WO2010029996A1 - 医薬組成物 - Google Patents
医薬組成物 Download PDFInfo
- Publication number
- WO2010029996A1 WO2010029996A1 PCT/JP2009/065912 JP2009065912W WO2010029996A1 WO 2010029996 A1 WO2010029996 A1 WO 2010029996A1 JP 2009065912 W JP2009065912 W JP 2009065912W WO 2010029996 A1 WO2010029996 A1 WO 2010029996A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- unsubstituted
- heterocyclic group
- pharmaceutical composition
- composition according
- Prior art date
Links
- 239000008194 pharmaceutical composition Substances 0.000 title claims abstract description 68
- 150000003839 salts Chemical class 0.000 claims abstract description 73
- 125000006615 aromatic heterocyclic group Chemical group 0.000 claims abstract description 55
- 230000000202 analgesic effect Effects 0.000 claims abstract description 52
- 125000006848 alicyclic heterocyclic group Chemical group 0.000 claims abstract description 49
- 125000003118 aryl group Chemical group 0.000 claims abstract description 49
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 34
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 31
- FDSSYPNUQHQSIQ-ONEGZZNKSA-N (2e)-penta-2,4-dienamide Chemical class NC(=O)\C=C\C=C FDSSYPNUQHQSIQ-ONEGZZNKSA-N 0.000 claims abstract description 20
- 208000002193 Pain Diseases 0.000 claims description 88
- 230000036407 pain Effects 0.000 claims description 86
- -1 tetrahydroisoquinolyl Chemical group 0.000 claims description 66
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 claims description 57
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 49
- 229910052757 nitrogen Inorganic materials 0.000 claims description 46
- UGJMXCAKCUNAIE-UHFFFAOYSA-N Gabapentin Chemical compound OC(=O)CC1(CN)CCCCC1 UGJMXCAKCUNAIE-UHFFFAOYSA-N 0.000 claims description 40
- 229960005181 morphine Drugs 0.000 claims description 28
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 27
- 125000000623 heterocyclic group Chemical group 0.000 claims description 26
- 239000000014 opioid analgesic Substances 0.000 claims description 26
- 239000000935 antidepressant agent Substances 0.000 claims description 25
- 229940005513 antidepressants Drugs 0.000 claims description 25
- 208000004296 neuralgia Diseases 0.000 claims description 25
- 208000021722 neuropathic pain Diseases 0.000 claims description 25
- 239000003795 chemical substances by application Substances 0.000 claims description 23
- 229960002870 gabapentin Drugs 0.000 claims description 20
- ZEUITGRIYCTCEM-KRWDZBQOSA-N (S)-duloxetine Chemical compound C1([C@@H](OC=2C3=CC=CC=C3C=CC=2)CCNC)=CC=CS1 ZEUITGRIYCTCEM-KRWDZBQOSA-N 0.000 claims description 17
- 230000001430 anti-depressive effect Effects 0.000 claims description 17
- 229960002866 duloxetine Drugs 0.000 claims description 17
- 230000002265 prevention Effects 0.000 claims description 16
- 125000000392 cycloalkenyl group Chemical group 0.000 claims description 15
- 230000001225 therapeutic effect Effects 0.000 claims description 15
- 239000004480 active ingredient Substances 0.000 claims description 14
- 125000003545 alkoxy group Chemical group 0.000 claims description 14
- 239000000730 antalgic agent Substances 0.000 claims description 13
- 229940005483 opioid analgesics Drugs 0.000 claims description 13
- 230000000069 prophylactic effect Effects 0.000 claims description 12
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 11
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 8
- OROGSEYTTFOCAN-DNJOTXNNSA-N codeine Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC OROGSEYTTFOCAN-DNJOTXNNSA-N 0.000 claims description 6
- XYYVYLMBEZUESM-UHFFFAOYSA-N dihydrocodeine Natural products C1C(N(CCC234)C)C2C=CC(=O)C3OC2=C4C1=CC=C2OC XYYVYLMBEZUESM-UHFFFAOYSA-N 0.000 claims description 6
- OROGSEYTTFOCAN-UHFFFAOYSA-N hydrocodone Natural products C1C(N(CCC234)C)C2C=CC(O)C3OC2=C4C1=CC=C2OC OROGSEYTTFOCAN-UHFFFAOYSA-N 0.000 claims description 6
- 125000005434 dihydrobenzoxazinyl group Chemical group O1N(CCC2=C1C=CC=C2)* 0.000 claims description 4
- LLPOLZWFYMWNKH-CMKMFDCUSA-N hydrocodone Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)CC(=O)[C@@H]1OC1=C2C3=CC=C1OC LLPOLZWFYMWNKH-CMKMFDCUSA-N 0.000 claims description 4
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 claims description 4
- 229960002813 lofepramine Drugs 0.000 claims description 4
- SAPNXPWPAUFAJU-UHFFFAOYSA-N lofepramine Chemical compound C12=CC=CC=C2CCC2=CC=CC=C2N1CCCN(C)CC(=O)C1=CC=C(Cl)C=C1 SAPNXPWPAUFAJU-UHFFFAOYSA-N 0.000 claims description 4
- 229960001233 pregabalin Drugs 0.000 claims description 4
- AYXYPKUFHZROOJ-ZETCQYMHSA-N pregabalin Chemical compound CC(C)C[C@H](CN)CC(O)=O AYXYPKUFHZROOJ-ZETCQYMHSA-N 0.000 claims description 4
- AHOUBRCZNHFOSL-YOEHRIQHSA-N (+)-Casbol Chemical compound C1=CC(F)=CC=C1[C@H]1[C@H](COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-YOEHRIQHSA-N 0.000 claims description 3
- GJJFMKBJSRMPLA-HIFRSBDPSA-N (1R,2S)-2-(aminomethyl)-N,N-diethyl-1-phenyl-1-cyclopropanecarboxamide Chemical compound C=1C=CC=CC=1[C@@]1(C(=O)N(CC)CC)C[C@@H]1CN GJJFMKBJSRMPLA-HIFRSBDPSA-N 0.000 claims description 3
- RTHCYVBBDHJXIQ-MRXNPFEDSA-N (R)-fluoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-MRXNPFEDSA-N 0.000 claims description 3
- TVYLLZQTGLZFBW-ZBFHGGJFSA-N (R,R)-tramadol Chemical compound COC1=CC=CC([C@]2(O)[C@H](CCCC2)CN(C)C)=C1 TVYLLZQTGLZFBW-ZBFHGGJFSA-N 0.000 claims description 3
- IJVCSMSMFSCRME-UHFFFAOYSA-N 3-methyl-2,4,4a,5,6,7,7a,13-octahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline-7,9-diol Chemical compound C12CCC(O)C3OC4=C5C32CCN(C)C1CC5=CC=C4O IJVCSMSMFSCRME-UHFFFAOYSA-N 0.000 claims description 3
- USSIQXCVUWKGNF-UHFFFAOYSA-N 6-(dimethylamino)-4,4-diphenylheptan-3-one Chemical compound C=1C=CC=CC=1C(CC(C)N(C)C)(C(=O)CC)C1=CC=CC=C1 USSIQXCVUWKGNF-UHFFFAOYSA-N 0.000 claims description 3
- GDLIGKIOYRNHDA-UHFFFAOYSA-N Clomipramine Chemical compound C1CC2=CC=C(Cl)C=C2N(CCCN(C)C)C2=CC=CC=C21 GDLIGKIOYRNHDA-UHFFFAOYSA-N 0.000 claims description 3
- HCYAFALTSJYZDH-UHFFFAOYSA-N Desimpramine Chemical compound C1CC2=CC=CC=C2N(CCCNC)C2=CC=CC=C21 HCYAFALTSJYZDH-UHFFFAOYSA-N 0.000 claims description 3
- GVGLGOZIDCSQPN-PVHGPHFFSA-N Heroin Chemical compound O([C@H]1[C@H](C=C[C@H]23)OC(C)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4OC(C)=O GVGLGOZIDCSQPN-PVHGPHFFSA-N 0.000 claims description 3
- JAQUASYNZVUNQP-USXIJHARSA-N Levorphanol Chemical compound C1C2=CC=C(O)C=C2[C@]23CCN(C)[C@H]1[C@@H]2CCCC3 JAQUASYNZVUNQP-USXIJHARSA-N 0.000 claims description 3
- UEQUQVLFIPOEMF-UHFFFAOYSA-N Mianserin Chemical compound C1C2=CC=CC=C2N2CCN(C)CC2C2=CC=CC=C21 UEQUQVLFIPOEMF-UHFFFAOYSA-N 0.000 claims description 3
- PHVGLTMQBUFIQQ-UHFFFAOYSA-N Nortryptiline Chemical compound C1CC2=CC=CC=C2C(=CCCNC)C2=CC=CC=C21 PHVGLTMQBUFIQQ-UHFFFAOYSA-N 0.000 claims description 3
- BRUQQQPBMZOVGD-XFKAJCMBSA-N Oxycodone Chemical compound O=C([C@@H]1O2)CC[C@@]3(O)[C@H]4CC5=CC=C(OC)C2=C5[C@@]13CCN4C BRUQQQPBMZOVGD-XFKAJCMBSA-N 0.000 claims description 3
- UQCNKQCJZOAFTQ-ISWURRPUSA-N Oxymorphone Chemical compound O([C@H]1C(CC[C@]23O)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O UQCNKQCJZOAFTQ-ISWURRPUSA-N 0.000 claims description 3
- AHOUBRCZNHFOSL-UHFFFAOYSA-N Paroxetine hydrochloride Natural products C1=CC(F)=CC=C1C1C(COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-UHFFFAOYSA-N 0.000 claims description 3
- ZTVQQQVZCWLTDF-UHFFFAOYSA-N Remifentanil Chemical compound C1CN(CCC(=O)OC)CCC1(C(=O)OC)N(C(=O)CC)C1=CC=CC=C1 ZTVQQQVZCWLTDF-UHFFFAOYSA-N 0.000 claims description 3
- 229960000836 amitriptyline Drugs 0.000 claims description 3
- KRMDCWKBEZIMAB-UHFFFAOYSA-N amitriptyline Chemical compound C1CC2=CC=CC=C2C(=CCCN(C)C)C2=CC=CC=C21 KRMDCWKBEZIMAB-UHFFFAOYSA-N 0.000 claims description 3
- RMRJXGBAOAMLHD-IHFGGWKQSA-N buprenorphine Chemical compound C([C@]12[C@H]3OC=4C(O)=CC=C(C2=4)C[C@@H]2[C@]11CC[C@]3([C@H](C1)[C@](C)(O)C(C)(C)C)OC)CN2CC1CC1 RMRJXGBAOAMLHD-IHFGGWKQSA-N 0.000 claims description 3
- 229960001736 buprenorphine Drugs 0.000 claims description 3
- IFKLAQQSCNILHL-QHAWAJNXSA-N butorphanol Chemical compound N1([C@@H]2CC3=CC=C(C=C3[C@@]3([C@]2(CCCC3)O)CC1)O)CC1CCC1 IFKLAQQSCNILHL-QHAWAJNXSA-N 0.000 claims description 3
- 229960001113 butorphanol Drugs 0.000 claims description 3
- 229960004606 clomipramine Drugs 0.000 claims description 3
- 229960004126 codeine Drugs 0.000 claims description 3
- 229960003914 desipramine Drugs 0.000 claims description 3
- XLMALTXPSGQGBX-GCJKJVERSA-N dextropropoxyphene Chemical compound C([C@](OC(=O)CC)([C@H](C)CN(C)C)C=1C=CC=CC=1)C1=CC=CC=C1 XLMALTXPSGQGBX-GCJKJVERSA-N 0.000 claims description 3
- 229960004193 dextropropoxyphene Drugs 0.000 claims description 3
- 229960002069 diamorphine Drugs 0.000 claims description 3
- RBOXVHNMENFORY-DNJOTXNNSA-N dihydrocodeine Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC RBOXVHNMENFORY-DNJOTXNNSA-N 0.000 claims description 3
- 229960000920 dihydrocodeine Drugs 0.000 claims description 3
- 229960002428 fentanyl Drugs 0.000 claims description 3
- PJMPHNIQZUBGLI-UHFFFAOYSA-N fentanyl Chemical compound C=1C=CC=CC=1N(C(=O)CC)C(CC1)CCN1CCC1=CC=CC=C1 PJMPHNIQZUBGLI-UHFFFAOYSA-N 0.000 claims description 3
- 229960002464 fluoxetine Drugs 0.000 claims description 3
- 229960004038 fluvoxamine Drugs 0.000 claims description 3
- CJOFXWAVKWHTFT-XSFVSMFZSA-N fluvoxamine Chemical compound COCCCC\C(=N/OCCN)C1=CC=C(C(F)(F)F)C=C1 CJOFXWAVKWHTFT-XSFVSMFZSA-N 0.000 claims description 3
- 229960000240 hydrocodone Drugs 0.000 claims description 3
- 229960004801 imipramine Drugs 0.000 claims description 3
- BCGWQEUPMDMJNV-UHFFFAOYSA-N imipramine Chemical compound C1CC2=CC=CC=C2N(CCCN(C)C)C2=CC=CC=C21 BCGWQEUPMDMJNV-UHFFFAOYSA-N 0.000 claims description 3
- 125000005956 isoquinolyl group Chemical group 0.000 claims description 3
- 229960003406 levorphanol Drugs 0.000 claims description 3
- 229960001797 methadone Drugs 0.000 claims description 3
- 229960003955 mianserin Drugs 0.000 claims description 3
- 229960000600 milnacipran Drugs 0.000 claims description 3
- 229960000805 nalbuphine Drugs 0.000 claims description 3
- NETZHAKZCGBWSS-CEDHKZHLSA-N nalbuphine Chemical compound C([C@]12[C@H]3OC=4C(O)=CC=C(C2=4)C[C@@H]2[C@]1(O)CC[C@@H]3O)CN2CC1CCC1 NETZHAKZCGBWSS-CEDHKZHLSA-N 0.000 claims description 3
- 229960001800 nefazodone Drugs 0.000 claims description 3
- VRBKIVRKKCLPHA-UHFFFAOYSA-N nefazodone Chemical compound O=C1N(CCOC=2C=CC=CC=2)C(CC)=NN1CCCN(CC1)CCN1C1=CC=CC(Cl)=C1 VRBKIVRKKCLPHA-UHFFFAOYSA-N 0.000 claims description 3
- 229960001158 nortriptyline Drugs 0.000 claims description 3
- 229960002085 oxycodone Drugs 0.000 claims description 3
- 229960002296 paroxetine Drugs 0.000 claims description 3
- VOKSWYLNZZRQPF-GDIGMMSISA-N pentazocine Chemical compound C1C2=CC=C(O)C=C2[C@@]2(C)[C@@H](C)[C@@H]1N(CC=C(C)C)CC2 VOKSWYLNZZRQPF-GDIGMMSISA-N 0.000 claims description 3
- 229960005301 pentazocine Drugs 0.000 claims description 3
- 125000004193 piperazinyl group Chemical group 0.000 claims description 3
- 125000004076 pyridyl group Chemical group 0.000 claims description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 3
- 229960003770 reboxetine Drugs 0.000 claims description 3
- CBQGYUDMJHNJBX-RTBURBONSA-N reboxetine Chemical compound CCOC1=CC=CC=C1O[C@H](C=1C=CC=CC=1)[C@@H]1OCCNC1 CBQGYUDMJHNJBX-RTBURBONSA-N 0.000 claims description 3
- 229960003394 remifentanil Drugs 0.000 claims description 3
- GGCSSNBKKAUURC-UHFFFAOYSA-N sufentanil Chemical compound C1CN(CCC=2SC=CC=2)CCC1(COC)N(C(=O)CC)C1=CC=CC=C1 GGCSSNBKKAUURC-UHFFFAOYSA-N 0.000 claims description 3
- 229960004739 sufentanil Drugs 0.000 claims description 3
- 125000005505 thiomorpholino group Chemical group 0.000 claims description 3
- 229960004380 tramadol Drugs 0.000 claims description 3
- TVYLLZQTGLZFBW-GOEBONIOSA-N tramadol Natural products COC1=CC=CC([C@@]2(O)[C@@H](CCCC2)CN(C)C)=C1 TVYLLZQTGLZFBW-GOEBONIOSA-N 0.000 claims description 3
- LLPOLZWFYMWNKH-UHFFFAOYSA-N trans-dihydrocodeinone Natural products C1C(N(CCC234)C)C2CCC(=O)C3OC2=C4C1=CC=C2OC LLPOLZWFYMWNKH-UHFFFAOYSA-N 0.000 claims description 3
- 229960002431 trimipramine Drugs 0.000 claims description 3
- ZSCDBOWYZJWBIY-UHFFFAOYSA-N trimipramine Chemical compound C1CC2=CC=CC=C2N(CC(CN(C)C)C)C2=CC=CC=C21 ZSCDBOWYZJWBIY-UHFFFAOYSA-N 0.000 claims description 3
- 229960004688 venlafaxine Drugs 0.000 claims description 3
- PNVNVHUZROJLTJ-UHFFFAOYSA-N venlafaxine Chemical compound C1=CC(OC)=CC=C1C(CN(C)C)C1(O)CCCCC1 PNVNVHUZROJLTJ-UHFFFAOYSA-N 0.000 claims description 3
- 229960002519 amoxapine Drugs 0.000 claims description 2
- QWGDMFLQWFTERH-UHFFFAOYSA-N amoxapine Chemical compound C12=CC(Cl)=CC=C2OC2=CC=CC=C2N=C1N1CCNCC1 QWGDMFLQWFTERH-UHFFFAOYSA-N 0.000 claims description 2
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 claims description 2
- 230000003449 preventive effect Effects 0.000 claims description 2
- 150000001875 compounds Chemical class 0.000 description 60
- 229940126062 Compound A Drugs 0.000 description 58
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 58
- 239000000203 mixture Substances 0.000 description 37
- 125000001424 substituent group Chemical group 0.000 description 32
- 238000000034 method Methods 0.000 description 25
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 24
- 238000009472 formulation Methods 0.000 description 21
- 241000700159 Rattus Species 0.000 description 20
- 206010058019 Cancer Pain Diseases 0.000 description 19
- 239000002904 solvent Substances 0.000 description 19
- 238000012360 testing method Methods 0.000 description 18
- 229910052736 halogen Inorganic materials 0.000 description 16
- 239000007864 aqueous solution Substances 0.000 description 15
- 150000002367 halogens Chemical class 0.000 description 15
- 238000002360 preparation method Methods 0.000 description 15
- 208000000114 Pain Threshold Diseases 0.000 description 14
- 206010039670 Sciatic nerve injury Diseases 0.000 description 14
- 230000037040 pain threshold Effects 0.000 description 14
- 230000000694 effects Effects 0.000 description 13
- 238000002347 injection Methods 0.000 description 13
- 239000007924 injection Substances 0.000 description 13
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 12
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 12
- 125000001589 carboacyl group Chemical group 0.000 description 12
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 12
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 12
- 239000008101 lactose Substances 0.000 description 12
- 235000019359 magnesium stearate Nutrition 0.000 description 12
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 12
- 229920001592 potato starch Polymers 0.000 description 11
- 239000001961 anticonvulsive agent Substances 0.000 description 10
- 125000004434 sulfur atom Chemical group 0.000 description 10
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- 238000007796 conventional method Methods 0.000 description 9
- 239000012153 distilled water Substances 0.000 description 9
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 9
- 230000006399 behavior Effects 0.000 description 7
- 125000004432 carbon atom Chemical group C* 0.000 description 7
- 229910052717 sulfur Inorganic materials 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- 238000000585 Mann–Whitney U test Methods 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- 241000699670 Mus sp. Species 0.000 description 5
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 5
- 125000003282 alkyl amino group Chemical group 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 239000003814 drug Substances 0.000 description 5
- 125000002950 monocyclic group Chemical group 0.000 description 5
- 125000004430 oxygen atom Chemical group O* 0.000 description 5
- 239000002504 physiological saline solution Substances 0.000 description 5
- 125000003107 substituted aryl group Chemical group 0.000 description 5
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 4
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 4
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 4
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 4
- 229940035676 analgesics Drugs 0.000 description 4
- 230000003556 anti-epileptic effect Effects 0.000 description 4
- 125000003710 aryl alkyl group Chemical group 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- 125000004093 cyano group Chemical group *C#N 0.000 description 4
- 238000011156 evaluation Methods 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 239000008187 granular material Substances 0.000 description 4
- 229920000609 methyl cellulose Polymers 0.000 description 4
- 239000001923 methylcellulose Substances 0.000 description 4
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 206010041349 Somnolence Diseases 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 125000005236 alkanoylamino group Chemical group 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- 125000004466 alkoxycarbonylamino group Chemical group 0.000 description 3
- 125000005115 alkyl carbamoyl group Chemical group 0.000 description 3
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 3
- 125000000304 alkynyl group Chemical group 0.000 description 3
- 125000002102 aryl alkyloxo group Chemical group 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 125000002619 bicyclic group Chemical group 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 3
- 230000002735 inhibitory effect on pain Effects 0.000 description 3
- 229910052751 metal Inorganic materials 0.000 description 3
- 239000002184 metal Substances 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 210000003497 sciatic nerve Anatomy 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 229940095064 tartrate Drugs 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 3
- 206010002091 Anaesthesia Diseases 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 208000001640 Fibromyalgia Diseases 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 208000004454 Hyperalgesia Diseases 0.000 description 2
- 206010065390 Inflammatory pain Diseases 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- 206010028813 Nausea Diseases 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- 208000001294 Nociceptive Pain Diseases 0.000 description 2
- QGMRQYFBGABWDR-UHFFFAOYSA-M Pentobarbital sodium Chemical compound [Na+].CCCC(C)C1(CC)C(=O)NC(=O)[N-]C1=O QGMRQYFBGABWDR-UHFFFAOYSA-M 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 206010036376 Postherpetic Neuralgia Diseases 0.000 description 2
- 208000032140 Sleepiness Diseases 0.000 description 2
- 102000003566 TRPV1 Human genes 0.000 description 2
- 125000002723 alicyclic group Chemical group 0.000 description 2
- 125000004656 alkyl sulfonylamino group Chemical group 0.000 description 2
- 125000004414 alkyl thio group Chemical group 0.000 description 2
- 206010053552 allodynia Diseases 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 230000006229 amino acid addition Effects 0.000 description 2
- 150000003863 ammonium salts Chemical class 0.000 description 2
- 230000037005 anaesthesia Effects 0.000 description 2
- 125000003435 aroyl group Chemical group 0.000 description 2
- 125000004104 aryloxy group Chemical group 0.000 description 2
- 210000001185 bone marrow Anatomy 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 125000000000 cycloalkoxy group Chemical group 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- 125000000723 dihydrobenzofuranyl group Chemical group O1C(CC2=C1C=CC=C2)* 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 238000001647 drug administration Methods 0.000 description 2
- 238000002651 drug therapy Methods 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 210000003141 lower extremity Anatomy 0.000 description 2
- 239000011259 mixed solution Substances 0.000 description 2
- 230000008693 nausea Effects 0.000 description 2
- 210000005036 nerve Anatomy 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 229960002275 pentobarbital sodium Drugs 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 2
- 125000004929 pyrrolidonyl group Chemical group N1(C(CCC1)=O)* 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 230000037321 sleepiness Effects 0.000 description 2
- 230000000638 stimulation Effects 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 238000001308 synthesis method Methods 0.000 description 2
- 125000005942 tetrahydropyridyl group Chemical group 0.000 description 2
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 206010044652 trigeminal neuralgia Diseases 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 1
- DTPWKUZPRRLULE-MRVPVSSYSA-N (3R)-5-amino-3-hydroxy-3,4-dihydro-1H-quinolin-2-one Chemical compound Nc1cccc2NC(=O)[C@H](O)Cc12 DTPWKUZPRRLULE-MRVPVSSYSA-N 0.000 description 1
- 0 *C(C(*)=C(*)I)=C(*)C(N(*)*)=O Chemical compound *C(C(*)=C(*)I)=C(*)C(N(*)*)=O 0.000 description 1
- 125000005871 1,3-benzodioxolyl group Chemical group 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- ZKAMEFMDQNTDFK-UHFFFAOYSA-N 1h-imidazo[4,5-b]pyrazine Chemical compound C1=CN=C2NC=NC2=N1 ZKAMEFMDQNTDFK-UHFFFAOYSA-N 0.000 description 1
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 1
- 125000000474 3-butynyl group Chemical group [H]C#CC([H])([H])C([H])([H])* 0.000 description 1
- PIGLNTUJPAEVJZ-UHFFFAOYSA-N 5-[4-(trifluoromethyl)phenyl]penta-2,4-dienoic acid Chemical compound OC(=O)C=CC=CC1=CC=C(C(F)(F)F)C=C1 PIGLNTUJPAEVJZ-UHFFFAOYSA-N 0.000 description 1
- 125000006043 5-hexenyl group Chemical group 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 208000006561 Cluster Headache Diseases 0.000 description 1
- 206010010774 Constipation Diseases 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 206010019233 Headaches Diseases 0.000 description 1
- 208000004404 Intractable Pain Diseases 0.000 description 1
- 239000001358 L(+)-tartaric acid Substances 0.000 description 1
- 235000011002 L(+)-tartaric acid Nutrition 0.000 description 1
- FEWJPZIEWOKRBE-LWMBPPNESA-N L-(+)-Tartaric acid Natural products OC(=O)[C@@H](O)[C@H](O)C(O)=O FEWJPZIEWOKRBE-LWMBPPNESA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 208000019695 Migraine disease Diseases 0.000 description 1
- 208000028389 Nerve injury Diseases 0.000 description 1
- 239000004727 Noryl Substances 0.000 description 1
- 229920001207 Noryl Polymers 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 208000004550 Postoperative Pain Diseases 0.000 description 1
- 208000004756 Respiratory Insufficiency Diseases 0.000 description 1
- 206010038678 Respiratory depression Diseases 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 206010039897 Sedation Diseases 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 238000000692 Student's t-test Methods 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 206010047700 Vomiting Diseases 0.000 description 1
- 208000021017 Weight Gain Diseases 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 125000004644 alkyl sulfinyl group Chemical group 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 1
- 230000036592 analgesia Effects 0.000 description 1
- 125000005428 anthryl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C3C(*)=C([H])C([H])=C([H])C3=C([H])C2=C1[H] 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 239000003416 antiarrhythmic agent Substances 0.000 description 1
- 229940125681 anticonvulsant agent Drugs 0.000 description 1
- 229960003965 antiepileptics Drugs 0.000 description 1
- 125000003725 azepanyl group Chemical group 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 125000004601 benzofurazanyl group Chemical group N1=C2C(=NO1)C(=CC=C2)* 0.000 description 1
- 125000004622 benzoxazinyl group Chemical group O1NC(=CC2=C1C=CC=C2)* 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 208000018912 cluster headache syndrome Diseases 0.000 description 1
- 229940125846 compound 25 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 125000002993 cycloalkylene group Chemical group 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000004090 cyclononenyl group Chemical group C1(=CCCCCCCC1)* 0.000 description 1
- 125000000522 cyclooctenyl group Chemical group C1(=CCCCCCC1)* 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000298 cyclopropenyl group Chemical group [H]C1=C([H])C1([H])* 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000004925 denaturation Methods 0.000 description 1
- 230000036425 denaturation Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- 125000004925 dihydropyridyl group Chemical group N1(CC=CC=C1)* 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 208000002173 dizziness Diseases 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 206010013781 dry mouth Diseases 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 229940034811 duloxetine 20 mg Drugs 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 229960004341 escitalopram Drugs 0.000 description 1
- WSEQXVZVJXJVFP-FQEVSTJZSA-N escitalopram Chemical compound C1([C@]2(C3=CC=C(C=C3CO2)C#N)CCCN(C)C)=CC=C(F)C=C1 WSEQXVZVJXJVFP-FQEVSTJZSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 125000003838 furazanyl group Chemical group 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 231100000869 headache Toxicity 0.000 description 1
- 125000000268 heptanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000003104 hexanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000002700 inhibitory effect on cancer Effects 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003384 isochromanyl group Chemical group C1(OCCC2=CC=CC=C12)* 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 210000000629 knee joint Anatomy 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 229960004090 maprotiline Drugs 0.000 description 1
- QSLMDECMDJKHMQ-GSXCWMCISA-N maprotiline Chemical compound C12=CC=CC=C2[C@@]2(CCCNC)C3=CC=CC=C3[C@@H]1CC2 QSLMDECMDJKHMQ-GSXCWMCISA-N 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 206010027599 migraine Diseases 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 229940035363 muscle relaxants Drugs 0.000 description 1
- 239000003158 myorelaxant agent Substances 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000008764 nerve damage Effects 0.000 description 1
- 210000004126 nerve fiber Anatomy 0.000 description 1
- 230000001272 neurogenic effect Effects 0.000 description 1
- 230000002981 neuropathic effect Effects 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 210000000929 nociceptor Anatomy 0.000 description 1
- 108091008700 nociceptors Proteins 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000005485 noradamantyl group Chemical group 0.000 description 1
- 229960002748 norepinephrine Drugs 0.000 description 1
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 1
- 230000000966 norepinephrine reuptake Effects 0.000 description 1
- 125000002801 octanoyl group Chemical group C(CCCCCCC)(=O)* 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 230000000010 osteolytic effect Effects 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000005961 oxazepanyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 125000000466 oxiranyl group Chemical group 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- RGCLLPNLLBQHPF-HJWRWDBZSA-N phosphamidon Chemical compound CCN(CC)C(=O)C(\Cl)=C(/C)OP(=O)(OC)OC RGCLLPNLLBQHPF-HJWRWDBZSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000001107 psychogenic effect Effects 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000005494 pyridonyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000036280 sedation Effects 0.000 description 1
- 230000001953 sensory effect Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 230000000697 serotonin reuptake Effects 0.000 description 1
- 229960002073 sertraline Drugs 0.000 description 1
- VGKDLMBJGBXTGI-SJCJKPOMSA-N sertraline Chemical compound C1([C@@H]2CC[C@@H](C3=CC=CC=C32)NC)=CC=C(Cl)C(Cl)=C1 VGKDLMBJGBXTGI-SJCJKPOMSA-N 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 125000005338 substituted cycloalkoxy group Chemical group 0.000 description 1
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 210000000225 synapse Anatomy 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical class C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000004588 thienopyridyl group Chemical group S1C(=CC2=C1C=CC=N2)* 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000002053 thietanyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 231100000027 toxicology Toxicity 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 210000000689 upper leg Anatomy 0.000 description 1
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 102000038650 voltage-gated calcium channel activity Human genes 0.000 description 1
- 108091023044 voltage-gated calcium channel activity Proteins 0.000 description 1
- 230000008673 vomiting Effects 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 230000004584 weight gain Effects 0.000 description 1
- 235000019786 weight gain Nutrition 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
Images



Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/02—Halogenated hydrocarbons
- A61K31/025—Halogenated hydrocarbons carbocyclic
- A61K31/03—Halogenated hydrocarbons carbocyclic aromatic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/045—Hydroxy compounds, e.g. alcohols; Salts thereof, e.g. alcoholates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/075—Ethers or acetals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
- A61K31/136—Amines having aromatic rings, e.g. ketamine, nortriptyline having the amino group directly attached to the aromatic ring, e.g. benzeneamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
- A61K31/137—Arylalkylamines, e.g. amphetamine, epinephrine, salbutamol, ephedrine or methadone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
- A61K31/167—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide having the nitrogen of a carboxamide group directly attached to the aromatic ring, e.g. lidocaine, paracetamol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/18—Sulfonamides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
- A61K31/197—Carboxylic acids, e.g. valproic acid having an amino group the amino and the carboxyl groups being attached to the same acyclic carbon chain, e.g. gamma-aminobutyric acid [GABA], beta-alanine, epsilon-aminocaproic acid or pantothenic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/38—Nitrogen atoms
Definitions
- the present invention relates to a pharmaceutical composition containing a pentadienamide derivative or a pharmaceutically acceptable salt thereof and an analgesic.
- pain is nociceptive pain caused by persistent stimulation of nociceptors by endogenous pain substances, etc., and neuropathic pain caused by abnormalities in the function of nerve fibers involved in nerve transmission , And psychogenic pain.
- various pains such as neuropathic pain, inflammatory pain and cancer pain as well as headaches such as migraine and cluster headache are known.
- Neuropathic pain is an intractable pain that is less effective with anti-inflammatory and opioid analgesics, such as postherpetic neuralgia, diabetic neuropathic pain, HIV-related pain, trigeminal neuralgia, pain associated with cancer chemotherapy, Fibromyalgia and the like are included.
- opioid analgesics such as postherpetic neuralgia, diabetic neuropathic pain, HIV-related pain, trigeminal neuralgia, pain associated with cancer chemotherapy, Fibromyalgia and the like are included.
- pain in which nociceptive pain and neuropathic pain are mixed includes postoperative pain, cancer pain, and the like.
- surgical therapy nerve block therapy, electrical stimulation therapy, drug therapy and the like are used.
- antidepressants, antidepressants, anticonvulsants, opioid analgesics, muscle relaxants, antiarrhythmic agents, etc. are used. In many cases, sufficient analgesic effect cannot be obtained.
- antidepressants are thought to exert analgesic effects by inhibiting central noradrenaline and serotonin reuptake and activating the descending pain suppression system, but nausea, nausea, dry mouth, sleepiness, etc. Side effects and may not be administered up to a dose sufficient to treat pain.
- opioid analgesics have been reported to be effective against neuropathic pain, where opioids have been considered to be ineffective, and the frequency of opioid analgesics for neuropathic pain will increase in the future. Is expected.
- opioid analgesics have side effects such as sedation, constipation, respiratory depression, vomiting, and drowsiness, and if the dose is reduced to avoid these side effects, the pain may not be relieved appropriately.
- Gabapentin and pregabalin which are also used as an antidepressant, are drugs that are marketed as neuropathic analgesics in the West. These drugs are thought to exert analgesic effects by controlling the release of neurotransmitters at synapses by binding to the ⁇ 2 ⁇ subunit of voltage-gated calcium channels.
- clinical effectiveness is not always sufficient (about 50%), and side effects such as dizziness, sleepiness, and weight gain are observed.
- TRPV1 Transient Receptor Potential Vanilloid 1 receptor antagonist containing a compound used in the present invention as an active ingredient is known (see Patent Document 1).
- An object of the present invention is to provide a pharmaceutical composition or the like useful as an agent for treating and / or preventing pain (pain).
- the present invention relates to the following (1) to (56).
- R 1 represents a substituted or unsubstituted aryl or a substituted or unsubstituted aromatic heterocyclic group
- R 2 represents substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted aryl, substituted or unsubstituted aromatic heterocyclic group or substituted or unsubstituted alicyclic heterocyclic group.
- R 3 represents a hydrogen atom, or together with R 4 and an adjacent nitrogen atom, forms a substituted or unsubstituted heterocyclic group
- R 4 represents substituted or unsubstituted lower alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted aromatic heterocyclic group or substituted or unsubstituted alicyclic heterocyclic group.
- R 5 , R 6 and R 7 are the same or different and each represents a hydrogen atom or methyl), and a pharmaceutically acceptable salt thereof and (b) an analgesic.
- R 5 , R 6 and R 7 are each a hydrogen atom.
- R 3 and R 4 together with the adjacent nitrogen atom form a substituted or unsubstituted heterocyclic group.
- R 3 and R 4 together with the adjacent nitrogen atom form a substituted or unsubstituted thiomorpholino.
- the antidepressant is duloxetine, amitriptyline, fluvoxatin, fluvoxamine, fluoxetine, nortriptyline, venlafaxine, paroxetine, reboxetine, milnacipran, imipramine, desipramine, clomipramine, trimipramine, amoxapine, lofepramine, estrothromrine, praspramel (24)
- the opioid analgesic is morphine, fentanyl, remifentanil, sufentanil, buprenorphine, hydromorphine, oxymorphine, pentazocine, tramadol, codeine, dihydrocodeine, oxycodone, methadone, heroin, nalbuphine, levorphanol, levalrphan, mepyridine, hydrocodone (25)
- the pharmaceutical composition according to (25) which is an opioid analgesic selected from the group consisting of propoxyphene and butorphanol.
- the pharmaceutical composition according to (26), wherein the antiepileptic agent is an antiepileptic agent selected from the group consisting of gabapentin and pregabalin.
- R 1 represents a substituted or unsubstituted aryl or a substituted or unsubstituted aromatic heterocyclic group
- R 2 represents substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted aryl, substituted or unsubstituted aromatic heterocyclic group or substituted or unsubstituted alicyclic heterocyclic group.
- R 3 represents a hydrogen atom, or together with R 4 and an adjacent nitrogen atom, forms a substituted or unsubstituted heterocyclic group
- R 4 represents substituted or unsubstituted lower alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted aromatic heterocyclic group or substituted or unsubstituted alicyclic heterocyclic group.
- R 5 , R 6 and R 7 are the same or different and each represents a hydrogen atom or methyl), or a pharmaceutically acceptable salt thereof and (b) an analgesic as an active ingredient Treatment and / or prevention agent.
- R 5 , R 6 and R 7 are the same or different and each represents a hydrogen atom or methyl), or a pharmaceutically acceptable salt thereof and (b) an analgesic as an active ingredient Treatment and / or prevention agent.
- R 1 represents a substituted or unsubstituted aryl or a substituted or unsubstituted aromatic heterocyclic group
- R 2 represents substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted aryl, substituted or unsubstituted aromatic heterocyclic group or substituted or unsubstituted alicyclic heterocyclic group.
- R 3 represents a hydrogen atom, or together with R 4 and an adjacent nitrogen atom, forms a substituted or unsubstituted heterocyclic group
- R 4 represents substituted or unsubstituted lower alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted aromatic heterocyclic group or substituted or unsubstituted alicyclic heterocyclic group.
- R 5 , R 6, and R 7 are the same or different and each represents a hydrogen atom or methyl)), or a pharmaceutically acceptable salt thereof, and (b) an analgesic, as an active ingredient A therapeutic and / or prophylactic agent for pain for administering (a) and (b) simultaneously or separately at intervals.
- R 5 , R 6, and R 7 are the same or different and each represents a hydrogen atom or methyl)), or a pharmaceutically acceptable salt thereof, and (b) an analgesic, as an active ingredient A therapeutic and / or prophylactic agent for pain for administering (a) and (b) simultaneously or separately at intervals.
- an analgesic as an active ingredient A therapeutic and / or prophylactic agent for pain for administering (a) and (b) simultaneously or separately at intervals.
- R 1 represents a substituted or unsubstituted aryl or a substituted or unsubstituted aromatic heterocyclic group
- R 2 represents substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted aryl, substituted or unsubstituted aromatic heterocyclic group or substituted or unsubstituted alicyclic heterocyclic group.
- R 3 represents a hydrogen atom, or together with R 4 and an adjacent nitrogen atom, forms a substituted or unsubstituted heterocyclic group
- R 4 represents substituted or unsubstituted lower alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted aromatic heterocyclic group or substituted or unsubstituted alicyclic heterocyclic group.
- R 5 , R 6 and R 7 are the same or different and each represents a hydrogen atom or methyl), a first component containing a pentadienamide derivative represented by the formula (I) or a pharmaceutically acceptable salt thereof; and (b) an analgesic. And a second component.
- R 5 , R 6 and R 7 are the same or different and each represents a hydrogen atom or methyl), a first component containing a pentadienamide derivative represented by the formula (I) or a pharmaceutically acceptable salt thereof; and (b) an analgesic.
- a second component is 3
- R 1 represents a substituted or unsubstituted aryl or a substituted or unsubstituted aromatic heterocyclic group
- R 2 represents substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted aryl, substituted or unsubstituted aromatic heterocyclic group or substituted or unsubstituted alicyclic heterocyclic group.
- R 3 represents a hydrogen atom, or together with R 4 and an adjacent nitrogen atom, forms a substituted or unsubstituted heterocyclic group
- R 4 represents substituted or unsubstituted lower alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted aromatic heterocyclic group or substituted or unsubstituted alicyclic heterocyclic group.
- R 5 , R 6 and R 7 are the same or different and each represents a hydrogen atom or methyl), a first component containing a pentadienamide derivative represented by the formula (I) or a pharmaceutically acceptable salt thereof; and (b) an analgesic.
- a kit for treating and / or preventing pain, comprising a second component. (34) The therapeutic and / or prophylactic agent for pain according to (30), wherein the pain is neuropathic pain. (35) The therapeutic and / or prophylactic agent for pain according to (31), wherein the pain is neuropathic pain.
- the pentadienamide derivative or a pharmaceutically acceptable salt thereof according to any one of (a) (1) to (23) and (b) an analgesic agent are administered simultaneously or separately at intervals.
- (42) (a) Treatment and / or prevention of pain containing the pentadienamide derivative or the pharmaceutically acceptable salt thereof according to any one of (1) to (23) and (b) an analgesic as an active ingredient Use of (a) for the manufacture of an agent.
- (43) (a) The pentadienamide derivative or the pharmaceutically acceptable salt thereof according to any one of (1) to (23) and (b) an analgesic agent as an active ingredient, (a) and (b) Use of (a) for the manufacture of a therapeutic and / or prophylactic agent for pain to be administered simultaneously or separately over time. (44) (a) a first component containing the pentadienamide derivative according to any one of (1) to (23) or a pharmaceutically acceptable salt thereof; and (b) a second component containing an analgesic agent; Use of (a) for producing a kit for the treatment and / or prevention of pain characterized by comprising: (45) The use according to (42), wherein the pain is neuropathic pain.
- the pentadienamide derivative or a pharmaceutically acceptable salt thereof according to any one of (a) (1) to (23) and (b) an analgesic are administered simultaneously or separately at intervals.
- a method for treating and / or preventing pain (53) The method according to (51), wherein the pain is neuropathic pain.
- the method according to (52), wherein the pain is neuropathic pain.
- the method according to (52), wherein the pain is cancer pain.
- the method according to (52), wherein the pain is cancer pain.
- the present invention provides, for example, a pharmaceutical composition containing (a) a pentadienamide derivative or a pharmaceutically acceptable salt thereof and (b) an analgesic.
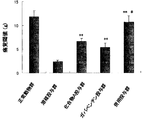
- FIG. 1 shows the pain threshold (g) after administration of Compound A and gabapentin. **: P ⁇ 0.01 [Wilcoxon rank sum test compared to solvent administration group]. #: P ⁇ 0.05 [Wilcoxon rank-sum test (Compound A and gabapentin administration group comparison)].
- FIG. 2 shows the pain threshold (g) after administration of Compound A and duloxetine. **: P ⁇ 0.01 [Wilcoxon rank sum test compared to solvent administration group].
- FIG. 3 shows the pain threshold (g) after administration of Compound A and morphine.
- FIG. 4 shows guarding behavior (pain protection behavior during walking) time (seconds) after administration of Compound A and morphine.
- each group of formula (I) examples include linear or branched alkyl having 1 to 10 carbon atoms, and more specifically, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert- Examples include butyl, pentyl, neopentyl, hexyl, heptyl, octyl, nonyl, decyl and the like.
- cycloalkyl examples include cycloalkyl having 3 to 10 carbon atoms, and more specifically, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclodecyl, noradamantyl, adamantyl, bicyclo [2.2. 1) heptyl, bicyclo [2.2.2] octyl, bicyclo [3.3.0] octyl, bicyclo [3.3.1] nonyl and the like.
- cycloalkenyl examples include cycloalkenyl having 3 to 10 carbon atoms, and more specific examples include cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclohexenyl, cycloheptenyl, cyclooctenyl, cyclononenyl, cyclodecenyl and the like.
- aryl examples include aryl having 6 to 14 carbon atoms, and more specifically include phenyl, naphthyl, indenyl, anthryl and the like.
- Aryl is, for example, aryl condensed with a cyclohexyl group, such as 1,2,3,4-tetrahydronaphthalene, or 1,2,3,4-tetrahydroquinolyl, 1,2,3,4-tetrahydroisoquino.
- Ril dihydroquinoxalinyl, tetrahydroquinoxalinyl, dihydrobenzofuranyl, chromanyl, isochromanyl, benzoxazinyl, dihydrobenzoxazinyl, cihydrobenzodioxinyl, 1,3-benzodioxolyl, etc.
- An aryl fused with an alicyclic heterocyclic group may be used.
- aromatic heterocyclic group for example, a 5-membered or 6-membered monocyclic aromatic heterocyclic group containing at least one atom selected from a nitrogen atom, an oxygen atom and a sulfur atom, and a 3- to 8-membered ring are condensed.
- Examples of the alicyclic heterocyclic group include a 5-membered or 6-membered monocyclic alicyclic heterocyclic group containing at least one atom selected from a nitrogen atom, an oxygen atom, and a sulfur atom, and a 3- to 8-membered ring. And a condensed alicyclic heterocyclic group containing at least one atom selected from a nitrogen atom, an oxygen atom and a sulfur atom, and more specifically, pyrrolidinyl, pyrrolidonyl, etc.
- the heterocyclic group formed together with the adjacent nitrogen atom includes an aromatic heterocyclic group formed together with the adjacent nitrogen atom or an alicyclic ring formed together with the adjacent nitrogen atom. And a formula heterocyclic group.
- the aromatic heterocyclic group formed together with the adjacent nitrogen atom include a 5-membered or 6-membered monocyclic aromatic heterocyclic group containing at least one nitrogen atom (the monocyclic aromatic group).
- a heterocyclic group may contain other nitrogen, oxygen or sulfur atoms), a bicyclic or tricyclic condensed 3- to 8-membered ring and containing at least one nitrogen atom
- Aromatic heterocyclic group (the condensed aromatic heterocyclic group may contain other nitrogen atom, oxygen atom or sulfur atom) and the like, more specifically pyrrolyl, imidazolyl, indolyl, Indazolyl, carbazolyl, pyrazolyl and the like can be mentioned.
- Examples of the alicyclic heterocyclic group formed together with the adjacent nitrogen atom include a 5- or 6-membered monocyclic alicyclic heterocyclic group containing at least one nitrogen atom (the monocyclic An alicyclic heterocyclic group may contain other nitrogen, oxygen or sulfur atoms), a bicyclic or tricyclic fused 3-8 membered ring and contains at least one nitrogen atom
- a condensed alicyclic heterocyclic group (the condensed alicyclic heterocyclic group may contain other nitrogen, oxygen or sulfur atoms), and more specifically pyrrolidinyl.
- the substituents in the substituted alicyclic heterocyclic group formed together with the adjacent nitrogen atom are the same or different, for example, 1 to 3 substituents, more specifically halogen, hydroxy, Amino, hydroxyimino, amidino, hydroxyamidino, nitro, mercapto, cyano, carboxy, methylenedioxy, ethylenedioxy, carbamoyl, sulfamoyl, sulfamoyloxy, lower alkenyl, cycloalkenyl, lower alkynyl, lower alkoxycarbonyl, mono or Di-lower alkylcarbamoyl, lower alkyl Rufinyl, lower alkyls
- Substituted, unsubstituted lower alkanoylamino may be the same or different, for example, more specifically 1 to 3 substituents.
- substituted or unsubstituted lower alkylsulfonyl substituted or unsubstituted lower alkylsulfonyl are the same or different, for example, having 1 to 3 substituents, more specifically, Halogen, hydroxy, cyano and the like
- substituted or unsubstituted lower alkoxy substituted or unsubstituted lower alkoxy [substituents in the substituted lower alkoxy are the same or different, for example, having 1 to 3 substituents, more specifically halogen, amino Mono or di (lower alkyl) amino, hydroxy, cyano, lower alkoxy, lower alkoxycarbonyl, lower alkoxycarbonylamino, lower alkanoyl, cycloalkyl, lower
- Substituted or unsubstituted lower alkanoyl [substituents in the substituted lower alkanoyl are the same or different and include, for example, 1 to 3 substituents, more specifically halogen and the like], substituted or non-substituted Substituted aryl (substituents in the substituted aryl are the same or different and include, for example, 1 to 3 substituents, more specifically halogen, nitro, hydroxy, amino, lower alkoxy, etc.), substituted or non-substituted Substituted aralkyl (substituents in the substituted aralkyl may be the same or different, examples For example, halogen, hydroxy, lower alkoxy, etc.), substituted or unsubstituted aromatic heterocyclic groups (substituents in the substituted aromatic heterocyclic groups are the same) Or differently, for example, having 1 to 3 substituents, more specifically, halogen, hydroxy,
- Substituents in the cyclic heterocyclic oxy are the same or different and include, for example, 1 to 3 substituents, more specifically halogen, hydroxy, lower alkyl, lower alkoxy, lower alkanoyl, etc.), substituted or non-substituted Substituted alicyclic heterocyclic alkyloxy (substituents in the substituted alicyclic heterocyclic alkyloxy are the same or different, for example, 1 to 3 substituents, more specifically halogen, hydroxy, lower alkyl, And lower alkoxy and lower alkanoyl).
- a substituted aryl, a substituted aromatic heterocyclic group, a substituted alicyclic heterocyclic group, a substituted aromatic heterocyclic group formed together with an adjacent nitrogen atom, and an adjacent nitrogen atom are formed.
- the substituent in the substituted alicyclic heterocyclic group is a substituted or unsubstituted lower alkyl (the substituents in the substituted lower alkyl are the same or different, for example, 1 to 3 substituents, more specifically halogen Amino, hydroxy, hydroxyimino, cyano, mono- or di-lower alkylamino, aromatic heterocyclic group, alicyclic heterocyclic group, and the like.
- the substituent in the substituted alicyclic heterocyclic group formed together with the substituted lower alkyl, the substituted alicyclic heterocyclic group and the adjacent nitrogen atom may be oxo.
- the substituent in the case where the substituted aryl is an aryl condensed with a substituted alicyclic heterocyclic group includes oxo.
- an alicyclic heterocyclic group, an aromatic heterocyclic group, an aromatic heterocyclic group formed together with an adjacent nitrogen atom, and a substituted alicyclic heterocyclic formed together with an adjacent nitrogen atom When the ring group has a nitrogen atom and / or a sulfur atom in the ring, the nitrogen atom and / or the sulfur atom may be oxidized.
- lower alkyl, cycloalkyl, cycloalkenyl, aryl, aromatic heterocyclic group and alicyclic heterocyclic group have the same meanings as described above.
- the halogen include fluorine, chlorine, bromine, and iodine atoms.
- the lower alkyl-substituted alicyclic heterocyclic group and the lower alkyl moiety of lower alkylthio have the same meaning as the lower alkyl.
- the two lower alkyl moieties in di-lower alkylamino and di-lower alkylcarbamoyl may be the same or different.
- the cycloalkylene moiety in the lower alkyl-substituted cycloalkyl has the same meaning as that obtained by removing one hydrogen atom from the cycloalkyl.
- the cycloalkyl moiety in cycloalkyloxy has the same meaning as the above cycloalkyl.
- the alkylene part in aralkyl, aralkyloxy, alicyclic heterocyclic alkyloxy and hydroxyalkyl has the same meaning as that obtained by removing one hydrogen atom from the lower alkyl.
- lower alkenyl examples include linear or branched alkenyl having 3 to 10 carbon atoms, and more specifically, allyl, 2-butenyl, 3-butenyl, 4-pentenyl, 5-hexenyl, Examples include 6-heptenyl, 6-octenyl, 2,6-octadienyl, 9-decenyl and the like.
- lower alkynyl examples include linear or branched alkynyl having 3 to 6 carbon atoms, and more specifically, propargyl, 3-butynyl, 3-hexynyl, 4-methyl-2-pentynyl and the like. Can be mentioned.
- Examples of the lower alkanoyl part of lower alkanoyl and lower alkanoylamino include linear or branched alkanoyl having 1 to 8 carbon atoms, and more specifically formyl, acetyl, propionyl, butyryl, isobutyryl, valeryl. , Isovaleryl, pivaloyl, hexanoyl, heptanoyl, octanoyl and the like.
- the aryl part of aralkyl, aryloxy, aralkyloxy and aroyl has the same meaning as the above aryl.
- the alicyclic heterocyclic group part in alicyclic heterocyclic oxy and alicyclic heterocyclic alkyloxy is synonymous with the said alicyclic heterocyclic group.
- the alicyclic heterocyclic group moiety in the lower alkyl-substituted alicyclic heterocyclic group has the same meaning as that obtained by removing one hydrogen atom from the alicyclic heterocyclic group.
- the aromatic heterocyclic group moiety in the aromatic heterocyclic aminosulfonyl has the same meaning as the aromatic heterocyclic group.
- compound (I) or a pharmaceutically acceptable salt thereof can be used, but preferably (i) R 5 , R 6 and R Compound (I) wherein 7 is a hydrogen atom or a pharmaceutically acceptable salt thereof, (ii) Compound (I) wherein R 1 and R 2 are substituted or unsubstituted aryl, or a pharmaceutically acceptable salt thereof Salt (iii) Compound (I) wherein R 3 is a hydrogen atom and R 4 is substituted or unsubstituted tetrahydroquinolyl or a pharmaceutically acceptable salt thereof can be used.
- the compound of the aspect which combined two or three said (i), (ii) and (iii) can be used. More preferably, compounds selected from compounds 188, 191, 229, 240, 257, 270, 310, 314, 332 and 333 can be used.
- Examples of the pharmaceutically acceptable salt of compound (I) include pharmaceutically acceptable metal salts, ammonium salts, organic amine addition salts, amino acid addition salts, acid addition salts, and the like.
- Examples of the pharmaceutically acceptable metal salt include alkali metal salts such as sodium salt and potassium salt, alkaline earth metal salts such as magnesium salt and calcium salt, aluminum salt and zinc salt, and the like.
- Examples of ammonium salts include ammonium and tetramethylammonium salts
- pharmaceutically acceptable organic amine addition salts include addition salts such as morpholine and piperidine, which are pharmaceutically acceptable.
- Examples of amino acid addition salts include addition salts of amino acids such as lysine, glycine, and phenylalanine.
- Examples of pharmaceutically acceptable acid addition salts include inorganic acid salts such as hydrochloride, sulfate, and phosphate, Examples thereof include organic acid salts such as acetate, maleate, fumarate, tartrate and citrate.
- the compound (I) and pharmaceutically acceptable salts thereof in the present invention may exist in the form of adducts with water or various solvents, and these adducts are also present in the pharmaceutical composition of the present invention, pain. It can be used for therapeutic and / or prophylactic agents, kits, pain treatment and / or prevention kits, pain treatment and / or prevention methods, and the like.
- the compound (I) may be purified as it is when it is obtained in the form of a salt. It may be isolated or purified by dissolving or suspending in an appropriate solvent and adding an acid or base to form a salt.
- Some compounds (I) in the present invention may have various stereoisomers, positional isomers, tautomers, enantiomers and the like.
- the pharmaceutical composition, the therapeutic and / or preventive agent for pain, the kit, the kit for treating and / or preventing pain, the method for treating and / or preventing pain, etc. include all these possible isomers and their isomers.
- a mixture can be used, and the mixing ratio may be any ratio.
- Compound (I) or a pharmaceutically acceptable salt thereof can be produced, for example, by the method described in WO2008 / 007780. Specific examples of the compounds used in the present invention are shown in Tables 1 to 6 below, but the compounds used in the present invention are not limited thereto.
- Intermediates and target compounds in the above production method are isolated and purified by subjecting them to separation and purification methods commonly used in organic synthetic chemistry, such as filtration, extraction, washing, drying, concentration, recrystallization, and various chromatography. Can do. The intermediate can be subjected to the next reaction without any particular purification.
- examples of the analgesic agent used for the pentadienamide derivative or a pharmaceutically acceptable salt thereof include antidepressants, opioid analgesics, antidepressants and the like.
- antidepressants include duloxetine, amitriptyline, fluvoxatin, fluvoxamine, fluoxetine, nortriptyline, venlafaxine, paroxetine, reboxetine, milnacipran, imipramine, desipramine, clomipramine, trimipramine, lofepramine, dressing, lofepramine, dressing Escitalopram, sertraline, maprotiline, mianserin, cetiptiline, nefazodone and the like. These may be used alone or in combination.
- opioid analgesics include morphine, fentanyl, remifentanil, sufentanil, buprenorphine, hydromorphine, oxymorphine, pentazocine, tramadol, codeine, dihydrocodeine, oxycodone, methadone, heroin, nalbuphine, levorphanol, Examples include levalorphan, mepyridine, hydrocodone, propoxyphene, butorphanol and the like. These may be used alone or in combination.
- the antiepileptic agent examples include ⁇ 2 ⁇ ligands such as gabapentin and pregabalin. These may be used alone or in combination.
- These analgesics are pharmaceutically acceptable salts (the pharmaceutically acceptable salts include those exemplified as the pharmaceutically acceptable salts of the compound (I)) or the like. Although it may exist as a hydrate, the pharmaceutical composition of the present invention, an agent for treating and / or preventing pain, a kit, a kit for treating and / or preventing pain, a method for treating and / or preventing pain, etc. These can also be used.
- analgesics such as the antidepressants, opioid analgesics, and antiepileptics exemplified above can be obtained as commercially available products or according to conventionally known methods.
- the pharmaceutical composition containing compound (I) or a pharmaceutically acceptable salt thereof and an analgesic can be used, for example, for the treatment and / or prevention of pain, and more specifically, neurogenic It can be used for treatment and / or prevention of pain, inflammatory pain, cancer pain and the like. More preferably, it can be used for treatment and / or prevention of neuropathic pain.
- neuropathic pain include postherpetic neuralgia, diabetic neuropathic pain, HIV-related pain, trigeminal neuralgia, pain associated with cancer chemotherapy, fibromyalgia and the like.
- the pharmaceutical composition of the present invention and the therapeutic and / or prophylactic agent for pain are single agents as long as they are formulated so as to contain Compound (I) or a pharmaceutically acceptable salt thereof and an analgesic. Although it can be used or administered as a (mixture) or a combination of a plurality of preparations, a combination of two or more preparations is preferred. When used or administered as a combination of multiple formulations, these multiple formulations can be used or administered simultaneously or separately over time. These preparations are preferably used in the form of tablets, injections and the like.
- the dose ratio (weight / weight) between compound (I) or a pharmaceutically acceptable salt thereof and the analgesic is the combination of compound (I) and analgesic used, and each of compound (I) and analgesic. It may be appropriately adjusted according to the efficacy and the like. Specifically, for example, 1/50 (compound (I) or a pharmaceutically acceptable salt / analgesic agent thereof) to 50,000 / 1, preferably 1/30 to 10,000. / 1, more preferably 1/20 to 5000/1, still more preferably 1/10 to 1000/1, still more preferably 1/10 to 100/1, even more preferably 1/10 to 10/1, most preferably Is a ratio between 1/5 and 5/1.
- a first component containing Compound (I) or a pharmaceutically acceptable salt thereof When administered as a combination of a plurality of preparations, for example, (a) a first component containing Compound (I) or a pharmaceutically acceptable salt thereof, and (b) a second component containing an analgesic agent.
- a first component containing Compound (I) or a pharmaceutically acceptable salt thereof When administered as a combination of a plurality of preparations, for example, (a) a first component containing Compound (I) or a pharmaceutically acceptable salt thereof, and (b) a second component containing an analgesic agent.
- a second component containing an analgesic agent When administered as a combination of a plurality of preparations, for example, (a) a first component containing Compound (I) or a pharmaceutically acceptable salt thereof, and (b) a second component containing an analgesic agent.
- Each of these can be formulated separately and prepared as a kit, and each component can be administered to the same
- kits There are no particular restrictions on the material, shape, etc. of the kit as long as it is a container that does not show, for example, denaturation of components that are contents due to external temperature or light during storage or elution of chemical components from the container. It consists of the above containers (for example, vials, bags, etc.) and contents, and has a form in which the first component and the second component, which are the contents, can be administered through separate routes (for example, tubes) or the same route. Things are used. Specific examples include kits such as tablets and injections.
- the method for treating and / or preventing pain according to the present invention can be carried out in the same manner as in the method for using or administering the compound (I) described above or a pharmaceutically acceptable salt thereof and an analgesic. That is, compound (I) or a pharmaceutically acceptable salt thereof and an analgesic are formulated so as to contain each active ingredient, and preferably two or more, for example, as a single agent or a combination of a plurality of formulations. It can be carried out by administering a combination of these preparations. When a plurality of preparations are administered in combination, these preparations can be administered simultaneously or separately over time, and can also be administered using a kit as described above.
- Test Example 1 Preparation of rat with sciatic nerve injury (neuropathic pain model) With reference to the method of Fisher et al. [Pain, 1998, Vol. 77, p. 59], sciatic nerve injured neuropathic pain A model was created.
- CD (SD) rats male, 5 weeks old, Charles River Japan, Inc. were exposed by exfoliating the sciatic nerve of the left hind limb under pentobarbital sodium anesthesia (50 mg / kg, intraperitoneal administration)
- the sciatic nerve was covered with a 2 mm long PE-60 polyethylene tube (trade name: Intramedic, size: PE-60, manufactured by Beckton Dickinson).
- rats that had passed 2-3 weeks after the operation were used.
- Von Frey filament (trade name: touch test sensory evaluator, model number: model 58011, manufactured by Muromachi Kikai Co., Ltd.) for the measurement of mechanically irritated allodynia commonly observed in neuropathic pain was used.
- an acrylic quadruple cage (width 390 mm ⁇ depth 210 mm ⁇ height 145 mm) covered with a metal mesh on the bottom and accustomed to the environment for about 20 minutes, the sole on the nerve injury side
- the von Frey filament was applied for about 4 seconds so that the von Frey filament was lightly bent, and the presence or absence of an escape reaction was observed. The result was shown as a pain threshold.
- Pain threshold (g) is determined by Dixon's up-down method [Annual Review of Pharmacology and Toxicology, 1980, Volume 20, p.441] Calculated by The pain threshold in normal rats was about 12 g, whereas the pain threshold in the sciatic nerve injury foot before drug administration was about 2 g, confirming a decrease in the pain threshold (g) recognized as allodynia.
- Test Example 2 Enhancement effect of gabapentin analgesic action in a rat pain model by Compound A Acrylic quadruple cage (width 390 mm x depth 210 mm x height) 145 mm), and gabapentin and Compound A were simultaneously administered by the following procedure, and pain threshold (g) was measured 3 hours later.
- gabapentin was dissolved in distilled water (Otsuka Pharmaceutical Factory) at a concentration of 20 mg / mL, orally administered at a dose of 100 mg / kg, and Compound A was 0.5% methylcellulose (0.6 mg / mL) It was suspended in 0.5% MC) aqueous solution and orally administered at a dose of 3 mg / kg.
- distilled water was orally administered to rats with sciatic nerve injury 5 mL / kg, and 0.5% MC aqueous solution was orally administered 5 mL / kg.
- gabapentin administration group gabapentin was orally administered to rats with sciatic nerve injury in the same manner as described above, and 0.5% MC aqueous solution was orally administered to 5 ⁇ mL / kg.
- compound A administration group 5 ⁇ mL / kg of distilled water was orally administered to rats with sciatic nerve injury, and 3 ⁇ mg / kg of compound A was orally administered in the same manner as described above. The test was conducted with 6 animals in each group. The results are shown in FIG.
- Test Example 3 Enhancement effect of duloxetine analgesia in rat pain model by compound A Effect of acrylic quadruplicate cage (width 390 mm ⁇ depth 210 mm ⁇ height) 145 mm), duloxetine and Compound A were simultaneously administered by the following procedure, and pain threshold (g) was measured 3 hours later.
- duloxetine was dissolved in distilled water (Otsuka Pharmaceutical Factory) at a concentration of 2 mg / mL, and orally administered at a dose of 10 mg / kg, and Compound A was 0.5% methylcellulose (0.6 mg / mL). It was suspended in 0.5% MC) aqueous solution and orally administered at a dose of 3 mg / kg.
- distilled water was orally administered to rats with sciatic nerve injury 5 mL / kg, and 0.5% MC aqueous solution was orally administered 5 mL / kg.
- duloxetine was orally administered to rats with sciatic nerve injury in the same manner as described above, and 0.5% MC aqueous solution was orally administered to 5 ⁇ mL / kg. Further, as a compound A administration group, 5 ⁇ mL / kg of distilled water was orally administered to rats with sciatic nerve injury, and 3 ⁇ mg / kg of compound A was orally administered in the same manner as described above. The test was conducted with 6 animals in each group. The results are shown in FIG.
- Test Example 4 Enhancement effect of morphine analgesic action in rat pain model by compound A Acrylic quadruple cage (width 390 mm ⁇ depth 210 mm ⁇ height) 145 mm), morphine was administered 2 hours after compound A was administered by the following procedure, and pain threshold (g) was measured 1 hour later.
- Compound A was suspended in 0.5% methylcellulose (0.5% MC) aqueous solution at a concentration of 0.6 mg / mL, and orally administered at a dose of 3 mg / kg, and morphine was added to physiological saline at a concentration of 0.25 mg / mL. It was dissolved in water (Otsuka Pharmaceutical Factory) and administered subcutaneously at a dose of 0.5 mg / kg (combination administration group).
- 0.5% MC aqueous solution was orally administered to rats with sciatic nerve injury and physiological saline was subcutaneously administered at 2 mL / kg.
- morphine administration group 0.5% ⁇ ⁇ ⁇ MC aqueous solution was orally administered to rats with sciatic nerve injury in the same manner as described above, and morphine was administered subcutaneously at 0.5 mg / kg. Furthermore, as a compound A administration group, compound A was orally administered to rats with sciatic nerve injury in the same manner as described above, and physiological saline was subcutaneously administered at 2 mg / kg. The test was conducted with 6 animals in each group. The results are shown in FIG.
- Test Example 5 Enhancement of morphine analgesic effect in mouse cancer pain model by compound A Honore et al. [Neuroscience, 2000, Vol. 98, p. 585] Developed a cancer pain model did. That is, C3H / He mice (male, 4 weeks old, Nippon Charles River) mice were pentobarbital sodium (50-60 mg / kg, administered intraperitoneally). The skin of the left knee joint was approximately 5 mm under anesthesia.
- mice on days 19-21 after tumor cell transplantation were used.
- the pain was evaluated by measuring the guarding behavior (pain-protecting behavior during walking) according to Honore et al. Report [Neuroscience, 2000, Vol. 98, p.585] IV. That is, the mouse was placed on the observation wire mesh and allowed to walk freely, and the guarding action time (the time for raising the left hind limb) during the 2-minute observation time was measured. Mice with a guarding action time of 5 seconds or longer before drug administration were regarded as pain behaviors and used for drug evaluation. Normal animals did not exhibit guarding behavior, but in cancer pain model animals, guarding behavior of about 10 seconds was observed.
- Compound A was suspended in an aqueous solution of 0.5% methylcellulose (0.5% MC) to a concentration of 1 mg / mL and orally administered at a dose of 10 mg / kg.
- Morphine was dissolved in physiological saline (Otsuka Pharmaceutical Factory) to a concentration of 1.5 mg / mL and administered subcutaneously at a dose of 3 mg / kg.
- physiological saline Otsuka Pharmaceutical Factory
- morphine was administered 1 hour before the evaluation.
- physiological saline (2 mL / kg) as the solvent is used instead of morphine
- 0.5% MC (10 mL / kg) as the solvent is administered instead of compound A. did.
- a compound A solvent and a morphine solvent were administered. The test was carried out with 10 animals in each group. The results are shown in FIG.
- the guarding action time was shortened more than the morphine administration group and the compound A administration group. That is, it was confirmed that administration of Compound A enhances the analgesic action of morphine in cancer pain mice. From the above test, it is clear that the use of compound (I) or a pharmaceutically acceptable salt thereof in combination with an opioid analgesic (for example, morphine) provides a strong inhibitory effect on cancer pain. It became. That is, by using a combination of compound (I) or a pharmaceutically acceptable salt thereof and an opioid analgesic, even if the opioid analgesic alone cannot sufficiently suppress cancer pain, sufficient control of cancer pain can be achieved. It was considered possible.
- an opioid analgesic for example, morphine
- Step 2 (2E, 4Z) -N-[(3R) -3-Hydroxy-2-oxo-1,2,3,4-tetrahydro-5-quinolyl] -5- (4-isopropoxyphenyl) -5
- 5- [4- (Trifluoromethyl) phenyl] -2,4-pentadienoic acid (6.00 g, 16.0 mmol) is dissolved in dimethylformamide (DMF, 120 mL), and compound a (3.21 g, 18.0 mmol), 1 -Ethyl-3- (3-dimethylaminopropyl) carbodiimide (EDC) hydrochloride (6.14 g, 32.0
- the pharmaceutical composition and the therapeutic and / or prophylactic agent for pain used in the present invention contain Compound (I) or a pharmaceutically acceptable salt thereof and each active ingredient of an analgesic. If it is formulated in such a manner, it can be used, administered or manufactured as a single agent or a combination of a plurality of formulations.
- These pharmaceutical compositions and agents for the treatment and / or prevention of pain are preferably in unit dosage forms suitable for oral administration such as tablets or parenteral administration such as injections.
- these plurality of preparations can be used or administered separately at the same time or at intervals.
- Each of these preparations is mixed with one or more pharmaceutically acceptable carriers (eg, diluents, solvents, excipients, etc.) in addition to the active ingredient, and is well known in the technical field of pharmaceutics. It is produced by any known method.
- tablets suitable for oral administration can be produced using excipients such as lactose, disintegrants such as starch, lubricants such as magnesium stearate, binders such as hydroxypropylcellulose, and the like.
- an injection suitable for parenteral administration can be produced using a diluent or a solvent such as a salt solution, a glucose solution, or a mixed solution of a saline solution and a glucose solution.
- a diluent or a solvent such as a salt solution, a glucose solution, or a mixed solution of a saline solution and a glucose solution.
- compound (I) or a pharmaceutically acceptable salt thereof and an analgesic When administered orally, for example, as tablets, compound (I) or a pharmaceutically acceptable salt thereof and an analgesic are each 0.01-1000 mg and 0.01-1000 mg, preferably 0.05-300, per adult. In the range of 0.05 to 300 mg, more preferably 0.05 to 100 mg and 0.05 to 100 mg, the dose is administered once or several times a day at the same time or separately at different times.
- Compound (I) or a pharmaceutically acceptable salt thereof and an analgesic are 0.001 to 1000 mg and 0.001 to 1000 mg, preferably 0.01 to 100 mg per adult. mg and 0.01 to 100 mg, more preferably 0.01 to 20 mg and 0.01 to 20 mg are administered once or several times a day, simultaneously or separately at different times.
- each dose and the number of administration are the efficacy of each active ingredient, although different depending on the dosage form, patient age, weight, symptoms, etc., it is preferable to prepare, use or administer as a single preparation at each dose when used or administered as a combination of the above plural preparations.
- Formulation Example 1 Tablet (Compound A) A tablet having the following composition is prepared by a conventional method. Compound A (40 g), lactose (286.8 g) and potato starch (60 g) are mixed, and 10% aqueous solution of hydroxypropylcellulose (120 g) is added thereto. The obtained mixture is kneaded by a conventional method, granulated and dried, and then sized to obtain granules for tableting. To this, 1.2 g of magnesium stearate is added and mixed, and tableting is performed with a tableting machine having a 8 mm diameter punch to obtain tablets (containing 20 mg of active ingredient per tablet). Formulation Compound A 20 mg Lactose 143.4 mg Potato starch 30 mg Hydroxypropylcellulose 6 mg Magnesium stearate 0.6 mg 200 mg
- Formulation Example 2 Tablet (compound A and gabapentin) A tablet having the following composition is prepared by a conventional method. 3 g of compound A, 100 g of gabapentin, 223.8 g of lactose and 60 g of potato starch are mixed, and 120 g of a 10% aqueous solution of hydroxypropylcellulose is added thereto. The obtained mixture is kneaded by a conventional method, granulated and dried, and then sized to obtain granules for tableting. To this, 1.2 g of magnesium stearate is added and mixed, and tableting is performed with a tableting machine having a 8 mm diameter punch to obtain tablets (containing 1.5 mg of Compound A and 50 mg of gabapentin per tablet). . Formulation Compound A 1.5 mg Gabapentin 50 mg Lactose 111.9 mg Potato starch 30 mg Hydroxypropylcellulose 6 mg Magnesium stearate 0.6 mg 200 mg
- Formulation Example 3 Tablet (compound A and duloxetine) A tablet having the following composition is prepared by a conventional method. 15 g of compound A, 50 g of duloxetine, 261.8 g of lactose and 60 g of potato starch are mixed, and 120 g of a 10% aqueous solution of hydroxypropylcellulose is added thereto. The obtained mixture is kneaded by a conventional method, granulated and dried, and then sized to obtain granules for tableting. 1.2 g of magnesium stearate is added and mixed, and tableting is performed with a tableting machine having a 8 mm diameter punch to obtain tablets (containing 7.5 mg of Compound A and 25 mg of duloxetine per tablet). . Formulation Compound A 7.5 mg Duloxetine 25 mg Lactose 130.9 mg Potato starch 30 mg Hydroxypropylcellulose 6 mg Magnesium stearate 0.6 mg 200 mg
- Formulation Example 4 Tablet (compound A and morphine alone)
- a tablet having the following composition is prepared by a conventional method.
- Compound A (60 g), morphine (10 g), lactose (256.8 g) and potato starch (60 g) are mixed, and hydroxypropylcellulose 10% aqueous solution (120 g) is added thereto.
- the obtained mixture is kneaded by a conventional method, granulated and dried, and then sized to obtain granules for tableting.
- 1.2 g of magnesium stearate is added and mixed, and tableting is performed with a tableting machine having a 8 mm diameter punch to obtain tablets (containing 30 mg of Compound A and 5 mg of morphine per tablet).
- Formulation Example 5 Injection (Compound A) An injection having the following composition is prepared by a conventional method. 1 mg of compound A is added to and mixed with distilled water for injection. Further, hydrochloric acid and aqueous sodium hydroxide solution are added to adjust the pH to 7, and then the total volume is made up to 1000 mL with distilled water for injection. The obtained mixed solution is aseptically filled into glass vials by 2 mL to obtain an injection (containing 2 ⁇ g of active ingredient per vial).
- Formulation Compound A 2 ⁇ g Hydrochloric acid appropriate amount Sodium hydroxide aqueous solution appropriate amount distilled water for injection appropriate amount 2.00 mL
- Formulation Example 6 Tablet (gabapentin) As in Example 1, tablets are prepared with the following formulation.
- Formulation Example 7 Tablet (Duloxetine) As in Example 1, tablets are prepared with the following formulation.
- Formulation Example 8 Tablet (morphine) As in Example 1, tablets are prepared with the following formulation. Prescription Morphine 20 mg Lactose 143.4 mg Potato starch 30 mg Hydroxypropylcellulose 6 mg Magnesium stearate 0.6 mg 200 mg
- the present invention can provide, for example, a pharmaceutical composition containing (a) a pentadienamide derivative or a pharmaceutically acceptable salt thereof and (b) an analgesic.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pain & Pain Management (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Rheumatology (AREA)
- Emergency Medicine (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
一般式(I)(式中、R1は置換もしくは非置換のアリール、または置換もしくは非置換の芳香族複素環基を表し、R2は置換もしくは非置換のアリール、または置換もしくは非置換の芳香族複素環基等を表し、R3は水素原子等を表し、R4は置換もしくは非置換の低級アルキル、置換もしくは非置換のアリール、または置換もしくは非置換の脂環式複素環基等を表し、R5、R6及びR7は、水素原子等を表す)で表されるペンタジエナミド誘導体またはその薬学的に許容される塩と(b)鎮痛剤とを含有する医薬組成物等を提供する。
Description
本発明は、ペンタジエナミド誘導体またはその薬学的に許容される塩と鎮痛剤とを含有する医薬組成物等に関する。
臨床的に痛みは、侵害受容器が内因性発痛物質等により持続的に刺激されるために生じる侵害受容性疼痛、神経伝達にかかわる神経線維の働きに異常をきたした結果生じる神経因性疼痛、及び心因性疼痛に分類される。
疼痛に分類される疾患として、神経因性疼痛、炎症性疼痛及び癌性疼痛のほか、偏頭痛や群発頭痛等の頭痛等の各種疼痛が知られている。
疼痛に分類される疾患として、神経因性疼痛、炎症性疼痛及び癌性疼痛のほか、偏頭痛や群発頭痛等の頭痛等の各種疼痛が知られている。
神経因性疼痛は、消炎性鎮痛剤やオピオイド鎮痛剤の効果が少ない難治性の疼痛で、帯状疱疹後神経痛、糖尿病性神経因性疼痛、HIV関連疼痛、三叉神経痛、癌化学療法に伴う疼痛、線維筋痛症等が含まれる。また、侵害受容性疼痛と神経因性疼痛が混在する疼痛には、術後痛や癌性疼痛等が含まれる。
神経因性疼痛の治療には、外科療法、神経ブロック療法、電気刺激療法、薬物療法等が用いられている。薬物療法としては、抗癲癇剤、抗うつ剤、抗けいれん剤、オピオイド鎮痛剤、筋弛緩剤、抗不整脈剤等が使用されているが、副作用の問題や発生機序の多様性等により単剤では十分な鎮痛効果が得られないことが多い。
神経因性疼痛の治療には、外科療法、神経ブロック療法、電気刺激療法、薬物療法等が用いられている。薬物療法としては、抗癲癇剤、抗うつ剤、抗けいれん剤、オピオイド鎮痛剤、筋弛緩剤、抗不整脈剤等が使用されているが、副作用の問題や発生機序の多様性等により単剤では十分な鎮痛効果が得られないことが多い。
例えば、抗うつ剤は、中枢におけるノルアドレナリン、セロトニン再取り込みを阻害し、下行性疼痛抑制系を賦活化することで、鎮痛作用を発揮すると考えられているが、悪心、吐き気、口渇、眠気等の副作用があり、疼痛治療に十分な用量まで投与ができない場合がある。オピオイド鎮痛剤は、近年、これまでオピオイドが効き難いとされてきた神経因性疼痛に対しても有効であることが報告され、今後、神経因性疼痛に対するオピオイド鎮痛剤の使用頻度は高くなることが予想される。しかしながら、オピオイド鎮痛剤には、鎮静、便秘、呼吸抑制、嘔吐、眠気等の副作用があり、こうした副作用を回避するために投与量を減らすと、疼痛が適切に緩和されなくなる可能性がある。抗癲癇剤としても使用されているガバペンチン及びプレガバリンは、欧米において神経因性鎮痛剤として上市されている薬剤である。これらの薬剤は、電位依存性カルシウムチャネルのα2δサブユニットに結合することにより、シナプスにおける神経伝達物質の放出を制御して鎮痛効果を発揮していると考えられている。しかしながら、臨床における有効性は必ずしも十分ではなく(50%程度)、めまい、眠気、体重増加等の副作用が認められる。
従って、これらの薬剤の鎮痛作用を増強することができる薬剤(治療方法)の開発が期待されている。
一方、本発明で用いられる化合物を有効成分として含有するTRPV1(Transient Receptor Potential Vanilloid 1)受容体拮抗剤が知られている(特許文献1参照)。
一方、本発明で用いられる化合物を有効成分として含有するTRPV1(Transient Receptor Potential Vanilloid 1)受容体拮抗剤が知られている(特許文献1参照)。
本発明の目的は、疼痛(痛み)の治療及び/または予防剤等として有用な医薬組成物等を提供することにある。
本発明は、以下の(1)~(56)に関する。
(1)
(a)式(I)
(1)
(a)式(I)

(式中、R1は置換もしくは非置換のアリールまたは置換もしくは非置換の芳香族複素環基を表し、
R2は置換もしくは非置換のシクロアルキル、置換もしくは非置換のシクロアルケニル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表し、
R3は水素原子を表すか、またはR4及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R4は置換もしくは非置換の低級アルキル、置換もしくは非置換のシクロアルキル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表すか、またはR3及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R5、R6及びR7は、同一または異なって水素原子またはメチルを表す)で表されるペンタジエナミド誘導体またはその薬学的に許容される塩と(b)鎮痛剤とを含有する医薬組成物。
(2)R5、R6及びR7が、それぞれ水素原子である(1)記載の医薬組成物。
(3)R3及びR4が、隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成する(1)または(2)記載の医薬組成物。
(4)R3及びR4が、隣接する窒素原子と一緒になって置換もしくは非置換のチオモルホリノを形成する(1)または(2)記載の医薬組成物。
(5)R3及びR4が、隣接する窒素原子と一緒になって置換もしくは非置換のピペリジノまたは置換もしくは非置換のピペラジニルを形成する(1)または(2)記載の医薬組成物。
(6)R3が、水素原子である(1)または(2)記載の医薬組成物。
(7)R4が、置換もしくは非置換のアリールである(1)、(2)または(6)記載の医薬組成物。
(8)R4が、置換もしくは非置換のジヒドロベンゾオキサジニルである(1)、(2)または(6)記載の医薬組成物。
(9)R4が、置換もしくは非置換のテトラヒドロイソキノリルである(1)、(2)または(6)記載の医薬組成物。
(10)R4が、置換もしくは非置換の1,2,3,4-テトラヒドロキノリルである(1)、(2)または(6)記載の医薬組成物。
(11)R4が、置換もしくは非置換の芳香族複素環基である(1)、(2)または(6)記載の医薬組成物。
(12)R4が、置換もしくは非置換のインダゾリルである(1)、(2)または(6)記載の医薬組成物。
(13)R4が、置換もしくは非置換のイソキノリルである(1)、(2)または(6)記載の医薬組成物。
(14)R1が、置換もしくは非置換のアリールである(1)~(13)のいずれかに記載の医薬組成物。
(15)R1が、4-(トリフルオロメチル)フェニルである(1)~(13)のいずれかに記載の医薬組成物。
(16)R2が、置換もしくは非置換のアリールである(1)~(15)のいずれかに記載の医薬組成物。
(17)R2が、置換もしくは非置換のフェニルである(1)~(15)のいずれかに記載の医薬組成物。
(18)R2が、置換もしくは非置換の低級アルコキシで置換されたフェニルである(1)~(15)のいずれかに記載の医薬組成物。
(19)R2が、4-(トリフルオロメチル)フェニルである(1)~(15)のいずれかに記載の医薬組成物。
(20)R2が、置換もしくは非置換の芳香族複素環基である(1)~(15)のいずれかに記載の医薬組成物。
(21)R2が、置換もしくは非置換のピリジルまたは置換もしくは非置換のピリミジニルである(1)~(15)のいずれかに記載の医薬組成物。
(22)(a)(2E,4Z)-N-[(3R)-3-ヒドロキシ-2-オキソ-1,2,3,4-テトラヒドロ-5-キノリル]-5-(4-イソプロポキシフェニル)-5-[4-(トリフルオロメチル)フェニル]-2,4-ペンタジエナミドと (b) 鎮痛剤とを含有する医薬組成物。
(23) (a) (2E,4Z)-5-(4-エトキシフェニル)-N-[(3R)-3-ヒドロキシ-2-オキソ-1,2,3,4-テトラヒドロ-5-キノリル]-5-[4-(トリフルオロメチル)フェニル]-2,4-ペンタジエナミドと (b) 鎮痛剤とを含有する医薬組成物。
(24)鎮痛剤が抗うつ剤である、(1)~(23)のいずれかに記載の医薬組成物。
(25)鎮痛剤がオピオイド鎮痛剤である、(1)~(23)のいずれかに記載の医薬組成物。
(26)鎮痛剤が抗癲癇剤である、(1)~(23)のいずれかに記載の医薬組成物。
(27)抗うつ剤がデュロキセチン、アミトリプチリン、フルボキサチン、フルボキサミン、フルオキセチン、ノルトリプチリン、ベンラファキシン、パロキセチン、レボキセチン、ミルナシプラン、イミプラミン、デシプラミン、クロミプラミン、トリミプラミン、アモキサピン、ロフェプラミン、ドレスピン、シタロプラム、エスシタロプラム、セルトラリン、マプロチリン、ミアンセリン、セチプチリン、及びネファゾドンからなる群から選ばれる抗うつ剤である(24)記載の医薬組成物。
(28)オピオイド鎮痛剤がモルヒネ、フェンタニル、レミフェンタニル、スフェンタニル、ブプレノルフィン、ヒドロモルフィン、オキシモルフィン、ペンタゾシン、トラマドール、コデイン、ジヒドロコデイン、オキシコドン、メサドン、ヘロイン、ナルブフィン、レボルファノール、レバロルファン、メピリジン、ヒドロコドン、プロポキシフェン、及びブトルファノールからなる群から選ばれるオピオイド鎮痛剤である(25)記載の医薬組成物。
(29)抗癲癇剤がガバペンチン及びプレガバリンからなる群から選ばれる抗癲癇剤である(26)記載の医薬組成物。
(30)(a)式(I)
R2は置換もしくは非置換のシクロアルキル、置換もしくは非置換のシクロアルケニル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表し、
R3は水素原子を表すか、またはR4及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R4は置換もしくは非置換の低級アルキル、置換もしくは非置換のシクロアルキル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表すか、またはR3及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R5、R6及びR7は、同一または異なって水素原子またはメチルを表す)で表されるペンタジエナミド誘導体またはその薬学的に許容される塩と(b)鎮痛剤とを含有する医薬組成物。
(2)R5、R6及びR7が、それぞれ水素原子である(1)記載の医薬組成物。
(3)R3及びR4が、隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成する(1)または(2)記載の医薬組成物。
(4)R3及びR4が、隣接する窒素原子と一緒になって置換もしくは非置換のチオモルホリノを形成する(1)または(2)記載の医薬組成物。
(5)R3及びR4が、隣接する窒素原子と一緒になって置換もしくは非置換のピペリジノまたは置換もしくは非置換のピペラジニルを形成する(1)または(2)記載の医薬組成物。
(6)R3が、水素原子である(1)または(2)記載の医薬組成物。
(7)R4が、置換もしくは非置換のアリールである(1)、(2)または(6)記載の医薬組成物。
(8)R4が、置換もしくは非置換のジヒドロベンゾオキサジニルである(1)、(2)または(6)記載の医薬組成物。
(9)R4が、置換もしくは非置換のテトラヒドロイソキノリルである(1)、(2)または(6)記載の医薬組成物。
(10)R4が、置換もしくは非置換の1,2,3,4-テトラヒドロキノリルである(1)、(2)または(6)記載の医薬組成物。
(11)R4が、置換もしくは非置換の芳香族複素環基である(1)、(2)または(6)記載の医薬組成物。
(12)R4が、置換もしくは非置換のインダゾリルである(1)、(2)または(6)記載の医薬組成物。
(13)R4が、置換もしくは非置換のイソキノリルである(1)、(2)または(6)記載の医薬組成物。
(14)R1が、置換もしくは非置換のアリールである(1)~(13)のいずれかに記載の医薬組成物。
(15)R1が、4-(トリフルオロメチル)フェニルである(1)~(13)のいずれかに記載の医薬組成物。
(16)R2が、置換もしくは非置換のアリールである(1)~(15)のいずれかに記載の医薬組成物。
(17)R2が、置換もしくは非置換のフェニルである(1)~(15)のいずれかに記載の医薬組成物。
(18)R2が、置換もしくは非置換の低級アルコキシで置換されたフェニルである(1)~(15)のいずれかに記載の医薬組成物。
(19)R2が、4-(トリフルオロメチル)フェニルである(1)~(15)のいずれかに記載の医薬組成物。
(20)R2が、置換もしくは非置換の芳香族複素環基である(1)~(15)のいずれかに記載の医薬組成物。
(21)R2が、置換もしくは非置換のピリジルまたは置換もしくは非置換のピリミジニルである(1)~(15)のいずれかに記載の医薬組成物。
(22)(a)(2E,4Z)-N-[(3R)-3-ヒドロキシ-2-オキソ-1,2,3,4-テトラヒドロ-5-キノリル]-5-(4-イソプロポキシフェニル)-5-[4-(トリフルオロメチル)フェニル]-2,4-ペンタジエナミドと (b) 鎮痛剤とを含有する医薬組成物。
(23) (a) (2E,4Z)-5-(4-エトキシフェニル)-N-[(3R)-3-ヒドロキシ-2-オキソ-1,2,3,4-テトラヒドロ-5-キノリル]-5-[4-(トリフルオロメチル)フェニル]-2,4-ペンタジエナミドと (b) 鎮痛剤とを含有する医薬組成物。
(24)鎮痛剤が抗うつ剤である、(1)~(23)のいずれかに記載の医薬組成物。
(25)鎮痛剤がオピオイド鎮痛剤である、(1)~(23)のいずれかに記載の医薬組成物。
(26)鎮痛剤が抗癲癇剤である、(1)~(23)のいずれかに記載の医薬組成物。
(27)抗うつ剤がデュロキセチン、アミトリプチリン、フルボキサチン、フルボキサミン、フルオキセチン、ノルトリプチリン、ベンラファキシン、パロキセチン、レボキセチン、ミルナシプラン、イミプラミン、デシプラミン、クロミプラミン、トリミプラミン、アモキサピン、ロフェプラミン、ドレスピン、シタロプラム、エスシタロプラム、セルトラリン、マプロチリン、ミアンセリン、セチプチリン、及びネファゾドンからなる群から選ばれる抗うつ剤である(24)記載の医薬組成物。
(28)オピオイド鎮痛剤がモルヒネ、フェンタニル、レミフェンタニル、スフェンタニル、ブプレノルフィン、ヒドロモルフィン、オキシモルフィン、ペンタゾシン、トラマドール、コデイン、ジヒドロコデイン、オキシコドン、メサドン、ヘロイン、ナルブフィン、レボルファノール、レバロルファン、メピリジン、ヒドロコドン、プロポキシフェン、及びブトルファノールからなる群から選ばれるオピオイド鎮痛剤である(25)記載の医薬組成物。
(29)抗癲癇剤がガバペンチン及びプレガバリンからなる群から選ばれる抗癲癇剤である(26)記載の医薬組成物。
(30)(a)式(I)

(式中、R1は置換もしくは非置換のアリールまたは置換もしくは非置換の芳香族複素環基を表し、
R2は置換もしくは非置換のシクロアルキル、置換もしくは非置換のシクロアルケニル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表し、
R3は水素原子を表すか、またはR4及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R4は置換もしくは非置換の低級アルキル、置換もしくは非置換のシクロアルキル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表すか、またはR3及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R5、R6及びR7は、同一または異なって水素原子またはメチルを表す)で表されるペンタジエナミド誘導体またはその薬学的に許容される塩と(b)鎮痛剤とを有効成分として含有する疼痛の治療及び/または予防剤。
(31)(a)式(I)
R2は置換もしくは非置換のシクロアルキル、置換もしくは非置換のシクロアルケニル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表し、
R3は水素原子を表すか、またはR4及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R4は置換もしくは非置換の低級アルキル、置換もしくは非置換のシクロアルキル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表すか、またはR3及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R5、R6及びR7は、同一または異なって水素原子またはメチルを表す)で表されるペンタジエナミド誘導体またはその薬学的に許容される塩と(b)鎮痛剤とを有効成分として含有する疼痛の治療及び/または予防剤。
(31)(a)式(I)

(式中、R1は置換もしくは非置換のアリールまたは置換もしくは非置換の芳香族複素環基を表し、
R2は置換もしくは非置換のシクロアルキル、置換もしくは非置換のシクロアルケニル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表し、
R3は水素原子を表すか、またはR4及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R4は置換もしくは非置換の低級アルキル、置換もしくは非置換のシクロアルキル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表すか、またはR3及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R5、R6及びR7は、同一または異なって水素原子またはメチルを表す)で表されるペンタジエナミド誘導体またはその薬学的に許容される塩と、(b)鎮痛剤と、を有効成分とする、(a)と(b)を同時にまたは時間を置いて別々に投与するための疼痛の治療及び/または予防剤。
(32)(a)式(I)
R2は置換もしくは非置換のシクロアルキル、置換もしくは非置換のシクロアルケニル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表し、
R3は水素原子を表すか、またはR4及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R4は置換もしくは非置換の低級アルキル、置換もしくは非置換のシクロアルキル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表すか、またはR3及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R5、R6及びR7は、同一または異なって水素原子またはメチルを表す)で表されるペンタジエナミド誘導体またはその薬学的に許容される塩と、(b)鎮痛剤と、を有効成分とする、(a)と(b)を同時にまたは時間を置いて別々に投与するための疼痛の治療及び/または予防剤。
(32)(a)式(I)

(式中、R1は置換もしくは非置換のアリールまたは置換もしくは非置換の芳香族複素環基を表し、
R2は置換もしくは非置換のシクロアルキル、置換もしくは非置換のシクロアルケニル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表し、
R3は水素原子を表すか、またはR4及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R4は置換もしくは非置換の低級アルキル、置換もしくは非置換のシクロアルキル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表すか、またはR3及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R5、R6及びR7は、同一または異なって水素原子またはメチルを表す)で表されるペンタジエナミド誘導体またはその薬学的に許容される塩を含有する第1成分と、(b)鎮痛剤を含有する第2成分とを有することを特徴とするキット。
(33)(a)式(I)
R2は置換もしくは非置換のシクロアルキル、置換もしくは非置換のシクロアルケニル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表し、
R3は水素原子を表すか、またはR4及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R4は置換もしくは非置換の低級アルキル、置換もしくは非置換のシクロアルキル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表すか、またはR3及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R5、R6及びR7は、同一または異なって水素原子またはメチルを表す)で表されるペンタジエナミド誘導体またはその薬学的に許容される塩を含有する第1成分と、(b)鎮痛剤を含有する第2成分とを有することを特徴とするキット。
(33)(a)式(I)

(式中、R1は置換もしくは非置換のアリールまたは置換もしくは非置換の芳香族複素環基を表し、
R2は置換もしくは非置換のシクロアルキル、置換もしくは非置換のシクロアルケニル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表し、
R3は水素原子を表すか、またはR4及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R4は置換もしくは非置換の低級アルキル、置換もしくは非置換のシクロアルキル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表すか、またはR3及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R5、R6及びR7は、同一または異なって水素原子またはメチルを表す)で表されるペンタジエナミド誘導体またはその薬学的に許容される塩を含有する第1成分と、(b)鎮痛剤を含有する第2成分とを有することを特徴とする疼痛の治療及び/または予防用キット。
(34)疼痛が神経因性疼痛である、(30)記載の疼痛の治療及び/または予防剤。
(35)疼痛が神経因性疼痛である、(31)記載の疼痛の治療及び/または予防剤。
(36)疼痛が癌性疼痛である、(30)記載の疼痛の治療及び/または予防剤。
(37)疼痛が癌性疼痛である、(31)記載の疼痛の治療及び/または予防剤。
(38)疼痛が神経因性疼痛である、(33)記載の疼痛の治療及び/または予防用キット。
(39)疼痛が癌性疼痛である、(33)記載の疼痛の治療及び/または予防用キット。
(40)(a)(1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩と、(b)鎮痛剤を投与して疼痛の治療及び/または予防に使用するための、(1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩。
(41)(a)(1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩と、(b)鎮痛剤と、を同時にまたは時間を置いて別々に投与して疼痛の治療及び/または予防に使用するための、(1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩。
(42)(a) (1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩と、(b)鎮痛剤を有効成分として含有する疼痛の治療及び/または予防剤を製造するための(a)の使用。
(43)(a) (1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩と、(b)鎮痛剤を有効成分とする、(a)と(b)を同時にまたは時間を置いて別々に投与するための疼痛の治療及び/または予防剤を製造するための(a)の使用。
(44)(a) (1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩を含有する第一成分と、(b)鎮痛剤を含有する第二成分とを有することを特徴とする疼痛の治療及び/または予防用キットを製造するための(a)の使用。
(45)疼痛が神経因性疼痛である、(42)記載の使用。
(46)疼痛が神経因性疼痛である、(43)記載の使用。
(47)疼痛が神経因性疼痛である、(44)記載の使用。
(48)疼痛が癌性疼痛である、(42)記載の使用。
(49)疼痛が癌性疼痛である、(43)記載の使用。
(50)疼痛が癌性疼痛である、(44)記載の使用。
(51)(a)(1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩と、(b)鎮痛剤を含有する組成物を投与する疼痛の治療及び/または予防方法。
(52)(a)(1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩と、(b)鎮痛剤とを、同時にまたは時間を置いて別々に投与する疼痛の治療及び/または予防方法。
(53)疼痛が神経因性疼痛である、(51)記載の方法。
(54)疼痛が神経因性疼痛である、(52)記載の方法。
(55)疼痛が癌性疼痛である、(51)記載の方法。
(56)疼痛が癌性疼痛である、(52)記載の方法。
R2は置換もしくは非置換のシクロアルキル、置換もしくは非置換のシクロアルケニル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表し、
R3は水素原子を表すか、またはR4及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R4は置換もしくは非置換の低級アルキル、置換もしくは非置換のシクロアルキル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表すか、またはR3及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R5、R6及びR7は、同一または異なって水素原子またはメチルを表す)で表されるペンタジエナミド誘導体またはその薬学的に許容される塩を含有する第1成分と、(b)鎮痛剤を含有する第2成分とを有することを特徴とする疼痛の治療及び/または予防用キット。
(34)疼痛が神経因性疼痛である、(30)記載の疼痛の治療及び/または予防剤。
(35)疼痛が神経因性疼痛である、(31)記載の疼痛の治療及び/または予防剤。
(36)疼痛が癌性疼痛である、(30)記載の疼痛の治療及び/または予防剤。
(37)疼痛が癌性疼痛である、(31)記載の疼痛の治療及び/または予防剤。
(38)疼痛が神経因性疼痛である、(33)記載の疼痛の治療及び/または予防用キット。
(39)疼痛が癌性疼痛である、(33)記載の疼痛の治療及び/または予防用キット。
(40)(a)(1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩と、(b)鎮痛剤を投与して疼痛の治療及び/または予防に使用するための、(1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩。
(41)(a)(1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩と、(b)鎮痛剤と、を同時にまたは時間を置いて別々に投与して疼痛の治療及び/または予防に使用するための、(1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩。
(42)(a) (1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩と、(b)鎮痛剤を有効成分として含有する疼痛の治療及び/または予防剤を製造するための(a)の使用。
(43)(a) (1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩と、(b)鎮痛剤を有効成分とする、(a)と(b)を同時にまたは時間を置いて別々に投与するための疼痛の治療及び/または予防剤を製造するための(a)の使用。
(44)(a) (1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩を含有する第一成分と、(b)鎮痛剤を含有する第二成分とを有することを特徴とする疼痛の治療及び/または予防用キットを製造するための(a)の使用。
(45)疼痛が神経因性疼痛である、(42)記載の使用。
(46)疼痛が神経因性疼痛である、(43)記載の使用。
(47)疼痛が神経因性疼痛である、(44)記載の使用。
(48)疼痛が癌性疼痛である、(42)記載の使用。
(49)疼痛が癌性疼痛である、(43)記載の使用。
(50)疼痛が癌性疼痛である、(44)記載の使用。
(51)(a)(1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩と、(b)鎮痛剤を含有する組成物を投与する疼痛の治療及び/または予防方法。
(52)(a)(1)~(23)のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩と、(b)鎮痛剤とを、同時にまたは時間を置いて別々に投与する疼痛の治療及び/または予防方法。
(53)疼痛が神経因性疼痛である、(51)記載の方法。
(54)疼痛が神経因性疼痛である、(52)記載の方法。
(55)疼痛が癌性疼痛である、(51)記載の方法。
(56)疼痛が癌性疼痛である、(52)記載の方法。
なお、(30)~(39)において、式(I)で表される化合物の範囲が、(2)~(23)のように限定された態様、及び/または(30)~(56)において、鎮痛剤の範囲が(24)~(29)のように限定された態様を採ることも可能である。
本発明により、例えば(a)ペンタジエナミド誘導体またはその薬学的に許容される塩と(b)鎮痛剤とを含有する医薬組成物等が提供される。
*:P<0.05 [溶媒投与群対比のウィルコクソン・ランク・サム・テスト(Wilcoxon rank sum test)]を示す。**: P<0.01[溶媒投与群対比のウィルコクソン・ランク・サム・テスト(Wilcoxon rank sum test)]を示す。#:P<0.05[化合物A投与群対比のウィルコクソン・ランク・サム・テスト(Wilcoxon rank sum test)]を示す。
図4は、化合物Aとモルヒネを投与後のguarding行動(歩行時の痛み防御行動)時間(秒)を示す。
*:P<0.05 [溶媒投与群対比のスチューデンツ・ティーテスト(Student's t-test)あるいはアスピンウエルチ・テスト(Aspin-Welch test]を示す。***: P<0.001[溶媒投与群対比のアスピンウエルチ・テスト(Aspin-Welch test)]を示す。#:P<0.001[化合物A投与群対比のスチューデンツ・ティーテスト(Student's t-test)]を示す。
以下、式(I)で表される化合物を化合物(I)という。
式(I)の各基の定義において、
低級アルキルとしては、例えば炭素数1~10の直鎖状または分枝鎖状のアルキルが挙げられ、より具体的には、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、ネオペンチル、ヘキシル、ヘプチル、オクチル、ノニル、デシル等が挙げられる。
式(I)の各基の定義において、
低級アルキルとしては、例えば炭素数1~10の直鎖状または分枝鎖状のアルキルが挙げられ、より具体的には、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、ネオペンチル、ヘキシル、ヘプチル、オクチル、ノニル、デシル等が挙げられる。
シクロアルキルとしては、例えば炭素数3~10のシクロアルキルが挙げられ、より具体的には、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、シクロデシル、ノルアダマンチル、アダマンチル、ビシクロ[2.2.1]ヘプチル、ビシクロ[2.2.2]オクチル、ビシクロ[3.3.0]オクチル、ビシクロ[3.3.1]ノニル等が挙げられる。
シクロアルケニルとしては、例えば炭素数3~10のシクロアルケニルが挙げられ、より具体的には、シクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニル、シクロヘプテニル、シクロオクテニル、シクロノネニル、シクロデセニル等が挙げられる。
アリールとしては、例えば炭素数6~14のアリールが挙げられ、より具体的にはフェニル、ナフチル、インデニル、アントリル等が挙げられる。なお、アリールは、例えば1,2,3,4-テトラヒドロナフタレン等の、シクロヘキシル基と縮合したアリール、または1,2,3,4-テトラヒドロキノリル、1,2,3,4-テトラヒドロイソキノリル、ジヒドロキノキサリニル、テトラヒドロキノキサリニル、ジヒドロベンゾフラニル、クロマニル、イソクロマニル、ベンゾオキサジニル、ジヒドロベンゾオキサジニル、シヒドロベンゾジオキシニル、1,3-ベンゾジオキソリル等の、脂環式複素環基と縮合したアリールであっても良い。
アリールとしては、例えば炭素数6~14のアリールが挙げられ、より具体的にはフェニル、ナフチル、インデニル、アントリル等が挙げられる。なお、アリールは、例えば1,2,3,4-テトラヒドロナフタレン等の、シクロヘキシル基と縮合したアリール、または1,2,3,4-テトラヒドロキノリル、1,2,3,4-テトラヒドロイソキノリル、ジヒドロキノキサリニル、テトラヒドロキノキサリニル、ジヒドロベンゾフラニル、クロマニル、イソクロマニル、ベンゾオキサジニル、ジヒドロベンゾオキサジニル、シヒドロベンゾジオキシニル、1,3-ベンゾジオキソリル等の、脂環式複素環基と縮合したアリールであっても良い。
芳香族複素環基としては、例えば窒素原子、酸素原子及び硫黄原子から選ばれる少なくとも1個の原子を含む5員または6員の単環性芳香族複素環基、3~8員の環が縮合した二環または三環性で窒素原子、酸素原子及び硫黄原子から選ばれる少なくとも1個の原子を含む縮環性芳香族複素環基等が挙げられ、より具体的にはピリジル、ピリドニル、ピラジニル、ピリミジニル、ピリダジニル、キノリル、イソキノリル、フタラジニル、キナゾリニル、キノキサリニル、ナフチリジニル、シンノリニル、ピロリル、ピラゾリル、イミダゾリル、トリアゾリル、テトラゾリル、チエニル、フリル、チアゾリル、オキサゾリル、インドリル、インダゾリル、ベンゾイミダゾリル、イソオキサゾリル、ベンゾトリアゾリル、ベンゾチアゾリル、ベンゾオキサゾリル、ベンゾオキサジアゾリル、プリニル、アクリジニル、カルバゾリル、ピラニル、フラザニル、ベンゾフラザニル、チアジアゾリル、オキサジアゾリル、チエノピリジル、チエノピラジニル、ピラゾロピリミジニル、イミダゾピラジン等が挙げられる。
脂環式複素環基としては、例えば窒素原子、酸素原子及び硫黄原子から選ばれる少なくとも1個の原子を含む5員または6員の単環性脂環式複素環基、3~8員の環が縮合した二環または三環性で窒素原子、酸素原子及び硫黄原子から選ばれる少なくとも1個の原子を含む縮環性脂環式複素環基等が挙げられ、より具体的にはピロリジニル、ピロリドニル、ピペリジノ、ピペリジル、ピペラジニル、モルホリノ、モルホリニル、チオモルホリノ、チオモルホリニル、ホモピペリジノ、ホモピペリジル、ホモピペラジニル、ジヒドロピリジル、テトラヒドロピリジル、テトラヒドロキノリル、テトラヒドロイソキノリル、テトラヒドロフラニル、テトラヒドロピラニル、ジヒドロベンゾフラニル、ジヒドロベンゾオキサジニル、インドリニル、イソインドリニル、ジヒドロキノリル、ジヒドロイソキノリル、ジヒドロピラニル、オキシラニル、オキセタニル、チエタニル、アジリジニル、アゼチジニル、アゼパニル、オキサゼパニル等が挙げられる。
隣接する窒素原子と一緒になって形成される複素環基としては、隣接する窒素原子と一緒になって形成される芳香族複素環基または隣接する窒素原子と一緒になって形成される脂環式複素環基が挙げられる。
隣接する窒素原子と一緒になって形成される芳香族複素環基としては、例えば少なくとも1個の窒素原子を含む5員または6員の単環性芳香族複素環基(該単環性芳香族複素環基は、他の窒素原子、酸素原子または硫黄原子を含んでいてもよい)、3~8員の環が縮合した二環または三環性で少なくとも1個の窒素原子を含む縮環性芳香族複素環基(該縮環性芳香族複素環基は、他の窒素原子、酸素原子または硫黄原子を含んでいてもよい)等が挙げられ、より具体的にはピロリル、イミダゾリル、インドリル、インダゾリル、カルバゾリル、ピラゾリル等が挙げられる。
隣接する窒素原子と一緒になって形成される芳香族複素環基としては、例えば少なくとも1個の窒素原子を含む5員または6員の単環性芳香族複素環基(該単環性芳香族複素環基は、他の窒素原子、酸素原子または硫黄原子を含んでいてもよい)、3~8員の環が縮合した二環または三環性で少なくとも1個の窒素原子を含む縮環性芳香族複素環基(該縮環性芳香族複素環基は、他の窒素原子、酸素原子または硫黄原子を含んでいてもよい)等が挙げられ、より具体的にはピロリル、イミダゾリル、インドリル、インダゾリル、カルバゾリル、ピラゾリル等が挙げられる。
隣接する窒素原子と一緒になって形成される脂環式複素環基としては、例えば少なくとも1個の窒素原子を含む5員または6員の単環性脂環式複素環基(該単環性脂環式複素環基は、他の窒素原子、酸素原子または硫黄原子を含んでいてもよい)、3~8員の環が縮合した二環または三環性で少なくとも1個の窒素原子を含む縮環性脂環式複素環基(該縮環性脂環式複素環基は、他の窒素原子、酸素原子または硫黄原子を含んでいてもよい)等が挙げられ、より具体的にはピロリジニル、ピロリドニル、ピペリジノ、ピペラジニル、モルホリノ、チオモルホリノ、ホモピペリジノ、ホモピペラジニル、テトラヒドロピリジル、テトラヒドロキノリル、テトラヒドロイソキノリル、インドリニル、イソインドリニル、ジヒドロキノリル、ジヒドロイソキノリル等が挙げられる。
置換低級アルキル、置換シクロアルキル、置換シクロアルケニル、置換アリール、置換低級アルコキシ、置換芳香族複素環基、置換脂環式複素環基、隣接する窒素原子と一緒になって形成される置換芳香族複素環基、隣接する窒素原子と一緒になって形成される置換脂環式複素環基における置換基としては、同一または異なって、例えば置換数1~3の、より具体的にはハロゲン、ヒドロキシ、アミノ、ヒドロキシイミノ、アミジノ、ヒドロキシアミジノ、ニトロ、メルカプト、シアノ、カルボキシ、メチレンジオキシ、エチレンジオキシ、カルバモイル、スルファモイル、スルファモイルオキシ、低級アルケニル、シクロアルケニル、低級アルキニル、低級アルコキシカルボニル、モノもしくはジ低級アルキルカルバモイル、低級アルキルスルフィニル、低級アルキルスルフィニルアミノ、低級アルキルスルホニルアミノ、トリフルオロメチルスルフィニル、低級アルキルチオ、ペンタフルオロチオ、アリールオキシ、アラルキルオキシ、アミノスルホニルオキシ、アロイル、低級アルコキシカルボニルアミノ、モノもしくはジ(置換もしくは非置換の低級アルキル)アミノ[該置換低級アルキルアミノ、低級アルキル(置換低級アルキル)アミノ及びジ置換低級アルキルアミノにおける置換基としては、同一または異なって、例えば置換数1~3の、より具体的にはハロゲン、ヒドロキシ、低級アルコキシ等が挙げられる]、置換もしくは非置換の低級アルカノイルアミノ(該置換低級アルカノイルアミノにおける置換基としては、同一または異なって、例えば置換数1~3の、より具体的にはハロゲン、ヒドロキシ、シアノ等が挙げられる)、置換もしくは非置換の低級アルキルスルホニル(該置換低級アルキルスルホニルにおける置換基としては、同一または異なって、例えば置換数1~3の、より具体的にはハロゲン、ヒドロキシ、シアノ等が挙げられる)、置換もしくは非置換の低級アルコキシ[該置換低級アルコキシにおける置換基としては、同一または異なって、例えば置換数1~3の、より具体的にはハロゲン、アミノ、モノもしくはジ(低級アルキル)アミノ、ヒドロキシ、シアノ、低級アルコキシ、低級アルコキシカルボニル、低級アルコキシカルボニルアミノ、低級アルカノイル、シクロアルキル、低級アルキル置換シクロアルキル、脂環式複素環基、低級アルキル置換脂環式複素環基等が挙げられる]、置換もしくは非置換のシクロアルキルオキシ(該置換シクロアルキルオキシにおける置換基としては、同一または異なって、例えば置換数1~3の、より具体的にはハロゲン、ヒドロキシ、低級アルキル、低級アルコキシ、低級アルカノイル等が挙げられる)、置換もしくは非置換の低級アルカノイル[該置換低級アルカノイルにおける置換基としては、同一または異なって、例えば置換数1~3の、より具体的にはハロゲン等が挙げられる]、置換もしくは非置換のアリール(該置換アリールにおける置換基としては、同一または異なって、例えば置換数1~3の、より具体的にはハロゲン、ニトロ、ヒドロキシ、アミノ、低級アルコキシ等が挙げられる)、置換もしくは非置換のアラルキル(該置換アラルキルにおける置換基としては、同一または異なって、例えば置換数1~3の、より具体的にはハロゲン、ヒドロキシ、低級アルコキシ等が挙げられる)、置換もしくは非置換の芳香族複素環基(該置換芳香族複素環基における置換基としては、同一または異なって、例えば置換数1~3の、より具体的にはハロゲン、ヒドロキシ、低級アルキル、ヒドロキシアルキル、低級アルコキシ、低級アルカノイル等が挙げられる)、置換もしくは非置換の芳香族複素環アミノスルホニル(該置換芳香族複素環アミノスルホニルにおける置換基としては、同一または異なって、例えば置換数1~3の、より具体的にはハロゲン、ヒドロキシ、低級アルキル、低級アルコキシ、低級アルカノイル等が挙げられる)、置換もしくは非置換の脂環式複素環基(該置換脂環式複素環基における置換基としては、同一または異なって、例えば置換数1~3の、より具体的にはハロゲン、ヒドロキシ、低級アルキル、低級アルコキシ、低級アルカノイル、アミノ等が挙げられる)、置換もしくは非置換の脂環式複素環オキシ(該置換脂環式複素環オキシにおける置換基としては、同一または異なって、例えば置換数1~3の、より具体的にはハロゲン、ヒドロキシ、低級アルキル、低級アルコキシ、低級アルカノイル等が挙げられる)、置換もしくは非置換の脂環式複素環アルキルオキシ(該置換脂環式複素環アルキルオキシにおける置換基としては、同一または異なって、例えば置換数1~3の、より具体的にはハロゲン、ヒドロキシ、低級アルキル、低級アルコキシ、低級アルカノイル等が挙げられる)等が挙げられる。なお、置換アリール、置換芳香族複素環基、置換脂環式複素環基、隣接する窒素原子と一緒になって形成される置換芳香族複素環基及び隣接する窒素原子と一緒になって形成される置換脂環式複素環基における置換基は、置換もしくは非置換の低級アルキル(該置換低級アルキルにおける置換基としては、同一または異なって、例えば置換数1~3の、より具体的にはハロゲン、アミノ、ヒドロキシ、ヒドロキシイミノ、シアノ、モノもしくはジ低級アルキルアミノ、芳香族複素環基、脂環式複素環基等が挙げられる)であってもよい。また、置換低級アルキル、置換脂環式複素環基及び隣接する窒素原子と一緒になって形成される置換脂環式複素環基における置換基はオキソであってもよい。また、置換アリールが置換された脂環式複素環基と縮合したアリールである場合の置換基もオキソを含む。さらに、脂環式複素環基、芳香族複素環基、隣接する窒素原子と一緒になって形成される芳香族複素環基及び隣接する窒素原子と一緒になって形成される置換脂環式複素環基が、その環内に窒素原子及び/または硫黄原子を有する場合、該窒素原子及び/または硫黄原子は酸化されていてもよい。
ここで、低級アルキル、シクロアルキル、シクロアルケニル、アリール、芳香族複素環基及び脂環式複素環基はそれぞれ前記と同義である。
ハロゲンとしては、フッ素、塩素、臭素、ヨウ素の各原子が挙げられる。
低級アルコキシ、低級アルコキシカルボニル、低級アルコキシカルボニルアミノ、モノもしくはジ低級アルキルアミノ、モノもしくはジ低級アルキルカルバモイル、低級アルキル置換シクロアルキル、低級アルキルスルフィニル、低級アルキルスルフィニルアミノ、低級アルキルスルホニル、低級アルキルスルホニルアミノ、低級アルキル置換脂環式複素環基及び低級アルキルチオの低級アルキル部分は、前記低級アルキルと同義である。なお、ジ低級アルキルアミノ及びジ低級アルキルカルバモイルにおける2つの低級アルキル部分は、それぞれ同一でも異なっていてもよい。
ハロゲンとしては、フッ素、塩素、臭素、ヨウ素の各原子が挙げられる。
低級アルコキシ、低級アルコキシカルボニル、低級アルコキシカルボニルアミノ、モノもしくはジ低級アルキルアミノ、モノもしくはジ低級アルキルカルバモイル、低級アルキル置換シクロアルキル、低級アルキルスルフィニル、低級アルキルスルフィニルアミノ、低級アルキルスルホニル、低級アルキルスルホニルアミノ、低級アルキル置換脂環式複素環基及び低級アルキルチオの低級アルキル部分は、前記低級アルキルと同義である。なお、ジ低級アルキルアミノ及びジ低級アルキルカルバモイルにおける2つの低級アルキル部分は、それぞれ同一でも異なっていてもよい。
低級アルキル置換シクロアルキルにおけるシクロアルキレン部分は、前記シクロアルキルから水素原子を1つ除いたものと同義である。
シクロアルキルオキシにおけるシクロアルキル部分は、前記シクロアルキルと同義である。
アラルキル、アラルキルオキシ、脂環式複素環アルキルオキシおよびヒドロキシアルキルにおけるアルキレン部分は、前記低級アルキルから水素原子を1つ除いたものと同義である。
シクロアルキルオキシにおけるシクロアルキル部分は、前記シクロアルキルと同義である。
アラルキル、アラルキルオキシ、脂環式複素環アルキルオキシおよびヒドロキシアルキルにおけるアルキレン部分は、前記低級アルキルから水素原子を1つ除いたものと同義である。
低級アルケニルとしては、例えば炭素数3~10の直鎖状または分枝鎖状のアルケニルが挙げられ、より具体的にはアリル、2-ブテニル、3-ブテニル、4-ペンテニル、5-へキセニル、6-へプテニル、6-オクテニル、2,6-オクタジエニル、9-デセニル等が挙げられる。
低級アルキニルとしては、例えば炭素数3~6の直鎖状または分枝鎖状のアルキニルが挙げられ、より具体的にはプロパルギル、3-ブチニル、3-ヘキシニル、4-メチル-2-ペンチニル等が挙げられる。
低級アルキニルとしては、例えば炭素数3~6の直鎖状または分枝鎖状のアルキニルが挙げられ、より具体的にはプロパルギル、3-ブチニル、3-ヘキシニル、4-メチル-2-ペンチニル等が挙げられる。
低級アルカノイル及び低級アルカノイルアミノの低級アルカノイル部分としては、例えば炭素数1~8の直鎖状または分枝鎖状のアルカノイルが挙げられ、より具体的にはホルミル、アセチル、プロピオニル、ブチリル、イソブチリル、バレリル、イソバレリル、ピバロイル、ヘキサノイル、ヘプタノイル、オクタノイル等が挙げられる。
アラルキル、アリールオキシ、アラルキルオキシ及びアロイルのアリール部分は、前記アリールと同義である。
アラルキル、アリールオキシ、アラルキルオキシ及びアロイルのアリール部分は、前記アリールと同義である。
脂環式複素環オキシ及び脂環式複素環アルキルオキシにおける脂環式複素環基部分は、前記脂環式複素環基と同義である。
低級アルキル置換脂環式複素環基における脂環式複素環基部分は、前記脂環式複素環基から水素原子を一つ除いたものと同義である。
芳香族複素環アミノスルホニルにおける芳香族複素環基部分は、前記芳香族複素環基と同義である。
低級アルキル置換脂環式複素環基における脂環式複素環基部分は、前記脂環式複素環基から水素原子を一つ除いたものと同義である。
芳香族複素環アミノスルホニルにおける芳香族複素環基部分は、前記芳香族複素環基と同義である。
本発明における医薬組成物、あるいは疼痛の治療及び/または予防剤として、化合物(I)またはその薬学的に許容される塩を用いることができるが、好ましくは(i)R5、R6及びR7がそれぞれ水素原子である化合物(I)またはその薬学的に許容される塩、(ii)R1及びR2が置換もしくは非置換のアリールである化合物(I)またはその薬学的に許容される塩、(iii)R3が水素原子であり、R4が置換もしくは非置換のテトラヒドロキノリルである化合物(I)またはその薬学的に許容される塩を用いることができる。さらに好ましくは、上記(i)、(ii)及び(iii)を2つまたは3つ組み合わせた態様の化合物を用いることができる。さらに好ましくは化合物188、191、229、240、257、270、310、314、332及び333から選ばれる化合物を用いることができる。
化合物(I)の薬学的に許容される塩としては、例えば薬学的に許容される金属塩、アンモニウム塩、有機アミン付加塩、アミノ酸付加塩、酸付加塩等が挙げられる。薬学的に許容される金属塩としては、例えばナトリウム塩、カリウム塩等のアルカリ金属塩、マグネシウム塩、カルシウム塩等のアルカリ土類金属塩、アルミニウム塩、亜鉛塩等が挙げられ、薬学的に許容されるアンモニウム塩としては、アンモニウム、テトラメチルアンモニウム等の塩が挙げられ、薬学的に許容される有機アミン付加塩としては、例えばモルホリン、ピペリジン等の付加塩が挙げられ、薬学的に許容されるアミノ酸付加塩としては、例えばリジン、グリシン、フェニルアラニン等のアミノ酸の付加塩が挙げられ、薬学的に許容される酸付加塩としては、例えば塩酸塩、硫酸塩、リン酸塩等の無機酸塩、酢酸塩、マレイン酸塩、フマル酸塩、酒石酸塩、クエン酸塩等の有機酸塩等が挙げられる。
本発明における化合物(I)及びその薬学的に許容される塩は、水または各種溶媒との付加物の形で存在することもあるが、これらの付加物も本発明の医薬組成物、疼痛の治療及び/または予防剤、キット、疼痛の治療及び/または予防用キット、疼痛の治療及び/または予防方法等に使用することができる。
本発明における化合物(I)の塩を取得したいとき、化合物(I)が塩の形で得られるときはそのまま精製すればよく、また、遊離の形で得られるときは、化合物(I)を適当な溶媒に溶解または懸濁し、酸または塩基を加えることにより塩を形成させて単離、精製すればよい。
本発明における化合物(I)の塩を取得したいとき、化合物(I)が塩の形で得られるときはそのまま精製すればよく、また、遊離の形で得られるときは、化合物(I)を適当な溶媒に溶解または懸濁し、酸または塩基を加えることにより塩を形成させて単離、精製すればよい。
本発明における化合物(I)の中には種々の立体異性体、位置異性体、互変異性体、鏡像異性体等が存在し得るものがある。本発明の医薬組成物、疼痛の治療及び/または予防剤、キット、疼痛の治療及び/または予防用キット、疼痛の治療及び/または予防方法等にはこれらの可能な全ての異性体及びそれらの混合物が使用でき、その混合比についても任意の比率でよい。
次に本発明で用いられる化合物(I)の製造法について説明する。化合物(I)またはその薬学的に許容される塩は、例えばWO2008/007780記載の方法により製造することができる。以下の表1~6に本発明に用いられる化合物の具体例を示すが、本発明に使用される化合物はそれらに限定されるものではない。
上記製造法における中間体及び目的化合物は、有機合成化学で常用される分離精製法、例えば、ろ過、抽出、洗浄、乾燥、濃縮、再結晶、各種クロマトグラフィー等に付して単離精製することができる。また、中間体においては特に精製することなく次の反応に供することも可能である。
上記製造法における中間体及び目的化合物は、有機合成化学で常用される分離精製法、例えば、ろ過、抽出、洗浄、乾燥、濃縮、再結晶、各種クロマトグラフィー等に付して単離精製することができる。また、中間体においては特に精製することなく次の反応に供することも可能である。


































本発明において、ペンタジエナミド誘導体またはその薬学的に許容される塩に対して用いられる鎮痛剤としては、抗うつ剤、オピオイド鎮痛剤、抗癲癇剤等が挙げられる。
抗うつ剤としては、具体的には例えばデュロキセチン、アミトリプチリン、フルボキサチン、フルボキサミン、フルオキセチン、ノルトリプチリン、ベンラファキシン、パロキセチン、レボキセチン、ミルナシプラン、イミプラミン、デシプラミン、クロミプラミン、トリミプラミン、アモキサピン、ロフェプラミン、ドレスピン、シタロプラム、エスシタロプラム、セルトラリン、マプロチリン、ミアンセリン、セチプチリン、ネファゾドン等が挙げられる。また、これらは単独でも組み合わされていてもよい。
抗うつ剤としては、具体的には例えばデュロキセチン、アミトリプチリン、フルボキサチン、フルボキサミン、フルオキセチン、ノルトリプチリン、ベンラファキシン、パロキセチン、レボキセチン、ミルナシプラン、イミプラミン、デシプラミン、クロミプラミン、トリミプラミン、アモキサピン、ロフェプラミン、ドレスピン、シタロプラム、エスシタロプラム、セルトラリン、マプロチリン、ミアンセリン、セチプチリン、ネファゾドン等が挙げられる。また、これらは単独でも組み合わされていてもよい。
オピオイド鎮痛剤としては、具体的には例えば、モルヒネ、フェンタニル、レミフェンタニル、スフェンタニル、ブプレノルフィン、ヒドロモルフィン、オキシモルフィン、ペンタゾシン、トラマドール、コデイン、ジヒドロコデイン、オキシコドン、メサドン、ヘロイン、ナルブフィン、レボルファノール、レバロルファン、メピリジン、ヒドロコドン、プロポキシフェン、ブトルファノール等が挙げられる。また、これらは単独でも組み合わされていてもよい。
抗癲癇剤としては、α2δリガンドであるガバペンチン、プレガバリン等が挙げられる。また、これらは単独でも組み合わされていてもよい。
これらの鎮痛剤は、薬学的に許容される塩(該薬学的に許容される塩としては、前記化合物(I)の薬学的に許容される塩として例示した塩等が挙げられる)またはこれらの水和物として存在することもあるが、本発明の医薬組成物、疼痛の治療及び/または予防剤、キット、疼痛の治療及び/または予防用キットならびに疼痛の治療及び/または予防方法等には、これらも使用することができる。
これらの鎮痛剤は、薬学的に許容される塩(該薬学的に許容される塩としては、前記化合物(I)の薬学的に許容される塩として例示した塩等が挙げられる)またはこれらの水和物として存在することもあるが、本発明の医薬組成物、疼痛の治療及び/または予防剤、キット、疼痛の治療及び/または予防用キットならびに疼痛の治療及び/または予防方法等には、これらも使用することができる。
また、上記で例示した抗うつ剤、オピオイド鎮痛剤及び抗癲癇剤などの鎮痛剤は、市販品として、または従来既知の方法に従って製造して得ることができる。
化合物(I)またはその薬学的に許容される塩と、鎮痛剤とを含有する医薬組成物は、例えば疼痛の治療及び/または予防に使用することができ、より具体的には、神経因性疼痛、炎症性疼痛、癌性疼痛等の治療及び/または予防に使用することができる。さらに好ましくは、神経因性疼痛の治療及び/または予防に使用することができる。神経因性疼痛としては、帯状疱疹後神経痛、糖尿病性神経因性疼痛、HIV関連疼痛、三叉神経痛、癌化学療法に伴う痛み、線維筋痛症等が挙げられる。
化合物(I)またはその薬学的に許容される塩と、鎮痛剤とを含有する医薬組成物は、例えば疼痛の治療及び/または予防に使用することができ、より具体的には、神経因性疼痛、炎症性疼痛、癌性疼痛等の治療及び/または予防に使用することができる。さらに好ましくは、神経因性疼痛の治療及び/または予防に使用することができる。神経因性疼痛としては、帯状疱疹後神経痛、糖尿病性神経因性疼痛、HIV関連疼痛、三叉神経痛、癌化学療法に伴う痛み、線維筋痛症等が挙げられる。
本発明の医薬組成物ならびに疼痛の治療及び/または予防剤は、化合物(I)またはその薬学的に許容される塩と、鎮痛剤とを含有するように製剤化したものであれば、単剤(合剤)としてでも複数の製剤の組み合わせとしてでも使用または投与することができるが、中でも2つ以上の製剤の組み合わせが好ましい。複数の製剤の組み合わせとして使用または投与する際には、これらの複数の製剤を、同時にまたは時間を置いて別々に使用または投与することができる。なお、これら製剤は、例えば錠剤、注射剤等の形態として用いることが好ましい。
化合物(I)またはその薬学的に許容される塩と鎮痛剤との用量比(重量/重量)は、使用する化合物(I)と鎮痛剤との組み合わせ、化合物(I)と鎮痛剤のそれぞれの効力等に応じて適宜調整すればよいが、具体的には例えば1/50(化合物(I)またはその薬学的に許容される塩/鎮痛剤)~50000/1、好ましくは1/30~10000/1、より好ましくは1/20~5000/1、さらに好ましくは1/10~1000/1、さらに好ましくは1/10~100/1、さらにより好ましくは1/10~10/1、最も好ましくは1/5~5/1の間の比である。
複数の製剤の組み合わせとして投与する際には、例えば(a)化合物(I)またはその薬学的に許容される塩を含有する第1成分と、(b)鎮痛剤を含有する第2成分とを、それぞれ別途製剤化し、キットとして作成しておき、このキットを用いてそれぞれの成分を同時にまたは時間を置いて、同一対象に対して同一経路または異なった経路で投与することもできる。
該キットとしては、例えば保存する際に外部の温度や光による内容物である成分の変性、容器からの化学成分の溶出等がみられない容器であれば材質、形状等は特に限定されない2つ以上の容器(例えばバイアル、バッグ等)と内容物からなり、内容物である上記第1成分と第2成分が別々の経路(例えばチューブ等)または同一の経路を介して投与可能な形態を有するものが用いられる。具体的には、錠剤、注射剤等のキットが挙げられる。
また、本発明の疼痛の治療及び/または予防方法は、上記で記載した化合物(I)またはその薬学的に許容される塩と、鎮痛剤の使用または投与方法と同様にして実施できる。すなわち、化合物(I)またはその薬学的に許容される塩と、鎮痛剤を、それぞれの有効成分を含有するように製剤化し、例えば単剤としてまたは複数の製剤の組み合わせとして、好ましくは2つ以上の製剤を組み合わせて投与することにより実施できる。複数の製剤を組み合わせて投与する際には、これら製剤は、同時にまたは時間を置いて別々に投与することができ、上記で記載したようなキットを用いて投与することもできる。
次に、化合物(I)またはその薬学的に許容される塩と鎮痛剤とを投与することによる疼痛の治療効果について試験例により具体的に説明する。化合物(I)またはその薬学的に許容される塩として、化合物A(化合物332)を用いたが、本発明はこれに限定されるものではない。
試験例1:坐骨神経損傷ラット(神経因性疼痛モデル)の作成
Fisherらの方法 [ペイン(Pain)、1998年、第77巻、p.59] を参考に、坐骨神経損傷性神経因性疼痛モデルを作製した。Crl:CD (SD) 系ラット(雄性、5週齢、日本チャールス・リバー社) を、ペントバルビタールナトリウム麻酔下 (50 mg/kg、腹腔内投与) で左後肢の坐骨神経を剥離し、露出した坐骨神経に長さ2 mmのPE-60ポリエチレンチューブ (商品名:Intramedic、サイズ:PE-60、Beckton Dickinson社製) を覆い被せた。実験には、手術後2-3週間経過したラットを用いた。
試験例1:坐骨神経損傷ラット(神経因性疼痛モデル)の作成
Fisherらの方法 [ペイン(Pain)、1998年、第77巻、p.59] を参考に、坐骨神経損傷性神経因性疼痛モデルを作製した。Crl:CD (SD) 系ラット(雄性、5週齢、日本チャールス・リバー社) を、ペントバルビタールナトリウム麻酔下 (50 mg/kg、腹腔内投与) で左後肢の坐骨神経を剥離し、露出した坐骨神経に長さ2 mmのPE-60ポリエチレンチューブ (商品名:Intramedic、サイズ:PE-60、Beckton Dickinson社製) を覆い被せた。実験には、手術後2-3週間経過したラットを用いた。
神経因性疼痛で一般的に認められる機械刺激性アロディニアの測定には、フォン・フレイ・フィラメント(von Frey filament(商品名:touch test sensory evaluator、型番:model 58011、室町機械(株)製))を用いた。ラットを底が金属性メッシュで覆われたアクリル製の4連ケージ (幅390 mm ×奥行き210 mm ×高さ145 mm) に入れ、約20分間環境に慣れさせた後、神経損傷側の足裏に垂直にフォン・フレイ・フィラメントを軽く曲がる程度に約4秒間適用し、逃避反応の有無を観察し、結果は痛覚閾値として示した。痛覚閾値(g)は、Dixonのアップ・ダウン(up down)法 [アニュアル・レビュー・オブ・ファーマコロジー・アンド・トキシコロジー(Annual Review of Pharmacology and Toxicology)、1980年、第20巻、p.441] により算出した。正常ラットの痛覚閾値は12 g程度であるのに対し、薬物投与前の坐骨神経損傷足での疼痛閾値は2 g程度で、アロディニアと認められる痛覚閾値(g)の低下が確認された。
試験例2:化合物Aによるラット疼痛モデルにおけるガバペンチン鎮痛作用の増強作用効果
上記の坐骨神経損傷ラットを底が金属性メッシュで覆われたアクリル製の4連ケージ (幅390 mm ×奥行き210 mm ×高さ145 mm) に入れ、以下の手順でガバペンチンと化合物Aをそれぞれ同時に投与し、その3時間後に痛覚閾値(g)を測定した。
試験例2:化合物Aによるラット疼痛モデルにおけるガバペンチン鎮痛作用の増強作用効果
上記の坐骨神経損傷ラットを底が金属性メッシュで覆われたアクリル製の4連ケージ (幅390 mm ×奥行き210 mm ×高さ145 mm) に入れ、以下の手順でガバペンチンと化合物Aをそれぞれ同時に投与し、その3時間後に痛覚閾値(g)を測定した。
併用投与群として、ガバペンチンを20 mg/mLの濃度で蒸留水(大塚製薬工場)に溶解し、100 mg/kgの用量で経口投与し、化合物Aを0.6 mg/mLの濃度で0.5%メチルセルロース(0.5% MC)水溶液に懸濁し、3 mg/kgの用量で経口投与した。溶媒投与群として、坐骨神経損傷ラットに蒸留水を5 mL/kg経口投与し、0.5% MC水溶液を5 mL/kg経口投与した。また、ガバペンチン投与群として、坐骨神経損傷ラットに上記と同様にガバペンチンを100 mg/kg経口投与し、0.5% MC水溶液を5 mL/kg経口投与した。さらに、化合物A投与群として、坐骨神経損傷ラットに蒸留水5 mL/kg経口投与し、上記と同様に化合物Aを3 mg/kg経口投与した。各群6匹で試験を実施した。結果を図1に示した。
化合物Aとガペンチンの併用投与群では、ガバペンチン投与群及び化合物A投与群と比べ、痛覚閾値(g)の増加が認められた。すなわち、化合物Aの投与により、坐骨神経損傷ラットにおけるガバペンチンの鎮痛作用が増強されることが確認された。
上記試験により、化合物(I)またはその薬学的に許容される塩を、抗癲癇剤(例えばガバペンチン等)と組み合わせて用いることにより、疼痛に対し、強力な抑制効果が得られることが明らかとなった。つまり、化合物(I)またはその薬学的に許容される塩と抗癲癇剤を組み合わせて用いることにより、抗癲癇剤単独では十分に疼痛を抑制できない場合でも、十分な疼痛の制御が可能と考えられた。また、従来用いられている抗癲癇剤の用量より少ない用量で同様の鎮痛効果が得られ、結果として抗癲癇剤の用量を削減でき、抗癲癇剤の有する副作用を軽減できると考えられた。
試験例3 化合物Aによるラット疼痛モデルにおけるデュロキセチン鎮痛作用の増強作用効果
上記の坐骨神経損傷ラットを底が金属性メッシュで覆われたアクリル製の4連ケージ (幅390 mm ×奥行き210 mm ×高さ145 mm) に入れ、以下の手順でデュロキセチンと化合物Aをそれぞれ同時に投与し、その3時間後に痛覚閾値(g)を測定した。
上記試験により、化合物(I)またはその薬学的に許容される塩を、抗癲癇剤(例えばガバペンチン等)と組み合わせて用いることにより、疼痛に対し、強力な抑制効果が得られることが明らかとなった。つまり、化合物(I)またはその薬学的に許容される塩と抗癲癇剤を組み合わせて用いることにより、抗癲癇剤単独では十分に疼痛を抑制できない場合でも、十分な疼痛の制御が可能と考えられた。また、従来用いられている抗癲癇剤の用量より少ない用量で同様の鎮痛効果が得られ、結果として抗癲癇剤の用量を削減でき、抗癲癇剤の有する副作用を軽減できると考えられた。
試験例3 化合物Aによるラット疼痛モデルにおけるデュロキセチン鎮痛作用の増強作用効果
上記の坐骨神経損傷ラットを底が金属性メッシュで覆われたアクリル製の4連ケージ (幅390 mm ×奥行き210 mm ×高さ145 mm) に入れ、以下の手順でデュロキセチンと化合物Aをそれぞれ同時に投与し、その3時間後に痛覚閾値(g)を測定した。
併用投与群として、デュロキセチンを2 mg/mLの濃度で蒸留水(大塚製薬工場)に溶解し、10 mg/kgの用量で経口投与し、化合物Aを0.6 mg/mLの濃度で0.5%メチルセルロース(0.5% MC)水溶液に懸濁し、3 mg/kgの用量で経口投与した。溶媒投与群として、坐骨神経損傷ラットに蒸留水を5 mL/kg経口投与し、0.5% MC水溶液を5 mL/kg経口投与した。また、デュロキセチン投与群として、坐骨神経損傷ラットに上記と同様にデュロキセチンを10 mg/kg経口投与し、0.5% MC水溶液を5 mL/kg経口投与した。さらに、化合物A投与群として、坐骨神経損傷ラットに蒸留水5 mL/kg経口投与し、上記と同様に化合物Aを3 mg/kg経口投与した。各群6匹で試験を実施した。結果を図2に示した。
化合物Aとデュロキセチンの併用投与群では、デュロキセチン投与群及び化合物A投与群と比べ、痛覚閾値(g)の増加が認められた。すなわち、化合物Aの投与により、坐骨神経損傷ラットにおけるデュロキセチンの鎮痛作用が増強されることが確認された。
上記試験により、化合物(I)またはその薬学的に許容される塩を、抗うつ剤(例えばデュロキセチン等)と組み合わせて用いることにより、疼痛に対し、強力な抑制効果が得られることが明らかとなった。つまり、化合物(I)またはその薬学的に許容される塩と抗うつ剤を組み合わせて用いることにより、抗うつ剤単独では十分に疼痛を抑制できない場合でも、十分な疼痛の制御が可能と考えられた。また、従来用いられている抗うつ剤の用量より少ない用量で同様の鎮痛効果が得られ、結果として抗うつ剤の用量を削減でき、抗うつ剤の有する副作用を軽減できると考えられた。
試験例4 化合物Aによるラット疼痛モデルにおけるモルヒネ鎮痛作用の増強作用効果
上記の坐骨神経損傷ラットを底が金属性メッシュで覆われたアクリル製の4連ケージ (幅390 mm ×奥行き210 mm ×高さ145 mm) に入れ、以下の手順で化合物Aを投与して2時間後にモルヒネを投与し、その1時間後に痛覚閾値(g)を測定した。
上記試験により、化合物(I)またはその薬学的に許容される塩を、抗うつ剤(例えばデュロキセチン等)と組み合わせて用いることにより、疼痛に対し、強力な抑制効果が得られることが明らかとなった。つまり、化合物(I)またはその薬学的に許容される塩と抗うつ剤を組み合わせて用いることにより、抗うつ剤単独では十分に疼痛を抑制できない場合でも、十分な疼痛の制御が可能と考えられた。また、従来用いられている抗うつ剤の用量より少ない用量で同様の鎮痛効果が得られ、結果として抗うつ剤の用量を削減でき、抗うつ剤の有する副作用を軽減できると考えられた。
試験例4 化合物Aによるラット疼痛モデルにおけるモルヒネ鎮痛作用の増強作用効果
上記の坐骨神経損傷ラットを底が金属性メッシュで覆われたアクリル製の4連ケージ (幅390 mm ×奥行き210 mm ×高さ145 mm) に入れ、以下の手順で化合物Aを投与して2時間後にモルヒネを投与し、その1時間後に痛覚閾値(g)を測定した。
併用投与群として、化合物Aを0.6 mg/mLの濃度で0.5%メチルセルロース(0.5% MC)水溶液に懸濁し、3 mg/kgの用量で経口投与し、モルヒネを0.25 mg/mLの濃度で生理食塩水(大塚製薬工場)に溶解し、0.5 mg/kgの用量で皮下投与した(併用投与群)。溶媒投与群として、坐骨神経損傷ラットに0.5% MC水溶液を5 mL/kg経口投与し、生理食塩水を2 mL/kg皮下投与した。また、モルヒネ投与群として、坐骨神経損傷ラットに上記と同様に0.5% MC水溶液を5 mL/kg経口投与し、モルヒネを0.5 mg/kg皮下投与した。さらに、化合物A投与群として、坐骨神経損傷ラットに上記と同様に化合物Aを3 mg/kg経口投与し、生理食塩水を2 mL/kg皮下投与した。各群6匹で試験を実施した。結果を図3に示した。
化合物Aとモルヒネの併用投与群では、モルヒネ投与群及び化合物A投与群と比べ、痛覚閾値(g)の増加が認められた。すなわち、化合物Aの投与により、坐骨神経損傷ラットにおけるモルヒネの鎮痛作用が増強されることが確認された。
上記試験により、化合物(I)またはその薬学的に許容される塩を、オピオイド鎮痛剤(例えばモルヒネ等)と組み合わせて用いることにより、疼痛に対し、強力な抑制効果が得られることが明らかとなった。つまり、化合物(I)またはその薬学的に許容される塩とオピオイド鎮痛剤を組み合わせて用いることにより、オピオイド鎮痛剤単独では十分に疼痛を抑制できない場合でも、十分な疼痛の制御が可能と考えられた。また、従来用いられているオピオイド鎮痛剤の用量より少ない用量で同様の鎮痛効果が得られ、結果としてオピオイド鎮痛剤の用量を削減でき、オピオイド鎮痛剤の有する副作用を軽減できると考えられた。
試験例5 化合物Aによるマウス癌性疼痛モデルにおけるモルヒネ鎮痛作用の増強作用効果
Honoreらの報告 [ニューロサイエンス(Neuroscience)、2000年、第98巻、p.585] を参考に癌性疼痛モデルを作製した。すなわち、C3H/He系マウス(雄性、4週齢、日本チャールス・リバー社)マウスをペントバルビタールナトリウム (50-60 mg/kg, 腹腔内投与) 麻酔下に左膝関節部の皮膚を約5 mm切開し、大腿骨遠位端より骨髄腔に26ゲージの注射針を刺入して、細胞を移植するための孔を開けた。27ゲージの注射針を接続したマイクロシリンジを用いて、2.5×104個の腫瘍細胞(NCTC 2472 osteolytic sarcoma cell)を含む細胞液 10 μLを骨髄腔内に注入した。その後、皮膚を縫合した。痛みの評価には、腫瘍細胞移植後の19-21日目のマウスを使用した。
上記試験により、化合物(I)またはその薬学的に許容される塩を、オピオイド鎮痛剤(例えばモルヒネ等)と組み合わせて用いることにより、疼痛に対し、強力な抑制効果が得られることが明らかとなった。つまり、化合物(I)またはその薬学的に許容される塩とオピオイド鎮痛剤を組み合わせて用いることにより、オピオイド鎮痛剤単独では十分に疼痛を抑制できない場合でも、十分な疼痛の制御が可能と考えられた。また、従来用いられているオピオイド鎮痛剤の用量より少ない用量で同様の鎮痛効果が得られ、結果としてオピオイド鎮痛剤の用量を削減でき、オピオイド鎮痛剤の有する副作用を軽減できると考えられた。
試験例5 化合物Aによるマウス癌性疼痛モデルにおけるモルヒネ鎮痛作用の増強作用効果
Honoreらの報告 [ニューロサイエンス(Neuroscience)、2000年、第98巻、p.585] を参考に癌性疼痛モデルを作製した。すなわち、C3H/He系マウス(雄性、4週齢、日本チャールス・リバー社)マウスをペントバルビタールナトリウム (50-60 mg/kg, 腹腔内投与) 麻酔下に左膝関節部の皮膚を約5 mm切開し、大腿骨遠位端より骨髄腔に26ゲージの注射針を刺入して、細胞を移植するための孔を開けた。27ゲージの注射針を接続したマイクロシリンジを用いて、2.5×104個の腫瘍細胞(NCTC 2472 osteolytic sarcoma cell)を含む細胞液 10 μLを骨髄腔内に注入した。その後、皮膚を縫合した。痛みの評価には、腫瘍細胞移植後の19-21日目のマウスを使用した。
痛みの評価は、Honoreらの報告[ニューロサイエンス(Neuroscience)、2000年、第98巻、p.585] に準じて、guarding行動(歩行時の痛み防御行動)時間を測定した。すなわち、マウスを観察用金網の上に置き自由に歩行させ、2分間の観察時間におけるguarding行動時間(左後肢をあげている時間)を測定した。薬物投与前のguarding行動時間が5秒以上のマウスを痛み行動が発症したものとみなし、薬物評価に用いた。正常動物はguarding行動を示さなかったが、癌性疼痛モデル動物では10秒程度のguarding行動が観察された。
化合物Aは、1 mg/mLの濃度になるように0.5%メチルセルロース(0.5% MC)水溶液に懸濁して、10 mg/kgの用量で経口投与した。モルヒネは、1.5 mg/mLの濃度になるように生理食塩水(大塚製薬工場)に溶解して、3 mg/kgの用量で皮下投与した。併用投与群では、評価の3時間前に化合物Aを、評価の1時間前にモルヒネを投与した。化合物A投与群では、モルヒネの代わりにその溶媒である生理食塩水(2 mL/kg)を、モルヒネ投与群では、化合物Aの代わりにその溶媒である0.5% MC(10 mL/kg)を投与した。溶媒投与群では、化合物Aの溶媒とモルヒネの溶媒を投与した。各群10匹で試験を実施した。結果を図4に示した。
併用投与群では、モルヒネ投与群及び化合物A投与群を上回るguarding行動時間の短縮が認められた。すなわち、化合物Aの投与により、癌性疼痛マウスにおけるモルヒネの鎮痛作用が増強されることが確認された。
上記試験により、化合物(I)またはその薬学的に許容される塩を、オピオイド鎮痛剤(例えばモルヒネ等)と組み合わせて用いることにより、癌性疼痛に対し、強力な抑制効果が得られることが明らかとなった。つまり、化合物(I)またはその薬学的に許容される塩とオピオイド鎮痛剤を組み合わせて用いることにより、オピオイド鎮痛剤単独では十分に癌性疼痛を抑制できない場合でも、十分な癌性疼痛の制御が可能と考えられた。また、従来用いられているオピオイド鎮痛剤の用量より少ない用量で同様の癌性鎮痛効果が得られ、結果としてオピオイド鎮痛剤の用量を削減でき、オピオイド鎮痛剤の有する副作用を軽減できると考えられた。
上記試験により、化合物(I)またはその薬学的に許容される塩を、オピオイド鎮痛剤(例えばモルヒネ等)と組み合わせて用いることにより、癌性疼痛に対し、強力な抑制効果が得られることが明らかとなった。つまり、化合物(I)またはその薬学的に許容される塩とオピオイド鎮痛剤を組み合わせて用いることにより、オピオイド鎮痛剤単独では十分に癌性疼痛を抑制できない場合でも、十分な癌性疼痛の制御が可能と考えられた。また、従来用いられているオピオイド鎮痛剤の用量より少ない用量で同様の癌性鎮痛効果が得られ、結果としてオピオイド鎮痛剤の用量を削減でき、オピオイド鎮痛剤の有する副作用を軽減できると考えられた。
(参考例1)化合物332の合成法
工程1: (3R)-5-アミノ-3-ヒドロキシ-2-オキソ-1,2,3,4-テトラヒドロキノリン(化合物a)の製造
論文[バイオオーガニック&メディシナル・ケミストリー・レターズ(Bioorganic and Medicinal Chemistry Letters)、2000年、第10巻、pp.1459-1462]記載の方法に準じて調製した5-アミノ-3-ヒドロキシ-2-オキソ-1,2,3,4-テトラヒドロキノリン(499 mg, 2.80 mmol)とL-(+)-酒石酸(420 mg, 2.80 mmol)をメタノール(50 mL)中に加え、70℃で1時間攪拌することにより完全に溶解させた。溶媒を減圧留去した後、メタノール(32 mL)を加え、70℃で1時間攪拌した。ジエチルエーテル(15 mL)を加えて、室温で終夜攪拌後、得られた結晶をろ過することにより、化合物a・0.5酒石酸塩(90%ee)(115 mg, 16%)を得た。
工程1: (3R)-5-アミノ-3-ヒドロキシ-2-オキソ-1,2,3,4-テトラヒドロキノリン(化合物a)の製造
論文[バイオオーガニック&メディシナル・ケミストリー・レターズ(Bioorganic and Medicinal Chemistry Letters)、2000年、第10巻、pp.1459-1462]記載の方法に準じて調製した5-アミノ-3-ヒドロキシ-2-オキソ-1,2,3,4-テトラヒドロキノリン(499 mg, 2.80 mmol)とL-(+)-酒石酸(420 mg, 2.80 mmol)をメタノール(50 mL)中に加え、70℃で1時間攪拌することにより完全に溶解させた。溶媒を減圧留去した後、メタノール(32 mL)を加え、70℃で1時間攪拌した。ジエチルエーテル(15 mL)を加えて、室温で終夜攪拌後、得られた結晶をろ過することにより、化合物a・0.5酒石酸塩(90%ee)(115 mg, 16%)を得た。
化合物a・0.5酒石酸塩(90%ee)(178 mg, 0.703 mmol)をメタノール(8.9 mL)中に加え、70℃で1時間攪拌することに完全に溶解させた。ジエチルエーテル(3.6 mL)を加えて、室温で終夜攪拌後、ろ過することにより得られた結晶を(クロロホルム:2-プロパノール=4:1)溶液に溶解し、飽和炭酸水素ナトリウム水溶液で洗浄後、無水硫酸マグネシウムで乾燥した。ろ過後、溶媒を減圧留去し、化合物a (99%ee)(25.8 mg, 27%)を得た。
1H NMR (DMSO-d6, δ ppm): 2.41 (dd, J = 12.2, 15.5 Hz, 1H), 3.00 (dd, J = 6.8, 15.5 Hz, 1H), 4.05 (dd, J = 6.8, 12.2 Hz, 1H), 5.02 (s, 2H), 5.28 (s, 1H), 6.09 (d, J = 7.8 Hz, 1H), 6.26 (d, J = 7.8 Hz, 1H), 6.80 (t, J = 7.8 Hz, 1H), 9.88 (br s, 1H).
ESIMS m/z: [M+H]+ 179.
工程2: (2E,4Z)-N-[(3R)-3-ヒドロキシ-2-オキソ-1,2,3,4-テトラヒドロ-5-キノリル]-5-(4-イソプロポキシフェニル)-5-[4-(トリフルオロメチル)フェニル]-2,4-ペンタジエナミド(化合物332)の製造
WO2008/007780記載の方法に準じて調製した(2E,4Z)-5-(4-イソプロポキシフェニル)-5-[4-(トリフルオロメチル)フェニル]-2,4-ペンタジエン酸(6.00 g, 16.0 mmol)をジメチルホルムアミド(DMF、120 mL)に溶解し、化合物a(3.21 g, 18.0 mmol)、1-エチル-3-(3-ジメチルアミノプロピル) カルボジイミド(EDC)塩酸塩(6.14 g, 32.0 mmol)及び1-ヒドロキシベンゾトリアゾール(HOBt)水和物(4.90 g, 32.0 mmol)を加え、50℃で8時間攪拌した。放冷後、反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。有機層を飽和重曹水で洗浄後、無水硫酸マグネシウムで乾燥した。ろ過後、溶媒を減圧留去し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン=2/8)で精製し、エタノール/水で再結晶することにより、化合物332(4.25 g, 49 %)を得た。
1H NMR (DMSO-d6, δ ppm): 1.31 (d, J = 6.0 Hz, 6H), 2.62 (dd, J = 12.0, 15.9 Hz, 1H), 3.02 (dd, J = 6.2, 15.9 Hz, 1H), 4.03-4.11 (m, 1H), 4.68 (sept, J = 6.0 Hz, 1H), 5.44 (d, J = 4.6 Hz, 1H), 6.60 (d, J = 14.8 Hz, 1H), 6.68-6.72 (m, 1H), 7.01-7.13 (m, 7H), 7.21 (dd, J = 11.6, 14.8 Hz, 1H), 7.54 (d, J = 8.4 Hz, 2H), 7.73 (d, J = 8.4 Hz, 2H), 9.71 (br s, 1H), 10.17 (br s, 1H).
ESIMS m/z: [M+H]+ 537.
1H NMR (DMSO-d6, δ ppm): 2.41 (dd, J = 12.2, 15.5 Hz, 1H), 3.00 (dd, J = 6.8, 15.5 Hz, 1H), 4.05 (dd, J = 6.8, 12.2 Hz, 1H), 5.02 (s, 2H), 5.28 (s, 1H), 6.09 (d, J = 7.8 Hz, 1H), 6.26 (d, J = 7.8 Hz, 1H), 6.80 (t, J = 7.8 Hz, 1H), 9.88 (br s, 1H).
ESIMS m/z: [M+H]+ 179.
工程2: (2E,4Z)-N-[(3R)-3-ヒドロキシ-2-オキソ-1,2,3,4-テトラヒドロ-5-キノリル]-5-(4-イソプロポキシフェニル)-5-[4-(トリフルオロメチル)フェニル]-2,4-ペンタジエナミド(化合物332)の製造
WO2008/007780記載の方法に準じて調製した(2E,4Z)-5-(4-イソプロポキシフェニル)-5-[4-(トリフルオロメチル)フェニル]-2,4-ペンタジエン酸(6.00 g, 16.0 mmol)をジメチルホルムアミド(DMF、120 mL)に溶解し、化合物a(3.21 g, 18.0 mmol)、1-エチル-3-(3-ジメチルアミノプロピル) カルボジイミド(EDC)塩酸塩(6.14 g, 32.0 mmol)及び1-ヒドロキシベンゾトリアゾール(HOBt)水和物(4.90 g, 32.0 mmol)を加え、50℃で8時間攪拌した。放冷後、反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。有機層を飽和重曹水で洗浄後、無水硫酸マグネシウムで乾燥した。ろ過後、溶媒を減圧留去し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン=2/8)で精製し、エタノール/水で再結晶することにより、化合物332(4.25 g, 49 %)を得た。
1H NMR (DMSO-d6, δ ppm): 1.31 (d, J = 6.0 Hz, 6H), 2.62 (dd, J = 12.0, 15.9 Hz, 1H), 3.02 (dd, J = 6.2, 15.9 Hz, 1H), 4.03-4.11 (m, 1H), 4.68 (sept, J = 6.0 Hz, 1H), 5.44 (d, J = 4.6 Hz, 1H), 6.60 (d, J = 14.8 Hz, 1H), 6.68-6.72 (m, 1H), 7.01-7.13 (m, 7H), 7.21 (dd, J = 11.6, 14.8 Hz, 1H), 7.54 (d, J = 8.4 Hz, 2H), 7.73 (d, J = 8.4 Hz, 2H), 9.71 (br s, 1H), 10.17 (br s, 1H).
ESIMS m/z: [M+H]+ 537.

(参考例2)化合物333の合成法
(2E,4Z)-5-(4-エトキシフェニル)-N-[(3R)-3-ヒドロキシ-2-オキソ-1,2,3,4-テトラヒドロ-5-キノリル]-5-[4-(トリフルオロメチル)フェニル]-2,4-ペンタジエナミド(化合物333)の製造
WO2008/007780記載の方法に準じて調製した(2E,4Z)-5-(4-エトキシフェニル)-5-[4-(トリフルオロメチル)フェニル]-2,4-ペンタジエン酸、および参考例1の工程1で得られた化合物aを用い、参考例1の工程2に記載の方法に準じて、化合物333を得た。
(2E,4Z)-5-(4-エトキシフェニル)-N-[(3R)-3-ヒドロキシ-2-オキソ-1,2,3,4-テトラヒドロ-5-キノリル]-5-[4-(トリフルオロメチル)フェニル]-2,4-ペンタジエナミド(化合物333)の製造
WO2008/007780記載の方法に準じて調製した(2E,4Z)-5-(4-エトキシフェニル)-5-[4-(トリフルオロメチル)フェニル]-2,4-ペンタジエン酸、および参考例1の工程1で得られた化合物aを用い、参考例1の工程2に記載の方法に準じて、化合物333を得た。
1H NMR (DMSO-d6, δppm): 1.36 (t, J = 7.0 Hz, 3H), 2.62 (dd, J = 12.1, 16.0 Hz, 1H), 3.02 (dd, J = 6.3, 16.0 Hz, 1H), 4.03-4.13 (m, 3H), 5.44 (d, J = 4.6 Hz, 1H), 6.60 (d, J = 14.8 Hz, 1H), 6.70 (dd, J = 3.1, 5.7 Hz, 1H), 7.03-7.14 (m, 7H), 7.20 (dd, J = 11.7, 14.3 Hz, 1H), 7.54 (d, J = 8.3 Hz, 2H), 7.73 (d, J = 8.3 Hz, 2H), 9.72 (br s, 1H), 10.17 (br s, 1H).
ESIMS m/z: [M+H]+ 523.
上述したように、本発明に使用される医薬組成物ならびに疼痛の治療及び/または予防剤は、化合物(I)またはその薬学的に許容される塩と、鎮痛剤のそれぞれの有効成分を含有するように製剤化したものであれば、単剤としてでも複数の製剤の組み合わせとしてでも使用、投与または製造することができる。これらの医薬組成物ならびに疼痛の治療及び/または予防剤は、錠剤等の経口的投与または注射剤等の非経口的投与に対して適する単位服用形態にあることが望ましい。また、複数の製剤の組み合わせとして使用または投与する際には、これらの複数の製剤を、同時にまたは時間を置いて別々に使用または投与することができる。
ESIMS m/z: [M+H]+ 523.
上述したように、本発明に使用される医薬組成物ならびに疼痛の治療及び/または予防剤は、化合物(I)またはその薬学的に許容される塩と、鎮痛剤のそれぞれの有効成分を含有するように製剤化したものであれば、単剤としてでも複数の製剤の組み合わせとしてでも使用、投与または製造することができる。これらの医薬組成物ならびに疼痛の治療及び/または予防剤は、錠剤等の経口的投与または注射剤等の非経口的投与に対して適する単位服用形態にあることが望ましい。また、複数の製剤の組み合わせとして使用または投与する際には、これらの複数の製剤を、同時にまたは時間を置いて別々に使用または投与することができる。
これら製剤は、それぞれ有効成分の他に製剤学的に許容される一種またはそれ以上の担体(例えば、希釈剤、溶剤、賦形剤等)と一緒に混合し、製剤学の技術分野においてよく知られている任意の方法により製造される。
経口投与に適当な、例えば錠剤等は、乳糖等の賦形剤、澱粉等の崩壊剤、ステアリン酸マグネシウム等の滑沢剤、ヒドロキシプロピルセルロース等の結合剤等を用いて製造できる。
経口投与に適当な、例えば錠剤等は、乳糖等の賦形剤、澱粉等の崩壊剤、ステアリン酸マグネシウム等の滑沢剤、ヒドロキシプロピルセルロース等の結合剤等を用いて製造できる。
非経口投与に適当な、例えば注射剤等は、塩溶液、ブドウ糖溶液または塩水とブドウ糖溶液の混合液等の希釈剤または溶剤等を用いて製造できる。
化合物(I)またはその薬学的に許容される塩と、鎮痛剤を複数の製剤の組み合わせとして使用または投与する場合には、それぞれの投与量及び投与回数は、それぞれの有効成分の効力、投与形態、患者の年齢、体重、治療すべき症状の性質もしくは重篤度等により異なるが、通常一日当たり、化合物(I)またはその薬学的に許容される塩と、鎮痛剤を、それぞれ以下の用量で投与するのが好ましい。
化合物(I)またはその薬学的に許容される塩と、鎮痛剤を複数の製剤の組み合わせとして使用または投与する場合には、それぞれの投与量及び投与回数は、それぞれの有効成分の効力、投与形態、患者の年齢、体重、治療すべき症状の性質もしくは重篤度等により異なるが、通常一日当たり、化合物(I)またはその薬学的に許容される塩と、鎮痛剤を、それぞれ以下の用量で投与するのが好ましい。
経口的に、例えば錠剤として投与する場合、化合物(I)またはその薬学的に許容される塩と、鎮痛剤を、成人一人あたり、それぞれ0.01~1000 mgと0.01~1000 mg、好ましくは0.05~300 mgと0.05~300 mg、さらに好ましくは0.05~100 mgと0.05~100 mgの範囲で、1日1回ないし数回にわけて、同時にまたは時間を置いて別々に投与する。
非経口的に、例えば注射剤として投与する場合、化合物(I)またはその薬学的に許容される塩と、鎮痛剤を、成人一人あたり0.001~1000 mgと0.001~1000 mg、好ましくは0.01~100 mgと0.01~100 mg、さらに好ましくは0.01~20 mgと0.01~20 mgを、1日1回ないし数回にわけて、同時にまたは時間を置いて別々に投与する。
非経口的に、例えば注射剤として投与する場合、化合物(I)またはその薬学的に許容される塩と、鎮痛剤を、成人一人あたり0.001~1000 mgと0.001~1000 mg、好ましくは0.01~100 mgと0.01~100 mg、さらに好ましくは0.01~20 mgと0.01~20 mgを、1日1回ないし数回にわけて、同時にまたは時間を置いて別々に投与する。
また上記の目的で、化合物(I)またはその薬学的に許容される塩と、鎮痛剤を、単剤として使用または投与する場合には、それぞれの用量及び投与回数はそれぞれの有効成分の効力、投与形態、患者の年齢、体重、症状等により異なるが、上記の複数の製剤の組み合わせとして使用または投与する場合のそれぞれの用量で1つの製剤として調製し、使用または投与するのが好ましい。
しかしながら、これら投与量及び投与回数に関しては、前述の種々の条件により変動する。
以下、本発明を実施例によりさらに具体的に説明するが、本発明の範囲はこれらの実施例に限定されることはない。
以下、本発明を実施例によりさらに具体的に説明するが、本発明の範囲はこれらの実施例に限定されることはない。
製剤例1:錠剤(化合物A)
常法により、次の組成からなる錠剤を調製する。化合物A 40g、乳糖286.8g及び馬鈴薯澱粉60gを混合し、これにヒドロキシプロピルセルロースの10%水溶液120gを加える。得られた混合物を常法により練合し、造粒して乾燥させた後、整粒し打錠用顆粒とする。これにステアリン酸マグネシウム1.2gを加えて混合し、径8mmの杵をもった打錠機で打錠を行って、錠剤(1錠あたり活性成分20mgを含有する)を得る。
処方 化合物A 20 mg
乳糖 143.4 mg
馬鈴薯澱粉 30 mg
ヒドロキシプロピルセルロース 6 mg
ステアリン酸マグネシウム 0.6 mg
200 mg
常法により、次の組成からなる錠剤を調製する。化合物A 40g、乳糖286.8g及び馬鈴薯澱粉60gを混合し、これにヒドロキシプロピルセルロースの10%水溶液120gを加える。得られた混合物を常法により練合し、造粒して乾燥させた後、整粒し打錠用顆粒とする。これにステアリン酸マグネシウム1.2gを加えて混合し、径8mmの杵をもった打錠機で打錠を行って、錠剤(1錠あたり活性成分20mgを含有する)を得る。
処方 化合物A 20 mg
乳糖 143.4 mg
馬鈴薯澱粉 30 mg
ヒドロキシプロピルセルロース 6 mg
ステアリン酸マグネシウム 0.6 mg
200 mg
製剤例2:錠剤(化合物Aとガバペンチンの単剤)
常法により、次の組成からなる錠剤を調製する。化合物A 3g、ガバペンチン 100g、乳糖223.8g及び馬鈴薯澱粉60gを混合し、これにヒドロキシプロピルセルロースの10%水溶液120gを加える。得られた混合物を常法により練合し、造粒して乾燥させた後、整粒し打錠用顆粒とする。これにステアリン酸マグネシウム1.2gを加えて混合し、径8mmの杵をもった打錠機で打錠を行って、錠剤(1錠あたり化合物A 1.5mg及びガバペンチン 50mgを含有する)を得る。
処方 化合物A 1.5 mg
ガバペンチン 50 mg
乳糖 111.9 mg
馬鈴薯澱粉 30 mg
ヒドロキシプロピルセルロース 6 mg
ステアリン酸マグネシウム 0.6 mg
200 mg
常法により、次の組成からなる錠剤を調製する。化合物A 3g、ガバペンチン 100g、乳糖223.8g及び馬鈴薯澱粉60gを混合し、これにヒドロキシプロピルセルロースの10%水溶液120gを加える。得られた混合物を常法により練合し、造粒して乾燥させた後、整粒し打錠用顆粒とする。これにステアリン酸マグネシウム1.2gを加えて混合し、径8mmの杵をもった打錠機で打錠を行って、錠剤(1錠あたり化合物A 1.5mg及びガバペンチン 50mgを含有する)を得る。
処方 化合物A 1.5 mg
ガバペンチン 50 mg
乳糖 111.9 mg
馬鈴薯澱粉 30 mg
ヒドロキシプロピルセルロース 6 mg
ステアリン酸マグネシウム 0.6 mg
200 mg
製剤例3:錠剤(化合物Aとデュロキセチンの単剤)
常法により、次の組成からなる錠剤を調製する。化合物A 15g、デュロキセチン 50g、乳糖261.8g及び馬鈴薯澱粉60gを混合し、これにヒドロキシプロピルセルロースの10%水溶液120gを加える。得られた混合物を常法により練合し、造粒して乾燥させた後、整粒し打錠用顆粒とする。これにステアリン酸マグネシウム1.2gを加えて混合し、径8mmの杵をもった打錠機で打錠を行って、錠剤(1錠あたり化合物A 7.5mg及びデュロキセチン 25mgを含有する)を得る。
処方 化合物A 7.5 mg
デュロキセチン 25 mg
乳糖 130.9 mg
馬鈴薯澱粉 30 mg
ヒドロキシプロピルセルロース 6 mg
ステアリン酸マグネシウム 0.6 mg
200 mg
常法により、次の組成からなる錠剤を調製する。化合物A 15g、デュロキセチン 50g、乳糖261.8g及び馬鈴薯澱粉60gを混合し、これにヒドロキシプロピルセルロースの10%水溶液120gを加える。得られた混合物を常法により練合し、造粒して乾燥させた後、整粒し打錠用顆粒とする。これにステアリン酸マグネシウム1.2gを加えて混合し、径8mmの杵をもった打錠機で打錠を行って、錠剤(1錠あたり化合物A 7.5mg及びデュロキセチン 25mgを含有する)を得る。
処方 化合物A 7.5 mg
デュロキセチン 25 mg
乳糖 130.9 mg
馬鈴薯澱粉 30 mg
ヒドロキシプロピルセルロース 6 mg
ステアリン酸マグネシウム 0.6 mg
200 mg
製剤例4:錠剤(化合物Aとモルヒネの単剤)
常法により、次の組成からなる錠剤を調製する。化合物A 60g、モルヒネ 10g、乳糖256.8g及び馬鈴薯澱粉60gを混合し、これにヒドロキシプロピルセルロースの10%水溶液120gを加える。得られた混合物を常法により練合し、造粒して乾燥させた後、整粒し打錠用顆粒とする。これにステアリン酸マグネシウム1.2gを加えて混合し、径8mmの杵をもった打錠機で打錠を行って、錠剤(1錠あたり化合物A 30mg及びモルヒネ 5mgを含有する)を得る。
処方 化合物A 30 mg
モルヒネ 5 mg
乳糖 128.4 mg
馬鈴薯澱粉 30 mg
ヒドロキシプロピルセルロース 6 mg
ステアリン酸マグネシウム 0.6 mg
200 mg
常法により、次の組成からなる錠剤を調製する。化合物A 60g、モルヒネ 10g、乳糖256.8g及び馬鈴薯澱粉60gを混合し、これにヒドロキシプロピルセルロースの10%水溶液120gを加える。得られた混合物を常法により練合し、造粒して乾燥させた後、整粒し打錠用顆粒とする。これにステアリン酸マグネシウム1.2gを加えて混合し、径8mmの杵をもった打錠機で打錠を行って、錠剤(1錠あたり化合物A 30mg及びモルヒネ 5mgを含有する)を得る。
処方 化合物A 30 mg
モルヒネ 5 mg
乳糖 128.4 mg
馬鈴薯澱粉 30 mg
ヒドロキシプロピルセルロース 6 mg
ステアリン酸マグネシウム 0.6 mg
200 mg
製剤例5:注射剤(化合物A)
常法により、次の組成からなる注射剤を調製する。化合物A 1mgを注射用蒸留水に添加して混合し、さらに塩酸及び水酸化ナトリウム水溶液を添加してpHを7に調整した後、注射用蒸留水で全量を1000mLとする。得られた混合液をガラスバイアルに2mLずつ無菌的に充填して、注射剤(1バイアルあたり活性成分2μgを含有する)を得る。
処方 化合物A 2 μg
塩酸 適量
水酸化ナトリウム水溶液 適量
注射用蒸留水 適量
2.00 mL
常法により、次の組成からなる注射剤を調製する。化合物A 1mgを注射用蒸留水に添加して混合し、さらに塩酸及び水酸化ナトリウム水溶液を添加してpHを7に調整した後、注射用蒸留水で全量を1000mLとする。得られた混合液をガラスバイアルに2mLずつ無菌的に充填して、注射剤(1バイアルあたり活性成分2μgを含有する)を得る。
処方 化合物A 2 μg
塩酸 適量
水酸化ナトリウム水溶液 適量
注射用蒸留水 適量
2.00 mL
製剤例6:錠剤(ガバペンチン)
実施例1と同様に以下の処方で錠剤を調製する。
処方 ガバペンチン 20 mg
乳糖 143.4 mg
馬鈴薯澱粉 30 mg
ヒドロキシプロピルセルロース 6 mg
ステアリン酸マグネシウム 0.6 mg
200 mg
実施例1と同様に以下の処方で錠剤を調製する。
処方 ガバペンチン 20 mg
乳糖 143.4 mg
馬鈴薯澱粉 30 mg
ヒドロキシプロピルセルロース 6 mg
ステアリン酸マグネシウム 0.6 mg
200 mg
製剤例7:錠剤(デュロキセチン)
実施例1と同様に以下の処方で錠剤を調製する。
処方 デュロキセチン 20 mg
乳糖 143.4 mg
馬鈴薯澱粉 30 mg
ヒドロキシプロピルセルロース 6 mg
ステアリン酸マグネシウム 0.6 mg
200 mg
実施例1と同様に以下の処方で錠剤を調製する。
処方 デュロキセチン 20 mg
乳糖 143.4 mg
馬鈴薯澱粉 30 mg
ヒドロキシプロピルセルロース 6 mg
ステアリン酸マグネシウム 0.6 mg
200 mg
製剤例8:錠剤(モルヒネ)
実施例1と同様に以下の処方で錠剤を調製する。
処方 モルヒネ 20 mg
乳糖 143.4 mg
馬鈴薯澱粉 30 mg
ヒドロキシプロピルセルロース 6 mg
ステアリン酸マグネシウム 0.6 mg
200 mg
実施例1と同様に以下の処方で錠剤を調製する。
処方 モルヒネ 20 mg
乳糖 143.4 mg
馬鈴薯澱粉 30 mg
ヒドロキシプロピルセルロース 6 mg
ステアリン酸マグネシウム 0.6 mg
200 mg
本発明により、例えば(a)ペンタジエナミド誘導体またはその薬学的に許容される塩と(b)鎮痛剤とを含有する医薬組成物等を提供することができる。
Claims (35)
- (a)式(I)

R2は置換もしくは非置換のシクロアルキル、置換もしくは非置換のシクロアルケニル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表し、
R3は水素原子を表すか、またはR4及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R4は置換もしくは非置換の低級アルキル、置換もしくは非置換のシクロアルキル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表すか、またはR3及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R5、R6及びR7は、同一または異なって水素原子またはメチルを表す)で表されるペンタジエナミド誘導体またはその薬学的に許容される塩と(b)鎮痛剤とを含有する医薬組成物。 - R5、R6及びR7が、それぞれ水素原子である請求項1記載の医薬組成物。
- R3及びR4が、隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成する請求項1または2記載の医薬組成物。
- R3及びR4が、隣接する窒素原子と一緒になって置換もしくは非置換のチオモルホリノを形成する請求項1または2記載の医薬組成物。
- R3及びR4が、隣接する窒素原子と一緒になって置換もしくは非置換のピペリジノまたは置換もしくは非置換のピペラジニルを形成する請求項1または2記載の医薬組成物。
- R3が、水素原子である請求項1または2記載の医薬組成物。
- R4が、置換もしくは非置換のアリールである請求項1、2または6記載の医薬組成物。
- R4が、置換もしくは非置換のジヒドロベンゾオキサジニルである請求項1、2または6記載の医薬組成物。
- R4が、置換もしくは非置換のテトラヒドロイソキノリルである請求項1、2または6記載の医薬組成物。
- R4が、置換もしくは非置換の1,2,3,4-テトラヒドロキノリルである請求項1、2または6記載の医薬組成物。
- R4が、置換もしくは非置換の芳香族複素環基である請求項1、2または6記載の医薬組成物。
- R4が、置換もしくは非置換のインダゾリルである請求項1、2または6記載の医薬組成物。
- R4が、置換もしくは非置換のイソキノリルである請求項1、2または6記載の医薬組成物。
- R1が、置換もしくは非置換のアリールである請求項1~13のいずれかに記載の医薬組成物。
- R1が、4-(トリフルオロメチル)フェニルである請求項1~13のいずれかに記載の医薬組成物。
- R2が、置換もしくは非置換のアリールである請求項1~15のいずれかに記載の医薬組成物。
- R2が、置換もしくは非置換のフェニルである請求項1~15のいずれかに記載の医薬組成物。
- R2が、置換もしくは非置換の低級アルコキシで置換されたフェニルである請求項1~15のいずれかに記載の医薬組成物。
- R2が、4-(トリフルオロメチル)フェニルである請求項1~15のいずれかに記載の医薬組成物。
- R2が、置換もしくは非置換の芳香族複素環基である請求項1~15のいずれかに記載の医薬組成物。
- R2が、置換もしくは非置換のピリジルまたは置換もしくは非置換のピリミジニルである請求項1~15のいずれかに記載の医薬組成物。
- 鎮痛剤が抗うつ剤である、請求項1~21のいずれかに記載の医薬組成物。
- 鎮痛剤がオピオイド鎮痛剤である、請求項1~21のいずれかに記載の医薬組成物。
- 鎮痛剤が抗癲癇剤である、請求項1~21のいずれかに記載の医薬組成物。
- 抗うつ剤がデュロキセチン、アミトリプチリン、フルボキサチン、フルボキサミン、フルオキセチン、ノルトリプチリン、ベンラファキシン、パロキセチン、レボキセチン、ミルナシプラン、イミプラミン、デシプラミン、クロミプラミン、トリミプラミン、アモキサピン、ロフェプラミン、ドレスピン、シタロプラム、エスシタロプラム、セルトラリン、マプロチリン、ミアンセリン、セチプチリン、及びネファゾドンからなる群から選ばれる抗うつ剤である請求項22記載の医薬組成物。
- オピオイド鎮痛剤がモルヒネ、フェンタニル、レミフェンタニル、スフェンタニル、ブプレノルフィン、ヒドロモルフィン、オキシモルフィン、ペンタゾシン、トラマドール、コデイン、ジヒドロコデイン、オキシコドン、メサドン、ヘロイン、ナルブフィン、レボルファノール、レバロルファン、メピリジン、ヒドロコドン、プロポキシフェン、及びブトルファノールからなる群から選ばれるオピオイド鎮痛剤である請求項23記載の医薬組成物。
- 抗癲癇剤がガバペンチン及びプレガバリンからなる群から選ばれる抗癲癇剤である請求項24記載の医薬組成物。
- (a)式(I)

R2は置換もしくは非置換のシクロアルキル、置換もしくは非置換のシクロアルケニル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表し、
R3は水素原子を表すか、またはR4及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R4は置換もしくは非置換の低級アルキル、置換もしくは非置換のシクロアルキル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表すか、またはR3及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R5、R6及びR7は、同一または異なって水素原子またはメチルを表す)で表されるペンタジエナミド誘導体またはその薬学的に許容される塩と(b)鎮痛剤とを有効成分として含有する疼痛の治療及び/または予防剤。 - (a)式(I)

R2は置換もしくは非置換のシクロアルキル、置換もしくは非置換のシクロアルケニル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表し、
R3は水素原子を表すか、またはR4及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R4は置換もしくは非置換の低級アルキル、置換もしくは非置換のシクロアルキル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表すか、またはR3及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R5、R6及びR7は、同一または異なって水素原子またはメチルを表す)で表されるペンタジエナミド誘導体またはその薬学的に許容される塩と、(b)鎮痛剤と、を有効成分とする、(a)と(b)を同時にまたは時間を置いて別々に投与するための疼痛の治療及び/または予防剤。 - (a)式(I)

R2は置換もしくは非置換のシクロアルキル、置換もしくは非置換のシクロアルケニル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表し、
R3は水素原子を表すか、またはR4及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R4は置換もしくは非置換の低級アルキル、置換もしくは非置換のシクロアルキル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表すか、またはR3及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R5、R6及びR7は、同一または異なって水素原子またはメチルを表す)で表されるペンタジエナミド誘導体またはその薬学的に許容される塩を含有する第1成分と、(b)鎮痛剤を含有する第2成分とを有することを特徴とするキット。 - (a)式(I)

R2は置換もしくは非置換のシクロアルキル、置換もしくは非置換のシクロアルケニル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表し、
R3は水素原子を表すか、またはR4及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R4は置換もしくは非置換の低級アルキル、置換もしくは非置換のシクロアルキル、置換もしくは非置換のアリール、置換もしくは非置換の芳香族複素環基または置換もしくは非置換の脂環式複素環基を表すか、またはR3及び隣接する窒素原子と一緒になって置換もしくは非置換の複素環基を形成し、
R5、R6及びR7は、同一または異なって水素原子またはメチルを表す)で表されるペンタジエナミド誘導体またはその薬学的に許容される塩を含有する第1成分と、(b)鎮痛剤を含有する第2成分とを有することを特徴とする疼痛の治療及び/または予防用キット。 - 疼痛が神経因性疼痛である、請求項28記載の疼痛の治療及び/または予防剤。
- 疼痛が神経因性疼痛である、請求項29記載の疼痛の治療及び/または予防剤。
- 疼痛が神経因性疼痛である、請求項31記載の疼痛の治療及び/または予防用キット。
- (a)請求項1~21のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩と、(b)鎮痛剤と、を同時にまたは時間を置いて別々に投与して疼痛の治療及び/または予防に使用するための、請求項1~21のいずれかに記載のペンタジエナミド誘導体またはその薬学的に許容される塩。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008-233398 | 2008-09-11 | ||
| JP2008233398 | 2008-09-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010029996A1 true WO2010029996A1 (ja) | 2010-03-18 |
Family
ID=42005246
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2009/065912 WO2010029996A1 (ja) | 2008-09-11 | 2009-09-11 | 医薬組成物 |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2010029996A1 (ja) |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005044786A1 (en) * | 2003-11-08 | 2005-05-19 | Bayer Healthcare Ag | Bicyclic amide, carbamate or urea derivatives as vanilloid receptor modulators |
| JP2006511535A (ja) * | 2002-12-13 | 2006-04-06 | ニューロジェン・コーポレーション | 疼痛治療用の併用療法 |
| JP2006515847A (ja) * | 2002-12-13 | 2006-06-08 | ニューロジェン・コーポレーション | カプサイシン受容体調節剤としてのカルボン酸、ホスフェート又はホスホネート置換キナゾリン−4−イルアミン類縁体 |
| JP2007523143A (ja) * | 2004-02-20 | 2007-08-16 | メルク シャープ エンド ドーム リミテッド | バニロイド−1受容体(vr1)の機能を調整する置換アミノヘテロ二環のプロドラッグ |
| WO2008007780A1 (fr) * | 2006-07-13 | 2008-01-17 | Kyowa Hakko Kirin Co., Ltd. | Dérivé du pentadiènamide |
| WO2008096755A1 (ja) * | 2007-02-07 | 2008-08-14 | Nippon Suisan Kaisha, Ltd. | バニロイド受容体(vr1)阻害剤及びその用途 |
-
2009
- 2009-09-11 WO PCT/JP2009/065912 patent/WO2010029996A1/ja active Application Filing
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006511535A (ja) * | 2002-12-13 | 2006-04-06 | ニューロジェン・コーポレーション | 疼痛治療用の併用療法 |
| JP2006515847A (ja) * | 2002-12-13 | 2006-06-08 | ニューロジェン・コーポレーション | カプサイシン受容体調節剤としてのカルボン酸、ホスフェート又はホスホネート置換キナゾリン−4−イルアミン類縁体 |
| WO2005044786A1 (en) * | 2003-11-08 | 2005-05-19 | Bayer Healthcare Ag | Bicyclic amide, carbamate or urea derivatives as vanilloid receptor modulators |
| JP2007523143A (ja) * | 2004-02-20 | 2007-08-16 | メルク シャープ エンド ドーム リミテッド | バニロイド−1受容体(vr1)の機能を調整する置換アミノヘテロ二環のプロドラッグ |
| WO2008007780A1 (fr) * | 2006-07-13 | 2008-01-17 | Kyowa Hakko Kirin Co., Ltd. | Dérivé du pentadiènamide |
| WO2008096755A1 (ja) * | 2007-02-07 | 2008-08-14 | Nippon Suisan Kaisha, Ltd. | バニロイド受容体(vr1)阻害剤及びその用途 |
Non-Patent Citations (4)
| Title |
|---|
| ACS, GEZA ET AL.: "Trifluoperazine Modulates [3H] Resiniferatoxin Binding by Human and Rat Vanilloid (Capsaicin) Receptors and Affects 45Ca Uptake by Adult Rat Dorsal Root Ganglion Neurones", JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS, vol. 274, no. 3, 1995, pages 1090 - 1098 * |
| JU MIZUNO ET AL.: "Q&A <Shitsumon> Mansei Totsu ni Taisuru Kakushu Koutsuyaku no Chiryo ni Tsuite", PAIN CLINIC, vol. 26, no. 4, April 2005 (2005-04-01), pages 576 - 577 * |
| VETTER, IRINA ET AL.: "Rapid, Opioid-sensitive Mechanisms Involved in Transient Receptor Potential Vaniloid 1 Sensitization", JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 283, no. 28, 11 July 2008 (2008-07-11), pages 19540 - 19550 * |
| WALKER, KATHARINE M ET AL.: "The VR1 antagonist Capsazepine reverses mechanical hyperalgesia in models of inflammatory and neuropathic pain", THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS, vol. 304, no. 1, 2003, pages 56 - 62 * |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2425041C2 (ru) | Кристаллические и другие формы солей, образованных из молочной кислоты и 4-амино-5-фтор-3-[6-(4-метилпиперазин-1-ил)-1н-бензимидазол-2-ил]-1н-хинолин-2-она | |
| KR101536023B1 (ko) | 조합된 sert, 5-ht3 및 5-ht1a 활성을 가진 화합물의 치료 용도 | |
| JP5371790B2 (ja) | 睡眠および認知に関するうつ病における残存症状の治療のための組み合わされたセロトニン再取り込み、5−ht3および5−ht1a活性を有する化合物としての1−[2−(2,4−ジメチルフェニルスルファニル)フェニル]ピペラジン | |
| JPWO2006088193A1 (ja) | 抗腫瘍剤 | |
| JP2020516591A (ja) | Morアゴニストおよびkorアゴニストを含有する医薬組成物、およびその用途 | |
| MXPA01011534A (es) | Metodos de preparar y utilizar n-desmetilzopiclona. | |
| US20200148681A1 (en) | Crystalline spirocyclic compound, a dosage form containing, a method for using in treatment of disease, and a method for recrystallizing | |
| JP2003535128A (ja) | 胃食道逆流性疾患の治療のための材料 | |
| CN102753540A (zh) | α4β2神经元烟碱型乙酰胆碱受体的配体 | |
| TW201305148A (zh) | 5-甲基-1-(萘-2-基)-1h-吡唑衍生物類 | |
| EP3661510A1 (en) | Methods of treating behavior alterations | |
| US20230382870A1 (en) | N-aromatic amide compounds, preparation methods and uses thereof | |
| JP2023531709A (ja) | セレブロン結合化合物、その組成物及びそれによる治療方法 | |
| KR20050114641A (ko) | 2,6-이치환된 스티릴을 갖는 질소 함유 헤테로환 유도체 | |
| KR20120099212A (ko) | 트라마돌 및 콕시브의 공결정 | |
| CA3119313A1 (en) | Pharmaceutical composition comprising histone deacetylase 6 inhibitors | |
| TW200922584A (en) | Organic compounds | |
| JPWO2006064780A1 (ja) | 便秘治療剤 | |
| EP2631233A1 (en) | Antagonist for mutated androgen receptor | |
| WO2020072675A1 (en) | Beta-carbolines as positive allosteric modulators of the human serotonin receptor 2c (5-ht2c) | |
| WO2010029996A1 (ja) | 医薬組成物 | |
| US20080242657A1 (en) | Treatment of Tremor with Histamine H3 Inverse Agonists or Hist Amine H3 Antagonists | |
| WO2010029995A1 (ja) | 疼痛治療剤 | |
| RU2615986C1 (ru) | Замещенные метил (2-{ 4-[3-(3-метансульфониламино-2-фтор-5-хлор-фенил)-1Н-пиразол-4-ил]пиримидин-2-иламино} -этил)карбаматы, способ их получения и применения | |
| WO2016039660A1 (ru) | ПРОИЗВОДНЫЕ 7-ФТОРО-8-ХЛОРО-5Н-ДИБЕНЗО [b, e] [1, 4] ДИАЗЕПИНА И ИХ ПРИМЕНЕНИЕ |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09813142 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09813142 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |