WO2010074110A1 - 環状アミノ安息香酸誘導体の製造方法 - Google Patents
環状アミノ安息香酸誘導体の製造方法 Download PDFInfo
- Publication number
- WO2010074110A1 WO2010074110A1 PCT/JP2009/071375 JP2009071375W WO2010074110A1 WO 2010074110 A1 WO2010074110 A1 WO 2010074110A1 JP 2009071375 W JP2009071375 W JP 2009071375W WO 2010074110 A1 WO2010074110 A1 WO 2010074110A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- acid derivative
- lower alkyl
- general formula
- alkyl group
- Prior art date
Links
- 0 CCC*C1=NC([Re])=C(C)*1 Chemical compound CCC*C1=NC([Re])=C(C)*1 0.000 description 1
- SUXHDZVIRIZEKN-UHFFFAOYSA-N Cc1c(C(NC(CCC2)CN2c2cccc(C(OC)=O)c2)=O)[s]c(-c(cc2)ccc2Cl)n1 Chemical compound Cc1c(C(NC(CCC2)CN2c2cccc(C(OC)=O)c2)=O)[s]c(-c(cc2)ccc2Cl)n1 SUXHDZVIRIZEKN-UHFFFAOYSA-N 0.000 description 1
- QVNUBXRMEWXQJM-KRWDZBQOSA-N Cc1c(C(N[C@@H](CCC2)CN2c(cc2C(OC)=O)ccc2N)=O)[s]c(-c(cc2)ccc2Cl)n1 Chemical compound Cc1c(C(N[C@@H](CCC2)CN2c(cc2C(OC)=O)ccc2N)=O)[s]c(-c(cc2)ccc2Cl)n1 QVNUBXRMEWXQJM-KRWDZBQOSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/56—Nitrogen atoms
Definitions
- the present invention relates to a method for producing a cyclic aminobenzoic acid derivative, particularly a 3-piperidinobenzoic acid derivative, having excellent agonist activity against human peroxisome proliferator activated receptor (PPAR).
- PPAR peroxisome proliferator activated receptor
- Ra represents a hydrogen atom or a lower alkyl group
- Rb represents a hydrogen atom, a lower alkyl group
- Rc represents a lower alkyl group, a lower alkoxy group, or a group represented by the general formula (Rd)
- Y represents an oxygen atom or a sulfur atom
- Re represents a lower alkyl group
- Rf represents a benzene ring or the like which may be substituted by a halogen atom
- Z represents an oxygen atom, a sulfur atom, — (CH 2 ) n— (n represents 0, 1, 2 )
- W represents a hydrogen atom, a halogen atom, or a lower alkyl optionally substituted with a halogen atom
- an aralkyl group which may have a substituent, an aryloxy group which may have a substituent, or
- the cyclic aminobenzoic acid derivative described in Patent Document 1 is produced by using a benzoic acid derivative having a W group and a leaving group as a raw material, and replacing the leaving group with a cyclic amino compound. Therefore, among the general formula (A) described in Patent Document 1, the following general formula (A ′)
- the method for producing a cyclic aminobenzoic acid derivative disclosed in Patent Document 1 requires that 1 to 3 equivalents of the phenylboronic acid derivative (B) and the copper reagent act on the piperidine derivative (C). Since the yield is less than 60% and a purification step by silica gel column chromatography is required, it is not suitable for industrial scale implementation.
- the present invention provides a production method suitable for industrial production by improving the operability, purification efficiency, yield, etc. of production in producing the cyclic aminobenzoic acid derivative represented by the general formula (A ′). With the goal.
- R 1 represents a hydrogen atom or a lower alkyl group
- R2 represents a hydrogen atom, a lower alkyl group or an aralkyl group
- R3 represents a lower alkyl group, a lower alkoxy group, an aralkyloxy group, an aryl group, or a general formula (3)
- Y represents an oxygen atom, a sulfur atom or methylene
- R4 represents a hydrogen atom or a lower alkyl group
- R5 is a) a halogen atom, b) benzene optionally having 1 to 3 substituents each independently selected from the group consisting of substituents consisting of a lower alkyl group optionally substituted with a halogen atom and c) a lower alkoxy group
- R1 is the same as defined above, and X represents a halogen atom, a trifluoromethanesulfonyloxy group, a paratoluenesulfonyloxy group, or a methanesulfonyloxy group]
- Formula (2) :
- R1 is a lower alkyl group
- X is a halogen atom
- R2 is a hydrogen atom
- R3 is a lower alkoxy group
- R4a represents a lower alkyl group, and R5a represents a benzene ring which 1 to 3 halogen atoms may be substituted
- R4a represents a lower alkyl group
- R5a represents a benzene ring which 1 to 3 halogen atoms may be substituted
- R3 represents the general formula (3a):
- the present invention is useful as an industrial production method for 3-piperidinobenzoic acid derivatives or hydrates thereof with few impurities, high yield, and easy purification.
- the “lower alkyl group” represented in the present specification means a linear or branched alkyl group having 1 to 6 carbon atoms, and examples thereof include a methyl group, an ethyl group, an n-propyl group, an iso- Examples include propyl group, n-butyl group, iso-butyl group, sec-butyl group, tert-butyl group, n-pentyl group, n-hexyl group and the like.
- the “halogen atom” represented in the present specification represents a fluorine atom, a chlorine atom, a bromine atom or an iodine atom.
- the “aralkyl group” represented in the present specification means a lower alkyl group substituted with 1 to 3 aryl groups, such as benzyl group, diphenylmethyl group, triphenylmethyl group, phenethyl group, phenylpropyl group. Groups and the like.
- the “lower alkoxy group” represented in the present specification means an alkoxy group having 1 to 6 carbon atoms which may be linear or branched.
- methoxy group, ethoxy group, n-propoxy group iso-
- examples thereof include a propoxy group, an n-butoxy group, an iso-butoxy group, a sec-butoxy group, a tert-butoxy group, an n-pentyloxy group, and an n-hexyloxy group.
- the “aralkyloxy group” represented in the present specification means a lower alkoxy group substituted with 1 to 3 aryl groups, such as a benzyloxy group, a diphenylmethyloxy group, a triphenylmethyloxy group, a phenethyl group. Examples thereof include an oxy group and a phenylpropyloxy group.
- the “aryl group” represented in the present specification means an aromatic hydrocarbon group having 6 to 10 carbon atoms, and examples thereof include a phenyl group and a naphthyl group.
- the term “lower alkyl group optionally substituted with a halogen atom” as used herein means a lower alkyl group optionally substituted with 1 to 3 halogen atoms, for example, trifluoromethyl Groups and the like.
- Nirous acid or a salt thereof represented in the present specification includes, for example, nitrous acid, sodium nitrite, potassium nitrite and the like.
- the “reducing agent” represented in the present specification is not limited as long as it can reductively remove a diazo group produced by nitrous acid or a salt thereof, such as formic acid, sodium borohydride, sulfuric acid. Iron (II), sodium bisulfite, hypophosphorous acid, phosphorous acid, etc. are mentioned.
- the structural formula of a compound is represented by a racemate for the sake of convenience, but the present invention includes all optical isomers based on asymmetric carbons that occur in the structure of the compound, and is limited to the formula for convenience. is not. Further, all salts of compounds and hydrates thereof are also included in the present invention.
- the present invention does not use a nitrite commonly used for the deamination reaction in the above deamination step, but by using nitrous acid or a salt thereof, the yield is remarkably improved and a high purity is obtained. It has been found that the target compound can be obtained.
- the 2-nitrobenzoic acid derivative represented by the general formula (1) is reacted with the piperidine derivative represented by the general formula (2), and the resulting 2-nitro-5-piperidinobenzoic acid derivative is obtained.
- the nitro group of (4) is reduced to give a 2-amino-5-piperidinobenzoic acid derivative (5), which is deaminated by using nitrous acid or a salt thereof and a reducing agent.
- This is a production method suitable for an industrial scale of a 3-piperidinobenzoic acid derivative represented by the general formula (6) or a hydrate thereof (see the following scheme).
- the 2-amino-5-piperidinobenzoic acid derivative (5) which is a synthetic intermediate of the present invention, can be produced by appropriately using the method described in Patent Document 1 (Step 1 and Step 2).
- the reduction of the nitro group of 2-nitro-5-piperidinobenzoic acid derivative (4) (step 2) can also be performed by a method using nickel chloride (II) or a hydrate thereof and sodium borohydride. This method is preferable because it is suitable for industrial production.
- Step 3 which is the most characteristic feature of the present invention, comprises deaminating the 2-amino-5-piperidinobenzoic acid derivative (5) by using nitrous acid or a salt thereof and a reducing agent, thereby producing 3-piperidino This is a step of derivatizing to a benzoic acid derivative (6).
- This step comprises the steps of reacting 2-amino-5-piperidinobenzoic acid derivative (5) with nitrous acid or a salt thereof to form a diazo compound, and reacting the diazo compound with a reducing agent to produce 3-piperidinobenzoic acid.
- Two steps of derivatization to the derivative (6) may be combined, or the 2-amino-5-piperidinobenzoic acid derivative (5) is stirred under reaction conditions in which nitrous acid or a salt thereof and a reducing agent are present simultaneously. By doing so, the 3-piperidinobenzoic acid derivative (6) may be led in one step.
- the 2-amino-5-piperidinobenzoic acid derivative (5) is stirred in a single step by stirring under reaction conditions in which nitrous acid or a salt thereof and a reducing agent are present simultaneously.
- -It is better to lead to piperidinobenzoic acid derivative (6).
- This step can be performed in the absence of a solvent or in the presence of a solvent, and the type of solvent is not particularly limited as long as it does not adversely affect the reaction.
- a solvent for example, N, N-dimethylformamide, dimethyl sulfoxide, acetonitrile, etc.
- Protic polar solvent, ether solvents such as tetrahydrofuran, 1,2-dimethoxyethane, 1,4-dioxane, halogen solvents such as chloroform and dichloromethane, alcohol solvents such as methanol, ethanol and 2-propanol, water solvents or These mixed solvents are mentioned.
- this step was performed at 5 to 10 ° C.
- the solvent used in this step is preferably an ether solvent or a mixed solvent containing an ether solvent, and particularly preferably a mixed solvent composed of N, N-dimethylformamide, tetrahydrofuran and water.
- Nirous acid or a salt thereof that can be used in this step includes, for example, nitrous acid, sodium nitrite, potassium nitrite and the like, and preferably sodium nitrite.
- the “reducing agent” is not particularly limited as long as it can reductively remove a diazo group produced by nitrous acid or a salt thereof.
- the deamination reaction in this step is carried out by a combination of these, preferably a combination of sodium nitrite and hypophosphorous acid, particularly preferably sodium nitrite, hypophosphorous acid, N, N-dimethylformamide. , Tetrahydrofuran and water.
- the amount of nitrous acid or a salt thereof used is preferably 1 to 10 equivalents, more preferably 2 to 4 equivalents, relative to the substrate.
- the reaction in this step may contain a catalyst.
- the catalyst include copper, copper (II) oxide, copper (II) chloride, and zinc.
- it is copper (II) oxide.
- the reaction temperature in this step is appropriately selected in the range from 0 ° C. to the reflux temperature of the solvent.
- this step is performed using sodium nitrite, hypophosphorous acid and N, N-dimethylformamide
- the reaction is completed in about 20 minutes when the reaction temperature is 25 to 30 ° C., but the reaction temperature is 5 If it is ⁇ 10 ° C., it takes about 120 minutes to complete the reaction.
- the reaction temperature in this step is preferably 10 ° C. or less, particularly preferably 5 to 10 ° C.
- 3-piperidinobenzoic acid having a high yield and high purity can be obtained by a simple operation by carrying out the above reaction and then performing purification such as hot suspension washing with a mixed solvent of water and 2-propanol.
- An acid derivative (6) or a hydrate thereof can be obtained.
- optically active piperidine derivative (2) by using the optically active piperidine derivative (2), optically active compounds (4), (5) and (6) having a corresponding configuration can be obtained.
- N, N-dimethylformamide (16.1 mL) was added to 5.35 g (28.9 mmol) of 5-fluoro-2-nitrobenzoic acid and 5.99 g (43.3 mmol) of potassium carbonate, and methyl iodide 8 while stirring. .20 g (57.8 mmol) was added dropwise, and the mixture was stirred at an external temperature of 25 to 30 ° C. for 1.5 hours, and then stirred at an external temperature of 35 to 40 ° C. for 2 hours.
- Step 2 (S) -2-Amino-5- ⁇ 3- [2- (4-chlorophenyl) -4-methylthiazole-5-carboxamide] piperidino ⁇ benzoic acid methyl ester
- ammonium chloride solution (prepared with 105.0 g of ammonium chloride and 947 mL of water) was added to the reaction solution, and then 2.10 L of ethyl acetate was added and dissolved by heating. After stirring at an internal temperature of 40 to 45 ° C. for 30 minutes, the organic layer was separated. It was washed sequentially with an ammonium chloride solution (prepared with 105.0 g of ammonium chloride and 945 mL of water) and an ammonium chloride solution (prepared with 75.0 g of ammonium chloride and 675 mL of water). 750 mL of N, N-dimethylformamide was added to the organic layer and concentrated under reduced pressure at an external temperature of 40 ° C. or lower.
- N, N-dimethylformamide 1.41 L was added with 170 g (920 mmol) of 5-fluoro-2-nitrobenzoic acid and 143 g (1.00 mol) of methyl iodide and dissolved with stirring, and 254 g of potassium carbonate ( 1.84 mol) was added, and the mixture was stirred at an internal temperature of 30 to 37 ° C. for 2 hours.
- the reaction solution was then subjected to (S) -3- [2- (4-chlorophenyl) -4-methylthiazole-5-carboxamide] piperidine 281 g (837 mmol) and N, N-dimethylformamide 1.41 at an internal temperature of 37 ° C.
- Step 2 (S) -2-Amino-5- (3- [2- (4-chlorophenyl) -4-methylthiazole-5-carboxamide] -piperidino) benzoic acid methyl ester
- Tetrahydrofuran 2.97 L was charged with (S) -5- (3- [2- (4-chlorophenyl) -4-methylthiazole-5-carboxamido] piperidino) -2-nitrobenzoic acid methyl ester 424 g (823 mmol). Then, 2.97 L of methanol and 39.1 g (165 mmol) of nickel (II) chloride hexahydrate were added. Under cooling, 93.4 g (2.47 mol) of sodium borohydride was slowly added at an internal temperature of 8 to 20 ° C., and the mixture was stirred at an internal temperature of 6 to 20 ° C. for 1.5 hours.
- Ethyl acetate 5.94 L and ammonium chloride aqueous solution (ammonium chloride 297 g + normal water 2.67 L) were added to the reaction solution, and after heating, the mixture was stirred at an internal temperature of 30 to 35 ° C. for 30 minutes.
- the organic layer was separated and washed successively with an aqueous ammonium chloride solution (297 g ammonium chloride + 2.67 L normal water) and an aqueous ammonium chloride solution (212 g ammonium chloride + 1.91 L normal water).
- 2.12 L of N, N-dimethylformamide was added and concentrated under reduced pressure at an external temperature of 40 ° C. or lower.
- Patent Document 1 has a low yield compared to the production method of the present invention. Moreover, since the manufacturing method of patent document 1 requires the refinement
- the production method of the present invention yielded the 3-piperidinobenzoic acid derivative (6) in a high yield as compared with the conventional production methods and the production methods that are widely used. -Since the piperidinobenzoic acid derivative (6) is also highly pure, purification is very easy. Therefore, the production method of the present invention is also suitable for industrial level production.
- the method for producing a cyclic aminobenzoic acid derivative of the present invention uses a 2-nitrobenzoic acid derivative (1) and a piperidine derivative (2) as raw materials and uses a reductive elimination reaction using a nitrous acid compound in the reaction step.
- the PPAR ⁇ agonist represented by the general formula (A ′) and hydrates thereof can be provided in good yield, which is useful as an industrial production method.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Hydrogenated Pyridines (AREA)
Abstract
ヒトペルオキシゾーム増殖薬活性化受容体(PPAR)の優れたアゴニストである環状アミノ安息香酸誘導体の、効率的かつ工業生産に適した製造方法を提供する。 2-ニトロ安息香酸誘導体(1)とピペリジン誘導体(2)を反応させ、得られた化合物のニトロ基を還元後、亜硝酸またはその塩と還元剤により脱アミノ化反応を行うことにより、PPARαアゴニストである3-ピペリジノ安息香酸誘導体又はその水和物(6)を製造する。
Description
本発明は、ヒトペルオキシゾーム増殖薬活性化受容体(PPAR)に対して優れたアゴニスト活性を有する環状アミノ安息香酸誘導体、特に3-ピペリジノ安息香酸誘導体の製造方法に関する。
下記一般式(A)

[式中、Raは水素原子または低級アルキル基を表し、Rbは水素原子、低級アルキル基などを表し、Rcは低級アルキル基、低級アルコキシ基、一般式(Rd)

(式中、Yは酸素原子または硫黄原子などを表し、Reは低級アルキル基などを表し、Rfはハロゲン原子が置換してもよいベンゼン環などを表す)で表される芳香族複素環基などを表し、Zは酸素原子、硫黄原子、-(CH2)n-(nは0,1,2を表す)を表し、Wは水素原子、ハロゲン原子、ハロゲン原子で置換されてもよい低級アルキル基、ハロゲン原子で置換されてもよい低級アルコキシ基、水酸基、ニトロ基、シアノ基、置換されてもよいアミノ基、置換基を有してもよいアリール基、置換基を有してもよい5員若しくは6員の芳香族複素環基及びその縮合環基、置換基を有してもよいアラルキル基、置換基を有してもよいアリールオキシ基又は置換基を有してもよいアラルキルオキシ基を表す]
で示される環状アミノ安息香酸誘導体は、ヒトペルオキシゾーム増殖薬活性化受容体(PPAR)に対して優れたアゴニストであり、PPARが関与する種々の病態(脂質代謝異常、糖尿病等)の治療等の医薬用途に有用であることが知られている(特許文献1)。
で示される環状アミノ安息香酸誘導体は、ヒトペルオキシゾーム増殖薬活性化受容体(PPAR)に対して優れたアゴニストであり、PPARが関与する種々の病態(脂質代謝異常、糖尿病等)の治療等の医薬用途に有用であることが知られている(特許文献1)。
特許文献1に記載された上記環状アミノ安息香酸誘導体は、W基及び脱離基を有する安息香酸誘導体を原料として用い、その脱離基を環状アミノ化合物に置換することにより製造するものであり、従って、特許文献1に記載された一般式(A)のうち、下記一般式(A’)

[式中、Ra、Rb、RcおよびZは、前記定義と同一]
で示される化合物の製造方法は、塩基及び酢酸銅存在下、一般式(B)
で示される化合物の製造方法は、塩基及び酢酸銅存在下、一般式(B)

[式中、Raは前記定義と同一]
で示されるフェニルボロン酸誘導体を一般式(C)
で示されるフェニルボロン酸誘導体を一般式(C)

[式中、Rb、RcおよびZは前記定義と同一]
で示されるピペリジン誘導体に対し2当量作用させるものである。
で示されるピペリジン誘導体に対し2当量作用させるものである。
特許文献1で開示されている環状アミノ安息香酸誘導体の製造方法は、上記の通り、ピペリジン誘導体(C)に対しフェニルボロン酸誘導体(B)および銅試薬を1~3当量作用させる必要があり、収率も60%に満たず、さらにシリカゲルカラムクロマトグラフィーによる精製工程が必要であることから、工業的規模の実施には適さない。
本発明は、一般式(A’)で示された環状アミノ安息香酸誘導体の製造に際し、製造の操作性、精製効率及び収率等を向上させ、工業的生産に適合する製造法を提供することを目的とする。
本発明は、一般式(A’)で示された環状アミノ安息香酸誘導体の製造に際し、製造の操作性、精製効率及び収率等を向上させ、工業的生産に適合する製造法を提供することを目的とする。
本発明者らは、上記課題を解決するため鋭意研究を重ねた結果、2-ニトロ安息香酸誘導体とピペリジン誘導体を用いることにより、効率的に一般式(A’)に包含される一般式(6)

[式中、R1は水素原子または低級アルキル基を表し;
R2は水素原子、低級アルキル基またはアラルキル基を表し;
R3は低級アルキル基、低級アルコキシ基、アラルキルオキシ基、アリール基または一般式(3)
R2は水素原子、低級アルキル基またはアラルキル基を表し;
R3は低級アルキル基、低級アルコキシ基、アラルキルオキシ基、アリール基または一般式(3)

(式中、Yは酸素原子、硫黄原子またはメチレンを表し;
R4は水素原子または低級アルキル基を表し;
R5は、
a)ハロゲン原子、
b)ハロゲン原子で置換されていてもよい低級アルキル基及び
c)低級アルコキシ基
からなる置換基群の中からそれぞれ独立して選ばれた1~3個の置換基を有していてもよいベンゼン環を表す)で表される芳香族複素環基を表す]で表される化合物を製造する方法を見出した。
R4は水素原子または低級アルキル基を表し;
R5は、
a)ハロゲン原子、
b)ハロゲン原子で置換されていてもよい低級アルキル基及び
c)低級アルコキシ基
からなる置換基群の中からそれぞれ独立して選ばれた1~3個の置換基を有していてもよいベンゼン環を表す)で表される芳香族複素環基を表す]で表される化合物を製造する方法を見出した。
すなわち本発明は、
1)一般式(5):
1)一般式(5):

[式中、R1、R2およびR3は、前記定義と同一である]で表される2-アミノ-5-ピペリジノ安息香酸誘導体を、亜硝酸またはその塩と還元剤を用いて脱アミノ化することを特徴とする一般式(6):
[式中、R1、R2およびR3は、前記定義と同一である]で表される3-ピペリジノ安息香酸誘導体又はその水和物の製造方法、
2) 一般式(1):

[式中、R1は前記定義と同一であり、Xはハロゲン原子、トリフルオロメタンスルホニルオキシ基、パラトルエンスルホニルオキシ基またはメタンスルホニルオキシ基を表す]で表される2-ニトロ安息香酸誘導体と、一般式(2):

[式中、R2及びR3は前記定義と同一である]で表されるピペリジン誘導体を反応させることにより、一般式(4):

[式中、R1、R2およびR3は、前記定義と同一である]で表される2-ニトロ-5-ピペリジノ安息香酸誘導体を得た後、次いでニトロ基を還元することによって、一般式(5):

[式中、R1、R2およびR3は、前記定義と同一である]で表される2-アミノ-5-ピペリジノ安息香酸誘導体を得た後、次いで亜硝酸またはその塩と還元剤を用いて脱アミノ化することを特徴とする一般式(6):

[式中、R1、R2およびR3は、前記定義と同一である]で表される3-ピペリジノ安息香酸誘導体又はその水和物の製造方法、
3)前記一般式(1)~(6)において、R1が低級アルキル基、Xがハロゲン原子、R2が水素原子、R3が低級アルコキシ基または一般式(3a) :

(式中、R4aは低級アルキル基を表し、R5aは1~3個のハロゲン原子が置換してもよいベンゼン環を表す)で表される芳香族複素環基である、1)又は2)記載の3-ピペリジノ安息香酸誘導体又はその水和物の製造方法、
4)前記一般式(1)~(6)において、R3が一般式(3a):

(式中、R4aおよびR5aは前記定義と同一である)で表される芳香族複素環基である、3)記載の3-ピペリジノ安息香酸誘導体又はその水和物の製造方法、
5)脱アミノ化工程において、亜硝酸またはその塩が亜硝酸ナトリウムである、1)~4)の何れかに記載の3-ピペリジノ安息香酸誘導体又はその水和物の製造方法、
6)脱アミノ化工程において、還元剤が次亜リン酸である、1)~5)の何れかに記載の3-ピペリジノ安息香酸誘導体又はその水和物の製造方法に関する。
本発明は、不純物が少なく、高収率でありかつ精製が簡便な3-ピペリジノ安息香酸誘導体またはその水和物の工業的製造方法として有用である。
本明細書中に表される「低級アルキル基」とは、炭素数1から6の直鎖若しくは分岐鎖状のアルキル基を意味し、例えば、メチル基、エチル基、n-プロピル基、iso-プロピル基、n-ブチル基、iso-ブチル基、sec-ブチル基、tert-ブチル基、n-ペンチル基、n-ヘキシル基などが挙げられる。
本明細書中に表される「ハロゲン原子」とは、フッ素原子、塩素原子、臭素原子またはヨウ素原子を表す。
本明細書中に表される「ハロゲン原子」とは、フッ素原子、塩素原子、臭素原子またはヨウ素原子を表す。
本明細書中に表される「アラルキル基」とは、1~3個のアリール基が置換した低級アルキル基を意味し、例えばベンジル基、ジフェニルメチル基、トリフェニルメチル基、フェネチル基、フェニルプロピル基などが挙げられる。
本明細書中に表される「低級アルコキシ基」とは、直鎖または分岐してもよい炭素数1から6のアルコキシ基を意味し、例えばメトキシ基、エトキシ基、n-プロポキシ基、iso-プロポキシ基、n-ブトキシ基、iso-ブトキシ基、sec-ブトキシ基、tert-ブトキシ基、n-ペンチルオキシ基、n-ヘキシルオキシ基などが挙げられる。
本明細書中に表される「低級アルコキシ基」とは、直鎖または分岐してもよい炭素数1から6のアルコキシ基を意味し、例えばメトキシ基、エトキシ基、n-プロポキシ基、iso-プロポキシ基、n-ブトキシ基、iso-ブトキシ基、sec-ブトキシ基、tert-ブトキシ基、n-ペンチルオキシ基、n-ヘキシルオキシ基などが挙げられる。
本明細書中に表される「アラルキルオキシ基」とは、1~3個のアリール基が置換した低級アルコキシ基を意味し、例えばベンジルオキシ基、ジフェニルメチルオキシ基、トリフェニルメチルオキシ基、フェネチルオキシ基、フェニルプロピルオキシ基などが挙げられる。
本明細書中に表される「アリール基」とは、炭素数6から10の芳香族炭化水素基を意味し、例えばフェニル基、ナフチル基などが挙げられる。
本明細書中に表される「ハロゲン原子で置換されていてもよい低級アルキル基」とは、1~3個のハロゲン原子で置換されていてもよい低級アルキル基を意味し、例えばトリフルオロメチル基などが挙げられる。
本明細書中に表される「アリール基」とは、炭素数6から10の芳香族炭化水素基を意味し、例えばフェニル基、ナフチル基などが挙げられる。
本明細書中に表される「ハロゲン原子で置換されていてもよい低級アルキル基」とは、1~3個のハロゲン原子で置換されていてもよい低級アルキル基を意味し、例えばトリフルオロメチル基などが挙げられる。
本明細書中に表される「亜硝酸またはその塩」とは、例えば亜硝酸、亜硝酸ナトリウム、亜硝酸カリウムなどが挙げられる。
本明細書中に表される「還元剤」とは、亜硝酸またはその塩により生成したジアゾ基を還元的に脱離させることができるものであれば良く、例えば蟻酸、水素化ホウ素ナトリウム、硫酸鉄(II)、亜硫酸水素ナトリウム、次亜リン酸、亜リン酸などが挙げられる。
本明細書中に表される「還元剤」とは、亜硝酸またはその塩により生成したジアゾ基を還元的に脱離させることができるものであれば良く、例えば蟻酸、水素化ホウ素ナトリウム、硫酸鉄(II)、亜硫酸水素ナトリウム、次亜リン酸、亜リン酸などが挙げられる。
なお、本明細書においては化合物の構造式は便宜上ラセミ体で表すが、本発明には化合物の構造上生ずる全ての、不斉炭素に基づく光学異性体を含み、便宜上の式に限定されるものではない。また、化合物の塩やその水和物も総て本発明に含まれる。
(製造方法)
従来技術において、前記一般式(A)で表される環状アミノ安息香酸誘導体のうちW基が水素原子である目的物質を製造する場合に、原料としてW基が水素原子である安息香酸誘導体を用いるところ、本発明は、ニトロ基の置換した2-ニトロ安息香酸誘導体を用いて2-ニトロ-5-ピペリジノ安息香酸誘導体を製造し、その後に、ニトロ基をアミノ基に還元し、更に脱アミノ化することにより、目的のW基が水素原子である3-ピペリジノ安息香酸誘導体を得るものであり、これにより工業的スケールに適した製造方法を提供できることを見出したものである。更に本発明は、上記脱アミノ化工程において、脱アミノ化反応に汎用される亜硝酸エステルを用いるのではなく、亜硝酸またはその塩を用いることにより、収率が格段に向上し、高純度の目的化合物が得られることを見出したものである。
従来技術において、前記一般式(A)で表される環状アミノ安息香酸誘導体のうちW基が水素原子である目的物質を製造する場合に、原料としてW基が水素原子である安息香酸誘導体を用いるところ、本発明は、ニトロ基の置換した2-ニトロ安息香酸誘導体を用いて2-ニトロ-5-ピペリジノ安息香酸誘導体を製造し、その後に、ニトロ基をアミノ基に還元し、更に脱アミノ化することにより、目的のW基が水素原子である3-ピペリジノ安息香酸誘導体を得るものであり、これにより工業的スケールに適した製造方法を提供できることを見出したものである。更に本発明は、上記脱アミノ化工程において、脱アミノ化反応に汎用される亜硝酸エステルを用いるのではなく、亜硝酸またはその塩を用いることにより、収率が格段に向上し、高純度の目的化合物が得られることを見出したものである。
具体的には、一般式(1)で表される2-ニトロ安息香酸誘導体に、一般式(2)で表されるピペリジン誘導体を反応させ、得られた2-ニトロ-5-ピペリジノ安息香酸誘導体(4)のニトロ基を還元して、2-アミノ-5-ピペリジノ安息香酸誘導体(5)とし、これを亜硝酸またはその塩と還元剤を用いることにより脱アミノ化することを特徴とする、一般式(6)で表される3-ピペリジノ安息香酸誘導体またはその水和物の工業的スケールに適した製造方法である(下記スキームを参照)。
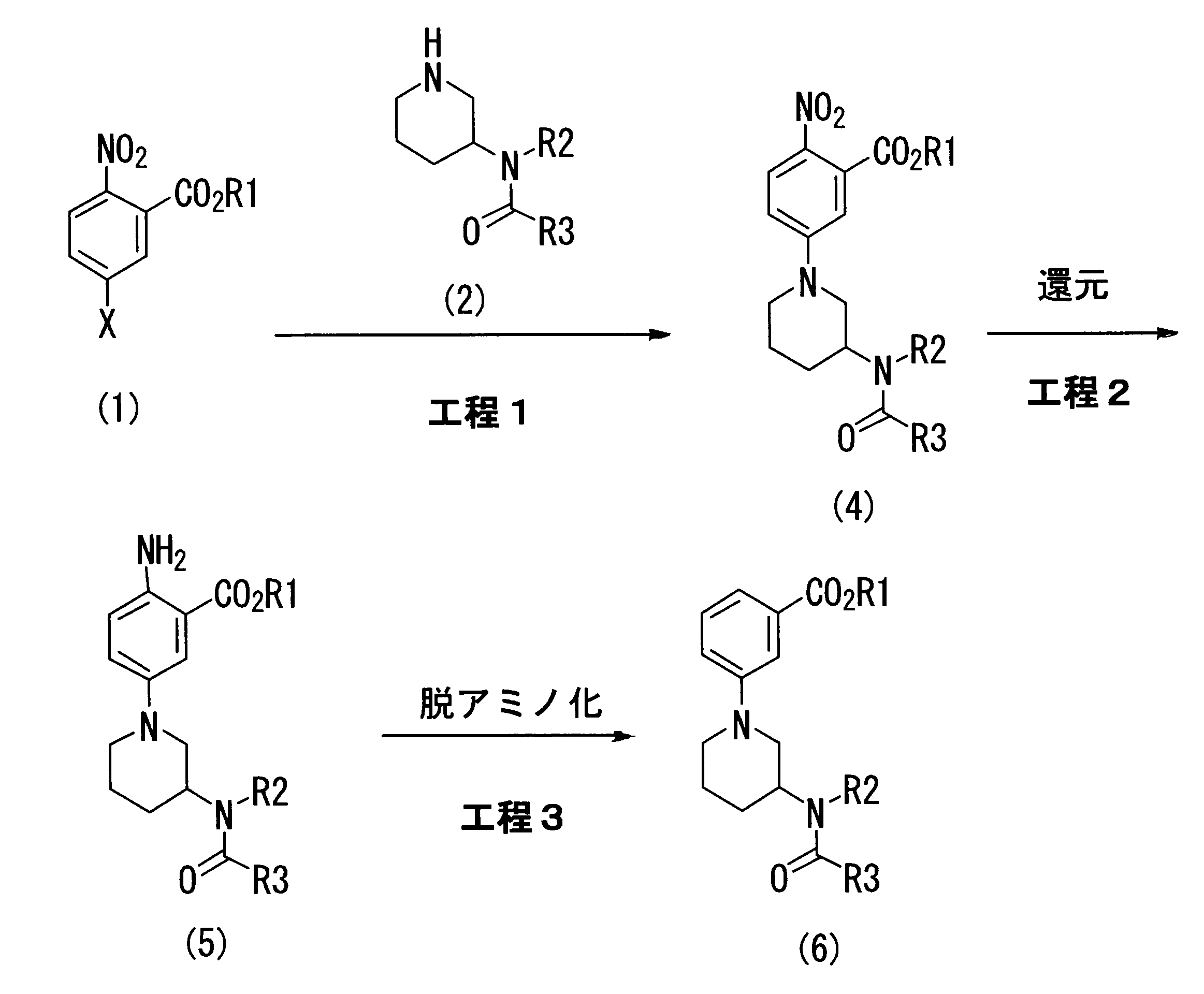
[式中、R1、R2、R3およびXは、前記定義と同一である]
本発明の合成中間体である2-アミノ-5-ピペリジノ安息香酸誘導体(5)は、特許文献1に記載の方法を適宜用いて製造することができる(工程1および工程2)。また、2-ニトロ-5-ピペリジノ安息香酸誘導体(4)のニトロ基の還元(工程2)は、塩化ニッケル(II)又はその水和物と水素化ホウ素ナトリウムを用いる方法によっても行うことができ、本方法は工業的生産に適しているため好ましい。
本発明の最も特徴的である工程3は、2-アミノ-5-ピペリジノ安息香酸誘導体(5)を、亜硝酸またはその塩と還元剤を用いることにより脱アミノ化反応させることにより、3-ピペリジノ安息香酸誘導体(6)へと誘導する工程である。
本工程は、2-アミノ-5-ピペリジノ安息香酸誘導体(5)を亜硝酸またはその塩と反応させジアゾ化合物を生成させる段階と、該ジアゾ化合物を還元剤と反応させることにより3-ピペリジノ安息香酸誘導体(6)へと誘導させる段階の2段階を組み合わせても良いし、2-アミノ-5-ピペリジノ安息香酸誘導体(5)を亜硝酸またはその塩と還元剤が同時に存在する反応条件下で撹拌することにより1段階で3-ピペリジノ安息香酸誘導体(6)へ導いても良い。好ましくは、安全面および操作の簡便さから、2-アミノ-5-ピペリジノ安息香酸誘導体(5)を亜硝酸またはその塩と還元剤が同時に存在する反応条件下で撹拌することにより1段階で3-ピペリジノ安息香酸誘導体(6)へ導くのが良い。
本工程は無溶媒または溶媒存在下で行うことができ、溶媒の種類としては、反応に悪影響を与えない限り特に制限はないが、例えば、N,N-ジメチルホルムアミド、ジメチルスルホキシド、アセトニトリル等の非プロトン性極性溶媒、テトラヒドロフラン、1,2-ジメトキシエタン、1,4-ジオキサン等のエーテル系溶媒、クロロホルム、ジクロロメタンなどのハロゲン系溶媒、メタノール、エタノール、2-プロパノールなどのアルコール系溶媒、水溶媒又はこれらの混合溶媒が挙げられる。例えば、亜硝酸ナトリウム及び次亜リン酸を用い5~10℃で本工程を行った場合、溶媒としてN,N-ジメチルホルムアミドを用いると約120分で反応が完了したが、溶媒としてN,N-ジメチルホルムアミド、テトラヒドロフラン及び水からなる混合溶媒を用いると反応の完了まで約240分を要した。本工程は反応の進行に伴い窒素ガスが発生するため、工業スケールで実施する場合、反応速度を制御することが安全面から好ましい。したがって、本工程で用いる溶媒としてはエーテル系溶媒又はエーテル系溶媒を含んだ混合溶媒が好ましく、特に好ましくは、N,N-ジメチルホルムアミド、テトラヒドロフラン及び水からなる混合溶媒である。
本工程に使用できる「亜硝酸またはその塩」とは、例えば亜硝酸、亜硝酸ナトリウム、亜硝酸カリウムなどが挙げられ、好ましくは亜硝酸ナトリウムが挙げられる。「還元剤」としては、亜硝酸またはその塩により生成したジアゾ基を還元的に脱離させることができるものであれば良く、例えば蟻酸、水素化ホウ素ナトリウム、硫酸鉄(II)、亜硫酸水素ナトリウム、次亜リン酸、亜リン酸などが挙げられ、好ましくは次亜リン酸が挙げられる。本工程の脱アミノ化反応はこれらの組み合わせにより実施され、好ましくは、亜硝酸ナトリウムと次亜リン酸の組み合わせであり、特に好ましくは、亜硝酸ナトリウム、次亜リン酸、N,N-ジメチルホルムアミド、テトラヒドロフラン及び水の組み合わせである。亜硝酸またはその塩の使用量は、好ましくは基質に対して1~10当量であり、より好ましくは2~4当量である。
本工程における反応は触媒を存在させても良く、触媒の種類としては、銅、酸化銅(II)、塩化銅(II)、亜鉛等が挙げられる。好ましくは、酸化銅(II)である。
本工程の反応温度は、0℃から溶媒の還流温度までの範囲で適宜選択される。例えば、亜硝酸ナトリウム、次亜リン酸及びN,N-ジメチルホルムアミドを用いて本工程を行った場合、反応温度を25~30℃とすると約20分で反応が完了するが、反応温度を5~10℃とすると反応の完了まで約120分を要する。前述のとおり、本工程は反応速度を制御することが安全面から好ましいことから、本工程の反応温度は10℃以下が好ましく、特に好ましくは5~10℃である。
さらに本工程においては、上記反応を実施後,水及び2-プロパノールの混合溶媒による熱時懸濁洗浄等の精製を行うことにより,簡便な操作で収率良く,かつ高純度の3-ピペリジノ安息香酸誘導体(6)又はその水和物を得ることができる。
一般に、芳香族一級アミンに亜硝酸およびその塩を作用させ生成するジアゾニウム化合物及びその塩は熱、衝撃等に不安定であり(化学プロセス安全ハンドブック、P16、田村昌三、朝倉書店、2000年)、反応途中において爆発の危険を伴うものであるが、本件発明の方法によれば、脱アミノ体を安全かつ収率よく製造できる。また、工程3における反応熱量を反応熱量計(ハイブリッドカロリーメータ)で測定し、総発熱量及び予想最高到達温度を評価した結果、工程3は熱的な危険性がないことも判明している。
なお、本発明の製造方法において、光学活性なピペリジン誘導体(2)を用いることで、それに対応した立体配置を有する光学活性な化合物(4)、(5)、(6)を得ることもできる。
次に本発明を具体例によって説明するが、これらの例によって本発明が限定されるものではない。
(参考例1)
5-フルオロ-2-ニトロ安息香酸メチルエステル
5-フルオロ-2-ニトロ安息香酸メチルエステル

5-フルオロ-2-ニトロ安息香酸 5.35g(28.9mmol)及び炭酸カリウム 5.99g(43.3mmol)にN,N-ジメチルホルムアミド(16.1mL)を加え、攪拌しながらヨウ化メチル 8.20g(57.8mmol)を滴下し、外温25~30℃で1.5時間攪拌した後、外温35~40℃で2時間攪拌した。水冷後、水 53.5mL及び酢酸エチル 53.5mLを加え抽出し、酢酸エチル層を分取し、得られた酢酸エチル層をチオ硫酸ナトリウム・五水和物 2.68gの水 53.5mL溶液で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧留去して淡褐色油状物の5-フルオロ-2-ニトロ安息香酸メチルエステル(5.64g、収率98%)を得た。
1H-NMR(400MHz, CDCl3)δ:3.95(3H, s), 7.31(1H, ddd, J = 9.0, 7.3, 2.7 Hz), 7.39(1H, dd, J = 7.8, 2.9 Hz), 8.03(1H, dd, J = 9.0, 4.6 Hz).
(実施例1)
工程1:
(S)-5-{3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ}-2-ニトロ安息香酸メチルエステル
工程1:
(S)-5-{3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ}-2-ニトロ安息香酸メチルエステル

(S)-3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジン 1.17 g (3.48 mmol)及び5-フルオロ-2-ニトロ安息香酸メチルエステル 691 mg (3.47 mmol)のN,N-ジメチルホルムアミド 9.7 mL溶液に、水1.17 g及び炭酸カリウム 288 mg (2.08 mmol)を加え、外温40~45℃で2時間撹拌した。反応液に水37 mLを加え晶析させた後、2 mol/L 塩酸 1.0 mLを加えて、30分間撹拌した。結晶を濾取し、水 50 mL、0.5 mol/L 塩酸 20 mL、水 50 mL及びエタノール 50 mLで順次洗浄した後、60℃で16時間送風乾燥して淡黄色粉末の(S)-5-{3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ}-2-ニトロ安息香酸メチルエステル(1.59 g、収率89%)を得た。
1H-NMR(400MHz, DMSO-d6)δ:1.52-1.60(1H, m), 1.67-1.76(1H, m), 1.82-1.84(1H, m), 1.95-1.99(1H, m), 2.57(3H, s), 3.16-3.24(2H, m), 3.34(3H, s), 3.89-3.93(2H, m), 3.99-4.02(1H, m), 7.08-7.12(2H, m), 7.59(2H, d, J = 8.8 Hz), 7.95(2H, d, J = 8.8 Hz), 8.02(1H, d, J = 9.3 Hz), 8.34(1H, d, J = 7.1 Hz).
なお、本工程中、(S)-3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジンは、特許文献1の実施例17の工程17bで得られる化合物と同一である。
なお、本工程中、(S)-3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジンは、特許文献1の実施例17の工程17bで得られる化合物と同一である。
工程2:
(S)-2-アミノ-5-{3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ}安息香酸メチルエステル
(S)-2-アミノ-5-{3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ}安息香酸メチルエステル

(S)-5-{3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ}-2-ニトロ安息香酸メチルエステル 150.0 g (291 mmol)のテトラヒドロフラン 1.05 L溶液に塩化ニッケル(II)六水和物13.85 g (58.3 mmol)のメタノール1.05 L溶液を加え、懸濁させた。冷却し、内温5~16℃で水素化ホウ素ナトリウム33.06 g (874 mmol)を撹拌下投入し,内温5~15℃で1時間撹拌した。反応液に塩化アンモニウム溶液(塩化アンモニウム105.0 g、水947 mLで調製)を加えた後、酢酸エチル2.10 Lを加え、加熱溶解した。内温40~45℃で30分間撹拌した後、有機層を分取した。塩化アンモニウム溶液(塩化アンモニウム105.0 gおよび水945 mLで調製)及び塩化アンモニウム溶液(塩化アンモニウム75.0 gおよび水675 mLで調製)で順次洗浄した。有機層にN,N-ジメチルホルムアミド 750 mLを投入し、外温40℃以下で減圧濃縮した。濃縮残留物にN,N-ジメチルホルムアミド 750 mLを投入し、加熱して溶解した後、内温50~56℃で常水300 mLを滴下し、30分間撹拌後、水1.20 Lを滴下した(晶析確認)。冷却して、内温8~25℃で30分間撹拌した。結晶をろ取し、水750 mLで洗浄して、湿潤粗結晶337.3 gを得た。
メタノール2.25 Lに湿潤粗結晶337.3 gを投入し、加熱した。内温50℃で水750 mLを投入し、内温50~60℃で30分間撹拌した。冷却し、内温9~25℃で30分間撹拌した後、結晶をろ取し、メタノール300 mLと水300 mLの混液で洗浄して、湿潤結晶303.5 gを得た。60℃で15時間送風乾燥して、(S)-2-アミノ-5-(3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ)安息香酸メチルエステル (126.8 g、収率:90%)を得た。
FAB-MS(positive) m/z:485[M + H]+.
1H-NMR(400MHz, DMSO-d6)δ:1.43-1.51(1H, m), 1.50-1.66(1H, m), 1.79-1.89(2H, m), 2.54-2.56(3H, m), 3.34(3H, s), 3.20-3.23(1H, m), 3.78(3H, s), 3.96-4.00(1H, m), 6.30(2H, s), 6.73(1H, d, J= 9.0 Hz), 7.11(1H, dd, J= 2.7, 8.8 Hz), 7.22(1H, d, J= 2.9 Hz), 7.59(2H, d, J = 8.5 Hz), 7.96(1H, d, J= 8.5 Hz), 8.23(2H, d, J = 7.8 Hz).
1H-NMR(400MHz, DMSO-d6)δ:1.43-1.51(1H, m), 1.50-1.66(1H, m), 1.79-1.89(2H, m), 2.54-2.56(3H, m), 3.34(3H, s), 3.20-3.23(1H, m), 3.78(3H, s), 3.96-4.00(1H, m), 6.30(2H, s), 6.73(1H, d, J= 9.0 Hz), 7.11(1H, dd, J= 2.7, 8.8 Hz), 7.22(1H, d, J= 2.9 Hz), 7.59(2H, d, J = 8.5 Hz), 7.96(1H, d, J= 8.5 Hz), 8.23(2H, d, J = 7.8 Hz).
工程3:
(S)-3-{3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ}安息香酸メチルエステル
(S)-3-{3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ}安息香酸メチルエステル

N,N-ジメチルホルムアミド 3.47 L及びテトラヒドロフラン 1.74 Lの混液に(S)-2-アミノ-5-{3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ}安息香酸メチルエステル 347 g(715 mmol)及び50%次亜リン酸1.21 Lを加え、10℃以下で亜硝酸ナトリウム98.7 g(1.43 mol)の水521 mL溶液を滴下した。10℃以下で1時間撹拌後、20~30℃で1時間撹拌した。加熱し、45~55℃で水3.47 Lを加えた。冷却し、25℃以下で30分間撹拌後、析出した結晶をろ取し、2-プロパノールと水の混液で洗浄して、湿潤粗結晶を得た。2-プロパノール2.78 Lと水694 mLの混液に湿潤粗結晶を加え、内温50~60℃で30分間撹拌後、冷却し、内温20~30℃で30分間撹拌した。析出した結晶をろ取し、2-プロパノールと水の混液で洗浄後、乾燥すると,茶褐色粉末の(S)-3-{3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ}安息香酸メチルエステル 286 g(609 mmol、収率85%)を得た。
FAB-MS(positive) m/z:470[M + H]+.
1H-NMR(CDCl3, 400 MHz)δ:1.80-1.91(4H, m), 2.72(3H, s), 3.05-3.09(1H, m), 3.29-3.41(3H, m), 3.91(3H, s), 4.41-4.42(1H, m), 6.32(1H, d, J= 7.8 Hz), 7.16(1H, dd, J= 2.4, 7.8 Hz), 7.34(1H, t, J= 7.8 Hz), 7.41(2H, d, J= 8.3 Hz), 7.57(1H, d, J = 7.8 Hz), 7.63(1H, d, J = 2.4 Hz), 7.87(2H, d, J = 8.3 Hz).
1H-NMR(CDCl3, 400 MHz)δ:1.80-1.91(4H, m), 2.72(3H, s), 3.05-3.09(1H, m), 3.29-3.41(3H, m), 3.91(3H, s), 4.41-4.42(1H, m), 6.32(1H, d, J= 7.8 Hz), 7.16(1H, dd, J= 2.4, 7.8 Hz), 7.34(1H, t, J= 7.8 Hz), 7.41(2H, d, J= 8.3 Hz), 7.57(1H, d, J = 7.8 Hz), 7.63(1H, d, J = 2.4 Hz), 7.87(2H, d, J = 8.3 Hz).
(実施例2)
工程1:
(S)-5-(3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ)-2-ニトロ安息香酸メチルエステル
工程1:
(S)-5-(3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ)-2-ニトロ安息香酸メチルエステル

N,N-ジメチルホルムアミド 1.41 Lに5-フルオロ-2-ニトロ安息香酸 170 g (920 mmol)およびヨウ化メチル143 g (1.00 mol)を加え撹拌下溶解させ、炭酸カリウム254 g (1.84 mol)を投入し、内温30~37℃で2時間撹拌した。次いで反応液に内温37℃で(S)-3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジン281 g (837 mmol)およびN,N-ジメチルホルムアミド 1.41 Lを投入し、内温40~49℃で3時間撹拌した。常水5.62 Lに内温40℃で反応液を撹拌下滴下して結晶化させ、N,N-ジメチルホルムアミド 562 mLおよび常水1.12 Lで順次洗い込み結晶化液に合一した。冷却後、内温14~25℃で30分間撹拌して、結晶をろ取し、常水1.41 Lで洗浄した。60℃で48時間送風乾燥して、黄色結晶性粉末の(S)-5-(3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ)-2-ニトロ安息香酸メチルエステル425 g (825 mmol、99%)を得た。
FAB-MS(positive) m/z:515[M + H]+.
1H-NMR(CDCl3, 400 MHz)δ:1.72-1.91(3H, m), 2.05-2.10(1H, m), 2.70(3H, s), 3.30-3.40(2H, m), 3.54-3.59(1H, m), 3.88(1H, dd, J= 2.9, 13.2 Hz), 3.92(3H, s), 4.22-4.26(1H, m), 5.83(1H, d, J=7.3 Hz), 6.91(1H, d, J= 2.9 Hz), 6.96(1H, dd, J= 2.9, 9.3 Hz), 7.42(2H, d, 8.8 Hz), 7.86(2H, d, J= 8.8 Hz), 8.02(1H, d, J= 9.3 Hz).
1H-NMR(CDCl3, 400 MHz)δ:1.72-1.91(3H, m), 2.05-2.10(1H, m), 2.70(3H, s), 3.30-3.40(2H, m), 3.54-3.59(1H, m), 3.88(1H, dd, J= 2.9, 13.2 Hz), 3.92(3H, s), 4.22-4.26(1H, m), 5.83(1H, d, J=7.3 Hz), 6.91(1H, d, J= 2.9 Hz), 6.96(1H, dd, J= 2.9, 9.3 Hz), 7.42(2H, d, 8.8 Hz), 7.86(2H, d, J= 8.8 Hz), 8.02(1H, d, J= 9.3 Hz).
工程2:
(S)-2-アミノ-5-(3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]-ピペリジノ)安息香酸メチルエステル
(S)-2-アミノ-5-(3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]-ピペリジノ)安息香酸メチルエステル

テトラヒドロフラン 2.97 Lに(S)-5-(3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ)-2-ニトロ安息香酸メチルエステル 424 g (823 mmol)を撹拌下投入して溶解させ、次いでメタノール2.97 Lおよび塩化ニッケル(II)六水和物39.1 g (165 mmol)を投入した。冷却下、内温8~20℃で水素化ホウ素ナトリウム93.4 g (2.47 mol)をゆっくり投入し、内温6~20℃で1.5時間撹拌した。反応液に酢酸エチル5.94 Lおよび塩化アンモニウム水溶液(塩化アンモニウム297 g +常水2.67 L)を投入し、加熱後、内温30~35℃で30分間撹拌した。有機層を分取し、塩化アンモニウム水溶液(塩化アンモニウム297 g +常水2.67 L)および塩化アンモニウム水溶液(塩化アンモニウム212 g +常水1.91 L)で順次洗浄した。有機層にN,N-ジメチルホルムアミド 2.12 Lを投入し、外温40℃以下で減圧濃縮した。濃縮残留物にN,N-ジメチルホルムアミド 2.12 Lを投入し、加熱して溶解した後、内温50~56℃で常水848 mLを滴下し、結晶化させた後、内温55~56℃で15分間撹拌した後、内温50~55℃で常水3.39 Lを滴下し、冷却して、内温10~25℃で30分間撹拌した。結晶をろ取し、常水2.12 Lで洗浄して、帯緑褐色の湿潤粗結晶を得た。
湿潤粗結晶をメタノール6.36 Lに撹拌下投入し、次いで常水2.12 Lを投入し、内温50~58℃で30分間撹拌した。冷却し、内温8~25℃で30分間撹拌した後、結晶をろ取し、メタノール848 mLと常水848 mLの混液で洗浄した。60℃で20時間送風乾燥して、灰緑色結晶性粉末の(S)-2-アミノ-5-(3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ)安息香酸メチルエステル 348 g (718 mmol、87%)を得た。
湿潤粗結晶をメタノール6.36 Lに撹拌下投入し、次いで常水2.12 Lを投入し、内温50~58℃で30分間撹拌した。冷却し、内温8~25℃で30分間撹拌した後、結晶をろ取し、メタノール848 mLと常水848 mLの混液で洗浄した。60℃で20時間送風乾燥して、灰緑色結晶性粉末の(S)-2-アミノ-5-(3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ)安息香酸メチルエステル 348 g (718 mmol、87%)を得た。
FAB-MS(positive) m/z:485[M + H]+.
1H-NMR(CDCl3, 400 MHz)δ:1.72-1.78(2H, m), 1.88-1.90(1H, m), 2.75(3H, s), 2.79-2.84(1H, m), 3.06-3.15(2H, m), 3.20-3.22(1H, m), 3.88(3H, s), 4.41(1H, brs), 5.49(2H, brs), 6.59(1H, d, J= 7.8 Hz), 6.65(1H, d, 8.8 Hz), 7.05(1H, dd, J= 2.4, 8.8 Hz), 7.42(2H, d, J= 8.8 Hz), 7.45(1H, d, J= 2.4 Hz), 7.88(2H, d, J = 8.8 Hz).
1H-NMR(CDCl3, 400 MHz)δ:1.72-1.78(2H, m), 1.88-1.90(1H, m), 2.75(3H, s), 2.79-2.84(1H, m), 3.06-3.15(2H, m), 3.20-3.22(1H, m), 3.88(3H, s), 4.41(1H, brs), 5.49(2H, brs), 6.59(1H, d, J= 7.8 Hz), 6.65(1H, d, 8.8 Hz), 7.05(1H, dd, J= 2.4, 8.8 Hz), 7.42(2H, d, J= 8.8 Hz), 7.45(1H, d, J= 2.4 Hz), 7.88(2H, d, J = 8.8 Hz).
(実施例3~実施例14)
実施例1の工程3の方法に準じ、2-アミノ-5-ピペリジノ安息香酸誘導体(5)から3-ピペリジノ安息香酸誘導体(6)への脱アミノ化反応(工程3)を各種条件下で行った。結果を表1にまとめる。
実施例1の工程3の方法に準じ、2-アミノ-5-ピペリジノ安息香酸誘導体(5)から3-ピペリジノ安息香酸誘導体(6)への脱アミノ化反応(工程3)を各種条件下で行った。結果を表1にまとめる。


上記検討結果から、本発明の製造方法によると、種々の構造を有する2-アミノ-5-ピペリジノ安息香酸誘導体(5)や、種々の反応溶媒を用いても、良好な収率で3-ピペリジノ安息香酸誘導体(6)が得られることがわかる。特に、還元剤として次亜リン酸を用い、テトラヒドロフランを含有する溶媒を用いた場合、高収率であった。
(比較例1)特許文献1記載の製造方法
従来知られていた3-ピペリジノ安息香酸誘導体(6)の製造方法として、特許文献1に記載の製造方法を表2に示す。
従来知られていた3-ピペリジノ安息香酸誘導体(6)の製造方法として、特許文献1に記載の製造方法を表2に示す。


特許文献1に記載の製造方法は、本発明の製造方法と比較して低収率であることがわかる。また、特許文献1の製造方法は、シリカゲル等による精製工程が必要であることから、精製効率が悪い。
(比較例2)亜硝酸エステルを用いた脱アミノ化反応
3-{3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ}安息香酸メチルエステル
3-{3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ}安息香酸メチルエステル

亜硝酸第三ブチル(0.0889 mL, 0.750 mmol)のテトラヒドロフラン溶液(0.5 mL)に、2-アミノ-5-{3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド]ピペリジノ}安息香酸メチルエステル 243 mg(0.500 mmol)のテトラヒドロフラン溶液(1.5 mL)を加えた後、加熱還流下1時間撹拌した。反応液を濃縮後、残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル = 10:1 → 2:1)で精製したところ無色粉末の3-{3-[2-(4-クロロフェニル)-4-メチルチアゾール-5-カルボキサミド] ピペリジノ}安息香酸メチルエステルを88.0 mg(37%)得た。
(比較例3)~(比較例12)
比較例2の方法に準じ、各種条件下で2-アミノ-5-ピペリジノ安息香酸誘導体(5)から3-ピペリジノ安息香酸誘導体(6)への脱アミノ化反応を行った。結果を表3にまとめる。
比較例2の方法に準じ、各種条件下で2-アミノ-5-ピペリジノ安息香酸誘導体(5)から3-ピペリジノ安息香酸誘導体(6)への脱アミノ化反応を行った。結果を表3にまとめる。


表3に示すように、汎用されている亜硝酸エステルを用いた脱アミノ化反応では、3-ピペリジノ安息香酸誘導体(6)の収率が低く、シリカゲル等による精製が必要であることから精製効率も十分ではなかった。
以上の結果から、従来の製造方法や汎用されている製造方法と比較して、本発明の製造方法は高収率で3-ピペリジノ安息香酸誘導体(6)が得られ、かつ、得られた3-ピペリジノ安息香酸誘導体(6)も高純度であることから、精製も極めて容易である。したがって、本発明の製造方法は工業レベルの製造にも適する。
本出願は、2008年12月25日出願の日本特許出願(特願2008-329224)、に基づくものであり、その内容はここに参照として取り込まれる。
本発明の環状アミノ安息香酸誘導体の製造方法は、2-ニトロ安息香酸誘導体(1)とピペリジン誘導体(2)を原料とし、亜硝酸化合物を用いた還元的な脱離反応を反応工程中に用いることによって、一般式(A’)で表されるPPARαアゴニスト及びその水和物を収率良く提供でき、その工業的製造方法として有用である。
Claims (6)
- 一般式(5):

R2は水素原子、低級アルキル基またはアラルキル基を表し;
R3は低級アルキル基、低級アルコキシ基、アラルキルオキシ基、アリール基または一般式(3):

R4は水素原子または低級アルキル基を表し;
R5は、
a)ハロゲン原子、
b)ハロゲン原子で置換されていてもよい低級アルキル基及び
c)低級アルコキシ基
からなる置換基群の中からそれぞれ独立して選ばれた1~3個の置換基を有していてもよいベンゼン環を表す)で表される芳香族複素環基を表す]で表される2-アミノ-5-ピペリジノ安息香酸誘導体を、亜硝酸またはその塩と還元剤を用いて脱アミノ化することを特徴とする一般式(6):

- 一般式(1):


R3は低級アルキル基、低級アルコキシ基、アラルキルオキシ基、アリール基または一般式(3):

R4は水素原子または低級アルキル基を表し;
R5は、
a)ハロゲン原子、
b)ハロゲン原子で置換されていてもよい低級アルキル基及び
c)低級アルコキシ基
からなる置換基群の中からそれぞれ独立して選ばれた1~3個の置換基を有していてもよいベンゼン環を表す)で表される芳香族複素環基を表す]で表されるピペリジン誘導体を反応させることにより、一般式(4):



- 前記一般式(1)~(6)において、R1が低級アルキル基、Xがハロゲン原子、R2が水素原子、R3が低級アルコキシ基または一般式(3a):

- 前記一般式(1)~(6)において、R3が一般式(3a):

- 脱アミノ化工程において、亜硝酸またはその塩が亜硝酸ナトリウムである、請求項1~4の何れかに記載の3-ピペリジノ安息香酸誘導体又はその水和物の製造方法。
- 脱アミノ化工程において、還元剤が次亜リン酸である、請求項1~5の何れかに記載の3-ピペリジノ安息香酸誘導体又はその水和物の製造方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008-329224 | 2008-12-25 | ||
| JP2008329224A JP2012051803A (ja) | 2008-12-25 | 2008-12-25 | 環状アミノ安息香酸エステル誘導体の製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010074110A1 true WO2010074110A1 (ja) | 2010-07-01 |
Family
ID=42287717
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2009/071375 WO2010074110A1 (ja) | 2008-12-25 | 2009-12-24 | 環状アミノ安息香酸誘導体の製造方法 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP2012051803A (ja) |
| WO (1) | WO2010074110A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6124012B2 (ja) * | 2011-05-26 | 2017-05-10 | 日産化学工業株式会社 | 1−(2−アミノ−置換フェニル)−2−ハロ−2,2−ジフルオロエタノン化合物及び1−(置換フェニル)−2−ハロ−2,2−ジフルオロエタノン化合物の製造方法 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005009969A1 (en) * | 2003-07-31 | 2005-02-03 | Sanofi-Aventis | Aminoquinoline derivatives and their use as adenosine a3 ligands |
| WO2005080388A1 (en) * | 2004-02-20 | 2005-09-01 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| WO2006010591A2 (en) * | 2004-07-27 | 2006-02-02 | Novartis Ag | Quinazoline derivatives |
| WO2006012577A2 (en) * | 2004-07-22 | 2006-02-02 | Bayer Pharmaceuticals Corporation | Quinazolinone derivatives useful for the regulation of glucose homeostasis and food intake |
| EP1780210A1 (en) * | 2004-08-11 | 2007-05-02 | Kyorin Pharmaceutical Co., Ltd. | Novel cyclic aminobenzoic acid derivative |
| WO2008113161A1 (en) * | 2007-03-19 | 2008-09-25 | Ulysses Pharmaceutical Products Inc. | Phosphate prodrugs of quinazolinyl nitrofurans, methods of obtaining, and use of same |
-
2008
- 2008-12-25 JP JP2008329224A patent/JP2012051803A/ja active Pending
-
2009
- 2009-12-24 WO PCT/JP2009/071375 patent/WO2010074110A1/ja active Application Filing
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005009969A1 (en) * | 2003-07-31 | 2005-02-03 | Sanofi-Aventis | Aminoquinoline derivatives and their use as adenosine a3 ligands |
| WO2005080388A1 (en) * | 2004-02-20 | 2005-09-01 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| WO2006012577A2 (en) * | 2004-07-22 | 2006-02-02 | Bayer Pharmaceuticals Corporation | Quinazolinone derivatives useful for the regulation of glucose homeostasis and food intake |
| WO2006010591A2 (en) * | 2004-07-27 | 2006-02-02 | Novartis Ag | Quinazoline derivatives |
| EP1780210A1 (en) * | 2004-08-11 | 2007-05-02 | Kyorin Pharmaceutical Co., Ltd. | Novel cyclic aminobenzoic acid derivative |
| WO2008113161A1 (en) * | 2007-03-19 | 2008-09-25 | Ulysses Pharmaceutical Products Inc. | Phosphate prodrugs of quinazolinyl nitrofurans, methods of obtaining, and use of same |
Non-Patent Citations (3)
| Title |
|---|
| ELDERFIELD R.C. ET AL: "Synthesis of Bz- polymethoxy-8-aminoquinolines and some derivatives thereof", JOURNAL OF ORGANIC CHEMISTRY, vol. 17, 1952, pages 358 - 370 * |
| FISHER T.H. ET AL: "Meta-substituent effects on benzyl free-radical stability", JOURNAL OF ORGANIC CHEMISTRY, vol. 55, no. 3, 1990, pages 1040 - 1043 * |
| OGAWA H. ET AL: "Studies on positive inotropic agents. VI. Synthesis of 1-aromatic ring substituted 4-(3, 4-dimethoxybenzoyl)piperazine derivatives", CHEMICAL & PHARMACEUTICAL BULLETIN, vol. 36, no. 7, 1988, pages 2401 - 2409 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6124012B2 (ja) * | 2011-05-26 | 2017-05-10 | 日産化学工業株式会社 | 1−(2−アミノ−置換フェニル)−2−ハロ−2,2−ジフルオロエタノン化合物及び1−(置換フェニル)−2−ハロ−2,2−ジフルオロエタノン化合物の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2012051803A (ja) | 2012-03-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5649971B2 (ja) | 2‐ヒドロキシ‐5‐フェニルアルキルアミノ安息香酸誘導体及びその塩の製造方法 | |
| EP3088391B1 (en) | Method for producing benzyl ester 2-aminonicotinate derivative | |
| WO2010074110A1 (ja) | 環状アミノ安息香酸誘導体の製造方法 | |
| JPWO2011001976A1 (ja) | スレオ−3−(3,4−ジヒドロキシフェニル)−l−セリンの製造法 | |
| KR20090064456A (ko) | 트리플루오로에톡시톨루엔의 제조방법 | |
| JP2008105970A (ja) | ジヒドロキノリン誘導体の製造方法及びその中間体 | |
| JP2773587B2 (ja) | O,o´−ジアシル酒石酸無水物の製造法 | |
| JP5380743B2 (ja) | 光学活性な4−アミノ−3−置換フェニルブタン酸の製造方法 | |
| CN106414414A (zh) | 三酮化合物的制造方法 | |
| KR100881890B1 (ko) | 사포그렐레이트 염산염의 제조방법 | |
| JPH01313457A (ja) | N‐(3’,4’‐ジメトキシ−シンナモイル)‐アンスラニル酸の製造方法 | |
| JP4245490B2 (ja) | 2−(ジクロロフェニル)−4−フェニルイミダゾール化合物 | |
| JP2003201281A (ja) | 4−(2−メチル−1−イミダゾリル)−2,2−ジフェニルブタンアミドの製造方法 | |
| JP5087059B2 (ja) | 4−(2−メチル−1−イミダゾリル)−2,2−ジフェニルブタンアミドの製造方法 | |
| EP0190524B1 (fr) | Nouveau procédé industriel de synthèse du N-[(1'-allyl 2'-pyrrolidinyl) méthyl] 2-méthoxy 4,5-azimido benzamide | |
| KR101004133B1 (ko) | 아세틸렌 화합물의 제조방법 | |
| JPS6327337B2 (ja) | ||
| US20070149606A1 (en) | Process for producing phenylacetic acid derivative | |
| JPH0140832B2 (ja) | ||
| JP2006298872A (ja) | 1−フルオロ−1−フェニルチオエテンの製造方法 | |
| JP4968602B2 (ja) | ベンズアミド誘導体の製造方法 | |
| JP2022025121A (ja) | プロリンアミド化合物の製造方法 | |
| JP2013035854A (ja) | テトラヒドロピラン化合物の製造方法 | |
| JPH05194336A (ja) | アミノアクリル酸誘導体 | |
| JPH06279380A (ja) | 芳香族アミド類の製造法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09834908 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09834908 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |