WO2010110409A1 - ピロンおよびピリドン誘導体の製造方法 - Google Patents
ピロンおよびピリドン誘導体の製造方法 Download PDFInfo
- Publication number
- WO2010110409A1 WO2010110409A1 PCT/JP2010/055316 JP2010055316W WO2010110409A1 WO 2010110409 A1 WO2010110409 A1 WO 2010110409A1 JP 2010055316 W JP2010055316 W JP 2010055316W WO 2010110409 A1 WO2010110409 A1 WO 2010110409A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituent group
- formula
- optionally substituted
- group
- compound
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 69
- 230000008569 process Effects 0.000 title claims abstract description 30
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical class OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 title abstract description 23
- ZPSJGADGUYYRKE-UHFFFAOYSA-N 2H-pyran-2-one Chemical compound O=C1C=CC=CO1 ZPSJGADGUYYRKE-UHFFFAOYSA-N 0.000 title abstract description 12
- 150000001875 compounds Chemical class 0.000 claims description 418
- 125000001424 substituent group Chemical group 0.000 claims description 305
- 239000000203 mixture Substances 0.000 claims description 115
- 125000000217 alkyl group Chemical group 0.000 claims description 113
- 125000002837 carbocyclic group Chemical group 0.000 claims description 94
- 125000000623 heterocyclic group Chemical group 0.000 claims description 91
- -1 cyano, hydroxy, carboxy, formyl Chemical group 0.000 claims description 88
- 125000003545 alkoxy group Chemical group 0.000 claims description 85
- 229910052739 hydrogen Inorganic materials 0.000 claims description 53
- 239000001257 hydrogen Substances 0.000 claims description 53
- 229910052736 halogen Inorganic materials 0.000 claims description 46
- 150000002367 halogens Chemical class 0.000 claims description 44
- 125000003282 alkyl amino group Chemical group 0.000 claims description 43
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 39
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 35
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 33
- 125000006239 protecting group Chemical group 0.000 claims description 29
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 28
- 239000013078 crystal Substances 0.000 claims description 27
- 238000004519 manufacturing process Methods 0.000 claims description 23
- 150000003839 salts Chemical class 0.000 claims description 18
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 17
- 125000005843 halogen group Chemical group 0.000 claims description 17
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 15
- 125000004414 alkyl thio group Chemical group 0.000 claims description 12
- 239000012453 solvate Substances 0.000 claims description 12
- 238000001228 spectrum Methods 0.000 claims description 12
- 125000003342 alkenyl group Chemical group 0.000 claims description 11
- 125000006323 alkenyl amino group Chemical group 0.000 claims description 9
- 125000006319 alkynyl amino group Chemical group 0.000 claims description 9
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims description 7
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 7
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 6
- 125000004043 oxo group Chemical group O=* 0.000 claims description 6
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 5
- 125000004448 alkyl carbonyl group Chemical group 0.000 claims description 5
- 125000004466 alkoxycarbonylamino group Chemical group 0.000 claims description 4
- 125000004457 alkyl amino carbonyl group Chemical group 0.000 claims description 4
- 125000003806 alkyl carbonyl amino group Chemical group 0.000 claims description 4
- 125000004390 alkyl sulfonyl group Chemical group 0.000 claims description 4
- 125000004656 alkyl sulfonylamino group Chemical group 0.000 claims description 4
- 150000001450 anions Chemical class 0.000 claims description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 claims description 3
- 206010022000 influenza Diseases 0.000 abstract description 21
- 239000003814 drug Substances 0.000 abstract description 15
- 229940079593 drug Drugs 0.000 abstract description 12
- 239000000543 intermediate Substances 0.000 abstract description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 249
- 239000000243 solution Substances 0.000 description 168
- 238000006243 chemical reaction Methods 0.000 description 122
- 239000002904 solvent Substances 0.000 description 120
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 111
- 238000005160 1H NMR spectroscopy Methods 0.000 description 102
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 96
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 87
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 86
- 239000007787 solid Substances 0.000 description 86
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 80
- 238000010898 silica gel chromatography Methods 0.000 description 74
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 72
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 71
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 70
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 66
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 66
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 55
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 51
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 51
- 229910052938 sodium sulfate Inorganic materials 0.000 description 51
- 235000011152 sodium sulphate Nutrition 0.000 description 51
- 239000012043 crude product Substances 0.000 description 50
- WORJEOGGNQDSOE-UHFFFAOYSA-N chloroform;methanol Chemical compound OC.ClC(Cl)Cl WORJEOGGNQDSOE-UHFFFAOYSA-N 0.000 description 41
- 239000011541 reaction mixture Substances 0.000 description 41
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 39
- 239000003921 oil Substances 0.000 description 39
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 37
- 238000001914 filtration Methods 0.000 description 35
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 33
- 235000017557 sodium bicarbonate Nutrition 0.000 description 33
- 239000010410 layer Substances 0.000 description 32
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 31
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 31
- 238000003756 stirring Methods 0.000 description 31
- 150000002431 hydrogen Chemical class 0.000 description 30
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 26
- 238000010511 deprotection reaction Methods 0.000 description 25
- 239000012044 organic layer Substances 0.000 description 25
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 24
- 238000006482 condensation reaction Methods 0.000 description 24
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 24
- 229920006395 saturated elastomer Polymers 0.000 description 24
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 23
- 238000004440 column chromatography Methods 0.000 description 23
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 23
- 239000000284 extract Substances 0.000 description 19
- 239000000126 substance Substances 0.000 description 19
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 18
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 18
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 18
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 17
- 125000004432 carbon atom Chemical group C* 0.000 description 17
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 17
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 17
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 16
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 16
- 230000018044 dehydration Effects 0.000 description 16
- 238000006297 dehydration reaction Methods 0.000 description 16
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 16
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 16
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 16
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 16
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 16
- CYSGHNMQYZDMIA-UHFFFAOYSA-N 1,3-Dimethyl-2-imidazolidinon Chemical compound CN1CCN(C)C1=O CYSGHNMQYZDMIA-UHFFFAOYSA-N 0.000 description 15
- 239000007864 aqueous solution Substances 0.000 description 15
- 239000003153 chemical reaction reagent Substances 0.000 description 15
- 238000005259 measurement Methods 0.000 description 15
- LBKJNHPKYFYCLL-UHFFFAOYSA-N potassium;trimethyl(oxido)silane Chemical compound [K+].C[Si](C)(C)[O-] LBKJNHPKYFYCLL-UHFFFAOYSA-N 0.000 description 15
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 14
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 14
- 125000003277 amino group Chemical group 0.000 description 14
- 238000002425 crystallisation Methods 0.000 description 14
- 230000008025 crystallization Effects 0.000 description 14
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 14
- 238000007254 oxidation reaction Methods 0.000 description 14
- 238000012360 testing method Methods 0.000 description 14
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 13
- 229910000024 caesium carbonate Inorganic materials 0.000 description 13
- 238000010992 reflux Methods 0.000 description 13
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 12
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 239000000706 filtrate Substances 0.000 description 12
- OXOZHAWWRPCVGL-UHFFFAOYSA-N lithium;trimethyl(oxido)silane Chemical compound [Li+].C[Si](C)(C)[O-] OXOZHAWWRPCVGL-UHFFFAOYSA-N 0.000 description 12
- 150000007522 mineralic acids Chemical class 0.000 description 12
- 229910000027 potassium carbonate Inorganic materials 0.000 description 12
- 238000000746 purification Methods 0.000 description 12
- 230000035484 reaction time Effects 0.000 description 12
- BWHDROKFUHTORW-UHFFFAOYSA-N tritert-butylphosphane Chemical compound CC(C)(C)P(C(C)(C)C)C(C)(C)C BWHDROKFUHTORW-UHFFFAOYSA-N 0.000 description 12
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 11
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 11
- 125000003172 aldehyde group Chemical group 0.000 description 11
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 11
- 239000012312 sodium hydride Substances 0.000 description 11
- 229910000104 sodium hydride Inorganic materials 0.000 description 11
- 239000000758 substrate Substances 0.000 description 11
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 10
- 102000004533 Endonucleases Human genes 0.000 description 10
- 108010042407 Endonucleases Proteins 0.000 description 10
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 10
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 10
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 10
- 241000700605 Viruses Species 0.000 description 10
- 239000007810 chemical reaction solvent Substances 0.000 description 10
- 239000003795 chemical substances by application Substances 0.000 description 10
- 238000000605 extraction Methods 0.000 description 10
- 239000007758 minimum essential medium Substances 0.000 description 10
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 10
- 239000008096 xylene Substances 0.000 description 10
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 9
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 9
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 9
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 9
- 230000001419 dependent effect Effects 0.000 description 9
- 230000002401 inhibitory effect Effects 0.000 description 9
- 238000002955 isolation Methods 0.000 description 9
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 8
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 8
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 8
- 125000005233 alkylalcohol group Chemical group 0.000 description 8
- 125000000304 alkynyl group Chemical group 0.000 description 8
- 125000003118 aryl group Chemical group 0.000 description 8
- 238000004821 distillation Methods 0.000 description 8
- 235000019253 formic acid Nutrition 0.000 description 8
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 8
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 8
- 229930040373 Paraformaldehyde Natural products 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 150000001408 amides Chemical class 0.000 description 7
- 238000001816 cooling Methods 0.000 description 7
- 230000000694 effects Effects 0.000 description 7
- 238000010828 elution Methods 0.000 description 7
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 7
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 7
- 229920002866 paraformaldehyde Polymers 0.000 description 7
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 6
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- 235000004694 Eucalyptus leucoxylon Nutrition 0.000 description 6
- 244000166102 Eucalyptus leucoxylon Species 0.000 description 6
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 6
- 238000006751 Mitsunobu reaction Methods 0.000 description 6
- 150000001299 aldehydes Chemical class 0.000 description 6
- 150000003973 alkyl amines Chemical class 0.000 description 6
- 238000007098 aminolysis reaction Methods 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- NPOMSUOUAZCMBL-UHFFFAOYSA-N dichloromethane;ethoxyethane Chemical compound ClCCl.CCOCC NPOMSUOUAZCMBL-UHFFFAOYSA-N 0.000 description 6
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 6
- REVROVKRCYLCAT-UHFFFAOYSA-N dimethyl 4-oxo-3-phenylmethoxypyran-2,5-dicarboxylate Chemical compound O1C=C(C(=O)OC)C(=O)C(OCC=2C=CC=CC=2)=C1C(=O)OC REVROVKRCYLCAT-UHFFFAOYSA-N 0.000 description 6
- 238000010931 ester hydrolysis Methods 0.000 description 6
- UREBWPXBXRYXRJ-UHFFFAOYSA-N ethyl acetate;methanol Chemical compound OC.CCOC(C)=O UREBWPXBXRYXRJ-UHFFFAOYSA-N 0.000 description 6
- 238000001704 evaporation Methods 0.000 description 6
- 230000002140 halogenating effect Effects 0.000 description 6
- 239000012535 impurity Substances 0.000 description 6
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 6
- 239000012299 nitrogen atmosphere Substances 0.000 description 6
- YLACRFYIUQZNIV-UHFFFAOYSA-N o-(2,4-dinitrophenyl)hydroxylamine Chemical compound NOC1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O YLACRFYIUQZNIV-UHFFFAOYSA-N 0.000 description 6
- 230000003647 oxidation Effects 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 125000005344 pyridylmethyl group Chemical group [H]C1=C([H])C([H])=C([H])C(=N1)C([H])([H])* 0.000 description 6
- 239000002994 raw material Substances 0.000 description 6
- 230000009257 reactivity Effects 0.000 description 6
- CGRKYEALWSRNJS-UHFFFAOYSA-N sodium;2-methylbutan-2-olate Chemical compound [Na+].CCC(C)(C)[O-] CGRKYEALWSRNJS-UHFFFAOYSA-N 0.000 description 6
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- RMNIZOOYFMNEJJ-UHFFFAOYSA-K tripotassium;phosphate;hydrate Chemical compound O.[K+].[K+].[K+].[O-]P([O-])([O-])=O RMNIZOOYFMNEJJ-UHFFFAOYSA-K 0.000 description 6
- 0 *C=C(C(CN)=O)C(C(*)=C)=O Chemical compound *C=C(C(CN)=O)C(C(*)=C)=O 0.000 description 5
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 5
- AUNNWHUVARSNRE-UHFFFAOYSA-N C(C)OC.C(Cl)(Cl)Cl Chemical compound C(C)OC.C(Cl)(Cl)Cl AUNNWHUVARSNRE-UHFFFAOYSA-N 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- LOMVENUNSWAXEN-UHFFFAOYSA-N Methyl oxalate Chemical compound COC(=O)C(=O)OC LOMVENUNSWAXEN-UHFFFAOYSA-N 0.000 description 5
- ZSXGLVDWWRXATF-UHFFFAOYSA-N N,N-dimethylformamide dimethyl acetal Chemical compound COC(OC)N(C)C ZSXGLVDWWRXATF-UHFFFAOYSA-N 0.000 description 5
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 5
- 150000001336 alkenes Chemical group 0.000 description 5
- 239000012298 atmosphere Substances 0.000 description 5
- 238000004364 calculation method Methods 0.000 description 5
- 125000000753 cycloalkyl group Chemical group 0.000 description 5
- UBJFKNSINUCEAL-UHFFFAOYSA-N lithium;2-methylpropane Chemical compound [Li+].C[C-](C)C UBJFKNSINUCEAL-UHFFFAOYSA-N 0.000 description 5
- 125000002757 morpholinyl group Chemical group 0.000 description 5
- 238000007248 oxidative elimination reaction Methods 0.000 description 5
- 229910052763 palladium Inorganic materials 0.000 description 5
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 5
- 241000712461 unidentified influenza virus Species 0.000 description 5
- QKWWDTYDYOFRJL-UHFFFAOYSA-N 2,2-dimethoxyethanamine Chemical compound COC(CN)OC QKWWDTYDYOFRJL-UHFFFAOYSA-N 0.000 description 4
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 4
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 4
- WUGQZFFCHPXWKQ-UHFFFAOYSA-N Propanolamine Chemical compound NCCCO WUGQZFFCHPXWKQ-UHFFFAOYSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 235000019270 ammonium chloride Nutrition 0.000 description 4
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 4
- OQROAIRCEOBYJA-UHFFFAOYSA-N bromodiphenylmethane Chemical compound C=1C=CC=CC=1C(Br)C1=CC=CC=C1 OQROAIRCEOBYJA-UHFFFAOYSA-N 0.000 description 4
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 4
- 238000010790 dilution Methods 0.000 description 4
- 239000012895 dilution Substances 0.000 description 4
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 4
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 4
- 239000013642 negative control Substances 0.000 description 4
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 4
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 4
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 4
- VSIVTUIKYVGDCX-UHFFFAOYSA-M sodium;4-[2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)tetrazol-2-ium-5-yl]benzene-1,3-disulfonate Chemical compound [Na+].COC1=CC([N+]([O-])=O)=CC=C1[N+]1=NC(C=2C(=CC(=CC=2)S([O-])(=O)=O)S([O-])(=O)=O)=NN1C1=CC=C([N+]([O-])=O)C=C1 VSIVTUIKYVGDCX-UHFFFAOYSA-M 0.000 description 4
- 239000013589 supplement Substances 0.000 description 4
- 125000004784 trichloromethoxy group Chemical group ClC(O*)(Cl)Cl 0.000 description 4
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 4
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 3
- GUGQQGROXHPINL-UHFFFAOYSA-N 2-oxobutanoyl chloride Chemical compound CCC(=O)C(Cl)=O GUGQQGROXHPINL-UHFFFAOYSA-N 0.000 description 3
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 3
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 3
- 239000005695 Ammonium acetate Substances 0.000 description 3
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 3
- 239000012981 Hank's balanced salt solution Substances 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 102000004142 Trypsin Human genes 0.000 description 3
- 108090000631 Trypsin Proteins 0.000 description 3
- 238000002441 X-ray diffraction Methods 0.000 description 3
- 238000007112 amidation reaction Methods 0.000 description 3
- 235000019257 ammonium acetate Nutrition 0.000 description 3
- 229940043376 ammonium acetate Drugs 0.000 description 3
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 3
- 125000000392 cycloalkenyl group Chemical group 0.000 description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 3
- 231100000135 cytotoxicity Toxicity 0.000 description 3
- 230000003013 cytotoxicity Effects 0.000 description 3
- 125000003493 decenyl group Chemical group [H]C([*])=C([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 3
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 3
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 3
- 235000019341 magnesium sulphate Nutrition 0.000 description 3
- 108020004999 messenger RNA Proteins 0.000 description 3
- XKJCHHZQLQNZHY-UHFFFAOYSA-N phthalimide Chemical compound C1=CC=C2C(=O)NC(=O)C2=C1 XKJCHHZQLQNZHY-UHFFFAOYSA-N 0.000 description 3
- 239000013641 positive control Substances 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- RMVRSNDYEFQCLF-UHFFFAOYSA-N thiophenol Chemical compound SC1=CC=CC=C1 RMVRSNDYEFQCLF-UHFFFAOYSA-N 0.000 description 3
- 239000012588 trypsin Substances 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- AGMZSYQMSHMXLT-SCSAIBSYSA-N (3r)-3-aminobutan-1-ol Chemical compound C[C@@H](N)CCO AGMZSYQMSHMXLT-SCSAIBSYSA-N 0.000 description 2
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 2
- HLVFKOKELQSXIQ-UHFFFAOYSA-N 1-bromo-2-methylpropane Chemical compound CC(C)CBr HLVFKOKELQSXIQ-UHFFFAOYSA-N 0.000 description 2
- WWILHZQYNPQALT-UHFFFAOYSA-N 2-methyl-2-morpholin-4-ylpropanal Chemical compound O=CC(C)(C)N1CCOCC1 WWILHZQYNPQALT-UHFFFAOYSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- 239000007821 HATU Substances 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 2
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 2
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 2
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 2
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 2
- 235000011130 ammonium sulphate Nutrition 0.000 description 2
- 239000004599 antimicrobial Substances 0.000 description 2
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 2
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 2
- 150000001244 carboxylic acid anhydrides Chemical class 0.000 description 2
- 239000012230 colorless oil Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 125000004212 difluorophenyl group Chemical group 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 238000006911 enzymatic reaction Methods 0.000 description 2
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 2
- 230000001747 exhibiting effect Effects 0.000 description 2
- 125000001207 fluorophenyl group Chemical group 0.000 description 2
- 239000006260 foam Substances 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 125000001072 heteroaryl group Chemical group 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 2
- 125000005956 isoquinolyl group Chemical group 0.000 description 2
- 229930027917 kanamycin Natural products 0.000 description 2
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 description 2
- 229960000318 kanamycin Drugs 0.000 description 2
- 229930182823 kanamycin A Natural products 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 238000000691 measurement method Methods 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 125000005981 pentynyl group Chemical group 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 2
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 2
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 2
- 125000004076 pyridyl group Chemical group 0.000 description 2
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 2
- 125000005493 quinolyl group Chemical group 0.000 description 2
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- UKLNMMHNWFDKNT-UHFFFAOYSA-M sodium chlorite Chemical compound [Na+].[O-]Cl=O UKLNMMHNWFDKNT-UHFFFAOYSA-M 0.000 description 2
- 229960002218 sodium chlorite Drugs 0.000 description 2
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 description 2
- 238000001308 synthesis method Methods 0.000 description 2
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 150000003512 tertiary amines Chemical class 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 238000005199 ultracentrifugation Methods 0.000 description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 2
- 229920002554 vinyl polymer Polymers 0.000 description 2
- 230000003612 virological effect Effects 0.000 description 2
- ARAIBEBZBOPLMB-UFGQHTETSA-N zanamivir Chemical compound CC(=O)N[C@@H]1[C@@H](N=C(N)N)C=C(C(O)=O)O[C@H]1[C@H](O)[C@H](O)CO ARAIBEBZBOPLMB-UFGQHTETSA-N 0.000 description 2
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- XYZIUGPCJRYTCP-INIZCTEOSA-N (2s)-1-n-benzyl-3-phenylpropane-1,2-diamine Chemical compound C([C@@H](N)CC=1C=CC=CC=1)NCC1=CC=CC=C1 XYZIUGPCJRYTCP-INIZCTEOSA-N 0.000 description 1
- ASWBNKHCZGQVJV-UHFFFAOYSA-N (3-hexadecanoyloxy-2-hydroxypropyl) 2-(trimethylazaniumyl)ethyl phosphate Chemical compound CCCCCCCCCCCCCCCC(=O)OCC(O)COP([O-])(=O)OCC[N+](C)(C)C ASWBNKHCZGQVJV-UHFFFAOYSA-N 0.000 description 1
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 1
- MPDDTAJMJCESGV-CTUHWIOQSA-M (3r,5r)-7-[2-(4-fluorophenyl)-5-[methyl-[(1r)-1-phenylethyl]carbamoyl]-4-propan-2-ylpyrazol-3-yl]-3,5-dihydroxyheptanoate Chemical compound C1([C@@H](C)N(C)C(=O)C2=NN(C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C2C(C)C)C=2C=CC(F)=CC=2)=CC=CC=C1 MPDDTAJMJCESGV-CTUHWIOQSA-M 0.000 description 1
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 description 1
- WOGITNXCNOTRLK-VOTSOKGWSA-N (e)-3-phenylprop-2-enoyl chloride Chemical compound ClC(=O)\C=C\C1=CC=CC=C1 WOGITNXCNOTRLK-VOTSOKGWSA-N 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- TYGPYXDUZUIJKY-UHFFFAOYSA-N 1,3-dioxan-2-ylmethanamine Chemical compound NCC1OCCCO1 TYGPYXDUZUIJKY-UHFFFAOYSA-N 0.000 description 1
- UBCHPRBFMUDMNC-UHFFFAOYSA-N 1-(1-adamantyl)ethanamine Chemical compound C1C(C2)CC3CC2CC1(C(N)C)C3 UBCHPRBFMUDMNC-UHFFFAOYSA-N 0.000 description 1
- NNEOYCMCJMLRSD-UHFFFAOYSA-N 1-benzyl-4-bromobenzene Chemical compound C1=CC(Br)=CC=C1CC1=CC=CC=C1 NNEOYCMCJMLRSD-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- 125000006507 2,4-difluorobenzyl group Chemical group [H]C1=C(F)C([H])=C(F)C(=C1[H])C([H])([H])* 0.000 description 1
- PAWQVTBBRAZDMG-UHFFFAOYSA-N 2-(3-bromo-2-fluorophenyl)acetic acid Chemical compound OC(=O)CC1=CC=CC(Br)=C1F PAWQVTBBRAZDMG-UHFFFAOYSA-N 0.000 description 1
- PAYROHWFGZADBR-UHFFFAOYSA-N 2-[[4-amino-5-(5-iodo-4-methoxy-2-propan-2-ylphenoxy)pyrimidin-2-yl]amino]propane-1,3-diol Chemical compound C1=C(I)C(OC)=CC(C(C)C)=C1OC1=CN=C(NC(CO)CO)N=C1N PAYROHWFGZADBR-UHFFFAOYSA-N 0.000 description 1
- VVCMGAUPZIKYTH-VGHSCWAPSA-N 2-acetyloxybenzoic acid;[(2s,3r)-4-(dimethylamino)-3-methyl-1,2-diphenylbutan-2-yl] propanoate;1,3,7-trimethylpurine-2,6-dione Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O.CN1C(=O)N(C)C(=O)C2=C1N=CN2C.C([C@](OC(=O)CC)([C@H](C)CN(C)C)C=1C=CC=CC=1)C1=CC=CC=C1 VVCMGAUPZIKYTH-VGHSCWAPSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- STVVMTBJNDTZBF-UHFFFAOYSA-N 2-amino-3-phenylpropan-1-ol Chemical compound OCC(N)CC1=CC=CC=C1 STVVMTBJNDTZBF-UHFFFAOYSA-N 0.000 description 1
- ASUDFOJKTJLAIK-UHFFFAOYSA-N 2-methoxyethanamine Chemical compound COCCN ASUDFOJKTJLAIK-UHFFFAOYSA-N 0.000 description 1
- 125000003229 2-methylhexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- QISAUDWTBBNJIR-UHFFFAOYSA-N 2-phenylmethoxyacetyl chloride Chemical compound ClC(=O)COCC1=CC=CC=C1 QISAUDWTBBNJIR-UHFFFAOYSA-N 0.000 description 1
- BGAJNPLDJJBRHK-UHFFFAOYSA-N 3-[2-[5-(3-chloro-4-propan-2-yloxyphenyl)-1,3,4-thiadiazol-2-yl]-3-methyl-6,7-dihydro-4h-pyrazolo[4,3-c]pyridin-5-yl]propanoic acid Chemical compound C1=C(Cl)C(OC(C)C)=CC=C1C1=NN=C(N2C(=C3CN(CCC(O)=O)CCC3=N2)C)S1 BGAJNPLDJJBRHK-UHFFFAOYSA-N 0.000 description 1
- AGMZSYQMSHMXLT-UHFFFAOYSA-N 3-aminobutan-1-ol Chemical compound CC(N)CCO AGMZSYQMSHMXLT-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- MCFBUIIRFZBRCU-UHFFFAOYSA-N 4-[1-[5-[6-(trifluoromethyl)-1h-benzimidazol-2-yl]pyridin-2-yl]piperidin-4-yl]oxycyclohexane-1-carboxylic acid Chemical compound C1CC(C(=O)O)CCC1OC1CCN(C=2N=CC(=CC=2)C=2NC3=CC(=CC=C3N=2)C(F)(F)F)CC1 MCFBUIIRFZBRCU-UHFFFAOYSA-N 0.000 description 1
- UDGUGZTYGWUUSG-UHFFFAOYSA-N 4-[4-[[2,5-dimethoxy-4-[(4-nitrophenyl)diazenyl]phenyl]diazenyl]-n-methylanilino]butanoic acid Chemical compound COC=1C=C(N=NC=2C=CC(=CC=2)N(C)CCCC(O)=O)C(OC)=CC=1N=NC1=CC=C([N+]([O-])=O)C=C1 UDGUGZTYGWUUSG-UHFFFAOYSA-N 0.000 description 1
- 125000004176 4-fluorobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1F)C([H])([H])* 0.000 description 1
- KJSJBKBZMGSIPT-UHFFFAOYSA-N 4-oxo-3-phenylmethoxypyran-2-carboxylic acid Chemical compound O1C=CC(=O)C(OCC=2C=CC=CC=2)=C1C(=O)O KJSJBKBZMGSIPT-UHFFFAOYSA-N 0.000 description 1
- LHEJDBBHZGISGW-UHFFFAOYSA-N 5-fluoro-3-(3-oxo-1h-2-benzofuran-1-yl)-1h-pyrimidine-2,4-dione Chemical compound O=C1C(F)=CNC(=O)N1C1C2=CC=CC=C2C(=O)O1 LHEJDBBHZGISGW-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 239000004254 Ammonium phosphate Substances 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- KMFZGSTXBRPYKL-UHFFFAOYSA-N CC(CO)CSCc1ccccc1 Chemical compound CC(CO)CSCc1ccccc1 KMFZGSTXBRPYKL-UHFFFAOYSA-N 0.000 description 1
- RYCVKDKYDRTCNC-UHFFFAOYSA-N CS(CC(COc1ccccc1)O)I Chemical compound CS(CC(COc1ccccc1)O)I RYCVKDKYDRTCNC-UHFFFAOYSA-N 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- HTJDQJBWANPRPF-UHFFFAOYSA-N Cyclopropylamine Chemical compound NC1CC1 HTJDQJBWANPRPF-UHFFFAOYSA-N 0.000 description 1
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- SIXHCCPAJIVTOY-UHFFFAOYSA-N Flutimide Natural products CC(C)CC1=NC(=CC(C)C)C(=O)N(O)C1=O SIXHCCPAJIVTOY-UHFFFAOYSA-N 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 206010035664 Pneumonia Diseases 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 206010057190 Respiratory tract infections Diseases 0.000 description 1
- OZBDFBJXRJWNAV-UHFFFAOYSA-N Rimantadine hydrochloride Chemical compound Cl.C1C(C2)CC3CC2CC1(C(N)C)C3 OZBDFBJXRJWNAV-UHFFFAOYSA-N 0.000 description 1
- 229910008051 Si-OH Inorganic materials 0.000 description 1
- 229910006358 Si—OH Inorganic materials 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 108010067390 Viral Proteins Proteins 0.000 description 1
- 108020000999 Viral RNA Proteins 0.000 description 1
- SPXSEZMVRJLHQG-XMMPIXPASA-N [(2R)-1-[[4-[(3-phenylmethoxyphenoxy)methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound C(C1=CC=CC=C1)OC=1C=C(OCC2=CC=C(CN3[C@H](CCC3)CO)C=C2)C=CC=1 SPXSEZMVRJLHQG-XMMPIXPASA-N 0.000 description 1
- QQIRAVWVGBTHMJ-UHFFFAOYSA-N [dimethyl-(trimethylsilylamino)silyl]methane;lithium Chemical compound [Li].C[Si](C)(C)N[Si](C)(C)C QQIRAVWVGBTHMJ-UHFFFAOYSA-N 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- SRVFFFJZQVENJC-IHRRRGAJSA-N aloxistatin Chemical compound CCOC(=O)[C@H]1O[C@@H]1C(=O)N[C@@H](CC(C)C)C(=O)NCCC(C)C SRVFFFJZQVENJC-IHRRRGAJSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 229960003805 amantadine Drugs 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- SWLVFNYSXGMGBS-UHFFFAOYSA-N ammonium bromide Chemical compound [NH4+].[Br-] SWLVFNYSXGMGBS-UHFFFAOYSA-N 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 229910000148 ammonium phosphate Inorganic materials 0.000 description 1
- 235000019289 ammonium phosphates Nutrition 0.000 description 1
- 229940035676 analgesics Drugs 0.000 description 1
- 239000000730 antalgic agent Substances 0.000 description 1
- 125000005428 anthryl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C3C(*)=C([H])C([H])=C([H])C3=C([H])C2=C1[H] 0.000 description 1
- 239000002259 anti human immunodeficiency virus agent Substances 0.000 description 1
- 229940124599 anti-inflammatory drug Drugs 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 239000000010 aprotic solvent Substances 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000002047 benzodioxolyl group Chemical group O1OC(C2=C1C=CC=C2)* 0.000 description 1
- 125000004618 benzofuryl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004619 benzopyranyl group Chemical group O1C(C=CC2=C1C=CC=C2)* 0.000 description 1
- 125000005874 benzothiadiazolyl group Chemical group 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 1
- IBLHIEZLGKFQRW-UHFFFAOYSA-N bis(3-chlorophenyl)methanol Chemical compound C=1C=CC(Cl)=CC=1C(O)C1=CC=CC(Cl)=C1 IBLHIEZLGKFQRW-UHFFFAOYSA-N 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000010531 catalytic reduction reaction Methods 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000034303 cell budding Effects 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 125000004775 chlorodifluoromethyl group Chemical group FC(F)(Cl)* 0.000 description 1
- QSKWJTXWJJOJFP-UHFFFAOYSA-N chloroform;ethoxyethane Chemical compound ClC(Cl)Cl.CCOCC QSKWJTXWJJOJFP-UHFFFAOYSA-N 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 125000004230 chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940127271 compound 49 Drugs 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000003678 cyclohexadienyl group Chemical group C1(=CC=CCC1)* 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000006547 cyclononyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000000298 cyclopropenyl group Chemical group [H]C1=C([H])C1([H])* 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 231100000517 death Toxicity 0.000 description 1
- 125000005070 decynyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C#C* 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- MNNHAPBLZZVQHP-UHFFFAOYSA-N diammonium hydrogen phosphate Chemical compound [NH4+].[NH4+].OP([O-])([O-])=O MNNHAPBLZZVQHP-UHFFFAOYSA-N 0.000 description 1
- 125000004987 dibenzofuryl group Chemical group C1(=CC=CC=2OC3=C(C21)C=CC=C3)* 0.000 description 1
- PQRPNAHMPIEDRO-UHFFFAOYSA-N dichloromethane;methoxyethane Chemical compound ClCCl.CCOC PQRPNAHMPIEDRO-UHFFFAOYSA-N 0.000 description 1
- 125000005433 dihydrobenzodioxinyl group Chemical group O1C(COC2=C1C=CC=C2)* 0.000 description 1
- 125000004598 dihydrobenzofuryl group Chemical group O1C(CC2=C1C=CC=C2)* 0.000 description 1
- 125000005434 dihydrobenzoxazinyl group Chemical group O1N(CCC2=C1C=CC=C2)* 0.000 description 1
- 125000004925 dihydropyridyl group Chemical group N1(CC=CC=C1)* 0.000 description 1
- 125000005056 dihydrothiazolyl group Chemical group S1C(NC=C1)* 0.000 description 1
- 125000000532 dioxanyl group Chemical group 0.000 description 1
- 125000005879 dioxolanyl group Chemical group 0.000 description 1
- DGODWNOPHMXOTR-UHFFFAOYSA-N dipotassium;dioxido(dioxo)osmium;dihydrate Chemical compound O.O.[K+].[K+].[O-][Os]([O-])(=O)=O DGODWNOPHMXOTR-UHFFFAOYSA-N 0.000 description 1
- 229940042406 direct acting antivirals neuraminidase inhibitors Drugs 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 235000013601 eggs Nutrition 0.000 description 1
- 239000012156 elution solvent Substances 0.000 description 1
- 230000007159 enucleation Effects 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- DQYBDCGIPTYXML-UHFFFAOYSA-N ethoxyethane;hydrate Chemical compound O.CCOCC DQYBDCGIPTYXML-UHFFFAOYSA-N 0.000 description 1
- RJCGNNHKSNIUAT-UHFFFAOYSA-N ethyl 3-aminopropanoate;hydron;chloride Chemical compound Cl.CCOC(=O)CCN RJCGNNHKSNIUAT-UHFFFAOYSA-N 0.000 description 1
- WDCDAAMJNUHOIY-UHFFFAOYSA-N ethyl acetate;2-propan-2-yloxypropane Chemical compound CCOC(C)=O.CC(C)OC(C)C WDCDAAMJNUHOIY-UHFFFAOYSA-N 0.000 description 1
- 229940028864 flumadine Drugs 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- SIXHCCPAJIVTOY-UITAMQMPSA-N flutimide Chemical compound CC(C)CC1=N\C(=C/C(C)C)C(=O)N(O)C1=O SIXHCCPAJIVTOY-UITAMQMPSA-N 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 238000012812 general test Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004634 hexahydroazepinyl group Chemical group N1(CCCCCC1)* 0.000 description 1
- MOTRZVVGCFFABN-UHFFFAOYSA-N hexane;2-propan-2-yloxypropane Chemical compound CCCCCC.CC(C)OC(C)C MOTRZVVGCFFABN-UHFFFAOYSA-N 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005980 hexynyl group Chemical group 0.000 description 1
- FUKUFMFMCZIRNT-UHFFFAOYSA-N hydron;methanol;chloride Chemical compound Cl.OC FUKUFMFMCZIRNT-UHFFFAOYSA-N 0.000 description 1
- HYOCSVGEQMCOGE-UHFFFAOYSA-N hydron;propane-1,3-diamine;dichloride Chemical compound Cl.Cl.NCCCN HYOCSVGEQMCOGE-UHFFFAOYSA-N 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 125000005945 imidazopyridyl group Chemical group 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 238000009776 industrial production Methods 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- XGZVUEUWXADBQD-UHFFFAOYSA-L lithium carbonate Chemical compound [Li+].[Li+].[O-]C([O-])=O XGZVUEUWXADBQD-UHFFFAOYSA-L 0.000 description 1
- 229910052808 lithium carbonate Inorganic materials 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 238000006198 methoxylation reaction Methods 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- GDOPTJXRTPNYNR-UHFFFAOYSA-N methyl-cyclopentane Natural products CC1CCCC1 GDOPTJXRTPNYNR-UHFFFAOYSA-N 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- REPVNSJSTLRQEQ-UHFFFAOYSA-N n,n-dimethylacetamide;n,n-dimethylformamide Chemical compound CN(C)C=O.CN(C)C(C)=O REPVNSJSTLRQEQ-UHFFFAOYSA-N 0.000 description 1
- WHQSYGRFZMUQGQ-UHFFFAOYSA-N n,n-dimethylformamide;hydrate Chemical compound O.CN(C)C=O WHQSYGRFZMUQGQ-UHFFFAOYSA-N 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- VHMAJOQMWXRUNK-UHFFFAOYSA-N n-methyl-1,3-dioxolan-2-amine Chemical compound CNC1OCCO1 VHMAJOQMWXRUNK-UHFFFAOYSA-N 0.000 description 1
- DLJPYODODWSDBI-UHFFFAOYSA-N n-methyl-2-nitrobenzenesulfonamide Chemical compound CNS(=O)(=O)C1=CC=CC=C1[N+]([O-])=O DLJPYODODWSDBI-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000005187 nonenyl group Chemical group C(=CCCCCCCC)* 0.000 description 1
- 125000005071 nonynyl group Chemical group C(#CCCCCCCC)* 0.000 description 1
- 238000007344 nucleophilic reaction Methods 0.000 description 1
- 125000005889 octahydrochromenyl group Chemical group 0.000 description 1
- 125000004365 octenyl group Chemical group C(=CCCCCCC)* 0.000 description 1
- 125000005069 octynyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C#C* 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 229960003752 oseltamivir Drugs 0.000 description 1
- NENPYTRHICXVCS-YNEHKIRRSA-N oseltamivir acid Chemical compound CCC(CC)O[C@@H]1C=C(C(O)=O)C[C@H](N)[C@H]1NC(C)=O NENPYTRHICXVCS-YNEHKIRRSA-N 0.000 description 1
- PGZUMBJQJWIWGJ-ONAKXNSWSA-N oseltamivir phosphate Chemical compound OP(O)(O)=O.CCOC(=O)C1=C[C@@H](OC(CC)CC)[C@H](NC(C)=O)[C@@H](N)C1 PGZUMBJQJWIWGJ-ONAKXNSWSA-N 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000005880 oxathiolanyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 125000000466 oxiranyl group Chemical group 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 1
- 125000005561 phenanthryl group Chemical group 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 125000004932 phenoxathinyl group Chemical group 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 125000005936 piperidyl group Chemical group 0.000 description 1
- 229920001467 poly(styrenesulfonates) Polymers 0.000 description 1
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 125000001844 prenyl group Chemical group [H]C([*])([H])C([H])=C(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 1
- 125000006324 propenyl amino group Chemical group 0.000 description 1
- 125000006320 propynyl amino group Chemical group 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 238000001028 reflection method Methods 0.000 description 1
- 229940061374 relenza Drugs 0.000 description 1
- 229960000888 rimantadine Drugs 0.000 description 1
- 238000006798 ring closing metathesis reaction Methods 0.000 description 1
- YBCAZPLXEGKKFM-UHFFFAOYSA-K ruthenium(iii) chloride Chemical compound [Cl-].[Cl-].[Cl-].[Ru+3] YBCAZPLXEGKKFM-UHFFFAOYSA-K 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 239000002911 sialidase inhibitor Substances 0.000 description 1
- 238000004513 sizing Methods 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- IIACRCGMVDHOTQ-UHFFFAOYSA-N sulfamic acid Chemical compound NS(O)(=O)=O IIACRCGMVDHOTQ-UHFFFAOYSA-N 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 229940061367 tamiflu Drugs 0.000 description 1
- QYJVBVKFXDHFPQ-UHFFFAOYSA-N tert-butyl n-(2-aminoethyl)-n-methylcarbamate Chemical compound NCCN(C)C(=O)OC(C)(C)C QYJVBVKFXDHFPQ-UHFFFAOYSA-N 0.000 description 1
- DKACXUFSLUYRFU-UHFFFAOYSA-N tert-butyl n-aminocarbamate Chemical compound CC(C)(C)OC(=O)NN DKACXUFSLUYRFU-UHFFFAOYSA-N 0.000 description 1
- 125000005063 tetradecenyl group Chemical group C(=CCCCCCCCCCCCC)* 0.000 description 1
- 125000005886 tetrahydrobenzothienyl group Chemical group 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- BSYVTEYKTMYBMK-UHFFFAOYSA-N tetrahydrofurfuryl alcohol Chemical compound OCC1CCCO1 BSYVTEYKTMYBMK-UHFFFAOYSA-N 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000005942 tetrahydropyridyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000005458 thianyl group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000004588 thienopyridyl group Chemical group S1C(=CC2=C1C=CC=N2)* 0.000 description 1
- 125000004587 thienothienyl group Chemical group S1C(=CC2=C1C=CS2)* 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000005505 thiomorpholino group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 125000005065 undecenyl group Chemical group C(=CCCCCCCCCC)* 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 230000006656 viral protein synthesis Effects 0.000 description 1
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 1
- 229960001028 zanamivir Drugs 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/79—Acids; Esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/79—Acids; Esters
- C07D213/80—Acids; Esters in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/79—Acids; Esters
- C07D213/803—Processes of preparation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/89—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members with hetero atoms directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/32—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/34—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D309/36—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with oxygen atoms directly attached to ring carbon atoms
- C07D309/38—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with oxygen atoms directly attached to ring carbon atoms one oxygen atom in position 2 or 4, e.g. pyrones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/34—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D309/36—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with oxygen atoms directly attached to ring carbon atoms
- C07D309/40—Oxygen atoms attached in positions 3 and 4, e.g. maltol
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/14—Ortho-condensed systems
Definitions
- the present invention relates to a pyrone derivative and a pyridone derivative, synthetic intermediates of novel anti-influenza drugs exhibiting cap-dependent endonuclease inhibitory activity, methods for producing them, and methods for using them.
- Influenza is an acute respiratory infection caused by influenza virus infection. There are millions of influenza-like patients reported every winter in Japan, and influenza is associated with high morbidity and mortality. This is a particularly important disease in high-risk groups such as infants and the elderly. The elderly have a high rate of pneumonia, and most of the deaths from influenza are occupied by the elderly.
- Anti-influenza drugs such as symmetrel (trade name: Amantadine) and flumadine (trade name: rimantadine), which inhibit the virus enucleation process, inhibit budding and release of virus from cells.
- Known neuraminidase inhibitors are Oseltamivir (trade name: Tamiflu) and Zanamivir (trade name: Relenza).
- Oseltamivir trade name: Tamiflu
- Zanamivir trade name: Relenza
- Cap-dependent endonuclease an influenza virus-derived enzyme, is essential for virus growth and is considered to be suitable as a target for anti-influenza drugs because it is a virus-specific enzyme activity that the host does not have .
- the cap-dependent endonuclease is an endonuclease activity that generates a fragment of 9 to 13 bases (including the base of the cap structure in the number) including the cap structure using the host mRNA precursor as a substrate. This fragment functions as a primer for viral RNA polymerase and is used to synthesize mRNA encoding the viral protein. That is, it is considered that a substance that inhibits cap-dependent endonuclease inhibits viral growth as a result of inhibiting viral protein synthesis by inhibiting viral mRNA synthesis.
- Patent Document 1 and Non-Patent Documents 1 and 2 As substances that inhibit the cap-dependent endonuclease, flutimide (Patent Document 1 and Non-Patent Documents 1 and 2), 4-substituted 2,4-dioxobutanoic acid (Non-Patent Documents 3 to 5) and the like have been reported. However, it has not yet been used clinically as an anti-influenza drug.
- Patent Documents 2 to 11 and Non-Patent Document 6 describe compounds having a structure similar to novel anti-influenza drugs exhibiting cap-dependent endonuclease inhibitory activity and methods for producing them. Further, pyrone derivatives and pyridone derivatives are disclosed in Non-Patent Documents 7 to 9.
- the object of the present invention is to provide novel pyrone and pyridone derivatives, methods for their production, and methods for their use, which are synthetic intermediates for novel anti-influenza drugs.
- formula (I), formula (II) or formula (III) useful as anti-influenza drugs (In the formula, R 1 is hydroxy, lower alkyloxy optionally substituted with substituent group E, carbocyclic lower alkyloxy optionally substituted with substituent group E, or substituted with substituent group E.
- Z is CR 12 R 13 , or a single bond;
- Y is CH 2 , an oxygen atom, or N—R 14 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 , R 11 , R 12 , R 13 , and R 14 are each independently hydrogen, substituent Lower alkyl optionally substituted with group E, lower alkenyl optionally substituted with substituent group E, lower alkynyl optionally substituted with substituent group E, optionally substituted with substituent group E Carbocyclic lower alkyl, or heterocyclic lower alkyl optionally substituted with substituent group E, R 3 and R 5 together form a carbocyclic group which may be substituted with
- R 7 and R 5 together may form a heterocyclic group optionally substituted with a 4-8 membered substituent group E;
- R 14 and R 10 may together form a heterocyclic group which may be substituted with a 4- to 8-membered substituent group E.
- Substituent group E halogen, cyano, hydroxy, carboxy, formyl, amino, oxo, nitro, lower alkyl, halogeno lower alkyl, lower alkyloxy, carbocyclic group optionally substituted by substituent group F, substituent Heterocyclic group optionally substituted with group F, carbocyclic lower alkyloxy optionally substituted with substituent group F, heterocyclic lower alkyloxy optionally substituted with substituent group F, substituent Carbocyclic lower alkylthio optionally substituted with group F, heterocyclic lower alkylthio optionally substituted with substituent group F, carbocyclic lower alkylamino optionally substituted with substituent group F, substituent group Heterocyclic lower alkylamino optionally substituted with F, carbocyclic oxy optionally substituted with substituent group F, heterocyclic oxy optionally substituted with substituent group F Carbocyclic carbonyl optionally substituted with substituent group F, heterocycl
- the present invention provides the following items.
- a heterocyclic group a carbocyclic lower alkyl optionally substituted with substituent group E, or a heterocyclic lower alkyl optionally substituted with substituent group E;
- R 2d is hydrogen, lower alkyl which may be substituted with substituent group E, carbocyclic lower alkyl which may be substituted with substituent group E, or heterocyclic ring which may be substituted with substituent group E
- Lower alkyl R 3d is hydrogen, lower alkyl optionally substituted with substituent group E, —N (R 3e ) 2 , or —OR 3e ;
- R 3e may each independently be lower alkyl optionally substituted with substituent group E, or may be taken together to form a heterocyclic group, and the wavy line represents E-form and / or Z-form Or a mixture thereof.
- Substituent group E halogen, cyano, hydroxy, carboxy, formyl, amino, oxo, nitro, lower alkyl, halogeno lower alkyl, lower alkyloxy, carbocyclic group optionally substituted by substituent group F, substituent Heterocyclic group optionally substituted with group F, carbocyclic lower alkyloxy optionally substituted with substituent group F, heterocyclic lower alkyloxy optionally substituted with substituent group F, substituent Carbocyclic lower alkylthio optionally substituted with group F, heterocyclic lower alkylthio optionally substituted with substituent group F, carbocyclic lower alkylamino optionally substituted with substituent group F, substituent group Heterocyclic lower alkylamino optionally substituted with F, carbocyclic oxy optionally substituted with substituent group F, heterocyclic oxy optionally substituted with substituent group F Carbocyclic carbonyl optionally substituted with substituent group F, heterocycl
- R 5d is hydrogen, halogen, lower alkyloxy optionally substituted with substituent group E, or —O—SO 2 —R 5e ;
- R 5e is a lower alkyl which may be substituted with the substituent group E, a carbocyclic group which may be substituted with the substituent group E, or a heterocyclic group which may be substituted with the substituent group E.
- (Item 2) Item 2. The method according to Item 1, wherein Step B and Step C are continuously performed.
- the compound represented by the formula (X2) is represented by the formula (X1): (In the formula, each symbol has the same meaning as item 1)
- the production method according to Item 1, 2, 3, 4, or Item 6 obtained by reacting with a compound represented by the formula:
- the production method according to the present invention is a novel anti-influenza drug, a compound encompassed by formula (I), formula (II), formula (III) or the like in a high yield, efficient and / or short process. It is a method that can be manufactured. Further, by carrying out the production method according to the present invention, it is possible to avoid the use of toxic reaction reagents, to avoid dangerous reactions, and to avoid the use of expensive reaction reagents, which are environmentally harmful reagents. And the use of solvents can be avoided.
- the pyrone derivative (X3), pyridone derivative (X4) and / or (X4 ′), which are the compounds of the present invention, are common intermediates in the production of the compounds included in the formula (I), the formula (II) or the formula (III). It is versatile and useful as a body. Furthermore, the pyrone derivative (X3) and / or the pyridone derivative (X4) can also be obtained as crystals. Since these crystals have low hygroscopicity and high stability to light and heat, they have advantages such as excellent storage and manufacturing handling.
- the production method and intermediates and crystals thereof according to the present invention are useful for industrial production of pharmaceuticals (anti-influenza drugs, anti-HIV drugs, anti-inflammatory drugs, analgesics, anti-tumor drugs, etc.). (Wherein each symbol is as defined above)
- 2 is a powder X-ray pattern of Compound 1D obtained in Example 1.
- the vertical axis represents peak intensity, and the horizontal axis represents diffraction angle (2 ⁇ ).
- 2 is a powder X-ray pattern of Compound 2D obtained in Example 2.
- the vertical axis represents peak intensity, and the horizontal axis represents diffraction angle (2 ⁇ ).
- 4 is a powder X-ray pattern of compound 9C ′ obtained in Example 9.
- the vertical axis represents peak intensity, and the horizontal axis represents diffraction angle (2 ⁇ ).
- halogen includes a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom.
- lower alkyl means a straight or branched chain having 1 to 15 carbon atoms, preferably 1 to 10 carbon atoms, more preferably 1 to 6 carbon atoms, and still more preferably 1 to 4 carbon atoms.
- Examples include n-octyl, isooctyl, n-nonyl and n-decyl.
- lower alkyl examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl and n-pentyl. Further preferred examples include methyl, ethyl, n-propyl, isopropyl and tert-butyl.
- lower alkenyl means 2 to 15 carbon atoms having one or more double bonds at an arbitrary position, preferably 2 to 10 carbon atoms, more preferably 2 to 6 carbon atoms, still more preferably. Includes straight-chain or branched alkenyl having 2 to 4 carbon atoms.
- Preferred embodiments of “lower alkenyl” include vinyl, allyl, propenyl, isopropenyl and butenyl.
- lower alkynyl is a straight chain or branched chain having 2 to 10 carbon atoms, preferably 2 to 8 carbon atoms, more preferably 3 to 6 carbon atoms, having one or more triple bonds at any position. Includes branched alkynyl. Specifically, ethynyl, propynyl, butynyl, pentynyl, hexynyl, heptynyl, octynyl, nonynyl, decynyl and the like are included. These may further have a double bond at an arbitrary position. Preferred embodiments of “lower alkynyl” include ethynyl, propynyl, butynyl and pentynyl.
- lower alkyloxy “lower alkyloxy”, “lower alkylcarbonyl”, “lower alkyloxycarbonyl”, “carbocyclic lower alkyl”, “heterocyclic lower alkyl”, “carbocyclic oxylower alkyl”, “heterocyclic oxy” “Lower alkyl”, “halogeno lower alkyl”, “carbocyclic lower alkyloxy”, “heterocyclic lower alkyloxy”, “carbocyclic lower alkylthio”, “heterocyclic lower alkylthio”, “carbocyclic lower alkylamino”, “hetero "Ring lower alkylamino”, “Halogeno lower alkyloxy”, "Lower alkyloxy lower alkyl”, “Lower alkyloxy lower alkyloxy”, “Lower alkylcarbonyl”, “Lower alkyloxycarbonyl”, “Lower alkylamino”, “ Lower alkylcarbonylamino
- the lower alkenyl part of “lower alkenylamino” is the same as the above “lower alkenyl”.
- the lower alkynyl moiety of “lower alkynylamino” is the same as the above “lower alkynyl”.
- the halogen part of “halogeno lower alkyl” and “halogeno lower alkyloxy” is the same as the above “halogen”.
- any position on the alkyl group of “lower alkyl” and “lower alkyloxy” may be substituted with the same or different halogen atoms.
- the “carbocyclic group” means a carbocyclic group having 3 to 20 carbon atoms, preferably 3 to 16 carbon atoms, more preferably 4 to 12 carbon atoms, and includes cycloalkyl, cycloalkenyl, Includes aryl and non-aromatic fused carbocyclic groups.
- cycloalkyl is a carbocyclic group having 3 to 16 carbon atoms, preferably 3 to 12 carbon atoms, more preferably 4 to 8 carbon atoms, such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclohexane.
- cycloalkenyl includes those having one or more double bonds at any position in the cycloalkyl ring, for example, cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclohexenyl, cyclo Examples include heptynyl, cyclooctynyl and cyclohexadienyl.
- aryl includes phenyl, naphthyl, anthryl, phenanthryl and the like, and phenyl is particularly preferable.
- non-aromatic fused carbocyclic group includes a group in which two or more cyclic groups selected from the above “cycloalkyl”, “cycloalkenyl” and “aryl” are condensed, for example, indanyl , Indenyl, tetrahydronaphthyl, fluorenyl, adamantyl, and the groups shown below Etc.
- Preferred embodiments of “carbocyclic group” include cycloalkyl, aryl and non-aromatic fused carbocyclic groups, specifically cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, phenyl Etc.
- Carbocyclic lower alkyl "Carbocyclic lower alkyloxy”, “Carbocyclic lower alkylthio", “Carbocyclic lower alkylamino”, “Carbocyclic oxy”, “Carbocyclic carbonyl”, and “Carbocyclic aminocarbonyl”
- the carbocyclic moiety is the same as the above “carbocyclic group”.
- heterocyclic group is a heteroaryl, non-aromatic heterocyclic group or bicyclic ring having one or more of the same or different heteroatoms arbitrarily selected from O, S and N in the ring.
- heterocyclic groups such as three-ring condensed heterocyclic groups.
- heteroaryl refers to pyrrolyl, imidazolyl, pyrazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazolyl, triazinyl, tetrazolyl, furyl, thienyl, isoxazolyl, oxazolyl, oxadiazolyl, isothiazolyl, thiazolyl, thiadiazolyl, etc. Membered aromatic cyclic groups.
- non-aromatic heterocyclic group means dioxanyl, thiylyl, oxiranyl, oxetanyl, oxathiolanyl, azetidinyl, thianyl, thiazolidinyl, pyrrolidinyl, pyrrolinyl, imidazolidinyl, imidazolinyl, pyrazolidinyl, pyrazolinyl, piperidyl, morpholinyl, morpholinyl, morpholinyl, morpholinyl, morpholinyl, morpholinyl , Thiomorpholinyl, thiomorpholino, dihydropyridyl, tetrahydropyridyl, tetrahydrofuryl, tetrahydropyranyl, dihydrothiazolyl, tetrahydrothiazolyl, tetrahydroisothiazolyl, dihydrooxazinyl,
- bicyclic fused heterocyclic group means indolyl, isoindolyl, indazolyl, indolizinyl, indolinyl, isoindolinyl, quinolyl, isoquinolyl, cinnolinyl, phthalazinyl, quinazolinyl, naphthyridinyl, quinoxalinyl, purinyl, pteridinyl, benzopyranyl, benzimidazolyl , Benzotriazolyl, benzisoxazolyl, benzoxazolyl, benzoxadiazolyl, benzoisothiazolyl, benzothiazolyl, benzothiadiazolyl, benzofuryl, isobenzofuryl, benzothienyl, benzotriazolyl, thienopyridyl, Thienopyrrolyl, thienopyrazolyl, thienopyrazinyl,
- the “tricyclic fused heterocyclic group” includes carbazolyl, acridinyl, xanthenyl, phenothiazinyl, phenoxathinyl, phenoxazinyl, dibenzofuryl, imidazoquinolyl, tetrahydrocarbazolyl, and the groups shown below Etc.
- the “heterocyclic group” include a 5- to 6-membered heteroaryl or non-aromatic heterocyclic group, and a 3-ring condensed heterocyclic group.
- Heterocyclic lower alkyl “heterocyclic lower alkyloxy”, “heterocyclic lower alkylthio”, “heterocyclic lower alkylamino”, “heterocyclic oxy”, “heterocyclic carbonyl” and “heterocyclic aminocarbonyl”
- the ring portion is the same as the above “heterocyclic group”.
- Step B and Step C means that after Step B is performed, Step C is performed without performing isolation operation or column chromatography purification of the product generated in Step B.
- the reaction vessel for performing step B and the reaction vessel for carrying out step C may be the same or different.
- the “lower alkyl optionally substituted with substituent group E” means that the “lower alkyl” is unsubstituted or selected from one or more chemically acceptable substituent groups E It means that the group is bonded. When a plurality of substituents are bonded, the plurality of substituents may be the same or different. For example, methyl, fluoromethyl, trifluoromethyl, chlorodifluoromethyl, and Etc.
- the “carbocyclic group optionally substituted with substituent group E” means that “carbocyclic group” is unsubstituted or one or more chemically acceptable substituent groups E. It means that the substituent selected from is bonded.
- the plurality of substituents may be the same or different.
- fluorophenyl, difluorophenyl, methoxyfluorophenyl and the like are included.
- “Carbocyclic lower alkyl optionally substituted with substituent group E” means that “carbocyclic group” and / or “lower alkyl” is unsubstituted or has one or more chemical groups. It means that a substituent selected from the substituent group E which is permissible is bonded.
- the plurality of substituents may be the same or different.
- Examples include 4-fluorobenzyl, 2,4-difluorobenzyl, 4-methoxy-2-fluorobenzyl, 4-methoxyphenyldifluoromethyl and the like.
- “Lower alkyloxy optionally substituted with substituent group E” “Carbocyclic lower alkyloxy optionally substituted with substituent group E”
- “Heterocycle optionally substituted with substituent group E” “Lower alkyloxy”, “lower alkenyl optionally substituted with substituent group E”, “amino optionally substituted with substituent group E”, “lower alkyl optionally substituted with substituent group E” “Amino”, “Lower alkenylamino optionally substituted with substituent group E”, “Lower alkynylamino optionally substituted with substituent group E”, “Carbon optionally substituted with substituent group E” “Ring lower alkylamino” and “heterocyclic lower alkylamino optionally substituted with substitu
- Carbocyclic group optionally substituted with substituent group F means that “carbocyclic group” is unsubstituted or one or more chemically acceptable substituent groups F. It means that the substituent selected from is bonded. When a plurality of substituents are bonded, the plurality of substituents may be the same or different. For example, fluorophenyl, difluorophenyl, methoxyfluorophenyl and the like are included. “Lower alkyloxy optionally substituted with substituent group F” means that the “carbocyclic group” moiety is unsubstituted or has one or more chemically acceptable substituent groups F. It means that the substituent selected from is bonded.
- the plurality of substituents may be the same or different.
- fluorobenzyloxy, difluorobenzyloxy, methoxyfluorobenzyloxy and the like are included.
- “Heterocyclic group optionally substituted with substituent group F” “Heterocyclic lower alkyloxy optionally substituted with substituent group F”, “Carbon optionally substituted with substituent group F” Ring lower alkylthio ”,“ heterocyclic lower alkylthio optionally substituted with substituent group F ”,“ carbocyclic lower alkylamino optionally substituted with substituent group F ”,“ substituted with substituent group F ”
- Optionally substituted heterocyclic lower alkylamino “ carbocyclic oxy optionally substituted with substituent group F ”,“ heterocyclic oxy optionally substituted with substituent group F ”,“ substituent group F A carbocyclic carbonyl optionally substituted with ", a" heterocyclic carbonyl
- heterocyclic group of the "-N (R 3e) 2 and R 3e together may form a heterocyclic group”
- the following formulas are included. Definition of “heterocyclic group” of the "-N (R 3e) 2, may also be R 3e together form a heterocyclic group” is also the same as described above.
- amino-protecting group may be a general protecting group of an amino group, and is exemplified as an amino-protecting group described in, for example, Protective Groups in Organic Synthesis, Theodora W Green (John Wiley & Sons). Of these, a tert-butyloxycarbonyl group and a benzyloxycarbonyl group are preferable.
- carboxyl protecting group may be a general amino protecting group, and is exemplified as a carboxyl protecting group described in, for example, Protective Groups in Organic Synthesis, Theodora W Green (John Wiley & Sons).
- Preferred examples include a methyl group, an ethyl group, a tert-butyl group, a methoxymethyl group, an allyl group, a benzyl group, and a p-methoxybenzyl group.
- the "counter anion of the ammonium cation" in X d, halogen -, CH 3 COO -, HCOO -, NO 3 -, BF 4 -, PF 6 -, HO -, Ph-SO 3 -, CH 3 -Ph- SO 3 ⁇ , CH 3 —SO 3 ⁇ , PO 4 3 ⁇ , SO 4 2 ⁇ , HSO 4 ⁇ and the like can be mentioned.
- halogen -, CH 3 COO -, NO 3 -, SO 4 2- When the anion is divalent or trivalent, the NH 4 + cation indicates an uncharged state by bonding two or three molecules, respectively.
- Specific examples of NH 4 + X d- include (NH 4 + ) 2 SO 4 2 ⁇ , (NH 4 + ) 3 PO 4 3 ⁇ and the like.
- the “leaving group” refers to a substituent that is eliminated by a nucleophilic reaction, and is halogen, —O—SO 2 —CH 3 , —O—SO 2 —CF 3 , —O—SO 2 —Ph or —O. -SO 2 -Ph-CH 3 and the like can be mentioned. Preferably, it is halogen.
- the “derivative of the formula (XA3)” refers to an aldehyde formed by oxidative cleavage of an olefin moiety to form an amide by a condensation reaction with an aldehyde group, a carboxyl group, or a carboxyl group.
- R b is hydrogen, lower alkyl optionally substituted with substituent group E, lower alkenyl optionally substituted with substituent group E, or lower alkyl optionally substituted with substituent group E.
- Ra is a lower alkylamino optionally substituted with the substituent group E, a lower alkenylamino optionally substituted with the substituent group E, In lower alkynylamino optionally substituted with substituent group E, carbocyclic lower alkylamino optionally substituted with substituent group E, or heterocyclic lower alkylamino optionally substituted with substituent group E A certain formula (XA4) is generated.
- Oxidative cleavage in “formation of an aldehyde group or a carboxyl group or an amide by a condensation reaction with a carboxyl group by oxidative cleavage of the olefin moiety of the formula (XA3)” is a generally known method
- the reaction can be performed under oxidation reaction conditions (for example, ozone oxidation reaction, RuCl 3 —NaIO 4 oxidation reaction, etc.).
- reaction product When the reaction product is an aldehyde form (XA3a), it can be led to a carboxyl group by subsequent general oxidation reaction conditions (for example, Cr 3 -pyridine, PCC oxidation, SO 3 -pyridine oxidation, NaClO 2 oxidation, etc.).
- general oxidation reaction for example, Cr 3 -pyridine, PCC oxidation, SO 3 -pyridine oxidation, NaClO 2 oxidation, etc.
- “Amidation by condensation reaction” is a general dehydration condensation reaction on a carboxyl group (Mitsunobu reaction, carboxylic acid halide, carboxylic anhydride, or condensing agent (eg, WSC, carbonyldiimidazole, dicyclohexylcarbodiimide), etc. Reaction using
- the “derivative of the formula (XA4 ′)” refers to an aldehyde formed by oxidative cleavage of an olefin moiety to form an amide by condensation reaction with an aldehyde group, a carboxyl group, or a carboxyl group.
- This step is a step of obtaining a solution containing the compound (X2) by reacting the compound (X1) and the compound (V1) as shown in the following reaction formula.
- the “solution” means a state in which the compound (X2) is dissolved, but a suspension in which the compound (X2) is not completely dissolved but dispersed, or a slurry state. Is also included.
- the “solution” in the present specification also includes those in a suspension state or a slurry state. (Wherein each symbol is as defined above)
- Compound (X1) is a commercially available reagent or can be obtained by a known method.
- R 1d is halogen
- the lower alcohol which may be substituted with the substituent group E, the carbocyclic lower alkyl alcohol which may be substituted with the substituent group E, or the substituent group E is substituted in a solvent.
- Lower alkyloxy which may be substituted, carbocyclic lower alkyloxy which may be substituted with substituent group E, heterocyclic lower alkyloxy which may be substituted with substituent group E, or —OSi (R 1e ) 3 (1) can also be obtained.
- Examples of “lower alkyloxy optionally substituted with substituent group E” in R 1d include methoxy, ethoxy, isopropoxy, trichloromethoxy, trifluoromethoxy and the like. Preferably it is methoxy.
- Examples of “carbocyclic lower alkyloxy optionally substituted with substituent group E” for R 1d include benzyloxy, phenethyloxy, 2,4-trifluorobenzyloxy, 4-methoxybenzyloxy and the like. . Benzyloxy is preferred.
- Examples of “heterocyclic lower alkyloxy optionally substituted with substituent group E” for R 1d include pyridylmethyloxy and the like.
- a preferred embodiment of R 1d is hydrogen, chloro, bromo, methoxy, benzyloxy.
- Examples of “lower alkyl optionally substituted with substituent group E” for R 2d include methyl, ethyl, n-propyl, iso-propyl, tert-butyl and the like.
- Examples of “carbocyclic lower alkyl optionally substituted with substituent group E” for R 2d include benzyl, 4-methoxybenzyl and the like.
- Examples of “heterocyclic lower alkyl optionally substituted with substituent group E” for R 2d include pyridylmethyl and the like. Preferred embodiments of R 2d are methyl, ethyl, n-propyl, iso-propyl, tert-butyl, benzyl and the like.
- R 1e are methyl, ethyl, n-propyl, iso-propyl, tert-butyl and the like.
- an aprotic solvent is preferable.
- examples include acetonitrile, tetrahydrofuran, dioxane, diethyl ether, dichloromethane, chloroform, toluene, xylene, ethyl acetate, N, N-dimethylformamide, N, N-dimethylacetamide, dimethylimidazolidinone and the like.
- the base may be any base that can deprotonate an alcohol reagent.
- n-butyllithium, tert-butyllithium, sodium-tert-butoxide, potassium-tert-butoxide, sodium-tert- Examples include pentoxide, sodium methoxide, sodium ethoxide, sodium hydride, lithium diisopropylamide, lithium bistrimethylsilylamide and the like.
- the amount of the base is about 1.0 to 3.0 molar equivalents relative to the compound (X1) in which R 1d is halogen.
- the amount of the alcohol reagent is about 0.5 to 1.5 molar equivalents relative to the compound (X1) in which R 1d is halogen.
- the reaction temperature is usually 0 ° C. to reflux temperature, preferably room temperature to 50 ° C.
- the reaction time is usually 10 minutes to 50 hours, preferably 1 to 4 hours.
- Compound (V1) can be obtained as a commercially available reagent or by a known method.
- Examples of "lower alkyl which may be substituted with a substituent group E" in P d is methyl, ethyl, trifluoromethyl and the like.
- a preferred embodiment of the P d is methyl.
- Examples of “lower alkyl optionally substituted with substituent group E” for R 3d include methyl, ethyl, trifluoromethyl and the like.
- Examples of “lower alkyl optionally substituted with substituent group E” for R 3e include methyl, ethyl, trifluoromethyl and the like.
- a preferred embodiment of R 3d is —N (CH 3 ) 2 , —OCH 3 , or pyrrolidinyl.
- reaction solvent for the reaction to obtain compound (X2) by reacting compound (X1) and compound (V1), for example, acetonitrile, tetrahydrofuran, dioxane, diethyl ether, dichloromethane, chloroform, toluene, xylene, ethyl acetate, N , N-dimethylformamide, N, N-dimethylacetamide, dimethylimidazolidinone and the like.
- the amount of compound (V1) to be used is about 1.0 to 3.0 molar equivalents relative to compound (X1), or compound (V1) may be used as a solvent.
- the reaction temperature is usually 0 ° C. to reflux temperature, preferably room temperature.
- the reaction time is usually 30 minutes to 50 hours, preferably 2 to 8 hours.
- Compound (X2) may be isolated by a general purification method (extraction, distillation, column chromatography, crystallization, etc.), but can also be used in the next reaction without isolation.
- Compound (X2) can also be obtained by the reaction shown below.
- R 7d is halogen, lower alkyloxy optionally substituted with substituent group E, or —O—SO 2 —R 5e , and other symbols are the same as above
- Compound (Z1) is a commercially available reagent or can be obtained by a known method.
- Examples of “lower alkyl optionally substituted with substituent group E” for R 2d include methyl, ethyl, n-propyl, iso-propyl, tert-butyl and the like.
- Examples of “carbocyclic lower alkyl optionally substituted with substituent group E” for R 2d include benzyl, 4-methoxybenzyl and the like.
- Examples of “heterocyclic lower alkyl optionally substituted with substituent group E” for R 2d include pyridylmethyl and the like. Preferred embodiments of R 2d are methyl, ethyl, n-propyl, iso-propyl, tert-butyl, benzyl and the like.
- Examples of “lower alkyl optionally substituted with substituent group E” for R 3d include methyl, ethyl, trifluoromethyl and the like.
- Examples of “lower alkyl optionally substituted with substituent group E” for R 3e include methyl, ethyl, trifluoromethyl and the like.
- a preferred embodiment of R 3d is —N (CH 3 ) 2 , —OCH 3 , or pyrrolidinyl.
- Examples of “lower alkyloxy optionally substituted with substituent group E” in R 1d include methoxy, ethoxy, isopropoxy, trichloromethoxy, trifluoromethoxy and the like. Preferably it is methoxy.
- Examples of “carbocyclic lower alkyloxy optionally substituted with substituent group E” for R 1d include benzyloxy, phenethyloxy, 2,4-trifluorobenzyloxy, 4-methoxybenzyloxy and the like. . Benzyloxy is preferred.
- Examples of “heterocyclic lower alkyloxy optionally substituted with substituent group E” for R 1d include pyridylmethyloxy and the like.
- a preferred embodiment of R 1d is hydrogen, chloro, bromo, methoxy, benzyloxy.
- Compound (Z1) is a commercially available reagent or can be obtained by a known method.
- Preferable embodiments of R 7d include chloro, bromo, methoxy, ethoxy, acetoxy, methanesulfonyloxy, trifluoromethanesulfonyloxy, paratoluenesulfonyloxy and the like.
- reaction solvent for the reaction to obtain compound (X2) by reacting compound (Z1) and compound (Z2), for example, acetonitrile, tetrahydrofuran, dioxane, diethyl ether, dichloromethane, chloroform, toluene, xylene, ethyl acetate, N , N-dimethylformamide, N, N-dimethylacetamide, dimethylimidazolidinone and the like.
- the amount of compound (Z2) to be used is about 1.0 to 3.0 molar equivalents relative to compound (Z1).
- the reaction temperature is usually ⁇ 10 ° C. to reflux temperature, preferably room temperature.
- the reaction time is usually 10 minutes to 10 hours, preferably 1 to 4 hours.
- Add tertiary amine if necessary. Examples of the tertiary amine include pyridine, triethylamine, dimethylaminopyridine, N-methylmorpholine and the like.
- Compound (X2) may be isolated by a general purification method (extraction, distillation, column chromatography, crystallization, etc.), but can also be used in the next reaction without isolation.
- This step is a step of obtaining a solution containing the compound (X3) by reacting the compound (X2) and the compound (V2) in the presence of a base as desired, as shown in the following reaction formula. (Wherein each symbol is as defined above)
- Examples of “lower alkyloxy optionally substituted with substituent group E” in R 1d include methoxy, ethoxy, isopropoxy, trichloromethoxy, trifluoromethoxy and the like. Preferably it is methoxy.
- Examples of “carbocyclic lower alkyloxy optionally substituted with substituent group E” for R 1d include benzyloxy, phenethyloxy, 2,4-trifluorobenzyloxy, 4-methoxybenzyloxy and the like. . Benzyloxy is preferred.
- Examples of “heterocyclic lower alkyloxy optionally substituted with substituent group E” for R 1d include pyridylmethyloxy and the like.
- a preferred embodiment of R 1d is hydrogen, chloro, bromo, methoxy, benzyloxy.
- Examples of “lower alkyl optionally substituted with substituent group E” for R 2d include methyl, ethyl, n-propyl, iso-propyl, tert-butyl and the like.
- Examples of “carbocyclic lower alkyl optionally substituted with substituent group E” for R 2d include benzyl, 4-methoxybenzyl and the like.
- Examples of “heterocyclic lower alkyl optionally substituted with substituent group E” for R 2d include pyridylmethyl and the like. Preferred embodiments of R 2d are methyl, ethyl, n-propyl, iso-propyl, tert-butyl and the like.
- Examples of “lower alkyl optionally substituted with substituent group E” for R 3d include methyl, ethyl, trifluoromethyl and the like.
- Examples of “lower alkyl optionally substituted with substituent group E” for R 3e include methyl, ethyl, trifluoromethyl and the like.
- a preferred embodiment of R 3d is —N (CH 3 ) 2 , —OCH 3 , or pyrrolidinyl.
- Compound (V2) can be obtained as a commercially available reagent or by a known method.
- Examples of “lower alkyl optionally substituted with substituent group E” for R 4d include methyl, ethyl, n-propyl, iso-propyl, tert-butyl and the like.
- Examples of “carbocyclic lower alkyl optionally substituted with substituent group E” for R 4d include benzyl, 4-methoxybenzyl and the like.
- heterocyclic lower alkyl optionally substituted with substituent group E” for R 4d include pyridylmethyl and the like.
- R 4d examples include methyl, ethyl, n-propyl, iso-propyl, tert-butyl, benzyl, 4-methoxybenzyl and the like.
- R 5d include chloro, bromo, methoxy, ethoxy, acetoxy, methanesulfonyloxy, trifluoromethanesulfonyloxy, paratoluenesulfonyloxy and the like. Particularly preferred are chloro, methoxy and ethoxy.
- reaction solvent examples include acetonitrile, tetrahydrofuran, dioxane, toluene, xylene, ethyl acetate, N, N-dimethylformamide, N, N-dimethylacetamide, dimethylimidazolidinone, N-methylmorpholine, N-methylpyrrolidinone and the like.
- base examples include n-butyllithium, tert-butyllithium, sodium-tert-butoxide, potassium-tert-butoxide, sodium-tert-pentoxide, sodium methoxide, sodium ethoxide, sodium hydride, lithium diisopropylamide, And lithium bistrimethylsilylamide.
- the amount of base used is about 1.0 to 5.0 molar equivalents relative to compound (X2), and the amount of compound (V2) used is about 1.5 to 5.0 relative to compound (X2).
- Molar equivalents or compound (V2) may be used with a solvent.
- the reaction temperature is usually ⁇ 80 ° C. to reflux temperature, preferably ⁇ 20 ° C. to 50 ° C.
- the reaction time is usually 30 minutes to 50 hours, preferably 2 to 12 hours.
- Compound (X3) may be isolated by a general purification method (extraction, distillation, column chromatography, crystallization, etc.), but can also be used in the next reaction without isolation. Preferably, it is isolated as a crystal from which impurities have been removed by crystallization.
- This step is a step of obtaining a solution containing the compound (XA3) by reacting the compound (X2) and the compound (VA2) in the presence of a base as desired, as shown in the following reaction formula. (Wherein each symbol is as defined above)
- R 1d , R 2d , R 3d , and R 5d in the formulas (X2) and (VA2) are the same as described above.
- Compound (VA2) can be obtained as a commercially available reagent or by a known method.
- Examples of “lower alkyl optionally substituted with substituent group E” for R 4e include methyl, ethyl and the like.
- Examples of the “carbocyclic group optionally substituted with substituent group E” for R 4e include phenyl and cyclohexyl.
- Examples of the “heterocyclic group optionally substituted with substituent group E” for R 4e include pyridyl and piperazyl.
- R 4e is preferably one in which one is hydrogen and the other is phenyl.
- the reaction solvent include acetonitrile, tetrahydrofuran, dioxane, toluene, xylene, ethyl acetate, N, N-dimethylformamide, N, N-dimethylacetamide, dimethylimidazolidinone, N-methylmorpholine, N-methylpyrrolidinone and the like. Can be mentioned.
- Examples of the base include n-butyllithium, tert-butyllithium, sodium-tert-butoxide, potassium-tert-butoxide, sodium-tert-pentoxide, sodium methoxide, sodium ethoxide, sodium hydride, lithium diisopropylamide, And lithium bistrimethylsilylamide.
- the amount of base used is about 1.0 to 5.0 molar equivalents relative to compound (XA2), and the amount of compound (VA2) used is about 1.0 to 2.0 relative to compound (X2). Molar equivalents.
- the reaction temperature is usually ⁇ 80 ° C. to 20 ° C., preferably ⁇ 80 ° C. to ⁇ 40 ° C.
- the reaction time is usually 5 minutes to 6 hours, preferably 15 minutes to 2 hours.
- Compound (XA3) is obtained.
- Compound (XA3) may be isolated by a general purification method (extraction, distillation, column chromatography, crystallization, etc.), but can also be used in the next reaction without isolation. Preferably, it is isolated as a crystal from which impurities have been removed by crystallization.
- This step is a step of obtaining compound (X4) by reacting compound (X3) and compound (V3) as shown in the following reaction formula. (Wherein each symbol is as defined above)
- Examples of “lower alkyloxy optionally substituted with substituent group E” in R 1d include methoxy, ethoxy, isopropoxy, trichloromethoxy, trifluoromethoxy and the like. Preferably it is methoxy.
- Examples of “carbocyclic lower alkyloxy optionally substituted with substituent group E” for R 1d include benzyloxy, phenethyloxy, 2,4-trifluorobenzyloxy, 4-methoxybenzyloxy and the like. . Benzyloxy is preferred.
- Examples of “heterocyclic lower alkyloxy optionally substituted with substituent group E” for R 1d include pyridylmethyloxy and the like.
- a preferred embodiment of R 1d is hydrogen, chloro, bromo, methoxy, benzyloxy.
- Examples of “lower alkyl optionally substituted with substituent group E” for R 2d include methyl, ethyl, n-propyl, iso-propyl, tert-butyl and the like.
- Examples of “carbocyclic lower alkyl optionally substituted with substituent group E” for R 2d include benzyl, 4-methoxybenzyl and the like.
- Examples of “heterocyclic lower alkyl optionally substituted with substituent group E” for R 2d include pyridylmethyl and the like. Preferred embodiments of R 2d are methyl, ethyl, n-propyl, iso-propyl, tert-butyl and the like.
- Examples of “lower alkyl optionally substituted with substituent group E” for R 4d include methyl, ethyl, n-propyl, iso-propyl, tert-butyl and the like.
- Examples of “carbocyclic lower alkyl optionally substituted with substituent group E” for R 4d include benzyl, 4-methoxybenzyl and the like.
- Examples of “heterocyclic lower alkyl optionally substituted with substituent group E” for R 4d include pyridylmethyl and the like.
- Preferred embodiments of R 4d include methyl, ethyl, n-propyl, iso-propyl, tert-butyl, benzyl, 4-methoxybenzyl and the like.
- Compound (V3) can be obtained as a commercially available reagent or by a known method.
- “lower alkyl optionally substituted with substituent group E” for R 6d include HC ( ⁇ O) —CH 2 —, CH (—OH) 2 —CH 2 —, MeO—CH (—OH ) —CH 2 —, dimethoxyethyl, diethoxyethyl, CH 2 ⁇ CH—CH 2 —, HO—CH 2 —CH (—OH) —CH 2 —, Or Etc.
- Examples of “amino optionally substituted with substituent E” in R 6d include methylamino, ethylamino, benzyloxycarbonylamino, tert-butoxycarbonylamino and the like.
- Examples of “carbocyclic group optionally substituted with substituent group E” for R 6d include Etc.
- Examples of the “heterocyclic group optionally substituted with substituent group E” in R 6d include Etc.
- reaction solvent examples include acetonitrile, tetrahydrofuran, dioxane, toluene, xylene, ethyl acetate, N, N-dimethylformamide, N, N-dimethylacetamide, dimethylimidazolidinone, N-methylmorpholine, N-methylpyrrolidinone, methanol. , Ethanol, isopropanol and the like.
- the amount of compound (V3) to be used is about 1.0 to 2.0 molar equivalents relative to compound (X3).
- the reaction temperature is usually 0 ° C. to reflux temperature, preferably 20 ° C. to 70 ° C.
- the reaction time is usually 30 minutes to 50 hours, preferably 2 to 12 hours.
- R 6d of the produced compound (X4) is an aldehyde group such as HC ( ⁇ O) —CH 2 —, MeO—CH (—OH) —CH 2 —, or CH (—OH) 2 —CH 2 —, or an equivalent thereof. If it is not a group having an isomer, a method for deprotecting the protecting group of the aldehyde group described in Protective Groups in Organic Synthesis, Theodora W Green (John Wiley & Sons), or International Publication No.
- Examples of the acid include, but are not limited to, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, p-toluenesulfonic acid, methanesulfonic acid, formic acid, acetic acid, trifluoroacetic acid, maleic acid, oxalic acid, etc. .
- the amount of the acid used is 2.0 to 10.0 molar equivalents relative to compound (X4).
- Acetic acid and formic acid may be used as a solvent, or may be used by mixing with the above acid.
- the reaction temperature is usually about 0 ° C. to 80 ° C., preferably 10 ° C. to 40 ° C.
- the reaction time is usually 30 minutes to 50 hours, preferably 2 to 12 hours.
- the protecting group of the amino group described in Protective Groups in Organic Synthesis, Theodora W Green (John Wiley & Sons) or a known method can be used.
- Deprotected compounds can also be obtained. The order in which the deprotection reactions are performed can be changed from time to time.
- Compound (X4) may be isolated by a general purification method (extraction, distillation, column chromatography, crystallization, etc.), but can also be used in the next reaction without isolation. Preferably, it is isolated as a crystal from which impurities have been removed by crystallization.
- This step is a step of obtaining a solution containing the compound (XA4) by reacting the compound (XA3) and the compound (V3) in the presence of a base as desired, as shown in the following reaction formula. (Wherein each symbol is as defined above)
- formulas (XA3) and (VA4) examples of R 1d , R 2d and R 4e and preferred embodiments thereof are the same as described above.
- Examples of “lower alkylamino optionally substituted with substituent group E” in R a in formula (XA4) include methylamino, ethylamino, isopropylamino, tert-butylamino, methoxymethylamino, methoxyethyl Amino, ethoxyethylamino, methoxypropylamino, hydroxyethylamino, piperazinylcarbonylethylamino, morpholinylcarbonylethylamino, methylaminoethylamino, methylsulfonylethylamino, tert-butylcarbonylaminoethylamino, isopropyloxycarbonylamino Examples include ethylamino, methylcarbonylaminoethylamino, aminoethylamino, tert-butyloxycarbonylaminoethylamino and the like.
- Examples of “lower alkenylamino optionally substituted with substituent group E” for R a include ethylenylamino, propenylamino and the like. Examples of “lower alkynylamino optionally substituted with substituent group E” for R a include propynylamino.
- Examples of the “carbocyclic lower alkylamino optionally substituted with substituent group E” for R a include benzylamino, difluorobenzylamino, chlorofluorobenzylamino, cyclopropylmethylamino, 4-fluorobenzylamino, cyclohexyl Examples include methylenylamino, cyclopropylamino, ethyloxycarbonylethylamino, carboxyethylamino, dimethylaminocarbonyl, 4-methoxybenzylamino, 4-methylbenzylamino and the like.
- heterocyclic lower alkylamino optionally substituted with substituent group E examples include pyridylmethylamino, tetrahydropyranylmethylenylamino, methylisoxazolylmethylenylamino and the like.
- Examples of “lower alkyl optionally substituted with substituent group E” for R 6d include HC ( ⁇ O) —CH 2 —, CH (—OH) 2 —CH 2 —, MeO—CH (—OH ) —CH 2 —, dimethoxyethyl, diethoxyethyl, CH 2 ⁇ CH—CH 2 —, HO—CH 2 —CH (—OH) —CH 2 —, Or
- R a is hydroxy, R 6d is
- R a is “lower alkylamino optionally substituted with substituent group E”, “lower alkenylamino optionally substituted with substituent group E”, “substituted with substituent group E”
- R 6d is Etc.
- Examples of “amino optionally substituted with substituent group E” in R 6d include methylamino, ethylamino, benzyloxycarbonylamino, tert-butoxycarbonylamino and the like.
- Examples of “carbocyclic group optionally substituted with substituent group E” for R 6d include Etc.
- Examples of the “heterocyclic group optionally substituted with substituent group E” in R 6d include Etc.
- R a is “lower alkylamino optionally substituted with substituent group E”, “lower alkenylamino optionally substituted with substituent group E”, “optionally substituted with substituent group E”
- a carboxyl group R a is hydroxy by general dehydration condensation reaction (Mitsunobu reaction, carboxylic acid halide, carboxylic anhydride, or a reaction using a condensing agent (for example, WSC, carbonyldiimidazole, dicyclohexylcarbodiimide)) Can do.
- reaction solvent examples include acetonitrile, tetrahydrofuran, dioxane, toluene, xylene, ethyl acetate, N, N-dimethylformamide, N, N-dimethylacetamide, dimethylimidazolidinone, N-methylmorpholine, N-methylpyrrolidinone, methanol. , Ethanol, isopropanol and the like.
- the amount of compound (V3) to be used is about 1.0 to 3.0 molar equivalents relative to compound (XA3).
- the reaction temperature is usually 0 ° C. to reflux temperature, preferably 20 ° C. to 70 ° C.
- the reaction time is usually 10 minutes to 50 hours, preferably 1 to 12 hours.
- R 6d of the produced compound (X4) is an aldehyde group such as HC ( ⁇ O) —CH 2 —, MeO—CH (—OH) —CH 2 —, or CH (—OH) 2 —CH 2 —, or an equivalent thereof.
- the group is not a group having a isomer
- HC ( ⁇ O) —CH 2 —, MeO—CH (—OH) —CH 2 — or CH which is a group having an aldehyde group or an equivalent thereof, is obtained by the same method as described above.
- (—OH) 2 —CH 2 — and the like is obtained by the same method as described above.
- the protecting group of the amino group described in Protective Groups in Organic Synthesis, Theodora W Green (John Wiley & Sons) or a known method can be used.
- Deprotected compounds can also be obtained. The order in which the deprotection reactions are performed can be changed from time to time.
- Compound (XA4) may be isolated by a general purification method (extraction, distillation, column chromatography, crystallization, etc.), but can also be used in the next reaction without isolation. Preferably, it is isolated as a crystal from which impurities have been removed by crystallization.
- This step is a step of obtaining compound (X4 ′) by reacting compound (X2) with compound (V2) and compound (V2 ′) in the presence of a base as desired, as shown in the following reaction formula. (Wherein each symbol is as defined above) Examples and preferred embodiments of R 1d , R 2d , R 3d , R 4d , and R 5d in formulas (X2) and (V2) are the same as described above.
- reaction solvent examples include acetonitrile, tetrahydrofuran, dioxane, toluene, xylene, ethyl acetate, N, N-dimethylformamide, N, N-dimethylacetamide, dimethylimidazolidinone, N-methylmorpholine, N-methylpyrrolidinone and the like.
- base examples include n-butyllithium, tert-butyllithium, sodium-tert-butoxide, potassium-tert-butoxide, sodium-tert-pentoxide, sodium methoxide, sodium ethoxide, sodium hydride, lithium diisopropylamide, And lithium bistrimethylsilylamide.
- the amount of base used is about 1.0 to 5.0 molar equivalents relative to compound (X2), and the amount of compound (V2) used is about 1.0 to 3.0 relative to compound (X2).
- Molar equivalents or compound (V2) may be used with a solvent.
- the reaction temperature is usually ⁇ 80 ° C. to reflux temperature, preferably ⁇ 20 ° C. to 30 ° C.
- the reaction time is usually 10 minutes to 10 hours, preferably 30 minutes to 4 hours. Subsequently, the compound (V2 ′) is added to the reaction solution and reacted.
- Examples of the compound (V2 ′) include ammonium acetate, ammonium chloride, ammonium bromide, ammonium sulfate, ammonium hydrogen sulfate, ammonium formate, ammonium nitrate, ammonium hydroxide, ammonium phosphate, NH 4 + BF 4 ⁇ , NH 4 + PF 6 ⁇ , NH 4 + Ph—SO 3 ⁇ , NH 4 + CH 3 —Ph—SO 3 ⁇ , NH 4 + CH 3 —SO 3 ⁇ , and the like.
- Ammonium acetate, ammonium chloride, ammonium sulfate, ammonium hydrogen sulfate, and ammonium formate are preferred.
- the amount of compound (V2 ′) to be used is about 1.0-3.0 molar equivalents with respect to compound (X2).
- the reaction temperature is usually 0 ° C. to reflux temperature, preferably 20 ° C. to 80 ° C.
- the reaction time is usually 10 minutes to 10 hours, preferably 30 minutes to 4 hours.
- Compound (X4 ′) may be isolated by a general purification method (extraction, distillation, column chromatography, crystallization, etc.), but can also be used in the next reaction without isolation. Preferably, it is isolated as a crystal from which impurities have been removed by crystallization.
- This step is a step of obtaining compound (X4) by reacting compound (X4 ′) and compound (V3 ′) in the presence of a base as desired, as shown in the following reaction formula.
- a base as desired
- Examples and preferred embodiments of R 1d , R 2d , R 4d and R 6d in the formulas (X4 ′) and (V3 ′) are the same as described above.
- the “leaving group” in L d is halogen, —O—SO 2 —CH 3 , —O—SO 2 —CF 3 , —O—SO 2 —Ph, —O—SO 2 —Ph—CH 3 or the like. Is mentioned. Preferably, it is halogen.
- R 6d of the produced compound (X4) is an aldehyde group such as HC ( ⁇ O) —CH 2 —, MeO—CH (—OH) —CH 2 —, or CH (—OH) 2 —CH 2 —, or an equivalent thereof.
- the method for deriving an aldehyde group or its equivalent when no body is present is the same as described above.
- reaction solvent examples include acetonitrile, ethyl acetate, N, N-dimethylformamide, N, N-dimethylacetamide, dimethylimidazolidinone, N-methylmorpholine, N-methylpyrrolidinone and the like.
- Examples of the base include potassium carbonate, cesium carbonate, sodium hydride, n-butyllithium, tert-butyllithium, sodium-tert-butoxide, potassium-tert-butoxide, sodium-tert-pentoxide, sodium methoxide, triethylamine, 4- Examples thereof include dimethylaminopyridine, diisopropylethylamine, DBU (1,8-diazabicyclo [5.4.0] undec-7-ene).
- the amount of the base used is about 1.0 to 5.0 molar equivalents relative to the compound (X4 ′), and the amount of the compound (V3 ′) used is about 1.0 to about 5.0 to the compound (X4 ′).
- reaction temperature is usually 0 ° C. to reflux temperature, preferably 20 ° C. to 80 ° C.
- the reaction time is usually 30 minutes to 24 hours, preferably 1 to 8 hours.
- Compound (X4) may be isolated by a general purification method (extraction, distillation, column chromatography, crystallization, etc.), but can also be used in the next reaction without isolation. Preferably, it is isolated as a crystal from which impurities have been removed by crystallization.
- R 6d in compound (X4) is —NH 2
- it can also be obtained by reacting compound (X4 ′) with O- (2,4-dinitrophenyl) hydroxylamine reagent in the presence of a base, if desired.
- the reaction solvent include N, N-dimethylformamide, N, N-dimethylacetamide, dimethylimidazolidinone, N-methylmorpholine, N-methylpyrrolidinone and the like.
- the base include potassium carbonate, cesium carbonate, sodium carbonate, lithium carbonate and the like.
- the amount of the base used is about 1.0 to 5.0 molar equivalents relative to compound (X4 ′), and the amount of the reagent used is about 1.0 to 4.0 relative to compound (X4 ′). Molar equivalents.
- the reaction temperature is usually 0 ° C. to reflux temperature, preferably 20 ° C. to 60 ° C.
- the reaction time is usually 30 minutes to 24 hours, preferably 1 to 8 hours.
- This step is a step of obtaining compound (I) when compound (X4A) has the following structure.
- R 15 is an amino protecting group, hydrogen, lower alkyl optionally substituted with substituent group E, lower alkenyl optionally substituted with substituent group E, or substituted with substituent group E.
- a ring-closed product can be obtained by causing an intramolecular dehydration condensation reaction (for example, Mitsunobu reaction) between the amide moiety (—CONH—R 15 ) and the hydroxy group of the compound (XA4).
- an intramolecular dehydration condensation reaction for example, Mitsunobu reaction
- R 15 is an amino protecting group
- the intramolecular dehydration condensation reaction can be performed after subjecting to a known deprotection reaction.
- R 7 is not hydrogen
- the target R 7 can be obtained by performing a known nucleophilic substitution reaction on the amino group.
- R 1d is lower alkyloxy optionally substituted with substituent group E, carbocyclic lower alkyloxy optionally substituted with substituent group E, heterocyclic lower ring optionally substituted with substituent group E
- alkyloxy or —OSi (R 1e ) 3 it can be led to a hydroxy group by subjecting to a known hydroxy deprotection reaction.
- R 1d is halogen, it can be led to a hydroxy group by reacting potassium trimethylsilanolate and lithium trimethylsilanolate and adding an aqueous solution of an inorganic acid.
- sodium hydride / water (Bioorganic Medicinal Chemistry Letters, 17, 1713, 2007), potassium hydroxide / tris (dibenzylideneacetone) dipalladium (Pd 2 dba 3 ) / di-tert-butylarylphosphine (Journal) of the American Chemical Society, 128, 10694, 2006), potassium phosphate hydrate (K 3 PO 4 .H 2 O) / tris (dibenzylideneacetone) dipalladium (Pd 2 dba 3 ) / tri-tert-butyl Phosphine (Tetrahedoron Letters, 48, 473, 2007) is also mentioned as a conversion reaction of halogen to a hydroxy group.
- R 1d of the raw material when R 1d of the raw material is halogen, it can be derivatized as it is, so that the reaction constant is reduced as compared with the method of carrying out alcohol protection and / or deprotection reaction. It is possible to construct a more advantageous industrial manufacturing method.
- R 1d is hydrogen
- R 1d is converted to halogen by reacting with a halogenating agent such as N-bromosuccinimide, N-chlorosuccinimide, or sulfuryl chloride, and then potassium trimethylsilanolate is similarly converted. It can also lead to a hydroxy group by reacting lithium trimethylsilanolate and adding an aqueous solution of an inorganic acid. Therefore, these R 1d can be appropriately selected according to the reactivity of the reaction substrate. In addition, in the said process, the order of each reaction can be changed suitably.
- This step is a step of obtaining compound (II) when compound (X4A) has the following structure.
- a ring-closed product can be obtained by causing an intramolecular dehydration condensation reaction (for example, Mitsunobu reaction) between the amide moiety (—CONH—R 15 ) and the hydroxy group of the compound (XA4).
- R 15 is an amino protecting group
- the intramolecular dehydration condensation reaction can be performed after subjecting to a known deprotection reaction.
- R 7 is not hydrogen
- the target R 7 can be obtained by performing a known nucleophilic substitution reaction on the amino group.
- R 1d is lower alkyloxy optionally substituted with substituent group E, carbocyclic lower alkyloxy optionally substituted with substituent group E, heterocyclic lower ring optionally substituted with substituent group E
- alkyloxy or —OSi (R 1e ) 3 it can be led to a hydroxy group by subjecting to a known hydroxy deprotection reaction.
- R 1d is halogen, it can be led to a hydroxy group by reacting potassium trimethylsilanolate and lithium trimethylsilanolate and adding an aqueous solution of an inorganic acid.
- sodium hydride / water (Bioorganic Medicinal Chemistry Letters, 17, 1713, 2007), potassium hydroxide / tris (dibenzylideneacetone) dipalladium (Pd 2 dba 3 ) / di-tert-butylarylphosphine (Journal) of the American Chemical Society, 128, 10694, 2006), potassium phosphate hydrate (K 3 PO 4 .H 2 O) / tris (dibenzylideneacetone) dipalladium (Pd 2 dba 3 ) / tri-tert-butyl Phosphine (Tetrahedoron Letters, 48, 473, 2007) is also mentioned as a conversion reaction of halogen to a hydroxy group.
- R 1d of the raw material when R 1d of the raw material is halogen, it can be derivatized as it is, so that the reaction constant is reduced as compared with the method of carrying out alcohol protection and / or deprotection reaction. It is possible to construct a more advantageous industrial manufacturing method.
- R 1d is hydrogen
- R 1d is converted to halogen by reacting with a halogenating agent such as N-bromosuccinimide, N-chlorosuccinimide, or sulfuryl chloride, and then potassium trimethylsilanolate is similarly converted. It can also lead to a hydroxy group by reacting lithium trimethylsilanolate and adding an aqueous solution of an inorganic acid. Therefore, these R 1d can be appropriately selected according to the reactivity of the reaction substrate. In addition, in the said process, the order of each reaction can be changed suitably.
- This step is a step of obtaining compound (I) when compound (X4A) has the following structure. (Wherein each symbol is as defined above) Obtaining a closed ring by causing an intramolecular dehydration condensation reaction (for example, Mitsunobu reaction, amidation reaction using a condensing agent, etc.) to the carboxyl group (—COOH) and amino group of compound (XA4). Can do.
- an intramolecular dehydration condensation reaction for example, Mitsunobu reaction, amidation reaction using a condensing agent, etc.
- R 15 is an amino protecting group
- the intramolecular dehydration condensation reaction can be performed after subjecting to a known deprotection reaction.
- R 7 is not hydrogen, the target R 7 can be obtained by performing a known nucleophilic substitution reaction on the amino group.
- R 1d is lower alkyloxy optionally substituted with substituent group E, carbocyclic lower alkyloxy optionally substituted with substituent group E, heterocyclic lower ring optionally substituted with substituent group E
- alkyloxy or —OSi (R 1e ) 3 it can be led to a hydroxy group by subjecting to a known hydroxy deprotection reaction.
- R 1d is halogen, it can be led to a hydroxy group by reacting potassium trimethylsilanolate and lithium trimethylsilanolate and adding an aqueous solution of an inorganic acid.
- sodium hydride / water (Bioorganic Medicinal Chemistry Letters, 17, 1713, 2007), potassium hydroxide / tris (dibenzylideneacetone) dipalladium (Pd 2 dba 3 ) / di-tert-butylarylphosphine (Journal) of the American Chemical Society, 128, 10694, 2006), potassium phosphate hydrate (K 3 PO 4 .H 2 O) / tris (dibenzylideneacetone) dipalladium (Pd 2 dba 3 ) / tri-tert-butyl Phosphine (Tetrahedoron Letters, 48, 473, 2007) is also mentioned as a conversion reaction of halogen to a hydroxy group.
- R 1d of the raw material when R 1d of the raw material is halogen, it can be derivatized as it is, so that the reaction constant is reduced as compared with the method of carrying out alcohol protection and / or deprotection reaction. It is possible to construct a more advantageous industrial manufacturing method.
- R 1d is hydrogen
- R 1d is converted to halogen by reacting with a halogenating agent such as N-bromosuccinimide, N-chlorosuccinimide, or sulfuryl chloride, and then potassium trimethylsilanolate is similarly converted. It can also lead to a hydroxy group by reacting lithium trimethylsilanolate and adding an aqueous solution of an inorganic acid. Therefore, these R 1d can be appropriately selected according to the reactivity of the reaction substrate. In addition, in the said process, the order of each reaction can be changed suitably.
- This step is a step of obtaining compound (II) when compound (X4A) has the following structure. (Wherein each symbol is as defined above) Obtaining a closed ring by causing an intramolecular dehydration condensation reaction (for example, Mitsunobu reaction, amidation reaction using a condensing agent, etc.) to the carboxyl group (—COOH) and amino group of compound (XA4). Can do.
- an intramolecular dehydration condensation reaction for example, Mitsunobu reaction, amidation reaction using a condensing agent, etc.
- R 15 is an amino protecting group
- the intramolecular dehydration condensation reaction is performed after subjecting to a known deprotection reaction.
- R 7 is not hydrogen, the target R 7 can be obtained by performing a known nucleophilic substitution reaction on the amino group.
- R 1d is lower alkyloxy optionally substituted with substituent group E, carbocyclic lower alkyloxy optionally substituted with substituent group E, heterocyclic lower ring optionally substituted with substituent group E
- alkyloxy or —OSi (R 1e ) 3 it can be led to a hydroxy group by subjecting to a known hydroxy deprotection reaction.
- R 1d is halogen, it can be led to a hydroxy group by reacting potassium trimethylsilanolate and lithium trimethylsilanolate and adding an aqueous solution of an inorganic acid.
- sodium hydride / water (Bioorganic Medicinal Chemistry Letters, 17, 1713, 2007), potassium hydroxide / tris (dibenzylideneacetone) dipalladium (Pd 2 dba 3 ) / di-tert-butylarylphosphine (Journal) of the American Chemical Society, 128, 10694, 2006), potassium phosphate hydrate (K 3 PO 4 .H 2 O) / tris (dibenzylideneacetone) dipalladium (Pd 2 dba 3 ) / tri-tert-butyl Phosphine (Tetrahedoron Letters, 48, 473, 2007) is also mentioned as a conversion reaction of halogen to a hydroxy group.
- R 1d of the raw material when R 1d of the raw material is halogen, it can be derivatized as it is, so that the reaction constant is reduced as compared with the method of carrying out alcohol protection and / or deprotection reaction. It is possible to construct a more advantageous industrial manufacturing method.
- R 1d is hydrogen
- R 1d is converted to halogen by reacting with a halogenating agent such as N-bromosuccinimide, N-chlorosuccinimide, or sulfuryl chloride, and then potassium trimethylsilanolate is similarly converted. It can also lead to a hydroxy group by reacting lithium trimethylsilanolate and adding an aqueous solution of an inorganic acid. Therefore, these R 1d can be appropriately selected according to the reactivity of the reaction substrate. In addition, in the said process, the order of each reaction can be changed suitably.
- This step is a step of obtaining compound (II) when compound (XA4) has the following structure.
- Ring closure by causing condensation reaction of the amide moiety (—CONH—R 15 ) and amino group (—NH—R 3 ) of compound (X4) with the compound (R 5 —CO—R 6 ) having a carbonyl group Can be obtained.
- R 5 —CO—R 6 is paraformaldehyde, where R 5 and R 6 are hydrogen.
- an acid is added as needed. Examples of the acid include acetic acid, formic acid, sulfuric acid and the like.
- the condensation reaction can be carried out after subjecting to a known deprotection reaction.
- R 7 is not hydrogen
- the target R 7 can be obtained by performing a known nucleophilic substitution reaction on the amino group.
- Known reaction -COOR 2d site if R 2d is not hydrogen (the ester hydrolysis reaction, deprotection reaction, such as a carboxyl protecting group) performs, after the R 2d and hydrogen, substituted with a substituent group E
- a known reaction such as a dehydration condensation reaction
- a carbocyclic lower alkylamine which may be substituted
- a lower alkyl alcohol which may be substituted with substituent group E, etc.
- R 2d is a lower alkyl group such as a methyl group or an ethyl group
- R 1d is lower alkyloxy optionally substituted with substituent group E, carbocyclic lower alkyloxy optionally substituted with substituent group E, heterocyclic lower ring optionally substituted with substituent group E
- alkyloxy or —OSi (R 1e ) 3 it can be led to a hydroxy group by subjecting to a known hydroxy deprotection reaction.
- R 1d When R 1d is halogen, it can be led to a hydroxy group by reacting potassium trimethylsilanolate and lithium trimethylsilanolate and adding an aqueous solution of an inorganic acid.
- sodium hydride / water Bioorganic Medicinal Chemistry Letters, 17, 1713, 2007
- potassium hydroxide / tris dibenzylideneacetone) dipalladium (Pd 2 dba 3 ) / di-tert-butylarylphosphine (Journal) of the American Chemical Society, 128, 10694, 2006
- potassium phosphate hydrate K 3 PO 4 .H 2 O
- tris dibenzylideneacetone dipalladium dipd 2 dba 3 ) / tri-tert-butyl Phosphine
- Tetrahedoron Letters, 48, 473, 2007 is also mentioned as a conversion reaction of halogen to a hydroxy group.
- R 1d of the raw material when R 1d of the raw material is halogen, it can be derivatized as it is, so that the reaction constant is reduced as compared with the method of carrying out alcohol protection and / or deprotection reaction. It is possible to construct a more advantageous industrial manufacturing method.
- R 1d is hydrogen
- R 1d is converted to halogen by reacting with a halogenating agent such as N-bromosuccinimide, N-chlorosuccinimide, or sulfuryl chloride, and then potassium trimethylsilanolate is similarly converted. It can also lead to a hydroxy group by reacting lithium trimethylsilanolate and adding an aqueous solution of an inorganic acid. Therefore, these R 1d can be appropriately selected according to the reactivity of the reaction substrate. In addition, in the said process, the order of each reaction can be changed suitably.
- This step is a step of obtaining compound (III) when compound (X4) has the following structure. (Wherein each symbol is as defined above) By reacting the aldehyde moiety of compound (X4) with the amino group of compound (V4) and —Y—H, a tricyclic compound can be obtained. When Z is a single bond, R 14 and R 10 may together form a heterocyclic group which may be substituted with a 4- to 8-membered substituent group E. In this case, the compound ( III) is a tetracyclic compound. Examples of compound (V4) include 3-aminobutanol.
- R 1d is lower alkyloxy optionally substituted with substituent group E, carbocyclic lower alkyloxy optionally substituted with substituent group E, heterocyclic lower ring optionally substituted with substituent group E
- alkyloxy or —OSi (R 1e ) 3 it can be led to a hydroxy group by subjecting to a known hydroxy deprotection reaction.
- R 1d is halogen, it can be led to a hydroxy group by reacting potassium trimethylsilanolate and lithium trimethylsilanolate and adding an aqueous solution of an inorganic acid.
- sodium hydride / water (Bioorganic Medicinal Chemistry Letters, 17, 1713, 2007), potassium hydroxide / tris (dibenzylideneacetone) dipalladium (Pd 2 dba 3 ) / di-tert-butylarylphosphine (Journal) of the American Chemical Society, 128, 10694, 2006), potassium phosphate hydrate (K 3 PO 4 .H 2 O) / tris (dibenzylideneacetone) dipalladium (Pd 2 dba 3 ) / tri-tert-butyl Phosphine (Tetrahedoron Letters, 48, 473, 2007) is also mentioned as a conversion reaction of halogen to a hydroxy group.
- R 1d of the raw material when R 1d of the raw material is halogen, it can be derivatized as it is, so that the reaction constant is reduced as compared with the method of carrying out alcohol protection and / or deprotection reaction. It is possible to construct a more advantageous industrial manufacturing method.
- R 1d is hydrogen
- R 1d is converted to halogen by reacting with a halogenating agent such as N-bromosuccinimide, N-chlorosuccinimide, or sulfuryl chloride, and then potassium trimethylsilanolate is similarly converted. It can also lead to a hydroxy group by reacting lithium trimethylsilanolate and adding an aqueous solution of an inorganic acid. Therefore, these R 1d can be appropriately selected according to the reactivity of the reaction substrate. In addition, in the said process, the order of each reaction can be changed suitably.
- N-dimethylformamide dimethyl acetal (4.9 ml, 36.5 mmol) was added dropwise to compound 3A (5.0 g, 30.4 mmol) at 0 ° C. with cooling.
- 100 ml of ethyl acetate was added to the reaction solution, followed by washing with 0.5N aqueous hydrochloric acid (50 ml).
- the aqueous layer was separated and extracted with ethyl acetate (50 ml).
- the organic layers were combined, washed successively with saturated aqueous sodium hydrogen carbonate and saturated brine, and dried over anhydrous sodium sulfate.
- Step 3 Add aminoacetaldehyde dimethyl acetal (0.13 ml, 1.20 mmol) to a solution of compound 3C (300 mg, 1.09 mmol) in ethanol (6 ml) at 0 ° C, 1 hour 30 minutes at 0 ° C, and 18 hours at room temperature. Then, the mixture was stirred at 60 ° C. for 4 hours.
- the reaction mixture was cooled to room temperature, diluted with chloroform, washed with saturated aqueous sodium hydrogen carbonate, and the aqueous layer was extracted with chloroform.
- the chloroform layers were combined, washed with saturated brine, and dried over anhydrous sodium sulfate.
- the solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (chloroform-methanol 100: 0 ⁇ 90: 10) to obtain 309.4 mg of compound 3F as a brown oil.
- N-dimethylformamide dimethyl acetal (12.2 ml, 92.2 mmol) was added dropwise to compound 4A (10.0 g, 76.8 mmol) at 0 ° C. with cooling. After stirring at 0 ° C. for 1 hour 30 minutes and then at room temperature for 2 hours 30 minutes, 100 ml of ethyl acetate was added to the reaction solution, and the solvent was distilled off. The obtained residue was purified by silica gel column chromatography (n-hexane-ethyl acetate 5: 5 ⁇ 0: 10 (v / v)) to obtain 12.45 g (yield 88%) of compound 4B as an oil.
- the reaction mixture was cooled to room temperature, diluted with chloroform, washed with saturated aqueous sodium hydrogen carbonate, and the aqueous layer was extracted with chloroform.
- the chloroform layers were combined, washed with saturated brine, and dried over anhydrous sodium sulfate.
- the solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (chloroform-methanol 100: 0 ⁇ 90: 10) to obtain 1.58 g of compound 4G as an oil.
- Second step Add cesium carbonate (73.3 mg, 0.23 mmol) and bromoacetaldehyde dimethyl acetal (38.0 mg, 0.23 mmol) to a solution of compound 6B (51.8 mg, 0.15 mmol) in N, N-dimethylformamide (1 ml) at room temperature. Stir overnight. Cesium carbonate (73.3 mg, 0.23 mmol) and bromoacetaldehyde dimethyl acetal (38.0 mg, 0.23 mmol) were added, and the mixture was further stirred at 100 ° C. for 20 minutes. Water was added to the reaction solution, followed by extraction with ethyl acetate. The organic layer was washed successively with water and saturated brine, and then dried over anhydrous sodium sulfate.
- Second Step 62% -H 2 SO 4 (316 mg, 2.0 mmol) was added to a solution of compound 6C (433.5 mg, 1.0 mmol) in formic acid (4 ml), and the mixture was stirred at room temperature for 3 hours.
- Methylene chloride was added to the reaction solution, washed with 0.5N aqueous sodium hydroxide solution (12 ml), and the aqueous layer was extracted with methylene chloride. The methylene chloride layers were combined, washed with saturated brine, and dried over anhydrous sodium sulfate. The solvent was distilled off to obtain 207.6 mg (yield 51%) of Compound 8C as a yellow foam.
- Step 3 Under a nitrogen stream, a solution of Compound 11C and ruthenium chloride (2.76 mg, 0.0133 mmol) in MeCN (5 ml) at room temperature with sodium periodate (625.8 mg, 2.93 mmol) and 96% sulfuric acid (287.4 mg, 2.93 mmol) aqueous solution (8 ml) was added dropwise over 10 minutes. After stirring the reaction solution at the same temperature for 5 minutes, ethyl acetate was added to separate the organic layer, and the aqueous layer was extracted twice with ethyl acetate. The combined extract was dried over sodium sulfate. The solvent was distilled off, and the resulting oil was purified by silica gel column chromatography.
- Step 5 To a solution of compound 11E (1.00 g, 3.14 mmol) in DMF (10 ml) at room temperature, add WSC HCl (1.20 g, 6.28 mmol) and HOBt (551.6 mg, 4.08 mmol), and stir at the same temperature for 90 minutes did. The reaction solution was cooled to 0 ° C., and a DMF (2 ml) solution of 2-methoxyethanamine (236.0 mg, 3.14 mmol) was added dropwise over 3 minutes. The reaction mixture was stirred at the same temperature for 1 hour, water was added, and the mixture was extracted 3 times with ethyl acetate. The extract was washed with water three times and dried over sodium sulfate.
- Step 6 Add dimethyl 3- (benzyloxy) -4-oxo-4H-pyran-2,5-dicarboxylate (974 mg, 3.06 mmol) and 12F (999 mg, 3.06 mmol) to toluene (10 ml) and add 110 ° C. For 5 hours. After the solvent was distilled off under reduced pressure, the obtained crude product was purified by silica gel column chromatography (chloroform-methanol, 98: 2, v / v) to obtain 1.51 g of compound 12G as a pale yellow solid.
- Trifluoroacetic acid (1 ml) was added to the compound 28H obtained in the seventh step and stirred at room temperature for 1 hour. After concentration under reduced pressure, the pH was adjusted to 3 with aqueous sodium bicarbonate and 2N hydrochloric acid, extracted with chloroform, and dried over sodium sulfate. After the solvent was distilled off under reduced pressure, chloroform-methanol-ethyl ether was added and the precipitated solid was collected by filtration to obtain 17 mg of compound 28 as a colorless solid.
- Step 6 Compound 49G (214 mg, 0.465 mmol) was dissolved in THF (4 ml), ethanol (2 ml) and methylene chloride (2 ml), and 2N aqueous sodium hydroxide solution (1.16 ml, 2.32 mmol) at room temperature. And stirred for 2.5 hours. 1N Hydrochloric acid was added, and the mixture was extracted with chloroform and dried over sodium sulfate. After the solvent was distilled off under reduced pressure, 158 mg of compound 49H was obtained as a yellow solid.
- Sixth Step 5 To a solution of compound 65F in DMF (0.5 mL), 2N aqueous sodium hydroxide solution (0.2 mL) was added, and the mixture was stirred at room temperature for 2 hours.
- Step 3 Suspend potassium carbonate (4.20 g, 30.42 mmol) in a DMF (10 ml) solution of the above compound 95C (2.62 g, 10.14 mmol) at room temperature and stir for 5 minutes, then O- (2,4-dinitrophenyl ) hydroxylamine (3.03 g, 15.21 mmol) was added and stirred at the same temperature for 3 hours. Water was added to the reaction solution, extracted with chloroform 5 times, and the extract was dried over sodium sulfate. After the solvent was distilled off, the resulting oil was purified by silica gel chromatography.
- Step 1 Add compound 95B (1.00 g, 3.55 mmol) and cyclopropanamine (0.492 ml, 7.10 mmol) to pyridine (20 ml), add 1-hydroxybenzotriazole (544 mg, 3.55 mmol) and 1- (3-dimethylaminopropyl) -3- Ethylcarbodiimide hydrochloride (1.36 g, 7.10 mmol) was added in order and stirred at room temperature for 18 hours.
- reaction solution was concentrated under reduced pressure, and the resulting crude product was subjected to silica gel column chromatography (chloroform-methanol, 97: 3 ⁇ 95: 5 ⁇ 90: 10, v / v), followed by amino column chromatography (chloroform-methanol). 97: 3, v / v), methylene chloride-ethyl ether was added, and the precipitated solid was collected by filtration to obtain 665 mg of compound 126C as a colorless solid.
- Step 1 Add compound 95B (2.40 g, 8.52 mmol) and ethyl 3-aminopropanoate hydrochloride (2.62 g, 17.0 mmol) to pyridine (30 ml), add 1-hydroxybenzotriazole (1.31 g, 8.52 mmol) and 1- (3- Dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (3.27 g, 17.0 mmol) was added in order, and the mixture was stirred at room temperature for 2 hours.
- Step 4 Compound 128C (1.00 g, 2.69 mmol) is dissolved in DMF (10 ml), and cesium carbonate (2.63 g, 8.08 mmol) and (bromomethylene) dibenzene (998 mg, 4.04 mmol) are added at 0 ° C to room temperature. For 18 hours. The reaction solution was poured into water and extracted with ethyl acetate, and the organic layer was washed with water and dried over sodium sulfate. The solvent was distilled off under reduced pressure, and the resulting crude product was purified by silica gel column chromatography (chloroform-methanol, 98: 2, v / v) to obtain 500 mg of compound 128D as a colorless gum.
- reaction solution was concentrated under reduced pressure, and the resulting crude product was subjected to silica gel column chromatography (chloroform-methanol, 97: 3 ⁇ 95: 5 ⁇ 90: 10, v / v), followed by amino column chromatography (chloroform-methanol). 97: 3, v / v) to give 721 mg of compound 135C as a colorless solid.
- Step 4 Bromodiphenylmethane (2.26 g) in a suspension of compound 165D (2.77 g, 7.50 mmol), potassium carbonate (2.23 g, 16.1 mmol) and sodium iodide (102 mg, 0.680 mmol) in acetonitrile (30 mL) at room temperature , 9.14 mmol) was added, and the mixture was stirred at 90 ° C. for 7 hours. The reaction mixture was poured into hydrochloric acid (2 N, 10 mL) and ice (20 g), extracted with chloroform (100 mL ⁇ 2), and dried over sodium sulfate.
- First stepCompound 49B (950 mg, 3.35 mmol), 3-aminopropan-1-ol (277 mg, 3.69 mmol) and sodium sulfate (1.91 g, 13.4 mmol) were added to toluene (25 ml), and 1 hour at room temperature.
- Boc2O (0.856 ml, 3.69 mmol) was added at room temperature and stirred for 18 hours. Further, Boc2O (0.400 ml, 1.72 mmol) was added at room temperature and stirred as it was for 60 hours.
- the reaction solution was filtered, and the filtrate was concentrated under reduced pressure.
- Trifluoroacetic acid (2 ml) was added to compound 171F (50 mg, 0.11 mmol) and stirred at room temperature for 1 hour. After concentration under reduced pressure, the pH was adjusted to 6 with aqueous sodium bicarbonate and 2N hydrochloric acid, extracted with chloroform, and dried over sodium sulfate. After the solvent was distilled off under reduced pressure, chloroform-methanol-ethyl ether was added and the precipitated solid was collected by filtration to obtain 24 mg of compound 171 as a colorless solid.
- Trifluoroacetic acid (1 ml) was added to the compound 175G obtained in the sixth step and stirred at room temperature for 1 hour. After concentration under reduced pressure, the pH was adjusted to 3 with aqueous sodium bicarbonate and 2N hydrochloric acid, extracted with chloroform, and dried over sodium sulfate. After evaporating the solvent under reduced pressure, chloroform-methanol-ethyl ether was added and the precipitated solid was collected by filtration to obtain 11 mg of compound 175 as a colorless solid.
- Powder X-ray diffraction measurement of the crystals obtained in each Example was performed under the following measurement conditions in accordance with the powder X-ray diffraction measurement method described in the general test method of the Japanese Pharmacopoeia. (apparatus) Bruker D-8 Discover (Method of operation) The sample was measured under the following conditions. Measurement method: Reflection method Light source type: Cu tube Use wavelength: CuK ⁇ ray tube current: 40 mA Tube voltage: 40Kv Sample plate: Glass X-ray incident angles: 3 ° and 12 °
- Test Example 1 Measurement of cap-dependent endonuclease (CEN) inhibitory activity 1) Preparation of substrate The 5′-terminal G was modified by phosphorylation, and the 2′-position hydroxyl group was modified by methoxylation. 30mer RNA (5'-pp- [m2'-O] GAA UAU (-Cy3) GCA UCA CUA GUA AGC UUU GCU CUA-BHQ2-3 ': U3 of Cy3 labeled and 3' end BHQ2 labeled: Nippon Bioservice And the cap structure was added using the ScriptCap system manufactured by EPICENTRE (product is m7G [5 ']-ppp- [5'] [m2'-O] GAA UAU (-Cy3 ) GCA UCA CUA GUA AGC UUU GCU CUA (-BHQ2) -3 ').
- RNP was prepared from virus particles according to a conventional method (reference: VIROLOGY (1976) 73, p327-338 OLGA M. ROCHOVANSKY). Specifically, A / WSN / 33 virus 1 ⁇ 10 3 PFU / mL, 200 ⁇ L was inoculated into 10-day-old chicken eggs and cultured at 37 ° C. for 2 days.
- the virus particles were purified by ultracentrifugation using 20% sucrose, solubilized using TritonX-100 and lysolecithin, and then the RNP fraction (50% by ultracentrifugation using a 30-70% glycerol density gradient). ⁇ 70% glycerol fraction) was collected and used as an enzyme solution (containing approximately 1 nM PB1, PB2, PA complex).
- Enzyme reaction solution (composition: 53 mM Tris-hydrochloride (pH 7.8), 1 mM MgCl 2 , 1.25 mM dithiothreitol, 80 mM NaCl, 12.5% glycerol, enzyme solution 0.15 ⁇ L in polypropylene 384-well plate ) was dispensed in 2.5 ⁇ L.
- 0.5 ⁇ L of DMSO was added to 0.5 ⁇ L of the test compound solution diluted stepwise with dimethyl sulfoxide (DMSO), positive control (PC) and negative control (NC) and mixed well.
- DMSO dimethyl sulfoxide
- PC positive control
- NC negative control
- reaction-stopped solution was heated at 85 ° C. for 5 minutes, rapidly cooled on ice for 2 minutes, and then analyzed with an ABI PRIZM 3730 genetic analyzer.
- Test Example 2 CPE inhibitory effect confirmation test ⁇ Material> ⁇ 2% FCS E-MEM (adjusted by adding kanamycin and FCS to MEM (Minimum Essential Medium) (Invitrogen)) ⁇ 0.5% BSA E-MEM (adjusted by adding kanamycin and BSA to MEM (Minimum Essential Medium) (Invitrogen)) ⁇ HBSS (hanks' Balanced Salt Solution) MDBK cells The cell number was adjusted to an appropriate number (3 ⁇ 10 5 / mL) with 2% FCS E-MEM. MDCK cells After washing twice with HBSS, it was adjusted to an appropriate cell number (5 ⁇ 10 5 / mL) with 0.5% BSA E-MEM.
- ⁇ Operation procedure> Dilution and dispensing of test sample
- 2% FCS E-MEM was used when MDBK cells were used, and 0.5% BSA E-MEM was used when MDCK cells were used.
- BSA E-MEM was used when MDCK cells were used.
- a test sample was diluted in advance to an appropriate concentration with a culture solution, and a 2- to 5-fold serial dilution series was prepared in a 96-well plate (50 ⁇ L / well). Two sheets for anti-Flu activity measurement and cytotoxicity measurement were prepared. Triplicate measurements were performed for each drug.
- influenza virus was diluted to an appropriate concentration with a culture solution, and dispensed into a 96-well plate containing a test sample at 50 ⁇ L / well.
- the culture solution was dispensed at 50 ⁇ L / well on the cytotoxicity measurement plate.
- ⁇ Dilution and dispensing of cells Cells adjusted to an appropriate number of cells were dispensed at 100 ⁇ L / well into a 96-well plate containing the test sample. The mixture was mixed with a plate mixer and cultured in a CO2 incubator.
- Both for anti-Flu activity measurement and cytotoxicity measurement were cultured for 3 days.
- ⁇ Dispensing of WST-8 The 96-well plate cultured for 3 days was observed with the naked eye and under a microscope to confirm the morphology of the cells and the presence or absence of crystals. The supernatant was removed so as not to suck cells from the plate.
- WST-8 Kit was diluted 10-fold with the culture solution, and 100 ⁇ L of this WST-8 solution was dispensed into each well. After mixing with a plate mixer, the cells were cultured for 1 to 3 hours in a CO2 incubator.
- Regarding the anti-Flu activity measurement plate after culturing, 10 ⁇ L of 10% SDS solution was dispensed into each well to inactivate the virus.
- ⁇ Measurement of absorbance The absorbance of the mixed 96-well plate was measured with EnVision at two wavelengths of 450 nm / 620 nm.
- the test compound showed a high cap-dependent endonuclease (CEN) inhibitory activity and a high CPE inhibitory effect.
- CEN cap-dependent endonuclease
- These compounds can be useful medicaments for treating and / or preventing symptoms and / or diseases induced by infection with influenza virus. Therefore, it can be said that the compounds and production methods according to the present invention are intermediate compounds and production methods useful for efficiently producing compounds that can be used as pharmaceuticals.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pyridine Compounds (AREA)
- Pyrane Compounds (AREA)
Abstract
Description
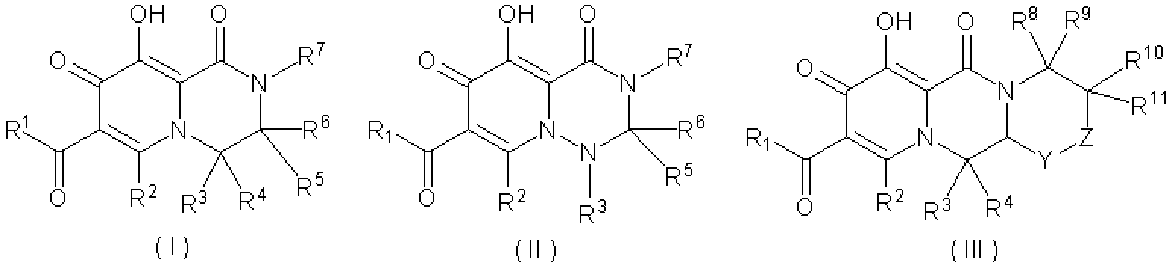
(式中、R1は、ヒドロキシ、置換基群Eで置換されていてもよい低級アルキルオキシ、置換基群Eで置換されていてもよい炭素環低級アルキルオキシ、置換基群Eで置換されていてもよい複素環低級アルキルオキシ、置換基群Eで置換されていてもよい炭素環低級アルキルアミノ、または置換基群Eで置換されていてもよい複素環低級アルキルアミノであり、
Zは、CR12R13、または単結合であり、
Yは、CH2、酸素原子、またはN-R14であり、
R2、R3、R4、R5、R6、R7、R8、R9、R10、R11、R12、R13、およびR14は、それぞれ独立して、水素、置換基群Eで置換されていてもよい低級アルキル、置換基群Eで置換されていてもよい低級アルケニル、置換基群Eで置換されていてもよい低級アルキニル、置換基群Eで置換されていてもよい炭素環低級アルキル、または置換基群Eで置換されていてもよい複素環低級アルキルであり、
R3およびR5は一緒になって4~8員の置換基群Eで置換されていてもよい炭素環式基または置換基群Eで置換されていてもよい複素環式基を形成していてもよく、またはR7およびR5は一緒になって4~8員の置換基群Eで置換されていてもよい複素環式基を形成していてもよく、
ここで、Zが単結合のとき、R14およびR10は一緒になって4~8員の置換基群Eで置換されていてもよい複素環式基を形成していてもよい。
置換基群E:ハロゲン、シアノ、ヒドロキシ、カルボキシ、ホルミル、アミノ、オキソ、ニトロ、低級アルキル、ハロゲノ低級アルキル、低級アルキルオキシ、置換基群Fで置換されていてもよい炭素環式基、置換基群Fで置換されていてもよい複素環式基、置換基群Fで置換されていてもよい炭素環低級アルキルオキシ、置換基群Fで置換されていてもよい複素環低級アルキルオキシ、置換基群Fで置換されていてもよい炭素環低級アルキルチオ、置換基群Fで置換されていてもよい複素環低級アルキルチオ、置換基群Fで置換されていてもよい炭素環低級アルキルアミノ、置換基群Fで置換されていてもよい複素環低級アルキルアミノ、置換基群Fで置換されていてもよい炭素環オキシ、置換基群Fで置換されていてもよい複素環オキシ、置換基群Fで置換されていてもよい炭素環カルボニル、置換基群Fで置換されていてもよい複素環カルボニル、置換基群Fで置換されていてもよい炭素環アミノカルボニル、置換基群Fで置換されていてもよい複素環アミノカルボニル、ハロゲノ低級アルキルオキシ、低級アルキルオキシ低級アルキル、低級アルキルオキシ低級アルキルオキシ、低級アルキルカルボニル、低級アルキルオキシカルボニル、低級アルキルオキシカルボニルアミノ、低級アルキルアミノ、低級アルキルカルボニルアミノ、低級アルキルアミノカルボニル、低級アルキルスルホニル、および低級アルキルスルホニルアミノ;
置換基群F:ハロゲン、ヒドロキシ、カルボキシ、アミノ、オキソ、ニトロ、低級アルキル、ハロゲノ低級アルキル、低級アルキルオキシ、およびアミノ保護基)
で例示される化合物等を効率的に製造することにある。
以下の工程:
(工程B)
式(X2):

(式中、R1dは、水素、ハロゲン、置換基群Eで置換されていてもよい低級アルキルオキシ、置換基群Eで置換されていてもよい炭素環低級アルキルオキシ、置換基群Eで置換されていてもよい複素環低級アルキルオキシ、または-OSi(R1e)3であり、
R1eは、それぞれ独立して、置換基群Eで置換されていてもよい低級アルキル、置換基群Eで置換されていてもよい炭素環式基、置換基群Eで置換されていてもよい複素環式基、置換基群Eで置換されていてもよい炭素環低級アルキルまたは置換基群Eで置換されていてもよい複素環低級アルキルであり、
R2dは、水素、置換基群Eで置換されていてもよい低級アルキル、置換基群Eで置換されていてもよい炭素環低級アルキル、または置換基群Eで置換されていてもよい複素環低級アルキルであり、
R3dは、水素、置換基群Eで置換されていてもよい低級アルキル、-N(R3e)2、または-OR3eであり、
R3eは、それぞれ独立して置換基群Eで置換されていてもよい低級アルキル、または一緒になって複素環式基を形成していてもよく、ならびに
波線は、E体および/もしくはZ体またはそれらの混合物を表わす。
置換基群E:ハロゲン、シアノ、ヒドロキシ、カルボキシ、ホルミル、アミノ、オキソ、ニトロ、低級アルキル、ハロゲノ低級アルキル、低級アルキルオキシ、置換基群Fで置換されていてもよい炭素環式基、置換基群Fで置換されていてもよい複素環式基、置換基群Fで置換されていてもよい炭素環低級アルキルオキシ、置換基群Fで置換されていてもよい複素環低級アルキルオキシ、置換基群Fで置換されていてもよい炭素環低級アルキルチオ、置換基群Fで置換されていてもよい複素環低級アルキルチオ、置換基群Fで置換されていてもよい炭素環低級アルキルアミノ、置換基群Fで置換されていてもよい複素環低級アルキルアミノ、置換基群Fで置換されていてもよい炭素環オキシ、置換基群Fで置換されていてもよい複素環オキシ、置換基群Fで置換されていてもよい炭素環カルボニル、置換基群Fで置換されていてもよい複素環カルボニル、置換基群Fで置換されていてもよい炭素環アミノカルボニル、置換基群Fで置換されていてもよい複素環アミノカルボニル、ハロゲノ低級アルキルオキシ、低級アルキルオキシ低級アルキル、低級アルキルオキシ低級アルキルオキシ、低級アルキルカルボニル、低級アルキルオキシカルボニル、低級アルキルオキシカルボニルアミノ、低級アルキルアミノ、低級アルキルカルボニルアミノ、低級アルキルアミノカルボニル、低級アルキルスルホニル、および低級アルキルスルホニルアミノ;
置換基群F:ハロゲン、ヒドロキシ、カルボキシ、アミノ、オキソ、ニトロ、低級アルキル、ハロゲノ低級アルキル、低級アルキルオキシ、およびアミノ保護基)
で表わされる化合物と、式(V2):
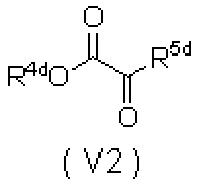
(式中、R4dは、置換基群Eで置換されていてもよい低級アルキル、置換基群Eで置換されていてもよい炭素環低級アルキル、または置換基群Eで置換されていてもよい複素環低級アルキルであり、
R5dは、水素、ハロゲン、置換基群Eで置換されていてもよい低級アルキルオキシ、または-O-SO2-R5eであり、
R5eは、置換基群Eで置換されていてもよい低級アルキル、置換基群Eで置換されていてもよい炭素環式基、または置換基群Eで置換されていてもよい複素環式基、置換基群Eで置換されていてもよい炭素環低級アルキル、または置換基群Eで置換されていてもよい複素環低級アルキルであり、ならびに
置換基群Eは前記と同意義である)
で表わされる化合物を反応させることにより、式(X3):

(式中、各記号は前記と同意義である)
で表わされる化合物を得る工程;
ならびに、
(工程C)
式(X3)で表わされる化合物と、式(V3):

(式中、R6dは、置換基群Eで置換されていてもよい低級アルキル、置換基群Eで置換されていてもよい低級アルケニル、置換基Eで置換されていてもよいアミノ、置換基Eで置換されていてもよい低級アルキルアミノ、置換基群Eで置換されていてもよい炭素環式基、または置換基群Eで置換されていてもよい複素環式基であり、
置換基群Eは前記と同意義である)
で表わされる化合物を反応させることにより、式(X4):

(式中、各記号は前記と同意義である)
で表される化合物を得る工程;
を含む、式(X4)で表わされる化合物またはその塩の製造方法。
工程Bおよび工程Cを連続して行なうことを特徴とする、項目1に記載の製造方法。
式(X2):

(式中、各記号は前記と同意義である)
で表わされる化合物と、式(VA2):

(式中、R4eは、それぞれ独立して水素、置換基群Eで置換されていてもよい低級アルキル、置換基群Eで置換されていてもよい炭素環式基、または置換基群Eで置換されていてもよい複素環式基、ならびに
R5dおよび置換基群Eは前記と同意義である)
で表される化合物を反応させる工程;ならびに
式(V3):

(式中、R6dは、前記と同意義である)
で表される化合物を反応させる工程を含む、式(XA4):

(式中、Raは、水素、ヒドロキシ、置換基群Eで置換されていてもよい低級アルキルアミノ、置換基群Eで置換されていてもよい低級アルケニルアミノ、置換基群Eで置換されていてもよい低級アルキニルアミノ、置換基群Eで置換されていてもよい炭素環低級アルキルアミノ、または置換基群Eで置換されていてもよい複素環低級アルキルアミノ、ならびに
各記号は、前記と同意義である)
で表わされる化合物またはその塩の製造方法。
式(X2):

(式中、各記号は項目1と同意義である)
で表わされる化合物と、式(V2)または式(VA2):
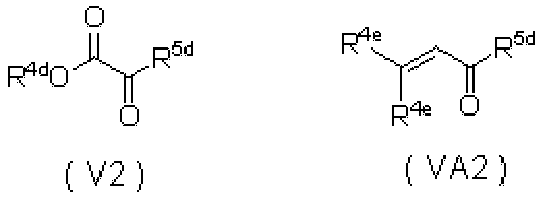
(式中、各記号は項目1と同意義である)
で表わされる化合物を反応させることを特徴とする、式(X3)または式(XA3):

(式中、各記号は項目1と同意義である)
で表わされる化合物またはその塩の製造方法。
式(X3)もしくは式(XA3):

(式中、各記号は項目1と同意義である)
で表わされる化合物、または式(XA3)の誘導体および、式(V3):

(式中、記号は項目1と同意義である)
で表わされる化合物を反応させる工程を含む、式(X4)もしくは式(XA4):

(式中、各記号は項目1および項目3と同意義である)
で表される化合物またはその塩の製造方法。
式(X2):

(式中、各記号は項目1と同意義である)
で表わされる化合物と、式(V2)または式(VA2):

(式中、各記号は項目1と同意義である)
で表わされる化合物、および式(V2’):
(式中、Xd-は、アンモニウムカチオンのカウンターアニオンである)
で表わされる化合物を反応させることを特徴とする、式(X4’)または式(XA4’):

(式中、各記号は項目1と同意義である)
で表わされる化合物またはその塩の製造方法。
項目6に記載の製造方法で得られる式(X4’)もしくは式(XA4’):

(式中、各記号は項目1と同意義である)
で表わされる化合物、または式(XA4’)の誘導体と、式(V3’):
(式中、R6dは項目1と同意義であり、
Ldは脱離基であり、ならびにPhはフェニル基を表わす)
で表わされる化合物を反応させる工程を含む、式(X4)もしくは式(XA4):
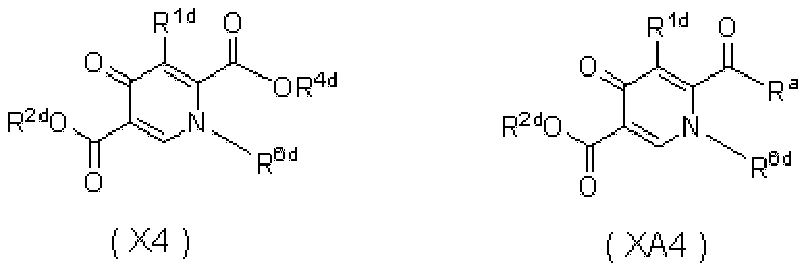
(式中、各記号は項目1または項目3と同意義である)
で表される化合物またはその塩の製造方法。
式(X2)で表わされる化合物が、式(X1):

(式中、各記号は項目1と同意義である)
で表わされる化合物と、式(V1):

(式中、Pdは、置換基群Eで置換されていてもよい低級アルキルであり、ならびにR3dおよび置換基群Eは項目1と同意義である)
で表わされる化合物とを反応させることによって得られる、項目1、2、3、4または項目6に記載の製造方法。
式(X2)で表わされる化合物が、式(Z1):

(式中、各記号は項目1と同意義である)
で表わされる化合物と、式(Z2):
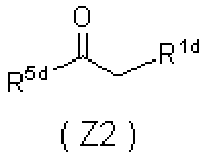
(式中、各記号は項目1と同意義である)
で表わされる化合物とを反応させることによって得られる、項目1、2、3、4または項目6に記載の製造方法。
式(X3):

(式中、各記号は項目1と同意義である)
表わされる化合物、もしくはその製薬上許容される塩またはそれらの溶媒和物。
式(X4):

(式中、各記号は項目1と同意義である)
表わされる化合物、もしくはその製薬上許容される塩またはそれらの溶媒和物。
式(X4’):
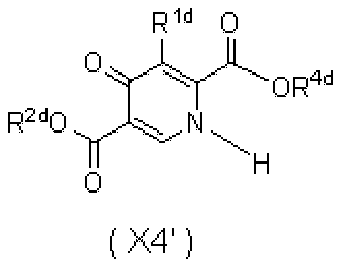
(式中、各記号は請求項1と同意義である)
表わされる化合物、もしくはその製薬上許容される塩またはそれらの溶媒和物。
粉末X線回折スペクトルにおいて、回折角度(2θ):7.9°±0.2°、10.0°±0.2°、11.5°±0.2°、20.0°±0.2°、23.4°±0.2°および34.0°±0.2°にピークを有する、式(1D):
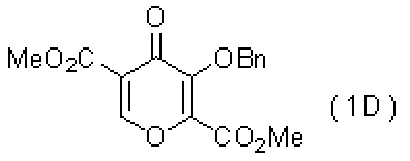
(式中、Meはメチル基、およびBnはベンジル基を表す)
で示される化合物またはその溶媒和物の結晶。
粉末X線回折スペクトルにおいて、回折角度(2θ):17.6°±0.2°、25.2°±0.2°、26.4°±0.2°および28.1°±0.2°にピークを有する、式(2D):

(式中、Meはメチル基、Et基はエチル基、およびBnはベンジル基を表す)
で示される化合物またはその溶媒和物の結晶。
粉末X線回折スペクトルにおいて、回折角度(2θ):14.2°±0.2°、16.0°±0.2°、22.0°±0.2°、22.2°±0.2°、24.4°±0.2°および25.9°±0.2°にピークを有する、式(9C’):

(式中、Meはメチル基、およびBnはベンジル基を表す)
で示される化合物またはその溶媒和物の結晶。
図1に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、項目13記載の結晶。
図2に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、項目14記載の結晶。
図3に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、項目15記載の結晶。

(式中、各記号は前記と同意義である)
本明細書中、「低級アルキル」とは、炭素数1~15、好ましくは炭素数1~10、より好ましくは炭素数1~6、さらに好ましくは炭素数1~4の直鎖又は分枝状のアルキルを包含し、例えばメチル、エチル、n-プロピル、イソプロピル、n-ブチル、イソブチル、sec-ブチル、tert-ブチル、n-ペンチル、イソペンチル、ネオペンチル、ヘキシル、イソヘキシル、n-へプチル、イソヘプチル、n-オクチル、イソオクチル、n-ノニル及びn-デシル等が挙げられる。「低級アルキル」の好ましい態様として、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、イソブチル、sec-ブチル、tert-ブチル、n-ペンチルが挙げられる。さらに好ましい態様として、メチル、エチル、n-プロピル、イソプロピル、tert-ブチルが挙げられる。
本明細書中、「低級アルケニル」とは、任意の位置に1以上の二重結合を有する炭素数2~15、好ましくは炭素数2~10、より好ましくは炭素数2~6、さらに好ましくは炭素数2~4の直鎖又は分枝状のアルケニルを包含する。具体的にはビニル、アリル、プロペニル、イソプロペニル、ブテニル、イソブテニル、プレニル、ブタジエニル、ペンテニル、イソペンテニル、ペンタジエニル、ヘキセニル、イソヘキセニル、ヘキサジエニル、ヘプテニル、オクテニル、ノネニル、デセニル、ウンデセニル、ドデセニル、トリデセニル、テトラデセニル、ペンタデセニル等を包含する。「低級アルケニル」の好ましい態様として、ビニル、アリル、プロペニル、イソプロペニル、ブテニルが挙げられる。
本明細書中、「低級アルキニル」とは、任意の位置に1以上の三重結合を有する炭素数2~10、好ましくは炭素数2~8、さらに好ましくは炭素数3~6の直鎖又は分枝状のアルキニルを包含する。具体的には、エチニル、プロピニル、ブチニル、ペンチニル、ヘキシニル、ヘプチニル、オクチニル、ノニニル、デシニル等を包含する。これらはさらに任意の位置に二重結合を有していてもよい。「低級アルキニル」の好ましい態様として、エチニル、プロピニル、ブチニル、ペンチニルが挙げられる。
本明細書中、「低級アルケニルアミノ」の低級アルケニル部分も上記「低級アルケニル」と同様である。
本明細書中、「低級アルキニルアミノ」の低級アルキニル部分も上記「低級アルキニル」と同様である。
本明細書中、「ハロゲノ低級アルキル」、および「ハロゲノ低級アルキルオキシ」のハロゲン部分も上記「ハロゲン」と同様である。ここでそれぞれ「低級アルキル」、および「低級アルキルオキシ」のアルキル基上の任意の位置が、同一または異なる1または複数個のハロゲン原子で置換されていてもよい。
具体的に「シクロアルキル」とは炭素数3~16、好ましくは炭素数3~12、より好ましくは炭素数4~8の炭素環式基であり、例えばシクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、シクロノニルおよびシクロデシル等を包含する。
具体的に「シクロアルケニル」とは、上記シクロアルキルの環中の任意の位置に1以上の二重結合を有しているものを包含し、例えばシクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニル、シクロへプチニル、シクロオクチニルおよびシクロヘキサジエニル等が挙げられる。
具体的に「アリール」とは、フェニル、ナフチル、アントリルおよびフェナントリル等を包含し、特にフェニルが好ましい。
具体的に「非芳香族縮合炭素環式基」とは、上記「シクロアルキル」、「シクロアルケニル」および「アリール」から選択される2個以上の環状基が縮合した基を包含し、例えばインダニル、インデニル、テトラヒドロナフチル、フルオレニル、アダマンチル、および以下に示される基
等が挙げられる。
「炭素環式基」の好ましい態様としては、シクロアルキル、アリールおよび非芳香族縮合炭素環式基が挙げられ、具体的には、シクロプロピル、シクロブチル、シクロペンチル、シクロへキシル、シクロへプチル、フェニル等が挙げられる。
「炭素環低級アルキル」、「炭素環低級アルキルオキシ」、「炭素環低級アルキルチオ」、「炭素環低級アルキルアミノ」、「炭素環オキシ」、「炭素環カルボニル」、および「炭素環アミノカルボニル」の炭素環部分も上記「炭素環式基」と同様である。
具体的に「ヘテロアリール」とは、ピロリル、イミダゾリル、ピラゾリル、ピリジル、ピリダジニル、ピリミジニル、ピラジニル、トリアゾリル、トリアジニル、テトラゾリル、フリル、チエニル、イソオキサゾリル、オキサゾリル、オキサジアゾリル、イソチアゾリル、チアゾリル、チアジアゾリル等の5~6員の芳香族環式基が挙げられる。
具体的に「非芳香族複素環式基」とは、ジオキサニル、チイラニル、オキシラニル、オキセタニル、オキサチオラニル、アゼチジニル、チアニル、チアゾリジニル、ピロリジニル、ピロリニル、イミダゾリジニル、イミダゾリニル、ピラゾリジニル、ピラゾリニル、ピペリジル、ピペラジニル、モルホリニル、モルホリノ、チオモルホリニル、チオモルホリノ、ジヒドロピリジル、テトラヒドロピリジル、テトラヒドロフリル、テトラヒドロピラニル、ジヒドロチアゾリル、テトラヒドロチアゾリル、テトラヒドロイソチアゾリル、ジヒドロオキサジニル、ヘキサヒドロアゼピニル、テトラヒドロジアゼピニル、テトラヒドロピリダジニル、ヘキサヒドロピリミジニル、ジオキソラニル等が挙げられる。
具体的に「2環の縮合複素環式基」とは、インドリル、イソインドリル、インダゾリル、インドリジニル、インドリニル、イソインドリニル、キノリル、イソキノリル、シンノリニル、フタラジニル、キナゾリニル、ナフチリジニル、キノキサリニル、プリニル、プテリジニル、ベンゾピラニル、ベンズイミダゾリル、ベンゾトリアゾリル、ベンズイソオキサゾリル、ベンズオキサゾリル、ベンズオキサジアゾリル、ベンゾイソチアゾリル、ベンゾチアゾリル、ベンゾチアジアゾリル、ベンゾフリル、イソベンゾフリル、ベンゾチエニル、ベンゾトリアゾリル、チエノピリジル、チエノピロリル、チエノピラゾリル、チエノピラジニル、フロピロリル、チエノチエニル、イミダゾピリジル、ピラゾロピリジル、チアゾロピリジル、ピラゾロピリミジニル、ピラゾロトリアニジル、ピリダゾロピリジル、トリアゾロピリジル、イミダゾチアゾリル、ピラジノピリダジニル、キナゾリニル、キノリル、イソキノリル、ナフチリジニル、ジヒドロチアゾロピリミジニル、テトラヒドロキノリル、テトラヒドロイソキノリル、ジヒドロベンゾフリル、ジヒドロベンズオキサジニル、ジヒドロベンズイミダゾリル、テトラヒドロベンゾチエニル、テトラヒドロベンゾフリル、ベンゾジオキソリル、ベンゾジオキソニル、クロマニル、クロメニル、オクタヒドロクロメニル、ジヒドロベンゾジオキシニル、ジヒドロベンゾオキセジニル、ジヒドロベンゾジオキセピニル、ジヒドロチエノジオキシニル等が挙げられる。
具体的に「3環の縮合複素環式基」とは、カルバゾリル、アクリジニル、キサンテニル、フェノチアジニル、フェノキサチイニル、フェノキサジニル、ジベンゾフリル、イミダゾキノリル、テトラヒドロカルバゾリル、および以下に示される基
等が挙げられる。
「複素環式基」の好ましい態様としては5~6員のヘテロアリールまたは非芳香族複素環式基、3環の縮合複素環式基が挙げられる。
「複素環低級アルキル」、「複素環低級アルキルオキシ」、「複素環低級アルキルチオ」、「複素環低級アルキルアミノ」、「複素環オキシ」、「複素環カルボニル」および「複素環アミノカルボニル」の複素環部分も上記「複素環式基」と同様である。
などを包含する。
「置換基群Eで置換されていてもよい炭素環式基」とは、「炭素環式基」が、無置換であるか、または1個もしくは複数個の化学的に許容できる置換基群Eから選ばれる置換基が結合していることを意味する。置換基が複数個結合している場合は、複数個の置換基は同一または異なっていてもよい。例えば、フルオロフェニル、ジフルオロフェニル、メトキシフルオロフェニルなどを包含する。
「置換基群Eで置換されていてもよい炭素環低級アルキル」とは、「炭素環式基」および/または「低級アルキル」が、無置換であるか、または1個もしくは複数個の化学的に許容できる置換基群Eから選ばれる置換基が結合していることを意味する。置換基が複数個結合している場合は、複数個の置換基は同一または異なっていてもよい。例えば、4-フルオロベンジル、2,4-ジフルオロベンジル、4-メトキシ2-フルオロベンジル、4-メトキシフェニルジフルオロメチルなどを包含する。
「置換基群Eで置換されていてもよい低級アルキルオキシ」、「置換基群Eで置換されていてもよい炭素環低級アルキルオキシ」、「置換基群Eで置換されていてもよい複素環低級アルキルオキシ」、「置換基群Eで置換されていてもよい低級アルケニル」、「置換基群Eで置換されていてもよいアミノ」、「置換基群Eで置換されていてもよい低級アルキルアミノ」、「置換基群Eで置換されていてもよい低級アルケニルアミノ」、「置換基群Eで置換されていてもよい低級アルキニルアミノ」、「置換基群Eで置換されていてもよい炭素環低級アルキルアミノ」、および「置換基群Eで置換されていてもよい複素環低級アルキルアミノ」も同様の意味である。
「置換基群Fで置換されていてもよい低級アルキルオキシ」とは、「炭素環式基」部分が、無置換であるか、または1個もしくは複数個の化学的に許容できる置換基群Fから選ばれる置換基が結合していることを意味する。置換基が複数個結合している場合は、複数個の置換基は同一または異なっていてもよい。例えば、フルオロベンジルオキシ、ジフルオロベンジルオキシ、メトキシフルオロベンジルオキシなどを包含する。
「置換基群Fで置換されていてもよい複素環式基」、「置換基群Fで置換されていてもよい複素環低級アルキルオキシ」、「置換基群Fで置換されていてもよい炭素環低級アルキルチオ」、「置換基群Fで置換されていてもよい複素環低級アルキルチオ」、「置換基群Fで置換されていてもよい炭素環低級アルキルアミノ」、「置換基群Fで置換されていてもよい複素環低級アルキルアミノ」、「置換基群Fで置換されていてもよい炭素環オキシ」、「置換基群Fで置換されていてもよい複素環オキシ」、「置換基群Fで置換されていてもよい炭素環カルボニル」、「置換基群Fで置換されていてもよい複素環カルボニル」、「置換基群Fで置換されていてもよい炭素環アミノカルボニル」、および「置換基群Fで置換されていてもよい複素環アミノカルボニル」も同様の意味である。
例えば、以下で示される式などを包含する。
「-N(R3e)2、のR3eが一緒になって複素環式基を形成していてもよく」の「複素環式基」の定義も上記と同様である。
「カルボキシル保護基」とは、一般的なアミノ基の保護基であればよく、例えば、Protective Groups in Organic Synthesis, Theodora W Green(John Wiley & Sons)に記載のカルボキシル保護基として例示される。好ましくは、メチル基、エチル基、tert-ブチル基、メトキシメチル基、アリル基、ベンジル基、p-メトキシベンジル基などが挙げられる。
Xdにおける「アンモニウムカチオンのカウンターアニオン」とは、ハロゲン-、CH3COO-、HCOO-、NO3 -、BF4 -、PF6 -、HO-、Ph-SO3 -、CH3-Ph-SO3 -、CH3-SO3 -、PO4 3-、SO4 2-またはHSO4 -などが挙げられる。好ましくは、ハロゲン-、CH3COO-、NO3 -、SO4 2-である。アニオンが2価または3価の場合は、NH4 +カチオンはそれぞれ2分子または3分子結合していることにより、帯電していない状態になっているものを指す。NH4 +Xd-の具体的例としては、(NH4 +)2SO4 2-、(NH4 +)3PO4 3-などが挙げられる。
「脱離基」とは、求核反応により脱離する置換基を示し、ハロゲン、-O-SO2-CH3、-O-SO2-CF3、-O-SO2-Phまたは-O-SO2-Ph-CH3などが挙げられる。好ましくは、ハロゲンである。

(式中、Rbは、水素、置換基群Eで置換されていてもよい低級アルキル、置換基群Eで置換されていてもよい低級アルケニル、置換基群Eで置換されていてもよい低級アルキニル、置換基群Eで置換されていてもよい炭素環低級アルキル、または置換基群Eで置換されていてもよい複素環低級アルキルであり、その他の各記号は項目1および項目3と同意義である)
に示される。上記の式(XA3a)が式(V3)と反応する場合は、Raが水素である式(XA4)が生成する。上記の式(XA3b)が式(V3)と反応する場合は、Raがヒドロキシである式(XA4)が生成する。上記の式(XA3b)が式(V3)と反応する場合は、Raがヒドロキシである式(XA4)が生成する。上記の式(XA3c)が式(V3)と反応する場合は、Raが置換基群Eで置換されていてもよい低級アルキルアミノ、置換基群Eで置換されていてもよい低級アルケニルアミノ、置換基群Eで置換されていてもよい低級アルキニルアミノ、置換基群Eで置換されていてもよい炭素環低級アルキルアミノ、または置換基群Eで置換されていてもよい複素環低級アルキルアミノである式(XA4)が生成する。
「式(XA3)のオレフィン部位が酸化開裂されることにより、アルデヒド基もしくはカルボキシル基または、カルボキシル基に対して縮合反応させることによりアミドを形成」における、「酸化開裂」は、一般的な公知の酸化反応条件(例えば、オゾン酸化反応、RuCl3-NaIO4酸化反応など)で行なうことができる。反応生成物がアルデヒド体(XA3a)である場合、続く一般的な酸化反応条件(例えば、Cr3-ピリジン、PCC酸化、SO3-ピリジン酸化、NaClO2酸化など)でカルボキシル基に導くことができる。
「縮合反応によりアミド化」は、カルボキシル基に対して一般的な脱水縮合反応(光延反応、カルボン酸ハロゲン化物、カルボン酸無水物、または縮合剤(例えば、WSC、カルボニルジイミダゾール、ジシクロヘキシルカルボジイミド)等を用いる反応)によって行うことができる。
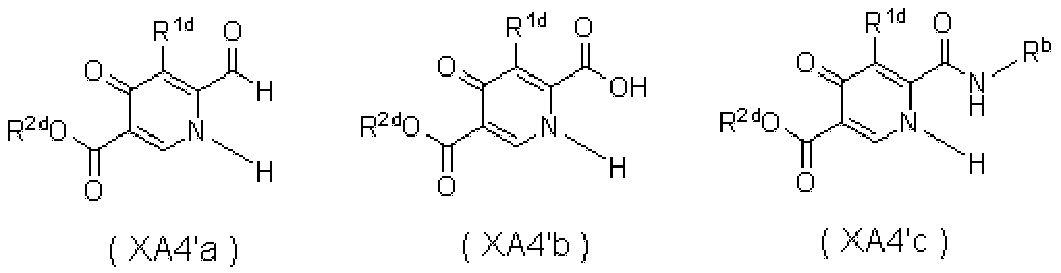
(式中、各記号は項目1、項目3および前記と同意義である)
に示される。上記の式(XA4’a)が式(V3’)と反応する場合は、Raが水素である式(XA4)が生成する。上記の式(XA4’b)が式(V3’)と反応する場合は、Raがヒドロキシである式(XA4)が生成する。上記の式(XA4’c)が式(V3’)と反応する場合は、Raが置換基群Eで置換されていてもよい低級アルキルアミノ、置換基群Eで置換されていてもよい低級アルケニルアミノ、置換基群Eで置換されていてもよい低級アルキニルアミノ、置換基群Eで置換されていてもよい炭素環低級アルキルアミノ、または置換基群Eで置換されていてもよい複素環低級アルキルアミノである式(XA4)が生成する。
「式(XA4’)のオレフィン部位が酸化開裂されることにより、アルデヒド基もしくはカルボキシル基または、カルボキシル基に対して縮合反応させることによりアミドを形成」も、前記(XA3)の場合と同様である。
(工程A)
本工程は、以下の反応式に示すように、化合物(X1)および化合物(V1)を反応させて、化合物(X2)を含む溶液を得る工程である。
ここで「溶液」とは、化合物(X2)が溶解した状態のものを意味するが、化合物(X2)が完全に溶解せず分散している懸濁液状態のもの、またはスラリー状態であるものも包含する。以下、本明細書中における「溶液」は、全て同様に懸濁液状態のもの、またはスラリー状態であるものも包含する。

(式中、各記号は前記と同意義である)
化合物(X1)は、市販の試薬であるか、公知の方法により得ることができる。
R1dがハロゲンの場合には、溶媒中、置換基群Eで置換されていてもよい低級アルコール、置換基群Eで置換されていてもよい炭素環低級アルキルアルコール、置換基群Eで置換されていてもよい複素環低級アルキルアルコール、または(R1e)3Si-OH等のアルコール試薬を加え、所望により塩基存在下で求核置換反応をすることによって、R1dが置換基群Eで置換されていてもよい低級アルキルオキシ、置換基群Eで置換されていてもよい炭素環低級アルキルオキシ、置換基群Eで置換されていてもよい複素環低級アルキルオキシ、または-OSi(R1e)3である化合物(X1)を得ることもできる。
R1dにおける「置換基群Eで置換されていてもよい炭素環低級アルキルオキシ」の例としては、ベンジルオキシ、フェネチルオキシ、2,4-トリフルオロベンジルオキシ、4-メトキシベンジルオキシ等が挙げられる。好ましくはベンジルオキシである。
R1dにおける「置換基群Eで置換されていてもよい複素環低級アルキルオキシ」の例としては、ピリジルメチルオキシ等が挙げられる。
R1dの好ましい態様は、水素、クロロ、ブロモ、メトキシ、ベンジルオキシである。
R2dにおける「置換基群Eで置換されていてもよい炭素環低級アルキル」の例としては、ベンジル、4-メトキシベンジル等が挙げられる。
R2dにおける「置換基群Eで置換されていてもよい複素環低級アルキル」の例としては、ピリジルメチル等が挙げられる。
R2dの好ましい態様は、メチル、エチル、n-プロピル、iso-プロピル、tert-ブチル、ベンジル等である。
上記の塩基としては、アルコール試薬の脱プロトン化が可能である塩基であれば良く、例えば、n-ブチルリチウム、tert-ブチルリチウム、ナトリウム-tert-ブトキシド、カリウム-tert-ブトキシド、ナトリウム-tert-ペントキシド、ナトリウムメトキシド、ナトリウムエトキシド、水素化ナトリウム、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド等が挙げられる。
塩基の量は、R1dがハロゲンである化合物(X1)に対して、約1.0~3.0モル等量である。
アルコール試薬の量は、R1dがハロゲンである化合物(X1)に対して、約0.5~1.5モル等量である。
反応温度は、通常、0℃~還流温度であり、好ましくは室温~50℃である。
反応時間は、通常、10分~50時間、好ましくは1~4時間である。
Pdにおける「置換基群Eで置換されていてもよい低級アルキル」の例としては、メチル、エチル、トリフルオロメチル等が挙げられる。Pdの好ましい態様は、メチルである。
R3dにおける「置換基群Eで置換されていてもよい低級アルキル」の例としては、メチル、エチル、トリフルオロメチル等が挙げられる。
R3eにおける「置換基群Eで置換されていてもよい低級アルキル」の例としては、メチル、エチル、トリフルオロメチル等が挙げられる。
R3dの好ましい態様は、-N(CH3)2、-OCH3、またはピロリジニルである。
化合物(V1)の使用量は、化合物(X1)に対して約1.0~3.0モル等量であるか、もしくは化合物(V1)を溶媒と使用してもよい。
反応温度は、通常、0℃~還流温度であり、好ましくは室温である。
反応時間は、通常、30分~50時間、好ましくは2~8時間である。
また、化合物(X2)は、下記に示す反応によっても得ることができる。

(式中、R7dは、ハロゲン、置換基群Eで置換されていてもよい低級アルキルオキシ、または-O-SO2-R5eであり、および
その他の各記号は上記と同様である)
化合物(Z1)は、市販の試薬であるか、公知の方法により得ることができる。
R2dにおける「置換基群Eで置換されていてもよい低級アルキル」の例としては、メチル、エチル、n-プロピル、iso-プロピル、tert-ブチル等が挙げられる。
R2dにおける「置換基群Eで置換されていてもよい炭素環低級アルキル」の例としては、ベンジル、4-メトキシベンジル等が挙げられる。
R2dにおける「置換基群Eで置換されていてもよい複素環低級アルキル」の例としては、ピリジルメチル等が挙げられる。
R2dの好ましい態様は、メチル、エチル、n-プロピル、iso-プロピル、tert-ブチル、ベンジル等である。
R3eにおける「置換基群Eで置換されていてもよい低級アルキル」の例としては、メチル、エチル、トリフルオロメチル等が挙げられる。
R3dの好ましい態様は、-N(CH3)2、-OCH3、またはピロリジニルである。
R1dにおける「置換基群Eで置換されていてもよい炭素環低級アルキルオキシ」の例としては、ベンジルオキシ、フェネチルオキシ、2,4-トリフルオロベンジルオキシ、4-メトキシベンジルオキシ等が挙げられる。好ましくはベンジルオキシである。
R1dにおける「置換基群Eで置換されていてもよい複素環低級アルキルオキシ」の例としては、ピリジルメチルオキシ等が挙げられる。
R1dの好ましい態様は、水素、クロロ、ブロモ、メトキシ、ベンジルオキシである。
R7dの好ましい態様としては、クロロ、ブロモ、メトキシ、エトキシ、アセトキシ、メタンスルホニルオキシ、トリフルオロメタンスルホニルオキシ、パラトルエンスルホニルオキシ等が挙げられる。
化合物(Z2)の使用量は、化合物(Z1)に対して約1.0~3.0モル等量である。
反応温度は、通常、-10℃~還流温度であり、好ましくは室温である。
反応時間は、通常、10分~10時間、好ましくは1~4時間である。
必要に応じて、3級アミンを添加する。3級アミンとしては、ピリジン、トリエチルアミン、ジメチルアミノピリジン、N-メチルモルホリン等が挙げられる。
本工程は、以下の反応式に示すように、所望により塩基存在下、化合物(X2)および化合物(V2)を反応させて、化合物(X3)を含む溶液を得る工程である。
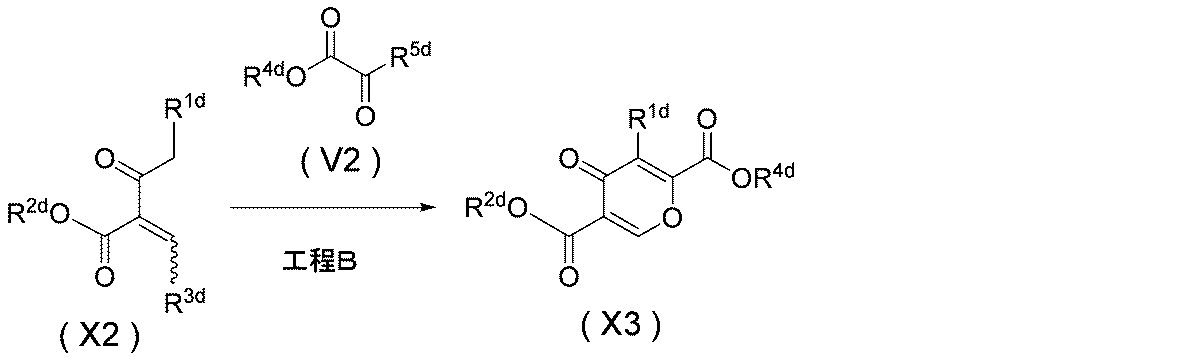
(式中、各記号は前記と同意義である)
R1dにおける「置換基群Eで置換されていてもよい炭素環低級アルキルオキシ」の例としては、ベンジルオキシ、フェネチルオキシ、2,4-トリフルオロベンジルオキシ、4-メトキシベンジルオキシ等が挙げられる。好ましくはベンジルオキシである。
R1dにおける「置換基群Eで置換されていてもよい複素環低級アルキルオキシ」の例としては、ピリジルメチルオキシ等が挙げられる。
R1dの好ましい態様は、水素、クロロ、ブロモ、メトキシ、ベンジルオキシである。
R2dにおける「置換基群Eで置換されていてもよい炭素環低級アルキル」の例としては、ベンジル、4-メトキシベンジル等が挙げられる。
R2dにおける「置換基群Eで置換されていてもよい複素環低級アルキル」の例としては、ピリジルメチル等が挙げられる。
R2dの好ましい態様は、メチル、エチル、n-プロピル、iso-プロピル、tert-ブチル等である。
R3dにおける「置換基群Eで置換されていてもよい低級アルキル」の例としては、メチル、エチル、トリフルオロメチル等が挙げられる。
R3eにおける「置換基群Eで置換されていてもよい低級アルキル」の例としては、メチル、エチル、トリフルオロメチル等が挙げられる。
R3dの好ましい態様は、-N(CH3)2、-OCH3、またはピロリジニルである。
R4dにおける「置換基群Eで置換されていてもよい低級アルキル」の例としては、メチル、エチル、n-プロピル、iso-プロピル、tert-ブチル等が挙げられる。
R4dにおける「置換基群Eで置換されていてもよい炭素環低級アルキル」の例としては、ベンジル、4-メトキシベンジル等が挙げられる。
R4dにおける「置換基群Eで置換されていてもよい複素環低級アルキル」の例としては、ピリジルメチル等が挙げられる。
R4dの好ましい態様としては、メチル、エチル、n-プロピル、iso-プロピル、tert-ブチル、ベンジル、4-メトキシベンジル等が挙げられる。
R5dの好ましい態様としては、クロロ、ブロモ、メトキシ、エトキシ、アセトキシ、メタンスルホニルオキシ、トリフルオロメタンスルホニルオキシ、パラトルエンスルホニルオキシ等が挙げられる。特に、クロロ、メトキシ、エトキシが好ましい。
反応溶媒としては、例えば、アセトニトリル、テトラヒドロフラン、ジオキサン、トルエン、キシレン、酢酸エチル、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、ジメチルイミダゾリジノン、N-メチルモルホリン、N-メチルピロリジノン等が挙げられる。
塩基としては、例えば、n-ブチルリチウム、tert-ブチルリチウム、ナトリウム-tert-ブトキシド、カリウム-tert-ブトキシド、ナトリウム-tert-ペントキシド、ナトリウムメトキシド、ナトリウムエトキシド、水素化ナトリウム、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド等が挙げられる。
塩基の使用量は、化合物(X2)に対して約1.0~5.0モル等量である
化合物(V2)の使用量は、化合物(X2)に対して約1.5~5.0モル等量であるか、もしくは化合物(V2)を溶媒と使用してもよい。
反応温度は、通常、-80℃~還流温度であり、好ましくは-20℃~50℃である。
反応時間は、通常、30分~50時間、好ましくは2~12時間である。
本工程は、以下の反応式に示すように、所望により塩基存在下、化合物(X2)および化合物(VA2)を反応させて、化合物(XA3)を含む溶液を得る工程である。
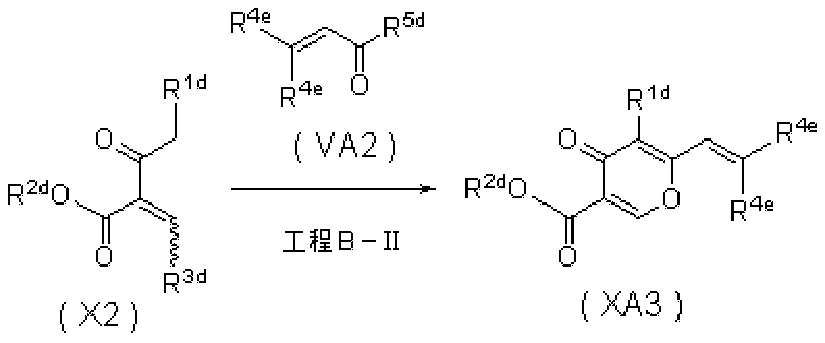
(式中、各記号は、前記と同意義である)
化合物(VA2)は、市販の試薬として、または公知の方法により得ることができる。
R4eの「置換基群Eで置換されていてもよい低級アルキル」の例としては、メチル、エチルなどが挙げられる。
R4eの「置換基群Eで置換されていてもよい炭素環式基」の例としては、フェニル、シクロヘキシルが挙げられる。
R4eの「置換基群Eで置換されていてもよい複素環式基」の例としては、ピリジル、ピペラジルが挙げられる。
R4eは、一方が水素であり、他方がフェニルであるものが好ましい。
反応溶媒としては、例えば、アセトニトリル、テトラヒドロフラン、ジオキサン、トルエン、キシレン、酢酸エチル、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、ジメチルイミダゾリジノン、N-メチルモルホリン、N-メチルピロリジノン等が挙げられる。
塩基としては、例えば、n-ブチルリチウム、tert-ブチルリチウム、ナトリウム-tert-ブトキシド、カリウム-tert-ブトキシド、ナトリウム-tert-ペントキシド、ナトリウムメトキシド、ナトリウムエトキシド、水素化ナトリウム、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド等が挙げられる。
塩基の使用量は、化合物(XA2)に対して約1.0~5.0モル等量である
化合物(VA2)の使用量は、化合物(X2)に対して約1.0~2.0モル等量である。
反応温度は、通常、-80℃~20℃であり、好ましくは-80℃~-40℃である。
反応時間は、通常、5分間~6時間、好ましくは15分間~2時間である。
本工程は、以下の反応式に示すように、化合物(X3)および化合物(V3)を反応させて、化合物(X4)を得る工程である。

(式中、各記号は前記と同意義である)
R1dにおける「置換基群Eで置換されていてもよい炭素環低級アルキルオキシ」の例としては、ベンジルオキシ、フェネチルオキシ、2,4-トリフルオロベンジルオキシ、4-メトキシベンジルオキシ等が挙げられる。好ましくはベンジルオキシである。
R1dにおける「置換基群Eで置換されていてもよい複素環低級アルキルオキシ」の例としては、ピリジルメチルオキシ等が挙げられる。
R1dの好ましい態様は、水素、クロロ、ブロモ、メトキシ、ベンジルオキシである。
R2dにおける「置換基群Eで置換されていてもよい炭素環低級アルキル」の例としては、ベンジル、4-メトキシベンジル等が挙げられる。
R2dにおける「置換基群Eで置換されていてもよい複素環低級アルキル」の例としては、ピリジルメチル等が挙げられる。
R2dの好ましい態様は、メチル、エチル、n-プロピル、iso-プロピル、tert-ブチル等である。
R4dにおける「置換基群Eで置換されていてもよい炭素環低級アルキル」の例としては、ベンジル、4-メトキシベンジル等が挙げられる。
R4dにおける「置換基群Eで置換されていてもよい複素環低級アルキル」の例としては、ピリジルメチル等が挙げられる。
R4dの好ましい態様としては、メチル、エチル、n-プロピル、iso-プロピル、tert-ブチル、ベンジル、4-メトキシベンジル等が挙げられる。
R6dにおける「置換基群Eで置換されていてもよい低級アルキル」の例としては、HC(=O)-CH2-、CH(-OH)2-CH2-、MeO-CH(-OH)-CH2-、ジメトキシエチル、ジエトキシエチル、CH2=CH-CH2-、HO-CH2-CH(-OH)-CH2-、

または、

等が挙げられる。
R6dにおける「置換基Eで置換されていてもよいアミノ」の例としては、メチルアミノ、エチルアミノ、ベンジルオキシカルボニルアミノ、tert-ブトキシカルボニルアミノ等が挙げられる。
R6dにおける「置換基群Eで置換されていてもよい炭素環式基」の例としては、

などが挙げられる。
R6dにおける「置換基群Eで置換されていてもよい複素環式基」の例としては、

などが挙げられる。
化合物(V3)の使用量は、化合物(X3)に対して約1.0~2.0モル等量である。
反応温度は、通常、0℃~還流温度であり、好ましくは20℃~70℃である。
反応時間は、通常、30分~50時間、好ましくは2~12時間である。
例えば、化合物(X4)のR6dが、ジメトキシエチルの場合、化合物(X4)を含む溶液に、酸を加えることによりHC(=O)-CH2-に導くことができる。酸としては特に限定されず、塩酸、臭化水素酸、硫酸、硝酸、リン酸、p-トルエンスルホン酸、メタンスルホン酸、ギ酸、酢酸、トリフルオロ酢酸、マレイン酸、シュウ酸等が例示される。酸の使用量は、化合物(X4)に対して2.0~10.0モル等量である。酢酸やギ酸は、溶媒として用いてもよく、上記の酸と混合して用いてもよい。
反応温度は、通常、約0℃~80℃、好ましくは10℃~40℃である。
反応時間は、通常、30分~50時間、好ましくは2~12時間である。
本工程は、以下の反応式に示すように、所望により塩基存在下、化合物(XA3)および化合物(V3)を反応させて、化合物(XA4)を含む溶液を得る工程である。

(式中、各記号は、前記と同意義である)
式(XA3)および(VA4)における、R1d、R2d、R4eの例ならびに好ましい態様は、それぞれ上記と同様である。
式(XA4)における、Raの「置換基群Eで置換されていてもよい低級アルキルアミノ」の例としては、メチルアミノ、エチルアミノ、イソプロピルアミノ、tert-ブチルアミノ、メトキシメチルアミノ、メトキシエチルアミノ、エトキシエチルアミノ、メトキシプロピルアミノ、ヒドロキシエチルアミノ、ピペラジニルカルボニルエチルアミノ、モルホリニルカルボニルエチルアミノ、メチルアミノエチルアミノ、メチルスルホニルエチルアミノ、tert-ブチルカルボニルアミノエチルアミノ、イソプロピルオキシカルボニルアミノエチルアミノ、メチルカルボニルアミノエチルアミノ、アミノエチルアミノ、tert-ブチルオキシカルボニルアミノエチルアミノなどが挙げられる。
Raの「置換基群Eで置換されていてもよい低級アルケニルアミノ」の例としては、エチレニルアミノ、プロペニルアミノなどが挙げられる。
Raの「置換基群Eで置換されていてもよい低級アルキニルアミノ」の例としては、プロピニルアミノが挙げられる。
Raの「置換基群Eで置換されていてもよい炭素環低級アルキルアミノ」の例としては、ベンジルアミノ、ジフルオロベンジルアミノ、クロロフルオロベンジルアミノ、シクロプロピルメチルアミノ、4-フルオロベンジルアミノ、シクロヘキシルメチレニルアミノ、シクロプロピルアミノ、エチルオキシカルボニルエチルアミノ、カルボキシエチルアミノ、ジメチルアミノカルボニル、4-メトキシベンジルアミノ、4-メチルベンジルアミノなどが挙げられる。
Raの「置換基群Eで置換されていてもよい複素環低級アルキルアミノ」の例としては、ピリジルメチルアミノ、テトラヒドロピラニルメチレニルアミノ、メチルイソキサゾリルメチレニルアミノなどが挙げられる。

または、
Raがヒドロキシの場合は、R6dは、

等が挙げられ、もしくは
Raが「置換基群Eで置換されていてもよい低級アルキルアミノ」、「置換基群Eで置換されていてもよい低級アルケニルアミノ」、「置換基群Eで置換されていてもよい低級アルキニルアミノ」、「置換基群Eで置換されていてもよい炭素環低級アルキルアミノ」、または「置換基群Eで置換されていてもよい複素環低級アルキルアミノ」の場合は、R6dは、

等が挙げられる。
R6dにおける「置換基群Eで置換されていてもよいアミノ」の例としては、メチルアミノ、エチルアミノ、ベンジルオキシカルボニルアミノ、tert-ブトキシカルボニルアミノ等が挙げられる。
R6dにおける「置換基群Eで置換されていてもよい炭素環式基」の例としては、

などが挙げられる。
R6dにおける「置換基群Eで置換されていてもよい複素環式基」の例としては、

などが挙げられる。
Raが、ヒドロキシの場合は、一般的な公知の酸化反応条件(例えば、オゾン酸化反応、RuCl3-NaIO4酸化反応など)で得られたアルデヒド(Raが水素)に対して、続く一般的な酸化反応条件(例えば、Cr3-ピリジン、PCC酸化、SO3-ピリジン酸化、NaClO2酸化など)で行うことができる。
Raが、「置換基群Eで置換されていてもよい低級アルキルアミノ」、「置換基群Eで置換されていてもよい低級アルケニルアミノ」、「置換基群Eで置換されていてもよい低級アルキニルアミノ」、「置換基群Eで置換されていてもよい炭素環低級アルキルアミノ」、または「置換基群Eで置換されていてもよい複素環低級アルキルアミノ」の場合は、カルボキシル基(Raがヒドロキシ)に対して一般的な脱水縮合反応(光延反応、カルボン酸ハロゲン化物、カルボン酸無水物、または縮合剤(例えば、WSC、カルボニルジイミダゾール、ジシクロヘキシルカルボジイミド)を用いる反応)によって行うことができる。
化合物(V3)の使用量は、化合物(XA3)に対して約1.0~3.0モル等量である。
反応温度は、通常、0℃~還流温度であり、好ましくは20℃~70℃である。
反応時間は、通常、10分~50時間、好ましくは1~12時間である。
本工程は、以下の反応式に示すように、所望により塩基存在下、化合物(X2)と化合物(V2)および化合物(V2’)を反応させて、化合物(X4’)を得る工程である。
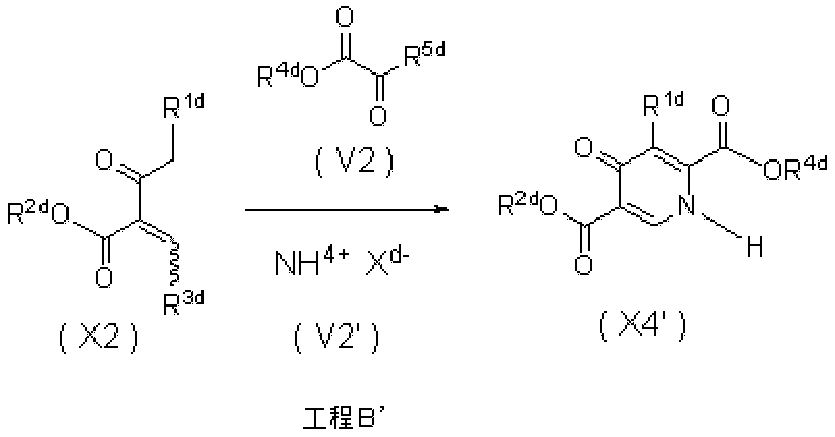
(式中、各記号は前記と同意義である)
式(X2)および(V2)における、R1d、R2d、R3d、R4d、およびR5dの例ならびに好ましい態様は、それぞれ上記と同様である。
反応溶媒としては、例えば、アセトニトリル、テトラヒドロフラン、ジオキサン、トルエン、キシレン、酢酸エチル、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、ジメチルイミダゾリジノン、N-メチルモルホリン、N-メチルピロリジノン等が挙げられる。
塩基としては、例えば、n-ブチルリチウム、tert-ブチルリチウム、ナトリウム-tert-ブトキシド、カリウム-tert-ブトキシド、ナトリウム-tert-ペントキシド、ナトリウムメトキシド、ナトリウムエトキシド、水素化ナトリウム、リチウムジイソプロピルアミド、リチウムビストリメチルシリルアミド等が挙げられる。
塩基の使用量は、化合物(X2)に対して約1.0~5.0モル等量である
化合物(V2)の使用量は、化合物(X2)に対して約1.0~3.0モル等量であるか、もしくは化合物(V2)を溶媒と使用してもよい。
反応温度は、通常、-80℃~還流温度であり、好ましくは-20℃~30℃である。
反応時間は、通常、10分~10時間、好ましくは30分~4時間である。
続いて上記の反応液に、化合物(V2’)を加えて反応させる。
化合物(V2’)の例としては、酢酸アンモニウム、塩化アンモニウム、臭化アンモニウム、硫酸アンモニウム、硫酸水素アンモニウム、ギ酸アンモニウム、硝酸アンモニウム、水酸化アンモニウム、リン酸アンモニウム、NH4 +BF4 -、NH4 +PF6 -、NH4 +Ph-SO3 -、NH4 +CH3-Ph-SO3 -、NH4 +CH3-SO3 -、等が挙げられる。好ましくは、酢酸アンモニウム、塩化アンモニウム、硫酸アンモニウム、硫酸水素アンモニウム、ギ酸アンモニウムである。
化合物(V2’)の使用量は、化合物(X2)に対して約1.0~3.0モル等量である。
反応温度は、通常、0℃~還流温度であり、好ましくは20℃~80℃である。
反応時間は、通常、10分~10時間、好ましくは30分~4時間である。
本工程により、化合物(X4’)を含む溶液が得られる。化合物(X4’)は一般的な精製方法(抽出、蒸留、カラムクロマトグラフィー、晶析等)により単離してもよいが、単離することなく次の反応に使用することもできる。好ましくは、晶析させることにより不純物を除いた結晶として単離する。
本工程は、以下の反応式に示すように、所望により塩基存在下、化合物(X4’)および化合物(V3’)を反応させて、化合物(X4)を得る工程である。

(式中、各記号は前記と同意義である)
式(X4’)および(V3’)における、R1d、R2d、R4dおよびR6dの例示ならびに好ましい態様は、それぞれ上記と同様である。
Ldにおける「脱離基」とは、ハロゲン、-O-SO2-CH3、-O-SO2-CF3、-O-SO2-Phまたは-O-SO2-Ph-CH3などが挙げられる。好ましくは、ハロゲンである。
塩基としては、炭酸カリウム、炭酸セシウム、水素化ナトリウム、n-ブチルリチウム、tert-ブチルリチウム、ナトリウム-tert-ブトキシド、カリウム-tert-ブトキシド、ナトリウム-tert-ペントキシド、ナトリウムメトキシド、トリエチルアミン、4-ジメチルアミノピリジン、ジイソプロピルエチルアミン、DBU(1,8-ジアザビシクロ[5.4.0]ウンデク-7-エン)等が挙げられる。
塩基の使用量は、化合物(X4’)に対して約1.0~5.0モル等量である
化合物(V3’)の使用量は、化合物(X4’)に対して約1.0~4.0モル等量であるか、もしくは化合物(V3’)を溶媒と使用してもよい。
反応温度は、通常、0℃~還流温度であり、好ましくは20℃~80℃である。
反応時間は、通常、30分~24時間、好ましくは1~8時間である。
反応溶媒としては、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、ジメチルイミダゾリジノン、N-メチルモルホリン、N-メチルピロリジノン等が挙げられる。
塩基としては、炭酸カリウム、炭酸セシウム、炭酸ナトリウム、炭酸リチウム等が挙げられる。
塩基の使用量は、化合物(X4’)に対して約1.0~5.0モル等量である
上記試薬の使用量は、化合物(X4’)に対して約1.0~4.0モル等量である。
反応温度は、通常、0℃~還流温度であり、好ましくは20℃~60℃である。
反応時間は、通常、30分~24時間、好ましくは1~8時間である。
本工程は、化合物(X4A)が以下の構造である場合に、化合物(I)を得る工程である。

(式中、R15は、アミノ保護基、水素、置換基群Eで置換されていてもよい低級アルキル、置換基群Eで置換されていてもよい低級アルケニル、置換基群Eで置換されていてもよい低級アルキニル、置換基群Eで置換されていてもよい炭素環低級アルキル、または置換基群Eで置換されていてもよい複素環低級アルキルであり、各記号は前記と同意義である)
化合物(XA4)のアミド部位(-CONH-R15)と、ヒドロキシ基に対して、分子内脱水縮合反応(例えば、光延反応)を生じさせることにより閉環体を得ることができる。R15が、アミノ保護基である場合は、公知の脱保護反応に付してから、前記分子内脱水縮合反応を行うことができる。
R7が水素でない場合は、アミノ基に対して、公知の求核置換反応を行うことにより、目的のR7を得ることができる。
-COOR2d部位はR2dが水素でない場合は公知の反応(エステル加水分解反応、カルボキシル保護基の脱保護反応など)を行ない、R2dを水素とした後、置換基群Eで置換されていてもよい炭素環低級アルキルアミン、置換基群Eで置換されていてもよい低級アルキルアルコール等と公知の反応(脱水縮合反応など)を行うことにより、-COR1に導くことができる。また、R2dがメチル基やエチル基などの低級アルキル基である場合は、アミノリシス反応を行うことにより、直接的に-COR1に導くこともできる。
R1dが、置換基群Eで置換されていてもよい低級アルキルオキシ、置換基群Eで置換されていてもよい炭素環低級アルキルオキシ、置換基群Eで置換されていてもよい複素環低級アルキルオキシ、または-OSi(R1e)3である場合は、公知のヒドロキシ脱保護反応に付すことにより、ヒドロキシ基に導くことができる。
R1dが、ハロゲンの場合、カリウムトリメチルシラノレート、リチウムトリメチルシラノレートを反応させて、無機酸の水溶液を加えることにより、ヒドロキシ基に導くことができる。別条件として、水素化ナトリウム/水(Bioorganic Medicinal Chemistry Letters, 17, 1713, 2007)、水酸化カリウム/トリス(ジベンジリデンアセトン)二パラジウム(Pd2dba3)/ジ-tert-ブチルアリールホスフィン(Journal of the American Chemical Society,128, 10694, 2006)、リン酸カリウム水和物(K3PO4・H2O)/トリス(ジベンジリデンアセトン)二パラジウム(Pd2dba3)/トリ-tert-ブチルホスフィン(Tetrahedoron Letters, 48, 473, 2007)も、ハロゲンのヒドロキシ基への変換反応として挙げられる。このように、原料のR1dがハロゲンの場合には、そのまま誘導体化することが可能になるので、アルコールの保護および/または脱保護反応を実施する方法と比較して、反応工定数が削減され、より有利な工業製法を構築可能となりえる。
R1dが、水素の場合、N-ブロモスクシイミド、N-クロロスクシイミド、塩化スルフリル等のハロゲン化剤を反応させることにより、R1dをハロゲンに変換した後、同様にカリウムトリメチルシラノレート、リチウムトリメチルシラノレートを反応させて、無機酸の水溶液を加えることにより、ヒドロキシ基に導くこともできる。よって、反応基質の反応性に応じて、適宜これらのR1dを選択することができる。
なお、上記工程においては、各反応の順序を適宜入れ替えることができる。
本工程は、化合物(X4A)が以下の構造である場合に、化合物(II)を得る工程である。

(式中、各記号は前記と同意義である)
化合物(XA4)のアミド部位(-CONH-R15)と、ヒドロキシ基に対して、分子内脱水縮合反応(例えば、光延反応)を生じさせることにより閉環体を得ることができる。R15が、アミノ保護基である場合は、公知の脱保護反応に付してから、前記分子内脱水縮合反応を行うことができる。
R7が水素でない場合は、アミノ基に対して、公知の求核置換反応を行うことにより、目的のR7を得ることができる。
-COOR2d部位はR2dが水素でない場合は公知の反応(エステル加水分解反応、カルボキシル保護基の脱保護反応など)を行ない、R2dを水素とした後、置換基群Eで置換されていてもよい炭素環低級アルキルアミン、置換基群Eで置換されていてもよい低級アルキルアルコール等と公知の反応(脱水縮合反応など)を行うことにより、-COR1に導くことができる。また、R2dがメチル基やエチル基などの低級アルキル基である場合は、アミノリシス反応を行うことにより、直接的に-COR1に導くこともできる。
R1dが、置換基群Eで置換されていてもよい低級アルキルオキシ、置換基群Eで置換されていてもよい炭素環低級アルキルオキシ、置換基群Eで置換されていてもよい複素環低級アルキルオキシ、または-OSi(R1e)3である場合は、公知のヒドロキシ脱保護反応に付すことにより、ヒドロキシ基に導くことができる。
R1dが、ハロゲンの場合、カリウムトリメチルシラノレート、リチウムトリメチルシラノレートを反応させて、無機酸の水溶液を加えることにより、ヒドロキシ基に導くことができる。別条件として、水素化ナトリウム/水(Bioorganic Medicinal Chemistry Letters, 17, 1713, 2007)、水酸化カリウム/トリス(ジベンジリデンアセトン)二パラジウム(Pd2dba3)/ジ-tert-ブチルアリールホスフィン(Journal of the American Chemical Society,128, 10694, 2006)、リン酸カリウム水和物(K3PO4・H2O)/トリス(ジベンジリデンアセトン)二パラジウム(Pd2dba3)/トリ-tert-ブチルホスフィン(Tetrahedoron Letters, 48, 473, 2007)も、ハロゲンのヒドロキシ基への変換反応として挙げられる。このように、原料のR1dがハロゲンの場合には、そのまま誘導体化することが可能になるので、アルコールの保護および/または脱保護反応を実施する方法と比較して、反応工定数が削減され、より有利な工業製法を構築可能となりえる。
R1dが、水素の場合、N-ブロモスクシイミド、N-クロロスクシイミド、塩化スルフリル等のハロゲン化剤を反応させることにより、R1dをハロゲンに変換した後、同様にカリウムトリメチルシラノレート、リチウムトリメチルシラノレートを反応させて、無機酸の水溶液を加えることにより、ヒドロキシ基に導くこともできる。よって、反応基質の反応性に応じて、適宜これらのR1dを選択することができる。
なお、上記工程においては、各反応の順序を適宜入れ替えることができる。
本工程は、化合物(X4A)が以下の構造である場合に、化合物(I)を得る工程である。

(式中、各記号は前記と同意義である)
化合物(XA4)のカルボキシル基(-COOH)と、アミノ基に対して、分子内脱水縮合反応(例えば、光延反応、縮合剤を用いたアミド化反応など)を生じさせることにより閉環体を得ることができる。R15が、アミノ保護基である場合は、公知の脱保護反応に付してから、前記分子内脱水縮合反応を行うことができる。
R7が水素でない場合は、アミノ基に対して、公知の求核置換反応を行うことにより、目的のR7を得ることができる。
-COOR2d部位はR2dが水素でない場合は公知の反応(エステル加水分解反応、カルボキシル保護基の脱保護反応など)を行ない、R2dを水素とした後、置換基群Eで置換されていてもよい炭素環低級アルキルアミン、置換基群Eで置換されていてもよい低級アルキルアルコール等と公知の反応(脱水縮合反応など)を行うことにより、-COR1に導くことができる。また、R2dがメチル基やエチル基などの低級アルキル基である場合は、アミノリシス反応を行うことにより、直接的に-COR1に導くこともできる。
R1dが、置換基群Eで置換されていてもよい低級アルキルオキシ、置換基群Eで置換されていてもよい炭素環低級アルキルオキシ、置換基群Eで置換されていてもよい複素環低級アルキルオキシ、または-OSi(R1e)3である場合は、公知のヒドロキシ脱保護反応に付すことにより、ヒドロキシ基に導くことができる。
R1dが、ハロゲンの場合、カリウムトリメチルシラノレート、リチウムトリメチルシラノレートを反応させて、無機酸の水溶液を加えることにより、ヒドロキシ基に導くことができる。別条件として、水素化ナトリウム/水(Bioorganic Medicinal Chemistry Letters, 17, 1713, 2007)、水酸化カリウム/トリス(ジベンジリデンアセトン)二パラジウム(Pd2dba3)/ジ-tert-ブチルアリールホスフィン(Journal of the American Chemical Society,128, 10694, 2006)、リン酸カリウム水和物(K3PO4・H2O)/トリス(ジベンジリデンアセトン)二パラジウム(Pd2dba3)/トリ-tert-ブチルホスフィン(Tetrahedoron Letters, 48, 473, 2007)も、ハロゲンのヒドロキシ基への変換反応として挙げられる。このように、原料のR1dがハロゲンの場合には、そのまま誘導体化することが可能になるので、アルコールの保護および/または脱保護反応を実施する方法と比較して、反応工定数が削減され、より有利な工業製法を構築可能となりえる。
R1dが、水素の場合、N-ブロモスクシイミド、N-クロロスクシイミド、塩化スルフリル等のハロゲン化剤を反応させることにより、R1dをハロゲンに変換した後、同様にカリウムトリメチルシラノレート、リチウムトリメチルシラノレートを反応させて、無機酸の水溶液を加えることにより、ヒドロキシ基に導くこともできる。よって、反応基質の反応性に応じて、適宜これらのR1dを選択することができる。
なお、上記工程においては、各反応の順序を適宜入れ替えることができる。
本工程は、化合物(X4A)が以下の構造である場合に、化合物(II)を得る工程である。

(式中、各記号は前記と同意義である)
化合物(XA4)のカルボキシル基(-COOH)と、アミノ基に対して、分子内脱水縮合反応(例えば、光延反応、縮合剤を用いたアミド化反応など)を生じさせることにより閉環体を得ることができる。R15が、アミノ保護基である場合は、公知の脱保護反応に付してから、前記分子内脱水縮合反応を行う。
R7が水素でない場合は、アミノ基に対して、公知の求核置換反応を行うことにより、目的のR7を得ることができる。
-COOR2d部位はR2dが水素でない場合は公知の反応(エステル加水分解反応、カルボキシル保護基の脱保護反応など)を行ない、R2dを水素とした後、置換基群Eで置換されていてもよい炭素環低級アルキルアミン、置換基群Eで置換されていてもよい低級アルキルアルコール等と公知の反応(脱水縮合反応など)を行うことにより、-COR1に導くことができる。また、R2dがメチル基やエチル基などの低級アルキル基である場合は、アミノリシス反応を行うことにより、直接的に-COR1に導くこともできる。
R1dが、置換基群Eで置換されていてもよい低級アルキルオキシ、置換基群Eで置換されていてもよい炭素環低級アルキルオキシ、置換基群Eで置換されていてもよい複素環低級アルキルオキシ、または-OSi(R1e)3である場合は、公知のヒドロキシ脱保護反応に付すことにより、ヒドロキシ基に導くことができる。
R1dが、ハロゲンの場合、カリウムトリメチルシラノレート、リチウムトリメチルシラノレートを反応させて、無機酸の水溶液を加えることにより、ヒドロキシ基に導くことができる。別条件として、水素化ナトリウム/水(Bioorganic Medicinal Chemistry Letters, 17, 1713, 2007)、水酸化カリウム/トリス(ジベンジリデンアセトン)二パラジウム(Pd2dba3)/ジ-tert-ブチルアリールホスフィン(Journal of the American Chemical Society,128, 10694, 2006)、リン酸カリウム水和物(K3PO4・H2O)/トリス(ジベンジリデンアセトン)二パラジウム(Pd2dba3)/トリ-tert-ブチルホスフィン(Tetrahedoron Letters, 48, 473, 2007)も、ハロゲンのヒドロキシ基への変換反応として挙げられる。このように、原料のR1dがハロゲンの場合には、そのまま誘導体化することが可能になるので、アルコールの保護および/または脱保護反応を実施する方法と比較して、反応工定数が削減され、より有利な工業製法を構築可能となりえる。
R1dが、水素の場合、N-ブロモスクシイミド、N-クロロスクシイミド、塩化スルフリル等のハロゲン化剤を反応させることにより、R1dをハロゲンに変換した後、同様にカリウムトリメチルシラノレート、リチウムトリメチルシラノレートを反応させて、無機酸の水溶液を加えることにより、ヒドロキシ基に導くこともできる。よって、反応基質の反応性に応じて、適宜これらのR1dを選択することができる。
なお、上記工程においては、各反応の順序を適宜入れ替えることができる。
本工程は、化合物(XA4)が以下の構造である場合に、化合物(II)を得る工程である。

(式中、各記号は前記と同意義である)
化合物(X4)のアミド部位(-CONH-R15)とアミノ基(-NH-R3)を、カルボニル基を有する化合物(R5-CO-R6)と縮合反応を生じさせることにより閉環体を得ることができる。R5-CO-R6の例としては、パラホルムアルデヒドが挙げられ、この場合は、R5およびR6が水素である。上記縮合反応を行う際、必要に応じて酸を加える。酸は、例えば、酢酸、ギ酸、硫酸などが挙げられる。
R15が、アミノ保護基である場合は、公知の脱保護反応に付してから、前記縮合反応を行うことができる。
R7が水素でない場合は、アミノ基に対して、公知の求核置換反応を行うことにより、目的のR7を得ることができる。
-COOR2d部位はR2dが水素でない場合は公知の反応(エステル加水分解反応、カルボキシル保護基の脱保護反応など)を行ない、R2dを水素とした後、置換基群Eで置換されていてもよい炭素環低級アルキルアミン、置換基群Eで置換されていてもよい低級アルキルアルコール等と公知の反応(脱水縮合反応など)を行うことにより、-COR1に導くことができる。また、R2dがメチル基やエチル基などの低級アルキル基である場合は、アミノリシス反応を行うことにより、直接的に-COR1に導くこともできる。
R1dが、置換基群Eで置換されていてもよい低級アルキルオキシ、置換基群Eで置換されていてもよい炭素環低級アルキルオキシ、置換基群Eで置換されていてもよい複素環低級アルキルオキシ、または-OSi(R1e)3である場合は、公知のヒドロキシ脱保護反応に付すことにより、ヒドロキシ基に導くことができる。
R1dが、ハロゲンの場合、カリウムトリメチルシラノレート、リチウムトリメチルシラノレートを反応させて、無機酸の水溶液を加えることにより、ヒドロキシ基に導くことができる。別条件として、水素化ナトリウム/水(Bioorganic Medicinal Chemistry Letters, 17, 1713, 2007)、水酸化カリウム/トリス(ジベンジリデンアセトン)二パラジウム(Pd2dba3)/ジ-tert-ブチルアリールホスフィン(Journal of the American Chemical Society,128, 10694, 2006)、リン酸カリウム水和物(K3PO4・H2O)/トリス(ジベンジリデンアセトン)二パラジウム(Pd2dba3)/トリ-tert-ブチルホスフィン(Tetrahedoron Letters, 48, 473, 2007)も、ハロゲンのヒドロキシ基への変換反応として挙げられる。このように、原料のR1dがハロゲンの場合には、そのまま誘導体化することが可能になるので、アルコールの保護および/または脱保護反応を実施する方法と比較して、反応工定数が削減され、より有利な工業製法を構築可能となりえる。
R1dが、水素の場合、N-ブロモスクシイミド、N-クロロスクシイミド、塩化スルフリル等のハロゲン化剤を反応させることにより、R1dをハロゲンに変換した後、同様にカリウムトリメチルシラノレート、リチウムトリメチルシラノレートを反応させて、無機酸の水溶液を加えることにより、ヒドロキシ基に導くこともできる。よって、反応基質の反応性に応じて、適宜これらのR1dを選択することができる。
なお、上記工程においては、各反応の順序を適宜入れ替えることができる。
本工程は、化合物(X4)が以下の構造である場合に、化合物(III)を得る工程である。

(式中、各記号は前記と同意義である)
化合物(X4)のアルデヒド部位が、化合物(V4)のアミノ基および-Y-Hと反応することにより、3環系の化合物を得ることができる。Zが単結合のとき、R14およびR10は一緒になって4~8員の置換基群Eで置換されていてもよい複素環式基を形成していてもよく、この場合は化合物(III)は、4環系の化合物となる。
化合物(V4)の例としては、3-アミノブタノール等が挙げられる。
-COOR2d部位はR2dが水素でない場合は公知の反応(エステル加水分解反応、カルボキシル保護基の脱保護反応など)を行ない、R2dを水素とした後、置換基群Eで置換されていてもよい炭素環低級アルキルアミン、置換基群Eで置換されていてもよい低級アルキルアルコール等と公知の反応(脱水縮合反応など)を行うことにより、-COR1に導くことができる。また、R2dがメチル基やエチル基などの低級アルキル基である場合は、アミノリシス反応を行うことにより、直接的に-COR1に導くこともできる。
R1dが、置換基群Eで置換されていてもよい低級アルキルオキシ、置換基群Eで置換されていてもよい炭素環低級アルキルオキシ、置換基群Eで置換されていてもよい複素環低級アルキルオキシ、または-OSi(R1e)3である場合は、公知のヒドロキシ脱保護反応に付すことにより、ヒドロキシ基に導くことができる。
R1dが、ハロゲンの場合、カリウムトリメチルシラノレート、リチウムトリメチルシラノレートを反応させて、無機酸の水溶液を加えることにより、ヒドロキシ基に導くことができる。別条件として、水素化ナトリウム/水(Bioorganic Medicinal Chemistry Letters, 17, 1713, 2007)、水酸化カリウム/トリス(ジベンジリデンアセトン)二パラジウム(Pd2dba3)/ジ-tert-ブチルアリールホスフィン(Journal of the American Chemical Society,128, 10694, 2006)、リン酸カリウム水和物(K3PO4・H2O)/トリス(ジベンジリデンアセトン)二パラジウム(Pd2dba3)/トリ-tert-ブチルホスフィン(Tetrahedoron Letters, 48, 473, 2007)も、ハロゲンのヒドロキシ基への変換反応として挙げられる。このように、原料のR1dがハロゲンの場合には、そのまま誘導体化することが可能になるので、アルコールの保護および/または脱保護反応を実施する方法と比較して、反応工定数が削減され、より有利な工業製法を構築可能となりえる。
R1dが、水素の場合、N-ブロモスクシイミド、N-クロロスクシイミド、塩化スルフリル等のハロゲン化剤を反応させることにより、R1dをハロゲンに変換した後、同様にカリウムトリメチルシラノレート、リチウムトリメチルシラノレートを反応させて、無機酸の水溶液を加えることにより、ヒドロキシ基に導くこともできる。よって、反応基質の反応性に応じて、適宜これらのR1dを選択することができる。
なお、上記工程においては、各反応の順序を適宜入れ替えることができる。
DMF:N,N-ジメチルホルムアミド
DMA:N,N-ジメチルアセトアミド
NMP:N-メチルピロリドン
DMI:ジメチルイミダゾリジノン
THF:テトラヒドロフラン
Ms:メタンスルホニル
Ts:パラトルエンスルホニル
Boc:tert-ブトキシカルボニル
DIBALH:ジイソブチルアルミニウムヒドリド
WSC:N-エチル-N’-(3-ジメチルアミノプロピル)カルボジイミド
HOBt:1-ヒドロキシベンゾトリアゾール
HATU:O-(7-アザベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウム ヘキサフルオロホスフェイト
NBS;N-ブロモスクシイミド
NCS:N-クロロスクシイミド
TEMPO:2,2,6,6-テトラメチルピペリジン-1-オキシルラジカル
PDC:ピリジニウムジクロロメート
DEAD:ジエチルアゾジカルボキシレート
DIAD:ジイソプロピルアゾジカルボキシレート
DMAP:4-ジメチルアミノピリジン
mCPBA:m-クロロ過安息香酸
DBU:1,8-ジアザビシクロ[5.4.0]-7-ウンデセン
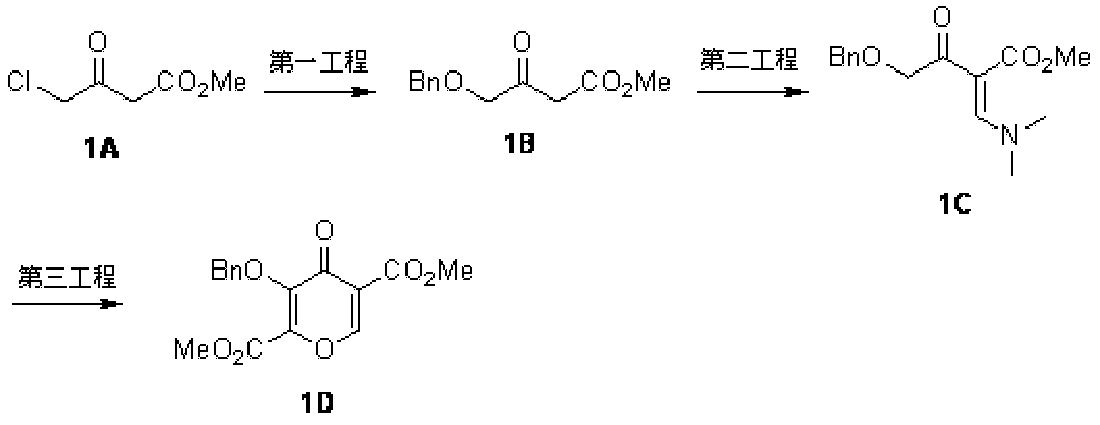
第一工程
Sodium tert-pentoxide (2.55 g, 23.2 mmol) のTHF (4 ml) 懸濁液にベンジルアルコール (1.00 g, 9.25 mmol) のTHF (3 ml) 溶液を窒素雰囲気下室温で加え、40℃で2時間撹拌した。この反応液を氷浴で冷却し、化合物1A (1.53 g, 10.2 mmol) のTHF (3 ml) 溶液を0-10℃で滴下した。反応液を室温で2時間撹拌後、2N塩酸 (15 ml)を加え、酢酸エチルで2回抽出した。合わせた抽出液を水、飽和重曹水、水、飽和食塩水で順次洗浄後、無水硫酸ナトリウムで乾燥した。溶媒を留去し、得られた油状物をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル 4:1, v/v) で精製し、化合物1B 1.89 g (収率92 %) を油状物として得た。
1H-NMR (CDCl3) δ: 3.56 (2H, s), 3.71 (3H, s), 4.14 (2H, s), 4.59 (2H, s), 7.27-7.42 (5H, m).
第二工程
化合物1B (1.80 g, 8.1 mmol) を 1,4-ジオキサン (18 mL) に溶解し、N,N-ジメチルホルムアミドジメチルアセタール (1.45 g, 12.2 mmol) を加えて室温で6 時間撹拌した。反応液を減圧下に濃縮後、残留物をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル 1:4, v/v) で精製し、化合物1C 1.77 g (収率79 %) を油状物として得た。
1H-NMR (CDCl3) δ: 2.90 (3H, br), 3.25 (3H, br), 3.69 (3H, s), 4.45 (2H, s), 4.59 (2H, s), 7.24-7.40 (5H, m), 7.73 (s, 1H).
第三工程
3頸フラスコに窒素雰囲気下Sodium tert-butoxide (2.55 g, 23.2 mmol)、シュウ酸ジメチル (639 mg, 5.41 mmol)、DMI (3 ml) を加え、これに化合物1C (0.50 g, 1.80 mmol) のDMI (2 ml) 溶液を25-30℃で滴下した。室温で7時間撹拌した後、2N塩酸 (10 ml)を加え、室温で15時間撹拌した。酢酸エチルで2回抽出し、合わせた抽出液を水、飽和重曹水、水、飽和食塩水で順次洗浄後、無水硫酸ナトリウムで乾燥した。溶媒を留去し、得られた残渣をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル 2:1から1:1, v/v) で精製し、化合物1D 488 mg (収率85 %) を白色結晶として得た。
1H-NMR (CDCl3) δ: 3.89 (3H, s), 3.93 (3H, s), 5.34 (2H, s), 7.32-7.40 (3H, m), 7.45-7.49 (2H, m), 8.50 (1H, s).

第一工程
Sodium tert-pentoxide (1.67 g, 15.2 mmol) のDMI (4 ml) 懸濁液にベンジルアルコール (0.66 g, 6.1 mmol) のDMI (3 ml) 溶液を窒素雰囲気下室温で加え、40℃で2時間撹拌した。この反応液を氷浴で冷却し、化合物2A (1.10 g, 6.68 mmol) のDMI (3 ml) 溶液を0-10℃で滴下した。反応液を0-5℃で2時間、室温で3時間撹拌後、2N塩酸 (15 ml)を加え、酢酸エチルで2回抽出した。合わせた抽出液を水、飽和重曹水、水、飽和食塩水で順次洗浄後、無水硫酸ナトリウムで乾燥した。溶媒を留去し、得られた油状物をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル 4:1, v/v) で精製し、化合物2B 1.29 g (収率90 %) を油状物として得た。
1H-NMR (CDCl3) δ: 1.25 (3H, t, J = 7.2 Hz), 3.54 (2H, s), 4.14 (2H, s), 4.17 (2H, q, J = 7.2Hz), 4.59 (2H, s), 7.28-7.40 (5H, m).
第二工程
化合物2B (9.73 g, 41.2 mmol) を トルエン (45 mL) に溶解し、N,N-ジメチルホルムアミドジメチルアセタール (7.36 g, 61.8 mmol) を加えて室温で5 時間撹拌した。反応液に水を加え、酢酸エチルで2回抽出した。合わせた抽出液を水、飽和食塩水で順次洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を留去し、得られた油状物をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル 1:1から3:7, v/v) で精製し、化合物2C 7.90 g (収率66 %) を油状物として得た。
1H-NMR (CDCl3) δ: 1.25 (3H, t, J = 7.2 Hz), 2.95 (3H, br), 3.22 (3H, br), 4.15 (2H, q, J = 7.2Hz), 4.45 (2H, s), 4.59 (2H, s), 7.22-7.40 (5H, m), 7.73 (1H, s).
第三工程
3頸フラスコに窒素雰囲気下Sodium tert-butoxide (495 mg, 5.15 mmol)、DMI (2 ml) を加え、これにシュウ酸ジメチル (608 mg, 5.15 mmol)、化合物2C (0.50 g, 1.72 mmol) のDMI (3 ml) 溶液を25-30℃で滴下した。室温で4時間撹拌した後、2N塩酸 (10 ml)を加え、室温で15時間撹拌した。トルエンで2回抽出し、合わせた抽出液を水、飽和重曹水、水、飽和食塩水で順次洗浄後、無水硫酸ナトリウムで乾燥した。溶媒を留去し、得られた残渣をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル 2:1, v/v) で精製し、化合物2D 420 mg (収率74 %) を白色結晶として得た。
1H-NMR (CDCl3) δ: 1.39 (3H, t, J = 7.2 Hz), 3.88 (3H, s), 4.39 (2H, q, J = 7.2Hz), 5.34 (2H, s), 7.30-7.41 (3H, m), 7.45-7.50 (2H, m), 8.48 (1H, s).
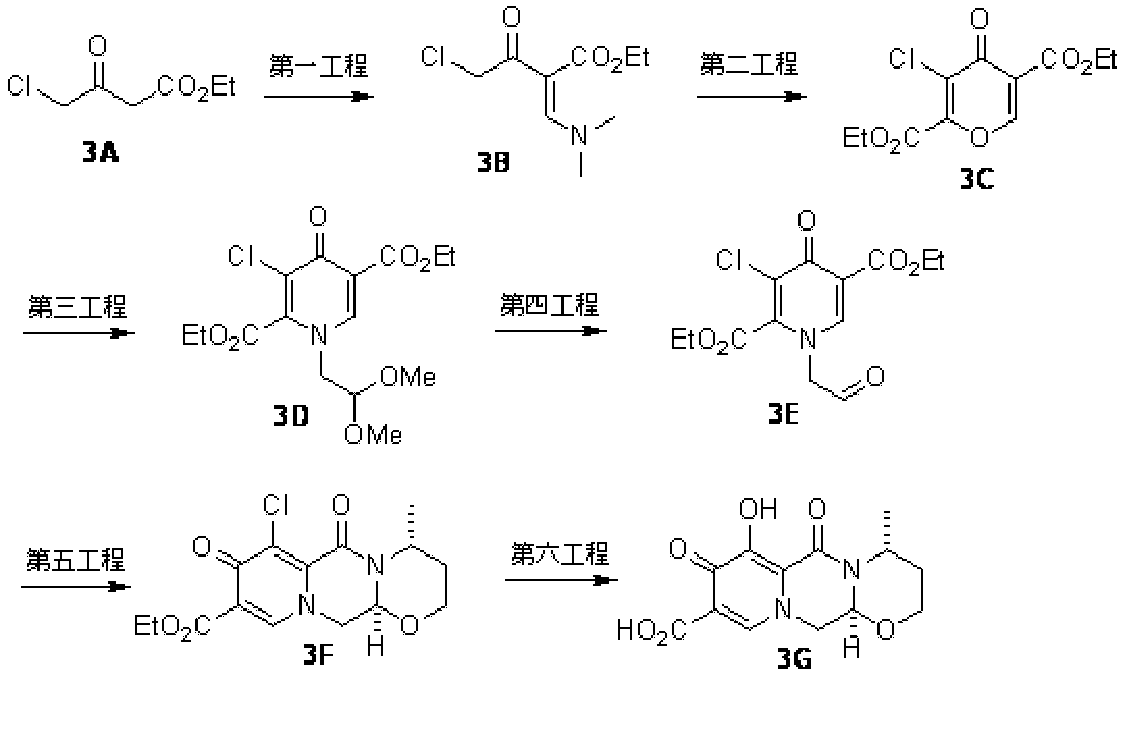
第一工程
化合物3A (5.0 g, 30.4 mmol) にN,N-dimethylformamide dimethyl acetal (4.9 ml, 36.5 mmol) を0℃にて冷却下滴下した。0℃で1時間攪拌したのち、反応液に酢酸エチル100mlを加え、0.5N塩酸水(50 ml)で洗浄した。水層を分液し酢酸エチル(50ml)で抽出した。有機層を合わせ、飽和重曹水、飽和食塩水で順次洗浄後、無水硫酸ナトリウムで乾燥した。溶媒を留去し、得られた残渣をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル 1:1 (v/v) → 酢酸エチル) で精製し、化合物3B 4.49 g (収率67 %) を油状物として得た。
1H-NMR (CDCl3)δ:1.32 (3H, t, J = 7.1 Hz), 2.90 (3H, br s), 3.29 (3H, br s), 4.23 (2H, q, J = 7.1 Hz), 4.54 (2H, s), 7.81 (1H, s).
第二工程
リチウムヘキサメチルジシラジド(1.0M トルエン溶液, 49 ml, 49.0 mmol)をテトラヒドロフラン (44 ml) で希釈し、これに-78℃にて冷却下、化合物3B (4.49 g, 20.4 mmol) のテトラヒドロフラン (10 ml) 溶液を滴下したのち、ethyl oxalyl chloride (3.35 g, 24.5 mmol) のテトラヒドロフラン (10 ml) 溶液を滴下した。-78℃で2時間攪拌後、0℃まで昇温した。反応液に2N塩酸を加えて20分間攪拌後、酢酸エチルで抽出し (200 ml x 2)、有機層を飽和重曹水、飽和食塩水で洗浄したのち、無水硫酸ナトリウムで乾燥した。溶媒を留去し、得られた残渣をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル 7:3→5:5→0:10 (v/v)) で精製し、化合物3C 1.77 g (収率31 %) を白色固体として得た。
1H-NMR (CDCl3)δ:1.36-1.46 (6H, m), 4.35-4.52 (8H, m), 8.53 (1H, s).
第三工程
化合物3C (300 mg, 1.09 mmol) のエタノール (6 ml) 溶液に aminoacetaldehyde dimethyl acetal (0.13 ml, 1.20 mmol) を0℃で加え、0℃にて1時間30分、室温にて18時間、次いで60℃にて4時間攪拌した。反応液を減圧下溶媒留去したのち、得られた残渣をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル 5:5→0:10 (v/v)) で精製し、化合物3D 252 mg (収率64 %) を油状物として得た。
1H-NMR (CDCl3)δ:1.36-1.47 (6H, m), 3.42 (6H, s), 3.90 (2H, d, J = 5.2 Hz), 4.37 (3H, q, J = 7.2 Hz), 4.50 (2H, q, J = 7.2 Hz), 8.16 (1H, s).
第四工程
化合物3D (1.02 g, 2.82 mmol) のギ酸 (10 ml) 溶液に62%-H2SO4 (892 mg, 5.64 mmol) を加え、室温で16時間攪拌した。減圧下ギ酸を溜去し残渣に塩化メチレンを加え、飽和重曹水を加えてpH=6.6に調整した。塩化メチレン層を分液し、水層を塩化メチレンで抽出した。塩化メチレン層を合わせ無水硫酸ナトリウムで乾燥した。溶媒を留去し、化合物3E 531.8 mg を黄色油状物として得た。
1H-NMR (CDCl3) δ: 1.28-1.49 (6H, m), 4.27-4.56 (4H, m), 4.84 (2H, s), 8.10 (1H, s), 9.72 (1H, s).
第五工程
化合物3E (531 mg, 1.68 mmol) のトルエン (5 ml) 溶液にメタノール(0.20 ml, 5.0 mmol)、(R)-3-アミノ-ブタン-1-オール (179 mg, 2.0 mmol)、酢酸 (0.096 ml, 1.70 mmol) を加え、4時間加熱還流した。反応液を室温まで冷却後クロロホルムで希釈したのち、飽和重曹水で洗浄し、水層をクロロホルムで抽出した。クロロホルム層を合わせ飽和食塩水で洗浄したのち、無水硫酸ナトリウムで乾燥した。溶媒を留去し、得られた残渣をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール 100:0→90:10) で精製し、化合物3F 309.4 mg を褐色油状物として得た。
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.1 Hz), 1.40 (3H, d, J = 7.1 Hz), 1.55-1.61 (1H, m), 2.19-2.27 (1H, m), 4.00 (1H, d, J = 1.5 Hz), 4.03 (1H, d, J = 2.5 Hz), 4.10 (1H, dd, J = 13.2, 6.3 Hz), 4.26 (1H, dd, J = 13.2, 3.8 Hz), 4.38 (2H, q, J = 7.1 Hz), 5.00-5.05 (1H, m), 5.31 (1H, dd, J = 6.4, 3.9 Hz), 8.10 (1H, s).
第六工程
化合物3F (159 mg, 0.47 mmol) の1,2-ジメトキシエタン(2 ml)溶液にカリウム トリメチルシラノレート(333 mg, 2.34 mmol)を加え、室温にて7時間攪拌した。反応液に1N-塩酸と飽和食塩水を加え、クロロホルムにて抽出した。クロロホルム層を合わせ、無水硫酸ナトリウムで乾燥した。溶媒を溜去し、化合物3G 34.4 mg (収率25%)を橙色粉末として得た。
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 3.5 Hz), 1.58-1.65 (1H, m), 2.26-2.30 (1H,m), 4.06-4.10 (2H, m), 4.31 (1H, dd, J = 13.8, 5.6 Hz), 4.48 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, t, J = 6.4 Hz), 5.36 (1H, dd, J = 5.5, 4.0 Hz), 8.44 (1H, s), 12.80 (1H, s), 14.90 (1H, s).

第一工程
化合物4A (10.0 g, 76.8 mmol) にN,N-dimethylformamide dimethyl acetal (12.2 ml, 92.2 mmol) を0℃にて冷却下滴下した。0℃で1時間30分、次いで室温で2時間30分攪拌したのち、反応液に酢酸エチル100mlを加え、溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル 5:5→0:10 (v/v)) で精製し、化合物4B 12.45 g (収率88 %) を油状物として得た。
1H-NMR (CDCl3)δ:1.32 (3H, t, J = 7.1 Hz), 2.33 (3H, s), 3.04 (6H, br s), 4.23 (2H, q, J = 7.2 Hz), 7.68 (1H, s).
第二工程
リチウムヘキサメチルジシラジド(1.0M トルエン溶液, 24 ml, 24.0 mmol)をテトラヒドロフラン (20 ml) で希釈し、これに-78℃にて冷却下、化合物4B (1.85 g, 10.0 mmol) のテトラヒドロフラン (5 ml) 溶液を滴下したのち、ethyl oxalyl chloride (1.34 ml, 12.0 mmol) のテトラヒドロフラン(5 ml) 溶液を滴下した。-78℃で2時間攪拌後、反応液に2N-塩酸を加えて室温にて20分間攪拌した。酢酸エチルで抽出し有機層を飽和重曹水、飽和食塩水で順次洗浄したのち、無水硫酸ナトリウムで乾燥した。溶媒を留去し、得られた残渣をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル 75:25→455:5 (v/v)) で精製し、化合物4C 1.03 g (収率43 %) を褐色油状物として得た。
1H-NMR (CDCl3)δ:1.38 (3H, t, J = 7.1 Hz), 1.42 (3H, t, J = 7.4 Hz), 4.33-4.47 (4H, m), 7.19 (1H, s), 8.54 (1H, s).
第三工程
化合物4C (680 mg, 2.83 mmol) のエタノール (6.8 ml) 溶液に aminoacetaldehyde dimethyl acetal (0.34 ml, 3.11 mmol) を0℃で加え、室温にて16時間静置した。反応液を減圧下溶媒留去したのち、得られた残渣をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル 90:10 (v/v)) で精製し、化合物4D 875 mg (収率94 %) を油状物として得た。
1H-NMR (CDCl3)δ:1.38 (3H, t, J = 7.1 Hz), 1.39 (3H, t, J = 7.1 Hz), 3.40 (6H, s), 4.33 (2H, d, J = 4.7 Hz), 4.37 (4H, q, J = 7.1 Hz), 4.49 (1H, t, J = 4.7 Hz), 7.06 (1H, s), 8.17 (1H, s).
第四工程
化合物4D (2.68 g, 8.18 mmol) のN,N-ジメチルホルムアミド (10 ml) 溶液に N-bromosuccinimide (1.46 g, 8.18 mmol) を加え、室温にて48時間攪拌した。反応液に飽和重曹水を加えたのち酢酸エチルで抽出し、有機層を水、飽和食塩水で順次洗浄後無水硫酸ナトリウムで乾燥した。溶媒を留去したのち、得られた残渣をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル 90:10 (v/v)) で精製し、化合物4E 2.83 g (収率85 %) を油状物として得た。
1H-NMR (CDCl3) δ: 1.41 (3H, t, J = 7.1 Hz), 1.48 (3H, t, J = 7.1 Hz), 3.42 (6H, s), 3.90 (2H, d, J = 5.0 Hz), 4.39 (2H, q, J = 7.1 Hz), 4.53 (3H, q, J = 14.3 Hz), 4.54 (3H, s), 4.57 (3H, t, J = 5.4 Hz), 8.19 (1H, s).
第五工程
化合物4E (2.23 g, 5.49 mmol) のギ酸 (15 ml) 溶液に62%-H2SO4 (1.74 g, 10.98 mmol) を加え、室温で8時間攪拌した。0.5N-水酸化ナトリウム水溶液(120 ml)を加え、塩化メチレンにて抽出した。塩化メチレン層を合わせ飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。溶媒を留去し、化合物4F 1.31 g を白色粉末として得た。
1H-NMR (CDCl3) δ: 1.31-1.46 (6H, m), 4.33-4.48 (4H, m), 4.82 (2H, s), 8.11 (1H, s), 9.71 (1H, s).
第六工程
化合物4F (1.31 g, 3.64 mmol) のトルエン (13 ml) 溶液にメタノール(0.44 ml, 10.9 mmol)、(R)-3-アミノ-ブタン-1-オール (389 mg, 4.36 mmol)、酢酸 (0.21 ml, 3.64 mmol) を加え、3時間加熱還流した。反応液を室温まで冷却後クロロホルムで希釈したのち、飽和重曹水で洗浄し、水層をクロロホルムで抽出した。クロロホルム層を合わせ飽和食塩水で洗浄したのち、無水硫酸ナトリウムで乾燥した。溶媒を留去し、得られた残渣をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール 100:0→90:10) で精製し、化合物4G 1.58 g を油状物として得た。
1H-NMR (CDCl3) δ: 1.40 (3H, d, J = 5.7 Hz), 1.56-1.60 (1H, m), 2.19-2.24 (1H, m), 3.99 (1H, d, J = 2.0 Hz), 4.02 (1H, d, J = 2.4 Hz), 4.11 (1H, dd, J = 13.3, 6.7 Hz), 4.28 (1H, dd, J = 13.3, 3.9 Hz), 4.36 (3H, q, J = 7.1 Hz), 4.49-4.56 (1H, m), 4.98-5.03 (1H, m), 5.34 (1H, dd, J = 6.6, 3.8 Hz), 8.07 (1H, s).
第七工程
化合物4G (300 mg, 0.78 mmol) の1,2-ジメトキシエタン(3 ml)溶液にカリウム トリメチルシラノレート(249 mg, 1.95 mmol)を加え、室温にて1時間攪拌した。カリウム トリメチルシラノレート(249 mg, 1.95 mmol)を追加し、さらに60℃にて1時間攪拌した。反応液に1N-塩酸と飽和食塩水を加え、クロロホルムにて抽出した。クロロホルム層を合わせ、無水硫酸ナトリウムで乾燥した。溶媒を溜去し、化合物3G 100.3 mg (収率43%)を黄色粉末として得た。
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 3.5 Hz), 1.58-1.65 (1H, m), 2.26-2.30 (1H,m), 4.06-4.10 (2H, m), 4.31 (1H, dd, J = 13.8, 5.6 Hz), 4.48 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, t, J = 6.4 Hz), 5.36 (1H, dd, J = 5.5, 4.0 Hz), 8.44 (1H, s), 12.80 (1H, s), 14.90 (1H, s).

第一工程
化合物5A (598 mg, 4.09 mmol) と N,N-dimethylformamide dimethyl acetal (488 mg, 4.09 mmol) をトルエン (1 ml) に溶解し、室温で11時間攪拌した。反応液を減圧下溶媒留去し、得られた残渣(化合物5Bを含む)は精製することなく、第二工程に使用した。
第二工程
Sodium tert-butoxide (400 mg, 4.16 mmol) をジメチルイミダゾリジノン (5 ml) に懸濁させ、これに第一工程で得られた粗生成物のジメチルイミダゾリジノン (5 ml) 溶液を加えたのち、dimethyl oxalate (983 mg, 8.32 mmol) のTHF (10 ml) 溶液を滴下し、室温で45分攪拌した。反応液を2N 塩酸-メタノール (20 ml) にあけ、0℃で20分攪拌した。水を加えたのち酢酸エチルで抽出し、有機層を水、飽和重曹水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。溶媒を留去したのち、得られた残渣をシリカゲルカラムクロマトグラフィーで精製し、化合物5C 222 mg (収率:5Aから22 %) を得た。
1H-NMR (CDCl3)δ:3.91 (3H, s), 3.97 (3H, s), 4.05 (3H, s), 8.50 (1H, s).

第一工程
リチウムヘキサメチルジシラジド(1.0M トルエン溶液, 12 ml, 12.0 mmol)をテトラヒドロフラン (11 ml) で希釈し、これに-78℃にて冷却下、化合物6A (1.46 g, 5.0 mmol) のテトラヒドロフラン (2 ml) 溶液を滴下したのち、ethyl oxalyl chloride (0.67 ml, 6.0 mmol) のテトラヒドロフラン (2 ml) 溶液を滴下した。-78℃で2時間攪拌後、反応液に酢酸アンモニウム (500 mg) と酢酸 (10 ml) を加え、65℃にて1時間30分攪拌した。反応液に水を加え酢酸エチルで抽出し、有機層を水、飽和重曹水で順次洗浄後無水硫酸ナトリウムで乾燥した。溶媒を留去し、得られた残渣をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル 55:45→45:55 (v/v)) で精製し、化合物6B 505.1 mg を黄色固体として得た。これをイソプロピルエーテル-ヘキサン (1:2) で洗い、減圧下乾燥することで、化合物6B 416.8 mg (収率24 %) を黄色結晶として得た。
1H-NMR (CDCl3)δ:1.35 (3H, t, J = 7.1 Hz), 1.46 (3H, t, J = 7.1 Hz), 4.40 (2H, q, J = 7.2 Hz), 4.50 (2H, q, J = 7.1 Hz), 5.20 (2H, s), 7.33-7.41 (3H, m), 7.49-7.52 (2H, m), 8.76 (1H, s), 11.61 (1H, br s).
第二工程
化合物6B (51.8 mg, 0.15 mmol) のN,N-ジメチルホルムアミド (1 ml) 溶液に炭酸セシウム (73.3 mg, 0.23 mmol) とbromoacetaldehyde dimethyl acetal (38.0 mg, 0.23 mmol) を加え、室温で終夜攪拌した。炭酸セシウム (73.3 mg, 0.23 mmol) およびbromoacetaldehyde dimethyl acetal (38.0 mg, 0.23 mmol) を追加し、さらに100℃にて20分間攪拌した。反応液に水を加えたのち酢酸エチルにて抽出し、有機層を水、飽和食塩水で順次洗浄後、無水硫酸ナトリウムで乾燥した。溶媒を留去し、得られた残渣をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル 50:50→30:70 (v/v)) で精製し、化合物6C 35.3 mg (収率54 %) を無色油状物として得た。
1H-NMR (CDCl3)δ:1.26 (3H, t, J = 7.1 Hz), 1.40 (3H, t, J = 7.1 Hz), 3.39 (6H, s), 3.91 (2H, d, J = 5.0 Hz), 4.29 (2H, q, J = 7.1 Hz), 4.40 (2H, q, J = 7.2 Hz), 4.50 (1H, t, J = 5.0 Hz), 5.30 (2H, s), 7.31-7.37 (3H, m), 7.43-7.46 (2H, m), 8.12 (1H, s).
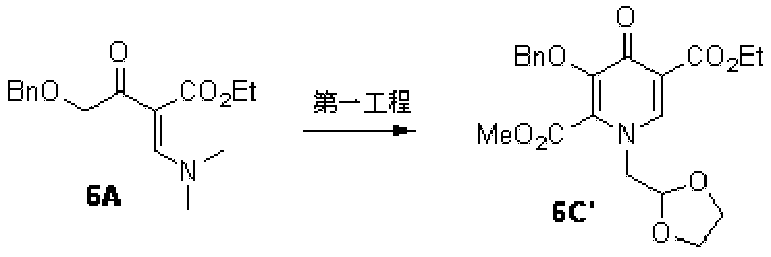
第一工程
化合物6A(291 mg, 1.0 mmol)とdimethyl oxalate (354 mg, 3.0 mmol)をジメチルイミダゾリジノン (1.4 ml) に溶解し、これにsodium methoxide (28%-メタノール溶液、0.30 ml , 1.5 mmol) を加え室温で2時間攪拌した。これに1,3-dioxolan-2-yl-methylamine (154 mg, 1.5 mmol) および酢酸(0.29 ml, 5.0 mmol)を加え、室温にて38時間攪拌した。反応液に飽和重曹水を加え、酢酸エチルにて抽出した。酢酸エチル層を合わせ、水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。溶媒を留去したのち、得られた残渣をシリカゲルカラムクロマトグラフィー (ヘキサン-酢酸エチル 33:67→15:85) で精製し、化合物6C’ 294.8 mg (収率70 %) を淡黄色油状物として得た。
1H-NMR (CDCl3) δ: 1.43 (3H, t, J = 7.1 Hz), 3.73-3.75 (2H, m), 3.81 (3H, s), 3.82-3.85 (2H, m), 4.21 (2H, d, J = 2.2 Hz), 4.42 (2H, q, J = 7.1 Hz), 5.14 (1H, t, J = 2.3 Hz), 5.32 (2H, s), 7.34-7.37 (3H, m), 7.44-7.46 (2H, m), 8.14 (1H, s).

第一工程
化合物8A (900 mg, 2.60 mmol) のエタノール (5 ml) 溶液に aminoacetaldehyde dimethyl acetal (7.80 mmol) を加え、室温にて22時間攪拌した。反応液に酢酸エチル (5 ml) と水 (5 ml)を加え、酢酸エチル (5ml) で抽出した。有機層を水 (10ml) で洗浄後溶媒を留去し、得られた残渣をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル 2:1) で精製し、化合物6C 0.37g (収率33 %) を無色油状物として得た。
1H-NMR (CDCl3) δ: 7.90 (1H, s), 7.45-7.43 (5H, m), 5.30 (2H, s), 4.51 (1H, t, J = 5.1 Hz), 4.40 (2H, q, J = 7.1 Hz), 4.30 (2H, q, J = 7.1 Hz), 3.91 (2H, d, J = 5.1 Hz), 3.46 (6H, s), 1.40 (3H, t, J = 7.1 Hz), 1.26 (3H, t, J = 7.1 Hz).
第二工程
化合物6C (433.5 mg, 1.0 mmol) のギ酸 (4 ml) 溶液に62%-H2SO4 (316 mg, 2.0 mmol) を加え、室温で3時間攪拌した。反応液に塩化メチレンを加え、0.5N-水酸化ナトリウム水溶液(12 ml)で洗浄し、水層を塩化メチレンで抽出した。塩化メチレン層を合わせ飽和食塩水で洗浄したのち、無水硫酸ナトリウムで乾燥した。溶媒を留去し、化合物8C 207.6 mg (収率51%)を黄色泡状物として得た。
1H-NMR (CDCl3) δ: 1.23 (3H, t, J = 7.1 Hz), 1.42 (3H, t, J = 7.1 Hz), 4.25 (2H, q, J = 7.2 Hz), 4.42 (2H, q, J = 7.1 Hz), 4.79 (2H, s), 5.34 (2H, s), 7.31-7.53 (5H, m), 8.05 (1H, s), 9.67 (1H, s).

第一工程
2頸フラスコに窒素雰囲気下化合物9C (291.3 mg, 10 mmol)をDMI (1.4 mL)に溶解し、シュウ酸ジメチル (354.3 mg, 3.0 mmol)、sodium methoxide (28%-メタノール溶液 0.3 mL, 1.5 mmol) を加え、室温にて2時間攪拌した。これに2-(aminomethyl)-1,3-dioxane (154.7 mg, 1.5 mmol) および酢酸 (0.29 mL, 5.0 mmol) を加え、室温にて5時間攪拌した。 反応液に酢酸エチル (50mL) を加え、水 (20 mL)、10%-塩化アンモニウム水溶液 (20mL)、水 (20mL)、飽和食塩水 (20 mL)で順次洗浄し、無水硫酸マグネシウムで乾燥した。 溶媒を留去し、得られた残渣をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル 1:1→1:3, v/v) で精製し、化合物9C’ 99.0 mg (収率25 %) を白色結晶として得た。
1H-NMR (CDCl3) δ: 8.14 (1H, s), 7.44-7.42 (5H, m), 5.29 (2H, s), 5.12 (1H, s), 4.19 (2H, s), 3.93 (3H, s), 3.83-3.70 (2H, m), 3.83 (2H, s).

第一工程
化合物1D(318.3 mg, 1.0 mmol)と酢酸 (0.023 mL, 0.4 mmol) をトルエン(2 mL) 溶解し、tert-butylcarbazate (132.2 mg, 1.0 mmol) のトルエン (2 mL) 溶液を65℃にて滴下し、65℃で5時間攪拌した。 反応液に飽和重曹水を加え、酢酸エチルにて抽出した。有機層を飽和食塩水にて洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を留去したのち、得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=50:50→33:67)で精製し、化合物10B 315.3 mg (収率72.9 %) を得た。
1H-NMR (DMSO-d6) δ: 1.36 (9H, s), 3.84 (6H, s), 5.11 (2H, s), 7.34-7.38 (5H, m), 8.33 (1H, s), 11.11 (1H, br s).
第二工程
化合物10B (310.3 mg, 0.72 mmol)をテトラヒドロフラン-メタノール (2 mL-1 mL) に1N-水酸化リチウム水溶液 (1.44 mL, 1.44 mmol) を加えて室温にて2時間攪拌した。氷水浴にて冷却して2N-塩酸を加えたのち、酢酸エチルと水を加えた。酢酸エチル層を分液し、水層を酢酸エチルで抽出した。酢酸エチル層を合わせ、飽和食塩水で洗浄し無水硫酸ナトリウムで乾燥した。溶媒を留去し、化合物10C 284.1 mg (収率94.3 %) を得た。
1H-NMR (CDCl3) δ: 1.43 (9H, s), 3.98 (3H, s), 5.34 (2H, s), 7.36-7.37 (5H, m), 8.44 (1H, s), 14.53 (1H, br s).
第一工程
化合物11A (12.8 g, 89.4 mmol) とピリジン(8.50 g, 107 mmol) のジクロロメタン (90 mL) 溶液を1-3℃に冷却し、同温度を保ちながら benzyloxyacetyl chloride (19.8 g, 107 mmol) のジクロロメタン (90 mL) 溶液を 50分間かけて滴下した。反応液を同温度で30分間撹拌後、徐々に60分間かけて15℃に上げ、氷水を加えた。ジクロロメタン層を分離し、水層をジクロロメタンで1回、抽出した。合わせた抽出液を3回水洗し、飽和食塩水で洗浄後、乾燥した。溶媒を留去し、得られた油状物をシリカゲルカラムクロマトグラフィーに付し、精製した。はじめに n-ヘキサンで溶出し、次いでn-ヘキサン-酢酸エチル (1:1, v/v) で溶出した。目的の画分を濃縮すると油状物として 22.2 g の化合物11Bを得た。
1H-NMR (CDCl3) δ: 1.25(3H, t, J=7.2Hz), 2.90(3H,brs), 3.24(3H, brs), 4.15(2H, q, J=7.2Hz), 4.45(2H, s), 4.58(2H, s), 7.25-7.38(5H, m), 7.72(1H, s).
第二工程
1規定のリチウムヘキサメチルジシラザンTHF溶液(4.29 ml, 4.29 mmol)を-78℃に冷却し、同温度を保ちながら化合物11B (500 mg, 1.72 mmol) とシンナモイルクロリド (343.2 mg, 2.06 mmol) のTHF溶液 (4 ml) を3分間かけて滴下した。反応液を同温度で 25 分間撹拌後、2規定塩酸(10 ml)を加え、更に 10 分間室温で撹拌した。反応溶液に酢酸エチルを加え有機層を分離し、水層を酢酸エチルで3回抽出した。合わせた抽出液を硫酸ナトリウムで乾燥した。溶媒を留去し、得られた油状物をシリカゲルカラムクロマトグラフィーに付し、精製した。n-ヘキサン-酢酸エチル (1:1, v/v) で溶出する画分から固体として364.3 mg(収率56%)の化合物11Cを得た。
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.2 Hz), 4.39 (2H, q, J = 7.2 Hz), 5.27 (2H, s), 6.99 (1H, d, J = 16.2 Hz), 7.23 (1H, d, J = 16.2), 7.26-7.48 (10H, m), 8.45 (1H, s).
第三工程
窒素気流下、化合物11Cと塩化ルテニウム(2.76 mg, 0.0133 mmol) のMeCN (5 ml)溶液に、室温下、過ヨウ素酸ナトリウム(625.8 mg, 2.93 mmol)と96% 硫酸 (287.4 mg, 2.93 mmol) の水溶液(8 ml)を10 分間かけて滴下した。反応液を同温度で5分間撹拌後、酢酸エチルを加え有機層を分離し、水層を酢酸エチルで2回抽出した。合わせた抽出液を硫酸ナトリウムで乾燥した。溶媒を留去し、得られた油状物をシリカゲルカラムクロマトグラフィーに付し、精製した。n-ヘキサン-酢酸エチル (1:1, v/v) で溶出する画分から油状物として303.2 mg (収率75%) の化合物11Dを得た。
1H-NMR (CDCl3) δ: 1.39 (3H, t, J = 6.9 Hz), 4.40 (2H, q, J = 6.9 Hz), 5.54 (2H, s), 7.37 (5H, s), 8.48 (1H, s), 9.85 (1H, s).
第四工程
化合物11D(1.00 g, 3.31 mmol)のMeCN (15 ml) 溶液に室温下、96% 硫酸 (421.7 mg, 4.30 mmol) と amidosululic acid (642.7 mg, 6.62 mmol) の水溶液 (10 ml) を加え撹拌し、同温度を保ちながら亜塩素酸ナトリウム (388.9 mg, 4.30 mmol) の水溶液 (10 ml) 5分間かけて滴下した。反応液を同温度で5分間撹拌後、飽和食塩水を加え、酢酸エチルで3回抽出した。合わせた抽出液を硫酸ナトリウムで乾燥した。溶媒を留去し、得られた油状物をシリカゲルカラムクロマトグラフィーに付し、精製した。はじめに クロロホルムで溶出し、次いでクロロホルム-MeOH (7:3, v/v) で溶出した。目的の画分を濃縮すると油状物として 748.8 mg (収率71%) の化合物11Eを得た。
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.2 Hz), 3.93 (1H, br s), 4.40 (2H, q, J = 7.2 Hz), 5.61 (2H, s), 7.38-7.44 (10 H, m), 8.52 (1H, s).
第五工程
化合物11E (1.00 g, 3.14 mmol)のDMF (10 ml)溶液に室温下、WSC HCl(1.20 g, 6.28 mmol) とHOBt (551.6 mg, 4.08 mmol) を加え、同温度で90分間撹拌した。反応液を0℃に冷却し、2-methoxyethanamine (236.0 mg, 3.14 mmol) のDMF (2 ml) 溶液を3分間かけて滴下した。反応液を同温度で1時間撹拌し、水を加え酢酸エチルで3回抽出した。抽出液を3回水洗し、硫酸ナトリウムで乾燥した。溶媒を留去し、得られた油状物をシリカゲルクロマトグラフィーに付し、精製した。はじめにn-ヘキサン-酢酸エチル (1:1, v/v)で溶出し、次いでn-ヘキサン-酢酸エチル (1:9, v/v)で溶出した。目的の画分を濃縮すると油状物として 928.5 mg (収率79%) の化合物11Fを得た。
1H-NMR (CDCl3) δ: 1.39 (3H, t, J = 7.2 Hz), 3.29 (3H, s), 3.41 (2H, t, J = 5.4 Hz), 3.47-3.53 (2H, m), 4.39 (2H, q, J = 7.2 Hz), 5.44 (2H, s), 7.36 (3H, m), 7.44-7.47 (2H, m), 8.07 (1H, br s), 8.54 (1H, s).
第六工程
化合物11F (500 mg, 1.33 mmol) と (S)-2-アミノ-3-フェニルプロパン-1-オール (604.2 mg, 4.0 mmol) のキシレン (2 ml) 溶液を120℃に加熱し、30分間撹拌した。反応溶液を室温まで冷却し、溶媒を留去後、得られた油状物をシリカゲルクロマトグラフィーに付し、精製した。はじめに クロロホルムで溶出し、次いでクロロホルム-MeOH (9:1, v/v) で溶出した。目的の画分を濃縮すると油状物として 487 mg (収率 72%) の化合物11G を得た。
1H-NMR (CDCl3) δ: 1.41 (3H, t, J = 6.9 Hz), 2.24-2.34 (1H, m), 2.24-3.00 (1H, m), 3.03-3.16 (1H, m), 3.05 (3H, m), 3.25-3.32 (2H, m), 4.13-4.19 (1H, m), 4.17-4.30 (1H, m), 4.36-4.47 (1H, m), 4.51-4.54 (1H, m), 4.55 (1H, d, J = 10.5 Hz), 5.78 (1H, t, J = 6.9 Hz), 7.17-7.26 (4H, m), 7.28-7.35 (5H, m), 7.49 (1H, t, J = 5.4 Hz), 6.32 (1H, s).
第七工程
化合物11G (2.86 g, 5.63 mmol) と トリフェニルホスフィン (2.21 g, 8.45 mmol)のTHF (6ml) 溶液に室温下、DEAD 40 wt% トルエン溶液 (3.68 g, 8.45 mmol) を3分間かけて滴下した。反応液を同温度で30分間撹拌し、溶媒を留去後、得られた油状物をシリカゲルクロマトグラフィーに付し、精製した。酢酸エチル-MeOH (9:1, v/v) で溶出する画分から油状物として 1.37 g (収率 50%) の化合物11H を得た。
1H-NMR (CDCl3) δ: 1.31 (3H, t, J = 7.2 Hz), 3.07 (2H, d, J = 6.9 Hz), 3.33 (3H, s), 3.57-3.80 (4H, m), 3.95 (1H, dd, J = 3.0 Hz, 6.6 Hz), 4.01-4.14 (1H, m), 4.16-4.34 (2H, m), 5.24 (1H, d, J = 9.9 Hz), 5.51 (1H, d, J = 9.9 Hz), 7.01-7.03 (2H, m), 7.21-7.37 (5H, m), 7.41-7.58 (1H, m), 7.64-7.69 (2H, m).
第八工程
化合物11H (1.0 g, 2.04 mmol) のEtOH (6 ml) 溶液に、2規定水酸化ナトリウム水溶液 (6 ml) を加え、室温下、30分間撹拌した。反応液を2規定塩酸で中和し、析出した固体を濾取し、乾燥すると、754 mg (収率 80%) の化合物11I を得た。
1H-NMR (CDCl3) δ: 3.10 (2H, d, J = 7.8 Hz), 3.33 (3H, s), 3.57-3.69 (4H, m), 3.82-3.90 (1H, m), 3.95 (1H, dd, J = 3.3 Hz, 13.8 Hz), 4.36 (1H, dd, J = 6.3 Hz, 7.5 Hz), 5.36 (1H, d, J = 10.2 Hz), 5.45 (1H, d, J = 10.2 Hz), 6.98-7.01 (2H, m), 7.28-7.39 (6H, m), 7.59 (2H, dd, J = 1.8 Hz, 8.1 Hz), 7.87 (1H, s).
第九工程
化合物11I (1.0 g, 2.16 mmol) をTHF (10 ml) に溶解し、10% Pd-C (200 mg) を加え、水素気流下、接触還元反応に付した。触媒を濾過により除去し、濾液を濃縮した。得られた残渣をエーテルで洗浄し、512 mg (収率 64%) の化合物11 を得た。
1H-NMR (CDCl3) δ: 6.24 (2H, d, J = 6.3 Hz), 3.36 (3H, s), 3.60-3.86 (5H, m), 4.14 (1H, d, J = 12.9 Hz), 4.47 (1H, s), 7.03-7.05 (2H, m), 7.30-7.35 (3H, m), 7.88 (1H, s), 12.68 (1H, s), 14.83 (1H, s).
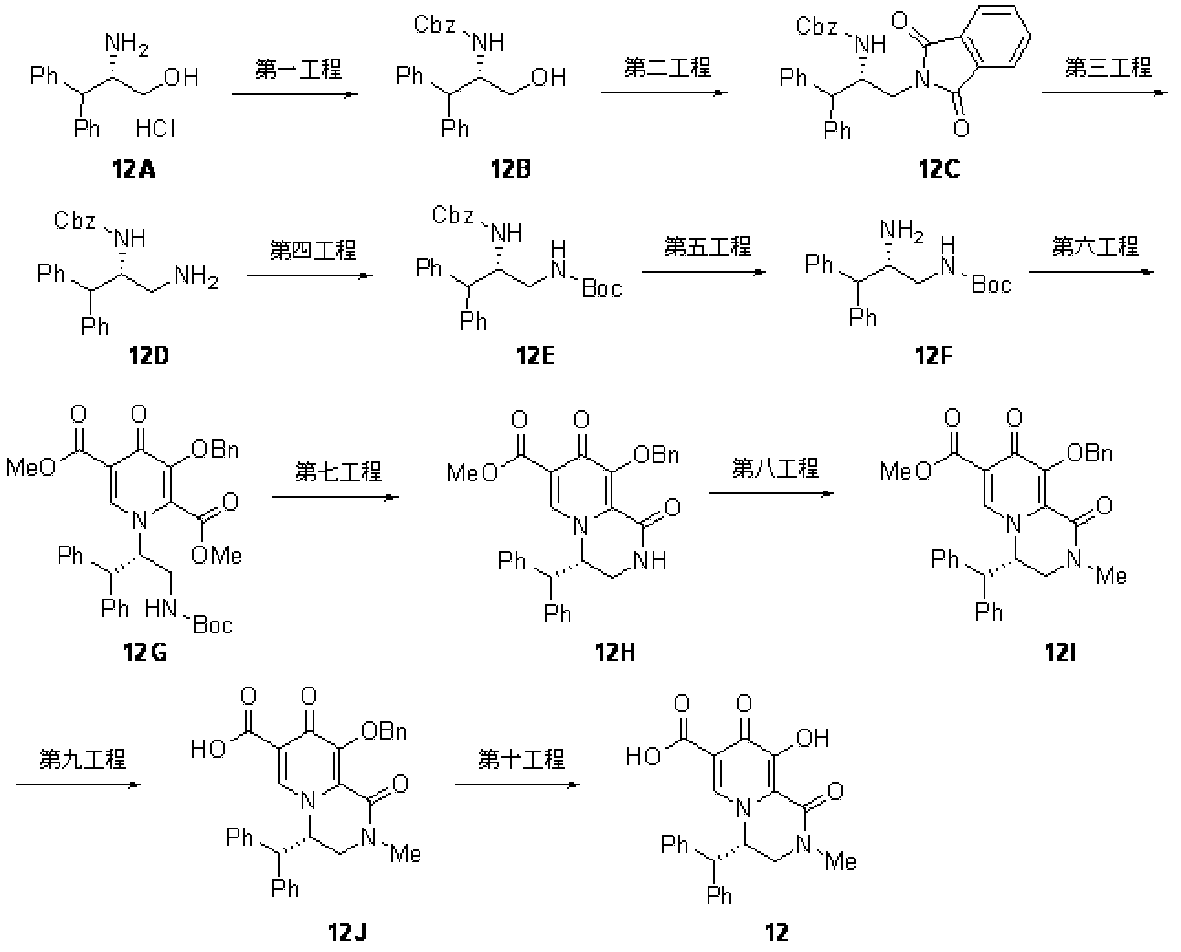
第一工程
化合物12A (1.53 g, 5.80 mmol) をTHF (6 ml) と水 (6 ml) に溶解し、炭酸カリウム (2.41 g,17.4 mmol) を加えて攪拌しておき、0℃でbenzyl chloroformate (1.09 g, 6.38 mmol) を滴下した。0℃で10分攪拌した後、室温で2時間攪拌した。反応液を重曹水にあけ、酢酸エチルで抽出した。抽出液を1規定塩酸と飽和食塩水で洗浄後、硫酸ナトリウムで乾燥した。溶媒を留去し、無色ガム状物質として 2.32 g の化合物12Bを得た。
1H-NMR (CDCl3) δ: 1.98 (1H, brs), 3.55 (1H, m), 3.75 (1H, m), 4.20 (1H, d, J = 10.5 Hz), 4.58 (1H, m), 4.83 (1H, brs), 5.07 (2H, s), 7.16-7.39 (15H, m).
第二工程
化合物12B (1.94 g, 5.37 mmol) とトリフェニルホスフィン (2.11 g, 8.05 mmol) とフタルイミド (948 mg, 6.44 mmol) をTHF (20 ml) に加え、diisopropyl azodicarboxylate (2.2M in toluene, 3.66 ml, 8.05 mmol) を室温で滴下した。室温で4時間攪拌した後、溶媒を減圧留去した。得られた粗生成物をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル, 1:1, v/v) で精製し、無色固体として 2.39 g の化合物12Cを得た。
1H-NMR (CDCl3) δ: 3.73 (2H, m), 4.05 (1H, d, J = 10.1Hz), 4.70 (1H, d, J = 9.6Hz), 4.77 (2H, d, J = 7.2 Hz) 5.02 (1H, m), 7.03-7.42 (15H, m), 7.68 (2H, dd, J = 5.7, 2.1 Hz), 7.78 (2H, dd, J = 5.7, 2.1Hz).
第三工程
化合物12C (2.39 g, 4.87 mmol) をTHF (20 ml) とメタノール (20 ml) に加え、hydrazine hydrate (4.88 g, 97.4 mmol) を加えて50℃で4時間攪拌した。白色沈殿物をろ過して除き、メタノールで洗った。ろ液を減圧留去した後、得られた粗生成物をアミノカラムクロマトグラフィー (クロロホルム-メタノール, 99:1, v/v) で精製し、無色固体として 1.41 g の化合物12Dを得た。
1H-NMR (CDCl3) δ: 2.63 (1H, dd, J = 13.2, 5.8 Hz), 2.86 (1H, d, J = 9.9 Hz), 4.07 (1H, d, J = 10.4 Hz), 4.53 (1H, m), 4.81 (1H, m), 5.00 (2H, d, 8.4Hz), 7.20-7.36 (10H, m).
第四工程
化合物12D (1.41 g, 3.91 mmol) をTHF (15 ml) に溶解し、室温でBoc2O (896 mg, 4.11 mmol)を加えた。1.5時間攪拌の後、溶媒を減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(n-ヘキサン-酢酸エチル, 1:1, v/v) で精製し、無色固体として 1.77 g の化合物12Eを得た。
1H-NMR (CDCl3) δ: 1.41 (9H, s), 3.23 (2H, brm), 3.97 (1H, d, J = 9.8 Hz), 4.58-4.80 (3H, m), 5.00 (2H, d, J = 9.8 Hz), 7.15-7.29 (10H, m).
第五工程
化合物12E (1.73 g, 3.76 mmol) とパラジウム‐活性炭素 (10%, wet, 200 mg) をメタノール (20ml) に加え、水素雰囲気下、室温で1時間攪拌した。セライトろ過した後、溶媒を減圧濃縮して無色オイル状物質12Fを1.01 g得た。
1H-NMR (CDCl3) δ: 1.44 (9H, s), 2.82 (1H, m), 3.31 (1H, m), 3.73 (2H, d, J = 6.9 Hz), 4.98 (1H, s), 7.18-7.39 (10H, m).
第六工程
dimethyl 3-(benzyloxy)-4-oxo-4H-pyran-2,5-dicarboxylate (974 mg, 3.06 mmol) と12F (999 mg,3.06 mmol) をトルエン (10 ml) に加え、110℃で5時間攪拌した。溶媒を減圧留去した後、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 98:2, v/v) で精製し、淡黄色固体として 1.51 g の化合物12Gを得た。
1H-NMR (CDCl3) δ: 1.36 (9H, s), 3.40 (1H, m), 3.53 (1H, m), 3.82 (3H, s), 3.91 (3H, s), 4.29 (1H, d, J = 11.3 Hz), 4.78 (1H, m), 4.82 (1H, m), 5.11 (1.9H, d, J = 7.5 Hz), 7.10-7.38 (10H, m), 8.27 (1H, s).
第七工程
化合物12G (1.45 g, 2.31 mmol) に4規定 HCl (酢酸エチル溶液, 20 ml) を加え、室温で1.5時間攪拌した。溶媒を減圧留去した後、飽和重曹水を加えて室温で1.5時間攪拌した。クロロホルムで抽出し、硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 95:5, v/v) で精製し、無色固体として1.01 g の化合物12Hを得た。
1H-NMR (CDCl3) δ: 3.40 (1H, dd, J = 13.6, 6.6 Hz), 3.78 (3H, s), 3.80 (1H, m), 4.37 (1H, d, J = 11.6 Hz), 4.59 (1H, d, J = 11.0 Hz), 5.43 (2H, d, J = 10.2 Hz), 5.93 (1H, d, J = 5.8 Hz), 7.03-7.21 (5H, m), 7.37 (9H, m), 7.63 (2H, m).
第八工程
化合物12H (50 mg, 0.10 mmol) をDMF (1 ml) に溶解し、炭酸セシウム (165 mg, 0.50 mmol)を加えた。室温で30分攪拌した後、ヨードメタン (0.032 ml, 0.50 mmol) を加えて室温で3.5時間攪拌した。反応液を水にあけ、酢酸エチルで抽出した後、硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 95:5, v/v) で精製し、無色固体として49 mg の化合物12Iを得た。
第九工程
化合物12I (49 mg, 0.096 mmol) をTHF (0.5 ml) とメタノール (0.5 ml) に溶解し、室温で2規定水酸化ナトリウム水溶液 (0.24 ml, 0.48 mmol) を加えてそのまま1.5時間攪拌した。1規定塩酸を加えてから酢酸エチルで抽出した後、硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、無色固体として54 mg の化合物12Jを得た。
MS: m/z = 481 [M+H]+.
第十工程
第九工程で得られた化合物12Jにトリフルオロ酢酸 (1 ml) を加えて、室温で1時間攪拌した。減圧濃縮した後、重曹水と2規定塩酸でpHを3に調節し、クロロホルムで抽出して硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、クロロホルム‐メタノール‐エチルエーテルを加えて析出した固体を濾取し、無色固体として26 mg の化合物12を得た。
1H-NMR (DMSO-d6) δ: 3.01 (3H, s), 3.26 (1H, t, J = 14.4 Hz), 4.23 (1H, dd, J = 13.5, 3.8 Hz), 4.57 (1H, d, J = 11.6 Hz), 5.78 (1H, d, J = 11.3 Hz), 7.16-7.70 (10H, m), 8.00 (1H, s), 13.00 (1H, s), 15.10 (1H, s).
MS: m/z = 405 [M+H]+.

第一工程
化合物28A (3.20 g, 17.1 mmol) をTHF (20 ml) に加え、トリエチルアミン (2.60 ml, 18.8 mmol) を加えて室温で10分攪拌した。Boc2O (4.09 g, 18.8 mmol) を室温で加えた後、そのまま2時間攪拌した。溶媒を減圧留去し、水を加えて酢酸エチルで抽出した。有機層を飽和食塩水で洗い、硫酸ナトリウムで乾燥した。溶媒を減圧留去して無色固体として5.17 g の化合物28Bを得た。
1H-NMR (CDCl3) δ: 1.52 (9H, s), 2.77 (2H, m), 3.03-3.12 (1H, m), 3.38 (1H, m), 3.90-3.98 (1H, m), 4.93 (1H, brs), 7.20-7.35 (5H, m).
第二工程
化合物28B (4.29 g, 17.1 mmol) とトリフェニルホスフィン (5.37 g, 20.5 mmol) とフタルイミド (2.76 g, 18.8 mmol) をTHF (60 ml) に加え、diethyl azodicarboxylate (2.2M in toluene, 11.6 ml, 25.6 mmol) を室温で滴下した。室温で1時間攪拌した後、溶媒を減圧留去した。得られた粗生成物をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル, 2:1, v/v) で精製し、無色固体として 6.13 g の化合物28Cを得た。
1H-NMR (CDCl3) δ: 1.30 (9H, s), 3.14 (1H, dd, J = 13.8, 6.2 Hz), 3.39 (2H, m), 3.87 (1H, m), 4.67 (1H, m), 4.81 (1H, brs), 7.16-7.19 (5H, m), 7.66 (2H, dd, J = 5.3, 3.1 Hz), 7.75 (2H, dd, J = 5.7, 3.0 Hz).
第三工程
化合物28C (1.00 g, 2.63 mmol) をTHF (7 ml) とメタノール (7 ml) に加え、hydrazine hydrate (2.63 g, 52.6 mmol) を加えて50℃で2時間攪拌した。白色沈殿物をろ過して除き、メタノールで洗った。ろ液を減圧留去した後、得られた粗生成物をアミノカラムクロマトグラフィー (クロロホルム-メタノール, 99:1, v/v) で精製し、無色固体として 249 mg の化合物28Dを得た。
1H-NMR (CDCl3) δ: 1.44 (9H, s), 1.95 (2H, brs), 2.55-3.31 (5H, m), 5.06 (1H, brs), 7.18-7.33 (5H, m).
第四工程
dimethyl 3-(benzyloxy)-4-oxo-4H-pyran-2,5-dicarboxylate (313 mg, 0.983 mmol) と28D (246 mg, 0.983 mmol) をトルエン (3 ml) に加え、100℃で2.5時間攪拌した。溶媒を減圧留去した後、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 98:2, v/v) で精製し、淡黄色ガム状物質として 320 mg の化合物28Eを得た。
1H-NMR (CDCl3) δ: 1.42 (9H, s), 3.07 (2H, m), 3.56 (2H, m), 3.68 (3H, s), 3.95 (3H, s), 4.26 (1H, s), 4.86 (1H, s), 5.18 (1H, d, J = 10.8 Hz), 5.22 (1H, d, J = 10.8 Hz), 7.01 (2H, m), 7.24-7.38 (8H, m), 8.22 (1H, s).
MS: m/z = 551 [M+H]+.
第五工程
化合物28E (315 mg, 0.572 mmol) に4規定 HCl (酢酸エチル溶液, 5 ml) を加え、室温で30分攪拌した。溶媒を減圧留去した後、飽和重曹水を加え、クロロホルムで抽出し、硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 95:5, v/v) で精製し、無色固体として210 mg の化合物28Fを得た。
1H-NMR (CDCl3) δ: 3.07-3.15 (2H, m), 3.34 (1H, dd, J = 13.2, 6.0 Hz), 3.74 (2H, m), 3.86 (3H, s), 4.12 (1H, m), 5.27 (1H, d, J = 10.1 Hz), 5.47 (1H, d, J = 10.1 Hz), 6.76 (1H, d, J = 6.4 Hz), 7.04 (2H, m), 7.32 (6H, m), 7.62 (2H, dd, J = 7.7, 1.4 Hz), 7.70 (1H, s).
MS: m/z = 419 [M+H]+.
第六工程
化合物28F (50 mg, 0.12 mmol) をDMF (1 ml) に溶解し、炭酸セシウム (195 mg, 0.597 mmol)を加えた。室温で30分攪拌した後、ヨードエタン (0.048 ml, 0.60 mmol) を加えて室温で3.5時間攪拌した。反応液を水にあけ、酢酸エチルで抽出した後、硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 95:5, v/v) で精製し、無色固体として47 mg の化合物28Gを得た。
1H-NMR (CDCl3) δ: 1.22 (3H, t, J = 7.2 Hz), 3.00-3.15 (2H, m), 3.28 (1H, dd, J = 13.6, 1.6 Hz), 3.48 (1H, m), 3.75 (1H, m), 3.85 (3H, s), 3.88 (1H, dd, J = 13.3, 3.2 Hz), 4.15 (1H, m), 5.25 (1H, d, J = 9.9 Hz), 5.50 (1H, d, J = 9.9 Hz), 7.04 (2H, m), 7.29-7.38 (6H, m), 7.60 (1H, s), 7.68 (2H, m).
MS: m/z = 447 [M+H]+.
第七工程
化合物28G (47 mg, 0.11 mmol) をTHF (0.5 ml) とメタノール (0.5 ml) に溶解し、室温で2規定水酸化ナトリウム水溶液 (0.26 ml, 0.53 mmol) を加えてそのまま1時間攪拌した。1規定塩酸を加えてから酢酸エチルで抽出した後、硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、無色固体として40 mg の化合物28Hを得た。
MS: m/z = 433 [M+H]+.
第八工程
第七工程で得られた化合物28Hにトリフルオロ酢酸 (1 ml) を加えて、室温で1時間攪拌した。減圧濃縮した後、重曹水と2規定塩酸でpHを3に調節し、クロロホルムで抽出して硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、クロロホルム‐メタノール‐エチルエーテルを加えて析出した固体を濾取し、無色固体として17 mg の化合物28を得た。
1H-NMR (DMSO-d6) δ: 1.17 (3H, t, J = 7.2 Hz), 3.08 (2H, m), 3.51-3.63 (3H, m), 4.08 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, brs), 7.21 (5H, m), 8.07 (1H, s), 12.98 (1H, s), 15.07 (1H, brs).
MS: m/z = 343 [M+H]+.

第一工程
化合物43A (2.00 g, 6.11 mmol) とトリフェニルホスフィン (2.40 g, 9.16 mmol) とフタルイミド (1.08 g, 7.33 mmol) をTHF (20 ml) に加え、diethyl azodicarboxylate (2.2M in toluene, 4.16 ml, 9.16 mmol) を室温で滴下した。室温で3時間攪拌した後、溶媒を減圧留去した。得られた粗生成物をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル, 1:1, v/v) で精製し、無色固体として 2.39 g の化合物43Bを得た。
1H-NMR (DMSO-d6) δ: 1.00 (9H, s), 3.30 (1H, m), 3.61 (1H, dd, J = 13.4, 10.2 Hz), 4.15 (1H, d, J = 12.2 Hz), 4.75 (1H, m), 6.79 (1H, d, J = 9.5 Hz), 7.25 (15H, m), 7.76-7.89 (4H, m).
第二工程
化合物43B (2.06 g, 4.51 mmol) をTHF (20 ml) とメタノール (20 ml) に加え、hydrazine hydrate (4.52 g, 90.2 mmol) を加えて60℃で5時間攪拌した。白色沈殿物をろ過して除き、メタノールで洗った。ろ液を減圧留去した後、得られた粗生成物をアミノカラムクロマトグラフィー (クロロホルム-メタノール, 99:1, v/v) で精製し、n-ヘキサンを加えて析出した固体を濾取し、無色固体として 1.25 g の化合物43Cを得た。
1H-NMR (CDCl3) δ: 1.32 (9H, s), 2.55 (1H, dd, J = 13.3, 6.0 Hz), 2.80 (1H, dd, J = 13.3, 3.5 Hz), 3.99 (1H, d, J = 10.1 Hz), 4.47 (2H, m), 7.13-7.33 (10H, m).
第三工程
dimethyl 3-(benzyloxy)-4-oxo-4H-pyran-2,5-dicarboxylate (488 mg, 1.53 mmol) と43C (500 mg, 1.53 mmol) をトルエン (8 ml) に加え、110℃で1時間攪拌した。溶媒を減圧留去した後、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 97:3→96:4→94:6, v/v) で精製し、淡黄色ガム状物質として 667 mg の化合物43Dを得た。
1H-NMR (CDCl3) δ: 1.28 (9H, s), 3.63 (3H, s), 3.80 (1H, m), 3.87 (3H, s), 4.02 (1H, dd, J = 14.5, 10.1 Hz), 4.21 (1H, d, J = 10.4 Hz), 4.47 (2H, m), 5.20 (1H, d, J = 10.8 Hz), 5.26 (1H, d, J = 10.7 Hz), 7.30 (15H, m), 8.05 (1H, s).
MS: m/z = 627 [M+H]+.
第四工程
化合物43D (664 mg, 1.06 mmol) に4規定 HCl (酢酸エチル溶液, 10 ml) を加え、室温で1時間攪拌した。溶媒を減圧留去した後、THFと飽和重曹水を加えて2.5時間攪拌した。クロロホルムで抽出し、硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、塩化メチレン-エチルエーテルを加えて析出した固体を濾取し、無色固体として458 mg の化合物43Eを得た。
1H-NMR (CDCl3) δ: 3.86 (3H, m), 3.92 (3H, s), 4.41-4.48 (1H, m), 5.32 (1H, d, J = 10.8 Hz), 5.42 (1H, d, J = 10.1 Hz), 5.92 (1H, s), 7.21-7.39 (13H, m), 7.59 (2H, m), 7.89 (1H, s).
MS: m/z = 495 [M+H]+.
第五工程
化合物43E (50 mg, 0.10 mmol) をDMF (1 ml) に溶解し、炭酸セシウム (165 mg, 0.51 mmol)を加えた。室温で30分攪拌した後、ヨードメタン (0.025 ml, 0.40 mmol) を加えて室温で1時間攪拌した。反応液を水にあけ、酢酸エチルで抽出した後、硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 97:3→95:5, v/v) で精製し、無色固体として60 mg の化合物43Fを得た。
1H-NMR (CDCl3) δ: 2.57 (3H, s), 3.75 (2H, d, J = 11.3 Hz), 3.93 (3H, s), 4.20-4.29 (2H, m), 5.25 (1H, d, J = 9.9 Hz), 5.57 (1H, d, J = 9.9 Hz), 7.15-7.41 (13H, m), 7.63 (1H, s), 7.72-7.76 (2H, m).
第六工程
第五工程で得られた化合物43FをTHF (0.5 ml) とメタノール (0.5 ml) に溶解し、室温で2規定水酸化ナトリウム水溶液 (0.25 ml, 0.50 mmol) を加えてそのまま1時間攪拌した。1規定塩酸を加えてから酢酸エチルで抽出した後、硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、無色ガム状の化合物43Gを得た。
第七工程
第六工程で得られた化合物43Gにトリフルオロ酢酸 (2 ml) を加えて、室温で1時間攪拌した。減圧濃縮した後、重曹水と2規定塩酸でpHを3に調節し、クロロホルムで抽出して硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、クロロホルム-エチルエーテルを加えて析出した固体を濾取し、無色固体として27 mg の化合物43を得た。
1H-NMR (DMSO-d6) δ: 2.53 (3H, s), 4.26 (1H, d, J = 10.9 Hz), 4.35 (1H, d, J = 13.3 Hz), 4.58 (1H, dd, J = 13.8, 3.5 Hz), 5.06 (1H, d, J = 10.9 Hz), 7.36 (10H,m), 8.36 (1H, s), 12.58 (1H, s), 15.62 (1H, s).
MS: m/z = 405 [M+H]+.

第一工程
Dess-Martin Periodinane (0.3M, 塩化メチレン溶液, 52.0 ml, 15.6 mmol) に化合物49A (2.97 g, 10.4 mmol) の塩化メチレン溶液 (20 ml) を0℃で滴下した。室温で3時間攪拌の後、1規定水酸化ナトリウム水溶液にあけ、エチルエーテルで抽出した。有機層を1規定水酸化ナトリウム水溶液と飽和食塩水で洗浄し、硫酸マグネシウムで乾燥した。溶媒を減圧留去した後、白色固体として化合物49Bを2.08 g得た。
1H-NMR (CDCl3) δ: 3.13 (2H, d, J = 6.6 Hz), 4.53 (1H, q, J = 6.7 Hz), 5.12 (2H, s), 5.28 (1H, brs), 7.26 (10H, m), 9.64 (1H, s).
第二工程
化合物49B (700 mg, 2.47 mmol) と2-aminoethanol (166 mg, 2.72 mmol) と硫酸ナトリウム (1.76 g, 12.4 mmol) をトルエン (20 ml) に加え、室温で1時間攪拌した。Boc2O (0.631 ml, 2.72 mmol) を室温で加え、そのまま18時間攪拌した。反応液をろ過し、ろ液を減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル, 1:1, v/v) で精製し、無色ガム状物質として49Cを893 mg得た。
第三工程
化合物49C (890 mg, 2.09 mmol) とパラジウム‐活性炭素 (10%, wet, 200 mg) をエタノール (20ml) に加え、水素雰囲気下、室温で2時間攪拌した。セライトろ過した後、溶媒を減圧濃縮して無色オイル状物質49Dを656 mg得た。
1H-NMR (CDCl3) δ: 1.40 (9H, s), 2.65-2.86 (2H, m), 3.32 (2H, m), 3.80 (2H, m), 4.03-4.12 (1H, m), 4.86 (1H, brs), 7.22 (5H, m).
第四工程
dimethyl 3-(benzyloxy)-4-oxo-4H-pyran-2,5-dicarboxylate (610 mg, 2.09 mmol) と49D (664 mg, 2.09 mmol) をトルエン (6 ml) に加え、100℃で4時間攪拌した。溶媒を減圧留去した後、得られた粗生成物をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル, 1:1, v/v) で精製し、淡黄色ガム状物質として 884 mg の化合物49Eを得た。
MS: m/z = 593 [M+H]+.
第五工程
化合物49E (860 mg, 1.45 mmol) に4規定 HCl (酢酸エチル溶液, 10 ml) を加えた。室温で30分攪拌した後、溶媒を減圧留去した。続いてトルエン (10 ml) と2-aminoethanol (0.175 ml, 2.90 mmol) を加え、80℃で30分攪拌した。溶媒を減圧留去した後、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 99:1→95:5→90:10, v/v) で精製し、無色ガム状物質の化合物49F 157 mgと黄色固体の化合物49G 217 mgを得た。
49F :1H-NMR (CDCl3) δ: 2.48 (1H, dd, J = 14.0, 11.4 Hz), 3.22 (1H, dd, J = 14.1, 3.3 Hz), 3.69 (1H, m), 3.77 (3H, s), 3.83-3.95 (1H, m), 4.08 (1H, m), 4.29 (1H, m), 4.41 (1H, m), 5.34 (2H, m), 5.48 (1H, d, J = 10.1 Hz), 6.86 (2H, m), 7.20-7.39 (7H, m), 7.64 (2H, m)
49G: 1H-NMR (DMSO-d6) δ: 3.70 (2H, t, J = 5.3 Hz), 3.73 (3H, s), 3.86 (2H, t, J = 5.3 Hz), 4.14 (2H, s), 4.98 (1H, t, J = 5.0 Hz), 5.06 (2H, s), 6.98 (1H, s), 7.35 (8H, m), 7.62 (2H, d, J = 7.1 Hz), 8.34 (1H, d, J = 0.8 Hz).
第六工程
化合物49G (214 mg, 0.465 mmol) をTHF (4 ml) とエタノール (2 ml) と塩化メチレン (2 ml)に溶解し、室温で2規定水酸化ナトリウム水溶液 (1.16 ml, 2.32 mmol) を加えてそのまま2.5時間攪拌した。1規定塩酸を加えてからクロロホルムで抽出した後、硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、黄色固体として158 mg の化合物49Hを得た。
1H-NMR (DMSO-d6) δ: 3.70 (2H, q, J = 5.2 Hz), 3.89 (2H, t, J = 5.3 Hz), 4.22 (2H, s), 4.97 (1H, t, J = 5.6 Hz), 5.12 (2H, s), 7.23-7.41 (9H, m), 7.60 (2H, m), 8.54 (1H, s).
第七工程
化合物49H (50.0 mg, 0.112 mmol) とパラジウム‐活性炭素 (10%, wet, 12 mg) をメタノール (1 ml) とDMF (3 ml) に加え、水素雰囲気下、室温で5時間攪拌した。セライトろ過した後、溶媒を減圧濃縮し、クロロホルム‐メタノール‐エチルエーテルを加えて析出した固体を濾取し、無色固体として9.0 mg の化合物49を得た。
1H-NMR (DMSO-d6) δ: 3.10 (2H, m), 3.51-3.69 (4H, m), 4.10 (1H, d, J = 10.7 Hz), 4.94 (2H, m), 7.11-7.26 (5H, m), 8.03 (1H, s), 12.94 (1H, brs), 15.30 (1H, brs).
MS: m/z = 359 [M+H]+.

第一工程
化合物50A (1.00 g, 3.98 mmol) とトリフェニルホスフィン (1.15 g, 4.48 mmol) とN-methyl-2-nitrobenzenesulfonamide (860 mg, 3.98 mmol) をTHF (10 ml) に加え、diethyl azodicarboxylate (2.2M in toluene, 1.99 ml, 4.38 mmol) を室温で滴下した。室温で3時間攪拌した後、溶媒を減圧留去した。得られた粗生成物をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル, 1:1, v/v) で精製し、無色ガム状物質として 710 mg の化合物50Bを得た。
第二工程
化合物50B (458 mg, 1.02 mmol) をアセトニトリルに溶解し、炭酸カリウム (422 mg, 3.06 mmol)とbenzenethiol (0.126 ml, 1.22 mmol) を加えて室温で5時間攪拌した。反応液を1規定水酸化ナトリウム水溶液にあけ、塩化メチレンで抽出し、硫酸ナトリウムで乾燥した。得られた粗生成物をアミノカラムクロマトグラフィー (クロロホルム-メタノール, 95:5, v/v) で精製し、無色オイル状物質として147 mgの化合物50Cを得た。
1H-NMR (CDCl3) δ: 1.36 (9H, s), 2.40 (3H, s), 2.51-2.89 (4H, m), 3.90 (1H, s), 4.69 (1H, s), 7.17-7.31 (5H, m).
第三工程
化合物50C (140 mg, 0.530 mmol) と3-(benzyloxy)-4-oxo-4H-pyran-2-carboxylic acid (WO2006/116764, 119mg, 0.482 mmol) をTHF (3 ml) に加え、1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (111 mg, 0.578 mmol) と1-hydroxybenzotriazole (65.1 mg, 0.482 mmol) を加えて室温で18時間攪拌した。反応液を重曹水にあけ、酢酸エチルで抽出し、硫酸ナトリウムで乾燥した。得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 97:3, v/v) で精製し、無色固体として 219 mg の化合物50Dを得た。
MS: m/z = 493 [M+H]+.
第四工程
化合物50D (216 mg, 0.439 mmol) に4規定 HCl (酢酸エチル溶液, 3 ml) を加えた。室温で1時間攪拌した後、溶媒を減圧留去した。続いてエタノール (4 ml) と飽和炭酸ナトリウム水溶液 (3 mll) を加え、60℃で2時間攪拌した。水を加え、酢酸エチルで抽出した後、硫酸ナトリウムで乾燥した。得られた粗生成物をアミノカラムクロマトグラフィー (クロロホルム-メタノール, 95:5, v/v) で精製し、淡黄色ガム状物質の化合物50E 108 mgを得た。
1H-NMR (CDCl3) δ: 3.00 (2H, m), 3.13 (3H, s), 3.18 (1H, m), 3.88 (1H, dd, J = 13.5, 3.4 Hz), 4.00-4.07 (1H, m), 5.26 (1H, d, J = 10.2 Hz), 5.46 (1H, d, J = 10.1 Hz), 6.25 (1H, d, J = 7.5 Hz), 6.73 (1H, d, J = 7.5 Hz), 6.99-7.02 (2H, m), 7.28-7.37 (6H, m), 7.63-7.67 (2H, m).
第五工程
化合物50E (105 mg, 0.280 mmol) にトリフルオロ酢酸 (2 ml) を加えて、室温で30分攪拌した。減圧濃縮した後、重曹水と2規定塩酸でpHを6に調節し、クロロホルムで抽出して硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、塩化メチレン‐メタノール‐エチルエーテルを加えて析出した固体を濾取し、無色固体として29 mg の化合物50を得た。
1H-NMR (DMSO-d6) δ: 2.99 (3H, s), 3.26-3.47 (3H, m), 4.07 (1H, d, J = 11.1 Hz), 4.80 (1H, m), 6.43 (1H, d, J = 6.9 Hz), 7.11-7.29 (5H, m), 7.50 (1H, d, J = 6.9 Hz).
MS: m/z = 285 [M+H]+.
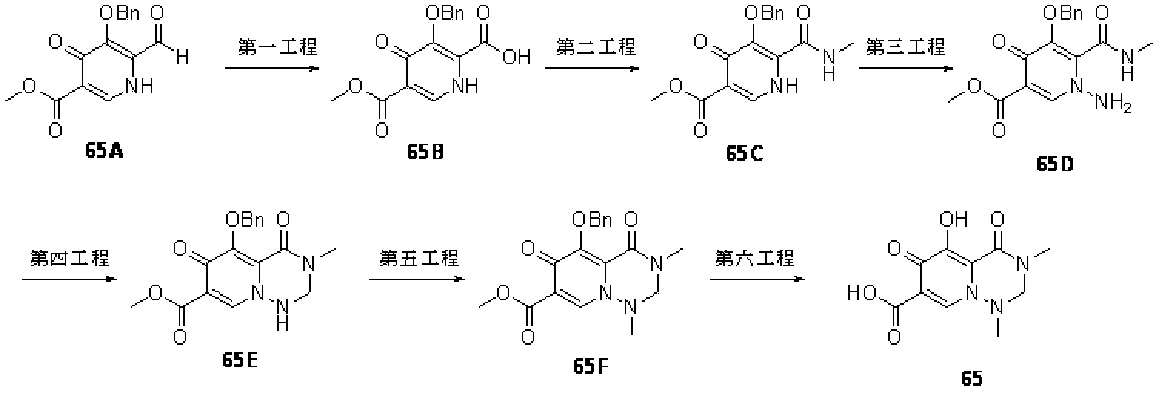
第一工程
化合物65A (WO2006/088173, 20.0 g, 69.6 mmol) のTHF (1.1 L) 溶液を水浴上で25℃に保ち、亜塩素酸ナトリウム (25.2 g, 278 mmol)、 アミド硫酸 (27.0 g, 278 mmol) の水 (378 mL) 溶液を、30分間かけて滴下した。反応液を同温度で1時間撹拌後、減圧濃縮した。残渣に、氷水 (100 mL)、 ジエチルエーテル (100 mL) を加え、析出した固体を濾過した。得られた粗精製物を、水およびジエチルエーテルで洗浄し、白色固体として 20.3 g の化合物65Bを得た。
1H-NMR (DMSO-d6) δ: 3.74 (3H, s), 5.11 (2H, s), 7.31-7.38 (3H,m), 7.48 (2H, d, J = 7.2 Hz), 8.11 (1H, s), 12.07(1H, brs).
第二工程
化合物65B (2.0 g, 6.59 mmol) を DMF (340 mL) に溶解し、HATU (2.76 g, 7.25 mmol)、メチルアミン (2 mol/L THF溶液, 3.63mL, 7.25mmol)、トリエチルアミン (9.89 mmol)を加えて室温で 5時間攪拌した。反応液を酢酸エチル-水に分配した。酢酸エチル層を分離し、水層を酢酸エチルで1回抽出した。合わせた抽出液を水、飽和食塩水で洗浄後、乾燥した。溶媒を留去し、白色固体として、1.66 gの化合物65Cの粗精製物を得た。
1H-NMR (DMSO-d6) δ: 3.38 (3H, brs), 3.75 (3H, s), 5.37 (2H, s), 7.34-7.44 (5H,m), 8.10 (1H, s), 8.38 (1H, s), 11.84(1H, brs).
第三工程
化合物65C (1.2g, 3.79 mmol) の DMF (20 mL) 溶液に、炭酸カリウム (1.04 g, 7.59 mmol)、O-(2,4-dinitrophenyl)hydroxylamine (831mg, 4.17 mmol)を加え、室温で3時間攪拌した。反応液に水を加え、析出した固体を濾取し、水で洗浄することにより、1.0 gの化合物 65Dの粗精製物を得た。
1H-NMR (DMSO-d6) δ: 3.74 (3H,s), 3.83 (3H, brs), 5.05 (2H, s), 6.46 (2H, brs), 7.31-7.38 (5H, m), 8.20 (1H, s), 8.52 (1H, brs).
第四工程
化合物65D (1.0 g, 3.02 mmol) の DMF (10 mL) 溶液に室温下、パラホルムアルデヒド (109 mg, 3.62 mmol)、酢酸 (0.017 ml, 0.302 mmol) を加え、 105℃で2時間撹拌した。反応液を0℃に冷却し、炭酸セシウム (3.44 g, 10.6 mmol) を加え、室温で1時間攪拌した。反応液に水を加え、酢酸エチル-水に分配した。有機層を飽和食塩水で洗浄、乾燥した。溶媒を留去し、120 mg の化合物65E を得た。
MS: m/z = 344 [M+H]+.
第五工程
化合物65E (17.0 mg, 0.05 mmol) の DMF (1 mL) 溶液に 炭酸セシウム (81.4 mg, 0.25 mmol) 、 メチルアミン (2 mol/L THF溶液, 0.125 ml, 0.25 mmol)を加え、室温で5 時間撹拌した。反応液を濾過し、濾液をLCMSで分取精製し、化合物65Fを得た。
MS: m/z = 358 [M+H]+.
第六工程
化合物65Fの DMF (0.5 mL) 溶液に、2N水酸化ナトリウム水溶液 (0.2 mL) を加え、室温で 2時間攪拌した。反応液にイオン交換樹脂DOWEX (50W-X8) を加え、濾過し、DMFで洗浄した。濾液を濃縮後、トリフルオロ酢酸 (0.5 mL) を加え、80℃で4時間攪拌した。反応液を濃縮後、水、クロロホルムを加えて有機層を分離した。有機層を濃縮後、LCMSで分取精製し、6.47 mg の化合物65を得た。
MS: m/z = 254 [M+H]+.

第一工程
化合物95A (WO2006/116764, 1g, 4.06 mmol) を28% アンモニア水に溶解させ、室温下、12時間撹拌した。反応液を濃縮後、得られた残渣を2規定塩酸で中和し、析出した固体を酢酸エチルに懸濁させ、濾取、乾燥すると、1.14 g (収率 100%)の化合物95B を得た。
1H-NMR (DMSO-d6) δ: 5.14 (2H, s), 7.31 (1H, d, J = 6.6 Hz), 7.34-7.41 (3H, m), 7.45-7.51 (2H, m), 8.17 (1H, d, J = 6.6 Hz).
第二工程
上記化合物95B (3.00 g, 10.65 mmol) のDMF (10 ml) 溶液に、室温下、WSC HCl (3.06 g, 15.98 mmol) と、HOBt (1.58 g, 11.7 mmol) を加え10分間撹拌し、メチルアミン 33wt.% エタノール溶液 (1.50 g, 15.98 mmol) を滴下した。反応液を同温度で2時間撹拌後、水を加え、クロロホルムで5回抽出した。抽出液を硫酸ナトリウムで乾燥し、溶媒を留去後、得られた油状物をシリカゲルクロマトグラフィーに付し、精製した。酢酸エチル-MeOH (6:4, v/v) で溶出する画分から固体として 2.62 g (収率 95%) の化合物95Cを得た。
1H-NMR (CDCl3) δ: 2.77 (3H, d, J = 4.8 Hz), 5.49 (2H, s), 6.57 (1H, d, J = 6.9 Hz), 7.25-7.43 (5H, m) ,7.48 (1H, t, J = 6.0 Hz), 8.23 (1H, brs), 9.77 (1H, brs).
第三工程
上記化合物95C (2.62 g, 10.14 mmol) のDMF (10 ml) 溶液に室温下、炭酸カリウム (4.20 g, 30.42 mmol)を懸濁させ5分間撹拌後、O-(2,4-dinitrophenyl)hydroxylamine (3.03 g, 15.21 mmol)を加え、同温度で3時間撹拌した。反応液に水を加え、クロロホルムで5回抽出し、抽出液を硫酸ナトリウムで乾燥した。溶媒を留去後、得られた油状物をシリカゲルクロマトグラフィーに付し、精製した。酢酸エチル-MeOH (6:4, v/v) で溶出する画分から固体として 1.41 g (収率 51%) の化合物95Dを得た。
1H-NMR (CDCl3) δ: 2.62 (3H, d, J = 5.1 Hz), 5.06 (2H, s), 5.22 (2H, s), 6.18 (1H, d, J = 7.8 Hz), 7.25-7.36 (5H, m), 5.89 (1H, d, J = 7.8 Hz), 7.57 (1H, q, J = 5.1 Hz).
第四工程
上記化合物95D (1.0 g, 3.66 mmol)のトルエン (10 ml) 溶液にパラホルムアルデヒド (109.9 mg, 3.66 mmol) と、酢酸 (22 mg, 0.37 mmol) を加え、100 ℃で40分間加熱撹拌した。冷却後、溶媒を留去し、残留物を精製することなくDMF (10 ml) に溶解し、氷冷下、炭酸セシウム (3.58 g, 10.98 mmol) を加え、10分間撹拌した。反応液へベンゾヒドリルブロミド (1.36 g, 5.49 mmol) を加え、室温下3時間撹拌後、水を加え酢酸エチルで3回抽出した。抽出液を3回水洗後、硫酸ナトリウムで乾燥した。溶媒を留去し、得られた油状物をシリカゲルクロマトグラフィーに付し、精製した。酢酸エチル-MeOH (9:1, v/v) で溶出する画分から固体として 1.26 g (収率 71%) の化合物95Eを得た。
1H-NMR (CDCl3) δ: 2.91 (3H, s), 4.26 (1H, d, J = 13.2 Hz), 4.77 (1H, d, J = 13.2 Hz), 5.12 (1H, s), 5.42 ( 1H, J = 13.2 Hz), 5.45 (1H, d, J = 13.2 Hz), 5.82 (1H, J = 7.5 Hz), 6.71 (1H, d, J = 7.5 Hz), 7.10-7.23 (5H, m), 7.27-7.46 (6H, m), 7.52 (2H, d, J = 6.9 Hz), 7.60-7.64 (2H, m).
第五工程
上記化合物95E (100 mg, 0.221 mmol) をトリフルオロ酢酸 (2 ml) に溶解させ、室温下1時間撹拌した。溶媒を留去し、残渣をジクロロメタン(2 ml) に溶解させ、飽和重曹水で中和した。得られた溶液をクエン酸水溶液で酸性にして、有機層を分離した。水層をジクロロメタンで1回抽出し、合わせた有機層を水洗後、硫酸ナトリウムで乾燥した。溶媒を留去後、得られた固体をジイソプロピルエーテルで洗浄し、50 mg (収率63%) の化合物95を得た。
1H-NMR (CDCl3) δ: 2.95 (3H, s), 4.36 (1H, d, J = 13.2 Hz), 4.95 (1H, d, J = 13.2 Hz), 5.22 (1H, s), 5.71 (1H, d, J = 7.8 Hz), 6.75 (1H, d, J = 7.8 Hz), 7.21 (5H, br s), 7.33-7.47 (4H, m), 7.55 (2H, d, J = 6.6 Hz).

第一工程
化合物95Dの合成法に準じて合成した化合物107A(3.0 g, 9.96 mmol) のDMF (30 ml) 溶液に、パラホルムアルデヒド (299 mg, 9.96 mmol)と、酢酸 (1 ml) を加え、120℃で4時間、加熱撹拌した。溶媒を留去後、残渣に酢酸エチル‐ジイソプロピルエーテルを加え析出した固体を濾取し、2.85 g (収率 91%) の化合物107Bを得た。
1H-NMR (CDCl3) δ: 1.19 (6H, J = 6.6 Hz), 4.34 (2H, J = 7.5 Hz), 4.72-4.86 (1H, m), 5.30 (2H, s), 5.49 (1H, t, J = 7.5 Hz), 6.36 (1H, d, J = 7.8 Hz), 7.26-7.35 (4H, m), 7.37 (1H, d, J = 7.8 Hz), 7.55-7.58 (2H, m).
第二工程
化合物107B (100 mg, 0.319 mmol) の酢酸 (2 ml) 溶液に室温下、96% 硫酸 (0.5 ml)と、bis(3-chlorophenyl)methanol (242.3 mg, 0.957 mmol) を加え、80℃で2時間撹拌した。反応液を室温まで冷却後、水を加え、酢酸エチルで3回抽出した。有機層を1回水洗後、硫酸ナトリウムで乾燥した。溶媒を留去後、残渣にジイソプロピルエーテルを加え、析出した固体を濾取し、42 mg (収率 29%)の化合物107を得た。
1H-NMR (CDCl3) δ: 0.953 (3H, d, J = 3.9 Hz), 1.12 (3H, d, J = 4.2 Hz), 4.51 (1H, 13.5 Hz), 4.83 (1H, d, J = 13.5 Hz), 4.83-4.92 (1H, m), 5.18 (1H, s), 5.74 (1H, d, J = 7.8 Hz), 6.73 (1H, d, J = 7.8 Hz), 6.90 (1H, d, J = 7.5 Hz), 7.12 (2H, dd, J = 7.2 Hz, 8.1 Hz), 7.19-7.22 (1H, m), 7.37-7.41 (3H, m), 7.55 (1H, s).

第一工程
化合物95B (1.00 g, 3.55 mmol) とcyclopropanamine (0.492 ml, 7.10 mmol) をピリジン (20ml)に加え、1-hydroxybenzotriazole (544 mg, 3.55 mmol) と1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (1.36 g, 7.10 mmol) を順に加えて室温で18時間攪拌した。溶媒を減圧留去し、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 95:5, v/v) 、続いてアミノカラムクロマトグラフィー (クロロホルム-メタノール, 99:1, v/v) で精製し、無色固体として1.19 g の化合物126Aを得た。
1H-NMR (CDCl3) δ: 0.22 (1H, m), 0.70 (2H, m), 2.76-2.83 (1H, m), 5.50 (2H, s), 6.59 (1H, dd, J = 7.0, 1.9 Hz), 7.44 (5H, d, J = 0.7 Hz), 7.53 (1H, dd, J = 6.9, 6.2 Hz), 8.30 (1H, brs), 9.71 (1H, brs).
第二工程
化合物126A (1.19 g, 4.19 mmol) をDMF (15 ml) に溶解し、炭酸カリウム (2.90 g, 20.1 mmol)を加えて室温で30分攪拌した。O-(2,4-dinitrophenyl)hydroxylamine (1.67 g, 8.38 mmol) を加えて室温で18時間攪拌した。反応液にクロロホルムを加えて析出した黄色沈殿物をろ過して除去し、ろ液を減圧濃縮した。得られた粗生成物をアミノカラムクロマトグラフィー (クロロホルム-メタノール, 97:3→95:5, v/v) で精製し、黄色固体として851 mg の化合物126Bを得た。
1H-NMR (CDCl3) δ: 0.41-0.46 (2H, m), 0.76 (2H, m), 2.73-2.81 (1H, m), 5.19 (2H, s), 5.61 (2H, s), 6.26 (1H, d, J = 7.2 Hz), 7.38 (5H, s), 7.44 (1H, d, J = 7.8 Hz), 7.70 (1H, s).
第三工程
化合物126B (847 mg, 2.83 mmol) とparaformaldehyde (255 mg, 8.49 mmol) をエタノール (12ml) に加え、マイクロウェーブ照射下、140℃で30分攪拌した。反応液を減圧濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 97:3→95:5→90:10, v/v) 、続いてアミノカラムクロマトグラフィー (クロロホルム-メタノール, 97:3, v/v) で精製し、塩化メチレン‐エチルエーテルを加えて析出した固体を濾取し、無色固体として665 mg の化合物126Cを得た。
1H-NMR (CDCl3) δ: 0.61-0.66 (2H, m), 0.87 (2H, m), 2.68-2.76 (1H, m), 4.32 (2H, d, J = 7.9 Hz), 5.28 (2H, s), 6.33 (1H, d, J = 7.7 Hz), 6.45 (1H, t, J = 7.7 Hz), 7.33 (3H, m), 7.38 (1H, d, J = 7.7 Hz), 7.52 (2H, m).
第四工程
化合物126C (100 mg, 0.321 mmol)をDMF (0.5 ml) に溶解し、炭酸セシウム (314 mg, 0.964 mmol) と(bromomethylene)dibenzene (119 mg, 0.482 mmol) を0℃で加えて室温で2時間攪拌した。反応液を水にあけ、酢酸エチルで抽出し、有機層を水で洗浄し、硫酸ナトリウムで乾燥した。溶媒を減圧留去し、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 97:3→95:5, v/v) で精製し、無色ガム状物質として124 mg の化合物126Dを得た。
1H-NMR (CDCl3) δ: 0.37-0.47 (2H, m), 0.74 (2H, m), 2.63-2.68 (1H, m), 4.35 (1H, d, J = 13.4 Hz), 4.65 (1H, d, J = 13.4 Hz), 5.07 (1H, s), 5.40 (1H, d, J = 10.7 Hz), 5.47 (1H, d, J = 10.5 Hz), 5.79 (1H, d, J = 7.6 Hz), 6.67 (1H, d, J = 7.8 Hz), 7.04-7.62 (15H, m).
第五工程
第四工程で得られた化合物126Dにトリフルオロ酢酸 (2 ml) を加えて、室温で1.5時間攪拌した。減圧濃縮した後、重曹水と2規定塩酸でpHを6に調節し、クロロホルムで抽出して硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、塩化メチレン‐エチルエーテルを加えて析出した固体を濾取し、無色固体として52 mg の化合物126を得た。
1H-NMR (DMSO-d6) δ: -0.19--0.06 (1H, m), 0.44-0.54 (1H, m), 0.82 (2H, m), 2.62-2.69 (1H, m), 4.21 (1H, d, J = 13.3 Hz), 5.11 (1H, d, J = 13.1 Hz), 5.32 (1H, s), 5.47 (1H, t, J = 11.1 Hz), 7.13 (1H, d, J = 7.6 Hz), 7.23 (3H, m), 7.28-7.47 (8H, m), 7.69 (2H, t, J = 8.5 Hz).
MS: m/z = 388 [M+H]+.

第一工程
化合物95B (2.40 g, 8.52 mmol) とethyl 3-aminopropanoate hydrochloride (2.62 g, 17.0 mmol) をピリジン (30 ml)に加え、1-hydroxybenzotriazole (1.31 g, 8.52 mmol) と1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (3.27 g, 17.0 mmol) を順に加えて室温で2時間攪拌した。溶媒を減圧留去し、得られた粗生成物をアミノカラムクロマトグラフィー (クロロホルム-メタノール, 95:5, v/v) で精製し、無色固体として1.90 g の化合物128Aを得た。
1H-NMR (CDCl3) δ: 1.29 (3H, t, J = 7.1 Hz), 2.48 (2H, t, J = 6.4 Hz), 3.58 (2H, q, J = 6.3 Hz), 4.17 (2H, q, J = 7.1 Hz), 5.59 (2H, s), 6.57 (1H, dd, J = 7.1, 1.6 Hz), 7.37-7.52 (6H, m), 8.73 (1H, brs), 9.72 (1H, brs).
第二工程
化合物128A (2.58 g, 7.49 mmol) をDMF (30 ml) に溶解し、炭酸カリウム (5.18 g, 37.5 mmol)を加えて室温で30分攪拌した。O-(2,4-dinitrophenyl)hydroxylamine (2.98 g, 15.0 mmol) を加えて室温で20時間攪拌した。反応液にクロロホルムを加えて析出した黄色沈殿物をろ過して除去し、ろ液を減圧濃縮した。得られた粗生成物をアミノカラムクロマトグラフィー (クロロホルム-メタノール, 97:3→95:5, v/v) 、続いてシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 95:5→92:8, v/v) で精製し、黄色固体として1.67 g の化合物128Bを得た。
1H-NMR (CDCl3) δ: 1.26 (3H, t, J = 7.2 Hz), 2.42 (2H, t, J = 6.6 Hz), 3.43 (2H, q, J = 6.4 Hz), 4.12 (2H, q, J = 7.1 Hz), 5.13 (2H, s), 5.53 (2H, s), 6.21 (1H, d, J = 7.6 Hz), 7.33 (5H, s), 7.39 (1H, d, J = 7.6 Hz), 7.85 (1H, t, J = 5.6 Hz).
第三工程
化合物128B (1.66 g, 4.62 mmol) とparaformaldehyde (416 mg, 13.9 mmol) をエタノール (20 ml) に加え、マイクロウェーブ照射下、140℃で30分攪拌した。反応液を減圧濃縮し、得られた粗生成物をアミノカラムクロマトグラフィー (クロロホルム-メタノール, 99:1→95:5, v/v) で精製し、無色固体として1.57 g の化合物128Cを得た。
1H-NMR (CDCl3) δ: 1.27 (3H, t, J = 7.2 Hz), 2.70 (2H, t, J = 5.7 Hz), 3.57 (2H, t, J = 5.8 Hz), 4.13 (2H, q, J = 7.1 Hz), 4.50 (2H, d, J = 7.9 Hz), 5.27 (2H, s), 5.87 (1H, t, J = 7.8 Hz), 6.32 (1H, d, J = 7.6 Hz), 7.31 (4H, m), 7.54 (2H, m).
第四工程
化合物128C (1.00 g, 2.69 mmol)をDMF (10 ml) に溶解し、炭酸セシウム (2.63 g, 8.08 mmol) と(bromomethylene)dibenzene (998 mg, 4.04 mmol) を0℃で加えて室温で18時間攪拌した。反応液を水にあけ、酢酸エチルで抽出し、有機層を水で洗浄し、硫酸ナトリウムで乾燥した。溶媒を減圧留去し、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 98:2, v/v) で精製し、無色ガム状物質として500 mg の化合物128Dを得た。
1H-NMR (CDCl3) δ: 1.25 (3H, t, J = 7.3 Hz), 2.46 (1H, m), 2.70-2.80 (1H, m), 2.87-2.96 (1H, m), 4.11 (2H, q, J = 7.3 Hz), 4.12 (1H, m), 4.48 (1H, d, J = 13.7 Hz), 4.85 (1H, d, J = 13.7 Hz), 5.10 (1H, s), 5.47 (2H, s), 5.83 (1H, d, J = 8.0 Hz), 6.73 (1H, d, J = 8.0 Hz), 7.37 (15H, m).
第五工程
化合物128D (40 mg, 0.074 mmol) にトリフルオロ酢酸 (1 ml) を加えて、室温で1時間攪拌した。減圧濃縮した後、重曹水と2規定塩酸でpHを6に調節し、クロロホルムで抽出して硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、塩化メチレン‐エチルエーテルを加えて析出した固体を濾取し、無色固体として20 mg の化合物128を得た。
1H-NMR (DMSO-d6) δ: 1.16 (3H, t, J = 7.1 Hz), 2.45-2.58 (3H, m), 3.70 (1H,m), 4.02 (2H, q, J = 7.1 Hz), 4.39 (1H, d, J = 13.4 Hz), 5.09 (1H, d, J = 13.3 Hz), 5.48 (1H, d, J = 3.2 Hz), 5.51 (1H, s), 7.19-7.38 (7H, m), 7.45 (2H, t, J = 7.3 Hz), 7.69 (2H, d, J = 7.2 Hz).
MS: m/z = 448 [M+H]+.

第一工程
化合物95B (1.50 g, 5.32 mmol) とtert-butyl 2-aminoethyl(methyl)carbamate (1.86 g, 10.7 mmol) をピリジン (20 ml)に加え、1-hydroxybenzotriazole (815 mg, 5.32 mmol) と1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (2.04 g, 10.7 mmol) を順に加えて室温で2時間攪拌した。反応液を1規定塩酸にあけ、酢酸エチルで抽出し、硫酸ナトリウムで乾燥した。溶媒を減圧留去し、得られた粗生成物をアミノカラムクロマトグラフィー (クロロホルム-メタノール, 95:5, v/v) で精製し、続いてシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 95:5, v/v) で精製し、無色ガム状物質として1.63 g の化合物135Aを得た。
1H-NMR (CDCl3) δ: 1.44 (9H, s), 2.82 (3H, s), 3.28 (4H, m), 5.59 (2H, s), 6.57 (1H, d, J = 6.0 Hz), 7.46 (6H, m), 8.46 (1H, m), 9.68 (1H, brs).
第二工程
化合物135A (1.05 g, 2.62 mmol) をDMF (15 ml) に溶解し、炭酸カリウム (1.81 g, 13.1 mmol)を加えて室温で30分攪拌した。O-(2,4-dinitrophenyl)hydroxylamine (1.04 g, 5.23 mmol) を加えて室温で18時間攪拌した。反応液にクロロホルムを加えて析出した黄色沈殿物をろ過して除去し、ろ液を減圧濃縮した。得られた粗生成物をアミノカラムクロマトグラフィー (クロロホルム-メタノール, 97:3→95:5, v/v) で精製し、淡黄色固体として887 mg の化合物135Bを得た。
1H-NMR (CDCl3) δ: 1.44 (9H, s), 2.84 (3H, s), 3.38 (4H, m), 5.33 (2H, s), 5.68 (1H, brs), 5.80 (1H, brs), 6.35 (1H, d, J = 7.6 Hz), 6.74 (1H, brs), 7.39 (5H, brm), 7.52 (1H, t, J = 9.5 Hz).
第三工程
化合物135B (880 mg, 2.11 mmol) とparaformaldehyde (190 mg, 6.34 mmol) をエタノール (18 ml) に加え、マイクロウェーブ照射下、140℃で30分攪拌した。反応液を減圧濃縮し、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 97:3→95:5→90:10, v/v) 、続いてアミノカラムクロマトグラフィー (クロロホルム-メタノール, 97:3, v/v) で精製し、無色固体として721 mg の化合物135Cを得た。
1H-NMR (CDCl3) δ: 1.29 (9H, s), 2.95 (3H, s), 4.38 (2H, brs), 5.33 (2H, brs), 6.36 (1H, d, J = 7.6 Hz), 6.85 (1H, t, J = 7.4 Hz), 7.33 (4H, m), 7.55 (2H, m).
MS: m/z = 429 [M+H]+.
第四工程
化合物135C (720 mg, 1.68 mmol)をDMF (3.5 ml) に溶解し、炭酸セシウム (1.64 g, 5.04 mmol) と(bromomethylene)dibenzene (623 mg, 2.52 mmol) を0℃で加えて室温で18時間攪拌した。反応液を水にあけ、酢酸エチルで抽出し、有機層を水で洗浄し、硫酸ナトリウムで乾燥した。溶媒を減圧留去し、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 97:3→95:5, v/v) で精製し、732 mg の化合物135Dを得た。
第五工程
化合物135D (727 mg, 1.22 mmol) に4規定 HCl (酢酸エチル溶液, 10 ml) を加えた。室温で1時間攪拌した後、溶媒を減圧留去した。飽和重曹水を加えてクロロホルムで抽出した後、硫酸ナトリウムで乾燥した。溶媒を減圧留去し、得られた粗生成物を塩化メチレン-エチルエーテルを加えて析出した固体を濾取し、無色固体として575 mg の化合物135Eを得た。
第六工程
化合物135E (50 mg, 0.10 mmol) にトリフルオロ酢酸 (2 ml) を加えて、室温で1.5時間攪拌した。減圧濃縮した後、重曹水と塩化アンモニウム水溶液でpHを6に調節し、クロロホルムで抽出して硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、塩化メチレン-エチルエーテルを加えて析出した固体を濾取し、無色固体として15 mg の化合物135を得た。
1H-NMR (DMSO-d6) δ: 2.40 (3H, s), 2.80 (1H, s), 3.12 (3H, m), 3.87 (1H, m), 4.37 (1H, d, J = 13.6 Hz), 5.10 (1H, d, J = 13.4 Hz), 5.52 (1H, s), 5.53 (1H, d, J = 5.5 Hz), 7.15-7.70 (11H, m).
MS: m/z = 405 [M+H]+.

第一工程
化合物165A (WO2006/088173, 37.0 g, 108 mmol) のDMF (370 mL) 溶液に室温で、炭酸カリウム (17.9 g, 129 mmol) およびヨウ化メチル (8.03 mL, 129 mmol) を順次加えた後、1.5時間攪拌した。反応液を塩化アンモニウム (20.8 g, 390 mmol) の水 (1110 mL) 溶液に氷冷下加え、析出した固体を濾取し、水洗することにより粗生成物 (33 g) を得た。また水層を食塩で塩析した後、酢酸エチルで抽出し、硫酸ナトリウムで乾燥した。溶媒を減圧下留去して得られた残さから粗生成物 (9 g) を得た。粗生成物を合わせてシリカゲルカラムクロマトグラフィー (酢酸エチル / n-ヘキサン = 50% → 100%) で精製し、化合物165B (36.5 g, 95 %) を白色固体として得た。
第二工程
化合物165B (36.5 g, 102 mmol) の1,4-ジオキサン (548 mL) 溶液に室温で、potassium osmate dihydrate (1.13 g, 3.06 mmol), 過ヨウ素酸ナトリウム (87.3 g, 408 mmol) および水 (365 mmol) を順次加えた後、6時間攪拌した。反応液を塩化メチレンで抽出し、硫酸ナトリウムで乾燥した。溶媒を減圧下留去して得られた残さをシリカゲルカラムクロマトグラフィー (酢酸エチル / n-ヘキサン = 50% → 100%) で精製し、化合物165C (33.0 g, 90 %) を褐色foamとして得た。
第三工程
化合物165C (1.38 g, 3.66 mmol) のトルエン (25 mL) 懸濁液に室温で、ethylenediamine (0.247 mL, 3.66 mmol) および酢酸 (0.0210 mL, 0.366 mmol) を順次加えた後、1時間攪拌し、さらに50℃で17時間攪拌した。析出した固体を濾取した後、エーテルで洗浄し、化合物165D (1.11 g, 100 %) を薄黄色固体として得た。
1HNMR (DMSO-d6) δ: 3.05 (2H, m), 3.26 (1H, m), 3.63 (2H, m), 3.75 (3H, s), 3.87 (1H, m), 4.52 (1H, dd, J = 3.3, 12.6 Hz), 4.69 (1H, m), 4.99 (1H, d, J = 10.4 Hz), 5.15 (1H, d, J = 10.4 Hz), 7.35 (3H, m), 7.54 (2H, m), 8.41 (1H, s).
第四工程
化合物165D (2.77 g, 7.50 mmol), 炭酸カリウム (2.23 g, 16.1 mmol) およびヨウ化ナトリウム (102 mg, 0.680 mmol) のアセトニトリル (30 mL) 懸濁液に室温で、bromodiphenylmethane (2.26 g, 9.14 mmol) を加え、90℃で7時間攪拌した。反応液を塩酸 (2 N, 10 mL) と氷 (20 g) にあけ、クロロホルム (100 mL x 2) で抽出し、硫酸ナトリウムで乾燥した。溶媒を減圧下留去して得られた残さをシリカゲルカラムクロマトグラフィー (クロロホルム / メタノール = 0% → 5%) で精製し、化合物165E (2.72 g, 68 %) を薄黄色固体として得た。
第五工程
化合物165E (2.72 g, 5.08 mmol) のエタノール (30 mL) 溶液に室温で、水酸化ナトリウム水溶液 (2 N, 10 mL) を加え、3日間攪拌した。反応液に室温で、塩酸 (1 N, 20 mL) を加え (pH = 1)、クロロホルム (100 mL x 2) で抽出し、硫酸ナトリウムで乾燥した。溶媒を減圧下留去して得られた残さをシリカゲルカラムクロマトグラフィー (クロロホルム / メタノール = 0% → 10%) で精製し、化合物165F (1.77 g, 67 %) を薄黄色固体として得た。
1HNMR (DMSO-d6) δ: 2.63 (1H, m), 3.16 (1H, m), 3.49 (1H, m), 3.73 (1H, m), 4.12 (2H, m), 4.56 (1H, m), 5.04 (1H, s), 5.09 (1H, d, J = 10.7 Hz), 5.19 (1H, d, J = 10.7 Hz), 7.28 - 7.53 (15H, m), 8.32 (1H, s), 8.39 (1H, s).
第六工程
化合物165F (1.77 g, 3.39 mmol) および塩化リチウム (0.515 g, 12.2 mmol) のN,N’-dimethylimidazolidinone (20 mL) 溶液を90℃で1時間攪拌した。反応液に室温で、水 (10 mL)、塩酸 (2 N, 10 mL) および水 (10 mL) を順次加えた。析出した固体を濾取し、エーテルで洗浄した後、DMF-水を加えて析出した固体を濾取し、化合物165 (599 mg, 41 %) を白色固体として得た。
1HNMR (DMSO-d6) δ: 2.60 (1H, m), 3.20 (1H, m), 3.64 (2H, m), 4.00 (2H, m), 4.55 (1H, m), 5.01 (1H, s), 7.28 - 7.47 (10H, m), 8.16 (1H, s), 11.97 (1H, brs).
MS: m/z = 432 [M+H]+.
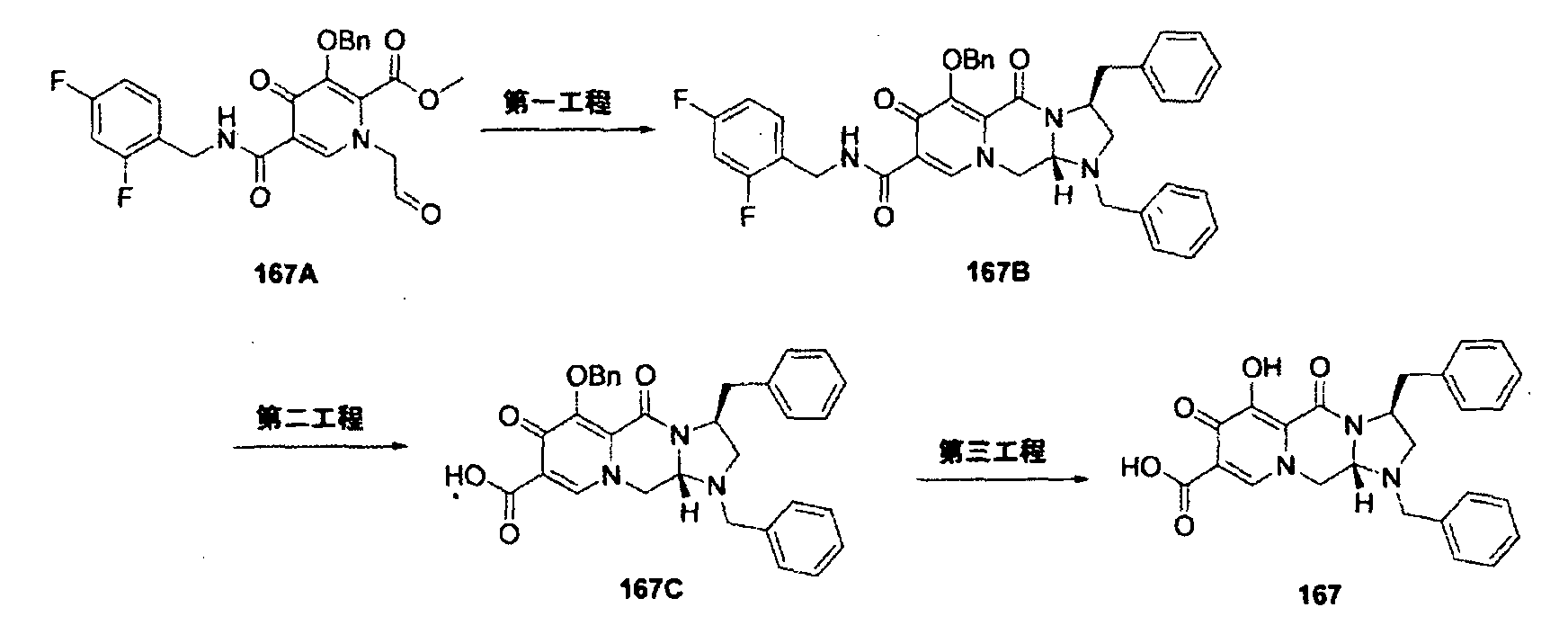
第一工程
化合物167A (WO2006/11674, 3.58 g, 7.61 mmol)のキシレン (30 ml)溶液に、(S)-N1-ベンジル-3-フェニルプロパン-1,2-ジアミン (Journal of the American Chemical Society; English; 127; 30; 2005; 10504, 1.83 g, 7.61 mmol) と酢酸 (0.5 ml) を加え、2時間還流した。室温まで冷却後、溶媒を留去し、得られた油状物をシリカゲルクロマトグラフィーに付し精製した。はじめにn-ヘキサン-酢酸エチル (9:1, v/v)で溶出し、次いでn-ヘキサン-酢酸エチル (1:1, v/v)で溶出した。目的の画分を濃縮すると油状物として 349 mg (収率7%) の化合物167Bを得た。
1HNMR (CDCl3) δ: 2.54 (1H, t, J = 9.6 Hz), 2.77 (1H, dd, J = 9.0 Hz, 13.2 Hz), 3.31 (1H, dd, J = 6.9 Hz, 9.6 Hz), 3.43-3.78 (5H, m), 4.04-4.15 (1H, m), 4.42-4.48 (1H, m), 4.62 (2H, d, J = 6.0 Hz), 5.29 (1H, d, J = 10.5 Hz), 5.43 (1H, d, J = 10.5 Hz), 6.77-6.85 (2H, m), 7.19-7.39 (14H, m), 7.60 (2H, d, J = 6.3 Hz), 8.05 (1H, s).
第二工程
化合物167B (968 mg, 1.47 mmol) のMeCN (10 ml) 溶液にBoc2O (3 ml) とDMAP (180 mg, 1.47 mmol)を加え、5時間加熱還流させた。反応液に、2規定水酸化ナトリウム水溶液を加えて反応を停止し、2規定塩酸を用いて中和した後に、酢酸エチルで3回抽出した。抽出液を飽和食塩水で洗浄後、溶媒を留去し、得られた油状物をシリカゲルクロマトグラフィーに付し精製した。はじめにn-ヘキサン-酢酸エチル (6:4, v/v)で溶出し、次いで酢酸エチルのみで溶出した。目的の画分を濃縮し固体として349 mg (収率 45%) の167Cを得た。
1HNMR (CDCl3) δ: 2.54 (1H, t = 9.0 Hz), 2.76 (1H, dd, J = 9.3 Hz, 16.5 Hz), 3.31 (1H, dd, J = 6.9 Hz, 9.6 Hz), 3.45 (1H, dd, J = 3.3 Hz, 12.6 Hz), 3.51-3.78 (4H, m), 4.04-4.13 (1H, m), 4.42-4.52 (1H, m), 4.61 (2H, d, J = 6.0 Hz), 2.79 (1H, d, J = 10.2 Hz), 5.29 (1H, d, J = 10.2 Hz), 5.43 (1H, d, J = 10.2 Hz), 6.76-7.39 (11H, m), 7.60 (2H, d, J = 6.6 Hz), 8.05 (1H, s), 10.42 (1H, t, J = 5.7 Hz).
第三工程
上記化合物167C (150 mg, 0.280 mmol) をトリフルオロ酢酸 (2 ml) に溶解させ、室温下1時間撹拌した。溶媒を留去し、残渣をジクロロメタン(2 ml) に溶解させ、飽和重曹水で中和した。得られた溶液をクエン酸水溶液で酸性にして、有機層を分離した。水層をジクロロメタンで1回抽出し、合わせた有機層を水洗後、硫酸ナトリウムで乾燥した。溶媒を留去後、得られた固体をジイソプロピルエーテルで洗浄し、71 mg (収率57%) の化合物167を得た。
1HNMR (CDCl3) δ: 2.65 (1H, dd, J = 8.4 Hz, 9.6 Hz), 2.97 (1H, dd, J = 9 Hz, 13.5 Hz), 3.43 (1J, dd, J = 7.2 Hz, 9.6 Hz), 3.55 (1H, dd, J = 3.0 Hz, 13.2 Hz), 3.61-3.80 (4H, m), 4.15 (1H, dd, J = 4.2 Hz, 9.9 Hz), 4.51-4.60 (1H, m), 7.15-7.18 (2H, m), 7.28-7.38 (8H, m), 8.02 (1H, s), 12.04 (1H, s).

第一工程
化合物95A (WO2006/116764, 500 mg, 2.03 mmol) の エタノール (5 mL) 溶液に2, 2-ジメトキシエタンアミン (0.49 ml, 4.47 mmol) を加え、80℃で3時間撹拌した。反応液を放冷した後、室温で酢酸 (0.27 ml, 4.69 mmol) を加え、減圧下で濃縮した。得られた残渣をDMF (5 mL) に溶解し、窒素雰囲気下、DBU (0.66 mL, 4.4 mmol) 続いてヨウ化メチル (1.02 mL, 16.2 mmol) を加え、室温で3時間攪拌した。反応液に飽和炭酸水素ナトリウム水溶液、酢酸エチルを加え、酢酸エチル層を分離し、水層を酢酸エチルで抽出した。合わせた抽出液に硫酸ナトリウムを加え、ろ過、濃縮し、得られた残渣をシリカゲルクロマトグラフィーに付し、精製した。クロロホルム-メタノール(9:1)で溶出し、目的の画分を濃縮すると茶色オイルとして258 mg の化合物169Aを得た。
1H-NMR (CDCl3) δ: 3.37 (6H, s), 3.80 (3H, s), 3.87 (2H, d, J = 4.8 Hz), 4.46 (1H, t, J = 4.8 Hz), 5.30 (2H, s), 6.75 (1H, d, J = 6.0 Hz), 7.30-7.41 (6H, m).
第二工程
化合物169A (1.00 g, 2.88 mmol) にギ酸 (31 mL) 続いて水 (5 mL) を加え、70℃で6.5時間攪拌した。反応混合物に水、酢酸エチルを加え、酢酸エチル層を分離し、水層を酢酸エチルで抽出した。合わせた抽出液を飽和炭酸水素ナトリウム水溶液で洗浄した後、硫酸ナトリウムを加え、ろ過、濃縮し、得られた残渣をシリカゲルクロマトグラフィーに付し、精製した。酢酸エチル-メタノールで溶出し、目的の画分を濃縮すると無色透明油状物としてアルデヒド水和物およびメチルアセタールの混合物を得た。得られた油状物をジクロロメタン (5 mL) に溶解し、1,3-ジアミノプロパン二塩酸塩 (354 mg, 2.41 mmol) 続いて酢酸 (0.069 ml, 1.2 mmol) を加え、室温で6時間攪拌した。反応液をジクロロメタンで希釈し、不溶物をろ過した後、減圧下で濃縮し、化合物169Bの粗精製物を得た。
MS: m/z =326.20 [M+H]+.
第三工程
化合物169B (391 mg, 1.20 mmol) のアセトニトリル (4 mL) 溶液に炭酸カリウム (498 mg, 3.61 mmol) 続いてブロモメチレンジベンゼン (890 mg, 3.61 mmol) を加えた。反応液を90℃で2時間撹拌後、反応液に水、酢酸エチル、brineを加え、酢酸エチル層を分離し、水層を酢酸エチルで1回、抽出した。合わせた抽出液を硫酸マグネシウムで乾燥させた後、ろ過、濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィーに付し、精製した。酢酸エチル-メタノールで溶出し、目的の画分を濃縮すると橙色固体として 106 mg の化合物169Cを得た。
MS: m/z = 492.15 [M+H] +.
第四工程
化合物169C (105 mg, 0.214 mmol) の DMI (2 mL) 溶液に塩化リチウム (27.2 mg, 0.641 mmol) を加え、90℃で3時間攪拌した。さらに塩化リチウム (27.2 mg, 0.641 mmol) を加え、90℃で1時間攪拌した。反応液を減圧下で濃縮し、得られた残渣をLCMS分取装置を用いて精製した。 溶出溶媒を留去し、残渣にジエチルエーテルを加え、析出した固体を濾取した。ジエチルエーテルで洗浄、乾燥することにより、 27 mgの化合物169を得た。
1H-NMR (CD3OD) δ: 1.63 (1H, dd, J = 13.4, 2.8 Hz), 1.84 (1H, br s), 2.55-2.64 (1H, m), 2.90-3.10 (2H, m), 4.30 (1H, dd, J = 14.5, 4.0 Hz), 4.52 (4H, dd, J = 14.5, 3.8 Hz), 4.63-4.75 (4H, m), 5.16 (1H, s), 6.16 (1H, d, J = 7.2 Hz), 6.78 (1H, d, J = 7.2 Hz), 7.16-7.32 (10H, m).
MS: m/z = 402.10 [M+H]+.

第一工程
化合物49B (950 mg, 3.35 mmol) と3-aminopropan-1-ol (277 mg, 3.69 mmol) と硫酸ナトリウム(1.91 g, 13.4 mmol)をトルエン (25 ml) に加え、室温で1時間攪拌した。Boc2O (0.856 ml, 3.69 mmol) を室温で加え、そのまま18時間攪拌した。さらにBoc2O (0.400 ml, 1.72 mmol) を室温で加え、そのまま60時間時間攪拌した。反応液をろ過し、ろ液を減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル, 1:1, v/v) で精製し、無色ガム状物質として171Aを1.02 g得た。
第二工程
化合物171A (1.01 g, 2.29 mmol) とパラジウム‐活性炭素 (10%, wet, 200 mg) をエタノール (20ml) に加え、水素雰囲気下、室温で1.5時間攪拌した。セライトろ過した後、溶媒を減圧濃縮して無色オイル状物質171Bを755 mg得た。
1H-NMR (CDCl3) δ: 1.42 (5H, s), 1.49 (4H, s), 1.56-1.92 (2H, m), 2.49 (0.4H, dd, J = 13.6, 9.8 Hz), 2.62 (0.6H, dd, J = 13.6, 8.5 Hz), 2.81 (0.4H, dd, J = 13.5, 3.6 Hz), 3.16 (1.6H, m), 3.60-4.14 (4H, m), 5.13 (0.6H, d, J = 8.8 Hz), 5.19 (0.4H, d, J = 8.5 Hz), 7.22-7.37 (5H, m).
第三工程
dimethyl 3-(benzyloxy)-4-oxo-4H-pyran-2,5-dicarboxylate (660 mg, 1.99 mmol) と171B (609 mg, 1.99 mmol) をトルエン (8 ml) に加え、100℃で1.5時間攪拌した。溶媒を減圧留去した後、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 99:1, v/v) で精製し、淡黄色ガム状物質として 1.02 g の化合物171Cを得た。
第四工程
化合物171C (991 mg, 1.60 mmol) に4規定 HCl (酢酸エチル溶液, 12 ml) を加えた。室温で1時間攪拌した後、溶媒を減圧留去した。続いてトルエン (12 ml) と3-aminopropan-1-ol (0.244 ml, 3.19 mmol) を加え、80℃で10分攪拌した。溶媒を減圧留去した後、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 99:1→95:5→90:10, v/v) で精製し、黄色ガム状物質の化合物171D 341 mgと無色固体の化合物171E 338 mgを得た。
171D: 1H-NMR (CDCl3) δ: 1.29 (3H, t, J = 7.1 Hz), 1.51 (1H, d, J = 13.7 Hz), 1.97 (1H, m), 2.91 (1H, dd, J = 13.8, 9.8 Hz), 2.99-3.10 (2H, m), 3.90 (1H, td, J = 12.1, 2.5 Hz), 4.12 (2H, m), 4.25 (2H, m), 4.83 (2H, m), 5.33 (1H, d, J = 10.1 Hz), 5.51 (1H, d, J = 10.1 Hz), 6.88 (2H, m), 7.23-7.40 (7H, m), 7.68 (2H, m)
171E: 1H-NMR (CDCl3) δ: 1.19 (3H, t, J = 7.2 Hz), 1.82-1.99 (2H, m), 2.73 (1H, dd, J = 14.0, 11.3 Hz), 3.13 (1H, m), 3.35 (1H, dd, J = 14.0, 3.4 Hz), 3.63 (1H, m), 3.90-4.26 (4H, m), 4.43 (1H, d, J = 13.6 Hz), 5.27 (1H, t, J = 3.5 Hz), 5.31 (2H, s), 6.78 (2H, dd, J = 6.3, 3.2 Hz), 7.01 (1H, d, J = 7.0 Hz), 7.18 (3H, t, J = 3.1 Hz), 7.28-7.39 (3H, m), 7.67 (2H, m).
第五工程
化合物171D (329 mg, 0.673 mmol) をエタノール (2 ml) とTHF (4 ml) に溶解し、2規定水酸化ナトリウム水溶液 (1.69 ml, 3.38 mmol) を加えて室温で1時間攪拌した。反応液に2規定塩酸を加えて酢酸エチルで抽出し、硫酸ナトリウムで乾燥した。溶媒を減圧濃縮することにより、無色固体として215 mg の化合物171Fを得た。
MS: m/z = 461 [M+H]+.
第六工程
化合物171F (50 mg, 0.11 mmol) にトリフルオロ酢酸 (2 ml) を加えて、室温で1時間攪拌した。減圧濃縮した後、重曹水と2規定塩酸でpHを6に調節し、クロロホルムで抽出して硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、クロロホルム‐メタノール‐エチルエーテルを加えて析出した固体を濾取し、無色固体として24 mg の化合物171を得た。
1H-NMR (DMSO-d6) δ: 1.63 (1H, d, J = 12.6 Hz), 1.83 (1H, m), 2.96-3.29 (3H, m), 4.05 (2H,m), 4.55 (1H, dd, J = 13.2, 4.4 Hz), 5.08 (1H, dd, J = 9.2, 5.4 Hz), 5.30 (1H, s), 7.19 (5H, m), 8.09 (1H, s), 12.84 (1H, brs).
MS: m/z = 371 [M+H]+.

第一工程
Dess-Martin Periodinane (0.3M, 塩化メチレン溶液, 25.0 ml, 7.50 mmol) に化合物2b (1.98 g, 5.48 mmol) の塩化メチレン溶液 (10 ml) を0℃で滴下した。室温で3時間攪拌の後、1規定水酸化ナトリウム水溶液にあけ、エチルエーテルで抽出した。有機層を1規定水酸化ナトリウム水溶液と飽和食塩水で洗浄し、硫酸マグネシウムで乾燥した。溶媒を減圧留去した後、シリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル, 2:1, v/v) で精製し、白色固体として化合物175Aを1.73 g得た。
1H-NMR (CDCl3) δ: 4.55 (1H, d, J = 7.3 Hz), 5.09 (2H, s), 5.14 (2H, m), 7.22-7.35 (15H, m), 9.62 (1H, s).
第二工程
化合物175A (1.30 g, 4.59 mmol) と3-aminopropan-1-ol (379 mg, 5.05 mmol) と硫酸ナトリウム (3.26 g, 22.4 mmol) をトルエン (40 ml) に加え、室温で1時間攪拌した。Boc2O (1.17 ml, 5.05 mmol) を室温で加え、そのまま18時間攪拌した。Boc2O (1.17 ml, 5.05 mmol) と硫酸ナトリウム (3.26 g, 22.4 mmol) を加え、60時間攪拌した。反応液をろ過し、ろ液を減圧濃縮した。得られた粗生成物をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル, 1:1, v/v) で精製し、無色固体として175Bを635 mg得た。
第三工程
化合物175B (632 mg, 1.22 mmol) とパラジウム‐活性炭素 (10%, wet, 100 mg) をエタノール (10ml) とTHF (5 ml) に加え、水素雰囲気下、室温で3時間攪拌した。セライトろ過した後、溶媒を減圧濃縮して無色オイル状物質175Cを502 mg得た。
1H-NMR (CDCl3) δ: 1.45 (9H, s), 1.77 (2H, m), 3.18-3.27 (1H, m), 3.43-3.51 (1H, m), 4.04 (4H, m), 4.92 (1H, d, J = 4.7 Hz), 7.28 (10H, m).
第四工程
dimethyl 3-(benzyloxy)-4-oxo-4H-pyran-2,5-dicarboxylate (390 mg, 1.22 mmol) と175C (468 mg, 1.22 mmol) をトルエン (5 ml) に加え、100℃で2時間攪拌した。溶媒を減圧留去した後、得られた粗生成物をシリカゲルカラムクロマトグラフィー (n-ヘキサン-酢酸エチル, 1:1, v/v) で精製し、淡黄色ガム状物質として 391 mg の化合物175Dを得た。
第五工程
化合物175D (388 mg, 0.568 mmol) に4規定 HCl (酢酸エチル溶液, 4 ml) を加えた。室温で1時間攪拌した後、溶媒を減圧留去した。続いてトルエン (4 ml) と3-aminopropan-1-ol (0.0870 ml, 1.14 mmol) を加え、80℃で5時間攪拌した。溶媒を減圧留去した後、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 98:2, v/v) で精製し、黄色ガム状物質の化合物175E 57 mgと茶色ガム状物質の化合物175F 44 mgを得た。
175E: 1H-NMR (CDCl3) δ: 1.91-2.00 (2H, m), 2.87 (1H, m), 3.78 (3H, s), 3.87 -4.15 (3H, m), 4.61 (1H, d, J = 12.1 Hz), 4.78 (2H, m), 5.33 (1H, d, J = 10.2 Hz), 5.63 (1H, d, J = 10.2 Hz), 6.95 (2H, m), 7.13-7.53 (12H, m), 7.76 (2H, m)
175F: 1H-NMR (CDCl3) δ: 1.83-1.97 (2H, m), 3.12-3.22 (1H, m), 3.50 (1H, m), 3.85 (3H, s), 3.90 (1H, m), 4.34-4.40 (1H, m), 4.74 (1H, d, J = 8.6 Hz), 4.84-4.89 (1H, m), 5.09 (1H, d, J = 3.3 Hz), 5.15 (1H, d, J = 9.9 Hz), 5.26 (1H, d, J = 9.6 Hz), 7.08-7.50 (13H, m), 7.65-7.77 (3H, m).
第六工程
化合物175E (57 mg, 0.10 mmol) をTHF (0.5 ml) とエタノール (0.5 ml) に溶解し、室温で2規定水酸化ナトリウム水溶液 (0.25 ml, 0.50 mmol) を加えてそのまま1時間攪拌した。1規定塩酸を加えてからクロロホルムで抽出した後、硫酸ナトリウムで乾燥した。溶媒を減圧留去し、得られた粗生成物をシリカゲルカラムクロマトグラフィー (クロロホルム-メタノール, 98:2, v/v) で精製し、化合物175Gを得た。
第七工程
第六工程で得られた化合物175Gにトリフルオロ酢酸 (1 ml) を加えて、室温で1時間攪拌した。減圧濃縮した後、重曹水と2規定塩酸でpHを3に調節し、クロロホルムで抽出して硫酸ナトリウムで乾燥した。溶媒を減圧留去した後、クロロホルム-メタノール-エチルエーテルを加えて析出した固体を濾取し、無色固体として11 mg の化合物175を得た。
1H-NMR (DMSO-d6) δ: 1.50 (1H, d, J = 13.1 Hz), 1.79 (1H, m), 3.17 (1H, m), 3.86 (1H, t, J = 11.0 Hz), 4.03 (1H, dd, J = 10.8, 4.1 Hz), 4.46 (1H, d, J = 12.0 Hz), 4.53 (1H, dd, J = 12.7, 4.2 Hz), 4.84 (1H, s), 5.85 (1H, d, J = 11.7 Hz), 7.22 (7H, m), 7.44 (2H, t, J = 7.6 Hz), 7.65 (2H, d, J = 7.3 Hz), 8.14 (1H, s), 12.75 (1H, s), 15.33 (1H, brs).
MS: m/z = 447 [M+H]+.
各実施例で得られた結晶の粉末X線回折測定は、日本薬局方の一般試験法に記載された粉末X線回折測定法に従い、以下の測定条件で行った。
(装置)
Bruker社製D-8Discover
(操作方法)
試料について、以下の条件で測定を行った。
測定法:反射法
光源の種類:Cu管球
使用波長:CuKα線
管電流:40mA
管電圧:40Kv
試料プレート:ガラス
X線の入射角:3°及び12°
1)基質の調製
5'末端のGを2リン酸化修飾、且つ2'位の水酸基をメトキシル化修飾し、5'末端から6番目のUをCy3標識、3'末端をBHQ2標識した30merRNA(5'‐pp-[m2'‐O]GAA UAU(‐Cy3) GCA UCA CUA GUA AGC UUU GCU CUA-BHQ2-3':日本バイオサービス社製)を購入し、EPICENTRE社製のスクリプトキャップ(ScriptCap)システムを使ってcap構造を付加した(産物はm7G [5’]-ppp-[5’] [m2’‐O]GAA UAU(-Cy3) GCA UCA CUA GUA AGC UUU GCU CUA(-BHQ2)-3’)。これを変性ポリアクリルアミドゲル電気泳動法にて分離・精製し、基質として使用した。
2)酵素の調製
RNPは定法に従いウイルス粒子から調製した(参考文献:VIROLOGY(1976) 73, p327-338 OLGA M. ROCHOVANSKY)。具体的にはA/WSN/33ウイルス1x103 PFU/mL、200μLを10日齢発育鶏卵に接種し、37℃で2日間培養後、鶏卵のしょう尿液を回収した。20%スクロースを用いた超遠心分離によりウイルス粒子を精製し、TritonX-100とリソレシチンを用いてウイルス粒子を可溶化後、30-70%グリセロール密度勾配を用いた超遠心分離によりRNP画分(50~70%グリセロール画分)を採取し、酵素液(約1nMのPB1・PB2・PA複合体を含む)として使用した。
3)酵素反応
ポリプロピレン製の384穴プレートに酵素反応液(組成: 53 mM Tris-塩酸塩 (pH7.8)、1mM MgCl2、1.25 mM ジチオスレイトール、80mM NaCl、12.5%グリセロール、酵素液0.15μL)を2.5μL分注した。次にジメチルスルホキシド(DMSO)で段階的に希釈した被検化合物溶液0.5μL、ポジティブコントロール(PC)及びネガティブコントロール(NC)には、DMSO 0.5μLを加え、よく混合した。次に基質溶液(1.4nM基質RNA、0.05%Tween20)2μLを加えて反応を開始し、室温で60分間インキュベートした後、反応液1μLを10μL のHi-Di Formamide溶液(サイジングマーカーとしてGeneScan 120 Liz Size Standardを含む:アプライドバイオシステム(ABI)社製。)に加え、反応を停止した。NCは反応開始前にEDTA(4.5mM)を加えることで予め反応を停止させた(表記濃度は全て終濃度である)。
3)阻害率(IC50値)の測定
反応停止させた溶液を85 ℃で5分間加熱し、氷上で2分間急冷後、ABI PRIZM 3730ジェネティックアナライザで分析した。解析ソフトABI Genemapperによりキャップ依存的エンドヌクレアーゼ産物のピークを定量し、PC、NCの蛍光強度をそれぞれ0%阻害、100%阻害として被検化合物のCEN反応阻害率(%)を求めた後、カーブフィッティング ソフトウェア (XLfit2.0:Model 205(IDBS社製)など)を使ってIC50値を求めた。被検物質のIC50値を表1に示す。
<材料>
・2% FCS E-MEM(MEM(Minimum Essential Medium)(Invitrogen)にカナマイシン及びFCSを添加して調整)
・0.5% BSA E-MEM(MEM(Minimum Essential Medium)(Invitrogen)にカナマイシン及びBSAを添加して調整)
・HBSS(hanks' Balanced Salt Solution)
・MDBK細胞
2% FCS E-MEMにて適当細胞数(3×105 /mL)に調整した。
・MDCK細胞
HBSSにて2回洗った後、0.5% BSA E-MEMにて適当細胞数(5×105 /mL)に調整した。
・Trypsin溶液
Trypsin from porcine pancreas(SIGMA)をPBS(-)にて溶解し、0.45μmのフィルターにてフィルトレーションした。
・EnVision(PerkinElmer)
・WST-8 Kit(キシダ化学)
・10% SDS溶液
・被験試料の希釈、分注
培養液として、MDBK細胞使用時には2% FCS E-MEMを使用し、MDCK細胞使用時には0.5% BSA E-MEMを用いた。以下、ウイルス・細胞・被験試料の希釈に対し、同様の培養液を使用した。
予め被験試料を培養液で適度な濃度に希釈し、96 wellプレートに2~5倍段階希釈系列を作製した(50μL/well)。抗Flu活性測定用、細胞毒性測定用の2枚作製した。各薬剤について3重測定を実施した。
MDCK細胞使用時には、抗Flu活性測定用にのみ、細胞にTrypsinを最終濃度3μg/mLとなるように添加した。
・インフルエンザウイルスの希釈、分注
予め、インフルエンザウイルスを培養液で適当な濃度に希釈し、被験試料が入った96 wellプレートに50μL/wellずつ分注した。細胞毒性測定用のプレートには、培養液を50μL/wellずつ分注した。
・細胞の希釈、分注
適当細胞数に調整した細胞を、被験試料が入った96 wellプレートに100μL/wellずつ分注した。
プレートミキサーで混和し、CO2インキュベーターで培養した。抗Flu活性測定用、細胞毒性測定用共に、3日間培養した。
・WST-8の分注
3日間培養した96 wellプレートを肉眼、顕微鏡下で観察し、細胞の形態・結晶の有無等を確認した。プレートから細胞を吸わないように上清を除いた。
WST-8 Kitを、培養液にて10倍希釈し、このWST-8溶液を各wellに100μLずつ分注した。プレートミキサーにて混和の後、CO2インキュベーターで1~3時間培養した。
抗Flu活性測定用プレートについては、培養後、各wellに10% SDS溶液を10μLずつ分注し、ウイルスを不活化した。
・吸光度の測定
混和した96wellプレートを、EnVisionで450 nm/620 nmの2波長で吸光度を測定した。
次の様な計算式に基づきMicrosoft Excelまたは同等の計算処理能力を有するプログラムを使用し算出した。
・50% インフルエンザ感染細胞死阻害濃度 (EC50)算出
EC50 = 10^ Z
Z = (50% - High %) / (High % -Low %) x {log(High conc.) - log(Low conc.)} + log(High conc.)
被検物質のIC50値を表1に示す。

被検化合物は、高いキャップ依存的エンドヌクレアーゼ(CEN)阻害活性を示し、高いCPE抑制効果を示した。これらの化合物はインフルエンザウイルスに感染することにより誘発される症状及び/又は疾患の治療及び/又は予防剤として有用な医薬となりうる。
したがって、本発明に係る化合物および製造方法は、医薬として使用しうる化合物を効率的に製造するために有用な中間体化合物および製造方法であるといえる。
Claims (18)
- 以下の工程:
(工程B)
式(X2):

(式中、R1dは、水素、ハロゲン、置換基群Eで置換されていてもよい低級アルキルオキシ、置換基群Eで置換されていてもよい炭素環低級アルキルオキシ、置換基群Eで置換されていてもよい複素環低級アルキルオキシ、または-OSi(R1e)3であり、
R1eは、それぞれ独立して、置換基群Eで置換されていてもよい低級アルキル、置換基群Eで置換されていてもよい炭素環式基、置換基群Eで置換されていてもよい複素環式基、置換基群Eで置換されていてもよい炭素環低級アルキルまたは置換基群Eで置換されていてもよい複素環低級アルキルであり、
R2dは、水素、置換基群Eで置換されていてもよい低級アルキル、置換基群Eで置換されていてもよい炭素環低級アルキル、または置換基群Eで置換されていてもよい複素環低級アルキルであり、
R3dは、水素、置換基群Eで置換されていてもよい低級アルキル、-N(R3e)2、または-OR3eであり、
R3eは、それぞれ独立して置換基群Eで置換されていてもよい低級アルキル、または一緒になって複素環式基を形成していてもよく、ならびに
波線は、E体および/もしくはZ体またはそれらの混合物を表わす。
置換基群E:ハロゲン、シアノ、ヒドロキシ、カルボキシ、ホルミル、アミノ、オキソ、ニトロ、低級アルキル、ハロゲノ低級アルキル、低級アルキルオキシ、置換基群Fで置換されていてもよい炭素環式基、置換基群Fで置換されていてもよい複素環式基、置換基群Fで置換されていてもよい炭素環低級アルキルオキシ、置換基群Fで置換されていてもよい複素環低級アルキルオキシ、置換基群Fで置換されていてもよい炭素環低級アルキルチオ、置換基群Fで置換されていてもよい複素環低級アルキルチオ、置換基群Fで置換されていてもよい炭素環低級アルキルアミノ、置換基群Fで置換されていてもよい複素環低級アルキルアミノ、置換基群Fで置換されていてもよい炭素環オキシ、置換基群Fで置換されていてもよい複素環オキシ、置換基群Fで置換されていてもよい炭素環カルボニル、置換基群Fで置換されていてもよい複素環カルボニル、置換基群Fで置換されていてもよい炭素環アミノカルボニル、置換基群Fで置換されていてもよい複素環アミノカルボニル、ハロゲノ低級アルキルオキシ、低級アルキルオキシ低級アルキル、低級アルキルオキシ低級アルキルオキシ、低級アルキルカルボニル、低級アルキルオキシカルボニル、低級アルキルオキシカルボニルアミノ、低級アルキルアミノ、低級アルキルカルボニルアミノ、低級アルキルアミノカルボニル、低級アルキルスルホニル、および低級アルキルスルホニルアミノ;
置換基群F:ハロゲン、ヒドロキシ、カルボキシ、アミノ、オキソ、ニトロ、低級アルキル、ハロゲノ低級アルキル、低級アルキルオキシ、およびアミノ保護基)
で表わされる化合物と、式(V2):

(式中、R4dは、置換基群Eで置換されていてもよい低級アルキル、置換基群Eで置換されていてもよい炭素環低級アルキル、または置換基群Eで置換されていてもよい複素環低級アルキルであり、
R5dは、水素、ハロゲン、置換基群Eで置換されていてもよい低級アルキルオキシ、または-O-SO2-R5eであり、
R5eは、置換基群Eで置換されていてもよい低級アルキル、置換基群Eで置換されていてもよい炭素環式基、または置換基群Eで置換されていてもよい複素環式基、置換基群Eで置換されていてもよい炭素環低級アルキル、または置換基群Eで置換されていてもよい複素環低級アルキルであり、ならびに
置換基群Eは前記と同意義である)
で表わされる化合物を反応させることにより、式(X3):
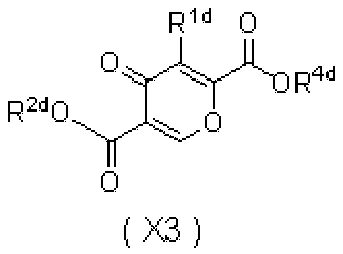
(式中、各記号は前記と同意義である)
で表わされる化合物を得る工程;
ならびに、
(工程C)
式(X3)で表わされる化合物と、式(V3):

(式中、R6dは、置換基群Eで置換されていてもよい低級アルキル、置換基群Eで置換されていてもよい低級アルケニル、置換基Eで置換されていてもよいアミノ、置換基Eで置換されていてもよい低級アルキルアミノ、置換基群Eで置換されていてもよい炭素環式基、または置換基群Eで置換されていてもよい複素環式基であり、
置換基群Eは前記と同意義である)
で表わされる化合物を反応させることにより、式(X4):

(式中、各記号は前記と同意義である)
で表される化合物を得る工程;
を含む、式(X4)で表わされる化合物またはその塩の製造方法。
- 工程Bおよび工程Cを連続して行なうことを特徴とする、請求項1に記載の製造方法。
- 式(X2):

で表わされる化合物と、式(VA2):
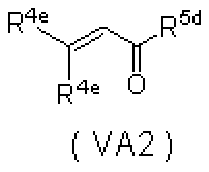
(式中、R4eは、それぞれ独立して水素、置換基群Eで置換されていてもよい低級アルキル、置換基群Eで置換されていてもよい炭素環式基、または置換基群Eで置換されていてもよい複素環式基、ならびに
R5dおよび置換基群Eは前記と同意義である)
で表わされる化合物を反応させる工程;ならびに
式(V3):
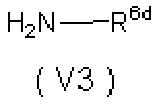
(式中、R6dは、前記と同意義である)
で表わされる化合物を反応させる工程を含む、式(XA4):

(式中、Raは、水素、ヒドロキシ、置換基群Eで置換されていてもよい低級アルキルアミノ、置換基群Eで置換されていてもよい低級アルケニルアミノ、置換基群Eで置換されていてもよい低級アルキニルアミノ、置換基群Eで置換されていてもよい炭素環低級アルキルアミノ、または置換基群Eで置換されていてもよい複素環低級アルキルアミノ、ならびに
各記号は、前記と同意義である)
で表わされる化合物またはその塩の製造方法。
- 式(X2):

(式中、各記号は請求項1と同意義である)
で表わされる化合物と、式(V2)または式(VA2):

(式中、各記号は請求項1と同意義である)
で表わされる化合物を反応させることを特徴とする、式(X3)または式(XA3):

(式中、各記号は請求項1と同意義である)
で表わされる化合物またはその塩の製造方法。
- 式(X3)もしくは式(XA3):

(式中、各記号は請求項1と同意義である)
で表わされる化合物、または式(XA3)の誘導体および、式(V3):

(式中、記号は請求項1と同意義である)
で表わされる化合物を反応させる工程を含む、式(X4)もしくは式(XA4):
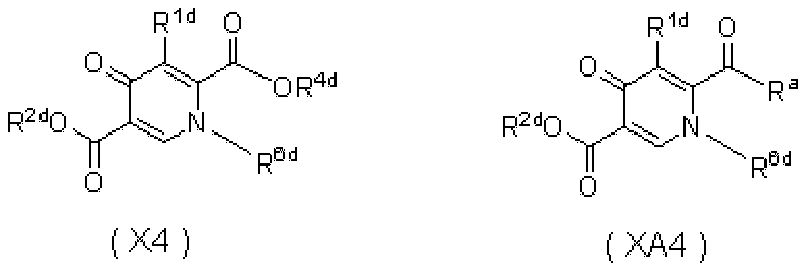
(式中、各記号は請求項1および請求項3と同意義である)
で表される化合物またはその塩の製造方法。
- 式(X2):

(式中、各記号は請求項1と同意義である)
で表わされる化合物と、式(V2)または式(VA2):

(式中、各記号は請求項1と同意義である)
で表わされる化合物、および式(V2’):

(式中、Xd-は、アンモニウムカチオンのカウンターアニオンである)
で表わされる化合物を反応させることを特徴とする、式(X4’)または式(XA4’):
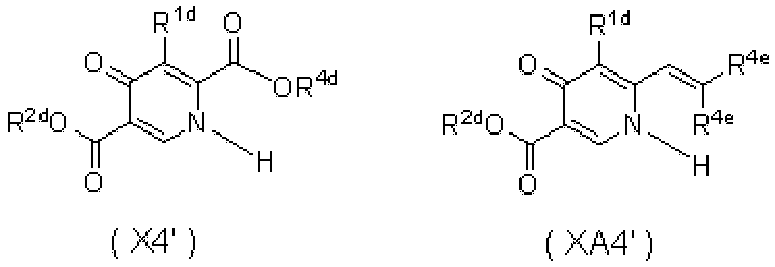
(式中、各記号は請求項1と同意義である)
で表わされる化合物またはその塩の製造方法。
- 請求項6に記載の製造方法で得られる式(X4’)もしくは式(XA4’):
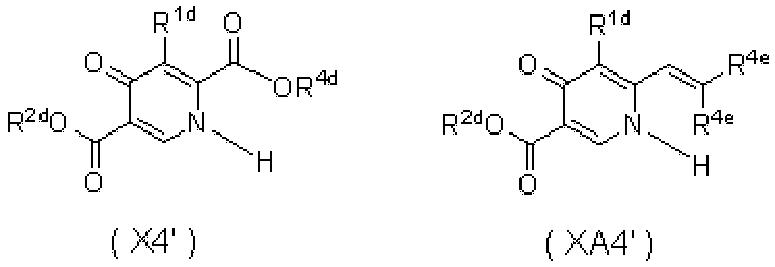
(式中、各記号は請求項1と同意義である)
で表わされる化合物、または式(XA4’)の誘導体と、式(V3’):

(式中、R6dは請求項1と同意義であり、
Ldは脱離基であり、ならびにPhはフェニル基を表わす)
で表わされる化合物を反応させる工程を含む、式(X4)もしくは式(XA4):
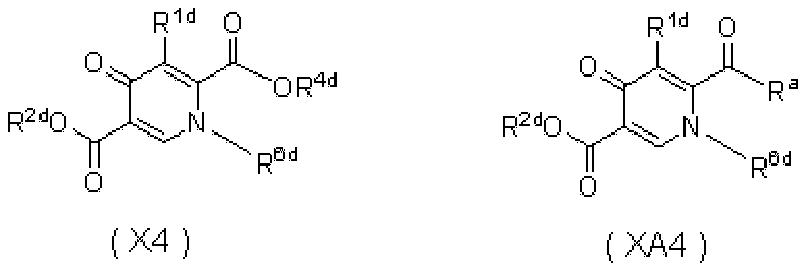
(式中、各記号は請求項1または請求項3と同意義である)
で表される化合物またはその塩の製造方法。
- 式(X2)で表わされる化合物が、式(X1):

(式中、各記号は請求項1と同意義である)
で表わされる化合物と、式(V1):

(式中、Pdは、置換基群Eで置換されていてもよい低級アルキルであり、ならびにR3dおよび置換基群Eは請求項1と同意義である)
で表わされる化合物とを反応させることによって得られる、請求項1、2、3、4または6に記載の製造方法。
- 式(X2)で表わされる化合物が、式(Z1):
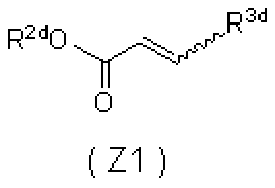
(式中、各記号は請求項1と同意義である)
で表わされる化合物と、式(Z2):
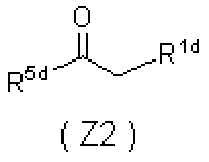
(式中、各記号は請求項1と同意義である)
で表わされる化合物とを反応させることによって得られる、請求項1、2、3、4または6に記載の製造方法。
- 式(X3):
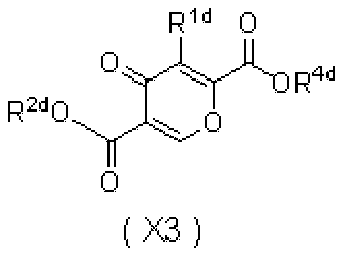
(式中、各記号は請求項1と同意義である)
表わされる化合物、もしくはその製薬上許容される塩またはそれらの溶媒和物。
- 式(X4):
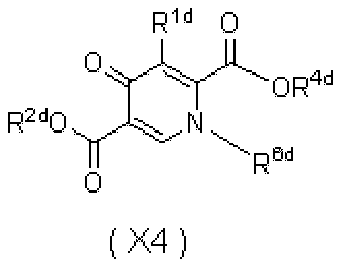
(式中、各記号は請求項1と同意義である)
表わされる化合物、もしくはその製薬上許容される塩またはそれらの溶媒和物。
- 式(X4’):
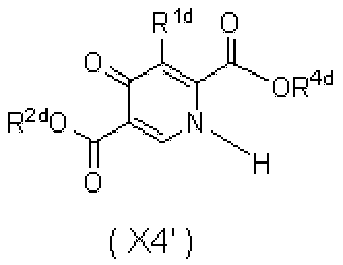
(式中、各記号は請求項1と同意義である)
表わされる化合物、もしくはその製薬上許容される塩またはそれらの溶媒和物。
- 粉末X線回折スペクトルにおいて、回折角度(2θ):7.9°±0.2°、10.0°±0.2°、11.5°±0.2°、20.0°±0.2°、23.4°±0.2°および34.0°±0.2°にピークを有する、式(1D):

(式中、Meはメチル基、およびBnはベンジル基を表す)
で示される化合物またはその溶媒和物の結晶。
- 粉末X線回折スペクトルにおいて、回折角度(2θ):17.6°±0.2°、25.2°±0.2°、26.4°±0.2°および28.1°±0.2°にピークを有する、式(2D):

(式中、Meはメチル基、Et基はエチル基、およびBnはベンジル基を表す)
で示される化合物またはその溶媒和物の結晶。
- 粉末X線回折スペクトルにおいて、回折角度(2θ):14.2°±0.2°、16.0°±0.2°、22.0°±0.2°、22.2°±0.2°、24.4°±0.2°および25.9°±0.2°にピークを有する、式(9C’):

(式中、Meはメチル基、およびBnはベンジル基を表す)
で示される化合物またはその溶媒和物の結晶。
- 図1に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、請求項13記載の結晶。
- 図2に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、請求項14記載の結晶。
- 図3に実質的に一致する粉末X線回折スペクトルにより特徴付けられる、請求項15記載の結晶。
Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| SG2011070208A SG174598A1 (en) | 2009-03-26 | 2010-03-26 | Process for producing pyrone and pyridone derivatives |
| JP2011506134A JP5632829B2 (ja) | 2009-03-26 | 2010-03-26 | ピロンおよびピリドン誘導体の製造方法 |
| KR1020117025156A KR101726888B1 (ko) | 2009-03-26 | 2010-03-26 | 피론 및 피리돈 유도체의 제조 방법 |
| ES10756205.0T ES2582211T3 (es) | 2009-03-26 | 2010-03-26 | Proceso para producir derivados de pirona y de piridona |
| EP10756205.0A EP2412709B1 (en) | 2009-03-26 | 2010-03-26 | Process for producing pyrone and pyridone derivatives |
| CN201080023795.8A CN102482219B (zh) | 2009-03-26 | 2010-03-26 | 吡喃酮和吡啶酮衍生物的制备方法 |
| US13/260,063 US8865907B2 (en) | 2009-03-26 | 2010-03-26 | Method of producing pyrone and pyridone derivatives |
| US14/478,685 US9260453B2 (en) | 2009-03-26 | 2014-09-05 | Method for producing pyrone and pyridone derivatives |
| US14/857,292 US9505783B2 (en) | 2009-03-26 | 2015-09-17 | Method of producing pyrone and pyridone derivatives |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009075290 | 2009-03-26 | ||
| JP2009-075290 | 2009-03-26 | ||
| JP2009142166 | 2009-06-15 | ||
| JP2009-142166 | 2009-06-15 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/260,063 A-371-Of-International US8865907B2 (en) | 2009-03-26 | 2010-03-26 | Method of producing pyrone and pyridone derivatives |
| US14/478,685 Division US9260453B2 (en) | 2009-03-26 | 2014-09-05 | Method for producing pyrone and pyridone derivatives |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010110409A1 true WO2010110409A1 (ja) | 2010-09-30 |
Family
ID=42781099
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/055316 WO2010110409A1 (ja) | 2009-03-26 | 2010-03-26 | ピロンおよびピリドン誘導体の製造方法 |
Country Status (9)
| Country | Link |
|---|---|
| US (3) | US8865907B2 (ja) |
| EP (2) | EP3109235A1 (ja) |
| JP (1) | JP5632829B2 (ja) |
| KR (1) | KR101726888B1 (ja) |
| CN (4) | CN105037259B (ja) |
| ES (1) | ES2582211T3 (ja) |
| SG (1) | SG174598A1 (ja) |
| TW (1) | TWI518084B (ja) |
| WO (1) | WO2010110409A1 (ja) |
Cited By (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012018065A1 (ja) * | 2010-08-05 | 2012-02-09 | 塩野義製薬株式会社 | Hivインテグラーゼ阻害活性を有する化合物の製造方法 |
| WO2013057251A2 (en) | 2011-10-21 | 2013-04-25 | F. Hoffmann-La Roche Ag | Heteroaryl hydroxamic acid derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| WO2013057253A1 (en) | 2011-10-21 | 2013-04-25 | F. Hoffmann-La Roche Ag | Pyrimidin-4-one derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| JP2013522366A (ja) * | 2010-03-23 | 2013-06-13 | ヴィーブ ヘルスケア カンパニー | カルバモイルピリドン誘導体及び中間体を調製するための方法 |
| US8552187B2 (en) | 2008-12-11 | 2013-10-08 | Shionogi & Co., Ltd. | Processes and intermediates for carbamoylpyridone HIV integrase inhibitors |
| US8580967B2 (en) | 2008-07-25 | 2013-11-12 | Shionogi & Co., Ltd. | Methyl 3-(benzyloxy)-1-(2,2-dihydroxyethyl)-4-oxo-1,4-dihydropyridine-2-carboxylate and processes for the preparation thereof |
| WO2013174931A1 (en) | 2012-05-23 | 2013-11-28 | Savira Pharmaceuticals Gmbh | 7-oxo-4,7 -dihydro- pyrazolo [1, 5 -a] pyrimidine derivatives which are useful in the treatment, amelioration or prevention of a viral disease |
| WO2013174930A2 (en) | 2012-05-23 | 2013-11-28 | Savira Pharmaceuticals Gmbh | 7-oxo-thiazolopyridine carbonic acid derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| US8624023B2 (en) | 2008-12-11 | 2014-01-07 | Shionogi & Co., Ltd. | Synthesis of carbamoylpyridone HIV integrase inhibitors and intermediates |
| WO2014023691A1 (en) | 2012-08-06 | 2014-02-13 | Savira Pharmaceuticals Gmbh | Dihydroxypyrimidine carbonic acid derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| WO2014108406A1 (en) | 2013-01-08 | 2014-07-17 | Savira Pharmaceuticals Gmbh | Pyrimidone derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| WO2014108408A1 (en) | 2013-01-08 | 2014-07-17 | Savira Pharmaceuticals Gmbh | Naphthyridinone derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| WO2014108407A1 (en) | 2013-01-08 | 2014-07-17 | Savira Pharmaceuticals Gmbh | Pyridone derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| WO2016005330A1 (en) | 2014-07-07 | 2016-01-14 | F. Hoffmann-La Roche Ag | Dihydropyridopyrazine-1,8-diones and their use in the treatment, amelioration or prevention of viral diseases |
| JP2016508134A (ja) * | 2012-12-21 | 2016-03-17 | ギリアード サイエンシーズ, インコーポレイテッド | 多環式カルバモイルピリドン化合物およびその薬学的用途 |
| WO2016128349A1 (en) | 2015-02-09 | 2016-08-18 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Antibodies specific to glycoprotein (gp) of ebolavirus and uses for the treatment and diagnosis of ebola virus infection |
| WO2017046362A1 (en) | 2015-09-18 | 2017-03-23 | F. Hoffmann-La Roche Ag | Pyrazolopyrazines and their use in the treatment, amelioration or prevention of a viral disease |
| WO2017046318A1 (en) | 2015-09-18 | 2017-03-23 | F. Hoffmann-La Roche Ag | Triazolones derivatives for use in the treatment, amelioration or prevention of a viral disease |
| WO2017046350A1 (en) | 2015-09-18 | 2017-03-23 | F. Hoffmann-La Roche Ag | Triazolones derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| WO2017060470A1 (en) | 2015-10-07 | 2017-04-13 | F. Hoffmann-La Roche Ag | Pyrrolopyrazine derivatives for use in the treatment, amelioration or prevention of influenza |
| US9682084B2 (en) | 2014-06-20 | 2017-06-20 | Gilead Sciences, Inc. | Crystalline forms of (2R,5S,13AR)-8-hydroxy-7,9,-dioxo-N-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide |
| KR20190017991A (ko) | 2016-06-20 | 2019-02-20 | 시오노기세야쿠 가부시키가이샤 | 치환된 다환성 피리돈 유도체의 제조 방법 및 그의 결정 |
| WO2019070059A1 (ja) | 2017-10-06 | 2019-04-11 | 塩野義製薬株式会社 | 置換された多環性ピリドン誘導体の立体選択的な製造方法 |
| US10456395B2 (en) | 2013-07-12 | 2019-10-29 | Gilead Sciences, Inc. | Substituted dipyrido[1,2-a:1′,2′-d]pyrazines for treating viral infections |
| US10519168B2 (en) | 2014-06-20 | 2019-12-31 | Gilead Sciences, Inc. | Synthesis of polycyclic-carbamoylpyridone compounds |
| US11098051B2 (en) | 2016-08-29 | 2021-08-24 | Novartis Ag | Fused tricyclic pyridazinone compounds useful to treat orthomyxovirus infections |
| WO2022107755A1 (ja) | 2020-11-17 | 2022-05-27 | 塩野義製薬株式会社 | 新規アクリジニウム塩およびその製造方法 |
| US11453677B2 (en) | 2016-03-08 | 2022-09-27 | Novartis Ag | Tricyclic compounds useful to treat orthomyxovirus infections |
| US11629149B2 (en) | 2018-02-28 | 2023-04-18 | Novartis Ag | 10-(di(phenyl)methyl)-4-hydroxy-8,9,9A,10-tetrahydro-7H-pyrrolo[1′,2′:4,5]pyrazino[1,2-b]pyridazine-3,5-dione derivatives and related compounds as inhibitors of the orthomyxovirus replication for treating influenza |
Families Citing this family (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI518084B (zh) * | 2009-03-26 | 2016-01-21 | 鹽野義製藥股份有限公司 | 哌喃酮與吡啶酮衍生物之製造方法 |
| PT3252058T (pt) | 2013-07-12 | 2021-03-09 | Gilead Sciences Inc | Compostos policíclicos-carbamoílpiridona e seu uso para o tratamento de infecções por hiv |
| US9856270B2 (en) | 2013-07-17 | 2018-01-02 | Ratiopharm Gmbh | Dolutegravir salts |
| EP3778603A1 (en) * | 2013-09-12 | 2021-02-17 | Janssen BioPharma, Inc. | 7,8-dihydro-3h-pyrazino[1,2-b]pyridazine-3,5(6h)-dione compounds and uses thereof |
| JP6360176B2 (ja) * | 2013-09-12 | 2018-07-18 | アリオス バイオファーマ インク. | ピリダジノン化合物およびその用途 |
| NO2717902T3 (ja) * | 2014-06-20 | 2018-06-23 | ||
| CA2953867A1 (en) * | 2014-07-07 | 2016-01-14 | F. Hoffmann-La Roche Ag | Pyridopyrazine compounds and their use in the treatment, amelioration or prevention of influenza |
| WO2016090545A1 (en) | 2014-12-09 | 2016-06-16 | Merck Sharp & Dohme Corp. | Spirocyclic heterocycle compounds useful as hiv integrate inhibitors |
| TWI738321B (zh) | 2014-12-23 | 2021-09-01 | 美商基利科學股份有限公司 | 多環胺甲醯基吡啶酮化合物及其醫藥用途 |
| US10730888B2 (en) * | 2015-02-06 | 2020-08-04 | Mylan Laboratories Limited | Process for the preparation of dolutegravir |
| CN107531717B (zh) | 2015-03-11 | 2021-07-27 | 詹森生物制药有限公司 | 氮杂-吡啶酮化合物及其用途 |
| ES2837383T3 (es) | 2015-04-02 | 2021-06-30 | Gilead Sciences Inc | Compuestos de carbamoilpiridonas policíclicos y su utilización farmacéutica |
| CN113004304B (zh) * | 2015-04-28 | 2022-03-18 | 盐野义制药株式会社 | 经取代的多环性吡啶酮衍生物及其前药 |
| LT3428170T (lt) * | 2015-04-28 | 2021-02-10 | Shionogi & Co., Ltd | Policiklinis piridono darinys nuo gripo ir jo provaistas |
| CN104860959B (zh) * | 2015-05-13 | 2017-01-18 | 中国科学院南海海洋研究所 | 一种α‑吡喃酮混源萜及其制备方法和应用 |
| WO2017109649A1 (en) | 2015-12-21 | 2017-06-29 | Lupin Limited | Process for the preparation of hiv integrase inhibitors |
| WO2017156194A1 (en) * | 2016-03-08 | 2017-09-14 | The Regents Of The University Of California | Compositions and methods for inhibiting influenza rna polymerase pa endonuclease |
| US10889556B2 (en) | 2016-03-08 | 2021-01-12 | The Regents Of The University Of California | Compositions and methods for inhibiting influenza RNA polymerase PA endonuclease |
| WO2018030463A1 (ja) | 2016-08-10 | 2018-02-15 | 塩野義製薬株式会社 | 置換された多環性ピリドン誘導体およびそのプロドラッグを含有する医薬組成物 |
| JOP20190130A1 (ar) | 2016-12-02 | 2019-06-02 | Merck Sharp & Dohme | مركبات حلقية غير متجانسة رباعية الحلقات مفيدة كمثبطات إنزيم مدمج لفيروس نقص المناعة البشرية (hiv) |
| CN109721615B (zh) | 2017-09-18 | 2020-12-29 | 广东东阳光药业有限公司 | 流感病毒复制抑制剂及其用途 |
| CN109867659A (zh) * | 2017-12-04 | 2019-06-11 | 江苏恒瑞医药股份有限公司 | 苯并哌啶类衍生物的制备方法 |
| WO2020112716A1 (en) * | 2018-11-26 | 2020-06-04 | Cocrystal Pharma, Inc. | Inhibitors of influenza virus replication |
| CN109912624B (zh) * | 2019-04-11 | 2021-05-11 | 杭州科巢生物科技有限公司 | 一种巴洛沙韦酯关键母核中间体的合成方法 |
| US20200398978A1 (en) | 2019-06-20 | 2020-12-24 | Bell Helicopter Textron Inc. | Low-drag rotor blade extension |
| CN115135646B (zh) * | 2019-12-23 | 2024-05-17 | 石家庄迪斯凯威医药科技有限公司 | 取代的多环化合物及其药物组合物和用途 |
| WO2021249522A1 (zh) * | 2020-06-12 | 2021-12-16 | 上海翰森生物医药科技有限公司 | 含吡啶酮稠环类衍生物抑制剂、其制备方法和应用 |
| CN112574155A (zh) * | 2020-11-25 | 2021-03-30 | 南京杰运医药科技有限公司 | 一种3-(苄氧基)-4-氧代-4h-吡喃-2-羧酸的合成方法 |
| CN112480007B (zh) * | 2020-12-08 | 2022-11-18 | 宿迁市科莱博生物化学有限公司 | 一种1,3-二甲基-1h-吡唑-4-羧酸的合成方法 |
| CN115572257B (zh) * | 2021-06-21 | 2024-07-09 | 江西帝劢药业有限公司 | 一种吡啶酮类化合物的合成方法 |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4769380A (en) * | 1983-04-29 | 1988-09-06 | Merrell Dow Pharmaceuticals Inc. | Cardiotonic 5-benzoyl-1,2-dihydro-2-oxo-3-pyridinecarboxylates |
| GB2280435A (en) | 1993-07-29 | 1995-02-01 | Merck & Co Inc | Anti-viral agent |
| WO2004024078A2 (en) | 2002-09-11 | 2004-03-25 | Merck & Co., Inc. | Dihydroxypyridopyrazine-1,6-dione compounds useful as hiv integrase inhibitors |
| WO2005016927A1 (ja) | 2003-08-13 | 2005-02-24 | Japan Tobacco Inc. | 含窒素縮合環化合物及びそのhivインテグラーゼ阻害剤としての利用 |
| WO2005087766A1 (en) | 2004-03-09 | 2005-09-22 | Istituto Di Ricerche Di Biologia Molecolare P Angeletti Spa | Hiv integrase inhibitors |
| WO2005092099A1 (en) | 2004-03-09 | 2005-10-06 | Merck & Co., Inc. | Hiv integrase inhibitors |
| WO2006011674A1 (en) | 2004-07-30 | 2006-02-02 | Matsushita Electric Works, Ltd. | Image processing device |
| WO2006030807A1 (ja) | 2004-09-15 | 2006-03-23 | Shionogi & Co., Ltd. | Hivインテグラーゼ阻害活性を有するカルバモイルピリドン誘導体 |
| WO2006066414A1 (en) | 2004-12-23 | 2006-06-29 | Virochem Pharma Inc. | Hydroxydihydropyridopy razine-1,8-diones and methods for inhibiting hiv integrase |
| WO2006088173A1 (ja) | 2005-02-21 | 2006-08-24 | Shionogi & Co., Ltd. | Hivインテグラーゼ阻害活性を有する2環性カルバモイルピリドン誘導体 |
| WO2006116764A1 (en) | 2005-04-28 | 2006-11-02 | Smithkline Beecham Corporation | Polycyclic carbamoylpyridone derivative having hiv integrase inhibitory activity |
| JP2006342115A (ja) | 2005-06-10 | 2006-12-21 | Shionogi & Co Ltd | Hivインテグラーゼ阻害活性を有する多環性化合物 |
| JP2007509850A (ja) * | 2003-10-23 | 2007-04-19 | バイエル・クロツプサイエンス・アクチエンゲゼルシヤフト | 2−ジハロアシル−3−アミノ−アクリル酸エステルおよび3−ジハロメチル−ピラゾール−4−カルボン酸エステルの製造方法 |
| WO2007049675A1 (ja) | 2005-10-27 | 2007-05-03 | Shionogi & Co., Ltd. | Hivインテグラーゼ阻害活性を有する多環性カルバモイルピリドン誘導体 |
Family Cites Families (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4210758A (en) * | 1979-03-21 | 1980-07-01 | Warner-Lambert Company | 1,5-Dihydro-1,5-dioxo-N-1H-tetrazol-5-yl-4H-[1]benzopyrano[3,4-b]pyridine-2-carboxamides |
| US4524149A (en) | 1982-03-15 | 1985-06-18 | Sterling Drug Inc. | 5-Alkanoyl-6-alkyl-2(1H)-pyridinones, their preparation and their cardiotonic use |
| US4735964A (en) | 1984-08-16 | 1988-04-05 | G. D. Searle & Co. | Kojic acid ether-ester derivatives |
| US4603144A (en) | 1984-08-16 | 1986-07-29 | G. D. Searle & Co. | Kojic acid ether-ester derivatives |
| US4812474A (en) | 1984-08-16 | 1989-03-14 | G. D. Searle & Co. | Kojic acid ether-ester derivatives |
| DE4405722A1 (de) * | 1994-02-23 | 1995-08-24 | Cassella Ag | Verfahren zur Herstellung von 6-Aryloxymethyl-1-hydroxy-4-methyl-2-pyridonen |
| US5849587A (en) * | 1995-06-09 | 1998-12-15 | Cornell Research Foundation, Inc. | Method of inhibiting viral replication in eukaryotic cells and of inducing apoptosis of virally-infected cells |
| DE59611243D1 (de) | 1995-09-29 | 2005-08-11 | Novartis Ag | Hydroxypyridinone |
| GB9711093D0 (en) | 1997-05-29 | 1997-07-23 | British Tech Group | Novel orally active iron (III) chelators |
| US6452008B2 (en) | 1998-02-25 | 2002-09-17 | Sumitomo Pharmaceuticals Company, Limited | Pyridone derivatives and process for preparing the same |
| US7256286B2 (en) | 1999-11-30 | 2007-08-14 | The Board Of Trustees Of The Leland Stanford Junior University | Bryostatin analogues, synthetic methods and uses |
| JP2005505525A (ja) | 2001-08-02 | 2005-02-24 | ニューロクライン バイオサイエンシーズ, インコーポレイテッド | 性腺刺激ホルモン放出ホルモンレセプターアンタゴニストおよびそれに関連する方法 |
| US6426418B1 (en) | 2001-11-02 | 2002-07-30 | Apotex, Inc. | Processes for the manufacturing of 3-hydroxy-N,1,6-trialkyl-4-oxo-1,4-dihydropyridine-2-carboxamide |
| DE10161978A1 (de) * | 2001-12-17 | 2003-06-26 | Bayer Ag | Verfahren zur Herstellung von 2-Halogenacyl-3-amino-acrylsäure-derivate |
| CA2379370A1 (en) | 2002-03-28 | 2003-09-28 | Apotex Inc. | Carboxylic acid derivatives of 3-hydroxy-4-oxo-1,4-dihydropyridine as iron chelators |
| AU2003301439A1 (en) | 2002-10-16 | 2004-05-04 | Gilead Sciences, Inc. | Pre-organized tricyclic integrase inhibitor compounds |
| EP1471063A1 (en) * | 2003-02-28 | 2004-10-27 | Exonhit Therapeutics S.A. | Compounds and methods of treating cell proliferative diseases, retinopathies and arthritis |
| JP2006232849A (ja) * | 2003-08-13 | 2006-09-07 | Japan Tobacco Inc | 含窒素縮合環化合物及びそのhivインテグラーゼ阻害剤としての利用 |
| AU2005249363A1 (en) | 2004-04-14 | 2005-12-15 | Zhenhong R. Cai | Phosphonate analogs of HIV integrase inhibitor compounds |
| CA2488034C (en) | 2004-11-19 | 2009-10-06 | Apotex Inc. | Process for the manufacture of 3-hydroxy-n-alkyl-1-cycloalkyl-6-alkyl-4-oxo-1,4-dihydropyridine-2-carboxamide and its related analogues |
| AR057023A1 (es) | 2005-05-16 | 2007-11-14 | Gilead Sciences Inc | Compuestos heterociclicos con propiedades inhibidoras de hiv-integrasa |
| EP1762250A1 (en) | 2005-09-12 | 2007-03-14 | Fresenius Kabi Deutschland GmbH | Conjugates of hydroxyalkyl starch and an active substance, prepared by chemical ligation via thiazolidine |
| US8314087B2 (en) | 2007-02-16 | 2012-11-20 | Amgen Inc. | Nitrogen-containing heterocyclyl ketones and methods of use |
| KR101695807B1 (ko) | 2008-07-25 | 2017-01-13 | 비이브 헬쓰케어 컴퍼니 | 화합물 |
| PT2320908E (pt) | 2008-07-25 | 2014-03-06 | Shionogi & Co | Pró-fármacos de dolutegravir |
| ES2449752T3 (es) | 2008-07-25 | 2014-03-21 | Viiv Healthcare Company | Proceso para la preparación de un derivado de pirido[1,2-a]pirrolo[1',2':3,4]imidazo[1,2-d]piracin-8-carboxamida |
| MX2011006241A (es) | 2008-12-11 | 2011-06-28 | Shionogi & Co | Sintesis de inhibidores de integrasa de vih de carbamoil-piridona e intermediarios. |
| ES2964383T3 (es) | 2008-12-11 | 2024-04-05 | Viiv Healthcare Co | Procesos e intermedios para inhibidores de la integrasa del VIH de carbamoilpiridona |
| TWI518084B (zh) * | 2009-03-26 | 2016-01-21 | 鹽野義製藥股份有限公司 | 哌喃酮與吡啶酮衍生物之製造方法 |
| JP5697163B2 (ja) * | 2009-03-26 | 2015-04-08 | 塩野義製薬株式会社 | 置換された3−ヒドロキシ−4−ピリドン誘導体 |
| WO2010147068A1 (ja) * | 2009-06-15 | 2010-12-23 | 塩野義製薬株式会社 | 置換された多環性カルバモイルピリドン誘導体 |
| TWI582097B (zh) | 2010-03-23 | 2017-05-11 | Viiv醫療保健公司 | 製備胺甲醯吡啶酮衍生物及中間體之方法 |
| CN103154004B (zh) | 2010-08-05 | 2016-07-06 | 盐野义制药株式会社 | 具有hiv整合酶抑制活性的化合物的制造方法 |
| US8902740B2 (en) * | 2011-11-10 | 2014-12-02 | At&T Intellectual Property I, L.P. | Methods, systems, and products for security services |
-
2010
- 2010-03-25 TW TW099108871A patent/TWI518084B/zh active
- 2010-03-26 US US13/260,063 patent/US8865907B2/en active Active
- 2010-03-26 KR KR1020117025156A patent/KR101726888B1/ko active IP Right Grant
- 2010-03-26 JP JP2011506134A patent/JP5632829B2/ja active Active
- 2010-03-26 CN CN201510349249.8A patent/CN105037259B/zh active Active
- 2010-03-26 SG SG2011070208A patent/SG174598A1/en unknown
- 2010-03-26 EP EP16164003.2A patent/EP3109235A1/en not_active Withdrawn
- 2010-03-26 CN CN201410403480.6A patent/CN104230795B/zh active Active
- 2010-03-26 WO PCT/JP2010/055316 patent/WO2010110409A1/ja active Application Filing
- 2010-03-26 CN CN201080023795.8A patent/CN102482219B/zh active Active
- 2010-03-26 EP EP10756205.0A patent/EP2412709B1/en active Active
- 2010-03-26 CN CN201410403411.5A patent/CN104151234B/zh active Active
- 2010-03-26 ES ES10756205.0T patent/ES2582211T3/es active Active
-
2014
- 2014-09-05 US US14/478,685 patent/US9260453B2/en active Active
-
2015
- 2015-09-17 US US14/857,292 patent/US9505783B2/en active Active
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4769380A (en) * | 1983-04-29 | 1988-09-06 | Merrell Dow Pharmaceuticals Inc. | Cardiotonic 5-benzoyl-1,2-dihydro-2-oxo-3-pyridinecarboxylates |
| GB2280435A (en) | 1993-07-29 | 1995-02-01 | Merck & Co Inc | Anti-viral agent |
| WO2004024078A2 (en) | 2002-09-11 | 2004-03-25 | Merck & Co., Inc. | Dihydroxypyridopyrazine-1,6-dione compounds useful as hiv integrase inhibitors |
| WO2005016927A1 (ja) | 2003-08-13 | 2005-02-24 | Japan Tobacco Inc. | 含窒素縮合環化合物及びそのhivインテグラーゼ阻害剤としての利用 |
| JP2007509850A (ja) * | 2003-10-23 | 2007-04-19 | バイエル・クロツプサイエンス・アクチエンゲゼルシヤフト | 2−ジハロアシル−3−アミノ−アクリル酸エステルおよび3−ジハロメチル−ピラゾール−4−カルボン酸エステルの製造方法 |
| WO2005087766A1 (en) | 2004-03-09 | 2005-09-22 | Istituto Di Ricerche Di Biologia Molecolare P Angeletti Spa | Hiv integrase inhibitors |
| WO2005092099A1 (en) | 2004-03-09 | 2005-10-06 | Merck & Co., Inc. | Hiv integrase inhibitors |
| WO2006011674A1 (en) | 2004-07-30 | 2006-02-02 | Matsushita Electric Works, Ltd. | Image processing device |
| WO2006030807A1 (ja) | 2004-09-15 | 2006-03-23 | Shionogi & Co., Ltd. | Hivインテグラーゼ阻害活性を有するカルバモイルピリドン誘導体 |
| WO2006066414A1 (en) | 2004-12-23 | 2006-06-29 | Virochem Pharma Inc. | Hydroxydihydropyridopy razine-1,8-diones and methods for inhibiting hiv integrase |
| WO2006088173A1 (ja) | 2005-02-21 | 2006-08-24 | Shionogi & Co., Ltd. | Hivインテグラーゼ阻害活性を有する2環性カルバモイルピリドン誘導体 |
| WO2006116764A1 (en) | 2005-04-28 | 2006-11-02 | Smithkline Beecham Corporation | Polycyclic carbamoylpyridone derivative having hiv integrase inhibitory activity |
| JP2006342115A (ja) | 2005-06-10 | 2006-12-21 | Shionogi & Co Ltd | Hivインテグラーゼ阻害活性を有する多環性化合物 |
| WO2007049675A1 (ja) | 2005-10-27 | 2007-05-03 | Shionogi & Co., Ltd. | Hivインテグラーゼ阻害活性を有する多環性カルバモイルピリドン誘導体 |
Non-Patent Citations (21)
| Title |
|---|
| ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, December 1994 (1994-12-01), pages 2827 - 2837 |
| ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, May 1996 (1996-05-01), pages 1304 - 1307 |
| BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 17, 2007, pages 5595 - 5599 |
| BIOORGANIC MEDICINAL CHEMISTRY LETTERS, vol. 17, 2007, pages 1713 |
| BIOORGANIC MEDICINAL CHEMISTRY LETTERS, vol. 17, 2027, pages 1713 |
| DEJOHN, D. ET AL.: "Functionalization of substituted 2(1H)- and 4(lH)-pyridones. III. The preparation of substituted 6-vinyl-1,2- dihydro-2-oxo and 1,4-dihydro-4-oxo-3- pyridinecarboxylic acids through the chemistry of pyridone dianions", JOURNAL OF HETEROCYCLIC CHEMISTRY, vol. 20, no. 5, 1983, pages 1295 - 1302, XP002944109 * |
| GROUNDWATER, P.W. ET AL.: "Synthesis and reactions of reduced flavones", JOURNAL OF CHEMICAL SOCIETY PARKIN TRANSACTION.1, 1997, pages 163 - 169, XP055089025 * |
| J. MED. CHEM., vol. 46, 2003, pages 1153 - 1164 |
| JOURNAL OF ORGANIC CHEMISTRY, vol. 25, 1960, pages 1052 - 1053 |
| JOURNAL OF ORGANIC CHEMISTRY, vol. 56, 1991, pages 4963 - 4967 |
| JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 128, 2006, pages 10694 |
| JOURNAL OF THE AMERICAN CHEMICAL SOCIETY; ENGLISH, vol. 127, no. 30, 2005, pages 10504 |
| KELLER, P.A.: "Product class 2: pyridinones and related systems", SCIENCE OF SYNTHESIS, vol. 15, 2005, pages 285 - 387, XP008111586 * |
| MCCOMBIE, S.W. ET AL.: "Generation and in situ acylation of enaminone anions: a convenient synthesis of 3-carbethoxy-4(lH)-pyridinones and -4-pyrones and related compounds", JOURNAL OF ORGANIC CHEMISTRY, vol. 56, no. 16, 1991, pages 4963 - 4967, XP055089020 * |
| OLGA M. ROCHOVANSKY, VIROLOGY, vol. 73, 1976, pages 327 - 338 |
| ROSS, W.J. ET AL.: "The Synthesis and Rearrangement of Epoxypyrones", TETRAHEDRON LETTERS, vol. 22, no. 23, 1981, pages 2207 - 2208, XP002628302 * |
| TETRAHEDORON LETTERS, vol. 48, 2007, pages 473 |
| TETRAHEDRON LETT, vol. 22, no. 23, 1981, pages 2207 |
| TETRAHEDRON LETT, vol. 36, no. 12, 1995 |
| THEODORA W GREEN: "Protective Groups in Organic Synthesis", JOHN WILEY & SONS |
| THEODORA W GREEN: "Protective Groups in Organic Synthesis", JOHN WILEY AND SONS |
Cited By (83)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8765965B2 (en) | 2008-07-25 | 2014-07-01 | Shionogi & Co., Ltd. | 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1,4-dihydro-2-pyridinecarboxylic acid of the formula P-6 and/or methyl 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1,4-dihydro-2-pyridinecarboxylate of the formula P-7 |
| US9012650B2 (en) | 2008-07-25 | 2015-04-21 | Shionogi & Co., Ltd. | Substituted (3S, 11aR)-N-[(2,4-difluorophenyl)methyl]-6-oxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamides of formula (I) useful as HIV agents |
| US8981129B2 (en) | 2008-07-25 | 2015-03-17 | Shionogi & Co., Ltd. | 2-(2-hydroxy-2-phenylethyl)-3-[(phenylmethyl)oxy]-4H-pyran-4-one of the formula P-3 and/or 2-[(E)-2-phenylethenyl]-3-[(phenylmethyl)oxy]-4H-Pyran-4-one of the formula P-4 |
| US8940912B2 (en) | 2008-07-25 | 2015-01-27 | Viiv Healthcare Company | 4-oxo-3-[(phenylmethyl)oxy]-4H-pyran-2-carboxylic acid |
| US9133216B2 (en) | 2008-07-25 | 2015-09-15 | Shionogi & Co., Ltd. | (3S,11aR)-6-[(phenylmethyl)oxy]-3-methyl-2,3,11,11a-tetrahydrooxazolo[3,2-a]pyrido[1,2-d]pyrazine-5,7-dione of the formula P-9 and/or (3S,11aR)-6-[(phenymethyl)oxy]-8-bromo-3-methyl-2,3,11,11a-tetrahydrooxazolo[3,2-a]pyrido[1,2-d]pyrazine-5,7-dione of the formula P-10 |
| US8580967B2 (en) | 2008-07-25 | 2013-11-12 | Shionogi & Co., Ltd. | Methyl 3-(benzyloxy)-1-(2,2-dihydroxyethyl)-4-oxo-1,4-dihydropyridine-2-carboxylate and processes for the preparation thereof |
| US9707246B2 (en) | 2008-07-25 | 2017-07-18 | Shionogi & Co., Ltd. | Substituted (3S,11AR)-N-[(2,4-difluorophenyl)methyl]-6-oxy-3-methyl-5,7-dioxo-2,3,5,7,11,11A-hexahydro[1,3]oxazolo[3,2-A]pyrido[1,2-D]pyrazine-8-carboxamides as HIV agents |
| US9242986B2 (en) | 2008-12-11 | 2016-01-26 | Shionogi & Co., Ltd. | Synthesis of carbamoylpyridone HIV integrase inhibitors and intermediates |
| US8552187B2 (en) | 2008-12-11 | 2013-10-08 | Shionogi & Co., Ltd. | Processes and intermediates for carbamoylpyridone HIV integrase inhibitors |
| US9365587B2 (en) | 2008-12-11 | 2016-06-14 | Shionogi & Co., Ltd. | Synthesis of carbamoylpyridone HIV integrase inhibitors and intermediates |
| US8754214B2 (en) | 2008-12-11 | 2014-06-17 | Shionogi & Co., Ltd. | Synthesis of carbamoylpyridone HIV integrase inhibitors and intermediates |
| US8624023B2 (en) | 2008-12-11 | 2014-01-07 | Shionogi & Co., Ltd. | Synthesis of carbamoylpyridone HIV integrase inhibitors and intermediates |
| US8669362B2 (en) | 2008-12-11 | 2014-03-11 | Shiongi & Co., Ltd. | Processes and Intermediates for carbamoylpyridone HIV integrase inhibitors |
| US10233196B2 (en) | 2010-03-23 | 2019-03-19 | Viiv Healthcare Company | Process for preparing substituted pyridinones as intermediates in the preparation of polycyclic carbamoylpyridone derivatives |
| US9938296B2 (en) | 2010-03-23 | 2018-04-10 | Viiv Healthcare Company | Process for preparing (4R,12aS)-N-(2,4-difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide |
| US9643981B2 (en) | 2010-03-23 | 2017-05-09 | Viiv Healthcare Company | Process for preparing (4R,12aS)-N-(2,4-difluorobenzyl)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12A-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide |
| US10174051B2 (en) | 2010-03-23 | 2019-01-08 | Viiv Healthcare Company | Substituted pyridinones as intermediates in the preparation of polycyclic carbamoylpyridone derivatives |
| US9120817B2 (en) | 2010-03-23 | 2015-09-01 | Viiv Healthcare Company | Process for preparing (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′ ,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxylic acid |
| US8889877B2 (en) | 2010-03-23 | 2014-11-18 | Viiv Healthcare Company | Processes for preparing pyridinone carboxylic acid aldehydes |
| JP2013522366A (ja) * | 2010-03-23 | 2013-06-13 | ヴィーブ ヘルスケア カンパニー | カルバモイルピリドン誘導体及び中間体を調製するための方法 |
| US10647728B2 (en) | 2010-03-23 | 2020-05-12 | Viiv Healthcare Company | Process for preparing (3S,11aR)-6-methoxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-Hexahydrooxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxylic acid |
| US10654870B2 (en) | 2010-03-23 | 2020-05-19 | Viiv Healthcare Company | Process for preparing substituted pyridinones as intermediates in the preparation of polycyclic carbamoylpyridone derivatives |
| US10654871B2 (en) | 2010-03-23 | 2020-05-19 | Viiv Healthcare Company | Process for preparing (3S,11aR)-N-(2,4-difluorobenzyl)-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydrooxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide |
| US9650394B2 (en) | 2010-08-05 | 2017-05-16 | Shionogi & Co., Ltd. | Methods of producing substituted (3S,11aR)-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-Hexahydrooxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamides |
| WO2012018065A1 (ja) * | 2010-08-05 | 2012-02-09 | 塩野義製薬株式会社 | Hivインテグラーゼ阻害活性を有する化合物の製造方法 |
| JPWO2012018065A1 (ja) * | 2010-08-05 | 2013-10-03 | 塩野義製薬株式会社 | Hivインテグラーゼ阻害活性を有する化合物の製造方法 |
| US10125147B2 (en) | 2010-08-05 | 2018-11-13 | Shionogi & Co., Ltd. | Crystalline methyl (4R,12aS)-7-(benzyloxy)-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxylate |
| US10125146B2 (en) | 2010-08-05 | 2018-11-13 | Shionogi & Co., Ltd. | Crystalline methyl (4R,12aS)-7-(benzyloxy)-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxylate |
| US9321789B2 (en) | 2010-08-05 | 2016-04-26 | Shinogi & Co., Ltd. | Methods of producing substituted (4R,12AS)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12A-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-B][1,3]oxazine-9-carboxamides |
| CN103154004A (zh) * | 2010-08-05 | 2013-06-12 | 盐野义制药株式会社 | 具有hiv整合酶抑制活性的化合物的制造方法 |
| CN106046022B (zh) * | 2010-08-05 | 2018-06-19 | 盐野义制药株式会社 | 具有hiv整合酶抑制活性的化合物的制造方法 |
| US10000508B2 (en) | 2010-08-05 | 2018-06-19 | Shionogi & Co., Ltd | Crystalline dimethyl 3-(benzyloxy)-1-(2,2-dimethoxyethyl)-4-oxo-1,4-dihydropyridine-2,5-dicarboxylate |
| CN106046022A (zh) * | 2010-08-05 | 2016-10-26 | 盐野义制药株式会社 | 具有hiv整合酶抑制活性的化合物的制造方法 |
| CN106083891A (zh) * | 2010-08-05 | 2016-11-09 | 盐野义制药株式会社 | 具有hiv整合酶抑制活性的化合物的制造方法 |
| EP3127908A3 (en) * | 2010-08-05 | 2017-02-15 | Shionogi & Co., Ltd | Process for preparing compound having hiv integrase inhibitory activity |
| US9969750B2 (en) | 2010-08-05 | 2018-05-15 | Shionogi And Co., Ltd. | Method of producing (3S,11aR)-6,8-dihalo-3-methyl-2,3,11,11a-tetrahydrooxazolo[3,2-a]pyrido[1,2-d]pyrazine-5,7-diones |
| JP5636054B2 (ja) * | 2010-08-05 | 2014-12-03 | 塩野義製薬株式会社 | Hivインテグラーゼ阻害活性を有する化合物の製造方法 |
| CN106083891B (zh) * | 2010-08-05 | 2018-03-23 | 盐野义制药株式会社 | 具有hiv整合酶抑制活性的化合物的制造方法 |
| US9802959B2 (en) | 2010-08-05 | 2017-10-31 | Shionogi & Co., Ltd. | Method of producing (4R,12aS)-7,9-dihalo-4-methyl-3,4,12,12a-tetrahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-6,8-diones |
| EP3456721A3 (en) * | 2010-08-05 | 2019-03-27 | Shionogi & Co., Ltd | Method of producing compounds having hiv integrase inhivitory activity |
| WO2013057253A1 (en) | 2011-10-21 | 2013-04-25 | F. Hoffmann-La Roche Ag | Pyrimidin-4-one derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| WO2013057251A2 (en) | 2011-10-21 | 2013-04-25 | F. Hoffmann-La Roche Ag | Heteroaryl hydroxamic acid derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| WO2013174930A2 (en) | 2012-05-23 | 2013-11-28 | Savira Pharmaceuticals Gmbh | 7-oxo-thiazolopyridine carbonic acid derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| WO2013174931A1 (en) | 2012-05-23 | 2013-11-28 | Savira Pharmaceuticals Gmbh | 7-oxo-4,7 -dihydro- pyrazolo [1, 5 -a] pyrimidine derivatives which are useful in the treatment, amelioration or prevention of a viral disease |
| WO2014023691A1 (en) | 2012-08-06 | 2014-02-13 | Savira Pharmaceuticals Gmbh | Dihydroxypyrimidine carbonic acid derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| JP2016179996A (ja) * | 2012-12-21 | 2016-10-13 | ギリアード サイエンシス インコーポレーテッド | 多環式カルバモイルピリドン化合物およびその薬学的用途 |
| US10689399B2 (en) | 2012-12-21 | 2020-06-23 | Gilead Sciences, Inc. | Substituted 3,4,5,6,8,10,14,14a-octahydro-2h-2,6-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazocines and methods for treating viral infections |
| US11548901B2 (en) | 2012-12-21 | 2023-01-10 | Gilead Sciences, Inc. | Substituted 1,4-methanopyrido[1′,2′:4,5]pyrazino[1,2-a]pyrimidines for treating viral infections |
| US10035809B2 (en) | 2012-12-21 | 2018-07-31 | Gilead Sciences, Inc. | Substituted 2,3,4,5,7,9,13,13a-octahydro-1,5-methanopyrido[1′,2′:4,5]pyrazino[1,2-a][1,3]diazepines and methods for treating viral infections |
| JP2016508134A (ja) * | 2012-12-21 | 2016-03-17 | ギリアード サイエンシーズ, インコーポレイテッド | 多環式カルバモイルピリドン化合物およびその薬学的用途 |
| US9732092B2 (en) | 2012-12-21 | 2017-08-15 | Gilead Sciences, Inc. | Substituted 2,3,4,5,7,9,13,13a-octahydropyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]OXAZEPINES and methods for treating viral infections |
| JP2019023204A (ja) * | 2012-12-21 | 2019-02-14 | ギリアード サイエンシス インコーポレーテッド | 多環式カルバモイルピリドン化合物およびその薬学的用途 |
| WO2014108406A1 (en) | 2013-01-08 | 2014-07-17 | Savira Pharmaceuticals Gmbh | Pyrimidone derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| WO2014108408A1 (en) | 2013-01-08 | 2014-07-17 | Savira Pharmaceuticals Gmbh | Naphthyridinone derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| WO2014108407A1 (en) | 2013-01-08 | 2014-07-17 | Savira Pharmaceuticals Gmbh | Pyridone derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| US11213523B2 (en) | 2013-07-12 | 2022-01-04 | Gilead Sciences, Inc. | Substituted pyrido[1,2-a]pyrrolo[1,2-d]pyrazines for treating viral infections |
| US10456395B2 (en) | 2013-07-12 | 2019-10-29 | Gilead Sciences, Inc. | Substituted dipyrido[1,2-a:1′,2′-d]pyrazines for treating viral infections |
| US11883397B2 (en) | 2013-07-12 | 2024-01-30 | Gilead Sciences, Inc. | Substituted pyrido[1,2-a]pyrrolo[1,2-d]pyrazines for treating viral infections |
| US10975096B2 (en) | 2014-06-20 | 2021-04-13 | Gilead Sciences, Inc. | Synthesis of polycyclic-carbamoylpyridone compounds |
| US11202780B2 (en) | 2014-06-20 | 2021-12-21 | Gilead Sciences, Inc. | Crystalline forms of (2R,5S,13aR)-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide |
| US10519168B2 (en) | 2014-06-20 | 2019-12-31 | Gilead Sciences, Inc. | Synthesis of polycyclic-carbamoylpyridone compounds |
| US10098886B2 (en) | 2014-06-20 | 2018-10-16 | Gilead Sciences, Inc. | Crystalline forms of (2R,5S,13AR)-8-hydroxy-7,9-dioxo-N-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13A- octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-B] [1,3] oxazepine-10-carboxamide |
| US9682084B2 (en) | 2014-06-20 | 2017-06-20 | Gilead Sciences, Inc. | Crystalline forms of (2R,5S,13AR)-8-hydroxy-7,9,-dioxo-N-(2,4,6-trifluorobenzyl)-2,3,4,5,7,9,13,13a-octahydro-2,5-methanopyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide |
| WO2016005330A1 (en) | 2014-07-07 | 2016-01-14 | F. Hoffmann-La Roche Ag | Dihydropyridopyrazine-1,8-diones and their use in the treatment, amelioration or prevention of viral diseases |
| WO2016128349A1 (en) | 2015-02-09 | 2016-08-18 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Antibodies specific to glycoprotein (gp) of ebolavirus and uses for the treatment and diagnosis of ebola virus infection |
| WO2017046318A1 (en) | 2015-09-18 | 2017-03-23 | F. Hoffmann-La Roche Ag | Triazolones derivatives for use in the treatment, amelioration or prevention of a viral disease |
| WO2017046350A1 (en) | 2015-09-18 | 2017-03-23 | F. Hoffmann-La Roche Ag | Triazolones derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| WO2017046362A1 (en) | 2015-09-18 | 2017-03-23 | F. Hoffmann-La Roche Ag | Pyrazolopyrazines and their use in the treatment, amelioration or prevention of a viral disease |
| WO2017060470A1 (en) | 2015-10-07 | 2017-04-13 | F. Hoffmann-La Roche Ag | Pyrrolopyrazine derivatives for use in the treatment, amelioration or prevention of influenza |
| US11453677B2 (en) | 2016-03-08 | 2022-09-27 | Novartis Ag | Tricyclic compounds useful to treat orthomyxovirus infections |
| EP4194459A1 (en) | 2016-06-20 | 2023-06-14 | Shionogi & Co., Ltd | Process for preparing substituted polycyclic pyridone derivative and crystal thereof |
| US11261198B2 (en) | 2016-06-20 | 2022-03-01 | Shionogi & Co., Ltd. | Process for preparing substituted polycyclic pyridone derivative and crystal thereof |
| US11807648B2 (en) | 2016-06-20 | 2023-11-07 | Shionogi & Co., Ltd. | Process for preparing substituted polycyclic pyridone derivative and crystal thereof |
| KR20220084425A (ko) | 2016-06-20 | 2022-06-21 | 시오노기세야쿠 가부시키가이샤 | 치환된 다환성 피리돈 유도체의 제조 방법 및 그의 결정 |
| KR20190017991A (ko) | 2016-06-20 | 2019-02-20 | 시오노기세야쿠 가부시키가이샤 | 치환된 다환성 피리돈 유도체의 제조 방법 및 그의 결정 |
| US11098051B2 (en) | 2016-08-29 | 2021-08-24 | Novartis Ag | Fused tricyclic pyridazinone compounds useful to treat orthomyxovirus infections |
| US11912715B2 (en) | 2016-08-29 | 2024-02-27 | Novartis Ag | Fused tricyclic pyridazinone compounds useful to treat orthomyxovirus infections |
| KR20200066644A (ko) | 2017-10-06 | 2020-06-10 | 시오노기세야쿠 가부시키가이샤 | 치환된 다환성 피리돈 유도체의 입체 선택적인 제조 방법 |
| US11286262B2 (en) | 2017-10-06 | 2022-03-29 | Shionogi & Co., Ltd. | Stereoselective process for preparing substituted polycyclic pyridone derivatives |
| WO2019070059A1 (ja) | 2017-10-06 | 2019-04-11 | 塩野義製薬株式会社 | 置換された多環性ピリドン誘導体の立体選択的な製造方法 |
| US11629149B2 (en) | 2018-02-28 | 2023-04-18 | Novartis Ag | 10-(di(phenyl)methyl)-4-hydroxy-8,9,9A,10-tetrahydro-7H-pyrrolo[1′,2′:4,5]pyrazino[1,2-b]pyridazine-3,5-dione derivatives and related compounds as inhibitors of the orthomyxovirus replication for treating influenza |
| US12071441B2 (en) | 2018-02-28 | 2024-08-27 | Novartis Ag | 10-(di(phenyl)methyl)-4-hydroxy-8,9,9A,10-tetrahydro-7H-pyrrolo[1′,2′:4,5]pyrazino[1,2-b]pyridazine-3,5-dione derivatives and related compounds as inhibitors of the orthomyxovirus replication for treating influenza |
| WO2022107755A1 (ja) | 2020-11-17 | 2022-05-27 | 塩野義製薬株式会社 | 新規アクリジニウム塩およびその製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN104230795A (zh) | 2014-12-24 |
| KR20110132462A (ko) | 2011-12-07 |
| EP2412709A4 (en) | 2013-11-27 |
| US20120022251A1 (en) | 2012-01-26 |
| EP3109235A1 (en) | 2016-12-28 |
| CN105037259A (zh) | 2015-11-11 |
| CN105037259B (zh) | 2018-03-13 |
| EP2412709A1 (en) | 2012-02-01 |
| TWI518084B (zh) | 2016-01-21 |
| ES2582211T3 (es) | 2016-09-09 |
| US20150031876A1 (en) | 2015-01-29 |
| JPWO2010110409A1 (ja) | 2012-10-04 |
| US20160002211A1 (en) | 2016-01-07 |
| KR101726888B1 (ko) | 2017-04-13 |
| SG174598A1 (en) | 2011-10-28 |
| US8865907B2 (en) | 2014-10-21 |
| CN104151234A (zh) | 2014-11-19 |
| JP5632829B2 (ja) | 2014-11-26 |
| US9505783B2 (en) | 2016-11-29 |
| CN102482219A (zh) | 2012-05-30 |
| TW201038570A (en) | 2010-11-01 |
| US9260453B2 (en) | 2016-02-16 |
| EP2412709B1 (en) | 2016-05-18 |
| CN102482219B (zh) | 2015-07-22 |
| CN104230795B (zh) | 2017-09-26 |
| CN104151234B (zh) | 2017-04-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5632829B2 (ja) | ピロンおよびピリドン誘導体の製造方法 | |
| JP6004552B2 (ja) | 置換された多環性カルバモイルピリドン誘導体 | |
| US9802959B2 (en) | Method of producing (4R,12aS)-7,9-dihalo-4-methyl-3,4,12,12a-tetrahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-6,8-diones | |
| CA2812363C (en) | Substituted polycyclic carbamoyl pyridone derivative prodrug |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080023795.8 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10756205 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011506134 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010756205 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13260063 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 6940/CHENP/2011 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 20117025156 Country of ref document: KR Kind code of ref document: A |