WO2011125587A1 - 永久磁石及び永久磁石の製造方法 - Google Patents
永久磁石及び永久磁石の製造方法 Download PDFInfo
- Publication number
- WO2011125587A1 WO2011125587A1 PCT/JP2011/057568 JP2011057568W WO2011125587A1 WO 2011125587 A1 WO2011125587 A1 WO 2011125587A1 JP 2011057568 W JP2011057568 W JP 2011057568W WO 2011125587 A1 WO2011125587 A1 WO 2011125587A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- magnet
- permanent magnet
- sintering
- organometallic compound
- powder
- Prior art date
Links
Images















Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01F—MAGNETS; INDUCTANCES; TRANSFORMERS; SELECTION OF MATERIALS FOR THEIR MAGNETIC PROPERTIES
- H01F41/00—Apparatus or processes specially adapted for manufacturing or assembling magnets, inductances or transformers; Apparatus or processes specially adapted for manufacturing materials characterised by their magnetic properties
- H01F41/02—Apparatus or processes specially adapted for manufacturing or assembling magnets, inductances or transformers; Apparatus or processes specially adapted for manufacturing materials characterised by their magnetic properties for manufacturing cores, coils, or magnets
-
- C—CHEMISTRY; METALLURGY
- C22—METALLURGY; FERROUS OR NON-FERROUS ALLOYS; TREATMENT OF ALLOYS OR NON-FERROUS METALS
- C22C—ALLOYS
- C22C33/00—Making ferrous alloys
- C22C33/02—Making ferrous alloys by powder metallurgy
- C22C33/0257—Making ferrous alloys by powder metallurgy characterised by the range of the alloying elements
- C22C33/0278—Making ferrous alloys by powder metallurgy characterised by the range of the alloying elements with at least one alloying element having a minimum content above 5%
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B22—CASTING; POWDER METALLURGY
- B22F—WORKING METALLIC POWDER; MANUFACTURE OF ARTICLES FROM METALLIC POWDER; MAKING METALLIC POWDER; APPARATUS OR DEVICES SPECIALLY ADAPTED FOR METALLIC POWDER
- B22F1/00—Metallic powder; Treatment of metallic powder, e.g. to facilitate working or to improve properties
- B22F1/16—Metallic particles coated with a non-metal
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B22—CASTING; POWDER METALLURGY
- B22F—WORKING METALLIC POWDER; MANUFACTURE OF ARTICLES FROM METALLIC POWDER; MAKING METALLIC POWDER; APPARATUS OR DEVICES SPECIALLY ADAPTED FOR METALLIC POWDER
- B22F3/00—Manufacture of workpieces or articles from metallic powder characterised by the manner of compacting or sintering; Apparatus specially adapted therefor ; Presses and furnaces
- B22F3/10—Sintering only
-
- C—CHEMISTRY; METALLURGY
- C22—METALLURGY; FERROUS OR NON-FERROUS ALLOYS; TREATMENT OF ALLOYS OR NON-FERROUS METALS
- C22C—ALLOYS
- C22C38/00—Ferrous alloys, e.g. steel alloys
- C22C38/002—Ferrous alloys, e.g. steel alloys containing In, Mg, or other elements not provided for in one single group C22C38/001 - C22C38/60
-
- C—CHEMISTRY; METALLURGY
- C22—METALLURGY; FERROUS OR NON-FERROUS ALLOYS; TREATMENT OF ALLOYS OR NON-FERROUS METALS
- C22C—ALLOYS
- C22C38/00—Ferrous alloys, e.g. steel alloys
- C22C38/005—Ferrous alloys, e.g. steel alloys containing rare earths, i.e. Sc, Y, Lanthanides
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01F—MAGNETS; INDUCTANCES; TRANSFORMERS; SELECTION OF MATERIALS FOR THEIR MAGNETIC PROPERTIES
- H01F1/00—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties
- H01F1/01—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials
- H01F1/03—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity
- H01F1/032—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials
- H01F1/04—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials metals or alloys
- H01F1/047—Alloys characterised by their composition
- H01F1/053—Alloys characterised by their composition containing rare earth metals
- H01F1/055—Alloys characterised by their composition containing rare earth metals and magnetic transition metals, e.g. SmCo5
- H01F1/057—Alloys characterised by their composition containing rare earth metals and magnetic transition metals, e.g. SmCo5 and IIIa elements, e.g. Nd2Fe14B
- H01F1/0571—Alloys characterised by their composition containing rare earth metals and magnetic transition metals, e.g. SmCo5 and IIIa elements, e.g. Nd2Fe14B in the form of particles, e.g. rapid quenched powders or ribbon flakes
- H01F1/0572—Alloys characterised by their composition containing rare earth metals and magnetic transition metals, e.g. SmCo5 and IIIa elements, e.g. Nd2Fe14B in the form of particles, e.g. rapid quenched powders or ribbon flakes with a protective layer
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01F—MAGNETS; INDUCTANCES; TRANSFORMERS; SELECTION OF MATERIALS FOR THEIR MAGNETIC PROPERTIES
- H01F1/00—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties
- H01F1/01—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials
- H01F1/03—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity
- H01F1/032—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials
- H01F1/04—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials metals or alloys
- H01F1/06—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials metals or alloys in the form of particles, e.g. powder
- H01F1/08—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials metals or alloys in the form of particles, e.g. powder pressed, sintered, or bound together
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01F—MAGNETS; INDUCTANCES; TRANSFORMERS; SELECTION OF MATERIALS FOR THEIR MAGNETIC PROPERTIES
- H01F1/00—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties
- H01F1/01—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials
- H01F1/03—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity
- H01F1/032—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials
- H01F1/04—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials metals or alloys
- H01F1/06—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials metals or alloys in the form of particles, e.g. powder
- H01F1/08—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials metals or alloys in the form of particles, e.g. powder pressed, sintered, or bound together
- H01F1/086—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials metals or alloys in the form of particles, e.g. powder pressed, sintered, or bound together sintered
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01F—MAGNETS; INDUCTANCES; TRANSFORMERS; SELECTION OF MATERIALS FOR THEIR MAGNETIC PROPERTIES
- H01F41/00—Apparatus or processes specially adapted for manufacturing or assembling magnets, inductances or transformers; Apparatus or processes specially adapted for manufacturing materials characterised by their magnetic properties
- H01F41/02—Apparatus or processes specially adapted for manufacturing or assembling magnets, inductances or transformers; Apparatus or processes specially adapted for manufacturing materials characterised by their magnetic properties for manufacturing cores, coils, or magnets
- H01F41/0253—Apparatus or processes specially adapted for manufacturing or assembling magnets, inductances or transformers; Apparatus or processes specially adapted for manufacturing materials characterised by their magnetic properties for manufacturing cores, coils, or magnets for manufacturing permanent magnets
- H01F41/0266—Moulding; Pressing
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B22—CASTING; POWDER METALLURGY
- B22F—WORKING METALLIC POWDER; MANUFACTURE OF ARTICLES FROM METALLIC POWDER; MAKING METALLIC POWDER; APPARATUS OR DEVICES SPECIALLY ADAPTED FOR METALLIC POWDER
- B22F2998/00—Supplementary information concerning processes or compositions relating to powder metallurgy
- B22F2998/10—Processes characterised by the sequence of their steps
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01F—MAGNETS; INDUCTANCES; TRANSFORMERS; SELECTION OF MATERIALS FOR THEIR MAGNETIC PROPERTIES
- H01F1/00—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties
- H01F1/01—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials
- H01F1/03—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity
- H01F1/032—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials
- H01F1/04—Magnets or magnetic bodies characterised by the magnetic materials therefor; Selection of materials for their magnetic properties of inorganic materials characterised by their coercivity of hard-magnetic materials metals or alloys
- H01F1/047—Alloys characterised by their composition
- H01F1/053—Alloys characterised by their composition containing rare earth metals
- H01F1/055—Alloys characterised by their composition containing rare earth metals and magnetic transition metals, e.g. SmCo5
- H01F1/057—Alloys characterised by their composition containing rare earth metals and magnetic transition metals, e.g. SmCo5 and IIIa elements, e.g. Nd2Fe14B
- H01F1/0571—Alloys characterised by their composition containing rare earth metals and magnetic transition metals, e.g. SmCo5 and IIIa elements, e.g. Nd2Fe14B in the form of particles, e.g. rapid quenched powders or ribbon flakes
- H01F1/0575—Alloys characterised by their composition containing rare earth metals and magnetic transition metals, e.g. SmCo5 and IIIa elements, e.g. Nd2Fe14B in the form of particles, e.g. rapid quenched powders or ribbon flakes pressed, sintered or bonded together
- H01F1/0577—Alloys characterised by their composition containing rare earth metals and magnetic transition metals, e.g. SmCo5 and IIIa elements, e.g. Nd2Fe14B in the form of particles, e.g. rapid quenched powders or ribbon flakes pressed, sintered or bonded together sintered
Definitions
- the present invention relates to a permanent magnet and a method for manufacturing the permanent magnet.
- Permanent magnet motors used in hybrid cars, hard disk drives, and the like have been required to be smaller, lighter, higher in output, and more efficient.
- the permanent magnet embedded in the permanent magnet motor is required to be thin and further improve the magnetic characteristics.
- Permanent magnets include ferrite magnets, Sm—Co magnets, Nd—Fe—B magnets, Sm 2 Fe 17 N x magnets, and Nd—Fe—B magnets with particularly high residual magnetic flux density. Used as a permanent magnet for a permanent magnet motor.
- a powder sintering method is generally used as a manufacturing method of the permanent magnet.
- the powder sintering method first, raw materials are coarsely pulverized, and magnet powder is manufactured by fine pulverization by a jet mill (dry pulverization). Thereafter, the magnet powder is put into a mold and press-molded into a desired shape while applying a magnetic field from the outside. Then, it is manufactured by sintering the solid magnet powder formed into a desired shape at a predetermined temperature (for example, 800 ° C. to 1150 ° C. for Nd—Fe—B magnets).
- a predetermined temperature for example, 800 ° C. to 1150 ° C. for Nd—Fe—B magnets.
- Nd-based magnets such as Nd—Fe—B have a problem that the heat-resistant temperature is low. Therefore, when an Nd magnet is used for a permanent magnet motor, if the motor is continuously driven, the coercive force and residual magnetic flux density of the magnet are gradually reduced. Therefore, when using an Nd magnet for a permanent magnet motor, in order to improve the heat resistance of the Nd magnet, Dy (dysprosium) or Tb (terbium) having high magnetic anisotropy is added, and the coercive force of the magnet is added. It is intended to further improve the above.
- the magnetic performance of a permanent magnet is basically improved by reducing the crystal grain size of the sintered body because the magnetic properties of the magnet are derived by the single domain fine particle theory.
- the crystal grain size of the sintered body it is necessary to reduce the grain size of the magnet raw material before sintering.
- a magnet raw material that has been finely pulverized into a fine particle size is molded and sintered, grain growth of the magnet particles occurs during sintering. It was larger than before sintering, and a fine crystal grain size could not be realized.
- the crystal grain size increases, the coercive force is remarkably lowered because the domain wall generated in the grain easily moves.
- a method of adding a material for suppressing the grain growth of the magnet particles to the magnet raw material before sintering can be considered.
- the surface of magnet particles before sintering is coated with a particle growth inhibitor such as a metal compound having a melting point higher than the sintering temperature, thereby suppressing the particle growth of the magnet particles during sintering.
- a particle growth inhibitor such as a metal compound having a melting point higher than the sintering temperature
- Japanese Patent No. 3298219 pages 4 and 5) Japanese Patent Laid-Open No. 2004-250781 (pages 10 to 12, FIG. 2)
- the grain growth inhibitor is added to the magnet powder in advance in the magnet raw material ingot as in Patent Document 2, the grain growth inhibitor is positioned on the surface of the magnet particles after sintering. Without diffusing into the magnet particles. As a result, the grain growth at the time of sintering cannot be sufficiently suppressed, and the residual magnetic flux density of the magnet is reduced. In addition, even if each sintered magnet particle can be made minute by suppressing grain growth, if each sintered magnet particle is in a dense state, the exchange interaction between each magnet particle May propagate. As a result, there is a problem that when a magnetic field is applied from the outside, the magnetization reversal of each magnet particle easily occurs and the coercive force decreases.
- the grain growth inhibitor is distributed unevenly with respect to the grain boundaries of the magnet by adding the grain growth inhibitor to the Nd magnet in a state where it is dispersed in an organic solvent.
- the C-containing material remains in the magnet even if the organic solvent is volatilized later by vacuum drying or the like.
- carbide is formed.
- the present invention has been made to solve the above-described conventional problems, and V, Mo, Zr, Ta, Ti, W, or Nb contained in the organometallic compound is effectively unevenly distributed with respect to the grain boundaries of the magnet. It is possible to reduce the activity of the calcined body activated by the calcining treatment, thereby preventing the magnet particles from subsequently binding with oxygen, and reducing the residual magnetic flux density and coercive force.
- An object of the present invention is to provide a permanent magnet and a method for manufacturing the permanent magnet.
- a permanent magnet according to the present invention includes a step of pulverizing a magnet raw material into magnet powder, and the pulverized magnet powder with the following structural formula M- (OR) x (wherein M is V, Mo, Zr, Ta, Ti, W or Nb, R is a hydrocarbon substituent, which may be linear or branched, and x is an arbitrary integer.)
- a step of attaching the organometallic compound to the particle surface of the magnet powder and a step of obtaining a calcined body by calcining the magnet powder having the organometallic compound attached to the particle surface in a hydrogen atmosphere.
- the permanent magnet according to the present invention is characterized in that the metal forming the organometallic compound is unevenly distributed at grain boundaries of the permanent magnet after sintering.
- the permanent magnet according to the present invention is characterized in that R in the structural formula M- (OR) x is an alkyl group.
- the permanent magnet according to the present invention is characterized in that R in the structural formula M- (OR) x is any one of an alkyl group having 2 to 6 carbon atoms.
- the permanent magnet according to the present invention is characterized in that the amount of carbon remaining after sintering is 0.15 wt% or less.
- the permanent magnet according to the present invention is characterized in that, in the step of calcining the molded body, the molded body is held in a temperature range of 200 ° C. to 900 ° C. for a predetermined time.
- the permanent magnet according to the present invention is characterized in that the dehydrogenation step holds the magnet powder for a predetermined time in a temperature range of 200 ° C. to 600 ° C. in a vacuum atmosphere.
- the method for producing a permanent magnet according to the present invention includes a step of pulverizing a magnet raw material into magnet powder, and the pulverized magnet powder with the following structural formula M- (OR) x (where M is V, Mo Zr, Ta, Ti, W, or Nb, R is an alkyl group, which may be linear or branched, and x is an arbitrary integer.)
- a step of attaching the organometallic compound to the particle surface of the magnet powder a step of calcining the magnet powder having the organometallic compound attached to the particle surface in a hydrogen atmosphere to obtain a calcined body, and the calcined body.
- the method for producing a permanent magnet according to the present invention is characterized in that R in the structural formula M- (OR) x is an alkyl group.
- R in the structural formula M- (OR) x is any one of an alkyl group having 2 to 6 carbon atoms.
- the method for producing a permanent magnet according to the present invention is characterized in that the step of calcining the magnet powder holds the magnet powder in a temperature range of 200 ° C. to 900 ° C. for a predetermined time.
- the method of manufacturing a permanent magnet according to the present invention is characterized in that the step of performing the dehydrogenation treatment holds the magnet powder for a predetermined time in a temperature range of 200 ° C. to 600 ° C. in a vacuum atmosphere.
- V, Mo, Zr, Ta, Ti, W, or Nb contained in the organometallic compound can be efficiently unevenly distributed with respect to the grain boundaries of the magnet.
- the addition amount of V, Mo, Zr, Ta, Ti, W, or Nb can be made small compared with the past, the fall of a residual magnetic flux density can be suppressed.
- the activity of the calcined body activated by the calcination treatment can be reduced.
- the magnet particles are prevented from being combined with oxygen thereafter, and the residual magnetic flux density and coercive force are not reduced.
- V, Mo, Zr, Ta, Ti, W or Nb which are high melting point metals, are unevenly distributed at the grain boundaries of the magnet after sintering.
- Mo, Zr, Ta, Ti, W or Nb suppresses the grain growth of the magnet particles during sintering, and also breaks the exchange interaction between the magnet particles after sintering, thereby reversing the magnetization of each magnet particle It is possible to improve the magnetic performance.
- the permanent magnet of the present invention since an organometallic compound composed of an alkyl group is used as the organometallic compound added to the magnet powder, when the magnet powder is calcined in a hydrogen atmosphere, the organometallic compound is used. It is possible to easily perform the thermal decomposition. As a result, the amount of carbon in the calcined body can be more reliably reduced.
- the magnet powder is calcined in a hydrogen atmosphere.
- the organometallic compound composed of an alkyl group having 2 to 6 carbon atoms
- the magnet powder is calcined in a hydrogen atmosphere.
- the pyrolysis of the organometallic compound can be more easily performed on the entire magnet powder. That is, the amount of carbon in the calcined body can be more reliably reduced by the calcining process.
- the amount of carbon remaining after sintering is 0.15 wt% or less, so that no gap is generated between the main phase of the magnet and the grain boundary phase, and the magnet It becomes possible to make the whole into the state sintered precisely, and it can prevent that a residual magnetic flux density falls. Further, a large number of ⁇ Fe is not precipitated in the main phase of the magnet after sintering, and the magnet characteristics are not greatly deteriorated.
- the step of calcining the molded body is performed by holding the molded body in a temperature range of 200 ° C. to 900 ° C. for a predetermined time, so that the organometallic compound is reliably pyrolyzed. It is possible to burn more than the necessary amount of carbon contained.
- the dehydrogenation process is performed by holding the magnet powder in a temperature range of 200 ° C. to 600 ° C. for a predetermined time. Even when NdH 3 having high activity is generated in the system magnet, it is possible to shift to NdH 2 having low activity without leaving it.
- a permanent magnet in which V, Mo, Zr, Ta, Ti, W or Nb contained in the organometallic compound is efficiently unevenly distributed with respect to the grain boundary of the magnet is obtained. It can be manufactured. As a result, in the manufactured permanent magnet, it is possible to suppress the grain growth of the magnet particles during sintering and to prevent the magnetization reversal of each magnet particle by breaking the exchange interaction between the magnet particles. The performance can be improved. Moreover, since the addition amount of V, Mo, Zr, Ta, Ti, W, or Nb can be made small compared with the past, the fall of a residual magnetic flux density can be suppressed.
- the activity of the calcined body activated by the calcining treatment can be reduced by performing the dehydrogenation treatment after the calcining treatment.
- the magnet particles are prevented from being combined with oxygen thereafter, and the residual magnetic flux density and coercive force are not reduced.
- the method for producing a permanent magnet according to the present invention since an organometallic compound composed of an alkyl group is used as the organometallic compound added to the magnet powder, when calcining the magnet powder in a hydrogen atmosphere, Thermal decomposition of the organometallic compound can be easily performed. As a result, the amount of carbon in the calcined body can be more reliably reduced.
- an organometallic compound composed of an alkyl group having 2 to 6 carbon atoms is used as the organometallic compound added to the magnet powder.
- the organometallic compound added to the magnet powder When calcination, it is possible to thermally decompose the organometallic compound at a low temperature. As a result, the pyrolysis of the organometallic compound can be more easily performed on the entire magnet powder. That is, the amount of carbon in the calcined body can be more reliably reduced by the calcining process.
- the step of calcining the molded body is performed by holding the molded body for a predetermined time in a temperature range of 200 ° C. to 900 ° C. More than the necessary amount of carbon contained by pyrolysis can be burned off.
- the dehydrogenation process is performed by holding the magnet powder for a predetermined time in a temperature range of 200 ° C. to 600 ° C. Even when NdH 3 having high activity is generated in the Nd-based magnet, it is possible to shift to NdH 2 having low activity without leaving it.
- FIG. 1 is an overall view showing a permanent magnet according to the present invention.
- FIG. 2 is an enlarged schematic view showing the vicinity of the grain boundary of the permanent magnet according to the present invention.
- FIG. 3 is a schematic diagram showing a magnetic domain structure of a ferromagnetic material.
- FIG. 4 is an enlarged schematic view showing the vicinity of the grain boundary of the permanent magnet according to the present invention.
- FIG. 5 is an explanatory view showing a manufacturing process in the first method for manufacturing a permanent magnet according to the present invention.
- FIG. 6 is an explanatory view showing a manufacturing process in the second method for manufacturing a permanent magnet according to the present invention.
- FIG. 7 is a diagram showing a change in the amount of oxygen when the calcination treatment in hydrogen is performed and when it is not performed.
- FIG. 1 is an overall view showing a permanent magnet according to the present invention.
- FIG. 2 is an enlarged schematic view showing the vicinity of the grain boundary of the permanent magnet according to the present invention.
- FIG. 3 is
- FIG. 8 is a graph showing the amount of carbon remaining in the permanent magnets of the permanent magnets of Examples 1 to 4 and Comparative Examples 1 and 2.
- FIG. 9 is a diagram showing an SEM photograph after sintering of the permanent magnet of Example 1 and the elemental analysis results of the grain boundary phase.
- FIG. 10 is a diagram showing an SEM photograph after sintering of the permanent magnet of Example 2 and the elemental analysis result of the grain boundary phase.
- FIG. 11 is a diagram in which the distribution state of the Nb element is mapped in the same field of view as the SEM photograph after sintering of the permanent magnet of Example 2 and the SEM photograph.
- FIG. 12 is a view showing an SEM photograph after sintering of the permanent magnet of Example 3 and the elemental analysis result of the grain boundary phase.
- FIG. 13 is a diagram in which the distribution state of the Nb element is mapped in the same field of view as the SEM photograph after sintering of the permanent magnet of Example 3 and the SEM photograph.
- FIG. 14 is a diagram showing an SEM photograph after sintering of the permanent magnet of Example 4 and the elemental analysis results of the grain boundary phase.
- FIG. 15 is a diagram in which the distribution state of the Nb element is mapped in the same field of view as the SEM photograph after sintering of the permanent magnet of Example 4 and the SEM photograph.
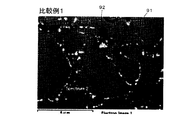
- 16 is a view showing an SEM photograph after sintering of the permanent magnet of Comparative Example 1.
- FIG. 17 is a view showing an SEM photograph after sintering of the permanent magnet of Comparative Example 2.
- FIG. 18 is a graph showing the carbon content in a plurality of permanent magnets manufactured by changing the calcination temperature conditions for the permanent magnets of Example 5 and Comparative Examples 3 and 4.
- FIG. 18 is a graph showing the carbon content in
- FIG. 1 is an overall view showing a permanent magnet 1 according to the present invention.
- 1 has a cylindrical shape, the shape of the permanent magnet 1 varies depending on the shape of the cavity used for molding.
- an Nd—Fe—B magnet is used as the permanent magnet 1 according to the present invention.
- Nb (niobium), V (vanadium), Mo (molybdenum), Zr (zirconium) for increasing the coercive force of the permanent magnet 1 are formed at the interfaces (grain boundaries) of the crystal grains forming the permanent magnet 1.
- Ta tantalum
- Ti titanium
- W tungsten
- each component is Nd: 25 to 37 wt%, Nb, V, Mo, Zr, Ta, Ti, W (hereinafter referred to as Nb etc.): 0.01 to 5 wt%, B: 1 to 2 wt%, Fe (electrolytic iron): 60 to 75 wt%. Further, in order to improve the magnetic characteristics, a small amount of other elements such as Co, Cu, Al and Si may be included.
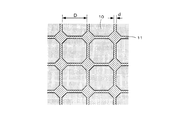
- a part of Nd is made of a refractory metal in the surface portion (outer shell) of the crystal grains of the Nd crystal particles 10 constituting the permanent magnet 1 as shown in FIG.
- a layer 11 hereinafter referred to as a refractory metal layer 11
- Nb or the like is unevenly distributed with respect to the grain boundaries of the Nd crystal particles 10.
- FIG. 2 is an enlarged view of the Nd crystal particles 10 constituting the permanent magnet 1.
- the refractory metal layer 11 is preferably nonmagnetic.
- substitution of Nb or the like is performed by adding an organometallic compound containing Nb or the like before forming a pulverized magnet powder as described later.
- Nd when sintering a magnet powder to which an organometallic compound containing Nb or the like is added, Nb or the like in the organometallic compound uniformly adhered to the particle surface of the Nd crystal particles 10 by wet dispersion is Nd.
- Replacement is performed by diffusing and penetrating into the crystal growth region of the crystal grains 10 to form the refractory metal layer 11 shown in FIG.
- the Nd crystal particles 10 are made of, for example, an Nd 2 Fe 14 B intermetallic compound, and the refractory metal layer 11 is made of, for example, an NbFeB intermetallic compound.
- M- (OR) x (wherein, M is V, Mo, Zr, Ta, Ti, W, or Nb, as described later), R is a substituent composed of hydrocarbon, It may be linear or branched, x is an arbitrary integer.)
- An organic metal compound containing Nb or the like (for example, niobium ethoxide, niobium n-propoxide, niobium n-butoxide, niobium n-hexoxide, etc.) ) Is added to the organic solvent and mixed with the magnet powder in a wet state.
- an organometallic compound containing Nb or the like can be dispersed in an organic solvent, and the organometallic compound containing Nb or the like can be uniformly attached to the surface of the Nd crystal particles 10.
- M- (OR) x (wherein M is V, Mo, Zr, Ta, Ti, W or Nb. R is a substituent composed of hydrocarbon, which may be linear or branched. And x is an arbitrary integer.)
- a metal alkoxide is an organometallic compound that satisfies the structural formula.
- the metal alkoxide is represented by a general formula M (OR) n (M: metal element, R: organic group, n: valence of metal or metalloid).
- metal or semimetal forming the metal alkoxide W, Mo, V, Nb, Ta, Ti, Zr, Ir, Fe, Co, Ni, Cu, Zn, Cd, Al, Ga, In, Ge, Sb, Y, lanthanide, etc. are mentioned.
- a refractory metal is particularly used.
- V, Mo, Zr, Ta, Ti, W or Nb among refractory metals in order to prevent mutual diffusion with the main phase of the magnet during sintering as will be described later.
- alkoxide is not particularly limited, and examples thereof include methoxide, ethoxide, propoxide, isopropoxide, butoxide, alkoxide having 4 or more carbon atoms, and the like.
- those having a low molecular weight are used for the purpose of suppressing residual coal by low-temperature decomposition as described later.
- methoxide having 1 carbon is easily decomposed and difficult to handle, ethoxide, methoxide, isopropoxide, propoxide, butoxide, etc., which are alkoxides having 2 to 6 carbon atoms contained in R, are used. It is preferable.
- M- (OR) x (wherein, M is V, Mo, Zr, Ta, Ti, W, or Nb as an organometallic compound to be added to the magnet powder.
- R is an alkyl group. May be linear or branched, x is an arbitrary integer), and more preferably M- (OR) x (wherein M is V, Mo, Zr, Ta, Ti).
- W or Nb R is any alkyl group having 2 to 6 carbon atoms, which may be linear or branched, and x is an arbitrary integer. desirable.
- the molded body formed by compacting is fired under appropriate firing conditions, it is possible to prevent Nb and the like from diffusing and penetrating (solid solution) into the Nd crystal particles 10.
- Nb etc. can be unevenly distributed only to a grain boundary after sintering.
- the core Nd 2 Fe 14 B intermetallic compound phase occupies a high volume ratio.
- the sintered Nd crystal particles 10 are in a dense state, it is considered that exchange interaction propagates between the Nd crystal particles 10.
- the non-magnetic refractory metal layer 11 coated on the surface of the Nd crystal particles 10 divides the exchange interaction between the Nd crystal particles 10, and each crystal even when a magnetic field is applied from the outside. Prevents magnetization reversal of particles.
- the refractory metal layer 11 coated on the surface of the Nd crystal particles 10 also functions as a means for suppressing so-called grain growth in which the average particle diameter of the Nd crystal particles 10 increases during sintering of the permanent magnet 1. .
- a mechanism for suppressing grain growth of the permanent magnet 1 by the refractory metal layer 11 will be described with reference to FIG.
- FIG. 3 is a schematic diagram showing a magnetic domain structure of a ferromagnetic material.
- a grain boundary which is a discontinuous boundary surface left between a crystal and another crystal, has excessive energy, grain boundary movement that attempts to reduce energy occurs at a high temperature. Therefore, when the magnet raw material is sintered at a high temperature (for example, 800 ° C. to 1150 ° C. for Nd—Fe—B magnets), the small magnet particles shrink and disappear, and the average particle size of the remaining magnet particles increases. So-called grain growth occurs.
- M- (OR) x (wherein M is V, Mo, Zr, Ta, Ti, W, or Nb.
- R is a substituent composed of hydrocarbon, which may be linear or branched.
- x is an arbitrary integer, Nb or the like, which is a refractory metal, is unevenly distributed at the interface of the magnet particles as shown in FIG. And this unevenly distributed refractory metal prevents the movement of grain boundaries generated at high temperatures, and can suppress grain growth.
- the particle diameter D of the Nd crystal particles 10 is 0.2 ⁇ m to 1.2 ⁇ m, preferably about 0.3 ⁇ m. Further, if the thickness d of the refractory metal layer 11 is about 2 nm, the growth of Nd magnet particles during sintering can be suppressed, and the exchange interaction between the Nd crystal particles 10 can be divided. However, if the thickness d of the refractory metal layer 11 becomes too large, the content of non-magnetic components that do not exhibit magnetism increases, so the residual magnetic flux density decreases.
- the refractory metal layer 11 does not need to be a layer composed of only an Nb compound, a V compound, a Mo compound, a Zr compound, a Ta compound, a Ti compound or a W compound (hereinafter referred to as a compound such as Nb). It may be a layer composed of a mixture of a compound and an Nd compound. In that case, a layer made of a mixture of a compound such as Nb and the Nd compound is formed by adding the Nd compound. As a result, liquid phase sintering during the sintering of the Nd magnet powder can be promoted.
- the Nd compounds to be added include NdH 2 , neodymium acetate hydrate, neodymium (III) acetylacetonate trihydrate, neodymium (III) 2-ethylhexanoate, neodymium (III) hexafluoroacetylacetonate Hydrates, neodymium isopropoxide, neodynium (III) phosphate n hydrate, neodymium trifluoroacetylacetonate, neodymium trifluoromethanesulfonate, and the like are desirable.
- FIG. 5 is an explanatory view showing a manufacturing process in the first manufacturing method of the permanent magnet 1 according to the present invention.
- an ingot made of a predetermined fraction of Nd—Fe—B (eg, Nd: 32.7 wt%, Fe (electrolytic iron): 65.96 wt%, B: 1.34 wt%) is manufactured. Thereafter, the ingot is roughly pulverized to a size of about 200 ⁇ m by a stamp mill or a crusher. Alternatively, the ingot is melted, flakes are produced by strip casting, and coarsely pulverized by hydrogen crushing.
- the coarsely pulverized magnet powder is either (a) in an atmosphere made of an inert gas such as nitrogen gas, Ar gas, or He gas having substantially 0% oxygen content, or (b) having an oxygen content of 0.0001.
- the oxygen concentration of substantially 0% is not limited to the case where the oxygen concentration is completely 0%, but may contain oxygen in such an amount that a very small amount of oxide film is formed on the surface of the fine powder. Means good.
- an organometallic compound solution to be added to the fine powder finely pulverized by the jet mill 41 is prepared.
- an organometallic compound containing Nb or the like is added in advance to the organometallic compound solution and dissolved.
- the organometallic compound to be dissolved is M- (OR) x (wherein M is V, Mo, Zr, Ta, Ti, W or Nb, and R is any alkyl group having 2 to 6 carbon atoms).
- x is an arbitrary integer
- niobium ethoxide, niobium n-propoxide, niobium n-butoxide, niobium n-hexoxide, etc. Is desirable.
- the amount of the organometallic compound containing Nb or the like to be dissolved is not particularly limited, but the content of Nb or the like with respect to the magnet after sintering is 0.001 wt% to 10 wt%, preferably 0.01 wt% to 5 wt%. An amount is preferred.
- the organometallic compound solution is added to the fine powder classified by the jet mill 41.
- the slurry 42 in which the fine powder of the magnet raw material and the organometallic compound solution are mixed is generated.
- the addition of the organometallic compound solution is performed in an atmosphere made of an inert gas such as nitrogen gas, Ar gas, or He gas.
- the produced slurry 42 is dried in advance by vacuum drying or the like before molding, and the dried magnet powder 43 is taken out. Thereafter, the dried magnet powder is compacted into a predetermined shape by the molding device 50.
- a dry method in which the dried fine powder is filled into the cavity
- a wet method in which the powder is filled into the cavity after slurrying with a solvent or the like.
- the dry method is used. Illustrate.
- the organometallic compound solution can be volatilized in the firing stage after molding.
- the molding apparatus 50 includes a cylindrical mold 51, a lower punch 52 that slides up and down with respect to the mold 51, and an upper punch 53 that also slides up and down with respect to the mold 51. And a space surrounded by them constitutes the cavity 54.
- the molding apparatus 50 has a pair of magnetic field generating coils 55 and 56 disposed above and below the cavity 54, and applies magnetic field lines to the magnet powder 43 filled in the cavity 54.
- the applied magnetic field is, for example, 1 MA / m.
- the dried magnet powder 43 is filled into the cavity 54. Thereafter, the lower punch 52 and the upper punch 53 are driven, and pressure is applied in the direction of the arrow 61 to the magnetic powder 43 filled in the cavity 54 to perform molding. Simultaneously with the pressurization, a pulse magnetic field is applied to the magnetic powder 43 filled in the cavity 54 by the magnetic field generating coils 55 and 56 in the direction of the arrow 62 parallel to the pressurization direction. Thereby orienting the magnetic field in the desired direction. Note that the direction in which the magnetic field is oriented needs to be determined in consideration of the magnetic field direction required for the permanent magnet 1 formed from the magnet powder 43.
- the slurry when using the wet method, the slurry may be injected while applying a magnetic field to the cavity 54, and wet molding may be performed by applying a magnetic field stronger than the initial magnetic field during or after the injection. Further, the magnetic field generating coils 55 and 56 may be arranged so that the application direction is perpendicular to the pressing direction.
- the compact 71 formed by compacting is held in hydrogen by holding it in a hydrogen atmosphere at 200 ° C. to 900 ° C., more preferably 400 ° C. to 900 ° C. (eg 600 ° C.) for several hours (eg 5 hours).
- the amount of hydrogen supplied during calcination is 5 L / min.
- decarbonization is performed in which the organometallic compound is thermally decomposed to reduce the amount of carbon in the calcined body.
- the calcination treatment in hydrogen is performed under the condition that the carbon content in the calcined body is 0.15 wt% or less, more preferably 0.1 wt% or less. Accordingly, the entire permanent magnet 1 can be densely sintered by the subsequent sintering process, and the residual magnetic flux density and coercive force are not reduced.
- the molded body 71 calcined by the above-described calcining treatment in hydrogen has a problem that NdH 3 exists and is easily combined with oxygen.
- the molded body 71 is preliminarily hydrogenated. Since it moves to the below-mentioned baking without making it contact with external air after baking, a dehydrogenation process becomes unnecessary. During the firing, hydrogen in the molded body is released.
- the sintering process which sinters the molded object 71 calcined by the calcination process in hydrogen is performed.
- a sintering method of the molded body 71 it is also possible to use pressure sintering which sinters in a state where the molded body 71 is pressed in addition to general vacuum sintering.
- the temperature is raised to about 800 ° C. to 1080 ° C. at a predetermined rate of temperature rise and held for about 2 hours. During this time, vacuum firing is performed, but the degree of vacuum is preferably 10 ⁇ 4 Torr or less. Thereafter, it is cooled and heat treated again at 600 ° C. to 1000 ° C. for 2 hours.
- the permanent magnet 1 is manufactured as a result of sintering.
- pressure sintering examples include hot press sintering, hot isostatic pressing (HIP) sintering, ultrahigh pressure synthetic sintering, gas pressure sintering, and discharge plasma (SPS) sintering.
- HIP hot isostatic pressing
- SPS discharge plasma
- the SPS is uniaxial pressure sintering that pressurizes in a uniaxial direction and is sintered by current sintering. Sintering is preferably used.
- FIG. 6 is an explanatory view showing a manufacturing process in the second manufacturing method of the permanent magnet 1 according to the present invention.
- the process until the slurry 42 is generated is the same as the manufacturing process in the first manufacturing method already described with reference to FIG.
- the produced slurry 42 is dried in advance by vacuum drying or the like before molding, and the dried magnet powder 43 is taken out. Thereafter, the dried magnet powder 43 is calcined in hydrogen by holding it in a hydrogen atmosphere at 200 ° C. to 900 ° C., more preferably 400 ° C. to 900 ° C. (eg 600 ° C.) for several hours (eg 5 hours).
- the amount of hydrogen supplied during calcination is 5 L / min.
- decarbonization is performed in which the remaining organometallic compound is thermally decomposed to reduce the amount of carbon in the calcined body.
- the calcination treatment in hydrogen is performed under the condition that the carbon content in the calcined body is 0.15 wt% or less, more preferably 0.1 wt% or less. Accordingly, the entire permanent magnet 1 can be densely sintered by the subsequent sintering process, and the residual magnetic flux density and coercive force are not reduced.
- dehydrogenation treatment is performed by holding the powder-like calcined body 82 calcined by calcination in hydrogen at 200 to 600 ° C., more preferably at 400 to 600 ° C. for 1 to 3 hours in a vacuum atmosphere. I do.
- the degree of vacuum is preferably 0.1 Torr or less.
- FIG. 7 shows the magnet powder with respect to the exposure time when the Nd magnet powder that has been calcined in hydrogen and the Nd magnet powder that has not been calcined in hydrogen are exposed to an atmosphere having an oxygen concentration of 7 ppm and an oxygen concentration of 66 ppm, respectively. It is the figure which showed the amount of oxygen in the inside.
- the oxygen content in the magnet powder increases from 0.4% to 0.8% in about 1000 seconds.
- the powder-like calcined body 82 subjected to the dehydrogenation treatment is compacted into a predetermined shape by the molding apparatus 50.
- the details of the molding apparatus 50 are the same as the manufacturing steps in the first manufacturing method already described with reference to FIG.
- a sintering process for sintering the formed calcined body 82 is performed.
- the sintering process is performed by vacuum sintering, pressure sintering, or the like, as in the first manufacturing method described above. Since the details of the sintering conditions are the same as those in the manufacturing process in the first manufacturing method already described, description thereof will be omitted. And the permanent magnet 1 is manufactured as a result of sintering.
- the first manufacturing method in which the magnet particles after molding are calcined in hydrogen are used.
- the pyrolysis of the organometallic compound can be more easily performed on the entire magnet particle. That is, it becomes possible to more reliably reduce the amount of carbon in the calcined body as compared with the first manufacturing method.
- the molded body 71 moves to firing without being exposed to the outside air after hydrogen calcination, so that a dehydrogenation step is unnecessary. Therefore, the manufacturing process can be simplified as compared with the second manufacturing method.
- the dehydrogenation step is not necessary when the firing is performed without contact with the outside air after the hydrogen calcination.
- Example 1 The alloy composition of the neodymium magnet powder of Example 1 is Nd more than the fraction based on the stoichiometric composition (Nd: 26.7 wt%, Fe (electrolytic iron): 72.3 wt%, B: 1.0 wt%).
- Nd / Fe / B 32.7 / 65.96 / 1.34 at wt%.
- 5 wt% of niobium ethoxide as an organometallic compound was added to the pulverized neodymium magnet powder.
- the calcination treatment was performed by holding the magnet powder before molding at 600 ° C. for 5 hours in a hydrogen atmosphere. The supply amount of hydrogen during calcination is 5 L / min. Further, the sintered calcined body was sintered by SPS sintering. The other steps are the same as those in [Permanent magnet manufacturing method 2] described above.
- Example 2 The organometallic compound to be added was niobium n-propoxide. Other conditions are the same as in the first embodiment.
- Example 3 The organometallic compound to be added was niobium n-butoxide. Other conditions are the same as in the first embodiment.
- Example 4 The organometallic compound to be added was niobium n-hexoxide. Other conditions are the same as in the first embodiment.
- Example 5 The molded calcined body was sintered by vacuum sintering instead of SPS sintering. Other conditions are the same as in the first embodiment.
- FIG. 8 is a graph showing the carbon content [wt%] in the permanent magnets of Examples 1 to 4 and Comparative Examples 1 and 2. As shown in FIG. 8, it can be seen that Examples 1 to 4 can greatly reduce the amount of carbon remaining in the magnet particles as compared with Comparative Examples 1 and 2. In particular, in Examples 1 to 4, the amount of carbon remaining in the magnet particles can be 0.15 wt% or less, and in Examples 2 to 4, the amount of carbon remaining in the magnet particles is 0.1 wt% or less. can do.
- Example 1 and Comparative Example 1 when the same organometallic compound is added, when the calcination treatment in hydrogen is performed, the calcination treatment in hydrogen is not performed.
- the amount of carbon in the magnet particles can be greatly reduced. That is, it can be seen that so-called decarbonization can be carried out by reducing the amount of carbon in the calcined body by thermally decomposing the organometallic compound by calcination in hydrogen. As a result, it is possible to prevent dense sintering of the entire magnet and a decrease in coercive force.
- M- (OR) x (wherein M is V, Mo, Zr, Ta, Ti, W or Nb. R is a hydrocarbon) A substituent, which may be linear or branched. X is an arbitrary integer.)
- an organometallic compound represented by (2) is added, the magnet is compared with the case where another organometallic compound is added. It can be seen that the amount of carbon in the particles can be greatly reduced. That is, the organometallic compound to be added is M- (OR) x (wherein M is V, Mo, Zr, Ta, Ti, W or Nb. R is a substituent composed of hydrocarbon, However, it may be branched.
- organometallic compound represented by (2) it is understood that decarbonization can be easily performed in the calcination treatment in hydrogen. As a result, it is possible to prevent dense sintering of the entire magnet and a decrease in coercive force. Further, when an organometallic compound composed of an alkyl group, more preferably an organometallic compound composed of an alkyl group having 2 to 6 carbon atoms, is used as the organometallic compound to be added, the magnet powder is calcined in a hydrogen atmosphere. In this case, it becomes possible to perform thermal decomposition of the organometallic compound at a low temperature. Thereby, the thermal decomposition of the organometallic compound can be more easily performed on the entire magnet particle.
- FIG. 9 is a diagram showing an SEM photograph after sintering of the permanent magnet of Example 1 and the elemental analysis results of the grain boundary phase.
- FIG. 10 is a diagram showing an SEM photograph after sintering of the permanent magnet of Example 2 and the elemental analysis results of the grain boundary phase.
- FIG. 11 is a diagram in which the distribution state of the Nb element is mapped in the same field of view as the SEM photograph after sintering of the permanent magnet of Example 2 and the SEM photograph.
- FIG. 9 is a diagram showing an SEM photograph after sintering of the permanent magnet of Example 1 and the elemental analysis results of the grain boundary phase.
- FIG. 10 is a diagram showing an SEM photograph after sintering of the permanent magnet of Example 2 and the elemental analysis results of the grain boundary phase.
- FIG. 11 is a diagram in which the distribution state of the Nb element is mapped in the same field of view as the SEM photograph after sintering of the permanent magnet of Example 2 and the SEM photograph.
- FIG. 12 is a view showing an SEM photograph after sintering of the permanent magnet of Example 3 and the elemental analysis results of the grain boundary phase.
- FIG. 13 is a diagram in which the Nb element distribution state is mapped in the same field of view as the SEM photograph and the SEM photograph after sintering of the permanent magnet of Example 3.
- FIG. 14 is an SEM photograph after sintering of the permanent magnet of Example 4 and the results of elemental analysis of the grain boundary phase.
- FIG. 15 is a diagram in which the Nb element distribution state is mapped in the same field of view as the SEM photograph after sintering of the permanent magnet of Example 4 and the SEM photograph. As shown in FIGS.
- Nb is detected from the grain boundary phase. That is, in the permanent magnets of Examples 1 to 4, it can be seen that in the grain boundary phase, a phase of NbFe-based intermetallic compound in which a part of Nd is substituted with Nb is generated on the surface of the main phase particle.
- the white portion indicates the distribution of the Nb element.
- the white portion of the mapping diagram (that is, the Nb element) is unevenly distributed around the main phase. That is, it can be seen that in the permanent magnet of Example 2, Nb is not diffused from the grain boundary phase to the main phase, and Nb is unevenly distributed at the grain boundaries of the magnet.
- the white portion indicates the distribution of the Nb element. Referring to the SEM photograph and mapping diagram of FIG. 13, the white portion (that is, Nb element) of the mapping diagram is unevenly distributed around the main phase.
- the white portion indicates the distribution of the Nb element. Referring to the SEM photograph and mapping diagram of FIG. 15, the white portion (that is, Nb element) of the mapping diagram is unevenly distributed around the main phase. That is, it can be seen that in the permanent magnet of Example 4, Nb is not diffused from the grain boundary phase to the main phase, and Nb is unevenly distributed at the grain boundaries of the magnet.
- FIG. 16 is a view showing an SEM photograph after sintering of the permanent magnet of Comparative Example 1.
- FIG. 17 is a view showing an SEM photograph after sintering of the permanent magnet of Comparative Example 2.
- a sintered permanent magnet is formed from a main phase (Nd 2 Fe 14 B) 91 of a neodymium magnet and a grain boundary phase 92 that looks like white spots.
- ⁇ Fe phase is also formed.
- Comparative Example 2 where the amount of residual carbon is larger than in Examples 1 to 4 and Comparative Example 1, in addition to the main phase 91 and the grain boundary phase 92, a large number of ⁇ Fe phases 93 that appear as black bands are formed. .
- ⁇ Fe is generated by carbide remaining during sintering. That is, since the reactivity between Nd and C is very high, if the C-containing material in the organometallic compound remains at a high temperature in the sintering process as in Comparative Example 2, carbide is formed. As a result, ⁇ Fe is precipitated in the main phase of the sintered magnet by the formed carbide, and the magnetic properties are greatly deteriorated.
- Examples 1 to 4 by using an appropriate organometallic compound as described above and carrying out a calcination treatment in hydrogen, the organometallic compound is thermally decomposed, and the contained carbon is burnt out in advance (the amount of carbon is reduced). Reduced).
- the contained carbon can be burned out more than necessary, and the carbon remaining in the magnet after sintering.
- the amount can be 0.15 wt% or less, more preferably 0.1 wt% or less.
- the organometallic compound to be added preferably has a low molecular weight (for example, one composed of an alkyl group having 2 to 6 carbon atoms). Used.
- FIG. 18 is a graph showing the carbon amount [wt%] in a plurality of permanent magnets manufactured by changing the calcination temperature conditions for the permanent magnets of Example 5 and Comparative Examples 3 and 4.
- FIG. 18 shows the result of maintaining the supply amounts of hydrogen and helium during calcination at 1 L / min for 3 hours. As shown in FIG. 18, it can be seen that the amount of carbon in the magnet particles can be greatly reduced when calcined in a hydrogen atmosphere as compared with calcining in a He atmosphere or a vacuum atmosphere. Also, from FIG.
- the carbon content is greatly reduced if the calcining temperature at the time of calcining the magnet powder in a hydrogen atmosphere is increased, and in particular, the carbon content is 0.15 wt. It can be seen that it is possible to make the value less than or equal to%.
- M- (OR) x (where M is V, Mo) with respect to the fine powder of the pulverized neodymium magnet.
- the organometallic compound solution is added to uniformly adhere the organometallic compound to the surface of the neodymium magnet particles. Thereafter, the green compact is subjected to a calcining treatment in hydrogen by holding it in a hydrogen atmosphere at 200 ° C. to 900 ° C. for several hours.
- the permanent magnet 1 is manufactured by performing vacuum sintering or pressure sintering.
- the added Nb or the like can be efficiently distributed on the grain boundaries of the magnet.
- the grain growth of magnet particles during sintering can be suppressed, and after sintering, the exchange interaction between the crystal particles is interrupted to prevent the magnetization reversal of each crystal particle and improve the magnetic performance. It becomes possible to make it.
- decarbonization can be easily performed as compared with the case where other organometallic compounds are added, and there is no possibility that the coercive force is reduced by the carbon contained in the sintered magnet. The whole can be sintered precisely.
- Nb or the like which is a high melting point metal
- Nb or the like which is a high melting point metal
- Nb or the like that is unevenly distributed at the grain boundaries suppresses the grain growth of the magnet particles during sintering, and the crystals after sintering By breaking the exchange interaction between particles, it is possible to prevent the magnetization reversal of each crystal particle and improve the magnetic performance.
- the addition amount of Nb etc. is small compared with the past, the fall of a residual magnetic flux density can be suppressed.
- a magnet to which an organometallic compound is added is calcined in a hydrogen atmosphere before sintering, so that the organometallic compound is thermally decomposed and carbon contained in the magnet particles is preliminarily burned out (the amount of carbon is reduced).
- the carbide is hardly formed in the sintering process.
- a large number of ⁇ Fe is not precipitated in the main phase of the magnet after sintering, and the magnet characteristics are not greatly deteriorated.
- the magnet powder or molded body can be produced in a hydrogen atmosphere.
- the thermal decomposition of the organometallic compound can be more easily performed on the entire magnet powder or the entire compact.
- the step of calcining the magnet powder or the molded body is performed by holding the molded body for a predetermined time in a temperature range of 200 ° C. to 900 ° C., more preferably 400 ° C.
- the amount of carbon remaining in the magnet after sintering is 0.15 wt% or less, more preferably 0.1 wt% or less, so that no voids are formed between the main phase of the magnet and the grain boundary phase, and It becomes possible to make the whole magnet into a densely sintered state, and it is possible to prevent the residual magnetic flux density from being lowered. Further, a large number of ⁇ Fe is not precipitated in the main phase of the magnet after sintering, and the magnet characteristics are not greatly deteriorated.
- the pyrolysis of the organometallic compound is performed in comparison with the case of calcining the molded magnet particles. This can be done more easily for the whole particle. That is, the amount of carbon in the calcined body can be reduced more reliably. Further, by performing the dehydrogenation treatment after the calcination treatment, the activity of the calcined body activated by the calcination treatment can be reduced. As a result, the magnet particles are prevented from being combined with oxygen thereafter, and the residual magnetic flux density and coercive force are not reduced. In addition, since the step of performing the dehydrogenation process is performed by holding the magnet powder in a temperature range of 200 ° C.
- NdH 3 having high activity is contained in the Nd-based magnet that has been subjected to the hydrogen calcining process. Even if is generated, it is possible to shift to NdH 2 having low activity without leaving any.
- this invention is not limited to the said Example, Of course, various improvement and deformation
- the pulverization conditions, kneading conditions, calcination conditions, dehydrogenation conditions, sintering conditions, etc. of the magnet powder are not limited to the conditions described in the above examples.
- niobium ethoxide, niobium n-propoxide, niobium n-butoxide and niobium n-hexoxide are used as organometallic compounds containing Nb and the like added to the magnet powder, but M- ( OR) x (wherein M is V, Mo, Zr, Ta, Ti, W or Nb. R is a substituent composed of hydrocarbon, which may be linear or branched. X is an arbitrary integer.
- organometallic compounds may be used as long as they are organometallic compounds represented by For example, an organometallic compound composed of an alkyl group having 7 or more carbon atoms or an organometallic compound composed of a substituent composed of a hydrocarbon other than an alkyl group may be used.
Landscapes
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Power Engineering (AREA)
- Mechanical Engineering (AREA)
- Metallurgy (AREA)
- Materials Engineering (AREA)
- Organic Chemistry (AREA)
- Manufacturing & Machinery (AREA)
- Crystallography & Structural Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Hard Magnetic Materials (AREA)
- Powder Metallurgy (AREA)
- Manufacturing Cores, Coils, And Magnets (AREA)
Abstract
Description
先ず、本発明に係る永久磁石1の構成について説明する。図1は本発明に係る永久磁石1を示した全体図である。尚、図1に示す永久磁石1は円柱形状を備えるが、永久磁石1の形状は成形に用いるキャビティの形状によって変化する。
本発明に係る永久磁石1としては例えばNd-Fe-B系磁石を用いる。また、永久磁石1を形成する各結晶粒子の界面(粒界)には、永久磁石1の保磁力を高める為のNb(ニオブ)、V(バナジウム)、Mo(モリブデン)、Zr(ジルコニウム)、Ta(タンタル)、Ti(チタン)又はW(タングステン)が偏在する。尚、各成分の含有量はNd:25~37wt%、Nb、V、Mo、Zr、Ta、Ti、Wのいずれか(以下、Nb等という):0.01~5wt%、B:1~2wt%、Fe(電解鉄):60~75wt%とする。また、磁気特性向上の為、Co、Cu、Al、Si等の他元素を少量含んでも良い。
次に、本発明に係る永久磁石1の第1の製造方法について図5を用いて説明する。図5は本発明に係る永久磁石1の第1の製造方法における製造工程を示した説明図である。
また、成形装置50には一対の磁界発生コイル55、56がキャビティ54の上下位置に配置されており、磁力線をキャビティ54に充填された磁石粉末43に印加する。印加させる磁場は例えば1MA/mとする。
また、湿式法を用いる場合には、キャビティ54に磁場を印加しながらスラリーを注入し、注入途中又は注入終了後に、当初の磁場より強い磁場を印加して湿式成形しても良い。また、加圧方向に対して印加方向が垂直となるように磁界発生コイル55、56を配置しても良い。
次に、本発明に係る永久磁石1の他の製造方法である第2の製造方法について図6を用いて説明する。図6は本発明に係る永久磁石1の第2の製造方法における製造工程を示した説明図である。
図7は水素中仮焼処理をしたNd磁石粉末と水素中仮焼処理をしていないNd磁石粉末とを、酸素濃度7ppm及び酸素濃度66ppmの雰囲気にそれぞれ暴露した際に、暴露時間に対する磁石粉末内の酸素量を示した図である。図7に示すように水素中仮焼処理した磁石粉末は、高酸素濃度66ppm雰囲気におかれると、約1000secで磁石粉末内の酸素量が0.4%から0.8%まで上昇する。また、低酸素濃度7ppm雰囲気におかれても、約5000secで磁石粉末内の酸素量が0.4%から同じく0.8%まで上昇する。そして、Nd磁石粒子が酸素と結び付くと、残留磁束密度や保磁力の低下の原因となる。
そこで、上記脱水素処理では、水素中仮焼処理によって生成された仮焼体82中のNdH3(活性度大)を、NdH3(活性度大)→NdH2(活性度小)へと段階的に変化させることによって、水素仮焼中処理により活性化された仮焼体82の活性度を低下させる。それによって、水素中仮焼処理によって仮焼された仮焼体82をその後に大気中へと移動させた場合であっても、Nd磁石粒子が酸素と結び付くことを防止し、残留磁束密度や保磁力を低下させることが無い。
一方、第1の製造方法では、成形体71は水素仮焼後に外気と触れさせることなく焼成に移るため、脱水素工程は不要となる。従って、前記第2の製造方法と比較して製造工程を簡略化することが可能となる。但し、前記第2の製造方法においても、水素仮焼後に外気と触れさせることがなく焼成を行う場合には、脱水素工程は不要となる。
(実施例1)
実施例1のネオジム磁石粉末の合金組成は、化学量論組成に基づく分率(Nd:26.7wt%、Fe(電解鉄):72.3wt%、B:1.0wt%)よりもNdの比率を高くし、例えばwt%でNd/Fe/B=32.7/65.96/1.34とする。また、粉砕したネオジム磁石粉末に有機金属化合物としてニオブエトキシドを5wt%添加した。また、仮焼処理は、成形前の磁石粉末を水素雰囲気において600℃で5時間保持することにより行った。そして、仮焼中の水素の供給量は5L/minとする。また、成形された仮焼体の焼結はSPS焼結により行った。尚、他の工程は上述した[永久磁石の製造方法2]と同様の工程とする。
添加する有機金属化合物をニオブn-プロポキシドとした。他の条件は実施例1と同様である。
添加する有機金属化合物をニオブn-ブトキシドとした。他の条件は実施例1と同様である。
添加する有機金属化合物をニオブn-ヘキソキシドとした。他の条件は実施例1と同様である。
成形された仮焼体の焼結をSPS焼結の代わりに真空焼結により行った。他の条件は実施例1と同様である。
添加する有機金属化合物をニオブエトキシドとし、水素中仮焼処理を行わずに焼結した。他の条件は実施例1と同様である。
添加する有機金属化合物をジルコニウムヘキサフルオロアセチルアセトナートとした。他の条件は実施例1と同様である。
仮焼処理を水素雰囲気ではなくHe雰囲気で行った。また、成形された仮焼体の焼結をSPS焼結の代わりに真空焼結により行った。他の条件は実施例1と同様である。
仮焼処理を水素雰囲気ではなく真空雰囲気で行った。また、成形された仮焼体の焼結をSPS焼結の代わりに真空焼結により行った。他の条件は実施例1と同様である。
図8は実施例1~4と比較例1、2の永久磁石の永久磁石中の残存炭素量[wt%]をそれぞれ示した図である。
図8に示すように、実施例1~4は比較例1、2と比較して磁石粒子中に残存する炭素量を大きく低減させることができることが分かる。特に、実施例1~4では、磁石粒子中に残存する炭素量を0.15wt%以下にでき、更に、実施例2~4では、磁石粒子中に残存する炭素量を0.1wt%以下とすることができる。
実施例1~4の永久磁石についてXMAによる表面分析を行った。図9は実施例1の永久磁石の焼結後のSEM写真及び粒界相の元素分析結果を示した図である。図10は実施例2の永久磁石の焼結後のSEM写真及び粒界相の元素分析結果を示した図である。図11は実施例2の永久磁石の焼結後のSEM写真及びSEM写真と同一視野でNb元素の分布状態をマッピングした図である。図12は実施例3の永久磁石の焼結後のSEM写真及び粒界相の元素分析結果を示した図である。図13は実施例3の永久磁石の焼結後のSEM写真及びSEM写真と同一視野でNb元素の分布状態をマッピングした図である。図14は実施例4の永久磁石の焼結後のSEM写真及び粒界相の元素分析結果を示した図である。図15は実施例4の永久磁石の焼結後のSEM写真及びSEM写真と同一視野でNb元素の分布状態をマッピングした図である。
図9、図10、図12、図14に示すように実施例1~4の各永久磁石では、粒界相からNbが検出されている。即ち、実施例1~4の永久磁石では、粒界相において、Ndの一部をNbで置換したNbFe系金属間化合物の相が主相粒子の表面に生成されていることが分かる。
以上の結果から、実施例1~4では、粒界相から主相へとNbが拡散しておらず、また、磁石の粒界にNbを偏在させることができていることが分かる。そして、焼結の際にNbが主相に固溶しないので、固相焼結により粒成長を抑制することが可能となる。
図16は比較例1の永久磁石の焼結後のSEM写真を示した図である。図17は比較例2の永久磁石の焼結後のSEM写真を示した図である。
また、実施例1~4と比較例1、2の各SEM写真を比較すると、残留炭素量が一定量以下(例えば0.2wt%以下)である実施例1~4や比較例1では、基本的にネオジム磁石の主相(Nd2Fe14B)91と白い斑点状に見える粒界相92から焼結後の永久磁石が形成されている。また、少量ではあるがαFe相についても形成されている。それに対して、実施例1~4や比較例1に比べて残留炭素量が多い比較例2は、主相91や粒界相92に加えて黒色帯状に見えるαFe相93が多数形成されている。ここで、αFeは焼結時において残留しているカーバイドによって生じるものである。即ち、NdとCとの反応性が非常に高いため、比較例2のように焼結工程において高温まで有機金属化合物中のC含有物が残ると、カーバイドを形成する。その結果、形成されたカーバイドによって焼結後の磁石の主相内にαFeが析出し、磁石特性を大きく低下させることとなる。
図18は実施例5と比較例3、4の永久磁石について、仮焼温度の条件を変更して製造した複数の永久磁石中の炭素量[wt%]を示した図である。尚、図18では仮焼中の水素及びヘリウムの供給量を1L/minとし、3時間保持した結果を示す。
図18に示すように、He雰囲気や真空雰囲気で仮焼した場合と比較して、水素雰囲気で仮焼した場合には磁石粒子中の炭素量をより大きく低減させることができることが分かる。また、図18からは、磁石粉末を水素雰囲気で仮焼する際の仮焼温度を高温にすれば炭素量がより大きく低減し、特に400℃~900℃とすることによって炭素量を0.15wt%以下とすることが可能であることが分かる。
更に、高融点金属であるNb等が焼結後に磁石の粒界に偏在するので、粒界に偏在されたNb等が焼結時の磁石粒子の粒成長を抑制するとともに、焼結後は結晶粒子間での交換相互作用を分断することによって各結晶粒子の磁化反転を妨げ、磁気性能を向上させることが可能となる。また、Nb等の添加量が従来に比べて少ないので、残留磁束密度の低下を抑制することができる。
また、有機金属化合物が添加された磁石を、焼結前に水素雰囲気で仮焼することにより、有機金属化合物を熱分解させて磁石粒子中に含有する炭素を予め焼失(炭素量を低減)させることができ、焼結工程でカーバイドがほとんど形成されることがない。その結果、焼結後の磁石の主相と粒界相との間に空隙を生じさせることなく、また、磁石全体を緻密に焼結することが可能となり、保磁力が低下することを防止できる。また、焼結後の磁石の主相内にαFeが多数析出することなく、磁石特性を大きく低下させることがない。
また、特に添加する有機金属化合物としてアルキル基から構成される有機金属化合物、より好ましくは炭素数2~6のアルキル基から構成される有機金属化合物を用いれば、水素雰囲気で磁石粉末や成形体を仮焼する際に、低温で有機金属化合物の熱分解を行うことが可能となる。それによって、有機金属化合物の熱分解を磁石粉末全体や成形体全体に対してより容易に行うことができる。
更に、磁石粉末や成形体を仮焼する工程は、特に200℃~900℃、より好ましくは400℃~900℃の温度範囲で成形体を所定時間保持することにより行うので、磁石粒子中に含有する炭素を必要量以上焼失させることができる。
その結果、焼結後に磁石に残存する炭素量が0.15wt%以下、より好ましくは0.1wt%以下となるので、磁石の主相と粒界相との間に空隙が生じることなく、また、磁石全体を緻密に焼結した状態とすることが可能となり、残留磁束密度が低下することを防止できる。また、焼結後の磁石の主相内にαFeが多数析出することなく、磁石特性を大きく低下させることがない。
また、特に第2の製造方法では、粉末状の磁石粒子に対して仮焼を行うので、成形後の磁石粒子に対して仮焼を行う場合と比較して、有機金属化合物の熱分解を磁石粒子全体に対してより容易に行うことができる。即ち、仮焼体中の炭素量をより確実に低減させることが可能となる。また、仮焼処理後に脱水素処理を行うことによって、仮焼処理により活性化された仮焼体の活性度を低下させることができる。それにより、その後に磁石粒子が酸素と結び付くことを防止し、残留磁束密度や保磁力を低下させることが無い。
また、脱水素処理を行う工程は、200℃~600℃の温度範囲で磁石粉末を所定時間保持することにより行うので、水素仮焼中処理を行ったNd系磁石中に活性度の高いNdH3が生成された場合であっても、残さずに活性度の低いNdH2へと移行させることが可能となる。
また、磁石粉末の粉砕条件、混練条件、仮焼条件、脱水素条件、焼結条件などは上記実施例に記載した条件に限られるものではない。
10 Nd結晶粒子
11 高融点金属層
12 高融点金属粒
91 主相
92 粒界相
93 αFe相
Claims (12)
- 磁石原料を磁石粉末に粉砕する工程と、
前記粉砕された磁石粉末に以下の構造式
M-(OR)x
(式中、MはV、Mo、Zr、Ta、Ti、W又はNbである。Rは炭化水素からなる置換基であり、直鎖でも分枝でも良い。xは任意の整数である。)
で表わされる有機金属化合物を添加することにより、前記磁石粉末の粒子表面に前記有機金属化合物を付着させる工程と、
前記有機金属化合物が粒子表面に付着された前記磁石粉末を水素雰囲気で仮焼して仮焼体を得る工程と、
前記仮焼体を真空雰囲気で加熱することにより脱水素処理を行う工程と、
前記脱水素処理された前記仮焼体を成形することにより成形体を形成する工程と、
前記成形体を焼結する工程と、
により製造されることを特徴とする永久磁石。 - 前記有機金属化合物を形成する金属が、焼結後に前記永久磁石の粒界に偏在していることを特徴とする請求項1に記載の永久磁石。
- 前記構造式中のRは、アルキル基であることを特徴とする請求項1又は請求項2に記載の永久磁石。
- 前記構造式中のRは、炭素数2~6のアルキル基のいずれかであることを特徴とする請求項3に記載の永久磁石。
- 焼結後に残存する炭素量が0.15wt%以下であることを特徴とする請求項1乃至請求項4のいずれかに記載の永久磁石。
- 前記磁石粉末を仮焼する工程は、200℃~900℃の温度範囲で前記磁石粉末を所定時間保持することを特徴とする請求項1乃至請求項5のいずれかに記載の永久磁石。
- 前記脱水素処理を行う工程は、真空雰囲気において200℃~600℃の温度範囲で前記磁石粉末を所定時間保持することを特徴とする請求項1乃至請求項6のいずれかに記載の永久磁石。
- 磁石原料を磁石粉末に粉砕する工程と、
前記粉砕された磁石粉末に以下の構造式
M-(OR)x
(式中、MはV、Mo、Zr、Ta、Ti、W又はNbである。Rは炭化水素からなる置換基であり、直鎖でも分枝でも良い。xは任意の整数である。)
で表わされる有機金属化合物を添加することにより、前記磁石粉末の粒子表面に前記有機金属化合物を付着させる工程と、
前記有機金属化合物が粒子表面に付着された前記磁石粉末を水素雰囲気で仮焼して仮焼体を得る工程と、
前記仮焼体を減圧雰囲気で加熱することにより脱水素処理を行う工程と、
前記脱水素処理された前記仮焼体を成形することにより成形体を形成する工程と、
前記成形体を焼結する工程と、
を有することを特徴とする永久磁石の製造方法。 - 前記構造式中のRは、アルキル基であることを特徴とする請求項8に記載の永久磁石の製造方法。
- 前記構造式中のRは、炭素数2~6のアルキル基のいずれかであることを特徴とする請求項9に記載の永久磁石の製造方法。
- 前記磁石粉末を仮焼する工程は、200℃~900℃の温度範囲で前記磁石粉末を所定時間保持することを特徴とする請求項8乃至請求項10のいずれかに記載の永久磁石の製造方法。
- 前記脱水素処理を行う工程は、真空雰囲気において200℃~600℃の温度範囲で前記磁石粉末を所定時間保持することを特徴とする請求項8乃至請求項11のいずれかに記載の永久磁石の製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11765487.1A EP2503568B1 (en) | 2010-03-31 | 2011-03-28 | Manufacturing method for permanent magnet |
| US13/499,560 US9005374B2 (en) | 2010-03-31 | 2011-03-28 | Permanent magnet and manufacturing method thereof |
| CN2011800039580A CN102549684A (zh) | 2010-03-31 | 2011-03-28 | 永久磁铁及永久磁铁的制造方法 |
| KR1020127007165A KR101189960B1 (ko) | 2010-03-31 | 2011-03-28 | 영구 자석 및 영구 자석의 제조 방법 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010084474 | 2010-03-31 | ||
| JP2010-084474 | 2010-03-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011125587A1 true WO2011125587A1 (ja) | 2011-10-13 |
Family
ID=44762536
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/057568 WO2011125587A1 (ja) | 2010-03-31 | 2011-03-28 | 永久磁石及び永久磁石の製造方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US9005374B2 (ja) |
| EP (1) | EP2503568B1 (ja) |
| JP (1) | JP4865100B2 (ja) |
| KR (1) | KR101189960B1 (ja) |
| CN (1) | CN102549684A (ja) |
| TW (1) | TW201212064A (ja) |
| WO (1) | WO2011125587A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110643989A (zh) * | 2019-09-30 | 2020-01-03 | 烟台正海磁性材料股份有限公司 | 一种钕铁硼磁体表面防腐处理方法 |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102576603B (zh) * | 2010-03-31 | 2014-04-16 | 日东电工株式会社 | 永久磁铁及永久磁铁的制造方法 |
| US8500921B2 (en) * | 2010-03-31 | 2013-08-06 | Nitto Denko Corporation | Permanent magnet and manufacturing method thereof |
| EP2503570B1 (en) * | 2010-03-31 | 2015-01-21 | Nitto Denko Corporation | Manufacturing method for permanent magnet |
| EP2503566B1 (en) * | 2010-03-31 | 2015-01-21 | Nitto Denko Corporation | Manufacturing method for permanent magnet |
| JP5011420B2 (ja) * | 2010-05-14 | 2012-08-29 | 日東電工株式会社 | 永久磁石及び永久磁石の製造方法 |
| JP5908247B2 (ja) * | 2011-09-30 | 2016-04-26 | 日東電工株式会社 | 永久磁石の製造方法 |
| JP6147505B2 (ja) * | 2012-03-12 | 2017-06-14 | 日東電工株式会社 | 希土類永久磁石の製造方法 |
| JP2015135935A (ja) * | 2013-03-28 | 2015-07-27 | Tdk株式会社 | 希土類磁石 |
| CN103215467B (zh) * | 2013-05-05 | 2015-07-08 | 沈阳中北真空磁电科技有限公司 | 一种高性能钕铁硼稀土永磁材料的制造方法 |
| JP6914617B2 (ja) * | 2016-05-11 | 2021-08-04 | Tdk株式会社 | 積層コイル部品 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH07211570A (ja) * | 1994-01-25 | 1995-08-11 | Tokin Corp | 希土類永久磁石の製造方法 |
| JPH1064746A (ja) * | 1996-08-23 | 1998-03-06 | Sumitomo Special Metals Co Ltd | 薄肉R−Fe−B系焼結磁石の製造方法 |
| JPH10214711A (ja) * | 1997-01-31 | 1998-08-11 | Isuzu Motors Ltd | 希土類系永久磁石の固化方法 |
| JP3298219B2 (ja) | 1993-03-17 | 2002-07-02 | 日立金属株式会社 | 希土類―Fe−Co−Al−V−Ga−B系焼結磁石 |
| JP2004250781A (ja) | 2002-10-08 | 2004-09-09 | Neomax Co Ltd | 焼結型永久磁石およびその製造方法 |
| JP2009259956A (ja) * | 2008-04-15 | 2009-11-05 | Nitto Denko Corp | 永久磁石及び永久磁石の製造方法 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0304054B1 (en) * | 1987-08-19 | 1994-06-08 | Mitsubishi Materials Corporation | Rare earth-iron-boron magnet powder and process of producing same |
| JP4525072B2 (ja) * | 2003-12-22 | 2010-08-18 | 日産自動車株式会社 | 希土類磁石およびその製造方法 |
| EP2506274B1 (en) * | 2010-03-31 | 2014-07-02 | Nitto Denko Corporation | Manufacturing method for permanent magnet |
| JP5011420B2 (ja) * | 2010-05-14 | 2012-08-29 | 日東電工株式会社 | 永久磁石及び永久磁石の製造方法 |
-
2011
- 2011-03-28 WO PCT/JP2011/057568 patent/WO2011125587A1/ja active Application Filing
- 2011-03-28 CN CN2011800039580A patent/CN102549684A/zh active Pending
- 2011-03-28 JP JP2011069076A patent/JP4865100B2/ja not_active Expired - Fee Related
- 2011-03-28 US US13/499,560 patent/US9005374B2/en not_active Expired - Fee Related
- 2011-03-28 EP EP11765487.1A patent/EP2503568B1/en not_active Not-in-force
- 2011-03-28 KR KR1020127007165A patent/KR101189960B1/ko not_active IP Right Cessation
- 2011-03-30 TW TW100111107A patent/TW201212064A/zh not_active IP Right Cessation
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3298219B2 (ja) | 1993-03-17 | 2002-07-02 | 日立金属株式会社 | 希土類―Fe−Co−Al−V−Ga−B系焼結磁石 |
| JPH07211570A (ja) * | 1994-01-25 | 1995-08-11 | Tokin Corp | 希土類永久磁石の製造方法 |
| JPH1064746A (ja) * | 1996-08-23 | 1998-03-06 | Sumitomo Special Metals Co Ltd | 薄肉R−Fe−B系焼結磁石の製造方法 |
| JPH10214711A (ja) * | 1997-01-31 | 1998-08-11 | Isuzu Motors Ltd | 希土類系永久磁石の固化方法 |
| JP2004250781A (ja) | 2002-10-08 | 2004-09-09 | Neomax Co Ltd | 焼結型永久磁石およびその製造方法 |
| JP2009259956A (ja) * | 2008-04-15 | 2009-11-05 | Nitto Denko Corp | 永久磁石及び永久磁石の製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2503568A4 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110643989A (zh) * | 2019-09-30 | 2020-01-03 | 烟台正海磁性材料股份有限公司 | 一种钕铁硼磁体表面防腐处理方法 |
| CN110643989B (zh) * | 2019-09-30 | 2023-01-10 | 烟台正海磁性材料股份有限公司 | 一种钕铁硼磁体表面防腐处理方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| TWI369701B (ja) | 2012-08-01 |
| TW201212064A (en) | 2012-03-16 |
| EP2503568A4 (en) | 2013-04-03 |
| US9005374B2 (en) | 2015-04-14 |
| KR101189960B1 (ko) | 2012-10-15 |
| KR20120048687A (ko) | 2012-05-15 |
| JP2011228668A (ja) | 2011-11-10 |
| EP2503568A1 (en) | 2012-09-26 |
| EP2503568B1 (en) | 2014-06-11 |
| JP4865100B2 (ja) | 2012-02-01 |
| CN102549684A (zh) | 2012-07-04 |
| US20120187327A1 (en) | 2012-07-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4865100B2 (ja) | 永久磁石及び永久磁石の製造方法 | |
| JP4923148B2 (ja) | 永久磁石及び永久磁石の製造方法 | |
| JP4865098B2 (ja) | 永久磁石及び永久磁石の製造方法 | |
| JP4923153B2 (ja) | 永久磁石及び永久磁石の製造方法 | |
| JP4923152B2 (ja) | 永久磁石及び永久磁石の製造方法 | |
| JP4865920B2 (ja) | 永久磁石及び永久磁石の製造方法 | |
| JP4923151B2 (ja) | 永久磁石及び永久磁石の製造方法 | |
| JP4923147B2 (ja) | 永久磁石及び永久磁石の製造方法 | |
| JP4865097B2 (ja) | 永久磁石及び永久磁石の製造方法 | |
| JP4865099B2 (ja) | 永久磁石及び永久磁石の製造方法 | |
| JP5908247B2 (ja) | 永久磁石の製造方法 | |
| JP4923149B2 (ja) | 永久磁石及び永久磁石の製造方法 | |
| JP4923150B2 (ja) | 永久磁石及び永久磁石の製造方法 | |
| JP5501830B2 (ja) | 永久磁石及び永久磁石の製造方法 | |
| JP5453154B2 (ja) | 永久磁石及び永久磁石の製造方法 | |
| JP2011216732A (ja) | 永久磁石及び永久磁石の製造方法 | |
| JP2011216724A (ja) | 永久磁石及び永久磁石の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180003958.0 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11765487 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20127007165 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2861/CHENP/2012 Country of ref document: IN Ref document number: 2011765487 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13499560 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |