WO2011158905A1 - ポリ乳酸の製造方法 - Google Patents
ポリ乳酸の製造方法 Download PDFInfo
- Publication number
- WO2011158905A1 WO2011158905A1 PCT/JP2011/063811 JP2011063811W WO2011158905A1 WO 2011158905 A1 WO2011158905 A1 WO 2011158905A1 JP 2011063811 W JP2011063811 W JP 2011063811W WO 2011158905 A1 WO2011158905 A1 WO 2011158905A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- lactide
- lactic acid
- racemic
- polylactic acid
- producing
- Prior art date
Links
- LJNPODXXBBBVMD-CRDPYYQKSA-N CC(C)(C)[Si](C)(C)c(cccc1/C=N/CC(C)(C)C/N=C/c2cccc([Si+](C)(C)C(C)(C)C)c2O)c1O Chemical compound CC(C)(C)[Si](C)(C)c(cccc1/C=N/CC(C)(C)C/N=C/c2cccc([Si+](C)(C)C(C)(C)C)c2O)c1O LJNPODXXBBBVMD-CRDPYYQKSA-N 0.000 description 1
Images











Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/78—Preparation processes
- C08G63/82—Preparation processes characterised by the catalyst used
- C08G63/84—Boron, aluminium, gallium, indium, thallium, rare-earth metals, or compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/02—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds
- C08G63/06—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds derived from hydroxycarboxylic acids
- C08G63/08—Lactones or lactides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/78—Preparation processes
- C08G63/82—Preparation processes characterised by the catalyst used
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/78—Preparation processes
- C08G63/82—Preparation processes characterised by the catalyst used
- C08G63/823—Preparation processes characterised by the catalyst used for the preparation of polylactones or polylactides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L101/00—Compositions of unspecified macromolecular compounds
- C08L101/16—Compositions of unspecified macromolecular compounds the macromolecular compounds being biodegradable
Definitions
- the present invention relates to a method for producing polylactic acid from glycerin, and the produced polylactic acid forms poly-L-lactic acid and poly-L-lactic acid by forming a eutectic of poly-L-lactic acid and poly-D-lactic acid.
- the present invention relates to a method which is a stereocomplex polylactic acid known to be a resin having higher heat resistance than D-lactic acid.
- bioplastics made from natural plants have attracted attention due to concerns about global warming.
- a major advantage of bioplastics is that they have carbon-neutral properties that do not affect the increase or decrease of carbon dioxide on the ground because they are originally planted on the ground. Because it is a plastic made by using materials derived from biomass that fixes carbon dioxide in the atmosphere by using biomass-derived materials, the balance of carbon dioxide is zero even if it is burned and discarded. It is said that it becomes.
- Polylactic acid is a polymer in which lactic acid is polymerized by ester bonds, and is one of the bioplastics that can be synthesized from plant-derived materials.
- lactic acid bacteria are allowed to act on glucose (glucose), sugar (sucrose) or the like, lactic acid is obtained by the fermentation action.
- the saccharide used as a raw material can be obtained in a large amount by allowing an enzyme (such as amylase) to act on starch obtained from potato, corn or the like, or extracting it from sugar cane or the like. From the viewpoint of “carbon neutral”, the demand for polylactic acid has increased in recent years.
- Lactic acid has one asymmetric carbon, and there are two types, L and D.
- a product obtained by polymerizing only the L-form is called poly-L-lactic acid
- a product obtained by polymerizing only the D-form is called poly-D-lactic acid.
- These are known to have a helical structure in the opposite direction due to their configuration, and the melting point is said to be about 175 ° C.
- Non-Patent Document 1 a method of producing cyanohydrin from acetaldehyde in the presence of an enzyme and subjecting it to acid hydrolysis (see Non-Patent Document 1), or a method of producing cyanohydrin from acetaldehyde and hydrocyanic acid, Is known to be esterified in the presence of hydrochloric acid or the like. According to such a method, racemic lactic acid in which equal amounts of L-lactic acid and D-lactic acid are mixed is produced.
- racemic lactic acid when racemic lactic acid is polymerized as it is, it becomes poly-DL-lactic acid in which L-lactic acid and D-lactic acid are mixed in one lactic acid polymer, and stereocomplex polylactic acid cannot be produced.
- the optical resolution method of racemic lactic acid is known to be separated by crystallization method or chromatography.
- these methods require complicated procedures and are expensive, and a large amount of compounds are separated. Had the disadvantage of being difficult.
- the present invention has been made in view of the above circumstances, and can use a carbon-neutral material that does not compete with foods such as sugars, and is complicated and expensive and difficult to mass-produce.
- An object of the present invention is to provide a method capable of producing stereocomplex polylactic acid without using an optical resolution method.
- FIG. 1 shows a process for producing stereocomplex polylactic acid from glycerin according to the present invention.
- the present invention is a method for producing stereocomplex polylactic acid, (1) producing a racemic sodium lactate aqueous solution by reacting glycerin with sodium hydroxide in high temperature and high pressure water; (2) recovering racemic lactic acid from the aqueous racemic sodium lactate solution; (3) producing a lactide mixture comprising mesolactide and racemic lactide by dimerizing the racemic lactic acid; (4) separating racemic lactide from meso lactide in the mixture; (5) A step of producing a stereocomplex type polylactic acid by polymerizing the racemic lactide using a salen type metal complex as a catalyst.
- glycerin and sodium hydroxide are dissolved in water to form an aqueous solution, which is maintained under high temperature and high pressure conditions of a temperature in the range of 250 to 350 ° C. and a pressure in the range of 5 to 15 MPa. Is done.
- the racemic lactic acid aqueous solution is acidified, and is brought into contact with an organic solvent to extract the racemic lactic acid into the organic solvent. Subsequently, the organic solvent is evaporated to recover the racemic lactic acid. Is done.
- the organic solvent is one selected from the group consisting of propanol, butanol, methyl acetate, triethylamine, and methyl ethyl ketone.
- an oligomer of lactic acid is produced as a precursor by dehydration condensation of racemic lactic acid, followed by depolymerization and cyclization to produce a lactide mixture of meso-lactide and racemic lactide, Is taken out of the reaction system as vapor.
- the conditions for generating the oligomer precursor are preferably a temperature of 100 to 200 ° C. and a pressure of 10 to 80 kPa.
- the conditions for producing the lactide are preferably a temperature of 150 to 250 ° C. and a pressure of 0.5 to 5 kPa.
- the step (4) is performed by a melt crystallization method using a melting point difference between racemic lactide having a melting point of 118 ° C. and meso lactide having a melting point of about 60 ° C.
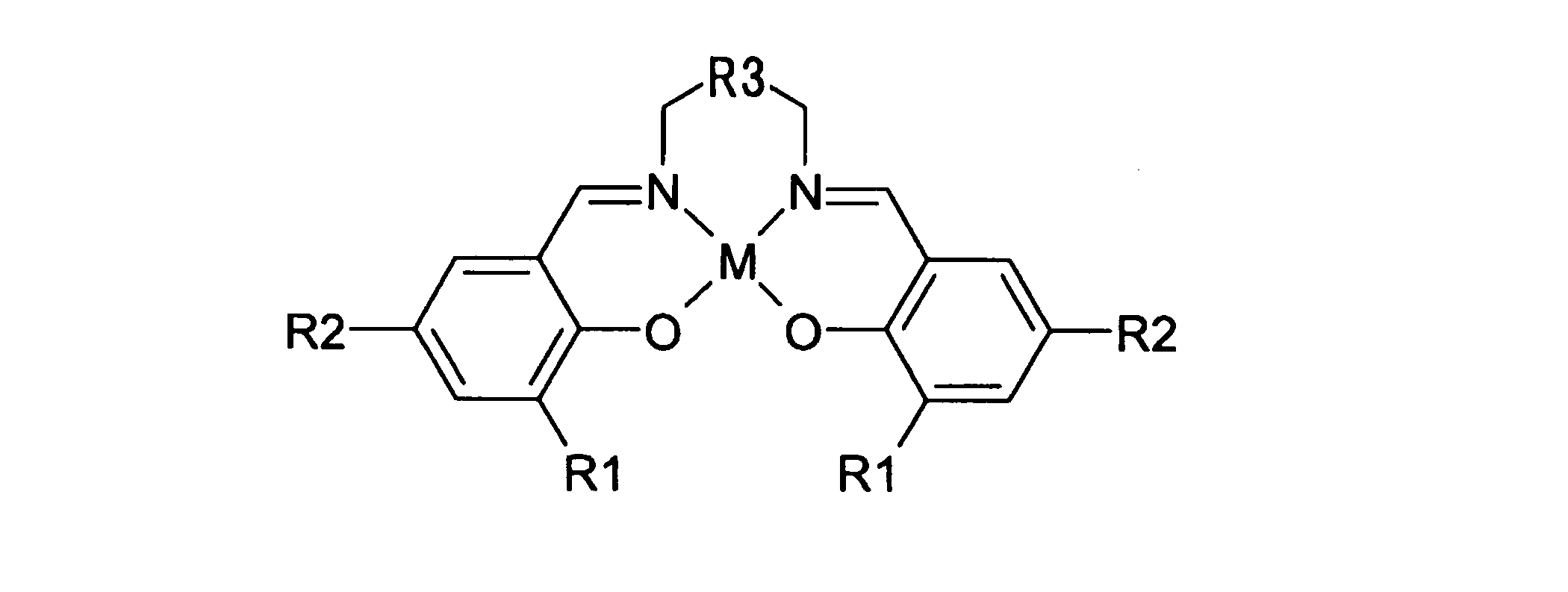
- the step (5) has the general formula:
- R1 and R2 are hydrogen, an alkyl group having 1 to 6 carbon atoms, an alkoxy group having 1 to 6 carbon atoms, a halogen group such as chlorine, bromine and fluorine, a silyl group, an aryl group having 6 to 18 nuclear carbon atoms, or methoxymethyl Represents a group.
- R3 represents a divalent aliphatic hydrocarbon group having 2 to 6 carbon atoms.
- M represents Al, Fe, Ti or Y.
- the present invention is a stereocomplex polylactic acid synthesized from glycerin produced by the method described in any one of the above.
- FIG. 1 a process for producing stereocomplex polylactic acid from conventional fermented lactic acid is shown in FIG.
- the fermentation method after producing poly-L-lactic acid and poly-D-lactic acid separately, they are mixed to produce stereocomplex polylactic acid.
- L-lactic acid or D-lactic acid is produced using different types of microorganisms.
- (2) and (7) purify lactic acid by separating the saccharide, medium, pH-adjusting alkaline substance and water used for fermentation.
- L-lactide or D-lactide is produced.
- impurities such as lactic acid, linear dimer and moisture in the lactide are removed.
- poly L lactic acid or poly D lactic acid is produced from lactide.
- poly-L-lactic acid and poly-D-lactic acid are mixed to produce stereocomplex polylactic acid. It can be seen that the conventional method shown in FIG. 2 requires more steps than the method of the present invention shown in FIG.
- Fig. 3 shows the process for producing stereocomplex polylactic acid from petroleum synthetic lactic acid.
- L-lactic acid and D-lactic acid can be simultaneously produced in equivalent amounts, but a step of separating L-lactic acid and D-lactic acid by an optical resolution method is required on the way.
- lactic acid is produced from petroleum as a raw material.
- the lactic acid produced here is racemic lactic acid in which L-lactic acid and D-lactic acid are mixed in an equivalent amount.
- impurities such as unreacted raw materials
- racemic lactic acid is removed.
- racemic lactic acid is separated into L-lactic acid and D-lactic acid.
- separation by crystallization method or chromatography is known.
- L-lactide or D-lactide is produced separately.
- impurities such as lactic acid, linear dimer and moisture in the lactide are removed.
- poly L lactic acid or poly D lactic acid is produced from lactide.
- poly-L-lactic acid and poly-D-lactic acid are mixed to produce stereocomplex polylactic acid. It can be seen that the conventional method shown in FIG. 3 requires more steps than the method of the present invention shown in FIG.
- the conventional raw material for polylactic acid was corn and potato as food, but the raw material for polylactic acid produced in the present invention is glycerin, an industrial by-product, so avoid competition with food. be able to. Since glycerin, a by-product in the fat and oil industry, is used as a raw material for lactic acid production, the supply of lactic acid to the plastics industry at a low cost and in large quantities can promote the spread of polylactic acid, which in turn contributes to the recycling society. It is expected to contribute to the formation.
- the present invention does not include a step of separating L-lactic acid and D-lactic acid or L-lactide and D-lactide by a so-called optical resolution method when producing a threo complex type polylactic acid. Since the optical resolution operation is very complicated and costly, it is industrially mainly used in the fields of expensive food products (amino acids and the like) and pharmaceuticals. For this reason, the ability to produce stereocomplex polylactic acid without including an optical resolution operation has a great industrial advantage. Therefore, in the present invention, it is possible to industrially supply stereocomplex polylactic acid having excellent thermal stability, which cannot be supplied in a large amount at a low cost by the conventional method.
- the polylactic acid produced by the present invention is a biomass plastic derived from animal and plant fats and oils, it is expected to contribute to measures against global warming from the viewpoint of carbon neutrality.
- FIG. 6 is a 1 H-NMR spectrum of the methine base region of polylactic acid obtained in Example 5a.
- 2 is a 1 H-NMR spectrum diagram of a methine base region of polylactic acid obtained in Comparative Example 1.
- FIG. 6 is a 1 H-NMR spectrum of the methine base region of polylactic acid obtained in Comparative Example 1.
- FIG. 4 is a flow sheet for explaining the method for producing the stereocomplex polylactic acid of the present invention.
- glycerin is used as a raw material.
- Glycerin is industrially produced as a by-product of soap, higher fatty acids or biodiesel fuel. Soap, higher fatty acids, and biodiesel fuel are produced using animal and vegetable fats and oils as raw materials. Since fats and oils are composed of glycerin and fatty acids, glycerin is always produced as a by-product in the production process. Soap, higher fatty acids, or diesel fuel are indispensable for daily life, and their production is increasing year by year. Accordingly, the production amount of glycerin as a by-product has increased, and in recent years, there has been a demand for its effective utilization method, and the present invention meets such social demands.
- the lactic acid production step (1) is a step of converting glycerin to lactic acid by the action of alkali and high-temperature high-pressure water.
- the raw material glycerin and an alkaline substance such as sodium hydroxide are mixed with water to form an aqueous solution, which is heated at a high temperature and a high pressure in a range of 250 to 350 ° C., for example, a temperature of 300 ° C.
- the glycerin can be converted to lactic acid with a yield of 70% or more by holding the sample for 10 to 200 minutes, for example, 60 minutes under the above conditions.
- the lactic acid purification step (2) is a step of taking out pure lactic acid by separating sodium and water from the sodium lactate aqueous solution obtained in step (1).
- alcohols such as propanol and butanol
- esters such as methyl acetate
- amines such as triethylamine
- ketones such as methyl ethyl ketone
- Lactic acid in an aqueous solution of sodium lactate is almost lactate (CH 3 -CH (OH) -COO -) is present as, but lactic acid by lowering the pH of the aqueous solution to 2.0 or less lactic acid (CH 3 -CH (OH ) -COOH).
- lactic acid becomes easy to be extracted to the organic solvent.
- the lactide production step (3) is a step of synthesizing lactide from racemic lactic acid.
- the lactide obtained by this process is a mixture of racemic lactide and meso lactide.
- Lactide refers to a cyclic compound formed by dehydration condensation of each other's hydroxyl group and carboxyl group in two molecules of lactic acid.
- L-lactide refers to a cyclic compound formed by dehydration condensation of two molecules of L-lactic acid.
- D-lactide refers to a cyclic compound formed by dehydration condensation of two molecules of D-lactic acid.
- Mesolactide refers to a cyclic compound formed by dehydration condensation of one molecule of L-lactic acid and one molecule of D-lactic acid.
- Racemic lactide refers to an equal mixture of L-lactide and D-lactide, that is, lactide with an optical purity of 0%.
- the oligomer refers to a lactic acid polymer composed of about 2 to 30 lactic acid molecules.
- lactic acid By heating to 200 ° C. under reduced pressure of 100 kPa or less, lactic acid can be dehydrated and condensed to produce a 10-20 mer oligomer.
- the stoichiometric formula of lactic acid condensation can be expressed as the following formula (2).
- the oligomer and lactide are in an equilibrium relationship as shown in the following formula (3).
- lactide Since the vapor pressure of lactide is higher than the vapor pressure of the oligomer, by heating under reduced pressure (210 ° C., 2 kPa), lactide can be reacted and evaporated and taken out of the system. When lactide is released from the system, the equilibrium of the formula proceeds in the direction in which lactide is generated. By repeating this, most of the oligomers can be converted to lactide.
- the lactide purification step (4) is a step of removing meso lactide from a mixture of racemic lactide and meso lactide.
- L-lactide and D-lactide other than the three-dimensional structure are exactly the same, but the physical properties (boiling point, melting point, solubility in solvents, etc.) of racemic lactide and meso-lactide, which are a mixture of L-lactide and D-lactide, are slightly different. Different. By utilizing this difference in physical properties, racemic lactide and meso lactide can be separated. For example, crystallization from a solvent, melt crystallization, or distillation.
- the melting point of racemic lactide is about 118 ° C.
- the melting point of meso lactide is about 60 ° C., so that the separation can be performed using the difference between these melting points.
- racemic lactide and meso lactide exist in the solid phase, but the abundance ratio of racemic lactide increases.
- racemic lactide in the solid phase is higher than before.
- the polylactic acid production step (5) is a step of producing stereocomplex polylactic acid by polymerizing racemic lactide.
- Racemic lactide can be stereoselectively ring-opened by using an aluminum complex having a salen type ligand having a specific substituent as a catalyst.
- Fig. 5 shows the principle of the reaction.
- an aluminum complex as a catalyst reacts with D-lactide or L-lactide to form a complex having asymmetry derived from each lactide.
- the monomers react one after another to grow the polymer, and the reacting monomer reacts while being sterically selected by the asymmetry derived from the monomer located at the polymer growth end. That is, when the polymer growth end is derived from, for example, D-lactide, the same D-lactide is also stereoselected for the next reacting monomer to react.
- FIG. 6 shows a flow of the lactic acid production apparatus according to the present invention.
- glycerin, sodium hydroxide and water are mixed to prepare a glycerin aqueous solution having a predetermined concentration.
- the amount of sodium hydroxide is desirably equimolar or more than glycerin.
- the glycerin aqueous solution is pressurized to a predetermined pressure and sent to the reactor at a predetermined flow rate.
- the liquid feeding amount is 20 L / hour.
- the pressure is adjusted depending on the reaction temperature, but is generally 6 to 20 MPa.
- the glycerin aqueous solution is heated to a predetermined temperature.
- the reaction temperature is 250-350 ° C.
- the reaction rate is slow, and when the reaction temperature is high, the produced lactic acid is decomposed. For this reason, approximately 280 to 320 ° C. is desirable.
- heating is performed by an electric heater, but a heat medium, water vapor, or other heat source may be used.
- the fluid at the outlet of the reactor (14) becomes a two-phase fluid of an aqueous sodium lactate solution and hydrogen gas.
- the volume of the reactor (14) is 20 L and the residence time is 1 hour.
- the residence time can be changed from 10 to 200 minutes by adjusting the amount of liquid delivered by the metering pump.
- the temperature of the two-phase fluid of sodium lactate aqueous solution and hydrogen gas is cooled to 100 ° C. or lower.
- the temperature of the two-phase fluid of sodium lactate aqueous solution and hydrogen gas is cooled to 100 ° C. or lower.
- the pressure control valve (16) reduces the pressure of the two-phase fluid of sodium lactate aqueous solution and hydrogen gas to atmospheric pressure.
- gas-liquid separator (17) the two-phase fluid of sodium lactate aqueous solution and hydrogen gas is separated into hydrogen gas and sodium lactate aqueous solution.
- gas-liquid separation is performed by a cyclone.
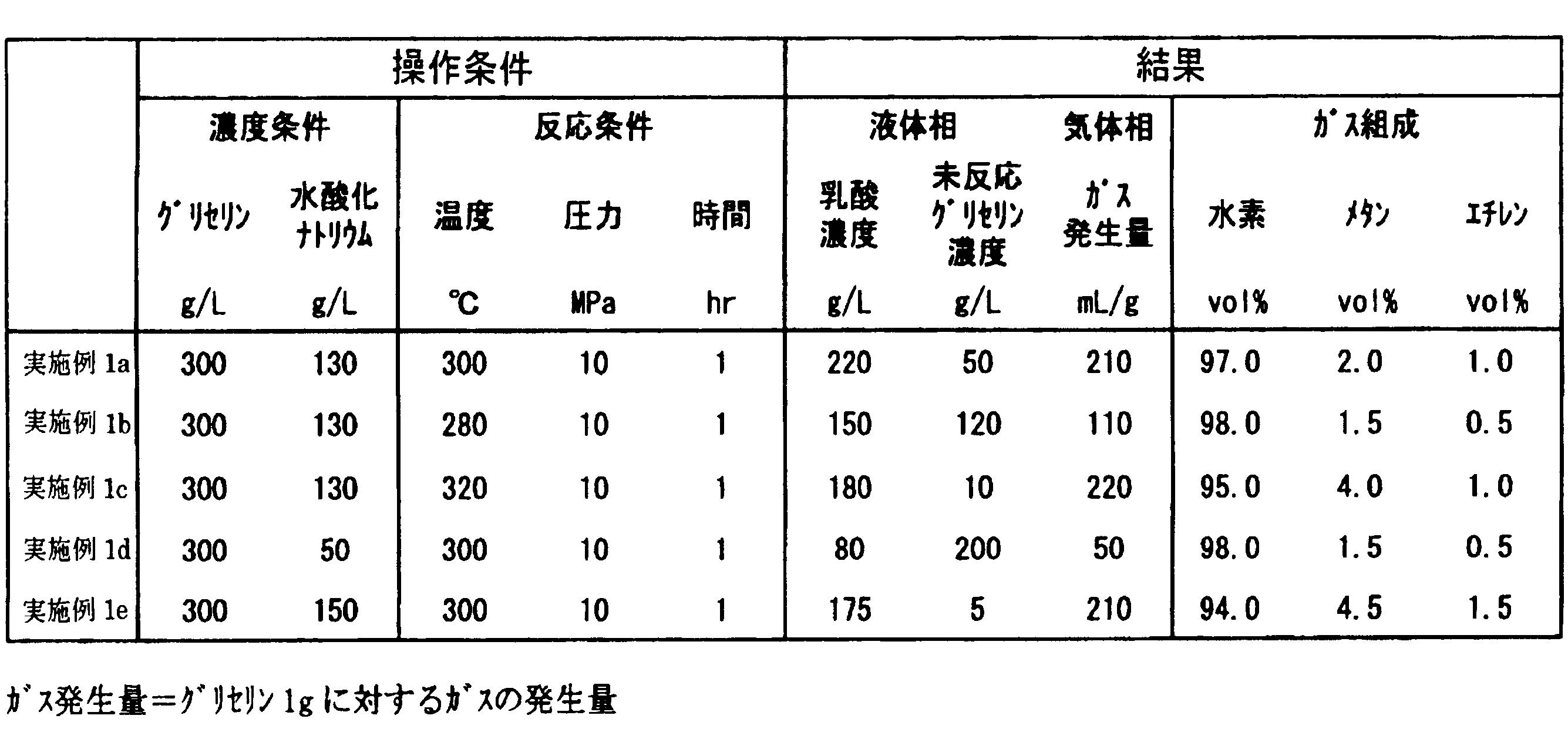
- Example 1a lactic acid was produced from an aqueous solution having a glycerin concentration of 300 g / L and a sodium hydroxide concentration of 130 g / L under conditions of a temperature of 300 ° C., a pressure of 10 MPa, and a time of 1 hour.
- the concentration of lactic acid in the reaction solution was 220 g / L
- the concentration of unreacted glycerol was 50 g / L
- the amount of gas generated was 210 mL / g-glycerol per 1 g of glycerol.
- the gas composition was 97 vol% hydrogen.
- Example 1b the temperature was lowered to 280 ° C., but the other conditions were the same as in Example 1a. Compared with Example 1a, the lactic acid concentration was as low as 150 g / L. This is considered to be because the reaction rate of glycerin was reduced by lowering the reaction temperature.
- Example 1c the temperature was raised to 320 ° C., but other conditions were the same as in Example 1a. Compared with Example 1a, the lactic acid concentration was as low as 180 g / L. This is considered to be because lactic acid produced by raising the reaction temperature was decomposed.
- Example 1d the sodium hydroxide concentration was lowered to 50 g / L, but the other conditions were the same as in Example 1a. Compared with Example 1a, the lactic acid concentration was as low as 80 g / L. This is considered to be because glycerin stopped reacting by lowering the sodium hydroxide concentration. As shown in the following formula (1), it is considered that sodium hydroxide should be added in an equimolar amount or more with glycerin.
- Example 1e the sodium hydroxide concentration was increased to 150 g / L, but the other conditions were the same as in Example 1a. Compared with Example 1a, the lactic acid concentration was as low as 175 g / L. This is probably because the lactic acid produced was easily decomposed by increasing the sodium hydroxide concentration.
- the lactic acid purification step (2) is a step of taking out only lactic acid by separating sodium and water from the sodium lactate aqueous solution obtained in the step (1).
- Table 2 shows the conditions for lactic acid purification. The operation procedure is shown below.
- Example 1a The aqueous sodium lactate solution obtained in Example 1a is adjusted to pH 1.0 with sulfuric acid.
- the aqueous phase and the organic phase are separated.
- the organic phase contains most of lactic acid and some water but very little sodium sulfate.
- the aqueous phase contains most of the sodium sulfate and some lactic acid and organic solvent.
- Example 2a methyl acetate was used as the extraction solvent.
- the total weight of the organic phase after extraction was 810 g, and the weight of lactic acid contained therein was 66 g.
- the amount of sodium sulfate contained in the organic phase was 0.6 g, and most of the sodium content could be removed.
- the organic phase contained water, glycerin and other impurities.
- the total weight of the residual liquid after distillation was 70 g
- the weight of lactic acid contained therein was 63 g.
- the solvent and water were evaporated from the organic phase by distillation, and lactic acid could be separated and recovered. By the above operation, lactic acid could be recovered from the sodium lactate aqueous solution.
- Example 2b propanol was used as the extraction solvent.
- the total weight of the organic phase after extraction was 1320 g, and the weight of lactic acid contained therein was 132 g. Further, the amount of sodium sulfate contained in the organic phase was 7.3 g, and most of the sodium content could be removed.
- the total weight of the residual liquid after distillation was 140 g, and the weight of lactic acid contained therein was 128 g.
- the amount of lactic acid recovered is larger than that of Example 2a, but the amount of sodium sulfate contained in lactic acid is also large. Also, since a large amount of water dissolves in the organic phase, energy is required for its evaporation, which is disadvantageous compared to Example 2a.
- Example 2c butanol was used as the extraction solvent.
- the total weight of the organic phase after extraction was 930 g, and the weight of lactic acid contained therein was 60 g.
- the amount of sodium sulfate contained in the organic phase was 3.0 g, and most of the sodium content could be removed.
- this organic phase was distilled with an evaporator, the total weight of the residual liquid after distillation was 60 g, and the weight of lactic acid contained therein was 55 g.
- Example 2d triethylamine was used as the extraction solvent.
- the total weight of the organic phase after extraction was 630 g, and the weight of lactic acid contained therein was 104 g.
- the amount of sodium sulfate contained in the organic phase was 0.07 g, and most of the sodium content could be removed.
- this organic phase was distilled with an evaporator, the residual liquid after distillation was 70 g, and the weight of lactic acid contained therein was 50 g.
- Sodium sulfate contained in lactic acid is less than in Example 2a, but there is much lactic acid loss in the solvent evaporation step.
- Example 2e methyl ethyl ketone was used as the extraction solvent.
- the total weight of the organic phase after extraction was 950 g, and the weight of lactic acid contained therein was 80 g.
- the amount of sodium sulfate contained in the organic phase was 1.6 g, and most of the sodium content could be removed.
- this organic phase was distilled with an evaporator, the total weight of the residual liquid after distillation was 85 g, and the weight of lactic acid contained therein was 78 g. Although the yield of lactic acid is larger than that of Example 2a, the amount of sodium sulfate contained in lactic acid is large.
- FIG. 7 shows a lactide production apparatus used in this process.
- Flask (21) is an eggplant-shaped flask having a volume of 200 mL, to which lactic acid as a raw material and tin octylate as a catalyst are supplied.
- the flask (21) is provided with a thermometer T1, and the sample temperature during heating can be measured.
- the flask (21) contains a stirrer (22) so that the sample can be well stirred during heating by the stirrer (23). Heating and temperature control of the flask (21) are performed by an oil bath (24). Further, a vacuum pump (25) is connected to the flask (21), and the pressure can be reduced to a predetermined pressure.
- Heating lactic acid under reduced pressure evaporates condensed water obtained by dehydration condensation of lactic acid. Lactic acid itself is converted to an oligomer. By further reducing the pressure of the generated oligomer and heating, lactide is generated from the oligomer, and the lactide is evaporated.
- Thermometer T2 measures the outlet temperature of the flask (21).
- the cooler (26) is a water-cooled or air-cooled glass tube that condenses evaporated water vapor or lactide vapor into a liquid state.
- the receiver (27) is a glass container with a scale, and can measure the amount of condensed water or lactide generated.
- the vacuum trap (28) collects water vapor and lactic acid vapor that could not be collected by the receiver. Cooled to -79 ° C with dry ice.
- the oil bath (24) is set to 130 ° C., and the pressure in the flask (21) is reduced to 80 kPa by the vacuum pump (25).
- Lactide is produced from the oligomer, and evaporation of lactide begins.
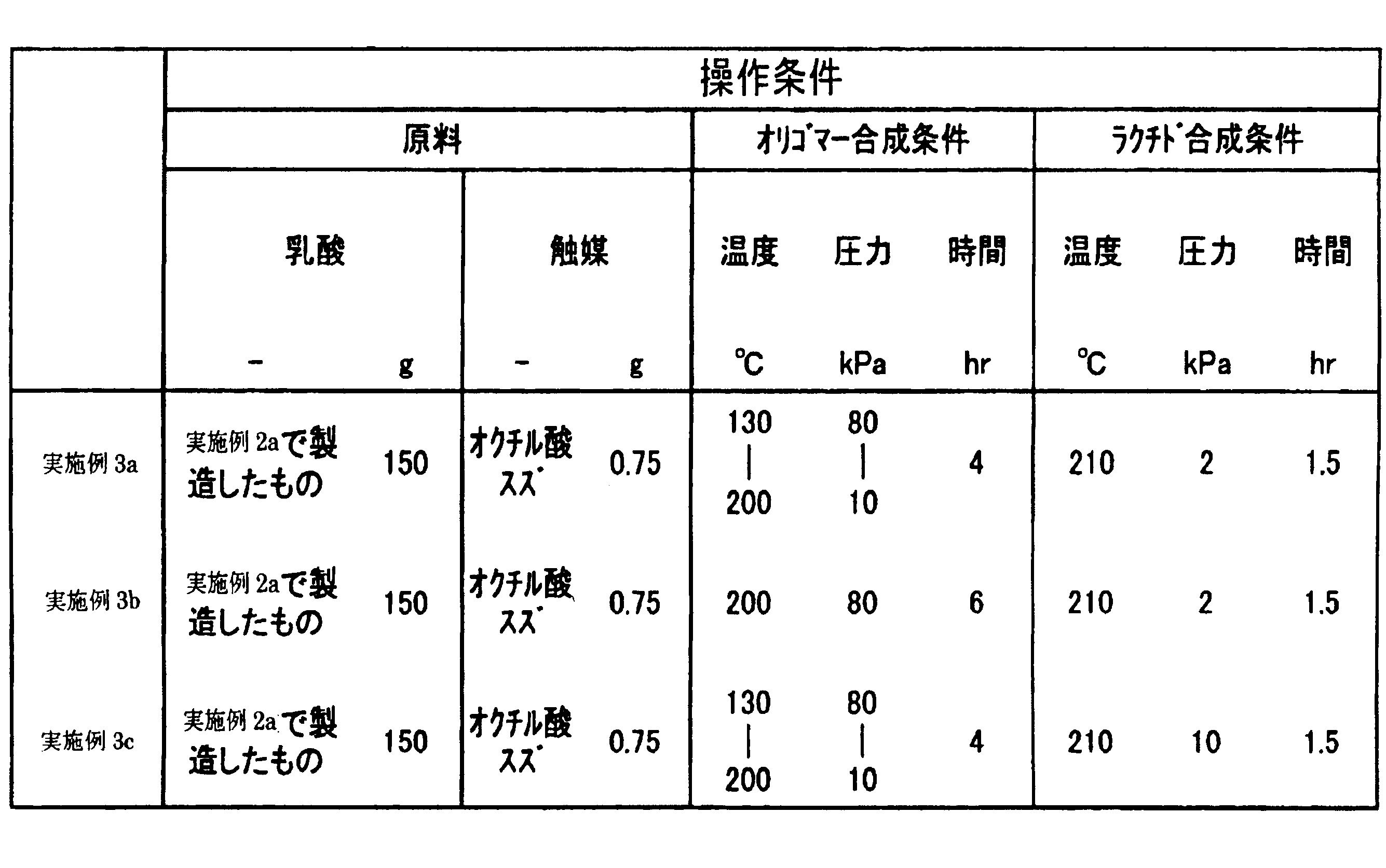
- Example 3a 150 g of lactic acid produced in Example 2a was used as a raw material.
- a catalyst 0.75 g of tin octylate was used.
- the synthesis conditions of the oligomer are a temperature of 130 to 200 ° C., a pressure of 80 to 10 kPa and a time of 4 hours.
- the lactide synthesis conditions are a temperature of 210 ° C., a pressure of 2 kPa, and a reaction time of 1.5 hours.
- the recovered amount of condensed water was 27 g. It was 1600 as a result of measuring the number average molecular weight of the obtained oligomer by the gel permeation chromatograph analyzer (GPC).
- the total amount of lactide recovered by the lactide synthesis experiment was 76 g.
- the composition of the lactide recovered by nuclear magnetic resonance analysis ( 1 H-NMR) it was found that it contained 34 g of racemic lactide and 27 g of meso lactide.
- the recovered lactide contained water, lactic acid, linear lactic acid dimer and the like as other impurities.
- Example 3b the oligomer synthesis conditions were a constant temperature of 200 ° C., a constant pressure of 80 kPa, and a reaction time of 6 hours. The other conditions are the same as in Example 3a.
- GPC gel permeation chromatograph
- Example 3c the lactide synthesis condition was a pressure of 10 kPa.
- the other conditions are the same as in Example 3a.
- the total amount of lactide recovered by the lactide synthesis experiment was 45 g, which was less than the lactide obtained in Example 3a.
- FIG. 8 shows a lactide purification apparatus used in the present lactide purification step.
- Table 6 shows the conditions and results of the lactide purification process. The operation procedure is shown below.
- the lactide obtained in Example 3a is used as a raw material. As shown in Table 6, the raw material lactide used was a mixture of racemic lactide and meso lactide. Moreover, lactic acid and a linear lactic acid dimer are contained as an impurity.
- the beaker (31) is immersed in the oil bath (32), and the lactide is heated to 130 ° C. to melt all the raw materials.
- the lactide temperature is measured with a thermometer T.
- the lactide is well stirred by rotating the stirring bar (34) with a stirrer (33).
- the temperature of the oil bath (32) is set to 75 ° C. and left for about 1 hour.
- the lactide used for purification was synthesized in Example 3a, and its composition was 45% by weight racemic lactide, 30% by weight meso lactide, and 20% by weight (water, lactic acid, linear lactic acid dimer, etc.).
- the raw material lactide was completely melted at 130 ° C., cooled to 65 ° C. and left for about 1 hour, and washed with propanol. Lactide recovered in the first melt crystallization operation was 65 g.
- Lactide recovered in the first melt crystallization operation was 65 g.
- the lactide recovered as a result of the second melt crystallization operation at 75 ° C. was 35 g.
- the lactide recovered as a result of the third melt crystallization operation at 95 ° C. was 25 g.
- Example 5 Polymerization process of stereocomplex type polylactic acid
- the racemic lactide purified in Example 4 was used as a raw material, and the salen-type aluminum complex having the structure shown in the following C1, C2, and C3 was used as a catalyst.
- FIG. 10 shows the 1 H-NMR spectrum of the methine base region of the obtained polylactic acid. A methine quadruple line is clearly observed. This also shows that the polymerized polylactic acid is a stereocomplex polylactic acid having a regular structure, which is composed of a polymer in which only L-lactic acid is bonded and a polymer in which only D-lactic acid is bonded.
- Table 7 summarizes the properties of the polylactic acid obtained in Example 5a.
- the weight yield of the obtained polylactic acid was 93% by weight. It was 14,000 as a result of measuring the number average molecular weight of the obtained polylactic acid with the gel permeation chromatography graph analyzer (GPC). It was 192 degreeC as a result of measuring melting
- the melting point of stereocomplex polylactic acid composed of pure L polylactic acid and D polylactic acid is said to be about 225 ° C., but the melting point of stereocomplex polylactic acid produced in Example 5a is lower than 225 ° C. This is because a small amount of D-lactic acid is mixed in the chain of the L polylactic acid molecule (or a small amount of L-lactic acid is mixed in the chain of the D polylactic acid molecule) and the stereoregularity is partially lost. Furthermore, it is considered that the melting point slightly decreased.
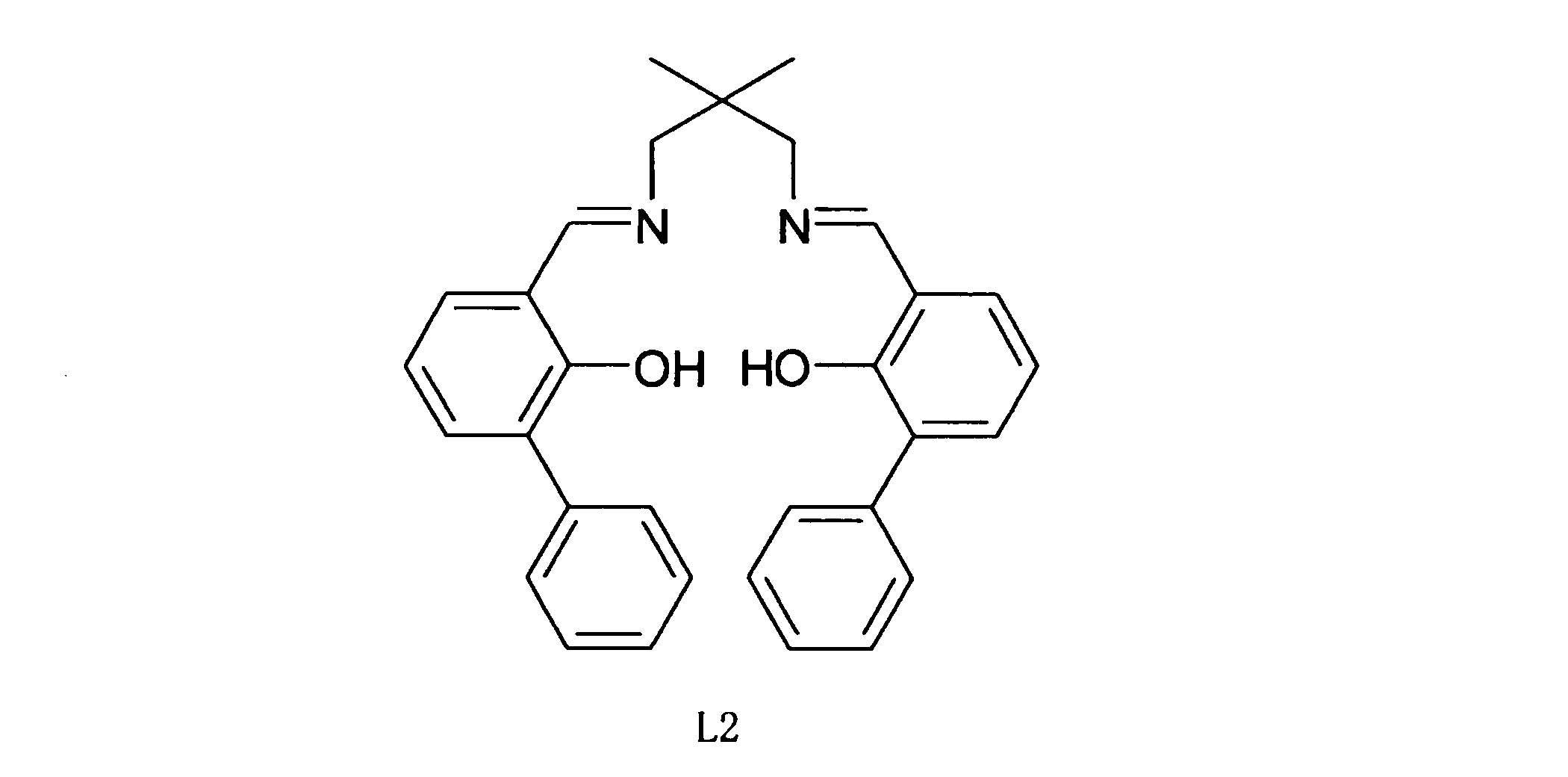
- Example 5b a ligand represented by Chemical Formula 8 L2 was used as a catalyst.
- the polymerization reaction time is 0.4 hours.
- Table 7 summarizes the properties of the polylactic acid obtained in Example 5b.
- the weight yield of the obtained polylactic acid was 95% by weight. It was 10,000 as a result of measuring the number average molecular weight of the obtained polylactic acid with a gel permeation chromatography analyzer (GPC). It was 171 degreeC as a result of measuring melting
- GPC gel permeation chromatography analyzer
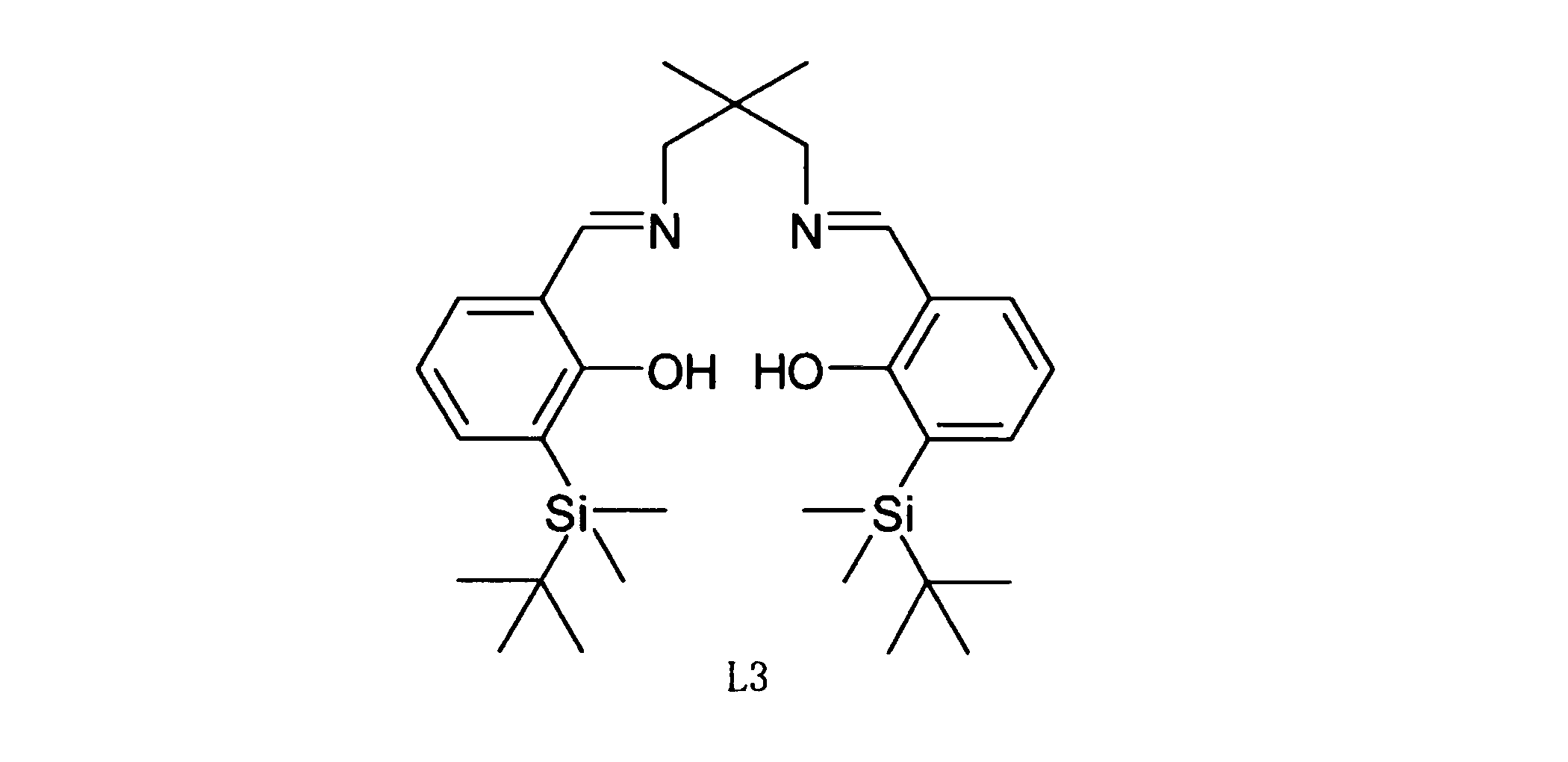
- Example 5c a ligand represented by Chemical Formula 9 L3 was used as a catalyst.
- the amount of catalyst C3 and initiator benzyl alcohol was reduced to 0.25 mmol and the polymerization reaction time was increased to 19 hours.
- Other conditions are the same as in Example 5a.
- Table 7 summarizes the properties of the polylactic acid obtained in Example 5c.
- the weight yield of the obtained polylactic acid was 90% by weight. It was 23,000 as a result of measuring the number average molecular weight of the obtained polylactic acid with the gel permeation chromatography graph analyzer (GPC). It was 207 degreeC as a result of measuring melting
- GPC gel permeation chromatography graph analyzer
- Example 5d so-called bulg polymerization was performed without using a toluene solvent.
- the toluene solvent was not used, C3 of the chemical formula 6 was used as the catalyst, the reaction temperature was 130 ° C., and the reaction time was 0.5 hours.
- Other conditions are the same as in Example 5a.
- Table 7 summarizes the properties of the polylactic acid obtained.
- the weight yield of the obtained polylactic acid was 98% by weight. It was 12,000 as a result of measuring the number average molecular weight of the obtained polylactic acid with the gel permeation chromatography graph analyzer (GPC). It was 160 degreeC as a result of measuring melting
- FIG. 11 shows a 1 H-NMR spectrum of the methine base region of the polylactic acid obtained. Since the quadruple line of the methine group is not clearly observed, it is assumed that the polymerized polylactic acid is a polylactic acid having a random structure in which only L-lactic acid and D-lactic acid are randomly bonded.
- the weight yield of the obtained polylactic acid was 88% by weight. It was 10,000 as a result of measuring the number average molecular weight of the obtained polylactic acid with the gel permeation chromatograph analyzer (GPC). Since the obtained polylactic acid is an amorphous (random structure) polymer, it did not show a melting point.
- Lactic acid production process 1 Lactic acid production process 2 Lactic acid purification process 3 Lactide production process 4 Lactide purification process 5 Polylactic acid production process
Abstract
Description
Chem. Commun., 2001, p1800
(1)高温高圧水中でグリセリンを水酸化ナトリウムと反応させることによって、ラセミ乳酸ナトリウム水溶液を製造する工程と、
(2)該ラセミ乳酸ナトリウム水溶液からラセミ乳酸を回収する工程と、
(3)該ラセミ乳酸を二量化することによりメソラクチドおよびラセミラクチドからなるラクチド混合物を生じさせる工程と、
(4)該混合物においてラセミラクチドをメソラクチドから分離する工程と、
(5)サレン型金属錯体を触媒として、該ラセミラクチドを重合することにより、ステレオコンプレックス型のポリ乳酸を製造する工程と
を包含することを特徴とする。
乳酸製造工程(1)は、アルカリと高温高圧水の作用によってグリセリンを乳酸に転換する工程である。

乳酸精製工程(2)は、工程(1)で得られた乳酸ナトリウム水溶液から、ナトリウムと水を分離して純粋な乳酸を取り出す工程である。
ラクチド製造工程(3)はラセミ乳酸からラクチドを合成する工程である。本工程により得られるラクチドは、ラセミラクチドとメソラクチドの混合物である。


ラクチド精製工程(4)は、ラセミラクチドとメソラクチドの混合物からメソラクチドを除去する工程である。
ポリ乳酸製造工程(5)は、ラセミラクチドを重合することによってステレオコンプレックス型ポリ乳酸を製造する工程である。
(操作手順)
図6に本発明に関わる乳酸製造装置のフローを示す。
表1に乳酸製造の結果をまとめる。
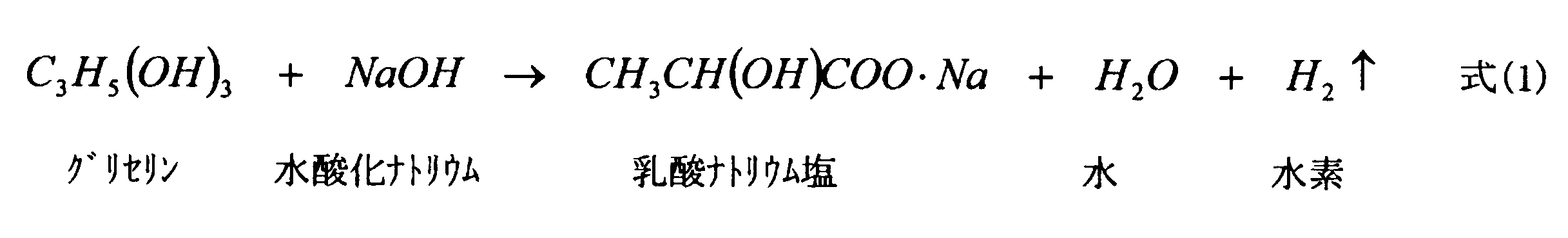
(操作手順)
乳酸精製工程(2)は、工程(1)で得られた乳酸ナトリウム水溶液から、ナトリウムと水を分離して乳酸のみを取り出す工程である。表2に乳酸精製時の条件を示す。また、以下に操作手順を示す。

表3に乳酸精製の結果をまとめる。

(装置)
図7に本工程に用いられるラクチド製造装置を示す。
・オリゴマーの合成
(1)実施例2aで得られた乳酸150gおよび触媒としてオクチル酸スズ0.75gを200mLのナス型フラスコ(21)に入れる。
(1)オイルバス(24)の温度を210℃に設定して、真空ポンプ(25)によりフラスコ内を2kPaに減圧する。
表4にラクチド合成時の条件を、表5にラクチド合成工程の結果をまとめる。

(操作手順)
図8に本ラクチド精製工程において用いられるラクチド精製装置を示す。表6にラクチド精製工程の条件と結果を示す。以下に操作手順を示す。
表6に条件と結果を示す。

実施例5aでは、実施例4で精製したラセミラクチドを原料として使用し、また、下記のC1、C2およびC3に示される構造のサレン型アルミニウム錯体を触媒として使用した。

窒素置換した試験管に、下記のL1で示される配位子0.5mmolおよびトルエンを5.0mL加え、配位子を溶解させる。この溶液を0℃に冷却後、トリメチルアルミニウムを0.7g(0.5mol)加え、室温に戻し約1時間攪拌して、触媒を調製した。

窒素置換した試験管に、ラセミラクチド7,200mg(50.0mmol)と開始剤としてベンジルアルコール0.5mmolを入れ、これにトルエン45mLを加えてよく攪拌して、ラセミラクチドを溶解した。この溶液に、触媒溶液を加えラクチドの重合を開始する。溶液は70℃に加熱して反応時間は6時間とした。
得られたポリ乳酸のX線回折スペクトルを図9に示す。図9より、2θ=12°、21°、24°にピークが観察される。ポリ乳酸のステレオコンプレックスは2θ=12°、21°、24°に特有のピークが検出されることが知られていることから、実施例5aで得られたポリ乳酸はステレオコンプレックスを形成していることが確認された(Ikeda, Y.; Jamshidi, K; Tuji, H; Hyon, S. H. Macromolecules 1987, 20, 904)。

2 乳酸精製工程
3 ラクチド製造工程
4 ラクチド精製工程
5 ポリ乳酸製造工程
Claims (10)
- ステレオコンプレックス型のポリ乳酸を製造する方法であって、
(1)高温高圧水中でグリセリンを水酸化ナトリウムと反応させることによって、ラセミ乳酸ナトリウム水溶液を製造する工程と、
(2)該ラセミ乳酸ナトリウム水溶液からナトリウムを分離してラセミ乳酸を回収する工程と、
(3)該ラセミ乳酸を二量化することによりメソラクチドおよびラセミラクチドからなるラクチド混合物を生じさせる工程と、
(4)該混合物からメソラクチドを分離してラセミラクチドを回収する工程と、
(5)サレン型金属錯体を触媒として、該ラセミラクチドを重合することにより、ステレオコンプレックス型のポリ乳酸を製造する工程と
を包含することを特徴とする、方法。 - 前記工程(1)は、グリセリンと水酸化ナトリウムを水に溶かして水溶液とし、これを250~350℃の範囲の温度および5~15MPaの範囲の圧力の高温高圧の条件下に保持することにより行われる、請求項1に記載の方法。
- 前記工程(2)は、ラセミ乳酸ナトリウム水溶液を酸性とし、これを有機溶媒と接触させることによりラセミ乳酸を有機溶媒に抽出し、続いて、有機溶媒を蒸発させることによりラセミ乳酸を回収することにより行われる、請求項1に記載の方法。
- 前記有機溶媒は、プロパノール、ブタノール、酢酸メチル、トリエチルアミンおよびメチルエチルケトンからなる群から選択される1種である、請求項3に記載の方法。
- 前記工程(3)は、ラセミ乳酸の脱水縮合により乳酸のオリゴマーを前駆体として生じさせ、続いて、これを解重合・環化することによってメソラクチドおよびラセミラクチドのラクチド混合物を生成させ、これを蒸気として反応系外に取り出すことにより行われる、請求項1に記載の方法。
- 前記オリゴマー前駆体を生じさせるための条件は、温度100~200℃、圧力10~80kPaである、請求項5に記載の方法。
- 前記ラクチドを生成させるための条件は、温度150~250℃、圧力0.5~5kPaである、請求項5または6に記載の方法。
- 前記工程(4)は、融点118℃のラセミラクチドと融点60℃のメソラクチドの融点差を利用する溶融晶析法により行われる、請求項1に記載の方法。
- 前記工程(5)は、一般式:
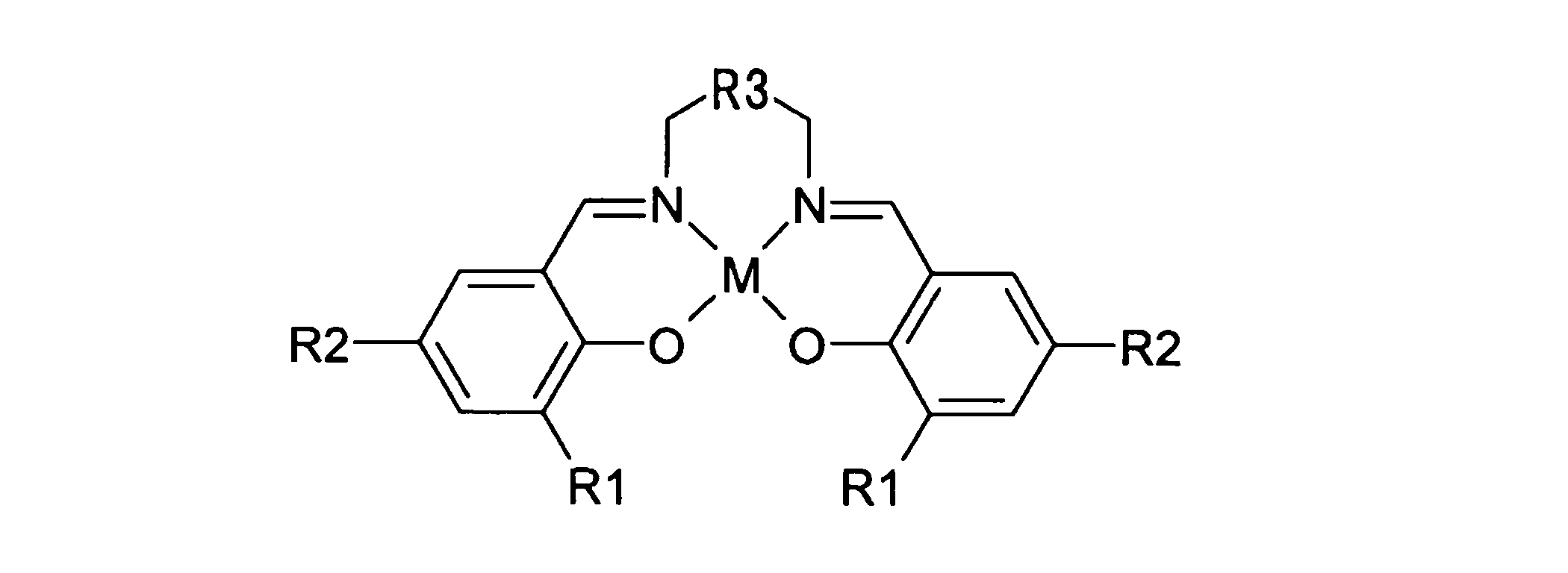
で表されるサレン型金属錯体を触媒として利用することにより行われる、請求項1に記載の方法。 - 前記請求項1~9のいずれか1つに記載の方法により製造される、グリセリンから合成されたステレオコンプレックス型ポリ乳酸。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13/704,793 US9056946B2 (en) | 2010-06-17 | 2011-06-16 | Method for producing polylactic acid |
| BR112012032124A BR112012032124A2 (pt) | 2010-06-17 | 2011-06-16 | método produzir ácido poliláctico stereocomplex, e ácido poliláctico stereocomplex sintetizado a partir de glicerina |
| EP11795807.4A EP2583989A4 (en) | 2010-06-17 | 2011-06-16 | PROCESS FOR PRODUCING POLY (LACTIC ACID) |
| CN2011800298000A CN103097432A (zh) | 2010-06-17 | 2011-06-16 | 聚乳酸的制备方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010137992A JP5679411B2 (ja) | 2010-06-17 | 2010-06-17 | ポリ乳酸の製造方法 |
| JP2010-137992 | 2010-06-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011158905A1 true WO2011158905A1 (ja) | 2011-12-22 |
Family
ID=45348298
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/063811 WO2011158905A1 (ja) | 2010-06-17 | 2011-06-16 | ポリ乳酸の製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9056946B2 (ja) |
| EP (1) | EP2583989A4 (ja) |
| JP (1) | JP5679411B2 (ja) |
| CN (1) | CN103097432A (ja) |
| BR (1) | BR112012032124A2 (ja) |
| WO (1) | WO2011158905A1 (ja) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013119959A1 (en) * | 2012-02-09 | 2013-08-15 | Novus International Inc. | Heteroatom containing cyclic dimers |
| LU91993B1 (de) * | 2012-05-03 | 2013-11-04 | D 01 P A C Holding | Verfahren zur Umsetzung von Glycerin zu organischen Salzen |
| CN103936974B (zh) * | 2014-03-26 | 2016-01-13 | 中国科学院长春应用化学研究所 | 一种稀土金属配合物催化剂及其制法和应用 |
| GB201406366D0 (en) | 2014-04-09 | 2014-05-21 | Plaxica Ltd | Biomass processing method |
| EP3056490A1 (en) * | 2015-02-13 | 2016-08-17 | PURAC Biochem BV | Method for manufacturing lactide |
| CN106957415B (zh) * | 2015-09-16 | 2019-07-12 | 武汉理工大学 | 一种用于二氧化碳、环氧丙烷和丙交酯三元共聚的复合催化剂及其制备方法 |
| CN106958051B (zh) * | 2017-05-10 | 2019-03-15 | 云南农业大学 | 一种组合植物的聚乳酸纤维的生产方法 |
| CN107417739B (zh) * | 2017-06-08 | 2019-11-15 | 中国科学院长春应用化学研究所 | 一种希夫碱铁化合物、其制备方法及其作为催化剂的应用 |
| CN107033193B (zh) * | 2017-06-08 | 2019-11-15 | 中国科学院长春应用化学研究所 | 一种希夫碱铁化合物、其制备方法及其作为催化剂的应用 |
| JP7417271B2 (ja) | 2018-09-20 | 2024-01-18 | バイオ燃料技研工業株式会社 | ポリ乳酸の製造方法 |
| CN111057223A (zh) * | 2019-12-17 | 2020-04-24 | 滨州市华康梦之缘生物科技有限公司 | 一种生产聚乳酸的方法 |
| CN113512181B (zh) * | 2021-08-09 | 2022-08-12 | 重庆大学 | 一种可低温加工的聚乳酸及其制备方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003064174A (ja) * | 2001-08-24 | 2003-03-05 | Nagoya Industrial Science Research Inst | ラクトンの開環重合用触媒、ポリエステルの製造方法、及びブロック共重合体の製造方法。 |
| JP2003511360A (ja) * | 1999-10-04 | 2003-03-25 | カーギル ダウ エルエルシー | 精製乳酸溶液の製造方法 |
| JP2003518476A (ja) * | 1999-10-12 | 2003-06-10 | プラク・ビオヘム・ベー・ブイ | 乳酸の連続的製造方法 |
| JP2006501213A (ja) * | 2002-08-06 | 2006-01-12 | ブリュッセルズ バイオテック | 乳酸又はその誘導体に於ける溶液からのポリラクチド生成方法 |
| WO2007001043A1 (ja) * | 2005-06-29 | 2007-01-04 | Hitachi Zosen Corporation | 乳酸の製造方法および乳酸製造装置 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5510526A (en) * | 1993-06-29 | 1996-04-23 | Cargill, Incorporated | Lactic acid production, separation and/or recovery process |
| GB0522154D0 (en) * | 2005-10-31 | 2005-12-07 | Univ Leeds | Novel catalytic materials and their use in the preparation of polymeric materials |
-
2010
- 2010-06-17 JP JP2010137992A patent/JP5679411B2/ja not_active Expired - Fee Related
-
2011
- 2011-06-16 US US13/704,793 patent/US9056946B2/en not_active Expired - Fee Related
- 2011-06-16 BR BR112012032124A patent/BR112012032124A2/pt not_active IP Right Cessation
- 2011-06-16 EP EP11795807.4A patent/EP2583989A4/en not_active Withdrawn
- 2011-06-16 WO PCT/JP2011/063811 patent/WO2011158905A1/ja active Application Filing
- 2011-06-16 CN CN2011800298000A patent/CN103097432A/zh active Pending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003511360A (ja) * | 1999-10-04 | 2003-03-25 | カーギル ダウ エルエルシー | 精製乳酸溶液の製造方法 |
| JP2003518476A (ja) * | 1999-10-12 | 2003-06-10 | プラク・ビオヘム・ベー・ブイ | 乳酸の連続的製造方法 |
| JP2003064174A (ja) * | 2001-08-24 | 2003-03-05 | Nagoya Industrial Science Research Inst | ラクトンの開環重合用触媒、ポリエステルの製造方法、及びブロック共重合体の製造方法。 |
| JP2006501213A (ja) * | 2002-08-06 | 2006-01-12 | ブリュッセルズ バイオテック | 乳酸又はその誘導体に於ける溶液からのポリラクチド生成方法 |
| WO2007001043A1 (ja) * | 2005-06-29 | 2007-01-04 | Hitachi Zosen Corporation | 乳酸の製造方法および乳酸製造装置 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2583989A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| BR112012032124A2 (pt) | 2016-11-16 |
| CN103097432A (zh) | 2013-05-08 |
| US9056946B2 (en) | 2015-06-16 |
| EP2583989A1 (en) | 2013-04-24 |
| EP2583989A4 (en) | 2014-09-03 |
| JP5679411B2 (ja) | 2015-03-04 |
| US20130178598A1 (en) | 2013-07-11 |
| JP2012001634A (ja) | 2012-01-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5679411B2 (ja) | ポリ乳酸の製造方法 | |
| Groot et al. | Production and purification of lactic acid and lactide | |
| Auras et al. | Poly (lactic acid): synthesis, structures, properties, processing, applications, and end of life | |
| KR101138240B1 (ko) | 재생가능한 원료로부터 폴리락트산(pla)의 생산 방법 | |
| CN111087381B (zh) | 乙交酯的精制处理方法 | |
| RU2631503C2 (ru) | Испытание качества полимеризуемой молочной кислоты и способ его осуществления | |
| JP2013503917A (ja) | カルボン酸のアルカリ金属塩から純粋アルキルエステルを調製するプロセス | |
| JP4048764B2 (ja) | 発酵乳酸を原料とするラクチドの製造方法及びポリ乳酸の製造方法 | |
| US8759545B2 (en) | Method of preparing lactide from lactate | |
| CA2975324C (en) | Method for manufacturing lactide | |
| KR101886434B1 (ko) | 유산으로부터 락타이드의 제조방법 | |
| CN105315155B (zh) | 制备聚乙醇酸低聚物的方法 | |
| JP2017521422A (ja) | 新規な乳酸回収方法 | |
| US9284403B2 (en) | Method to produce semi-crystalline polylactides | |
| CN110105324A (zh) | 异辛酸锌催化合成丙交酯的方法 | |
| KONOPLEV et al. | Purification of Crude Lactide to Polymerization Grade Purity by Molt Recrystallization Method. | |
| KR101809663B1 (ko) | 알킬 프로피오네이트와 물 혼합용매를 이용한 광학순도가 향상된 락타이드의 제조방법 | |
| BRPI1003073A2 (pt) | processo de purificação do ácido láctico para sìntese do lactato |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180029800.0 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11795807 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1201006547 Country of ref document: TH Ref document number: 12012502480 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011795807 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13704793 Country of ref document: US |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012032124 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112012032124 Country of ref document: BR Kind code of ref document: A2 Effective date: 20121217 |