffizienten von Bi(tfa)3, Bi(pFBz)3 und Bi ( 3 , StfmBz ) 3 in THF gegen die Wellenlänge; die UV-Absorptionsspektren von BUPH1, Bi(tfa)3 und mit unterschiedlich Abscheidungsraten durch Ko- Evaporation hergestellter Bi (tfa) 3 : BUPHl-Schichten; die UV-Absorptionsspektren von BUPH1 und mit unterschiedlich Abscheidungsraten durch Ko-Evaporation hergestellter Bi (pFBz) 3 : BUPHl-Schichten; das normierte Photolumineszenzspektrum von BUPH1 und mit unterschiedlich Abscheidungsraten durch Ko- Evaporation hergestellter Bi (tfa) 3 : BUPHl-Schichten; das normierte Photolumineszenzspektrum von BUPH1 und mit unterschiedlich Abscheidungsraten durch Ko- Evaporation hergestellter Bi (pFBz) 3 : BUPHl- Schichten; das XRD-Spektrum eines Bi ( tfa) 3 : BUPH1 (1:1) -Films; die UV-Absorptionsspektren im Bereich zwischen 250 und 475 nm von SnCl2-BUPHl-Addukten oder -Komplexen mit unterschiedlicher Zusammensetzung. Dargestellt sind die UV-Absorptionsspektren von 1:1, 1:2 und 1:3 Addukten oder Komplexen in THF bei Raumtemperatur; die UV-Absorptionsspektren im Bereich zwischen 350 und 475 nm von SnCl2-BUPHl -Addukten oder -Komplexen mit unterschiedlicher Zusammensetzung, Dargestellt sind die UV-Absorptionsspektren von 1:1, 1:2 und
1:3 Addukten oder Komplexen in THF bei Raumtemperatur. Die Daten entsprechen den Ergebnissen aus Figur 14 und sind nur vergrößert und ausschnittsweise dargestell ;
Fig. 16 das normierte Photolumineszenz-Spektrum von SnCl2- BÜPH1 -Addukten oder -Komplexen mit unterschiedlicher Zusammensetzung (1:1, 1:2 und 1:3} in THF bei Raumtemperatur. Die Anregungswellenlänge betrug 410 nm.
Fig. 17 das zeitkorrelierte Einzelphotonenzählungs- (TCSPC) - Spektrum von 1:1 SnCl2-BUPHl -Addukten oder - Komplexen in THF bei Raumtemperatur. Dargestellt ist zudem die mathematische Anpassung;
Fig. 18 das zeitkorrelierte Einzelphotonenzählungs- (TCSPC) - Spektrum von 1:2 SnCl2-BUPHl -Addukten oder - Komplexen in THF bei Raumtemperatur. Dargestellt ist zudem die mathematische Anpassung;
Fig. 19 das zeitkorrelierte Einzelphotonenzählungs- (TCSPC) - Spektrum von 1:3 SnCl2-BUPHl -Addukten oder - Komplexen in THF bei Raumtemperatur. Dargestellt ist zudem die mathematische Anpassung;
Fig. 20 die UV-Absorptionsspektren im Bereich zwischen 250 und 475 nm von PbTFA-BUPHl -Addukten oder -Komplexen mit unterschiedlicher Zusammensetzung, Dargestellt sind die UV-Absorptionsspektren von 1:1, 1:2 und
1:3 Addukten oder Komplexen in THF bei Raumtemperatur;
Fig. 21 die UV-Absorptionsspektren im Bereich zwischen 350 und 430 nm von PbTFA.BUPHl -Addukten oder -Komplexen mit unterschiedlicher Zusammensetzung. Dargestellt sind die UV-Absorptionsspektren von 1:1, 1:2 und 1 : 3 Addukten oder Komplexen in THF bei Raumtempera-
tur. Die Daten entsprechen den Ergebnissen aus Figur 20 und sind nur vergrößert und ausschnittsweise dargestellt ; Fig. 22 das normierte Photolumineszenz -Spektrum von PbTFA- BUPHl-Addukten oder -Komplexen mit unterschiedlicher Zusammensetzung (1:1, 1:2 und 1:3) in THF bei Raumtemperatur. Die Anregungswellenlänge betrug 410 nm.
Fig. 23 das zeitkorrelierte Einzelphotonenzählungs- (TCSPC) - Spektrum von 1:1 PbTFA.BUPHl-Addukten oder - Komplexen in THF bei Raumtemperatur. Dargestellt ist zudem die mathematische Anpassung;
Fig. 24 das zeitkorrelierte Einzelphotonenzählungs- (TCSPC) - Spektrum von 1:2 PbTFA.BUPHl-Addukten oder - Komplexen in THF bei Raumtemperatur. Dargestellt ist zudem die mathematische Anpassung
Fig. 25 das zeitkorrelierte Einzelphotonenzählungs- (TCSPC) ~
Spektrum von 1:3 PbTFA.BUPHl-Addukten oder - Komplexen in THF bei Raumtemperatur. Dargestellt ist zudem die mathematische Anpassung,
Beispiele :
Im Rahmen des erfindungsgemäßen Verfahrens wird ein organi- scher Fluoreszenz-Emitter durch Wechselwirkung mit einem schweren Hauptgruppenmetall (In, Tl, Sn, Pb, Sb, Bi) innerhalb einer Schicht oder in Lösung zu einer auf Phosphoreszenz basierenden Aussendung oder Aufnahme von Licht befähigt. Zur Veranschaulichung des Prinzips werden Verbindungen mit den Hauptgruppenmetallen Bi, Pb und Sn vorgestellt.
Als organischer Fluoreszenz-Emitter wird 4, 7-di ( 9H-carbazol- 9-yl) -1, 10-phenanthrolin (BUPH1) eingesetzt.
Charakterisierung des organischen Fluoreszen -Emitters
BUPHl ist ein ungeladenes, neutrales organisches Molekül fol- genden Aufbaus
wobei ein elektronentransportierender Phenanthrolin-Kern mit zwei Löcher-transportierenden Carbazol-Einheiten verbunden ist. Die Verbindung ist eine bidentate Lewis-Base und die beiden Stickstoffe des Kerns können, ebenso wie die n- Elektronensysteme der Aromaten mit geladenen Metallen wechselwirken .
In Figur 3 sind die Spektren des festen BUPHl und des BUPHl gelöst in Tetrahydrofuran (THF) dargestellt. Das BUPHl zeigt bei Raumtemperatur eine blaue Photolumineszenz mit einer maximalen Emission des festen Materials bei 420 nm und der in THF-gelösten Verbindung bei 425 nm. Die Wellenlängenverschiebung von 5 nm ergibt sich aufgrund der Polarität des gewählten Lösungsmittels.
In Figur 4 ist das Photolumineszenz-Spektrum von BUPHl in 2- Methyl-THF bei 77 K gezeigt. Die Verbindung zeigt 5 Emissi- onsmaxima bei 410, 475, 510, 550 und 600 nm. Aufgrund der Tatsache, dass die Maxima bei 475, 510, 550 und 600 nm deutlich tiefer liegen als bei 410 nm kann darauf geschlossen werden, dass diese Emissionen aus dem Triplett-Zustand erfol- gen und folglich von Phosphoreszenzübergängen (T1->S0) herrühren. Allgemein ist diese Quantenausbeute geringer, da dieser Übergang Spin-verboten ist. Desweiteren spricht auch die Sto- kes-Verschiebung der Emissionen bei 475, 510, 550 und 600 nm für das Vorliegen eines Phosphoreszenzüberganges des BUPHl
bei tiefen Temperaturen. Phosphoreszente Übergänge liegen, verglichen mit Fluoreszenzübergängen, immer in der Energie tiefer. Desweiteren spricht die Tatsache, dass die 475, 510, 550 und 600 nm Emissionen im Raumtemperaturspektrum fehlen auch für das vorliegen von Triplett-Emissionen. Daraus folgt, dass das erste Emissionsmaximum einem Fluoreszenz- und die weiteren Übergänge Phosphoreszenz -Übergängen zugeordnet werden können. Die Energie des Triplett-Überganges liegt im Vergleich mit anderen Lewis-Basen erstaunlicherweise mit 2,6 eV (475 nm) sehr hoch.
Die Photolumineszenz Quantenausbeute (englisch
photoluminescence quantum yield - PLQY) von BUPH1 wurde in THF Lösung bestimmt. PLQY eines Fluorophors (Emitter) gibt das Verhältnis zwischen der Anzahl der emittierten und absorbierten Photonen an. Eine Methode zur Berechnung wird zum Beispiel in Albert M. Brouwer Pure Ap l . Chem., Vol. 83, No . 12, pp. 2213-2228, 2011 angegeben. Die BUPH1-PLQY in THF liegt, unter Verwendung von 9 , lODiphenylantharacene in
Cyclohexan als Referenz, bei 29,8 %.
Figur 5 zeigt das zeitkorrelierte Einzelphotonenzählungs- (TCSPC) -Spektrum von BUPH1 in THF zur Bestimmung der Fluoreszenz-Lebensdauer. Die Figur zeigt den exponentiellen Photolu- mineszenz -Abfall als Funktion der Zeit. Das Experiment wurde in inerter Atmosphäre mit Äex 295 nm, Aem 420nm und TAC 50ns durchgeführt. Der TAC („time to amplitude", Zeit/Amplituden- Umwandler) ist ein Teil des TCSPC Spektrometer, das ein Aus- gangssignal mit einer Amplitude proportional zum Zeitinter- vall zwischen Eingangs „Start-" und „Stopp- "Impulsen erzeugt. Die Amplitudenverteilung der AusgangsImpulse wird dann von einem Multichannel-Analyzer aufgezeichnet. Sie ist somit ein Maß für die Verteilung der Zeitintervalle zwischen den Start- und Stopp- Impulsen und wird oft als „Zeitspektrum11 bezeich- net. Die Daten können mit einer einzelnen Exponentialfunktion angepasst werden, woraus sich eine Lebensdauer von 7,06*10~9 s für BUPH1 in THF bei Raumtemperatur ergibt (CHISQ = 1.14). Dies bedeutet, dass die Lebensdauer des angeregten Zustandes
bei Raumtemperatur auf einer Nanosekunden-Zeitskala liegt, welches einem fluoreszenten Übergang, mit einer üblichen Lebensdauer von 1CT9 bis zu 1CT7 s, entspricht, In Figur 6 ist das Cyclo-Voltagramm von BUPH1 in Acetonitril dargestellt. Die Messung wurde mit Pt/Pt Elektroden gegen eine Ag/AgCl Referenzelektrode mit einer Rate von 20 mV/s aufgenommen. Aus dem Oxidationspotential von BUPH1 (nach BUPH1+) von 1,43 V lässt sich die Energie des HOMO-Levels berechnen. Man erhält eine HOMO-Lage des BUPH1 von 5,82 eV.
A, Beispiele unter Einsatz des schweren Hauptgruppenmetalls Bi I. Charakterisierung der eingesetzten Metall-Komplexe
In Figur 7 sind die UV-Absorptionsspektren von Bi(tfa)3, Bi(pFBz)3 und Bi (3 , StfmBz) 3 dargestellt. Die Strukturformeln der Verbindungen sind weiter unten aufgeführt.
Die einzelnen Spektren zeigen eine starke n-n* und n-n* Absorption, welche auf die Carboxylat -Liganden und das Bi- Zentralatoin zurückgeführt werden kann. Die Bi -Carboxylate ab- sorbieren Licht nur in der UV-Region des Spektrums, deshalb erscheinen sie als weißer Festkörper. Der eingeschobene Graph zeigt eine Taue -Auftragung mit welcher der optische Bandabstand der einzelnen Verbindungen bestimmt werden kann. Die Graphen zeigen ein lineares Regime, welches den Startpunt der Absorption markiert. Extrapoliert man dieses lineare Regime zur Abzisse, so erhält man die Energie des optischen Bandabstandes. Für Bi(tfa)3, Bi(pFBz)3 und Bi (3 , StfmBz) 3 ergibt sich eine relativ große Bandlücke im Bereich von 4,46, 4,32 und 4,34 eV. Damit ergibt sich, dass die Bi-Carboxylate Isolator- Eigenschaften zeigen.
Die Figur 8 zeigt die Auftragung der molaren Extinktionskoeffizienten von Bi(tfa)3, Bi(pFBz)3 und Bi (3 , StfmBz) 3 in THF gegen die Wellenlänge. Bei 265 nrrt beträgt der Exintinktions- koeffizient für Bi(tfa)3, Bi(pFBz)3 und Bi (3 , StfmBz) 3 243, 3065 und 2200 Lmol^cm"1. Im Falle des Bi(pFBz)3 und des
Bi {3 , StfmBz) 3 sind die n-π* und die n-n* Übergänge erlaubt, welches zu Extinktionskoeffizienten von über 1000 Lmol^crrf1 führt .
II. Herstellung der Schichten
Ein vorgefertigtes Quarzsubstrat wird für 10 Minuten mittels eines Sauerstoffplasmas behandelt und schnellstmöglich in einen Evaporator überführt, welcher sich innerhalb einer Argon gefüllten Glovebox mit einem Wassergehalt kleiner 2 ppm be- findet.
Das thermische Verdampfen erfolgt bei einem Basisdruck kleiner als 2xl0~6 mbar, welcher während des gesamten Bedamp- fungsschrittes beibehalten wird,
Der Metallkomplex und der organische Emitter werden gleichzeitig auf eine Temperatur kurz unterhalb ihres Verdampfungs- punktes aufgeheizt. Anschließend wird der Metallkomplex solange weiter aufgeheizt, bis eine konstante Verdampfungsrate erzielt ist. Ebenso verfährt man mit dem organischen Emitter und bei beidseitig konstanten Verdampfungsratenraten wird der Schieber des Evaporators geöffnet.
Die Abscheiderate beider Substanzen wird auf 1 Ä/s einge- stellt, wobei die Konzentration des Bismuth- Komplexes in Abhängigkeit zum gewünschten Bi :BUPH1 -Verhältnis eingeregelt wird, z.B. eine Konzentration von 50% wird mit einer BUPH1 Abscheiderate von 0,5 Ä/s und einer Bi-Abscheiderate von 0,5 Ä/s erreicht. Dies entspricht einem Verhältnis von 1:1.
Nach erfolgter Bedampfung werden beide Quellen auf unter 40 °C herab gekühlt und der Verdampfer mit trockenem Argon geflutet. Es wurden eine Reihe Bi : BUPH1-Filme mit unterschiedlicher Zusammensetzung über das oben beschriebene Ko-Evaporations- verfahren hergestellt. Damit wurden 200 nm dicke Bi:BUPHl- Filme als Emitterschichten auf einem Quarzglas abgeschieden. Es wurden folgende Verhältnisse eingestellt (die Abkürzungen von Bismuth (III) trifluoroacetat Bi(tfa)3 und die von
Bi (III) Pentafluorobenzoat Bi(pFBz}3) :
Verbindung Verhältnis
Bi(tfa)3 : BUPHl 1 1
Bi(tfa)3 : BUPHl 1 2
Bi(tfa)3 : BUPHl 1 3
Bi(tfa)3 : BUPHl 1 4
Bi{tfa}3 : BUPHl 0 1
Bi{tfa)3 : BUPHl 3 1
Bi (pFBz) 3 : BUPHl 1 1
Bi (pFBz) 3 : BUPHl 1 2
Bi (pFBz) 3 : BUPHl 1 3
III. Charakterisierung der durch Ko-Evaporation hergestellten Schichten
Ill.a UV-Absorption
Die Figur 9 zeigt die UV-Absorptionsspektren von BUPHl, Bi(tfa)3 und mit unterschiedlich Abscheidungsraten durch Ko- Evaporation hergestellter Bi (tfa) 3 : BUPHl-Schichten . Die
Schichtdicke beträgt 200 nm. Es sind für die Filme zwei Haupt-Absorptionsbanden sichtbar. Die dominierende Absorptionsbande liegt zwischen 230-350 nm und kann einem Spinerlaubten n-n* Übergang am BUPHl zugeordnet werden. Im Vergleich zu dem reinen BUPHl-Film fehlt in den Komposit-
Schichten (Metall + Emitter) der n-n* Übergang des BUPHl bei 335 nm. Dies deutet auf eine Koordination der freien Stickstoff Elektronenpaare an das Bismut hin. Daraus folgt, dass das BUPHl unter Bildung eines Adduktes oder Komplexes mit dem Bismuth wechselwirkt. Desweiteren zeigt das Absorptionsspektrum der Komposit-Filme eine niedrig liegende Absorptionsbande von 350 nm - 500 nm, welche bis in den sichtbaren Bereich hineinreicht. Diese Bande kann wahrscheinlich einem
intraliganden Ladungstransfer zugeordnet werden,
In Figur 10 sind die UV-Absorptionsspektren von BUPH1 und mit unterschiedlich Abscheidungsraten durch Ko-Evaporation hergestellter Bi (pFBz) 3 : BUPH1-Schichten dargestellt. Die Schichtdicke beträgt jeweils 200 nm. Im Vergleich zu dem reinen BUPHl-Film fehlt in den Komposit-Schichten der n-n* Übergang des BUPH1 bei 335 nm. Dies deutet auf eine Koordination des freien Stickstoff Elektronenpaares an das Bismut hin. Daraus folgt, dass das BUPH1 unter Bildung eines Adduktes oder Komplexes an das Bismuth bindet/koordiniert. Desweiteren zeigt das Absorptionsspektrum der Komposit -Filme eine niedrig liegende Absorptionsbande von 350 nm - 500 nm, welche bis in den sichtbaren Bereich hineinreicht. Diese Bande kann wahrscheinlich einem intraliganden Ladungstransfer zugeordnet werden. Ill.b Photolumineszenz
Figur 11 zeigt das normierte Photolumineszenzspektrum von BUPH1 und mit unterschiedlich Abscheidungsraten durch Ko- Evaporation hergestellter Bi (tfa) 3 : BUPH1 -Schichten. Die
Schichtdicke betrug jeweils 200 nm. Die Emissionsspektren sind auf die maximale Intensität des BUPH1- Films hin normiert. Das Emissionsmaximum liegt für BUPH1 bei 420 und für die Komposit-Schichten bei 585 nm. BUPHl-Film Xex: 365 nm, Xem 420 nm; Bi (tfa ) 3 : BUPH1 = 1:1 Äex: 410 nm, Xem 585 nm;
Bi (tfa) 3:BUPH1 = 1:2 Xex: 410 nm, Xem 580 nm; Bi (tfa ) 3 : BUPH1 = 1:3 Xex: 410 nm, Xem 585 nm; Bi { tfa) 3 : BUPH1 = 1:4 Xex: 410 nm, Xem 585 nm; Bi ( tfa) 3 : BUPH1 = 3:1 Xex: 410 nm, Xem 585 nm. Im Vergleich mit dem Tieftemperatur-Emissionsspektrum von BUPH1, welche Emissionsmaxima bei 520, 550 und 5600 nm auf- wies, zeigt das Anlagerungsaddukt oder der -Komplex nur eine breite Phosphoreszenz-Emissionsbande bei Raumtemperatur.
Die Figur 12 zeigt das normierte Photolumineszenzspektrum von BUPH1 und mit unterschiedlich Abscheidungsraten durch Ko- Evaporation hergestellter Bi (pFBz) 3 : BUPH1-Schichten . Die
Schichtdicke beträgt jeweils 200 nm. BUPH1 Xex: 335 nm Xem 420 nm; Bi (pFBz ) 3 : BUPHl = 1:1 Xex: 395 nm, Xem 535 nm;
Bi (pFBz) 3 :BUPH1 = 1:2 Xex: 395 nm, Xem 542 nm;
Bi (pFBz) 3: BUPHl = 1:3 λβχ:395 nm, Aem 540 nm. Die omposit- Schichten zeigen eine maximale Emission bei 550 nm. Durch Vergleich mit Figur 11 ergibt sich, dass die Emission bei höheren Wellenlängen zwischen den unterschiedlichen Bi- Komplexen unterschiedlich ist. Dies wahrscheinlich aufgrund der Tatsache, dass die Metall-Liganden (Trifluoracetat oder Pentafluoracetat) die Emissionswellenlängen des erhaltenen Bi : BUPHl-Adduktes oder Komplexes beeinflussen können. Im Vergleich mit dem Tieftemperatur-Emissionsspektrum des BUPHl, welche Emissionsmaxima bei 520, 550 und 5600 nm aufwies, zeigt der Komplex nur eine breite Phosphoreszenz- Emissionsbande bei Raumtemperatur.
III.c Photolumineszenz Quantenausbeute (PLQY)
Die Photolumineszenz Quantenausbeute (PLQY) von Kompositschichten konnte nicht in solid State bestimmt werden. Aus diesem Grund wurden die Schichten von Bi { tfa) 3 : BUPHl 1:2 und Bi (pFBz) 3 : BUPHl 1:2 in Dichlormethan gelöst und die PLQY in Lösung gemessen. Die PLQY von Bi {tfa} 3 : BUPHl 1:2 beträgt 3,7% (Coumarin 153 in Ethanol als Referenz). Für Bi (pFBz) 3 : BUPHl liegt die PLQY bei 6% (Coumarin 153 in Ethanol als Referenz) . Diese Werte der PLQY liegen deutlich über den PLQY anderer Bismut Komplexe aus der Literatur, welche in der Regel unter- halb 1 % liegen. Beispielsweise genannt sei eine PLQY in Höhe von 0,2 % für Dithienobismol , erhalten nach der WO 2011
111621 AI.
Ill.d Zeitkorrelierte Einzelphotonenzählung TCSPC
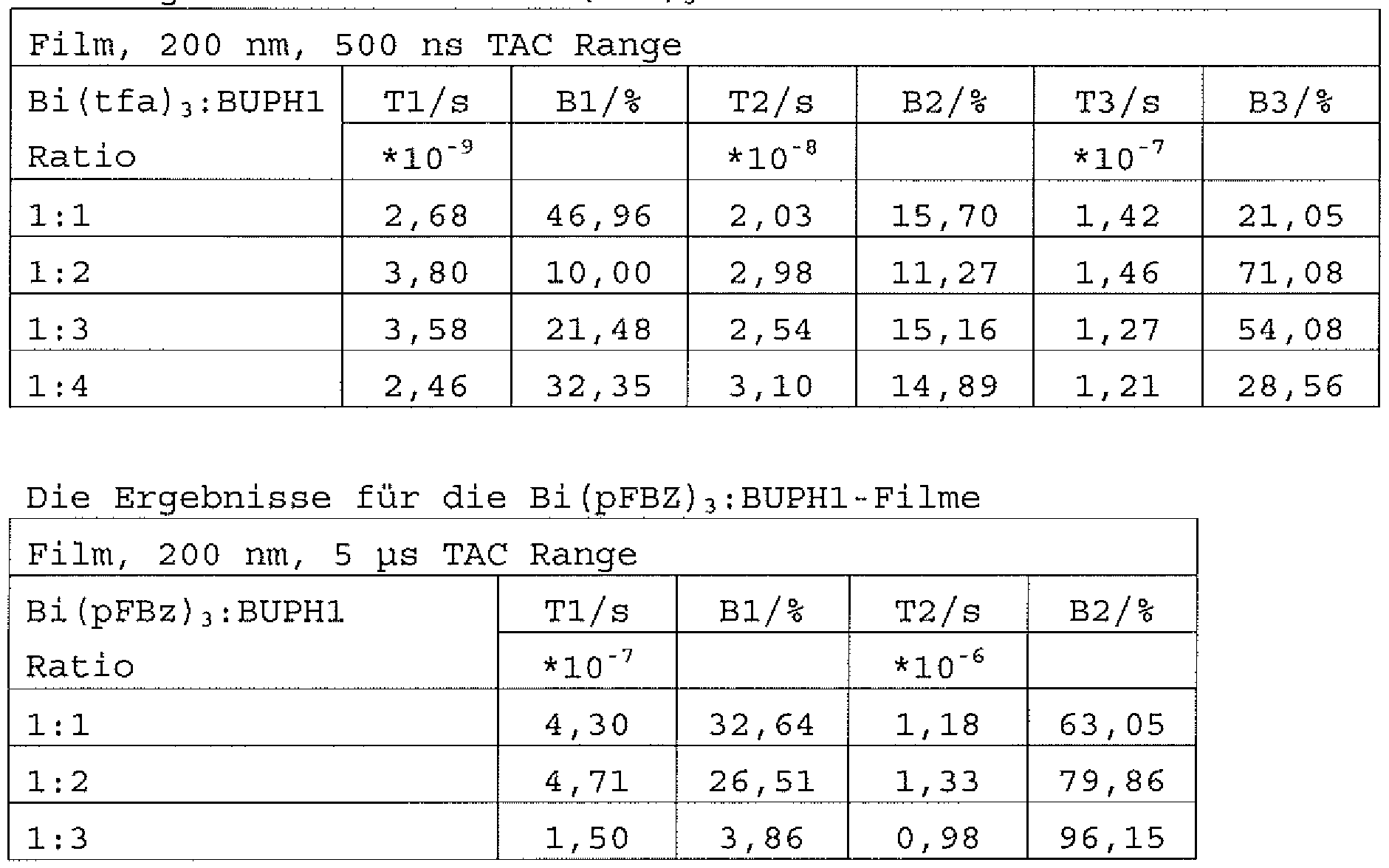
Die mit unterschiedlichen Zusammenset ungen abgeschiedenen Bi (tfa) 3 : BUPHl- und Bi (pFBz) 3 : BUPHl-Filme wurden mittels TCSPC-Messungen (time-correlated single photon counting) in einer inerten Atmosphäre untersucht. Bei Raumtemperatur lie- fert die TCSCP-Messung für die Bi : BUPHl-Filme komplexe Lebensdauern im ikrosekunden-Bereich. Dies ist ein klarer Hinweis für das Vorliegen eines Phosphoreszenzüberganges. Die
Ergebnisse der Messungen an den einzelnen Filmen sind in den folgenden Tabellen dargestellt.
Ergebnisse für die Bi {tfa} 3 : BUPHl-Filme
Wie aus den Graphen ersichtlich ist offenbar die Strahlungs- lebensdauer der Emitter in den Bi {tfa) 3 : BUPHl-Filmen im Ver- gleich zu den Emittern in den Bi (pFBz) 3 : BUPHl-Filmen etwas kürzer. Anscheinend führt der Einsatz von Bi{tfa)3 zu einer stärkeren Spin-Bahnkopplung des organischen Emitters, welches den Phosphoreszenz-Übergang des BUPH1 in diesem Komplex quantenmechanisch stärker erlaubt. Dies wiederum führt im Ver- gleich zu Bi(pFBz)3 zu einer kürzeren Strahlungslebensdauer. Der Lewis-Säurecharakter von Bi(tfa)3 ist im Vergleich zu Bi(pFBz)3 höher, was in diesem Beispiel zu einer verstärkten Wechselwirkung führt. Anzumerken ist weiterhin, dass die Strahlungslebensdauer der Emitter in den Bi (tfa) 3 : BUPHl-Filmen mittels einer tri- Exponentialfunktion, die der Bi (pFBz) 3 : BUPH1 Filme mit einer di-Exponentialfunktion, angepasst wurden. Dies kann darauf
hindeuten, dass ein ganzes Ensemble von Molekülen im Film aktiv ist und an der Emission teilnimmt.
Ill.e XRD-Spetrum
In Figur 13 ist das XRD-Spektrum eines Bi ( tfa) 3 : BUPH1 (1:1)- Films dargestellt. Die Schichtdicke des Bi (tfa) 3 : BUPH1 (1:1) Filmes beträgt 2 μττι. Der Film erzeugt im Röntgen- diffraktogramm nur einen breiten Kalo über einen weiten 2-
Theta Bereich, welches für eine amorphe Anordnung der Metallkomplex-Emitter-Verbindungen spricht. Dies bedeutet, dass die einzelnen phosphoreszenten Ensembles unregelmäßig, amorph, in der Schicht angeordnet sind. Dies kann darauf zurückzuführen sein, dass es sich um gemischt stöchiometrische Verbindungen im Film handelt oder, dass sich aufgrund der gewählten Herstellmethodik keine Fernordnung zwischen den einzelnen Emitter-Ensembles einstellen kann. B. Beispiele unter Einsatz des schweren Hauptgruppenmetalls
Sn
Als weiteres Beispiel von Emittern, die ein schweres Hauptgruppenmetall enthalten und bei Raumtemperatur Phosphoreszenz aufweisen werden Sn-Verbindungen eingesetzt. Als Metallausgangsstoff wurde Zinn(II) -Chlorid (SnCl2) verwendet. BUPH1 ist ein organisch fluoreszenter Emitter, welcher durch schwache elektrostatische und/oder π-Wechselwirkungen mit dem Schweratom wechselwirkt. Durch den Einfluss des Schwermetalls werden bisher spin-verbotene elektronische Übergänge von
BUPH1 quantenmechanisch erlaubt und es wird ein nennenswerter Phosphoreszenz-Beitrag bei Raumtemperatur erhalten. Diese Ergebnisse zeigen die prinzipielle Eignung dieser Verbindungsklasse auch für den Einsatz in Schichten organisch elektri- scher Bauelemente.
I. Herstellung von Sn-BUPHl-Komposit Lösungen in THF
In 3 ml Tetrahydrofuran (THF) Lösung lässt man SnCl2 und BUPH1 in einem Verhältnis von 1:1, 1:2 oder 1:3 reagieren und untersucht diese Lösung dann spektroskopisch. Es wurden 5 μΐ SnCl2 (10~2 M in THF) und 5 μΐ BUPH1 (10~2 M in THF) in 3 ml THF eingesetzt, um ein Stoffmengenverhältnis von 1:1 zu erhalten.
II. Charakterisierung der hergestellten Lösungen
Il.a UV-Absorption
Die Figur 14 zeigt die UV-Absorptionsspektren von SnCl2-BUPHl in THF in unterschiedlichen Stoffmengenverhältnissen (1:1, 1:2 und 1:3). Die Absorptionsbanden in den Komposit-THF- Lösungen von SnCl2-BUPHl zwischen 250-375 nm sind identisch mit den optischen Übergängen von BUPH1 {vergleiche Fig. 3) in diesem Bereich. Die Intensitätserhöhung der Absorptionsbanden von SnCl2-BUPHl ist proportional zu der Konzentration an BUPHl in der Lösung. Interessanterweise zeigt das Absorptionsspektrum der Komposit-THF-Lösungen zusätzlich eine niedrig liegende Absorptionsbande im Bereich von 375 - 450 nm, welche bis in den sichtbaren Bereich hineinreicht. Dieser Bereich ist in Figur 15 vergrößert dargestellt. Diese Bande kann wahrscheinlich einem Intraliganden-Ladungstransfer des gebildeten BUPHl-Sn-Adduktes oder -Komplexes zugeordnet werden.
Il.b Photolumineszenz Die Figur 16 zeigt die PhotolumineszenzSpektren von SnCl2- BUPHl in THF Lösung in einem Stoffmengenverhältnis von 1:1, 1:2 und 1:3. Die Spektren wurden mit einer Wellenlänge von 410 nm angeregt, da wahrscheinlich bei dieser Wellenlänge der Intraligand-Ladungstransfer stattfindet, welcher für die Phosphoreszenz-Emissionsbande verantwortlich zeichnet. Die Komposit-THF-Lösungen zeigen eine maximale Emission bei 580 nm und die Intensität der Banden ändert sich kaum mit der Erhöhung der BUPHl Konzentration in Lösung. Ohne durch die The-
orie gebunden zu sein, wird die Emission hauptsächlich durch BUPH1 verursacht, welches aufgrund des Einflusses des Schwermetalls Zinn auf die spin-verbotenen elektronischen Übergänge zur Phosphoreszenz befähigt wird. Im Vergleich mit dem Tief- temperatur-Emissionsspektrum von BUPH1 (vgl. Fig. 4), welches Emissionsmaxima bei 520, 550 und 600 nm aufweist, zeigt das SnCl2-BUPHl Anlagerungsaddukt oder der -Komplex eine breite Phosphoreszenz -Emissionsbande bei Raumtemperatur. II . c Zeitkorrelierte Einzelphotonenzählung TCSPC
Die mit unterschiedlichen Stoffmengenverhältnissen hergestellten SnCl2-BüPHl Lösungen wurden mittels zeitkorrelierter Einzelphotonenzählung-Messungen in einer inerten Atmosphäre (Argon) untersucht. Bei Raumtemperatur liefert die TCSCP-
Messung für die SnCl2-BUPHl Anlagerungsaddukte oder -Komplexe Lebensdauern im Mikrosekunden-Bereich {Mengenverhältnis 1:1 in Figur 17, 1:2 in Figur 18 und 1:3 in Figur 19). Dies ist ein klarer Hinweis für das Vorliegen eines Phosphoreszenz- Überganges. Die Ergebnisse der Messungen an den einzelnen Proben sind in unten stehender Tabelle dargestellt.
Die Spektren wurden auf einer 5 Ξ Zeitskala gemessen, wobei die Phosphoreszenz -Lebensdauer der untersuchten Komposit-THF Lösungen sich im Bereich von 0,76 bis 1,12 Mikrosekunden bewegen.
C. Beispiele unter Einsatz des schweren Hauptgruppenmetalls Pb
Als weiteres Beispiel von Emittern, die ein schweres Hauptgruppenmetall enthalten und bei Raumtemperatur Phosphoreszenz aufweisen werden Pb-Verbindungen eingesetzt. Diese Ergebnisse zeigen die prinzipielle Eignung dieser Verbindungsklasse auch für den Einsatz in Schichten organisch elektrischer Bauelemente ,
I Herstellung von Pb-BUPHl-Komposit Lösungen in THF 3 ml THF Lösung lässt man PbTFA (Blei-Trifluoroacetat) und
BUPH1 in einem Verhältnis von 1:1, 1:2 oder 1:3 reagieren und untersucht die Lösung dann spektroskopisch. Es wurden 5 μΐ PbTFA (10~2 M in THF) und 5 μΐ BUPH1 (1CT2 M in THF) in 3 ml THF eingesetzt, um ein Stoffmengenverhältnis von 1:1 zu er- halten.
II. Charakterisierung der hergestellten Lösungen Il.a UV-Absorption
Die Figuren 20 und 21 zeigen die UV-VIS Absorptionsspektren von PbTFA-BUPHl in THF in einem Stoffmengenverhältnis von 1:1, 1:2 und 1:3, Die Absorptionsbanden in den Komposit-THF-Lösungen von PbTFA- BUPHl zwischen 250-375 nm sind identisch mit den optischen Übergängen von BUPH1 in diesem Bereich. Die Intensitätserhöhung der Absorptionsbanden von PbTFA-BUPHl ist proportional zu der Konzentration von BUPH1 in der Lösung. Interessanter- weise zeigt das Absorptionsspektrum der Komposit-THF-Lösungen zusätzlich eine niedrig liegende Absorptionsbande im Bereich von 375 nm - 415 nm (Figur 21) , welche sich bis in den sichtbaren Bereich erstreckt. Diese Bande kann wahrscheinlich einem intraliganden Ladungstransfer des gebildeten BUPHl-Pb- Adduktes oder -Komplexes zugeordnet werden.
Il.b Photolumineszenz
Figur 22 zeigt das Photolumineszenz -Spektrum von PbTFA-BUPHl in THF Lösung mit einem Stoffmengenverhältnis von 1:1, 1:2 und 1:3. Die Verbindungen wurden mit einer Wellenlänge von 410 nm angeregt, da wahrscheinlich bei dieser Wellenlänge der Intraligand-Ladungstransfer stattfindet, welcher für die Phosphoreszenz -Emissionsbande verantwortlich zeichnet. Die Komposit-THF-Lösungen zeigen eine maximale Emission bei 536 nm und die Intensität der Banden ändert sich kaum mit der Erhöhung der BUPH1 Konzentration in Lösung, Ohne durch die The- orie gebunden zu sein, wird die Emission hauptsächlich durch BUPH1 verursacht, welches aufgrund des Einflusses des Schwer¬ metalls Blei auf die spin-verbotenen elektronischen Übergänge zur Phosphoreszenz befähigt wird. Im Vergleich mit dem Tief- temperatur-Emissionsspektrum von BUPH1 (siehe Figur 3) , wel- che Emissionsmaxima bei 520, 550 und 600 nm aufweist, zeigt das PbTFA-BUPHl Anlagerungsaddukt oder der -Komplex eine breite Phosphoreszenz -Emissionsbande bei Raumtemperatur.
II.c Zeitkorrelierte Einzelphotonenzählung TCSPC
Die mit unterschiedlichen Stoffmengenverhältnissen hergestellten PbTFA-BUPHl Lösungen wurden mittels zeitkorrelierter Einzelphotonenzählung-Messungen in einer inerten Atmosphäre (Argon) untersucht. Bei Raumtemperatur liefert die TCSCP- Messung für die PbTFA-BUPHl Anlagerungsaddukte oder -Komplexe
Lebensdauern im Mikrosekunden-Bereich (Mengenverhältnis 1:1 in Figur 23, 1:2 in Figur 24 und 1:3 in Figur 25) . Dies ist ein klarer Hinweis für das Vorliegen eines Phosphoreszenz- Überganges. Die Ergebnisse der Messungen an den einzelnen Proben sind in unten stehender Tabelle dargestellt.
PbTFA : BUPH1
Stoffmengenverhältnis Tl/ ps
1:1 2,61
1:2 9, 58
1:3 52, 56
Die Spektren wurden auf einer 20 ps Zeitskala gemessen, wobei die Phosphoreszenz-Lebensdauern der untersuchten Komposit-THF Lösungen sich im Bereich von 2,61 bis 52,56 Mikrosekunden bewegen.
Obwohl die Erfindung im Detail durch das bevorzugte Ausführungsbeispiel näher illustriert und beschrieben wurde, so ist die Erfindung nicht durch die offenbarten Beispiele eingeschränkt und andere Variationen können vom Fachmann hieraus abgeleitet werden, ohne den Schutzumfang der Erfindung zu verlassen .