4-取代基 -2-羟基吗啉 -3-酮及其制备方法 技术领域
本发明涉及药物化学领域,具体涉及阿瑞吡坦的中间体式 I化合 物 4-取代基 -2-羟基吗啉 -3-酮及其制备方法。 背景技术
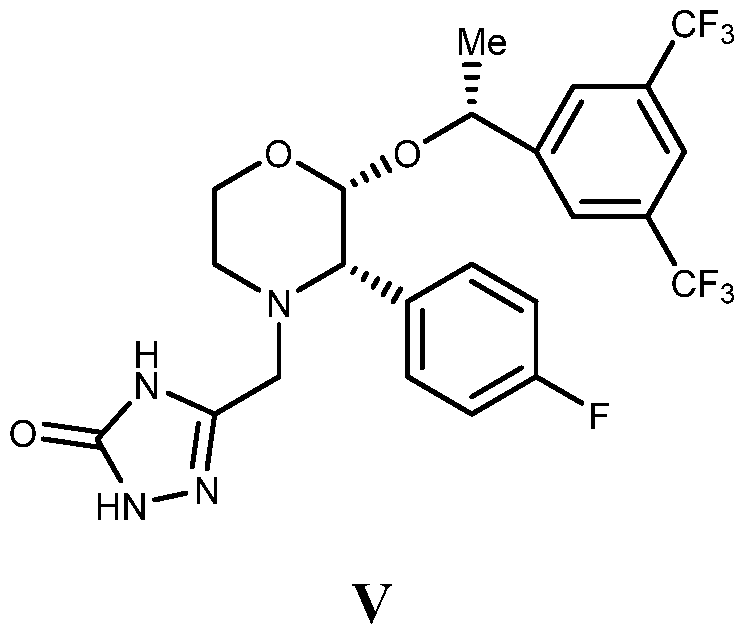
阿瑞吡坦 (Aprepitant),化学名为 5-[[(2 35)-2-[( ?)-1-[3,5-双(三 氟曱基苯基) 乙氧基】-3- ( 4-氟苯基) -4-吗啉基】曱基】 -1,2-二氢 -3H-1,2,4-三峻 -3-酮, 其结构式如下式 V所示:
阿瑞吡坦是美国默克公司研发的第一个神经激肽 -l(NK-l)受体 拮抗剂。 该药物可通过血脑屏障, 高选择性地拮抗 NK-1受体, 达到 阻断 P物质的作用, 且与 NK-2和 NK-3受体的亲和力较低。 该药品 临床用于预防化疗恶心、呕吐症状和术后恶心、呕吐症状。 阿瑞吡坦 于 2003年在美国上市, 商品名 Emend。
Karel M. J. Brands等人在 J. AM. CHEM. SOC. 2003, 125,
2129-2135中报道的合成路线中, 式 VII化合物是其重要中间体, 其 合成方法为以式 VI化合物为原料,和乙醛酸水溶液经缩合反应制得, 反应方程式如下:
另外 Todd D. Nelson在 Tetrahedron Letters 45 (2004) 8917-8920 中也报道了式 VII化合物的制备方法, 该方法通过式 VI化合物和草 酸二乙酯反应得式 VIII化合物,式 VIII化合物经三仲丁基硼氢化锂 还原得式 VII化合物。该方法收率低,且需要用到易燃、具有腐蚀性 和价格昂贵的还原试剂三仲丁基硼氢化锂, 不利于工业生产。 H
本发明以式 IV化合物为原料, 和乙醛酸水溶液经缩合反应制得 4-取代基 -2-羟基吗啉 -3-酮。 尤其是当苯环上连有吸电子取代基时, 可以明显提高 4-取代基 -2-羟基吗 ~3-酮的反应收率。 发明内容
本发明提供了一类具有神经活性的分子, 尤其是神经激肽 -1 受
体拮抗剂阿瑞吡坦的中间体 4-取代基 -2-羟基吗啉 -3-酮( I )及其制 备方法。当芳基上连有吸电子取 J ^时,与 J. AM. CHEM. SOC. 2003 125, 2129-2135中报道的相比,可以明显提高 4-取代基 -2-羟基吗啉 -3- 酮的收率并且缩短反应时间。
一类 4-取代基 -2-羟基吗 ~3-酮 物,其结构如下式 I所示:
其中, G选自:
II
其中,
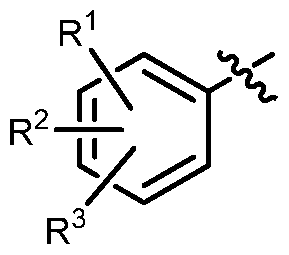
R\ R2、 R3独立的选自: 氢、 取代或未取代的 d-C6烷基、 取代或未取代的 c2-c6链烯基、 c2-c6炔基、 取代或未取代的苯基、 卤代、 -CN、 -CX3 ( X选自 Cl,Br,F )、 -N02、 -SR6、 -SOR6、 -S02R6、 -(CH2)m-NR4R5、 -NR4COR5、 -NR4C02R5、 -CONR4R5、 -COR4, -C02R4、 羟基、 d-C6烷氧基; d-C6烷基和 C2-C6链烯基上的取代 基可独立的选自: 羟基、 氧代、 d-C6烷氧基、 苯基、 -CN、 -N02、 卤代、 -NR4R5、 -NR4COR5、 -NR4C02R5、 -CONR4R5、 -COR4, -C02R4; 苯基上的取代基可选自: 羟基、 d-C6烷基、 d-C6烷氧基、 C2-C6链
烯基、 卤代、 -CN、 -N02、 -CF3、 -(CH2)m-NR4R5、 -NR4COR5、 -NR4C02R5、 -CONR4R5、 -COR4, -C02R4; 且 R1, R2、 R3不能同 时为氢;
优选 R1, R2、 R3独立的选自: 氢、 未取代的 d-C6烷基、 d-Ci 烷氧基、 卤素、 -N02、 -CN、 -CX3 ( X选自 Cl,Br,F ); 且 R1 R2、 R3不能同时为氢;
进一步优选 R1, R2、 R3独立的选自: 氢、 -N02, -CI, -CF3; 且
R R2、 R3不能同时为氢;
R4和 R5独立地选自:氢、 d-C6烷基、单羟基取代的 d-C6烷基、 苯基;
R6为氢或(^-( 6烷基;
m选自 1, 2, 3, 4;
R
7、 R
8、 R
9独立的选自: 氢、 取代或未取代的 d-C
6烷基、 取代或未取代的 C
2-C
6链烯基、 C
2-C
6炔基、 取代或未取代的苯基、 卤代、 -CN、 -CX
3 ( X选自 Cl,Br,F; )、 -N0
2、 -SR
12、 -SOR
12、 -S0
2R
12、 -(CH
2)
m-NR
10R
u、 -NR
10COR
U、 -NR
10CO
2R
u、 -CONR
10R
U、 -COR
10, -C0
2R
1G、 羟基、 d-C
6烷氧基; d-C
6烷基和 C
2-C
6链烯基上的取代
基各自独立的选自: 羟基、 氧代、 d_
6烷氧基、 苯基、 -CN、 -N0
2、 卤代、 -NRWR
11, -NR
10COR
N,
-CONR
10R
N, -COR
10, -C0
2R
10; 苯基上的取代基可选自: 羟基、 d-C
6烷基、 d-C
6烷氧基、 C
2-C
6链烯基、卤代、 -CN、 -N0
2、 -CF
3、 -(CH m-NRWR
11, -NR
10COR
n, -NR
10CO
2R
u、 -CONR
10R
U、 -COR
10, -C0
2R
10;
优选 R7、 R8、 R9独立的选自: 氢、 未取代的 d-C6烷基、 d-Ci 烷氧基、 卤素、 -N02、 -CN、 -CX3 ( X选自 Cl,Br,F ); 进一步优选 R7、 R8、 R9都为氢。
R10和 R11独立地选自: 氢、 d-C6烷基、单羟! ^代 d-C6烷基、 苯基;
R12为氢或 CH^烷基;
m选自 1,2,3,4。
上述式 I化合物具体优选自下列化合物:
本发明还包括了式 I化合物 4-取代基 -2-羟基吗啉 -3-酮的制备方 法,该方法以如下式 IV化合物为原料,和乙醛酸水溶液反应, 制得相 应的式 I化合物 4-取代基 -2-羟基吗啉 -3-酮;
其中,
G选自:
II
其中,
R R2、 R3独立的选自: 氢、 取代或未取代的 d-C6烷基、 取 代或未取代的 c2-c6链烯基、 c2-c6炔基、 取代或未取代的苯基、 卤 代、 -CN、 -CX3 ( X选自 Cl, Br, F )、 -N02、 -SR6、 -SOR6、 -S02R6、 -(CH2)m-NR4R5、 -NR4COR5、 -NR4C02R5、 -CONR4R5、 -COR4, -C02R4、 羟基、 d-C6烷氧基; d-C6烷基和 C2-C6链烯基上的取代
基可独立的选自: 羟基、 氧代、 d-C6烷氧基、 苯基、 -CN、 -N02 卤代、 -NR4R5、 -NR4COR5、 -NR4C02R5、 -CONR4R5、 -COR4, -C02R4; 苯基上的取代基可选自: 羟基、 d-C6烷基、 d-C6烷氧基、 c2-c6链 烯基、 卤代、 -CN、 -N02、 -CF3、 -(CH2)m-NR4R5、 -NR4COR5、 -NR4C02R5、 -CONR4R5、 -COR4, -C02R4; 且 R1, R2、 R3不能同 时为氢;
R4和 R5独立地选自:氢、 d-C6烷基、单羟基取代的 d-C6烷基、 苯基;
R6为氢或 烷基;
m选自 1, 2,3或 4;
R
7、 R
8、 R
9独立的选自: 氢、 取代或未取代的 d-C
6烷基、 取 代或未取代的 C
2-C
6链烯基、 C
2-C
6炔基、 取代或未取代的苯基、 卤 代、 -CN、 -CX
3 ( X选自 Cl,Br,F )、 -N0
2、 -SR
12、 -SOR
12、 -S0
2R
12、 -(CH
2)
m-NR
10R
u、 -NR
10COR
U、 -NR
10CO
2R
u、 -CONR
10R
U、 -COR
10, -C0
2R
1G、 羟基、 d-C
6烷氧基; d-C
6烷基和 d-C
6链烯基上的取代 基各自独立的选自: 羟基、氧代、 d-C
6烷氧基、苯基、 -CN、 -N0
2、 卤代、 -NRWR
11, -NR
10COR
N,
-CONR
10R
N, -COR
10,
-C0
2R
10; 苯基上的取代基可选自: 羟基、 d-C
6烷基、 d-C
6烷氧基、 C
2-C
6链烯基、卤代、 -CN、 -N0
2、 -CF
3、 -(CH m-NRWR
11, -NR
10COR
n, -NR
10CO
2R
u、 -CONR
10R
U、 -COR
10, -C0
2R
10;
R10和 R11独立地选自: 氢、 d-C6烷基、 单羟基取代的 d-C6烷基、 苯基;
R12为氢或 烷基;
m选自 1, 2,3或 4。
在该方法中, 所示式 II基团上的 R1, R2、 R3吸电子效应越强, 4-取代基 -2-羟基吗 ~3-酮的反应收率越高。
该反应的溶剂选自:乙酸乙酯、 C6-C12的烷烃、 苯、 曱苯、 对二 曱苯、 氯苯、 邻二氯苯丙酮、 二氯曱烷、 氯仿、 硝基曱烷、 N,N-二曱 基曱酰胺、 二曱亚砜、 2-丁酮、 d-C6醇、 1,3-二氧六环、 1,4-二氧六 环、 四氢呋喃、 乙腈、 1,2-二曱基乙醚、 水以及它们的混合物。
该反应的溶剂优选自可以和水混溶的溶剂, 包括丙酮、 2-丁酮、 N,N-二曱基曱酰胺、 二曱亚砜、 d-C3醇、 1,3-二氧六环、 1,4-二氧六 环、 四氢呋喃、 乙腈、 1,2-二曱基乙醚以及它们的混合物。
该反应的溶剂优选自四氢呋喃、 乙腈以及它们的混合物。
该反应的溶剂特别优选自四氢呋喃。
该反应的温度选自 30~100 °C, 优选 60~70 °C。
该反应的乙醛酸水溶液可选自乙醛酸一水合物水溶液,质量比为
1~99%的乙醛酸水溶液, 其中优选质量比为 50%的乙醛酸水溶液。
本发明还提供了一种制备具有如下结构的阿瑞匹坦关键中间体 式 IX化合物的方法, 其特征在于, 以式 I化合物为原料, 可制得到 如下式 IX化合物:
IX
具体反应步骤如下:
i )使式 I化合物与三氟醋酐反应制得如下式 X化合物:
.0. .OCOCF3
G' ii )使式 X化合物与如 XI化合物,
在三氟化硼的催化下反应得如下式 XII化合物:
iii )使式 XII化合物在四氢芳樟醇钾的作用下经手性转化得式 XIII 化合物:
iv )使式 XIII化合物与对氟苯基溴化镁反应后, 再用钯 /碳催化氢 化得式 IX化合物。
由 I化合物制备 IX化合物的反 方程如下所示:
IX 本发明还包括了所述式 I化合物在制备阿瑞吡坦中的应用。
本发明釆用当芳基上连有吸电子取代基时, 与 J. AM. CHEM. SOC. 2003, 125, 2129-2135中报道的相比,在制备 N-取代 -2-羟基吗啉 -3-酮中, 可以极大的提高反应收率, 缩短反应时间。 在制备阿瑞吡 坦的应用中, 可以明显的降低生产成本。
具体实施方式
实施例 1: 2-羟基 -4-(4-硝基苄基)吗 ~3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (46.4ml, 0.408mol), ( 50wt% 表示乙醛酸的质量比为 50%, 下同), 四氢呋喃 (31ml), 升温至回流, 緩慢滴加 2-(4-硝基苄基氨基)乙醇 (40g, 0.204mol)的四氢呋喃溶液 (71ml), 滴毕, 继续回流 8小时。 加入水(102ml ), 减压蒸除四氢呋 喃。 残留物升温至溶解, 降温结晶, 得淡黄色固体 2-羟基 -4-(4-硝基 苄基)吗 "3-酮 (47.2g, 91.7%);
TOF-MS (m/z): 253.1 [M+l] ;
^-NMR (400MHz, CDC13) δ: 8.21 (d, =8.8, 2H), 7.52 (d, =8.8,
2H), 7.11 (d, =6.4, 1H), 5.06 (d, =6.4, 1H), 4.65 (dd, =43.6, 15.6, 2H), 4.15 (ddd, =11.6, 11.2, 3.6, 1H), 3.73 (ddd, =12.0, 4.4, 2.0, 1H), 3.43 (ddd, =14.8, 12.0, 4.8, 1H), 3.19 (ddd, =10.8, 3.6, 1.6, 1H)。
实施例 2: 2-羟基 -4-(3-硝基苄基)吗 "3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (46.4ml, 0.408mol), 四氢呋 喃 (31ml), 升温至回流, 緩慢滴加 2-(3-硝基苄基氨基)乙醇 (40g, 0.204mol)的四氢呋喃溶液 (71ml), 滴毕, 继续回流 8 小时。 加入水 ( 102ml ), 减压蒸除四氢呋喃。 残留物升温至溶解, 降温结晶, 得 淡黄色固体 2-羟基 -4-(3-硝基苄基)吗啉 -3-酮 (46.5g, 90.3%);
TOF-MS (m/z): 253.1 [M+l];
^-NMR (400MHz, CDC13) δ: 8.11 (d, m, 2H), 7.75 (d, m, 2H), 7.18 (d, =6.4, 1H), 5.10 (d, =6.4, 1H), 4.55 (dd, =65.6, 15.6, 2H), 4.21 (ddd, =12.0, 11.0, 3.6, 1H), 3.79 (ddd, =12.0, 4.6, 2.0, 1H), 3.46 (ddd, =16.0, 12.0, 4.8, 1H), 3.22 (ddd, =11.2, 3.6, 1.6, 1H)。
实施例 3: 2-羟基 -4-(2-硝基苄基)吗 ~3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (46.4ml, 0.408mol), 四氢呋 喃(31ml), 升温至回流, 緩慢滴加 2-(2-硝基苄基氨基)乙醇 (40g,0.204mol)的四氢呋喃溶液 (71ml), 滴毕, 继续回流 8小时。加入 水(102ml ), 减压蒸除四氢呋喃。 残留物升温至溶解, 降温结晶, 得淡黄色固体 2-羟基 -4-(2-硝基苄基)吗啉 -3-酮 (46.1g,89.5%);
TOF-MS (m/z): 253.1 [M+l] ;
^-NMR (400MHz, CDC13) δ: 8.06 (dd, =8.0, 1.2, 1H), 7.76 (ddd, =8.0, 7.2, 0.8, 1H), 7.56 (ddd, =8.4, 7.2, 0.4, 1H), 7.38 (d, =8.0, 1H), 7.15 (d, =6.4, 1H), 5.08 (d, =6.0, 1H), 4.82 (dd, =71.2, 15.6, 2H), 4.20 (ddd, =15.2, 7.6, 4.0,1H), 3.76 (ddd, =9.6, 7.2, 2.4, 1H), 3.47 (ddd, =12.0, 10.8, 4.8, 1H), 3.21 (ddd, =12.4, 3.6, 2.4, 1H)。
实施例 4: 2-羟基 -4-(2-氯苄基)吗啉 -3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (49.1ml, 0.431mol), 四氢呋 喃 (32ml),升温至回流,緩慢滴加 2-(2-氯苄基 基)乙醇 (40g, 0.215mol) 的四氢呋喃溶液 (76ml), 滴毕, 继续回流 8小时。 加入水(108ml ), 减压蒸除四氢呋喃。 残留物升温溶解, 降温结晶, 得白色固体 2-羟 基 -4_(2_氯苄基)吗 酮 (41.¾, 78.9%);
TOF-MS (m/z): 242.2 [M+l];
^-NMR (400MHz, CDC13) δ: 7.47 (dd, =7.6, 1.6, 1H), 7.35 (ddd,
14.4, 7.6, 5.6, 2H), 7.25 (m, 1H), 7.09 (d, =6.4, 1H), 5.07 (d, =6.4, 1H), 4.60 (dd, =42.8, 16.0, 2H), 4.18 (ddd, =14.8, 11.2, 3.6, IH), 3.74 (ddd, =12.0, 4.8, 2.0, 1H), 3.42 (ddd, =15.2, 10.4, 4.4, 1H), 3.17 (ddd, =12.4, 4.0, 2.4, 1H)。
实施例 5: 2-羟基 -4-(3-氯苄基)吗啉 -3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (49.1ml, 0.431mol) , 四氢呋 喃 (32ml),升温至回流,緩慢滴加 2-(3-氯苄基氨基)乙醇 (40g,0.215mol) 的四氢呋喃溶液 (75ml), 滴毕, 继续回流 12小时。 加入水(108ml ), 减压蒸除四氢呋喃。 残留物升温溶解, 降温结晶, 得白色固体 2-羟 基 -4-(3-氯苄基)吗啉 -3-酮(40.2g, 76.9%);
TOF-MS (m/z): 242.2 [M+l] ;
^-NMR (400MHz, CDC13) δ: 7.51 (m, 2H), 7.33 (m, 2H), 7.12 (d, J=6.2, 1H), 5.08 (d, J=6.2, 1H), 4.54 (dd, =44.0, 12.2, 2H), 4.14 (ddd, =16.0, 11.6, 4.0, 1H), 3.69 (ddd, =12.4, 4.8, 2.4, 1H), 3.38 (ddd, =12.8, 11.0, 4.8, 1H), 3.20 (ddd, =12.4, 3.6, 2.4, IH) ;
实施例 6: 2-羟基 -4-(4-氯苄基)吗啉 -3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (49.1ml, 0.431mol) , 四氢呋 喃 (32ml),升温至回流,緩慢滴加 2-(4-氯苄基氨基)乙醇 (40g,0.215mol) 的四氢呋喃溶液 (75ml), 滴毕, 继续回流 12小时。 加入水(108ml ), 减压蒸除四氢呋喃。 残留物升温溶解, 降温结晶, 得白色固体 2-羟 基 _4-(4-氯苄基)吗 ~3-酮 (40.8g,78.1%);
TOF-MS (m/z): 242.2 [M+l] ;
^-NMR (400MHz, CDC13) δ: 7.34 (dd, =60.8, 8.0, 4H), 7.06 (d, =6.0, 1H), 5.04 (d, =6.0, 1H), 4.50 (dd, =52.0, 15.2, 2H), 4.11 (ddd, =15.6, 11.2, 4.0, 1H), 3.69 (ddd, =12.0, 4.8, 2.0, 1H), 3.36 (ddd, =12.4, 10.8, 4.8, 1H), 3.17 (ddd, =12.8, 3.6, 2.4, 1H)。
实施例 7: 2-羟基 -4-(2,4-二氯苄基)吗啉 -3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (41.3ml, 0.363mol) , 四氢呋
喃 (27ml), 升温至回流, 緩慢滴加 2-(2,4-二氯苄基氨基)乙醇 (40g, 0.182mol)的四氢呋喃溶液 (64ml), 滴毕, 继续回流 8 小时。 加入水 ( 91ml ), 减压蒸除四氢呋喃。 残留物升温溶解, 降温结晶, 得白色 固体 2-羟基 -4-(2,4-二氯苄基)吗啉 -3-酮 (41.4g, 82.4%);
TOF-MS (m/z): 276.2 [M+l] ;
^-NMR (400MHz, CDC13) δ: 7.63 (d, =2.4, 1H), 7.45 (dd, =8.4, 2.0, 1H), 7.27 (d, =8.4, 1H), 7.11 (d, =6.4, 1H), 5.06 (d, =5.6, 1H), 4.57 (dd, =47.2, 15.6, 2H), 4.17 (ddd, =15.6, 10.8, 3.6, 1H), 3.73 (ddd, =12.0, 4.8, 2.0, IH), 3.42 (ddd, =15.6 10.8, 4.8, IH), 3.18 (ddd, =12·4,4·0,2·4, 1H)。
实施例 8: 2-羟基 -4-(4-三氟曱基苄基)吗 ~3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (41.5ml, 0.364mol), 四氢呋 喃 (27ml), 升温至回流, 緩慢滴加 2-(4-三氟曱基苄基 基)乙醇 (40g, 0.182mol)的四氢呋喃溶液 (64ml), 滴毕, 继续回流 8小时。反应结束 后, 加入水(91ml ), 减压蒸除四氢呋喃。 残留物升温溶解, 降温结 晶,得白色固体 2-羟基 -4-(4-三氟曱基苄基)吗 ~3-酮 (47.8g,75.2%);
TOF-MS (m/z): 276.0 [M+l];
^-NMR (400MHz, CDC13) δ: 7.71 (d, =8.8, 1H), 7.47 (d, =8.0, 1H), 7.07 (d, =6.4, 1H), 5.06 (d, =6.4, 1H), 4.60 (dd, =41.6, 15.2, 2H), 4.14 (ddd, =15.6, 10.8, 3.6, 1H), 3.71 (ddd, =12.0, 4.8, 2.4, 1H), 3.40 (ddd, =15.6 10.8, 4.8, 1H), 3.26 (ddd, =12.8, 4.0, 2.4, 1H)。
实施例 9: 2-羟基 -4-(3-三氟曱基苄基)吗 ~3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (41.5ml, 0.364mol), 四氢呋 喃 (27ml), 升温至回流, 緩慢滴加 2-(3-三氟曱基苄基 基)乙醇 (40g,
0.182mol)的四氢呋喃溶液 (64ml), 滴毕, 继续回流 8小时。反应结束 后, 加入水(91ml ), 减压蒸除四氢呋喃。 残留物升温溶解, 降温结 晶,得白色固体 2-羟基 -4-(3-三氟曱基苄基)吗啉 -3-酮 (48.6g, 76.8%);
TOF-MS (m/z): 276.0 [M+l] ;
^-NMR (400MHz, CDC13) δ: 7.80 (m, 2H), 7.51 (m, 2H), 7.10 (d, =6.4, 1H), 5.12 (d, =6.4, 1H), 4.62 (dd, =54.6, 15.2, 2H), 4.16 (ddd, =15.8, 11.2, 3.6, 1H), 3.74 (ddd, =12.2, 4.8, 2.4, 1H), 3.44 (ddd, =15.8, 11.0, 4.8, 1H), 3.28 (ddd, =12.8, 4.4, 2.4, 1H)。
实施例 10: 2-羟基 -4-(2-三氟曱基苄基)吗啉 -3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (41.5ml, 0.364mol), 四氢呋 喃 (27ml), 升温至回流, 緩慢滴加 2-(2-三氟曱基苄基 基)乙醇 (40g, 0.182mol)的四氢呋喃溶液 (64ml), 滴毕, 继续回流 8小时。反应结束 后, 加入水(91ml ), 减压蒸除四氢呋喃。 残留物升温溶解, 降温结 晶,得白色固体 2-羟基 _4_(2_三氟曱基苄基)吗 ~3_酮(52. , 82.9%); TOF-MS (m/z): 276.0 [M+l];
^-NMR (400MHz, CDC13) δ: 7.96 (m, 2H), 7.72 (m, 2H), 7.21 (d, =6.4, 1H), 5.34 (d, =6.4, 1H), 4.62 (dd, =64.2, 14.4, 2H), 4.16 (ddd, =16.2, 11.0, 4.4, 1H), 3.74 (ddd, =12.4, 4.6, 2.4, 1H), 3.44 (ddd, =15.8, 11.0, 4.8, 1H), 3.28 (ddd, =12.6, 4.6, 2.4, 1H)。
实施例 11: 2-羟基 -4-(4-萘 -2-基曱基)吗 ~3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (45.3ml, 0.398mol), 四氢呋 喃(30ml), 升温至回流, 緩慢滴加 2- (萘 -2-基曱基氨基)乙醇 (40g,0.199mol)的四氢呋喃溶液 (70ml), 滴毕, 继续回流 24小时。 加 入水(100ml ), 减压蒸除四氢呋喃。 残留物升温溶解, 降温结晶,
得白色固体 2-羟基 -4-(4-萘 -2-基曱基)吗啉 -3-酮 (40.3g,78.8%);
TOF-MS (m/z): 258.1 [M+l] ;
^-NMR (400MHz, CDC13) δ: 7.84 (m, 3H), 7.54 (s, 1H), 7.50 (m, 2H), 7.40 (dd, =8.4, 1.6, 1H), 7.07 (d, =6.0, 1H), 5.06 (d, =5.6, 1H), 4.69 (dd, =31.6, 14.8, 2H), 4.13 (ddd, =13.5, 9.3, 3.6, 1H), 3.70 (ddd, =12.0, 4.8, 2.0, 1H), 3.39 (ddd, =15.6 10.4, 4.8, 1H), 3.26 (ddd, =12.0, 3.6, 2.4, 1H)。
实施例 12: 2-羟基 -4-(3-氡基苄基)吗 "3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (51.7ml, 0.454mol), 四氢呋 喃 (34ml), 升温至回流, 緩慢滴加 2-(3-氰基苄基氨基)乙醇 (40g, 0.227mol)的四氢呋喃溶液 (79ml), 滴毕, 继续回流 12小时。 加入水 ( 113ml ), 减压蒸除四氢呋喃。 残留物升温溶解, 降温结晶,得白色 固体 2-羟基 -4-(3-^J^苄基)吗啉 -3-酮 (37.6g, 71.4%);
TOF-MS (m/z): 233.1 [M+l];
^-NMR (400MHz, CDC13) δ: 7.88 (m, 2H), 7.50 (m, 2H), 7.22 (d, =8.4, 1H), 5.12 (d, =6.4, 1H), 4.62 (dd, =68.8, 15.2, 2H), 4.24 (ddd, =12.0, 11.2, 4.0, 1H), 3.78 (ddd, =12.4, 4.8, 2.0, 1H), 3.51 (ddd, =12.6, 10.6, 4.8, 1H), 3.22 (ddd, =12.8, 3.6, 2.4, IH) 。
实施例 13: 2-羟基 -4-(2-氡基苄基)吗 ~3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (51.7ml, 0.454mol), 四氢呋 喃 (34ml), 升温至回流, 緩慢滴加 2-(2-氰基苄基氨基)乙醇 (40g, 0.227mol)的四氢呋喃溶液 (79ml), 滴毕, 继续回流 12小时。 加入水 ( 113ml ), 减压蒸除四氢呋喃。 残留物升温溶解, 降温结晶,得白色 固体 2-羟基 -4-(2-^J^苄基)吗啉 -3-酮 (39.9g, 75.8%);
TOF-MS (m/z): 233.1 [M+l] ;
^-NMR (400MHz, CDC13) δ: 8.02 (m, 2H), 7.72 (m, 2H), 7.36 (d, =8.6, 1H), 5.22 (d, =6.8, 1H), 4.74 (dd, =72.6, 15.8, 2H), 4.36 (ddd, •7=12.4, 11.4, 4.0, 1H), 3.94 (ddd, =12.0, 4.6, 2.0, 1H), 3.69 (ddd, =12.4, 10.6, 4.2, 1H), 3.31 (ddd, =12.6, 3.4, 2.4, IH) 。
实施例 14: 2-羟基 -4-(4-曱基苄基)吗 ~3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (55.1ml, 0.484mol), 四氢呋 喃(36ml), 升温至回流, 緩慢滴加 2-(4-曱基苄基氨基)乙醇 (40g,0.242mol)的四氢呋喃溶液 (85ml), 滴毕, 继续回流 18小时。 加 入水(121ml ), 减压蒸除四氢呋喃。 残留物升温溶解, 降温结晶, 得白色固体 2-羟基 -4-(4-曱基苄基)吗啉 -3-酮 (31.2g,58.3%);
TOF-MS (m/z): 222.1 [M+l] ;
^-NMR (400MHz, CDC13) δ: 7.13 (dd, =19.6, 8.4, 4H), 7.02 (s, 1H), 5.03 (s, 1H), 4.47 (dd, =44.0, 14.4, 2H), 4.08 (ddd, =12.0, 11.6, 4.0, 1H), 3.68 (ddd, =9.6, 4.8, 2.4, 1H), 3.30 (ddd, =12.4, 10.8, 5.2, 1H), 3.08 (ddd, =10.4, 3.6, 2.0, 1H)。
实施例 15: 2-羟基 -4-(4-曱氧基苄基)吗啉 -3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (50.3ml, 0.442mol), 四氢呋 喃 (33ml), 升温至回流, 緩慢滴加 2-(4-曱氧基苄基氨基)乙醇 (40g,0.221mol)的四氢呋喃溶液 (78ml), 滴毕, 继续回流 18小时。 加 入水(100ml ), 减压蒸除四氢呋喃。 残留物升温溶解, 降温结晶, 得白色固体 2-羟基 -4-(4-曱氧基苄基)吗啉 -3-酮 (23.4g,44.7%);
TOF-MS (m/z): 238.1 [M+l] ;
^-NMR (400MHz, CDC13) δ: 7.18 (d, =8.8, 2H), 7.06 (d, =1.6,
1H), 6.90 (d, =8.8, 2H), 5.01 (d, =6.4, 1H), 4.43 (dd, =22.8, 14.4, 2H), 4.06 (ddd, =11.6, 11.2, 4.0, 1H), 3.73 (s, 3H), 3.67 (ddd, =12.0, 4.8, 2.0, 1H), 3.30 (ddd, =12.4, 11.2, 4.8, 1H), 3.08 (ddd, =12.4, 3.6, 2.0, 1H)。
实施例 16: 2-羟基 -4-(2-曱氧基苄基)吗啉 -3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (50.3ml, 0.442mol), 四氢呋 喃 (33ml), 升温至回流, 緩慢滴加 2-(2-曱氧基苄基氨基)乙醇 (40g,0.221mol)的四氢呋喃溶液 (78ml), 滴毕, 继续回流 18小时。 加 入水(100ml ), 减压蒸除四氢呋喃。 残留物升温溶解, 降温结晶, 得白色固体 2-羟基 -4-(2-曱氧基苄基)吗啉 -3-酮 (14.3g,27.2%);
TOF-MS (m/z): 238.1 [M+l];
^-NMR (400MHz, CDC13) δ: 7.25 (m, 1H), 7.07 (d, =7.6, 1H), 6.99 (d, =8.0, 1H), 6.91 (t, =7.6, 1H), 5.01 (s, 1H), 4.43 (dd, =38.8, 15.2, 2H), 4.10 (ddd, J=11.6, 11.2, 4.0, 1H), 3.77 (s, 3H), 3.67 (ddd, =12.0, 3.6, 2.0, 1H), 3.36 (ddd, =12.8, 10.4, 5.2, 1H), 3.12 (ddd, =10.0, 6.0 2.4, 1H)。
实施例 17: 2-羟基 -4-(3-曱基苄基)吗啉 -3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (55.1ml, 0.484mol), 四氢呋 喃(36ml), 升温至回流, 緩慢滴加 2-(3-曱基苄基氨基)乙醇 (40g,0.242mol)的四氢呋喃溶液 (85ml), 滴毕, 继续回流 24小时。 加 入水(121ml ), 减压蒸除四氢呋喃。 残留物升温溶解, 降温结晶, 得白色固体 2-羟基 -4-(3-曱基苄基)吗啉 -3-酮 (10.3g,19.3%);
TOF-MS (m/z): 222.1 [M+l] ;
^-NMR (400MHz, CDC13) δ: 7.24 (m, 2H), 7.02 (m, 1H), 5.33 (s,
1H), 4.57 (dd, =80.4, 14.8, 2H), 4.23 (ddd, =14.0, 10.4, 4.0, 1H), 4.12 (s, 1H), 3.67 (ddd, =11.6, 4.4, 3.2, 1H), 3.42 (ddd, =12.4, 10.4, 4.4, 1H), 3.27 (s, 1H), 3.08 (ddd, =10.8, 4.8, 3.6, 1H)。
实施例 18: 2-羟基 -4-(2,4-二曱 苄基)吗 ~3-酮的制备
反应瓶中加入 50wt%乙醛酸水溶液 (43.1ml, 0.379mol), 四氢呋 喃 (28ml), 升温至回流, 緩慢滴加 2-(2,4-二曱氧基苄基氨基)乙醇 (40g,0.189mol)的四氢呋喃溶液 (67ml), 滴毕, 继续回流 24小时。 加 入水(95ml ), 减压蒸除四氢呋喃。 残留物升温溶解, 降温结晶, 得 白色固体 2-羟基 -4-(2,4-二曱氧基苄基)吗 ~3-酮 (9.75g, 19.3%) ;
TOF-MS (m/z): 268.1 [M+1] ;
^-NMR (400MHz, CDC13) δ: 7.19 (d, =8.8, 1H), 6.46 (m, 2H), 5.25 (d, =2.8, IH), 4.57 (dd, =30.8, 14.0, 2H), 4.18 (ddd, =14.0, 10.0, 4.0, 1H), 3.80 (s, 6H), 3.67 (ddd, J=10.8, 6.2, 4.0, 1H), 3.42 (ddd, J=12.8, 9.6, 4.4, 1H), 3. 19 (ddd, J=10.0, 6.0, 4.0, 1H)。
实例 19: 2-羟基 -4-(4-硝基苄基)吗啉 -3-酮的制备
反应瓶中加入 10wt%乙醛酸水溶液 (232.0ml, 0.408mol), 四氢呋 喃 (31ml), 升温至回流, 緩慢滴加 2-(4-硝基苄基氨基)乙醇 (40g, 0.204mol)的四氢呋喃溶液 (71ml), 滴毕, 继续回流 8 小时。 加入水 ( 102ml ), 减压蒸除四氢呋喃。 残留物升温至溶解, 降温结晶, 得 淡黄色固体 2-羟基 -4-(4-硝基苄基)吗啉 -3-酮 (40.3g, 78.3%)。
TOF-MS (m/z): 253.1 [M+1] ;
^-NMR (400MHz, CDC13) δ: 8.22 (d, =8.8, 2H), 7.51 (d, =8.8, 2H), 7.11 (d, =6.4, 1H), 5.08 (d, =6.4, 1H), 4.65 (dd, =43.6, 15.6, 2H), 4.15 (ddd, =11.6, 11.2, 3.6, 1H), 3.72 (ddd, =12.0, 4.4, 2.0, 1H),
3.42 (ddd, =14.8, 12.0, 4.8, 1H), 3.19 (ddd, =10.8, 3.6, 1.6, 1H)。
实例 20: 2-羟基 -4-(4-硝基苄基)吗啉 -3-酮的制备
反应瓶中加入 30wt%乙醛酸水溶液 (77.3ml, 0.408mol), 四氢呋 喃 (31ml), 升温至回流, 緩慢滴加 2-(4-硝基苄基氨基)乙醇 (40g, 0.204mol)的四氢呋喃溶液 (71ml), 滴毕, 继续回流 8 小时。 加入水 ( 102ml ), 减压蒸除四氢呋喃。 残留物升温至溶解, 降温结晶, 得 淡黄色固体 2-羟基 -4-(4-硝基苄基)吗啉 -3-酮 (43.0g, 83.6%) 0
TOF-MS (m/z): 253.1 [M+l] ;
^-NMR (400MHz, CDC13) δ: 8.20 (d, =8.8, 2H), 7.51 (d, =8.8, 2H), 7.11 (d, =6.4, 1H), 5.08 (d, =6.4, 1H), 4.66 (dd, =43.6, 15.6, 2H), 4.15 (ddd, =11.6, 11.2, 3.6, 1H), 3.70 (ddd, =12.0, 4.4, 2.0, 1H), 3.40 (ddd, =14.8, 12.0, 4.8, 1H), 3.20 (ddd, =10.8, 3.6, 1.6, 1H)。
实例 21: 2-羟基 -4-(4-硝基苄基)吗啉 -3-酮的制备
反应瓶中加入 70wt%乙醛酸水溶液 (33.1ml, 0.408mol), 四氢呋 喃 (31ml), 升温至回流, 緩慢滴加 2-(4-硝基苄基氨基)乙醇 (40g, 0.204mol)的四氢呋喃溶液 (71ml), 滴毕, 继续回流 8 小时。 加入水 ( 102ml ), 减压蒸除四氢呋喃。 残留物升温至溶解, 降温结晶, 得 淡黄色固体 2-羟基 -4-(4-硝基苄基)吗啉 -3-酮 (41.5g, 80.7%)。
TOF-MS (m/z): 253.1 [M+l];
H-NMR (400MHz, CDC13) δ: 8.21 (d, =8.8, 2H), 7.52 (d, =8.8,
2H), 7.13 (d, =6.4, 1H), 5.08 (d, =6.4, 1H), 4.65 (dd, =43.8, 15.6, 2H), 4.17 (ddd, =11.6, 11.2, 3.6, 1H), 3.72 (ddd, =12.0, 4.4, 2.0, 1H), 3.42 (ddd, =14.6, 12.0, 4.8, 1H), 3.17 (ddd, =10.8, 3.6, 1.6, 1H)。
实例 22: 2-羟基 -4-(4-硝基苄基)吗啉 -3-酮的制备
反应瓶中加入 90wt%乙醛酸水溶液 (25.8ml, 0.408mol), 四氢呋 喃 (31ml), 升温至回流, 緩慢滴加 2-(4-硝基苄基氨基)乙醇 (40g, 0.204mol)的四氢呋喃溶液 (71ml), 滴毕, 继续回流 8 小时。 加入水 ( 102ml ), 减压蒸除四氢呋喃。 残留物升温至溶解, 降温结晶, 得 淡黄色固体 2-羟基 -4-(4-硝基苄基)吗啉 -3-酮 (37.8g, 73.4%) 0
TOF-MS (m/z): 253.1 [M+l];
^-NMR (400MHz, CDC13) δ: 8.20 (d, =8.6, 2H), 7.51 (d, =8.8, 2H), 7.14 (d, =6.4, 1H), 5.08 (d, =6.4, 1H), 4.66 (dd, =43.6, 15.6, 2H), 4.14 (ddd, =11.6, 11.2, 3.6, 1H), 3.72 (ddd, =12.0, 4.4, 2.0, 1H), 3.42 (ddd, =14.8, 12.0, 4.8, 1H), 3.19 (ddd, =10.8, 3.6, 1.6, 1H)。