WO2013067666A1 - 制备齐多夫定及其中间体的方法 - Google Patents
制备齐多夫定及其中间体的方法 Download PDFInfo
- Publication number
- WO2013067666A1 WO2013067666A1 PCT/CN2011/081856 CN2011081856W WO2013067666A1 WO 2013067666 A1 WO2013067666 A1 WO 2013067666A1 CN 2011081856 W CN2011081856 W CN 2011081856W WO 2013067666 A1 WO2013067666 A1 WO 2013067666A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- reaction
- formula
- compound
- trityl
- group
- Prior art date
Links
- 0 CC(C(N1)=O)=C*(C2)([C@]2(CC2)OC2(C2)[C@]2O)C1=O Chemical compound CC(C(N1)=O)=C*(C2)([C@]2(CC2)OC2(C2)[C@]2O)C1=O 0.000 description 3
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/073—Pyrimidine radicals with 2-deoxyribosyl as the saccharide radical
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the invention belongs to the technical field of medicinal chemistry, and in particular relates to a method for preparing zidovudine and an intermediate for preparing zidovudine. Background technique
- Zidovudine is the world's first anti-AIDS drug approved by the US FDA. Because of its exact efficacy, it is the most basic combination of "cocktail" therapy. To date, zidovudine remains one of the first choices for the treatment of AIDS in many developing countries. Its structural formula is as follows: The current method for producing zidovudine is mainly the route disclosed by US Pat. No. 5,214,442.
- This by-product makes the purification of zidovudine products difficult and the total yield is difficult to improve.
- Another object of the invention is to provide an intermediate for the preparation of zidovudine.
- a novel method of preparing zidovudine is provided, the method comprising the steps of:
- X is a halogen, preferably chlorine or bromine
- Is a hydroxy protecting group preferably a fluorenyl group or a C 3 -6 fluorenylcarbonyl group, more preferably a trityl group, a pivaloyl group or a trimethylpropionyl group;
- P 2 is a decylsulfonyl group, a fluorinated d 4 fluorenylsulfonyl group, an arylsulfonyl group or a -CS-R, wherein R is a d 4 fluorenyl group; preferably a methylsulfonyl group, a trifluoromethanesulfonyl group, a p-toluene group Sulfonyl or -CS-R, wherein R is methyl.
- the compound of the formula (VI) and the compound of the formula (III) can be directly subjected to the next reaction without separation, thereby realizing a three-pot and one-pot frying process.
- the compound of the formula (I), the compound of the formula (VI) and the compound of the formula (III) can be directly subjected to the next reaction without isolation, thereby realizing a four-pot and one-pot frying process.
- 5'-trityl- 2'-haloththymidine is methanesulfonylated at the 3'-position to give 5'-trityl-3'-methanesulfonyl-2'-halothymidine;
- the reaction temperature of the step 1) is 20-80 ° C, preferably 40-70 ° C; and the reaction solvent is a basic organic solvent, preferably pyridine.
- the reaction reagent of the step 2) is methanesulfonyl chloride; the reaction temperature is 0-5 ° C, and the reaction solvent is a halogenated hydrocarbon solvent, preferably dichloromethane.
- the hydrogenation dehydrogenation reagent of the step 3) is Raney nickel/triethylamine and hydrogen, and the reaction temperature is 20-60 ° C, preferably 30-50 ° C; the reaction solvent is an alcohol solvent, preferably A Alcohol.
- the alkaline condition described in the step 4) is selected from the group consisting of alkali metal/DMSO, alcohol solution of sodium alkoxide or potassium alkoxide, alcohol solution of sodium hydroxide or potassium hydroxide, sodium carbonate, potassium carbonate or lithium carbonate.
- the reaction solvent is an alcohol solvent, preferably methanol or ethanol
- the reaction temperature is 20 -80 ° C, preferably 50-70 ° C.
- the reaction reagent of step 5) is an azide, preferably lithium azide, or sodium azide, or sodium azide/anhydrous lithium chloride/ammonium chloride (a mole between the three) The ratio is 2-3: 0.8-1.2: 1), the reaction solvent is DMF; the reaction temperature is 60-120 ° C, preferably 80-110. C.
- the acidic condition described in the step 6) is selected from the group consisting of aqueous hydrochloric acid, aqueous sulfuric acid or acetic acid, p-toluenesulfonic acid, preferably aqueous hydrochloric acid or p-toluenesulfonic acid;
- the reaction solvent is an alcohol solvent, preferably methanol; It is 10-50 ° C, preferably 25-40 ° C.
- the reaction temperature of the step 1) is 20-80 ° C;
- the reaction solvent is a basic organic solvent;
- the reaction reagent of the step 2) is methanesulfonyl chloride; 0-5 ° C, the reaction solvent is a halogenated hydrocarbon solvent;
- the hydrogenation dehydrogenation reagent of step 3) is Raney nickel / triethylamine and hydrogen, the reaction temperature is 20-60 ° C ;
- the reaction solvent is an alcohol solvent
- the alkaline condition described in the step 4) is selected from the group consisting of an alkali metal/DMSO, an alcohol solution of sodium alkoxide or potassium alkoxide, an alcohol solution of sodium hydroxide or potassium hydroxide, an aqueous solution of sodium carbonate, potassium carbonate or lithium carbonate, An aqueous solution of sodium sulfonate, an aqueous solution of sodium p-toluenesulfonate, triethylamine or DBU;
- the reaction solvent
- the reaction temperature is 60-120 ° C ; and the acidic condition described in the step 6) is selected from the aqueous solution of hydrochloric acid, aqueous sulfuric acid or acetic acid or p-toluenesulfonic acid; the reaction solvent is an alcohol solvent; the reaction temperature is 10-50 ° C .
- the reaction temperature of step 1) is 40-70 .
- the reaction solvent is pyridine; the reaction reagent of the step 2) is methanesulfonyl chloride; the reaction temperature is 0-5.
- the reaction solvent is methylene chloride;
- the hydrogenation dehydrogenation reagent of step 3 is Raney nickel / triethylamine and hydrogen, the reaction temperature is 30-50 ° C ;
- the reaction solvent is methanol;
- the alkaline condition is selected from an aqueous solution of sodium carbonate, potassium carbonate or lithium carbonate;
- the reaction solvent is methanol or ethanol;
- the reaction temperature is 50-70 ° C ;
- the reaction reagent of the step 5) is lithium azide or azide.
- X is a halogen, preferably chlorine or bromine;
- P 2 is a decylsulfonyl group, a fluorinated d 4 fluorenylsulfonyl group, an arylsulfonyl group or a -CS-R, wherein R is a d 4 fluorenyl group; preferably a methylsulfonyl group, a trifluoromethanesulfonyl group, a p-toluene group Sulfonyl or -CS-R, wherein R is methyl.
- X is chlorine or bromine; is trityl, pivaloyl or trimethylpropanoyl; and P 2 is methylsulfonyl or p-toluenesulfonyl.
- X is halogen, preferably chlorine or bromine; It is a hydroxy protecting group, preferably a fluorenyl group or a C 3 -6 fluorenylcarbonyl group, more preferably a trityl group, a pivaloyl group or a trimethylpropionyl group.
- X is chlorine or bromine
- Pi is trityl, pivaloyl or trimethylpropanoyl.
- X is chlorine or bromine
- P1 is trityl or pivaloyl.
- the method of the invention can avoid the production of the 3',5'-dihydroxy protecting agent, thereby greatly improving the total yield of zidovudine, and at the same time, the refining process of the product is simplified due to the large reduction of impurities. It is easier to increase the purity.
- the technical features of each of the preferred technical solutions and the more preferred technical solutions may be combined with each other to form a new technical solution unless otherwise stated. For the purpose of brevity, the applicant has omitted the detailed description of these combinations in the specification, however, all the technical solutions combined with these technical features should be considered to be written in the specification in a clear manner.
- A/B as used in the specification and claims means that both A and B are present.
- palladium carbon/sodium acetate means the simultaneous presence of palladium carbon and sodium acetate
- alkali metal/DMSO means simultaneous use of alkali metal and DMSO.
- Lithium azide, or sodium azide/anhydrous lithium chloride/ammonium chloride means lithium azide, or both sodium azide, anhydrous lithium chloride and ammonium chloride.
- Ronkon/triethylamine means the simultaneous use of Raney nickel and triethylamine.
Abstract
一种制备齐多夫定(B)的方法,所述方法包括如下歩骤:1)以2'-卤代胸苷(A)为原料,将其5'-位羟基进行保护,得到式(I)化合物;2)式(I)化合物经3'-位羟基酰化得到式(VI)化合物;3)式(VI)化合物经脱卤得到式(111)化合物;4)式(III)化合物消除得到得到式(IV)化合物,5)式(IV)化合物经叠氮化反应得到式(V)化合物;6)式(V)化合物脱保护得到齐多夫定(B);具体的反应式如下(C)。式中:X为卤素;P1为羟基保护基;P2为C1-4烷基磺酰基、氟代的C1-4烷基磺酰基、芳基磺酰基或-CS-R,其中R为C1-4烷基。
Description
制备齐多夫定及其中间体的方法 技术领域
本发明属于药物化学技术领域,具体涉及制备齐多夫定的方法 以及用于制备齐多夫定的中间体。 背景技术
齐多夫定是世界上第一个获得美国 FDA批准生产的抗艾滋 药品, 因其疗效确切, 成为"鸡尾酒 "疗法最基本的组合成分。 迄 为止, 齐多夫定仍是许多发展中国家治疗艾滋病的首选药之一 其结构式如下:
 目前生产齐多夫定的方法主要是美国专利 US5124442公开的 路线
目前生产齐多夫定的方法主要是美国专利 US5124442公开的 路线

Scheme 1
所用原料 β_胸苷目前生产上多采用化学合成法,而化学合成法 中最常用的是以 5-甲基尿苷为原料的工艺路线 (见 《药学进展》 , 2005

Scheme 2 在实验中发明人发现, 由 β-胸苷选择性保护 5'-位羟基时有 10%-15%的 3',5'-二羟基保

这个副产物使得齐多夫定的产品提纯比较困难,总收率也很难 再得到提高。
因此,本领域需要一条更有效的、产率更高的合成齐多夫定的 方法。 发明内容
本发明的一个目的是提供一种齐多夫定的新合成方法, 以降
低 3',5'-二羟基保护物的百分比, 提高产率。
本发明的另一个目的是提供一种制备齐多夫定的中间体。 在本发明的第一方面,提供一种新的制备齐多夫定的方法,所 方法包括如下歩骤:
1 )以 2'-卤代胸苷为原料,将其 5'-位羟基进行保护,得到式(I)
2) 式 (I) 化合物经 3'-位羟基酰化得到式 (VI) 化合物;
3 ) 式 (VI) 化合物经脱卤得到式 (111)化合物;
4) 式 (III) 化合物消除得到得到式 (IV)化合物,
5 ) 式 (IV) 化合物经叠氮化反应得到式 (V) 化合物;
6) 式 (V) 化合物脱保护得到齐多夫定;
具体的反应式如下:
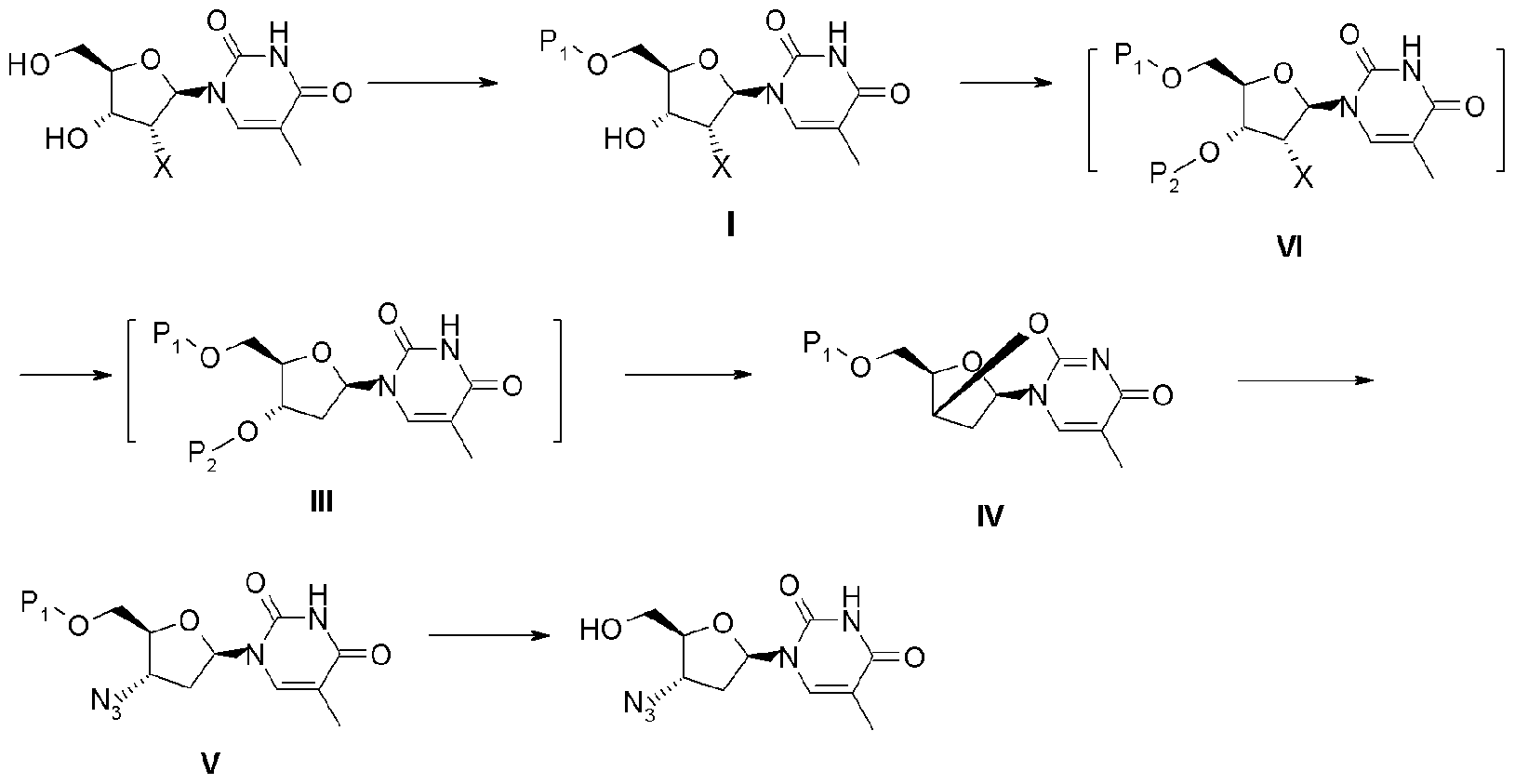
Scheme 3 式中: X为卤素, 优选为氯或溴; ?为羟基保护基, 优选为垸 基或 C3_6垸基羰基,更优选为三苯甲基、特戊酰基或三甲基丙酰基;
P2为 垸基磺酰基、氟代的 d_4垸基磺酰基、芳基磺酰基或 -CS-R, 其中 R为 d_4垸基; 优选为甲磺酰基、 三氟甲磺酰基、 对甲苯磺酰 基或 -CS-R, 其中 R为甲基。
在一优选的实施方式中, 式 (VI) 化合物和式 (III) 化合物 均可不经分离直接进行下一歩反应, 实现三歩一锅炒的工艺。
在一优选的实施方式中, 式 (I) 化合物、 式 (VI) 化合物和 式 (III) 化合物可不经分离直接进行下一歩反应, 从而实现四歩 一锅炒的工艺。
在一优选的实施方式中, 具体歩骤可描述如下:
1 ) 以 2'-卤代胸苷为原料, 与三苯甲基氯甲垸反应, 得到 5'- 三苯甲基 _2'_卤胸苷;
2 ) 5'-三苯甲基 -2'-卤胸苷经 3'-位甲磺酰化得到 5'-三苯甲基 -3'-甲磺酰基 -2'-卤胸苷;
3 ) 5'-三苯甲基 -3'-甲磺酰基 -2'-卤胸苷经氢化脱卤得到 5'-三苯 甲基 -3'-甲磺酰基胸苷;
4 ) 5'-三苯甲基 -3'-甲磺酰基胸苷在碱性条件下消除得到 5'-三 苯甲基 -2,3'-脱水胸苷;
5 ) 5'-三苯甲基 -2,3'-脱水胸苷经叠氮化反应得到 5'-三苯甲基 -3'-叠氮胸苷;
6 ) 5'-三苯甲基 -3'-叠氮胸苷在酸性条件下脱保护得到齐多夫 定。 原料 2'-卤代胸苷可参考 US4914233报道的方法进行制备。 在优选的实施方式 ( l )中:
较佳地, 歩骤 1 ) 的反应温度为 20-80 °C, 优选 40-70 °C ; 反应 溶剂为碱性有机溶剂, 优选吡啶。
较佳地, 歩骤 2 ) 的反应试剂为甲磺酰氯; 反应温度为 0-5 °C, 反应溶剂为卤代烃溶剂, 优选二氯甲垸。
较佳地, 歩骤 3 ) 的氢化脱氢的试剂为雷尼镍 /三乙胺和氢气, 反应温度为 20-60°C, 优选 30-50°C ; 反应溶剂为醇类溶剂, 优选甲
醇。
较佳地, 歩骤 4) 所述的碱性条件选自碱金属 /DMSO, 醇钠或 醇钾的醇溶液, 氢氧化钠或氢氧化钾的醇溶液, 碳酸钠、 碳酸钾 或碳酸锂的水溶液, 甲磺酸钠水溶液, 对甲苯磺酸钠水溶液, 三 乙胺或者 DBU, 优选碳酸钠、 碳酸钾或碳酸锂的水溶液; 反应溶 剂为醇类溶剂,优选为甲醇或乙醇;反应温度为 20-80°C,优选 50-70 °C。
较佳地, 歩骤 5 ) 的反应试剂为叠氮化物, 优选叠氮化锂, 或 叠氮化钠, 或叠氮化钠 /无水氯化锂 /氯化铵 (三者之间的摩尔比为 2-3: 0.8-1.2: 1) , 反应溶剂为 DMF; 反应温度为 60-120 °C, 优选 80-110。C。 较佳地, 歩骤 6 ) 所述的酸性条件选自盐酸水溶液、 硫酸水溶 液或醋酸、 对甲苯磺酸, 优选盐酸水溶液或对甲苯磺酸; 反应溶 剂为醇类溶剂, 优选为甲醇; 反应温度为 10-50°C, 优选 25-40°C。 在优选的实施方式 (l)中, 优选地, 歩骤 1 )的反应温度为 20-80 °C ; 反应溶剂为碱性有机溶剂; 歩骤 2 ) 的反应试剂为甲磺酰氯; 反应温度为 0-5°C, 反应溶剂为卤代烃溶剂; 歩骤 3 ) 的氢化脱氢的 试剂为雷尼镍 /三乙胺和氢气, 反应温度为 20-60°C ; 反应溶剂为醇 类溶剂; 歩骤 4 ) 所述的碱性条件选自碱金属 /DMSO , 醇钠或醇钾 的醇溶液, 氢氧化钠或氢氧化钾的醇溶液, 碳酸钠、 碳酸钾或碳 酸锂的水溶液, 甲磺酸钠水溶液, 对甲苯磺酸钠水溶液, 三乙胺 或者 DBU; 反应溶剂为醇类溶剂; 反应温度为 20-80°C ; 歩骤 5 ) 的 反应试剂为叠氮化物, 反应溶剂为 DMF; 反应温度为 60-120 °C ; 以及歩骤 6 ) 所述的酸性条件选自盐酸水溶液、 硫酸水溶液或醋酸 或对甲苯磺酸; 反应溶剂为醇类溶剂; 反应温度为 10-50°C。 在优选的实施方式 (l)中,更优选地,歩骤 1 )的反应温度为 40-70
。C ; 反应溶剂为吡啶; 歩骤 2) 的反应试剂为甲磺酰氯; 反应温度 为 0-5。C, 反应溶剂为二氯甲垸; 歩骤 3) 的氢化脱氢的试剂为雷尼 镍 /三乙胺和氢气, 反应温度为 30-50°C; 反应溶剂为甲醇; 歩骤 4) 所述的碱性条件选自碳酸钠、 碳酸钾或碳酸锂的水溶液; 反应溶 剂为甲醇或乙醇; 反应温度为 50-70°C; 歩骤 5) 的反应试剂为叠氮 化锂,或叠氮化钠 /无水氯化锂 /氯化铵,且三者之间的摩尔比为 2-3: 0.8-1.2: 1, 反应溶剂为 DMF; 反应温度为 80-110°C; 以及歩骤 6) 所述的酸性条件为盐酸水溶液或对甲苯磺酸; 反应溶剂为甲醇; 反应温度为 25-40 °C。 在本发明的第二方面,提供一种制备齐多夫定的中间体,如以 下式 (VI)所示:

VI 式中 X为卤素, 优选为氯或溴; ?为羟基保护基, 优选为垸基 或 C3_6垸基羰基, 更优选为三苯甲基、 特戊酰基或三甲基丙酰基;
P2为 垸基磺酰基、氟代的 d_4垸基磺酰基、芳基磺酰基或 -CS-R, 其中 R为 d_4垸基; 优选为甲磺酰基、 三氟甲磺酰基、 对甲苯磺酰 基或 -CS-R, 其中 R为甲基。
在一优选的实施例中, X为氯或溴; 为三苯甲基、 特戊酰基 或三甲基丙酰基; P2为甲磺酰基或对甲苯磺酰基。
在一更优选的实施例中, X为氯或溴; P1为三苯甲基; P2为甲 磺酰基或对甲苯磺酰基。 在本发明的第三方面,提供一种制备齐多夫定的中间体,如以 下式 (I)所示:

I
式中, X为卤素, 优选氯或溴; ?为羟基保护基, 优选为垸基 或 C3_6垸基羰基, 更优选为三苯甲基、 特戊酰基或三甲基丙酰基。
在一优选的实施例中, X为氯或溴; Pi为三苯甲基、 特戊酰基 或三甲基丙酰基。
在一更优选的实施例中, X为氯或溴; P1为三苯甲基或特戊酰 基。 本发明的方法可避免产生将 3',5'-二羟基保护物, 从而大大提 高了齐多夫定的总收率, 同时因为杂质的大为减少, 使得产品的 精制工艺变得简单, 产品更容易提高纯度。 在本说明书中, 除非有其他说明,各个优选技术方案和更优选 技术方案的技术特征可以相互组合形成新的技术方案。 为了简要 目的, 申请人在说明书中省略了这些组合的具体描述, 然而, 所 有这些技术特征组合后的技术方案均应当被认为以明确的方式书 面记载于本说明书中。 说明书和权利要求书中所用的 "A/B " 表示 A和 B同时存在, 例如"钯碳 /醋酸钠 "表示同时存在钯碳和醋酸钠, "碱金属 /DMSO" 表示同时使用碱金属和 DMSO; "叠氮化锂, 或叠氮化钠 /无水氯 化锂 /氯化铵"表示叠氮化锂, 或者同时使用叠氮化钠、 无水氯化 锂和氯化铵。 "雷尼镍 /三乙胺"表示同时使用雷尼镍和三乙胺。 下面结合具体实施例, 进一歩阐述本发明。 应理解, 这些实施 例仅用于说明本发明而不用于限制本发明的范围。 下列实施例中
未注明具体条件的实验方法, 通常按照常规条件, 或按照制造厂 商所建议的条件。 除非另外说明, 否则百分比和份数按重量计算。 实施例 1
5,-三苯甲基 -2,-溴胸苷的制备
室温下在反应瓶中加入 300ml吡啶, 2,-溴胸苷(50.0g,0.16mol, 参考 US4914233报道的方法进行制备) , 三苯基氯甲垸 (54.0g, 0.19mol ) 。 升温至 60 °C, TLC跟踪反应直至原料反应完全。 加入 水终止反应。 减压浓缩至粘稠状, 将残留物溶解于 600ml二氯甲垸 中, 用水洗涤 2次, 无水硫酸镁干燥。 过滤, 减压浓缩至干, 得到 100g白色泡沬状固体。 ifi-NMR: δ 1.43(s, 3H), 3.03(d, IH), 3.45(d, IH), 3.58(d, IH), 4.28(s, IH), 4.49(d, IH), 4.64(t, IH), 6.26(d, IH), 7.28(m, 3H), 7.36(m, 6H), 7.38(m, 6H), 7.57(s, IH), 9.56(s, IH).
5,-三苯甲基 -3,-甲磺酰基 -2,-溴胸苷的制备
室温下加入 40ml二氯甲垸, 5,-三苯甲基 -2,-溴胸苷 (10.0g, 0.018mol ) , 冰浴冷却至 0 °C。 同时分别滴入甲磺酰氯 (2.0ml, 0.026mol )和 4ml吡啶, 控制反应温度低于 5 °C。 TLC跟踪原料反应 完全后, 滤去不溶物。 滤液减压浓缩至干, 残留物直接投入下歩 反应。 产品可通过柱层析 (乙酸乙酯:正己垸 =1: 2 ) , 得到白色 固体。 ^-NMR: δ 1.43(s, 3H), 3.16(s, 3H), 3.50(d, IH), 3.63(d, IH), 4.55(d, IH), 4.70(t, IH), 5.55(m, IH), 6.26(d, IH), 7.36-7.48(m, 15H): 7.57(s, IH), 9.09(s, IH).
5,-三苯甲基 -3,-甲磺酰基胸苷的制备
室温下在上述残留物中加入 50ml甲醇, 15g雷尼镍, 2.0ml三乙 胺, 常压下通入氢气。搅拌下升温至 40 °C, TLC跟踪原料反应完全 后停止通氢气。 抽滤, 减压浓缩干, 所得浆状残留物直接投入下 歩反应。 产品可通过柱层析 (乙酸乙酯:正己垸 =1: 3 ) , 得到白 色固体。 ifi-NMR: δ 1.43(s, 3H), 2.45(m, IH), 2.65(m, IH), 3.03(s,
3H), 3.48(m, 2H), 4.30(m, 1H), 5.39(m, 1H), 6.40(m, 1H) 7.28-7.38(m, 15H), 7.56(s, 1H), 8.65(s, 1H).
5,-三苯甲基 -2,3,-脱水胸苷的制备
在上述残留物中加入 50ml甲醇, 15ml饱和碳酸钠水溶液, 加 热至回流。 TLC跟踪原料反应完全后, 降温至 40°C, 减压浓缩至粘 稠状。 残留物加入 20ml水, 用二氯甲垸 100ml分 3次萃取, 合并有 机相。 有机相用饱和食盐水洗涤, 无水硫酸镁干燥。 过滤, 滤液 减压浓缩至干,得到 8.0g类白色固体,收率 95%。 ^-NMR: δ 1.90(s, 3H), 2.35(m, 1H), 2.64(m, 1H), 3.38(m, 2H), 4.28(m, 1H), 5.13(m, 1H), 5.45(d, 1H), 6.81(m, 1H), 7.30-7.48(m, 15H).
5,-三苯甲基 -3,-叠氮胸苷的制备
搅拌下依次加入 250ml DMF、 叠氮化锂 ( 19.0g, 0.39mol) , 5,-三苯甲基 -2,3,-脱水胸苷 ( 60.0g, 0.13mol) , 缓慢升温至 100°C 反应。 TLC跟踪原料反应完全后, 冷至室温, 滤去不溶物。 搅拌下 往滤液中缓慢滴入 500ml水, 滴完后继续打浆 2小时。 抽滤, 鼓风 干燥得到 62.9g 类白色固体, 收率 96%。 ifi-NMR: δ 1.55(s, 3H), 2.48(m, 1H), 2.66(m, 1H), 4.18(m, 1H), 4.31(m, 1H), 4.58(m, 2H), 6.20(m, 1H), 7.15(s, 1H), 7.25-7.36(m, 15H), 8.83(s, 1H). 齐多夫定的制备
搅拌下加入 250ml甲醇、 5,-三苯甲基 -3,-叠氮胸苷 (50.0g, 0.098mol)和 2ml浓盐酸, 室温下反应 3小时。 TLC跟踪原料反应完 全后, 加入 0.8g氢氧化钠终止反应。 减压浓缩至粘稠状, 残留物中 加入 250ml水, 加热至 75 °C, 搅拌 1小时。 趁热过滤除去不溶物。 滤液减压浓缩至干, 加入 300ml乙酸乙酯溶解, 加热至 60°C, 活性 炭脱色, 趁热过滤, 滤液减压浓缩至干, 得到 26.5g 类白色固体。 粗品用异丙醇重结晶得到纯度为 99.8%的齐多夫定 24.8g, 收率 95%。 MS: m/z 267 (M+) 。
实施例 2
5'-特戊酰基 -2'-氯胸苷的制备
室温下在反应瓶中加入 100ml吡啶, 2,-氯胸苷 ( 14.0g, 0.052mol ) , 特戊酰氯 (8.0ml, 0.065mol ) 。 升温至 40 °C, TLC 跟踪反应直至原料反应完全。 加入水终止反应。 减压浓缩至粘稠 状, 将残留物溶解于 200ml二氯甲垸中, 用水洗涤 2次, 无水硫酸 镁干燥。 过滤, 减压浓缩至干, 得到 18g类白色泡沬。 ^-NMR: δ 1.20(s, 9H), 1.45(s, 3H), 3.41(d, 1H), 3.62(t, 1H), 4.14(s, 1H), 4.36(d, 1H), 4.53(t, 1H), 5.02(m, 1H), 6.19(d, 1H), 7.56(s, 1H), 9.47(s, 1H).
5'-特戊酰基 -3'-甲磺酰基 -2'-氯胸苷的制备
反应瓶中加入 50ml二氯甲垸, 上述 18g白色泡沬状固体, 搅拌 溶解, 冰浴冷却至 0°C。 同时分别滴入甲磺酰氯(4.8ml, 0.062mol ) 和 10ml吡啶, 控制反应温度低于 5 °C。 TLC跟踪原料反应完全后, 缓慢滴入 30ml饱和碳酸钠水溶液终止反应。 有机层减压浓缩回收 二氯甲垸, 残留物直接投入下歩反应。
5'-特戊酰基 -3'-甲磺酰基胸苷的制备
室温下在上述残留物中加入 100ml甲醇, 1.8g 5%的钯炭, 醋酸 钠 (5.1g, 0.062mol ) , 搅拌, 常压下通入氢气。 TLC跟踪原料反 应完全后停止通氢气。 抽滤, 减压浓缩干, 真空干燥, 得到 14.7g 浅黄色固体, 收率 70%。 ifi-NMR: δ 1.18(s, 9H), 2.05-2.20(d, 2H), 2.35(s, 3H), 3.15(s, 3H), 4.05-4.25(m, 2H), 4.55(m, 1H), 5.05(m, 1H), 5.46(m, 1H), 7.45(s, 1H), 9.28(s, 1H).
5'-特戊酰基 -2,3'-脱水胸苷的制备
在反应瓶中加入 5'-特戊酰基 -3'-甲磺酰基胸苷 ( 10.0g, 0.025mol ) , 75ml乙醇, 15ml饱和碳酸钠水溶液, 加热至回流。 TLC跟踪原料反应完全后, 降温至 40 °C, 减压浓缩至粘稠状。残留 物加入 50ml水, 用二氯甲垸 75ml分 3次萃取, 合并有机相。 有机相
用饱和食盐水洗涤, 无水硫酸镁干燥。 过滤, 滤液减压浓缩至干, 得到 7.0g类白色固体粗品, 收率 91%。 ifi-NMR : δ 1.18(s, 9H), 2.05-2.20(d, 2H), 2.35(s, 3H), 3.45(m, IH), 4.05-4.25(m, 2H), 4.48(m: IH), 4.55(m, IH), 6.48(s, IH).
5'-特戊酰基 -3'-叠氮胸苷的制备
搅拌下依次加入 30ml DMF, 叠氮化钠 ( 4.5g, 0.069mol ) , 1.5g无水氯化锂 (1.5g, 0.035mol) , 氯化铵 (1.5g, 0.028mol) , 5,-特戊酰基 -3,,2-脱水胸苷 ( 5.0g, 0.016mol) , 缓慢升温至 110°C 反应。 TLC跟踪原料反应完全后, 冷至室温, 滤去不溶物。 搅拌下 往滤液中缓慢滴入 50ml水, 滴完后继续打浆 30分钟。 抽滤, 鼓风 干燥得到 5.1g 浅黄色固体, 收率 91%。 ifi-NMR: δ 1.20(s, 9H), 1.68(s, 3H), 2.10-2.30(m, 2H), 4.15(m, IH), 4.28(m, IH), 4.47(m, 1H): 4.54(m, IH), 6.18(t, IH), 7.19(s, IH), 8.39(s, IH). 齐多夫定的制备
搅拌下加入 65ml甲醇, 5,-特戊酰基 -3,-叠氮胸苷 (5.0g, 0.014mol), 6.5ml 25%甲醇钠的甲醇溶液,室温下搅拌 1小时。 TLC 跟踪原料反应完全后, 用强酸性树脂 (Dowex 50-200*8 ) 中和, 调 pH至 6左右, 过滤回收树脂, 甲醇洗涤。合并滤液, 活性炭脱色, 减压浓缩至干, 得到的类白色固体再用异丙醇重结晶, 纯度为 99.5%的齐多夫定3.2 收率 86%。 MS: m/z 267 (M+) 。 实施例 3
5,-三苯甲基 -3,-甲磺酰基 -2,-氯胸苷的制备
室温下在反应瓶中加入2001111吡啶,2-氯胸苷(50.(^,0.1811101), 三苯基氯甲垸 (60.0g, 0.21mol) 。 升温至 60°C, TLC跟踪反应直 至原料反应完全。 撤去油浴, 用冰浴冷却至 0 °C, 滴入甲磺酰氯 ( 16.5ml, 0.22mol) , 控制反应温度低于 5 °C。 TLC跟踪原料反应 完全后, 加入水终止反应。 减压浓缩至粘稠状, 将残留物溶解于 1000ml二氯甲垸中, 用食盐水洗涤 2次, 无水硫酸镁干燥。 过滤,
减压浓缩至干,得到 l lOg白色泡沬状固体。 ^-NMR: δ 1.41(s, 3H), 3.15(s, 3H), 3.50(d, IH), 3.63(d, IH), 4.53(d, IH), 4.68(t, IH), 5.54(m: IH), 6.25(d, IH), 7.35-7.45(m, 15H), 7.51(s, IH), 9.06(s, IH). 实施例 4
5,-三甲基丙酰基 -3,-乙硫酰基 -2,-氯胸苷的制备
室温下在反应瓶中加入 100ml二氯甲垸, 2'-氯胸苷 (14.0g, 0.052mol ) , 三甲基丙酰氯 (8.0ml, 0.065mol ) , 10ml吡啶。 升 温至 40 °C, TLC跟踪反应直至原料反应完全。冷却至 10 °C, 滴入乙 硫酰氯 (3.0ml, 0.042mol ) , 控制反应温度低于 15 °C。 TLC跟踪 原料反应完全后, 缓慢滴入 30ml饱和碳酸钠水溶液终止反应。 减 压浓缩回收二氯甲垸, 残留物直接投入下歩反应。 1H-NMR: δ 1.03(m, 9H), 1.12(s, 3H), 1.33(m, 2H), 1.45(s, 3H), 3.39(d, IH), 3.60(t, IH), 4.12(s, IH), 4.34(d, IH), 4.52(t, IH), 4.95(m, IH), 6.16(d IH), 7.52(s, IH), 9.31(s, IH).
5,-三甲基丙酰基 -3,-乙硫酰基胸苷的制备
室温下在上述残留物中加入 100ml甲醇, 2g 5%的钯炭, 醋酸 钠 (6.0g, 0.073mol ) , 搅拌, 常压下通入氢气。 TLC跟踪原料反 应完全后停止通氢气。 抽滤, 减压浓缩至干, 所得浆状残留物直 接投入下歩反应。
5,-三甲基丙酰基 -2,3, -脱水胸苷的制备
在上歩残留物中加入 100ml乙腈, 碳酸钾 (8.6g, 0.062mol ) , 加热至回流。 TLC跟踪原料反应完全后, 降温至 40 °C, 减压浓缩至 粘稠状。 残留物加入 50ml水, 用二氯甲垸 100ml分 3次萃取, 合并 有机相。 有机相用饱和食盐水洗涤, 无水硫酸镁干燥。 过滤, 滤 液减压浓缩至干, 得到 7. lg类浅黄色泡沬。 1H-NMR: δ 1.01(m, 9H), 1.95(m, 2H), 2.01-2.15(d, 2H), 2.31(s, 3H), 3.43(m, IH), 4.01-4.15(m, 2H), 4.45(m, IH), 4.53(m, IH), 6.50(s, IH).
5,-三甲基丙酰基 -3,-叠氮胸苷的制备
搅拌下依次加入 30ml DMF, 叠氮化钠 (6.5g, O.lOmol) , 上 歩产品的 DMF 10ml溶液, 缓慢升温至 110°C反应。 TLC跟踪原料反 应完全后, 冷至室温, 滤去不溶物。搅拌下往滤液中缓慢滴入 80ml 水, 滴完后继续打浆 1小时。 抽滤, 鼓风干燥得到 5.6g黄色固体。 1H-NMR: δ 1.05(m, 9H), 1.58(m, 2H), 1.62(s, 3H), 1.75(m, 1H), 1.88(m, 1H), 2.12(m, 1H), 4.05-4.15(d, 2H), 4.17(m, 1H), 5.35(m, 1H), 7.53(s, 1H), 9.67(s, 1H). 齐多夫定的制备
搅拌下加入 65ml甲醇, 5,-三甲基丙酰基 -3,-叠氮胸苷 (5.0g, 0.014mol), 6.5ml 25%甲醇钠的甲醇溶液,室温下搅拌 1小时。 TLC 跟踪原料反应完全后, 用强酸性树脂 (Dowex 50-200*8 ) 中和, 调 pH至 6左右, 过滤回收树脂, 甲醇洗涤。合并滤液, 活性炭脱色, 减压浓缩至干, 得到的类白色固体再用异丙醇重结晶, 可得到纯 度为 99%的齐多夫定 2.2g。 MS: m/z 267 (M+) 。 在本发明提及的所有文献都在本申请中引用作为参考,就如同 每一篇文献被单独引用作为参考那样。 此外应理解, 在阅读了本 发明的上述内容之后, 本领域技术人员可以对本发明作各种改动 或修改, 这些等价形式同样落于本申请所附权利要求书所限定的 范围。
Claims
1. 一种制备齐多夫定的方法, 所述方法包括如下歩骤:
1 )以 2'-卤代胸苷为原料,将其 5'-位羟基进行保护,得到式(I) 化合物;
2) 式 (I) 化合物经 3'-位羟基酰化得到式 (VI) 化合物;
3 ) 式 (VI) 化合物经脱卤得到式 (111)化合物;
4) 式 (III) 化合物消除得到得到式 (IV)化合物,
5 ) 式 (IV) 化合物经叠氮化反应得到式 (V) 化合物;
6) 式 (V) 化合物脱保护得到齐多夫定;
具体的反应式如下:

2. 如权利要求 1所述的方法, 其特征在于, X为氯或溴; ?1为 三苯甲基、 特戊酰基或三甲基丙酰基; P2为甲磺酰基、 三氟甲磺酰 基、 对甲苯磺酰基或 -CS-R, 其中 R为甲基。
3. 如权利要求 1所述的方法, 其特征在于, 反应式中式 (VI ) 化合物和式 (III)的化合物不经分离直接进行下一歩反应, 实现三歩 一锅炒的工艺。
4. 如权利要求 1所述的方法, 其特征在于, 反应式中式 (I)化合
物、 式 (VI) 化合物和式 (III)的化合物不经分离直接进行下一歩反 应, 实现四歩一锅炒的工艺。
5. 如权利要求 1所述的方法, 其特征在于, 所述方法可以具体 地描述为包含以下歩骤:
1) 以 2'-卤代胸苷为原料, 与三苯甲基氯甲垸反应, 得到 5'- 三苯甲基 _2'_卤胸苷;
2) 5'-三苯甲基 -2'-卤胸苷经 3'-位甲磺酰化得到 5'-三苯甲基 -3'-甲磺酰基 -2'-卤胸苷;
3) 5'-三苯甲基 -3'-甲磺酰基 -2'-卤胸苷经氢化脱卤得到 5'-三苯 甲基 -3'-甲磺酰基胸苷;
4) 5'-三苯甲基 -3'-甲磺酰基胸苷在碱性条件下消除得到 5'-三 苯甲基 -2,3'-脱水胸苷;
5) 5'-三苯甲基 -2,3'-脱水胸苷经叠氮化反应得到 5'-三苯甲基 -3'-叠氮胸苷;
6) 5'-三苯甲基 -3'-叠氮胸苷在酸性条件下脱保护得到齐多夫 定。
6. 如权利要求 5所述的方法, 其特征在于,
歩骤 1) 的反应温度为 20-80°C; 反应溶剂为碱性有机溶剂; 歩骤 2) 的反应试剂为甲磺酰氯; 反应温度为 0-5°C, 反应溶剂 为卤代烃溶剂;
歩骤 3) 的氢化脱氢的试剂为雷尼镍 /三乙胺和氢气, 反应温度 为 20-60°C; 反应溶剂为醇类溶剂;
歩骤 4) 所述的碱性条件选自碱金属 /DMSO, 醇钠或醇钾的醇 溶液, 氢氧化钠或氢氧化钾的醇溶液, 碳酸钠、 碳酸钾或碳酸锂 的水溶液, 甲磺酸钠水溶液, 对甲苯磺酸钠水溶液, 三乙胺或者 DBU; 反应溶剂为醇类溶剂; 反应温度为 20-80 °C;
歩骤 5) 的反应试剂为叠氮化物, 反应溶剂为 DMF; 反应温度 为 60-120°C; 以及
歩骤 6) 所述的酸性条件选自盐酸水溶液、 硫酸水溶液或醋酸
或对甲苯磺酸; 反应溶剂为醇类溶剂; 反应温度为 10-50°C。
7. 如权利要求 5所述的方法, 其特征在于,
歩骤 1) 的反应温度为 40-70°C; 反应溶剂为吡啶;
歩骤 2) 的反应试剂为甲磺酰氯; 反应温度为 0-5°C, 反应溶剂 为二氯甲垸;
歩骤 3) 的氢化脱氢的试剂为雷尼镍 /三乙胺和氢气, 反应温度 为 30-50°C; 反应溶剂为甲醇;
歩骤 4) 所述的碱性条件选自碳酸钠、 碳酸钾或碳酸锂的水溶 液; 反应溶剂为甲醇或乙醇; 反应温度为 50-70°C;
歩骤 5) 的反应试剂为叠氮化锂, 或叠氮化钠 /无水氯化锂 /氯 化铵且三者之间的摩尔比为 2-3: 0.8-1.2: 1, 反应溶剂为 DMF; 反 应温度为 80-110°C; 以及
歩骤 6) 所述的酸性条件为盐酸水溶液或对甲苯磺酸; 反应溶 剂为甲醇; 反应温度为 25-40°C。
8. —种制备齐多 式 (VI)所示:
 式中 X为卤素; ?为羟基保护基; P2为 垸基磺酰基、氟代的 垸基磺酰基、 芳基磺酰基或 -CS-R, 其中 R为 d_4垸基。
式中 X为卤素; ?为羟基保护基; P2为 垸基磺酰基、氟代的 垸基磺酰基、 芳基磺酰基或 -CS-R, 其中 R为 d_4垸基。
9. 如权利要求 12所述的中间体, 其特征在于 X为氯或溴; Pi 为三苯甲基、 特戊酰基或三甲基丙酰基; P2为甲磺酰基或对甲苯磺 酰基。
10. 如权利要求 12所述的中间体, 其特征在于 X为氯或溴; P1 为三苯甲基; P2为甲磺酰基或对甲苯磺酰基。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201180002522.XA CN103443095B (zh) | 2011-11-07 | 2011-11-07 | 制备齐多夫定及其中间体的方法 |
| PCT/CN2011/081856 WO2013067666A1 (zh) | 2011-11-07 | 2011-11-07 | 制备齐多夫定及其中间体的方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/CN2011/081856 WO2013067666A1 (zh) | 2011-11-07 | 2011-11-07 | 制备齐多夫定及其中间体的方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013067666A1 true WO2013067666A1 (zh) | 2013-05-16 |
Family
ID=48288430
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2011/081856 WO2013067666A1 (zh) | 2011-11-07 | 2011-11-07 | 制备齐多夫定及其中间体的方法 |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN103443095B (zh) |
| WO (1) | WO2013067666A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113461759A (zh) * | 2021-07-02 | 2021-10-01 | 华东理工大学 | 基于连续流微反应技术合成齐多夫定叠氮化中间体的方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK161788A (da) * | 1987-03-25 | 1988-09-26 | Wellcome Found | Fremgangsmaade til fremstilling af zidovudin |
| FR2653771B1 (fr) * | 1989-10-27 | 1994-09-23 | Univ Paris Curie | Procede de preparation de l'azt (azido-3'-desoxy-3'-thymidine) et de composes apparentes. |
| JP3677790B2 (ja) * | 1993-08-04 | 2005-08-03 | 味の素株式会社 | ヌクレオシド誘導体とその製造方法 |
| CN101190934B (zh) * | 2006-11-27 | 2010-12-15 | 上海迪赛诺化学制药有限公司 | 相转移法制备齐多夫定叠氮中间体的方法 |
| CN101376667B (zh) * | 2007-08-27 | 2011-01-12 | 上海迪赛诺医药发展有限公司 | 一种合成齐多夫定的中间体及其制备方法和该中间体在合成齐多夫定中的应用 |
-
2011
- 2011-11-07 WO PCT/CN2011/081856 patent/WO2013067666A1/zh active Application Filing
- 2011-11-07 CN CN201180002522.XA patent/CN103443095B/zh active Active
Non-Patent Citations (2)
| Title |
|---|
| CHEN, BANG-CHI ET AL.: "A new Synthesis of the Anti-AIDS Drug AZT from 5-Methyluridine", TETRAHEDRON LETTERS, vol. 36, no. 44, 1995, pages 7961 - 7964, XP055067152 * |
| HUANG, JAI-TUNG ET AL.: "Fluorinated Sugar Analogues of Potential Anti-HIV-1 Nucleosides", JOURNAL OF MEDICINAL CHEMISTRY, vol. 34, no. 5, 1991, pages 1640 - 1646, XP002970744 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113461759A (zh) * | 2021-07-02 | 2021-10-01 | 华东理工大学 | 基于连续流微反应技术合成齐多夫定叠氮化中间体的方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN103443095A (zh) | 2013-12-11 |
| CN103443095B (zh) | 2016-01-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3712130B1 (en) | Method for synthesis of roxadustat and intermediate compounds thereof | |
| WO2013049605A1 (en) | Processes for the preparation of an intermediate in the synthesis of eltrombopag | |
| WO2008144980A1 (fr) | Procédé de préparation et intermédiaires de la capécitabine | |
| KR101308258B1 (ko) | 엔독시펜의 신규한 제조 방법 | |
| WO2009094847A1 (fr) | Dérivé hydroxyle de capécitabine, procédés de préparation et d' utilisation de la capécitabine | |
| CN110386918B (zh) | 一种5-ht1f激动剂化合物的制备方法 | |
| WO2009082846A1 (fr) | Dérivé hydroxyle de la capécitabine, ses procédés de préparation et ses utilisations pour préparer la capécitabine | |
| WO2013067669A1 (zh) | 制备齐多夫定及其中间体的方法 | |
| US20200102291A1 (en) | Process for the preparation of a sulfonamide structured kinase inhibitor | |
| WO2013067664A1 (zh) | 制备齐多夫定及其中间体的方法 | |
| WO2013067666A1 (zh) | 制备齐多夫定及其中间体的方法 | |
| US6512125B1 (en) | Preparation of 1H-indol-1-amines | |
| WO2009084037A2 (en) | Novel process for preparation of o-desmethylvenlafaxine | |
| RU2204564C2 (ru) | Способ получения производного дезоксиуридина | |
| EP3356372B1 (en) | Novel process for preparing thienopyrimidine compound and intermediates used therein | |
| WO2014157021A1 (ja) | ピリダジノン化合物の製造方法 | |
| CN104098499B (zh) | 5-苄氧基-2-(4-苄氧基苯基)-3-甲基-1h-吲哚的制备方法 | |
| JP2003212861A (ja) | ピリミジニルアルコール誘導体の製造方法及びその合成中間体 | |
| EP3609875B1 (en) | An improved process for the preparation of n-(3-ethynylphenyl)-7-methoxy-6-(3-morpholinopropoxy) quinazolin -4-amine dihydrochloride | |
| EP4359384A1 (en) | Process for the preparation of a cyp11a1 inhibitor and intermediates thereof | |
| KR100377578B1 (ko) | 온단세트론 및 그의 염의 제조방법 | |
| JPH09263584A (ja) | 7−アミノ−2,3−ジヒドロ−2−オキソ−ピリド [2,3−d] ピリミジン及びその製造法 | |
| WO2022202814A1 (ja) | ピリミジン化合物の製造方法 | |
| CN115785081A (zh) | 一种雷替曲塞的制备方法 | |
| CN116496180A (zh) | 一种生产制备林扎戈利中间体的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11875511 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11875511 Country of ref document: EP Kind code of ref document: A1 |