Verfahren zur Herstellung von ABS-Zusammensetzungen mit verbesserter Oberfläche
Die vorliegende Erfindung betrifft ein Verfahren zur Herstellung von Zusammensetzungen enthaltend im Emulsionspolymersiationsverfahren hergestellte vinylaromatische Copolymere mit herstellungsbedingten Salzinklusionen, die sich durch eine verbesserte Oberflächenqualität auszeichnen, nachdem die Granulate durch Kontakt mit flüssigem Wasser angefeuchtet wurden. Diese Zusammensetzungen eignen sich insbesondere zur Herstellung von Formkörpern mit alterungsstabil defekt-freier Class A-Oberfläche. Insbesondere betrifft die Erfindung ein Verfahren, in dem die Copolymere durch Kontakt mit flüssigem Wasser angefeuchtet und in diesem Wasser bei definierter Temperatur und für eine definierte Zeit gelagert und nachfolgend vom nicht oberflächlich anhaftendem Wasser getrennt und optional auch oberflächlich getrocknet werden. Zur Befeuchtung wird beispielsweise ein Aufbau eingesetzt, der einem Klärbecken ähnelt. Das so vorbehandelte Granulat wird im Anschluss in einem Compoundierungsschritt verarbeitet.
Die vorliegende Erfindung betrifft darüber hinaus die nach dem erfindungsgemäßen Verfahren hergestellten Zusammensetzungen und ihre Verwendung zur Herstellung von Formkörpern mit Class- A-Oberflächen-Anforderung und partiellem oder vollständigem Hochglanz-Finish, welche gegebenenfalls partiell oder vollständig einem weiteren Oberflächenbehandlungsschritt durch beispielsweise Lackierung, Folienhinterspritzung, Metallisierung durch Vakuumbedampfung oder Galvanisierung unterzogen sein können.
Im Emulsionspolymerisationsverfahren hergestellte vinylaromatische Copolymere enthaltende Zusammensetzungen, die herstellungsbedingt Salzinklusionen enthalten, sind literaturbekannt. Quellen für solche herstellungsbedingten Salzinklusionen sind mannigfaltig, beispielsweise im Emulsionspolymerisationsverfahren als Hilfsstoffe eingesetzte Emulgatorlösungen, Polymerisationsinitiatorlösungen, Pufferlösungen und Fällmittellösungen, welche bei der Aufarbeitung des Polymerisats je nach Verfahren im Material verbleiben oder aber nur unvollständig aus dem Material wieder entfernt werden. Insbesondere die in traditionellen Verfahren, wie sie zum Beispiel in EP 459 161 Bl , DE 2 021 398 und DE 28 15 098 in der Regel durchgeführte Fällung von in Emulsionspolymerisation hergestellten Vinylpolymerisatlatices mittels Zugabe von Säuren und/oder Salzen trägt in erheblichem Ausmaß zu der Salzfracht des finalen Polymers bei, da diese Salze im Allgemeinen durch nachgeschaltete Verfahrensschritte (Wäschen) nur unzureichend beziehungsweise mit hohem Aufwand (Energie und Wasser/ Abwasser) wieder aus dem Produkt entfernt werden können. Als Koagulantien werden beispielsweise und bevorzugt wässrige Lösungen von wasserlöslichen Salzen wie beispielsweise Alkali-, Erdalkali- oder Aluminiumchloride, -Sulfate, -nitrate, -phosphate, -acetate, -
formiate, -aluminate oder -carbonate, besonders bevorzugt Aluminiumchlorid, Calciumchlorid und Magnesiumsulfat-Lösungen, gegebenenfalls in Kombination mit anorganischen oder organischen Säuren wie beispielsweise Salzsäure, Schwefelsäure, Phosphorsäure, Borsäure, Ameisensäure, Essigsäure, Propionsäure und Zitronsäure verwendet. In der Literatur wird beschrieben, dass solche Salzinklusionen in vinylaromatische Copolymere enthaltenden Zusammensetzungen zu unerwünschten Effekten führen können.
WO 2009/071537 beispielsweise offenbart, dass Magnesium- und/oder Calciumverbindungen in schlagzähmodifizierten vinylaromatischen Copolymeren ausgewählt aus der Gruppe der Acrylnitril- Butadien-Styrol-Copolymere (ABS), Acrylnitril-Styrol-Acrylat-Copolymere (ASA) und Methacrylat- Acrylnitril-Butadien-Styrol-Copolymere (MABS), optional enthaltend Polycarbonat und Zusatzstoffe, bei der thermoplastischen Formgebung durch Spritzguss oder Extrusion zu unerwünschter B elagsbildung am F ormgebungswerkzeug führen und b eansprucht ins ofern s olche Zusammensetzungen mit einem Gehalt an Magnesium- und/oder Calciumverbindungen von 0 mg/kg bis 100 mg/kg. Die in diesen Zusammensetzungen zum Einsatz kommenden Emulsionspolymerisate werden statt durch Zugabe von Magnesiumsulfatlösung wie traditionell üblich durch Gefrierfällung auf einer Scherbeneismaschine gefällt.
WO 98/28344 offenbart ein Verfahren zur kontinuierlichen Koagulation von wässrigen Dispersionen von Pfropfkautschuken durch Scherung, welches den bekannten Nachteil der Fällung mittels Säuren und/oder Salzen als Koagulantien, dass oft Verunreinigungen in den aufgearbeiteten Polymeren verbleiben, die zu einer Beeinträchtigung der Produkteigenschaften führen können, überwindet.
Ein Problem von thermoplastischen Zusammensetzungen enthaltend im Emulsionspolymersiations- verfahren hergestellte vinylaromatische Copolymere mit herstellungsbedingten Salzinklusionen ist es, dass aus ihnen hergestellte Formteile bei Exposition gegenüber Feuchtigkeit (beispielsweise Kondenswasser oder Luftfeuchte), insbesondere bei erhöhten Temperaturen, zur unerwünschten Ausbildung von Oberflächendefekten (Bläschenbildung) neigen, welche den Einsatz solcher Zusammensetzungen in Formteilen mit Hochglanz-Finish und Class A-Oberflächenanforderung einschränken.
EP 2 398 842 AI offenbart ein Compoundierungsverfahren zur Herstellung schlagzähmodifizierter Polycarbonatzusammensetzungen mit reduziertem Gehalt an flüchtigen organischen Verbindungen, in welchem dem als Schlagzähmodifikator eingesetzten pulverförmigen Pfropfpolymersiat 2 bis 40 Gew.- %, bezogen auf die Summe aus Schlagzähmodifikator und Wasser, an flüssigem Wasser zugesetzt wird
und die so hergestellte Vormischung bei der Compoundierung der schlagzähmodifizierter Polycarbonatzusammensetzungen zum Einsatz kommt. Dieses Verfahren entspricht einem Vergleichsbeispiel in dieser Anmeldung.
In JP2010110935 werden Granulate mit Wasser gemischt mit dem Ziel, Staub-/ oder Feinanteile von den Granulaten zu entfernen. Über einen porösen Körper wird das Wasser im Anschluss wieder entfernt. WO2010052872 beschreibt ebenfalls die Vermischung von Granulaten zur oberflächlichen Reinigung der Granulate. Diese Verfahrensweisen ermöglichen jedoch nicht eine erfindungsgemässe Behandlung.
EP2072203 beschreibt die Behandlung von Granulaten mit Wasser zu dem Zweck, Restmonomere zu entfernen. Dazu werden die Granulate für 15 min bis 6 h in Wasser oder anderen Flüssigkeiten gekocht, um Restmonomere zu entfernen.
WO2008090674 offenbart eine Methode zur Abkühlung von PC Granulaten nach Stranggranulierung. Nach dem Granulator gibt es einen zweiten Abkühlschritt in Wasser mit vorgegebenem
Temperaturpro fil. DE102004053929 und DE 202004017275 beschreibt die Thermische Behandlung nach
Unterwassergranulierung. Granulat wird nach Granulierung und Zentrifuge erneut mit Kühlwasser vermischt und dann in einem Trockner getrocknet.
Die Aufgabe der vorliegenden Erfindung bestand somit darin, ein verbessertes Verfahren bereitzustellen, welches die Herstellung thermoplastischen Zusammensetzungen erlaubt, enthaltend im Emulsionspolymersiationsverfahren hergestellte vinylaromatische Copolymere mit herstellungsbedingten Salzinklusionen, die sich durch eine verbesserte Oberflächenqualität nach Wärme-Feucht- Lagerung auszeichnen und insofern zur Herstellung von Formkörpern mit alterungsstabil visuell Defekt-freier Class A-Oberfläche eignen.
Unter„visuell Defekt-freie Class A-Oberflächen" im Rahmen der vorliegenden Erfindung sind dabei Oberflächen zu verstehen, die keine Bläschen mit einer mit bloßem Auge als störend empfundenen
Anzahl und Durchmesser aufweisen. Vorzugsweise weisen solche„visuell Defekt-freie Class A- Oberflächen" eine relative Fläche an Defekten mit Blasentopographie bezogen auf die untersuchte Oberflächengröße (Arei) von kleiner als 50 ppm, bevorzugt von kleiner als 30 ppm, besonders bevorzugt von kleiner als 20 ppm auf.
Weiterhin weisen diese Oberflächen in bevorzugter Ausführungsform nach einer Wärme-Feucht- Behandlung (Schwitzwassertest gemäß DIN EN ISO 6270-2, Prüfdauer 72 h) keine Bläschen mit einem Durchmesser von größer als 300 μιη auf.
Solche visuell Defekt-freien Class A-Oberflächen weisen jedoch dennoch häufig Bläschen auf, die mit optischen Hilfsmitteln, z. B. Lupe oder Mikroskop, sichtbar sind. Die relative Fläche an Defekten mit Blasentopographie bezogen auf die untersuchte Oberflächengröße (Arei) beträgt bevorzugt 0, 1 bis 50 ppm, besonders bevorzugt 1 bis 30 ppm, besonders bevorzugt 3 bis 20 ppm. Die maximale Defektgröße, das heißt der Durchmesser der größten auf solchen visuell Defekt-freien Class-A- Oberflächen gefundenen Defekte mit Bläschentopografie, liegt vorzugsweise in einem Bereich von 10 μιη Μ8 300 μιη.
Überraschenderweise wurde nun gefunden, dass diese Aufgabe erfüllt wird durch ein Verfahren, zur Herstellung von Zusammensetzungen enthaltend
A) 0 bis 98 Gew.-Teile, bevorzugt 1 bis 95 Gew. -Teile, insbesondere 30 bis 85 Gew. -Teile, bezogen auf die Summe aus A und B, eines oder eine Mischung mehrerer thermoplastischer Polymere verschieden von B und
B) 2 bis 100 Gew.-Teile, bevorzugt 5 bis 99 Gew.-Teile, besonders bevorzugt 15 bis 70 Gew.- Teile, bezogen auf die Summe aus A und B, aus
Bl ) mindestens einem im Emulsionspolymersiationsverfahren hergestellten Pfropfpolymerisat, B2) optional mindestens einem im Masse-, Suspensions- oder Lösungspolymeriationsverfahren hergestellten Pfropfpolymerisat,
B3) optional mindestens einem kautschukfreien Vinyl(co)polymerisat, und
C) 0 bis 30 Gew.-Teile, bevorzugt 0,1 bis 20 Gew.-Teile, insbesondere 0,3 bis 7 Gew.-Teile, bezogen auf die Summe auf A und B, mindestens eines handelsüblichen Polymeradditivs, wobei sich die Summe der Gewichtsteile A und B zu 100 addiert, dadurch gekennzeichnet, dass a) in einem ersten Verfahrensschritt die gesamte(n) das Salz enthaltende(n) Komponente(n) aus B, optional mit einem Teil oder der Gesamtmenge der restlichen Komponenten aus B, A und C, vollständig durch Kontakt mit flüssigem Wasser in einem Tauchbecken, Silo oder Mischer mit Wasser
beaufschlagt und darin für eine Zeit von mindestens 24 h, bevorzugt mindestens 48 h, besonders bevorzugt mindestens 72 h gelagert wird, b) in einem zweiten Verfahrensschritt das Granulat von dem nicht oberflächig anhaftendem Wasser getrennt wird und optional auch oberflächig getrocknet wird und in diesem Verfahrensschritt ein Granulat mit einer inneren Feuchte von 0,3 bis 2 Gew.-%, bevorzugt 0,5 bis 1,8 Gew.-%, besonders bevorzugt von 0,6 bis 1,6 Gew.-% , b ezogen auf di e Summe an Wass er und der der Wasserbeaufschlagung unterzogenen Komponenten, erzeugt wird, c) in einem dritten Verfahrensschritt die so mit Wasser beaufschlagte(n) Komponente(n) aufgeschmolzen und im aufgeschmolzenen Zustand geknetet werden und/oder d) in einem vierten Verfahrensschritt die so präparierte(n) Komponente(n) mit den restlichen Komponenten der Zusammensetzung gemischt, die Mischung erneut aufgeschmolzen, geknetet und die Komponenten der Mischung dadurch ineinander dispergiert werden, wobei mindestens in einem der Schritte c) oder d) ein Unterdruck von bevorzugt mindestens 200 mbar, weiter bevorzugt von mindestens 500 mbar, besonders bevorzugt von mindestens 800 mbar angelegt wird und dadurch das im Verfahrensschritt a) in das Verfahren eingebrachte Wasser wieder aus dem Produkt entfernt wird.
Die Bestimmung der inneren Feuchte erfolgt nach oberflächlicher Granulattrocknung mittels Karl- Fischer Titration. Das Granulat wird mit einer IR- Waage bis zur Gewichtskonstanz auf eine
Temperatur von 80 °C erhitzt, um die oberflächlich anhaftende Feuchte zu entfernen. Die auf diese Art und Weise entfernte Wassermenge bezogen auf das Granulatgewicht wird als Oberflächenfeuchte bezeichnet. Im Anschluss wird das oberflächlich getrocknete Granulat einer Karl-Fischer Titration unterzogen. Als innere Feuchte wird die Wassermenge bezeichnet, die, bezogen auf das
Granulatgewicht, mittels Karl-Fischer Titration ermittelt wird. Die Gesamtfeuchte eines Granulats entspricht der Summe aus innerer Feuchte und Oberflächenfeuchte.
Die Beaufschlagung mit flüssigem Wasser kann kontinuierlich oder in einem alternativen Verfahren diskontinuierlich erfolgen.
In einer bevorzugten Ausführungsform erfolgt die Beaufschlagung mit flüssigem Wasser diskontinuierlich in einem oder mehreren Tauchbecken, wie aus der Kläranlagentechnik bekannt ist. Die Temperaturen in den verschiedenen Becken können unterschiedlich sein. In einer bevorzugten Ausführungsform werden zwei Tauchbecken verwendet.
In den Tauchbecken können Rechen, Paddel oder andersartige mechanische Rührer eingebracht sein. Ein Durchströmen des Tauchbeckens mit oder ohne eingebrachte Mischelemente kann ebenfalls vorteilhaft für eine gute Durchmischung sein. In einer bevorzugten Ausführungsform werden in den Tauchbecken Krählwerke zur stressfreien Umwälzung der Granulate eingesetzt. Dies gewährleistet eine homogene Durchmischung der Granulate. Die Becken sind in einer bevorzugten Ausführungsform außerdem mit Bodenräum Einrichtungen zum Ausräumen der Granulate versehen. Weiterhin können die Tauchbecken mit einer Rinne zur Aufnahme und Ausschleusung von Schwimmmaterial versehen sein.
Die Beaufschlagung mit flüssigem Wasser erfolgt im Temperaturbereich von 5 bis 95 °C, bevorzugt, von 10 bis 90 °C, besonders bevorzugt von 20 bis 85 °C.
Dabei sind die Vorzugsbereiche für die Prozesstemperaturen im Verfahrensschritt a) nach oben dadurch begrenzt, dass oberhalb 85°C, die Granulate mit zunehmender Temperatur in zunehmendem Maße zur Erweichung und dadurch zur Verklebung neigen und dadurch ihre Dosierfähigkeit in den weiteren Verfahrensschritten b) und gegebenenfalls c) nachteilig beeinflusst wird oder aber weitere Verfahrensschritte notwendig sind, um die Granulate wieder in eine dosierfähige Form zu bringen.
Die Beaufschlagung mit flüssigem Wasser erfolgt unter Normaldruck.
Der Transport der Granulate aus den Becken kann in einer bevorzugten Ausführungsform mit Hilfe einer Strahlpumpe realisiert werden.
Alternative Ausführungsformen sind wasserbefüllte Silos oder Mischer, wie beispielsweise Taumelmischer. In einem Taumelmischer kann eine gute Durchmischung durch Drehbewegungen des Mischers von außen und damit Verwirb elungen im Produkt erzielt werden.
In einer bevorzugten Ausführungsform ist der Befeuchtungsapparatur eine Apparatur zur Abtrennung des oberflächlichen Wassers von den ausgetragenen Granulaten nachgeschaltet. Eine solche Apparatur kann beispielsweise ein Bandtrockner, ein Zentrifugaltrockner, ein Fließbetttrockner oder ein Stromtrockner, ein Umlufttrockenschrank oder ein Schachttrockner sein. Eine bevorzugte
Ausführungsform ist ein Zentrifugaltrockner. Weiterhin kann eine Förderung, beispielsweise eine Flugförderung mit trockener und/ oder beheizter Luft zur Oberflächentrocknung nachgeschaltet sein. Dieser Schritt sichert eine einfache Transportfähigkeit, Lagerfähigkeit und Dosierbarkeit der Granulate in nachfolgenden Verarbeitungsschritten. Das ausgeschleuste Wasser kann im Kreis gefahren und den Tauchbecken wieder zugeführt werden.
Von der Trocknungsapparatur können die Granulate in ein Silo transportiert werden.
Die mittlere Verweilzeit der Granulate in der Befeuchtungsapparatur beträgt mindestens 24 h, bevorzugt mindestens 48 h, besonders bevorzugt mindestens 72 h. Hierbei sollen besonders bevorzugt maximal 10 % der Granulate eine Verweilzeit von weniger als 72 h erfahren und maximal 1 % der Granulate eine Verweilzeit von weniger als 24 h erfahren. In der bevorzugten Ausführungsform beträgt die mittlere Verweilzeit nicht länger als 1000 h, bevorzugt nicht länger als 700 h, besonders bevorzugt nicht länger als 500 h, ganz besonders bevorzugt nicht länger als 200h.
Vorzugsweise beträgt die Expositionzeit der das Salz enthaltenden Komponente(n) B oder nur der das Salz enthaltenden Komponente Bl , oder des das Salz enthaltende Präcompound aus Komponente Bl mit mindestens einer der Komponenten B2 und B3 oder mit einer Teilmenge mindestens einer der Komponenten B2 und B3, mit Wasser mindestens 24h , bevorzugt mindestens 48 h, besonders bevorzugt mindestens 72 h. In ebenfalls bevorzugter Ausführungsform beträgt die Expositionszeit nicht länger als 1000 h, bevorzugt nicht länger als 700 h, besonders bevorzugt nicht länger als 500 h, ganz bevorzugt nicht länger als 200 h. Bevorzugt wird die das Salz enthaltende Komponente(n) B, oder nur die das Salz enthaltende Komponente B l , oder der das Salz enthaltende Präcompound aus Komponente B l mit mindestens einer der Komponenten B2 und B3 oder mit einer Teilmenge mindestens einer der Komponenten B2 und B3 als Granulat eingesetzt.
Unter„Granulat" im Sinne der Erfindung wird eine Komponente oder eine Mischung aus mehreren Komponenten verstanden, die im festen Aggregatszustand vorliegt. Die Größe der Granulate beträgt 2- 5 mm, besonders bevorzugt 2,5-4 mm. Die Granulatkörner können beliebige Form aufweisen, beispielsweise Linsenform, Kugelform oder Zylinderform.
Unter„Pulver" oder„pulverförmig" im Sinne der Erfindung wird eine Komponente oder eine Mischung aus mehreren Komponenten verstanden, die im festen Aggregatszustand vorliegt und bei denen die Partikel Teilchengrößen von kleiner als 2 mm, bevorzugt von kleiner als 1 mm, insbesondere von kleiner als 0,5 mm aufweisen. Optional können zwischen der Anfeuchtung und der Compoundierung weitere Schritte beispielsweise zur Lagerung, Abfüllung, Transport oder Ähnlichem erfolgen.
In einer alternativen und bevorzugten Ausführungsform wird die Gesamtheit oder eine Teilmenge der Komponenten A und C sowie die Restmengen der Komponente B der Zusammensetzung bereits im Verfahrensschritt (b) zugesetzt, durch den Knetvorgang ineinander dispergiert und das im Verfahrensschritt a) in das Verfahren eingebrachte Wasser durch Anlegen eines Unterdrucks von
bevorzugt von mindestens 200 mbar, weiter bevorzugt von mindestens 500 mbar, besonders bevorzugt von mindestens 800 mbar wieder aus dem Produkt entfernt wird.
In einem letzten Schritt e) wird die Zusammensetzung generell anschließend wieder abgekühlt und granuliert. Erfindungsgemäß enthält die Komponente B, vorzugsweise die Komponente Bl, mindestens ein anorganisches Salz bestehend aus einem Kation ausgewählt aus der Gruppe der Alkalimetalle, Erdalkalimetalle und Aluminium und einem Anion ausgewählt aus der Gruppe bestehend aus Chlorid, Sulfat, Nitrat, Phosphat, Acetat und Formiat.
Bevorzugt ist das Salz ein Alkali-, Erdalkali- oder Aluminiumchlorid oder ein Alkali-, Erdalkali- oder Aluminiumsulfat, oder eine Mischung daraus, besonders bevorzugt ist das Salz ausgewählt aus der Gruppe bestehend aus Aluminiumchlorid, Calciumchlorid und Magnesiumsulfat, oder Mischungen daraus, ganz besonders bevorzugt ist das Salz Magnesiumsulfat.
In einer bevorzugten Ausführungsform besteht die Zusammensetzung nur aus den Komponenten A, B und C. In einer weiteren bevorzugten Ausführungsform besteht die Komponente B aus mindestens zwei Komponenten ausgewählt aus der Gruppe bestehend aus Bl , B2 und B3, weiter bevorzugt aus den Komponenten B 1 und B3, besonders bevorzugt aus Bl, B2 und B3.
Das anorganische Salz wird bevorzugt über die Komponente B 1 in die Zusammensetzung eingebracht, welches das Salz bevorzugt als herstellungsbedingte Verunreinigung enthält. Besonders bevorzugt liegt das Salz in Form von herstellungsbedingten Salzinklusionen in der Komponente B 1 vor.
Die Komponente B, vorzugsweise die Komponente Bl, enthält das Salz in einer Konzentration von 100 bis 5000 mg/kg, bevorzugt von 150 bis 3000 mg/kg, besonders bevorzugt von 200 bis 1500 mg/kg, bezogen auf die Zusammensetzung.
Der Gehalt an anorganischem Salz wird über die Anionengehalte an Chlorid, Sulfate, Nitrat, Phosphat, Acetat, oder Formiat, bevorzugt Chlorid oder Sulfat, besonders bevorzugt Sulfat ermittelt. Eine solche B estimmung erfolgt nach geeignetem Materialaufschluss ionenchromatographisch über Leitfähigkeitsmessung gemäß dem in den Beispielen beschriebenen Verfahren zur Bestimmung des Magnesiumsulfatgehaltes.
Vorteilhaft bei diesem Verfahren ist einerseits die einfachere weitgehende, in bevorzugter Ausführungsform ausschließliche Handhabung der Komponente B und deren Bestandteile in Form von Granulaten gegenüber Pulvern, die zu Verklebungen neigen und auch Explosionsneigung aufweisen, andererseits auch die Möglichkeit, ABS in Granulatform mit hoher herstellungsbedingter Salzfracht ohne weitere aufwendige Reinigungsschritte wie Waschen oder Schmelzefiltration zur
Herstellung von Class A-Oberflächenbauteilen zu verwenden.
Komponente A
Als Komponente A kommen grundsätzlich alle Arten von Komponente B verschiedener thermoplastischer Polymere oder Mischungen aus zwei oder mehr als zwei solcher thermoplastischer Polymere in Frage.
Beispielhaft seien hier genannt Polyolefine (wie Polyethylen und Polypropylen), thermoplastische Polyurethane, Polyacetale (wie Polyoxymethylen und Polyphenylenether), Polyamide, Polyimide, Polycarbonate, Polyester, Polyestercarbonate, Polysulfone, Polyarylate, Polyarylether, Polyphenylenether, Polyarylsulfone, Polyarylsulfide, Polyethersulfone, Polyphenylensulfid, Polyetherketone, Polyamidimide, Polyetherimide und Polyesterimide.
Besonders bevorzugt kommt als Komponente A mindestens ein Polymer ausgewählt aus der Gruppe bestehend aus Polycarbonat, Polyestercarbonat und Polyester, besonders bevorzugt mindestens ein Polymer ausgewählt aus der Gruppe bestehend aus aromatischem Polycarbonat, aromatischem Polyestercarbonat und aromatischem Polyester, ganz besonders bevorzugt ein Polymer ausgewählt aus der Gruppe bestehend aus aromatischem Polycarbonat und aromatischem Polyestercarbonat zum Einsatz.
Erfindungsgemäß geeignete aromatische Polycarbonate und/oder aromatische Polyestercarbonate gemäß Komponente A sind literaturbekannt oder nach literaturbekannten Verfahren herstellbar (zur Herstellung aromatischer Polycarbonate siehe beispielsweise Schnell, "Chemistry and Physics of Polycarbonates", Interscience Publishers, 1964 sowie die DE-AS 1 495 626, DE-A 2 232 877, DE-A 2 703 376, DE-A 2 714 544, DE-A 3 000 610, DE-A 3 832 396; zur Herstellung aromatischer Polyestercarbonate, z. B. DE-A 3 077 934).
Die Herstellung aromatischer Polycarbonate erfolgt z.B. durch Umsetzung von Diphenolen mit Kohlensäurehalogeniden, vorzugsweise Phosgen und/oder mit aromatischen Dicarbonsäuredihaloge-
niden, vorzugsweise Benzoldicarbonsäuredihalogeniden, nach dem Phasengrenzflächenverfahren, gegebenenfalls unter Verwendung von Kettenabbrechern, beispielsweise Monophenolen und gegebenenfalls unter Verwendung von trifunktionellen oder mehr als trifunktionellen Verzweigern, beispiels- weise Triphenolen oder Tetraphenolen. Ebenso ist eine Herstellung über ein Schmelzepolymerisationsverfahren durch Umsetzung von Diphenolen mit beispielsweise Diphenylcarbonat möglich.
Diphenole zur Herstellung der aromatischen Polycarbonate und/oder aromatischen Polyestercarbonate sind vorzugsweise solche der Formel (I)
A eine Einfachbindung, Cl bis C5-Alk len, C2 bis C5-Alk liden, C5 bis C6-Cycloalkyliden, -O- , -SO-, -CO-, -S-, -S02-, C6 bis C12-Arylen, an das weitere aromatische gegebenenfalls Heteroatome enthaltende Ringe kondensiert sein können, oder ein Rest der Formel (II) oder (III)
B jeweils Cl bis C12-Alkyl, vorzugsweise Methyl, Halogen, vorzugsweise Chlor und/oder Brom
x jeweils unabhängig voneinander 0, 1 oder 2, p 1 oder 0 sind, und
R5 und R6 für jedes XI individuell wählbar, unabhängig voneinander Wasserstoff oder Cl bis C6- Alkyl, vorzugsweise Wasserstoff, Methyl oder Ethyl, XI Kohlenstoff und m eine ganze Zahl von 4 bis 7, bevorzugt 4 oder 5 bedeuten, mit der Maßgabe, dass an mindestens einem Atom XI, R5 und R6 gleichzeitig Alkyl sind.
Bevorzugte Diphenole sind Hydrochinon, Resorcin, Dihydroxydiphenole, Bis-(hydroxyphenyl)- Cl -C5-alkane, Bis-(hydroxyphenyl)-C5-C6-cycloalkane, Bis-(hydroxyphenyl)-ether, Bis-(hydroxy- phenyl)-sulfoxide, Bis-(hydroxyphenyl)-ketone, Bis-(hydroxyphenyl)-sulfone und a,a-Bis-(hydroxy- phenyl)-diisopropyl-benzole sowie deren kernbromierte und/oder kernchlorierte Derivate.
Besonders bevorzugte Diphenole sind 4,4'-Dihydroxydiphenyl, Bisphenol- A, 2,4-Bis(4-hydroxy- phenyl)-2-methylbutan, 1 ,1 -Bis-(4-hydroxyphenyl)-cyclohexan, 1 , 1 -Bis-(4-hydroxyphenyl)-3.3.5 - trimethylcyclohexan, 4,4'-Dihydroxydiphenylsulfid, 4,4'-Dihydroxydiphenylsulfon sowie deren di- und tetrabromierten oder chlorierten Derivate wie beispielsweise 2,2-Bis(3-Chlor-4-hydroxyphenyl)- propan, 2,2-Bis-(3,5-dichlor-4-hydroxyphenyl)-propan oder 2,2-Bis-(3,5-dibrom-4-hydroxyphenyl)- propan. Insbesondere bevorzugt ist 2,2-Bis-(4-hydroxyphenyl)-propan (Bisphenol-A).
Es können die Diphenole einzeln oder als beliebige Mischungen eingesetzt werden. Die Diphenole sind literaturbekannt oder nach literaturbekannten Verfahren erhältlich. Für die Herstellung der thermoplastischen, aromatischen Polycarbonate geeignete Kettenabbrecher sind beispielsweise Phenol, p-Chlorphenol, p-tert.-Butylphenol oder 2,4,6-Tribromphenol, aber auch langkettige Alkylphenole, wie 4-[2-(2,4,4-Trimethylpentyl)]-phenol, 4-(l,3-Tetramethylbutyl)-phenol gemäß DE-A 2 842 005 oder Monoalkylphenol oder Dialkylphenole mit insgesamt 8 bis 20 Kohlenstoffatomen in den Alkylsubstituenten, wie 3,5-di-tert.-Butylphenol, p-iso-Octylphenol, p-ter - Octylphenol, p-Dodecylphenol und 2-(3,5-Dimethylheptyl)-phenol und 4-(3,5-Dimethylheptyl)- phenol. Die Menge an einzusetzenden Kettenabbrechern beträgt im allgemeinen zwischen 0,5 Mol%, und 10 Mol%, bezogen auf die Molsumme der jeweils eingesetzten Diphenole.
Die thermoplastischen, aromatischen Polycarbonate haben bevorzugt mittlere Gewichtsmittelmolekulargewichte (Mw, gemessen durch Gelpermeationschromatographie in Methylenchlorid bei
25°C mit Polycarbonat als Standard) von 20.000 bis 40.000 g/mol, vorzugsweise 22.000 bis 35.000 g/mol, besonders bevorzugt 24.000 bis 32.000 g/mol.
Die thermoplastischen, aromatischen Polycarbonate können in bekannter Weise verzweigt sein, und zwar vorzugsweise durch den Einbau von 0,05 bis 2,0 Mol%, bezogen auf die Summe der eingesetzten Diphenole, an dreifunktionellen oder mehr als dreifunktionellen Verbindungen, beispielsweise solchen mit drei und mehr phenolischen Gruppen.
Geeignet sind sowohl Homopolycarbonate als auch Copolycarbonate. Zur Herstellung erfindungsgemäßer Copolycarbonate gemäß Komponente A können auch 1 bis 25 Gew.%, vorzugsweise 2,5 bis 25 Gew.%, bezogen auf die Gesamtmenge an einzusetzenden Diphenolen, Polydiorganosiloxane mit Hydroxyaryloxy-Endgruppen eingesetzt werden. Diese sind bekannt (US 3 419 634) und nach literaturbekannten Verfahren herstellbar . Di e H erst ellung Polydiorganosiloxanhaltiger Copolycarbonate ist in der DE-A 3 334 782 beschrieben.
Bevorzugte Polycarbonate sind neben den Bisphenol-A-Homopolycarbonaten die Copolycarbonate von Bisphenol-A mit bis zu 15 Mol%, bezogen auf die Molsummen an Diphenolen, anderen als bevorzugt oder besonders bevorzugt genannten Diphenolen, insbesondere 2,2-Bis(3,5-dibrom-4- hydroxyphenyl)-propan.
Aromatische Dicarbonsäuredihalogemde zur Herstellung von aromatischen Polyestercarbonaten sind vorzugsweise die Disäuredichloride der Isophthalsäure, Terephthalsäure, Diphenylether-4,4'- dicarbonsäure und der Naphthalin-2,6-dicarbonsäure. Besonders bevorzugt sind Gemische der Disäuredichloride der Isophthalsäure und der Terephthalsäure im Verhältnis zwischen 1 :20 und 20: 1.
Bei der Herstellung von Polyestercarbonaten wird zusätzlich ein Kohlensäurehalogenid, vorzugsweise Phosgen, als bifunktionelles Säurederivat mit verwendet.
Als Kettenabbrecher für die Herstellung der aromatischen Polyestercarbonate kommen außer den bereits genannten Monophenolen noch deren Chlorkohlensäureester sowie die Säurechloride von aromatischen Monocarbonsäuren, die gegebenenfalls durch Cl bis C22-Alkylgruppen oder durch Halogenatome substituiert sein können, sowie aliphatische C2 bis C22-Monocarbonsäurechloride in Betracht.
Die Menge an Kettenabbrechern beträgt jeweils 0,1 bis 10 Mol%, bezogen im Falle der phenolischen Kettenabbrecher auf Mol Diphenol und im Falle von Monocarbonsäurechlorid-Kettenabbrecher auf Mol Dicarbonsäuredichlorid.
Die aromatischen Polyestercarbonate können auch aromatische Hydroxycarbonsäuren eingebaut enthalten.
Die aromatischen Polyestercarbonate können sowohl linear als auch in bekannter Weise verzweigt sein (siehe dazu DE-A 2 940 024 und DE-A 3 007 934).
Als Verzweigungsmittel können beispielsweise drei- oder mehrfunktionelle Carbonsäurechloride, wie Trimesinsäuretrichlorid, Cyanursäuretrichlorid, 3 ,3 '-,4,4'-B enzophenon-tetracarbonsäuretetrachlorid, 1,4,5,8-Napthalintetracarbon-säuretetrachlorid oder Pyromellithsäuretetrachlorid, in Mengen von 0,01 bis 1,0 Mol% (bezogen auf eingesetzte Dicarbonsäuredichloride) oder drei- oder mehrfunktionelle Phenole, wie Phloroglucin, 4,6-Dimethyl-2,4,6-tri-(4-hydroxyphenyl)-hept-2-en, 4,6-Dimethyl-2,4-6- tri-(4-hydroxyphenyl)-heptan, 1 ,3 ,5 -Tri-(4-hydroxyphenyl)-benzol, 1,1,1 -Tri-(4-hydroxyphenyl)-ethan, Tri-(4-hydroxyphenyl)-phenylmethan, 2,2-Bis[4,4-bis(4-hydroxy-phenyl)-cyclohexyl]-propan, 2,4- Bis(4-hydroxyphenyl-isopropyl)-phenol, Tetra-(4-hydroxyphenyl)-methan, 2,6-Bis(2-hydroxy-5- methyl-benzyl)-4-methyl-phenol, 2-(4-Hydroxyphenyl)-2-(2,4-dihydroxyphenyl)-propan, Tetra-(4-[4- hydroxyphenyl-isopropyl]-phenoxy)-methan, 1 ,4-Bis[4,4'-dihydroxytri-phenyl)-methyl]-benz o 1 , in Mengen von 0,01 bis 1,0 Mol% bezogen auf eingesetzte Diphenole verwendet werden. Phenolische Verzweigungsmittel können mit den Diphenolen vorgelegt, Säurechlorid-Verzweigungsmittel können zusammen mit den Säuredichloriden eingetragen werden.
In den thermoplasti schen, aromati schen P olyesterc arb onaten kann der Anteil an Carbonatstruktureinheiten beliebig variieren. Vorzugsweise beträgt der Anteil an Carbonatgruppen bis zu 100 Mol%, insbesondere bis zu 80 Mol%, besonders bevorzugt bis zu 50 Mol%, bezogen auf die Summe an Estergruppen und Carbonatgruppen. Sowohl der Ester- als auch der Carbonatanteil der aromatischen Polyestercarbonate kann in Form von Blöcken oder statistisch verteilt im Polykondensat vorliegen.
Die thermoplastischen, aromatischen Polycarbonate und Polyestercarbonate können allein oder im beliebigen Gemisch eingesetzt werden.
Komponente Bl
Bei der Komp onente B l handelt es sich um P fropfp olymeri sate, hergestellt im Emulsionspolymersiationsverfahren, von in bevorzugter Ausführungsform,
Bl .l) 5 bis 95 Gew.-%, vorzugsweise 10 bis 70 Gew.-%, besonders bevorzugt 20 bis 60 Gew.-%, bezogen auf die Komponente B 1 , einer Mischung aus
Bl .l .1) 65 bis 85 Gew.-%, bevorzugt 70 bis 80 Gew.-%, bezogen auf B l . l , mindestens eines Monomeren ausgewählt aus der Gruppe der Vinylaromaten (wie beispielsweise Styrol, a-Methyl- styrol), kernsubstituierten Vinylaromaten (wie beispielsweise p-Methylstyrol, p-Chlorstyrol) und Methacrylsäure-(C1-C8)-Alkylester (wie beispielsweise Methylmethacrylat, Ethylmethacrylat) und Bl .l .2) 15 bis 35 Gew.-%, bevorzugt 20 bis 30 Gew.-%, bezogen auf Bl .l, mindestens eines Monomeren ausgewählt aus der Gruppe der Vinylcyanide (wie beispielsweise ungesättigte Nitrile wie Acrylnitril und Methacrylnitril), (Meth)Acrylsäure-(Cl-C8)-Alkylester (wie beispielsweise Methylmethacrylat, n-Butylacrylat, tert.-Butylacrylat) und Derivate (wie beispielsweise Anhydride und Imide) ungesättigter Carbonsäuren (beispielsweise Maleinsäureanhydrid und N-Phenyl-Maleinimid) auf
B1.2) 95 bis 5 Gew.-%, vorzugsweise 90 bis 30 Gew.-%, besonders bevorzugt 80 bis 40 Gew.-%, bezogen auf die Komponente B 1 , wenigstens einer elastomeren Pfropfgrundlage.
Die Pfropfgrundlage hat bevorzugt eine Glasübergangstemperatur < 0°C, weiter bevorzugt < -20°C, besonders bevorzugt <-60°C. Glasübergangstemperaturen werden, soweit in der vorliegenden Erfindung nicht anders angegeben, mittels dynamischer Differenzkalorimetrie (DSC) gemäß der Norm DIN EN 61006 bei einer Heizrate von 10 K/min mit Definition der Tg als Mittelpunkttemperatur (Tangentenmethode) und Stickstoff als Schutzgas bestimmt.
Die Pfropfpartikel in der Komponente Bl weisen bevorzugt eine mittlere Teilchengröße (D50-Wert) von 0,05 bis 5 μιη, vorzugsweise von 0,1 bis 1,0 μιη, besonders bevorzugt von 0,2 bis 0,5 μιη auf.
Die mittlere Teilchengröße D50 ist der Durchmesser, oberhalb und unterhalb dessen jeweils 50 Gew.- % der Teilchen liegen. Sie wird, soweit in der vorliegenden Anmeldung nicht explizit anders
angegeben, mittels Ultrazentrifugenmessung (W. Scholtan, H. Lange, Kolloid, Z. und Z. Polymere 250 (1972), 782-1796) bestimmt.
Bevorzugte Monomere B 1.1.1 sind ausgewählt aus mindestens einem der Monomere Styrol, a- Methylstyrol und Methylmethacrylat, bevorzugte Monomere B l .1.2 sind ausgewählt aus mindestens einem der Monomere Acrylnitril, Maleinsäureanhydrid und Methylmethacrylat.
Besonders bevorzugte Monomere sind B 1.1.1 Styrol und Bl .1.2 Acrylnitril.
Für die Pfropfpolymerisate Bl geeignete Pfropfgrundlagen B 1.2 sind beispielsweise Dienkautschuke, Dien-Vinyl-Blockcopolymer-Kautschuke, EP(D)M-Kautschuke, a l s o s o l c h e a u f B a s i s Ethylen/Propylen und gegebenenfalls Dien, Acrylat-, Polyurethan-, Silikon-, Chloropren- und Ethylen/Vinylacetat-Kautschuke sowie Mischungen aus solchen Kautschuken bzw. Silikon-Acrylat- Kompositkautschuke, in denen die Silikon- und die Acrylatkomponenten chemisch miteinander (z.B. durch Pfropfung) miteinander verknüpft sind.
Bevorzugte Pfropfgrundlagen B1.2 sind Dienkautschuke (z.B. auf Basis von Butadien oder Isopren), Dien-Vinyl-Blockcopolymer-Kautschuke (z.B. auf Basis von Butadien- und Styrolblöcken), Copolymerisate von Dienkautschuken mit weiteren copolymerisierbaren Monomeren (z.B. gemäß
B 1.1.1 und B l.1.2) und Mischungen aus den zuvor genannten Kautschuktypen. Besonders bevorzugt sind reiner Polybutadienkautschuk und Styrol-Butadien-Blockcopolymerkautschuk.
Der Gelanteil der Pfropfpolymerisate beträgt mindestens 40 Gew.-%, bevorzugt mindestens 60 Gew.- %, besonders bevorzugt mindestens 75 Gew.-% (gemessen in Aceton). Der Gelgehalt der Pfropfpolymerisate wird, soweit in der vorliegenden Erfindung nicht anders angegeben, bei 25°C als in Aceton als Lösungsmittel unlöslicher Anteil bestimmt (M. Hoffmann, H. Krömer, R. Kuhn, Polymeranalytik I und II, Georg Thieme-Verlag, Stuttgart 1977).
Die Pfropfpolymerisate B 1 werden hergestellt durch radikalische Polymerisation.
Das Pfropfpolymerisat B 1 umfasst herstellungsbedingt im Allgemeinen freies, d.h. nicht chemisch an die Kautschukgrundlage gebundenes Copolymerisat aus B 1.1.1 und B l .1.2, welches sich dadurch auszeichnet, dass es in geeigneten Lösungsmitteln (z.B. Aceton) gelöst werden kann.
Bevorzugt enthält die Komponente Bl ein freies Copolymerisat aus B 1.1.1 und B l .1.2, welches ein gewichtsgemitteltes Molekulargewicht (Mw), bestimmt per Gelpermeationschromatographie mit
Polystyrol als Standard, von bevorzugt 30000 bis 150000 g/mol, besonders bevorzugt von 40000 bis 120000 g/mol aufweist.
Komponente B2
Als Komp onente B2 können die erfindungsgemäß en Zusammensetzungen optional Pfropfpolymerisate, hergestellt im Masse-, Lösungs- oder Suspensionspolymerisationsverfahren enthalten. Hierbei handelt es sich in bevorzugter Ausführungsform um Pfropfpolymerisate von
B2.1) 5 bis 95 Gew.-%, vorzugsweise 80 bis 93 Gew.-%, besonders bevorzugt 85 bis 92 Gew.-%, ganz besonders bevorzugt 87 bis 93 Gew.-%, bezogen auf die Komponente B2, einer Mischung aus
B2.1.1) 65 bis 85 Gew.-%, bevorzugt 70 bis 80 Gew.-%, bezogen auf die Mischung B.2.1, mindestens eines Monomeren ausgewählt aus der Gruppe der Vinylaromaten (wie beispielsweise Styrol, a- Methylstyrol), kernsubstituierten Vinylaromaten (wie beispielsweise p-Methylstyrol, p -Chlor styrol) und Methacrylsäure-(C1 -C8)-Alkylester (wie beispielsweise Methylmethacrylat, Ethylmethacrylat) und
B2.1.2) 15 bis 35 Gew.-%, bevorzugt 20 bis 30 Gew.-% bezogen auf die Mischung B2.1, mindestens eines Monomeren ausgewählt aus der Gruppe der Vinylcyanide (wie beispielsweise ungesättigte
Nitrile wie Acrylnitril und Methacrylnitril), (Meth)Acrylsäure-(Cl-C8)-Alkylester (wie beispielsweise Methylmethacrylat, n-Butylacrylat, tert.-Butylacrylat) und Derivate (wie beispielsweise Anhydride und Imide) ungesättigter Carbonsäuren (beispielsweise Maleinsäureanhydrid und N-Phenyl-Maleinimid) auf B2.2) 95 bis 5 Gew.-%, vorzugsweise 20 bis 7 Gew.-%, besonders bevorzugt 15 bis 8 Gew.-%, ganz besonders bevorzugt 13 bis 7 Gew.-%, bezogen auf die Komponente B2, wenigstens einer Pfropfgrundlage.
Die Pfropfgrundlage hat vorzugsweise eine Glasübergangstemperatur < 0°C, bevorzugt < -20°C, besonders bevorzugt < -60°C. Die Pfropfpartikel in der Komponente B2 weisen bevorzugt eine mittlere Teilchengröße (D50-Wert) von 0,1 bis 10 μηι, vorzugsweise von 0,2 bis 2 μηι, besonders bevorzugt von 0,3 bis 1,0 μηι, ganz besonders bevorzugt von 0,3 bis 0,6 μηι auf.
Bevorzugte Monomere B2.1.1 sind ausgewählt aus mindestens einem der Monomere Styrol, a- Methylstyrol und Methylmethacrylat, bevorzugte Monomere B2.1.2 sind ausgewählt aus mindestens einem der Monomere Acrylnitril, Maleinsäureanhydrid und Methylmethacrylat.
Besonders bevorzugte Monomere sind B2.1.1 Styrol und B2.1.2 Acrylnitril. Für die Pfropfpolymerisate B2 geeignete Pfropfgrundlagen B2.2 sind beispielsweise Dienkautschuke, Dien-Vinyl-Blockcopolymer-Kautschuke, EP(D)M-Kautschuke, also solche auf Basis Ethylen/- Propylen und gegebenenfalls Dien, Acrylat-, Polyurethan-, Silikon-, Chloropren- und Ethylen/Vi- nylacetat-Kautschuke sowie Mischungen aus solchen Kautschuken bzw. Silikon-Acrylat-Komposit- kautschuke, in denen die Silikon- und die Acrylatkomponenten chemisch miteinander (z.B. durch Pfropfung) miteinander verknüpft sind .
Bevorzugte Pfropfgrundlagen B2.2 sind Dienkautschuke (z.B. auf Basis von Butadien oder Isopren), Dien-Vinyl-Blockcopolymer-Kautschuke (z.B. auf Basis von Butadien- und Styrolblöcken), Copolymerisate von Dienkautschuken mit weiteren copolymerisierbaren Monomeren (z.B. gemäß B2.1.1 und B2.1.2) und Mischungen aus den zuvor genannten Kautschuktypen. Besonders bevorzugt als Pfropfgrundlage B2.2 sind Styrol-Butadien-Blockcopolymerkautschuke und Mischungen von Styrol-Butadien-Blockcopolymerkautschuken mit reinem Polybutadienkautschuk.
Der Gelanteil der Pfropfpolymerisate B2 beträgt vorzugsweise 10 bis 35 Gew.-%, besonders bevorzugt 15 bis 30 Gew.-%, ganz besonders bevorzugt 17 bis 23 Gew.-% (gemessen in Aceton).
Besonders bevorzugte Polymerisate B2 sind z.B. ABS-Polymerisate hergestellt durch radikalische Polymerisation, welche in bevorzugter Ausführungsform bis zu 10 Gew.-%, besonders bevorzugt bis zu 5 Gew.-%, besonders bevorzugt 2 bis 5 Gew.-%, jeweils bezogen auf das Pfropfpolymerisat B2, an n-Butylacrylat enthalten.
Das Pfropfpolymerisat B2 umfasst im Allgemeinen herstellungsbedingt freies, d.h. nicht chemisch an die Kautschukgrundlage gebundenes Copolymerisat aus B2.1.1 und B2.1.2, welches sich dadurch auszeichnet, dass es in geeigneten Lösungsmittel (z.B. Aceton) gelöst werden kann.
Bevorzugt enthält die Komponente B2 freies Copolymerisat aus B2.1.1 und B2.1.2, welches ein gewichtsgemitteltes Molekulargewicht (Mw), bestimmt per Gelpermeationschromatographie mit Polystyrol als Standard, von bevorzugt 50000 bis 200000 g/mol, besonders bevorzugt von 70000 bis 150000 g/mol, besonders bevorzugt von 80000 bis 120000 g/mol aufweist.
Komponente B3
Die Zusammensetzung kann als weitere Komponente B3 optional (Co)Polymerisate von mindestens einem Monomeren aus der Gruppe der Vinylaromaten, Vinylcyanide (ungesättigte Nitrile), (Meth)- Acrylsäure-(C1 bis C8)-Alkylester, ungesättigte Carbonsäuren sowie Derivate (wie Anhydride und Imide) ungesättigter Carbonsäuren enthalten.
Insbesondere geeignet sind als Komponente B3 (Co)Polymerisate aus
B3.1 50 bis 99 Gew.-%, bevorzugt 65 bis 85 Gew.-%, besonders bevorzugt 70 bis 80 Gew.-% bezogen auf das (Co)Polymerisat B3 mindestens eines Monomeren ausgewählt aus der Gruppe der Vinylaromaten (wie beispielsweise Styrol, α-Methylstyrol), kernsubstituierten Vinylaromaten (wie beispielsweise p-Methylstyrol, p-Chlorstyrol) und (Meth)Acrylsäure-(Cl-C8)-Alkylester (wie beispielsweise Methylmethacrylat, n-Butylacrylat, tert.-Butylacrylat) und
B3.2 1 bis 50 Gew.-%, bevorzugt 15 bis 35 Gew.-%, besonders bevorzugt 20 bis 30 Gew.-% bezogen auf das (Co)Polymerisat B3 mindestens eines Monomeren ausgewählt aus der Gruppe der Vinylcyanide (wie beispielsweise ungesättigte Nitrile wie Acrylnitril und Methacrylnitril), (Meth)Acrylsäure-(C 1 -C8)-Alkylester (wie beispielsweise Methylmethacrylat, n-Butylacrylat, tert.-
Butylacrylat), ungesättigte Carbonsäuren und Derivate ungesättigter Carbonsäuren (beispielsweise Maleinsäureanhydrid und N-Phenyl-Maleinimid).
Diese (Co)Polymerisate B3 sind harzartig, thermoplastisch und kautschukfrei. Besonders bevorzugt ist das Copolymerisat aus B3.1 Styrol und B3.2 Acrylnitril. Derartige (Co)Polymerisate B3 sind bekannt und lassen sich durch radikalische Polymerisation, insbesondere durch Emulsions-, Suspensions-, Lösungs- oder Massepolymerisation herstellen.
Die (Co)Polymerisate B3 besitzen ein gewichtsgemitteltes Molekulargewicht (Mw), bestimmt per Gelpermeationschromatographie mit Polystyrol als Standard, von bevorzugt 50000 bis 200000 g/mol, besonders bevorzugt von 70000 bis 150000 g/mol, besonders bevorzugt von 80000 bis 130000 g/mol. Komponente C
Die Zusammensetzung kann als Komponente C optional weiterhin handelsübliche Polymeradditive enthalten.
Als handelsübliche Polymeradditive gemäß Komponente C kommen Additive wie beispielsweise Flammschutzmittel (beispielsweise Phosphor- oder Halogenverbindungen), Flammschutzsynergisten (beispielsweise nanoskalige Metalloxide), rauchhemmende Additive (beispielsweise Borsäure oder Borate), Antidrippingmittel (beispielsweise Verbindungen der Substanzklassen der fluorierten Polyolefine, der Silikone sowie Aramidfasern), interne und externe Gleit- und Entformungsmittel (b ei sp i elswei s e P entaerythrittetrastearat , Montanwachs o der P o lyethylenwax) , Fließfähigkeitshilfsmittel (beispielsweise niedermolekulare Vinyl(co)polymerisate), Antistatika (beispielsweise Blockcopolymere aus Ethylenoxid und Propylenoxid, andere Polyether oder Polyhydroxyether, Poletheramide, Polyesteramide oder Sulfonsäuresalze), Leitfähigkeitsadditive (beispielsweise Leitruß oder Carbon Nanotubes), Stabilisatoren (beispielsweise UV/Licht- Stabilisatoren, Thermostabilisatoren, Antioxidantien, Umesterungsinhibitoren, Hydrolyseschutzmittel), antibakteriell wirkende Additive (beispielsweise Silber oder Silbersalze), kratzfestigkeitsverbessernde Additive (beispielsweise Silikonöle oder harte Füllstoffe wie Keramik(hohl)kugeln oder Quarzpulver), IR-Absorbentien, optische Aufheller, fluoreszierende Additive, Füll- und Verstärkungsstoffe (z.B. Talk, gemahlene Glas- oder Karbonfasern, Glas- oder Keramik(hohl)kugeln, Glimmer, Kaolin, CaCCb und Glas schuppen), Säuren sowie Farbstoffe und Pigmente (beispielsweise Ruß, Titandioxid oder Eisenoxid), oder aber Mischungen mehrerer der genannten Additive in Frage.
In bevorzugter Ausführungsform enthalten die erfindungsgemäßen Zusammensetzungen als Komponente C mindestens je eine Komponente ausgewählt aus der Gruppe der Entformungsmittel und Stabilisatoren. In besonders bevorzugter Ausführungsform kommt als Entformungsmittel Pentaerythrittetrastearat zum Einsatz. In besonders bevorzugter Ausführungsform kommt als Stabilisator mindestens eine Verbindung ausgewählt aus der Gruppe der sterisch gehinderten Phenole, der organischen Phosphite und der Brönstedt-sauren Verbindungen zum Einsatz.
Als Komponente C können die erfindungsgemäßen Zusammensetzungen insbesondere auch Flammschutzmittel, beispielsweise halogenierte organische Verbindungen bzw. phosphorhaltige Flammschutzmittel enthalten. Letztgenannte kommen bevorzugt zum Einsatz.
Phosphorhaltige Flammschutzmittel im erfindungsgemäßen Sinne sind bevorzugt ausgewählt aus den Gruppen der mono- und oligomeren Phosphor- und Phosphonsäureester, Phosphonatamine und Phosphazene, wobei auch Mischungen von mehreren Verbindungen ausgewählt aus einer oder verschiedenen dieser Gruppen als Flammschutzmittel zum Einsatz kommen können. Auch andere hier nicht speziell erwähnte halogenfreie Phosphorverbindungen können alleine oder in beliebiger Kombination mit anderen halogenfreien Phosphorverbindungen eingesetzt werden.
Bevorzugte mono- und oligomere Phosphor- bzw. Phosphonsäureester sind Phosphorverbindungen der allgemeinen Formel (IV)
Rl , R2, R3 und R4, unabhängig voneinander jeweils gegebenenfalls halogeniertes Cl bis C8-Alkyl, jeweils gegebenenfalls durch Alkyl, vorzugsweise Cl bis C4-Alkyl, und/oder Halogen, vorzugsweise Chlor, Brom, substituiertes C5 bis C6-Cycloalkyl, C6 bis C20-Aryl oder C7 bis C12-Aralkyl, n unabhängig voneinander, 0 oder 1 q 0 bis 30 und X einen ein- oder mehrkernigen aromatischen Rest mit 6 bis 30 C-Atomen, oder einen linearen oder verzweigten aliphatischen Rest mit 2 bis 30 C-Atomen, der OH-substituiert sein und bis zu 8 Etherbindungen enthalten kann, bedeuten.
Bevorzugt stehen Rl , R2, R3 und R4 unabhängig voneinander für Cl bis C4-Alkyl, Phenyl, Naphthyl oder Phenyl-Cl-C4-alkyl. Die aromatischen Gruppen Rl , R2, R3 und R4 können ihrerseits mit Halogen- und/oder Alkylgruppen, vorzugsweise Chlor, Brom und/oder Cl bis C4-Alkyl substituiert sein. Besonders bevorzugte Aryl-Reste sind Kresyl, Phenyl, Xylenyl, Propylphenyl oder Butylphenyl sowie die entsprechenden bromierten und chlorierten Derivate davon.
X in der Formel (IV) bedeutet bevorzugt einen ein- oder mehrkernigen aromatischen Rest mit 6 bis 30 C-Atomen. Dieser leitet sich bevorzugt von Diphenolen der Formel (I) ab. n in der Formel (IV) kann, unabhängig voneinander, 0 oder 1 sein, vorzugsweise ist n gleich 1. q steht für Werte von 0 bis 30. Bei Einsatz von Mischungen verschiedener Komponenten der Formel (IV) können Mischungen vorzugsweise zahlengemittelte q- Werte von 0,3 bis 10, besonders bevorzugt 0,5 bis 10, insbesondere 1,05 bis 1,4 verwendet werden.
X steht besonders bevorzugt für
oder deren chlorierte oder bromierte Derivate, insbesondere leitet sich X von Resorcin, Hydrochinon, Bisphenol A oder Diphenylphenol ab. Besonders bevorzugt leitet sich X von Bisphenol A ab.
Der Einsatz von oligomeren Phosphorsäureestern der Formel (IV), die sich vom Bisphenol A ableiten, ist besonders vorteilhaft, da die mit dieser Phosphorverbindung ausgerüsteten Zusammensetzungen eine besonders hohe Spannungsriss- und Hydrolysebeständigkeit sowie eine besonders geringe Neigung zur Belagsbildung bei der Spritzgussverarbeitung aufweisen. Des Weiteren lässt sich mit diesen Flammschutzmitteln eine besonders hohe Wärmeformbeständigkeit erzielen.
Als erfindungsgemäße Komponente C können Monophosphate (q=0), Oligophosphate (q=l-30) oder Mischungen aus Mono- und Oligophosphaten eingesetzt werden.
Monophosphorverbindungen der Formel (IV) sind insbesondere Tributylphosphat, Tris-(2-chlorethyl)- phosphat, Tris-(2,3-dibromprobyl)-phosphat, Triphenylphosphat, Trikresylphosphat, Diphenylkresylphosphat, Diphenyloctylphosphat, Diphenyl-2-ethylkresylphosphat, Tri-(isopropyl- phenyl)-phosphat, halogensubstituierte Arylphosphate, Methylphosphonsäuredimethylester, Methyl- phosphensäurediphenylester, Phenylphosphonsäurediethylester, Triphenylphosphinoxid oder Trikresyl- phosphinoxid.
Die Phosphorverbindungen gemäß Formel (IV) sind bekannt (vgl. z.B. EP-A 363 608, EP-A 640 655) oder lassen sich nach bekannten Methoden in analoger Weise herstellen (z.B. Ullmanns Enzyklopädie der technischen Chemie, Bd. 18, S. 301 ff. 1979; Houben-Weyl, Methoden der organischen Chemie, Bd. 12/1 , S. 43; Beilstein Bd. 6, S. 177).
Die mittleren q-Werte können bestimmt werden, indem mittels geeigneter Methode (Gaschromatographie (GC), High Pressure Liquid Chromatography (HP LC ) ,
Gelpermeationschromatographie (GPC)) die Zusammensetzung der Phosphat-Mischung (Molekulargewichtsverteilung) bestimmt wird und daraus die Mittelwerte für q berechnet werden.
Phosphonatamine sind vorzugsweise Verbindungen der Formel (V)
A3-y-NBly (V) in welcher
A für einen Rest der Formel (Va)
RH und R12 unabhängig voneinander für unsubstituiertes oder substituiertes Cl -ClO-Alkyl oder für unsubstituiertes oder substituiertes C6 bis C10-Aryl, stehen,
R13 und R14 unabhängig voneinander für unsubstituiertes oder substituiertes Cl bis ClO-Alkyl oder unsubstituiertes oder substituiertes C6 bis C10-Aryl stehen oder
R13 und R14 zusammen für unsubstituiertes oder substituiertes C3 bis ClO-Alkylen stehen, y die Zahlenwerte 0, 1 oder 2 bedeuten und
Bl unabhängig für Wasserstoff, gegebenenfalls halogeniertes C2 bis C8-Alkyl, unsubstituiertes oder substituiertes C6 bis C10-Aryl steht.
Bl steht vorzugsweise unabhängig für Wasserstoff, für Ethyl, n- oder iso-Propyl, welche durch Halogen substituiert sein können, unsubstituiertes oder durch Cl bis C4-Alkyl und/oder Halogen substituiertes C6 bis ClO-Aryl, insbesondere Phenyl oder Naphthyl.
Alkyl in RH, R12, R13 und R14 steht unabhängig vorzugsweise für Methyl, Ethyl, n-Propyl, iso- Propyl, n-, iso-, sek. oder tert.-Butyl, Pentyl oder Hexyl.
Substituiertes Alkyl in RH, R12, R13 und R14 steht unabhängig vorzugsweise für durch Halogen substituiertes Cl bis ClO-Alkyl, insbesondere für ein- oder zweifach substituiertes Methyl, Ethyl, n- Propyl, iso-Propyl, n-, iso-, sek. oder tert.-Butyl, Pentyl oder Hexyl.
C6 bis ClO-Aryl steht in Rl 1, R12, R13 und R14 unabhängig vorzugsweise für Phenyl, Naphthyl oder Binaphthyl, insbesondere o-Phenyl, o-Naphthyl, o-Binaphthyl, welche durch Halogen (im allgemeinen ein-, zwei- oder dreifach) substituiert sein können.
R13 und R14 können zusammen mit den Sauerstoffatomen, an die sie direkt gebunden sind, und dem Phosphoratom eine Ringstruktur bilden.
B ei spi elhaft und vorzugsweise werden genannt: 5,5,5',5 ',5",5"-Hexamethyltris(l ,3,2- dioxaphosphorinan-methan)amino-2,2',2„-trioxid der Formel (Va-1)
l,3,2-Dioxaphosphorinan-2-m e t h a n a m i n , N-butyl-N[(5,5-dimethyl-l,3,2-dioxaphosphorinan-2- yl)methyl] -5 ,5 -dimethyl-, P,2-dioxide; 1 ,3 ,2-Dioxaphosphorinane-2-methanamin, N-[ [5„5 -dimethyl- 1,3,2 -dioxaphosphorinan-2 -yl)methyl] -5 ,5 -dimethyl-N-phenyl-, P ,2 -dioxid; 1 ,3 ,2 -Dioxaphosphorinan- 2-methanamin, N,N-dibutyl-5,5-dimethyl-, 2-oxid, l,3,2-Dioxaphosphorinan-2-methanimin, N-[(5,5- dimethyl- 1 ,3 ,2-dioxaphosphorinan-2-yl)methyl] -N-ethyl-5 ,5 -dimethyl-, P,2-dioxid, 1 ,3 ,2-Dioxaphos- phorinan-2-methanamin, N-butyl-N-[(5,5-dichloromethyl-l,3,2-dioxaphosphorinan-2-yl)-methyl]-5,5- di-chloromethyl-, P,2-dioxid, 1 ,3 ,2 -Dioxaphosphorinan-2 -methanamin, N-[(5,5-di-chloromethyl-l ,3,2- dioxoaphosphorinan-2-yl)methyl]-5,5-di-chloromethyl-N-phenyl-, P,2-dioxid; 1 ,3,2-
Dioxaphosphorinan-2-methanamin, N,N-di-(4-chlorobutyl)-5,5-dimethyl-2-oxide; 1 ,3,2-
Dioxaphosphorinan-2-methanimin, N-[(5,5-dimethyl-l ,3,2-dioxaphosphorinan-2-yl)methan]-N-(2- chloroethyl)-5,5-di(chloromethyl)-, P2-dioxid.
Bevorzugt sind weiterhin:
Verbindungen der Formel (Va-2) oder (Va-3)
RH, R12, R13 und R14 die oben angegebenen Bedeutungen haben.
Besonders bevorzugt sind Verbindungen der Formel (Va-2) und (Va-1). Die Herstellung der Phosphonatamine ist beispielsweise in US-PS 5 844 028 beschrieben.
Phosphazene sind Verbindungen der Formeln (Via) und (VIb)
worin
R jeweils gleich oder verschieden ist und für Amino, jeweils gegebenenfalls halogeniertes, vorzugsweise mit Fluor halogeniertes Cl bis C8-Alk l, oder Cl bis C8-Alkoxy, jeweils gegebenenfalls durch Alkyl, vorzugsweise Cl bis C4-Alkyl, und/oder Halogen, vorzugsweise Chlor und/oder Brom, substituiertes C5 bis C6-Cycloalkyl, C6 bis C20-Aryl, vorzugsweise Phenyl oder Naphthyl, C6 bis C20-Aryloxy, vorzugsweise Phenoxy, Naphthyloxy, oder C7 bis C12-Aralk l, vorzugsweise Phenyl-
Cl -C4-alkyl, steht, k für 0 oder eine Zahl von 1 bis 15, vorzugsweise für eine Zahl von 1 bis 10 steht.
Beispielhaft seien Propoxyphosphazen, Phenoxyphosphazen, Methylphenoxyphosphazen, Aminophosphazen und Fluoralkylphosphazene genannt. Bevorzugt ist Phenoxyphosphazen. Die Phosphazene können allein oder als Mischung eingesetzt werden. Der Rest R kann immer gleich sein oder 2 oder mehre Reste in den Formeln (Via) und (VIb) können verschieden sein. Phosphazene und deren Herstellung sind beispielsweise in EP-A 728 811 , DE-A 1 961668 und WO 97/40092 beschrieben.
Die Flammschutzmittel können allein oder in beliebiger Mischung untereinander oder in Mischung mit anderen Flammschutzmitteln eingesetzt werden.
Darüber hinaus enthalten flammgeschütze Zusammensetzungen in bevorzugter Ausführungsform die zuvorgenannten Flammschutzmittel in Kombination mit mindestens einem Antidrippingmittel ausgewählt aus den Substanzklassen der fluorierten Polyolefine, der Silikone sowie Aramidfasern. Besonders bevorzugt kommen Polytetrafluorethylen-Polymere als Antidrippingmittel zum Einsatz. Die nach dem erfindungsgemäßen Verfahren hergestellten Formmassen können zur Herstellung von Formkörpern jeder Art verwendet werden. Diese können durch Spritzguss, Extrusion und Blasformverfahren hergestellt werden. Eine weitere Form der Verarbeitung ist die Herstellung von Formkörpern durch Tiefziehen aus zuvor hergestellten Platten oder Folien.
Beispiele für solche Formkörper sind Folien, Profile, Gehäuseteile jeder Art, z.B. für Haushaltsgeräte wie Saftpressen, Kaffeemaschinen, Mixer; für Büromaschinen wie Monitore, Flatscreens, Notebooks, Drucker, Kopierer; Platten, Rohre, Elektroinstallationskanäle, Fenster, Türen und weitere Profile für den Bausektor (Innenausbau und Außenanwendungen) sowie Elektro- und Elektronikteile wie Schalter, Stecker und Steckdosen sowie Karrosserie- bzw. Innenbauteile für Nutzfahrzeuge, insbesondere für den Automobilbereich.
Insbesondere können die nach dem erfindungsgemäßen Verfahren hergestellten Formmassen beispielsweise auch zur Herstellung von folgenden Formkörpern oder Formteilen verwendet werden: Innenausbauteile für Schienenfahrzeuge, Schiffe, Flugzeuge, Busse und andere Kraftfahrzeuge, Gehäuse von Kleintransformatoren enthaltenden Elektrogeräten, Gehäuse für Geräte zur Informationsverarbeitung und -Übermittlung, Gehäuse und Verkleidung von medizinischen Geräten, Massagegeräte und Gehäuse dafür, Spielfahrzeuge für Kinder, flächige Wandelemente, Gehäuse für Sicherheitseinrichtungen, wärmeisolierte Transportbehältnisse, Formteile für Sanitär- und Badausrüstungen, Abdeckgitter für Lüfteröffnungen und Gehäuse für Gartengeräte.
Die nach dem erfindungsgemäßen Verfahren hergestellten Formmassen eignen sich besonders auch zur Herstellung von Formkörpern oder Formteilen mit Class-A-Oberflächen- Anforderung und Hochglanz- Finish, welche gegebenenfalls partiell oder vollständig einem weiteren Oberflächenbehandlungsschritt durch beispielsweise Lackierung, Folienhinterspritzung, Metallisierung durch Vakuumbedampfung oder Galvanisierung unterzogen wurden.
Unter„hochglänzend" verstanden wird im Sinne der vorliegenden Erfindung ein Glanzgrad ermittelt in Reflexion gemäß DIN 67530 bei einem Messwinkel von 60° von mindestens 95, bevorzugt von mindestens 97, besonders bevorzugt von mindestens 99. Gegenstand der Erfindung sind somit auch Formkörper oder Formteile aus den erfindungsgemäßen Zusammensetzungen mit vollständigem oder partiellen Hochglanz-Finish, welche gegebenenfalls partiell oder vollständig einem weiteren Oberflächenbehandlungsschritt durch beispielsweise Lackierung, Folienhinterspritzung, Metallisierung durch Vakuumbedampfung oder Galvanisierung unterzogen wurden.
Gegenstand der Erfindung sind somit auch Formkörper oder Formteile aus den Zusammensetzungen hergestellt nach dem erfindungsgemäßen Verfahren mit vollständigem oder partiellen Hochglanz- Finish, welche gegebenenfalls partiell oder vollständig einem weiteren Oberflächenbehandlungsschritt durch beispielsweise Lackierung, Folienhinterspritzung, Metallisierung durch Vakuumbedampfung oder Galvanisierung unterzogen wurden.
Beispiele Komponente AI
Lineares Polycarbonat auf Basis Bisphenol-A mit einem gewichtsgemittelten Molekulargewicht Mw von 28 kg/mol (bestimmt durch GPC in Methylenchlorid bei 25 °C mit Polycarbonat als Standard). Komponente Bl
Präcompound, vorliegend als Granulat, aus 50 Gew.-% eines Pfropfpolymerisats vom ABS-Typ, hergestellt im Emulsionspolymersiationsverfahren, mit einem A:B:S-Verhältnis von 12:50:38 Gew.-% und 50 Gew.-% eines Styrol-Acrylnitril-Copolymerisats, h erg e st el lt im Massepolymersiationsverfahren, mit einem Styrol-Acrylnitril- Verhältnis von 76:24 Gew.-% und mit einem per GPC mit Polystyrol als Standard in Dimethylformamid bei 20°C gemessenen gewichtsgemittelten Molekulargewicht Mw von 100 kg/mol. Die Komponente B l enthält herstellungsbedingt 900 mg/kg des in der Koagulation des Pfropfpolymerisats eingesetzten Fällmittels Magnesiumsulfat. Dieses Magnesiumsulfat liegt gemäß Nachweis durch Rasterelektronenmikroskopie (REM) gekoppelt mit energiedispersiver Röntgenspektroskopie (EDX) in kristallinen Domänen mit einer Dimension von zum Teil bis über 100 μηι vor.
Die Bestimmung des Magnesiumsulfatgehaltes in Komponente B l erfolgte über eine quantitative Bestimmung des Sulfationengehaltes und dessen Umrechnung auf Magnesiumsulfat, da eine Ermittlung aus dem Magnesiumgehalt aufgrund fehlender Selektivität auf MgSCu nicht möglich ist. Hierzu wurde ca. 1 g der Komponente Bl genau eingewogen, mit 25 ml Aceton, p.A. versetzt und das Gemisch für 30 Minuten im Ultraschallbad behandelt. Die entstandene Suspension wurde mit Milliporewasser auf 200 ml aufgefüllt und durchgeschüttelt. Die so behandelte Suspension wurde membranfiltriert. Die Bestimmung des Sulfationen-Gehaltes erfolgte im Filtrat ionenchromatographisch unter Verwendung eines Ionenchromatographen DIONEX DX 600 (Fa. DIONEX) (Trennsäule: IonPac AS 11, 4x250mm (Fa.DIONEX); mobile Phase: Gradient NaOH, c= 0,004/0,076 mol/L; Flußrate: 1,8 ml/min; Autosamplertemperatur: 23°C; Säulentemperatur: 35°C;
Suppression: elektrochemisch, ASRS 300, 4mm; Detektion: Leitfähigkeit).
Komponente B2 n-Butylacrylat-modifiziertes Pfropfpolymerisat vom ABS-Typ, h erge st ellt im Massepolymersiationsverfahren, mit einem A:B:S-Verhältnis von 21 :10:65 Gew.-% und mit einem n- Butylacrylatgehalt von 4 Gew.-%. Der D50-Wert der Pfropfpartikeldurchmesser bestimmt durch Ultrazentrifugation beträgt 0,5 μηι. Die dem Pfropfpolymersiat zugrunde liegende Pfropfgrundlage ist ein Styrol-Butadien-Blockcopolymer-Kautschuk (SBR). Der Gelgehalt des Pfropfpolymerisats gemessen in Aceton liegt bei 20 Gew.-%. Das per GPC mit Polystyrol als Standard in Dimethylformamid bei 20°C gemessene gewichtsgemittelte Molekulargewicht Mw des freien, d.h. nicht chemisch an den Kautschuk gebundenen bzw. in den Kautschukpartikeln in für Aceton unlöslicher Form inkludierten n-Butylacrylat-modifizierten SANs beträgt 110 kg/mol.
Komponente B3
Styrol-Acrylnitril-Copolymerisat, hergestellt im Massepolymersiationsverfahren, mit einem Styrol- Acrylnitril-Verhältnis von 76:24 Gew.-% und mit einem per GPC mit Polystyrol als Standard in Dimethylformamid bei 20°C gemessenen gewichtsgemittelten Molekulargewicht Mwvon 100 kg/mol. Komponente Cl
Pentaerythrittetrastearat als Gleit-/Entformungsmittel Komponente C2
Phosphorigsäureester des Bis-(2-hydroxy-3-cyclohexyl-5-methyl-phenyl)-methans mit der Formel
Thermostabilisator, Irganox 1076, BASF (Ludwigshafen, Deutschland)
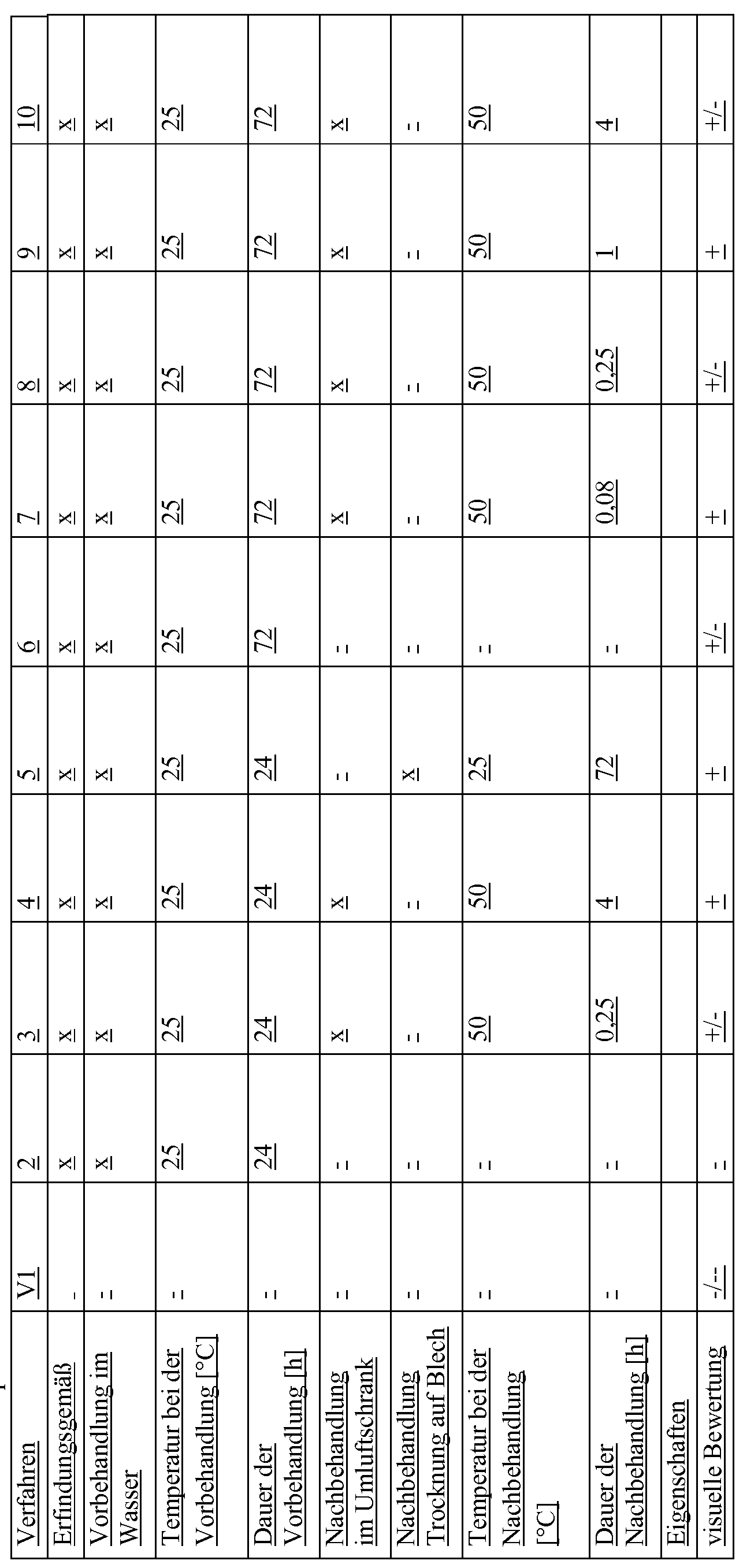
Die Zusammensetzungen der in Tabelle 1 aufgeführten Beispiele und Vergleichsbeispiele VI , 2, 3, 4, 5, 6, 7, 8, 9 und 10 enthalten alle 60,35 Gew.-Teile der Komponente AI
23,16 .-Teile der Komponente Bl
8,90 Gew.-Teile der Komponente B2
6,53. -Teile der Komponente B3
0,74 Gew.-Teile der Komponente Cl 0,12 Gew.-Teile der Komponente C2
0,20 Gew.-Teile der Komponente C3 und unterscheiden sich ausschließlich im zur Herstellung verwendeten Verfahren.
Herstellung der Zusammensetzungen und Prüfung
Die Herstellung der Zusammensetzungen VI, 2, 3, 4, 5, 6, 7, 8, 9 und 10 erfolgte auf einem
Zweiwellenextruder ZSK25WLE der Firma Coperion mit Verhältnis Länge zu Durchmesser L/D=48 bei einer Massetemperatur von 260 bis 270°C und unter Vakuumentgasung bei einem Druck von 70 mbar (absolut). Die Komponente B 1 wurde zum einen unbehandelt eingesetzt (VI), zum anderen in Wasser vorbehandelt (2-10). Die Vorbehandlung fand in einem mit Wasser gefüllten Gefäß bei 25 °C statt. In den Beispielen 2 und 6 wurden die Granulate nach der Behandlung im Wasser abgesiebt und dann oberflächenbenetzt im finalen Compoundierungsschritt eingesetzt. In den Beispielen 3, 4, 7, 8, 9 und 10 wurden die Granulate im Anschluss in einem Umlufttrockenschrank für eine bestimmte Zeit bei 50 °C getrocknet. Im Beispiel 5 wurden die Granulate im Anschluss an die Vorbehandlung im Wasser noch für 72 h auf einem Blech bei 25 °C getrocknet. Details zu Vorbehandlung in Wasser und zur Nachbehandlung der Granulate können Tabelle 1 entnommen werden. In den Fällen, in denen das
Granulat nach der Feuchtebehandlung getrocknet wurde, war das Granulat oberflächentrocken und wurde auch so im finalen Compoundierungsschritt eingesetzt.
Die aus der j eweiligen Compoundierung resultierenden Granulate wurden auf einer Spritzgussmaschine (Fa. Arburg) bei Schmelzetemperaturen von 260°C und einer Werkzeugtemperatur von 80°C zu Platten der Abmessung 150 mm x 105 mm x 2 mm verarbeitet. Hierbei kam ein hochglanzpoliertes Werkzeug zum Einsatz. Diese Platten wurden für 3 Tage bei 40°C einer Luftatmosphäre mit einer relativen Luftfeuchtigkeit von 95% ausgesetzt. Danach erfolgte eine visuelle Begutachtung durch 3 unabhängige Gutachter gemäß folgender Bewertungsgrundlage:
++ keinerlei Blasen oder nur vereinzelte sehr kleine Blasen
+ einige sehr kleine, noch nicht störende Blasen viele sehr kleine Blasen und/oder nur vereinzelte größere Blasen viele größere Blasen
Die Beispiele und Vergleichsbeispiele sind in der Tabelle 1 zusammengefasst. Die Daten zeigen, dass nur diejenigen nach dem erfindungsgemäßen Verfahren hergestellten Formmassen die verbesserten Oberflächeneigenschaften nach Wärme-Feucht-Lagerung gemäß der Aufgabe dieser Erfindung aufweisen und sich insofern zur Herstellung von Formkörpern mit alterungsstabil visuell Defekt-freier Class A-Oberfläche eignen.
abelle 1 : Beispiele