WO2012073639A1 - 水素生成用炭素触媒及びその製造方法並びにこれを用いて水素を生成する方法 - Google Patents
水素生成用炭素触媒及びその製造方法並びにこれを用いて水素を生成する方法 Download PDFInfo
- Publication number
- WO2012073639A1 WO2012073639A1 PCT/JP2011/075188 JP2011075188W WO2012073639A1 WO 2012073639 A1 WO2012073639 A1 WO 2012073639A1 JP 2011075188 W JP2011075188 W JP 2011075188W WO 2012073639 A1 WO2012073639 A1 WO 2012073639A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydrogen
- carbon catalyst
- catalyst
- carbon
- gas
- Prior art date
Links
Images




Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/74—Iron group metals
- B01J23/745—Iron
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/74—Iron group metals
- B01J23/755—Nickel
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/18—Carbon
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/02—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the alkali- or alkaline earth metals or beryllium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/32—Manganese, technetium or rhenium
- B01J23/34—Manganese
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/74—Iron group metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/74—Iron group metals
- B01J23/75—Cobalt
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/78—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with alkali- or alkaline earth metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/06—Washing
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/08—Heat treatment
- B01J37/082—Decomposition and pyrolysis
- B01J37/084—Decomposition of carbon-containing compounds into carbon
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B3/00—Hydrogen; Gaseous mixtures containing hydrogen; Separation of hydrogen from mixtures containing it; Purification of hydrogen
- C01B3/02—Production of hydrogen or of gaseous mixtures containing a substantial proportion of hydrogen
- C01B3/22—Production of hydrogen or of gaseous mixtures containing a substantial proportion of hydrogen by decomposition of gaseous or liquid organic compounds
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B3/00—Hydrogen; Gaseous mixtures containing hydrogen; Separation of hydrogen from mixtures containing it; Purification of hydrogen
- C01B3/02—Production of hydrogen or of gaseous mixtures containing a substantial proportion of hydrogen
- C01B3/22—Production of hydrogen or of gaseous mixtures containing a substantial proportion of hydrogen by decomposition of gaseous or liquid organic compounds
- C01B3/24—Production of hydrogen or of gaseous mixtures containing a substantial proportion of hydrogen by decomposition of gaseous or liquid organic compounds of hydrocarbons
- C01B3/26—Production of hydrogen or of gaseous mixtures containing a substantial proportion of hydrogen by decomposition of gaseous or liquid organic compounds of hydrocarbons using catalysts
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/02—Processes for making hydrogen or synthesis gas
- C01B2203/0266—Processes for making hydrogen or synthesis gas containing a decomposition step
- C01B2203/0277—Processes for making hydrogen or synthesis gas containing a decomposition step containing a catalytic decomposition step
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/10—Catalysts for performing the hydrogen forming reactions
- C01B2203/1041—Composition of the catalyst
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/12—Feeding the process for making hydrogen or synthesis gas
- C01B2203/1205—Composition of the feed
- C01B2203/1211—Organic compounds or organic mixtures used in the process for making hydrogen or synthesis gas
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/12—Feeding the process for making hydrogen or synthesis gas
- C01B2203/1205—Composition of the feed
- C01B2203/1211—Organic compounds or organic mixtures used in the process for making hydrogen or synthesis gas
- C01B2203/1217—Alcohols
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/12—Feeding the process for making hydrogen or synthesis gas
- C01B2203/1205—Composition of the feed
- C01B2203/1211—Organic compounds or organic mixtures used in the process for making hydrogen or synthesis gas
- C01B2203/1217—Alcohols
- C01B2203/1223—Methanol
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/12—Feeding the process for making hydrogen or synthesis gas
- C01B2203/1205—Composition of the feed
- C01B2203/1211—Organic compounds or organic mixtures used in the process for making hydrogen or synthesis gas
- C01B2203/1217—Alcohols
- C01B2203/1229—Ethanol
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/12—Feeding the process for making hydrogen or synthesis gas
- C01B2203/1205—Composition of the feed
- C01B2203/1211—Organic compounds or organic mixtures used in the process for making hydrogen or synthesis gas
- C01B2203/1235—Hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/12—Feeding the process for making hydrogen or synthesis gas
- C01B2203/1205—Composition of the feed
- C01B2203/1211—Organic compounds or organic mixtures used in the process for making hydrogen or synthesis gas
- C01B2203/1235—Hydrocarbons
- C01B2203/1241—Natural gas or methane
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/12—Feeding the process for making hydrogen or synthesis gas
- C01B2203/1205—Composition of the feed
- C01B2203/1211—Organic compounds or organic mixtures used in the process for making hydrogen or synthesis gas
- C01B2203/1235—Hydrocarbons
- C01B2203/1247—Higher hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/12—Feeding the process for making hydrogen or synthesis gas
- C01B2203/1205—Composition of the feed
- C01B2203/1211—Organic compounds or organic mixtures used in the process for making hydrogen or synthesis gas
- C01B2203/1235—Hydrocarbons
- C01B2203/1252—Cyclic or aromatic hydrocarbons
Definitions
- the present invention relates to a carbon catalyst for producing hydrogen, a method for producing the same, and a method for producing hydrogen using the same, and particularly relates to the provision of a carbon catalyst for producing hydrogen that exhibits excellent catalytic activity.
- metal catalysts such as nickel and iron are mainly used for the thermal decomposition reaction of methane.
- carbon is deposited on the metal catalyst as hydrogen is generated, and as a result, the metal catalyst is deactivated.
- Patent Document 1 Patent Document 1
- Patent Document 2 Patent Document 2
- Non-Patent Document 1 Since the carbon catalyst itself is a carbon material, it is difficult to deactivate even if carbon deposition occurs due to decomposition of methane.
- the present invention has been made in view of the above problems, and it is an object of the present invention to provide a carbon catalyst for hydrogen generation exhibiting excellent catalytic activity, a method for producing the same, and a method for producing hydrogen using the same. I will.
- a carbon catalyst for hydrogen generation according to an embodiment of the present invention for solving the above problems is a carbon catalyst obtained by carbonization of a raw material containing an organic substance and a transition metal, and is a hydrocarbon compound and / or oxygen-containing It is used for hydrogen production by thermal decomposition of organic compounds.
- ADVANTAGE OF THE INVENTION According to this invention, the carbon catalyst for hydrogen production which shows the outstanding catalyst activity can be provided.
- the carbon catalyst for hydrogen generation may be obtained by supporting an alkaline earth metal on the carbonized material generated by the carbonization.
- the hydrogen generating carbon catalyst is a mixed gas of hydrogen gas, deuterium gas, and argon gas (hydrogen) in a hydrogen-deuterium exchange reaction using a reaction tube filled with a predetermined weight of the carbon catalyst for hydrogen generation.
- Flow rate 10 mL / min
- deuterium flow rate 10 mL / min
- argon flow rate 30 mL / min
- the hydrogen dissociation activity calculated by dividing the total decrease by the predetermined weight may be 10 mmol / g or more.
- a method for producing a hydrogen generation catalyst includes carbonization of a raw material containing an organic substance and a transition metal, and the carbonized material generated by the carbonization is alkaline earth. It is characterized by carrying a metal. ADVANTAGE OF THE INVENTION According to this invention, the manufacturing method of the carbon catalyst for hydrogen production which shows the outstanding catalyst activity can be provided.
- a method according to an embodiment of the present invention for solving the above-mentioned problem is to generate hydrogen by thermally decomposing a hydrocarbon compound and / or an oxygen-containing organic compound using any one of the above-described carbon catalysts for hydrogen generation. It is characterized by doing.
- ADVANTAGE OF THE INVENTION According to this invention, the method of producing
- the carbon catalyst for hydrogen generation according to the present embodiment (hereinafter referred to as “the present catalyst”) is a carbon catalyst obtained by carbonization of a raw material containing an organic substance and a transition metal, and is a hydrocarbon compound and / or oxygen-containing material. It is a carbon catalyst used for hydrogen production by thermal decomposition of organic compounds.
- the organic substance used as a raw material for the catalyst is not particularly limited as long as it can be carbonized, and any one or more kinds can be used.
- an organic substance containing a nitrogen atom can be preferably used.
- an organic compound containing a nitrogen atom can be used.
- the nitrogen-containing organic compound is not particularly limited as long as it contains a nitrogen atom in its molecule.
- a high molecular weight organic compound for example, a resin such as a thermosetting resin or a thermoplastic resin
- a low molecular weight organic compound One or both of these can be used. Biomass can also be used.
- a ligand capable of coordinating with a metal can be preferably used. That is, in this case, an organic compound containing one or more coordination atoms in the molecule is used. More specifically, for example, an organic compound containing, as a coordination atom, one or more selected from the group consisting of a nitrogen atom, a phosphorus atom, an oxygen atom, and a sulfur atom in the molecule can be used. In addition, for example, an organic compound containing one or more selected from the group consisting of an amino group, a phosphino group, a carboxyl group, and a thiol group in the molecule can also be used as a coordination group.
- the organic substance can also contain, for example, one or more selected from the group consisting of a boron atom, a phosphorus atom, an oxygen atom, and a sulfur atom as a component that improves the activity of the catalyst.
- organic substances include phenol resin, polyfurfuryl alcohol, furan, furan resin, phenol formaldehyde resin, melamine, melamine resin, epoxy resin, chelate resin, polyamideimide resin, pyrrole, polypyrrole, polyvinylpyrrole, 3- Methyl polypyrrole, acrylonitrile, polyacrylonitrile, polyacrylonitrile-polymethacrylic acid copolymer, polyvinylidene chloride, thiophene, oxazole, thiazole, pyrazole, vinylpyridine, polyvinylpyridine, pyridazine, pyrimidine, piperazine, pyran, morpholine, imidazole, 1- Methylimidazole, 2-methylimidazole, quinoxaline, aniline, polyaniline, succinic acid dihydrazide, adipic acid dihydrazide Polysulfone, polyamino bismaleimide, polyimide,
- the transition metal is not particularly limited as long as it does not inhibit the activity of the present catalyst, and any one or more of transition metals (Group 3 to Group 12 in the periodic table) can be used. Transition metals belonging to the 4th period of group 12 to group 12 can preferably be used.
- the transition metal can be used as a simple substance of the transition metal or a compound of the metal.
- the metal compound include metal salts, metal oxides, metal hydroxides, metal nitrides, metal sulfides, metal carbides, metal complexes, metal salts, metal oxides, metal sulfides, Metal complexes can be preferably used.
- a metal complex is formed in the raw material.
- the total amount of the transition metal relative to the raw material is not particularly limited as long as the present catalyst has a desired characteristic, and can be, for example, 0.1 to 50% by mass, and 0.5 to 30% by mass. 1 to 20% by mass.
- the raw material may further contain other components. That is, the raw material may include, for example, a carbon material.
- the carbon material is not particularly limited, and any one or more kinds can be used. That is, as the carbon material, for example, a carbon material having no catalytic activity by itself can be used.
- carbon black, carbon nanotube, carbon nanohorn, carbon fiber, carbon fibril, graphite powder, activated carbon, glassy carbon, mesoporous carbon, carbon fiber, fullerene, onion-like carbon, graphene, charcoal, coal char, biomass One or more selected from the group consisting of char, organic substances and carbonized substances can be used.
- the raw materials containing the organic substance and the transition metal described above are mixed.
- the method for mixing the raw materials is not particularly limited, and for example, a mortar or a stirring device can be used.
- one or more mixing methods such as powder mixing in which an organic substance and a transition metal are mixed in powder form, or solvent mixing in which a solvent is added and mixed can also be used.
- this catalyst is obtained by carbonizing the raw material prepared as mentioned above.
- carbonization a raw material is heated and held at a predetermined temperature (carbonization temperature) at which the raw material can be carbonized.
- the carbonization temperature is not particularly limited as long as the raw material can be carbonized, and can be, for example, 300 ° C. or higher. More specifically, the carbonization temperature can be, for example, 300 ° C. or higher and 1500 ° C. or lower, preferably 400 ° C. or higher and 1200 ° C. or lower, more preferably 500 ° C. or higher and 1100 ° C. or lower. It can be.
- the heating rate when heating the raw material to the carbonization temperature is not particularly limited and can be, for example, 0.5 ° C./min or more and 300 ° C./min or less.
- the time for holding the raw material at the carbonization temperature is not particularly limited as long as the raw material can be carbonized, and can be, for example, 5 minutes or longer. More specifically, the carbonization time can be, for example, 5 minutes or more and 240 minutes or less, preferably 20 minutes or more and 180 minutes or less.
- Carbonization is preferably performed under an inert gas such as nitrogen (for example, under the flow of an inert gas).
- a carbonized material produced by carbonization of the raw material may be obtained as it is as the present catalyst.
- the present catalyst may be a fine particle carbon catalyst obtained by pulverizing a carbonized material.
- the method for pulverizing the carbonized material is not particularly limited, and for example, a pulverizing apparatus such as a ball mill or a bead mill can be used.
- the average particle size of the carbonized material after pulverization can be, for example, 150 ⁇ m or less, and preferably 100 ⁇ m or less.
- the present catalyst may be a carbon catalyst obtained by supporting an alkaline earth metal on a carbonized material generated by carbonization. That is, the present catalyst is produced by carbonizing a raw material containing an organic substance and a transition metal, and supporting an alkaline earth metal on the carbonized material generated by the carbonization.
- the catalyst contains an alkaline earth metal supported after carbonization.
- This alkaline earth metal is mainly supported on the surface of the present catalyst.
- the activity of the catalyst can be effectively improved as compared with the case where the alkaline earth metal is not supported.
- the improvement of the catalytic activity by the support of the alkaline earth metal is a result of repeated investigations by the inventors of the present invention, and as a result, focusing on the hydrogen dissociation activity of the carbon catalyst as shown in the examples described later. It is what I found.
- the method for supporting the alkaline earth metal on the carbonized material is not particularly limited.
- the carbon supported on the alkaline earth metal by mixing the powdered carbonized material and the powdered alkaline earth metal.
- the present catalyst comprising the chemical material can be obtained.
- a mortar or a stirring device can be used.
- the present catalyst can be obtained by supporting an alkaline earth metal on a carbonized material by an impregnation supporting method, an ion exchange supporting method, a sol-gel method, or a coprecipitation method.
- the alkaline earth metal is not particularly limited, and one or more selected from the group consisting of beryllium (Be), magnesium (Mg), calcium (Ca), strontium (Sr), and barium (Ba) can be used.
- Be beryllium
- Mg magnesium
- Ca calcium
- Sr strontium
- Ba barium
- One or more selected from the group consisting of magnesium (Mg), calcium (Ca) and barium (Ba) can be preferably used.
- the amount of the alkaline earth metal supported by the catalyst is not particularly limited as long as the activity of the catalyst is improved. That is, the amount of the alkaline earth metal contained in the catalyst is, for example, 0.1 to 50% by weight with respect to the carbonized material supporting the alkaline earth metal (alkaline earth metal with respect to 100 parts by weight of the carbonized material). Metal) of 0.1 to 50 parts by weight, preferably 0.5 to 30% by weight, more preferably 1 to 20% by weight.
- the present catalyst may be a carbon catalyst obtained by subjecting a carbonized material generated by carbonization to further treatment. That is, the present catalyst may be a carbon catalyst obtained by subjecting the carbonized material to a metal removal treatment, for example. Furthermore, in this case, the present catalyst may be a carbon catalyst obtained by, for example, supporting an alkaline earth metal on a carbonized material that has been subjected to metal removal treatment. By subjecting the carbonized material to a metal removal treatment, the transition metal can be removed from the carbonized material, and the active points of the carbon structure can be exposed.
- the metal removal process is a process for removing transition metals contained in the carbonized material.
- the metal removal treatment is not particularly limited as long as it can remove the transition metal contained in the carbonized material or reduce the amount of the transition metal, and for example, an acid cleaning treatment or an electrolytic treatment can be performed.
- the acid used for the acid cleaning treatment is not particularly limited as long as the effect of the metal removal treatment can be obtained, and any one or more of them can be used. That is, for example, one or more selected from the group consisting of hydrochloric acid (for example, concentrated hydrochloric acid), nitric acid (for example, concentrated nitric acid), and sulfuric acid (for example, concentrated sulfuric acid) can be used.
- hydrochloric acid for example, concentrated hydrochloric acid
- nitric acid for example, concentrated nitric acid
- sulfuric acid for example, concentrated sulfuric acid
- a mixed acid prepared by mixing at a volume ratio can be used.
- the method of the acid cleaning treatment is not particularly limited, and for example, a method of immersing and holding the carbonized material in a solution containing an acid can be used.
- this catalyst when this catalyst is obtained through a metal removal process, this catalyst may be substantially free of transition metals or may contain residual transition metals.
- the transition metal remaining in the catalyst can be confirmed by a method such as elemental analysis.
- the present catalyst may be a carbon catalyst obtained by introducing (doping) a nitrogen atom or a boron atom into a carbonized material generated by carbonization of a raw material.
- the carbonized material can be doped with nitrogen atoms or boron atoms in any step.
- a gas phase doping method such as an ammoxidation method or a CVD method, a liquid phase doping method, or a gas phase-liquid phase doping method can be used.
- a nitrogen source such as ammonia, melamine, or acetonitrile or a boron source such as boric acid or sodium borohydride is mixed with a carbonized material, and the resulting mixture is an inert gas such as nitrogen, argon, or helium.
- Nitrogen atoms can be introduced into the surface of the carbonized material by holding at a temperature of 550 ° C. or more and 1200 ° C. or less for 5 minutes or more and 180 minutes or less in an atmosphere.
- the present catalyst is applied to a carbonized material produced by carbonization of raw materials, activation treatment such as carbon dioxide activation, phosphoric acid activation, alkali activation, hydrogen activation, ammonia activation, nitric oxide activation, electrolytic activation, and / or It is good also as a carbon catalyst obtained by performing liquid phase oxidation, such as nitric acid oxidation, mixed acid oxidation, and hydrogen peroxide oxidation.
- activation treatment such as carbon dioxide activation, phosphoric acid activation, alkali activation, hydrogen activation, ammonia activation, nitric oxide activation, electrolytic activation, and / or It is good also as a carbon catalyst obtained by performing liquid phase oxidation, such as nitric acid oxidation, mixed acid oxidation, and hydrogen peroxide oxidation.
- the specific surface area determined by the nitrogen adsorption BET method of the present catalyst can be, for example, 10 m 2 / g or more, preferably 100 m 2 / g or more. More specifically, the specific surface area of the present catalyst can be, for example, 200 m 2 / g or more and 3000 m 2 / g or less, preferably 300 m 2 / g or more and 3000 m 2 / g or less.
- this catalyst is used for hydrogen generation by thermal decomposition of hydrocarbon compounds and / or oxygen-containing organic compounds. That is, this catalyst has an activity of catalyzing a reaction in which a hydrocarbon compound and / or an oxygen-containing organic compound is thermally decomposed to generate hydrogen.
- the hydrogen dissociation activity calculated by dividing the total decrease by the predetermined weight may be 10 mmol / g or more.
- the method according to this embodiment is a method for generating hydrogen by thermally decomposing a hydrocarbon compound and / or an oxygen-containing organic compound using the present catalyst.
- the hydrocarbon compound and the oxygen-containing organic compound are not particularly limited as long as they generate hydrogen by thermal decomposition. That is, as the hydrocarbon compound, for example, one or more selected from the group consisting of aliphatic hydrocarbons, alicyclic hydrocarbons, and aromatic hydrocarbons can be used.
- aliphatic hydrocarbon for example, those having 1 to 20 carbon atoms can be preferably used, and those having 1 to 12 carbon atoms can be particularly preferably used. Specifically, for example, one or more selected from the group consisting of methane, ethane, ethylene, propane, propylene, and butane can be used.
- alicyclic hydrocarbon for example, those having 3 to 12 carbon atoms can be preferably used. Specifically, for example, one or more selected from the group consisting of cyclopropane, cyclobutane, cyclopentane and cyclohexane can be used.
- aromatic hydrocarbon for example, those having 5 to 16 carbon atoms can be preferably used. Specifically, for example, one or more selected from the group consisting of benzene, toluene, xylene, ethylbenzene, and tetralin can be used.
- oxygen-containing organic compound for example, one or more selected from the group consisting of alcohols, ethers, esters and ketones can be used.
- alcohols for example, those having 1 to 12 carbon atoms can be preferably used.
- ethers for example, those having 2 to 12 carbon atoms can be preferably used.
- dimethyl ether, ethyl methyl ether, diethyl ether, oxacyclopentane, and crown ether can be used.
- esters for example, those having 2 to 12 carbon atoms can be preferably used. Specifically, for example, methyl formate, methyl acetate, ethyl acetate, propyl acetate, methyl propionate, methyl butyrate, ethyl butyrate, methyl acrylate, ethyl acrylate, butyl acrylate, methyl methacrylate, butyl methacrylate
- One or more selected from the group can be used.
- ketones for example, those having 3 to 6 carbon atoms can be preferably used. Specifically, for example, one or more selected from the group consisting of propanone, pentanone, butanone and cyclohexanone can be used.
- hydrogen is generated by thermally decomposing a hydrocarbon compound and / or an oxygen-containing organic compound in the presence of the present catalyst. That is, in this method, the hydrocarbon compound and / or the oxygen-containing organic compound and the present catalyst are brought into contact under heating.
- the hydrocarbon compound and / or the oxygen-containing organic compound those that are gaseous or liquid can be preferably used, and the gaseous carbon-hydrogen compound and / or oxygen-containing organic compound can be particularly preferably used.
- a mixture of a hydrocarbon compound and / or an oxygen-containing organic compound and other components may be brought into contact with the present catalyst. That is, when a gaseous hydrocarbon compound and / or oxygen-containing organic compound is used, for example, a mixture containing the hydrocarbon compound and / or oxygen-containing organic compound and an inert gas such as argon, nitrogen, and helium It is good also as making gas contact with this catalyst.
- the present catalyst may be brought into contact with a biomass gas containing a hydrocarbon compound and / or an oxygen-containing organic compound.
- the biomass gas may contain other components such as moisture and carbon dioxide, for example.
- Organic pyrolysis obtained by pyrolyzing organic substances such as synthetic resin (polyethylene, polystyrene, polyester, thermosetting resin, phenol resin, epoxy resin, bakelite resin, polycarbonate, etc.), petroleum, kerosene, heavy oil, etc. It is good also as making gas and this catalyst contact.
- synthetic resin polyethylene, polystyrene, polyester, thermosetting resin, phenol resin, epoxy resin, bakelite resin, polycarbonate, etc.
- petroleum kerosene, heavy oil, etc. It is good also as making gas and this catalyst contact.
- the temperature at which the present catalyst is brought into contact with the hydrocarbon compound and / or the oxygen-containing organic compound is not particularly limited as long as the hydrocarbon compound and / or the oxygen-containing organic compound is thermally decomposed to generate hydrogen. 300 ° C. or higher, preferably 500 ° C. or higher. More specifically, this temperature can be, for example, 300 to 1100 ° C., preferably 500 to 1000 ° C., and more preferably 600 to 1000 ° C.
- Carbon catalyst CA (Fe) 0.4 g of polyvinyl pyridine, 0.45 g of iron (III) chloride hexahydrate, and 0.5 g of ketjen black (ECP600JD, manufactured by Lion Corporation) are put in a mortar and mixed uniformly.
- the raw material was prepared.
- the obtained raw material was put into a horizontal image furnace, heated in a nitrogen gas atmosphere at a heating rate of 50 ° C./min, held at a carbonization temperature of 900 ° C. for 1 hour, and carbonized. And the carbonization material produced
- the carbon catalyst CA (Fe) had a BET specific surface area of 630 m 2 / g.
- Carbon catalyst CA (Fe) AW Carbon catalyst CA (Fe) AW
- the metal removal treatment by acid cleaning was performed on the carbon catalyst CA (Fe). That is, 100 mL of concentrated hydrochloric acid was added to 1 g of the carbon catalyst CA (Fe) and stirred for 1 hour. After precipitating the carbon catalyst and removing the solution, 100 mL of a solution prepared by mixing concentrated hydrochloric acid and distilled water at 1: 1 (volume ratio) was added and stirred for 1 hour. After the carbon catalyst was precipitated and the solution was removed, 100 mL of distilled water was added and stirred for 1 hour.
- the solution containing the carbon catalyst was filtered using a filtration membrane (pore size: 1.0 ⁇ m, manufactured by Millipore), and washed with distilled water until the filtrate became neutral.
- the collected carbon catalyst was vacuum-dried at 60 ° C. for 12 hours.
- a carbon catalyst CA (Fe) AW subjected to the metal removal treatment was obtained.
- the carbon catalyst CA (Fe) AW had a BET specific surface area of 690 m 2 / g.
- Carbon catalyst CA (Co) A carbon catalyst CA (Co) was obtained in the same manner as the above-described carbon catalyst CA (Fe) except that cobalt chloride hexahydrate was used instead of iron (III) chloride hexahydrate.
- the carbon catalyst CA (Co) had a BET specific surface area of 670 m 2 / g.
- Carbon catalyst CA (Ni) A carbon catalyst CA (Ni) was obtained in the same manner as the above-described carbon catalyst CA (Fe) except that nickel chloride hexahydrate was used instead of iron (III) chloride hexahydrate.
- the carbon catalyst CA (Ni) had a BET specific surface area of 650 m 2 / g.
- Carbon catalyst CA (Mn) 1.5 g of polyacrylonitrile-polymethacrylic acid copolymer was dissolved in 30 g of dimethylformamide. Thereafter, 1.25 g of manganese chloride tetrahydrate and 1.5 g of 2-methylimidazole were added and stirred for 2 hours to obtain a solution.
- Ketjen black (EC600JD, manufactured by Lion Corporation) was added to the obtained solution so that the content in the precursor composition described later was 67% by weight, and mixed using a mortar. Further, this mixture was dried under reduced pressure at 60 ° C. and 6.4 ⁇ 10 ⁇ 2 Pa for 12 hours to remove dimethylformamide. A precursor composition was thus obtained.
- the precursor composition was set in a forced circulation dryer. Then, in the atmosphere, the temperature in the dryer was raised from room temperature to 150 ° C. over 30 minutes, and then raised from 150 ° C. to 220 ° C. over 2 hours. Thereafter, the precursor composition was held at 220 ° C. for 3 hours. Thus, the precursor composition was infusible to obtain a carbonization raw material.
- the carbonization material was pulverized. That is, a silicon nitride ball having a diameter of 10 mm was set in a planetary ball mill (P-7, manufactured by Fritsch Japan Co., Ltd.), and the carbonized material was pulverized for 50 minutes at a rotational speed of 650 rpm. The pulverized carbonized material was taken out and classified with a sieve having an aperture of 106 ⁇ m. The carbonized material that passed through the sieve was obtained as the carbon catalyst CA (Mn). The carbon catalyst CA (Mn) had a BET specific surface area of 900 m 2 / g.
- Comparative sample KB A commercially available ketjen black (ECP600JD, manufactured by Lion Corporation), which was also used as a raw material for the carbon catalyst, was used as a comparative sample KB.
- the comparative sample KB had a BET specific surface area of 1200 m 2 / g.
- Comparative sample BP Commercially available carbon black (Black Pearls 2000, manufactured by CABOT) was used as a comparative sample BP.
- the comparative sample BP had a BET specific surface area of 1500 m 2 / g.
- Comparative sample Fe / BP was prepared by supporting iron on comparative sample BP. That is, first, about 0.1 g of iron (III) nitrate nonahydrate was placed in a recovery flask and dissolved in 100 mL of distilled water. Subsequently, the comparative sample BP was added to this iron nitrate aqueous solution. Furthermore, about 5 mL of methanol was added, and it stirred for 10 minutes with the ultrasonic wave. After stirring, the eggplant flask was placed on an evaporator and rotated under reduced pressure for 20 minutes, and then placed in a 60 ° C. hot water bath and dried under reduced pressure.
- pretreatment (reduction treatment) was performed at a heating rate of 50 ° C./min in a hydrogen atmosphere and held at 350 ° C. for 1 hour. )
- the reaction tube was heated from room temperature to 900 ° C. at a rate of temperature increase of 10 ° C./min to carry out a thermal decomposition reaction of methane. After the temperature reached 900 ° C., the reaction tube was kept at 900 ° C. for 20 minutes while continuing to flow the mixed gas.
- a blank was measured. That is, a quartz reaction tube not filled with a carbon catalyst and a comparative sample is installed in a catalyst analyzer (manufactured by Nippon Bell Co., Ltd.), and argon gas is circulated at a flow rate of 50 mL / min for 30 minutes, so that the gas phase in the system Was replaced with argon. Furthermore, the pretreatment which heated the reaction tube at the temperature increase rate of 50 degree-C / min in argon atmosphere and hold
- the hydrogen gas concentration during the temperature rising process was analyzed by a quadrupole mass spectrometer (Q-mass), and the amount of hydrogen gas decreased at each temperature was determined.
- reaction tube was installed in a commercially available catalyst analyzer (manufactured by Nippon Bell Co., Ltd.), and argon gas was circulated at a flow rate of 50 mL / min for 30 minutes to replace the gas phase in the system with argon. Furthermore, the pretreatment which heated the reaction tube at the temperature increase rate of 50 degree-C / min in argon atmosphere and hold
- the hydrogen gas concentration during the temperature rising process was analyzed by a quadrupole mass spectrometer (Q-mass), and the amount of hydrogen gas decreased at each temperature was determined. Then, at each temperature, a value obtained by subtracting the hydrogen gas reduction amount obtained by the blank measurement from the hydrogen gas reduction amount obtained by using the sample was obtained by actually using the sample. Calculated as the amount of decrease in hydrogen gas.
- Q-mass quadrupole mass spectrometer
- the calculated hydrogen gas decrease amount was plotted against the temperature, and a curve showing the correlation between the hydrogen gas decrease amount and the temperature was created. From the prepared curve, the total reduction amount of hydrogen gas from 40 ° C. to 600 ° C. was calculated. Then, the value obtained by dividing the total reduction amount of hydrogen gas thus calculated by the weight of the used carbon catalyst or comparative sample (20 mg) is the hydrogen dissociation activity (mmol / g) per weight of the carbon catalyst or comparative sample. ).
- FIG. 1 shows the result of evaluating the hydrogen generation rate in hydrogen generation by thermal decomposition of methane using any of the carbon catalyst and the comparative sample described above.
- the horizontal axis indicates the temperature (° C.) at which methane was thermally decomposed
- the vertical axis indicates the hydrogen production rate ( ⁇ mol / (min ⁇ m 2 )) per specific surface area of the carbon catalyst or the comparative sample at each temperature. Indicates.
- black circles indicate carbon catalyst CA (Fe)
- black triangles indicate carbon catalyst CA (Co)
- black diamonds indicate carbon catalyst CA (Ni)
- black squares indicate carbon catalyst CA (Mn).
- Half-black diamonds are carbon catalyst CA (Fe) AW
- white circles are comparative samples Fe / BP
- white squares are comparative samples BP
- white diamonds are comparative samples KB. Each is shown.
- the hydrogen generation rate at least at 600 to 900 ° C. in the presence of the carbon catalyst is equal to or higher than that in the presence of the comparative sample.
- the carbon catalyst CA (Fe) the carbon catalyst CA ( The rate of hydrogen production when using Co) and the carbon catalyst CA (Ni) was significantly higher.
- FIG. 2 shows the amount of hydrogen produced ( ⁇ mol) in 20 minutes when the reaction temperature is maintained at 900 ° C., the rate of decrease in catalyst activity (when the carbon catalyst CA (Fe) or the comparative sample Fe / BP is used). %), And the results of evaluating the amount of hydrogen production ( ⁇ mol /%) per catalyst activity reduction rate are shown.
- the amount of hydrogen produced was calculated as the amount of hydrogen gas produced between the time when the temperature reached 900 ° C. and the time when 20 minutes had passed since the temperature was maintained at 900 ° C.
- the rate of decrease in catalyst activity is the difference between the hydrogen generation rate when the temperature reaches 900 ° C. and the hydrogen generation rate when 20 minutes have passed after maintaining the temperature at 900 ° C., and the former hydrogen generation rate is 100 Calculated as a percentage.
- the hydrogen production amount per catalyst activity reduction rate was calculated by dividing the hydrogen production amount calculated as described above by the catalyst activity reduction rate.
- the amount of hydrogen generated per catalyst activity decrease rate represents the amount of hydrogen generated while the catalyst activity decreases by 1%. Therefore, the larger the amount of hydrogen generated per catalyst activity decrease rate, the more hydrogen is generated before the catalyst activity of the carbon catalyst or comparative sample decreases by a predetermined percentage, that is, the carbon catalyst or comparative sample is deactivated. The amount of hydrogen produced until this is large.
- the amount of hydrogen produced when the carbon catalyst CA (Fe) was used was significantly larger than that when the comparative sample Fe / BP was used.
- the rate of decrease in the catalytic activity of the carbon catalyst CA (Fe) was smaller than that of the comparative sample Fe / BP. That is, the catalytic activity of the carbon catalyst CA (Fe) was less likely to be lower than that of the comparative sample Fe / BP.
- the hydrogen production amount per catalyst activity decreasing rate of the carbon catalyst CA (Fe) was significantly larger than that of the comparative sample Fe / BP.
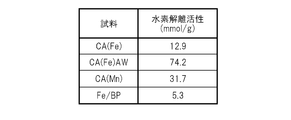
- FIG. 3 shows hydrogen dissociation activity (mmol) in a hydrogen-deuterium exchange reaction using any one of the carbon catalyst CA (Fe), the carbon catalyst CA (Fe) AW, the carbon catalyst CA (Mn), and the comparative sample Fe / BP. / G) shows the result of evaluation.
- the hydrogen dissociation activities of the three types of carbon catalysts were all higher than that of the comparative sample. Although not shown, it was also confirmed that when a carbon catalyst was used, dissociation of hydrogen began to occur at a lower temperature than when a comparative sample was used.
- the carbon catalyst was considered to have higher catalytic activity for dissociating hydrogen than the comparative sample. Based on this result, the inventors of the present invention have come up with the idea of supporting an alkaline earth metal such as magnesium or calcium suitable for hydrogen storage on a carbon catalyst, as will be described later.
- Carbon catalyst Mg / CA (Fe) Carbon catalyst CA (Fe) and magnesium hydroxide were placed in an agate mortar and mixed. Thus, a carbon catalyst Mg / CA (Fe) supporting 3% by weight of magnesium (3 parts by weight of magnesium with respect to 100 parts by weight of the carbon catalyst CA (Fe)) with respect to the carbon catalyst CA (Fe) was obtained.
- Carbon catalyst Mg / CA (Fe) AW for the carbon catalyst CA (Fe) AW in the same manner as the above-mentioned carbon catalyst Mg / CA (Fe) except that the carbon catalyst CA (Fe) AW was used instead of the carbon catalyst CA (Fe).
- a carbon catalyst Mg / CA (Fe) AW carrying magnesium by weight was obtained.
- Carbon catalyst Mg / CA (Mn) 3% by weight with respect to the carbon catalyst CA (Mn) in the same manner as the above-mentioned carbon catalyst Mg / CA (Fe) except that the carbon catalyst CA (Mn) was used instead of the carbon catalyst CA (Fe).
- a carbon catalyst Mg / CA (Mn) supporting magnesium was obtained.
- Carbon catalyst Ca / CA (Mn) Carbon carrying 3% by weight of calcium with respect to the carbon catalyst CA (Mn) in the same manner as the above-mentioned carbon catalyst Ca / CA (Mn) except that calcium hydroxide was used instead of magnesium hydroxide. Catalyst Ca / CA (Mn) was obtained.
- FIG. 4 shows the results of evaluating the hydrogen production rate in hydrogen production by thermal decomposition of methane using either a carbon catalyst supporting an alkaline earth metal or a comparative sample.
- the horizontal axis represents the temperature (° C.) at which methane was thermally decomposed
- the vertical axis represents the hydrogen production rate ( ⁇ mol / (min ⁇ m 2 )) per specific surface area of the carbon catalyst or comparative sample at each temperature. Indicates.
- Black triangles indicate carbon catalyst Mg / CA (Mn), black squares indicate carbon catalyst Ca / CA (Mn), black reverse triangles indicate carbon catalyst Mg / CA (Fe), and black diamonds indicate carbon catalyst Mg / CA (Fe) AW
- white triangles indicate the results of using the comparative sample Mg / Fe / BP
- white squares indicate the results of using the comparative sample Mg / BP.
- the results of using the comparative sample Fe / BP not supporting the alkaline earth metal shown in FIG. 1 are indicated by white circles.
- FIG. 5 shows the hydrogen production rate ( ⁇ mol / (min ⁇ m) at 900 ° C. for each of the carbon catalyst CA (Fe), the carbon catalyst CA (Fe) AW, the carbon catalyst CA (Mn), and the comparative sample Fe / BP. 2 )) shows the result of comparing before and after loading of magnesium.
- the hydrogen production rate in the presence of the carbon catalyst supporting magnesium or calcium was significantly higher than that in the presence of the comparative sample. Further, as is apparent from comparison between FIG. 5 and FIG. 4 and FIG. 1, the hydrogen production rate when the carbon catalyst was used was remarkably increased by loading magnesium on the carbon catalyst.
- the carbon catalyst having higher hydrogen dissociation activity in FIG. 3 improves the catalytic activity by loading magnesium (in the “after Mg loading / before Mg loading” column in FIG. 5). The increase rate (%) of the hydrogen production rate shown was also large.
- the hydrogen production rate in the presence of the comparative sample Mg / Fe / BP was lower than that in the presence of the comparative sample Fe / BP. That is, as shown in FIG. 5, the hydrogen generation rate when the comparative sample Fe / BP was used was decreased by loading magnesium on the comparative sample Fe / BP. Further, even when magnesium was supported on the carbon catalyst BP not supporting iron, the hydrogen generation rate was hardly changed compared to before supporting magnesium (see FIGS. 1 and 4).
- the high catalytic activity of the carbon catalyst supporting the alkaline earth metal is such that the specific carbon structure of the carbon catalyst obtained by carbonization of the raw material containing the organic substance and the transition metal, and the alkaline earth metal This was thought to be due to a specific synergistic effect of the properties possessed.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Materials Engineering (AREA)
- Inorganic Chemistry (AREA)
- Combustion & Propulsion (AREA)
- General Health & Medical Sciences (AREA)
- Health & Medical Sciences (AREA)
- Physics & Mathematics (AREA)
- Thermal Sciences (AREA)
- Catalysts (AREA)
- Hydrogen, Water And Hydrids (AREA)
Abstract
Description
0.4gのポリビニルピリジンと、0.45gの塩化鉄(III)六水和物と、0.5gのケッチェンブラック(ECP600JD、ライオン株式会社製)と、を乳鉢に入れ、均一に混合し、原料を調製した。得られた原料を横型イメージ炉に入れ、窒素ガス雰囲気下、50℃/分の昇温速度で加熱し、炭素化温度900℃にて1時間保持し、炭素化した。そして、炭素化により生成された炭素化材料を炭素触媒CA(Fe)として得た。この炭素触媒CA(Fe)のBET比表面積は630m2/gであった。
炭素触媒CA(Fe)に、酸洗浄による金属除去処理を施した。すなわち、炭素触媒CA(Fe)1gに100mLの濃塩酸を加え、1時間攪拌した。炭素触媒を沈殿させ、溶液を除去した後、濃塩酸と蒸留水とを1:1(体積比)で混合した溶液を100mL加え、1時間攪拌した。炭素触媒を沈殿させ、溶液を除去した後、蒸留水を100mL加え、1時間攪拌した。この炭素触媒を含有する溶液を、ろ過膜(孔径1.0μm、Millipore製)を使用してろ過し、ろ液が中性になるまで蒸留水で洗浄した。回収された炭素触媒を60℃で12時間、真空乾燥させた。こうして、金属除去処理が施された炭素触媒CA(Fe)AWを得た。この炭素触媒CA(Fe)AWのBET比表面積は690m2/gであった。
塩化鉄(III)六水和物に代えて、塩化コバルト六水和物を使用したこと以外は、上述の炭素触媒CA(Fe)と同様にして、炭素触媒CA(Co)を得た。この炭素触媒CA(Co)のBET比表面積は670m2/gであった。
塩化鉄(III)六水和物に代えて、塩化ニッケル六水和物を使用したこと以外は、上述の炭素触媒CA(Fe)と同様にして、炭素触媒CA(Ni)を得た。この炭素触媒CA(Ni)のBET比表面積は650m2/gであった。
1.5gのポリアクリロニトリル-ポリメタクリル酸共重合体を30gのジメチルホルムアミドに溶解させた。その後、1.25gの塩化マンガン四水和物と1.5gの2-メチルイミダゾールとを加え、2時間攪拌して溶液を得た。得られた溶液に、ケッチェンブラック(EC600JD、ライオン株式会社製)を、後述の前駆体組成物における含有量が67重量%となるように加え、乳鉢を用いて混合した。さらに、この混合物を、60℃、6.4×10-2Paで12時間減圧乾燥し、ジメチルホルムアミドを除去した。こうして前駆体組成物を得た。
炭素触媒の原料にも使用した市販のケッチェンブラック(ECP600JD、ライオン株式会社製)を比較試料KBとして使用した。この比較試料KBのBET比表面積は1200m2/gであった。
市販のカーボンブラック(Black Pearls 2000、CABOT社製)を比較試料BPとして使用した。この比較試料BPのBET比表面積は1500m2/gであった。
比較試料BPに鉄を担持させることにより比較試料Fe/BPを調製した。すなわち、まず、約0.1gの硝酸鉄(III)九水和物をナスフラスコへ入れて100mLの蒸留水に溶解した。次いで、この硝酸鉄水溶液に比較試料BPを加えた。さらに、約5mLのメタノールを加え、超音波で10分間攪拌した。攪拌後、ナスフラスコをエバポレーターに設置して減圧下で20分間回転させ、次いで60℃の湯浴につけて減圧乾燥させた。
炭化水素化合物としてメタンを使用し、上述した炭素触媒及び比較試料のいずれかの存在下で、メタンの熱分解による水素の生成を実施した。すなわち、30mgの炭素触媒又は比較試料を内径1cmの石英製反応管に充填した。次いで、この反応管を縦型イメージ炉へ設置し、アルゴン雰囲気下で10℃/分の昇温速度で加熱し、700℃で1時間保持する前処理を行った。なお、比較試料Fe/BPを使用した場合には、上述の前処理に代えて、水素雰囲気下で50℃/分の昇温速度で加熱し、350℃で1時間保持する前処理(還元処理)を行った。
炭素触媒及び比較試料の特性の一つとして、水素分子を水素原子に解離させる触媒活性を、水素(H2)-重水素(D2)交換反応に基づき評価した。すなわち、水素ガス(H2)及び重水素ガス(D2)を含む混合ガスを炭素触媒又は比較試料と接触させた場合における解離した水素ガスの量を、TPR(Temperature Programmed Reaction)法にて評価した。
図1には、上述した炭素触媒及び比較試料のいずれかを使用したメタンの熱分解による水素生成において、水素生成速度を評価した結果を示す。図1において、横軸はメタンの熱分解を行った温度(℃)を示し、縦軸は各温度における炭素触媒又は比較試料の比表面積あたりの水素生成速度(μmol/(min・m2))を示す。
炭素触媒CA(Fe)と、水酸化マグネシウムと、をメノウ乳鉢へ入れて混合した。こうして、炭素触媒CA(Fe)に対して3重量%のマグネシウム(炭素触媒CA(Fe)100重量部に対して3重量部のマグネシウム)を担持した炭素触媒Mg/CA(Fe)を得た。
炭素触媒CA(Fe)に代えて、炭素触媒CA(Fe)AWを使用したこと以外は、上述の炭素触媒Mg/CA(Fe)と同様にして、炭素触媒CA(Fe)AWに対して3重量%のマグネシウムを担持した炭素触媒Mg/CA(Fe)AWを得た。
炭素触媒CA(Fe)に代えて、炭素触媒CA(Mn)を使用したこと以外は、上述の炭素触媒Mg/CA(Fe)と同様にして、炭素触媒CA(Mn)に対して3重量%のマグネシウムを担持した炭素触媒Mg/CA(Mn)を得た。
水酸化マグネシウムに代えて、水酸化カルシウムを使用したこと以外は、上述の炭素触媒Ca/CA(Mn)と同様にして、炭素触媒CA(Mn)に対して3重量%のカルシウムを担持した炭素触媒Ca/CA(Mn)を得た。
炭素触媒CA(Fe)に代えて、比較試料BPを使用したこと以外は、上述の炭素触媒Mg/CA(Fe)と同様にして、比較試料BPに対して3重量%のマグネシウムを担持した比較試料Mg/BPを得た。
比較試料BPに代えて、比較試料Fe/BPを使用したこと以外は、上述の比較試料Mg/BPと同様にして、比較試料Fe/BPに対して3重量%のマグネシウムを担持した比較試料Mg/Fe/BPを得た。
上述の実施例1と同様にして、アルカリ土類金属を担持した炭素触媒及び比較試料のいずれかの存在下で、メタンの熱分解による水素生成を実施した。なお、前処理としては、上述の実施例1における前処理に代えて、炭素触媒又は比較試料を水素雰囲気下で50℃/分の昇温速度で加熱し、650℃で1時間保持する前処理(還元処理)を行った。
図4には、アルカリ土類金属を担持した炭素触媒及び比較試料のいずれかを使用したメタンの熱分解による水素生成において、水素生成速度を評価した結果を示す。図4において、横軸はメタンの熱分解を行った温度(℃)を示し、縦軸は各温度における炭素触媒又は比較試料の比表面積あたりの水素生成速度(μmol/(min・m2))を示す。
Claims (5)
- 有機物と遷移金属とを含む原料の炭素化により得られる炭素触媒であって、
炭化水素化合物及び/又は含酸素有機化合物の熱分解による水素生成に使用される
ことを特徴とする水素生成用炭素触媒。 - 前記炭素化により生成された炭素化材料にアルカリ土類金属を担持して得られる
ことを特徴とする請求項1に記載された水素生成用炭素触媒。 - 所定重量の前記水素生成用炭素触媒を充填した反応管を用いた水素-重水素交換反応において、水素ガスと重水素ガスとアルゴンガスとの混合ガス(水素流量=10mL/分、重水素流量=10mL/分、アルゴン流量=30mL/分)下で前記反応管を10℃/分の昇温速度で40℃から600℃まで加熱した際の前記水素ガスの総減少量を前記所定重量で除して算出される水素解離活性が10mmol/g以上である
ことを特徴とする請求項1又は2に記載された水素生成用炭素触媒。 - 有機物と遷移金属とを含む原料を炭素化し、
前記炭素化により生成された炭素化材料にアルカリ土類金属を担持する
ことを特徴とする水素生成用炭素触媒の製造方法。 - 請求項1又は2に記載された水素生成用炭素触媒を使用して、炭化水素化合物及び/又は含酸素有機化合物を熱分解して水素を生成する
ことを特徴とする方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020137013378A KR101335712B1 (ko) | 2010-11-29 | 2011-11-01 | 수소 생성용 탄소 촉매 및 그 제조방법, 그리고 이것을 이용해서 수소를 생성하는 방법 |
| CA2819092A CA2819092C (en) | 2010-11-29 | 2011-11-01 | Carbon catalyst for hydrogen production, method for producing catalyst, and method for producing hydrogen using catalyst |
| US13/988,609 US9050583B2 (en) | 2010-11-29 | 2011-11-01 | Carbon catalyst for hydrogen production, method for producing catalyst, and method for producing hydrogen using catalyst |
| CN201180057398.7A CN103249482B (zh) | 2010-11-29 | 2011-11-01 | 用于氢气生产的碳催化剂、制备催化剂的方法、和采用催化剂生产氢气的方法 |
| EP11844573.3A EP2647428B1 (en) | 2010-11-29 | 2011-11-01 | Use of a carbon catalyst for hydrogen production, carbon catalyst and method for producing hydrogen |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010265334A JP5193274B2 (ja) | 2010-11-29 | 2010-11-29 | 水素生成用炭素触媒及びその製造方法並びにこれを用いて水素を生成する方法 |
| JP2010-265334 | 2010-11-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012073639A1 true WO2012073639A1 (ja) | 2012-06-07 |
Family
ID=46171589
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/075188 WO2012073639A1 (ja) | 2010-11-29 | 2011-11-01 | 水素生成用炭素触媒及びその製造方法並びにこれを用いて水素を生成する方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US9050583B2 (ja) |
| EP (1) | EP2647428B1 (ja) |
| JP (1) | JP5193274B2 (ja) |
| KR (1) | KR101335712B1 (ja) |
| CN (1) | CN103249482B (ja) |
| CA (1) | CA2819092C (ja) |
| WO (1) | WO2012073639A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015079955A1 (ja) * | 2013-11-29 | 2015-06-04 | 日清紡ホールディングス株式会社 | 固体塩基触媒並びにこれに関する方法及び反応装置 |
| CN110813311A (zh) * | 2019-11-08 | 2020-02-21 | 成都理工大学 | 一种催化NaBH4水解制氢的高效率Ru/Co催化剂的制备方法 |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5193274B2 (ja) | 2010-11-29 | 2013-05-08 | 国立大学法人群馬大学 | 水素生成用炭素触媒及びその製造方法並びにこれを用いて水素を生成する方法 |
| JP5889613B2 (ja) * | 2011-11-25 | 2016-03-22 | 国立大学法人群馬大学 | 金属担持用担体、金属担持触媒、メタネーション反応装置及びこれらに関する方法 |
| US11465113B2 (en) * | 2015-02-13 | 2022-10-11 | Thermo Fisher Scientific (Bremen) Gmbh | Use of a reactor, methods, and device for quantitatively obtaining molecular hydrogen from substances |
| CN105289614B (zh) * | 2015-03-06 | 2017-11-17 | 深圳市国创新能源研究院 | 一种用于制氢的镍‑碳基催化剂材料的制备方法 |
| KR102079120B1 (ko) | 2018-06-18 | 2020-02-19 | 한국과학기술연구원 | 칼슘염에 담지된 금속 촉매, 이의 제조방법 및 이를 이용한 함산소 화합물의 수첨탈산소 반응방법 |
| CN110538629A (zh) * | 2019-08-06 | 2019-12-06 | 广东工业大学 | 一种利用手机保护膜粘合剂制备吸附材料的方法及其制得的吸附材料和应用 |
| CN110649272A (zh) * | 2019-09-29 | 2020-01-03 | 先进储能材料国家工程研究中心有限责任公司 | 质子交换膜燃料电池用催化剂的制备工艺 |
| WO2021087405A1 (en) * | 2019-10-30 | 2021-05-06 | West Virginia University | Methods and compositions for production of co2-free hydrogen and carbon nanomaterials by methane decomposition |
| CN111689466A (zh) * | 2020-05-27 | 2020-09-22 | 深圳市中科纳米科技有限公司 | 有机废弃物的综合处理方法及其处理系统 |
| CN114570429B (zh) * | 2020-11-30 | 2023-09-05 | 浙江工业大学 | 一种单原子负载共价有机框架材料及其制备与在光解水制氢中的应用 |
| JP7408614B2 (ja) | 2021-12-01 | 2024-01-05 | 日清紡ホールディングス株式会社 | 水素製造用触媒及び水素製造方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08165101A (ja) | 1994-12-14 | 1996-06-25 | Agency Of Ind Science & Technol | 水素の製造方法 |
| JP2009521387A (ja) * | 2005-12-21 | 2009-06-04 | ヴァイレント エナジー システムズ インク. | 触媒、および含酸素化合物の製造方法 |
| JP2010083789A (ja) * | 2008-09-30 | 2010-04-15 | Hokkaido Univ | 固体塩基触媒、その製造方法及びこれを使用する方法 |
| JP2010184906A (ja) * | 2009-02-13 | 2010-08-26 | Tokyo Institute Of Technology | 芳香族アルコールの酸化方法 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6689711B2 (en) * | 2001-10-09 | 2004-02-10 | Metallic Power, Inc. | Methods of producing oxygen reduction catalyst |
| JP3798300B2 (ja) | 2001-11-13 | 2006-07-19 | 東邦瓦斯株式会社 | 水素の製造方法 |
| CA2476255C (en) * | 2002-02-14 | 2012-07-31 | Monsanto Technology Llc | Oxidation catalyst and process for its preparation and process for oxidation using it |
| AU2003289357A1 (en) * | 2002-12-17 | 2004-07-09 | Asahi Kasei Chemicals Corporation | Electrode catalyst for oxygen reduction and gas diffusion electrode |
| JP4174564B2 (ja) * | 2003-03-04 | 2008-11-05 | 株式会社日本製鋼所 | 無担持炭化水素直接分解触媒の製造方法ならびに炭化水素直接分解による水素と炭素の製造方法 |
| PL1623957T3 (pl) * | 2005-02-10 | 2008-06-30 | Bestrong Int Ltd | Sposób i urządzenie do wytwarzania wodoru |
| JP5481646B2 (ja) | 2008-06-04 | 2014-04-23 | 清蔵 宮田 | 炭素触媒、燃料電池、蓄電装置 |
| WO2010064556A1 (ja) * | 2008-12-02 | 2010-06-10 | 日清紡ホールディングス株式会社 | 炭素触媒及びその製造方法、これを用いた電極及び電池 |
| US8372781B2 (en) * | 2009-11-05 | 2013-02-12 | Nisshinbo Holdings, Inc. | Carbon catalyst and use thereof |
| JP5193274B2 (ja) | 2010-11-29 | 2013-05-08 | 国立大学法人群馬大学 | 水素生成用炭素触媒及びその製造方法並びにこれを用いて水素を生成する方法 |
-
2010
- 2010-11-29 JP JP2010265334A patent/JP5193274B2/ja active Active
-
2011
- 2011-11-01 EP EP11844573.3A patent/EP2647428B1/en active Active
- 2011-11-01 KR KR1020137013378A patent/KR101335712B1/ko active IP Right Grant
- 2011-11-01 WO PCT/JP2011/075188 patent/WO2012073639A1/ja active Application Filing
- 2011-11-01 CN CN201180057398.7A patent/CN103249482B/zh active Active
- 2011-11-01 CA CA2819092A patent/CA2819092C/en active Active
- 2011-11-01 US US13/988,609 patent/US9050583B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08165101A (ja) | 1994-12-14 | 1996-06-25 | Agency Of Ind Science & Technol | 水素の製造方法 |
| JP2009521387A (ja) * | 2005-12-21 | 2009-06-04 | ヴァイレント エナジー システムズ インク. | 触媒、および含酸素化合物の製造方法 |
| JP2010083789A (ja) * | 2008-09-30 | 2010-04-15 | Hokkaido Univ | 固体塩基触媒、その製造方法及びこれを使用する方法 |
| JP2010184906A (ja) * | 2009-02-13 | 2010-08-26 | Tokyo Institute Of Technology | 芳香族アルコールの酸化方法 |
Non-Patent Citations (2)
| Title |
|---|
| N. MURADOV ET AL., CATALYSIS TODAY, vol. 102-103, 2005, pages 225 - 223 |
| See also references of EP2647428A4 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015079955A1 (ja) * | 2013-11-29 | 2015-06-04 | 日清紡ホールディングス株式会社 | 固体塩基触媒並びにこれに関する方法及び反応装置 |
| CN110813311A (zh) * | 2019-11-08 | 2020-02-21 | 成都理工大学 | 一种催化NaBH4水解制氢的高效率Ru/Co催化剂的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US9050583B2 (en) | 2015-06-09 |
| EP2647428A1 (en) | 2013-10-09 |
| CA2819092C (en) | 2014-01-21 |
| JP2012115725A (ja) | 2012-06-21 |
| JP5193274B2 (ja) | 2013-05-08 |
| EP2647428A4 (en) | 2014-06-11 |
| CN103249482A (zh) | 2013-08-14 |
| CA2819092A1 (en) | 2012-06-07 |
| KR20130077894A (ko) | 2013-07-09 |
| KR101335712B1 (ko) | 2013-12-04 |
| US20130243687A1 (en) | 2013-09-19 |
| CN103249482B (zh) | 2014-11-26 |
| EP2647428B1 (en) | 2022-04-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5193274B2 (ja) | 水素生成用炭素触媒及びその製造方法並びにこれを用いて水素を生成する方法 | |
| Zhu et al. | Carbon‐supported single metal site catalysts for electrochemical CO2 reduction to CO and beyond | |
| Bulut et al. | MnO x-promoted PdAg alloy nanoparticles for the additive-free dehydrogenation of formic acid at room temperature | |
| JP5889613B2 (ja) | 金属担持用担体、金属担持触媒、メタネーション反応装置及びこれらに関する方法 | |
| Pendem et al. | Zeolitic imidazolate framework-mediated synthesis of Co3O4 nanoparticles encapsulated in N-doped graphitic carbon as an efficient catalyst for selective oxidation of hydrocarbons | |
| JP4890623B2 (ja) | 水素吸蔵炭素材料 | |
| Fang et al. | Sacrificial sucrose strategy achieved enhancement of ammonia synthesis activity over a ceria-supported Ru catalyst | |
| Saka | Very efficient dehydrogenation of methanolysis reaction with nitrogen doped Chlorella Vulgaris microalgae carbon as metal-free catalysts | |
| Bai et al. | Preparation of nitrogen doped biochar-based iron catalyst for enhancing gasoline-range hydrocarbons production | |
| Luan et al. | Effect of oxygen vacancy of lignite-char-supported Co catalysts doped with In on efficient dry reforming of methane | |
| Jiang et al. | Molybdenum Carbide for Catalytic De/hydrogenation Process: Synthesis, Modulation and Applications | |
| Zhang et al. | Carbon modified active pairs of Co-Co2C for CO2 hydrogenation to alcohols | |
| Cheng et al. | Enhanced hydrogen storage and CO2 capture capacities on carbon aerogels from Ni-N co-doping | |
| WO2023100626A1 (ja) | 水素製造用触媒及び水素製造方法 | |
| Du et al. | Nitrogen Defective Engineering of a Metal-Free Carbon Catalyst for Ammonia Electrosynthesis from Nitrate | |
| Luan et al. | Construction of metal-anchored and defect-rich N-doped lignite-char supported cobalt catalysts for pressurized dry reforming of methane | |
| Wang et al. | Defect-Domianted Intrinsic Activity of Carbon Governs Pd Nanoparticles for Boosted Formic Acid Dehydrogenation | |
| Galallah et al. | Optimizing Mo2C-based Catalytic System for Efficient CO2 Conversion and CO Selectivity through Carbon-nitrogen Supporting and Potassium Promotion | |
| Krishan et al. | Functionalized Metal-Free Carbon Nanosphere Catalyst for the Selective C–N Bond Formation under Open-Air Conditions | |
| CN116669847A (zh) | 再生碳和再活化催化剂的方法 | |
| Rivera‐Cárcamo et al. | Preparation of Supported Metal Single‐Atom Catalysts on Carbon Supports |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11844573 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13988609 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 20137013378 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2819092 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |