WO2010058751A1 - [1,3,4]オキサジアゾール化合物の薬理上許容される塩又はその水和物 - Google Patents
[1,3,4]オキサジアゾール化合物の薬理上許容される塩又はその水和物 Download PDFInfo
- Publication number
- WO2010058751A1 WO2010058751A1 PCT/JP2009/069431 JP2009069431W WO2010058751A1 WO 2010058751 A1 WO2010058751 A1 WO 2010058751A1 JP 2009069431 W JP2009069431 W JP 2009069431W WO 2010058751 A1 WO2010058751 A1 WO 2010058751A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acceptable salt
- pharmacologically acceptable
- acid
- compound
- hydrate
- Prior art date
Links
- 150000003839 salts Chemical class 0.000 title claims abstract description 80
- -1 [1,3,4]oxadiazole compound Chemical class 0.000 title claims description 27
- 150000001875 compounds Chemical class 0.000 claims abstract description 100
- 239000013078 crystal Substances 0.000 claims abstract description 56
- 238000000034 method Methods 0.000 claims abstract description 12
- KFDCQAWKWQKJRS-JBLDHEPKSA-N N'-[(1S,2R,4S)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]-4-(1,3,4-oxadiazol-2-yl)cyclohexyl]oxamide Chemical compound CN1CCc2nc(sc2C1)C(=O)N[C@@H]1C[C@H](CC[C@@H]1NC(=O)C(N)=O)c1nnco1 KFDCQAWKWQKJRS-JBLDHEPKSA-N 0.000 claims description 59
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 claims description 42
- 239000002253 acid Substances 0.000 claims description 41
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 claims description 39
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 38
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 33
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 33
- 238000004519 manufacturing process Methods 0.000 claims description 23
- 239000000243 solution Substances 0.000 claims description 21
- 239000002904 solvent Substances 0.000 claims description 19
- 238000003756 stirring Methods 0.000 claims description 17
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 claims description 14
- 239000003814 drug Substances 0.000 claims description 13
- 238000001914 filtration Methods 0.000 claims description 13
- 208000007536 Thrombosis Diseases 0.000 claims description 11
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 claims description 11
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 claims description 10
- YIKSCQDJHCMVMK-UHFFFAOYSA-N Oxamide Chemical compound NC(=O)C(N)=O YIKSCQDJHCMVMK-UHFFFAOYSA-N 0.000 claims description 10
- 230000015572 biosynthetic process Effects 0.000 claims description 10
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 10
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 10
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 10
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 10
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 claims description 9
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 claims description 9
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 claims description 9
- 238000000354 decomposition reaction Methods 0.000 claims description 9
- 239000011976 maleic acid Substances 0.000 claims description 9
- 229940124597 therapeutic agent Drugs 0.000 claims description 9
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 claims description 8
- 238000002076 thermal analysis method Methods 0.000 claims description 8
- 208000005189 Embolism Diseases 0.000 claims description 7
- 238000010438 heat treatment Methods 0.000 claims description 7
- 229940049920 malate Drugs 0.000 claims description 7
- 230000000069 prophylactic effect Effects 0.000 claims description 7
- 239000000706 filtrate Substances 0.000 claims description 6
- 239000000047 product Substances 0.000 claims description 6
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 claims description 4
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 claims description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 claims description 4
- 206010051055 Deep vein thrombosis Diseases 0.000 claims description 4
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 claims description 4
- 108010074860 Factor Xa Proteins 0.000 claims description 4
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 claims description 4
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 claims description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 4
- 229910019142 PO4 Inorganic materials 0.000 claims description 4
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 claims description 4
- 206010047249 Venous thrombosis Diseases 0.000 claims description 4
- 230000023555 blood coagulation Effects 0.000 claims description 4
- 239000001530 fumaric acid Substances 0.000 claims description 4
- 239000001630 malic acid Substances 0.000 claims description 4
- 235000011090 malic acid Nutrition 0.000 claims description 4
- 238000002844 melting Methods 0.000 claims description 4
- 230000008018 melting Effects 0.000 claims description 4
- 239000011259 mixed solution Substances 0.000 claims description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 claims description 4
- 239000010452 phosphate Substances 0.000 claims description 4
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 claims description 4
- 229940095064 tartrate Drugs 0.000 claims description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 4
- 206010002383 Angina Pectoris Diseases 0.000 claims description 3
- 206010008088 Cerebral artery embolism Diseases 0.000 claims description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 claims description 3
- 208000001435 Thromboembolism Diseases 0.000 claims description 3
- 229940077388 benzenesulfonate Drugs 0.000 claims description 3
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 claims description 3
- 239000003130 blood coagulation factor inhibitor Substances 0.000 claims description 3
- 206010008118 cerebral infarction Diseases 0.000 claims description 3
- 208000026106 cerebrovascular disease Diseases 0.000 claims description 3
- 201000010849 intracranial embolism Diseases 0.000 claims description 3
- 208000010125 myocardial infarction Diseases 0.000 claims description 3
- 230000000250 revascularization Effects 0.000 claims description 3
- 230000004580 weight loss Effects 0.000 claims description 3
- 206010003658 Atrial Fibrillation Diseases 0.000 claims description 2
- 208000010378 Pulmonary Embolism Diseases 0.000 claims description 2
- 239000003112 inhibitor Substances 0.000 claims description 2
- 239000008194 pharmaceutical composition Substances 0.000 claims description 2
- 201000010099 disease Diseases 0.000 abstract description 7
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 7
- 230000003073 embolic effect Effects 0.000 abstract description 6
- 230000001732 thrombotic effect Effects 0.000 abstract description 6
- 230000002401 inhibitory effect Effects 0.000 abstract description 5
- 230000002265 prevention Effects 0.000 abstract description 3
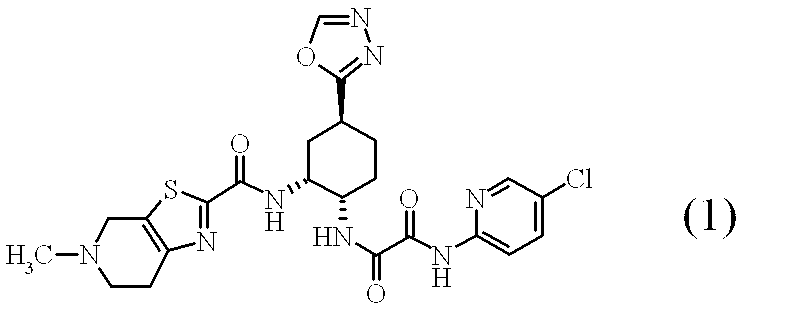
- GRFOFWKESSPXRZ-DUVNUKRYSA-N n'-(5-chloropyridin-2-yl)-n-[(1s,2r,4s)-2-[(5-methyl-6,7-dihydro-4h-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]-4-(1,3,4-oxadiazol-2-yl)cyclohexyl]oxamide Chemical compound N([C@H]1CC[C@@H](C[C@H]1NC(=O)C1=NC=2CCN(CC=2S1)C)C=1OC=NN=1)C(=O)C(=O)NC1=CC=C(Cl)C=N1 GRFOFWKESSPXRZ-DUVNUKRYSA-N 0.000 abstract 1
- 238000003860 storage Methods 0.000 description 20
- 239000000203 mixture Substances 0.000 description 11
- 230000007774 longterm Effects 0.000 description 10
- 239000007857 degradation product Substances 0.000 description 9
- 238000002360 preparation method Methods 0.000 description 8
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- 238000009472 formulation Methods 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 238000010521 absorption reaction Methods 0.000 description 4
- 239000008186 active pharmaceutical agent Substances 0.000 description 4
- 238000003795 desorption Methods 0.000 description 4
- 229940088679 drug related substance Drugs 0.000 description 4
- 150000002688 maleic acid derivatives Chemical class 0.000 description 4
- 238000005406 washing Methods 0.000 description 4
- FKASFBLJDCHBNZ-UHFFFAOYSA-N 1,3,4-oxadiazole Chemical class C1=NN=CO1 FKASFBLJDCHBNZ-UHFFFAOYSA-N 0.000 description 3
- 0 CN(CC1)CC(*2)=C1N=C2C(N=C)=[Cl-] Chemical compound CN(CC1)CC(*2)=C1N=C2C(N=C)=[Cl-] 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- 238000010586 diagram Methods 0.000 description 3
- 150000004683 dihydrates Chemical class 0.000 description 3
- 238000009776 industrial production Methods 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 208000010718 Multiple Organ Failure Diseases 0.000 description 2
- 206010038563 Reocclusion Diseases 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000002198 insoluble material Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 230000003449 preventive effect Effects 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- 208000034486 Multi-organ failure Diseases 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 208000006193 Pulmonary infarction Diseases 0.000 description 1
- 206010043540 Thromboangiitis obliterans Diseases 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- 239000003377 acid catalyst Substances 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 206010061592 cardiac fibrillation Diseases 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 230000015271 coagulation Effects 0.000 description 1
- 238000005345 coagulation Methods 0.000 description 1
- 238000011978 dissolution method Methods 0.000 description 1
- 238000010981 drying operation Methods 0.000 description 1
- 238000000921 elemental analysis Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 230000002600 fibrillogenic effect Effects 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 210000002837 heart atrium Anatomy 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 150000004701 malic acid derivatives Chemical class 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 208000029744 multiple organ dysfunction syndrome Diseases 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 238000004321 preservation Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 238000011085 pressure filtration Methods 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 230000007575 pulmonary infarction Effects 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
Images






Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Definitions
- the present invention exhibits an inhibitory action on activated blood coagulation factor X (FXa), has high water solubility and excellent long-term storage stability, and is useful as a prophylactic / therapeutic agent for thrombotic and / or embolic diseases.
- the present invention relates to a pharmacologically acceptable salt of compound (1) which is a [1,3,4] oxadiazole derivative, a hydrate thereof, or a crystal thereof, and an industrial production method thereof.
- the following compound (1) is a compound that exhibits a strong FXa inhibitory action and is useful as a prophylactic / therapeutic agent for thrombotic and / or embolic diseases, as disclosed in Patent Document 1.
- An object of the present invention is to provide a compound (1) useful as a pharmaceutical compound for the prevention and treatment of thrombotic and / or embolic diseases, which has high water solubility, high quality, and long-term storage stability required as a pharmaceutical product. It is an object to provide a drug substance form having a high molecular weight and an industrial production method for the drug substance form.
- the present inventors examined not only the pharmacological action but also the final drug substance form of a compound useful as a pharmaceutical in terms of solubility in water and long-term storage stability.
- basic compounds are known to form acid addition salts with acids for preparing pharmacologically acceptable salts, but for preparing pharmacologically acceptable salts with compound (1).
- an acid decomposition product (A) accompanied by decomposition of [1,3,4] oxadiazole was obtained, and [1,3,4] oxadiazole was obtained. It became clear that there was a big problem in manufacture of the acid addition salt of the compound (1) having a ring.
- Step D a step of filtering crystals precipitated by the stirring operation of (Step C) above;
- the present invention provides the following pharmaceutical (16): a pharmaceutical comprising the compound according to any one of (1) to (9); (17): an activated blood coagulation factor X (FXa) inhibitor comprising the compound according to any one of (1) to (9); (18): a blood coagulation inhibitor containing the compound according to any one of (1) to (9); (19): A prophylactic and / or therapeutic agent for thrombus or embolus containing the compound according to any one of (1) to (9); (20): After cerebral infarction, cerebral embolism, myocardial infarction, angina pectoris, deep vein thrombosis, deep vein thrombosis, artificial valve / joint replacement containing the compound according to any one of (1) to (9) Thrombus formation, Thrombus formation and reocclusion after revascularization, Non-valvular atrial fibrillation (NVAF) preventive and / or
- NVAF Non-valvular atrial fibrillation
- the pharmacologically acceptable salt of the compound (1) of the present invention or a hydrate thereof, or a crystal thereof, in particular, the 1-maleate crystal (1-a) of the compound (1) has good water solubility. Because there is no change in weight due to moisture absorption and desorption, there are few concerns about formulation as a pharmaceutical, excellent long-term storage stability, and excellent FXa inhibitory action. is there. Therefore, the 1-maleate crystal (1-a) of compound (1) is useful as a pharmaceutical compound for the prevention / treatment of thrombotic and / or embolic diseases.
- FIG. 3 is a diagram showing a powder X-ray diffraction pattern of 1 ⁇ maleate salt (1-a) of compound (1).
- FIG. 5 is a graph showing the moisture absorption / desorption property of 1 ⁇ maleate (1-a) of compound (1).
- FIG. 4 is a table showing the solubility of 1 ⁇ maleate salt (1-a) of compound (1) in water, JP 1 liquid and JP 2 liquid at 37 ° C. : Is a table showing the long-term storage stability prediction by Arrhenius plot based on the actual measurement value of the 1-maleate salt (1-a) of compound (1) when stored for 2 months.
- Examples of pharmacologically acceptable salts in the present invention include inorganic acid addition salts such as hydrochloride, hydrobromide, sulfate, phosphate and nitrate, acetate, propionate, glycolate, and succinic acid.
- Organic acid addition salts such as salts, lactate, oxalate, maleate, tartrate, malonate, succinate, fumarate, mesylate, tosylate, ascorbate .
- Examples of the acid addition salt of compound (1) include hydrochloride, hydrobromide, sulfate, mesylate, benzenesulfonate, tosylate, phosphate, acetate, glycolate, lactate, Malonate, tartrate, malate, maleate, citrate, succinate or hydrates thereof are preferred; hydrochloride and maleate are more preferred; preferable.
- the pharmacologically acceptable salt of the above compound (1) or a hydrate thereof is preferably a crystal; hydrochloride, hydrobromide, maleate, malate, fumarate and glycolate or Their hydrate crystals are more preferred; hydrochloride crystals and maleate crystals are more preferred; 1 ⁇ maleate crystals are particularly preferred.
- the diffraction angle (2 ⁇ ) by powder X-ray diffraction is 7.4, 12.6, 14.7, 18.3, 20.9, 22.7. , 24.2, 24.7, and 25.6 degrees ( ⁇ 0.2 degrees) are preferred.
- the diffraction angle (2 ⁇ ) by powder X-ray diffraction is 6.1, 8.3, 12.3, 13.2, 16.0, 17.8, 19 Those having characteristic peaks at 0.0, 20.9, and 23.6 degrees ( ⁇ 0.2 degrees) are preferred.
- the crystal of the malate salt of the compound (1) has a diffraction angle (2 ⁇ ) by powder X-ray diffraction of 5.2, 8.0, 8.4, 15.5, and 15.9 degrees ( ⁇ 0. Those having a characteristic peak at 2 degrees are preferred.
- the diffraction angle (2 ⁇ ) by powder X-ray diffraction is 5.5, 8.4, 13.2, 18.5, 19.2, 22.3, 29. Those having characteristic peaks at 0.0 and 29.5 degrees ( ⁇ 0.2 degrees) are preferred.
- the maleate salt of compound (1) the following formula (1-a)
- the crystals of compound (1-a) are 8.4, 13.9, 15.4, 16.7, 17.2, 17.7, 18.8 as the diffraction angle (2 ⁇ ) by powder X-ray diffraction.
- the crystal of the compound (1-a) is 8.4, 13.9, 15.4, 16.7, 17.2, 17.7, 18 as the diffraction angle (2 ⁇ ) by powder X-ray diffraction. .8, 19.4, 22.3, 22.1, 23.0 and 27.9 degrees ( ⁇ 0.2 degrees) with characteristic peaks, and a) temperatures between 220 ° C. and 225 ° C. Thermal analysis (DTA) profile with endothermic peaks in b) Weight loss at temperatures between 210 ° C. and 230 ° C.
- DTA thermal analysis
- melting point Is preferably characterized by:
- a method of producing a pharmacologically acceptable salt of the above compound (1) or a hydrate crystal thereof in order to suppress the formation of a degradation product [acid degradation product (A)] in the production process, It is preferred to add in portions the acid to prepare the acceptable salt.
- a method for producing a crystal of compound (1) in a high yield while suppressing the formation of decomposition products in the production process it corresponds to about half the amount of compound (1) and 1 mol of compound (1).
- the acid for preparing 0.498 to 0.502 mol of a pharmacologically acceptable salt is preferably added in two divided portions.
- a more specific method for producing a pharmacologically acceptable salt of compound (1) or a hydrate thereof, or a crystal thereof includes the following (Step A) to (Step D). .
- Step A N 1- (5-chloropyridin-2-yl) -N 2 -[(1S, 2R, 4S) -2- ⁇ [(5-methyl-4,5,6,7- Tetrahydrothiazolo [5,4-c] pyridin-2-yl) carbonyl] amino ⁇ -4-([1,3,4] oxadiazol-2-yl) cyclohexyl] ethanediamide (1) and compound (1 ) Adding an acid for preparing 0.498 to 0.502 mol of a pharmacologically acceptable salt with respect to 1 mol of), heating in a solvent and stirring; (Step B): A step of filtering the mixed solution obtained in (Step A) and filtering off insoluble matters while hot; (Step C): In order to prepare the pharmacologically acceptable salt of 0.498 to 0.502 mol with respect to 1 mol of compound (1) by heating the filtrate of (Step B).
- Step D a step of filtering crystals precipitated by the stirring operation of (Step C) above.
- the solvent in (Step A) water-containing methanol, water-containing ethanol, water-containing propanol, water-containing isopropanol, water-containing acetone or the like is preferable; More preferably hydrous ethanol or hydrous isopropanol; Ethanol containing 10 to 25% by volume of water is particularly preferred.
- the amount of the solvent in (Step A) is preferably 5 to 15 times (volume / weight) relative to the weight of the compound (1); more preferably 8 to 12 times (volume / weight).
- the time for heating and stirring so that the internal temperature of the mixed solution in (Step A) is 60 to 70 ° C. is preferably 30 minutes to 1 hour.
- the filter paper used in the step of hot filtration in (Step B) may be a commercially available one that is usually used, and filtration may be performed under normal pressure filtration or reduced pressure.
- the insoluble material is preferably washed with the same solvent as in (Step A) heated to 60 to 70 ° C .; the amount of the solvent used for washing is compound (1) and a pharmacologically acceptable salt. It is preferably 1 to 3 times (volume / weight) relative to the weight of the acid to be used; more preferably 2 times (volume / weight).
- the acid for preparing the pharmacologically acceptable salt is re-added, so that the acid for preparing the pharmacologically acceptable salt is added dropwise in a solution using a suitable solvent.
- a suitable solvent and its use amount, and a temperature for preparing an acid solution for preparing a pharmacologically acceptable salt include pharmacology. If the acid to prepare the top acceptable salt is an organic acid, it is preferable to select a solvent and dissolution method that prevents side reactions such as esterification with its own self-acid catalyst.
- Solvents for preparing acid solutions for preparing pharmacologically acceptable salts include water-containing C1-C3 alcohols; C1-C3 alcohols containing 3-7% by volume of water are preferred; More preferably, ethanol containing 7% by volume or isopropanol containing 3-7% by volume of water; More preferred is an ethanol solution containing 3-7% by volume of water: An ethanol solution containing 5% by volume of water is particularly preferred.
- the amount of the solvent used to prepare the acid solution for preparing the pharmacologically acceptable salt used in (Step C) is preferably 5 to 15 times (volume / weight) with respect to compound (1); 10 times (capacity / weight) is more preferable.
- the temperature at which the acid solution for preparing a pharmacologically acceptable salt in (Step C) is added dropwise is preferably 40 to 50 ° C. which is the temperature of the filtrate, and the addition time is about 10 minutes. preferable.
- Stirring after completion of the dropping in (Step C) gradually lowers the temperature of the solution to complete crystallization.
- the temperature and time for stirring after completion of the dropping in (Step C) are 40 to 50 ° C. for 1.5 to 3 hours, 20 to 30 ° C. for 1.5 to 3 hours, and 0 to 10 ° C. for 10 to 10 hours. It is preferable to stir for 15 hours.
- (Step D) is a step of filtering crystals precipitated by the stirring operation of (Step C).
- the operation of filtering the precipitated crystals in (Step D) is preferably performed at 0 to 10 ° C.
- the filtered crystals of (Step D) may be washed or washed, and the washing solvent for the washing operation is preferably ethanol containing 10 to 20% by volume of water that has been cooled to 0 to 10 ° C. in advance; Ethanol containing 15% by volume of water that has been cooled to 0 to 10 ° C. in advance is more preferable.
- the amount of the washing solvent used is preferably 2 to 5 times (volume / weight) relative to compound (1); more preferably 3 times (volume / weight).
- the drying operation is preferably carried out at 40-60 ° C.
- N 1- (5-chloropyridin-2-yl) -N 2 -[(1S, 2R, 4S) -2- ⁇ [(5-methyl-4,5,6,7) of compound (1) -Tetrahydrothiazolo [5,4-c] pyridin-2-yl) carbonyl] amino ⁇ -4-([1,3,4] oxadiazol-2-yl) cyclohexyl] ethanediamide (1) (3.00 g 5.50 mmol), C1 to C3 alcohol containing 10 to 25% by volume of water is added 8 to 12 times (volume / weight).
- the wet body is dried under reduced pressure at 40 ° C. to give the corresponding hydrochloride, hydrobromide, maleate, malate, fumarate or glycolate.
- the thus obtained salt of compound (1), hydrate thereof, or crystal thereof, particularly 1 ⁇ maleate salt (1-a) has good water solubility, high purity and storage stability. It is useful as an excellent medicine.
- compound (1-a) exhibits a highly activated blood coagulation factor X (FXa) inhibitory action, it is useful as a blood coagulation inhibitor, a prophylactic and / or therapeutic agent for thrombus or embolism.
- the compound (1-a) is used for various diseases causing thrombus or embolism, such as cerebral infarction, cerebral embolism, myocardial infarction, angina pectoris, pulmonary infarction, pulmonary embolism, Buerger's disease, deep vein thrombosis, generalized Intravascular coagulation syndrome, thrombus formation after prosthetic valve / joint replacement, thrombus formation and reocclusion after revascularization, multiple organ failure (MODS), thrombus formation during extracorporeal circulation or blood coagulation during blood collection, non-valvular atria It is useful as a prophylactic and / or therapeutic agent for thromboembolism based on fibrillation (NVAF).
- NVAF non-valvular atria
- the medicament of the present invention can be administered by various methods including oral administration. Moreover, when it is set as an injection, it can be administered by any method such as intravenous injection, intramuscular injection, and subcutaneous injection. About the preparation method of this formulation, a suitable formulation can be selected according to the administration method, and the preparation method of the various formulation normally used can be used. Examples of oral preparations include tablets, powders, granules, capsules, solutions, syrups, elixirs, oily or aqueous suspensions, and the like. As injections, stabilizers, preservatives, and solubilizing agents may be used in the preparation.
- the medicament containing the compound (1) of the present invention is preferably administered once per day per adult as the compound (1) and repeated at appropriate intervals.
- the dose is in the range of 0.01 mg to 1000 mg, preferably in the range of 0.1 mg to 200 mg, more preferably 1 mg to 100 mg.
- Example 1 The moisture absorption / desorption properties of the compound (1) and various salts of the compound (1) were evaluated. The amount of moisture adsorbed / desorbed was evaluated by measuring the change in weight over time in a relative humidity (RH) range of 10 to 90% in about 20 mg of crystals in a microbalance (automatic water vapor adsorption apparatus).
- RH relative humidity
- the 1-maleate salt (1-a) of compound (1) had a weight change of plus or minus 0.5% at 40 to 60% RH.
- the 1-maleate salt (1-a) of the compound (1) shows that the free form of the crystal of the compound (1) shows a change in weight at a relative humidity of 40-60%, and between the anhydride and hydrate. It became clear to solve the problem of mutual conversion.
- Test Example 2 According to the test method of the Japanese Pharmacopoeia, the solubility in water (37 ° C.), JP 1 liquid and 2 liquid was evaluated. As a result, as shown in the table in FIG. 6, the solubility of compound (1-a) in water was good.
- Test Example 4 The thermal analysis (TG / DTA) of various salts of compound (1) such as compound (1-a) and the free form of compound (1) was carried out.
- Test Example 5 After 2 months of refrigerated storage (5 ° C.) of various salts of compound (1) such as compound (1-a) and room temperature storage (25 ° C.), the acid degradation product (A) and The increase in total impurities was measured. Further, the increase in acid degradation products (A) and total impurities from the time point of 2 months was calculated by Arrhenius plot, and the long-term storage stability of each salt crystal was predicted. As a result, as shown in the table in FIG.
- the increase rate of the acid degradation product (A) after 2 months storage of the hydrochloride salt of the compound (1) was refrigerated (5 ° C.). Increased by 0.10% and stored at room temperature (25 ° C) by 0.54%. Moreover, in the increase prediction of the acid degradation product (A) at the time of 36 months by Arrhenius plot based on the actual measurement value at the time of storage for 2 months, 0.58% at refrigerated storage (5 ° C), at room temperature (25 ° C) The increase was 3.25%. On the other hand, as shown in the table of [FIG. 7], the actually measured values of the acid decomposition product (A) after the storage of 1-maleate salt (1-a) of compound (1) for 2 months are 5 ° C.
- the insoluble material was filtered while hot, and washed with ethanol (6 mL) containing 25% water that had been heated to 70 ° C. in advance.
- the internal temperature of the filtrate was kept at 45 ° C., and a solution of maleic acid (321.64 mg, 2.77 mmol) in 5% water containing ethanol (30 mL) was added dropwise over 10 minutes. After 5 minutes, crystallization started.
- the mixture was stirred at the same temperature for 2 hours and then allowed to cool. After stirring for 2 hours at an internal temperature of 21 to 28 ° C., the mixture was stirred for 12 hours in a low temperature room (room temperature 3-5 ° C.). Further, the mixture was cooled with ice water and stirred at an internal temperature of 2 ° C.
- the pharmacologically acceptable salt of the compound (1) of the present invention or a hydrate thereof, or a crystal thereof, in particular, the 1-maleate crystal (1-a) of the compound (1) has good water solubility. Since there is no weight change due to moisture absorption and desorption, there is no concern in the formulation process as a pharmaceutical, and it has excellent long-term storage stability, so that it can be used as a pharmaceutical compound for thrombotic and / or embolic diseases. It is useful as a preventive / therapeutic agent.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Pulmonology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
血栓性及び/又は塞栓性疾患の予防・治療用の医薬化合物として有用なFXa阻害作用を持つ化合物(1)の薬理上許容される塩、その水和物、又はそれらの結晶、及びそれらの工業的な製造方法を提供する。下記の式(1)で表される、N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミドの薬理上許容される塩、その水和物、又はそれらの結晶。
Description
本発明は、活性化血液凝固第X因子(FXa)の阻害作用を示し、水溶性が高く、かつ長期保存安定性にも優れ、血栓性及び/又は塞栓性疾患の予防・治療薬として有用な[1,3,4]オキサジアゾール誘導体である化合物(1)の薬理上許容される塩、その水和物、又はそれらの結晶、及びそれらの工業的な製造法に関する。
下記の化合物(1)は、特許文献1に開示されているように、強力なFXa阻害作用を示し、血栓性及び/又は塞栓性疾患の予防・治療薬として有用な化合物である。

これまでに、医薬化合物として有用である化合物(1)の薬理上許容される塩、その水和物、又はそれらの結晶に関する報告はない。また、化合物(1)の薬理上許容される塩、その水和物、又はそれらの結晶を工業的に製造する方法に関する報告はない。
本発明の課題は、血栓性及び/又は塞栓性疾患の予防・治療用の医薬化合物として有用である化合物(1)の、医薬品として求められる、高水溶性、高品質、かつ長期の保存安定性の高い原薬形態、及びその原薬形態の工業的な製造方法を提供することにある。
そこで本発明者らは、薬理作用だけでなく、水に対する溶解性や長期保存安定性等においても医薬品として有用な化合物の最終の原薬形態を見出すべく検討した。
一般に、塩基性化合物は薬理上許容される塩を調製するための酸との酸付加塩を形成することは知られているが、化合物(1)と薬理上許容される塩を調製するための酸との酸付加塩の製造を試み場合、[1,3,4]オキサジアゾールの分解を伴った酸分解物(A)を与えることが判明し、[1,3,4]オキサジアゾール環を持つ化合物(1)の酸付加塩の製造に大きな問題点があることが明らかになった。そこで、医薬品化合物として有望な化合物(1)の酸付加塩を製造する方法を獲得するために種々検討を行った。
その結果、化合物(1)の酸付加塩を製造する工業的な製造方法を見出し、高純度かつ高収率で医薬品化合物の最終の原薬形態としての良好な水溶性、長期保存安定性に優れる、化合物(1)の薬理上許容される塩又はその水和物、又はそれらの塩、特に化合物(1)の1・マレイン酸塩の結晶(1-a)を見いだし、本発明を完成するに至った。
すなわち、本発明は
(1):下記の式(1)
一般に、塩基性化合物は薬理上許容される塩を調製するための酸との酸付加塩を形成することは知られているが、化合物(1)と薬理上許容される塩を調製するための酸との酸付加塩の製造を試み場合、[1,3,4]オキサジアゾールの分解を伴った酸分解物(A)を与えることが判明し、[1,3,4]オキサジアゾール環を持つ化合物(1)の酸付加塩の製造に大きな問題点があることが明らかになった。そこで、医薬品化合物として有望な化合物(1)の酸付加塩を製造する方法を獲得するために種々検討を行った。
その結果、化合物(1)の酸付加塩を製造する工業的な製造方法を見出し、高純度かつ高収率で医薬品化合物の最終の原薬形態としての良好な水溶性、長期保存安定性に優れる、化合物(1)の薬理上許容される塩又はその水和物、又はそれらの塩、特に化合物(1)の1・マレイン酸塩の結晶(1-a)を見いだし、本発明を完成するに至った。
すなわち、本発明は
(1):下記の式(1)

で表される、N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミドの薬理上許容される塩又はその水和物;
(2):薬理上許容される塩が、塩酸塩、臭化水素酸塩、硫酸塩、メシル酸塩、ベンゼンスルホン酸塩、トシル酸塩、リン酸塩、酢酸塩、グリコール酸塩、乳酸塩、マロン酸塩、酒石酸塩、リンゴ酸塩、マレイン酸塩、クエン酸塩、コハク酸塩である(1)に記載の塩又はその水和物;及び
(3):薬理上許容される塩が、塩酸塩又はマレイン酸塩である(1)に記載の塩又はその水和物;
に関するものである。
また、本発明は、下記の
(4):下記の式(1)
(2):薬理上許容される塩が、塩酸塩、臭化水素酸塩、硫酸塩、メシル酸塩、ベンゼンスルホン酸塩、トシル酸塩、リン酸塩、酢酸塩、グリコール酸塩、乳酸塩、マロン酸塩、酒石酸塩、リンゴ酸塩、マレイン酸塩、クエン酸塩、コハク酸塩である(1)に記載の塩又はその水和物;及び
(3):薬理上許容される塩が、塩酸塩又はマレイン酸塩である(1)に記載の塩又はその水和物;
に関するものである。
また、本発明は、下記の
(4):下記の式(1)

で表される、N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミドの薬理上許容される塩又はその水和物の結晶;
(5):薬理上許容される塩が、塩酸塩、臭化水素酸塩、マレイン酸塩、りんご酸塩、フマル酸塩又はグリコール酸塩、又はそれらの水和物である(4)に記載の結晶;
(6):薬理上許容される塩が、塩酸塩又はマレイン酸塩、又はそれらの水和物である(4)に記載の結晶;
(7):下記の式(1-a)
(5):薬理上許容される塩が、塩酸塩、臭化水素酸塩、マレイン酸塩、りんご酸塩、フマル酸塩又はグリコール酸塩、又はそれらの水和物である(4)に記載の結晶;
(6):薬理上許容される塩が、塩酸塩又はマレイン酸塩、又はそれらの水和物である(4)に記載の結晶;
(7):下記の式(1-a)

で表される、N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド・1・マレイン酸塩の結晶である(4)に記載の結晶;
(8):粉末X線回折による回折角(2θ)として、8.4、13.9、15.4、16.7、17.2、17.7、18.8、19.4、20.3、22.1、23.0及び27.9度(±0.2度)に特徴的ピークを有する(7)に記載の結晶;及び
(9):さらに、
(a)220℃~225℃に吸熱量ピークを有する熱分析(DTA)プロフィール;
(b)210℃~230℃に重量減少を示す熱分析(TG)プロフィール;
(c)相対湿度40~60%における重量変化の増減がプラスマイナス0.5%以内;又は
(d)融点(分解)として202℃~204℃(未補正);
で特徴付けられる、(8)に記載の結晶;
に関するものである。
また、本発明は、下記の
(10):下記の式(1)
(8):粉末X線回折による回折角(2θ)として、8.4、13.9、15.4、16.7、17.2、17.7、18.8、19.4、20.3、22.1、23.0及び27.9度(±0.2度)に特徴的ピークを有する(7)に記載の結晶;及び
(9):さらに、
(a)220℃~225℃に吸熱量ピークを有する熱分析(DTA)プロフィール;
(b)210℃~230℃に重量減少を示す熱分析(TG)プロフィール;
(c)相対湿度40~60%における重量変化の増減がプラスマイナス0.5%以内;又は
(d)融点(分解)として202℃~204℃(未補正);
で特徴付けられる、(8)に記載の結晶;
に関するものである。
また、本発明は、下記の
(10):下記の式(1)
で表される、N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミドの薬理上許容される塩又はその水和物、又はそれらの結晶の製造方法であって、
薬理上許容される塩を調製するための酸を分割添加することを特徴とし、製造過程における分解物の生成を抑制する製造方法;
(11):(工程A):N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド(1)、及び化合物(1)の1モルに対して0.498~0.502モルの薬理上許容される塩を調製するための酸を添加し、溶媒中で加温して撹拌する工程;
(工程B):(工程A)で得られた混合液をろ過して、不溶物を熱時ろ別する工程;
(工程C):(工程B)のろ液を、ろ液を加温し、化合物(1)の1モルに対して0.498~0.502モルの薬理上許容される塩を調製するための酸の溶液を滴下し、滴下終了後、攪拌する工程;及び
(工程D):上記の(工程C)の攪拌操作で析出した結晶をろ過する工程;
上記の(工程A)~(工程D)からなる(10)に記載の製造方法;
(12):薬理上許容される塩を調製するための酸が、塩酸、臭化水素酸、マレイン酸、りんご酸、フマル酸又はグリコール酸である(10)又は(11)の製造方法;
(13):薬理上許容される塩を調製するための酸が、塩酸又はマレイン酸である(9)又は(10)の製造方法;
(14):(工程A)の溶媒が、含水C1~C3アルコールである(10)の製造方法;及び
(15):薬理上許容される塩を調製するための酸の溶液を調製する溶媒が、水を3~7容量%含むC1~C3アルコール溶液である(10)の製造方法;
に関するものである。
さらに本発明は、以下の
(16):(1)~(9)のいずれか1項に記載の化合物を含有する医薬;
(17):(1)~(9)のいずれか1項に記載の化合物を含有する活性化血液凝固第X因子(FXa)阻害剤;
(18):(1)~(9)のいずれか1項に記載の化合物を含有する血液凝固抑制剤;
(19):(1)~(9)のいずれか1項に記載の化合物を含有する血栓又は塞栓の予防及び/又は治療剤;
(20):(1)~(9)のいずれか1項に記載の化合物を含有する脳梗塞、脳塞栓、心筋梗塞、狭心症、肺塞栓、深部静脈血栓症、人工弁/関節置換後の血栓形成、血行再建後の血栓形成及び再閉塞、非弁膜性心房細動(NVAF)に基づく血栓塞栓症の予防剤及び/又は治療剤;及び
(21):(1)~(9)のいずれか1項に記載の化合物、及び薬理上許容される担体を含有する医薬組成物;
に関するものである。
薬理上許容される塩を調製するための酸を分割添加することを特徴とし、製造過程における分解物の生成を抑制する製造方法;
(11):(工程A):N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド(1)、及び化合物(1)の1モルに対して0.498~0.502モルの薬理上許容される塩を調製するための酸を添加し、溶媒中で加温して撹拌する工程;
(工程B):(工程A)で得られた混合液をろ過して、不溶物を熱時ろ別する工程;
(工程C):(工程B)のろ液を、ろ液を加温し、化合物(1)の1モルに対して0.498~0.502モルの薬理上許容される塩を調製するための酸の溶液を滴下し、滴下終了後、攪拌する工程;及び
(工程D):上記の(工程C)の攪拌操作で析出した結晶をろ過する工程;
上記の(工程A)~(工程D)からなる(10)に記載の製造方法;
(12):薬理上許容される塩を調製するための酸が、塩酸、臭化水素酸、マレイン酸、りんご酸、フマル酸又はグリコール酸である(10)又は(11)の製造方法;
(13):薬理上許容される塩を調製するための酸が、塩酸又はマレイン酸である(9)又は(10)の製造方法;
(14):(工程A)の溶媒が、含水C1~C3アルコールである(10)の製造方法;及び
(15):薬理上許容される塩を調製するための酸の溶液を調製する溶媒が、水を3~7容量%含むC1~C3アルコール溶液である(10)の製造方法;
に関するものである。
さらに本発明は、以下の
(16):(1)~(9)のいずれか1項に記載の化合物を含有する医薬;
(17):(1)~(9)のいずれか1項に記載の化合物を含有する活性化血液凝固第X因子(FXa)阻害剤;
(18):(1)~(9)のいずれか1項に記載の化合物を含有する血液凝固抑制剤;
(19):(1)~(9)のいずれか1項に記載の化合物を含有する血栓又は塞栓の予防及び/又は治療剤;
(20):(1)~(9)のいずれか1項に記載の化合物を含有する脳梗塞、脳塞栓、心筋梗塞、狭心症、肺塞栓、深部静脈血栓症、人工弁/関節置換後の血栓形成、血行再建後の血栓形成及び再閉塞、非弁膜性心房細動(NVAF)に基づく血栓塞栓症の予防剤及び/又は治療剤;及び
(21):(1)~(9)のいずれか1項に記載の化合物、及び薬理上許容される担体を含有する医薬組成物;
に関するものである。
本発明の化合物(1)の薬理上許容される塩又はその水和物、又はそれらの結晶、特に化合物(1)の1・マレイン酸塩の結晶(1-a)は,良好な水溶性を有し、かつ吸脱湿性による重量変化がないことから、医薬品としての製剤化などでの懸念点が少なく、長期の保存安定性にも優れ、かつ優れたFXaの阻害作用を示すことから有用である。
したがって、化合物(1)の1・マレイン酸塩の結晶(1-a)は、医薬品化合物として血栓性及び/又は塞栓性疾患の予防・治療薬として有用である。
したがって、化合物(1)の1・マレイン酸塩の結晶(1-a)は、医薬品化合物として血栓性及び/又は塞栓性疾患の予防・治療薬として有用である。
以下に、本発明を詳細に説明する。
下記の式(1)
下記の式(1)

で表される、N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド(1)の薬理上許容される塩としては、薬理上許容される塩であれば特に限定はなく、それらの塩は水和物を形成してもよい。
本発明における薬理上許容される塩の例としては、塩酸塩、臭化水素塩、硫酸塩、リン酸塩及び硝酸塩等の無機酸付加塩、酢酸塩、プロピオン酸塩、グリコール酸塩、コハク酸塩、乳酸塩、シュウ酸塩、マレイン酸塩、酒石酸塩、マロン酸塩、コハク酸塩、フマル酸塩、メシル酸塩、トシル酸塩、アスコルビン酸塩等の有機酸付加塩を挙げることができる。
化合物(1)の酸付加塩としては、塩酸塩、臭化水素酸塩、硫酸塩、メシル酸塩、ベンゼンスルホン酸塩、トシル酸塩、リン酸塩、酢酸塩、グリコール酸塩、乳酸塩、マロン酸塩、酒石酸塩、リンゴ酸塩、マレイン酸塩、クエン酸塩、コハク酸塩、又はそれらの水和物が好ましく;塩酸塩、及びマレイン酸塩がより好ましく;1・マレイン酸塩が特に好ましい。
本発明における薬理上許容される塩の例としては、塩酸塩、臭化水素塩、硫酸塩、リン酸塩及び硝酸塩等の無機酸付加塩、酢酸塩、プロピオン酸塩、グリコール酸塩、コハク酸塩、乳酸塩、シュウ酸塩、マレイン酸塩、酒石酸塩、マロン酸塩、コハク酸塩、フマル酸塩、メシル酸塩、トシル酸塩、アスコルビン酸塩等の有機酸付加塩を挙げることができる。
化合物(1)の酸付加塩としては、塩酸塩、臭化水素酸塩、硫酸塩、メシル酸塩、ベンゼンスルホン酸塩、トシル酸塩、リン酸塩、酢酸塩、グリコール酸塩、乳酸塩、マロン酸塩、酒石酸塩、リンゴ酸塩、マレイン酸塩、クエン酸塩、コハク酸塩、又はそれらの水和物が好ましく;塩酸塩、及びマレイン酸塩がより好ましく;1・マレイン酸塩が特に好ましい。
上記の化合物(1)の薬理上許容される塩又はその水和物としては、結晶が好ましく;塩酸塩、臭化水素酸塩、マレイン酸塩、りんご酸塩、フマル酸塩及びグリコール酸塩又はそれらの水和物の結晶がより好ましく;塩酸塩の結晶及びマレイン酸塩の結晶がさらに好ましく;1・マレイン酸塩の結晶が特に好ましい。
化合物(1)の臭化水素酸塩の結晶としては、粉末X線回折による回折角(2θ)として、7.4、12.6、14.7、18.3、20.9、22.7、24.2、24.7、及び25.6度(±0.2度)に特徴的ピークを有するものが好ましい。
化合物(1)のグリコール酸塩の結晶としては、粉末X線回折による回折角(2θ)として、6.1、8.3、12.3、13.2、16.0、17.8、19.0、20.9、及び23.6度(±0.2度)に特徴的ピークを有するものが好ましい。
化合物(1)のりんご酸塩の結晶としては、粉末X線回折による回折角(2θ)として、5.2、8.0、8.4、15.5、及び15.9度(±0.2度)に特徴的ピークを有するものが好ましい。
化合物(1)のフマル酸塩の結晶としては、粉末X線回折による回折角(2θ)として、5.5、8.4、13.2、18.5、19.2、22.3、29.0、及び29.5度(±0.2度)に特徴的ピークを有するものが好ましい。
化合物(1)のマレイン酸塩の結晶としては、下記の式(1-a)
化合物(1)のフマル酸塩の結晶としては、粉末X線回折による回折角(2θ)として、5.5、8.4、13.2、18.5、19.2、22.3、29.0、及び29.5度(±0.2度)に特徴的ピークを有するものが好ましい。
化合物(1)のマレイン酸塩の結晶としては、下記の式(1-a)

で表される、N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド・1・マレイン酸塩の結晶が特に好ましい。
化合物(1-a)の結晶としては、粉末X線回折による回折角(2θ)として、8.4、13.9、15.4、16.7、17.2、17.7、18.8、19.4、20.3、22.1、23.0及び27.9度(±0.2度)に特徴的ピークを有する結晶が好ましい。
また、化合物(1-a)の結晶としては、粉末X線回折による回折角(2θ)として、8.4、13.9、15.4、16.7、17.2、17.7、18.8、19.4、20.3、22.1、23.0及び27.9度(±0.2度)に特徴的ピークを有し、かつ下記の
a)220℃~225℃の温度に吸熱量ピークを有する熱分析(DTA)プロフィール;
b)熱分析(TG)における、210℃~230℃の温度での重量減少;
c)相対湿度40~60%における重量変化の増減がプラスマイナス0.5%以内;又は
d)融点(分解)として202℃~204℃(未補正);
で特徴付けられるものが好ましい。
上記の化合物(1)の薬理上許容される塩又はその水和物の結晶を製造する方法としては、製造過程における分解物[酸分解物(A)]の生成を抑制するために、薬理上許容される塩を調製するための酸を分割添加することが好ましい。
製造過程での分解物の生成を抑制し、化合物(1)の結晶を高収率で製造する方法としては、化合物(1)、及び化合物(1)の1モルに対して約半量に相当する、0.498~0.502モルの薬理上許容される塩を調製するための酸を、2回に分けて分割添加することが好ましい。
より具体的な化合物(1)の薬理上許容される塩又はその水和物、又はそれらの結晶を製造する方法としては、以下の(工程A)~(工程D)から構成されるものである。
すなわち、(工程A):N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド(1)、及び化合物(1)の1モルに対して0.498~0.502モルの薬理上許容される塩を調製するための酸を添加し、溶媒中で加温して撹拌する工程;
(工程B):(工程A)で得られた混合液をろ過して、不溶物を熱時ろ別する工程;
(工程C):(工程B)のろ液を、ろ液を加温し、化合物(1)の1モルに対して0.498~0.502モルの薬理上許容される塩を調製するための酸の溶液を滴下し、滴下終了後、攪拌する工程;及び
(工程D):上記の(工程C)の攪拌操作で析出した結晶をろ過する工程;である。
(工程A)の溶媒としては、含水メタノール、含水エタノール、含水プロパノール、含水イソプロパノール又は含水アセトン等が好ましく;
含水エタノール又は含水イソプロパノールがより好ましく;
水を10~25容量%含むエタノールが特に好ましい。
(工程A)の溶媒の量としては、化合物(1)の重量に対して5~15倍(容量/重量)が好ましく;8~12倍(容量/重量)がより好ましい。
(工程A)の混合液の内温を60~70℃になるように加温して撹拌する時間としては、30分~1時間が好ましい。
(工程B)の、熱時ろ過する工程に用いるろ紙としては、通常使用する市販のものでよく、ろ過は常圧ろ過又は減圧を行えばよい。
不溶物は、60~70℃に加温した、(工程A)と同じ溶媒で洗浄するのが好ましく;洗浄に使用する溶媒の量としては、化合物(1)及び薬理上許容される塩を調製するための酸の重量に対して1~3倍(容量/重量)が好ましく;2倍(容量/重量)がより好ましい。
(工程C)では、薬理上許容される塩を調製するための酸の再添加をするため、薬理上許容される塩を調製するための酸は、好適な溶媒を用いた溶液で滴下するのが好ましい。薬理上許容される塩を調製するための酸の溶液を調製するときの、好適な溶媒及びその使用量、薬理上許容される塩を調製するための酸の溶液を調製する温度としては、薬理上許容される塩を調製するための酸が有機酸である場合、それ自身の自己酸触媒でのエステル化などの副反応を防止する溶媒及び溶解方法を選択するのが好ましい。
薬理上許容される塩を調製するための酸の溶液を調製する溶媒としては含水C1~C3アルコールを挙げることができ;水を3~7容量%含むC1~C3アルコールが好ましく;水を3~7容量%含むエタノール又は水を3~7容量%含むイソプロパノールがより好ましく;
水を3~7容量%含むエタノール溶液がさらに好ましく:
水を5容量%含むエタノール溶液が特に好ましい。
(工程C)で使用する薬理上許容される塩を調製するための酸の溶液を調製する溶媒の使用量としては、化合物(1)に対して5~15倍(容量/重量)が好ましく;10倍(容量/重量)がより好ましい。
(工程C)の、薬理上許容される塩を調製するための酸の溶液を滴下する温度としては、ろ液の保温の温度である40~50℃が好ましく;滴下時間としては10分程度が好ましい。
(工程C)の、滴下終了後の攪拌は、徐々に溶液の温度を下降させ、結晶化を完結させるものである。(工程C)の、滴下終了後の攪拌時の温度と時間としては、40~50℃で1.5~3時間、20~30℃で1.5~3時間、0~10℃で10~15時間攪拌するのが好ましい。
(工程D)は、(工程C)の攪拌操作で析出した結晶をろ過する工程である。
(工程D)の、析出した結晶をろ過する操作は、0~10℃で実施するのが好ましい。(工程D)の、ろ過した結晶は洗浄しても洗浄してもよく、洗浄する操作の洗浄溶媒としては、予め0~10℃に冷却した、水を10~20容量%含むエタノールが好ましく;予め0~10℃に冷却した、水を15容量%含むエタノールがより好ましい。
洗浄溶媒の使用量としては、化合物(1)に対して2~5倍(容量/重量)が好ましく;3倍(容量/重量)がより好ましい。
(工程D)では、ろ過した結晶を乾燥するのが好ましい。乾燥操作は、減圧下で40~60℃で実施するのが好ましく;減圧下で40~45℃で行うのがより好ましい。
さらに、上記製造方法の具体的な態様としては、以下の(1)から(8)の操作を挙げることができる。
(1)化合物(1)のN1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド(1)(3.00g、5.50mmol)に、水を10~25容量%含むC1~C3アルコールを、8~12倍(容量/重量)を添加する。
(2)塩酸、臭化水素酸、マレイン酸、りんご酸、フマル酸又はグリコール酸を、2.76mmol添加する。
(3)混合液の内温を60~70℃になるように加温し、混合液を0分~1時間撹拌する。
(4)不溶物を熱時ろ過する。
(5)予め60~70℃に加温しておいた水を3~7容量%含むC1~C3アルコールを使用して、化合物(1)及びマレイン酸の重量に対して1~3倍(容量/重量)量で洗浄する。
(6)ろ液の内温を40~50℃に保温し、塩酸、臭化水素酸、マレイン酸、りんご酸、フマル酸又はグリコール酸の2.76mmol量を、水を3~7容量%含むC1~C3アルコール溶液[溶液量は、化合物(1)に対して5~15倍(容量/重量)]を10分程度で滴下する。
(7)晶析が開始された場合は、反応混合物の内温を40~50℃に保ち、同温度で1.5~3時間、20~30℃で1.5~3時間、0~10℃で10~15時間攪拌する。
(8)析出結晶をろ過し、予め予め0~10℃に冷却した水を10~20容量%含むエタノールで洗浄する。湿体を40℃で減圧乾燥して、対応する塩酸塩、臭化水素酸塩、マレイン酸塩、りんご酸塩、フマル酸塩又はグリコール酸塩が得られる。
このようにして得られた化合物(1)の塩、その水和物、又はそれらの結晶、特に1・マレイン酸塩(1-a)は、水溶性が良好であり、高純度かつ保存安定性に優れた医薬として有用である。
また化合物(1-a)は、高い活性化血液凝固第X因子(FXa)阻害作用を示すことから、血液凝固抑制剤、血栓又は塞栓の予防及び/又は治療剤として有用である。
さらに化合物(1-a)は、血栓又は塞栓の原因となる各種疾患、例えば、脳梗塞、脳塞栓、心筋梗塞、狭心症、肺梗塞、肺塞栓、バージャー病、深部静脈血栓症、汎発性血管内凝固症候群、人工弁/関節置換後の血栓形成、血行再建後の血栓形成及び再閉塞、多臓器不全(MODS)、体外循環時の血栓形成又は採血時の血液凝固、非弁膜性心房細動(NVAF)に基づく血栓塞栓症等の予防剤及び/又は治療剤として有用である。
本発明の医薬は、経口投与を始めとして種々の方法によって投与することができる。また、注射剤とする場合には静脈内注射、筋肉内注射、皮下注射等の何れの方法によっても投与できる。
かかる製剤の調製方法については、投与法に応じ適当な製剤を選択し、通常用いられている各種製剤の調製法が使用できる。
経口製剤としては、例えば錠剤、散剤、顆粒剤、カプセル剤や、溶液剤、シロップ剤、エリキシル剤、油性ないし水性の懸濁液等を例示できる。注射剤としては製剤中に安定剤、防腐剤、溶解補助剤を使用することもあり、これらの補助剤等を含むこともある溶液を容器に収納後、所望によって凍結乾燥等によって固形製剤として用時調製の製剤としてもよい。また、液体製剤としては溶液、懸濁液、乳液剤等を挙げることができるが、これらの製剤を調製する際、添加剤として懸濁化剤、乳化剤等を使用することもできる。
本発明化合物(1)を含有する医薬は、化合物(1)として成人1人1日当り一回投与し、適当な間隔で繰り返すのが望ましい。投与量は0.01mg~1000mgの範囲、好ましくは0.1mg~200mgの範囲であり、より好ましくは1mg~100mgである。
化合物(1-a)の結晶としては、粉末X線回折による回折角(2θ)として、8.4、13.9、15.4、16.7、17.2、17.7、18.8、19.4、20.3、22.1、23.0及び27.9度(±0.2度)に特徴的ピークを有する結晶が好ましい。
また、化合物(1-a)の結晶としては、粉末X線回折による回折角(2θ)として、8.4、13.9、15.4、16.7、17.2、17.7、18.8、19.4、20.3、22.1、23.0及び27.9度(±0.2度)に特徴的ピークを有し、かつ下記の
a)220℃~225℃の温度に吸熱量ピークを有する熱分析(DTA)プロフィール;
b)熱分析(TG)における、210℃~230℃の温度での重量減少;
c)相対湿度40~60%における重量変化の増減がプラスマイナス0.5%以内;又は
d)融点(分解)として202℃~204℃(未補正);
で特徴付けられるものが好ましい。
上記の化合物(1)の薬理上許容される塩又はその水和物の結晶を製造する方法としては、製造過程における分解物[酸分解物(A)]の生成を抑制するために、薬理上許容される塩を調製するための酸を分割添加することが好ましい。
製造過程での分解物の生成を抑制し、化合物(1)の結晶を高収率で製造する方法としては、化合物(1)、及び化合物(1)の1モルに対して約半量に相当する、0.498~0.502モルの薬理上許容される塩を調製するための酸を、2回に分けて分割添加することが好ましい。
より具体的な化合物(1)の薬理上許容される塩又はその水和物、又はそれらの結晶を製造する方法としては、以下の(工程A)~(工程D)から構成されるものである。
すなわち、(工程A):N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド(1)、及び化合物(1)の1モルに対して0.498~0.502モルの薬理上許容される塩を調製するための酸を添加し、溶媒中で加温して撹拌する工程;
(工程B):(工程A)で得られた混合液をろ過して、不溶物を熱時ろ別する工程;
(工程C):(工程B)のろ液を、ろ液を加温し、化合物(1)の1モルに対して0.498~0.502モルの薬理上許容される塩を調製するための酸の溶液を滴下し、滴下終了後、攪拌する工程;及び
(工程D):上記の(工程C)の攪拌操作で析出した結晶をろ過する工程;である。
(工程A)の溶媒としては、含水メタノール、含水エタノール、含水プロパノール、含水イソプロパノール又は含水アセトン等が好ましく;
含水エタノール又は含水イソプロパノールがより好ましく;
水を10~25容量%含むエタノールが特に好ましい。
(工程A)の溶媒の量としては、化合物(1)の重量に対して5~15倍(容量/重量)が好ましく;8~12倍(容量/重量)がより好ましい。
(工程A)の混合液の内温を60~70℃になるように加温して撹拌する時間としては、30分~1時間が好ましい。
(工程B)の、熱時ろ過する工程に用いるろ紙としては、通常使用する市販のものでよく、ろ過は常圧ろ過又は減圧を行えばよい。
不溶物は、60~70℃に加温した、(工程A)と同じ溶媒で洗浄するのが好ましく;洗浄に使用する溶媒の量としては、化合物(1)及び薬理上許容される塩を調製するための酸の重量に対して1~3倍(容量/重量)が好ましく;2倍(容量/重量)がより好ましい。
(工程C)では、薬理上許容される塩を調製するための酸の再添加をするため、薬理上許容される塩を調製するための酸は、好適な溶媒を用いた溶液で滴下するのが好ましい。薬理上許容される塩を調製するための酸の溶液を調製するときの、好適な溶媒及びその使用量、薬理上許容される塩を調製するための酸の溶液を調製する温度としては、薬理上許容される塩を調製するための酸が有機酸である場合、それ自身の自己酸触媒でのエステル化などの副反応を防止する溶媒及び溶解方法を選択するのが好ましい。
薬理上許容される塩を調製するための酸の溶液を調製する溶媒としては含水C1~C3アルコールを挙げることができ;水を3~7容量%含むC1~C3アルコールが好ましく;水を3~7容量%含むエタノール又は水を3~7容量%含むイソプロパノールがより好ましく;
水を3~7容量%含むエタノール溶液がさらに好ましく:
水を5容量%含むエタノール溶液が特に好ましい。
(工程C)で使用する薬理上許容される塩を調製するための酸の溶液を調製する溶媒の使用量としては、化合物(1)に対して5~15倍(容量/重量)が好ましく;10倍(容量/重量)がより好ましい。
(工程C)の、薬理上許容される塩を調製するための酸の溶液を滴下する温度としては、ろ液の保温の温度である40~50℃が好ましく;滴下時間としては10分程度が好ましい。
(工程C)の、滴下終了後の攪拌は、徐々に溶液の温度を下降させ、結晶化を完結させるものである。(工程C)の、滴下終了後の攪拌時の温度と時間としては、40~50℃で1.5~3時間、20~30℃で1.5~3時間、0~10℃で10~15時間攪拌するのが好ましい。
(工程D)は、(工程C)の攪拌操作で析出した結晶をろ過する工程である。
(工程D)の、析出した結晶をろ過する操作は、0~10℃で実施するのが好ましい。(工程D)の、ろ過した結晶は洗浄しても洗浄してもよく、洗浄する操作の洗浄溶媒としては、予め0~10℃に冷却した、水を10~20容量%含むエタノールが好ましく;予め0~10℃に冷却した、水を15容量%含むエタノールがより好ましい。
洗浄溶媒の使用量としては、化合物(1)に対して2~5倍(容量/重量)が好ましく;3倍(容量/重量)がより好ましい。
(工程D)では、ろ過した結晶を乾燥するのが好ましい。乾燥操作は、減圧下で40~60℃で実施するのが好ましく;減圧下で40~45℃で行うのがより好ましい。
さらに、上記製造方法の具体的な態様としては、以下の(1)から(8)の操作を挙げることができる。
(1)化合物(1)のN1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド(1)(3.00g、5.50mmol)に、水を10~25容量%含むC1~C3アルコールを、8~12倍(容量/重量)を添加する。
(2)塩酸、臭化水素酸、マレイン酸、りんご酸、フマル酸又はグリコール酸を、2.76mmol添加する。
(3)混合液の内温を60~70℃になるように加温し、混合液を0分~1時間撹拌する。
(4)不溶物を熱時ろ過する。
(5)予め60~70℃に加温しておいた水を3~7容量%含むC1~C3アルコールを使用して、化合物(1)及びマレイン酸の重量に対して1~3倍(容量/重量)量で洗浄する。
(6)ろ液の内温を40~50℃に保温し、塩酸、臭化水素酸、マレイン酸、りんご酸、フマル酸又はグリコール酸の2.76mmol量を、水を3~7容量%含むC1~C3アルコール溶液[溶液量は、化合物(1)に対して5~15倍(容量/重量)]を10分程度で滴下する。
(7)晶析が開始された場合は、反応混合物の内温を40~50℃に保ち、同温度で1.5~3時間、20~30℃で1.5~3時間、0~10℃で10~15時間攪拌する。
(8)析出結晶をろ過し、予め予め0~10℃に冷却した水を10~20容量%含むエタノールで洗浄する。湿体を40℃で減圧乾燥して、対応する塩酸塩、臭化水素酸塩、マレイン酸塩、りんご酸塩、フマル酸塩又はグリコール酸塩が得られる。
このようにして得られた化合物(1)の塩、その水和物、又はそれらの結晶、特に1・マレイン酸塩(1-a)は、水溶性が良好であり、高純度かつ保存安定性に優れた医薬として有用である。
また化合物(1-a)は、高い活性化血液凝固第X因子(FXa)阻害作用を示すことから、血液凝固抑制剤、血栓又は塞栓の予防及び/又は治療剤として有用である。
さらに化合物(1-a)は、血栓又は塞栓の原因となる各種疾患、例えば、脳梗塞、脳塞栓、心筋梗塞、狭心症、肺梗塞、肺塞栓、バージャー病、深部静脈血栓症、汎発性血管内凝固症候群、人工弁/関節置換後の血栓形成、血行再建後の血栓形成及び再閉塞、多臓器不全(MODS)、体外循環時の血栓形成又は採血時の血液凝固、非弁膜性心房細動(NVAF)に基づく血栓塞栓症等の予防剤及び/又は治療剤として有用である。
本発明の医薬は、経口投与を始めとして種々の方法によって投与することができる。また、注射剤とする場合には静脈内注射、筋肉内注射、皮下注射等の何れの方法によっても投与できる。
かかる製剤の調製方法については、投与法に応じ適当な製剤を選択し、通常用いられている各種製剤の調製法が使用できる。
経口製剤としては、例えば錠剤、散剤、顆粒剤、カプセル剤や、溶液剤、シロップ剤、エリキシル剤、油性ないし水性の懸濁液等を例示できる。注射剤としては製剤中に安定剤、防腐剤、溶解補助剤を使用することもあり、これらの補助剤等を含むこともある溶液を容器に収納後、所望によって凍結乾燥等によって固形製剤として用時調製の製剤としてもよい。また、液体製剤としては溶液、懸濁液、乳液剤等を挙げることができるが、これらの製剤を調製する際、添加剤として懸濁化剤、乳化剤等を使用することもできる。
本発明化合物(1)を含有する医薬は、化合物(1)として成人1人1日当り一回投与し、適当な間隔で繰り返すのが望ましい。投与量は0.01mg~1000mgの範囲、好ましくは0.1mg~200mgの範囲であり、より好ましくは1mg~100mgである。
以下、実施例により本発明を具体的に説明するが、本発明はなんらこれに限定されるものではない。
(試験例1)
実施例の化合物(1)、化合物(1)の各種の塩について吸脱湿性を評価した。結晶約20mgをマイクロバランス(自動水蒸気吸着装置)中、相対湿度(RH)範囲10~90%での経時的重量変化を測定することにより水分の吸脱着量を評価した。
(試験例1)
実施例の化合物(1)、化合物(1)の各種の塩について吸脱湿性を評価した。結晶約20mgをマイクロバランス(自動水蒸気吸着装置)中、相対湿度(RH)範囲10~90%での経時的重量変化を測定することにより水分の吸脱着量を評価した。
その結果、[図5]に示すように、化合物(1)の1・マレイン酸塩(1-a)は、40~60%RHにおいて、プラスマイナス0.5%の重量変化であった。
化合物(1)の1・マレイン酸塩(1-a)は、フリー体である化合物(1)の結晶が、相対湿度40-60%で重量変化を示し、無水物と水和物の間で相互変換が起きる問題点を解決することが明らかになった。
(試験例2)
日本薬局方の試験方法に従って、水(37℃)、日局1液及び2液に対する溶解度を評価した。
その結果、[図6]の表に示すように、化合物(1-a)の水への溶解性は良好であった。フリ-体である化合物(1)の結晶の溶解性に比べて大きく改善されていることが明らかになった。
(試験例3)
化合物(1-a)等、化合物(1)の各種の塩、及び化合物(1)のフリー体の結晶を調製し、これらについて粉末X線測定装置を用いて、結晶形の測定を実施した。
その結果、また、[図1]に示すように、化合物(1)の塩酸塩においては、化合物(1)の塩酸塩・2水和物と化合物(1)の塩酸塩・2.5水和物が存在することが明らかになった。一方、[図3]に化合物(1)の1・マレイン酸塩(1-a)の粉末X線解析図を示す。
(試験例4)
化合物(1-a)等の化合物(1)の各種の塩及び化合物(1)のフリー体の熱分析(TG/DTA)の測定を実施した。
(試験例5)
化合物(1-a)等、化合物(1)の各種の塩の冷蔵保存(5℃)及び、室温保存時(25℃)の2ヶ月経過時点で、開始時からの酸分解物(A)及び総不純物の増加を測定した。また、2ヶ月経過時点からの酸分解物(A)及び総不純物の増加をArrhenius plotによって算出し、それぞれの塩の結晶の長期保存安定性を予測した。
その結果、[図2]の表に示すように、化合物(1)の塩酸塩・2水和物の2ヶ月保存経過時の酸分解物(A)の増加率は、冷蔵保存(5℃)では0.10%、室温保存時(25℃)では0.54%の増加を示した。また、2ヶ月保存経過時点の実測値に基づくArrhenius plotによる36ヶ月経過時の酸分解物(A)の増加予測では、冷蔵保存(5℃)で0.58%、室温保存時(25℃)では3.25%の増加であった。一方、[図7]の表に示すように、化合物(1)の1・マレイン酸塩(1-a)の2ヶ月保存経過時の酸分解物(A)の実測値は、5℃及び25℃ともに、開始時から0.08%の増加と低値であり、2ヶ月保存経過時の酸分解物(A)の増加率に基づくArrhenius plotによる36ヶ月経過時の酸分解物(A)の増加予測では、冷蔵保存(5℃)で0.1%以下、室温保存時(25℃)では0.7%以下と、長期保存安定性が良好であった。
[実施例1] N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド・1・マレイン酸塩(1-a)
化合物(1)の1・マレイン酸塩(1-a)は、フリー体である化合物(1)の結晶が、相対湿度40-60%で重量変化を示し、無水物と水和物の間で相互変換が起きる問題点を解決することが明らかになった。
(試験例2)
日本薬局方の試験方法に従って、水(37℃)、日局1液及び2液に対する溶解度を評価した。
その結果、[図6]の表に示すように、化合物(1-a)の水への溶解性は良好であった。フリ-体である化合物(1)の結晶の溶解性に比べて大きく改善されていることが明らかになった。
(試験例3)
化合物(1-a)等、化合物(1)の各種の塩、及び化合物(1)のフリー体の結晶を調製し、これらについて粉末X線測定装置を用いて、結晶形の測定を実施した。
その結果、また、[図1]に示すように、化合物(1)の塩酸塩においては、化合物(1)の塩酸塩・2水和物と化合物(1)の塩酸塩・2.5水和物が存在することが明らかになった。一方、[図3]に化合物(1)の1・マレイン酸塩(1-a)の粉末X線解析図を示す。
(試験例4)
化合物(1-a)等の化合物(1)の各種の塩及び化合物(1)のフリー体の熱分析(TG/DTA)の測定を実施した。
(試験例5)
化合物(1-a)等、化合物(1)の各種の塩の冷蔵保存(5℃)及び、室温保存時(25℃)の2ヶ月経過時点で、開始時からの酸分解物(A)及び総不純物の増加を測定した。また、2ヶ月経過時点からの酸分解物(A)及び総不純物の増加をArrhenius plotによって算出し、それぞれの塩の結晶の長期保存安定性を予測した。
その結果、[図2]の表に示すように、化合物(1)の塩酸塩・2水和物の2ヶ月保存経過時の酸分解物(A)の増加率は、冷蔵保存(5℃)では0.10%、室温保存時(25℃)では0.54%の増加を示した。また、2ヶ月保存経過時点の実測値に基づくArrhenius plotによる36ヶ月経過時の酸分解物(A)の増加予測では、冷蔵保存(5℃)で0.58%、室温保存時(25℃)では3.25%の増加であった。一方、[図7]の表に示すように、化合物(1)の1・マレイン酸塩(1-a)の2ヶ月保存経過時の酸分解物(A)の実測値は、5℃及び25℃ともに、開始時から0.08%の増加と低値であり、2ヶ月保存経過時の酸分解物(A)の増加率に基づくArrhenius plotによる36ヶ月経過時の酸分解物(A)の増加予測では、冷蔵保存(5℃)で0.1%以下、室温保存時(25℃)では0.7%以下と、長期保存安定性が良好であった。
[実施例1] N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド・1・マレイン酸塩(1-a)

N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド(1)(3.00g、5.50mmol)にエタノール(22.5mL)及びH2O(7.5mL)を添加した後、マレイン酸(320.36mg、2.76mmol)を添加した。混合液の内温を65℃に昇温し、40分間攪拌した。不溶物を熱時ろ過し、予め70℃に加温しておいた25%の水を含むエタノール(6mL)で洗浄した。ろ液の内温を45℃に保温し、マレイン酸(321.64mg、2.77mmol)の5%の水を含むエタノール(30mL)溶液を10分間かけて滴下した。5分後、晶析が始まった。同温度で2時間攪拌後、放冷した。内温21~28℃で2時間攪拌した後、低温室(室温3-5℃)で12時間攪拌した。さらに、氷水で冷却し、内温2℃で1時間攪拌した。析出している結晶をろ過し、予め0℃に冷却しておいた85%EtOH(9mL)で洗浄した。湿体を40℃で減圧乾燥し、標題化合物(2.91g、4.34mmol、78.9%)を得た。
融点(分解):202℃~204℃;
元素分析:C23H25ClN8O4S・C4H4O4
理論値:C;49.05,H;4.42,N;16.95,Cl;5.36, S;4.85.
実測値:C;49.21,H;4.35,N;16.97,Cl;5.17, S;5.00;
1H-NMR(400MHz,DMSO-d6)δ:10.31(s,1H),9.20-9.27(d,J=8.3Hz,1H),9.15(s,1H),8.81-8.87(d,J=7.3Hz,1H),8.44-8.49(dd,J=2.2,1.0Hz,1H),7.99-8.08(m,2H),6.08(s,2H),4.38-4.54(m,3H),4.10-4.20(m,1H),3.40-3.56(m,2H),3.28-3.39(m,1H),3.05-3.23(m,2H),2.88(s,3H),2.38-2.50(m,1H),2.06-2.24(m,2H),1.90-2.02(m,1H),1.66-1.85(m,2H).
融点(分解):202℃~204℃;
元素分析:C23H25ClN8O4S・C4H4O4
理論値:C;49.05,H;4.42,N;16.95,Cl;5.36, S;4.85.
実測値:C;49.21,H;4.35,N;16.97,Cl;5.17, S;5.00;
1H-NMR(400MHz,DMSO-d6)δ:10.31(s,1H),9.20-9.27(d,J=8.3Hz,1H),9.15(s,1H),8.81-8.87(d,J=7.3Hz,1H),8.44-8.49(dd,J=2.2,1.0Hz,1H),7.99-8.08(m,2H),6.08(s,2H),4.38-4.54(m,3H),4.10-4.20(m,1H),3.40-3.56(m,2H),3.28-3.39(m,1H),3.05-3.23(m,2H),2.88(s,3H),2.38-2.50(m,1H),2.06-2.24(m,2H),1.90-2.02(m,1H),1.66-1.85(m,2H).
本発明の化合物(1)の薬理上許容される塩又はその水和物、又はそれらの結晶、特に化合物(1)の1・マレイン酸塩の結晶(1-a)は、良好な水溶性を有し、かつ吸脱湿性による重量変化がないことから医薬品としての製剤化工程での懸念点がなく、長期の保存安定性にも優れることから、医薬品化合物として血栓性及び/又は塞栓性疾患の予防・治療薬として有用である。
Claims (21)
- 下記の式(1)

- 薬理上許容される塩が、塩酸塩、臭化水素酸塩、硫酸塩、メシル酸塩、ベンゼンスルホン酸塩、トシル酸塩、リン酸塩、酢酸塩、グリコール酸塩、乳酸塩、マロン酸塩、酒石酸塩、リンゴ酸塩、マレイン酸塩、クエン酸塩、コハク酸塩である請求項1に記載の塩又はその水和物。
- 薬理上許容される塩が、塩酸塩又はマレイン酸塩である請求項1に記載の塩又はその水和物。
- 下記の式(1)

- 薬理上許容される塩が、塩酸塩、臭化水素酸塩、マレイン酸塩、りんご酸塩、フマル酸塩又はグリコール酸塩、又はそれらの水和物である請求項4に記載の結晶。
- 薬理上許容される塩が、塩酸塩又はマレイン酸塩、又はそれらの水和物である請求項4に記載の結晶。
- 下記の式(1-a)

- 粉末X線回折による回折角(2θ)として、8.4、13.9、15.4、16.7、17.2、17.7、18.8、19.4、20.3、22.1、23.0及び27.9度(±0.2度)に特徴的ピークを有する請求項7に記載の結晶。
- さらに、
(a)220℃~225℃に吸熱量ピークを有する熱分析(DTA)プロフィール;
(b)210℃~230℃に重量減少を示す熱分析(TG)プロフィール;
(c)相対湿度40~60%における重量変化の増減がプラスマイナス0.5%以内;又は
(d)融点(分解)として202℃~204℃(未補正);
で特徴付けられる、請求項8に記載の結晶。 - 下記の式(1)

薬理上許容される塩を調製するための酸を分割添加することを特徴とし、製造過程における分解物の生成を抑制する製造方法。 - (工程A):N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド(1)、及び化合物(1)の1モルに対して0.498~0.502モルの薬理上許容される塩を調製するための酸を添加し、溶媒中で加温して撹拌する工程;
(工程B):(工程A)で得られた混合液をろ過して、不溶物を熱時ろ別する工程;
(工程C):(工程B)のろ液を、ろ液を加温し、化合物(1)の1モルに対して0.498~0.502モルの薬理上許容される塩を調製するための酸の溶液を滴下し、滴下終了後、攪拌する工程;及び
(工程D):上記の(工程C)の攪拌操作で析出した結晶をろ過する工程;
上記の(工程A)~(工程D)からなる請求項10に記載の製造方法。 - 薬理上許容される塩を調製するための酸が、塩酸、臭化水素酸、マレイン酸、りんご酸、フマル酸又はグリコール酸である請求項10又は11の製造方法。
- 薬理上許容される塩を調製するための酸が、塩酸又はマレイン酸である請求項10又は11の製造方法。
- (工程A)の溶媒が、含水C1~C3アルコールである請求項11の製造方法。
- 薬理上許容される塩を調製するための酸の溶液を調製する溶媒が、水を3~7容量%含むC1~C3アルコール溶液である請求項11の製造方法。
- 請求項1~9のいずれか1項に記載の化合物を含有する医薬。
- 請求項1~9のいずれか1項に記載の化合物を含有する活性化血液凝固第X因子阻害剤。
- 請求項1~9のいずれか1項に記載の化合物を含有する血液凝固抑制剤。
- 請求項1~9のいずれか1項に記載の化合物を含有する血栓又は塞栓の予防及び/又は治療剤。
- 請求項1~9のいずれか1項に記載の化合物を含有する脳梗塞、脳塞栓、心筋梗塞、狭心症、肺塞栓、深部静脈血栓症、人工弁/関節置換後の血栓形成、血行再建後の血栓形成及び再閉塞、非弁膜性心房細動に基づく血栓塞栓症の予防剤及び/又は治療剤。
- 請求項1~9のいずれか1項に記載の化合物、及び薬理上許容される担体を含有する医薬組成物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008-293985 | 2008-11-18 | ||
| JP2008293985 | 2008-11-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010058751A1 true WO2010058751A1 (ja) | 2010-05-27 |
Family
ID=42198191
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2009/069431 WO2010058751A1 (ja) | 2008-11-18 | 2009-11-16 | [1,3,4]オキサジアゾール化合物の薬理上許容される塩又はその水和物 |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW201022282A (ja) |
| WO (1) | WO2010058751A1 (ja) |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004058715A1 (ja) * | 2002-12-25 | 2004-07-15 | Daiichi Pharmaceutical Co., Ltd. | ジアミン誘導体 |
| WO2008156159A1 (ja) * | 2007-06-21 | 2008-12-24 | Daiichi Sankyo Company, Limited | ジアミン誘導体の製造法 |
-
2009
- 2009-11-16 WO PCT/JP2009/069431 patent/WO2010058751A1/ja active Application Filing
- 2009-11-17 TW TW098138917A patent/TW201022282A/zh unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004058715A1 (ja) * | 2002-12-25 | 2004-07-15 | Daiichi Pharmaceutical Co., Ltd. | ジアミン誘導体 |
| WO2008156159A1 (ja) * | 2007-06-21 | 2008-12-24 | Daiichi Sankyo Company, Limited | ジアミン誘導体の製造法 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW201022282A (en) | 2010-06-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2020183408A (ja) | {[5−(3−クロロフェニル)−3−ヒドロキシピリジン−2−カルボニル]アミノ}酢酸の固体形態、組成物、及びその使用 | |
| JP5416408B2 (ja) | 5−クロロ−n−({(5s)−2−オキソ−3−[4−(3−オキソ−4−モルホリニル)−フェニル]−1,3−オキサゾリジン−5−イル}−メチル)−2−チオフェンカルボキサミドの新規多形および無定形 | |
| JP2009520742A (ja) | Dpp−iv阻害剤の新規な塩および多形 | |
| JP5305421B2 (ja) | ジアミン誘導体の製造方法 | |
| JP5268902B2 (ja) | ピロロピリミジノン誘導体の塩およびその製造方法 | |
| WO2012031017A1 (en) | CRYSTALLINE FORMS OF A FACTOR Xa INHIBITOR | |
| TW201514165A (zh) | D-葡萄糖醇,1-脫氧-1-(甲胺基)-,-1-(6-胺基-3,5-二氟吡啶-2-基)-8-氯-6-氟-1,4-二氫-7-(3-羥基氮雜環丁烷-1-基)-4-側氧基-3-喹啉羧酸的結晶形態與其製備程序 | |
| KR20240093542A (ko) | 리나프라잔 글루레이트의 염산염의 다형체 | |
| WO2010082531A1 (ja) | 活性化血液凝固因子阻害剤 | |
| WO2017160703A1 (en) | Solid state forms of nilotinib salts | |
| JP2023521411A (ja) | (9r,13s)-13-{4-[5-クロロ-2-(4-クロロ-1h-1,2,3-トリアゾール-1-イル)フェニル]-6-オキソ-1,6-ジヒドロピリミジン-1-イル}-3-(ジフルオロメチル)-9-メチル-3,4,7,15-テトラアザトリシクロ[12.3.1.02,6]オクタデカ-1(18),2(6),4,14,16-ペンタエン-8-オンの結晶体 | |
| JP2019517494A (ja) | 腎髄質外層カリウムチャネル阻害剤としての医薬的に許容される塩 | |
| WO2010058751A1 (ja) | [1,3,4]オキサジアゾール化合物の薬理上許容される塩又はその水和物 | |
| JP6393310B2 (ja) | 5−クロロ−チオフェン−2−カルボン酸[(s)−2−[メチル−3−(2−オキソ−ピロリジン−1−イル)−ベンゼンスルホニルアミノ]−3−(4−メチル−ピペラジン−1−イル)−3−オキソ−プロプリル]アミドの酒石酸塩 | |
| JP6357100B2 (ja) | 6−(ピペリジン−4−イルオキシ)−2h−イソキノリン−1−オン塩酸塩の結晶性溶媒和物 | |
| EP2782575B1 (en) | Crystalline salts of r)-3-[6-amino-pyridin-3-yl]-2-(1-cyclohexyl-1h-imidazol-4-yl)-propionic acid | |
| EP2782576B1 (en) | Sodium salt of (r)-3-[6-amino-pyridin-3-yl]-2-(1-cyclohexyl-1h-imidazol-4-yl)-propionic acid | |
| WO2024169765A1 (zh) | 一种二肽类化合物的盐、其制备方法和用途 | |
| JP2008505075A (ja) | 1−(インドール−6−カルボニル−d−フェニルグリシニル)−4−(1−メチルピペリジン−4−イル)ピペラジンd−酒石酸塩 | |
| WO2016127962A1 (en) | An amorphous solid form of suvorexant with sulphuric acid | |
| JP2014518236A (ja) | 6−(ピペリジン−4−イルオキシ)−2h−イソキノリン−1−オン塩酸塩の多形体 | |
| KR20240093547A (ko) | 리나프라잔 글루레이트의 메실레이트 염의 다형체 | |
| CA2574326A1 (en) | Novel form of celecoxib | |
| TW200845971A (en) | Novel heteroaryl carboxamides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09827532 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09827532 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |