WO2012073635A1 - α-メチル-β-ケトエステルの新規製造法 - Google Patents
α-メチル-β-ケトエステルの新規製造法 Download PDFInfo
- Publication number
- WO2012073635A1 WO2012073635A1 PCT/JP2011/075090 JP2011075090W WO2012073635A1 WO 2012073635 A1 WO2012073635 A1 WO 2012073635A1 JP 2011075090 W JP2011075090 W JP 2011075090W WO 2012073635 A1 WO2012073635 A1 WO 2012073635A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- toluene
- aqueous solution
- ethyl
- compound represented
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/333—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton
- C07C67/343—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/66—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety
- C07C69/67—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of saturated acids
- C07C69/716—Esters of keto-carboxylic acids or aldehydo-carboxylic acids
Definitions
- the present invention relates to a novel process for producing ⁇ -methyl- ⁇ -ketoester useful as a raw material for various chemical products.
- ⁇ -Methyl- ⁇ -ketoesters having a methyl group at the ⁇ -position of ⁇ -ketoesters are known to be useful as raw materials for various useful chemicals such as pharmaceuticals, agricultural chemicals, and fragrances.
- ethyl 2-methylacetoacetate (EMA) is a ⁇ -ketoester compound having a methyl group at the ⁇ -position of ethyl acetoacetate (EAA), but is used as a synthetic intermediate for perfume.
- ⁇ -Methyl- ⁇ -ketoethyl ester is used as a synthetic intermediate for agricultural and horticultural fungicides.
- ⁇ -ketoester such as ethyl acetoacetate is produced by reaction with methyl halide such as methyl bromide or methyl iodide in the presence of a base.
- methyl halide such as methyl bromide or methyl iodide
- acetic anhydride in the presence of ethyl 2-bromopropionate and zinc.
- Non-Patent Document 1 describes that dimethyl sulfate is used for ethyl acetoacetate as a synthesis method of ethyl 2-methylacetoacetate. The details of the law are not described at all.
- Patent Document 1 ethyl acetoacetate is condensed with formaldehyde, and the resulting alkylidene compound is hydrogenated in the presence of a palladium catalyst to produce the desired ⁇ -methyl- ⁇ -ketoester.
- a method is disclosed.
- the yield is about 40%, and there is a problem of industrial waste because zinc chloride is used.
- Patent Document 2 discloses a method in which ethyl acetoacetate is condensed with formaldehyde in the presence of acetic anhydride, and the produced 2-acetoxymethylacetoacetate is hydrogenolyzed in the presence of a palladium catalyst.
- a method for producing ethyl 2-methylacetoacetate is disclosed. However, this method requires a step of heating to 80 ° C. or higher under a high pressure of 50 atm, and is not necessarily a general method.
- ⁇ -ketoester is used as a starting material, solid paraformaldehyde is dissolved in ⁇ -ketoester and acetic anhydride, and reacted in the presence of a lower alcohol.
- a method for obtaining ⁇ -methyl- ⁇ -ketoester by hydrogenolysis is disclosed. However, in this method, palladium is used at the time of hydrogenolysis, and although it is not a harsh condition as disclosed in Swiss Patent CH560176, a special apparatus is used for reduction under pressure. Cost.
- WO2009 / 020221 (patent document 4), 2-ethyl acetoacetate is dissolved in an aprotic solvent, and then a methylating agent in which dimethyl sulfate and methyl iodide are mixed in the presence of an inorganic base is used.
- a method for producing ethyl methylacetoacetate is disclosed. However, in this method, expensive methyl iodide is still used. Therefore, a method that can be manufactured at a lower cost is desired.
- WO2009 / 020211 Patent Document 4 describes a reference example using dimethyl sulfate without a solvent, but only describes that the production ratio was 48%. The yield of isolated ethyl 2-methylacetoacetate is not described at all.
- the inventors of the present invention provide a method for producing ⁇ -methyl- ⁇ -ketoester simply and in a high yield by reacting ⁇ -ketoester as a starting material with a methylating agent comprising inexpensive dimethyl sulfate in the presence of a base. I found it. Further, the inventors have found a method for obtaining high-purity ⁇ -methyl- ⁇ -ketoester by extracting and removing the raw ⁇ -ketoester with a basic aqueous solution. The present invention is based on these findings.
- the object of the present invention is to provide a method capable of producing highly pure ⁇ -methyl- ⁇ -ketoester simply and inexpensively.
- the compound represented by the formula consists of toluene, ethylbenzene, ethanol, methanol, acetone, ethyl acetate, xylene, tetrahydrofuran, cyclohexane, N, N-dimethylformamide, N-methyl-2-pyrrolidone, and N, N-dimethylacetamide.
- a method comprising a step of reacting with a methylating agent comprising dimethyl sulfate in an organic solvent in which one or more selected from the group is combined in the
- a mixture of the compound represented by the formula (I) and the compound represented by the formula (II), or the compound represented by the formula (I) and the formula (II) A method comprising a step of washing with a basic aqueous solution a solution in which the compound represented by) is dissolved in one or more organic solvents selected from toluene, ethylbenzene, ethyl acetate, xylene, and cyclohexane. Is done.
- the production method according to the present invention may be an organic solvent, either a protic solvent or an aprotic solvent, and these solvents can be used alone or in combination. It is advantageous in that ⁇ -methyl- ⁇ -ketoester can be produced without using expensive methyl halide, which consists of dimethyl sulfate. Further, the production method according to the present invention does not require a special apparatus, and is advantageous in that ⁇ -methyl- ⁇ -ketoester can be produced very easily and inexpensively as compared with the conventional method. Furthermore, it is advantageous in that a highly pure ⁇ -methyl- ⁇ -ketoester can be obtained by washing with a basic aqueous solution.
- the “C 1-6 alkyl group” means a linear or branched alkyl group having 1 to 6 carbon atoms.
- methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, i-butyl group, s-butyl group, t-butyl group, n-pentyl group, n-hexyl group and the like can be mentioned.
- the C 1-6 alkyl group is preferably a C 1-4 alkyl group, more preferably a C 1-2 alkyl group.
- the “optionally substituted alkyl group” means that one or more hydrogen atoms on the alkyl group are substituted by one or more substituents (which may be the same or different).
- An alkyl group or an unsubstituted alkyl group It will be apparent to those skilled in the art that the maximum number of substituents can be determined depending on the number of substitutable hydrogen atoms on the alkyl.
- examples of the “alkyl group optionally substituted by a halogen atom” include a chloromethyl group, a fluoromethyl group, and a trifluoromethyl group.
- the “methylating agent comprising dimethyl sulfate” is characterized in that dimethyl sulfate is used alone as the methylating agent.
- a solvent In a specific use mode, a solvent, It goes without saying that salts and the like may be used.
- the ⁇ -methyl- ⁇ -ketoester which is the object of the production method according to the present invention is a compound represented by the formula (I).
- R 1 is a C 1-6 alkyl group which may be substituted with a halogen atom, preferably a C 1-4 alkyl group, more preferably a C 1-2 alkyl group. And most preferably a methyl group.
- R 2 is a C 1-6 alkyl group which may be substituted with a halogen atom, preferably a C 1-4 alkyl group, more preferably a C 1-2 alkyl group. And most preferably an ethyl group.
- preferred examples of the compound of the formula (I) include compounds in which R 1 is a methyl group and R 2 is a C 1-4 alkyl group, particularly preferably 2-methylacetate. Examples include ethyl acetate or methyl 2-methylacetoacetate.
- the method comprises the step of reacting a ⁇ -ketoester with a methylating agent comprising dimethyl sulfate in an organic solvent in the presence of a base. It is provided to produce a compound represented by I).
- the ⁇ -ketoester used as a raw material is a compound represented by the formula (II).
- R 1 is a C 1-6 alkyl group which may be substituted with a halogen atom, preferably a C 1-4 alkyl group, more preferably a C 1-2 alkyl group. And most preferably a methyl group.
- R 2 is a C 1-6 alkyl group which may be substituted with a halogen atom, preferably a C 1-4 alkyl group, more preferably a C 1-2 alkyl group. And most preferably an ethyl group.
- preferred examples of the compound of the formula (II) include compounds in which R 1 is a methyl group and R 2 is a C 1-4 alkyl group, particularly preferably ethyl acetoacetate. Or methyl acetoacetate is mentioned.
- the compound represented by the formula (II) can be used by dissolving in a solvent.
- Solvents used for dissolving the compound represented by the formula (II) include toluene, ethylbenzene, acetone, ethyl acetate, xylene, tetrahydrofuran, cyclohexane, N, N-dimethylformamide, N-methyl-2-pyrrolidone, In addition to aprotic organic solvents such as N, N-dimethylacetamide, protic organic solvents such as ethanol and methanol can be used. These solvents may be used alone or in combination of two or more. Preferred is toluene or a single solvent of ethanol, or a mixed solvent of ethanol and toluene or methanol and toluene, and more preferred is a mixed solvent of ethanol and toluene.
- the amount of the solvent is not particularly limited as long as the compound represented by the formula (II) can be dissolved.
- the amount of the solvent is 2 to 20 v / v% based on the compound represented by the formula (II). it can.
- the amount of dimethyl sulfate can be 0.5 to 2.0 equivalents, preferably 0.8 to 1.25 equivalents, relative to the compound represented by the formula (II).
- examples of the “base” used in the reaction together with the methylating agent include alkali metal or alkaline earth metal hydroxides, carbonates, alcohol salts, and the like.
- lithium hydroxide, sodium hydroxide , Potassium hydroxide, cesium hydroxide, lithium carbonate, sodium carbonate, potassium carbonate, cesium carbonate, sodium bicarbonate, potassium bicarbonate, sodium ethoxide, sodium methoxide, and the like preferably sodium hydroxide, Potassium hydroxide or sodium ethoxide is preferable, and sodium hydroxide is more preferable.
- the amount of the base used in the reaction together with the methylating agent is not particularly limited as long as the effect of the present invention is exhibited, but it is used in an amount of 0.8 to 1.5 equivalents relative to the compound represented by the formula (II). it can.
- the addition of the methylating agent comprising dimethyl sulfate to the compound represented by the formula (II) is preferably carried out gradually over several minutes to several hours.
- the methylating agent may be added in small portions or continuously.
- the time required for the addition depends on the amount of the methylating agent and the amount of the compound represented by formula (II). Can be determined as appropriate.
- the methylating agent can be added preferably in the range of 40 to 75 ° C, more preferably in the range of 40 to 65 ° C.
- the time for which the compound represented by the formula (II) is reacted with dimethyl sulfate is 1 to 24 hours.
- the compound of formula (II) dissolved in an organic solvent is reacted with a methylating agent consisting of dimethyl sulfate in the presence of a base. It is a manufacturing method of the compound represented. By reacting with the methylating agent, the compound represented by the formula (II) is methylated.
- a compound represented by the formula (II) which is toluene or a single solvent of ethanol or dissolved in a mixed solvent of ethanol and toluene, methanol and toluene is hydroxylated.
- a method for producing a compound represented by formula (I) comprising a step of reacting with a methylating agent comprising dimethyl sulfate in the presence of sodium, potassium hydroxide or sodium ethoxide. By reacting with the methylating agent, the compound represented by the formula (II) is methylated.
- the obtained compound of the formula (I) is preferably purified by washing with a basic aqueous solution.
- the “base” used for washing with the basic aqueous solution may be the same as the “base” used for the reaction together with the methylating agent.
- a method for producing a compound represented by the formula (I) further comprising a washing step with a basic aqueous solution after the step of reacting a methylating agent.
- the amount of the “base” in the basic aqueous solution used in the washing step is 0.1 to 5 times the molar equivalent of the amount of the compound represented by the formula (II) present in the organic solvent. Preferably, it is 0.5 to 2 times the molar equivalent, and more preferably 0.5 to 1.5 times the molar equivalent.
- the volume of the basic aqueous solution used in the washing step can be 0.01 to 10 times the volume of the amount of the organic solvent, preferably 0.1 to 3 times the volume. Preferably, a capacity of 0.1 to 1 times can be used.
- the basic aqueous solution may further contain 1% by weight to a saturated amount of sodium chloride.
- the number of times of washing with the basic aqueous solution is not particularly limited, but may be multiple times, preferably 1 to 3 times.
- one or two selected from the group consisting of toluene, ethylbenzene, ethyl acetate, xylene, and cyclohexane after the step of washing with the basic aqueous solution and after the step of reacting the methylating agent are performed.
- a method for producing a compound represented by formula (I) which comprises a step of performing solvent substitution with an organic solvent obtained by combining the above.
- the O-methyl compound as a by-product is decomposed into a ⁇ -ketoester, and the basic aqueous solution is obtained. It can be removed by washing with.
- the method for producing the compound of formula (I) further comprises a step of treating with an acidic aqueous solution after a step of reacting with a methylating agent before a washing step with a basic aqueous solution.
- a step of treating with an acidic aqueous solution is preferably performed after the solvent substitution.
- acids such as hydrochloric acid, sulfuric acid, phosphoric acid and the like can be mentioned, and hydrochloric acid is preferred.
- the concentration of the “acid” in the washing with the acidic aqueous solution can be 0.001 to 12 N, preferably 0.12 to 2.4 N, and more preferably 0.48 to 1.92 N. It is.
- the solution reacted with the methylating agent after washing with the basic aqueous solution and after the step of reacting with the methylating agent is added at 0.degree. It is preferable to further include a step for 5 to 48 hours, preferably at 60 to 70 ° C. for 0.5 to 4 hours. By adding this step, the yield of the compound represented by formula (I) can be improved. Moreover, the yield of the compound represented by the formula (I) can be further improved by reacting with a methylating agent comprising dimethyl sulfate again in the presence of a base after this step.
- Present means that the liquid reacted with the methylating agent for 0.5 to 48 hours, preferably 0.5 to 4 hours may be allowed to stand, but is more preferably stirred. Further, when reacting again with the methylating agent, the base is 0.1 to 0.4 equivalent and the dimethyl sulfate is 0.1 to 0.4 equivalent relative to the compound represented by the formula (II). Is preferably reacted.
- the method for producing a compound represented by the formula (I), which is selected and used between the step of reacting with a methylating agent again and the step of treating with the above acidic aqueous solution. Can be provided. By-product generation can be suppressed by either the step of reacting again with the methylating agent or the step of treating with an acidic aqueous solution.
- the above-mentioned solvent replacement can be performed, followed by washing with a basic aqueous solution.
- the obtained high purity compound represented by the formula (I) can be used as a synthetic intermediate for agricultural and horticultural fungicides (for example, compounds described in WO2001 / 92231) and fragrances.
- a process for producing a compound represented by formula (I) Compounds represented by the formula (II) are converted to toluene, ethylbenzene, ethanol, methanol, acetone, ethyl acetate, xylene, tetrahydrofuran, cyclohexane, N, N-dimethylformamide, N-methyl-2-pyrrolidone, and N, N— Reaction with a methylating agent consisting of dimethyl sulfate in the presence of a base in an organic solvent combining one or more selected from the group consisting of dimethylacetamide, A step of washing with a basic aqueous solution (preferably, sodium hydroxide as a base),
- the amount of the base in the basic aqueous solution is 0.5 to 1.5 times the molar equivalent of the compound of the formula (II) present in the organic solvent.
- a process for producing a compound represented by formula (I) Compounds represented by the formula (II) are converted to toluene, ethylbenzene, ethanol, methanol, acetone, ethyl acetate, xylene, tetrahydrofuran, cyclohexane, N, N-dimethylformamide, N-methyl-2-pyrrolidone, and N, N— Reaction with a methylating agent consisting of dimethyl sulfate in the presence of a base in an organic solvent combining one or more selected from the group consisting of dimethylacetamide, A step of washing with a basic aqueous solution (preferably, sodium hydroxide as a base), Here, the volume of the basic aqueous solution is 0.1 to 1 times the amount of the organic solvent.
- a method for producing the compound represented by formula (I) is provided.
- a process for producing a compound represented by formula (I) Compounds represented by the formula (II) are converted to toluene, ethylbenzene, ethanol, methanol, acetone, ethyl acetate, xylene, tetrahydrofuran, cyclohexane, N, N-dimethylformamide, N-methyl-2-pyrrolidone, and N, N— Reaction with a methylating agent consisting of dimethyl sulfate in the presence of a base in an organic solvent combining one or more selected from the group consisting of dimethylacetamide, A step of washing with a basic aqueous solution (preferably, sodium hydroxide as a base),
- the amount of the base in the basic aqueous solution is 0.5 to 1.5 times the molar equivalent of the compound of the formula (II) present in the organic solvent, and the volume of the basic aqueous solution is 0.1
- a methylating agent consisting of dimethyl sulfate is reacted in the presence of a base, Solvent replacement with one or more organic solvents selected from the group consisting of toluene, ethylbenzene, ethyl acetate, xylene, and cyclohexane,
- a method for producing a compound represented by the formula (I) which comprises a step of washing with a basic aqueous solution (preferably, sodium hydroxide as a base).
- a methylating agent consisting of dimethyl sulfate is reacted in the presence of a base, Solvent replacement with one or more organic solvents selected from the group consisting of toluene, ethylbenzene, ethyl acetate, xylene, and cyclohexane,
- Solvent replacement with one or more organic solvents selected from the group consisting of toluene, ethylbenzene, ethyl acetate, xylene, and cyclohexane A step of washing with a basic aqueous solution (preferably, sodium hydroxide as a base),
- the amount of the base in the basic aqueous solution is 0.5 to 1.5 times the molar equivalent of the compound of the formula (II) present in the organic solvent
- the volume of the basic aqueous solution is 0.1 to 1 times the amount of
- a method for producing the compound represented by formula (I) is provided.
- a method comprising a step of washing with a neutral aqueous solution is provided, and preferably a method for purifying the compound represented by formula (I) is provided.
- the compound represented by the formula (I) is an ⁇ -methyl- ⁇ -ketoester represented by the formula (II).
- the compound is a ⁇ -ketoester, and R 1 and R 2 in the formulas (I) and (II) have the same meaning as defined above, and the same applies to the preferred embodiments thereof.
- Examples of the “base” used for washing with a basic aqueous solution include alkali metal or alkaline earth metal hydroxides, carbonates, and ammonia, and examples thereof include lithium hydroxide, sodium hydroxide, and potassium hydroxide.
- Cesium hydroxide, lithium carbonate, sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogen carbonate, potassium hydrogen carbonate, and ammonia water base preferably sodium hydroxide.
- the amount of the “base” in the washing with the basic aqueous solution is 0.1 to 5 times the molar equivalent of the compound represented by the formula (II) present in the organic solvent (remaining in the reaction solution).
- the molar equivalent is preferably 0.5 to 2 times, and more preferably 0.5 to 1.5 times the molar equivalent.
- the volume of the aqueous solution in the washing with the basic aqueous solution can be 0.01 to 10 times, preferably 0.1 to 3 times, more preferably the amount of the organic solvent. Can be used in a volume of 0.1 to 1 times.
- the basic aqueous solution may further contain 1% by weight to a saturated amount of sodium chloride.
- the compound represented by the formula (I) and the compound represented by the formula (II) are dissolved in toluene with respect to the compound of the formula (II). It is represented by the formula (I) including a step of washing the toluene layer with a basic aqueous solution containing 0.5 to 1.5 times molar equivalent sodium hydroxide and 0.1 to 1 times the volume of the toluene layer.
- a method for purifying the compound can be mentioned.
- the compound represented by the formula (II) can be selectively extracted and removed from the aqueous layer.
- the number of times of washing with the basic aqueous solution is not particularly limited, but may be multiple times, preferably 1 to 3 times.
- the GC area% described in the examples indicates the area percentage of the target substance when measured under the conditions of gas chromatography (GC) (manufactured by Shimadzu Corporation) shown below.
- GC conditions gas chromatography
- Detector FID Column: DB-5 (0.25 mm ⁇ 30 m, film thickness: 0.25 ⁇ m)
- Carrier gas He Inlet temperature: 150 ° C
- Detector temperature 200 ° C
- Example 1 Synthesis of ethyl 2-methylacetoacetate Sodium hydroxide (40 g) was dissolved in a mixed solvent of ethanol (400 mL) and toluene (400 mL) by heating to 70 ° C. After cooling to 44 ° C., ethyl acetoacetate (130 g) was added to the solution. The reaction temperature was kept at 44 ° C. to 60 ° C., and dimethyl sulfate (126 g) was gradually added over 15 minutes, followed by stirring at 44 ° C. for 2.5 hours.
- an aqueous solution (400 mL) containing 1.1 moles of sodium hydroxide (9.9 g) and 5% by weight of sodium chloride was added to the raw material ethyl acetoacetate remaining in the toluene layer, and the toluene layer was washed. did.
- an aqueous solution (400 mL) containing 1.25 times moles of sodium hydroxide (1.86 g) and 5% by weight of sodium chloride was added to the raw material ethyl acetoacetate remaining in the toluene layer, and the toluene layer was added. Washed.
- the obtained toluene layer was concentrated under reduced pressure (40 ° C., 70 mmHg) to obtain 535 mL (yield 61.7%) of a toluene solution containing 16.6% by weight of the title compound.
- the 1 H-NMR spectrum of the obtained compound coincided with that of a commercially available product (manufactured by Tokyo Chemical Industry Co., Ltd.).
- the toluene solution contained 0.35% by weight of EAA.
- Example 2 Synthesis of methyl 2-methylacetoacetate Sodium hydroxide (4 g) was dissolved in a mixed solvent of methanol (25 mL) and toluene (45 mL) by heating to 70 ° C. After cooling to 40 ° C., methyl acetoacetate (11.6 g) was added to the solution. Further, 20 mL of methanol was added, the reaction temperature was kept at 40 ° C. to 50 ° C., and dimethyl sulfate (12.6 g) was gradually added over 15 minutes, followed by stirring at 40 ° C. for 2 hours.
- methyl 2-methylacetoacetate, methyl acetoacetate, methyl 2,2-dimethylacetoacetate, and methyl-3-methylbut-2-enoate were 71.3%, 14.4%, and 2.1, respectively. %, 9.4% (GC area%).
- the solvent in the obtained reaction solution was distilled off under reduced pressure (35 ° C./110 mmHg), and 15 mL of toluene was added to the residue. To this was added 50 mL of 1.2N hydrochloric acid containing 15% by weight of sodium chloride, and the mixture was stirred at room temperature for 6 hours.
- an aqueous solution (40 mL) containing 1.2 times moles of sodium hydroxide (0.776 g) and 10% by weight of sodium chloride with respect to the raw material methyl acetoacetate remaining in the toluene layer was added to wash the toluene layer. did. Subsequently, 25 mL of an aqueous solution containing 1 mol of sodium hydroxide (0.08 g) with respect to the raw material methyl acetoacetate remaining in the toluene layer and containing 10% by weight of sodium chloride was added to wash the toluene layer.
- the toluene layer was concentrated under reduced pressure (40 ° C., 50 mmHg) to obtain 12.8 mL of toluene solution containing 45.2% by weight of the title compound (yield 44.5%).
- the 1 H-NMR spectrum of the obtained compound coincided with that of a commercially available product (manufactured by Tokyo Chemical Industry Co., Ltd.).
- the toluene solution contained 0.2% by weight of methyl acetoacetate.
- Example 3 Synthesis of ethyl 2-methylacetoacetate Sodium ethoxide (6.8 g) was dissolved in ethanol (30 mL), and toluene (50 mL) was added thereto . After the ethanol was distilled off under reduced pressure, toluene was added to make a constant volume of 100 mL. Ethyl acetoacetate (13 g) was added at room temperature, dimethyl sulfate (7.56 g) was further added, and the mixture was stirred at room temperature for 1 hour.
- ethyl 2-methylacetoacetate, ethyl acetoacetate, ethyl 2,2-dimethylacetoacetate, and ethyl-3-methoxybut-2-enoate were 75.4%, 15.1%, 0.8, respectively. %, 6.1% (GC area%).
- the insoluble matter produced was filtered, and the filtrate was washed with toluene and combined with the filtrate. After concentration under reduced pressure to 76 mL, saturated brine (45 mL) was added, and the mixture was stirred at room temperature for 21 hr.
- Example 4 Synthesis of ethyl 2-methylacetoacetate Sodium hydroxide (4 g) was dissolved in ethanol (80 mL) by heating at 65 ° C. After cooling to 45 ° C., ethyl acetoacetate (13 g) was added to the solution. The reaction temperature was maintained at 45 ° C. to 60 ° C., and dimethyl sulfate (12.6 g) was gradually added over 10 minutes, followed by stirring at 45 ° C. for 2.5 hours.
- an aqueous solution (containing 5% sodium chloride) (40 mL) containing 1.1 moles of sodium hydroxide (0.857 g) with respect to the raw material ethyl acetoacetate remaining in the toluene layer was added, and the toluene layer was washed. did.
- an aqueous solution (containing 5% sodium chloride) (40 mL) containing 1.25 times moles of sodium hydroxide (0.192 g) relative to the raw material ethyl acetoacetate remaining in the toluene layer was added, and the toluene layer was washed. did.
- the obtained toluene layer was concentrated under reduced pressure (40 ° C., 70 mmHg) to obtain a toluene solution (42 mL) (yield 51%) containing 17.5 wt% of the title compound.
- the 1 H-NMR spectrum of the obtained compound coincided with that of a commercially available product (manufactured by Tokyo Chemical Industry Co., Ltd.).
- the toluene solution contained 0.34% by weight of EAA.
- Example 5 Synthesis of ethyl 2-methylacetoacetate Potassium hydroxide (6.6 g) was dissolved in a mixed solvent of ethanol (25 mL) and toluene (25 mL) by heating at 65 ° C. After cooling to 44 ° C., ethyl acetoacetate (13 g) was added to the solution. The reaction temperature was kept at 45 ° C. to 65 ° C., and dimethyl sulfate (12.6 g) was gradually added over 15 minutes, followed by stirring at 45 ° C. for 2.5 hours.
- an aqueous solution (25 mL) containing 1.1 moles of sodium hydroxide (1.1 g) and 5% by weight of sodium chloride is added to the raw material ethyl acetoacetate remaining in the toluene layer, and the toluene layer is washed. did.
- an aqueous solution (25 mL) containing 1.25 times moles of sodium hydroxide (0.23 g) and 5% by weight of sodium chloride is added to the raw material ethyl acetoacetate remaining in the toluene layer, and the toluene layer is washed. did.
- the obtained toluene layer was concentrated under reduced pressure (40 ° C., 70 mmHg) to obtain a toluene solution (33 mL) (yield 51%) containing 22.3% by weight of the title compound.
- the 1 H-NMR spectrum of the obtained compound coincided with that of a commercially available product (manufactured by Tokyo Chemical Industry Co., Ltd.).
- the toluene solution contained 0.27% by weight of EAA.
- Example 6 Synthesis of ethyl 2-methylacetoacetate Sodium hydroxide (4 g) was suspended in toluene (80 mL). After adding ethyl acetoacetate (13 g) to the solution at 44 ° C., toluene (40 mL) was further added. Dimethyl sulfuric acid (12.6 g) was gradually added over 5 minutes and then stirred at 45 ° C. for 2.5 hours. During the reaction, toluene (40 mL) was further added.
- an aqueous solution (40 mL) containing 1.1 moles of sodium hydroxide (2.62 g) and 5% by weight of sodium chloride was added to the raw material ethyl acetoacetate remaining in the toluene layer, and the toluene layer was washed. did.
- an aqueous solution (40 mL) containing 1.25 times moles of sodium hydroxide (0.32 g) and 5% by weight of sodium chloride is added to the raw material ethyl acetoacetate remaining in the toluene layer, and the toluene layer is washed.
- Example 7 Synthesis of ethyl 2-methylacetoacetate Sodium hydroxide (2 g) was dissolved in ethanol (20 mL) by heating to 60 ° C. Toluene (20 mL) was added thereto, and after cooling to 45 ° C., ethyl acetoacetate (6.5 g) was added to the solution. The reaction temperature was kept at 45 ° C. to 55 ° C., and dimethyl sulfate (6.3 g) was gradually added over 5 minutes, followed by stirring at 45 ° C. for 1 hour. Water (0.8 mL) was added to the reaction solution, stirred at 68 ° C. for 2.25 hours, and then cooled to 45 ° C.
- Example 8 Purification of ethyl 2-methylacetoacetate Toluene (50 mL) containing ethyl 2-methylacetoacetate (7.2 g) and ethyl acetoacetate (2.6 g) contains sodium hydroxide (400 mg) and is chlorinated. An aqueous solution (50 mL) containing 5% by weight of sodium was added and partitioned. The contents of ethyl 2-methylacetoacetate and ethylacetoacetate in the toluene layer were quantified by gas chromatography (GC).
- GC gas chromatography
- Example 9 Purification of ethyl 2-methylacetoacetate Toluene (50 mL) containing ethyl 2-methylacetoacetate (7.2 g) and ethyl acetoacetate (2.6 g) contains sodium hydroxide (800 mg) and is chlorinated. An aqueous solution (50 mL) containing 5% by weight of sodium was added and partitioned. The contents of ethyl 2-methylacetoacetate and ethylacetoacetate in the toluene layer were quantified by GC.
- Example 10 Purification of ethyl 2-methylacetoacetate Into toluene (50 mL) containing ethyl 2-methylacetoacetate (7.2 g) and ethyl acetoacetate (2.6 g), sodium hydroxide (1.2 g) was added. An aqueous solution (50 mL) containing 5% by weight of sodium chloride was added and partitioned. The contents of ethyl 2-methylacetoacetate and ethylacetoacetate in the toluene layer were quantified by GC.
- Example 11 Purification of ethyl 2-methylacetoacetate Into toluene (50 mL) containing ethyl 2-methylacetoacetate (7.2 g) and ethyl acetoacetate (2.6 g), sodium hydroxide (1.6 g) was added. An aqueous solution (50 mL) containing 5% by weight of sodium chloride was added and partitioned. The contents of ethyl 2-methylacetoacetate and ethylacetoacetate in the toluene layer were quantified by GC.
- Example 12 Purification of ethyl 2-methylacetoacetate Toluene (50 mL) containing ethyl 2-methylacetoacetate (7.2 g) and ethyl acetoacetate (2.6 g) contains sodium hydroxide (800 mg) and is chlorinated. An aqueous solution (5 mL) containing 5% by weight of sodium was added and partitioned. The contents of ethyl 2-methylacetoacetate and ethylacetoacetate in the toluene layer were quantified by GC.
- Example 13 Purification of ethyl 2-methylacetoacetate Toluene (50 mL) containing ethyl 2-methylacetoacetate (7.2 g) and ethyl acetoacetate (2.6 g) contains sodium hydroxide (800 mg) and is chlorinated. An aqueous solution (10 mL) containing 5% by weight of sodium was added and partitioned. The contents of ethyl 2-methylacetoacetate and ethylacetoacetate in the toluene layer were quantified by GC.
- Example 14 Purification of ethyl 2-methylacetoacetate Toluene (50 mL) containing ethyl 2-methylacetoacetate (7.2 g) and ethyl acetoacetate (2.6 g) contains sodium hydroxide (800 mg) and is chlorinated. An aqueous solution (25 mL) containing 5% by weight of sodium was added and partitioned. The contents of ethyl 2-methylacetoacetate and ethylacetoacetate in the toluene layer were quantified by GC.
- Example 15 Purification of ethyl 2-methylacetoacetate Toluene (50 mL) containing ethyl 2-methylacetoacetate (7.2 g) and ethyl acetoacetate (2.6 g) contains sodium hydroxide (800 mg) and is chlorinated. An aqueous solution (100 mL) containing 5% by weight of sodium was added and partitioned. The ethyl 2-methylacetoacetate and the content of ethyl acetoacetate in the toluene layer were quantified by GC.
- Example 16 Purification of ethyl 2-methylacetoacetate Toluene (50 mL) containing ethyl 2-methylacetoacetate (7.2 g) and ethyl acetoacetate (2.6 g) contains sodium hydroxide (800 mg) and is chlorinated. An aqueous solution (150 mL) containing 5% by weight of sodium was added and partitioned. The contents of ethyl 2-methylacetoacetate and ethylacetoacetate in the toluene layer were quantified by GC.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description

R1は、ハロゲン原子で置換されていてもよいC1-6アルキル基を表し、
R2は、ハロゲン原子で置換されていてもよいC1-6アルキル基を表す]、
下記式(II):

で表される化合物を、トルエン、エチルベンゼン、エタノール、メタノール、アセトン、酢酸エチル、キシレン、テトラヒドロフラン、シクロヘキサン、N,N-ジメチルホルムアミド、N-メチル-2-ピロリドン、およびN,N-ジメチルアセトアミドからなる群から選択される一または二以上を組み合わせた有機溶媒中、塩基存在下で、ジメチル硫酸からなるメチル化剤と反応させる工程を含んでなる方法が提供される。
本発明による製造法の目的物であるα-メチル-β-ケトエステルは、前記式(I)で表される化合物である。
本発明によれば、β-ケトエステルを、有機溶媒中、塩基存在下で、ジメチル硫酸からなるメチル化剤と反応させる工程を含んでなる、式(I)で表される化合物を製造することが提供される。
式(II)で表される化合物を、トルエン、エチルベンゼン、エタノール、メタノール、アセトン、酢酸エチル、キシレン、テトラヒドロフラン、シクロヘキサン、N,N-ジメチルホルムアミド、N-メチル-2-ピロリドン、およびN,N-ジメチルアセトアミドからなる群から選択される一または二以上を組み合わせた有機溶媒中、塩基存在下で、ジメチル硫酸からなるメチル化剤と反応させ、
塩基性水溶液(好ましくは、塩基としては水酸化ナトリウム)により洗浄する工程を含み、
ここで、塩基性水溶液中の塩基の量が、有機溶媒中に存在する式(II)の化合物に対して0.5~1.5倍モル当量である、
式(I)で表される化合物の製造方法が提供される。
式(II)で表される化合物を、トルエン、エチルベンゼン、エタノール、メタノール、アセトン、酢酸エチル、キシレン、テトラヒドロフラン、シクロヘキサン、N,N-ジメチルホルムアミド、N-メチル-2-ピロリドン、およびN,N-ジメチルアセトアミドからなる群から選択される一または二以上を組み合わせた有機溶媒中、塩基存在下で、ジメチル硫酸からなるメチル化剤と反応させ、
塩基性水溶液(好ましくは、塩基としては水酸化ナトリウム)により洗浄する工程を含み、
ここで、塩基性水溶液の容量が、有機溶媒の量に対して0.1~1倍量である、
式(I)で表される化合物の製造方法が提供される。
式(II)で表される化合物を、トルエン、エチルベンゼン、エタノール、メタノール、アセトン、酢酸エチル、キシレン、テトラヒドロフラン、シクロヘキサン、N,N-ジメチルホルムアミド、N-メチル-2-ピロリドン、およびN,N-ジメチルアセトアミドからなる群から選択される一または二以上を組み合わせた有機溶媒中、塩基存在下で、ジメチル硫酸からなるメチル化剤と反応させ、
塩基性水溶液(好ましくは、塩基としては水酸化ナトリウム)により洗浄する工程を含み、
ここで、塩基性水溶液中の塩基の量が、有機溶媒中に存在する式(II)の化合物に対して0.5~1.5倍モル当量であり、塩基性水溶液の容量が、有機溶媒の量に対して0.1~1倍量である、
式(I)で表される化合物の製造方法が提供される。
式(II)で表される化合物を、エタノールまたはトルエンの単独溶媒、またはエタノールおよびトルエンもしくはメタノールおよびトルエンの混合溶媒中、塩基存在下で、ジメチル硫酸からなるメチル化剤と反応させ、
メチル化剤で反応させた液を40~75℃中で0.5~48時間存在させ、
さらに塩基存在下でジメチル硫酸からなるメチル化剤を反応させ、
トルエン、エチルベンゼン、酢酸エチル、キシレン、およびシクロヘキサンからなる群から選択される一または二以上を組み合わせた有機溶媒で溶媒置換を行い、
塩基性水溶液(好ましくは、塩基としては水酸化ナトリウム)により洗浄する工程を含む
式(I)で表される化合物の製造方法が提供される。
式(II)で表される化合物を、エタノールまたはトルエンの単独溶媒、またはエタノールおよびトルエンもしくはメタノールおよびトルエンの混合溶媒中、塩基存在下で、ジメチル硫酸からなるメチル化剤と反応させ、
メチル化剤で反応させた液を60~70℃中で0.5~4時間存在させ、
さらに塩基存在下でジメチル硫酸からなるメチル化剤を反応させ、
トルエン、エチルベンゼン、酢酸エチル、キシレン、およびシクロヘキサンからなる群から選択される一または二以上を組み合わせた有機溶媒で溶媒置換を行い、
塩基性水溶液(好ましくは、塩基としては水酸化ナトリウム)により洗浄する工程を含み、
ここで、塩基性水溶液中の塩基の量が、有機溶媒中に存在する式(II)の化合物に対して0.5~1.5倍モル当量であり、塩基性水溶液の容量が、有機溶媒の量に対して0.1~1倍量である、
式(I)で表される化合物の製造方法が提供される。
本発明の一つの態様によれば、式(I)で表される化合物と、式(II)で表される化合物との混合物、または式(I)で表される化合物および式(II)で表される化合物がトルエン、エチルベンゼン、酢酸エチル、キシレン、およびシクロヘキサンからなる群から選択される一または二以上を組み合わせた有機溶媒に溶解した溶液を塩基性水溶液で洗浄する工程を含んでなる方法が提供され、好ましくは式(I)で表される化合物の精製方法が提供される。例えば、上記式(I)で表される化合物の製造方法において、式(II)で表される化合物が残存していた場合に、本方法を用いることにより、残存した式(II)で表される化合物を除去し、式(I)で表される化合物を精製することができる。
(GCの条件)
検出器:FID
カラム:DB-5(0.25mm×30m、膜厚:0.25μm)
キャリアガス:He
注入口温度:150℃ 検出器温度:200℃
カラム温度:40℃ 1分→(8℃/分昇温)→100℃ 3分→(10℃/分昇温)→120℃ 1分
水酸化ナトリウム(40g)をエタノール(400mL)とトルエン(400mL)の混合溶媒に70℃に加温して溶解した。44℃まで冷却後、当該溶液にアセト酢酸エチル(130g)を加えた。反応温度を44℃~60℃に保ち、ジメチル硫酸(126g)を15分間かけて徐々に加えた後、44℃で2.5時間撹拌した。この反応液中には、2-メチルアセト酢酸エチル、アセト酢酸エチル、2,2-ジメチルアセト酢酸エチル、ethyl-3-methoxybut-2-enoateがそれぞれ73.1%、17.3%、2.7%、4.6%含まれていた(GC area%)。得られた反応液中の溶媒を減圧留去(40℃/90mmHg)し、残渣にトルエン(400mL)を加えた。これに15重量%の塩化ナトリウムを含む1.2N-塩酸(400mL)を加え、室温で6時間撹拌した。続いて、トルエン層に残留する原料アセト酢酸エチルに対し1.1倍モルの水酸化ナトリウム(9.9g)を含みかつ5重量%の塩化ナトリウムを含む水溶液(400mL)を加え、トルエン層を洗浄した。続いて、トルエン層に残留する原料のアセト酢酸エチルに対し1.25倍モルの水酸化ナトリウム(1.86g)を含みかつ5重量%の塩化ナトリウムを含む水溶液(400mL)を加え、トルエン層を洗浄した。得られたトルエン層を減圧濃縮(40℃、70mmHg)し、標題化合物を16.6重量%含むトルエン溶液535mL(収率61.7%)を得た。得られた化合物の1H-NMRスペクトルは市販品(東京化成株式会社製)のものと一致した。なお、このトルエン溶液中にはEAAを0.35重量%含有していた。
水酸化ナトリウム(4g)をメタノール(25mL)とトルエン(45mL)の混合溶媒に70℃に加温して溶解した。40℃まで冷却後、当該溶液にアセト酢酸メチル(11.6g)を加えた。更にメタノール20mLを加え、反応温度を40℃~50℃に保ち、ジメチル硫酸(12.6g)を15分間かけて徐々に加えた後、40℃で2時間撹拌した。この反応液中には、2-メチルアセト酢酸メチル、アセト酢酸メチル、2,2-ジメチルアセト酢酸メチル、methyl-3-methoxybut-2-enoateがそれぞれ71.3%、14.4%、2.1%、9.4%含まれていた(GC area%)。得られた反応液中の溶媒を減圧留去(35℃/110mmHg)し、残渣にトルエン15mLを加えた。これに15重量%の塩化ナトリウムを含む1.2N-塩酸50mLを加え、室温で6時間撹拌した。続いて、トルエン層に残留する原料アセト酢酸メチルに対し1.2倍モルの水酸化ナトリウム(0.776g)を含みかつ10重量%の塩化ナトリウムを含む水溶液(40mL)を加え、トルエン層を洗浄した。続いて、トルエン層に残留する原料アセト酢酸メチルに対し1倍モルの水酸化ナトリウム(0.08g)を含みかつ10重量%の塩化ナトリウムを含む水溶液25mLを加え、トルエン層を洗浄した。トルエン層を減圧濃縮(40℃、50mmHg)し、標題化合物を45.2重量%含むトルエン溶液12.8mL(収率44.5%)を得た。得られた化合物の1H-NMRスペクトルは市販品(東京化成株式会社社製)のものと一致した。なお、このトルエン溶液中にはアセト酢酸メチルを0.2重量%含有していた。
ナトリウムエトキシド(6.8g)をエタノール(30mL)に溶解し、これにトルエン(50mL)を加えた。エタノールを減圧留去した後、トルエンを加え、100mL定容とした。室温でアセト酢酸エチル(13g)を加えた後、ジメチル硫酸(7.56g)を更に加え、室温で1時間撹拌した。この反応液中には、2-メチルアセト酢酸エチル、アセト酢酸エチル、2,2-ジメチルアセト酢酸エチル、ethyl-3-methoxybut-2-enoateがそれぞれ75.4%、15.1%、0.8%、6.1%含まれていた(GC area%)。生じた不溶物をろ過し、ろ物をトルエンで洗浄し、ろ液と合わせた。76mLまで減圧濃縮した後、飽和食塩水(45mL)を加えて21時間室温で撹拌した。続いて、トルエン層に残留する原料アセト酢酸エチルに対し1倍モルの水酸化ナトリウム(0.6g)を含む水溶液(50mL)を加え、トルエン層を洗浄した。トルエン層を減圧濃縮(40℃、25mmHg)し、標題化合物5.35g(純度90%、収率33.4%)を得た。得られた化合物の1H-NMRスペクトルは市販品(東京化成株式会社社製)のものと一致した。なお、この生成物中にはEAAを2.4%含有していた。
水酸化ナトリウム(4g)をエタノール(80mL)に65℃で加温して、溶解した。45℃まで冷却後、当該溶液にアセト酢酸エチル(13g)を加えた。反応温度を45℃~60℃に保ち、ジメチル硫酸(12.6g)を10分間かけて徐々に加えた後、45℃で2.5時間撹拌した。この反応液中には、2-メチルアセト酢酸エチル、アセト酢酸エチル、2,2-ジメチルアセト酢酸エチル、ethyl-3-methoxybut-2-enoateがそれぞれ70.4%、16.1%、5.6%、6.0%含まれていた(GC area%)。得られた反応液中の溶媒を減圧留去(40℃/85mmHg)し、残渣にトルエン40mLを加えた。これに1.2N-塩酸(15%の塩化ナトリウムを含む)(40mL)を加え、室温で6時間撹拌した。続いて、トルエン層に残留する原料アセト酢酸エチルに対し1.1倍モルの水酸化ナトリウム(0.857g)を含む水溶液(5%の塩化ナトリウムを含む)(40mL)を加え、トルエン層を洗浄した。続いて、トルエン層に残留する原料アセト酢酸エチルに対し1.25倍モルの水酸化ナトリウム(0.192g)を含む水溶液(5%の塩化ナトリウムを含む)(40mL)を加え、トルエン層を洗浄した。得られたトルエン層を減圧濃縮(40℃、70mmHg)し、標題化合物を17.5重量%含むトルエン溶液(42mL)(収率51%)を得た。得られた化合物の1H-NMRスペクトルは市販品(東京化成株式会社社製)のものと一致した。なお、このトルエン溶液中にはEAAを0.34重量%含有していた。
水酸化カリウム(6.6g)をエタノール(25mL)とトルエン(25mL)の混合溶媒に65℃で加温して溶解した。44℃まで冷却後、当該溶液にアセト酢酸エチル(13g)を加えた。反応温度を45℃~65℃に保ち、ジメチル硫酸(12.6g)を15分間かけて徐々に加えた後、45℃で2.5時間撹拌した。この反応液中には、2-メチルアセト酢酸エチル、アセト酢酸エチル、2,2-ジメチルアセト酢酸エチル、ethyl-3-methoxybut-2-enoateがそれぞれ64.4%、14.9%、6.6%、9.6%含まれていた(GC area%)。得られた反応液中の溶媒を減圧留去(40℃/86mmHg)し、残渣にトルエン(40mL)を加えた。これに15重量%の塩化ナトリウムを含む1.2N-塩酸(40mL)を加え、室温で6時間撹拌した。続いて、トルエン層に残留する原料アセト酢酸エチルに対し1.1倍モルの水酸化ナトリウム(1.1g)を含みかつ5重量%の塩化ナトリウムを含む水溶液(25mL)を加え、トルエン層を洗浄した。続いて、トルエン層に残留する原料アセト酢酸エチルに対し1.25倍モルの水酸化ナトリウム(0.23g)を含みかつ5重量%の塩化ナトリウムを含む水溶液(25mL)を加え、トルエン層を洗浄した。得られたトルエン層を減圧濃縮(40℃、70mmHg)し、標題化合物を22.3重量%含むトルエン溶液(33mL)(収率51%)を得た。得られた化合物の1H-NMRスペクトルは市販品(東京化成株式会社社製)のものと一致した。なお、このトルエン溶液中にはEAAを0.27重量%含有していた。
水酸化ナトリウム(4g)をトルエン(80mL)に懸濁した。44℃で、当該溶液にアセト酢酸エチル(13g)を加えた後、更にトルエン(40mL)を加えた。ジメチル硫酸(12.6g)を5分間かけて徐々に加えた後、45℃で2.5時間撹拌した。反応の途中、トルエン(40mL)を更に加えた。この反応液中には、2-メチルアセト酢酸エチル、アセト酢酸エチル、2,2-ジメチルアセト酢酸エチル、ethyl-3-methoxybut-2-enoateがそれぞれ67.3%、26.1%、1.0%、3.2%含まれていた(GC area%)。これに15重量%の塩化ナトリウムを含む1.2N-塩酸(80mL)を加え、室温で6時間撹拌した。続いて、トルエン層に残留する原料アセト酢酸エチルに対し1.1倍モルの水酸化ナトリウム(2.62g)を含みかつ5重量%の塩化ナトリウムを含む水溶液(40mL)を加え、トルエン層を洗浄した。続いて、トルエン層に残留する原料アセト酢酸エチルに対し1.25倍モルの水酸化ナトリウム(0.32g)を含みかつ5重量%の塩化ナトリウムを含む水溶液(40mL)を加え、トルエン層を洗浄し、標題化合物を2.6重量%含むトルエン溶液(160mL)(収率29%)を得た。得られた化合物の1H-NMRスペクトルは市販品(東京化成株式会社社製)のものと一致した。なお、このトルエン溶液中にはEAAを0.18重量%含有していた。
水酸化ナトリウム(2g)をエタノール(20mL)に60℃に加温して溶解した。これにトルエン(20mL)を加え、45℃まで冷却後、当該溶液にアセト酢酸エチル(6.5g)を加えた。反応温度を45℃~55℃に保ち、ジメチル硫酸(6.3g)を5分間かけて徐々に加えた後、45℃で1時間撹拌した。この反応液に水0.8mLを加え、68℃で2.25時間撹拌した後、45℃まで冷却した。この溶液に水酸化ナトリウム(0.4g)を60℃で溶解したエタノール(4mL)を加え、更にジメチル硫酸(1.26g)を加えて、45℃で2.75時間撹拌した。この反応液中には、2-メチルアセト酢酸エチル、アセト酢酸エチル、2,2-ジメチルアセト酢酸エチル、ethyl-3-methoxybut-2-enoateがそれぞれ80.5%、7.7%、5.4%、1.4%含まれていた(GC area%)。続いて、10重量%の食塩水(40mL)を加えた。更に、この溶液中に含まれる溶媒を減圧留去(100mHg、40℃)した後、トルエン(40mL)を加えて撹拌した。トルエン層を分液した後、続いて、トルエン層に残留する原料アセト酢酸エチルに対して1.25倍モルの水酸化ナトリウム(0.32g)を含みかつ、5重量%の食塩を含む水溶液(45mL)を加え、トルエン層を洗浄した。こうして得られたトルエン層(45mL)は、標題化合物を10.3重量%含有しており(収率64.3%)、EAAの残留は0.21重量%であった。得られた化合物の1H-NMRスペクトルは市販品(東京化成株式会社社製)のものと一致した。
2-メチルアセト酢酸エチル(7.2g)と、アセト酢酸エチル(2.6g)とを含むトルエン(50mL)に水酸化ナトリウム(400mg)を含み、かつ塩化ナトリウム5重量%を含む水溶液(50mL)を加え、分配した。トルエン層の2-メチルアセト酢酸エチルと、アセト酢酸エチルとの含量をガスクロマトグラフィー(GC)で定量した。
2-メチルアセト酢酸エチル(7.2g)と、アセト酢酸エチル(2.6g)とを含むトルエン(50mL)に水酸化ナトリウム(800mg)を含み、かつ塩化ナトリウム5重量%を含む水溶液(50mL)を加え、分配した。トルエン層の2-メチルアセト酢酸エチルと、アセト酢酸エチルとの含量をGCで定量した。
2-メチルアセト酢酸エチル(7.2g)と、アセト酢酸エチル(2.6g)とを含むトルエン(50mL)に水酸化ナトリウム(1.2g)を含み、かつ塩化ナトリウム5重量%を含む水溶液(50mL)を加え、分配した。トルエン層の2-メチルアセト酢酸エチルと、アセト酢酸エチルとの含量をGCで定量した。
2-メチルアセト酢酸エチル(7.2g)と、アセト酢酸エチル(2.6g)とを含むトルエン(50mL)に水酸化ナトリウム(1.6g)を含み、かつ塩化ナトリウム5重量%を含む水溶液(50mL)を加え、分配した。トルエン層の2-メチルアセト酢酸エチルと、アセト酢酸エチルとの含量をGCで定量した。
2-メチルアセト酢酸エチル(7.2g)と、アセト酢酸エチル(2.6g)とを含むトルエン(50mL)に水酸化ナトリウム(800mg)を含み、かつ塩化ナトリウム5重量%を含む水溶液(5mL)を加え、分配した。トルエン層の2-メチルアセト酢酸エチルと、アセト酢酸エチルとの含量をGCで定量した。
2-メチルアセト酢酸エチル(7.2g)と、アセト酢酸エチル(2.6g)とを含むトルエン(50mL)に水酸化ナトリウム(800mg)を含み、かつ塩化ナトリウム5重量%を含む水溶液(10mL)を加え、分配した。トルエン層の2-メチルアセト酢酸エチルと、アセト酢酸エチルとの含量をGCで定量した。
2-メチルアセト酢酸エチル(7.2g)と、アセト酢酸エチル(2.6g)とを含むトルエン(50mL)に水酸化ナトリウム(800mg)を含み、かつ塩化ナトリウム5重量%を含む水溶液(25mL)を加え、分配した。トルエン層の2-メチルアセト酢酸エチルと、アセト酢酸エチルとの含量をGCで定量した。
2-メチルアセト酢酸エチル(7.2g)と、アセト酢酸エチル(2.6g)とを含むトルエン(50mL)に水酸化ナトリウム(800mg)を含み、かつ塩化ナトリウム5重量%を含む水溶液(100mL)を加え、分配した。トルエン層の2-メチルアセト酢酸エチルと、アセト酢酸エチルの含量とをGCで定量した。
2-メチルアセト酢酸エチル(7.2g)と、アセト酢酸エチル(2.6g)とを含むトルエン(50mL)に水酸化ナトリウム(800mg)を含み、かつ塩化ナトリウム5重量%を含む水溶液(150mL)を加え、分配した。トルエン層の2-メチルアセト酢酸エチルと、アセト酢酸エチルとの含量をGCで定量した。

Claims (12)
- 下記式(I)で表される化合物の製造方法であって:
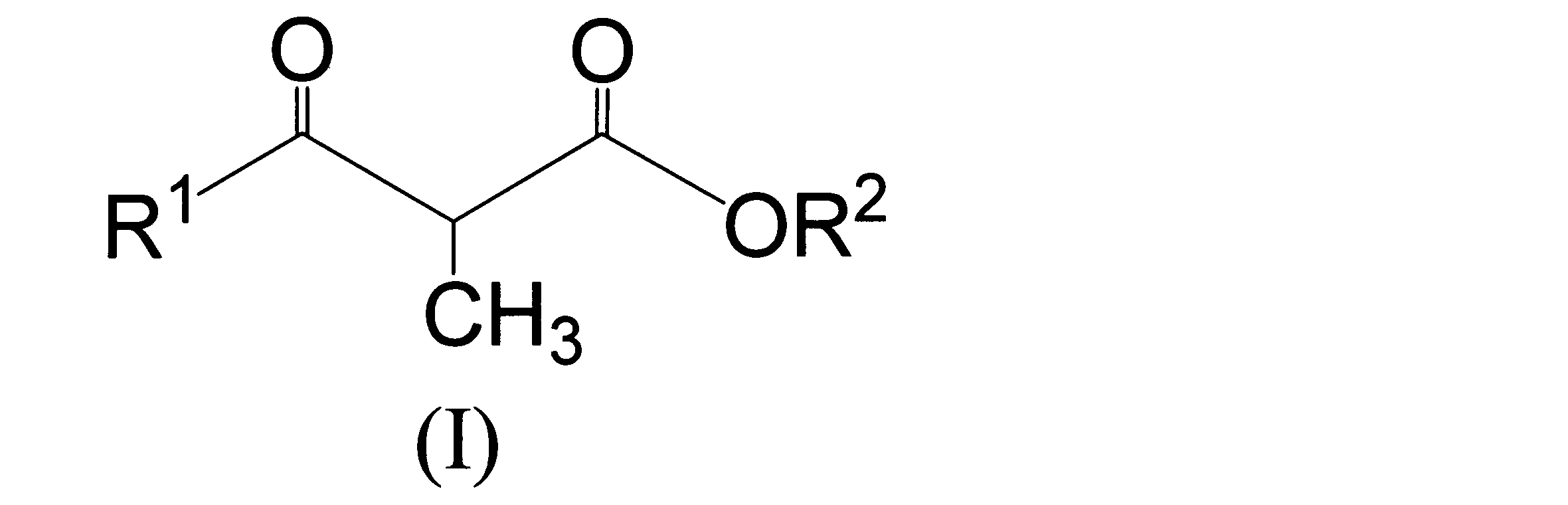
R1は、ハロゲン原子で置換されていてもよいC1-6アルキル基を表し、
R2は、ハロゲン原子で置換されていてもよいC1-6アルキル基を表す]、
下記式(II):

で表される化合物を、トルエン、エチルベンゼン、エタノール、メタノール、アセトン、酢酸エチル、キシレン、テトラヒドロフラン、シクロヘキサン、N,N-ジメチルホルムアミド、N-メチル-2-ピロリドン、およびN,N-ジメチルアセトアミドからなる群から選択される一または二以上を組み合わせた有機溶媒中、塩基存在下で、ジメチル硫酸からなるメチル化剤と反応させる工程を含んでなる、方法。 - 有機溶媒がエタノールまたはトルエンの単独溶媒であるか、またはエタノールおよびトルエンもしくはメタノールおよびトルエンの混合溶媒である、請求項1に記載の方法。
- 請求項1に記載のメチル化剤を反応させる工程の後に、塩基性水溶液による洗浄工程をさらに含む、請求項1または2に記載の方法。
- 請求項3に記載の洗浄工程の前、請求項1に記載のメチル化剤を反応させる工程の後に、トルエン、エチルベンゼン、酢酸エチル、キシレン、およびシクロヘキサンからなる群から選択される一または二以上を組み合わせた有機溶媒で溶媒置換を行う工程を含む、請求項3に記載の方法。
- 請求項3に記載の洗浄工程の前、請求項1に記載のメチル化剤を反応させる工程の後に、酸性水溶液で処理する工程をさらに含む、請求項3または4に記載の方法。
- 請求項3に記載の洗浄工程の前、請求項1に記載のメチル化剤を反応させる工程の後に、メチル化剤で反応させた液を40~75℃中で0.5~48時間存在させる工程をさらに含む、請求項3~5のいずれか一項に記載の方法。
- 塩基性水溶液中の塩基の量が、有機溶媒中に存在する式(II)の化合物に対して、0.5~1.5倍モル当量である、請求項3~6のいずれか一項に記載の方法。
- 塩基性水溶液の容量が、有機溶媒の量に対して0.1~1倍量である、請求項3~7のいずれか一項に記載の方法。
- R1が、メチル基であって、R2が、C1-4アルキル基である、請求項1に記載の方法。
- R2がメチル基またはエチル基である、請求項1または9に記載の方法。
- 請求項1に記載の式(I)で表される化合物と、式(II)で表される化合物との混合物、または
請求項1に記載の式(I)で表される化合物および式(II)で表される化合物がトルエン、エチルベンゼン、酢酸エチル、キシレン、およびシクロヘキサンからなる群から選択される一または二以上を組み合わせた有機溶媒に溶解した溶液
を塩基性水溶液で洗浄する工程を含んでなる、方法。 - 塩基性水溶液中の塩基の量が、有機溶媒に存在する式(II)の化合物に対して0.5~1.5倍モル当量である、請求項11に記載の方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020137015269A KR20130132854A (ko) | 2010-11-29 | 2011-10-31 | α-메틸-β-케토에스테르의 신규 제조 방법 |
| JP2012546746A JPWO2012073635A1 (ja) | 2010-11-29 | 2011-10-31 | α−メチル−β−ケトエステルの新規製造法 |
| CN2011800572895A CN103228610A (zh) | 2010-11-29 | 2011-10-31 | α-甲基-β-酮酯的新型制备方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010-264886 | 2010-11-29 | ||
| JP2010264886 | 2010-11-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012073635A1 true WO2012073635A1 (ja) | 2012-06-07 |
Family
ID=46171585
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/075090 WO2012073635A1 (ja) | 2010-11-29 | 2011-10-31 | α-メチル-β-ケトエステルの新規製造法 |
Country Status (5)
| Country | Link |
|---|---|
| JP (1) | JPWO2012073635A1 (ja) |
| KR (1) | KR20130132854A (ja) |
| CN (1) | CN103228610A (ja) |
| TW (1) | TW201233672A (ja) |
| WO (1) | WO2012073635A1 (ja) |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009020211A1 (ja) * | 2007-08-08 | 2009-02-12 | Meiji Seika Kaisha, Ltd. | α-メチル-β-ケトエステルの新規製造法 |
-
2011
- 2011-10-31 JP JP2012546746A patent/JPWO2012073635A1/ja not_active Ceased
- 2011-10-31 CN CN2011800572895A patent/CN103228610A/zh active Pending
- 2011-10-31 KR KR1020137015269A patent/KR20130132854A/ko not_active Application Discontinuation
- 2011-10-31 WO PCT/JP2011/075090 patent/WO2012073635A1/ja active Application Filing
- 2011-10-31 TW TW100139578A patent/TW201233672A/zh unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009020211A1 (ja) * | 2007-08-08 | 2009-02-12 | Meiji Seika Kaisha, Ltd. | α-メチル-β-ケトエステルの新規製造法 |
Non-Patent Citations (1)
| Title |
|---|
| CHEMICAL ABSTRACTS, vol. 32, 1938 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN103228610A (zh) | 2013-07-31 |
| KR20130132854A (ko) | 2013-12-05 |
| TW201233672A (en) | 2012-08-16 |
| JPWO2012073635A1 (ja) | 2014-05-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2015191706A1 (en) | Terpene-derived compounds and methods for preparing and using same | |
| CN109232212B (zh) | 一种由异戊烯醇合成甲基庚烯酮的方法 | |
| JP5689321B2 (ja) | 2−アミノ−4−トリフルオロメチルピリジン類の製造方法 | |
| JP6702623B2 (ja) | メデトミジンの合成に有用な3−アリールブタナールなどの化合物の調製方法 | |
| JP4303792B2 (ja) | キノロン誘導体の製造方法 | |
| WO2012073635A1 (ja) | α-メチル-β-ケトエステルの新規製造法 | |
| JP5188475B2 (ja) | 2−(3−ニトロベンジリデン)アセト酢酸イソプロピルの製造方法 | |
| JP4967613B2 (ja) | テトラフルオロテレフタル酸ジフルオライドの製造方法 | |
| JPH0517214B2 (ja) | ||
| FR3011839A1 (fr) | Procede de preparation de derives d'acetamidophenyle | |
| JP5275235B2 (ja) | α−メチル−β−ケトエステルの新規製造法 | |
| JP3823385B2 (ja) | 2,4,5−トリフルオロ−3−ヨ−ド安息香酸およびそのエステル類の製造方法 | |
| JP5029000B2 (ja) | ハロゲン置換ベンゼンジメタノールの製造法 | |
| JP2007254293A (ja) | α−メチレン−β−アルキル−γ−ブチロラクトンの製造法 | |
| JP2007031437A (ja) | 任意選択で置換されていてもよい臭化トリフルオロメチルフェナシルの調製方法 | |
| JP2007153823A (ja) | アレンカルボン酸エステル類の製造法 | |
| JP2011037724A (ja) | ピリミジニルアルコール誘導体の製造方法及びその合成中間体 | |
| JP6434261B2 (ja) | ヨードベンズアミド型アルコール酸化触媒 | |
| JP2004149440A (ja) | ベンジルカルバゼート化合物の製造法 | |
| JP2009079024A (ja) | 2−ナフトール誘導体の製造方法 | |
| JP4560309B2 (ja) | 光学活性なアミノ酸誘導体の製造方法 | |
| WO2008001826A1 (fr) | Procédé de fabrication d'un benzènediméthanol substitué par halogène | |
| EP3845518A1 (en) | Preparation method for (1r,3s)-3-amino-1-cyclopentanol and salts thereof | |
| JP2001011015A (ja) | 酒石酸低級アルキルジエステルの製造法 | |
| JP2000063321A (ja) | 光学純度の高い長鎖β−ヒドロキシカルボン酸の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11845202 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2012546746 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20137015269 Country of ref document: KR Kind code of ref document: A |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11845202 Country of ref document: EP Kind code of ref document: A1 |