WO2009103880A2 - Catalyseur comprenant au moins une zéolithe particuliere et au moins une silice-alumine et procédé d'hydrocraquage de charges hydrocarbonées utilisant un tel catalyseur - Google Patents
Catalyseur comprenant au moins une zéolithe particuliere et au moins une silice-alumine et procédé d'hydrocraquage de charges hydrocarbonées utilisant un tel catalyseur Download PDFInfo
- Publication number
- WO2009103880A2 WO2009103880A2 PCT/FR2008/001721 FR2008001721W WO2009103880A2 WO 2009103880 A2 WO2009103880 A2 WO 2009103880A2 FR 2008001721 W FR2008001721 W FR 2008001721W WO 2009103880 A2 WO2009103880 A2 WO 2009103880A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- catalyst
- advantageously
- zeolite
- alumina
- silica
- Prior art date
Links
- 239000003054 catalyst Substances 0.000 title claims abstract description 177
- 239000010457 zeolite Substances 0.000 title claims abstract description 144
- HNPSIPDUKPIQMN-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical compound O=[Si]=O.O=[Al]O[Al]=O HNPSIPDUKPIQMN-UHFFFAOYSA-N 0.000 title claims abstract description 96
- 229910021536 Zeolite Inorganic materials 0.000 title claims abstract description 94
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 title claims abstract description 77
- 238000004517 catalytic hydrocracking Methods 0.000 title claims abstract description 73
- 238000000034 method Methods 0.000 title claims abstract description 71
- 229930195733 hydrocarbon Natural products 0.000 title abstract description 11
- 150000002430 hydrocarbons Chemical class 0.000 title abstract description 11
- 239000004215 Carbon black (E152) Substances 0.000 title abstract description 10
- 229910052751 metal Inorganic materials 0.000 claims abstract description 54
- 239000002184 metal Substances 0.000 claims abstract description 54
- 150000002739 metals Chemical class 0.000 claims abstract description 20
- 239000000203 mixture Substances 0.000 claims description 46
- 230000008569 process Effects 0.000 claims description 39
- 238000006243 chemical reaction Methods 0.000 claims description 33
- -1 VIB metals Chemical class 0.000 claims description 30
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 28
- 239000011148 porous material Substances 0.000 claims description 26
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 25
- 239000001257 hydrogen Substances 0.000 claims description 18
- 229910052739 hydrogen Inorganic materials 0.000 claims description 18
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 17
- 239000003921 oil Substances 0.000 claims description 15
- 238000002459 porosimetry Methods 0.000 claims description 15
- 229910052757 nitrogen Inorganic materials 0.000 claims description 14
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 claims description 13
- 229910052753 mercury Inorganic materials 0.000 claims description 13
- 239000000377 silicon dioxide Substances 0.000 claims description 12
- 230000003197 catalytic effect Effects 0.000 claims description 11
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 claims description 10
- 239000000945 filler Substances 0.000 claims description 10
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 claims description 10
- 239000007789 gas Substances 0.000 claims description 9
- VILCJCGEZXAXTO-UHFFFAOYSA-N 2,2,2-tetramine Chemical compound NCCNCCNCCN VILCJCGEZXAXTO-UHFFFAOYSA-N 0.000 claims description 8
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 8
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 8
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 claims description 7
- 229910052750 molybdenum Inorganic materials 0.000 claims description 7
- 239000011733 molybdenum Substances 0.000 claims description 7
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 claims description 7
- 229910017052 cobalt Inorganic materials 0.000 claims description 5
- 239000010941 cobalt Substances 0.000 claims description 5
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 claims description 5
- 229910052759 nickel Inorganic materials 0.000 claims description 5
- 229910052697 platinum Inorganic materials 0.000 claims description 5
- 229910052721 tungsten Inorganic materials 0.000 claims description 5
- 239000010937 tungsten Substances 0.000 claims description 5
- 229910052742 iron Inorganic materials 0.000 claims description 4
- 229910052763 palladium Inorganic materials 0.000 claims description 4
- 230000007704 transition Effects 0.000 claims description 3
- 238000002441 X-ray diffraction Methods 0.000 claims description 2
- 239000000295 fuel oil Substances 0.000 claims description 2
- 239000001993 wax Substances 0.000 claims description 2
- 239000002699 waste material Substances 0.000 claims 1
- 239000000758 substrate Substances 0.000 abstract 1
- 239000000243 solution Substances 0.000 description 24
- 239000002253 acid Substances 0.000 description 22
- 229910052796 boron Inorganic materials 0.000 description 19
- 238000005470 impregnation Methods 0.000 description 19
- 229910052710 silicon Inorganic materials 0.000 description 19
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 18
- 150000001875 compounds Chemical class 0.000 description 18
- 238000002360 preparation method Methods 0.000 description 18
- 239000010703 silicon Substances 0.000 description 17
- 239000011159 matrix material Substances 0.000 description 16
- 229910052698 phosphorus Inorganic materials 0.000 description 15
- 239000002243 precursor Substances 0.000 description 15
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 14
- 239000011574 phosphorus Substances 0.000 description 14
- 238000011282 treatment Methods 0.000 description 14
- 238000001354 calcination Methods 0.000 description 13
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 13
- 238000001125 extrusion Methods 0.000 description 12
- 150000003839 salts Chemical class 0.000 description 12
- 229910052717 sulfur Inorganic materials 0.000 description 11
- 239000000725 suspension Substances 0.000 description 11
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 10
- 239000000843 powder Substances 0.000 description 10
- 239000000047 product Substances 0.000 description 10
- 238000007493 shaping process Methods 0.000 description 10
- 239000011593 sulfur Substances 0.000 description 10
- 229910052782 aluminium Inorganic materials 0.000 description 9
- 239000006185 dispersion Substances 0.000 description 9
- FAHBNUUHRFUEAI-UHFFFAOYSA-M hydroxidooxidoaluminium Chemical compound O[Al]=O FAHBNUUHRFUEAI-UHFFFAOYSA-M 0.000 description 9
- 239000007787 solid Substances 0.000 description 9
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 8
- 230000002378 acidificating effect Effects 0.000 description 8
- 229910001593 boehmite Inorganic materials 0.000 description 7
- 239000000499 gel Substances 0.000 description 7
- 238000002156 mixing Methods 0.000 description 7
- 229910000510 noble metal Inorganic materials 0.000 description 7
- 238000000926 separation method Methods 0.000 description 7
- 239000011734 sodium Substances 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 6
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 6
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 6
- QGZKDVFQNNGYKY-UHFFFAOYSA-O ammonium group Chemical group [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 6
- 239000007864 aqueous solution Substances 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 229910017604 nitric acid Inorganic materials 0.000 description 6
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 5
- 238000010335 hydrothermal treatment Methods 0.000 description 5
- 239000012535 impurity Substances 0.000 description 5
- 238000004519 manufacturing process Methods 0.000 description 5
- 239000012071 phase Substances 0.000 description 5
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 5
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 4
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 4
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 4
- VXAUWWUXCIMFIM-UHFFFAOYSA-M aluminum;oxygen(2-);hydroxide Chemical compound [OH-].[O-2].[Al+3] VXAUWWUXCIMFIM-UHFFFAOYSA-M 0.000 description 4
- 150000003863 ammonium salts Chemical class 0.000 description 4
- 239000007900 aqueous suspension Substances 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 4
- 229960002645 boric acid Drugs 0.000 description 4
- 235000010338 boric acid Nutrition 0.000 description 4
- 125000002091 cationic group Chemical group 0.000 description 4
- 238000009826 distribution Methods 0.000 description 4
- 238000001035 drying Methods 0.000 description 4
- 239000011737 fluorine Substances 0.000 description 4
- 229910052731 fluorine Inorganic materials 0.000 description 4
- 229910021472 group 8 element Inorganic materials 0.000 description 4
- 150000002894 organic compounds Chemical class 0.000 description 4
- 239000003208 petroleum Substances 0.000 description 4
- 235000011007 phosphoric acid Nutrition 0.000 description 4
- 238000004064 recycling Methods 0.000 description 4
- 150000003377 silicon compounds Chemical class 0.000 description 4
- DDFHBQSCUXNBSA-UHFFFAOYSA-N 5-(5-carboxythiophen-2-yl)thiophene-2-carboxylic acid Chemical compound S1C(C(=O)O)=CC=C1C1=CC=C(C(O)=O)S1 DDFHBQSCUXNBSA-UHFFFAOYSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- MXRIRQGCELJRSN-UHFFFAOYSA-N O.O.O.[Al] Chemical compound O.O.O.[Al] MXRIRQGCELJRSN-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- MHCAFGMQMCSRGH-UHFFFAOYSA-N aluminum;hydrate Chemical compound O.[Al] MHCAFGMQMCSRGH-UHFFFAOYSA-N 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- QGAVSDVURUSLQK-UHFFFAOYSA-N ammonium heptamolybdate Chemical compound N.N.N.N.N.N.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.[Mo].[Mo].[Mo].[Mo].[Mo].[Mo].[Mo] QGAVSDVURUSLQK-UHFFFAOYSA-N 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 239000004327 boric acid Substances 0.000 description 3
- 239000000084 colloidal system Substances 0.000 description 3
- 239000000470 constituent Substances 0.000 description 3
- 239000000446 fuel Substances 0.000 description 3
- 229910001679 gibbsite Inorganic materials 0.000 description 3
- 238000000265 homogenisation Methods 0.000 description 3
- 229910052500 inorganic mineral Inorganic materials 0.000 description 3
- 238000005342 ion exchange Methods 0.000 description 3
- 238000004898 kneading Methods 0.000 description 3
- 239000011707 mineral Substances 0.000 description 3
- 235000010755 mineral Nutrition 0.000 description 3
- 239000002808 molecular sieve Substances 0.000 description 3
- DDTIGTPWGISMKL-UHFFFAOYSA-N molybdenum nickel Chemical compound [Ni].[Mo] DDTIGTPWGISMKL-UHFFFAOYSA-N 0.000 description 3
- 235000012239 silicon dioxide Nutrition 0.000 description 3
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 3
- 229910018072 Al 2 O 3 Inorganic materials 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- BOTDANWDWHJENH-UHFFFAOYSA-N Tetraethyl orthosilicate Chemical compound CCO[Si](OCC)(OCC)OCC BOTDANWDWHJENH-UHFFFAOYSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 229910052910 alkali metal silicate Inorganic materials 0.000 description 2
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical group [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 2
- XFBXDGLHUSUNMG-UHFFFAOYSA-N alumane;hydrate Chemical class O.[AlH3] XFBXDGLHUSUNMG-UHFFFAOYSA-N 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- ANBBXQWFNXMHLD-UHFFFAOYSA-N aluminum;sodium;oxygen(2-) Chemical compound [O-2].[O-2].[Na+].[Al+3] ANBBXQWFNXMHLD-UHFFFAOYSA-N 0.000 description 2
- APUPEJJSWDHEBO-UHFFFAOYSA-P ammonium molybdate Chemical compound [NH4+].[NH4+].[O-][Mo]([O-])(=O)=O APUPEJJSWDHEBO-UHFFFAOYSA-P 0.000 description 2
- 239000011609 ammonium molybdate Substances 0.000 description 2
- 229940010552 ammonium molybdate Drugs 0.000 description 2
- 235000018660 ammonium molybdate Nutrition 0.000 description 2
- 125000000129 anionic group Chemical group 0.000 description 2
- 229940111121 antirheumatic drug quinolines Drugs 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- 150000001638 boron Chemical class 0.000 description 2
- 238000004523 catalytic cracking Methods 0.000 description 2
- 150000001805 chlorine compounds Chemical class 0.000 description 2
- 238000000975 co-precipitation Methods 0.000 description 2
- 238000005345 coagulation Methods 0.000 description 2
- 230000015271 coagulation Effects 0.000 description 2
- WHDPTDWLEKQKKX-UHFFFAOYSA-N cobalt molybdenum Chemical compound [Co].[Co].[Mo] WHDPTDWLEKQKKX-UHFFFAOYSA-N 0.000 description 2
- 239000012084 conversion product Substances 0.000 description 2
- BUACSMWVFUNQET-UHFFFAOYSA-H dialuminum;trisulfate;hydrate Chemical compound O.[Al+3].[Al+3].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O BUACSMWVFUNQET-UHFFFAOYSA-H 0.000 description 2
- VLXBWPOEOIIREY-UHFFFAOYSA-N dimethyl diselenide Natural products C[Se][Se]C VLXBWPOEOIIREY-UHFFFAOYSA-N 0.000 description 2
- WQOXQRCZOLPYPM-UHFFFAOYSA-N dimethyl disulfide Chemical compound CSSC WQOXQRCZOLPYPM-UHFFFAOYSA-N 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 229910001657 ferrierite group Inorganic materials 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 150000004679 hydroxides Chemical class 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 239000003350 kerosene Substances 0.000 description 2
- 239000007791 liquid phase Substances 0.000 description 2
- 150000002736 metal compounds Chemical class 0.000 description 2
- MOWMLACGTDMJRV-UHFFFAOYSA-N nickel tungsten Chemical compound [Ni].[W] MOWMLACGTDMJRV-UHFFFAOYSA-N 0.000 description 2
- 150000002823 nitrates Chemical class 0.000 description 2
- 230000008520 organization Effects 0.000 description 2
- 238000005453 pelletization Methods 0.000 description 2
- 230000000737 periodic effect Effects 0.000 description 2
- 229920001296 polysiloxane Polymers 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 150000003141 primary amines Chemical class 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 150000003248 quinolines Chemical class 0.000 description 2
- 239000011347 resin Substances 0.000 description 2
- 229920005989 resin Polymers 0.000 description 2
- 150000003335 secondary amines Chemical class 0.000 description 2
- 229910001388 sodium aluminate Inorganic materials 0.000 description 2
- NTHWMYGWWRZVTN-UHFFFAOYSA-N sodium silicate Chemical compound [Na+].[Na+].[O-][Si]([O-])=O NTHWMYGWWRZVTN-UHFFFAOYSA-N 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 230000003068 static effect Effects 0.000 description 2
- 238000005987 sulfurization reaction Methods 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- 239000012808 vapor phase Substances 0.000 description 2
- 238000004876 x-ray fluorescence Methods 0.000 description 2
- BNGXYYYYKUGPPF-UHFFFAOYSA-M (3-methylphenyl)methyl-triphenylphosphanium;chloride Chemical compound [Cl-].CC1=CC=CC(C[P+](C=2C=CC=CC=2)(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 BNGXYYYYKUGPPF-UHFFFAOYSA-M 0.000 description 1
- ZZBAGJPKGRJIJH-UHFFFAOYSA-N 7h-purine-2-carbaldehyde Chemical compound O=CC1=NC=C2NC=NC2=N1 ZZBAGJPKGRJIJH-UHFFFAOYSA-N 0.000 description 1
- 239000004254 Ammonium phosphate Substances 0.000 description 1
- 238000004438 BET method Methods 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- RWSOTUBLDIXVET-UHFFFAOYSA-N Dihydrogen sulfide Chemical compound S RWSOTUBLDIXVET-UHFFFAOYSA-N 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- GYHNNYVSQQEPJS-UHFFFAOYSA-N Gallium Chemical compound [Ga] GYHNNYVSQQEPJS-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- YZCKVEUIGOORGS-UHFFFAOYSA-N Hydrogen atom Chemical group [H] YZCKVEUIGOORGS-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- OTRAYOBSWCVTIN-UHFFFAOYSA-N OB(O)O.OB(O)O.OB(O)O.OB(O)O.OB(O)O.N.N.N.N.N.N.N.N.N.N.N.N.N.N.N Chemical compound OB(O)O.OB(O)O.OB(O)O.OB(O)O.OB(O)O.N.N.N.N.N.N.N.N.N.N.N.N.N.N.N OTRAYOBSWCVTIN-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 1
- 229910004298 SiO 2 Inorganic materials 0.000 description 1
- 239000004115 Sodium Silicate Substances 0.000 description 1
- FKNQFGJONOIPTF-UHFFFAOYSA-N Sodium cation Chemical compound [Na+] FKNQFGJONOIPTF-UHFFFAOYSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- QZYDAIMOJUSSFT-UHFFFAOYSA-N [Co].[Ni].[Mo] Chemical compound [Co].[Ni].[Mo] QZYDAIMOJUSSFT-UHFFFAOYSA-N 0.000 description 1
- PCBMYXLJUKBODW-UHFFFAOYSA-N [Ru].ClOCl Chemical compound [Ru].ClOCl PCBMYXLJUKBODW-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 239000003929 acidic solution Substances 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 238000005054 agglomeration Methods 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- RREGISFBPQOLTM-UHFFFAOYSA-N alumane;trihydrate Chemical compound O.O.O.[AlH3] RREGISFBPQOLTM-UHFFFAOYSA-N 0.000 description 1
- 150000004645 aluminates Chemical class 0.000 description 1
- 229910000323 aluminium silicate Inorganic materials 0.000 description 1
- ZRIUUUJAJJNDSS-UHFFFAOYSA-N ammonium phosphates Chemical class [NH4+].[NH4+].[NH4+].[O-]P([O-])([O-])=O ZRIUUUJAJJNDSS-UHFFFAOYSA-N 0.000 description 1
- 235000019289 ammonium phosphates Nutrition 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 239000003637 basic solution Substances 0.000 description 1
- 229910001680 bayerite Inorganic materials 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 230000001588 bifunctional effect Effects 0.000 description 1
- 230000002902 bimodal effect Effects 0.000 description 1
- 229920001222 biopolymer Polymers 0.000 description 1
- 229910052810 boron oxide Inorganic materials 0.000 description 1
- 150000001649 bromium compounds Chemical class 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 239000006229 carbon black Substances 0.000 description 1
- 235000019241 carbon black Nutrition 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 238000005341 cation exchange Methods 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- JPNWDVUTVSTKMV-UHFFFAOYSA-N cobalt tungsten Chemical compound [Co].[W] JPNWDVUTVSTKMV-UHFFFAOYSA-N 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910052593 corundum Inorganic materials 0.000 description 1
- 239000010431 corundum Substances 0.000 description 1
- 238000005336 cracking Methods 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- JKWMSGQKBLHBQQ-UHFFFAOYSA-N diboron trioxide Chemical compound O=BOB=O JKWMSGQKBLHBQQ-UHFFFAOYSA-N 0.000 description 1
- 229910001882 dioxygen Inorganic materials 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 239000002019 doping agent Substances 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000011066 ex-situ storage Methods 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000008394 flocculating agent Substances 0.000 description 1
- 150000002222 fluorine compounds Chemical class 0.000 description 1
- 229910052733 gallium Inorganic materials 0.000 description 1
- 239000003502 gasoline Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 238000005469 granulation Methods 0.000 description 1
- 230000003179 granulation Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 230000036571 hydration Effects 0.000 description 1
- 238000006703 hydration reaction Methods 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 238000007654 immersion Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229910052741 iridium Inorganic materials 0.000 description 1
- GKOZUEZYRPOHIO-UHFFFAOYSA-N iridium atom Chemical compound [Ir] GKOZUEZYRPOHIO-UHFFFAOYSA-N 0.000 description 1
- KWUUWVQMAVOYKS-UHFFFAOYSA-N iron molybdenum Chemical compound [Fe].[Fe][Mo][Mo] KWUUWVQMAVOYKS-UHFFFAOYSA-N 0.000 description 1
- GXBKELQWVXYOPN-UHFFFAOYSA-N iron tungsten Chemical compound [W][Fe][W] GXBKELQWVXYOPN-UHFFFAOYSA-N 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 230000001050 lubricating effect Effects 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 229910052976 metal sulfide Inorganic materials 0.000 description 1
- 238000003801 milling Methods 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- KBJMLQFLOWQJNF-UHFFFAOYSA-N nickel(ii) nitrate Chemical compound [Ni+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O KBJMLQFLOWQJNF-UHFFFAOYSA-N 0.000 description 1
- 229910017464 nitrogen compound Inorganic materials 0.000 description 1
- 150000002830 nitrogen compounds Chemical class 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002897 organic nitrogen compounds Chemical class 0.000 description 1
- 229910052762 osmium Inorganic materials 0.000 description 1
- SYQBFIAQOQZEGI-UHFFFAOYSA-N osmium atom Chemical compound [Os] SYQBFIAQOQZEGI-UHFFFAOYSA-N 0.000 description 1
- 239000001301 oxygen Chemical group 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 238000001935 peptisation Methods 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- DHRLEVQXOMLTIM-UHFFFAOYSA-N phosphoric acid;trioxomolybdenum Chemical compound O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.OP(O)(O)=O DHRLEVQXOMLTIM-UHFFFAOYSA-N 0.000 description 1
- IYDGMDWEHDFVQI-UHFFFAOYSA-N phosphoric acid;trioxotungsten Chemical compound O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.O=[W](=O)=O.OP(O)(O)=O IYDGMDWEHDFVQI-UHFFFAOYSA-N 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- KVOIJEARBNBHHP-UHFFFAOYSA-N potassium;oxido(oxo)alumane Chemical compound [K+].[O-][Al]=O KVOIJEARBNBHHP-UHFFFAOYSA-N 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 239000012429 reaction media Substances 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000007670 refining Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- 239000010948 rhodium Substances 0.000 description 1
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 1
- 229910052707 ruthenium Inorganic materials 0.000 description 1
- 238000010008 shearing Methods 0.000 description 1
- CGFYHILWFSGVJS-UHFFFAOYSA-N silicic acid;trioxotungsten Chemical compound O[Si](O)(O)O.O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1.O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1.O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1.O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1 CGFYHILWFSGVJS-UHFFFAOYSA-N 0.000 description 1
- ABTOQLMXBSRXSM-UHFFFAOYSA-N silicon tetrafluoride Chemical compound F[Si](F)(F)F ABTOQLMXBSRXSM-UHFFFAOYSA-N 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229910001415 sodium ion Inorganic materials 0.000 description 1
- 235000019795 sodium metasilicate Nutrition 0.000 description 1
- 229910052911 sodium silicate Inorganic materials 0.000 description 1
- 238000001694 spray drying Methods 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000003784 tall oil Substances 0.000 description 1
- 238000007669 thermal treatment Methods 0.000 description 1
- 125000000101 thioether group Chemical group 0.000 description 1
- 150000003568 thioethers Chemical group 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000004627 transmission electron microscopy Methods 0.000 description 1
- 238000010977 unit operation Methods 0.000 description 1
- 239000010913 used oil Substances 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites
- B01J29/06—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof
- B01J29/80—Mixtures of different zeolites
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites
- B01J29/06—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof
- B01J29/70—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of types characterised by their specific structure not provided for in groups B01J29/08 - B01J29/65
- B01J29/72—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of types characterised by their specific structure not provided for in groups B01J29/08 - B01J29/65 containing iron group metals, noble metals or copper
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites
- B01J29/06—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof
- B01J29/70—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of types characterised by their specific structure not provided for in groups B01J29/08 - B01J29/65
- B01J29/72—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of types characterised by their specific structure not provided for in groups B01J29/08 - B01J29/65 containing iron group metals, noble metals or copper
- B01J29/74—Noble metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites
- B01J29/06—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof
- B01J29/70—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of types characterised by their specific structure not provided for in groups B01J29/08 - B01J29/65
- B01J29/72—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of types characterised by their specific structure not provided for in groups B01J29/08 - B01J29/65 containing iron group metals, noble metals or copper
- B01J29/76—Iron group metals or copper
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites
- B01J29/06—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof
- B01J29/70—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of types characterised by their specific structure not provided for in groups B01J29/08 - B01J29/65
- B01J29/78—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of types characterised by their specific structure not provided for in groups B01J29/08 - B01J29/65 containing arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/0009—Use of binding agents; Moulding; Pressing; Powdering; Granulating; Addition of materials ameliorating the mechanical properties of the product catalyst
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G47/00—Cracking of hydrocarbon oils, in the presence of hydrogen or hydrogen- generating compounds, to obtain lower boiling fractions
- C10G47/02—Cracking of hydrocarbon oils, in the presence of hydrogen or hydrogen- generating compounds, to obtain lower boiling fractions characterised by the catalyst used
- C10G47/10—Cracking of hydrocarbon oils, in the presence of hydrogen or hydrogen- generating compounds, to obtain lower boiling fractions characterised by the catalyst used with catalysts deposited on a carrier
- C10G47/12—Inorganic carriers
- C10G47/16—Crystalline alumino-silicate carriers
- C10G47/20—Crystalline alumino-silicate carriers the catalyst containing other metals or compounds thereof
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/12—Silica and alumina
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2229/00—Aspects of molecular sieve catalysts not covered by B01J29/00
- B01J2229/10—After treatment, characterised by the effect to be obtained
- B01J2229/20—After treatment, characterised by the effect to be obtained to introduce other elements in the catalyst composition comprising the molecular sieve, but not specially in or on the molecular sieve itself
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2229/00—Aspects of molecular sieve catalysts not covered by B01J29/00
- B01J2229/30—After treatment, characterised by the means used
- B01J2229/42—Addition of matrix or binder particles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites
- B01J29/06—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof
- B01J29/65—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of the ferrierite type, e.g. types ZSM-21, ZSM-35 or ZSM-38, as exemplified by patent documents US4046859, US4016245 and US4046859, respectively
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites
- B01J29/06—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof
- B01J29/70—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of types characterised by their specific structure not provided for in groups B01J29/08 - B01J29/65
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites
- B01J29/06—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof
- B01J29/70—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of types characterised by their specific structure not provided for in groups B01J29/08 - B01J29/65
- B01J29/7042—TON-type, e.g. Theta-1, ISI-1, KZ-2, NU-10 or ZSM-22
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites
- B01J29/06—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof
- B01J29/70—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of types characterised by their specific structure not provided for in groups B01J29/08 - B01J29/65
- B01J29/7046—MTT-type, e.g. ZSM-23, KZ-1, ISI-4 or EU-13
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/60—Catalysts, in general, characterised by their form or physical properties characterised by their surface properties or porosity
Definitions
- the present invention relates to a catalyst comprising at least one hydrodehydrogenating metal selected from the group consisting of Group VIB metals and Group VIII metals and a carrier comprising at least one silica-alumina, and at least one COK-7 zeolite alone, or in admixture with at least one zeolite ZBM-30.
- the invention also relates to a process for hydroconversion of hydrocarbon feeds using said catalyst. More particularly hydroconversion is understood to mean the hydrocracking of hydrocarbon feeds. The invention makes it particularly possible to obtain improved yields of middle distillates.
- Hydrocracking of heavy oil cuts is a very important process of refining which makes it possible to produce, from excessively heavy and unremarkable heavy loads, the lighter fractions such as gasolines, fuels and light gas oils that the refiner seeks in order to adapt his production to the structure of the request.
- Certain hydrocracking processes also make it possible to obtain a highly purified residue that can constitute excellent bases for oils.
- the advantage of catalytic hydrocracking is to provide middle distillates, jet fuels and gas oils, of very good quality.
- the gasoline produced has a much lower octane number than that resulting from catalytic cracking.
- One of the great interests of hydrocracking is to present a great flexibility at various levels: flexibility in the catalysts used, which brings a flexibility of the charges to be treated and the level of the products obtained.
- flexibility in the catalysts used which brings a flexibility of the charges to be treated and the level of the products obtained.
- One of the parameters that can be controlled is the acidity of the catalyst support.
- the catalysts used in hydrocracking are all of the bifunctional type associating an acid function with a hydrogenating function.
- the acid function is provided by supports with large surface areas (generally 150 to 800 m 2 / g) with superficial acidity, such as halogenated aluminas (chlorinated or fluorinated in particular), silica-aluminas and zeolites.
- the hydrogenating function is provided either by one or more metals of group VIII of the periodic table of elements, or by a combination of at least one metal of group VIB of the periodic table and at least one metal of group VIII.
- catalysts for catalytic hydrocracking are, for the most part, constituted by weakly acidic supports, such as amorphous silica-aluminas by example. These systems are more particularly used to produce middle distillates of very good quality.
- hydrocracking market catalysts are based on silica-alumina associated with either a Group VIII metal or, preferably, when the heteroatomic content of the feedstock to be treated exceeds 0.5 wt. combination of Group VIB and VIII metal sulphides. These systems have a very good selectivity in middle distillates, and the products formed are of good quality. These catalysts, for the less acidic of them, can also produce lubricating bases. The disadvantage of all these catalytic systems based on amorphous support is, as we said, their low activity.
- Catalysts comprising zeolite Y of the FAU structural type, or beta type catalysts in turn have a higher catalytic activity than those of silica-aluminas, but have selectivities of light products, unwanted, higher.
- the zeolites used for the preparation of hydrocracking catalysts are characterized by several quantities such as their Si / Al molar ratio. framework, their crystalline parameter, their porous distribution, their specific surface, their capacity of recovery in sodium ion, or their capacity of adsorption of water vapor.
- EP 0 544 766 claims a hydrocracking process for the production of middle distillates using a large-pore hydrocracking catalyst and a catalyst comprising an intermediate pore aluminophosphate molecular sieve to improve the cold properties of the distillates means.
- the hydrocracking catalyst has a hydrodehydrogenating activity and a crunchy support selected from the group formed by silica-alumina, silica-alumina-titanium, clays, zeolitic molecular sieves such as faujasites, zeolites X, Y, taken alone or in situ. mixture, the support being preferably non-zeolitic.
- the intermediate-pore aluminophosphate molecular sieve is selected from SAPO-11, SAPO-31 and SAPO-41 silicoaluminophosphates.
- a catalyst comprising at least one hydro-dehydrogenating metal selected from the group consisting of Group VIB metals and Group VIII metals and a carrier comprising at least one silica-alumina, and at least one COK-7 zeolite alone, or in admixture with at least one ZBM-30 zeolite, leads to unexpected catalytic performance in terms of hydrocracking hydrocarbon feedstocks and more particularly makes it possible to achieve yields of middle distillates (kerosene and gas oil) significantly improved over the catalysts known in the prior art and / or improved product qualities.
- middle distillates kerosene and gas oil
- the invention thus relates to such a catalyst as well as a process for hydrocracking hydrocarbon feeds using said catalyst.
- the subject of the invention is a catalyst comprising at least one hydro-dehydrogenating metal selected from the group consisting of Group VIB metals and Group VIII metals and a support comprising at least one silica-alumina, and at least one a COK-7 zeolite alone, or in admixture with at least one zeolite ZBM-30.
- the invention also relates to a hydrocracking process using said catalyst.
- the support of the catalyst according to the present invention comprises at least one COK-7 zeolite alone, or in admixture with at least one zeolite ZBM-30.
- Zeolite ZBM-30 is described in patent EP-A-46 504, and zeolite COK-7 is described in patent applications EP 1 702 888 A1 or FR 2 882 744 A1.
- the COK-7 zeolite used in the catalyst according to the present invention is synthesized in the presence of the organic triethylenetetramine structurant.
- the ZBM-30 zeolite used in the catalyst according to the present invention is synthesized in the presence of the organic triethylenetetramine structurant.
- the support of the catalyst according to the present invention comprises at least one COK-7 zeolite, synthesized in the presence of the organic template triethylenetetramine, in admixture with at least one zeolite ZBM-30 synthesized in the presence of the organic template triethylenetetramine.
- the proportion of each of the zeolites in the mixture of the two zeolites is advantageously between 20 and 80 % by weight relative to the total weight of the mixture of the two zeolites, and preferably the proportion of each of the zeolites in the mixture of the two zeolites is between 30 and 70% by weight relative to the total weight of the mixture of the two zeolites.
- the support of the catalyst according to the present invention may also comprise at least one zeolite chosen from the group formed by zeolites of structural type TON, FER, MTT
- the zeolite of structural type TON which can also be used in the composition of the support of the catalyst according to the present invention is advantageously chosen from the group formed by the zeolites Theta-1, ISI-1, NU-10, KZ-2 and ZSM-22 described. in the "Atlas of Zeolite Structure Types", cited above, and in the case of zeolite ZSM-22, in US Pat. Nos. 4,566,477 and 4,902,406, and in the case of zeolite NU-10, in EP-65400 and EP-77624.
- the zeolite of structural type FER which can also enter the composition of the support of the catalyst according to the present invention is advantageously chosen from the group formed by zeolites ZSM-35, ferrierite, FU-9 and ISI-6, described in the book " Atlas of Zeolite
- the MTT structural type zeolite which can also be used in the composition of the catalyst support according to the present invention is advantageously chosen from the group formed by zeolites ZSM-23, EU-13, ISI-4 and KZ-1 described in the book. "Atlas of Zeolite
- zeolites of structural type TON which can also be used in the composition of the catalyst support according to the present invention, zeolites ZSM-22 and NU-10 are preferred.
- zeolites of FER structural type that can also be included in the composition of the catalyst support according to the present invention, zeolites ZSM-35 and ferrierite are preferred.
- the support of the catalyst according to the invention contains a mixture of zeolite COK-7 with at least one zeolite chosen from the group formed by the zeolites of structural type TON, FER, MTT, the COK-7 zeolite. optionally being mixed with zeolite ZBM-30.
- the support of the catalyst according to the invention contains a mixture of two zeolites and, preferably, a mixture of the COK-7 zeolite with the ZSM-22 zeolite or the NIMO zeolite.
- the proportion of each of the zeolites in the mixture of the two zeolites is advantageously between 20 and 80% by weight relative to the total weight of the mixture of the two zeolites, and preferably the proportion of each of the zeolites in the mixture of the two zeolites is 50 % by weight relative to the total weight of the mixture of the two zeolites.
- the zeolites present in the support of the catalyst according to the invention advantageously comprise silicon and at least one element T chosen from the group formed by aluminum, iron, gallium, phosphorus and boron, and preferably said element T is aluminum
- the overall Si / Al ratio of the zeolites used in the composition of the catalyst support according to the invention as well as the chemical composition of the samples are determined by X-ray fluorescence and atomic absorption.
- the Si / Al ratios of the zeolites described above are advantageously those obtained in the synthesis according to the procedures described in the various documents cited or obtained after post-synthesis dealumination treatments well known to those skilled in the art. such as and not limited to hydrothermal treatments followed or not acid attacks or even direct acid attacks by solutions of mineral or organic acids.
- the zeolites used in the composition of the support of the catalyst according to the invention are advantageously calcined and exchanged by at least one treatment with a solution of at least one ammonium salt so as to obtain the ammonium form of the zeolites which, once calcined, leads to to the hydrogen form of said zeolites.
- the zeolites used in the composition of the support of the catalyst according to the invention are advantageously at least partly, preferably almost completely, in acid form, that is to say in acid form (H + ).
- the atomic ratio Na / T is generally advantageously less than 0.1 and preferably less than 0.5 and even more preferably less than 0.01. Silica-alumina.
- the support of the catalyst according to the invention also comprises at least one silica-alumina.
- Silica-aluminas can not be considered as aluminosilicates as close to ideality as zeolites. It is possible to obtain silica-aluminas in the complete range of composition ranging from 0 to 100% Al 2 O 3 , but the degree of association of the two Si and Al elements and therefore the homogeneity of the solid are highly dependent on the method of preparation.
- the silica-alumina is homogeneous on a micrometer scale and contains an amount greater than 5% by weight and less than or equal to 95% by weight of silica (SiO 2 ), said silica-alumina having the characteristics following: a mean pore diameter, measured by mercury porosimetry, of between 20 and 140 ⁇ , a total pore volume, measured by mercury porosimetry, of between 0.1 ml / g and 0.5 ml / g, a porous volume total, measured by nitrogen porosimetry, between 0.1 ml / g and 0.5 ml / g, a BET specific surface area of between 100 and 550 m 2 / g, a pore volume, measured by mercury porosimetry, included in FIGS.
- an X-ray diffraction pattern which contains at least the principal characteristic lines of at least one of the transition aluminas included in the group consisting of alpha, rho, chi, eta, gamma, kappa, theta and delta alumina.
- said silica-alumina contains:
- silica mass content of between 10 and 80% by weight, preferably a silica content of greater than 20% by weight and less than 80% by weight, and even more preferably greater than 25% by weight and less than 75% by weight, the silica content is advantageously between 10 and 50% by weight, this silica content is measured using X-ray fluorescence.
- a content of cationic impurities for example Na + ) of less than 0.1% by weight, preferably less than 0.05% by weight and even more preferably less than 0.025% by weight.
- the content of cationic impurities means the total content of alkali and alkaline earth.
- an anionic impurities content (e.g., SO 4 2 ', IC ) less than 1% by weight, preferably less than 0.5% by weight and even more preferably less than 0.1% weight.
- the catalyst further comprises a hydrogenating function, that is to say, at least one hydro-dehydrogenating element chosen from the group consisting of Group VIII and Group VIB metals, taken alone or as a mixture.
- the group VIII elements are chosen from iron, cobalt, nickel, ruthenium, rhodium, palladium, osmium, iridium or platinum, taken alone or as a mixture.
- the elements of group VIII are chosen from the noble metals of group VIII, the elements of group VIII are advantageously chosen from platinum and palladium.
- the elements of group VIII are chosen from non-noble metals of group VIII, the elements of group VIII are advantageously chosen from iron, cobalt and nickel.
- the group VIB elements of the catalyst according to the present invention are selected from tungsten and molybdenum.
- the hydrogenating function comprises a group VIII element and a group VIB element
- the following metal combinations are preferred: nickel-molybdenum, cobalt-molybdenum, iron-molybdenum, iron-tungsten, nickel-tungsten, cobalt- tungsten, and very preferably: nickel-molybdenum, cobalt-molybdenum, nickel-tungsten. It is also possible to use combinations of three metals such as nickel-cobalt-molybdenum example. When these combinations of metals are employed, these metals are preferably used in their sulfurized form.
- the content of the hydro-dehydrogenating element of said catalyst according to the present invention chosen from the group formed by the metals of group VIB and of group VIII is between 0.1 and 60% by weight relative to the total mass of said catalyst, preferably between 0 , 1 to 50% by weight and very preferably between 0.1 to 40% by weight.
- the catalyst preferably contains a noble metal content of less than 5% by weight, more preferably less than 2% by weight relative to the total weight of said catalyst.
- the noble metals are preferably used in their reduced form.
- the catalyst of the present invention also comprises at least one amorphous or poorly crystallized porous mineral matrix of oxide type selected from aluminas, aluminates and silicas.
- a matrix containing alumina in all its forms known to those skilled in the art, and very preferably gamma alumina, is used.
- the catalyst also contains at least one doping element selected from the group consisting of boron, silicon and phosphorus, and preferably boron and / or silicon.
- the doping element chosen from the group formed by boron, silicon and / or phosphorus may advantageously be in the matrix, the zeolite, the silica-alumina or preferably may be deposited on the catalyst and in this case be located mainly on the matrix.
- the doping element introduced, and in particular silicon, mainly located on the matrix of the support can advantageously be characterized by techniques such as the Castaing microprobe (distribution profile of the various elements), transmission electron microscopy coupled with an analysis. X catalyst components, or even by establishing a distribution map of the elements present in the catalyst by electron microprobe.
- the catalyst also contains at least one group VIIA element, preferably chlorine and fluorine, and optionally also at least one group VIIB element.
- Catalyst composition preferably chlorine and fluorine, and optionally also at least one group VIIB element.
- the catalyst according to the present invention advantageously contains generally in% by weight with respect to the total mass of the catalyst:
- said catalyst also contains from 0.1 to 99%, preferably from 0.2 to 99.8%, very preferably from 0.5 to 90%, and more preferably from 1 to 80% by weight.
- at least one COK-7 zeolite alone, or in the case where the COK-7 zeolite is used as a mixture with at least one zeolite ZBM-30 the proportion of each of the zeolites in the mixture of the two zeolites COK-7 and ZBM- 30 is advantageously between 20 and 80% by weight relative to the total weight of the mixture of the two zeolites, and preferably the proportion of each of the zeolites in the mixture of the two zeolites is between 30 and 70% by weight relative to the total weight of the mixture of the two zeolites;
- said catalyst optionally containing:
- zeolites from 0 to 60%, preferably from 5 to 40%, of at least one zeolite chosen from the group formed by zeolites of structural type TON, FER, MTT,
- the Group VIB 1 metals of Group VIII of the catalyst of the present invention are advantageously present in whole or in part in the metal and / or oxide and / or sulfide form.
- the catalysts used in the process according to the invention can be prepared according to all methods well known to those skilled in the art, from the carrier based on silico-aluminum matrix and based on at least one COK-7 zeolite alone or in admixture with at least one zeolite ZBM-30.
- the catalyst further contains a hydrogenating phase.
- any method of silica-alumina synthesis known to those skilled in the art leading to a homogeneous silica-alumina at the micrometer scale and in which the cationic impurities (for example Na + ) can be reduced to less than 0.1% preferably at a content of less than 0.05% by weight and even more preferably less than 0.025% by weight and in which the anionic impurities (for example SO 4 2 " , CI " ) can be reduced to less than 1% and more preferably less than 0.05% by weight is suitable for preparing the supports that can be used in the process for preparing the silica-alumina used in the catalyst according to the invention.
- the silico-aluminum matrices advantageously obtained from a mixture at any stage of a compound of alumina which is partially soluble in an acidic medium with a totally soluble silica compound or with a totally soluble combination of alumina and silica hydrated, shaping followed by a hydrothermal or thermal treatment to homogenize micrometric scale, or even nanoscale, allowed to obtain a particularly active catalyst.
- partially soluble in acidic medium the applicant understands that bringing the alumina compound into contact before any addition of the totally soluble silica compound or the combination with an acidic solution, for example of nitric acid or sulfuric acid, causes them to react. partial dissolution.
- the silica compounds used according to the invention may advantageously have been chosen from the group formed by silicic acid, silicic acid sols, water-soluble alkali silicates, cationic silicon salts, for example sodium metasilicate hydrate, Ludox® in ammoniacal form or in alkaline form, quaternary ammonium silicates.
- the silica sol may advantageously be prepared according to one of the methods known to those skilled in the art.
- a solution of decationized orthosilicic acid is prepared from a water-soluble alkali silicate by ion exchange on a resin.
- the totally soluble hydrous silica-aluminas used according to the invention can advantageously be prepared by true coprecipitation under controlled stationary conditions (pH, concentration, temperature, average residence time) by reaction of a basic solution containing the silicon, for example under sodium silicate form, optionally aluminum, for example in the form of sodium aluminate with an acid solution containing at least one aluminum salt, for example aluminum sulphate. At least one carbonate or CO 2 may optionally be added to the reaction medium.
- the applicant intends a process by which at least one fully soluble aluminum compound in basic or acid medium as described below, at least one silicon compound as described below are contacted simultaneously or sequentially in the presence of at least one precipitant and / or coprecipitant compound so as to obtain a mixed phase consisting essentially of silica-hydrated alumina which is optionally homogenized by intense stirring, shearing, colloid milling or by combination of these unit operations.
- the alumina compounds used according to the invention are advantageously partially soluble in acid medium. They are advantageously chosen wholly or partly from the group of alumina compounds of general formula AI 2 O 3 , nH 2 O.
- hydrated alumina compounds may be used, such as: hydrargillite, gibbsite, bayerite, boehmite, pseudo-boehmite and amorphous or essentially amorphous alumina gels. It is also advantageous to use the dehydrated forms of these compounds which consist of transition aluminas and which comprise at least one of the phases taken from the group: rho, khi, eta, gamma, kappa, theta, and delta, which differ essentially in the organization of their crystalline structure.
- the alpha alumina commonly called corundum can advantageously be incorporated in a small proportion in the support according to the invention.
- Boehmite is generally described as an aluminum monohydrate of formula AI 2 O 3 , nH 2 O which in fact encompasses a wide continuum of materials of variable degree of hydration and organization with more or less well defined boundaries: most hydrated gelatinous boehmite, with n being greater than 2, pseudo-boehmite or microcrystalline boehmite with n between 1 and 2, then crystalline boehmite and finally well crystallized boehmite in large crystals with n close to 1
- the morphology of aluminum monohydrate can vary within wide limits between these two acicular or prismatic extreme forms. A whole set of variable shapes can be used between these two forms: chain, boats, interwoven plates.
- Relatively pure aluminum hydrates can advantageously be used in powder form, amorphous or crystallized or crystallized containing an amorphous part.
- the aluminum hydrate can also advantageously be introduced in the form of aqueous suspensions or dispersions.
- the aqueous suspensions or dispersions of aluminum hydrate used according to the invention may advantageously be gelable or coagulable.
- the aqueous dispersions or suspensions may also advantageously be obtained as is well known to those skilled in the art by peptization in water or acidulated water of aluminum hydrates.
- the aluminum hydrate dispersion may advantageously be carried out by any method known to those skilled in the art: in a "batch" reactor, a continuous mixer, a kneader, a colloid mill. Such a mixture may advantageously also be carried out in a plug flow reactor and, in particular, in a static mixer. Lightnin reactors can be mentioned.
- alumina having been previously subjected to a treatment that may improve its degree of dispersion.
- a treatment that may improve its degree of dispersion.
- homogenization it is advantageous to use at least one of the homogenization treatments described in the text that follows.
- aqueous dispersions or suspensions of alumina that can be used are advantageously aqueous suspensions or dispersions of fine or ultra-fine boehmites which are composed of particles having colloidal dimensions.
- the fine or ultra-fine boehmites used according to the present invention may advantageously have been obtained according to the French patent FR-B-1 261 182 and FR-B-1 381 282 or in the European patent application EP-A-15. 196.
- aqueous suspensions or dispersions obtained from pseudo-boehmite, amorphous alumina gels, aluminum hydroxide gels or ultra-fine hydrargillite It is also advantageous to use aqueous suspensions or dispersions obtained from pseudo-boehmite, amorphous alumina gels, aluminum hydroxide gels or ultra-fine hydrargillite.
- Aluminum monohydrate may advantageously be purchased from a variety of commercial sources of alumina such as in particular PURAL®, CATAPAL®, DISPERAL®, DISPAL® marketed by SASOL or HIQ® marketed by ALCOA 1 or according to the methods Known to those skilled in the art: it can be prepared by partial dehydration of aluminum trihydrate by conventional methods or it can advantageously be prepared by precipitation. When these aluminas are in the form of a gel, they are advantageously peptized with water or an acidulated solution. In precipitation, the acid source may advantageously be for example chosen from at least one of the following compounds: aluminum chloride, aluminum sulphate, aluminum nitrate.
- the basic source of aluminum may advantageously be chosen from basic aluminum salts such as sodium aluminate and potassium aluminate.
- the zeolites used in the catalyst according to the invention are advantageously commercial zeolites or zeolites synthesized according to the procedures described in the patents mentioned above.
- the zeolites used in the composition of the catalyst according to the invention are advantageously at least partly, preferably almost completely, in acid form, that is to say in hydrogen (H + ) form.
- the matrix according to the invention may advantageously be prepared according to all methods well known to those skilled in the art from the supports prepared as described above.
- the zeolite can advantageously be introduced according to any method known to those skilled in the art and at any stage of the preparation of the support or catalyst.
- a preferred method of preparing the catalyst according to the present invention comprises the following steps:
- the zeolite may advantageously be introduced during the preparation of the silica-alumina.
- the zeolite may advantageously be, without limitation, for example in the form of powder, milled powder, suspension, suspension having undergone deagglomeration treatment.
- the zeolite can advantageously be slurried acidulated or not at a concentration adjusted to the final zeolite content referred to the support.
- This suspension commonly called a slip is advantageously then mixed with the precursors of the silica-alumina at any stage of its synthesis as described above.
- the zeolite can advantageously also be introduced during the shaping of the support with the elements which constitute the matrix with possibly at least one binder
- the zeolite may advantageously be, without being limiting, in the form of powder, ground powder, suspension, suspension having undergone deagglomeration treatment.
- the preparation and treatment (s) and the shaping of the zeolite can thus advantageously constitute a step in the preparation of these catalysts.
- the zeolite / silica-alumina matrix is obtained by mixing the silica-alumina and the zeolite, and the mixture is then shaped.
- the zeolite / silica-alumina matrix may advantageously be shaped by any technique known to those skilled in the art.
- the shaping can advantageously be carried out for example by extrusion, by pelletization, by the method of drop coagulation ("oil-drop"), by rotating plate granulation or by any other method well known to those skilled in the art. .
- the shaping can advantageously also be carried out in the presence of the various constituents of the catalyst and extrusion of the obtained mineral paste, by pelletizing, shaped into beads at the rotating bezel or drum, drop coagulation, "oil-drop” , “oil-up”, or any other known method of agglomeration of a powder containing alumina and optionally other ingredients selected from those mentioned above.
- the catalysts used according to the invention advantageously have the shape of spheres or extrudates. It is however advantageous that the catalyst is in the form of extruded with a diameter of between 0.5 and 5 mm and more particularly between 0.7 and 2.5 mm.
- the shapes are advantageously cylindrical (which may be hollow or not), cylindrical twisted, multilobed (2, 3, 4 or 5 lobes for example), rings.
- the cylindrical shape is preferably used in a preferred manner, but any other form may be used.
- these supports implemented according to the present invention may advantageously have been treated as is well known to those skilled in the art by additives to facilitate the shaping and / or improve the final mechanical properties of the supports to base of silico-aluminum matrices.
- additives there may be mentioned in particular cellulose, carboxymethylcellulose, carboxy-ethylcellulose, tall oil, xanthan gums, surfactants, flocculating agents such as polyacrylamides, carbon black, starches, stearic acid, polyacrylic alcohol, polyvinyl alcohol, biopolymers, glucose, polyethylene glycols, etc.
- the shaping may advantageously be carried out using the catalyst shaping techniques known to those skilled in the art, such as, for example: extrusion, coating, spray drying or tabletting.
- Water may be advantageously added or removed to adjust the viscosity of the paste to be extruded. This step can advantageously be carried out at any stage of the kneading step.
- a predominantly solid compound and preferably an oxide or a hydrate.
- a hydrate is preferably used and even more preferably an aluminum hydrate. The loss on ignition of this hydrate is preferably greater than 15%.
- the acid content added to the kneading before forming is advantageously less than 30%, preferably between 0.5 and 20% by weight of the anhydrous mass of silica and alumina involved in the synthesis.
- Extrusion can advantageously be performed by any conventional tool, commercially available.
- the paste resulting from the mixing is advantageously extruded through a die, for example by means of a piston or a single screw or twin extrusion screw.
- This extrusion step may advantageously be carried out by any method known to those skilled in the art.
- the support extrusions according to the invention generally have advantageously a crush strength of at least 70 N / cm and preferably greater than or equal to 100 N / cm. Calcination of zeolite / silica-alumina support
- Drying is carried out by any technique known to those skilled in the art.
- calcine preferably in the presence of molecular oxygen, for example by conducting a sweep of air, at a temperature of less than or equal to 1100 ° C.
- At least one calcination can advantageously be performed after any of the steps of the preparation.
- This treatment for example, can advantageously be carried out in a traversed bed, in a licked bed or in a static atmosphere.
- the furnace used may be a rotating rotary kiln or a vertical kiln with radial traversed layers.
- the calcination conditions depend mainly on the maximum temperature of use of the catalyst.
- the preferred calcination conditions are advantageously between more than one hour at 200 ° C.
- the calcination can advantageously be carried out in the presence of water vapor.
- the final calcination may optionally be carried out in the presence of an acidic or basic vapor.
- the calcination can be carried out under partial pressure of ammonia.
- Post-synthesis treatments may advantageously be carried out so as to improve the properties of the catalyst.
- the zeolite / silica-alumina support can thus be optionally subjected to a hydrothermal treatment in a confined atmosphere.
- Hydrothermal treatment in a confined atmosphere means treatment by autoclaving in the presence of water at a temperature above room temperature.
- the support can advantageously be treated.
- the support can advantageously be impregnated, prior to its autoclaving, the autoclaving being done either in the vapor phase or in the liquid phase, this vapor or liquid phase of the autoclave possibly being acidic or not.
- This impregnation, prior to autoclaving may be acidic or not.
- This impregnation, prior to autoclaving may advantageously be carried out dry or by immersion of the support in an acidic aqueous solution. Dry impregnation means contacting the support with a solution volume less than or equal to the total pore volume of the support. Preferably, the impregnation is carried out dry.
- the autoclave is preferably a rotary basket autoclave such as that defined in patent application EP-A-0 387 109.
- the temperature during autoclaving may advantageously be between 100 and 250 ° C for a period of time between 30 minutes and 3 hours.
- the hydro-dehydrogenating element may advantageously be introduced at any stage of the preparation, very preferably after forming the zeolite / silica-alumina support.
- the shaping is advantageously followed by calcination, the hydrogenating element can also be advantageously introduced before or after this calcination.
- the preparation generally ends with a calcination at a temperature of 250 to 600 ° C.
- Another of the preferred methods according to the present invention advantageously consists in shaping the support after kneading thereof, then passing the dough thus obtained to through a die to form extrudates with a diameter of between 0.4 and 4 mm.
- the hydrogenating function can advantageously be then introduced in part only or in full, at the time of mixing.
- the support is impregnated with an aqueous solution.
- the impregnation of the support is preferably carried out by the "dry" impregnation method well known to those skilled in the art.
- the impregnation may advantageously be carried out in a single step by a solution containing all the constituent elements of the final catalyst.
- the hydrogenating function may also advantageously be introduced by one or more ion exchange operations on the calcined support constituted by a zeolite as previously described, dispersed in the chosen matrix, using solutions containing the precursor salts of the chosen metals. .
- the hydrogenating function may advantageously be introduced by one or more impregnation operations of the shaped and calcined support, with a solution containing at least one precursor of at least one oxide of at least one metal chosen from the group formed by the metals of groups VIII and group VIB metals one (s) precursor (s) of at least one oxide of at least one metal from group VIII being preferably introduced (s) after those of group VIB or simultaneously the latter, if the catalyst contains at least one Group VIB metal and at least one Group VIII metal.
- the catalyst advantageously contains at least one element of group VIB, for example molybdenum
- the impregnation of molybdenum may advantageously be facilitated by the addition of phosphoric acid in the ammonium paramolybdate solutions, which also makes it possible to introduce the phosphorus so as to promote the catalytic activity.
- the catalyst contains as dopant at least one element selected from silicon, boron and phosphorus. These elements are advantageously introduced on a support already containing at least one COK-7 zeolite alone, or in admixture with at least one zeolite ZBM-30, at least one silica-alumina, as defined above, and at least one selected metal. in the group consisting of Group VIB metals and Group VIII metals.
- the catalyst contains boron, silicon and phosphorus and optionally the element selected from group VIIA halide ions, these elements can advantageously be introduced into the catalyst at various levels of the preparation and in various ways.
- Impregnation of the metal is preferably carried out by the so-called “dry” impregnation method well known to those skilled in the art.
- the impregnation may advantageously be carried out in a single step by a solution containing all the constituent elements of the final catalyst.
- the P, B, Si and the element chosen from the group VIIA halide ions can advantageously be introduced by one or more impregnation operations with excess of solution on the calcined precursor.
- a preferred method according to the invention consists in preparing an aqueous solution of at least one boron salt such as ammonium biborate or ammonium pentaborate in an alkaline medium and in the presence of of oxygenated water and to carry out a so-called dry impregnation, in which the pore volume of the precursor is filled with the solution containing boron.
- boron salt such as ammonium biborate or ammonium pentaborate
- the catalyst contains silicon
- a solution of a silicon-type silicon compound is advantageously used.
- the deposition of boron and silicon can advantageously also be carried out simultaneously using a solution containing a boron salt and a silicon-type silicon compound.
- a solution containing a boron salt and a silicon-type silicon compound for example in the case where for example the precursor is a nickel-molybdenum type catalyst supported on a support containing zeolite and alumina, it is possible to impregnate this precursor with the aqueous solution of ammonium biborate and Rhodorsil E1P silicone from the company Rhône Poulenc, to carry out a drying, for example at 80 ° C.
- the catalyst contains at least one group VIIA element, preferably fluorine
- a solution of ammonium fluoride to dry, for example at 80 ° C. C, and carry out a calcination for example and preferably in air in crossed bed, for example at 500 ° C for 4 hours.
- the catalyst contains phosphorus
- a step of intermediate drying of the catalyst is generally advantageously carried out at a temperature generally between 60 and 250 ° C and an intermediate calcination step of the catalyst is generally advantageously carried out at a temperature of temperature between 250 and 600 0 C.
- the wet solid is advantageously allowed to stand under a humid atmosphere at a temperature of between 10 and 80 ° C., and then the wet solid obtained is dried at a temperature of between 60 and 150 ° C., and finally the solid obtained is calcined at a temperature of between 150 and 800 ° C.
- the sources of Group VIB elements which can advantageously be used are well known to those skilled in the art.
- oxides and hydroxides, molybdic and tungstic acids and their salts in particular ammonium salts such as ammonium molybdate, ammonium heptamolybdate.
- ammonium tungstate phosphomolybdic acid, phosphotungstic acid and their salts, silicomolybdic acid, acid silicotungstic and their salts.
- Oxides and ammonium salts such as ammonium molybdate, ammonium heptamolybdate and ammonium tungstate are preferably used.
- group VIII elements which can advantageously be used are well known to those skilled in the art.
- non-noble metals nitrates, sulphates, phosphates, halides, for example chlorides, bromides and fluorides, carboxylates, for example acetates, hydroxides and carbonates
- noble metals halides are advantageously used, for example chlorides, nitrates, acids such as chloroplatinic acid, oxychlorides such as ammoniacal ruthenium oxychloride.
- cationic complexes such as ammonium salts when it is desired to deposit the platinum on the zeolite by cation exchange.
- the preferred phosphorus source is orthophosphoric acid H 3 PO 4, but its salts and esters such as ammonium phosphates are also suitable.
- the phosphorus may for example be introduced in the form of a mixture of phosphoric acid and a basic organic compound containing nitrogen such as ammonia, primary and secondary amines, cyclic amines, compounds of the family of pyridine and quinolines and compounds of the pyrrole family.
- ethyl orthosilicate Si (OEt) 4 siloxanes, polysiloxanes, halide silicates such as ammonium fluorosilicate (NH / SiFg) or sodium fluorosilicate Na2SiF ⁇ .
- silicomolybdic acid and its salts, silicotungstic acid and its salts can also advantageously be used Silicon can be added, for example by impregnation of ethyl silicate in solution in a water / alcohol mixture Silicon can be added, for example by impregnation a silicone-type silicon compound suspended in water.
- the boron source may advantageously be boric acid, preferably orthoboric acid H 3 BO 3 , ammonium biborate or pentaborate, boron oxide, boric esters.
- Boron may for example be introduced in the form of a mixture of boric acid, hydrogen peroxide and a basic organic compound containing nitrogen such as ammonia, primary and secondary amines, cyclic amines, compounds of the family of pyridine and quinolines and compounds of the pyrrole family. Boron may advantageously be introduced for example by a boric acid solution in a water / alcohol mixture.
- the sources of group VIIA elements which can advantageously be used are well known to those skilled in the art.
- the fluoride anions may advantageously be introduced in the form of hydrofluoric acid or its salts. These salts are formed with alkali metals, ammonium or an organic compound. In the latter case, the salt is advantageously formed in the reaction mixture by reaction between the organic compound and the hydrofluoric acid. It is also possible to use hydrolysable compounds which release fluoride anions in water, such as ammonium fluorosilicate (NH SiF ⁇ , silicon tetrafluoride SiF 4 or sodium Na2SiFg.
- the fluorine can advantageously be introduced e.g. by impregnation with an aqueous solution of hydrofluoric acid or ammonium fluoride.
- the catalysts thus obtained, in oxide form after calcination, may optionally be brought at least partly into the metal or sulphide form.
- the catalysts obtained by the present invention are advantageously shaped into grains of different shapes and sizes. They are advantageously used in general in the form of cylindrical or multi-lobed extrusions such as bilobed, trilobed, straight-lobed or twisted, but may optionally be manufactured and used in the form of crushed powder, tablets, rings, balls, wheels. They have a specific surface area measured by nitrogen adsorption according to the BET method (Brunauer, Emmett, Teller, J. Am Chem Soc., Vol 60, 309-316 (1938)) of between 50 and 600 m 2 / g. , a pore volume measured by mercury porosimetry of between 0.2 and 1.5 cm 3 / g and a pore size distribution that can be monomodal, bimodal or polymodal.
- BET method Brunauer, Emmett, Teller, J. Am Chem Soc., Vol 60, 309-316 (1938)
- the catalysts thus obtained are used in conversion reactions of hydrocarbon feeds (in the broad sense of transformation) and in particular hydrocracking reactions.
- the catalysts described above are used in hydrocracking reactions of hydrocarbon feedstocks such as petroleum cuts.
- the feedstocks advantageously employed in the process are gasolines, kerosenes, gas oils, vacuum gas oils, atmospheric residues, vacuum residues, atmospheric distillates, vacuum distillates, heavy fuels, oils and the like. , waxes and paraffins, used oils, residues or deasphalted crudes, fillers derived from thermal or catalytic conversion processes and their mixtures. They contain heteroatoms such as sulfur, oxygen and nitrogen and possibly metals. Charges from the Fischer-Tropsch process are excluded.
- the catalysts of the invention are used in the hydrocracking process according to the invention and preferably in a hydrocracking process of heavy hydrocarbon cuts of vacuum distillate type, deasphalted or hydrotreated residues or the like.
- the heavy cuts preferably consist of at least 80% by volume of compounds whose boiling points are at least 350 ° C. and preferably between 350 and 580 ° C. (that is to say corresponding to compounds containing at least 15 to 20 carbon atoms). They usually contain heteroatoms such as sulfur and nitrogen. The nitrogen content is usually between 1 and 5000 ppm by weight and the sulfur content between 0.01 and 5% by weight.
- the catalysts used in the hydrocarbon feedstock hydrocracking process according to the invention are preferably subjected to a sulphurization treatment which makes it possible, at least in part, to convert the metal species into sulphide before they come into contact with the feedstock. treat.
- This activation treatment by sulphurisation is well known to those skilled in the art and can be performed by any method already described in the literature.
- a conventional sulphurization method well known to those skilled in the art consists in heating the catalyst in the presence of hydrogen sulphide at a temperature of between 150 and 800 ° C., preferably between 250 and 600 ° C., generally in a reaction zone at crossed bed.
- the catalyst of the present invention can be advantageously used in the hydrocracking of vacuum distillate type cuts heavily loaded with sulfur and nitrogen.
- the desired products are middle distillates and / or oils.
- the hydrocracking is used in combination with a prior hydrotreatment step in a process for the improved production of middle distillates together with the production of oil bases having a viscosity number between 95 and 150.
- the invention also relates to hydrocracking processes using the hydrocracking catalysts according to the invention.
- the conditions of the hydrocracking such as temperature, pressure, hydrogen recycling rate, hourly space velocity, may be very variable depending on the nature of the load, the quality of the desired products and the facilities available to the refiner.
- the temperature is generally advantageously greater than 200 ° C. and preferably between 250 and 480 ° C.
- the pressure is advantageously greater than 0.1 MPa and preferably greater than 1 MPa.
- the hydrogen recycling rate is advantageously at least 50 and preferably between 80 and 5000 normal liters of hydrogen per liter of filler.
- the hourly volume velocity is advantageously between 0.1 and 20 volumes of filler per volume of catalyst and per hour.
- the hydrocracking processes according to the invention advantageously cover the pressure and conversion ranges from mild hydrocracking to high pressure hydrocracking.
- mild hydrocracking is meant a hydrocracking advantageously leading to moderate conversions, generally less than 55% and preferably less than 40%, and operating at low pressure, generally between 2 MPa and 12 MPa and preferably between 2 MPa and 6 MPa. .
- High-pressure hydrocracking is understood to mean hydrocracking advantageously leading to high conversions, generally greater than 55%, and operating at high pressure, generally greater than 6 MPa.
- the catalyst of the present invention may advantageously be used alone, in one or more catalytic beds, in one or more reactors, in a so-called one-step hydrocracking scheme, with or without liquid recycling of the unconverted fraction, optionally in combination with a hydrorefining catalyst located upstream of the catalyst of the present invention.
- the catalyst according to the present invention is advantageously used in the second reaction zone, in one or more beds, in one or more reactors, in association or otherwise with a hydrorefining catalyst located upstream of the catalyst of the present invention.
- the hydrocracking in one step advantageously comprises firstly and generally a high hydrorefining which is intended to carry out a hydrodenitrogenation and a desulphurization of the feed before it is sent to the hydrocracking catalyst proper. , especially in the case where it comprises a zeolite.
- This extensive hydrorefining of the charge results in only a limited conversion of the charge, in lighter fractions, which remains insufficient and must be completed on the more active hydrocracking catalyst.
- no separation occurs between the two types of catalysts.
- the entire effluent at the outlet of the reactor is advantageously injected onto the hydrocracking catalyst proper and only then is separation of the products formed carried out.
- This version of the hydrocracking also called "Once Through” has a variant that advantageously has a recycling of the unconverted fraction to the reactor for further conversion of the charge.
- the conversion level is advantageously less than 55% and preferably less than 40%.
- the catalyst according to the invention is advantageously employed at a temperature which is generally or equal to 230 0 C and preferably at 300 0 C, generally at most 480 C C, and often between 350 and 450 0 C.
- the pressure is advantageously greater than 2 MPa and preferably 3 MPa, it is less than 12 MPa and preferably less than 10 MPa.
- the amount of hydrogen is preferably at least 100 normal liters of hydrogen per liter of filler and preferably between 200 and 3000 normal liters of hydrogen per liter of filler.
- the hourly volume velocity is advantageously between 0.15 and 10 h -1 " Under these conditions, the catalysts according to the present invention have a better activity in conversion, hydrodesulfurization and hydrodenitrogenation than commercial catalysts.
- the hydrocracking is carried out at high pressure (total pressure greater than 6 MPa), the conversion level is then advantageously greater than 55%.
- the method according to the invention then operates at a temperature preferably greater than or equal to 230 0 C and preferably between 300 and 480 ° C and very preferably between 300 and 440 0 C 1 5 MPa to a higher pressure and preferably greater than 7 MPa, very preferably greater than 10 MPa and more preferably greater than 12 MPa, with a hydrogen amount of at least 100NI / I of charge and preferably between 200 and 3000N / l of hydrogen per liter of feed and at an hourly space velocity is generally between 0.15 and 10 h "1.
- Embodiment Two-step process
- the hydrocracking in two stages advantageously comprises a first stage whose objective, as in the "one stage” process, is to carry out hydrorefining of the charge, but also to achieve a conversion of the latter of the order in general from 40 to 60%.
- the effluent from the first step then advantageously undergoes separation (distillation), which is usually called intermediate separation, which aims to separate the conversion products from the unconverted fraction.
- separation distillation
- intermediate separation which aims to separate the conversion products from the unconverted fraction.
- the second step of a two-stage hydrocracking process only the fraction of the unconverted feedstock in the first step is processed. This separation allows a two-stage hydrocracking process to be more selective in middle distillate (kerosene + diesel) than a one-step process.
- the intermediate separation of the conversion products avoids their "over cracking" in naphtha and gas in the second step on the hydrocracking catalyst.
- the unconverted fraction of the feedstock treated in the second stage generally contains very low levels of sulfur and NH 3 as well as organic nitrogen compounds, generally less than 20 ppm by weight or less 10 ppm weight.
- the catalysts used in the second stage of the two-stage hydrocracking processes are preferably noble group VIII catalysts, even more preferably platinum and / or palladium catalysts.
- the catalysts according to the invention are advantageously used in the second stage.
- the process of the present invention may advantageously be used for partial hydrocracking, that is to say mild or moderate, advantageously under moderate pressure conditions, for example of vacuum distillate-type cuts. loaded with sulfur and nitrogen which have been previously hydrotreated.
- the conversion level is less than 55% and preferably less than 40%.
- the catalyst of the first step may advantageously be any hydrotreatment catalyst known to those skilled in the art.
- This hydrotreatment catalyst advantageously comprises a matrix preferably based on alumina and at least one metal having a hydrogenating function.
- the hydrotreatment function is provided by at least one metal or metal compound, alone or in combination, chosen from Group VIII and Group VIB metals, such as chosen from nickel, cobalt, molybdenum and tungsten, in particular .
- this catalyst may optionally contain phosphorus and optionally boron.
- the first step advantageously takes place at a temperature of 350-460 ° C., preferably 360-450 ° C., a total pressure of at least 2 MPa; and preferably 3 MPa 1 hourly space velocity of 0.1 -5 Ir " and preferably 0.2-2 h " and with a hydrogen amount of at least 100 NI / NI filler, and preferably 260 -3000 NI / NI charge.
- the temperatures are advantageously greater than or equal to 230 ° C. and often between 300 and 480 ° C., preferably between 330 ° C. and 450 ° C.
- the pressure is advantageously at least 2 MPa and preferably 3 MPa, it is less than 12 MPa and preferably less than 10 MPa.
- the amount of hydrogen is advantageously at least 100 Nl / l of filler and preferably between 200 and 3000 Nl / l of hydrogen per liter of filler.
- the hourly volume velocity is advantageously generally between 0.15 and 10 h -1 .
- the catalysts of the present invention have a better activity in conversion, hydrodesulfurization, hydrodenitrogenation and a better selectivity in middle distillates than commercial catalysts The service life of the catalysts is also improved in the moderate pressure range.
- the catalyst according to the present invention can be used for hydrocracking under high pressure conditions of at least 6 MPa.
- the treated sections are, for example, of the vacuum distillate type which are heavily loaded with sulfur and nitrogen which have been previously hydrotreated.
- the conversion level is greater than 55%.
- the petroleum fraction conversion process advantageously takes place in two stages, the catalyst according to the invention being used in the second stage.
- the catalyst of the first step may advantageously be any hydrotreatment catalyst known to those skilled in the art.
- This hydrotreatment catalyst advantageously comprises a matrix preferably based on alumina and at least one metal having a hydrogenating function.
- the hydrotreatment function is provided by at least one metal or metal compound, alone or in combination, chosen from Group VIII and Group VIB metals, such as chosen from nickel, cobalt, molybdenum and tungsten, in particular .
- this catalyst may optionally contain phosphorus and optionally boron.
- the first step advantageously takes place at a temperature of 350-460 0 C 1 preferably 360-450 0 C, a pressure higher than 3 MPa, an hourly space velocity of 0.1-5 h "'and preferably 0, 2-2 h '' 'with a quantity of hydrogen of at least 100 NI / NI of filler, and preferably 260-3000 NI / NI load.
- the temperatures are advantageously greater than or equal to 230 ° C. and often between 300 and 480 ° C. and preferably between 300 and 440 ° C.
- the pressure is greater than 5 MPa and preferably greater than 7 MPa, more preferably greater than 10 MPa and more preferably greater than 12 MPa.
- the amount of hydrogen is advantageously at least 10 ONI / l of charge and preferably between 200 and 3000 Nl / I of hydrogen per liter of feedstock.
- the hourly volume velocity is advantageously generally between 0.15 and 10 h " ".
- the catalysts of the present invention have a better conversion activity and a better selectivity in middle distillates than commercial catalysts, even for zeolite contents considerably lower than those of commercial catalysts.
- hydrocracking catalyst C1 (according to the invention) containing a COK-7 zeolite and a silica-alumina
- hydrocracking catalyst C2 (in accordance with the invention) containing a COK-7 zeolite, a ZBM-30 zeolite and a silica-alumina
- a catalyst C4 (not in accordance with the invention) containing a zeolite Y and a silica-alumina
- the COK-7 zeolite is synthesized according to patent EP 1 702 888 A1 with the organic structuring agent triethylenetetramine. Then it is calcined at 550 ° C. under a stream of dry air for 12 hours.
- the zeolite H-COK-7 (acid form) thus obtained has an Si / Al ratio of 52 and an Na / Al ratio of less than 0.002.
- the zeolite ZBM-30 is synthesized according to the patent BASF EP-A-46504 with the organic structuring triethylenetetramine. Then it is calcined at 550 ° C. under a stream of dry air for 12 hours.
- the zeolite H-ZBM-30 (acid form) thus obtained has an Si / Al ratio of 45 and an Na / Al ratio of less than 0.001.
- a silica-alumina precursor SA1 is prepared in the following manner: An alumina hydrate is prepared according to the teachings of US-A-3,124,418. After filtration, the freshly prepared precipitate (P1) is mixed with a sodium hydroxide solution. silicic acid prepared by exchange on decationizing resin. The proportions of the two solutions are adjusted to achieve a composition of 70% Al 2 O 3 - 30% SiO 2 on the final solid. This mixture is rapidly homogenized in a commercial colloid mill in the presence of nitric acid so that the nitric acid content of the suspension at the mill outlet is 8% based on the mixed silica-alumina solid. Then, the suspension (P2) is conventionally dried in an atomizer in a conventional manner of 300 ° C.
- the powder thus prepared is shaped in a Z-shaped arm in the presence of 8% of nitric acid relative to anhydrous product.
- the extrusion is carried out by passing the paste through a die provided with orifices of diameter 1, 4 mm.
- E1 extruded containing 100% silica-alumina thus obtained are dried at 150 0 C 1 then calcined at 550 0 C, then calcined at 750 0 C in the presence of water vapor.
- zeolite COK-7 3 g of zeolite COK-7, 2 g of ZBM-30 described above and 15 g of the precursor of the silica-alumina P2 described above are then mixed. This mixing is done before introduction into the extruder.
- the zeolite powder is first wetted and added to the matrix suspension in the presence of 66% nitric acid (7% by weight of acid per gram of dry gel) and then kneaded for 15 minutes. At the end of this mixing, the paste obtained is passed through a die having cylindrical orifices of diameter equal to 1.4 mm.
- the extrudates are then dried overnight at 120 ° C. under air and then calcined at 550 ° C. under air.
- the E3 extrudates contain 20% by weight of zeolite (60% COK-7 + 40% ZBM-30) and 80% silica-alumina.
- extrusions E1.E2 and E3 are then impregnated dry with an aqueous solution of a mixture of ammonium heptamolybdate, nickel nitrate and orthophosphoric acid, dried overnight at 120 ° C. under air and finally calcined under air at 550 ° C.
- the oxide weight contents of the catalysts C1, C2 and C3 thus obtained are 3.0% NiO, 14.0% MoO 3 and 4.6% P 2 O 5 .
- Catalyst C4 is identical to catalyst C1 with zeolite Y instead of zeolite COK-7.
- the zeolite Y used is a reference commercial zeolite CBV780 (Zeolyst International). It has a Si / Al ratio of 43.5 and an Na / Al ratio of less than 0.004.
- Catalysts C1, C2, C3 and C4 are evaluated by hydrocracking a vacuum distillate under the conditions of high conversion hydrocracking (60-100%).
- the petroleum feed is a hydrotreated vacuum distillate whose main characteristics are as follows:
- This feedstock was obtained by hydrotreating a vacuum distillate over an HR448 catalyst sold by AXENS comprising a group VIB element and a group VIII element deposited on alumina.
- a sulfur-containing precursor compound of H 2 S (DMDS) and a nitrogen compound precursor of NH 3 (aniline) are added to the hydrotreated feed in order to simulate the partial pressures of H 2 S and NH 3 present in the second stage of hydrocracking.
- the load is thus enriched with 2.5% sulfur and 1400 ppm nitrogen.
- the feed thus prepared is injected into the hydrocracking test unit which comprises a fixed bed reactor with up-flow of the feed ("up-flow") into which 50 ml of catalyst C1, C2 or C3.
- the catalyst is sulfurized mixture by a gas oil + DMDS + aniline up to 320 0 C. It should be noted that any method of sulfurization in-situ or ex-situ is suitable. Once the sulfurization is complete, the charge described above can be transformed.
- the operating conditions of the test unit are as follows:
- the catalytic performances are expressed by the temperature which makes it possible to reach a crude conversion level of 80% and by the crude distillate average selectivity of 150-380 ° C. These catalytic performances are measured on the catalyst after a stabilization period. , usually at least 48 hours, has been respected.
- the gross conversion CB is taken equal to:
- the gross selectivity SB in middle distillate is taken as equal to: weight of the fraction (150 - 380 ° C) of the effluent oc3 - 1 UU x weight of the fraction 380 ° C ⁇ of the effluent
- the middle distillates obtained are composed of products having a boiling point of between 150 and 380 ° C.
- Table 1 shows that the addition of COK-7 to the silica-alumina makes it possible to improve both the activity of the catalyst and the selectivity in DM.
- Table 1 Catalytic activities of catalysts in hydrocracking high gross conversion (80%)
- Table 2 shows that the addition of COK-7 to silica-alumina in comparison with zeolite Y makes it possible to improve the iso-conversion middle distillate selectivity.
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Crystallography & Structural Chemistry (AREA)
- Materials Engineering (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Inorganic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Catalysts (AREA)
- Production Of Liquid Hydrocarbon Mixture For Refining Petroleum (AREA)
- Silicates, Zeolites, And Molecular Sieves (AREA)
Abstract
Description

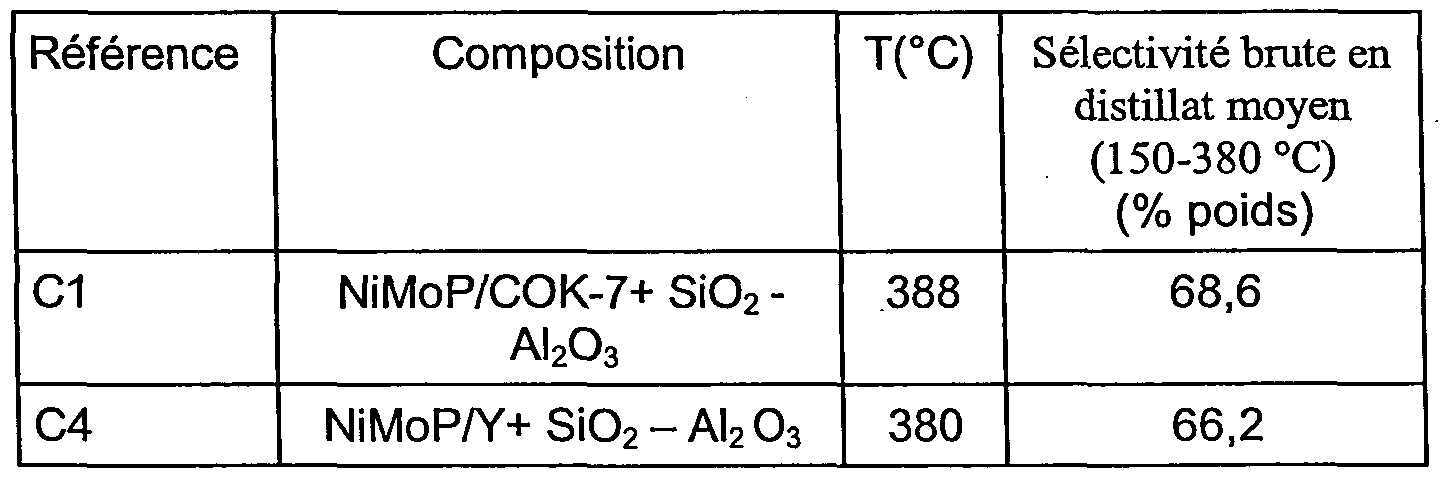
Claims
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08872627A EP2234721A2 (fr) | 2008-01-04 | 2008-12-10 | Catalyseur comprenant au moins une zéolithe particuliere et au moins une silice-alumine et procédé d'hydrocraquage de charges hydrocarbonées utilisant un tel catalyseur |
| BRPI0821825-0A BRPI0821825A2 (pt) | 2008-01-04 | 2008-12-10 | Catalisador que inclui pelo menos uma zeólita em particular e pelo menos uma sílica-alumina e processo para o hidrocraqueamento de estoque de alimentação de hidrocarboneto que usa tal catalisador |
| US12/811,636 US20110042270A1 (en) | 2008-01-04 | 2008-12-10 | Catalyst comprising at least one particular zeolite and at least one silica-alumina, and process for hydrocracking hydrocarbon feeds using said catalyst |
| CN2008801238113A CN101909751A (zh) | 2008-01-04 | 2008-12-10 | 包含至少一种特定沸石和至少一种二氧化硅-氧化铝的催化剂,和使用所述催化剂加氢裂化烃进料的方法 |
| JP2010541077A JP2011508667A (ja) | 2008-01-04 | 2008-12-10 | 少なくとも1種の特定のゼオライトと少なくとも1つのシリカ−アルミナとを含む触媒、および前記触媒を用いた炭化水素供給原料の水素化分解方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR0800055A FR2926028B1 (fr) | 2008-01-04 | 2008-01-04 | Catalyseur comprenant au moins une zeolithe particuliere et au moins une silice-alumine et procede d'hydrocraquage de charges hydrocarbonees utilisant un tel catalyseur |
| FR08/00055 | 2008-01-04 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009103880A2 true WO2009103880A2 (fr) | 2009-08-27 |
| WO2009103880A3 WO2009103880A3 (fr) | 2009-12-03 |
Family
ID=39595569
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/FR2008/001721 WO2009103880A2 (fr) | 2008-01-04 | 2008-12-10 | Catalyseur comprenant au moins une zéolithe particuliere et au moins une silice-alumine et procédé d'hydrocraquage de charges hydrocarbonées utilisant un tel catalyseur |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US20110042270A1 (fr) |
| EP (1) | EP2234721A2 (fr) |
| JP (1) | JP2011508667A (fr) |
| KR (1) | KR20100110854A (fr) |
| CN (1) | CN101909751A (fr) |
| BR (1) | BRPI0821825A2 (fr) |
| FR (1) | FR2926028B1 (fr) |
| WO (1) | WO2009103880A2 (fr) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20120022224A1 (en) | 2010-07-22 | 2012-01-26 | Geraldine Tosin | Particles Including Zeolite Catalysts And Their Use In Oligomerization Processes |
| KR101743293B1 (ko) * | 2010-10-22 | 2017-06-05 | 에스케이이노베이션 주식회사 | 다환방향족 탄화수소로부터 고부가의 경방향족 탄화수소를 제조하는 수소화 분해 촉매 |
| KR102008954B1 (ko) * | 2011-10-24 | 2019-08-08 | 토탈 라피나쥬 프랑스 | 메조기공-함유 촉매의 제조방법, 이에 따라 획득된 촉매 및 이의 수소첨가전환 공정에서의 용도 |
| RU2502787C1 (ru) * | 2012-08-27 | 2013-12-27 | Федеральное государственное бюджетное учреждение науки Институт проблем переработки углеводородов Сибирского отделения Российской академии наук | Способ уменьшения вязкости мазута |
| FR3003563B1 (fr) * | 2013-03-21 | 2015-03-20 | IFP Energies Nouvelles | Procede de conversion de charges issues de sources renouvelables mettant en oeuvre un catalyseur comprenant une zeolithe nu-10 et une silice alumine |
| KR101554265B1 (ko) | 2013-12-19 | 2015-09-18 | 에쓰대시오일 주식회사 | 비결정질 실리카알루미나-제올라이트 복합체 및 이의 제조방법 |
| WO2016029387A1 (fr) * | 2014-08-27 | 2016-03-03 | 中国石油天然气集团公司 | Catalyseur de transfert de mercaptan bimétallique utilisé dans l'élimination à basse température de mercaptans contenus dans le gaz de pétrole liquéfié |
| KR102444820B1 (ko) * | 2016-12-21 | 2022-09-20 | 사우디 아라비안 오일 컴퍼니 | 수소화분해 공정을 위해 촉매 적재를 최적화하는 방법 |
| US11185850B2 (en) * | 2019-12-02 | 2021-11-30 | Saudi Arabian Oil Company | Dual functional composite catalyst for olefin metathesis and cracking |
| US11577235B1 (en) * | 2021-08-13 | 2023-02-14 | Chevron U.S.A. Inc. | Layered catalyst reactor systems and processes for hydrotreatment of hydrocarbon feedstocks |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0046504A1 (fr) * | 1980-08-21 | 1982-03-03 | BASF Aktiengesellschaft | Zéolite métalsilicate cristalline ZBM-30 et procédé pour sa préparation |
| FR2852864A1 (fr) * | 2003-03-24 | 2004-10-01 | Inst Francais Du Petrole | Catalyseur comprenant au moins une zeolithe choisie parmi zbm-30, zsm-48, eu-2 et eu-11 et au moins une zeolithe y et procede d'hydroconversion de charges hydrocarbonees utilisant un tel catalyseur |
| FR2863913A1 (fr) * | 2003-12-23 | 2005-06-24 | Inst Francais Du Petrole | Catalyseur zeolithique,support a base de matrice silico-aluminique et de zeolithe, et procede d'hydrocraquage de charges hydrocarbonees |
| EP1702888A1 (fr) * | 2005-03-07 | 2006-09-20 | Institut Francais Du Petrole | Solide cristallise COK-7, procécé de préparation et utilisation pour la transformation d'hydrocarbures |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2846574B1 (fr) * | 2002-10-30 | 2006-05-26 | Inst Francais Du Petrole | Catalyseur et procede d'hydrocraquage de charges hydrocarbonees |
| FR2852865B1 (fr) * | 2003-03-24 | 2007-02-23 | Inst Francais Du Petrole | Catalyseur et son utilisation pour l'amelioration du point d'ecoulement de charges hydrocarbonnees |
| US7402236B2 (en) * | 2004-07-22 | 2008-07-22 | Chevron Usa | Process to make white oil from waxy feed using highly selective and active wax hydroisomerization catalyst |
| FR2874837B1 (fr) * | 2004-09-08 | 2007-02-23 | Inst Francais Du Petrole | Catalyseur dope et procede ameliore de traitement de charges hydrocarbonees |
| FR2881128B1 (fr) * | 2005-01-24 | 2007-03-23 | Inst Francais Du Petrole | Nouvelle methode de synthese de la zeolithe zbm-30 a partir d'un melange de composes amines |
-
2008
- 2008-01-04 FR FR0800055A patent/FR2926028B1/fr not_active Expired - Fee Related
- 2008-12-10 EP EP08872627A patent/EP2234721A2/fr not_active Withdrawn
- 2008-12-10 CN CN2008801238113A patent/CN101909751A/zh active Pending
- 2008-12-10 JP JP2010541077A patent/JP2011508667A/ja active Pending
- 2008-12-10 BR BRPI0821825-0A patent/BRPI0821825A2/pt not_active IP Right Cessation
- 2008-12-10 KR KR1020107017268A patent/KR20100110854A/ko not_active Application Discontinuation
- 2008-12-10 US US12/811,636 patent/US20110042270A1/en not_active Abandoned
- 2008-12-10 WO PCT/FR2008/001721 patent/WO2009103880A2/fr active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0046504A1 (fr) * | 1980-08-21 | 1982-03-03 | BASF Aktiengesellschaft | Zéolite métalsilicate cristalline ZBM-30 et procédé pour sa préparation |
| FR2852864A1 (fr) * | 2003-03-24 | 2004-10-01 | Inst Francais Du Petrole | Catalyseur comprenant au moins une zeolithe choisie parmi zbm-30, zsm-48, eu-2 et eu-11 et au moins une zeolithe y et procede d'hydroconversion de charges hydrocarbonees utilisant un tel catalyseur |
| FR2863913A1 (fr) * | 2003-12-23 | 2005-06-24 | Inst Francais Du Petrole | Catalyseur zeolithique,support a base de matrice silico-aluminique et de zeolithe, et procede d'hydrocraquage de charges hydrocarbonees |
| EP1702888A1 (fr) * | 2005-03-07 | 2006-09-20 | Institut Francais Du Petrole | Solide cristallise COK-7, procécé de préparation et utilisation pour la transformation d'hydrocarbures |
Also Published As
| Publication number | Publication date |
|---|---|
| FR2926028A1 (fr) | 2009-07-10 |
| JP2011508667A (ja) | 2011-03-17 |
| BRPI0821825A2 (pt) | 2015-06-16 |
| US20110042270A1 (en) | 2011-02-24 |
| FR2926028B1 (fr) | 2010-02-12 |
| WO2009103880A3 (fr) | 2009-12-03 |
| KR20100110854A (ko) | 2010-10-13 |
| EP2234721A2 (fr) | 2010-10-06 |
| CN101909751A (zh) | 2010-12-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2590108C (fr) | Catalyseur zeolithique a teneur controlee en element dopant et procede ameliore de traitement de charges hydrocarbonees | |
| EP1415712B1 (fr) | Catalyseur et procédé d'hydrocraquage de charges hydrocarbonées | |
| EP1634643B1 (fr) | Catalyseur dopé sur support silice-alumine et procédé de traitement de charges hydrocarbonées | |
| EP2313344B1 (fr) | Procede de production de distillats moyens par hydrocraquage de charges issues du procédé fischer-tropsch en presence d'un catalyseur comprenant un solide izm-2 | |
| WO2009103880A2 (fr) | Catalyseur comprenant au moins une zéolithe particuliere et au moins une silice-alumine et procédé d'hydrocraquage de charges hydrocarbonées utilisant un tel catalyseur | |
| EP1590424B1 (fr) | Procede de production de distillats moyens par hydroisomerisation et hydrocraquage de charges issues du procede fischer-tropsch | |
| CA2858049C (fr) | Catalyseur comprenant au moins une zeolithe nu-86, au moins une zeolithe usy et une matrice minerale poreuse et procede d'hydroconversion de charges hydrocarbonees utilisant ce catalyseur | |
| FR2863913A1 (fr) | Catalyseur zeolithique,support a base de matrice silico-aluminique et de zeolithe, et procede d'hydrocraquage de charges hydrocarbonees | |
| CA2605400C (fr) | Procede de production de distillats moyens par hydroisomerisation et hydrocraquage de charges issues du procede fischer-tropsch | |
| EP2794102B1 (fr) | Procede de preparation d'un catalyseur utilisable en hydroconversion comprenant au moins une zéolithe nu-86 | |
| WO2009144411A2 (fr) | Procédé de production de distillats moyens par hydrocraquage de charges issues du procédé fischer-tropsch avec un catalyseur a base d'un materiau cristallise | |
| FR2846664A1 (fr) | Procede flexible de production de bases huiles et de distillats moyens avec une etape de pretraitement convertissant suivie d'un deparaffinage catalytique | |
| WO2009144412A2 (fr) | Procédé de production de distillats moyens par hydrocraquage de charges issues du procédé fischer-tropsch avec un catalyseur a base d'un materiau amorphe | |
| WO2009106705A2 (fr) | Procede de production de distillats moyens par hydroisomerisation et hydrocraquage de charges issues du procede fischer-tropsch | |
| FR2981943A1 (fr) | Procede d'hydrocraquage oligomerisant d'une charge paraffinique issue de la synthese fischer tropsch mettant en oeuvre un catalyseur a base de zeolithe beta dispersee | |
| WO2013153318A1 (fr) | Procede d'hydrotraitement et de deparaffinage d'une charge distillats moyens utilisant un catalyseur a base de zeolithe izm-2 | |
| WO2013153317A1 (fr) | Procédé de deparaffinage de charges hydrocarbonées utilisant un catalyseur a base de zeolithe izm-2 | |
| WO2011045484A1 (fr) | Procede de production de distillat moyen a partir de cires fischer tropsch utilisant un catalyseur a base de zeolithe modifiee | |
| WO2023117475A1 (fr) | Catalyseur comprenant un support à base de matrice silico-aluminique et de zéolithe, sa préparation et procédé d'hydrocraquage de charges hydrocarbonées |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880123811.3 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08872627 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008872627 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3982/CHENP/2010 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010541077 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20107017268 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12811636 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0821825 Country of ref document: BR Kind code of ref document: A2 Effective date: 20100702 |