WO2014167966A1 - ラクチド-ラクトン共重合体の製造方法 - Google Patents
ラクチド-ラクトン共重合体の製造方法 Download PDFInfo
- Publication number
- WO2014167966A1 WO2014167966A1 PCT/JP2014/057531 JP2014057531W WO2014167966A1 WO 2014167966 A1 WO2014167966 A1 WO 2014167966A1 JP 2014057531 W JP2014057531 W JP 2014057531W WO 2014167966 A1 WO2014167966 A1 WO 2014167966A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- lactide
- group
- complex
- lactone
- dioxomolybdenum
- Prior art date
Links
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/78—Preparation processes
- C08G63/82—Preparation processes characterised by the catalyst used
- C08G63/823—Preparation processes characterised by the catalyst used for the preparation of polylactones or polylactides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/02—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds
- C08G63/06—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds derived from hydroxycarboxylic acids
- C08G63/08—Lactones or lactides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/66—Polyesters containing oxygen in the form of ether groups
- C08G63/664—Polyesters containing oxygen in the form of ether groups derived from hydroxy carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/42—Polycondensates having carboxylic or carbonic ester groups in the main chain
- C08G18/4266—Polycondensates having carboxylic or carbonic ester groups in the main chain prepared from hydroxycarboxylic acids and/or lactones
- C08G18/4269—Lactones
- C08G18/4277—Caprolactone and/or substituted caprolactone
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/42—Polycondensates having carboxylic or carbonic ester groups in the main chain
- C08G18/4266—Polycondensates having carboxylic or carbonic ester groups in the main chain prepared from hydroxycarboxylic acids and/or lactones
- C08G18/428—Lactides
Definitions
- the present invention relates to a method for producing a lactide-lactone copolymer.
- polylactide which is a homopolymer of lactide (LA, a cyclic compound formed by dehydration condensation of two molecules of lactic acid), that is, polylactic acid, has 1) fast degradation and absorption in vivo, and 2) drug permeability. Low, 3) low biodegradability in soil, 4) hydrophilic properties, etc.
- poly ( ⁇ -caprolactone) which is a homopolymer of ⁇ -caprolactone (CL), which is one of lactones, is 1) slowly decomposed and absorbed in vivo, 2) has high drug permeability 3) It has high biodegradability in soil and 4) is hydrophobic.
- Patent Document 2 a carbon compound of molybdenum is used as a catalyst in a method for producing a homopolymer of lactone such as ⁇ -caprolactone.
- an LA-CL copolymer is produced using an aluminum-salen type complex (Al-salen) as a catalyst.
- Al-salen aluminum-salen type complex
- the ligand used for the catalyst requires a multi-step synthesis and is complicated to prepare. It is expensive and time consuming.
- the preparation of the Al-salen catalyst uses an ignitable water-free reagent, which is problematic in terms of safety.
- an Al-salen catalyst is also inferior in storage stability, preparation at the time of use is required.
- Patent Document 2 although a catalyst of molybdenum carbon compound is used in the production of a lactone homopolymer such as ⁇ -caprolactone, only a lactone such as ⁇ -caprolactone is described as a constituent monomer. Production of the polymer is not envisaged.
- One object of the present invention is to provide a simple method for producing a lactide-lactone copolymer in which the distribution of each monomer is controlled in the copolymer.
- Another object of the present invention is to provide a safe method for producing such a lactide-lactone copolymer.
- the present inventors have found that a lactide-lactone copolymer can be produced using a predetermined molybdenum compound as a catalyst, and the present invention has been completed. That is, the present invention is as follows.
- Item 1 A method for producing a lactide-lactone copolymer, wherein lactide and lactone are copolymerized using a molybdenum compound as a catalyst.
- Item 2 The method for producing a lactide-lactone copolymer according to Item 1, wherein the lactone is ⁇ -caprolactone.
- Item 3 The lactide-lactone copolymer according to Item 1 or 2, wherein the molybdenum compound is selected from a chelate compound of molybdenum, a polyoxomolybdenum salt, an alkoxide of molybdenum, a salt of molybdenum ion and an organic acid, or a carbonyl compound of molybdenum. Production method.
- Item 4 The method for producing a lactide-lactone copolymer according to Item 1 or 2, wherein the molybdenum compound is a hexavalent molybdenum complex.
- Item 5 The method for producing a lactide-lactone copolymer according to Item 4, wherein the hexavalent molybdenum complex is a dioxomolybdenum (VI) complex or a polyoxomolybdenum salt.
- the hexavalent molybdenum complex is a dioxomolybdenum (VI) complex or a polyoxomolybdenum salt.
- Item 6. The lactide according to Item 5, wherein the dioxomolybdenum (VI) complex is selected from a dioxomolybdenum (VI) acetylacetonate complex, a dioxomolybdenum (VI) salen complex, or a dioxomolybdenum (VI) salicylaldehyde complex.
- a method for producing a lactone copolymer is selected from a dioxomolybdenum (VI) acetylacetonate complex, a dioxomolybdenum (VI) salen complex, or a dioxomolybdenum (VI) salicylaldehyde complex.
- the dioxomolybdenum (VI) salen complex is an oxomolybdenum (VI) salen complex represented by the following general formula (1):
- R 1 is a divalent aliphatic hydrocarbon group having 2 to 7 carbon atoms
- R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 and R 9 are independently hydrogen, an alkyl group having 1 to 4 carbon atoms, an alkoxy group having 1 to 4 carbon atoms, Item 7.
- R 1 represents — (CH 2 ) 2 —, — (CH 2 ) 3 —, —CH 2 CH (CH 3 ) —, —CH 2 —C (CH 3 ) 2 —CH 2 —, — (CH 2 ) 4 -, or 1,2-cyclohexyl, 1,3-cyclohexyl, 1,4-cyclohexyl, 1,2-cyclopentyl group, a 1,2-cycloheptyl group
- R 2, R 4, R 7 , R 9 is hydrogen or —CH 3
- R 5 and R 6 are hydrogen, —CH 3 , —CH 2 CH 3 , —C (CH 3 ) 3 , —OCH 3 , —Cl, —Br, or — Item 8 wherein R 3 and R 8 are hydrogen, —CH 3 , —CH 2 CH 3 , —C (CH 3 ) 3 , —OCH 3 , —Cl, —Br, or —Item 8
- the dioxomolybdenum (VI) salicylaldehyde complex is a dioxomolybdenum (VI) salicylaldehyde complex represented by the following structural formula (6):
- R 1 , R 2 , R 3 and R 4 are independently hydrogen, —Cl, —Br, —I, —NO 2 , —OH, —COOH, linear or branched having 1 to 6 carbon atoms.
- R 1 , R 2 , R 3 and R 4 are each independently hydrogen, —Cl, —Br, —I, —OCH 3 , —OCH 2 CH 3 , —NO 2 , —OH, —COOH, —CH 3, or lactide according to claim 9 is -CH 2 CH 3 - method of manufacturing a lactone copolymer.
- the polyoxomolybdenum salt includes [Mo 3 O 10 ] 2 ⁇ , [Mo 6 O 20 ] 4 ⁇ , [Mo 7 O 24 ] 6 ⁇ , [Mo 8 O 26 ] 4 ⁇ , and [Mo 10 O 34 ].
- Item 6 A lactide-lactone copolymer according to Item 5, which is a salt of an anion selected from the group consisting of 8- and a cation selected from the group consisting of ammonium ion, alkylammonium ion and alkali metal ion. Method.
- the catalyst molybdenum compound can be produced by a simple process, or a commercially available product can be used. Molybdenum is also an essential trace metal in living organisms and has low toxicity or nontoxicity. Therefore, it is possible to produce a lactide-lactone copolymer simply, inexpensively and / or safely. In addition, the molecular weight and molecular weight distribution of the lactide-lactone copolymer and the distribution of each monomer in the copolymer can be easily controlled.
- the present invention provides a method for producing a lactide-lactone copolymer, wherein lactide and lactone are copolymerized using a molybdenum compound as a catalyst.
- monomers lactide and lactone commercially available products may be used, or they may be synthesized by known methods.
- the lactide may be LL-lactide, DD-lactide, DL-lactide (mesolactide), or a mixture of one or more thereof, and LL-lactide is particularly preferable from the viewpoints of economy and practicality.
- the lactone is a lactone which is unsubstituted or substituted with an alkyl group, for example, ⁇ -valerolactone (VL), ⁇ -ethyl- ⁇ -valerolactone, ⁇ -caprolactone (CL), ⁇ -methyl- ⁇ -caprolactone, ⁇ -methyl- ⁇ -caprolactone, ⁇ -methyl- ⁇ -caprolactone, ⁇ , ⁇ -dimethyl- ⁇ -caprolactone, 3,3,5-trimethyl- ⁇ -caprolactone, enanthlactone (7-heptanolide), and dodecanolactone (12-dodecanolide). More preferably, the lactone is ⁇ -valerolactone (VL), ⁇ -caprolactone (CL), or one or more mixtures thereof. Particularly preferably, the lactone is ⁇ -caprolactone (CL).
- the catalyst used in the method for producing a lactide-lactone copolymer of the present invention is a molybdenum compound.
- the molybdenum compound comprises (i) a chelate of molybdenum, (ii) a polyoxomolybdenum salt, (iii) an alkoxide of molybdenum, (iv) a salt of molybdenum ions and organic acids, and (v) a carbonyl of molybdenum. Selected from compounds.
- the chelate compound of molybdenum is a molybdenum metal chelate compound of molybdenum and acetylacetone, benzoylacetone, trifluoroacetylacetone, ethyl acetoacetate, salicylaldehyde, acetylacetoneimine, salicylaldehydeimine and the like.
- the chelate compound of molybdenum includes a dioxomolybdenum (VI) complex described later.
- the polyoxomolybdenum salt is a salt composed of an anion polyoxomolybdenum ion (polyacid) and a cation, and preferably [Mo 3 O 10 ] 2 ⁇ , [Mo 6 O 20 ] 4 ⁇ , [ An anion selected from the group consisting of Mo 7 O 24 ] 6 ⁇ , [Mo 8 O 26 ] 4- , and [Mo 10 O 34 ] 8-, and a group consisting of ammonium ions, alkylammonium ions, and alkali metal ions It is a salt with a more selected cation.
- the alkoxide of molybdenum is represented by the general formula Mo (OR) 4 , and R represents a residue obtained by removing OH from an alcohol, such as an alkyl group such as methyl, ethyl, propyl, or butyl.
- organic acids in the case of salts of molybdenum ions and organic acids are carboxylic acids, especially naphthenic acid, citric acid, oxalic acid, benzoic acid, etc., and the resulting salts are molybdenum naphthenate, molybdenum citrate, A method for producing such a salt of molybdenum ion and organic acid is described in, for example, JP-A-62-219990.
- the carbonyl compound of molybdenum is a molybdenum compound containing a carbonyl group such as Mo (CO) 6 , (CO) 5 Mo (C 5 H 5 ), and (C 5 H 5 ) Mo (CO) 3 H.
- the molybdenum compound is a hexavalent molybdenum complex, preferably a hexavalent molybdenum complex having ring-opening polymerization activity.
- Hexavalent molybdenum has many application examples as a catalyst such as a ring-opening metathesis catalyst, and is stable and easily available.
- the hexavalent molybdenum complex is preferably a dioxomolybdenum (VI) complex or a polyoxomolybdenum salt.
- dioxomolybdenum (VI) complex examples include a dioxomolybdenum (VI) acetylacetonate complex (MoO 2 (acac) 2 ) known as a ring-opening polymerization catalyst, a bidentate ligand, a tridentate ligand, Examples include dioxomolybdenum (VI) complexes obtained by reacting various coordination ligands such as tetradentate ligands with MoO 2 (acac) 2 .
- the MoO 2 (acac) 2 complex itself is a complex having a bidentate ligand.
- a dioxomolybdenum (VI) complex using a bidentate ligand and a synthesis method thereof are disclosed in, for example, JP-A-2006-50064; A.0064Sakakura et al., Adv. Synth. Catal. 349,6461641-1646, 2007; Sakakura et al., 65, 2102-2109, 2009; M. Gomez et al., Eur. J. Inorg. Chem., 1071-1076, 2001.
- Dioxomolybdenum (VI) complexes using tridentate ligands and methods for their synthesis are described, for example, in L. Casella, et al., Inorg. Chim. Acta, 14,89-97, 1988; Y. Li, et al Chem. Commun., 1551-1552, 2000; H. Zhang et al., Inorg. Chem. 45,1745-1753, 2006; Fr.Demande, Patent, 43, 2003.
- Dioxomolybdenum (VI) complexes using tetradentate ligands and methods for their synthesis are described, for example, in K. Yamanouchi et al., Inorg. Chim. Acta, 9, -161-164, 1974; W. E. Hill, et al., Inorg. Chim. Acta, 35, 35-41, 1979; 41Behzad Zeynizadeh et al., Bull. Chem. Soc. Jpn, 78, 307-315, 2005; E. Y. Tshuva et al., Organometallics, 20, 3017-3028, 2001; Y. Wong, et al., Dalton Trans, 39, 4602-4611, 2010.
- the dioxomolybdenum (VI) complex is dioxomolybdenum (VI) acetylacetonate complex (MoO2 (acac) 2), dioxomolybdenum (VI) salen complex, dioxomolybdenum (VI) salicylaldehyde complex , Dioxomolybdenum (VI) imine complex, or dioxomolybdenum (VI) tropolone complex, preferably dioxomolybdenum (VI) acetylacetonato complex, dioxomolybdenum (VI) salen complex, or dioxomolybdenum (VI ) Salicylaldehyde complex.
- silica means N, N′-Bis (salicylidene) ethylene-1,2-diamine, its homologues, and Schiff base ligands that are derivatives thereof.
- the dioxomolybdenum (VI) salen complex is represented by the following general formula (1).
- R 1 is a divalent aliphatic hydrocarbon group having 2 to 7 carbon atoms
- R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 and R 9 are independently hydrogen (—H), an alkyl group having 1 to 4 carbon atoms, or 1 to 4 carbon atoms.
- alkyl group having 1 to 4 carbon atoms examples include methyl group, ethyl group, propyl group, isopropyl group, n-butyl group, sec-butyl group, isobutyl group, and t-butyl group.
- aryl group examples include an aryl group having 6 to 18 carbon atoms, particularly a phenyl group.
- R 1 is — (CH 2 ) 2 —, — (CH 2 ) 3 —, —CH 2 CH (CH 3 ) —, —CH 2 —C (CH 3 ) 2 —CH 2 —, — (CH 2) 4 -, 1,2 cyclohexyl, 1,3-cyclohexyl, 1,4-cyclohexyl, 1,2-cyclopentyl, or 1,2 cycloheptyl group.
- R 2 , R 3 , R 4 , R 7 , R 8 , and R 9 are independently hydrogen, an alkyl group having 1 to 4 carbon atoms, —OCH 3 , —Cl, —Br, or — I.
- R 5 and R 6 are independently hydrogen, an alkyl group having 1 to 4 carbon atoms, a phenyl group, a triisopropylsilyl group, a triisobutylsilyl group, a triphenylsilyl group, —OCH 3 , —Cl. , -Br, or -I.
- R 2 and R 9 , R 3 and R 8 , R 4 and R 7 , and R 5 and R 6 are the same.
- R 1 is — (CH 2 ) 2 —, — (CH 2 ) 3 —, —CH 2 —C (CH 3 ) 2 —CH 2 —, or — (CH 2 ) 4 —
- R 2 , R 4 , R 7 , R 9 are hydrogen or —CH 3
- R 5 and R 6 are hydrogen, —CH 3 , —CH 2 CH 3 , —C (CH 3 ) 3 , —OCH 3 , —Cl, —Br, or —I
- R 3 and R 8 are hydrogen, —CH 3 , —CH 2 CH 3 , —C (CH 3 ) 3 , —OCH 3 , —Cl, —Br, or — I.
- the dioxomolybdenum (VI) salen complex is represented by the following formula (1a).
- R 1 is — (CH 2 ) 2 —, — (CH 2 ) 3 —, —CH 2 —C (CH 3 ) 2 —CH 2 —, or — (CH 2 ) 4 —
- R 5 and R 6 Is hydrogen, —CH 3 , —CH 2 CH 3 , —C (CH 3 ) 3 , —OCH 3 , —Cl, —Br, or —I
- R 3 and R 8 are hydrogen, —CH 3 , — CH 2 CH 3 , —C (CH 3 ) 3 , —OCH 3 , —Cl, —Br, or —I.
- the dioxomolybdenum (VI) salen complex can be synthesized by a known method. For example, K. Yamanouchi et al., Inorg. Chim. Acta, 9, 161-164, 1974 or WE Hill, et al., Inorg. Chim. Acta, 35, 35-41, 1979 etc.
- a ligand (4) was synthesized from diamine (2) and salicylaldehyde derivative (3) as shown in the following chemical formula, and a certain amount, but not limited, of 1 to 4 equivalents of MoO is usually used for this ligand (4).
- the dioxomolybdenum (VI) salen complex produces cis- ⁇ and / or cis- ⁇ stereoisomers by appropriately changing the solvent, but cis- ⁇ is more advantageous in terms of the high ring-opening polymerization activity. preferable.
- the produced complex is identified by a known method such as NMR.
- the dioxomolybdenum (VI) salicylaldehyde complex is represented by the following general formula (6).
- R 1 , R 2 , R 3 and R 4 are independently hydrogen, —Cl, —Br, —I, —NO 2 , —OH, —COOH, linear or branched having 1 to 6 carbon atoms.
- alkyl group examples include methyl group, ethyl group, propyl group, isopropyl group, n-butyl group, sec-butyl group, isobutyl group, t-butyl group, propyl group, or pentyl group, preferably methyl group , Ethyl group, propyl group, or isopropyl group.
- the aryl group is preferably a 5- to 7-membered aryl group, and the aryloxy group is a 5- to 7-membered aryloxy group.
- the alkoxy group is preferably an alkoxy group having 1 to 6 carbon atoms, more preferably 1 to 4 carbon atoms, and further preferably a methoxy group (—OCH 3 ) or an ethoxy group (—OCH 2 CH 3 ). .
- the alkanesulfonamide group is preferably an alkanesulfonamide group in which the alkyl chain has 1 to 6 carbon atoms.
- R 1 , R 2 , R 3 and R 4 are each independently hydrogen, —Cl, —Br, —I, —OCH 3 , —OCH 2 CH 3 , —NO 2 , —OH, — COOH, —CH 3 , or —CH 2 CH 3 .
- R 1 is hydrogen, —CH 3 , or —CH 2 CH 3
- R 2 is hydrogen, —Cl, —Br, —I, —OCH 3 , —OCH 2 CH 3 , —NO. 2 , —CH 3 , or —CH 2 CH 3
- R 3 is hydrogen, —CH 3 , or —CH 2 CH 3
- R 4 is hydrogen, —OCH 3 , —OCH 2 CH 3 , —CH 3 or —CH 2 CH 3 .
- Dioxomolybdenum (VI) salicylaldehyde complex has been synthesized as a reaction intermediate of many complexes such as imine complexes (K. Yamanouchi et al., Inorg. Chim. Acta, 9, 83-86, 1974). As shown in the reaction formula, a precipitate formed by neutralizing ammonium molybdate (7) and salicylaldehyde derivative (8) with hydrochloric acid in methanol-H 2 O can be obtained by filtration (product (6 )). The formed complex is identified by a known method such as CHN elemental analysis.
- the dioxomolybdenum (VI) imine complex is synthesized by adding an amine to an ethanol solution of MoO 2 [(3-OMe) salad] 2 complex and refluxing for 30 minutes. See, for example, K. Yamanouchi, et al., Inorg. Chim. Acta, 1974, 9, 83-86.
- the dioxomolybdenum (VI) tropolone complex is synthesized by adding an ethanol solution of tropolone to an aqueous solution of ammonium molybdate and then stirring for 30 minutes in an acidified system by adding sulfuric acid. See for example WPGriffith, et al., Polyhedron, 6 (5), 891, 1987.
- the polyoxomolybdenum salt is preferably [Mo 3 O 10 ] 2 ⁇ , [Mo 6 O 20 ] 4 ⁇ , [Mo 7 O 24 ] 6 ⁇ , [Mo 8 O 26 ] 4 ⁇ , and [Mo 10 O 34].
- a salt of an anion selected from the group consisting of 8- and a cation selected from the group consisting of ammonium ion, alkylammonium ion and alkali metal ion, both of which are known and produced by known methods Is possible.
- the polyoxomolybdenum salt is a salt of the above anion and ammonium ion, and particularly preferably the polyoxomolybdenum salt is (NH 4 ) 8 [Mo 10 O 34 ].
- (NH 4 ) 8 [Mo 10 O 34 ] is obtained by thermally decomposing ammonium molybdate (NH 4 ) 6 Mo 7 O 24 ⁇ 4H 2 O at 150 ° C. for 3 hours under reduced pressure. See, for example, J. E. Baez et al., Polymer, 46, 12118-12129, 2005.
- an initiator may be further added. Even if there is no initiator, copolymerization occurs with atmospheric moisture as an initiator, but the polymerization degree and reaction can be controlled by adding the initiator.
- the initiator include metal alkoxides (see, for example, JP-A-2001-2763) or alcohols.
- alcohols include aliphatic alcohols including higher alcohols having 6 or more carbon atoms having a high boiling point such as lauryl alcohol, cyclododecanol. And aromatic alcohols such as alicyclic alcohol or benzyl alcohol, and polyhydric alcohols.
- the polymerization mode in the case of copolymerizing lactide and lactone using the above molybdenum compound as a catalyst is not particularly limited.
- any of solution polymerization method, slurry polymerization method, and bulk polymerization method is adopted.
- the organic solvent used in this case include aromatic hydrocarbons such as toluene, benzene, and xylene, chlorine-based hydrocarbons such as chloroform and trichloroethylene, and THF.
- the amount of the catalyst used for the polymerization varies depending on the kind of the solvent, the conditions under which the polymerization reaction is performed, etc., but is generally in the range of 0.0005 to 1.0 mol% of the raw material monomer, and usually 0.001 to 0.00. Even if it is about 1 mol%, a polymer can be obtained in a short time.
- the temperature at which the copolymer production method according to the present invention is carried out is appropriately determined according to the type or amount of the catalyst and monomer used, or the molecular weight of the polymer to be produced, etc., but generally the reaction temperature is 50 ° C. ⁇ 200 ° C is preferred.
- the reaction time of the polymerization reaction in the method for producing a copolymer according to the present invention is also appropriately determined according to the type or amount of the catalyst and monomer used, or the molecular weight of the polymer to be produced. Minute to several days, preferably 5 minutes to 3 days, and it is practical that the reaction is completed within 2 days (48 hours).
- the copolymer obtained by the production method of the present invention includes any copolymer, and among them, a random copolymer is advantageously obtained.
- the term “random copolymer” refers to a monomer charge composition different from an alternating copolymer, a periodic copolymer, a block copolymer, and a graft copolymer among the copolymers ( It means a copolymer in which the monomers are arranged randomly according to the molar ratio.
- lactide to lactone are copolymerized at a ratio of 1: 1, the average chain length of lactide (also referred to as average chain length) and the average chain length of lactone are both close to an ideal random copolymer.
- a lactide-lactone copolymer with an approximate distribution of lactide and lactone close to 2 in the polymer molecule is advantageously obtained.
- lactide and lactone are almost uniformly distributed in the polymer molecule means that in the lactide-lactone copolymer, both the average chain length of lactide and the average chain length of lactone are 1. It means 0 to 3.5.
- Their average chain length is preferably 1.5 to 3.0, more preferably 1.7 to 2.5.
- the arrangement of each monomer, and the molecular weight and molecular weight distribution of the copolymer are advantageously controlled.
- the random copolymer is not necessarily a copolymer strictly according to Bernoulli statistics.
- the average chain length of lactide and ⁇ -caprolactone is calculated in the literature (J. Kasperczyk et al., “Coodination polymerization of lactides, 4 The role of transesterification in the copolymerization of L, L-lactide and ⁇ -caprolactone ", Die Makromolekulare Chemie Volume 194, Issue 3, pages 913-925, March 1993).
- the average chain length of lactide: L LA is obtained from the following formula (A)
- the average chain length: L CL of ⁇ -caprolactone is calculated from the following formula (B).
- C represents an ⁇ -caprolactone unit in the copolymer
- LL represents a lactide unit in the copolymer
- [] represents the integrated intensity of the 13 C NMR corresponding peak of each triad.
- the molecular weight and molecular weight distribution of the obtained copolymer and the distribution of each monomer in the copolymer can be easily controlled, and the obtained lactide-lactone copolymer can be easily obtained.
- the coalescence can be used for biomedical material applications.
- the average molecular weight of the lactide-lactone copolymer can be controlled by the amount of the initiator, and is usually several thousand to several hundred thousand.
- the charged molar ratio of lactide and lactone can be appropriately set.
- the charged molar ratio of lactide and lactone is usually 1:99 to 99: 1, more preferably 10:90 to 90:10, for example, 25:75 to 75:25.
- a polymer is advantageously obtained.
- the molar ratio of lactide to lactone in the obtained copolymer is also usually 1:99 to 99: 1, more preferably 10:90 to 90:10, for example 25:75 to 75:25.
- the properties of the obtained polymer are evaluated by the following measuring method.
- the reaction rate of lactide and the reaction rate of lactone were determined by carrying out 1 H-NMR measurement on the extracted polymerization solution, and the residual amount of lactide and lactone obtained by this, the production amount of lactide and lactone (monomer conversion amount), and It is calculated from the ratio. Further, the content ratio of lactide and lactone in the copolymer is calculated from the result of 1 H-NMR measurement performed on the copolymer contained in the extracted polymerization solution.
- the number average molecular weight (Mn) and the molecular weight distribution (Mw / Mn) are calculated from the results of chromatography, for example, gel permeation chromatography or size exclusion chromatography, on the copolymer contained in the extracted polymerization solution.
- the average chain length of lactide and the average chain length of lactone were determined by using the result of 13 C-NMR measurement on the copolymer contained in the extracted polymerization solution, and the above Die Makromolekulare Chemie Volume 194, Issue 3, pages 913-925. , March 1993 based on the above formulas (A) and (B).
- NMR NMR was measured using a BRUKER DRX500 spectrometer (manufactured by Bruker). The solvent used was CDCl 3 and DMSO-d 6 containing 0.03 vol% TMS as a standard substance.
- GPC Gel permeation chromatography
- LC-10ADVP Shimadzu Corporation
- RID-10A Shimadzu Corporation
- column Shodex GPC KF-804L Showa Denko Co., Ltd.
- oven temperature 40 ° C THF Measurement was performed at a flow rate of 0.5 ml / min.
- a sample was prepared with 0.5 ml of THF for 1.8 mg of polymer, and 10 ⁇ l was injected for measurement.
- the molecular weight Mn, Mw and the molecular weight ratio Mw / Mn were calculated by preparing a calibration curve using polystyrene as a standard substance.
- DSC Differential scanning calorimeter
- diamine (2) was added to an ethanol solution of the corresponding salicylaldehyde derivative (3) so that the equivalent amount of the salicylaldehyde derivative was 2 equivalents to the diamine, and the reaction was carried out for 2 to 5 hours with heating under reflux. After completion of the reaction, the reaction mixture was ice-cooled, and the resulting precipitate was filtered, washed several times with ethanol, and dried to obtain the corresponding ligand (4) in Table 2.
- reaction rate and degree of polymerization were measured by NMR using CDCl 3 as a solvent.
- Example 4 Dioxomolybdenum salicylaldehyde complex According to the synthesis method described in K. Yamanouchi, S. Yamada, Inorg. Chim. Acta, 9, 83-86, 1974, a salicylaldehyde derivative and ammonium molybdate were converted into Methanol-H 2 Dissolve in O and slowly add 1 ml of 12M HCl. Then, it was made to react at room temperature for 4 hours, the produced
- Example 5 Measurement of Ring-Opening Polymerization Activity of Dioxomolybdenum Salicylaldehyde Complex
- the ring-opening polymerization activity of ⁇ -caprolactone (CL) was examined by reacting at 80 ° C. in mesitylene using cyclododecanol as an initiator, and found 15 for MoO 2 [(5-OMe) salad] 2. It was shown that 90% or more of the reaction progressed in about minutes, the highest activity, and the other complexes also almost completed the reaction in 30 minutes (Table 8).
- Example 7 Synthesis of ⁇ -CL-LA Copolymer with Various Molybdenum Catalysts MoO 2 [(5-OMe) salad] 2 , MoO 2 [(3-OMe) Di-Me-saltn] (cis- ⁇ type), Using four kinds of catalysts of MoO 2 (acac) 2 and (NH 4 ) 8 [Mo 10 O 34 ], lactide and ⁇ -caprolactone were copolymerized at a molar ratio of 50/50, and the obtained ⁇ -CL -The randomness of the LA copolymer was investigated.
- MoO 2 [(5-OMe) salad] 2 is obtained by the method described in K. Yamanouchi et al., Inorg. Chim. Acta, 1974, 9, 83-86, and MoO 2 [(3-OMe) Di-Me- [Saltn] was prepared by the method described in Example 1, and (NH 4 ) 8 [Mo 10 O 34 ] was prepared by the method described in J. E. Baez et al., Polymer, 46, 12118-12129, 2005.
- [LA + CL]: [Initiator]: [Catalyst] 100: 1: 0.05 (molar ratio), the initiator was cyclododecanol, the solvent was mesitylene, and polymerization was performed at 110 ° C. Yield, average chain lengths L LA and L CL of lactide and caprolactone are measured by 13 C-NMR, molecular weight and molecular weight ratio are measured by GPC, and glass transition temperature (Tg) is measured by differential scanning calorimeter (DSC). (Table 10). As a result, the average chain lengths L LA and L CL were both lower than 2 and no melting point (Tm) was shown in DSC in any of the catalysts. It was found that it was generated.
Abstract
Description
本願は、2013年4月12日に出願した特願2013-084129号明細書(その全体が参照により本明細書中に援用される)の優先権の利益を主張するものである。
本発明は、ラクチド-ラクトン共重合体の製造方法に関する。
すなわち、本発明は以下の通りである。

R1は、炭素数2~7の二価の脂肪族炭化水素基であり、
R2、R3、R4、R5、R6、R7、R8及びR9は、独立して、水素、炭素数が1~4のアルキル基、炭素数1~4のアルコキシ基、シリル基、アリール基、メトキシメチル基、-Cl、-Br、又は-Iである、項6に記載のラクチド-ラクトン共重合体の製造方法。

ジオキソモリブデン(VI)サレン錯体は、下記の一般式(1)で表される。

R1は、炭素数2~7の二価の脂肪族炭化水素基であり、
R2、R3、R4、R5、R6、R7、R8及びR9は、独立して、水素(-H)、炭素数が1~4のアルキル基、炭素数1~4のアルコキシ基、シリル基、アリール基、メトキシメチル基、-Cl、-Br、又は-Iである。
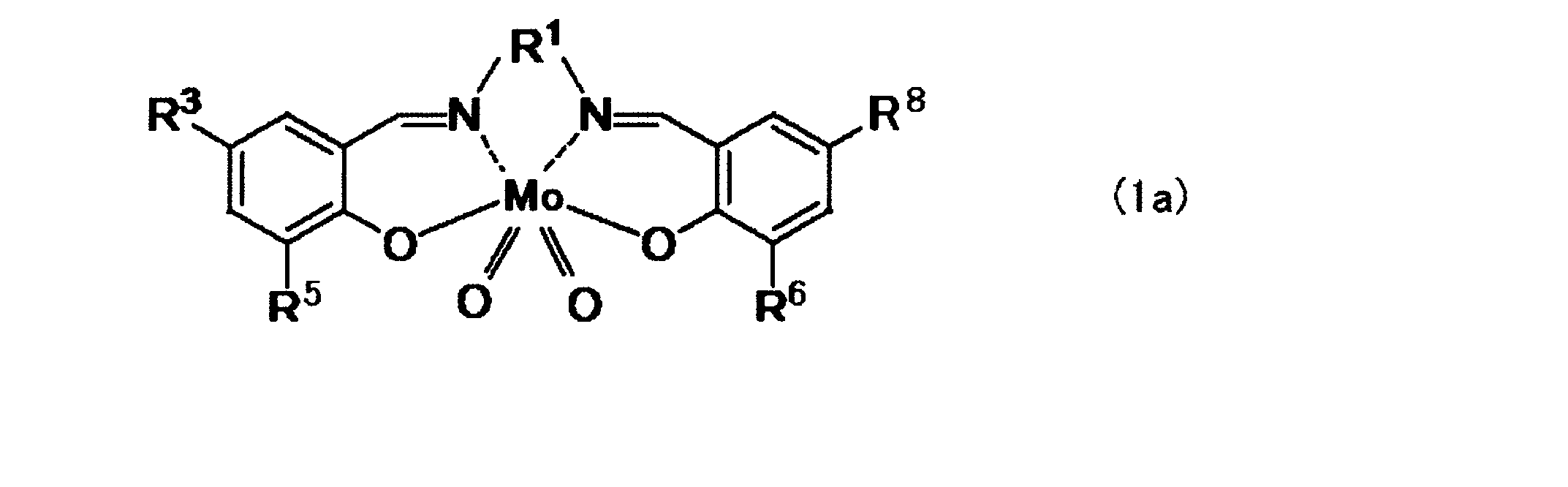
R3及びR8、並びにR5及びR6はそれぞれ同一であり、
R1は-(CH2)2-、-(CH2)3-、-CH2-C(CH3)2-CH2-、又は-(CH2)4-であり、R5及びR6は水素、-CH3、-CH2CH3、-C(CH3)3、-OCH3、-Cl、-Br、又は-Iであり、R3及びR8は水素、-CH3、-CH2CH3、-C(CH3)3、-OCH3、-Cl、-Br、又は-Iである。





以下の実施例において、別段明記がない限り、NMR、GPC及びDSCは以下の条件で行った。
NMRはBRUKER DRX500 spectrometer (Bruker社製)を用いて測定した。溶媒には標準物質としてTMSを0.03vol%含むCDCl3, DMSO-d6を用いた。
(開始剤消費率)=a/(a+d)
(重合度)=(c+e)/e

GPCはポンプ(LC-10ADVP、株式会社島津製作所)及びRI検出器(RID-10A、株式会社島津製作所)を用い、カラムShodex GPC KF-804L(昭和電工株式会社製)でオーブン温度40℃、THF流速0.5ml/分にて測定した。ポリマー1.8mgに対してTHF0.5mlで試料を作成し、10μl注入して測定した。分子量Mn, Mw及び分子量比Mw/Mnはポリスチレンを標準物質として検量線を作成して計算した。
DSCはDSC 2920(TA Instrument 社製)を用いてHeating rate10℃/minで-80~200℃の範囲でガラス転移温度(Tg)を測定した。通常測定を行い放冷した後、再び測定しデータとした。
実施例1 ジオキソモリブデンサレン錯体の製造
K. Yamanouchi et al., Inorg. Chim. Acta, 9, 161-164, 1974又はW. E. Hill et al., Inorg. Chim. Acta, 35, 35-41, 1979等を参考にして、下記の表2の反応式におけるようにジアミン(2)とサリチルアルデヒド誘導体(3)からリガンド(4)を合成した。


実施例1で得られたジオキソモリブデンサレン錯体のうち、MoO2(3-OMe)salen、MoO2(3-OMe)saltn、MoO2(3-OMe)Di-Me-saltn、及びMoO2(3-OMe)saltetのcis-α立体異性型について、ε-カプロラクトン:開始剤:触媒=100:10:1(モル比)の条件で、開始剤としてベンジルアルコールを用いてトルエン還流下で1時間反応させてε-カプロラクトン(CL)の開環重合活性を調べたところ(ε-カプロラクトン110μl、ベンジルアルコール10μl、触媒0.1mmol、及びトルエン1mL)、表4のように、いずれの錯体の触媒活性も高かった。反応率及び重合度はCDCl3を溶媒に用いてNMRで測定した。


ε-カプロラクトン:開始剤:触媒=100:1:0.05(モル比)の条件で、開始剤としてシクロドデカノールを用いてメシチレン中110℃で15分反応したところ、表6の様にMoO2(3-OMe)Di-Me-saltnの活性が最も高いことが分かった。

K. Yamanouchi, S. Yamada, Inorg. Chim. Acta, 9, 83-86, 1974に記載された合成方法に従い、サリチルアルデヒド誘導体とモリブデン酸アンモニウムをMethanol-H2Oに溶解し、12M HCl 1mlをゆっくり加えた。その後、室温で4時間反応させ、生じた沈殿をろ過し、各種ジオキソモリブデンサリチルアルデヒド錯体を合成した(下記の化7の反応式、表7)。各錯体の合成方法は以下の通りである。また、同定は元素分析により行った。


Salicylaldehyde 2.2009g (18.02mmol)のMethanol溶液5mlに七モリブデン酸アンモニウム((NH4)6Mo7O24・4H2O) 1.2392g (1.003mmol; salicylaldehyde: Mo = 2.5:1) の水溶液15 mlを加え、撹拌しながら12M HCl 1mlをゆっくりと加えた。そのまま室温で4時間反応させ、沈殿をろ過し、Methanolと水のそれぞれで洗浄した後に減圧下で乾燥し、2.4210g (6.540mmol)の黄色結晶を得た。(収率93.2%)
mp=250-267℃ (分解), Anal. Calcd For C14H10O6Mo: C, 45.4; H, 2.72. Found: C, 45.7, H, 2.80.
o-vanillin 2.7393g (18.00mmol) のMethanol溶液5mlに七モリブデン酸アンモニウム((NH4)6Mo7O24・4H2O) 1.2347g (0.999mmol)の水溶液15mlを加え、撹拌しながら12M HCl 1mlをゆっくりと加えた。そのまま室温で4時間反応させ、沈殿をろ過し、Methanolと水のそれぞれで洗浄した後に減圧下で乾燥し、2.8222g (6.560mmol)の黄色結晶を得た。(収率93.8%)
mp=160-227℃ (分解), Anal. Calcd For C16H14O8Mo: C, 44.7; H, 3.3. Found: C, 45, H, 3.3.
3-MethylSalicylaldehyde 1.2280g (9.019mmol)のMethanol溶液2.5mlに七モリブデン酸アンモニウム((NH4)6Mo7O24・4H2O) 0.6181g (0.5000mmol)の水溶液7.5mLを加え、撹拌しながら12M HCl 0.5 mlをゆっくりと加えた。そのまま室温で4時間反応させ、沈殿をろ過し、Methanolと水のそれぞれで洗浄した後に減圧下で乾燥することで、1.2824g (3.220mmol)の黄色結晶を得た。(収率92.0%)
mp=265-285℃ (分解), Anal. Calcd For C16H14O6Mo: C, 48.3; H, 3.54. Found: C, 49.2, H, 4.0
5-MethoxylSalicylaldehyde 1.3613g (8.950mmol)のMethanol溶液2.5mlに七モリブデン酸アンモニウム((NH4)6Mo7O24・4H2O) 0.6183g (0.5002mmol)の水溶液7.5mlを加え、撹拌しながら12M HCl 0.5mlをゆっくりと加えた。そのまま室温で4時間反応させ、沈殿をろ過し、Methanolと水のそれぞれで洗浄した後に減圧下で乾燥することで、1.2032g (2.797mmol)の黄色結晶を得た。(収率79.9%)
mp=160-240℃ (分解), Anal. Calcd For C16H14O8Mo: C, 44.7; H, 3.3. Found: C, 43.4, H, 3.4
5-MethylSalicylaldehyde 1.2337g (9.061mmol)のMethanol溶液20mlに七モリブデン酸アンモニウム((NH4)6Mo7O24・4H2O) 0.6234g (0.5044mmol)の水溶液5mlを加え、撹拌しながら12M HCl 0.5mlをゆっくりと加えた。そのまま室温で4時間反応させ、沈殿をろ過し、Methanolと水のそれぞれで洗浄した後に減圧下で乾燥することで、0.8222g (2.065mmol)の黄色結晶を得た。(収率58.5%)
mp=180-250℃ (分解), Anal. Calcd For C16H14O6Mo: C, 48.3; H, 3.54. Found: C, 45.5, H, 3.3
2-Hydroxy-1-naphthaldehyde 1.5537g (9.024mmol)のMethanol溶液2.5mlに七モリブデン酸アンモニウム((NH4)6Mo7O24・4H2O) 0.192g (0.5010mmol)の水溶液7.5mlを加え、撹拌しながら12M HCl 0.5mlをゆっくりと加えた。そのまま室温で4時間反応させ、沈殿をろ過し、Methanolと水のそれぞれで洗浄した後に減圧下で乾燥することで、0.9991g (2.1245mmol)の黄色結晶を得た。(収率60.7%)
実施例4で得られたジオキソモリブデンサリチルアルデヒド錯体を、ε-カプロラクトン:開始剤:触媒=100:1:0.05(モル比)の条件で、開始剤としてシクロドデカノールを用いてメシチレン中80℃で反応させてε-カプロラクトン(CL)の開環重合活性を調べたところ、MoO2[(5-OMe)salad]2では15分程度で90%以上の反応が進行し、最も活性が高く、他の錯体も30分でほぼ反応が終了し、活性が高いことが示された(表8)。

MoO2[(5-OMe)salad]2を触媒として用い、ラクチド(LA)とε-カプロラクトン(CL)を種々の割合で加えて重合させた。[LA+CL]:[開始剤]:[触媒]=100:1:0.05(モル比)とし、溶媒はメシチレンとし、110℃で重合させた。反応率、生成したポリマーのモノマー比率はNMRにより測定した(表9)。生じたポリマーの分子量はGPCにて測定した。

MoO2[(5-OMe)salad]2、MoO2[(3-OMe)Di-Me-saltn](cis-α型)、MoO2(acac) 2、及び(NH4)8[Mo10O34]の4種類の触媒を用いて、ラクチドとε-カプロラクトンをモル比50/50で共重合させ、得られたε-CL-LA共重合体のランダム性について調べた。

Claims (11)
- ラクチドとラクトンとを、モリブデン化合物を触媒として使用して、共重合させることを特徴とする、ラクチド-ラクトン共重合体の製造方法。
- 前記ラクトンはε-カプロラクトンである請求項1に記載のラクチド-ラクトン共重合体の製造方法。
- 前記モリブデン化合物は、モリブデンのキレート化合物、ポリオキソモリブデン塩、モリブデンのアルコキシド、モリブデンイオンと有機酸の塩類、又はモリブデンのカルボニル化合物から選択される請求項1又は2に記載のラクチド-ラクトン共重合体の製造方法。
- 前記モリブデン化合物は六価モリブデンの錯体である請求項1又は2に記載のラクチド-ラクトン共重合体の製造方法。
- 前記六価モリブデンの錯体は、ジオキソモリブデン(VI)錯体又はポリオキソモリブデン塩である請求項4に記載のラクチド-ラクトン共重合体の製造方法。
- 前記ジオキソモリブデン(VI)錯体は、ジオキソモリブデン(VI)アセチルアセトナト錯体、ジオキソモリブデン(VI)サレン錯体、又はジオキソモリブデン(VI)サリチルアルデヒド錯体から選択される請求項5に記載のラクチド-ラクトン共重合体の製造方法。
- 前記ジオキソモリブデン(VI)サレン錯体は、下記の一般式(1)で表されるオキソモリブデン(VI)サレン錯体であり、

R1は、炭素数2~7の二価の脂肪族炭化水素基であり、
R2、R3、R4、R5、R6、R7、R8及びR9は、独立して、水素、炭素数が1~4のアルキル基、炭素数1~4のアルコキシ基、シリル基、アリール基、メトキシメチル基、-Cl、-Br、又は-Iである、請求項6に記載のラクチド-ラクトン共重合体の製造方法。 - 前記R1は-(CH2)2-、-(CH2)3-、-CH2CH(CH3)-、-CH2-C(CH3)2-CH2-、-(CH2)4-、1,2-シクロヘキシル基、1,3-シクロヘキシル基、1,4-シクロヘキシル基、1,2-シクロペンチル基、又は1,2-シクロヘプチル基であり、R2、R4、R7、R9は水素又は-CH3であり、R5及びR6は水素、-CH3、-CH2CH3、-C(CH3)3、-OCH3、-Cl、-Br、又は-Iであり、R3及びR8は水素、-CH3、-CH2CH3、-C(CH3)3、-OCH3、-Cl、-Br、又は-Iである、項7に記載のラクチド-ラクトン共重合体の製造方法。
- 前記ジオキソモリブデン(VI)サリチルアルデヒド錯体は、下記構造式(6)で表されるジオキソモリブデン(VI)サリチルアルデヒド錯体であり、

- 前記R1、R2、R3及びR4はそれぞれ独立して水素、-Cl、-Br、-I、-OCH3、-OCH2CH3、-NO2、-OH、-COOH、-CH3、又は-CH2CH3である請求項9に記載のラクチド-ラクトン共重合体の製造方法。
- 前記ポリオキソモリブデン塩は、[Mo3 O10 ]2- 、[Mo6 O20 ]4- 、[Mo7 O24 ]6- 、[Mo8 O26 ]4- 、及び[Mo10O34 ]8- からなる群より選ばれた陰イオンと、アンモニウムイオン、アルキルアンモニウムイオン及びアルカリ金属イオンからなる群より選ばれた陽イオンとの塩である請求項5に記載のラクチド-ラクトン共重合体の製造方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015511172A JP6242382B2 (ja) | 2013-04-12 | 2014-03-19 | ラクチド−ラクトン共重合体の製造方法 |
| EP14782275.3A EP2985302B1 (en) | 2013-04-12 | 2014-03-19 | Method for producing lactide-lactone copolymer |
| US14/783,640 US9527955B2 (en) | 2013-04-12 | 2014-03-19 | Method for producing lactide-lactone copolymer |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013084129 | 2013-04-12 | ||
| JP2013-084129 | 2013-04-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014167966A1 true WO2014167966A1 (ja) | 2014-10-16 |
Family
ID=51689378
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/057531 WO2014167966A1 (ja) | 2013-04-12 | 2014-03-19 | ラクチド-ラクトン共重合体の製造方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US9527955B2 (ja) |
| EP (1) | EP2985302B1 (ja) |
| JP (1) | JP6242382B2 (ja) |
| WO (1) | WO2014167966A1 (ja) |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS55149320A (en) * | 1979-05-09 | 1980-11-20 | Nippon Ester Co Ltd | Polyester preparation |
| JPS5649728A (en) | 1979-09-28 | 1981-05-06 | Daicel Chem Ind Ltd | Production of lactone high polymer |
| JP2001002763A (ja) | 1999-06-18 | 2001-01-09 | Daicel Chem Ind Ltd | ラクチド/ラクトン共重合体及びその製造方法 |
| JP2005517062A (ja) * | 2002-02-06 | 2005-06-09 | ポリガニックス ビー. ブイ. | DL−ラクチド−ε−カプロラクトンコポリマー |
| JP2005220333A (ja) * | 2004-02-03 | 2005-08-18 | Bmg Inc | 生体内分解吸収性高分子及びその製造方法 |
| JP2005306999A (ja) * | 2004-04-21 | 2005-11-04 | Jms Co Ltd | ラクチド−カプロラクトン共重合体 |
| JP2006050064A (ja) | 2004-08-02 | 2006-02-16 | Omron Entertainment Kk | 写真撮影編集方法及びその装置 |
| JP2008007608A (ja) * | 2006-06-28 | 2008-01-17 | Gunze Ltd | 金属触媒の含有量が少ない生体内分解吸収性高分子の製法 |
| JP2009132769A (ja) * | 2007-11-29 | 2009-06-18 | Gunze Ltd | 医療用インプラント用ラクチド/ε−カプロラクトン共重合体 |
| WO2010110460A1 (ja) | 2009-03-27 | 2010-09-30 | 国立大学法人名古屋大学 | ラクチド・ε-カプロラクトン共重合体の製造方法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5575422A (en) * | 1978-11-30 | 1980-06-06 | Daicel Chem Ind Ltd | Preparation of lactonepolyester |
| JP2006022282A (ja) * | 2004-07-09 | 2006-01-26 | Regulus Co Ltd | 導電性樹脂組成物および導電性樹脂成形物 |
| KR101102475B1 (ko) | 2006-06-28 | 2012-01-05 | 군제 가부시키가이샤 | 금속 촉매의 함유량이 적은 생체내 분해흡수성 고분자 및 그 제조 방법 |
| EP2218466A4 (en) | 2007-11-29 | 2013-01-16 | Gunze Kk | COPOLYMER OF LACTIDE / EPSILON-CAPROLACTONE FOR MEDICAL IMPLANT, PROCESS FOR PRODUCTION OF LACTIDE / EPSILON-CAPROLACTONE COPOLYMER FOR MEDICAL IMPLANT, MEDICAL IMPLANT AND ARTIFICIAL HORSE-MOTHER |
| US8143369B2 (en) * | 2009-06-02 | 2012-03-27 | International Business Machines Corporation | Polymers bearing pendant pentafluorophenyl ester groups, and methods of synthesis and functionalization thereof |
| WO2012065711A1 (en) * | 2010-11-18 | 2012-05-24 | Saudi Basic Industries Corporation | Process for preparing a polyester |
-
2014
- 2014-03-19 JP JP2015511172A patent/JP6242382B2/ja not_active Expired - Fee Related
- 2014-03-19 US US14/783,640 patent/US9527955B2/en active Active
- 2014-03-19 WO PCT/JP2014/057531 patent/WO2014167966A1/ja active Application Filing
- 2014-03-19 EP EP14782275.3A patent/EP2985302B1/en not_active Not-in-force
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS55149320A (en) * | 1979-05-09 | 1980-11-20 | Nippon Ester Co Ltd | Polyester preparation |
| JPS5649728A (en) | 1979-09-28 | 1981-05-06 | Daicel Chem Ind Ltd | Production of lactone high polymer |
| JP2001002763A (ja) | 1999-06-18 | 2001-01-09 | Daicel Chem Ind Ltd | ラクチド/ラクトン共重合体及びその製造方法 |
| JP2005517062A (ja) * | 2002-02-06 | 2005-06-09 | ポリガニックス ビー. ブイ. | DL−ラクチド−ε−カプロラクトンコポリマー |
| JP2005220333A (ja) * | 2004-02-03 | 2005-08-18 | Bmg Inc | 生体内分解吸収性高分子及びその製造方法 |
| JP2005306999A (ja) * | 2004-04-21 | 2005-11-04 | Jms Co Ltd | ラクチド−カプロラクトン共重合体 |
| JP2006050064A (ja) | 2004-08-02 | 2006-02-16 | Omron Entertainment Kk | 写真撮影編集方法及びその装置 |
| JP2008007608A (ja) * | 2006-06-28 | 2008-01-17 | Gunze Ltd | 金属触媒の含有量が少ない生体内分解吸収性高分子の製法 |
| JP2009132769A (ja) * | 2007-11-29 | 2009-06-18 | Gunze Ltd | 医療用インプラント用ラクチド/ε−カプロラクトン共重合体 |
| WO2010110460A1 (ja) | 2009-03-27 | 2010-09-30 | 国立大学法人名古屋大学 | ラクチド・ε-カプロラクトン共重合体の製造方法 |
Non-Patent Citations (17)
| Title |
|---|
| A. SAKAKURA ET AL., ADV. SYNTH. CATAL., vol. 349, 2007, pages 1641 - 1646 |
| A. SAKAKURA ET AL., ADV. SYNTH. CATAL., vol. 65, 2009, pages 2102 - 2109 |
| BEHZAD ZEYNIZADEH ET AL., BULL. CHEM. SOC. JPN, vol. 78, 2005, pages 307 - 315 |
| DIE MAKROMOLEKULARE CHEMIE, vol. 194, no. 3, March 1993 (1993-03-01), pages 913 - 925 |
| E. Y. TSHUVA ET AL., ORGANOMETALLICS, vol. 20, 2001, pages 3017 - 3028 |
| H. ZHANG ET AL., INORG. CHEM., vol. 45, 2006, pages 1745 - 1753 |
| J. E. BAEZ ET AL., POLYMER, vol. 46, 2005, pages 12118 - 12129 |
| J. KASPERCZYK ET AL.: "Coordination polymerization of lactides, 4. The role of transesterification in the copolymerization of L,L-lactide and 2-caprolactone", DIE MAKROMOLEKULARE CHEMIE, vol. 194, no. 3, March 1993 (1993-03-01), pages 913 - 925, XP000348955, DOI: doi:10.1002/macp.1993.021940315 |
| K. YAMANOUCHI ET AL., INORG. CHIM. ACTA, vol. 9, 1974, pages 161 - 164 |
| K. YAMANOUCHI ET AL., INORG. CHIM. ACTA, vol. 9, 1974, pages 83 - 86 |
| K. YAMANOUCHI; S. YAMADA, INORG. CHIM. ACTA, vol. 9, 1974, pages 83 - 86 |
| L. CASELLA ET AL., INORG. CHIM. ACTA, vol. 14, 1988, pages 89 - 97 |
| M. GOMEZ ET AL., EUR. J. INORG. CHEM., 2001, pages 1071 - 1076 |
| W. E. HILL ET AL., INORG. CHIM. ACTA, vol. 35, 1979, pages 35 - 41 |
| W.P. GRIFFITH ET AL., POLYHEDRON, vol. 6, no. 5, 1987, pages 891 |
| Y. LI ET AL., CHEM. COMMUN., 2000, pages 1551 - 1552 |
| Y. WONG ET AL., DALTON TRANS, vol. 39, 2010, pages 4602 - 4611 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2014167966A1 (ja) | 2017-02-16 |
| EP2985302A1 (en) | 2016-02-17 |
| US9527955B2 (en) | 2016-12-27 |
| EP2985302A4 (en) | 2016-11-30 |
| EP2985302B1 (en) | 2018-01-10 |
| JP6242382B2 (ja) | 2017-12-06 |
| US20160053049A1 (en) | 2016-02-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Sung et al. | Zinc and magnesium complexes incorporated by bis (amine) benzotriazole phenoxide ligand: Synthesis, characterization, photoluminescent properties and catalysis for ring-opening polymerization of lactide | |
| Darensbourg et al. | Ring-opening polymerization of cyclic esters and trimethylene carbonate catalyzed by aluminum half-salen complexes | |
| Kan et al. | Aluminum methyl, alkoxide and α-alkoxy ester complexes supported by 6, 6′-dimethylbiphenyl-bridged salen ligands: synthesis, characterization and catalysis for rac-lactide polymerization | |
| Liu et al. | Ring‐opening polymerization of β‐butyrolactone catalyzed by efficient magnesium and zinc complexes derived from tridentate anilido‐aldimine ligand | |
| CN109694471B (zh) | 一种吡啶基脲催化剂及其在开环聚合中的应用 | |
| WO2010110460A1 (ja) | ラクチド・ε-カプロラクトン共重合体の製造方法 | |
| Kong et al. | Synthesis of N, N, O-chelate zinc and aluminum complexes and their catalysis in the ring-opening polymerization of ε-caprolactone and rac-lactide | |
| WO2008128548A2 (en) | Catalyst and method for polymerization and copolymerization of lactide | |
| Xiao et al. | Bulk ring-opening polymerization (ROP) of L-lactide catalyzed by Ni (ii) and Ni (ii)-Sm (iii) complexes based on a salen-type schiff-base ligand | |
| Routaray et al. | Synthesis and structural studies of copper (II) complex supported by–ONNO–tetradentate ligand: Efficient catalyst for the ring-opening polymerization of lactide | |
| Wei et al. | Aluminum complexes with Schiff base bridged bis (indolyl) ligands: synthesis, structure, and catalytic activity for polymerization of rac-lactide | |
| Daneshmand et al. | Catalytic-site-mediated chain-end control in the polymerization of rac-lactide with copper iminopyrrolide complexes | |
| Chuang et al. | Synthesis, characterization, and catalytic activity of sodium ketminiate complexes toward the ring-opening polymerization of l-lactide | |
| Collins et al. | Rare-earth metal complexes derived from the acids Ph2C (X) CO2H (X= OH, NH2): Structural and ring opening polymerization (ROP) studies | |
| Trofymchuk et al. | Synthesis and structures of N-arylcyano-β-diketiminate zinc complexes and adducts and their application in ring‐opening polymerization of L-lactide | |
| Pang et al. | Enolic Schiff-base aluminum complexes and their application in lactide polymerization | |
| CN107417739B (zh) | 一种希夫碱铁化合物、其制备方法及其作为催化剂的应用 | |
| Hu et al. | Enolic Schiff base zinc amide complexes: highly active catalysts for ring-opening polymerization of lactide and ε-caprolactone | |
| Jiang et al. | Phenoxy-imine/-amide aluminum complexes with pendant or coordinated pyridine moieties: Solvent effects on structural type and catalytic capability for the ROP of cyclic esters | |
| CN104710447A (zh) | 一种含有吡咯基团的手性铝配合物及其制备方法和聚乳酸的制备方法 | |
| Duan et al. | Ring‐opening polymerization of lactide catalyzed by bimetallic salen‐type titanium complexes | |
| JP6242382B2 (ja) | ラクチド−ラクトン共重合体の製造方法 | |
| US9777023B2 (en) | Dinuclear indium catalysts and their use for (Co)polymerization of cyclic esters | |
| KR101864005B1 (ko) | 고리형 에스테르기를 가진 모노머의 중합 반응의 촉매 및 이를 이용한 폴리머의 제조 방법 | |
| JP3431561B2 (ja) | 非晶性2,5−ジ置換フェノール酸化重合体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14782275 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015511172 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14783640 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014782275 Country of ref document: EP |