WO2008028590A1 - Neue substituierte bipyridin-derivate und ihre verwendung als adenosin rezeptor liganden - Google Patents
Neue substituierte bipyridin-derivate und ihre verwendung als adenosin rezeptor liganden Download PDFInfo
- Publication number
- WO2008028590A1 WO2008028590A1 PCT/EP2007/007572 EP2007007572W WO2008028590A1 WO 2008028590 A1 WO2008028590 A1 WO 2008028590A1 EP 2007007572 W EP2007007572 W EP 2007007572W WO 2008028590 A1 WO2008028590 A1 WO 2008028590A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- formula
- hydroxy
- alkyl
- alkylamino
- Prior art date
Links
- 0 **C(**1)=C*(*)C=C1c(c(C#N)c(nc1N)Sc2ccccc2)c1C#N Chemical compound **C(**1)=C*(*)C=C1c(c(C#N)c(nc1N)Sc2ccccc2)c1C#N 0.000 description 4
- RXSLHYTZMIUANX-UHFFFAOYSA-N CC(Nc(cc1)ncc1C(O)=O)=O Chemical compound CC(Nc(cc1)ncc1C(O)=O)=O RXSLHYTZMIUANX-UHFFFAOYSA-N 0.000 description 1
- VVDNQGICIUVXTD-UHFFFAOYSA-N CC(Nc1ncc(C=O)cc1)=O Chemical compound CC(Nc1ncc(C=O)cc1)=O VVDNQGICIUVXTD-UHFFFAOYSA-N 0.000 description 1
- SGBVTSMREKMKRA-UHFFFAOYSA-N CC(Nc1ncc(CO)cc1)=O Chemical compound CC(Nc1ncc(CO)cc1)=O SGBVTSMREKMKRA-UHFFFAOYSA-N 0.000 description 1
- VGNDGMBSYVSQOO-FQEVSTJZSA-N CC1(C)O[C@@H](COc(cc2)ncc2-c(c(C#N)c(nc2N)SCc3c[o]c(-c(cc4)ccc4Cl)n3)c2C#N)CO1 Chemical compound CC1(C)O[C@@H](COc(cc2)ncc2-c(c(C#N)c(nc2N)SCc3c[o]c(-c(cc4)ccc4Cl)n3)c2C#N)CO1 VGNDGMBSYVSQOO-FQEVSTJZSA-N 0.000 description 1
- BGGPUCCJQGIJRL-UHFFFAOYSA-N ClCc1c[o]c(-c(cc2)ccc2Cl)n1 Chemical compound ClCc1c[o]c(-c(cc2)ccc2Cl)n1 BGGPUCCJQGIJRL-UHFFFAOYSA-N 0.000 description 1
- RHRWTBORHGIPTG-UHFFFAOYSA-N Fc(c(F)c1)ccc1-c1nc(CCl)c[o]1 Chemical compound Fc(c(F)c1)ccc1-c1nc(CCl)c[o]1 RHRWTBORHGIPTG-UHFFFAOYSA-N 0.000 description 1
- NDUMNYHIXOJHPM-UHFFFAOYSA-N Nc(nc(c(C#N)c1-c(cc2)cnc2OCCO)SCc2c[s]c(-c(cc3)ccc3Cl)n2)c1C#N Chemical compound Nc(nc(c(C#N)c1-c(cc2)cnc2OCCO)SCc2c[s]c(-c(cc3)ccc3Cl)n2)c1C#N NDUMNYHIXOJHPM-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/84—Nitriles
- C07D213/85—Nitriles in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the present application relates to novel substituted 2,4'- and 3,4'-bipyridine derivatives, processes for their preparation, their use for the treatment and / or prophylaxis of diseases and their use for the preparation of medicaments for the treatment and / or prophylaxis of Diseases, preferably for the treatment and / or prevention of hypertension and other cardiovascular diseases.
- Adenosine a purine nucleoside
- Adenosine is present in all cells and is released from a variety of physiological and pathophysiological stimuli.
- Adenosine is produced intracellularly by the degradation of adenosine 5'-monophosphate (AMP) and S-adenosyl homocysteine as an intermediate, but can be released from the cell and then functions as a hormone-like substance or neurotransmitter through binding to specific receptors.
- AMP adenosine 5'-monophosphate
- S-adenosyl homocysteine as an intermediate, but can be released from the cell and then functions as a hormone-like substance or neurotransmitter through binding to specific receptors.
- adenosine Under normoxic conditions, the concentration of free adenosine in the extracellular space is very low. However, the extracellular concentration of adenosine in the affected organs increases dramatically under both ischemic and hypoxic conditions. For example, it is known that adenosine inhibits platelet aggregation and increases blood flow in the coronary arteries. It also affects blood pressure, heart rate, neurotransmitter release and lymphocyte differentiation. In adipocytes, adenosine is able to inhibit lipolysis and thus reduce the concentration of free fatty acids and triglycerides in the blood.
- adenosine aim to increase the supply of oxygen in the affected organs or to reduce the metabolism of these organs in order to achieve an adaptation of the organ metabolism to the organ perfusion under ischemic or hypoxic conditions.
- adenosine receptor-selective ligands are substances which bind selectively to one or more subtypes of the adenosine receptors and either mimic the action of adenosine (adenosine agonists) or block its action (adenosine antagonists).
- adenosine receptors are mediated intracellularly by the messenger cAMP.
- an activation of the membrane-bound adenylate cyclase leads to an increase in the intracellular cAMP, while the binding of adenosine to the Al or A3 receptors causes a decrease in the intracellular cAMP content via adenylate cyclase inhibition.
- adenosine receptors In the cardiovascular system, the main effects of activation of adenosine receptors are bradycardia, negative inotropy and protection of the heart from ischemia (preconditioning) via Al receptors, dilation of the vessels via A2a and A2b receptors, as well as inhibition of fibroblasts and smooth muscle cell proliferation via A2b receptors.
- A2b receptors by adenosine or specific A2b agonists leads to a blood pressure reduction via the dilation of vessels. Lowering blood pressure is accompanied by a reflex increase in heart rate. Heart rate increase can be reduced by activation of Al receptors by specific Al agonists.
- the above-mentioned receptor selectivity can be determined by the action of the substances on cell lines expressing the respective receptor subtypes after stable transfection with the corresponding cDNA (see in this regard the document ME Olah, H. Ren, J. Ostrowski, KA Jacobson, GL Stiles , "Cloning, expression, and characterization of the unique bovine al adenosine receptor.” Studies on the ligand binding site by site-directed mutagenesis ", J. Biol. Chem. 267 (1992), pp. 10764-10770, the disclosure of which is hereby incorporated by reference Scope is incorporated by reference).
- adenosine receptor-specific valid ligands are mainly derivatives based on the natural adenosine [S.-A. Poulsen and R.J. Quinn, "Adenosine Receptors: New Opportunities for Future Drugs", Bioorganic and Medicinal Chemistry 6 (1998), pages 619-641].
- these known from the prior art adenosine ligands usually have the disadvantage that they do not really act receptor specific, are less effective than the natural adenosine or after oral administration are only very weakly effective. Therefore, they are mainly used only for experimental purposes.
- WO 01/25210 and WO 02/070485 describe substituted 2-thio-3,5-dicyano-4-aryl-6-amino-pyridines as adenosine receptor ligands for the treatment of diseases.
- 2-thio-3,5-dicyano-4-phenyl-6-aminopyridines are disclosed as selective ligands of the adenosine A1 receptor
- phenylaminothiazole derivatives are described as dual adenosine A1 / A2b agonists claimed for the treatment of hypertension and other cardiovascular diseases.
- these compounds have, in some cases only very limited solubility in water and other physiological media, which, for example, complicates their formability or even a parenteral application.
- WO 01/62233 discloses various pyridine and pyrimidine derivatives and their use as adenosine receptor modulators. Substituted 3,5-dicyanopyridines as calcium-dependent potassium channel openers for the treatment of urological diseases are claimed in EP 1 302 463-A1.
- WO 2004/054505 claims the use of aminocyanopyridine derivatives as MK 2 inhibitors for the treatment of TNF-mediated diseases.
- the use of 4-aryl- or 4-heteroaryl-substituted aminocyanopyridines as androgen receptor modulators is described in US 2005/0182105.
- WO 02/50071 discloses amino thiazole derivatives as tyrosine kinase inhibitors for the treatment of cancer as well as immunological and allergic diseases.
- the object of the present invention is to provide novel compounds which are selective agonists of the adenosine A1 receptor, as selective agonists of the adenosine A2b receptor or as selective dual agonists of the adenosine A1 and A2b receptor, as such for the treatment and / or prevention in particular of hypertension and other cardiovascular diseases, the metabolic syndrome, diabetes and dyslipidemias and organ protection in transplants and surgical procedures are suitable and beyond have improved solubility in water and physiological media.
- the present invention relates to compounds of the formula (I)
- one of the two ring members X and Y is N and the other is CR 6 , wherein
- R 6 is hydrogen or (C 1 -C 4 ) -alkyl
- Z is NR 7 or O, in which
- R 7 is hydrogen or (CrC 4) alkyl which may be substituted with hydroxy or (C r C 4) alkoxy,
- R 1 and R 2 are identical or different and are each independently of the other hydrogen or (C 1 -C 6 ) -alkyl which is monosubstituted or disubstituted, identical or different, with hydroxyl, (C 1 -C 4 ) -alkoxy,
- Amino, mono- (Ci-C 4 ) -alkylamino, di- (C r C 4 ) alkylamino, carboxyl, (Ci-C 4 ) alkoxycarbonyl and / or a 4- to 7-membered heterocycle may be substituted , stand,
- heterocycle contains one or two ring heteroatoms from the series N, O and / or S and in turn once or twice, identically or differently, with (Ci-C 4 ) - alkyl, hydroxy, oxo and / or (Ci -C 4 ) -alkoxy may be substituted,
- R 1 and R 2 together with the nitrogen atom to which they are attached form a 4- to 7-membered heterocycle containing one further ring heteroatom from the series N, O or S and one or two times, same or different , may be substituted by (C 1 -C 4 ) -alkyl, hydroxyl, oxo and / or (C 1 -C 4 ) -alkoxy,
- R 3 is hydrogen or (Ci-Ce) -alkyl, which is mono- or disubstituted by identical or different (C 3 -C 6) cycloalkyl, oxo, hydroxy, (C r C4) alkoxy, carboxyl, amino , mono- (C, -C 4) - alkylamino and / or di- (C iC 4) alkylamino may be substituted, or (C 4 -Ce) -CyCIo- alkyl,
- the mentioned cycloalkyl radicals in turn up to two times by identical or different, may be substituted with (C r C 4) alkyl, hydroxy, oxo and / or (C r C 4) alkoxy, and these cycloalkyl groups one ring -CH 2 group can be exchanged for an O atom,
- R 4 is hydrogen, halogen, (C r C4) alkyl or (C r C 4) alkoxy, wherein alkyl and alkoxy can be up to trisubstituted by fluorine,
- R 5 is (C 6 -C 10) -aryl or 5- to 10-membered heteroaryl having up to three ring heteroatoms from the series N, O and / or S, which in each case
- (U) may be substituted with pyrrolidines, piperidino, morpholino, piperazino, N '- (Ci-C 4 ) alkylpiperazino or a group of the formula -LR 8 , wherein
- L is a bond, NH or O means
- R 8 is phenyl or 5- or 6-membered heteroaryl having up to three ring heteroatoms from the series N, O and / or S, which in each case one to three times, identically or differently, with halogen, nitro, cyano, (Ci -C) -Allyl, trifluoromethyl, hydroxy, (C 1 -C 6 ) -alkoxy, difluoromethoxy, trifluoromethoxy, amino, mono- (Ci-C 6) alkylamino, di- (Ci-C 6) alkylamino, (C r C6) -alkoxycarbonyl and / or carboxyl can be substituted,
- Compounds according to the invention are the compounds of the formula (I) and their salts, solvates and solvates of the salts comprising the compounds of the formulas below and their salts, solvates and solvates of the salts encompassed by formula (I) and those of the formula (I), hereinafter referred to as exemplary compounds and their salts, solvates and solvates of the salts, as far as the compounds of formula (I), the compounds mentioned below are not already salts, solvates and solvates of the salts.
- the compounds of the invention may exist in stereoisomeric forms (enantiomers, diastereomers).
- the invention therefore includes the enantiomers or diastereomers and their respective mixtures. From such mixtures of enantiomers and / or diastereomers, the stereoisomerically uniform components can be isolated in a known manner.
- the present invention encompasses all tautomeric forms.
- Salts used in the context of the present invention are physiologically acceptable salts of the compounds according to the invention. Also included are salts which are themselves unsuitable for pharmaceutical applications but can be used, for example, for the isolation or purification of the compounds of the invention.
- Physiologically acceptable salts of the compounds of the invention include acid addition salts of mineral acids, carboxylic acids and sulfonic acids, e.g. Salts of hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, benzenesulfonic acid, naphthalene disulfonic acid, acetic acid, trifluoroacetic acid, propionic acid, lactic acid, tartaric acid, malic acid, citric acid, fumaric acid, maleic acid and benzoic acid.
- salts of hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, benzenesulfonic acid, naphthalene disulfonic acid acetic acid, trifluoroacetic acid, propi
- Physiologically acceptable salts of the compounds according to the invention also include salts of customary bases, such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts) and ammonium salts derived from ammonia or organic amines having 1 to 16 carbon atoms, as exemplified and preferably ethylamine, diethylamine, triethylamine, ethyldiisopropylamine, monoethanolamine, diethanolamine, Triethanolamine, dicyclohexylamine, dimethylaminoethanol, procaine, dibenzylamine, N-methylmorpholine, arginine, lysine, ethylenediamine and N-methylpiperidine.
- customary bases such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts) and ammoni
- solvates are those forms of the compounds according to the invention which form a complex in the solid or liquid state by coordination with solvent molecules. Hydrates are a special form of solvates that coordinate with water. As solvates, hydrates are preferred in the context of the present invention.
- the present invention also includes prodrugs of the compounds of the invention.
- prodrugs includes compounds which may themselves be biologically active or inactive, but during their residence time in the body are converted to compounds of the invention (for example metabolically or hydrolytically).
- (C 1 -Q) -AlkVl and (C 1 -C 1 VAIkVl in the context of the invention are a straight-chain or branched alkyl radical having 1 to 6, 1 to 4 or 1 to 3 carbon atoms.
- a straight-chain or branched one is preferred Alkyl radical having from 1 to 4, more preferably from 1 to 3, carbon atoms, by way of example and by preference: methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, 1 Ethylpropyl, n-pentyl and n-hexyl.
- (C7-Cfi) -alkenyl in the context of the invention is a straight-chain or branched alkenyl radical having 2 to 6 carbon atoms and one or two double bonds. Preference is given to a straight-chain or branched alkenyl radical having 2 to 4 carbon atoms and one double bond.
- Examples which may be mentioned are: methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, tert-butoxy, n-pentoxy and n-hexoxy.
- (QC alkoxycarbonyl and (C j -QVAlkoxycarbonyl are in the context of the invention a straight-chain or branched alkoxy radical having from 1 to 6 or 1 to 4 carbon atoms, which is linked via a carbonyl group.
- a linear or branched alkoxy carbonyl group is preferred with From 1 to 4 carbon atoms in the alkoxy group, by way of example and by preference: methoxycarbonyl, ethoxycarbonyl, n-propoxycarbonyl, isopropoxycarbonyl and tert-butoxycarbonyl.
- Mono-CC j -QValkylamino and mono-fCVCO-alkylamino stand for the purposes of the invention for an amino group having a straight-chain or branched alkyl substituent which has 1 to 6, 1 to 4 or 1 to 3 carbon atoms. Preference is given to a straight-chain or branched monoalkylamino radical having 1 to 4, particularly preferably 1 to 3, carbon atoms.
- Examples which may be mentioned are: methylamino, ethylamino, n-propylamino, isopropylamino, n-butylamino, tert-butylamino, n-pentylamino and n-hexylamino.
- Mono (C 2 -CW) -alkenylamino in the context of the invention represents an amino group having a straight-chain or branched alkenyl substituent which has 2 to 6 carbon atoms. Preference is given to a straight-chain or branched monoalkenylamino radical having 1 to 4 carbon atoms.
- allylamino 1-methylprop-2-en-1-ylamino, 2-methylprop-2-en-1-ylamino, but-2-en-1-ylamino and 3-but-1-enyl ylamino.
- DHCpCaValkylamino and di-fCVCO-alkylamino in the context of the invention represent an amino group having two identical or different straight-chain or branched alkyl substituents, each having 1 to 6, 1 to 4 or 1 to 3 carbon atoms. Preference is given to straight-chain or branched dialkylamino radicals each having 1 to 4, particularly preferably 1 to 3, carbon atoms in each case.
- N N-dimethylamino, N, N-diethylamino, N-ethyl-N-methylamino, N-methyl-Nn-propylamino, N-isopropyl-Nn-propylamino, N, N-diisopropylamino, Nn Butyl-N-methylamino, N-tert-butyl-N-methylamino, N-ethyl-Nn-pentylamino and Nn-hexyl-N-methylamino.
- (Cfi-Cjn) -aryl represents in the context of the invention an aromatic carbocycle having 6 or 10 ring carbon atoms.
- Preferred aryl radicals are phenyl and ⁇ aphthyl.
- a 4- to 7-membered heterocycle in the context of the invention is a saturated heterocycle having a total of 4 to 7 ring atoms which contains one or two ring heteroatoms from the series ⁇ , O and / or S and via a ring carbon atom or optionally a ring nitrogen atom is linked.
- Preferred is a 5- or 6-membered heterocycle having one or two ring heteroatoms from the series ⁇ and / or O.
- 5- to 10-membered heteroaryl is in the context of the invention for a mono- or optionally bicyclic aromatic heterocycle (heteroaromatic) having a total of 5 to 10 ring atoms, up to three identical or different ring heteroatoms from the series N, O and contains S and is linked via a ring carbon atom or optionally via a ring nitrogen atom.
- aromatic heterocycle heteromatic
- Examples which may be mentioned are: furyl, pyrrolyl, thienyl, pyrazolyl, imidazolyl, thiazolyl, oxazolyl, isoxazolyl, isothiazolyl, triazolyl, oxadiazolyl, thiadiazolyl, pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl, triazinyl, benzofuranyl, benzothienyl, benzimidazolyl, benzoxa zolyl, benzothiazolyl, benzotriazolyl, indolyl, indazolyl, quinolinyl, isoquinolinyl, naphthyridinyl, quinazolinyl, quinoxalinyl, phthalazinyl, pyrazolo [3,4-b] pyridinyl.
- Halogen in the context of the invention includes fluorine, chlorine, bromine and iodine. Preference is given to chlorine or fluorine.
- radicals are substituted in the compounds according to the invention, the radicals can, unless otherwise specified, be monosubstituted or polysubstituted. In the context of the present invention, the meaning is independent of each other for all radicals which occur repeatedly. Substitution with one, two or three identical or different substituents is preferred. Most preferably, the substitution with one or two identical or different substituents.
- one of the two ring members X and Y is N and the other is CH,
- Z is NR 7 or O, in which
- R 7 is hydrogen or methyl
- R 1 is hydrogen or (C r C 4) alkyl substituted with hydroxy, (C r C4) alkoxy, amino, mono- (C r C 4) alkylamino, di- (Ci-C 4) alkylamino , Carboxyl, (C 1 -C 4 ) -alkoxycarbonyl or a 5- or 6-membered heterocycle may be substituted, wherein said heterocycle contains one or two ring heteroatoms from the series N and / or O and in turn may be monosubstituted or disubstituted, identically or differently, by methyl, ethyl, hydroxy, methoxy and / or ethoxy,
- R 2 is hydrogen or methyl
- R 1 and R 2 together with the nitrogen atom to which they are attached form a 5- or 6-membered heterocycle containing a further ring heteroatom from the series N and O and one or two times, identical or different, with Methyl, ethyl, hydroxy, methoxy and / or ethoxy may be substituted,
- R 3 is (Ci-C 4) -alkyl which is mono- or disubstituted by identical or different (C 3 -C 5) cycloalkyl, oxo, hydroxy, (C r C 3) alkoxy, amino, mono- (Ci-C 3) alkylamino and / or di- (C r C 3) - alkylamino may be substituted, or represents cyclopentyl or cyclohexyl,
- R 4 is hydrogen, fluorine or chlorine
- R 5 is phenyl or 5- or 6-membered heteroaryl having up to three ring heteroatoms from the series N, O and / or S, which in each case
- (U) may be substituted with morpholino, N '- (Ci-C 4 ) alkylpiperazino or a group of the formula -LR 8 , wherein
- L is a bond or NH
- R 8 is phenyl or 5- or 6-membered heteroaryl having up to three ring heteroatoms from the series N, O and / or S, which in each case one to three times, identically or differently, with fluorine, chlorine, cyano, (C r C 4 ) -alkyl, trifluoromethyl, (C 1 -C 4 ) -alkoxy, trifluoromethoxy and / or carboxyl may be substituted,
- one of the two ring members X and Y is N and the other is CH,
- Z is NH or O
- R 1 is hydrogen or (Ci-C 4) alkyl substituted with hydroxy, (C 1 -Q) -alkoxy, amino, mono- (C r C 4) -alkylamino or di- (C r C 4) alkylamino may be substituted,
- R 2 is hydrogen or methyl
- R 1 and R 2 together with the nitrogen atom to which they are attached form a pyrrolidino, piperidino, morpholino or piperazino ring, each one or two times, identically or differently, with methyl, ethyl, hydroxy, methoxy and / or ethoxy may be substituted,
- R 3 is (Q-GO-alkyl which may be monosubstituted or disubstituted by identical or different substituents, oxo, hydroxy, methoxy, ethoxy and / or amino,
- R 4 is hydrogen
- R 5 is phenyl or 5- or 6-membered heteroaryl having up to two ring heteroatoms from the series N, O and / or S, which in each case
- (//) may be substituted with a group of the formula -LR 8 wherein L is a bond or NH
- R 8 is phenyl or pyridyl, which in each case may be monosubstituted or disubstituted, identical or different, with fluorine, chlorine, cyano, methyl, trifluoromethyl and / or methoxy,
- R 1 is hydrogen or (C 1 -C 4 ) -alkyl which is hydroxyl, (C 1 -C 4 ) -alkoxy, amino, mono- (Q-C 4 ) -alkylamino or di- (C 1 -C 4 ) -alkylamino may be substituted,
- R 2 is hydrogen or methyl
- R 1 and R 2 together with the nitrogen atom to which they are attached form a pyrrolidino, piperidino, morpholino, piperazino or N'-methylpiperazino ring,
- R 3 is 2-hydroxyethyl, 2-hydroxy-1-methylethyl, 2-hydroxypropyl, 2-hydroxy-2-methylpropyl, 3-hydroxypropyl or 2,3-dihydroxypropyl,
- R 4 is hydrogen
- R 5 is pyrazolyl, oxazolyl, thiazolyl, pyridyl or pyrimidinyl, each of which
- (/) may be substituted with methyl, ethyl or amino
- R 8 is phenyl or pyridyl, which in each case may be monosubstituted or disubstituted, identical or different, with fluorine, chlorine, cyano, methyl and / or methoxy,
- one of the two ring members X and Y is N and the other is CH,
- Z is NH or O
- R 1 and R 2 are each hydrogen
- R 3 is 2-hydroxyethyl, 2-hydroxy-1-methylethyl, 2-hydroxypropyl, 2-hydroxy-2-methylpropyl, 3-hydroxypropyl, 2,3-dihydroxypropyl or acetyl,
- R 4 is hydrogen
- R 5 is oxazolyl, thiazolyl or pyridyl, each of which may be substituted with methyl, ethyl, amino or a group of the formula -LR 8 wherein
- L is a bond or NH
- R 8 is phenyl or pyridyl, which in each case may be monosubstituted or disubstituted, identical or different, with fluorine, chlorine, cyano, methyl and / or methoxy,
- Another object of the present invention is a process for the preparation of the compounds of the formula (I) according to the invention, which comprises reacting a compound of the formula (D)
- Q is a suitable leaving group, preferably halogen, in particular chlorine, bromine or iodine, or mesylate, tosylate or triflate,
- Suitable solvents for the process according to the invention are all organic solvents which are inert under the reaction conditions. These include alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol and tert-butanol, ketones such as acetone and methyl ethyl ketone, acyclic and cyclic ethers such as diethyl ether, 1- methyl tert-butyl ether, 1,2- Dimethoxyethane, tetrahydrofuran and dioxane, esters such as ethyl acetate or butyl acetate, hydrocarbons such as benzene, toluene, xylene, hexane and cyclohexane, chlorinated hydrocarbons such as dichloromethane, trichloromethane and chlorobenzene, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DM
- Suitable bases are the customary inorganic or organic bases. These include preferably alkali metal hydroxides such as, for example, lithium, sodium or potassium hydroxide, alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate, alkali hydrogen carbonates such as sodium or potassium bicarbonate, alkali metal alcoholates such as sodium or potassium methoxide, sodium or potassium ethanolate or potassium hydroxide.
- alkali metal hydroxides such as, for example, lithium, sodium or potassium hydroxide
- alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate
- alkali hydrogen carbonates such as sodium or potassium bicarbonate
- alkali metal alcoholates such as sodium or potassium methoxide, sodium or potassium ethanolate or potassium hydroxide.
- amides such as sodium amide, lithium, sodium or potassium bis (trimethylsilyl) amide or lithium diisopropylamide
- organometallic compounds such as butyllithium or phenyllithium
- organic amines such as triethylamine, diisopropylethylamine, pyridine, 1,8-diazabicyclo [5.4 .0] undec-7-ene (DBU) or 1,5-diazabicyclo [4.3.0] non-5-ene (DBN).
- DBU 1,8-diazabicyclo [5.4 .0] undec-7-ene
- DBN 1,5-diazabicyclo [4.3.0] non-5-ene
- the base may in this case be used in an amount of 1 to 10 mol, preferably from 1 to 5 mol, in particular from 1 to 4 mol, based on 1 mol of the compound of the formula (IT).
- the reaction is generally carried out in a temperature range from -78 ° C to +140 0 C, forthcoming Trains t in the range from -20 0 C to +60 0 C, especially at 0 0 C to +40 0 C.
- the reaction can in normal, elevated or reduced pressure (eg in the range of 0.5 to 5 bar). Generally, one works at normal pressure.
- the alkali metal sulfide used is preferably sodium sulfide in an amount of from 1 to 10 mol, preferably from 1 to 5 mol, in particular from 1 to 4 mol, based on 1 mol of the compound of the formula (V).
- Suitable solvents are all organic solvents which are inert under the reaction conditions. These include preferably dimethylformamide, N-methylpyrrolidinone, pyridine and acetonitrile. It is likewise possible to use mixtures of the abovementioned solvents. Particularly preferred is dimethylformamide.
- the reaction is generally carried out in a temperature range of +20 0 C to +140 0 C, forthcoming Trains t in the range of +20 0 C to +120 0 C, in particular at +60 0 C to +100 0 C.
- the reaction can be carried out at normal, elevated or reduced pressure (eg in the range of 0.5 to 5 bar). Generally, one works at normal pressure.
- the compounds of the formula (V) can be prepared analogously to processes described in the literature [cf. eg Kambe et al., Synthesis, 531-533 (1981); Elnagdi et al., Z. Naturforsch. 47b. 572-578 (1991); Reddy et al., J. Med. Chem. 49, 607-615 (2006); Evdokimov et al., Org. LeU. 8, 899-902 (2006)].
- Compounds of the formula (II) in which at least one of the two radicals R 1 and R 2 is not hydrogen may be prepared by first compounding compounds of the formula (V) with copper (II) chloride and isoamyl nitrite in a suitable solvent of the formula (VI)
- R 1A has the abovementioned meaning of R 1 ,
- R> 2A has the abovementioned meaning of R
- the process step (V) - »(VI) is generally carried out with a molar ratio of 2 to 12 moles of copper ( ⁇ ) chloride and 2 to 12 moles of isoamyl nitrite based on 1 mol of the compound of formula (V).
- Suitable solvents for this process step are all organic solvents which are inert under the reaction conditions. These include acyclic and cyclic ethers such as diethyl ether and tetrahydrofuran, esters such as ethyl acetate or butyl acetate, hydrocarbons such as benzene, toluene, xylene, hexane and cyclohexane, chlorinated hydrocarbons such as dichloromethane, 1, 2-dichloroethane and chlorobenzene, or other solvents such as Dimethylformamide, acetonitrile or pyridine. It is likewise possible to use mixtures of the abovementioned solvents.
- acyclic and cyclic ethers such as diethyl ether and tetrahydrofuran
- esters such as ethyl acetate or butyl acetate
- hydrocarbons such as benzene, toluene, xylene,
- Preferred solvents are acetonitrile and dimethylformamide.
- the reaction is generally carried out in a temperature range from -78 ° C to +180 0 C, preferably in the range of +20 0 C to +100 0 C, in particular at +20 0 C to +60 0 C.
- the reaction can be carried out under normal , increased or decreased pressure (eg in the range of 0.5 to 5 bar). Generally, one works at normal pressure.
- the process step (VI) + (VII) -> (VOT) is generally carried out with a molar ratio of 1 to 8 mol of the compound of formula (VIT) based on 1 mol of the compound of formula (VI).
- Suitable solvents for this process step are all organic solvents which are inert under the reaction conditions. These include alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol and tert-butanol, ketones such as acetone and methyl ethyl ketone, acyclic and cyclic ethers such as diethyl ether, 1,2-dimethoxyethane, tetrahydrofuran and dioxane, esters such as ethyl acetate or Butyl acetate, hydrocarbons such as benzene, toluene, xylene, hexane and cyclohexane, chlorinated hydrocarbons such as dichloromethane, 1,2-dichloroethane and chlorobenzene, or other solvents such as dimethylformamide, acetonitrile, pyridine or dimethyl sulfoxide. Water is also suitable as a
- the reaction is generally carried out in a temperature range from 0 ° C to +180 0 C, preferably in the range of +20 0 C to +120 0 C, in particular at +20 0 C to +100 0 C.
- the reaction may be at atmospheric, increased or decreased pressure (eg in the range of 0.5 to 5 bar). Generally, one works at normal pressure.
- the process step (VHT) - »(H) is generally carried out with a molar ratio of 1 to 8 MoI sodium sulfide based on 1 mol of the compound of formula (VIET).
- Suitable solvents for this process step are all organic solvents which are inert under the reaction conditions. These include alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol and tert-butanol, ketones such as acetone and methyl ethyl ketone, acyclic and cyclic ethers such as diethyl ether, 1,2-dimethoxyethane, tetrahydrofuran and dioxane, esters such as ethyl acetate or Butyl acetate, hydrocarbons such as benzene, toluene, xylene, hexane and cyclohexane, chlorinated hydrocarbons such as dichloromethane, 1,2-dichloroethane and chlorobenzene, or other solvents such as dimethylformamide, acetonitrile, pyridine, dimethyl sulfoxide or N-methylpyrrolidin
- the reaction is generally carried out in a temperature range of 0 0 C to +180 0 C, preferably in the range of +20 0 C to +120 0 C, in particular at +40 0 C to +100 0 C.
- the reaction may be at atmospheric, increased or decreased pressure (eg in the range of 0.5 to 5 bar). Generally, one works at normal pressure.
- the compounds of the formula (VII) are either commercially available, known to the person skilled in the art or can be prepared by customary methods.
- reaction parameters previously described for the sequence (V) - »(VI) -> (VIE), such as solvents, reaction temperatures and molar ratios find application in an analogous manner.
- the compounds of formula (HI) are likewise commercially available, known from the literature or can be prepared by methods known from the literature.
- amides, thioamides or thiourea derivatives with a 1,3-dihaloacetone, 2-substituted oxazole and thiazole derivatives of the formula (III-A), (DI-B) or (DI-C) obtained (see Scheme 6):
- NCS N-chlorosuccinimide
- Oxazole derivatives of the formula (II) substituted in the 5-position can be obtained, for example, by reduction and subsequent halogenation of corresponding oxazole-4-carboxylic acid esters, which in turn are accessible by acylation of ⁇ -isocyanatoacetates (see Scheme 9): Scheme 9
- [DBU 1, 8-diazabicyclo [5.4.0] undec-7-ene; see. e.g. M. Suzuki et al., J. Org. Chem. 1973, 38, 3571-3575].
- Cat. Catalyst; see. eg, Finch, N., et al., J. Med. Chem. 1980, 23, 1405-1410; ibid. 1978, 21, 1269-1274].
- NMM N-methylmorpholine
- ⁇ MMO N-methylmorpholine-N-oxide
- Pr n-propyl
- the compounds of the invention show an unpredictable, valuable pharmacological activity spectrum and are therefore particularly suitable for the prophylaxis and / or treatment of diseases.
- the substances according to the invention have an improved solubility in water and other physiological media compared with the compounds of the prior art, which is advantageous, for example, for formability and / or parenteral administration.
- the pharmaceutical activity of the compounds according to the invention can be explained by their action as potent, selective ligands on adenosine A1 and / or A2b receptors. They act as selective Al, selective A2b or as selective dual Al / A2b agonists.
- adenosine receptor ligands on adenosine A1 and / or A2b receptors those adenosine receptor ligands are referred to, in which on the one hand a significant effect on Al and / or A2b adenosine receptor subtypes and on the other hand no or a clear weaker effect (factor 10 or higher) on A2a and A3 adenosine receptor subtypes, with respect to the test methods for the selectivity of activity, reference is made to those described in section BI. described tests.
- the compounds of the formula (J) are suitable, alone or in combination with one or more other active substances, for the prophylaxis and / or treatment of various diseases, for example, in particular in hypertension and other diseases of the cardiovascular system (cardiovascular diseases) and for cardioprotection.
- diseases of the cardiovascular system or of cardiovascular diseases, in addition to hypertension are to be understood as meaning, in particular, the following diseases: peripheral and cardiac vascular diseases, coronary heart disease, coronary restenosis, e.g. Restenosis after balloon dilatation of peripheral blood vessels, acute coronary syndrome, stable and unstable angina pectoris, heart failure, tachycardia, arrhythmias, atrial and ventricular fibrillation and peripheral circulatory disorders.
- peripheral and cardiac vascular diseases e.g. Restenosis after balloon dilatation of peripheral blood vessels, acute coronary syndrome, stable and unstable angina pectoris, heart failure, tachycardia, arrhythmias, atrial and ventricular fibrillation and peripheral circulatory disorders.
- the compounds according to the invention are also particularly suitable for the reduction of the myocardium affected by an infarct and for the prophylaxis of secondary infarcts.
- the compounds according to the invention are particularly suitable for the prophylaxis and / or treatment of thromboembolic disorders and ischaemias such as myocardial infarction, stroke and transient ischemic attacks as well as for the protection of organs during transplantations and surgical interventions, for example at the heart.
- the compounds according to the invention are, for example, in particular the prophylaxis and / or treatment of diseases of the genitourinary area, such as irritable bladder, erectile dysfunction and female sexual dysfunction, but also the prophylaxis and / or treatment of inflammatory diseases, such as asthma and inflammatory dermatoses, neuroinflammatory diseases of the central nervous system, such as, for example, conditions after cerebral infarction, Alzheimer's disease, neurodegenerative diseases, as well as pain, cancer and nausea and vomiting associated with cancer treatment.
- diseases of the genitourinary area such as irritable bladder, erectile dysfunction and female sexual dysfunction
- inflammatory diseases such as asthma and inflammatory dermatoses
- neuroinflammatory diseases of the central nervous system such as, for example, conditions after cerebral infarction, Alzheimer's disease, neurodegenerative diseases, as well as pain, cancer and nausea and vomiting associated with cancer treatment.
- a further area of indication are, for example, in particular the prophylaxis and / or treatment of respiratory diseases such as, for example, asthma, chronic bronchitis, emphysema of the lungs, bronchiectasis, cystic fibrosis (cystic fibrosis) and pulmonary hypertension.
- respiratory diseases such as, for example, asthma, chronic bronchitis, emphysema of the lungs, bronchiectasis, cystic fibrosis (cystic fibrosis) and pulmonary hypertension.
- the compounds of the invention for example, in particular for the prophylaxis and / or treatment of diabetes, especially diabetes mellitus, diabetic sequelae such. Nephropathy and neuropathy, the metabolic syndrome and dyslipidaemias.
- Another object of the present invention is the use of the compounds of the invention for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases.
- Another object of the present invention is the use of the compounds of the invention for the manufacture of a medicament for the treatment and / or prophylaxis of Erkran- kungen, in particular the aforementioned diseases.
- Another object of the present invention is a method for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases, using an effective amount of at least one of the compounds of the invention.
- the compounds of the invention may be used alone or as needed in combination with other agents.
- Another object of the present invention are pharmaceutical compositions containing at least one of the compounds of the invention and one or more other active ingredients, in particular for the treatment and / or prophylaxis of the aforementioned diseases.
- Suitable combination active ingredients which may be mentioned by way of example and preferably include lipid metabolism-changing active ingredients, antidiabetics, hypotensive agents, circulation-promoting and / or antithrombotic agents, antioxidants, chemokine receptor antagonists, p38 kinase inhibitors, NPY agonists, orexin Agonists, anorectics, PAF-AH inhibitors, anti-phlogists (COX inhibitors, LTB 4 receptor antagonists) and analgesics such as aspirin.
- the present invention particularly relates to combinations of at least one of the compounds according to the invention with at least one active substance which alters the lipid metabolism. substance, an antidiabetic agent, a hypotensive agent and / or an antithrombotic agent.
- the compounds of the invention may preferably be with one or more
- the lipid metabolism-changing active substances by way of example and preferably from the group of HMG-CoA reductase inhibitors, inhibitors of HMG-CoA reductase expression,
- Squalene synthesis inhibitors include ACAT inhibitors, LDL receptor inducers, cholesterol absorption inhibitors, polymeric bile acid adsorbers, bile acid reabsorption inhibitors, MTP inhibitors, lipase inhibitors, LpL activators, fibrates, niacin, CETP inhibitors, PPAR- ⁇ -, PPAR- ⁇ and / or PPAR- ⁇ agonists, RXR modulators, FXR modulators, LXR modulators, thyroid hormones and / or thyroid mimetics, ATP citrate lyase inhibitors,
- Lp (a) antagonists cannabinoid receptor 1 antagonists, leptin receptor agonists, bomberin receptor agonists, histamine receptor agonists and the antioxidants / free radical scavengers;
- Antidiabetic agents mentioned in Red List 2004 / H, Chapter 12 and by way of example and preferably those from the group of sulfonylureas, biguanides, meglitinide derivatives,
- Glucosidase inhibitors oxadiazolidinones, thiazolidinediones, GLP1 receptor agonists, glutagon antagonists, insulin sensitizers, CCK1 receptor agonists, leptin receptor agonists, inhibitors of liver enzymes involved in the stimulation of gluconeogenesis and / or or glycogenolysis, modulators of glucose uptake and potassium channel openers, such as those disclosed in WO 97/26265 and WO 99/03861;
- hypotensive agents by way of example and preferably from the group of calcium antagonists, angiotensin Aue antagonists, ACE inhibitors, beta-receptor blockers, alpha-receptor blockers, diuretics, phosphodiesterase inhibitors, sGC stimulators, enhancers of the cGMP levels, aldosterone antagonists, mineralocorticoid receptor antagonists, ECE inhibitors and the vasopeptidase inhibitors, and / or
- Antithrombotic agents by way of example and preferably from the group of platelet aggregation inhibitors or anticoagulants
- lipid metabolism-changing active compounds are preferably compounds from the group of HMG-Co A reductase inhibitors, squalene synthesis inhibitors, ACAT inhibitors,
- Cholesterol absorption inhibitors MTP inhibitors, lipase inhibitors, thyroid hormones and / or
- Thyroid mimetics niacin receptor agonists, CETP inhibitors, PPAR- ⁇ agonists, PPAR- ⁇ - Agonists, PPAR- ⁇ agonists, polymeric bile acid adsorbers, bile acid reabsorption inhibitors, antioxidants / radical scavengers, and the cannabinoid receptor 1 antagonists.
- the compounds according to the invention are administered in combination with an HMG-CoA reductase inhibitor from the class of statins, such as by way of example and preferably lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, rosuvastatin, cerivastatin or pitavastatin.
- statins such as by way of example and preferably lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, rosuvastatin, cerivastatin or pitavastatin.
- the compounds according to the invention are administered in combination with a squalene synthesis inhibitor, such as by way of example and preferably BMS-188494 or TAK-475.
- a squalene synthesis inhibitor such as by way of example and preferably BMS-188494 or TAK-475.
- the compounds according to the invention are administered in combination with an ACAT inhibitor, such as by way of example and preferably avasimibe, melinamide, pactimibe, eflucimibe or SMP-797.
- an ACAT inhibitor such as by way of example and preferably avasimibe, melinamide, pactimibe, eflucimibe or SMP-797.
- the compounds according to the invention are administered in combination with a cholesterol absorption inhibitor, such as by way of example and preferably ezetimibe, tiqueside or pamaqueside.
- a cholesterol absorption inhibitor such as by way of example and preferably ezetimibe, tiqueside or pamaqueside.
- the compounds according to the invention are administered in combination with an MTP inhibitor such as, for example and preferably, implitapide, BMS-201038, R-103757 or JTT-130.
- an MTP inhibitor such as, for example and preferably, implitapide, BMS-201038, R-103757 or JTT-130.
- the compounds according to the invention are administered in combination with a lipase inhibitor, such as, for example and preferably, orlistat.
- a lipase inhibitor such as, for example and preferably, orlistat.
- the compounds according to the invention are administered in combination with a thyroid hormone and / or thyroid mimetic, such as by way of example and preferably D-thyroxine or 3,5,3'-triiodothyronine (T3).
- a thyroid hormone and / or thyroid mimetic such as by way of example and preferably D-thyroxine or 3,5,3'-triiodothyronine (T3).
- the compounds according to the invention are administered in combination with an agonist of the Niac ⁇ i receptor, such as by way of example and preferably niacin, Acipimox, Agravecol.
- the compounds according to the invention are administered in combination with a CETP inhibitor, such as, by way of example and by way of preference, torcetrapib, JTT-705, BAY 60-5521, BAY 78-7499 or CETP vaccine (Avant).
- a CETP inhibitor such as, by way of example and by way of preference, torcetrapib, JTT-705, BAY 60-5521, BAY 78-7499 or CETP vaccine (Avant).
- the compounds according to the invention are administered in combination with a PPAR- ⁇ agonist, by way of example and preferably pioglitazone or rosiglitazone.
- the compounds according to the invention are administered in combination with a PPAR- ⁇ agonist, such as by way of example and preferably GW-501516 or BAY 68-5042.
- a PPAR- ⁇ agonist such as by way of example and preferably GW-501516 or BAY 68-5042.
- the compounds according to the invention are administered in combination with a polymeric bile acid adsorbent, such as, by way of example and by way of preference, cholestyramine, colestipol, colesolvam, cholesta gel or colestimide.
- a polymeric bile acid adsorbent such as, by way of example and by way of preference, cholestyramine, colestipol, colesolvam, cholesta gel or colestimide.
- ASBT BAT
- AZD-7806 S-8921
- AK-105 AK-105
- BARI-1741 AK-105
- SC-435 SC-635.
- the compounds according to the invention are administered in combination with an antioxidant / free-radical scavenger, such as, for example, Ubiaft and preferably probucol, AGI-1067, BO-653 or AEOL-10150.
- an antioxidant / free-radical scavenger such as, for example, Ubiaft and preferably probucol, AGI-1067, BO-653 or AEOL-10150.
- the compounds according to the invention are administered in combination with a cannabinoid receptor 1 antagonist, such as by way of example and preferably rimonabant or SR-147778.
- a cannabinoid receptor 1 antagonist such as by way of example and preferably rimonabant or SR-147778.
- Antidiabetic agents are preferably understood as meaning insulin and insulin derivatives as well as orally active hypoglycemic agents.
- Insulin and insulin derivatives here include both insulins of animal, human or biotechnological origin as well as mixtures thereof.
- the orally active hypoglycans are preferably sulfonylureas, biguanides, meglitinide derivatives, glucosidase inhibitors and PPAR- ⁇ agonists.
- the compounds according to the invention are administered in combination with insulin.
- the compounds according to the invention are administered in combination with a sulphonylurea, such as, by way of example and by way of preference, tolbutamide, glibenclamide, glimepiride, glipizide or gliclazide.
- a sulphonylurea such as, by way of example and by way of preference, tolbutamide, glibenclamide, glimepiride, glipizide or gliclazide.
- the compounds according to the invention are administered in combination with a biguanide, by way of example and preferably metformin.
- the compounds according to the invention are administered in combination with a meglitinide derivative, such as by way of example and preferably repaglinide or nateglinide.
- a meglitinide derivative such as by way of example and preferably repaglinide or nateglinide.
- the compounds according to the invention are administered in combination with a glucosidase inhibitor, such as by way of example and preferably migolith or acarbose.
- the compounds according to the invention are administered in combination with a PPAR- ⁇ agonist, for example from the class of thiazolidinediones, such as, by way of example and by way of preference, pioglitazone or rosiglitazone.
- a PPAR- ⁇ agonist for example from the class of thiazolidinediones, such as, by way of example and by way of preference, pioglitazone or rosiglitazone.
- the blood pressure lowering agents are preferably understood as meaning compounds from the group of calcium antagonists, angiotensin AH antagonists, ACE inhibitors, beta-receptor blockers, alpha-receptor B-relaxers and diuretics.
- the compounds according to the invention are administered in combination with a calcium antagonist, such as, by way of example and by way of preference, nifedipine, amlodipine, verapamil or diltiazem.
- a calcium antagonist such as, by way of example and by way of preference, nifedipine, amlodipine, verapamil or diltiazem.
- the compounds according to the invention are administered in combination with an angiotensin AH antagonist, such as by way of example and preferably losartan, valsartan, candesartan, embusartan, olmesartan or telmisartan.
- angiotensin AH antagonist such as by way of example and preferably losartan, valsartan, candesartan, embusartan, olmesartan or telmisartan.
- the compounds according to the invention are administered in combination with an ACE inhibitor, such as by way of example and preferably enalapril, captopril, lisinopril, ramipril, delapril, fosinopril, quinopril, perindopril or trandopril.
- an ACE inhibitor such as by way of example and preferably enalapril, captopril, lisinopril, ramipril, delapril, fosinopril, quinopril, perindopril or trandopril.
- the compounds according to the invention are used in combination with a beta-receptor blocker such as, by way of example and by way of preference, propranolol, atenolol, timolol, pindolol, alprenolol, oxprenolol, penbutolol, bupranolol, metipropanol, nadolol, mepindolol, carazalol, Sotalol, metoprolol, betaxolol, celiprolol, bisoprolol, Carteolol, esmolol, labetalol, carvedilol, adaprolol, landiolol, nebivolol, epanolol or bucine dolol administered.
- a beta-receptor blocker such as, by way of example and by way of preference, propranolol, atenolol
- the compounds according to the invention are used in combination with a diuretic, such as by way of example and preferably furosemide, bumetanide, torsemide, bendroflumethiazide, chlorothiazide, hydrochlorothiazide, hydroflumethiazide, methyclothiazide, polythiazide, trichloromethiazide, chlorthalidone, indapamide, metolazone, quineth- azon, acetazolamide, dichlorophenamide, methazolamide, glycerol, isosorbide, mannitol, amiloride or triamterene.
- a diuretic such as by way of example and preferably furosemide, bumetanide, torsemide, bendroflumethiazide, chlorothiazide, hydrochlorothiazide, hydroflumethiazide, methyclothiazide, polythiazide, trich
- the compounds according to the invention are administered in combination with antisympathotonics such as reserpine, clonidine or alpha-methyl-dopa, with potassium channel agonists such as minoxidil, diazoxide, dihydralazine or hydralazine, or with nitric oxide-releasing substances such as glyceryl nitrate or nitroprusside sodium.
- antisympathotonics such as reserpine, clonidine or alpha-methyl-dopa
- potassium channel agonists such as minoxidil, diazoxide, dihydralazine or hydralazine
- nitric oxide-releasing substances such as glyceryl nitrate or nitroprusside sodium.
- Antithrombotic agents are preferably understood as meaning compounds from the group of platelet aggregation inhibitors or anticoagulants.
- the compounds according to the invention are administered in combination with a platelet aggregation inhibitor, such as, by way of example and by way of preference, aspirin, clopidogrel, ticlopidine or dipyridamole.
- a platelet aggregation inhibitor such as, by way of example and by way of preference, aspirin, clopidogrel, ticlopidine or dipyridamole.
- the compounds according to the invention are administered in combination with a thrombin inhibitor, such as, by way of example and by way of preference, ximelagatran, melagatran, bivalirudin or Clexane.
- a thrombin inhibitor such as, by way of example and by way of preference, ximelagatran, melagatran, bivalirudin or Clexane.
- the compounds according to the invention are administered in combination with a GPUb / IHa antagonist, such as, by way of example and by way of preference, tirofiban or abciximab.
- a GPUb / IHa antagonist such as, by way of example and by way of preference, tirofiban or abciximab.
- the compounds according to the invention are used in combination with a factor Xa inhibitor, such as by way of example and preferably rivaroxaban (BAY 59-7939), DU-176b, apixaban, otamixaban, fidexaban, razaxaban, fondaparinux, idraparinux, PMD No. 3112, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 or SSR-128428.
- the compounds according to the invention are administered in combination with heparin or a low molecular weight (LMW) heparin derivative.
- LMW low molecular weight
- the compounds according to the invention are administered in combination with a vitamin K antagonist, such as by way of example and preferably coumarin.
- compositions containing at least one compound of the invention usually together with one or more inert, non-toxic, pharmaceutically suitable excipients, and their use for the purposes mentioned above.
- the compounds according to the invention can act systemically and / or locally.
- they may be applied in a suitable manner, e.g. oral, parenteral, pulmonary, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctivae otic or as an implant or stent.
- the compounds according to the invention can be administered in suitable administration forms.
- the compounds of the invention rapidly and / or modified donating application forms containing the compounds of the invention in crystalline and / or amorphized and / or dissolved form, such.
- Tablets uncoated or coated tablets, for example with enteric or delayed-release or insoluble coatings which control the release of the compound of the invention
- Parenteral administration can be accomplished by bypassing a resorption step (e.g., intravenous, intraarterial, intracardiac, intraspinal, or intralumbar) or by resorting to absorption (e.g., intramuscular, subcutaneous, intracutaneous, percutaneous, or intraperitoneal).
- a resorption step e.g., intravenous, intraarterial, intracardiac, intraspinal, or intralumbar
- absorption e.g., intramuscular, subcutaneous, intracutaneous, percutaneous, or intraperitoneal.
- parenteral administration are suitable as application forms u.a. Injection and infusion preparations in the form of solutions, suspensions, emulsions, lyophilisates or sterile powders.
- inhalation medicaments eg powder inhalers, nebulizers
- nasal drops solutions or sprays
- lingual, sublingual or buccal are suitable.
- the compounds according to the invention can be converted into the stated administration forms. This can be done in a conventional manner by mixing with inert, non-toxic, pharmaceutically suitable excipients.
- These adjuvants include, among others. Carriers (for example microcrystalline cellulose, lactose, mannitol), solvents (for example liquid polyethylene glycols), emulsifiers and dispersants or wetting agents (for example sodium dodecyl sulfate, polyoxysorbitanoleate), binders (for example polyvinylpyrrolidone), synthetic and natural polymers (for example albumin ), Stabilizers (eg antioxidants such as ascorbic acid), dyes (eg inorganic pigments such as iron oxides) and flavor and / or odoriferous agents.
- Carriers for example microcrystalline cellulose, lactose, mannitol
- solvents for example liquid polyethylene glycols
- emulsifiers and dispersants or wetting agents for example sodium dodec
- the dosage is about 0.01 to 100 mg / kg, preferably about 0.01 to 20 mg / kg and most preferably 0.1 to 10 mg / kg body weight.
- Device type MS Micromass ZQ
- Device type HPLC Waters Alliance 2795; Column: Phenomenex Synergi 2 ⁇ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 l of water + 0.5 ml of 50% formic acid, eluent B: 1 l of acetonitrile + 0.5 ml of 50% formic acid; Gradient: 0.0 min 90% A - »2.5 min 30% A ⁇ 3.0 min 5% A ⁇ 4.5 min 5% A; Flow: 0.0 min 1 ml / min ⁇ 2.5 min / 3.0 min / 4.5 min 2 ml / min; Oven: 5O 0 C; UV detection: 210 nm.
- Device type MS Micromass ZQ
- Device type HPLC HP 1100 Series
- UV DAD Column: Phenomenex Synergi 2 ⁇ Hydro-RP Mercury 20 mm x 4 mm
- Eluent A 1 l of water + 0.5 ml of 50% formic acid
- eluent B 1 l of acetonitrile + 0.5 ml of 50% formic acid
- Flow 0.0 min 1 ml / min ⁇ 2.5 min / 3.0 min / 4.5 min 2 ml / min
- Oven 5O 0 C
- UV detection 210 nm.
- Device type MS Micromass ZQ
- Device type HPLC Waters Alliance 2795; Column: Merck Chromolith SpeedROD RP-18e 100 mm x 4.6 mm; Eluent A: water + 500 ⁇ l 50% formic acid / 1, eluent B: acetonitrile + 500 ⁇ l 50% formic acid / 1; Gradient: 0.0 min 10% B ⁇ 7.0 min 95% B ⁇ 9.0 min 95% B; Oven: 35 ° C; Flow: 0.0 min 1.0 ml / min ⁇ 7.0 min 2.0 ml / min ⁇ 9.0 min 2.0 ml / min; UV detection: 210 nm.
- Device type MS Micromass ZQ
- Device type HPLC HP 1100 Series
- UV DAD Column: Phenomenex Gemini 3 ⁇ 30 mm x 3.00 mm
- Eluent A 1 l of water + 0.5 ml of 50% formic acid
- eluent B 1 l of acetonitrile + 0.5 ml of 50% formic acid
- Flow 0.0 min 1 ml / min ⁇ 2.5 min / 3.0 min / 4.5 min 2 ml / min
- Oven 50 ° C .
- UV detection 210 nm.
- Device Type MS Waters ZQ
- Device type HPLC Waters Alliance 2795
- Eluent A 1 l of water + 0.5 ml of 50% formic acid
- eluent B 1 l of acetonitrile + 0.5 ml of 50% formic acid
- Flow 2 ml / min
- Oven 40 ° C
- UV detection 210 nm.
- Device type MS Micromass ZQ
- Device type HPLC Waters Alliance 2795; Column: Phenomenex syn ergi 2.5 ⁇ MAX-RP 100A Mercury 20mm x 4mm; Eluent A: 1 l of water + 0.5 ml of 50% formic acid, eluent B: 1 l of acetonitrile + 0.5 ml of 50% formic acid; Gradient: 0.0 min 90% A ⁇ 0.1 min 90% A ⁇ 3.0 min 5% A ⁇ 4.0 min 5% A ⁇ 4.01 min 90% A; Flow: 2 ml / min; Oven: 50 ° C .; UV detection: 210 nm.
- the aqueous phase is extracted three times with 20 ml of ethyl acetate each time. After combining the organic phases, the solvent is removed on a rotary evaporator and the residue is purified chromatographically on silica gel 60 (mobile phase: gradient cyclohexane / ethyl acetate 10: 1 ⁇ 2: 1).
- the title compound is prepared analogously to PK Mahata et al., Tetrahedron 59, 2631-2639 (2003): 1.5 g (5.3 mmol) of the compound from Example 13A and 263 mg (5.26 mmol) of hydrazine hydrate are dissolved in 40 ml of ethanol and heated to reflux for 2 h. The solvent is removed on a rotary evaporator and the residue is purified chromatographically on silica gel 60 (mobile phase: gradient cyclohexane / ethyl acetate 5: 1 ⁇ 2: 1).
- reaction mixture is stirred for 8 h while slowly warming to RT.
- the reaction solution is then cooled again to 0 0 C, carefully mixed with 0.4 ml of water and 0.8 ml of 1 N sodium hydroxide solution and stirred after heating to RT for 8 h at this temperature.
- the mixture is filtered and the filtrate is concentrated on a rotary evaporator. The remaining crude product is used without further purification in the subsequent reaction.
- the phases are separated and the organic phase is washed twice with 30 ml of water each time.
- the organic phase is dried over magnesium sulfate and the solvent is removed on a rotary evaporator. removed.
- the residue is purified by column chromatography on silica gel 60 (mobile phase gradient: cyclohexane / ethyl acetate 50: 1 ⁇ 2: 1). The product thus obtained is used without further purification in the subsequent stage.
- Example 32A The title compound is prepared analogously to Example 32A starting from S - (-) - 2,3-dimethyl-1,3-dioxolane-4-methanol.
- the product fraction obtained is washed with sat. Sodium bicarbonate solution (5 ml) and extracted with ethyl acetate (three times 5 ml each). The combined organic phases are dried over magnesium sulfate and the solvent is removed on a rotary evaporator.
- Example 42 The title compound is prepared analogously to Example 42 starting from 81 mg (0.14 mmol) of the compound from Example 33A.
- the purification of the crude product is likewise carried out as described in Example 42.
- Cells of the permanent line CHO are stably transfected with the cDNA for the adenosine receptor subtypes Al, A2a and A2b.
- the adenosine Al receptors are coupled to adenylate cyclase s proteins of the Gj on proteins, while the adenosine A2a and A2b receptors via G. Accordingly, cAMP production in the cell is inhibited or stimulated. Via a cAMP-dependent promoter, the expression of the luciferase is then modulated.
- the luciferase test is optimized with the aim of high sensitivity and reproducibility, low variance and suitability for implementation on a robotic system by varying several test parameters, such as cell density, growing phase and test incubation, forskolin concentration and medium composition.
- test parameters such as cell density, growing phase and test incubation, forskolin concentration and medium composition.
- the stock cultures are grown in DMEM / F12 medium with 10% FCS (fetal calf serum) at 37 ° C under 5% CO 2 and split 1:10 each after 2-3 days.
- Test cultures are seeded at 2,000 cells per well in 384-well plates and grown at 37 ° C for approximately 48 hours. Then the medium is replaced by a physiological saline solution (130 mM sodium chloride, 5 mM potassium chloride, 2 mM calcium chloride, 20 mM HEPES, 1 mM magnesium chloride hexahydrate, 5 mM sodium hydrogen carbonate, pH 7.4).
- DMSO substances to be tested are in a dilution series of 5 x 10 '11 M to 3 x 10 "6 M (final concentration) to the test cultures pipetted (maximum DMSO final concentration in test mixture: 0.5%). 10 minutes later, forskolin to All cultures are then incubated for four hours at 37 ° C. Thereafter, 35 ⁇ l of a solution consisting of 50% lysis reagent (30 mM disodium hydrogenphosphate, 10% glycerol, 3% triton XIOO) are added to the test cultures.
- 50% lysis reagent (30 mM disodium hydrogenphosphate, 10% glycerol, 3% triton XIOO) are added to the test cultures.
- luciferase substrate solution 25 mM TrisHCl, 2 mM dithiothreitol (DTT), pH 7.8) and 50% luciferase substrate solution (2.5 mM ATP, 0.5 mM luciferin, 0.1 mM coenzyme A, 10 mM tricine, 1.35 mM magnesium sulfate, 15 mM DTT, pH 7.8), shaken for about 1 minute and the luciferase activity measured with a camera system.
- the EC 50 values ie the concentrations at which the Al cell inhibits 50% of the luciferase response or in the case of the A2b and A2a cells 50% of the maximum stimulability mi t of the corresponding substance are reached.
- the reference compound used in these experiments is the adenosine-analogous compound NECA (5-N-ethylcarboxamido-adenosine), which binds with high affinity to all adenosine receptor subtypes and a has an agonistic activity [Klotz, KN, Hessling, J., Hegler, J., Owman, C, KuIl, B., Fredholm, BB, Lohse, MJ, "Comparative pharmacology of human adenosine receptor subtypes - characterization of stably transfected receptors in CHO cells ", Naunyn Schmiedeberg's Arch. Pharmacol. 357. 1-9 (1998)].
- NECA adenosine-analogous compound
- Table 1 lists the EC 50 values of representative embodiments for the receptor stimulation on adenosine A1, A2a and A2b receptor subtypes:
- the caudal artery is prepared and clamped in a conventional apparatus for measuring isolated vessels.
- the vessels are perfused in a heat bath and contracted with phenylephrine.
- the degree of contraction is determined by a contraction knife.
- test substances are given and measured the decrease in the contraction of the vessels.
- a decrease in contraction corresponds to a dilatation of the vessels.
- the EC 50 value of a test substance with regard to its relaxing properties is the concentration at which the contraction of the vessels is reduced by 50%.
- Guards Claw monkeys carrying an internal transmitter which can permanently measure both blood pressure and heart rate (telemetric detection of hemodynamic parameters), test substances are administered orally in various concentrations. Subsequently, blood pressure and heart rate and their changes are recorded over 6-24 hours.
- PBS buffer pH 7.4 90.00 g NaCl pa (for example from Merck, Item No. 1.06404.1000), 13.61 g KH 2 PO 4 pa (for example from Merck, Item No. 1.04873.1000) and 83.35 g of 1N NaOH (eg Fa. Bernd
- Acetate buffer pH 4.6 Weigh out 5.4 g of sodium acetate x 3 H 2 O pa (eg from Merck, Item No. 1.06267.0500) into a 100 ml volumetric flask, dissolve in 50 ml of water, add 2.4 g of glacial acetic acid, make up to 100 ml with water, check the pH and adjust to pH 4.6 if necessary;
- Dimethylsulfoxide e.g., Baker Co., Art No. 71572500
- Preparation of the starting solution for calibration solutions (stock solution): Approximately 0.5 mg of the test substance is weighed exactly into a 2 ml Eppendorf Safe-Lock tube (Eppendorf, Item No. 0030 120,094) to a concentration of 600 ⁇ g / ml mixed with DMSO (eg 0.5 mg of substance + 833 ⁇ l of DMSO) and shaken to complete dissolution by means of a vortexer.
- Calibration solution 1 (20 ⁇ g / ml): Mix 34.4 ⁇ l of the stock solution with 1000 ⁇ l of DMSO and homogenize.
- Calibration solution 2 (2.5 ⁇ g / ml): 100 ⁇ l of the calibration solution 1 are mixed with 700 ⁇ l of DMSO and homogenized.
- Sample solution for solubility up to 10 g / l in PBS buffer pH 7.4 Approximately 5 mg of the test substance are weighed exactly into a 2 ml Eppendorf Safe-Lock tube (Eppendorf, Art Concentration of 5 g / l with PBS buffer pH 7.4 added (eg, 5 mg of substance + 500 ul PBS buffer pH 7.4).
- Sample solution for solubility up to 10 g / l in acetate buffer pH 4.6 Approximately 5 mg of the test substance are weighed exactly into a 2 ml Eppendorf-Safe-Lock tube (Eppendorf, Art No. 0030 120,094) and added to a concentration of 5 g / l with acetate buffer pH 4.6 added (eg 5 mg of substance + 500 ul acetate buffer pH 4.6).
- Sample solution for solubility up to 10 g / l in water Approximately 5 mg of the test substance are weighed exactly into a 2 ml Eppendorf Safe-Lock tube (Eppendorf, article No. 0030 120,094) and added to a concentration of 5 g / l mixed with water (eg 5 mg substance + 500 ul water).
- the sample solutions thus prepared are shaken for 24 hours at 1400 rpm in a temperature shaker (for example, Fa. Eppendorf Thermomixer comfort No. 5355 000.011 with shift block Ref. 5362.000.019) at 20 0 C. Of these solutions are each 180 ul and transferred to Beckman Polyallomer Centrifuge Tubes (Item No. 343621). These solutions are centrifuged for 1 hour at about 223,000 xg (eg Beckman Optima L-90K Ultracentrifuge with Type 42.2 Ti rotor at 42,000 rpm).
- 100 ⁇ l of the supernatant are taken from each sample solution and diluted 1: 5, 1: 100 and 1: 1000 with the respectively used solvent (water, PBS buffer 7.4 or acetate buffer pH 4.6). Each dilution is bottled in a suitable vessel for HPLC analysis.
- Agilent 1100 with DAD (G1315A), quat. Pump (G1311A), autosampler CTC HTS PAL, degasser (G1322A) and column thermostat (G1316A); Column: Phenomenex Gemini C18, 50 mm x 2 mm, 5 ⁇ ; Temperature: 40 ° C .; Eluent A: water / phosphoric acid pH 2; Eluent B: acetonitrile; Flow rate: 0.7 ml / min; Gradient: 0-0.5 min 85% A, 15% B; Ramp: 0.5-3 min 10% A, 90% B; 3-3.5 min 10% A, 90% B; Ramp: 3.5-4 min 85% A, 15% B; 4-5 minutes 85% A, 15% B.
- Agilent 1100 with DAD (G1315A), quat. Pump (G1311A), autosampler CTC HTS PAL, degasser (G1322A) and column thermostat (G1316A); Column: VDSoptilab Kromasil 100 Cl 8, 60 mm x 2.1 mm, 3.5 ⁇ ; Temperature: 30 ° C .; Eluent A: water + 5 ml perchloric acid / l; Eluent B: acetonitrile; Flow rate: 0.75 ml / min; Gradient: 0-0.5 min 98% A, 2% B; Ramp: 0.5-4.5 min 10% A, 90% B; 4.5-6 min 10% A, 90% B; Ramp: 6.5-6.7 min 98% A, 2% B; 6.7-7.5 min 98% A, 2% B.
- the compounds according to the invention can be converted into pharmaceutical preparations as follows:
- the mixture of compound of the invention, lactose and starch is granulated with a 5% solution (m / m) of the PVP in water.
- the granules are mixed after drying with the magnesium stearate for 5 minutes.
- This mixture is compressed with a conventional tablet press (for the tablet format see above).
- a pressing force of 15 kN is used as a guideline for the compression.
- a single dose of 100 mg of the compound of the invention corresponds to 10 ml of oral suspension.
- the rhodigel is suspended in ethanol, the compound according to the invention is added to the suspension. While stirring, the addition of water. Until the completion of the swelling of Rhodigels is stirred for about 6 h.
- the compound of the invention is suspended in the mixture of polyethylene glycol and polysorbate with stirring. The stirring is continued until complete dissolution of the erf ⁇ ndungswashen connection.
- the compound of the invention is dissolved in a concentration below saturation solubility in a physiologically acceptable solvent (e.g., isotonic saline, glucose solution 5% and / or PEG 400 solution 30%).
- a physiologically acceptable solvent e.g., isotonic saline, glucose solution 5% and / or PEG 400 solution 30%.
- the solution is sterile filtered and filled into sterile and pyrogen-free injection containers.
Abstract
Description







































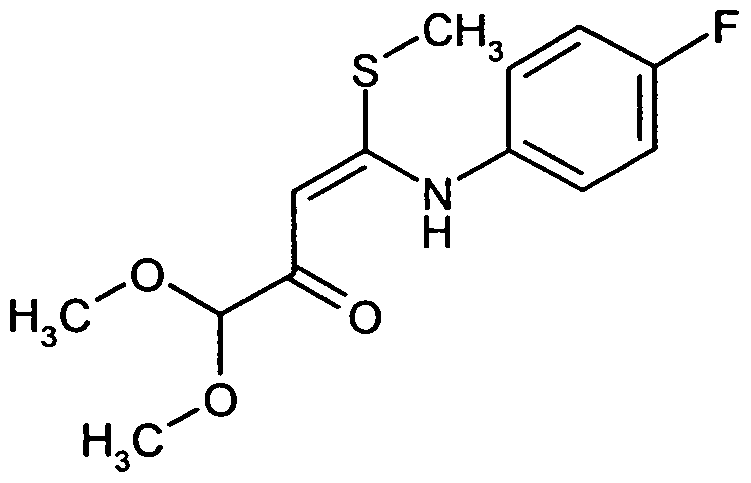






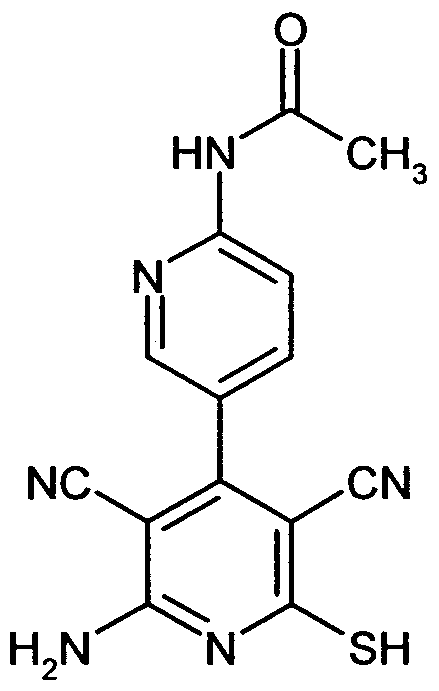









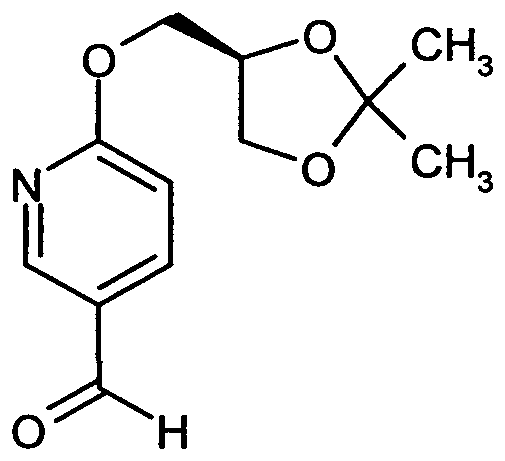













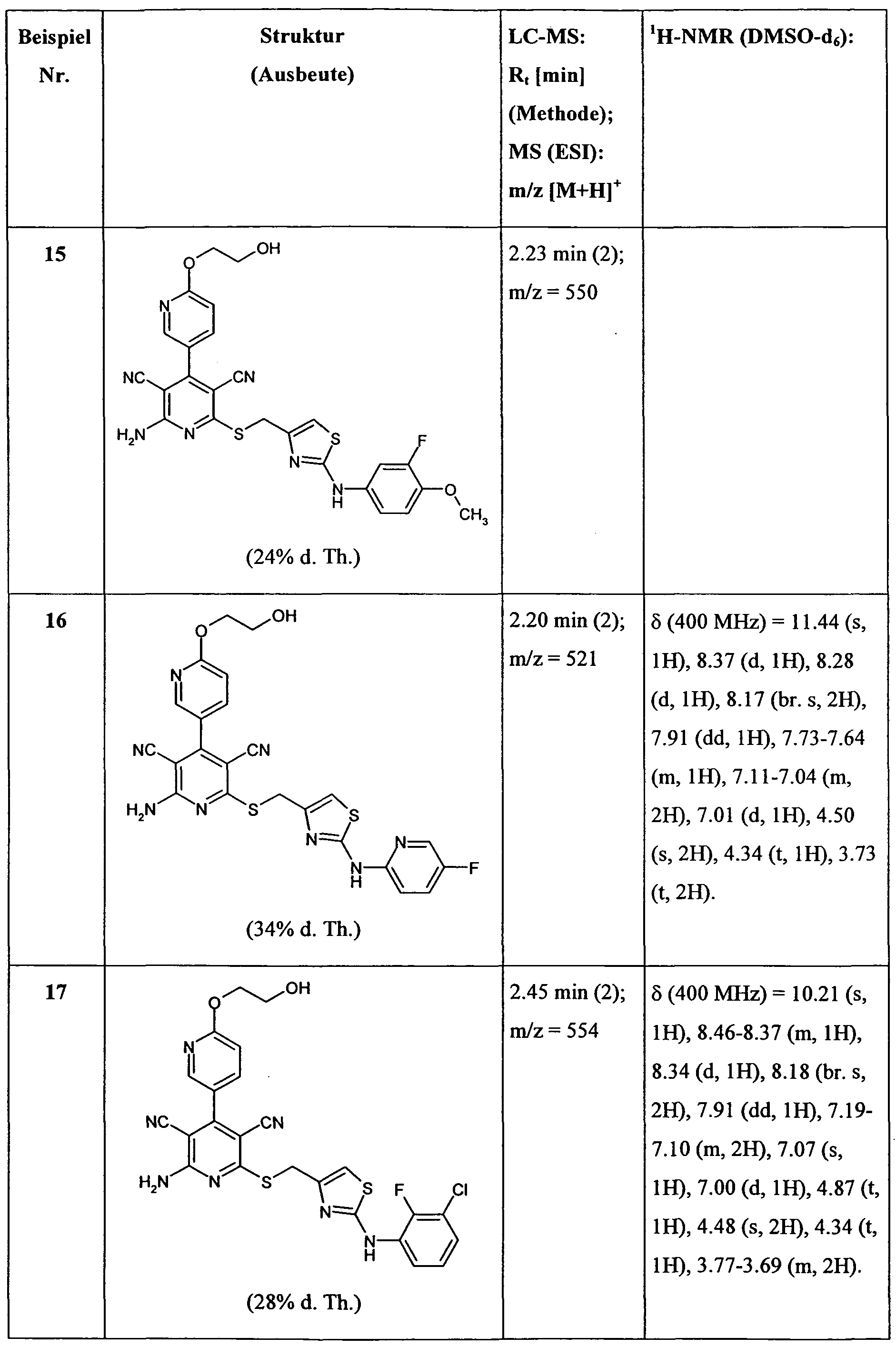




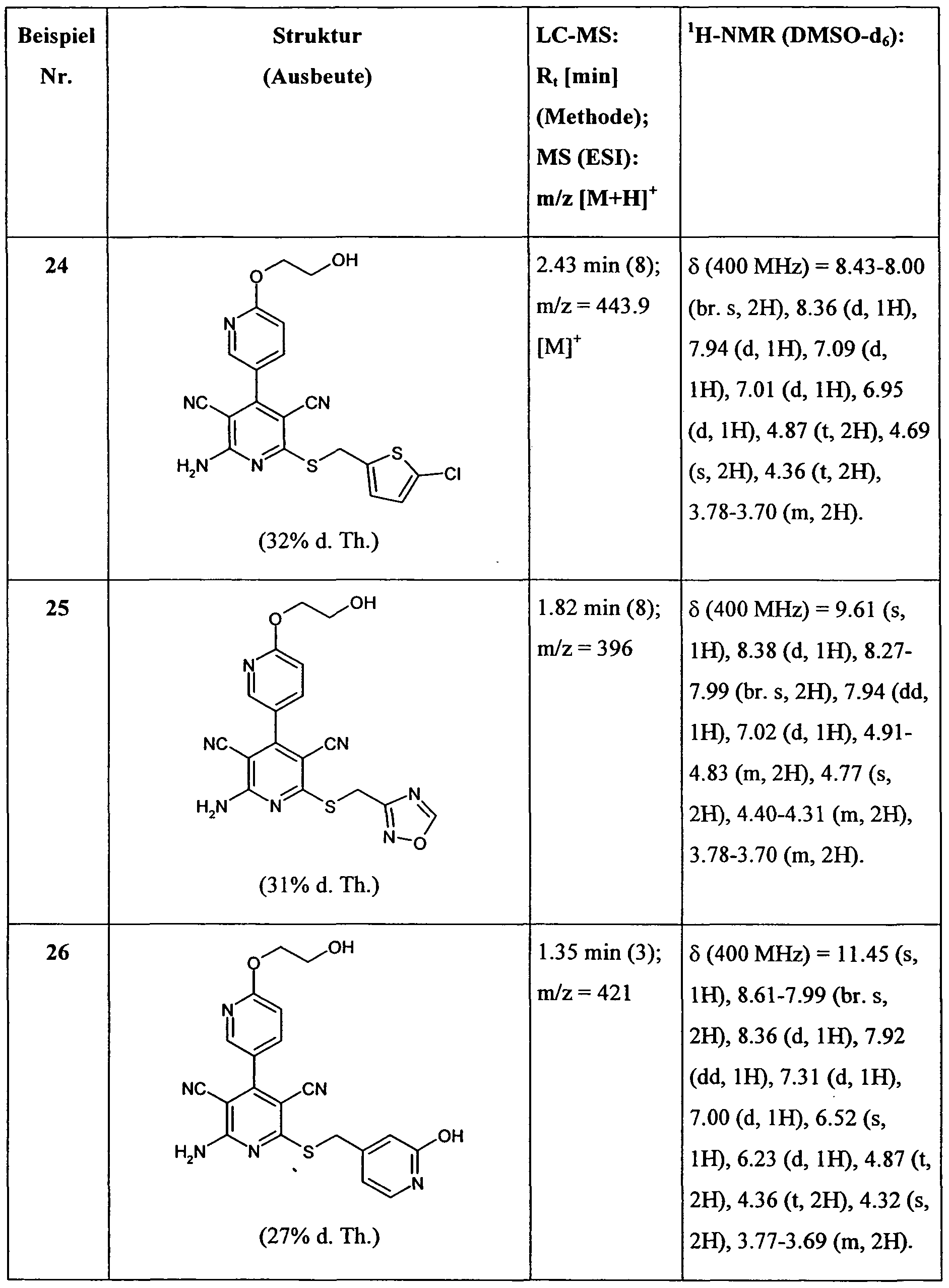









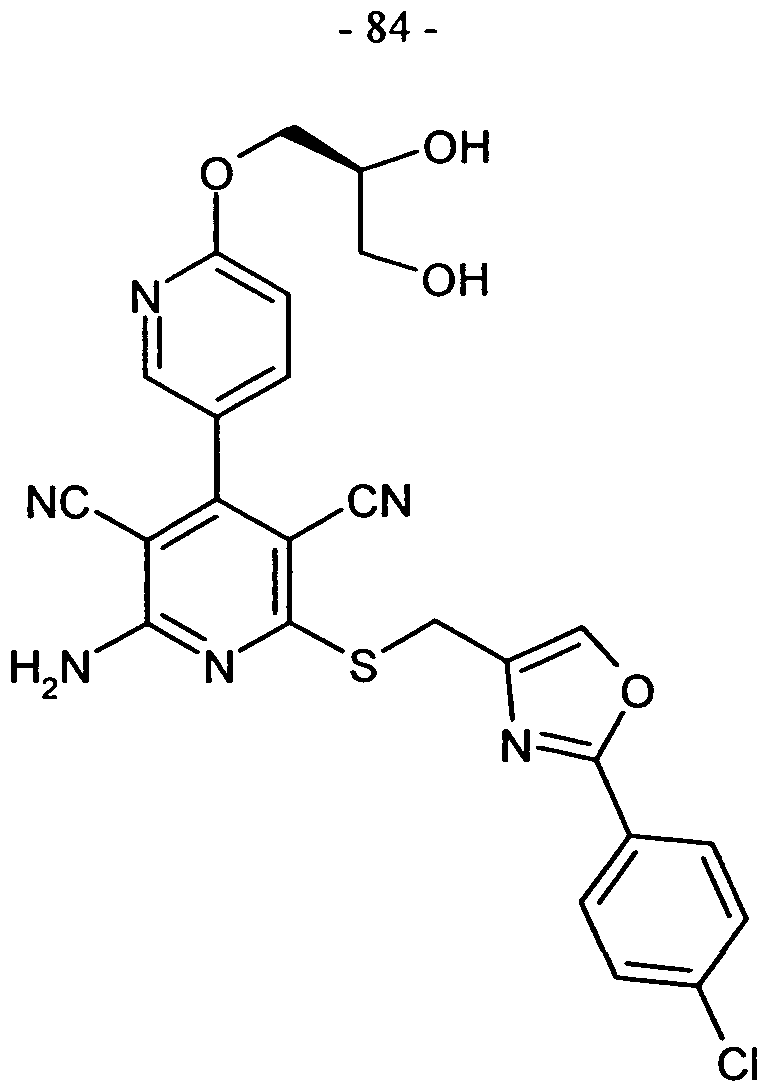






Claims
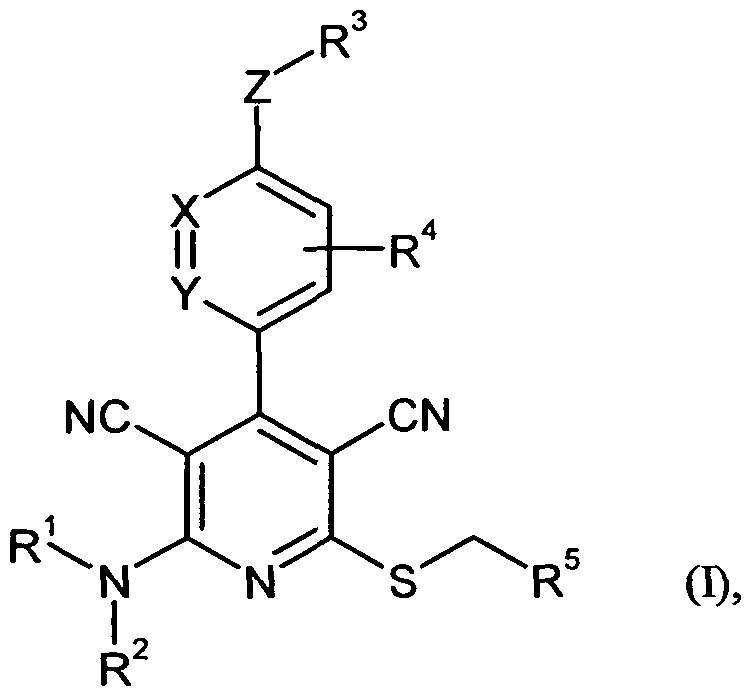

Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009527038A JP5331693B2 (ja) | 2006-09-08 | 2007-08-30 | 新規置換ビピリジン誘導体およびアデノシン受容体リガンドとしてのそれらの使用 |
| EP07801993A EP2066637A1 (de) | 2006-09-08 | 2007-08-30 | Neue substituierte bipyridin-derivate und ihre verwendung als adenosin rezeptor liganden |
| US12/440,423 US8653109B2 (en) | 2006-09-08 | 2007-08-30 | Substituted bipyridine derivatives and their use as adenosine receptor ligands |
| CA2662728A CA2662728C (en) | 2006-09-08 | 2007-08-30 | Substituted bipyridine derivatives and their use |
| US14/175,719 US20140155401A1 (en) | 2006-09-08 | 2014-02-07 | Novel substituted bipyridine derivatives and their use as adenosine receptor ligands |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102006042143.4 | 2006-09-08 | ||
| DE102006042143A DE102006042143A1 (de) | 2006-09-08 | 2006-09-08 | Neue substituierte Bipyridin-Derivate und ihre Verwendung |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/440,423 A-371-Of-International US8653109B2 (en) | 2006-09-08 | 2007-08-30 | Substituted bipyridine derivatives and their use as adenosine receptor ligands |
| US14/175,719 Continuation US20140155401A1 (en) | 2006-09-08 | 2014-02-07 | Novel substituted bipyridine derivatives and their use as adenosine receptor ligands |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008028590A1 true WO2008028590A1 (de) | 2008-03-13 |
Family
ID=38896950
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2007/007572 WO2008028590A1 (de) | 2006-09-08 | 2007-08-30 | Neue substituierte bipyridin-derivate und ihre verwendung als adenosin rezeptor liganden |
Country Status (11)
| Country | Link |
|---|---|
| US (2) | US8653109B2 (de) |
| EP (1) | EP2066637A1 (de) |
| JP (1) | JP5331693B2 (de) |
| AR (1) | AR062630A1 (de) |
| CA (1) | CA2662728C (de) |
| CL (1) | CL2007002610A1 (de) |
| DE (1) | DE102006042143A1 (de) |
| PE (1) | PE20080772A1 (de) |
| TW (1) | TW200823209A (de) |
| UY (1) | UY30578A1 (de) |
| WO (1) | WO2008028590A1 (de) |
Cited By (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2010086101A1 (de) | 2009-01-29 | 2010-08-05 | Bayer Schering Pharma Aktiengesellschaft | Alkylamino-substituierte dicyanopyridine und deren aminosäureester-prodrugs |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2012000945A1 (de) | 2010-06-30 | 2012-01-05 | Bayer Pharma Aktiengesellschaft | Substituierte dicyanopyridine und ihre verwendung |
| EP2567959A1 (de) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridin-4-Carbonsäureamid-Derivate als Kinaseinhibitoren |
| WO2013063526A1 (en) | 2011-10-28 | 2013-05-02 | Lumena Pharmaceuticals, Inc. | Bile acid recycling inhibitors for treatment of hypercholemia and cholestatic liver disease |
| WO2014060375A2 (en) * | 2012-10-18 | 2014-04-24 | Bayer Pharma Aktiengesellschaft | 5-fluoro-n-(pyridin-2-yl)pyridin-2-amine derivatives containing a sulfone group |
| WO2014144485A1 (en) | 2013-03-15 | 2014-09-18 | Lumena Pharmaceuticals, Inc. | Bile acid recycling inhibitors for treatment of barrett's esophagus and gastroesophageal reflux disease |
| WO2014144650A2 (en) | 2013-03-15 | 2014-09-18 | Lumena Pharmaceuticals, Inc. | Bile acid recycling inhibitors for treatment of primary sclerosing cholangitis and inflammatory bowel disease |
| EP2995317A1 (de) | 2010-05-26 | 2016-03-16 | Satiogen Pharmaceuticals, Inc. | Gallensäurerückflusshemmer zur behandlung von diabetes, adipositas und entzündlichen magen-darm-erkrankungen |
| US9650340B2 (en) | 2012-11-15 | 2017-05-16 | Bayer Pharma Aktiengesellschaft | 5-fluoro-N-(pyridin-2-yl)pyridin-2-amine derivatives containing a sulfoximine group |
| US9650361B2 (en) | 2012-11-15 | 2017-05-16 | Bayer Pharam Aktiengesellschaft | N-(pyridin-2-yl)pyrimidin-4-amine derivatives containing a sulfoximine group |
| US9669034B2 (en) | 2011-05-24 | 2017-06-06 | Bayer Intellectual Property Gmbh | 4-aryl-N-phenyl-1,3,5-triazin-2-amines containing a sulfoximine group |
| US9708293B2 (en) | 2012-10-18 | 2017-07-18 | Bayer Pharma Aktiengesellschaft | N-(pyridin-2-yl)pyrimidin-4-amine derivatives containing a sulfone group |
| US9770445B2 (en) | 2013-07-04 | 2017-09-26 | Bayer Pharma Aktiengesellschaft | Sulfoximine substituted 5-fluoro-N-(pyridin-2-yl)pyridin-2-amine derivatives and their use as CDK9 kinase inhibitors |
| US9790189B2 (en) | 2014-04-01 | 2017-10-17 | Bayer Pharma Aktiengesellschaft | Disubstituted 5-fluoro pyrimidine derivatives containing a sulfondiimine group |
| US9856242B2 (en) | 2014-03-13 | 2018-01-02 | Bayer Pharma Aktiengesellscaft | 5-fluoro-N-(pyridin-2-yl)pyridin-2-amine derivatives containing a sulfone group |
| EP3266457A1 (de) | 2011-10-28 | 2018-01-10 | Lumena Pharmaceuticals LLC | Gallensäurerückflusshemmer zur behandlung von cholestase-erkrankungen im kindesalter |
| US9884849B2 (en) | 2014-10-16 | 2018-02-06 | Bayer Pharma Aktiengesellschaft | Fluorinated benzofuranyl-pyrimidine derivatives containing a sulfoximine group |
| US9902716B2 (en) | 2014-10-16 | 2018-02-27 | Bayer Pharma Aktiengesellschaft | Fluorinated benzofuranyl-pyrimidine derivatives containing a sulfone group |
| US9963464B2 (en) | 2014-04-11 | 2018-05-08 | Bayer Pharma Aktiengesellschaft | Macrocyclic compounds |
| WO2018153897A1 (de) | 2017-02-22 | 2018-08-30 | Bayer Pharma Aktiengesellschaft | Selektive partielle adenosin a1 rezeptor-agonisten in kombination mit hcn-kanal-hemmern |
| WO2018153900A1 (de) | 2017-02-22 | 2018-08-30 | Bayer Aktiengesellschaft | Selektive partielle adenosin a1 rezeptor-agonisten in kombination mit sglt-2-hemmern |
| WO2018153898A1 (de) | 2017-02-22 | 2018-08-30 | Bayer Pharma Aktiengesellschaft | Selektive partielle adenosin a1 rezeptor-agonisten in kombination mit mineralocorticoid-rezeptor-antagonisten |
| WO2018153899A1 (de) | 2017-02-22 | 2018-08-30 | Bayer Pharma Aktiengesellschaft | Selektive partielle adenosin a1 rezeptor-agonisten in kombination mit stimulatoren und/oder aktivatoren der löslichen guanylatcyclase (sgc) |
| WO2018153895A1 (de) | 2017-02-22 | 2018-08-30 | Bayer Pharma Aktiengesellschaft | Selektive partielle adenosin a1 rezeptor-agonisten in kombination mit einem inhibitor der neutralen endopeptidase und/oder einem angiotensin ii rezeptor-antagonisten |
| US11242356B2 (en) | 2017-03-28 | 2022-02-08 | Bayer Aktiengesellschaft | PTEFb inhibiting macrocyclic compounds |
| US11254690B2 (en) | 2017-03-28 | 2022-02-22 | Bayer Pharma Aktiengesellschaft | PTEFb inhibiting macrocyclic compounds |
| US11701347B2 (en) | 2018-02-13 | 2023-07-18 | Bayer Aktiengesellschaft | Use of 5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine for treating diffuse large B-cell lymphoma |
| EP4241840A2 (de) | 2019-02-12 | 2023-09-13 | Mirum Pharmaceuticals, Inc. | Verfahren zur behandlung von cholestase |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102004042607A1 (de) | 2004-09-03 | 2006-03-09 | Bayer Healthcare Ag | Substituierte Phenylaminothiazole und ihre Verwendung |
| DE102006056740A1 (de) | 2006-12-01 | 2008-06-05 | Bayer Healthcare Ag | Cyclisch substituierte 3,5-Dicyano-2-thiopyridine und ihre Verwendung |
| DE102006056739A1 (de) | 2006-12-01 | 2008-06-05 | Bayer Healthcare Ag | Substituierte 4-Amino-3,5-dicyano-2-thiopyridine und ihre Verwendung |
| DE102007035367A1 (de) | 2007-07-27 | 2009-01-29 | Bayer Healthcare Ag | Substituierte Aryloxazole und ihre Verwendung |
| DE102007036076A1 (de) | 2007-08-01 | 2009-02-05 | Bayer Healthcare Aktiengesellschaft | Dipeptoid-Produgs und ihre Verwendung |
| DE102007061764A1 (de) * | 2007-12-20 | 2009-06-25 | Bayer Healthcare Ag | Anellierte Cyanopyridine und ihre Verwendung |
| DE102007061763A1 (de) * | 2007-12-20 | 2009-06-25 | Bayer Healthcare Ag | Substituierte azabicyclische Verbindungen und ihre Verwendung |
| DE102008013587A1 (de) * | 2008-03-11 | 2009-09-17 | Bayer Schering Pharma Aktiengesellschaft | Heteroaryl-substituierte Dicyanopyridine und ihre Verwendung |
| JP2011520997A (ja) * | 2008-05-29 | 2011-07-21 | バイエル・シェーリング・ファルマ・アクチェンゲゼルシャフト | 2−アルコキシ置換ジシアノピリジン類およびそれらの使用 |
| DE102008062567A1 (de) | 2008-12-16 | 2010-06-17 | Bayer Schering Pharma Aktiengesellschaft | Dipeptoid-Prodrugs und ihre Verwendung |
| AU2015272011C1 (en) * | 2009-01-29 | 2017-12-14 | Bayer Intellectual Property Gmbh | Alkylamine-substituted dicyanopyridine and amino acid ester prodrugs thereof |
| US20120058983A1 (en) | 2010-09-02 | 2012-03-08 | Bayer Pharma Aktiengesellschaft | Adenosine A1 agonists for the treatment of glaucoma and ocular hypertension |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001025210A2 (de) * | 1999-10-01 | 2001-04-12 | Bayer Aktiengesellschaft | Substituierte 2-thio-3,5-dicyano-4-aryl-6-aminopyridine und ihre verwendung als adenosin-rezeptorliganden |
| WO2001062233A2 (en) * | 2000-02-25 | 2001-08-30 | F. Hoffmann La Roche Ag | Adenosine receptor modulators |
| WO2003053441A1 (de) * | 2001-12-11 | 2003-07-03 | Bayer Healthcare Ag | Substituierte 2-thio-3,5-dicyano-4-phenyl-6-aminopyridine und ihre verwendung |
| WO2006027142A1 (de) * | 2004-09-03 | 2006-03-16 | Bayer Healthcare Ag | Substituierte phenylaminothiazole und ihre verwendung |
Family Cites Families (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4052510A (en) | 1974-12-18 | 1977-10-04 | Sandoz, Inc. | 4-alkyl-2,6-di(secondary or tertiary alkylamino) pyridines, compositions thereof and methods for treating diabetes and obesity |
| TW299333B (de) | 1992-12-29 | 1997-03-01 | Takeda Pharm Industry Co Ltd | |
| CA2192820C (en) | 1994-06-16 | 2000-11-28 | Yuhpyng L. Chen | Pyrazolo and pyrrolopyridines |
| DE4430638A1 (de) | 1994-08-29 | 1996-03-07 | Bayer Ag | Verwendung von substituierten 4-Phenyl-6-amino-nicotinsäurederivaten als Arzneimittel |
| JPH09132529A (ja) | 1995-11-09 | 1997-05-20 | Ono Pharmaceut Co Ltd | 一酸化窒素合成酵素阻害剤 |
| AU727775B2 (en) | 1996-01-17 | 2000-12-21 | Novo Nordisk A/S | Fused 1,2,4-thiadiazine and fused 1,4-thiazine derivatives, their preparation and use |
| EP2311806A3 (de) | 1996-01-29 | 2011-08-10 | The United States of America, Represented by the Secretary, Department of Health and Human Services | Dihydropyridin-, Pyridin-, Benzopyranon- und Triazolchinazolinderivate, ihre Herstellung und Verwendung als Adenosinrezeptorantagonisten |
| JPH10324687A (ja) | 1997-02-19 | 1998-12-08 | Nippon Soda Co Ltd | ピロール化合物、製法および農園芸用殺菌剤 |
| JP2002507197A (ja) | 1997-05-30 | 2002-03-05 | ビーエーエスエフ アクチェンゲゼルシャフト | 置換チオピリジン |
| BR9810592A (pt) | 1997-07-16 | 2000-09-12 | Novo Nordisk As | Composto, processos para preparar um composto, para tratar ou prevenir doenças do sistema endócrino e para a fabricação de um medicamento, composição farmacêutica, e, uso de um composto |
| US6632823B1 (en) | 1997-12-22 | 2003-10-14 | Merck & Co., Inc. | Substituted pyridine compounds useful as modulators of acetylcholine receptors |
| DE19834044A1 (de) | 1998-07-29 | 2000-02-03 | Bayer Ag | Neue substituierte Pyrazolderivate |
| DE19834047A1 (de) | 1998-07-29 | 2000-02-03 | Bayer Ag | Substituierte Pyrazolderivate |
| DE19943636A1 (de) | 1999-09-13 | 2001-03-15 | Bayer Ag | Neuartige Dicarbonsäurederivate mit pharmazeutischen Eigenschaften |
| DE19943634A1 (de) | 1999-09-13 | 2001-04-12 | Bayer Ag | Neuartige Dicarbonsäurederivate mit pharmazeutischen Eigenschaften |
| DE19943635A1 (de) | 1999-09-13 | 2001-03-15 | Bayer Ag | Neuartige Aminodicarbonsäurederivate mit pharmazeutischen Eigenschaften |
| PL359416A1 (en) | 2000-07-18 | 2004-08-23 | Yamanouchi Pharmaceutical Co, Ltd. | Medicine comprising dicyanopyridine derivative |
| AR031176A1 (es) | 2000-11-22 | 2003-09-10 | Bayer Ag | Nuevos derivados de pirazolpiridina sustituidos con piridina |
| EP1341772A2 (de) | 2000-12-11 | 2003-09-10 | E. I. du Pont de Nemours and Company | Chinalozinone und pyridinylpyrimidinone zur bekämpfung wirbelloser schädlinge |
| DK1347971T3 (da) | 2000-12-21 | 2006-05-15 | Bristol Myers Squibb Co | Thiazolylinhibitorer af tyrosinkinaser fra Tec-familien |
| DE10110438A1 (de) | 2001-03-05 | 2002-09-19 | Bayer Ag | Substituierte 2-Oxy-3,5-dicyano-4-aryl-6-aminopyridine und ihre Verwendung |
| DE10110750A1 (de) | 2001-03-07 | 2002-09-12 | Bayer Ag | Neuartige Aminodicarbonsäurederivate mit pharmazeutischen Eigenschaften |
| DE10110754A1 (de) | 2001-03-07 | 2002-09-19 | Bayer Ag | Substituierte 2-Thio-3,5-dicyano-4-aryl-6-aminopyridine und ihre Verwendung |
| DE10110749A1 (de) | 2001-03-07 | 2002-09-12 | Bayer Ag | Substituierte Aminodicarbonsäurederivate |
| DE10110747A1 (de) | 2001-03-07 | 2002-09-12 | Bayer Ag | Substituierte 2,6-Diamino-3,5-dicyano-4-aryl-pyridine und ihre Verwendung |
| DE10115922A1 (de) | 2001-03-30 | 2002-10-10 | Bayer Ag | Cyclisch substituierte 2-Thio-3,5-dicyano-4-aryl-6-aminopyridine und ihre Verwendung |
| DE10115945A1 (de) | 2001-03-30 | 2002-10-02 | Bayer Ag | Substituierte 2-Carba-3,5-dicyano-4-aryl-6-aminopyridine und ihre Verwendung |
| DE10134481A1 (de) | 2001-07-16 | 2003-01-30 | Bayer Ag | Substituierte 2-Thio-3,5-dicyano-4-phenyl-6-aminopyridine und ihre Verwendung |
| JP2003183254A (ja) | 2001-12-20 | 2003-07-03 | Yamanouchi Pharmaceut Co Ltd | 2−アシルアミノ−3,5−ジシアノピリジン誘導体又はその塩 |
| EP1506189A1 (de) | 2002-04-26 | 2005-02-16 | Vertex Pharmaceuticals Incorporated | Pyrrol-derivate als inhibitoren von erk2 und deren verwendung |
| DE10220570A1 (de) | 2002-05-08 | 2003-11-20 | Bayer Ag | Carbamat-substituierte Pyrazolopyridine |
| EP1388342A1 (de) | 2002-08-07 | 2004-02-11 | Aventis Pharma Deutschland GmbH | Acylierte, Heteroaryl-kondensierte Cycloalkenylamine und deren Verwendung als Arzneimittel |
| WO2004014368A1 (en) | 2002-08-12 | 2004-02-19 | Sugen, Inc. | 3-pyrrolyl-pyridopyrazoles and 3-pyrrolyl-indazoles as novel kinase inhibitors |
| JP2006512338A (ja) | 2002-12-12 | 2006-04-13 | ファルマシア・コーポレーション | マイトジェン活性化プロテインキナーゼ−活性化プロテインキナーゼ−2阻害剤としてのアミノシアノピリジンの使用法 |
| AR042667A1 (es) | 2002-12-26 | 2005-06-29 | Taisho Pharmaceutical Co Ltd | Derivados de pirrolopirimidina y pirrolopiridina sustituidos con un grupo amino ciclico |
| TW200418829A (en) | 2003-02-14 | 2004-10-01 | Avanir Pharmaceutics | Inhibitors of macrophage migration inhibitory factor and methods for identifying the same |
| AR045047A1 (es) | 2003-07-11 | 2005-10-12 | Arena Pharm Inc | Derivados arilo y heteroarilo trisustituidos como moduladores del metabolismo y de la profilaxis y tratamiento de desordenes relacionados con los mismos |
| CN1856307A (zh) | 2003-09-23 | 2006-11-01 | 默克公司 | 喹啉钾通道抑制剂 |
| WO2005046603A2 (en) | 2003-11-10 | 2005-05-26 | Synta Pharmaceuticals, Corp. | Pyridine compounds |
| US20050182105A1 (en) | 2004-02-04 | 2005-08-18 | Nirschl Alexandra A. | Method of using 3-cyano-4-arylpyridine derivatives as modulators of androgen receptor function |
| JP2007161585A (ja) | 2004-06-25 | 2007-06-28 | Taisho Pharmaceut Co Ltd | 環状アミノ基で置換されているピロロピリミジン及びピロロピリジン誘導体 |
| JP2008503443A (ja) | 2004-06-25 | 2008-02-07 | 大正製薬株式会社 | テトラヒドロピリジンで置換されたピロロピリミジン及びピロロピリジンの誘導体 |
| DE102004032651A1 (de) | 2004-07-06 | 2006-02-16 | Bayer Healthcare Ag | Verwendung von substituierten 2-Thio-3,5-dicyano-4-phenyl-6-aminopyriden bei der Behandlung von Übelkeit und Erbrechen |
| ATE494895T1 (de) | 2004-09-20 | 2011-01-15 | Xenon Pharmaceuticals Inc | Pyridin-derivate zur hemmung der menschlichen stearoyl-coa-desaturase |
| WO2006099958A1 (en) | 2005-03-24 | 2006-09-28 | Bayer Healthcare Ag | Use of substituted 2-thio-3,5-dicyano-4-phenyl-6-aminopyridines for the treatment of reperfusion injury and reperfusion damage |
| US7750015B2 (en) | 2005-05-17 | 2010-07-06 | Schering Corporation | Nitrogen-containing heterocyclic compounds and methods of use thereof |
| WO2007073855A1 (en) | 2005-12-23 | 2007-07-05 | Bayer Healthcare Ag | Use of adenosine a1 receptor agonists for the protection of renal cells against toxic effects caused by aminoglycosides during treatment of infectious diseases |
| DE102006009813A1 (de) | 2006-03-01 | 2007-09-06 | Bayer Healthcare Ag | Verwendung von A2b/A1 Rezeptor Agonisten zur Modulation der Lipidspiegel |
| EP2019827A1 (de) | 2006-04-28 | 2009-02-04 | Avexa Limited | Integraseinhibitoren 3 |
| DE102006056739A1 (de) | 2006-12-01 | 2008-06-05 | Bayer Healthcare Ag | Substituierte 4-Amino-3,5-dicyano-2-thiopyridine und ihre Verwendung |
| DE102006056740A1 (de) | 2006-12-01 | 2008-06-05 | Bayer Healthcare Ag | Cyclisch substituierte 3,5-Dicyano-2-thiopyridine und ihre Verwendung |
| DE102007035367A1 (de) | 2007-07-27 | 2009-01-29 | Bayer Healthcare Ag | Substituierte Aryloxazole und ihre Verwendung |
| DE102007036076A1 (de) | 2007-08-01 | 2009-02-05 | Bayer Healthcare Aktiengesellschaft | Dipeptoid-Produgs und ihre Verwendung |
| DE102008013587A1 (de) | 2008-03-11 | 2009-09-17 | Bayer Schering Pharma Aktiengesellschaft | Heteroaryl-substituierte Dicyanopyridine und ihre Verwendung |
| DE102008062567A1 (de) | 2008-12-16 | 2010-06-17 | Bayer Schering Pharma Aktiengesellschaft | Dipeptoid-Prodrugs und ihre Verwendung |
| DE102009006602A1 (de) | 2009-01-29 | 2010-08-05 | Bayer Schering Pharma Aktiengesellschaft | Alkylamino-substituierte Dicyanopyridine und deren Aminosäureester-Prodrugs |
-
2006
- 2006-09-08 DE DE102006042143A patent/DE102006042143A1/de not_active Withdrawn
-
2007
- 2007-08-30 CA CA2662728A patent/CA2662728C/en not_active Expired - Fee Related
- 2007-08-30 WO PCT/EP2007/007572 patent/WO2008028590A1/de active Application Filing
- 2007-08-30 US US12/440,423 patent/US8653109B2/en not_active Expired - Fee Related
- 2007-08-30 JP JP2009527038A patent/JP5331693B2/ja not_active Expired - Fee Related
- 2007-08-30 EP EP07801993A patent/EP2066637A1/de not_active Withdrawn
- 2007-09-03 AR ARP070103888A patent/AR062630A1/es not_active Application Discontinuation
- 2007-09-07 UY UY30578A patent/UY30578A1/es not_active Application Discontinuation
- 2007-09-07 PE PE2007001207A patent/PE20080772A1/es not_active Application Discontinuation
- 2007-09-07 TW TW096133320A patent/TW200823209A/zh unknown
- 2007-09-07 CL CL200702610A patent/CL2007002610A1/es unknown
-
2014
- 2014-02-07 US US14/175,719 patent/US20140155401A1/en not_active Abandoned
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001025210A2 (de) * | 1999-10-01 | 2001-04-12 | Bayer Aktiengesellschaft | Substituierte 2-thio-3,5-dicyano-4-aryl-6-aminopyridine und ihre verwendung als adenosin-rezeptorliganden |
| WO2001062233A2 (en) * | 2000-02-25 | 2001-08-30 | F. Hoffmann La Roche Ag | Adenosine receptor modulators |
| WO2003053441A1 (de) * | 2001-12-11 | 2003-07-03 | Bayer Healthcare Ag | Substituierte 2-thio-3,5-dicyano-4-phenyl-6-aminopyridine und ihre verwendung |
| WO2006027142A1 (de) * | 2004-09-03 | 2006-03-16 | Bayer Healthcare Ag | Substituierte phenylaminothiazole und ihre verwendung |
Cited By (55)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| JP2012516290A (ja) * | 2009-01-29 | 2012-07-19 | バイエル・ファルマ・アクチェンゲゼルシャフト | アルキルアミノ置換ジシアノピリジンおよびそのアミノ酸エステルプロドラッグ |
| CN102388040B (zh) * | 2009-01-29 | 2015-05-13 | 拜耳知识产权有限责任公司 | 烷基氨基-取代的二氰基嘧啶和它们的氨基酸酯药物前体 |
| KR20110108377A (ko) * | 2009-01-29 | 2011-10-05 | 바이엘 파마 악티엔게젤샤프트 | 알킬아민-치환된 디시아노피리딘 및 그의 아미노산 에스테르 전구약물 |
| TWI511968B (zh) * | 2009-01-29 | 2015-12-11 | Bayer Ip Gmbh | 經烷基胺基-取代之二氰基吡啶類及其胺基酸酯前藥 |
| CN104844687B (zh) * | 2009-01-29 | 2019-04-02 | 拜耳知识产权有限责任公司 | 烷基氨基-取代的二氰基嘧啶和它们的氨基酸酯药物前体 |
| EP2743270A1 (de) | 2009-01-29 | 2014-06-18 | Bayer Intellectual Property GmbH | Alkylamino-substituierte Dicyanopyridine und deren Aminosäureester-Prodrugs |
| DE102009006602A1 (de) | 2009-01-29 | 2010-08-05 | Bayer Schering Pharma Aktiengesellschaft | Alkylamino-substituierte Dicyanopyridine und deren Aminosäureester-Prodrugs |
| JP2015120727A (ja) * | 2009-01-29 | 2015-07-02 | バイエル・インテレクチュアル・プロパティ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツングBayer Intellectual Property GmbH | アルキルアミノ置換ジシアノピリジンおよびそのアミノ酸エステルプロドラッグ |
| EA019275B1 (ru) * | 2009-01-29 | 2014-02-28 | Байер Интеллектуэль Проперти Гмбх | Алкиламинозамещенные дицианопиридины и их пролекарства в виде сложных эфиров с аминокислотами |
| KR101833188B1 (ko) | 2009-01-29 | 2018-02-27 | 바이엘 인텔렉쳐 프로퍼티 게엠베하 | 알킬아민-치환된 디시아노피리딘 및 그의 아미노산 에스테르 전구약물 |
| CN104844687A (zh) * | 2009-01-29 | 2015-08-19 | 拜耳知识产权有限责任公司 | 烷基氨基-取代的二氰基嘧啶和它们的氨基酸酯药物前体 |
| WO2010086101A1 (de) | 2009-01-29 | 2010-08-05 | Bayer Schering Pharma Aktiengesellschaft | Alkylamino-substituierte dicyanopyridine und deren aminosäureester-prodrugs |
| KR101717305B1 (ko) * | 2009-01-29 | 2017-03-16 | 바이엘 인텔렉쳐 프로퍼티 게엠베하 | 알킬아민-치환된 디시아노피리딘 및 그의 아미노산 에스테르 전구약물 |
| EP4137137A1 (de) | 2010-05-26 | 2023-02-22 | Satiogen Pharmaceuticals, Inc. | Gallensäurerecyclinghemmer und satiogene zur behandlung von diabetes, adipositas und entzündlichen magen-darm-erkrankungen |
| EP2995317A1 (de) | 2010-05-26 | 2016-03-16 | Satiogen Pharmaceuticals, Inc. | Gallensäurerückflusshemmer zur behandlung von diabetes, adipositas und entzündlichen magen-darm-erkrankungen |
| EP3593802A2 (de) | 2010-05-26 | 2020-01-15 | Satiogen Pharmaceuticals, Inc. | Gallensäurerückflusshemmer zur behandlung von diabetes, adipositas und entzündlichen magen-darm-erkrankungen |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2012000945A1 (de) | 2010-06-30 | 2012-01-05 | Bayer Pharma Aktiengesellschaft | Substituierte dicyanopyridine und ihre verwendung |
| DE102010030688A1 (de) | 2010-06-30 | 2012-01-05 | Bayer Schering Pharma Aktiengesellschaft | Substituierte Dicyanopyridine und ihre Verwendung |
| US9669034B2 (en) | 2011-05-24 | 2017-06-06 | Bayer Intellectual Property Gmbh | 4-aryl-N-phenyl-1,3,5-triazin-2-amines containing a sulfoximine group |
| US9962389B2 (en) | 2011-05-24 | 2018-05-08 | Bayer Intellectual Property Gmbh | 4-aryl-N-phenyl-1,3,5-triazin-2-amines containing a sulfoximine group |
| EP2567959A1 (de) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridin-4-Carbonsäureamid-Derivate als Kinaseinhibitoren |
| EP3266457A1 (de) | 2011-10-28 | 2018-01-10 | Lumena Pharmaceuticals LLC | Gallensäurerückflusshemmer zur behandlung von cholestase-erkrankungen im kindesalter |
| WO2013063526A1 (en) | 2011-10-28 | 2013-05-02 | Lumena Pharmaceuticals, Inc. | Bile acid recycling inhibitors for treatment of hypercholemia and cholestatic liver disease |
| US11376251B2 (en) | 2011-10-28 | 2022-07-05 | Shire Human Genetic Therapies, Inc. | Bile acid recycling inhibitors for treatment of pediatric cholestatic liver diseases |
| US11229661B2 (en) | 2011-10-28 | 2022-01-25 | Shire Human Genetic Therapies, Inc. | Bile acid recycling inhibitors for treatment of pediatric cholestatic liver diseases |
| US10512657B2 (en) | 2011-10-28 | 2019-12-24 | Lumena Pharmaceutials Llc | Bile acid recycling inhibitors for treatment of pediatric cholestatic liver diseases |
| EP3278796A1 (de) | 2011-10-28 | 2018-02-07 | Lumena Pharmaceuticals LLC | Gallensäurerückflusshemmer zur behandlung von hypercholämie und cholestatischer lebererkrankung |
| US9708293B2 (en) | 2012-10-18 | 2017-07-18 | Bayer Pharma Aktiengesellschaft | N-(pyridin-2-yl)pyrimidin-4-amine derivatives containing a sulfone group |
| US9670161B2 (en) | 2012-10-18 | 2017-06-06 | Bayer Pharma Aktiengesellschaft | 5-fluoro-N-(pyridin-2-yl)pyridin-2-amine derivatives containing a sulfone group |
| WO2014060375A3 (en) * | 2012-10-18 | 2014-08-07 | Bayer Pharma Aktiengesellschaft | 5-fluoro-n-(pyridin-2-yl)pyridin-2-amine derivatives containing a sulfone group |
| WO2014060375A2 (en) * | 2012-10-18 | 2014-04-24 | Bayer Pharma Aktiengesellschaft | 5-fluoro-n-(pyridin-2-yl)pyridin-2-amine derivatives containing a sulfone group |
| US9650361B2 (en) | 2012-11-15 | 2017-05-16 | Bayer Pharam Aktiengesellschaft | N-(pyridin-2-yl)pyrimidin-4-amine derivatives containing a sulfoximine group |
| US9877954B2 (en) | 2012-11-15 | 2018-01-30 | Bayer Pharma Aktiengesellschaft | 5-fluoro-N-(pyridin-2-yl)pyridin-2-amine derivatives containing a sulfoximine group |
| US9650340B2 (en) | 2012-11-15 | 2017-05-16 | Bayer Pharma Aktiengesellschaft | 5-fluoro-N-(pyridin-2-yl)pyridin-2-amine derivatives containing a sulfoximine group |
| WO2014144650A2 (en) | 2013-03-15 | 2014-09-18 | Lumena Pharmaceuticals, Inc. | Bile acid recycling inhibitors for treatment of primary sclerosing cholangitis and inflammatory bowel disease |
| WO2014144485A1 (en) | 2013-03-15 | 2014-09-18 | Lumena Pharmaceuticals, Inc. | Bile acid recycling inhibitors for treatment of barrett's esophagus and gastroesophageal reflux disease |
| US9770445B2 (en) | 2013-07-04 | 2017-09-26 | Bayer Pharma Aktiengesellschaft | Sulfoximine substituted 5-fluoro-N-(pyridin-2-yl)pyridin-2-amine derivatives and their use as CDK9 kinase inhibitors |
| US9856242B2 (en) | 2014-03-13 | 2018-01-02 | Bayer Pharma Aktiengesellscaft | 5-fluoro-N-(pyridin-2-yl)pyridin-2-amine derivatives containing a sulfone group |
| US9790189B2 (en) | 2014-04-01 | 2017-10-17 | Bayer Pharma Aktiengesellschaft | Disubstituted 5-fluoro pyrimidine derivatives containing a sulfondiimine group |
| US9963464B2 (en) | 2014-04-11 | 2018-05-08 | Bayer Pharma Aktiengesellschaft | Macrocyclic compounds |
| US9902716B2 (en) | 2014-10-16 | 2018-02-27 | Bayer Pharma Aktiengesellschaft | Fluorinated benzofuranyl-pyrimidine derivatives containing a sulfone group |
| US9884849B2 (en) | 2014-10-16 | 2018-02-06 | Bayer Pharma Aktiengesellschaft | Fluorinated benzofuranyl-pyrimidine derivatives containing a sulfoximine group |
| WO2018153898A1 (de) | 2017-02-22 | 2018-08-30 | Bayer Pharma Aktiengesellschaft | Selektive partielle adenosin a1 rezeptor-agonisten in kombination mit mineralocorticoid-rezeptor-antagonisten |
| WO2018153895A1 (de) | 2017-02-22 | 2018-08-30 | Bayer Pharma Aktiengesellschaft | Selektive partielle adenosin a1 rezeptor-agonisten in kombination mit einem inhibitor der neutralen endopeptidase und/oder einem angiotensin ii rezeptor-antagonisten |
| WO2018153899A1 (de) | 2017-02-22 | 2018-08-30 | Bayer Pharma Aktiengesellschaft | Selektive partielle adenosin a1 rezeptor-agonisten in kombination mit stimulatoren und/oder aktivatoren der löslichen guanylatcyclase (sgc) |
| WO2018153900A1 (de) | 2017-02-22 | 2018-08-30 | Bayer Aktiengesellschaft | Selektive partielle adenosin a1 rezeptor-agonisten in kombination mit sglt-2-hemmern |
| WO2018153897A1 (de) | 2017-02-22 | 2018-08-30 | Bayer Pharma Aktiengesellschaft | Selektive partielle adenosin a1 rezeptor-agonisten in kombination mit hcn-kanal-hemmern |
| US11242356B2 (en) | 2017-03-28 | 2022-02-08 | Bayer Aktiengesellschaft | PTEFb inhibiting macrocyclic compounds |
| US11254690B2 (en) | 2017-03-28 | 2022-02-22 | Bayer Pharma Aktiengesellschaft | PTEFb inhibiting macrocyclic compounds |
| US11691986B2 (en) | 2017-03-28 | 2023-07-04 | Bayer Aktiengesellschaft | PTEFB inhibiting macrocyclic compounds |
| US11701347B2 (en) | 2018-02-13 | 2023-07-18 | Bayer Aktiengesellschaft | Use of 5-fluoro-4-(4-fluoro-2-methoxyphenyl)-N-{4-[(S-methylsulfonimidoyl)methyl]pyridin-2-yl}pyridin-2-amine for treating diffuse large B-cell lymphoma |
| EP4241840A2 (de) | 2019-02-12 | 2023-09-13 | Mirum Pharmaceuticals, Inc. | Verfahren zur behandlung von cholestase |
| EP4245367A2 (de) | 2019-02-12 | 2023-09-20 | Mirum Pharmaceuticals, Inc. | Verfahren zur behandlung von cholestase |
Also Published As
| Publication number | Publication date |
|---|---|
| DE102006042143A1 (de) | 2008-03-27 |
| JP5331693B2 (ja) | 2013-10-30 |
| EP2066637A1 (de) | 2009-06-10 |
| JP2010502659A (ja) | 2010-01-28 |
| PE20080772A1 (es) | 2008-08-07 |
| UY30578A1 (es) | 2008-05-02 |
| AR062630A1 (es) | 2008-11-19 |
| US8653109B2 (en) | 2014-02-18 |
| US20100093728A1 (en) | 2010-04-15 |
| CA2662728A1 (en) | 2008-03-13 |
| CA2662728C (en) | 2015-03-31 |
| US20140155401A1 (en) | 2014-06-05 |
| TW200823209A (en) | 2008-06-01 |
| CL2007002610A1 (es) | 2008-02-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008028590A1 (de) | Neue substituierte bipyridin-derivate und ihre verwendung als adenosin rezeptor liganden | |
| EP2099781B1 (de) | Substituierte 4-amino-3,5-dicyano-2-thiopyridine und ihre verwendung | |
| EP2099788B1 (de) | Cyclisch substituierte 3,5-dicyano-2-thiopyridine und ihre verwendung | |
| EP2274299B1 (de) | Heteroaryl-substituierte dicyanopyridine und ihre verwendung zur behandlung kardiovaskulärer erkrankungen | |
| EP2185550B1 (de) | Substituierte aryloxazole und ihre verwendung | |
| EP2235011B1 (de) | Substituierte pyrrolo[2, 3-b]- und pyrazolo[3, 4-b]pyridine als adenosin rezeptor liganden | |
| EP2297104B1 (de) | 2-alkoxy-substituierte dicyanopyridine und ihre verwendung | |
| DE102006009813A1 (de) | Verwendung von A2b/A1 Rezeptor Agonisten zur Modulation der Lipidspiegel | |
| WO2009100827A1 (de) | Cycloalkoxy-substituierte 4-phenyl-3,5-dicyanopyridine und ihre verwendung | |
| EP2556831B1 (de) | 3,4-Dihydro-4-oxo-5-aryl-pyrido[2,3-d]pyrimidin-6-carbonitrile als Adenosin Rezeptor Liganden zur Behandlung von cardiovasculären Erkrankungen |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07801993 Country of ref document: EP Kind code of ref document: A1 |
|
| REEP | Request for entry into the european phase |
Ref document number: 2007801993 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007801993 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2662728 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2009527038 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12440423 Country of ref document: US |