WO2013189266A1 - 喜树碱类化合物及其制备和用途 - Google Patents
喜树碱类化合物及其制备和用途 Download PDFInfo
- Publication number
- WO2013189266A1 WO2013189266A1 PCT/CN2013/077336 CN2013077336W WO2013189266A1 WO 2013189266 A1 WO2013189266 A1 WO 2013189266A1 CN 2013077336 W CN2013077336 W CN 2013077336W WO 2013189266 A1 WO2013189266 A1 WO 2013189266A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- amino
- alkyl
- mmol
- compound
- pharmaceutically acceptable
- Prior art date
Links
- WAVYZKPFLMIKCX-UHFFFAOYSA-N CC(C=C(C(CC1)=O)N1C1=O)=C1C#N Chemical compound CC(C=C(C(CC1)=O)N1C1=O)=C1C#N WAVYZKPFLMIKCX-UHFFFAOYSA-N 0.000 description 1
- XEEKVQZYBCXAIK-UHFFFAOYSA-N CCOC(C(C(C=C(C1(CC2)OCCO1)N2C1=O)=C1C#N)OC(C(CCC1)N1S(c1ccc(C)cc1)(=O)=O)=O)=O Chemical compound CCOC(C(C(C=C(C1(CC2)OCCO1)N2C1=O)=C1C#N)OC(C(CCC1)N1S(c1ccc(C)cc1)(=O)=O)=O)=O XEEKVQZYBCXAIK-UHFFFAOYSA-N 0.000 description 1
- AMRJRUZWECXLRL-WOKNPCPGSA-N CC[C@@](C(OCC)=O)(C(C=C(C1(CC2)OCCO1)N2C1=O)=C1C#N)OC(C(CCC1)N1S(c1ccc(C)cc1)(=O)=O)=O Chemical compound CC[C@@](C(OCC)=O)(C(C=C(C1(CC2)OCCO1)N2C1=O)=C1C#N)OC(C(CCC1)N1S(c1ccc(C)cc1)(=O)=O)=O AMRJRUZWECXLRL-WOKNPCPGSA-N 0.000 description 1
- MYTICZHIIGDKOF-FQEVSTJZSA-N CC[C@](C(C=C1N2Cc3cc(cc(c(F)c4)O)c4nc13)=C(CO1)C2=O)(C1=O)O Chemical compound CC[C@](C(C=C1N2Cc3cc(cc(c(F)c4)O)c4nc13)=C(CO1)C2=O)(C1=O)O MYTICZHIIGDKOF-FQEVSTJZSA-N 0.000 description 1
- GCLITCVVSRIYHJ-UHFFFAOYSA-N COC(c(c(N)ccn1)c1OC)=O Chemical compound COC(c(c(N)ccn1)c1OC)=O GCLITCVVSRIYHJ-UHFFFAOYSA-N 0.000 description 1
- OGQNKGQFMLAEFH-UHFFFAOYSA-N COc(cc(C(N)=O)c([N+]([O-])=O)c1)c1F Chemical compound COc(cc(C(N)=O)c([N+]([O-])=O)c1)c1F OGQNKGQFMLAEFH-UHFFFAOYSA-N 0.000 description 1
- HJKRTVPQSWKCNF-INIZCTEOSA-N COc(ccc(cc1CN23)c4nc1C2=CC([C@@H](C(OC1)=O)O)=C1C3=O)c4O Chemical compound COc(ccc(cc1CN23)c4nc1C2=CC([C@@H](C(OC1)=O)O)=C1C3=O)c4O HJKRTVPQSWKCNF-INIZCTEOSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/22—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains four or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
Definitions
- the present invention relates to a novel class of camptothecin compounds having antitumor activity, pharmaceutical compositions thereof and uses thereof. Background technique
- Camptothecin (s)-4-ethyl-4-hydroxy-1H-pyran[3'4':6,7]pyridazine[l,2-b]quina Porphyrin-3,14(4H,12H)-dione) was first introduced in 1966 by Wall et al. [J. Am. Chem. Soc, 1966, 88(16), 3888-990] from Chinese paulownia plants. An alkaloid extracted from a tree (camptotheca acuminate). Tests have shown that it has antitumor activity against leukemia and many solid tumors and is a natural cytotoxic compound.
- camptothecin derivatives In order to obtain a camptothecin derivative with high activity and low toxicity, structural modification and optimization were carried out, and a series of camptothecin derivatives were synthesized by semi-synthesis and total synthesis.
- the US FDA approved the listing of the camptothecin drug Irinotecan (CPT-11) for the treatment of colorectal cancer.
- Topotecan was approved for marketing for ovarian cancer, and the US FDA approved it as a second-line treatment for small cell lung cancer (SCLC) in 1999.
- SCLC small cell lung cancer
- Camptothecin is the second anticancer drug extracted from plants after paclitaxel. Since camptothecin has a remarkable therapeutic effect, wide anti-tumor spectrum, and no cross-resistance with other anti-tumor drugs, it has become one of the most widely used anti-tumor drugs in clinical use since its listing. However, existing camptothecin compounds still have significant side effects. Therefore, increasing the activity of camptothecin derivatives and reducing their toxicity have been the focus of research on such compounds. Summary of the invention
- the invention relates to a compound of formula I or a pharmaceutically acceptable salt, solvate, polymorph, tautomer or prodrug thereof:
- ⁇ is CR 5 or N
- At least one of X, Y, and Z is N;
- Ri R 2 and R 3 , R 4 and R 5 are each independently selected from the group consisting of hydrogen, halogen, hydroxy, alkoxy, amino, mono(alkyl)amino, di(alkyl)amino, cycloalkylamino, fluorenyl, An alkylthio group or a cyano group;
- Both R 3 and R 4 or both R 4 and R 5 or R 5 and both together with the carbon atom to which they are attached constitute a five to seven membered non-aromatic ring.
- the invention relates to a compound of formula I or a pharmaceutically acceptable salt, solvate, polymorph, tautomer or prodrug thereof:
- X is CR 3 ;
- Y is CR 4 ;
- Z is CR 5 ;
- Both R 3 and R 4 or both R 4 and R 5 or R 5 and both together with the carbon atom to which they are attached constitute a five to seven member non-aromatic containing one hetero atom selected from nitrogen, oxygen or sulfur.
- the remaining groups in the ring, and R 2 , R 3 , R 4 , R 5 are each independently selected from the group consisting of hydrogen, halogen, hydroxy, alkoxy, amino, mono(alkyl)amino, di(alkyl)amino , cycloalkylamino, decyl, alkylthio or cyano; or
- Ri R 2 and R 3 are each hydrogen, and R 4 and R 5 are different from each other and are each independently selected from a halogen, a hydroxyl group or an alkoxy group;
- Ri R 3 is hydrogen
- R 2 is amino or alkyl
- R 4 and R 5 are each independently selected from halogen, hydroxy, alkoxy, amino, mono(alkyl)amino or di(alkyl)amino.
- the invention in another aspect, relates to a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the invention, or a pharmaceutically acceptable salt, solvate, polymorph, tautomer or prodrug thereof, and a pharmaceutically acceptable carrier .
- the invention relates to a compound of the invention, or a pharmaceutically acceptable salt, solvate, polymorph, tautomer or prodrug thereof, or a pharmaceutical composition of the invention for use in prevention and/or Or treat a tumor.
- the invention relates to a compound of the invention, or a pharmaceutically acceptable salt, solvate, polymorph, tautomer or prodrug thereof, or a pharmaceutical composition of the invention, in the manufacture of an antineoplastic agent application.
- the invention relates to a method of treating and/or preventing a tumor in a mammal, in particular a human, comprising administering to a mammal, in particular a human, in need thereof a therapeutically effective amount of a compound of the invention or a pharmaceutical thereof Acceptable Salts, solvates, polymorphs, tautomers or prodrugs or pharmaceutical compositions of the invention.
- the manufacturer's instructions for use of the kit can be utilized, or the reaction can be carried out and purified according to methods well known in the art or as described in the present invention.
- the above techniques and methods can generally be carried out according to conventional methods well known in the art, as described in the various summaries and more specific references cited and discussed in this specification.
- the group and its substituents can be selected by those skilled in the art to provide stable structural moieties and compounds.
- substituent When a substituent is described by a conventional chemical formula written from left to right, the substituent also includes the chemically equivalent substituent obtained when the structural formula is written from right to left.
- substituent -CH 2 0- is equivalent to -OCH 2 -.
- d- 6 alkyl refers to an alkyl group as defined below having a total of from 1 to 6 carbon atoms.
- the total number of carbon atoms in the simplified symbol does not include carbon that may be present in the substituents of the group.
- halogen means fluoro, chloro, bromo or iodo.
- Haldroxy means an -OH group.
- Cyano means -CN.
- Amino means -NH 2 .
- alkyl means consisting only of carbon atoms and hydrogen atoms, and is not unsaturated.
- a bond a linear or branched hydrocarbon chain group having, for example, 1 to 12 (preferably 1 to 8, more preferably 1 to 6) carbon atoms and bonded to the remainder of the molecule by a single bond.
- alkyl groups include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, 2-methylbutyl, 2 , 2-dimethylpropyl, n-hexyl, heptyl, 2-methylhexyl, 3-methylhexyl, octyl, decyl and decyl.
- alkoxy refers to a radical of the formula -OR a where R a is alkyl as defined above.
- alkoxy groups include, but are not limited to, methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, isobutoxy, sec-butoxy, tert-butoxy, and the like.
- alkylthio refers to a radical of the formula -81, wherein R a is alkyl as defined above.
- alkoxy groups include, but are not limited to, methylthio, ethylthio, propylthio, isopropylthio, and the like.
- mono(indenyl)amino refers to a radical of the formula -NHR a , wherein is alkyl as defined above.
- mono(alkyl)amino groups include, but are not limited to, methylamino, ethylamino, isopropylamino, and the like.
- di(alkyl)amino refers to a radical of the formula -NR a R b wherein each of the radicals and the radicals are independently alkyl as defined above.
- examples of the di(alkyl)amino group include, but are not limited to, dimethylamino, diethylamino, dipropylamino, methylethylamino and the like.
- cycloalkyl as a group or part of another group means a stable non-aromatic monocyclic or polycyclic hydrocarbon group consisting solely of carbon atoms and hydrogen atoms, which may include condensing a ring system, a bridged ring system or a spiro ring system having from 3 to 15 carbon atoms, preferably from 3 to 10 carbon atoms, more preferably from 3 to 8 carbon atoms, and which is saturated or unsaturated and may be suitably employed
- the carbon atom is connected to the rest of the molecule by a single bond.
- a carbon atom in a cycloalkyl group may be optionally oxidized.
- cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, cyclohexadienyl, cycloheptyl, cyclooctyl, 1H- Indenyl, 2,3-indanyl, 1,2,3,4-tetrahydro-naphthyl, 5,6,7,8-tetrahydro-naphthyl, 8,9-dihydro-7H-benzene And cyclohepten-6-yl, 6,7,8,9-tetrahydro-5H-benzocycloheptenyl, 5,6,7,8,9,10-hexahydro-benzocyclooctenyl , fluorenyl, bicyclo [2.2.1] heptyl, 7,7-dimethyl-bicyclo[2.2.1]hept
- cycloalkylamino refers to a radical of the formula -NHR e , wherein R e is cycloalkyl as defined above.
- heterocyclyl as a group or part of another group means a stable consisting of 2 to 14 carbon atoms and 1 to 6 hetero atoms selected from nitrogen, oxygen and sulfur.
- a heterocyclic group may be a monocyclic, bicyclic, tricyclic or more cyclic ring system, which may include a fused ring system, a bridged ring system or a spiro ring system;
- the nitrogen, carbon or sulfur atom may optionally be oxidized; the nitrogen atom may optionally be quaternized; and the heterocyclic group may be partially or fully saturated.
- the cyclic group can be attached to the remainder of the molecule via a carbon atom or a hetero atom and through a single bond.
- one or more of the rings may be an aryl or heteroaryl group as defined hereinafter, provided that the point of attachment to the rest of the molecule is a non-aromatic ring atom.
- a heterocyclic group is preferably a stable 4 to 11 membered non-aromatic monocyclic, bicyclic, bridged or spiro group containing from 1 to 3 heteroatoms selected from nitrogen, oxygen or sulfur.
- heterocyclic groups include, but are not limited to, pyrrolidinyl, morpholinyl, piperazinyl, homopiperazinyl, piperidinyl, thiomorpholinyl, 2,7-diaza-spiro[3.5]fluorene.
- Alkan-7-yl 2-oxa-6-aza-spiro[3.3]heptane-6-yl, 2,5-diaza-bicyclo[2.2.1]heptan-2-yl, aza Cyclobutane, pyranyl, tetrahydropyranyl, thiopyranyl, tetrahydrofuranyl, oxazinyl, dioxocyclopentyl, tetrahydroisoquinolinyl, decahydroisoquinolinyl, imidazolinyl, Imidazolidinyl, quinazolidinyl, thiazolidinyl, isothiazolidinyl, isoxazolidinyl, indanyl, octahydroindenyl, octahydroisodecyl, pyrrolidinyl, pyrazolidinyl , phthalimido and the like.
- fused refers to a ring structure in which two or more rings have one or more bonds in common.
- non-aromatic ring includes cycloalkyl and heterocyclyl rings as defined above.
- the non-aromatic ring of the present application may contain an unsaturated bond, but the unsaturated bond does not cause the ring to produce aromaticity.
- aromatic refers to a delocalized electronic conjugate system having 4n + 2 electrons in the ring portion containing one or more rings, wherein n is an integer.
- optionally substituted alkyl means that the alkyl group is substituted or unsubstituted, i.e., the description includes both substituted alkyl and unsubstituted alkyl.
- a chemical moiety is generally considered to be a chemical entity that is embedded or attached to a molecule.
- the compound of the present invention contains an olefinic double bond
- the compound of the present invention is intended to contain E- and Z-geometric isomers unless otherwise stated.
- Tautomer refers to an isomer formed by the transfer of a proton from one atom of a molecule to another atom of the same molecule. All tautomeric forms of the compounds of the invention will also be embraced within the scope of the invention.
- pharmaceutically acceptable salt includes pharmaceutically acceptable acid addition salts and pharmaceutically acceptable base addition salts.
- “Pharmaceutically acceptable acid addition salt” means a salt formed with an inorganic or organic acid which retains the bioavailability of the free base without other side effects.
- Inorganic acid salts include, but are not limited to, hydrochlorides, hydrobromides, sulfates, nitrates, phosphates, and the like; organic acid salts include, but are not limited to, formate, acetate, 2,2-dichloroacetate , trifluoroacetate, propionate, hexanoate, octoate, decanoate, ⁇ -monoenoate, glycolate, gluconate, lactate, sebacate, hexane Acid salt, glutarate, malonate, oxalate, maleate, succinate, fumarate, tartrate, citrate, palmitate, stearate, oleate , cinnamate, laurate, malate, glutamate, pyroglutamate, aspartate, benzoate,
- “Pharmaceutically acceptable base addition salt” means a salt formed with an inorganic base or an organic base capable of maintaining the bioavailability of the free acid without other side effects.
- Salts derived from inorganic bases include, but are not limited to, sodium salts, potassium salts, lithium salts, ammonium salts, calcium salts, magnesium salts, iron salts, zinc salts, copper salts, manganese salts, aluminum salts, and the like.
- Preferred inorganic salts are ammonium salts, sodium salts, potassium salts, calcium salts and magnesium salts.
- Salts derived from organic bases include, but are not limited to, the following salts: primary amines, secondary amines and tertiary amines, substituted amines, including naturally substituted amines, cyclic amines, and basic ion exchange resins.
- ammonia isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, ethanolamine, diethanolamine, triethanolamine, dimethylethanolamine, 2-dimethylaminoethanol, 2-diethylaminoethanol, bicyclo Hexylamine, lysine, arginine, histidine, caffeine, procaine, choline, betaine, ethylenediamine, glucosamine, methylglucamine, theobromine, hydrazine, piperazine, piperazine Pyridine, N-ethylpiperidine, polyamine resin, and the like.
- Preferred organic bases include isopropylamine, diethylamine, ethanolamine, trimethylamine, dicyclohexylamine, choline, and caffeine.
- Polymorph refers to a different solid crystalline phase of certain compounds of the invention resulting from the presence of two or more different molecular arrangements in a solid state. Certain compounds of the invention may exist in more than one crystal form, and the invention is intended to include various crystal forms and mixtures thereof.
- solvate refers to an aggregate comprising one or more molecules of the compound of the invention and one or more solvent molecules.
- the solvent may be water, and the solvate in this case is a hydrate.
- the solvent may be an organic solvent.
- the compounds of the present invention may exist as hydrates, including monohydrates, dihydrates, hemihydrates, sesquihydrates, trihydrates, tetrahydrates, and the like, as well as the corresponding solvated forms.
- the compounds of the present invention form true solvates, but in some cases, it is also possible to retain only a variable amount of water or a mixture of water and a portion of the indefinite solvent.
- the compound of the present invention can be reacted in a solvent or precipitated or crystallized from a solvent. Solvates of the compounds of the invention are also included within the scope of the invention.
- the invention also includes prodrugs of the above compounds.
- prodrug means a compound which can be converted into a biologically active compound of the invention under physiological conditions or by solvolysis.
- prodrug refers to a pharmaceutically acceptable metabolic precursor of a compound of the invention.
- the prodrug may be inactive when administered to an individual in need thereof, but is converted in vivo to the active compound of the invention.
- Prodrugs are typically rapidly converted in vivo to produce the parent compound of the invention, for example by hydrolysis in blood.
- Prodrug compounds generally provide the advantage of solubility, histocompatibility or sustained release in mammalian organisms.
- Prodrugs include known amino protecting groups and carboxy protecting groups.
- pharmaceutical composition refers to a formulation of a compound of the present invention and a medium generally accepted in the art for delivery of a biologically active compound to a mammal (e.g., a human).
- the medium includes a pharmaceutically acceptable carrier.
- the purpose of the pharmaceutical composition is to promote the administration of the organism, and to facilitate the absorption of the active ingredient to exert biological activity.
- pharmaceutically acceptable refers to a substance that does not affect the biological activity or properties of the compounds of the present invention.
- composition e. carrier or diluent
- relatively non-toxic ie the substance can be applied to the individual without causing adverse biological reactions or A poor way to interact with any of the components contained in the composition.
- pharmaceutically acceptable carrier includes, but is not limited to, any adjuvants, carriers, excipients, glidants, sweeteners approved by the relevant government authorities for acceptable use by humans or domestic animals. , diluents, preservatives, dyes/colorants, flavoring agents, surfactants, wetting agents, dispersing agents, suspending agents, stabilizers, isotonic agents, solvents or emulsifiers.
- subject refers to an individual having a disease or condition, and the like, including mammals and non-mammals.
- mammals include, but are not limited to, any member of the mammalian class: humans, non-human primates (eg, chimpanzees and other mites and monkeys); livestock, such as cattle, horses, sheep, goats, pigs; domestic animals For example, rabbits, dogs and cats; laboratory animals, including rodents such as rats, mice and guinea pigs.
- non-mammals include, but are not limited to, birds and fish.
- the mammal is a human.
- preventing include the possibility of reducing the occurrence or deterioration of a disease or condition by a patient.
- treatment and other similar synonyms as used herein includes the following meanings:
- an "effective amount” for treatment refers to at least one agent or compound that, after administration, is sufficient to alleviate one or more symptoms of the disease or condition being treated to some extent.
- the amount. The result can be the reduction and/or alleviation of signs, symptoms or causes, or any other desired changes in the biological system.
- an "effective amount” for treatment is the amount of a composition comprising a compound disclosed herein that is required to provide a significant conditional relief effect in the clinic.
- An effective amount suitable for any individual case can be determined using techniques such as dose escalation testing.
- administering refers to a method of delivering a compound or composition to a desired site for biological action. These methods include, but are not limited to, oral routes, duodenal routes, parenteral injections (including intravenous, subcutaneous, intraperitoneal, intramuscular, intraarterial injection or infusion), topical administration, and rectal administration.
- pharmaceutical combination means a medical treatment obtained by mixing or combining more than one active ingredient, It includes both fixed and unfixed combinations of active ingredients.
- fixed combination refers to the simultaneous administration of at least one compound described herein and at least one synergistic agent to a patient in the form of a single entity or a single dosage form.
- unfixed combination refers to the simultaneous administration, combination or sequential administration of at least one of the compounds described herein to a patient in the form of separate entities and At least one synergistic formulation. These also apply to cocktail therapy, for example the administration of three or more active ingredients.
- the invention relates to a compound of formula I or a pharmaceutically acceptable salt, solvate, polymorph, tautomer or prodrug thereof:
- X is CR 3 or N
- Y is CR 4 or N
- Z is CR 5 or N
- At least one of X, Y, and ⁇ is ⁇ ;
- Ri R 2 and R 3 , R 4 and R 5 are each independently selected from the group consisting of hydrogen, halogen, hydroxy, alkoxy, amino, mono(alkyl)amino, di(alkyl)amino, cycloalkylamino, fluorenyl, An alkylthio group or a cyano group;
- Both R 3 and R 4 or both R 4 and R 5 or both R 5 and R together with the carbon atom to which they are attached constitute a five to seven membered non-aromatic ring.
- the invention relates to a compound of formula I or a pharmaceutically acceptable salt, solvate, polymorph, tautomer or prodrug thereof, wherein:
- Ri R 2 and R 3 , F and R 5 are each independently selected from the group consisting of hydrogen, halogen, hydroxy, d- 6 alkoxy, amino, mono(d- 6 alkyl)amino, di(d- 6 alkyl)amino, C 3 _ 8 cycloalkylamino, fluorenyl, d 6 alkylthio or cyano; or
- Both R 3 and R 4 or both R 4 and R 5 or both R 5 and R together with the carbon atom to which they are attached constitute five to seven heteroatoms containing from 0 to 3 selected from nitrogen, oxygen or sulfur. Yuan non-aromatic ring.
- the invention relates to a compound of formula I or a pharmaceutically acceptable salt, solvate, polymorph, tautomer or prodrug thereof, wherein:
- Ri R 2 and R 3 , R 4 , R 5 are each independently selected from hydrogen, hydroxy, d- 6 alkoxy, amino, mono(d- 6 alkyl)amino, di(d- 6 alkyl)amino, fluorenyl or d_ 6 alkylthio group.
- the invention relates to a compound of formula I or a pharmaceutically acceptable salt, solvate, polymorph, tautomer or prodrug thereof, wherein:
- Ri R 2 is hydrogen
- R 3 , R 4 , R 5 are each independently selected from hydrogen, hydroxy, d- 6 alkoxy, amino, mono(d- 6 alkyl)amino or Two (C 1-6 yard base) amino groups.
- the invention relates to a compound of formula I or a pharmaceutically acceptable salt thereof, dissolved
- X is CR 3 ;
- Y is CR 4 ;
- Z is CR 5 ;
- Both R 3 and R 4 or both R 4 and R 5 or R 5 and both together with the carbon atom to which they are attached constitute a five to seven member non-aromatic containing one hetero atom selected from nitrogen, oxygen or sulfur.
- a ring, and the remaining groups of Ri, R 2 , R 3 , R 4 , R 5 are each independently selected from the group consisting of hydrogen, halogen, hydroxy, alkoxy, amino, mono(alkyl)amino, di(alkyl) An amino group, a cycloalkylamino group, a decyl group, an alkylthio group or a cyano group; or
- Ri R 2 and R 3 are each hydrogen, and R 4 and R 5 are different from each other and are each independently selected from a halogen, a hydroxyl group or an alkoxy group;
- Ri R 3 is hydrogen
- R 2 is amino or alkyl
- R 4 and R 5 are each independently selected from halogen, hydroxy, alkoxy, amino, mono(alkyl)amino or di(alkyl)amino.
- the invention relates to a compound of formula I or a pharmaceutically acceptable salt, solvate, polymorph, tautomer or prodrug thereof, wherein:
- R 4 and R 5 together with the carbon atom to which they are attached constitute a five- to seven-membered non-aromatic ring containing one hetero atom selected from nitrogen, oxygen or sulfur, and R 2 and R 3 are each independently selected from hydrogen, halogen, hydroxy, d_ 6 alkoxy, amino, mono (d_ 6 alkyl) amino, di (d_ 6 alkyl) amino or C 3 _ 8 cycloalkyl group.
- the invention relates to a compound of formula I or a pharmaceutically acceptable salt, solvate, polymorph, tautomer or prodrug thereof, wherein:
- Both R 4 and R 5 together with the carbon atom to which they are attached constitute a hetero atom containing one selected from the group consisting of nitrogen, oxygen and sulfur.
- a five-membered non-aromatic ring, and Ri, R 2 , and R 3 are all hydrogen.
- the invention relates to a compound of formula I or a pharmaceutically acceptable salt, solvate, polymorph, tautomer or prodrug thereof, wherein:
- Ri R 2 and R 3 are each hydrogen, and R 4 and R 5 are different from each other and are each independently selected from a halogen, a hydroxyl group or a C alkoxy group.
- the invention relates to a compound of formula I or a pharmaceutically acceptable salt, solvate, polymorph, tautomer or prodrug thereof, wherein:
- Ri R 3 is hydrogen, R 2 is amino or d 6 alkyl, and R 4 and R 5 are each independently selected from halogen, hydroxy or
- the invention relates to a compound of formula I or a pharmaceutically acceptable salt thereof,
- the invention in another aspect, relates to a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the invention, or a pharmaceutically acceptable salt, solvate, polymorph, tautomer or prodrug thereof, and a pharmaceutically acceptable carrier .
- the pharmaceutical composition of the present invention can be formulated into solid, semi-solid, liquid or gaseous preparations such as tablets, capsules, powders, granules, ointments, solutions, suppositories, injections, inhalants, gels, microspheres and Aerosol.
- compositions of the present invention can be prepared by methods well known in the pharmaceutical art.
- a pharmaceutical composition intended for administration by injection can be prepared by combining a compound of the present invention or a pharmaceutically acceptable salt or prodrug thereof with sterilized distilled water to form a solution.
- Surfactants may be added to promote the formation of a homogeneous solution or suspension.
- Actual methods of preparing pharmaceutical compositions are known to those skilled in the art, for example see The Science and Practice of Pharmacy (Wo Pharmaceutical Science and Practice of the bucket), 20 th Edition (Philadelphia College of Pharmacy and Science, 2000).
- dosage forms suitable for oral administration include capsules, tablets, granules And syrup and the like.
- the compound of the present invention contained in these preparations may be a solid powder or granule; a solution or suspension in an aqueous or non-aqueous liquid; a water-in-oil or oil-in-water emulsion or the like.
- the above dosage forms can be prepared from the active compound with one or more carriers or excipients via conventional pharmaceutical methods.
- non-toxic carriers include, but are not limited to, mannitol, lactose, starch, magnesium stearate, cellulose, glucose, sucrose, and the like.
- Carriers for liquid preparations include, but are not limited to, water, physiological saline, aqueous dextrose, ethylene glycol, polyethylene glycol, and the like.
- the active compound can form a solution or suspension with the above carriers. The particular mode of administration and dosage form will depend on the physicochemical properties of the compound itself, as well as the severity of the disease being applied.
- the invention relates to a compound of the invention, or a pharmaceutically acceptable salt, solvate, polymorph, tautomer or prodrug thereof, or a pharmaceutical composition of the invention for use in prevention and/or Or treat a tumor.
- the invention relates to a compound of the invention, or a pharmaceutically acceptable salt, solvate, polymorph, tautomer or prodrug thereof, or a pharmaceutical composition of the invention, in the manufacture of an antineoplastic agent application.
- the invention in another aspect, relates to a method of treating and/or preventing a tumor in a mammal, in particular a human, comprising administering to a mammal, in particular a human, in need thereof a therapeutically effective amount of a compound of the invention or a pharmaceutical thereof Acceptable salts, solvates, polymorphs, tautomers or prodrugs or pharmaceutical compositions of the invention.
- tumor includes but is not limited to: leukemia, gastrointestinal stromal tumor, histiocytic lymphoma, non-small cell lung cancer, small cell lung cancer, pancreatic cancer, lung cancer, lung squamous cell carcinoma, lung adenocarcinoma, Breast cancer, prostate cancer, liver cancer, skin cancer, epithelial cell carcinoma, cervical cancer, ovarian cancer, colon cancer, colon cancer, nasopharyngeal cancer, brain cancer, bone cancer, esophageal cancer, melanoma, kidney cancer, oral cancer, etc. .
- a therapeutically effective daily dose is from about 0.001 mg/kg to about 100 mg/kg; a preferred therapeutically effective dose is from about 0.01 mg/kg to about 50 mg/kg; a more preferred therapeutically effective dose is about 1 mg/kg Up to approx. 25 mg/kg
- the compounds of the invention may be used in combination or in combination with one or more other compounds of the invention or one or more other anti-cancer drugs to treat and/or prevent tumors.
- Drugs that can be used in combination with the compounds of the invention include, but are not limited to, docetaxel, gemcitabine, cisplatin, carboplatin, Gleevec, temozolomide, doxorubicin, dacarbazine, trococaine, etoposide, soft red And cytarabine and the like.
- the intermediate compound functionality may need to be protected by a suitable protecting group.
- suitable protecting groups include trialkylsilyl or diarylalkylsilyl groups (e.g., tert-butyldimethylsilyl, tert-butyldiphenylsilyl or trimethylsilyl) , tetrahydropyranyl, benzyl, and the like.
- Suitable protecting groups for amino, mercapto and fluorenyl include t-butoxycarbonyl, benzyloxycarbonyl and the like.
- Suitable thiol protecting groups include -C(0)-R" (wherein R" is alkyl, aryl or arylalkyl), p-methoxybenzyl, trityl and the like.
- Suitable carboxy protecting groups include alkyl, aryl or aralkyl esters.
- protecting groups are detailed in Greene, TW and PGM Wuts, Protective Groups in Organi Synthesis , (1999), 4 th Ed., Wiley medium.
- the protecting group can also be a polymeric resin.
- the compound of the formula I of the present invention can be produced by one or two of the following synthetic routes.
- Test conditions a. p-toluenesulfonic acid (catalytic amount), toluene, reflux. Synthetic route two
- Test conditions a. p-toluenesulfonic acid (catalytic amount), toluene, reflux.
- the compound 2 used in the synthetic routes 1 and 2 can be produced by the following method.
- Step 4 7'-Methyl-5'-oxo-3',5'-dihydro-2cyclopentane-2,1'-pyridazine]-6'-carbonitrile
- Step 5 2-(6'-Cyano-5'-oxo-3',5'-dihydro-2 ⁇ -spiro[[1,3]dioxolane-2,1'-pyridazine ]-7'-yl)ethyl acetate
- Step 6 Ethyl bromide-[6-cyano-1,1-(ethylenedioxy-tetrahydropyridazin-7-yl)acetate
- Step 7 2-[N-p-Toluenesulfonyl-(R)-valine-yl]-2-[6-cyano-U-(ethylenedioxy)-5-oxo-1,2, 3,5-tetrahydropyridazin-7-yl]ethyl acetate
- Step 8 (S)-2-[N-p-Toluenesulfonyl-(R)-valine-yl]-2-[6-cyano-U-(ethylenedioxy)-5-oxo- 1,2,3,5-tetrahydropyridazin-7-yl]butyric acid B
- Step 9 (S)-2-[N-p-toluenesulfonyl-(R)-valine-based]-2-[6-(acetamidomethyl)-l,l-(ethylenedioxy) -5-oxo-1,2,3,5-tetrahydropyridazin-7-yl]
- Step 10 (S)-2-[N-p-Toluenesulfonyl-(R)-valine-based]-2-[6-(acetoxymethyl)-l,l-(ethylenedioxy) -5-oxo-1,2,3,5-tetrahydropyridazin-7-yl] -
- Step 11 (S)-4-Ethyl-6,6-(ethylenedioxy)-l,4,7,8-tetrahydro-4-hydroxypyrano[3,4-f]indole Pyrazine-3,10(6H)-dione
- Step 12 (S)-4-Ethyl-4-hydroxy-7,8-dihydro-1H-pyrano[3,4-f]pyridazine-3,6,10(4H)-trione (title compound)
- the instrument used for nuclear magnetic resonance spectroscopy was a Varian VNMRS-400 resonance spectrometer.
- the recording format of the NMR data is: proton number, peak type (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet), coupling constant (in Hertz) ).
- reaction mixture was refluxed overnight, cooled, washed with saturated sodium sulfate and brine, dried and evaporated. The residue was dissolved in MeOH (10 mL). The reaction system was stirred under a 1 atmosphere of hydrogen atmosphere for 2 hours, and the catalyst was removed by filtration and concentrated in vacuo to remove solvent.
- Step 1 4-[(di-tert-butoxycarbonyl)amino]-2-chloropyridine
- n-Butyllithium (11.3 mL, 2.4 M, 27 mmol) was added dropwise to a solution of diisopropylamine C2.74 g (27 mmol) in dry tetrahydrofurane (50 mL). After stirring for 30 minutes, it was cooled to -60 ° C, and a solution of 4-[(di-tert-butoxycarbonyl)amino]-2-chloropyridine (2.55 g, 7.8 mmol) in tetrahydrofuran was added dropwise. Stirring was continued for 1 hour, and a saturated ammonium chloride solution was added. The mixture was extracted with EtOAc. EtOAc was evaporated. The residue was purified by EtOAc EtOAc EtOAc (EtOAc) -8.20 (2H, m), 1.64 (9H, s), 1.53 (9H, s).
- 9-Hydroxy-10-aza-20CS)-camptothecin is a by-product obtained by the synthesis of 9-methoxy-10-aza-20CS)-camptothecin (Example 2, Step 5).
- Step 2 2-(Dimethylamino)-4-aminonicotinic acid tert-butyl
- Methyl 6-methoxy-3-nitropicolinate (1.03 g, 4.8 mmol), iron powder (1.08 g, 19 mmol), ammonium chloride (389 mg, 7.3 mmol) and ethanol/water (50 mL) The mixture of 4:1) was stirred at 80 ° C for 4 hours. After cooling, it was filtered, and the filtrate was poured into water and extracted with ethyl acetate. The organic phase was washed with EtOAc EtOAc m.
- 1,4-Dibromo-2-fluorobenzene (6.86 g, 27 mmol), ethylene glycol (35 mL), N-methylpyrrolidone (35 mL) and potassium t-butoxide (1 1.2 g, 95 mmol)
- the mixture was stirred at 100 ° C under nitrogen overnight. Cool to room temperature and slowly add water (15 mL) over 30 minutes. After filtration, the filter cake was washed with ethylene glycol, water was added to the filtrate, and the mixture was stirred for 30 minutes. Cool to 15 ° C and let stand for 1 hour.
- 2,3-Dihydrobenzofuran-6-carboxaldehyde (249 mg, 1.7 mmol) was added to concentrated nitric acid (5 mL), stirred at room temperature for 2 hrs, then poured into ice water, extracted with ethyl acetate and washed with brine , concentrated in vacuo. A mixture of ethanol/water (12 mL, 5:1), iron powder (290 mg, 5.2 mmol) and ammonium chloride (104 mg, 1.9 mmol) was added to the residue and the mixture was stirred at 80 ° C for 2 hours. , filtered.
- Step 5 7-Amino-9-methoxy-10-fluoro-20-(S)-camptothecin (title compound)
- Human lung cancer cell A549 was cultured in DMEM (Gibco®) medium supplemented with 10% fetal bovine serum (Hyclone®); human colon cancer cells SW620, HT-29 and human prostate cancer cell PC-3 were supplemented with 10 % fetal calf serum (Hyclone®) in RPMI-1640 (Gibco®) medium was cultured.
- the culture conditions of the above cells were 37 ° C, 95% air and 5% C0 2 , cultured in a 25 cm 2 or 75 cm 2 plastic tissue culture flask (Coming®), and subcultured 2 to 3 times a week.
- Cells were 1 ⁇ 10 3 cells/well (A549), 2 ⁇ 10 3 cells/well (SW620 and HT29), 3 ⁇ 10 3 fine
- the density of cells/well (PC-3) was seeded in 96-well cell culture plates (Coming®) at 195 ⁇ /well and at 37 °C. C, 95% air and 5% C0 2 were cultured.
- the test compound was added, and the compound was serially diluted with 10% DMSO from 10 mM (dissolved in 100% DMSO), and 4 ⁇ M each was added to 96 ⁇ L of serum-free medium.
- the compound diluted in 5 ⁇ of the medium was added to the plate inoculated with the cells.
- the final concentration of DMSO in the cell culture medium was 0.1%, and the final concentration of the test compound was 0.3 nM to 10 ⁇ M.
- the cells were incubated for 3 days at 37 °C.
Abstract
本发明涉及式I的化合物、其药物组合物及其作为抗肿瘤药的用途。
Description
喜树碱类化合物及其制备和用途 技术领域
本发明涉及一类新的具有抗肿瘤活性的喜树碱类化合物, 其药物组合物及其用 途。 背景技术
喜树碱 (camptothecin, CPT, IUPAC 命名: (s)-4-乙基 -4-羟基 -1H-吡喃 [3'4' :6,7] 吲哚嗪并 [l,2-b]喹啉 -3,14(4H,12H)-二酮)是在 1966年首先由 Wall等人 [J. Am. Chem. Soc, 1966, 88(16), 3888-990]从中国珙桐科植物喜树 (camptotheca acuminate)中提取出 来的一种生物碱。 试验表明, 它对白血病和许多实体瘤具有抗肿瘤活性, 是一种天然 的细胞毒性化合物。 到 20世纪 70年代, 已经对 CPT进行了 I期和 II期临床试验, 发现尽管其具有抗肿瘤活性, 但存在着许多的副作用。 副作用包括骨髓抑制、 胃肠道 毒性、 出血性膀胱炎、 脱发、 腹泻、 恶心、 呕吐等。
为了得到活性高、 毒性小的喜树碱衍生物, 人们进行了结构改造和优化, 通过半 合成及全合成方法合成了一系列喜树碱衍生物。 1994年, 美国 FDA批准喜树碱类药 物伊立替康 (Irinotecan , CPT-11)上市, 用于治疗结直肠癌。 1996 年, 拓扑替康 (Topotecan)获准上市用于治疗卵巢癌, 美国 FDA又于 1999年批准其作为小细胞肺癌 (SCLC)的二线治疗药物。 目前尚有十余种喜树碱类药物处于临床试验阶段。
喜树碱是继紫杉醇后的第二个从植物中提取的抗癌药物。由于喜树碱类药物疗效 显著、 抗肿瘤谱广、 与其它抗肿瘤药物无交叉耐药性, 因此自其上市以来, 已成为临 床上使用最多的抗肿瘤药物之一。 但现有的喜树碱类化合物依然存在明显的副作用。 因此提高喜树碱类衍生物的活性, 降低其毒性一直是这类化合物研究的重点。 发明内容
一方面, 本发明涉及式 I的化合物或其药学上可接受的盐、溶剂化物、多晶型物、 互变异构体或前药:

其巾:
X为 CR3或 N;
Y为 CR4或 Ν;
Ζ为 CR5或 N;
X、 Y、 Z中至少有一个为 N;
Ri R2以及 R3、 R4、 R5各自独立地选自氢、 卤素、 羟基、 烷氧基、 氨基、 单 (烷 基)氨基、 二 (烷基)氨基、 环烷基氨基、 巯基、 烷硫基或氰基;
或者
R3和 R4二者或 R4和 R5二者或 R5和 二者与它们所连接的碳原子共同组成五 至七元非芳香环。
另一方面, 本发明涉及式 I的化合物或其药学上可接受的盐、 溶剂化物、 多晶型 物、 互变异构体或前药:

其巾:
X为 CR3;
Y为 CR4 ;
Z为 CR5;
R3和 R4二者或 R4和 R5二者或 R5和 二者与它们所连接的碳原子共同组成含 有 1个选自氮、 氧或硫的杂原子的五至七元非芳香环, 且 、 R2、 R3、 R4、 R5中的 其余基团各自独立地选自氢、 卤素、 羟基、 烷氧基、 氨基、 单 (烷基)氨基、 二 (烷基) 氨基、 环烷基氨基、 巯基、 烷硫基或氰基; 或者
Ri R2、 R3均为氢, 且 R4、 R5互相不同且各自独立地选自卤素、 羟基或烷氧基; 或者
Ri R3均为氢, R2为氨基或烷基, 且 R4、 R5各自独立地选自卤素、 羟基、 烷氧 基、 氨基、 单 (烷基)氨基或二 (烷基)氨基。
另一方面, 本发明涉及药物组合物, 其包含本发明的化合物或其药学上可接受的 盐、 溶剂化物、 多晶型物、 互变异构体或前药, 以及药学上可接受的载体。
另一方面,本发明涉及本发明的化合物或其药学上可接受的盐、溶剂化物、多晶型物、 互变异构体或前药或者本发明的药物组合物, 其用于预防和 /或治疗肿瘤。
另一方面, 本发明涉及本发明的化合物或其药学上可接受的盐、 溶剂化物、 多晶 型物、 互变异构体或前药或者本发明的药物组合物在制备抗肿瘤药中的应用。
另一方面, 本发明涉及治疗和 /或预防哺乳动物、 特别是人的肿瘤的方法, 所述方法 包括对有需要的哺乳动物、特别是人给予治疗有效量的本发明的化合物或其药学上可接受
的盐、 溶剂化物、 多晶型物、 互变异构体或前药或者本发明的药物组合物。 具体实施方式
定义
除非另有定义,否则本文所有科技术语具有的涵义与权利要求主题所属领域技术人员 通常理解的涵义相同。 除非另有说明, 本文全文引用的所有专利、专利申请、 公开材料通 过引用方式整体并入本文。
应理解,上述简述和下文的详述为示例性且仅用于解释,而不对本发明主题作任何限 制。 在本申请中, 除非另有具体说明, 否则使用单数时也包括复数。 还应注意, 除非另有 说明, 否则所用 "或"、 "或者"表示 "和 /或"。 此外, 所用术语"包括"以及其它形式, 例 如 "包含"、 "含"和 "含有"并非限制性。
可在参考文献 (包括 Carey and Sundberg "ADVANCED ORGANIC CHEMISTRY, 4th ED." Vols. A (2000) and B (2001), Plenum Press, New York) 中找到对标准化学术语的定义。 除非另有说明, 否则采用本领域技术范围内的常规方法, 如质谱、 NMR、 IR和 UV/Vis 光谱法和药理学方法。 除非提出具体定义, 否则本文在分析化学、有机合成化学以及药物 和药物化学的有关描述中采用的术语是本领域已知的。可在化学合成、化学分析、药物制 备、制剂和递送, 以及对患者的治疗中使用标准技术。例如, 可利用厂商对试剂盒的使用 说明,或者按照本领域公知的方式或本发明的说明来实施反应和进行纯化。通常可根据本 说明书中引用和讨论的多个概要性和较具体的文献中的描述,按照本领域熟知的常规方法 实施上述技术和方法。在本说明书中,可由本领域技术人员选择基团及其取代基以提供稳 定的结构部分和化合物。
当通过从左向右书写的常规化学式描述取代基时,该取代基也同样包括从右向左书写 结构式时所得到的在化学上等同的取代基。 举例而言, -CH20-等同于 -OCH2-。
本文所用的章节标题仅用于组织文章的目的,而不应被解释为对所述主题的限制。本 申请中引用的所有文献或文献部分包括但不限于专利、专利申请、 文章、 书籍、操作手册 和论文, 均通过引用方式整体并入本文。
在本文中定义的某些化学基团前面通过简化符号来表示该基团中存在的碳原子 总数。 例如, d_6烷基是指具有总共 1至 6个碳原子的如下文所定义的烷基。 简化符 号中的碳原子总数不包括可能存在于所述基团的取代基中的碳。
除前述以外, 当用于本申请的说明书及权利要求书中时, 除非另外特别指明, 否 则以下术语具有如下所示的含义。
在本申请中, 术语"卤素"是指氟、 氯、 溴或碘。
"羟基"是指 -OH基团。
"巯基"是指 -SH基团。
"氧代 "是指 =0基团。
"氰基"是指 -CN。
"氨基"是指 -NH2。
在本申请中,作为基团或是其它基团的一部分 (例如用在卤素取代的烷基等基团中), 术语"烷基"意指仅由碳原子和氢原子组成、 不含不饱和键、 具有例如 1至 12个 (优 选为 1至 8个, 更优选为 1至 6个)碳原子且通过单键与分子的其余部分连接的直链 或支链的烃链基团。烷基的实例包括但不限于甲基、 乙基、 正丙基、异丙基、 正丁基、 异丁基、 仲丁基、 叔丁基、 正戊基、 2-甲基丁基、 2,2-二甲基丙基、 正己基、 庚基、 2- 甲基己基、 3-甲基己基、 辛基、 壬基和癸基等。
在本申请中, 术语 "烷氧基"是指式 -ORa基团, 其中 Ra为如上文所定义的烷基。 烷氧基的实例包括但不限于甲氧基、 乙氧基、 丙氧基、 异丙氧基、 正丁氧基、 异丁氧 基、 仲丁氧基和叔丁氧基等。
在本申请中, 术语 "烷硫基"是指式 -81^基团, 其中 Ra为如上文所定义的烷基。 烷氧基的实例包括但不限于甲硫基、 乙硫基、 丙硫基、 异丙硫基等。
在本申请中, 术语 "单 (浣基)氨基"是指式 -NHRa基团, 其中 为如上文所定义 的烷基。 单 (烷基)氨基的实例包括但不限于甲氨基、 乙氨基、 异丙氨基等。
在本申请中, 术语 "二 (烷基)氨基" 是指式 -NRaRb基团, 其中 ^和 ^各自独立 地为如上文所定义的烷基。 二 (烷基)氨基的实例包括但不限于二甲氨基、 二乙氨基、 二丙氨基、 甲基乙基氨基等。
在本申请中, 作为基团或是其它基团的一部分, 术语 "环烷基 " 意指仅由碳原子 和氢原子组成的稳定的非芳香族单环或多环烃基, 其可包括稠合环体系、桥环体系或 螺环体系, 具有 3至 15个碳原子, 优选具有 3至 10个碳原子, 更优选具有 3至 8个 碳原子,且其为饱和或不饱和并可经由任何适宜的碳原子通过单键与分子的其余部分 连接。 除非本说明书中另外特别指明, 环烷基中的碳原子可以任选地被氧化。 环烷基 的实例包括但不限于环丙基、 环丁基、 环戊基、 环戊烯基、 环己基、 环己烯基、 环己 二烯基、 环庚基、 环辛基、 1H-茚基、 2,3-二氢化茚基、 1,2,3,4-四氢-萘基、 5,6,7,8-四 氢-萘基、 8,9-二氢 -7H-苯并环庚烯 -6-基、 6,7,8,9-四氢 -5H-苯并环庚烯基、 5,6,7,8,9,10- 六氢-苯并环辛烯基、芴基、二环 [2.2.1]庚基、 7,7-二甲基 -二环 [2.2.1]庚基、二环 [2.2.1] 庚烯基、 二环 [2.2.2]辛基、 二环 [3.1.1]庚基、 二环 [3.2.1]辛基、 二环 [2.2.2]辛烯基、 二 环 [3.2.1]辛烯基、 金刚烷基、 八氢 -4,7-亚甲基 -1H-茚基和八氢 -2,5-亚甲基 -并环戊二烯 基等。
在本申请中, 术语"环烷基氨基"是指式 -NHRe基团, 其中 Re为如上文所定义的 环烷基。
在本申请中, 作为基团或是其它基团的一部分, 术语 "杂环基"意指由 2至 14个 碳原子以及 1至 6个选自氮、 氧和硫的杂原子组成的稳定的 3元至 20元非芳香族环 状基团。 除非本说明书中另外特别指明, 否则杂环基可以为单环、 双环、 三环或更多 环的环体系, 其可包括稠合环体系、 桥环体系或螺环体系; 其杂环基中的氮、 碳或硫 原子可任选地被氧化; 氮原子可任选地被季铵化; 且杂环基可为部分或完全饱和。 杂
环基可以经由碳原子或者杂原子并通过单键与分子其余部分连接。在包含稠环的杂环 基中, 一个或多个环可以是下文所定义的芳基或杂芳基, 条件是与分子其余部分的连 接点为非芳香族环原子。 就本发明的目的而言, 杂环基优选为包含 1至 3个选自氮、 氧或硫的杂原子的稳定的 4元至 11元非芳香性单环、 双环、 桥环或螺环基团, 更优 选为包含 1个选自氮、氧或硫的杂原子的稳定的 5元至 7元非芳香性单环基团。 杂环 基的实例包括但不限于: 吡咯烷基、 吗啉基、 哌嗪基、 高哌嗪基、 哌啶基、 硫代吗啉 基、 2,7-二氮杂-螺 [3.5]壬烷 -7-基、 2-氧杂 -6-氮杂-螺 [3.3]庚烷 -6-基、 2,5-二氮杂 -双环 [2.2.1]庚烷 -2-基、 氮杂环丁烷基、 吡喃基、 四氢吡喃基、 噻喃基、 四氢呋喃基、 噁嗪 基、 二氧环戊基、 四氢异喹啉基、 十氢异喹啉基、 咪唑啉基、 咪唑烷基、 喹嗪基、 噻 唑烷基、 异噻唑烷基、 异噁唑烷基、 二氢吲哚基、 八氢吲哚基、 八氢异吲哚基、 吡咯 烷基、 吡唑烷基、 邻苯二甲酰亚氨基等。
本文单独或组合使用的术语 "稠合 "是指其两个或多个环共同具有一个或多个键 的环结构。
在本申请中, 术语 "非芳香环"包括上文所定义的环烷基和杂环基环。 本申请的 非芳香环可包含不饱和键, 但该不饱和键并不导致该环产生芳香性。
本文中使用的术语"芳香性 "是指包含一个或多个环的环部分具有 4n+2个电子的 离域化电子共扼体系, 其中 n为整数。
在本申请中, "任选的"或 "任选地"表示随后描述的事件或状况可能发生也可能 不发生, 且该描述同时包括该事件或状况发生和不发生的情况。 例如, "任选地被取 代的烷基"表示烷基被取代或未被取代, 即该描述同时包括被取代的烷基与未被取代 的烷基。
本文所用术语 "部分"、 "结构部分"、 "化学部分"、 "基团"、 "化学基团"是指分子中 的特定片段或官能团。 化学部分通常被认为是嵌入或附加到分子上的化学实体。
当本发明的化合物中含有烯双键时, 除非另有说明, 否则本发明的化合物旨在包含 E-和 Z-几何异构体。
"互变异构体"是指质子从分子的一个原子转移至相同分子的另一个原子而形成的异 构体。 本发明的化合物的所有互变异构形式也将包含在本发明的范围内。
在本申请中, 术语"药学上可接受的盐"包括药学上可接受的酸加成盐和药学上可 接受的碱加成盐。
"药学上可接受的酸加成盐"是指能够保留游离碱的生物有效性而无其它副作用的, 与无机酸或有机酸所形成的盐。无机酸盐包括但不限于盐酸盐、氢溴酸盐、硫酸盐、硝酸 盐、 磷酸盐等; 有机酸盐包括但不限于甲酸盐、 乙酸盐、 2,2-二氯乙酸盐、 三氟乙酸盐、 丙酸盐、 己酸盐、 辛酸盐、 癸酸盐、 ^一碳烯酸盐、 乙醇酸盐、 葡糖酸盐、 乳酸盐、 癸 二酸盐、 己二酸盐、 戊二酸盐、 丙二酸盐、 草酸盐、 马来酸盐、 琥珀酸盐、 富马酸盐、 酒石酸盐、 柠檬酸盐、 棕榈酸盐、 硬脂酸盐、 油酸盐、 肉桂酸盐、 月桂酸盐、 苹果酸 盐、 谷氨酸盐、 焦谷氨酸盐、 天冬氨酸盐、 苯甲酸盐、 甲磺酸盐、 苯磺酸盐、 对甲苯磺
酸盐、 海藻酸盐、 抗坏血酸盐、 水杨酸盐、 4-氨基水杨酸盐、 萘二磺酸盐等。 这些盐 可通过本领域已知的方法制备。
"药学上可接受的碱加成盐"是指能够保持游离酸的生物有效性而无其它副作用的、 与无机碱或有机碱所形成的盐。衍生自无机碱的盐包括但不限于钠盐、钾盐、锂盐、铵盐、 钙盐、 镁盐、 铁盐、 锌盐、 铜盐、 锰盐、 铝盐等。 优选的无机盐为铵盐、 钠盐、 钾盐、 钙 盐及镁盐。衍生自有机碱的盐包括但不限于以下的盐: 伯胺类、仲胺类及叔胺类, 被取代 的胺类, 包括天然的被取代胺类、 环状胺类及碱性离子交换树脂, 例如氨、 异丙胺、三甲 胺、 二乙胺、 三乙胺、 三丙胺、 乙醇胺、 二乙醇胺、 三乙醇胺、 二甲基乙醇胺、 2-二甲氨 基乙醇、 2-二乙氨基乙醇、 二环己胺、 赖氨酸、 精氨酸、 组氨酸、 咖啡因、 普鲁卡因、 胆 碱、 甜菜碱、 乙二胺、 葡萄糖胺、 甲基葡萄糖胺、 可可碱、 嘌呤、 哌嗪、 哌啶、 N-乙基 哌啶、 聚胺树脂等。 优选的有机碱包括异丙胺、 二乙胺、 乙醇胺、 三甲胺、 二环己基胺、 胆碱及咖啡因。 这些盐可通过本专业已知的方法制备。
"多晶型物"是指本发明的某些化合物在固体状态下由于存在两种或两种以上不同分 子排列而产生的不同固体结晶相。本发明的某些化合物可以存在一种以上的晶型,本发明 旨在包括各种晶型及其混合物。
通常,结晶化作用会产生本发明化合物的溶剂化物。本发明中使用的术语 "溶剂化物" 是指包含一个或多个本发明化合物分子与一个或多个溶剂分子的聚集体。 溶剂可以是水, 该情况下的溶剂化物为水合物。 或者, 溶剂可以是有机溶剂。 因此, 本发明的化合物可以 以水合物存在, 包括单水合物、 二水合物、 半水合物、 倍半水合物、 三水合物、 四水合物 等, 以及相应的溶剂化形式。本发明化合物可形成真实的溶剂化物, 但在某些情况下, 也 可以仅保留不定的水或者水加上部分不定溶剂的混合物。本发明的化合物可以在溶剂中反 应或者从溶剂中沉淀析出或结晶出来。本发明化合物的溶剂化物也包含在本发明的范围之 内。
本发明还包括上述化合物的前药。 在本申请中, 术语 "前药" 表示可在生理学条 件下或通过溶剂分解而被转化成本发明的生物活性化合物的化合物。 因此, 术语 "前 药"是指本发明的化合物的药学上可接受的代谢前体。 当被给予有需要的个体时, 前 药可以不具有活性, 但在体内被转化成本发明的活性化合物。前药通常在体内迅速转 化, 而产生本发明的母体化合物, 例如通过在血液中水解来实现。 前药化合物通常在 哺乳动物生物体内提供溶解度、 组织相容性或缓释的优点。 前药包括已知的氨基保护 基和羧基保护基。 具体的前药制备方法可参照 Saulnier, M. G., et al., Bioorg. Med. Chem. Lett. 1994, 4, 1985-1990; Greenwald, R. B., et al., J. Med. Chem. 2000, 43, 475。
在本申请中, "药物组合物" 是指本发明化合物与本领域通常接受的用于将生物 活性化合物输送至哺乳动物 (例如人) 的介质的制剂。 该介质包括药学上可接受的载 体。 药物组合物的目的是促进生物体的给药, 利于活性成分的吸收进而发挥生物活性。
本文所用术语 "药学上可接受的"是指不影响本发明化合物的生物活性或性质的物质
(如载体或稀释剂),并且相对无毒, 即该物质可施用于个体而不造成不良的生物反应或以
不良方式与组合物中包含的任意组分相互作用。
在本申请中, "药学上可接受的载体" 包括但不限于任何被相关的政府管理部门 许可为可接受供人类或家畜使用的佐剂、 载体、 赋形剂、 助流剂、 增甜剂、 稀释剂、 防腐剂、 染料 /着色剂、 矫味剂、 表面活性剂、 润湿剂、 分散剂、 助悬剂、 稳定剂、 等渗剂、 溶剂或乳化剂。
本文所用的有关术语 "受试者"、 "患者"或 "个体"是指患有疾病或病症等的个体, 包括哺乳动物和非哺乳动物。 哺乳动物的实例包括但不限于哺乳动物纲的任何成员: 人, 非人的灵长类动物(例如黑猩猩和其它猿类和猴); 家畜, 例如牛、 马、 绵羊、 山羊、 猪; 家养动物, 例如兔、 狗和猫; 实验室动物, 包括啮齿类动物, 例如大鼠、 小鼠和豚鼠等。 非哺乳动物的实例包括但不限于鸟类和鱼类等。在本文提供的一个有关方法和组合物的实 施方案中, 所述哺乳动物为人。
本文所用术语 "预防的"、 "预防"和 "防止"包括使病患减少疾病或病症的发生或恶 化的可能性。
本文所用的术语 "治疗"和其它类似的同义词包括以下含义:
(i)预防疾病或病症在哺乳动物中出现,特别是当这类哺乳动物易患有该疾病或病症, 但尚未被诊断为已患有该疾病或病症时;
(ii) 抑制疾病或病症, 即遏制其发展;
(iii) 缓解疾病或病症, 即, 使该疾病或病症的状态消退; 或者
(iv) 减轻该疾病或病症所造成的症状。
本文所使用术语 "有效量"、 "治疗有效量"或 "药学有效量"是指服用后足以在某种 程度上缓解所治疗的疾病或病症的一个或多个症状的至少一种药剂或化合物的量。其结果 可以为迹象、 症状或病因的消减和 /或缓解, 或生物系统的任何其它所需变化。 例如, 用 于治疗的"有效量"是在临床上提供显著的病症缓解效果所需的包含本文公开化合物的组 合物的量。 可使用诸如剂量递增试验的技术测定适合于任意个体病例中的有效量。
本文所用术语 "服用 "、 "施用 "、 "给药"、 "给予"等是指能够将化合物或组合物递送 到进行生物作用的所需位点的方法。 这些方法包括但不限于口服途径、 经十二指肠途径、 胃肠外注射 (包括静脉内、 皮下、 腹膜内、 肌内、 动脉内注射或输注)、 局部给药和经直 肠给药。本领域技术人员熟知可用于本文所述化合物和方法的施用技术,例如在 Goodman and Gilman, The Pharmacological Basis of Therapeutics, current ed.; Pergamon; and Remington's, Pharmaceutical Sciences (current edition), Mack Publishing Co., Easton, Pa中讨 论的那些。 在优选的实施方案中, 本文讨论的化合物和组合物通过口服施用。
本文所使用术语 "药物组合"、 "药物联用"、 "联合用药"、 "施用其它治疗"、 "施用其 它治疗剂"等是指通过混合或组合不止一种活性成分而获得的药物治疗,其包括活性成分 的固定和不固定组合。术语"固定组合"是指以单个实体或单个剂型的形式向患者同时施 用至少一种本文所述的化合物和至少一种协同药剂。术语 "不固定组合"是指以单独实体 的形式向患者同时施用、合用或以可变的间隔时间顺次施用至少一种本文所述的化合物和
至少一种协同制剂。 这些也应用到鸡尾酒疗法中, 例如施用三种或更多种活性成分 本发明的具体实施方式
一方面, 本发明涉及式 I的化合物或其药学上可接受的盐、溶剂化物、多晶型物、 互变异构体或前药:

其巾:
X为 CR3或 N;
Y为 CR4或 N;
Z为 CR5或 N;
X、 Y、 Ζ中至少有一个为 Ν;
Ri R2以及 R3、 R4、 R5各自独立地选自氢、 卤素、 羟基、 烷氧基、 氨基、 单 (烷 基)氨基、 二 (烷基)氨基、 环烷基氨基、 巯基、 烷硫基或氰基;
或者
R3和 R4二者或 R4和 R5二者或 R5和 R 二者与它们所连接的碳原子共同组成五 至七元非芳香环。
在一个优选的实施方式中, 本发明涉及式 I的化合物或其药学上可接受的盐、 溶 剂化物、 多晶型物、 互变异构体或前药, 其中:
Ri R2以及 R3、 F 、 R5各自独立地选自氢、 卤素、 羟基、 d_6烷氧基、 氨基、 单 (d_6烷基)氨基、 二 (d_6烷基)氨基、 C3_8环烷基氨基、 巯基、 d_6烷硫基或氰基; 或者
R3和 R4二者或 R4和 R5二者或 R5和 R 二者与它们所连接的碳原子共同组成含 有 0至 3个选自氮、 氧或硫的杂原子的五至七元非芳香环。
在一个更优选的实施方式中, 本发明涉及式 I的化合物或其药学上可接受的盐、 溶剂化物、 多晶型物、 互变异构体或前药, 其中:
Ri R2以及 R3、 R4、 R5各自独立地选自氢、 羟基、 d_6烷氧基、 氨基、 单 (d_6 烷基)氨基、 二 (d_6烷基)氨基、 巯基或 d_6烷硫基。
在一个更优选的实施方式中, 本发明涉及式 I的化合物或其药学上可接受的盐、 溶剂化物、 多晶型物、 互变异构体或前药, 其中:
Ri R2均为氢; 且
R3、 R4、 R5各自独立地选自氢、 羟基、 d_6烷氧基、 氨基、 单 (d_6烷基)氨基或
二 (C1-6院基)氨基。
在一个优选的实施方式中, 本发明涉及式 I的化合物或其药学上可接受的盐、 溶

其巾:
X为 CR3;
Y为 CR4 ;
Z为 CR5;
R3和 R4二者或 R4和 R5二者或 R5和 二者与它们所连接的碳原子共同组成含 有 1个选自氮、 氧或硫的杂原子的五至七元非芳香环, 且 Ri、 R2、 R3、 R4、 R5中的 其余基团各自独立地选自氢、 卤素、 羟基、 烷氧基、 氨基、 单 (烷基)氨基、 二 (烷基) 氨基、 环烷基氨基、 巯基、 烷硫基或氰基; 或者
Ri R2、 R3均为氢, 且 R4、 R5互相不同且各自独立地选自卤素、 羟基或烷氧基; 或者
Ri R3均为氢, R2为氨基或烷基, 且 R4、 R5各自独立地选自卤素、 羟基、 烷氧 基、 氨基、 单 (烷基)氨基或二 (烷基)氨基。
在一个优选的实施方式中, 本发明涉及式 I的化合物或其药学上可接受的盐、 溶 剂化物、 多晶型物、 互变异构体或前药, 其中:
R4和 R5二者与它们所连接的碳原子共同组成含有 1个选自氮、 氧或硫的杂原子 的五至七元非芳香环, 且 、 R2、 R3各自独立地选自氢、 卤素、 羟基、 d_6烷氧基、 氨基、 单 (d_6烷基)氨基、 二 (d_6烷基)氨基或 C3_8环烷基氨基。
在一个更优选的实施方式中, 本发明涉及式 I的化合物或其药学上可接受的盐、 溶剂化物、 多晶型物、 互变异构体或前药, 其中:
R4和 R5二者与它们所连接的碳原子共同组成含有 1个选自氮、 氧和硫的杂原子
的五元非芳香环, 且 Ri、 R2、 R3均为氢。
在另一个优选的实施方式中, 本发明涉及式 I的化合物或其药学上可接受的盐、 溶剂化物、 多晶型物、 互变异构体或前药, 其中:
Ri R2、 R3均为氢, 且 R4、 R5互相不同且各自独立地选自卤素、 羟基或 C^烷 氧基。
在另一个优选的实施方式中, 本发明涉及式 I的化合物或其药学上可接受的盐、 溶剂化物、 多晶型物、 互变异构体或前药, 其中:
Ri R3均为氢, R2为氨基或 d_6烷基, 且 R4、 R5各自独立地选自卤素、 羟基或
Cl_6院氧基。
在一个更优选的实施方式中, 本发明涉及式 I的化合物或其药学上可接受的盐、

另一方面, 本发明涉及药物组合物, 其包含本发明的化合物或其药学上可接受的 盐、 溶剂化物、 多晶型物、 互变异构体或前药, 以及药学上可接受的载体。
本发明的药物组合物可以被配制为固态、 半固态、 液态或气态制剂, 如片剂、 胶囊、 粉剂、 颗粒剂、 膏剂、 溶液剂、 栓剂、 注射剂、 吸入剂、 凝胶剂、 微球及气溶胶。
本发明的药物组合物可以通过制药领域中公知的方法制备。例如, 旨在注射给药的药 物组合物可以通过将本发明的化合物或其药学上可接受的盐或前药与灭菌的蒸馏水组合 来制备, 从而形成溶液剂。可添加表面活性剂以促进形成均匀溶液或混悬液。制备药物组 合物的实际方法为本领域技术人员所已知, 例如可参见 The Science and Practice of Pharmacy (制药禾斗学与实践), 20th Edition (Philadelphia College of Pharmacy and Science, 2000)。
本发明的药物组合物的给药途径包括但不限于口服、局部、经皮、肌肉、静脉、吸入、 肠胃外、 舌下、 直肠、 阴道及鼻内。 例如, 适合口服给药的剂型包括胶囊、 片剂、 颗粒剂
以及糖浆等。这些制剂中包含的本发明的化合物可以是固体粉末或颗粒;水性或非水性液 体中的溶液或是混悬液;油包水或水包油的乳剂等。上述剂型可由活性化合物与一种或多 种载体或辅料经由通用的药剂学方法制成。 上述的载体需要与活性化合物或其他辅料兼 容。 对于固体制剂, 常用的无毒载体包括但不限于甘露醇、 乳糖、 淀粉、 硬脂酸镁、 纤维 素、 葡萄糖、 蔗糖等。 用于液体制剂的载体包括但不限于水、 生理盐水、 葡萄糖水溶液、 乙二醇和聚乙二醇等。活性化合物可与上述载体形成溶液或是混悬液。具体的给药方式和 剂型取决于化合物本身的理化性质以及所应用疾病的严重程度等。本领域技术人员能够根 据上述因素并结合其自身具有的知识来确定具体的给药途径。 例如可参见: 李俊, 《临床 药理学》, 人民卫生出版社, 2008.06; 丁玉峰, 论临床剂型因素与合理用药, 医药导报, 26(5), 2007; Howard C. Ansel, Loyd V. Allen, Jr., Nicholas G. Popovich著,江志强主译,《药 物剂型与给药体系》, 中国医药科技出版社, 2003.05。
另一方面,本发明涉及本发明的化合物或其药学上可接受的盐、溶剂化物、多晶型物、 互变异构体或前药或者本发明的药物组合物, 其用于预防和 /或治疗肿瘤。
另一方面, 本发明涉及本发明的化合物或其药学上可接受的盐、 溶剂化物、 多晶 型物、 互变异构体或前药或者本发明的药物组合物在制备抗肿瘤药中的应用。
另一方面, 本发明涉及治疗和 /或预防哺乳动物、 特别是人的肿瘤的方法, 所述方法 包括对有需要的哺乳动物、特别是人给予治疗有效量的本发明的化合物或其药学上可接受 的盐、 溶剂化物、 多晶型物、 互变异构体或前药或者本发明的药物组合物。
在本申请中, 术语"肿瘤"包括但不限于: 白血病、 胃肠间质瘤、组织细胞性淋巴瘤、 非小细胞肺癌、 小细胞肺癌、胰腺癌、肺癌、肺鳞癌、肺腺癌、 乳腺癌、前列腺癌、肝癌、 皮肤癌、 上皮细胞癌、 宫颈癌、 卵巢癌、 肠癌、 结肠癌、 鼻咽癌、 脑癌、 骨癌、 食道癌、 黑色素瘤、 肾癌、 口腔癌等疾病。
通常, 治疗有效的日剂量为约 0.001 mg/kg至约 100 mg/kg; 优选的治疗有效剂量为 约 0.01 mg/kg至约 50 mg/kg; 更优选的治疗有效剂量为约 1 mg/kg至约 25 mg/kg
本文中所提供的有效剂量的范围并非意图限制本发明的范围,而是代表优选的剂量范 围。但是, 最优选的剂量可针对个别个体而进行调整, 这是本领域技术人员所了解且可决 定的 (例如参阅 Berkow等人编著, Merck手册, 第 16版, Merck公司, Rahway, N.J., 1992)。
本发明化合物可以与一种或多种其它的本发明化合物或者一种或多种其它抗癌药物 联合或组合使用, 以治疗和 /或预防肿瘤。 可以与本发明化合物联用的药物包括但不限于 多西他赛、 吉西他滨、 顺铂、 卡铂、 格列卫、 替莫唑胺、 阿霉素、 达卡巴嗪、 特罗凯、 依 托泊苷、 柔红霉素以及阿糖胞苷等。 实验部分
以下反应方案示例性地说明本发明化合物的制备方法。
本领域技术人员应当理解,在以下描述中,只有当取代基的组合可以得到稳定的化合
物时, 这类取代基的组合才是允许的。
本领域技术人员还应当理解,在下文所述的方法中, 中间体化合物官能团可能需要由 适当的保护基保护。 这样的官能团包括羟基、氨基、巯基及羧酸。合适的羟基保护基包括 三烷基甲硅烷基或二芳基烷基甲硅烷基(例如叔丁基二甲基甲硅烷基、叔丁基二苯基甲硅 烷基或三甲基甲硅烷基)、 四氢吡喃基、 苄基等。 合适的氨基、 脒基及胍基的保护基包括 叔丁氧羰基、 苄氧羰基等。 合适的巯基保护基包括 -C(0)-R" (其中 R"为烷基、 芳基或芳 烷基)、 对甲氧基苄基、 三苯甲基等。 合适的羧基保护基包括烷基、 芳基或芳烷基酯类。
保护基可根据本领域技术人员已知的和如本文所述的标准技术来引入和除去。
保护基的使用详述于 Greene, T. W.与 P. G. M. Wuts, Protective Groups in Organi Synthesis, (1999), 4th Ed., Wiley中。 保护基还可为聚合物树脂。
本发明通式 I所示化合物可通过以下的合成路线一或二来制备。
合成路线

试验条件: a. 对甲苯磺酸 (催化量), 甲苯, 回流。 合成路线二

试验条件: a. 对甲苯磺酸 (催化量), 甲苯, 回流。
可通过以下的方法制备合成路线一和二中所用的化合物 2。
(S)-4-乙基 -4-羟基 -1,8-二氢 -1H-吡 [3,4-f]吲哚嗪 -3,6,10(4H)-三酮

步骤 1 : 2,4-二氧代戊酸乙酯

在 30分钟内, 向无水乙醇 (600 mL)中加入金属钠 (25.3 g, 1.1 mol), 将反应液在 室温下搅拌 2小时。在 1小时的时间里,向所得的醇钠溶液中缓慢滴加草酸二乙酯 (146 g, 1.0 mol)和干燥的丙酮 (58 g, 1.0 mol)的混合液, 滴加完毕后将反应液室温下继续 搅拌 1小时。 将反应液过滤, 将滤饼转入冰 (500 g)、 水 (1 L)混合物中, 用浓硫酸酸化 至 pH值为 1, 再用乙酸乙酯萃取。 将乙酸乙酯相用盐水洗, 用无水硫酸钠干燥, 真 空浓缩, 得到标题所示化合物 (95 g, 60 %)。 1H NMR (CDC13): δ 6.37 (1Η, s), 4.35 (2H, q, J = 7.2 Hz), 2.26 (3H, s), 1.38 (3H, t, J = 7.2 Hz)。
步骤 2: 6-氰基 -1-羟基 -7-甲基 -5- 嗪 -2-羧酸甲酯

将 2,4-二氧代戊酸乙酯 (95 g, 0.60 mol), 原甲酸三乙酯(177.6 g, 1.2 mol), 一水 合对甲苯磺酸 (2.28g, 12 mmol)与无水甲醇(150 mL)的混合液在 40°C下搅拌 1小时, 然后加入氰基乙酰胺 (55.0 g, 0.66 mol), 碳酸钾 (91 g, 0.66 mol)和二甲亚砜 (2 L)。 将 反应液加热至 70°C, 搅拌 3小时。 然后在 2小时内, 向混合液中滴加丙烯酸甲酯 (413 g, 4.8 mol), 并将反应液在 70°C下搅拌过夜。 将反应液冷至室温, 倾入水 (10 L)中, 用浓硫酸酸化至 pH值为 1, 并继续在室温下搅拌 1小时。 将混合液过滤, 将滤饼用 甲醇洗涤并干燥,得到标题所示化合物 (45.5 g, 31 %)o 1H NMR (DMSO-d6): δ 6.79 (1Η, s), 4.62 (2H, s), 3.73 (3H, s), 2.43 (3H, s)。
步骤 3 : 7-甲基 -1,5-二氧代 -1,2,3,5-四 -甲腈
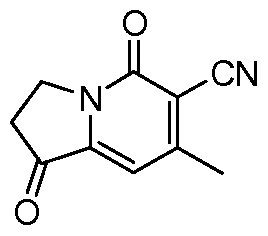
将 6-氰基 -1-羟基 -7-甲基 -5-氧代 -3,5-二氢吲哚嗪 -2-羧酸甲酯 (45.5 g, 185 mmol) 加入至 1 : 1的浓盐酸与醋酸的混合液 (500 mL)中, 将反应液加热至 100°C, 搅拌 2小 时,冷却到室温。真空浓缩,得到标题所示化合物 (34.0 g, 97.8 %)o 1H NMR (DMSO-d6): δ 6.84 (1Η, s), 4.08 (2Η, t, J = 6.4 Hz), 2.85 (2H, t, J = 6.4 Hz), 2.46 (3H, s)。
步骤 4: 7'-甲基 -5'-氧代 -3',5'-二氢 -2 环戊烷 -2,1'-吲哚嗪 ]-6'-甲腈

将 7-甲基 -1,5-二氧代 -1,2,3,5-四氢吲哚嗪 -6-甲腈 (34.0 g, 181 mmol)溶于二氯甲烷 (1.5 L)中, 向此溶液中加入乙二醇 (22.4 g, 361 mmol)和三甲基氯硅烷 (78.1 g, 723
mmol) o 将反应液在 25°C、 氮气保护下搅拌 72小时。 将反应液过滤除去黑色悬浮物, 然后用 1 M的氢氧化钠溶液洗涤。 将有机相用盐水洗, 用无水硫酸钠干燥, 将真空浓 缩的残余物通过柱层析纯化, 得到标题所示化合物 (32.0 g, 76 %)。 1H NMR (CDC13): δ 6.19 (1H, s), 4.19-4.11 (6H, m), 2.48 (3H, s), 2.39 (2H, t, J = 7.8 Hz)。
步骤 5 : 2-(6'-氰基 -5'-氧代 -3',5'-二氢 -2Ή-螺 [[1,3]二氧环戊烷 -2,1'-吲哚嗪 ]-7'-基)乙酸 乙酯

向 7'-甲基 -5'-氧代 -3',5'-二氢 -2Ή-螺 ,3]二氧环戊烷 -2J'—吲哚嗪 ]-6'-甲腈 (32.0 g, 138 mmol), 碳酸二乙酯 (32.6 g, 276 mmol)与干燥甲苯 (500 mL)的混合液中, 加入氢 氧化钠(1 1.0 g, 276 mmol, 60 %)和无水乙醇 (5 mL)。 将反应液加热至 100°C, 搅拌 3 小时。 然后除去溶剂, 将残余物倾入水中, 并用二氯甲烷萃取。 将有机相用盐水洗, 用无水硫酸钠干燥后真空浓缩, 并用柱层析纯化, 得到标题所示化合物 (30.2 g, 72 %)。 1H NMR (CDCI3): δ 6.33 (1Η, s), 4.22-4.13 (8H, m), 3.78 (2H, s), 2.41 (2H, t, J = 7.8 Hz), 1.29 (3H, q, J = 7.8 Hz)。
步骤 6: 溴 -[6-氰基 -1,1- (亚乙二氧 -四氢吲哚嗪 -7-基]乙酸乙酯

¾E 30°C 下, 向 2-(6'-氰基 -5'-氧代 -3',5'-二氢 -2Ή-螺 [[1,3]二氧环戊烷 -2,1'-吲哚 嗪] -7'-基)乙酸乙酯 (30.2 g, 99 mmol), 氢氧化钠 (3.96 g, 99 mmol, 60 %)与干燥的乙 二醇二甲醚 (300 mL)的混合液中滴加冷的干燥的液溴(1.56 g, 99 mmol)的乙二醇二甲 醚 (20 mL)溶液。将反应液保持在该温度下搅拌 30分钟,随后一次加入冰水混合物(100 mL)。 真空浓缩除去有机溶剂, 将残余物倾入水中, 并用二氯甲烷萃取。 将有机相用 盐水洗, 用无水硫酸钠干燥,真空浓缩, 得到标题所示化合物 (33.6 g, 89 %)。 1H NMR (CDCI3): δ 6.68 (1H, s), 5.60 (1H, s), 4.31-4.14 (8H, m), 2.43 (2H, t, J = 7.8 Hz), 1.26 (3H, q, J = 7.8 Hz)。
步骤 7: 2-[N-对甲苯磺酰基 -(R)-脯氨酸基] -2-[6-氰基 -U- (亚乙二氧基 )-5-氧代 -1,2,3,5- 四氢吲哚嗪 -7-基]乙酸乙酯


向干燥的 N,N-二甲基甲酰胺(100 mL)中加入 2-[N-对甲苯磺酰基 -(R)-脯氨酸 基] -2-[6-氰基 -1 ,1- (亚乙二氧基 )-5-氧代 -1,2,3,5-四氢吲哚嗪 -7-基]乙酸乙酯 (11.42 g, 20 mmol), 碘乙烷 (3.43 g, 22 mmol)和氢氧化钠(840 mg, 21 mmol, 60 %)。 将反应液加 热至 30°C, 搅拌 3小时, 随后加入水 (30 mL)。 浓缩除去溶剂, 将残余物倾入水中, 用二氯甲烷萃取。将有机相用盐水洗,用无水硫酸钠干燥,真空浓缩并用柱层析纯化, 得白色固体 (6.10 g)。 在 80°C下, 将此固体溶于异丙醇 (450 mL), 然后缓慢降温至室 温, 有晶体析出。 过滤, 将母液浓缩得到标题所示化合物 (2.10 g, 17 %, e.e. > 95 %)。 1H NMR (CDCI3): δ 7.78 (2Η, d, J = 8.4 Hz), 7.34 (2H, d, J = 8.0 Hz), 6.60 (1H, s), 4.51-4.47 (1H, m), 4.33-4.05 (8H, m), 3.56-3.51 (1H, m), 3.25-3.19 (1H, m), 2.59 (2H, q, J = 7.6 Hz), 2.45-2.41 (5H, m), 2.16-1.92 (3H, m), 1.73-1.65 (1H, m), 1.32 (3H, t, J = 7.2 Hz), 0.90 (3H, t, J = 7.6 Hz
步骤 9: (S)-2-[N-对甲苯磺酰基 -(R)-脯氨酸基] -2-[6- (乙酰胺甲基 )-l,l- (亚乙二氧基 )-5- 氧代 -1,2,3,5-四氢吲哚嗪 -7-基]丁

将 (S)-2-[N-对甲苯磺酰基 -(R)-脯氨酸基] -2-[6-氰基 - 1,1 - (亚乙二氧基 )-5-氧代
-1,2,3,5-四氢吲哚嗪 -7-基]丁酸乙酯(1.0 g, 1.7 mmol), 雷尼镍 (1.0 g), 醋酸酐(15 mL) 和醋酸 (5 mL)的混合液在氢气(1.5 atm)气氛下加热至 40°C, 搅拌 4小时。 过滤, 将滤 液浓缩并用柱层析纯化,得到标题所示化合物 (890 mg, 82 %)。 1H NMR (CDC13): δ 7.76 (2Η, d, J = 8.4 Hz), 7.32 (2H, d, J = 8.0 Hz), 7.03 (1H, t, J = 4.8 Hz), 6.70 (1H, s), 4.78-4.73 (IH, m), 4.45-4.02 (10H, m), 3.55-3.50 (IH, m), 3.26-3.20 (IH, m), 2.73-2.64 (IH, m), 2.59-2.51 (IH, m), 2.43-2.40 (5H, m), 2.05-1.92 (6H, m), 1.74-1.67 (IH, m), 1.28 (3H, t, J = 7.2 Hz), 0.88 (3H, t, J = 7.2 Hz)。
步骤 10: (S)-2-[N-对甲苯磺酰基 -(R)-脯氨酸基] -2-[6- (乙酰氧甲基 )-l,l- (亚乙二氧基 )-5- 氧代 -1,2,3,5-四氢吲哚嗪 -7-基] -

将 (S)-2-[N-对甲苯磺酰基 -(R)-脯氨酸基] -2-[6- (乙酰胺甲基 )-1,1- (亚乙二氧基 )-5- 氧代 -1,2,3,5-四氢吲哚嗪 -7-基]丁酸乙酯 (890 mg, 1.4 mmol)溶于醋酸酐 (15 mL)和醋酸 5 mL)的混合溶液中。 在 0°C下, 向反应液中加入亚硝酸钠 (380 mg, 5.5 mmol), 并 保持在该温度下搅拌 4小时。 随后过滤, 向滤液中加入四氯化碳 (30 mL), 加热回流 过夜。浓缩除去溶剂后, 将残余物倾入水中, 并用乙酸乙酯萃取。将有机相用盐水洗, 用无水硫酸钠干燥, 真空浓缩后用柱层析纯化, 得到标题所示化合物 (660 mg, 74%) 1H NMR (CDCI3): δ 7.75 (2Η, d, J = 8.0 Hz), 7.34 (2H, d, J = 8.0 Hz), 6.78 (IH, s), 5.27 (IH, d, J = 11.2 Hz), 5.22 (1H, d, J = 11.2 Hz), 4.39-4.06 (9H, m), 3.61-3.56 (1H, m), 3.27-3.22 (1H, m), 2.68-2.62 (1H, m), 2.45-2.33 (6H, m), 2.05-1.92 (6H, m), 1.71-1.67 (IH, m), 1.24 (3H, t, J = 7.2 Hz), 0.90 (3H, t, J = 7.6 Hz)。
步骤 11 : (S)-4-乙基 -6,6- (亚乙二氧基)-l,4,7,8-四氢 -4-羟基吡喃并 [3,4-f]吲哚嗪 -3,10(6H)-二酮

向甲醇 C6 mL)和水 C2 mL)的混合液中加入 CS)-2-[N-对甲苯磺酰基 -(R)-脯氨酸 基] -2-[6- (乙酰氧甲基 )-1,1- (亚乙二氧基 )-5-氧代 -1,2,3,5-四氢吲哚嗪 -7-基] -丁酸乙酯 (660 mg, 1.0 mmol), 水合氢氧化锂(171 mg, 4.1 mmol), 并在室温下搅拌 2小时。 随 后真空浓缩除去甲醇, 向反应液中加入醋酸 (l mL)和二氯甲烷 (20 mL)。将混合液在室 温下搅拌 10小时。 真空浓缩, 残余物用柱层析纯化, 得到标题所示化合物 (270 mg,
86 %)。 Ή NMR (CDC13): δ 6.58 (1H, s), 5.60 (1H, d, J = 16.4 Hz), 5.17 (1H, d, J = 16.4 Hz), 4.21-4.11 (6H, m), 3.77 (1H, s), 2.42 (2H, t, J = 6.8 Hz), 1.85-1.75 (2H, m), 0.98 (3H, t, J = 7.6 Hz)。
步骤 12: (S)-4-乙基 -4-羟基 -7,8-二氢 -1H-吡喃并 [3,4-f]吲哚嗪 -3,6,10(4H)-三酮 (标题 化合物)
将 (S)-4-乙基 -6,6- (亚乙二氧基 )-1,4,7,8-四氢 -4-羟基吡喃并 [3,4-f]吲哚嗪 -3,10(6H)- 二酮 (270 mg, 0.88 mmol)与三氟醋酸 (2 mL)和水 (0.5 mL)的混合液在氮气保护下室温 搅拌 3小时。真空浓缩,将残余物用柱层析纯化,得到标题所示化合物 (202 mg, 87 %)。 1H NMR (CDCI3): δ 7.22 (1H, s), 5.68 (1Η, d, J = 17.2 Hz), 5.24 (1H, d, J = 17.2 Hz), 4.36-4.32 (2H, m), 3.65 (1H, s), 2.99-2.95 (2H, m), 1.84-1.78 (2H, m), 0.98 (3H, t, J = 7.6 Hz)。 实施例
涉及对水和 /或氧气敏感的实验的所有操作都在预干燥的玻璃仪器中于氮气氛下 进行。 除非另有说明, 所有原料均为商业原料, 并且在使用前未作进一步纯化。
柱层析采用的是青岛海洋化工所生产的硅胶 (200-300 目)。 薄层色谱采用默克公 司(E. Merck)的预涂色谱板 (;硅胶 60PF254, 0.25毫米
核磁共振光谱分析使用的仪器为瓦里安 VNMRS-400共振光谱仪。化学位移以四 甲基硅烷 (TMS = S 0.00)为内标。 核磁共振氢谱数据的记录格式为: 质子数, 峰型 (s, 单峰; d, 双重峰; t, 三重峰; q, 四重峰; m, 多重峰), 耦合常数(以赫兹为单位)。
液质联用使用的是安捷伦 LC 1200系列 (5微米 C18色谱柱)仪器。 -甲氧基 -11 -氮杂 -20CS)-喜树碱

0°C下, 向 2-甲氧基异烟酸 C5.0 g, 33 mmol)的四氢呋喃 C40 mL)溶液中分批加入 四氢铝锂(1.9 g, 49 mmol) o 反应液在 0°C下继续搅拌 1小时, 然后缓慢滴加饱和硫 酸钠溶液。 过滤, 所得滤液用乙酸乙酯萃取, 有机相饱和食盐水洗, 真空浓缩。 将所 得残余物溶于二氯甲烷 C30 mL)中, 加入三氧化铬吡啶 C10.6 g, 49 mmol)。 反应混合物 室温下搅拌 2小时后倾倒于硅胶短柱上并用乙酸乙酯淋洗,所得溶液真空浓缩去除溶
剂。通过柱层析 (石油醚: 乙酸乙酯 =10 : 1)对所得残余物纯化得到标题所示化合物 (646 mg, 14%)。 1H NMR (CDC13): δ 10.01 (1H, s), 8.36 (1H, d, J = 5.2 Hz), 7.29 (1H, dd, J = 1.2 Hz, 5.2 Hz), 7.14 (1H, d, J = 1.2 Hz), 3.99 (3H, s)。
步骤 2: 4-(l,3-二氧戊环 -2-基) -6-甲 -3-胺

室温下, 向 2-甲氧基吡啶 -4-甲醛 (137 mg, 1.0 mmol)的三氟乙酸酐 (2 mL)溶液中 滴加发烟硝酸 (1 mL)。 反应液在 25°C下搅拌 3天后倾倒于冰水混合物中, 加入饱和 碳酸钠溶液调节 pH值到大于 10。 所得混合物用乙酸乙酯萃取, 有机相用盐水洗, 干 燥并真空去除溶剂。 将所得残余物溶于甲苯 (5 mL), 并加入对甲苯磺酸 (5 mg)和乙二 醇 (105 mg, 1.7 mmol)。 使反应混合液回流过夜, 冷却后用饱和碳酸钠和食盐水洗, 干燥, 并真空去除溶剂。 将残余物溶于甲醇 (10 mL), 加入钯 /碳催化剂 (20 mg)并氢气 置换。 反应体系在 1大气压的氢气氛下搅拌 2小时, 过滤去除催化剂并真空浓缩去除 溶剂。 通过柱层析对所得残余物纯化, 得到标题所示化合物 (15 mg, 8 %)。 1H NMR (DMSO-de): δ 9.31 (1Η, s), 8.56 (1H, s), 7.42 (1H, s), 7.34 (1H, s), 6.55 (1H, s), 5.43 (2H, s), 5.30 (2H, s), 4.02 (3H, s), 2.04-1.91 (2H, m), 0.90-0.85 (3H, m)。
步骤 3 : 10-甲氧基 -11 -氮杂 -20(S)-喜树碱 (标题化合物)
向 4-CU-二氧戊环 -2-基) -6-甲氧基吡啶 -3-胺 Cl l mg, 0.05 mmol)的甲苯溶液中加 入(S) 4-乙基- 4-羟基- 1 ,8-二氢- 1 H- P比喃并 [3、4- f|吲垛嗪 - 3,6, : )(4Η .Ξ:嗣(1 3 mg, 0.05 nimoi)和对甲苯磺酸 (2 mg)并氮气置换。 使反应液在氮气氛下加热回流≡小时, 真空 浓縮去除溶荆。 通过高效液相色谱将历得残余物纯化, 得到标题所示化合物 (3 mg, 16% 1H NMR (DMSO-de): δ 9.31 (1H, s), 8.56 (1H, s), 7.42 (1H, s), 7.34 (1H, s), 6.55 (1H, s), 5.43 (2H, s), 5.30 (2H, s), 4.02 (3H, s), 2.04-1.91 (2H, m), 0.90-0.85 (3H, m)。 实施例 2: 9-甲氧基 -10-氮杂 -20C

步骤 1 : 4- [(二叔丁氧羰基)氨基] -2-氯吡啶

步骤 2: 2-氯 -4- (叔丁氧羰基氨基)烟

在 0°C和氮气保护下, 向二异丙胺 C2.74 g, 27 mmol)的干燥四氢呋喃 C50 mL)溶液 中滴加正丁基锂(11.3 mL, 2.4 M, 27 mmol)。 继续搅拌 30分钟后冷却至 -60°C, 并在 此温度下滴加 4- [(二叔丁氧羰基)氨基] -2-氯吡啶 (2.55 g, 7.8 mmol)的四氢呋喃溶液。 继续搅拌 1小时候, 加入饱和氯化铵溶液。 将所得混合物用乙酸乙酯萃取, 有机相用 盐水洗, 干燥并真空浓缩去除溶剂。通过柱层析 (石油醚:乙酸乙酯 = 10: 1)将所得残余 物纯化,得到标题所示化合物(1.85 g, 73 %) o 1H NMR (CDCI3): δ 8.72 (IH, s), 8.24-8.20 (2H, m), 1.64 (9H, s), 1.53 (9H, s)。
步骤 3 : 2-甲氧基 -4-氨基烟酸甲酯

向 2-氯 -4 叔丁氧羰基氨基)烟酸叔丁酯 330 mg, 1 mmol)的干燥甲苯 (15 mL)溶液 中加入氢化钠 (60%, 120 mg, 3.0 mmol)和甲醇 (约 0.5 mL)。 将混合物在封管中加热 到 100°C并搅拌 6小时。 冷却后, 将反应液倾倒于硅胶短柱上并用乙酸乙酯淋洗, 真 空浓缩去除溶剂。 通过柱层析将所得残余物纯化, 得到标题所示化合物 (155 mg, 92 %)。 1H NMR (CDCI3): δ 7.77 (IH, d, J = 6.0 Hz), 6.18 (IH, d, J = 6.0 Hz), 6.09 (2H, br), 3.95 (3H, s), 3.89 (3H, s)。
步骤 4: 2-甲氧基 -4-氨基 -3吡啶醛

在 0°C下, 向 2-甲氧基 -4-氨基烟酸甲酯 (30 mg, 0.18 mmol)的四氢呋喃 (;3 mL)溶 液中滴加硼烷的四氢呋喃溶液 (0.9 mL, 1 M, 0.9 mmol), 室温搅拌 4小时。 滴加大约
1 mL水, 并用乙酸乙酯萃取。 有机相用食盐水洗, 干燥并真空浓缩去除溶剂。 将残
余物溶于四氢呋喃 (5 mL)溶液中, 加入二氧化锰 (42 mg, 0.48 mmol), 室温搅拌过夜。 过滤, 将滤液真空浓缩去除溶剂。 通过柱层析 (石油醚:乙酸乙酯 = 3: 1)将所得残余物 纯化, 得到标题所示化合物 (22 mg, 81 1H NMR (CDC13): δ 10.31 (IH, s), 7.81 (IH, d, J = 6.4 Hz), 6.15 (1H, d, J = 6.4 Hz), 3.98 (3H, s)。
步骤 5 : 9-甲氧基 -10-氮杂 -20(S)-喜树碱 (标题化合物)
以类似于实施例 1步骤 3中所描述的方法, 由 2-甲氧基 -4-氨基 -3吡啶醛和 (S)-4- 乙基 -4-羟基 -1,8-二氢 -1H-吡喃并 [3,4-f]吲哚嗪 -3,6,10(4H)-三酮反应, 得到标题所示化 合物。 NMR (CD3OD): δ 8.86 (1H, s), 7.63 (1H, d, J =7.6 Hz), 7.54 (1H, s), 6.81 (1H, d, J = 7.6 Hz), 5.49 (1H, d, J = 16.4 Hz), 5.30 (1H, d, J = 16.4 Hz), 5.22 (2H, s), 3.56 (3H, s), 1.87-1.84 (2H, m), 0.92-0.88 (3H, m)。 实施例 3 : 9-羟基 -10-氮杂 -20(S

9-羟基 -10-氮杂 -20CS)-喜树碱为合成 9-甲氧基 -10-氮杂 -20CS)-喜树碱 (实施例 2, 步骤 5)时所得副产物。 1H NMR (DMSO-de): δ 11.70 (1H, br), 8.86 (1H, s), 7.54 (1H, dd, J = 6.0 Hz, 7.2 Hz), 7.30 (1H, s), 6.75 (1H, d, J = 7.2 Hz), 6.54 (1H, s), 5.43 (2H, s), 5.24 (2H, s), 1.89-1.82 (2H, m), 0.88-0.84 (3H, m)。 实施例 4: 9- (;二甲氨基 10-氮杂
步骤 1 : 2- (二甲氨基) -4- (叔丁氧

向 2-氯 -4- (;叔丁氧羰基氨基)烟酸叔丁酯 (657 mg, 2.0 mmol)的 DME (10 mL)溶液 中加入二甲胺盐酸盐 (490 mg, 6.0 mmol)和三乙胺(1.01 g, 10 mmol)。 将反应液在封 管中加热到 110°C, 搅拌过夜。 冷却后, 将反应液倾倒于饱和碳酸钠溶液中, 用乙酸 乙酯萃取, 合并有机相并用饱和食盐水洗, 真空浓缩去除溶剂。 通过柱层析 (石油醚:
乙酸乙酯 = 8: 1)将所得残余物纯化, 得到标题所示化合物 (670 mg, 96 %)。 1H NMR (CDC13): δ 9.22 (1H, br), 8.03 (1H, d, J = 6.4 Hz), 7.55 (1H, d, J = 6.4 Hz), 3.00 (6H, s), 1.60 (9H, s), 1.51 (9H, s)。
步骤 2: 2- (二甲氨基) -4-氨基烟酸叔丁

以类似于实施例 2步骤 3中所描述的方法, 由 2- (二甲氨基) -4- (叔丁氧羰基氨基) 烟酸叔丁酯合成标题所示化合物。 1H NMR (CDC13): δ 7.75 (1Η, d, J = 6.4 Hz), 5.92 (1H, d, J = 6.4 Hz), 5.45 (2H, br), 3.01 (6H, s), 1.59 (9H, s)。
步骤 3 : 2- (二甲氨基) -4-氨基 -3吡啶醛

以类似于实施例 2步骤 4中所描述的方法, 由 2- (二甲氨基) -4-氨基烟酸叔丁酯合 成标题所示化合物。 1H NMR (CDC13): δ 9.74 (1Η, s), 7.84 (1H, d, J = 6.0 Hz), 6.02 (1H, d, J= 6.0 Hz), 3.08 (6H, s)。
步骤 4: 9- (二甲氨基) -10-氮杂 -20(S)-喜树碱 (标题化合物)
以类似于实施例 1 步骤 3 中所描述的方法, 由 2- (二甲氨基) -4-氨基 -3吡啶醛和 (S)-4-乙基 -4-羟基 -1,8-二氢 -1H-吡喃并 [3,4-f]吲哚嗪 -3,6,10(4H)-三酮反应,得到标题所 示化合物。 NMR (CD3OD): δ 9.16 (1H, s), 7.92 (1Η, d, J = 6.8 Hz), 7.66 (1H, s), 7.48 (1H, d, J = 6.8 Hz), 5.58 (1H, d, J = 8.8 Hz), 5.41-5.36 (3H, m), 3.56 (6H, s), 1.96-1.93 (2H,m), 1.00-0.96 (3H, m)。 实施例 5 : 9-氮杂 -10-甲氧基 -2 -CS)-喜树碱

步骤 1 : 6-氯 -3-硝基吡啶腈

将 2,6-二氯 -3-硝基吡啶 (3.00 g, 16 mmol), 氰化亚铜 (2.87 g, 32 mmol)和 N-甲基吡咯 烷酮 (10 mL)的混合液在 180°C下搅拌 3小时。 随后冷却至室温, 过滤。 将滤饼用乙酸乙 酯 (30 mL)洗涤两次, 将所得的乙酸乙酯合并, 真空浓缩得到标题所示化合物 (1.10 g, 39
%)。 Ή NMR (CDCI3): δ 8.58 (1H, d, J = 8.8 Hz), 7.79 (1H, d, J
步骤 2: 6-甲氧基 -3-硝基吡啶亚胺

向甲醇 (30 mL)中加入氢氧化钠 (560 mg, 14 mmol), 室温搅拌 30分钟后, 加入 6-氯 -3-硝基吡啶腈(1.00 g, 5.6 mmol)。 将反应液在室温下继续搅拌 3小时, 然后倾入 水中,用二氯甲烷萃取。有机相用盐水洗,干燥,真空浓缩,得到标题所示化合物 (1.10 g, 93 %)。 1H NMR (CDCI3): δ 8.28 (1H, br), 8.08 (1Η, d, J = 8.8 Hz), 6.88 (1H, d, J = 8.8 Hz), 4.05 (3H, s), 3.92 (3H, s)。
步骤 3 : 6-甲氧基 -3-硝基吡啶甲酸

将 6-甲氧基 -3-硝基吡啶亚胺甲酯(1.10 g, 5.2 mmol)溶于甲醇(10 mL), 加入浓盐 酸 (3 mL)。 将反应液室温搅拌 6小时后, 用碳酸钠溶液中和。 所得混合液用乙酸乙酯 萃取, 有机相用盐水洗, 干燥, 真空浓缩, 得到标题所示化合物 (1.03 g, 93 %)。 1H NMR (CDCI3): δ 8.34 (1H, d, J = 9.2 Hz), 6.92 (1H, d, J = 9.2 Hz), 4.06 (3H, s), 4.03 (3H, s)。
步骤 4: 3-氨基 -6-甲氧基吡啶甲酸

将 6-甲氧基 -3-硝基吡啶甲酸甲酯(1.03 g, 4.8 mmol), 铁粉(1.08 g, 19 mmol), 氯 化铵 (389 mg, 7.3 mmol)和乙醇 /水 (50 mL, 4: 1)的混合液在 80°C下搅拌 4小时。 冷却 后过滤, 将滤液倾入水中, 用乙酸乙酯萃取。 有机相用水洗, 干燥, 真空浓缩, 所得 残余物经柱层析纯化, 得到标题所示化合物 (420 mg, 48 %)。 1H NMR (CDCI3): δ 7.05 (1Η, d, J = 8.8 Hz), 6.81 (1H, d, J = 8.8 Hz), 5.46 (2H, br), 3.94 (3H, s), 3.92 (3H, s)。 步骤 5 : (3-氨基 -6-甲氧基吡啶 -2-

0°C下, 向 3-氨基 -6-甲氧基吡啶甲酸甲酯 (420 mg, 2.3 mmol)的四氢呋喃(10 mL) 溶液中加入四氢铝锂 (262 mg, 6.9 mmol)。 将反应液在 0°C下搅拌 1小时, 然后滴加 饱和硫酸钠溶液, 混合液继续搅拌 2小时, 然后过滤。 将滤液用乙酸乙酯萃取, 将有 机相用盐水洗,干燥,真空浓缩。所得残余物经柱层析纯化,得到标题所示化合物 (170
mg, 48 %)。 Ή NMR (CDC13): δ 7.04-7.02 (1H, m), 6.56 (1H, d, J = 8.4 Hz), 4.61 (2H, s)
3.90 (3H, s), 3.39 (2H, br)。
步骤 6: 3-氨基 -6-甲氧基吡啶甲醛

将 (3-氨基 -6-甲氧基吡啶 -2-基)甲醇(154 mg, 1.0 mmol), 二氧化锰 (522 mg, 6.0 mmol)和二氯甲烷 (10 mL)的混合液在室温下搅拌 6小时。 然后过滤, 将滤饼用二氯甲 烷洗涤, 合并有机相, 真空浓缩。所得残余物经柱层析纯化, 得到标题所示化合物 (28 mg, 18 %)。 1H NMR (CDC13): δ 9.92 (1H, s), 7.01 (1Η, d, J = 8.8 Hz), 6.79 (1H, d, J = 8.8 Hz), 5.82 (2H, br), 3.90 (3H, s)。
步骤 7: 9-氮杂 -10-甲氧基 -20-(S)-喜树碱 (标题化合物)
将 3-氨基 -6-甲氧基吡啶甲醛(10 mg, 0.066 mmol), (S)-4-乙基 -4-羟基 -7,8-二氢 -1H- 吡喃并 [3,4-f]吲哚嗪 -3,6,1(K4H 三酮(17 mg, 0.066 mmol), 对甲苯磺酸 (2 mg)与干燥 的甲苯 (5 mL)的混合液在氮气保护下回流 3小时。 然后将反应液真空浓缩, 向所得残 余物加入甲醇 (10 mL), 室温搅拌 30分钟, 过滤, 固体用甲醇洗涤, 干燥得到标题所 示化合物(12 mg, 49 %)。 1H NMR (DMSO-d6): δ 8.49 (1Η, s), 8.42 (1Η, d, J = 9.2 Hz), 7.38 (1H, d, J = 9.2 Hz), 7.28 (1H, s), 6.51 (1H, s), 5.42 (2H, s), 5.28 (2H, s), 4.05 (3H, s), 1.89-1.82 (2H, m), 0.88-0.85 (3H, m)。 实施例 6: 2,3-二氢呋喃并 [2,3- -2(HS 喜树碱

步骤 1 : 2-(2,5-二溴苯氧基)乙醇

将 1,4-二溴 -2-氟苯 (6.86 g, 27 mmol), 乙二醇 (35 mL), N-甲基吡咯烷酮 (35 mL) 和叔丁醇钾(1 1.2 g, 95 mmol)的混合液在 100°C、 氮气保护下搅拌过夜。 冷却至室温, 在 30分钟内慢慢加入水(15 mL)。 过滤, 将滤饼用乙二醇洗涤, 向滤液中加入水, 搅 拌 30分钟。 冷却至 15°C, 静置 1小时。 将析出的固体过滤, 用水洗涤, 干燥, 得到 标题所示化合物 (4.70 g, 59%)。 1H NMR (CDCI3): δ 7.40 (1H, d, J = 8.4 Hz), 7.05 (1H, d, J = 2.4 Hz), 7.02-6.70 (1H, m), 4.14 (2H, t, J = 4.4 Hz), 4.02-3.98 (2H, m), 2.13 (1H, t, J = 6.4 Hz)。
步骤 2: 1,4-二溴 -2-(2-溴乙氧基)苯

向 2-(2,5-二溴苯氧基)乙醇 (2.00 g, 6.8 mmol)的甲苯 (20 mL)溶液中加入三溴化磷 (2.01 g, 7.4 mmol)。 将混合液加热至 90°C, 搅拌 10小时, 然后冷却至室温。 缓慢加 入 1 N的氢氧化钠溶液猝灭反应。 将有机相用水洗, 干燥, 真空浓缩, 得到标题所示 化合物 (2.13 g, 88 %)。 1H NMR (CDC13): δ 7.41 (1Η, d, J = 9.2 Hz), 7.04-7.01 (2H, m), 4.33 (2H, t, J = 6.4 Hz), 3.68 (2H, t, J = 6.4 Hz)。
步骤 3 : 6-溴 -2,3-二氢苯并呋喃

在 -78°C氮气保护下, 向 1,4-二溴 -2-(2-溴乙氧基)苯 (2.13 g, 5.9 mmol)的苯 /四氢 呋喃 C28mL, 2:5)溶液中滴加正丁基锂 (2.5 mL, 6.1 mmol)。 将混合液在 -78°C下搅拌 1 小时, 然后用醋酸猝灭。 加入水, 分离有机相, 干燥, 真空浓缩, 得到标题所示化合 物(1.00 g, 85 %)。 1H NMR (CDC13): δ 7.03 (1H, d, J = 8.0 Hz), 6.91-6.93 (2H, m), 4.58 (2H, t, J = 4.8 Hz), 3.15 (2H, t, J = 4.8 Hz)。
步骤 4: 2,3-二氢苯并呋喃 -6-甲醛

将 6-溴 -2,3-二氢苯并呋喃(1.00 g, 5.0 mmol)的干燥的四氢呋喃 (20 mL)溶液冷却 至 -78°C, 在氮气保护下滴加正丁基锂 (2.3 mL, 2.4 M, 5.5 mmol)。 将反应液在 -78°C 下搅拌 30分钟, 然后滴加干燥的 N,N-二甲基甲酰胺 C731 mg, 10 mmol)。 将反应液在 -78°C下继续搅拌 1小时, 然后用饱和氯化铵溶液猝灭。 将混合液用乙酸乙酯萃取, 分离有机相, 干燥浓缩得到标题所示化合物 (384 mg, 52 %)。 1H NMR (CDCI3): δ 9.92 (IH, s), 7.40-7.33 (2H, m), 4.64 (2H, t, J = 8.8 Hz), 3.31-3.26 (2H, m)。
步骤 5 : 5-氨基 -2,3-二氢苯并呋喃 -6-

将 2,3-二氢苯并呋喃 -6-甲醛 (249 mg, 1.7 mmol)加入浓硝酸 (5 mL)中, 室温搅拌 2 小时, 然后倾入冰水中, 用乙酸乙酯萃取, 用盐水洗, 真空浓缩。 向残余物中加入乙 醇 /水(12 mL, 5: 1)混合液, 铁粉 (290 mg, 5.2 mmol)和氯化铵(104 mg, 1.9 mmol) 将 混合液在 80°C下搅拌 2小时, 过滤。 将滤液浓缩, 将残余物用柱层析纯化, 得到标 题所示化合物 (56 mg, 27 %)。 1H NMR (CDCI3): δ 9.75 (IH, s), 6.84 (IH, s), 6.57 (IH, s), 5.89 (2H, br), 4.52 (2H, t, J = 8.4 Hz), 3.19-3.15 (2H, m)。
步骤 6: 2,3-二氢呋喃并 [2,3-j]-20-(S)-喜树碱 (标题化合物)
向干燥的甲苯 (5 mL)中加入 5-氨基 -2,3-二氢苯并呋喃 -6-甲醛 (35 mg, 0.22 mmol),
(S)-4-乙基 -4-羟基 -7,8-二氢 -IH-吡喃并 [3,4-f]吲哚嗪 -3,6,10(4H)-三酮(37 mg , 0.14 mmol)和对甲苯磺酸 (3 mg), 使反应液在氮气保护下回流 3小时, 真空浓缩。 所得残 余物经制备型高效液相色谱分离, 得到标题所示化合物 (7 mg , 13 %)。 1H NMR (DMSO-de): δ 8.45 (IH, s), 8.00 (IH, s), 7.33 (IH, s), 7.26 (IH, s), 6.51 (IH, s), 5.41 (2H, s), 5.23 (2H, s), 4.71 (2H, t, J = 8.0 Hz), 3.47 (2H, t, J = 8.0 Hz), 1.99-1.86 (2H, m), 0.90-0.85 (3H, m)。 实施例 7: 10-甲氧基 -11-羟基 - -(S 喜树碱

步骤 1 : 4- (苄氧基) -3-甲氧基苯甲醛

将 4-羟基 -3-甲氧基苯甲醛 (2.0 g, 12.8 mmol), 苄溴 (2.2 g, 12.8 mmol)和碳酸钾 (880 mg, 6.4 mmol)加入丙酮 (30 mL)中, 使反应液回流过夜。 加入水, 过滤, 将滤饼 用水洗涤, 在乙醇中重结晶, 得到标题所示化合物 (3.1 g, 99 %)。 1H NMR (CDC13): δ 9.84 (1H, s), 7.45-7.31 (7H, m), 6.99 (1H, d, J = 8.4 Hz), 5.25 (2H, s), 3.95 (3H, s)。
步骤 2: 4- (苄氧基) -5-甲氧基 -2-硝

向浓硝酸 C3 mL)中加入 4- (;苄氧基 3-甲氧基苯甲醛 (242 mg, 1.0 mmol), 在 0°C 下搅拌 1小时, 过滤。 将所得固体水洗, 干燥, 得到标题所示化合物 (280 mg, 97 %)。 1H NMR (CDCI3): δ 10.45 (1H, s), 7.68 (1Η, s), 7.48-7.35 (7Η, m), 5.28 (2H, s), 4.03 (3H, s)。
步骤 3 : 2-胺基 -4-羟基 -5-甲氧基苯

将 4- (苄氧基) -5-甲氧基 -2-硝基苯甲醛 (280 mg, 0.97 mmol)和醋酸 (3 mL)的混合物 加热至 85°C, 然后加入氢溴酸 (48 %, 1 mL)。 将反应液在 85°C下搅拌 1小时。 冷却, 加入水, 用乙酸乙酯萃取, 将有机相用盐水洗, 干燥, 真空浓缩。 向所得残余物中加 入乙醇 /水(10 mL, 4: 1), 铁粉 (212 mg, 3.8 mmol)和氯化铵 (76 mg, 1.4 mmol)。 将混 合液在 80°C下搅拌 30分钟, 过滤。 向滤液中加入水, 用乙酸乙酯萃取。 将有机相用 盐水洗, 干燥, 真空浓缩。 所得残余物经柱层析纯化, 得到标题所示化合物 (62 mg,
52 %)。 Ή NMR (CDCI3): δ 9.67 (1H, s), 6.86 (1H, s), 6.22 (1H, s), 6.20 (1H, br), 6.03 (2H, br), 3.89 (3H, s)。
步骤 4: 10-甲氧基 -11-羟基 -20-(S)-喜树碱 (标题化合物)
将 2-胺基 -4-羟基 -5-甲氧基苯甲醛(19 mg, 0.11 mmol), (S)-4-乙基 -4-羟基 -7,8-二 氢 -1H-卩比喃并 [3,4-f]吲哚嗪 -3,6,10(4H)-三酮 (26 mg, 0.1 mmol)和水合对甲苯磺酸(1 mg)与干燥的甲苯 (3 mL)的混合液在氮气保护下加热至 90°C, 搅拌 2小时。 真空浓缩 以除去溶剂, 向残余物中加入甲醇 (10 mL), 在室温下搅拌 30分钟, 过滤, 将固体用 甲醇洗涤,干燥,得到标题所示化合物(19 mg, 48 %)。 1H NMR (DMSO-d6): δ 10.30 (IH, br), 8.41 (1H, s), 7.44 (1H, s), 7.37 (1H, s), 7.23 (1H, s), 6.49 (1H, s), 5.40 (2H, s), 5.21 (2H, s), 3.94 (3H, s), 1.97-1.84 (2H, m), 0.89-0.82 (3H, m)。 实施例 8: 10-羟基 -11-氟 -20-(S

步骤 1 : (4-氟 -3-甲氧基苯基)甲醇

在 0°C下, 向 4-氟 -3-甲氧基苯甲酸甲酯 (8.3 g, 45 mmol)的四氢呋喃(;30 mL)溶液 中加入四氢铝锂 (3.42 g, 90 mmol 将反应液在 0°C下搅拌 1小时, 然后滴加饱和硫 酸钠溶液。 过滤, 将滤液真空浓缩, 得到标题所示化合物 (7.1 g, 100 1H NMR (CDCI3): δ 7.06-7.00 (2Η, m), 6.81-6.83 (1Η, m), 4.63 (2Η, s), 3.89 (3H, s), 1.90 (1H, s)。 步骤 2: 4-氟 -3-甲氧基苯甲醛

向 (4-氟 -3-甲氧基苯基)甲醇 (1.56 g, 10 mmol)的二氯甲烷 (40 mL)溶液中加入三氧 化铬吡啶 (4.31 g, 20 mmol)。 将混合液在室温下搅拌 30分钟, 过滤。 将滤液倾入硅 胶短柱中, 用二氯甲烷洗脱。 将所得溶液真空浓缩, 得到标题所示化合物 (1.25 g, 81 %)。 1H NMR (CDCI3): δ 9.92 (1H, s), 7.52 (1Η, dd, J = 2.0 Hz, 8.4 Hz), 7.47-7.43 (1H, m), 7.26-7.22 (IH, m), 3.96 (3H, s)。
步骤 3 : 2-氨基 -4-氟 -5-羟基苯甲醛

在 0°C下, 向 4-氟 -3-甲氧基苯甲醛 (770 mg, 5.0 mmol)的浓硫酸(ΙΟ mL)溶液中滴
加发烟硝酸 C95%, 315 mg, 4.8 mmol)。 将反应液在室温下搅拌 1小时, 倾入冰水中, 过滤, 将滤饼用水洗, 干燥。 将所得残余物溶于 N,N-二甲基甲酰胺 (20 mL)中, 加入 氯化锂(1.06 g, 25 mmol), 回流 4小时, 倾入水中, 滴加浓盐酸酸化至 pH < 4, 然后 用乙酸乙酯萃取。 将有机相用盐水洗, 干燥, 真空浓缩。 向所得残余物中加入乙醇 / 水 (25 mL, 4: 1)混合液, 铁粉(1.21 g, 22 mmol)和氯化铵 (433 mg, 8.1 mmol)。 将混合 液加热至 80°C搅拌 2小时, 过滤。 向滤液中加入水, 用乙酸乙酯萃取。 将有机相用 盐水洗, 干燥, 真空浓缩, 所得残余物经柱层析纯化, 得到标题所示化合物 (125 mg, 16 %)。 1H NMR (CDC13): δ 9.75 (1H, s), 7.14 (1H, d, J = 9.6 Hz), 6.41 (1H, d, J = 12.0 Hz), 5.96 (2H, br), 4.58 (1H, d, J = 3.6 Hz)。
步骤 4: 10-羟基 -11-氟 -20-(S)-喜树碱 (标题化合物)
使 2-氨基 -4-氟 -5-羟基苯甲醛 (33 mg, 0.21 mmol), (S)-4-乙基 -4-羟基 -7,8-二氢 -1H- 吡喃并 [3,4-f]吲哚嗪 -3,6,10(4H)-三酮 (37 mg, 0.14 mmol), 对甲苯磺酸 (3 mg)与干燥甲 苯 (5 mL)的混合液在氮气保护下回流 3小时。 然后将反应液真空浓缩, 残余物经制备 型高效液相色谱分离,得到标题所示化合物 (5 mg, 10 %)。 1H NMR (DMSO-d6): δ 11.01 (1H, s), 8.53 (1Η, s), 7.93 (1Η, d, J = 12.4 Hz), 7.49 (1H, d, J = 9.6 Hz), 7.27 (1H, s), 6.51 (1H, s), 5.42 (2H, s), 5.24 (2H, s), 1.86-1.84 (2H, m), 0.89-0.86 (3H, m)。 实施例 9: 10-甲氧基 -11-氟 -20

步骤 1 : 2-氨基 -4-氟 -5-甲氧基苯甲

0°C下, 向 4-氟 -3-甲氧基苯甲醛 ( 70 mg, 5.0 mmol)的浓硫酸 (ΙΟ mL)溶液中滴加 发烟硝酸 C95 %, 315 mg, 4.8 mmol)。 将反应液在室温下搅拌 1小时后, 倾入冰水中, 过滤, 将滤饼水洗, 干燥。 向所得残余物中加入乙醇 /水 (25 mL, 4: 1), 铁粉 (1.12 g, 20 mmol)和氯化铵 (404 mg, 7.6 mmol)。 将反应液在 80°C搅拌 2小时, 过滤。 向滤液 中加入水, 用乙酸乙酯萃取。 将有机相用盐水洗, 干燥, 真空浓缩。 所得残余物经柱 层析纯化, 得到标题所示化合物(180 mg, 21 %)。 1H NMR (CDC13): δ 9.78 (IH, s), 7.04 (1H, d, J = 9.2 Hz), 6.41 (1H, d, J = 1 1.6 Hz), 6.03 (2H, br), 3.86 (1H, s)。
步骤 2: 10-甲氧基 -11-氟 -20-(S)-喜树碱 (标题化合物)
使 2-氨基 -4-氟 -5-甲氧基苯甲醛 (34 mg, 0.20 mmol), (S)-4-乙基 -4-羟基 -7,8-二氢
-1H-吡喃并 [3,4-f]吲哚嗪 -3,6,10(4H)-三酮 (34 mg, 0.13 mmol), 对甲苯磺酸 (3 mg)与干
燥的甲苯 (6 mL)的混合液在氮气保护下回流 3小时。 将反应液真空浓缩, 将所得残余 物用甲醇洗涤,干燥,得到标题所示化合物 (40 mg, 78 %)o 1H NMR (DMSO-d6): δ 8.58 (IH, s), 7.96 (IH, d, J = 12.4 Hz), 7.74 (IH, d, J = 8.8 Hz), 7.27 (IH, s), 6.51 (IH, s), 5.40 (2H, s), 5.26 (2H, s), 1.86-1.82 (2H, m), 0.87-0.83 (3H, m)。 实施例 10: 11-甲氧基 -12-羟 -20-(S 喜树碱

步骤 1 : 2-氨基 -3-羟基 -4-甲氧基苯甲醛

向 3-羟基 -4-甲氧基苯甲醛 C456 mg, 3.0 mmol)的醋酸 (;10 mL)溶液中滴加浓硝酸
(65 %, 290 mg, 3.0 mmol), 将反应液在室温下搅拌 2小时后, 倾入水中, 用乙酸乙 酯萃取。将有机相用盐水洗,干燥,真空浓缩。向所得残余物中加入乙醇 /水 (50 mL, 4: 1) 混合液, 铁粉 (672 mg, 12 mmol)和氯化铵 (636 mg, 12 mmol), 加热至 80°C, 搅拌 1 小时。 过滤, 向滤液中加入水, 用乙酸乙酯萃取。 将有机相用盐水洗, 干燥, 真空浓 缩,所得残余物经柱层析纯化,得到标题所示化合物 (140 mg, 28 %)。 1H NMR (CDC13): δ 9.77 (1H, s), 7.09 (1H, d, J = 8.8 Hz), 6.40 (1H, d, J = 8.8 Hz), 5.37 (2H, br), 5.06 (1H, br), 3.95 (3H, s)。
步骤 2: 11-甲氧基 -12-羟基 -20-(s)-喜树碱 (标题化合物)
将 2-氨基 -3-羟基 -4-甲氧基苯甲醛(19 mg, 0.11 mmol), (S)-4-乙基 -4-羟基 -7,8-二 氢 -1H-吡喃并 [3,4-f]吲哚嗪 -3,6,10(4H)-三酮 (26 mg, 0.1 mmol), 对甲苯磺酸(l mg)与 干燥的甲苯 (5 mL)的混合液在氮气保护下加热至 90°C, 搅拌 2小时。将反应液真空浓 缩, 向残余物中加入甲醇 (10 mL), 在室温下搅拌 30分钟。 过滤, 所得固体用甲醇洗 涤,干燥,得到标题所示化合物(17 mg,43 %)o 1H NMR (DMSO-d6): δ 9.50 (IH, br), 8.54 (IH, s), 7.56-7.55 (2H, m), 6.54-6.53 (1H, m), 5.41 (2H, s), 5.23 (2H, s), 3.94 (3H, s), 1.97-1.84 (2H, m), 0.89-0.82 (3H, m)。 实施例 11 : 7-氨基 -9-甲氧基 -10-氟 -20-(S 喜树碱

步骤 1 : 4-氟 -5-甲氧基 -2-硝基苯甲

0°C下, 向 4-氟 -3-甲氧基苯甲酸 C3.40 g, 20 mmol)的浓硫酸 C25 mL)溶液中加入硝 酸钾 C202 mg, 20 mmol), 将反应液在 40°C下搅拌 2小时。 冷至室温后, 倾入冰水中, 用乙酸乙酯萃取。 将有机相用盐水洗, 干燥, 真空浓缩。 所得残余物经柱层析纯化, 得到标题所示化合物(1.81 g, 42 %)。 1H NMR (DMSO-d6): δ 8.08-8.05 (1Η, m), 7.49-7.47 (1H, m), 3.95 (3H,s)。
步骤 2: 4-氟 -5-甲氧基 -2-硝基苯甲

向二氯亚砜 /氯仿 CIO mL, 1 :2)混合液中加入 4-氟 -5-甲氧基 -2-硝基苯甲酸 (860 mg, 4.0 mmol), 将反应液加热至 65°C, 搅拌过夜。 真空浓缩, 将所得残余物溶于干燥的 乙酸乙酯 EtOAc (30 mL), 在 0°C下通入氨气 15分钟。然后将混合液用饱和碳酸钠溶 液洗涤,将有机相用盐水洗, 干燥, 真空浓缩, 得到标题所示化合物 (856 mg, 100 %)。 1H NMR (CDC13): δ 8.08-8.02 (1H, m), 7.70 (1Η, br), 7.33-7.31 (1Η, m), 3.93 (3H,s)。 步骤 3 : 4-氟 -5-甲氧基 -2-硝基苯腈

向三氟醋酸酐 /二氯甲烷 (10 mL, 1 :2)混合液中加入 4-氟 -5-甲氧基 -2-硝基苯甲酰 胺 (856 mg, 4.0 mmol), 将反应液在室温下搅拌 4小时。 然后加入水, 用二氯甲烷萃 取。 将有机相用盐水洗, 干燥, 真空浓缩。 所得残余物经柱层析纯化, 得到标题所示 化合物 (675 mg, 86 %)。 1H NMR (DMSO-d6): δ 8.12-8.09 (1Η, m), 7.40-7.38 (1Η, m), 4.11 (3H,s
步骤 4: 2-氨基 -4-氟 -5-甲氧基苯腈

向乙醇 /水 (30 mL, 1 : 1)混合液中加入 4-氟 -5-甲氧基 -2-硝基苯腈(196 mg, 1.0 mmol)和保险粉 (522 mg, 3.0 mmol), 将反应液加热至 80°C, 搅拌 2小时。 真空浓缩
后, 加入水, 用乙酸乙酯萃取。 将有机相用盐水洗, 干燥, 真空浓缩。 所得残余物用 柱层析纯化, 得到标题所示化合物(130 mg, 78 %)。 1H NMR (CDC13): δ 6.95-6.93 (IH, m), 6.53-6.50 (1H, m), 4.23 (2H, br), 3.82 (3H,s)。
步骤 5 : 7-氨基 -9-甲氧基 -10-氟 -20-(S)-喜树碱 (标题化合物)
使 2-氨基 -4-氟 -5-甲氧基苯腈(18 mg, 0.11 mmol), (S)-4-乙基 -4-羟基 -7,8-二氢 -1H- 吡喃并 [3,4-f]吲哚嗪 -3,6,1(K4H 三酮 (26 mg, 0.10 mmol), 对甲苯磺酸(1 mg)和干燥的 甲苯 (5 mL)的混合液在氮气保护下回流 24小时。 真空浓缩, 所得残余物经制备型高 效液相色谱分离, 得到标题所示化合物 (3 mg, 7 %)。 1H NMR (DMSO-d6): δ 8.10 (IH, br), 7.91-7.89 (1H, m), 7.68-7.65 (1H, m), 7.31 (1H, s), 6.50 (1H, br), 5.39 (2H, s), 5.01 (2H, m), 3.85 (3H, s), 1.97-1.84 (2H, m), 0.89-0.82 (3H, m)。 实施例 12: 10-甲氧基 -11-氟 -7- -200¾-喜树碱

步骤 1 : 4-氟 -N,3-二甲氧基 -N-甲基

室温下,向 4-氟 -3-甲氧基苯甲酸 (1.7 g, 10 mmol), N,O-二甲基羟胺盐酸盐 (1.17 g, 12 mmol)的二氯甲烷 (20 mL)溶液中加入三乙胺 (7.0 mmol, 50 mmol), EDCI (2.11 g, 11 mmol)。 反应液在室温下搅拌 36小时, 然后倾入 30 mL水中, 用二氯甲烷萃取。 将有机相用饱和食盐水洗, 真空浓缩。所得残余物通过柱层析纯化得到标题所示化合 物 (628 mg, 29%) 1H NMR (CDCI3): δ 7.37-7.35 (1H, m), 7.32-7.28 (1Η, m), 7.10-7.05 (IH, m), 3.91 (3H, s), 3.55 (3H, s), 3.36 (3H, s)。
步骤 2: 4-氟 -3-甲氧基苯乙酮

将 4-氟 -N,3-二甲氧基 甲基苯甲酰胺 C628 mg, 2.9 mmol)的四氢呋喃 (;5 mL)溶液 冷却至 0°C。 在氮气保护下, 滴加甲基溴化镁溶液 (3.2 mL, 4.4 mmol)。 在 0°C搅拌 15 分钟后, 用饱和氯化铵溶液猝灭。 所得混合物用乙酸乙酯萃取。 将有机相用盐水 洗, 干燥并真空去除溶剂得到标题所示化合物 (465 mg, 94 %)。 1H NMR (CDCI3): δ 7.64-7.61 (1H, m), 7.55-7.52 (1Η, m), 7.17-7.12 (1Η, m), 3.96 (3Η, s), 2.60 (3H, s)。
步骤 3: 4-氟 -5-甲氧基 -2-硝基苯乙酮

将 4-氟 -3-甲氧基苯乙酮 (465 mg, 2.8 mmol)溶于浓硫酸 (3 mL)中, 冷却至 0°C, 缓慢滴加发烟硝酸 (183 mg, 2.8 mmol)。 反应液在 0°C继续搅拌半小时后, 倾入水中。 所得混合物用乙酸乙酯萃取。将有机相用盐水洗, 干燥并真空去除溶剂得到标题所示 化合物 (435 mg, 74 %)。1H NMR (CDC13): δ 7.94 (1Η, d,J= 10.4 Hz), 6.87 (1Η, d, J= 8.0 Hz), 4.02 (3H, s), 2.54 (3H, s)。
步骤 4: 2-氨基 -4-氟 -5-甲氧基苯乙酮

将 4-氟 -5-甲氧基 -2-硝基苯乙酮 (435 mg, 2.0 mmol)、 氯化铵(164 mg, 3.0 mmol) 和铁粉 (456mg, 8.2 mmol)在乙醇 /水 (20 mL, 4:1)混合溶液中回流 3小时, 然后过滤。 将滤液倾入水 (30 mL)中, 用乙酸乙酯萃取。 将有机相用饱和食盐水洗, 真空浓缩。 所得残余物通过柱层析纯化得到标题所示化合物 (271 mg, 72%) 1HNMR (CDCI3): δ 7.26 (1Η, d, J= 9.2 Hz), 6.39 (1H, d, J= 12.8 Hz), 6.18 (2H, br), 3.85 (3H, s), 2.54 (3H, s)。
步骤 5: 10-甲氧基 -11-氟 -7-甲基 -200S)-喜树碱 (标题化合物)
向 2-氨基 -4-氟 -5-甲氧基苯乙酮(13 mg, 0.071 mmol)的甲苯溶液中加入 (5)-4-乙基 -4-羟基 -1,8-二氢 -1H-吡喃并 [3,4-f]吲哚嗪 -3,6,10(4H)-三酮(13 mg, 0.05 mmol)和对甲 苯磺酸 (2 mg)并氮气置换。将反应液在氮气氛下加热回流三小时,真空浓缩去除溶剂。 通过高效液相色谱对所得残余物纯化得到标题所示化合物 (6 mg, 30%) 1H NMR (OMSO-d6): δ 7.92 (1Η, d, J= 12.4 Hz), 7.61 (1H, d, J= 9.2 Hz), 7.24 (1H, s), 6.51 (1H, s), 5.41 (2H, s), 5.21 (2H, s), 4.03 (3H, s), 2.74 (3H, s), 1.87-1.83 (2H, m), 0.88-0.84 (3H, m)。 体外活性实验:
将人肺癌细胞 A549用补充有 10%胎牛血清 (Hyclone®) 的 DMEM (Gibco®) 培养基进行培养; 将人结肠癌细胞 SW620, HT-29和人前列腺癌细胞 PC-3用补充有 10%胎牛血清 (Hyclone®) 的 RPMI-1640 (Gibco®)培养基进行培养。 上述细胞的培 养条件是 37°C, 95%空气和 5% C02, 培养于 25 cm2或 75 cm2塑料组织培养瓶 (Coming®) 中, 一周传代培养 2〜3次。
将细胞以 1X103细胞 /孔 (A549), 2X 103细胞 /孔 ( SW620和 HT29), 3X103细
胞 /孔 (PC-3 ) 的密度接种在 96孔细胞培养板 (Coming®) 中, 195 μΐ/孔, 并在 37 。C , 95%空气和 5% C02中进行培养。 24小时后加入待测化合物, 将化合物从 10 mM (溶于 100 % DMSO中) 开始用 100% DMSO进行 3倍梯度稀释, 每个浓度取 4 μΐ加 入到 96 μΐ的无血清培养基中, 最后取 5 μΐ培养基稀释后的化合物加入到接种有细胞 的培养板中。 细胞培养液中 DMSO终浓度为 0.1%, 所试化合物的终浓度是 0.3 nM〜 10 μΜ。 将所述细胞于 37°C温育 3天。
3天后, 根据 ATP和萤光素酶的反应用发光计测量活细胞的活力水平 (Cell Titre Glo®萤光试剂盒, Pramega 在该方法中, 可以根据所述化合物对细胞生长的影响 来判断化合物的效力。由产生的数据生成剂量响应曲线,并通过 Prism程序计算 IC50。 本发明代表性化合物的生物活性:

Claims
权利要求书:
1 . 式 I 的化合物或其药学上可接受的盐、 溶剂化物、 多晶型物、 互变异构体或 前药:

其中:
X为 CR3或 N;
Y为 CR4或 N;
Z为 CR5或 N;
X、 Y、 Ζ中至少有一个为 Ν;
Ri R2以及 R3、 R4、 R5各自独立地选自氢、 卤素、 羟基、 烷氧基、 氨基、 单 (烷 基)氨基、 二 (烷基)氨基、 环烷基氨基、 巯基、 烷硫基或氰基;
或者
R3和 R4二者或 R4和 R5二者或 R5和 R 二者与它们所连接的碳原子共同组成五 至七元非芳香环。
2. 如权利要求 1所述的化合物或其药学上可接受的盐、 溶剂化物、 多晶型物、 互变异构体或前药, 其中:
Ri R2以及 R3、 R4、 R5各自独立地选自氢、 卤素、 羟基、 d_6烷氧基、 氨基、 单 (d_6烷基)氨基、 二 (d_6烷基)氨基、 C3_8环烷基氨基、 巯基、 d_6烷硫基或氰基; 或者
和 R4二者或 和 R5二者或 和 二者与它们所连接的碳原子共同组成含 有 0至 3个选自氮、 氧或硫的杂原子的五至七元非芳香环。
3. 如权利要求 1或 2所述的化合物或其药学上可接受的盐、 溶剂化物、 多晶型 物、 互变异构体或前药, 其中:
Ri R2以及 R3、 F 、 R5各自独立地选自氢、 羟基、 d_6烷氧基、 氨基、 单 (C^ 烷基)氨基、 二 (d_6烷基)氨基、 巯基或 d_6烷硫基。
4. 如权利要求 1至 3中任一项所述的化合物或其药学上可接受的盐、溶剂化物、 多晶型物、 互变异构体或前药, 其中:
Ri、 R2均为氢;
R3、 R4、 R5各自独立地选自氢、 羟基、 d_6烷氧基、 氨基、 单 (d_6烷基)氨基或 二 (Cw烷基)氨基。
5. 如权利要求 4所述的化合物或其药学上可接受的盐、 溶剂化物、 多晶型物、

其巾:
X为 CR3;
Y为 CR4 ;
Z为 CR5;
R3和 R4二者或 R4和 R5二者或 R5和 二者与它们所连接的碳原子共同组成含 有 1个选自氮、 氧或硫的杂原子的五至七元非芳香环, 且 、 R2、 R3、 R4、 R5中的 其余基团各自独立地选自氢、 卤素、 羟基、 烷氧基、 氨基、 单 (烷基)氨基、 二 (烷基) 氨基、 环烷基氨基、 巯基、 烷硫基或氰基; 或者
Ri R2、 R3均为氢, 且 R4、 R5互相不同且各自独立地选自卤素、 羟基或烷氧基; 或者
Ri R3均为氢, R2为氨基或烷基, 且 R4、 R5各自独立地选自卤素、 羟基、 烷氧 基、 氨基、 单 (烷基)氨基或二 (烷基)氨基。
7. 如权利要求 6所述的化合物或其药学上可接受的盐、 溶剂化物、 多晶型物、 互变异构体或前药, 其中:
和 R5二者与它们所连接的碳原子共同组成含有 1个选自氮、 氧和硫的杂原子 的五至七元非芳香环, 且 Ri、 R2、 R3各自独立地选自氢、 卤素、 羟基、 d_6烷氧基、 氨基、 单 (d_6烷基)氨基、 二 (d_6烷基)氨基和 C3_8环烷基氨基。
8. 如权利要求 6或 7所述的化合物或其药学上可接受的盐、 溶剂化物、 多晶型 物、 互变异构体或前药, 其中:
R4和 R5二者与它们所连接的碳原子共同组成含有 1个选自氮、 氧或硫的杂原子 的五元非芳香环, 且 Ri、 R2、 R3均为氢。
9. 如权利要求 6所述的化合物或其药学上可接受的盐、 溶剂化物、 多晶型物、 互变异构体或前药, 其中:
Ri R2、 R3均为氢, 且 R4、 R5互相不同且各自独立地选自卤素、 羟基或 C^烷 氧基。
10. 如权利要求 6所述的化合物或其药学上可接受的盐、 溶剂化物、 多晶型物、 互变异构体或前药, 其中:
Ri R3均为氢, R2为氨基或 d_6烷基, 且 R4、 R5各自独立地选自卤素、 羟基或
Cl_6院氧基。
1 1 . 如权利要求 6所述的化合物或其药学上可接受的盐、 溶剂化物、 多晶型物、 互

12. 一种药物组合物, 其包含: 权利要求 1-11 中任一项所述的化合物或其药学 上可接受的盐、溶剂化物、多晶型物、互变异构体或前药, 以及药学上可接受的载体。
13. 权利要求 1-11 中任一项所述的化合物或其药学上可接受的盐、 溶剂化物、 多晶型物、 互变异构体或前药或者权利要求 12所述的药物组合物在制备抗肿瘤药中 的应用。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13806765.7A EP2862571A4 (en) | 2012-06-18 | 2013-06-17 | CAMPTOTHECIN COMPOUND, AND ITS MANUFACTURE AND USE |
| US14/409,284 US9458170B2 (en) | 2012-06-18 | 2013-06-17 | Compound of camptothecin and preparation and use thereof |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201210200616 | 2012-06-18 | ||
| CN201210200616.4 | 2012-06-18 | ||
| CN201210280442.7 | 2012-08-08 | ||
| CN201210280442 | 2012-08-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013189266A1 true WO2013189266A1 (zh) | 2013-12-27 |
Family
ID=49768110
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2013/077336 WO2013189266A1 (zh) | 2012-06-18 | 2013-06-17 | 喜树碱类化合物及其制备和用途 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US9458170B2 (zh) |
| EP (1) | EP2862571A4 (zh) |
| CN (2) | CN103509030A (zh) |
| WO (1) | WO2013189266A1 (zh) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1990003169A1 (en) * | 1988-09-28 | 1990-04-05 | Research Triangle Institute | Synthesis of camptothecin and analogs thereof |
| WO1991005556A1 (en) * | 1989-10-23 | 1991-05-02 | Research Triangle Institute | Camptothecin analogs as potent inhibitors of human colorectal cancer |
| GB2280674A (en) * | 1993-08-03 | 1995-02-08 | Glaxo Inc | Pyrido[3,4-b]-pyridine antitumor compounds |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5106742A (en) * | 1987-03-31 | 1992-04-21 | Wall Monroe E | Camptothecin analogs as potent inhibitors of topoisomerase I |
| US5364858A (en) * | 1987-03-31 | 1994-11-15 | Research Triangle Institute | Camptothecin analogs as potent inhibitors of topoisomerase I |
| US4894456A (en) * | 1987-03-31 | 1990-01-16 | Research Triangle Institute | Synthesis of camptothecin and analogs thereof |
| JPH0615547B2 (ja) * | 1988-01-20 | 1994-03-02 | 株式会社ヤクルト本社 | 新規なカンプトテシン誘導体 |
| JP2684104B2 (ja) * | 1990-02-09 | 1997-12-03 | 株式会社ヤクルト本社 | 新規なカンプトテシン誘導体 |
| US8575188B2 (en) * | 2009-06-17 | 2013-11-05 | Threshold Pharmaceuticals, Inc. | Camptothecin derivatives |
-
2013
- 2013-06-17 CN CN201310239097.7A patent/CN103509030A/zh active Pending
- 2013-06-17 WO PCT/CN2013/077336 patent/WO2013189266A1/zh active Application Filing
- 2013-06-17 CN CN201710447065.4A patent/CN107216336A/zh active Pending
- 2013-06-17 EP EP13806765.7A patent/EP2862571A4/en not_active Withdrawn
- 2013-06-17 US US14/409,284 patent/US9458170B2/en not_active Expired - Fee Related
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1990003169A1 (en) * | 1988-09-28 | 1990-04-05 | Research Triangle Institute | Synthesis of camptothecin and analogs thereof |
| WO1991005556A1 (en) * | 1989-10-23 | 1991-05-02 | Research Triangle Institute | Camptothecin analogs as potent inhibitors of human colorectal cancer |
| GB2280674A (en) * | 1993-08-03 | 1995-02-08 | Glaxo Inc | Pyrido[3,4-b]-pyridine antitumor compounds |
Non-Patent Citations (16)
| Title |
|---|
| "ADVANCED ORGANIC CHEMISTRY", vol. B, 2001, PLENUM PRESS |
| "Remington's, Pharmaceutical Sciences", MACK PUBLISHING CO. |
| "The Merck Manual", 1992, MERCK AND CO. |
| "The Science and Practice of Pharmacy", 2000, PHILADELPHIA COLLEGE OF PHARMACY AND SCIENCE |
| CAREY; SUNDBERG: "ADVANCED ORGANIC CHEMISTRY", vol. A, 2000 |
| DING YUFENG: "Discussion on Clinical Dosage Form Factors and Drug Rational Use in Hospital", HERALD OF MEDICINE, vol. 26, no. 5, 2007 |
| GOODMAN; GILMAN: "The Pharmacological Basis of Therapeutics", PERGAMON |
| GREENE, T.W.; P. G. M. WUTS: "Greene's Protective Groups in Organic Synthesis", 1999, WILEY |
| GREENWALD, R. B. ET AL., J MED. CHEM., vol. 43, 2000, pages 475 |
| HOWARD C. ANSEL; LOYD V. ALLEN, JR.; NICHOLAS G. POPOVICH: "Drug Dosage Forms and Drug Delivery System", May 2003, CHINESE MEDICAL SCIENCE AND TECHNOLOGY PRESS |
| LI JUN: "Clinical Pharmacology", June 2008, PEOPLE'S MEDICAL PUBLISHING HOUSE |
| LU, A.J. ET AL.: "3D-QSAR study of 20(s)-camptothecin analogs", ACTA PHARMACOL SIN, vol. 28, no. 2, February 2007 (2007-02-01), pages 307 - 314, XP055182355 * |
| MU, YAOSONG ET AL.: "Advance in Research of Structure Modification and Structure-Activity Relation of Camptothecin and Its Analogues", CHIN PHARM J, vol. 41, no. 11, June 2006 (2006-06-01), pages 804 - 806, XP008172728 * |
| SAULNIER, M. G., BIOORG. MED. CHEM. LETT., vol. 4, 1994, pages 1985 - 1990 |
| See also references of EP2862571A4 * |
| WALL ET AL., J AM. CHEM. SOC., vol. 88, no. 16, 1966, pages 3888 - 990 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN107216336A (zh) | 2017-09-29 |
| CN103509030A (zh) | 2014-01-15 |
| US20150166559A1 (en) | 2015-06-18 |
| EP2862571A4 (en) | 2016-03-16 |
| US9458170B2 (en) | 2016-10-04 |
| EP2862571A1 (en) | 2015-04-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN113767103B (zh) | 新型螺环类K-Ras G12C抑制剂 | |
| JP6877407B2 (ja) | Ntrk関連障害の治療に有用な化合物および組成物 | |
| JP6644792B2 (ja) | ベンザゼピンスルホンアミド化合物 | |
| CN115448923B (zh) | 嘧啶并环化合物及其制备方法和应用 | |
| ES2833576T3 (es) | Inhibidores de glutaminasa novedosos | |
| WO2021129824A1 (zh) | 新型K-Ras G12C抑制剂 | |
| ES2928169T3 (es) | Derivados de 4-(3H-indol-5-il)-N-(piridin-2-il)pirimidin-2-amina como inhibidores de proteína cinasa, y método de preparación y uso médico de los mismos | |
| WO2017084640A1 (zh) | 一种含氮杂环化合物及其制备方法与在抑制激酶活性中的应用 | |
| CN112300153B (zh) | 一种杂环化合物、药物组合物和用途 | |
| CN112745335A (zh) | 一种三并杂环化合物及其用途 | |
| AU2018329047B2 (en) | Cycloolefin substituted heteroaromatic compounds and their use | |
| KR20200078610A (ko) | 아미노치환 질소함유 축합고리 화합물 및 그의 제조방법과 용도 | |
| CN113527299B (zh) | 一类含氮稠环类化合物、制备方法和用途 | |
| CN112851663A (zh) | 一种并杂环化合物及其用途 | |
| CN112707905A (zh) | 一种三并杂环化合物及其制备方法和用途 | |
| WO2020038458A1 (zh) | 一类稠环三氮唑类化合物、制备方法和用途 | |
| WO2019192031A1 (zh) | 雷公藤内酯醇衍生物及其制备方法和应用 | |
| WO2018019301A1 (zh) | 一种氟代雷公藤内酯醇衍生物 | |
| WO2022171088A1 (zh) | 吡唑并[3,4-d]嘧啶-3-酮衍生物 | |
| WO2013189266A1 (zh) | 喜树碱类化合物及其制备和用途 | |
| CN102229563A (zh) | 4-氨基喹啉衍生物及其制备方法和用途 | |
| WO2012007619A1 (es) | Proceso de obtención de derivados solubles en agua de 20(s)camptotecina como agentes antitumorales | |
| TW200408642A (en) | Hexacyclic compounds | |
| CN115701429B (zh) | 4-(1h-吲哚-1-基)嘧啶-2-氨基衍生物及其制备方法和应用 | |
| CN117447455A (zh) | 一种含氮杂环化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13806765 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14409284 Country of ref document: US |