WO2007122970A1 - 核内受容体に結合するリガンド - Google Patents
核内受容体に結合するリガンド Download PDFInfo
- Publication number
- WO2007122970A1 WO2007122970A1 PCT/JP2007/056780 JP2007056780W WO2007122970A1 WO 2007122970 A1 WO2007122970 A1 WO 2007122970A1 JP 2007056780 W JP2007056780 W JP 2007056780W WO 2007122970 A1 WO2007122970 A1 WO 2007122970A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- general formula
- agent according
- represented
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/34—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having five-membered rings with one oxygen as the only ring hetero atom, e.g. isosorbide
- A61K31/341—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having five-membered rings with one oxygen as the only ring hetero atom, e.g. isosorbide not condensed with another ring, e.g. ranitidine, furosemide, bufetolol, muscarine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Definitions
- the present invention relates to a human peroxisome proliferator-activated ⁇ receptor gamma (hereinafter abbreviated as PPAR ⁇ ) agonist.
- PPAR ⁇ human peroxisome proliferator-activated ⁇ receptor gamma
- PPAR y is a ligand-dependent transcription factor belonging to the nuclear receptor superfamily, and is highly expressed in adipose tissue, immune cells, adrenal gland, spleen and the like.
- PPARRT has been reported to be deeply involved in adipocyte differentiation (see Non-Patent Document 1), and research on PPAR y is accelerating as a factor linking type 2 diabetes and obesity.
- PPAR y is also expressed in vascular smooth muscle and endothelial cells, and PPAR y agonists are thought to act on PPAR y in blood vessels in an anti-arteriosclerotic manner. It has also been shown that PPAR y agonists may be used clinically in addition to treating type II diabetes.
- thiazolidinediones that are ligands of PPAR y described later have a blood lipid lowering action (see, for example, Patent Document 1). More recent studies have reported that PPAR y affects monocyte mobilization and foam cell force cholesterol excretion, which are important events in the development of atherosclerosis (2). reference). Furthermore, troglitazone, a PPAR y agonist, has been reported to suppress vascular smooth muscle cell proliferation and intimal thickening (see Non-Patent Document 3). From these facts, PPAR y agonists are considered to be clinically usable as therapeutic agents for suppressing arteriosclerosis and vascular stenosis.
- the body is metabolically and chemically unstable and cannot be used as a medicine.
- thiazolidine derivatives have been reported as ligands for PPAR ⁇ .
- examples of such thiazolidine derivatives include 4 alkoxybenzylthiazolyl.
- Thiazolidinediones such as gin 2, 4 dione derivatives (for example, see Patent Documents 1 to 9), monocyclic benzamide derivatives (for example, see Patent Documents 10 to 12), indole derivatives (for example, see Patent Documents 13 and 14)
- Benzoisoxazole derivatives see, for example, Patent Document 15
- benzofuran derivatives see, for example, Patent Document 16.
- these compounds have been reported to have side effects such as compounds and heart failure that cause liver damage. These compounds are also different from the compounds of the present invention.
- the present inventor pays attention to a nuclear receptor, examines a method for identifying a compound that modulates the nuclear receptor, and includes a step of determining a compound that covalently binds to cysteine in the nuclear receptor.
- a method for identifying a compound that modulates a nuclear compound has been clarified (see Patent Document 17).
- Patent Document 17 a PPAR y agonist that is still fully satisfactory has not been obtained.
- Patent Document 1 JP-A-7-173158
- Patent Document 2 Japanese Patent Laid-Open No. 55-22636
- Patent Document 3 Japanese Patent Application Laid-Open No. 60-51189
- Patent Document 4 Japanese Patent Laid-Open No. 61-85372
- Patent Document 5 Japanese Patent Laid-Open No. 61-286376
- Patent Document 6 Japanese Patent Laid-Open No. 1 131169
- Patent Document 7 JP-A-2-83384
- Patent Document 8 Japanese Patent Laid-Open No. 5-213913
- Patent Document 9 Japanese Patent Laid-Open No. 4-210977
- Patent Document 10 JP-T 6-502146
- Patent Document 11 JP-A-8-333355
- Patent Document 12 JP-A-9-48771
- Patent Document 13 JP-A-9 169746
- Patent Document 14 JP-A-9 176162
- Patent Document 15 Japanese Patent Application Laid-Open No. 2004-67697
- Patent Document 16 Japanese Patent Laid-Open No. 10-237049
- Patent Document 17 Japanese Patent Laid-Open No. 2006-30037
- Non-Patent Document 1 Tontonoz, BP, et al., Cell, 1994, 7th 9 ⁇ , P. 1147-1156
- Non-Patent Document 2 Chinetti, G. et al., Circulation, 2000, No. 101, P. 2411-2417
- Non-patent document 3 Law, R. et al., The 'Journal' of 'Tari-Cal' Invest., 1996, No. 98, P. 1897-1905
- Non-Patent Document 4 Lehmann, J. M. et al., The 'Journal', Biol. Chem., 1995, No. 270, P. 12953—
- Non-Patent Document 5 Forman, B. M. et al., Cell, 1995, 83rd, P. 803-812
- Non-Patent Document 6 Kliewer, SA et al., Cell, 1995, 83rd, P. 813-819
- An object of the present invention is to provide a novel PPAR y agonist having a novel structure.
- PPAR y has a wide variety of endogenous ligands with large ligand-binding pockets, making it difficult to design new ligands. Therefore, the present inventor performed nuclear screening from various compounds by in silico screening based on computer-based simulation according to the method for identifying a compound that modulates a nuclear receptor described in JP-A-2006-30037. Compounds that covalently bound to cysteine in the internal receptor were selected. Specifically, first, compounds having ⁇ , / 3-unsaturated ketones were extracted by motif search (database: Namiki Shoji Co., Ltd.). Among the extracted compounds having ⁇ -unsaturated ketone, compounds having ⁇ -unsaturated ketone on the straight chain were selected.
- a ring represents a heterocyclic ring
- R 1 represents a hydrogen atom or an optionally substituted carboxyalkyl group
- R 2 represents — COR 5 (wherein R 5 has a substituent) C carry
- R 4 represents a hydrogen atom or a C alkyl group. Or a salt thereof or a pro thereof
- PPAR y agonist characterized in that it contains a drug
- R 1 ′ represents an optionally esterified carboxyalkyl group
- R 3 ′ represents a hydrogen atom or a halogen atom
- R 5 has a substituent, and may be a C aryl group. Or place
- the present invention also relates to an insulin-resistant pathological condition characterized by administering a PPAR ⁇ agonist containing a compound represented by the general formula (I), a salt thereof, or a prodrug thereof to mammals including humans.
- the present invention relates to a method for preventing or treating.
- the present invention relates to the use of a compound represented by the general formula (I) or a salt thereof or a prodrug thereof for producing a prophylactic or therapeutic agent for an insulin resistant disease state.
- the invention's effect [0009]
- the PPAR y agonist according to the present invention can be covalently bound to PPAR y to increase the transcriptional activity of the gene in the nuclear receptor.
- the PPAR y agonist according to the present invention can be used as an agent for preventing or treating an insulin-resistant pathological condition such as type II diabetes, obesity, hyperlipidemia or arteriosclerosis.
- an insulin-resistant pathological condition such as type II diabetes, obesity, hyperlipidemia or arteriosclerosis.
- the PPAR y agonist according to the present invention is useful as a preventive or therapeutic agent for type II diabetes, obesity or hyperlipidemia, a plurality of diseases such as type II diabetes, obesity or hyperlipidemia are simultaneously observed. Conditions that develop, i.e. metabolic syndrome and lifestyle-related diseases can also be prevented, treated or ameliorated.
- FIG. 1 shows the results of SDS-PAGE of PPAR y LBD labeled with rhodamine maleimide.
- FIG. 2 is a graph showing the luciferase activity of a ligand in cultured COS-7 cells. (Example 2)
- the heterocyclic ring represented by the A ring includes, for example, an oxygen atom, a nitrogen atom, and 1 to 5 heteroatoms selected from Z or a sulfur atom. Including 3- to 6-membered heterocycles which may be partially or fully saturated.
- heterocyclic ring examples include aziridine, azetidine, imidazole, imidazoline, imidazolidine, thiazole, isothiazole, thiazoline, thiazolidine, oxazole, isoxazole, oxazolidine, isoxazolidine, pyran, pyrazine, Pyrazole, pyrazoline, virazolidine, piperazine, piperidine, pyridine, pyrimidine, pyridazine, pyrrole, pyrroline, pyrrolidine, furan, furazane, triazine, morpholine, thiomorpholine, thiopyran or thiophene ring are preferred.
- the heterocyclic ring represented by the A ring is particularly preferably a furan ring or a pyrazole ring, which is preferably a 5-membered heterocyclic ring containing one oxygen atom or a 5-membered heterocyclic ring containing 1 or 2 nitrogen atoms.
- the carboxyalkyl group represented by R 1 is preferably a C carboxyalkyl group.
- carboxymethyl, carboxyethyl, carboxypropyl, strong carboxybutyl, carboxypentyl and the like are preferable, and carboxyethyl is particularly preferable.
- the carboxyl group in the carboxyalkyl group may be esterified, or the “optionally esterified carboxyl group” may be represented by the formula —COOR (where R is a hydrogen atom or substituted, And a group represented by (representing a hydrocarbon group).
- R is a hydrogen atom or substituted
- a group represented by Representing a hydrocarbon group
- lower alkoxycarbonyl examples include methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, isopropoxycarbonyl, butoxycarbonyl, isobutoxycarbonyl, sec butoxycarbonyl, tert butoxycarbonyl, pentyloxycarbonyl, isopentyloxy And C alkoxycarbons such as carbo- yl and neopentyloxy carbo yl.
- aryloxyballs include C-aryloxyballs such as phenoxycarbon, 1-naphthoxyball, 2-naphthoxycarbol, etc.
- aralkyloxycarbol examples include C aralkyloxycarbol such as benzyloxycarbol, phenoxycarboxyl and the like (preferably
- COR 5 represented by R 2 the C aryl group represented by R 5 is a phenol group.
- a naphthyl group, an anthracenyl group, etc., and a phenyl group is preferred.
- the C carry
- Examples of the substituent that the 6-14 group may have include, for example, a C alkyl group and a C alkoxy group.
- C alkyl group includes straight or branched C
- 1-6 1-6 kill groups such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec butyl, tert-butyl, n pentyl, isopentyl, 2-methylbutyl, neopentyl, 1 ethylpropyl , N-hexyl, isohexyl, 4-methylpentyl, 3-methylpentyl, 2-methylpentyl, 1-methylpentyl, 3,3 dimethylbutyl, 2,2-dimethylbutyl, 1,1-dimethylbutyl, 1,2— Examples thereof include dimethylbutyl, 1,3 dimethylbutyl, 2,3 dimethylbutyl, 1-ethylbutyl, and 2-ethylbutyl.
- C alkoxy group includes straight or branched C alkoxy groups
- the heterocyclic group represented by R 5 is, for example, a heterocyclic ring containing 1 to 5 heteroatoms selected from an oxygen atom, a nitrogen atom, and Z or a sulfur atom. Group.
- heterocyclic group examples include 1 pyrrolyl, 2 pyrrolyl, 3-pyrrolyl, pyrrolidyl, 1 imidazolyl, 2 imidazolyl, 4 imidazolyl, 1-pyrazolyl, 3 pyrazolyl, 4 pyrazolyl, virazolinyl, 2 thiazolyl, 4 Thiazolyl, 5 thiazolyl, 3 isothiazolyl, 4 isothiazolyl, 5 isothiazolyl, 2 oxazolyl, 4-oxazolyl, 5-oxazolyl, 3-isoxazolyl, 4 isoxazolyl, 5 isoxazolyl, piperidino, 2 piperidyl, 4 piperidyl, 4 pyridyl, 4 3 pyridyl, 4 pyridyl, pyrajur, 2 pyrimidyl, 4 pyrimidyl, 5 pyrimidyl, 3 pyridazil, 4
- heterocyclic group represented by R 5 a 5-membered heterocyclic group containing one sulfur atom is preferred, and a 2-chenyl group or a 3-chyl group is more preferred, and a 2-chyl group is more preferred.
- substituent that the heterocyclic group may have may include the same group as the substituent that the C aryl group represented by R 5 may have.
- the halogen atom represented by R 3 for example, a chlorine atom, a fluorine atom, and bromine atom.
- Esterification represented by R 3 may be the same as the carboxyl group in the carboxyalkyl group represented by R 1 above as “esterified! /, May! /, Carboxyl group”. Groups.
- the C alkyl group represented by R 4 includes the C aryl group represented by R 5 described above.
- a pharmaceutically acceptable salt is preferable.
- a metal salt an ammonium salt, a salt with an organic base, a salt with an inorganic acid, an organic salt.
- examples include salts with acids, salts with basic or acidic amino acids, and the like.
- the metal salt include alkali metal salts such as sodium salt and potassium salt; alkaline earth metal salts such as calcium salt, magnesium salt and barium salt; aluminum salt and the like.
- salts with organic bases For example, trimethylamine, triethylamine, pyridine, picoline, 2,6-lutidine, ethanolamine, diethanolamine, triethanolamine, cyclohexylamine, dicyclohexylamine, N, ⁇ '-dibenzylethylene And salts with diamine and the like.
- the salt with inorganic acid include salts with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like.
- Preferable examples of the salt with an organic acid include formic acid, acetic acid, trifluoroacetic acid, phthalic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, succinic acid, succinic acid, malic acid, methanesulfonic acid, and benzenesulfone. And salts with acid, ⁇ -toluenesulfonic acid and the like.
- Preferable examples of the salt with basic amino acid include salts with arginine, lysine, orthine and the like, and preferable examples of the salt with acidic amino acid include, for example, aspartic acid, glutamic acid and the like. Salt.
- the prodrug of the compound represented by the general formula (I) includes a compound that is converted into a compound represented by the general formula (I) by a reaction with an enzyme, gastric acid, or the like in a living body.
- a prodrug of the compound represented by the general formula (I) for example, when the compound represented by the general formula (I) has a carboxyl group, the compound in which the carboxyl group is esterified or amidated, for example,
- Examples of the compound represented by the general formula (I) include, but are not limited to, compounds in which the carboxyl group of the compound represented by the general formula (I) is ethyl ester, phenyl ester, carboxymethyl ester, or methylamidated. .
- the prodrug of the compound represented by the general formula (I) has a general formula under physiological conditions as described in Yodogawa Shoten, 1990, “Drug Development”, Vol. 7, “Molecular Design”, pages 163-198. It may be changed to the compound represented by (I). Salts or prodrugs of the compounds represented by the general formula (I) include hydrates and non-hydrates.
- the compound represented by the general formula (I) includes the general formula (II)
- R 1 ′ represents an optionally esterified carboxyalkyl group
- R 3 ′ represents a hydrogen atom or a halogen atom
- R 5 has the same meaning as described above
- R 3 ′′ represents an optionally carboxyl group, and R 4 has the same meaning as described above).
- the carboxyalkyl group which may be esterified represented by R 1 ′ in the general formula ( ⁇ ) has the same meaning as the carboxyalkyl group represented by R 1 above.
- carboxyethyl is preferable.
- the halogen atom represented by R 3 ′ has the same meaning as the halogen atom represented by R 3 .
- Examples of the compound represented by the general formula ( ⁇ ) include the following compounds (1) to (12) (hereinafter referred to as Compound 1, Compound 2, Compound 3, Compound 4, Compound 5, Compound 6, Compound 7, respectively).
- Etc. are preferable.
- esterification represented by R 3 ′′ may be used, and the carboxyl group may be used.
- the esterification represented by R 3 may have the same meaning as L and a carboxyleno group.
- R 3 may be esterified, but the carboxyl group is preferably carboxy! /.
- Examples of the compound represented by the general formula (III) include:
- Compound 13 (Hereinafter referred to as Compound 13) and the like are preferred.
- the compounds 1 to 13 are known compounds, and the compound represented by the general formula (I) can be easily produced based on a known method or a method known per se.
- optical isomers of the above compounds can be produced by a method known per se.
- optically active synthetic intermediates are used, or the final racemate is optically resolved according to a conventional method. Can be obtained.
- optical resolution method include methods known per se, such as fractional recrystallization method, chiral column method, diastereomer method and the like.
- the compound represented by the general formula (I) or a salt or prodrug thereof used in the PPAR y agonist according to the present invention (hereinafter sometimes simply referred to as the compound of the general formula (I)) .) Can be covalently bound to cysteine present in the nuclear receptor. Confirmation of the covalent bond with cysteine can be carried out, for example, by the method described in JP-A-2006-30037.
- the PPAR ⁇ agonist according to the present invention can be covalently bound to a receptor that activates proliferation of peroxisomes in a cell to activate (activate) the receptor.
- PPAR ⁇ When PPAR ⁇ is activated (activated) by a ligand, glucose uptake can be activated in, for example, skeletal muscle, fat or liver. Therefore, it is possible to prevent or treat a state in which glucose uptake is suppressed, for example, an insulin resistant disease state.
- Insulin resistance includes a state in which the action of insulin that promotes glucose uptake from blood in skeletal muscle, fat, liver, or the like is reduced or suppressed in these tissues. Examples of insulin resistance pathologies include type II diabetes (insulin-independent diabetes), obesity, hyperlipidemia, arteriosclerosis and the like. Insulin-resistant conditions include type II diabetes
- the PPAR ⁇ agonist according to the present invention can be used for the prevention or treatment of type II diabetes (insulin-independent diabetes), obesity, hyperlipidemia or arteriosclerosis.
- the prevention or treatment includes prevention, treatment or improvement of metabolic syndrome and lifestyle-related diseases.
- type II diabetes includes abnormal glucose tolerance as a symptom before onset of type II diabetes.
- Prevention or treatment of type 2 diabetes includes lowering blood glucose levels.
- glucose tolerance Includes delay or prevention of progression to disease, and also includes delay or prevention of progression from insulin-independent ⁇ -type diabetes to insulin-dependent type I diabetes, and diabetics associated with diabetes This includes delaying or preventing the progression of complications such as diabetic retinopathy, diabetic nephropathy or diabetic neuropathy.
- obesity includes visceral fat accumulation type obesity.
- Obesity is a condition in which fat is excessively accumulated in the body, exhibits insulin resistance, and can induce type II diabetes and hypertension. For this reason, prevention or treatment of obesity includes prevention or prevention of type II diabetes and hypertension.
- blood total cholesterol, LDL (low density lipoprotein) cholesterol, neutral fat (TG), free fatty acid value or remnant-like lipoprotein cholesterol ( RLP-cholesterol) or the like shows a high value
- prevention, treatment or improvement of a state where blood HDL cholesterol shows a low value or prevention, treatment or improvement of a state where blood HDL cholesterol shows a low value.
- the prevention of hyperlipidemia includes suppressing or preventing arteriosclerosis in the heart and brain caused by hyperlipidemia.
- Arteriosclerosis is accompanied by stenosis or occlusion in the lumen of the arterial wall, accompanied by decreased blood flow, for example, transient ischemic attack, cerebral infarction, myocardial infarction, angina, etc. obtain.
- Atherosclerosis includes atherosclerosis.
- Prevention or treatment of arteriosclerosis includes, for example, suppressing or preventing cerebral artery disease such as cerebral infarction, coronary artery disease such as angina pectoris, and myocardial infarction.
- the PPAR y agonist according to the present invention is used not only for humans but also for mammals other than humans (for example, monkeys, horses, horses, pigs, hidges, dogs, cats, rats, mice, chimpanzees, etc.). Is also applicable.
- the PPAR y agonists according to the present invention include human PPAR y agonists, and human PPAR y agonists are preferred when the PPAR y agonist is applied to humans.
- the PPAR ⁇ agonist according to the present invention can be used for oral administration or parenteral administration by mixing the compound of general formula (I) as it is or by adding a pharmacologically acceptable carrier to the compound of general formula (I).
- the dosage form can be used.
- the dosage form for oral administration include tablets (including sugar-coated tablets and film-coated tablets), pills, granules, powders, capsules (including soft capsules and microcapsules), syrups, and emulsions. Or suspension agents etc. It is.
- dosage forms for parenteral administration include injections, infusions, drops, suppositories, and the like.
- compositions include, for example, butyric acid polymers, glycolic acid polymers, butyric acid-glycolic acid copolymers, mixtures of butyric acid polymers and glycolic acid polymers, or bases such as polyglycerol fatty acid esters. Combinations can be made into sustained-release preparations.
- the content of the compound of the general formula (I) in the preparation in the PPAR y agonist according to the present invention is preferably selected as appropriate according to the form of the preparation, but is usually about 1! 99% by mass is preferred.
- the PPAR y agonist according to the present invention can be produced by using the compound of the general formula (I) as a raw material, for example, the method described in the 14th revised Japanese Pharmacopoeia, General Rules for Preparations or generally used in this field. It can be carried out according to known production methods or according to known production methods. In addition, when the PPAR y agonist is produced in the above-mentioned dosage form, it is preferable to produce it by appropriately adding an appropriate amount of additives usually used in pharmaceutical preparations.
- the additive examples include excipients (eg, lactose, sucrose, glucose, starch, sucrose, microcrystalline cellulose, licorice powder, mannitol, sodium hydrogen carbonate, calcium phosphate, calcium sulfate, etc.), binders (eg, Gum arabic, carmelol or salts thereof, gelatin, carboxymethylcellulose, hydroxypropylcellulose, popidone, etc.), disintegrant (eg carmellose, starch, calcium carbonate, crystalline cellulose, low substituted hydroxypropylcellulose, etc.), lubricant ( For example, magnesium silicate, stearic acid, magnesium stearate, calcium stearate, talc, plasma paraffin, macrogol, etc.), surfactant (eg, sodium lauryl sulfate, polysorbate 80, sorbitan monofatty acid ester) Steal, polyoxyl stearate 40, polyoxyethylene coin castor oil, etc.), suspension or thickener (eg, gum arab
- the PPAR ⁇ agonist according to the present invention is produced into, for example, a tablet, for example, the above-mentioned excipient, binder, disintegrant, lubricant and the like are added to the compound of the general formula (I).
- the compound of general formula (I) can be produced by containing the above-mentioned excipient, binder, disintegrant and the like.
- the above-mentioned excipients and the like are added to the compound of general formula (I), and in the case of producing syrups, for example, the compound of general formula (I) is used.
- a suspending agent, a surfactant or an emulsifier and a solvent are added to the compound of the general formula (I) as an injection.
- a solvent and a soothing agent are added to the compound of general formula (I)
- a suppository base is added to the compound of general formula (I), for example. It can be made to contain.
- the tablet or capsule may be coated with an enteric coating agent such as methacrylic acid copolymer, hydroxypropylmethylcellulose acetate succinate or hydroxypropylmethylcellulose phthalate. As the capsule, an enteric capsule may be used.
- the PPARy agonist according to the present invention can be used safely with stability and low toxicity.
- the daily dose varies depending on the patient's condition, body weight, type of compound, route of administration, etc.For example, in the case of oral administration to patients with type II diabetes, adult (body weight approximately 60 kg)
- the amount is about 1 ⁇ g to 500 mg, preferably about 10 ⁇ g to 50 mg as an active ingredient (a compound represented by the general formula (I) or a salt thereof or a prodrug thereof). It is preferable to administer in 3 divided doses.
- the PPAR y agonist according to the present invention may be used as long as it does not inhibit the action of the present invention, For example, it can be used in combination with anti-obesity agents, anti-diabetic agents, anti-hyperlipidemic agents, antihypertensive agents and the like.
- anti-obesity agent examples include leptin, dextroamphetamine, amphetamine, fenfluramine, dexfenfluramine, sibutramine, mazindol, phentermine and the like.
- Antidiabetic drugs include insulin, sulfonylurea (eg, tolptamide, darribenclamide, daripizide, etc.), biguanide (eg, metformin, etc.), meglitide (eg, repaglinide, etc.), ⁇ -darcosidase Inhibitors (for example, miglitonole, akanolevose, etc.), nateglinide, troglitazone, rosiglitazone, pioglitazone and the like can be mentioned.
- sulfonylurea eg, tolptamide, darribenclamide, daripizide, etc.
- biguanide eg, metformin, etc.
- meglitide eg, repaglinide, etc.
- ⁇ -darcosidase Inhibitors for example, miglitonole, akanolevose, etc.
- nateglinide for
- Antihyperlipidemic agents include, for example, cholestyramine, colestipol, clofibrate, gemfib mouth gil, fenofibrate, tesaglitazar, atorvastatin, flupastatin, oral pastatin, pravastatin, simpastatin, cerivastatin, and assipimottas , Probucol or dextrothyroxine.
- antihypertensive agents include 13 blockers such as alprenolol, atenolol, timolol, pindolol or propranolol; angiotensin converting enzyme such as benazepril, captopril, enalabril, fuocinopril, lisinopril, quinapril or ramipril.
- Inhibitors -fedipine, ferrodipine,-cardipine, isradipine,-calcium channel blockers such as modipine, diltiazem or veranomil; and 1 blocker such as doxazosin, urapidil, prazosin or terazosin.
- the present invention also provides the use of a compound represented by the general formula (I) or a salt thereof or a prodrug thereof for producing an agent for preventing or treating an insulin-resistant pathological condition.
- a compound represented by the general formula (I) or a salt thereof or a prodrug thereof for producing an agent for preventing or treating an insulin-resistant pathological condition.
- the use of the compound represented by the general formula (I) or a salt thereof or a prodrug thereof for producing a therapeutic agent for type 2 diabetes, obesity, hyperlipidemia or arteriosclerosis are the same as described above.
- the compound represented by the general formula (I) or a salt thereof, a prodrug thereof or a production method thereof is the same as described above.
- the present invention further relates to a method for preventing or treating an insulin-resistant pathological condition wherein a compound represented by the general formula (I) or a salt thereof or a prodrug thereof is administered to mammals including humans. There is also.
- the compound represented by formula (I) and preferred forms thereof are the same as described above.
- the compound represented by the general formula (I) or a salt thereof, a prodrug thereof or a production method thereof is the same as described above.
- the prevention or treatment method of the present invention includes mammals including humans, that is, mammals other than humans, such as monkeys, mice, horses, pigs, hidges, nu, cats, rats. It can also be applied to mice, chimpanzees, etc. That is, an insulin-resistant pathological condition is prevented or treated by administering a compound represented by the general formula (I) or a salt thereof or a prodrug thereof to these mammals.
- the prevention or treatment method of the present invention is preferably applied to a patient exhibiting an insulin-resistant pathological condition that is preferably applied to a human. In addition, among insulin-resistant pathologies, it is preferably applied to patients with type II diabetes, obesity, hyperlipidemia, or arteriosclerosis. When the prevention or treatment method of the present invention is applied to humans, it is preferable to administer human PPARy agonists.
- the method of administering the compound represented by the general formula (I) or a salt thereof or a prodrug thereof to a human or the like is not particularly limited, and may be administered by an appropriate administration route according to the formulation form. it can.
- the compound represented by the general formula (I) or a salt thereof or a prodrug thereof and other drugs such as the above-mentioned anti-obesity agent, anti-diabetic drug, A hyperlipidemia agent, an antihypertensive agent or the like can also be used in combination.
- Novagen's pET28b NdelZBamHI site contains DNA with human PPARy (SEQ ID NO: 1) and BD (aa. 195-477) N-end and C-end restriction enzyme sites (Ndel and BamHI) added. Cloning was performed, and the nucleotide sequence was confirmed by sequencing. E.
- coli BL 21 (DE3) was transformed into 1 L LB (Luria-Bertani) medium (1% by mass Bacto Tripton 0.5 Incubate at 37 ° C until the absorbance (OD600nm) reaches 0.8 with 1% by weight Yeast Extract, 1% by weight NaCl), then cool to 18 ° C and ImM IPTG (Isopropyl-1-thio- ⁇ — D-galatatopyranoside) was added and cultured for 18 hours. E. coli was collected by centrifugation, suspended in 20 mM Hepes (pH 7.4), 200 mM NaCl, 25 mM imidazole solution, and stored at ⁇ 20 ° C.
- the cells were sonicated and the insoluble protein was removed by centrifugation. After that, the cells were subjected to affinity chromatography using a nickel ram (eluent: 20 mM Tris (pH7.4), 250 mM NaCl, 250 mM imidazole).
- PPAR y LBD was purified. Further, the buffer eluate was replaced with 20 mM Tris (pH 7.4) and 150 mM NaCl at the same time as purification by Superdex 200 gel filtration chromatography. The obtained PPAR y LBD was concentrated by ultrafiltration until the concentration of PPAR y LBD was about 5 mgZmL to obtain a PPAR y LBD solution.
- ligand Compound 12
- DMSO dimethylsulfoxide
- SDS sodium dodecyl sulfate
- TCEP tris [2-carboxyethyl] phosphine
- GW9662 (2-black mouth 5-nitro 1-phenol benzamide; PPAR y antagonist; Biochemistry; 2002; 41 (21); 6640-6650) was used.
- FIG. 1 shows that the signal of PPAR ⁇ LBD labeled with rhodamine maleimide is weak, indicating that the ligand is covalently bound.
- BRL49653 When MCC555 and PPAR y LBD solution were incubated, the signal of P PAR ⁇ LBD was as strong as that of the control, indicating that PPAR y LBD was not covalently bound to BRL49653 or MCC555.
- the PPAR y LBD signal is very weak compared to the control.
- 15d-PGj or GW9662 is covalently bound to PPAR y LBD and the ligand.
- COS-7 cells were cultured in a culture solution [Dulbecco's modified Eagle medium (DMEM) containing 10% FCS (child fetus serum)]. COS-7 cells cultivated the day before transfection are seeded in 24-well plates at 5 x 10 4 cells, and the next day, UA S -tk-Luc, pEYFP-Cl, pCMX-gal- PPAR ⁇ The plasmid (shiraki, T. et al., J. Biol.
- Fig. 2 shows the luciferase activity of the ligand (compounds 1 to 13). Both compounds show that luciferase activity has higher transcriptional activity than the control.
- the PPAR ⁇ agonist of the present invention is useful as an agent for preventing or treating an insulin-resistant pathological condition.
Landscapes
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Diabetes (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Epidemiology (AREA)
- Child & Adolescent Psychology (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description
明 細 書
核内受容体に結合するリガンド
技術分野
[0001] 本発明は、ヒトペルォキシゾーム増殖剤活性ィ匕受容体ガンマ(以下、 PPAR γと略 記する。)作動剤に関する。
背景技術
[0002] PPAR yは核内受容体スーパーファミリーに属するリガンド依存性の転写因子であ り、脂肪組織、免疫細胞、副腎、脾臓等に高発現している。脂肪細胞の分化に PPA R Tが深く関与していることが報告され (非特許文献 1参照)、 Π型糖尿病と肥満をつ なぐ因子として PPAR yの研究が加速している。また、最近では血管平滑筋、内皮細 胞にも PPAR yの発現が認められ、 PPAR y作動剤は血管の PPAR yに抗動脈硬 化的に作用すると考えられている。また、 PPAR y作動剤は II型糖尿病の治療以外 にも臨床使用できる可能性があることが示されている。例えば、後記する PPAR yの リガンドであるチアゾリジンジオン類は血中脂質低下作用を有する (例えば、特許文 献 1参照)。さらに最近の研究によれば、 PPAR yは、ァテローム性動脈硬化症の発 症に重要な事象である単球動員及び泡沫細胞力 のコレステロール排出に影響を 及ぼすと報告されている(非特許文献 2参照)。さらに、 PPAR y作動剤のトログリタゾ ンは、血管平滑筋細胞増殖と血管内膜の肥厚を抑制させるとの報告がある(非特許 文献 3参照)。これらのことから、 PPAR y作動剤は、動脈硬化症や血管狭窄を抑制 するための治療剤として臨床使用できると考えられている。
[0003] 近年、 II型糖尿病に効果のあるチアゾリジン誘導体力 SPPAR γのリガンド (非特許 文献 4、 5参照)であることが分かると同時に 15 デォキシ一 Δ12'14 プロスタグランジ ン j (15d PGj )が内在性の PPAR γのリガンドとして報告された (非特許文献 6参
2 2
照)。しかし内因性の PPAR γのリガンドである 15d— PGj等の不飽和脂肪酸誘導
2
体は代謝的にも化学的にも不安定であり、医薬として供する事はできない。
このため、 PPAR γのリガンドとして新たなチアゾリジン誘導体が種々報告されてい る。そのようなチアゾリジン誘導体としては、例えば、 4 アルコキシベンジルチアゾリ
ジン 2, 4 ジオン誘導体等のチアゾリジンジオン類 (例えば、特許文献 1〜9参照) 、単環のベンズアミド誘導体 (例えば、特許文献 10〜 12参照)、インドール誘導体( 例えば、特許文献 13, 14参照)、ベンゾイソキサゾール誘導体 (例えば、特許文献 1 5参照)又はべンゾフラン誘導体 (例えば、特許文献 16参照)等が挙げられる。しかし 、これら化合物は、肝障害をきたすィ匕合物や心不全等の副作用が報告されている。 また、これら化合物は、本発明の化合物とは異なる。
一方、本発明者は、核内受容体に注目し、核内受容体を調節する化合物を同定す る方法につき検討し、核内受容体中のシスティンと共有結合する化合物を判定する 工程を含む核内化合物を調節する化合物の同定方法を明らかにした (特許文献 17 参照)。しかし、未だ十分満足する PPAR y作動剤は得られていな力つた。
特許文献 1 :特開平 7— 173158号公報
特許文献 2:特開昭 55 - 22636号公報
特許文献 3:特開昭 60 - 51189号公報
特許文献 4:特開昭 61— 85372号公報
特許文献 5:特開昭 61— 286376号公報
特許文献 6:特開平 1 131169号公報
特許文献 7:特開平 2— 83384号公報
特許文献 8:特開平 5— 213913号公報
特許文献 9 :特開平 4— 210977号公報
特許文献 10:特表平 6— 502146号公報
特許文献 11 :特開平 8— 333355号公報
特許文献 12 :特開平 9— 48771号公報
特許文献 13 :特開平 9 169746号公報
特許文献 14:特開平 9 176162号公報
特許文献 15:特開 2004- 67697号公報
特許文献 16:特開平 10— 237049号公報
特許文献 17:特開 2006- 30037号公報
非特許文献 1 :トントンッ 'ビ一'ピー(Tontonoz, BP)ら、細胞(Cell)、 1994年、第 7
9卷、 P. 1147 - 1156
非特許文献 2 :チネッティ'ジ一(Chinetti, G. )ら、サーキュレーション(Circulation )、 2000年、第 101卷、 P. 2411 - 2417
非特許文献 3 :口一'アール(Law, R. )ら、ザ'ジャーナル'ォブ 'タリ-カル 'インべス ティゲーシヨン (J. Clin. Invest. ) , 1996年、第 98卷、 P. 1897—1905
非特許文献 4 :レーマン 'ジェ^ ~ ·ェム(Lehmann, J. M. )ら、ザ'ジャーナル'ォブ ィォロジカル.ケミストリー (J. Biol. Chem. ) , 1995年、第 270卷、 P. 12953—
12956
非特許文献 5 :フォーマン'ビ一'ェム(Forman, B. M. )ら、細胞(Cell)、 1995年、 第 83卷、 P. 803 - 812
非特許文献 6 :タリーヮ一'エス'エー(Kliewer, S . A. )ら、細胞(Cell)、 1995年、 第 83卷、 P. 813 - 819
発明の開示
発明が解決しょうとする課題
[0005] 本発明は、新 ヽ構造を持った新規 PPAR y作動剤を提供することを目的とする。
課題を解決するための手段
[0006] PPAR yはリガンド結合ポケットが大きぐ内在性リガンドも多岐に渡っており、新規 リガンドの設計が困難である。そこで、本発明者は、特開 2006-30037号公報に記 載の核内受容体を調節する化合物を同定する方法に従い、種々の化合物から、コン ピュータ利用によるシミュレーションに基づく in silicoスクリーニングにより、核内受容 体中のシスティンと共有結合する化合物を選択した。具体的には、まず α , /3—不 飽和ケトンを有する化合物をモチーフ検索 (データベース:ナミキ商事株式会社)によ り抽出した。前記抽出した , β 不飽和ケトンを有する化合物のうち、直鎖上に , β 不飽和ケトンを有する化合物を選択した。次いで、類似する構造式によるクラ スター分類を行い、薬物一タンパク質ドッキングシミュレーションプログラム(DOCK 及び GOLD)で核内受容体の形状と高 、相補性のある薬物をグラフトポロジカルに 探索をおこなった。選択したィ匕合物について、核内受容体中の遺伝子の転写活性を 測定したところ、該遺伝子の転写活性を促進する化合物に共通する構造を有するこ
とを知見した。本発明者は、該知見に基づきさらに研究を進め、本発明を完成するに いたった。
すなわち、本発明は、
[1]一般式 (I)
[化 1]

(式中、 A環は複素環を示し、 R1は水素原子又はエステルイ匕していてもよいカルボキ シアルキル基を示し、 R2は— COR5 (式中、 R5は置換基を有していてもよい C ァリー
6-14 ル基又は置換基を有していてもよい複素環基を示す。 )又は— NOを示し、 R3は水
2
素原子、ハロゲン原子又はエステルイ匕していてもよいカルボキシル基を示し、 R4は水 素原子又は C アルキル基を示す。 )で表される化合物もしくはその塩又はそのプロ
1-6
ドラッグを含有することを特徴とする PPAR y作動剤、
[2]A環がフラン環又はピラゾール環である前記 [1]に記載の剤
[3]化合物が、一般式 (II)
[化 2]

(式中、 R1'はエステル化していてもよいカルボキシアルキル基を示し、 R3'は水素原 子又はハロゲン原子を示し、 R5は置換基を有して 、てもよ 、C ァリール基又は置
6-14
換基を有して ヽてもよ ヽ複素環基を示す。 )で表される化合物である前記 [ 1 ]に記載 の剤、
[4]R5で示されるァリール基がフ ニル基である前記 [3]に記載の剤、
[5]R5で示される複素環基が 2—チェニル基である前記 [3]に記載の剤

である前記 [3]に記載の剤,
[7]化合物が、一般式 (III)
[化 4]

(式中、 R3はエステルイ匕していてもよいカルボキシル基を示し、 R4は水素原子又は C アルキル基を示す。)で表される化合物である前記 [1]に記載の剤、
1-6
[8]化合物が、
[化 5]

[9]インスリン抵抗性の病態の予防又は治療剤である前記 [1]〜 [8]の 、ずれかに 記載の剤、
[10]インスリン抵抗性の病態力 1型糖尿病、肥満、高脂血症又は動脈硬化症である 前記 [9]に記載の剤、
に関する。
また、本発明は、ヒトを含む哺乳動物に一般式 (I)で表される化合物もしくはその塩 又はそのプロドラッグを含有する PPAR γ作動剤を投与することを特徴とするインスリ ン抵抗性の病態を予防又は治療する方法に関する。さらに本発明は、インスリン抵抗 性の病態の予防又は治療剤を製造するための一般式 (I)で表される化合物もしくは その塩又はそのプロドラッグの使用に関する。
発明の効果
[0009] 本発明に係る PPAR y作動剤は、 PPAR yに共有結合して、核内受容体中の遺 伝子の転写活性を上昇させることができる。本発明に係る PPAR y作動剤は、インス リン抵抗性の病態、例えば II型糖尿病、肥満、高脂血症又は動脈硬化症の予防又は 治療剤として使用できる。さらに、本発明に係る PPAR y作動剤は、 II型糖尿病、肥 満又は高脂血症の予防又は治療剤として有用であるので、 II型糖尿病、肥満又は高 脂血症等の病気が複数同時に発症する状態、すなわちメタボリックシンドロームや生 活習慣病も予防、治療又は改善し得る。
図面の簡単な説明
[0010] [図 1]図 1は、ローダミンマレイミドによりラベルされた PPAR y LBDの SDS— PAGE の結果を示す図である。(実施例 1)
[図 2]図 2は、培養 COS— 7細胞におけるリガンドのルシフェラーゼ活性を示す図であ る。(実施例 2)
発明を実施するための最良の形態
[0011] 一般式 (I)で表される化合物において、 A環で示される複素環としては、例えば酸 素原子、窒素原子及び Z又は硫黄原子から選択される 1〜5個のへテロ原子を含む 、一部又は全部飽和されていてもよい 3〜6員の複素環が挙げられる。該複素環とし ては具体的には、例えば、アジリジン、ァゼチジン、イミダゾール、イミダゾリン、イミダ ゾリジン、チアゾール、イソチアゾール、チアゾリン、チアゾリジン、ォキサゾール、イソ ォキサゾール、ォキサゾリジン、イソォキサゾリジン、ピラン、ピラジン、ピラゾール、ピ ラゾリン、ビラゾリジン、ピぺラジン、ピぺリジン、ピリジン、ピリミジン、ピリダジン、ピロ ール、ピロリン、ピロリジン、フラン、フラザン、トリアジン、モルホリン、チオモルホリン、 チォピラン又はチォフェン環等が好ましく挙げられる。中でも、 A環で示される複素環 としては、酸素原子を 1個含む 5員の複素環又は窒素原子を 1もしくは 2個含む 5員の 複素環が好ましぐフラン環又はピラゾール環がとりわけ好ましい。
[0012] R1で示されるカルボキシアルキル基としては、 C カルボキシアルキル基が好ましく
2-6
、具体的には、例えばカルボキシメチル、カルボキシェチル、カルボキシプロピル、力 ルボキシブチル及びカルボキシペンチル等が好ましく挙げられ、カルボキシェチルが とりわけ好ましい。
[0013] 該カルボキシアルキル基におけるカルボキシル基はエステル化されていてもよぐ「 エステル化されていてもよいカルボキシル基」としては、式— COOR(Rは水素原子 又は置換されて 、てもよ 、炭化水素基を示す)で表される基等が挙げられる。なかで も、カルボキシ、低級アルコキシカルボ-ル、ァリールォキシカルボ-ル又はァラルキ ルォキシカルボ-ル等が好ましく挙げられる。「低級アルコキシカルボ-ル」としては、 例えばメトキシカルボニル、エトキシカルボニル、プロポキシカルボニル、イソプロポキ シカルボニル、ブトキシカルボニル、イソブトキシカルボニル、 sec ブトキシカルボ二 ル、 tert ブトキシカルボニル、ペンチルォキシカルボニル、イソペンチルォキシカル ボ -ル、ネオペンチルォキシカルボ-ル等の C アルコキシカルボ-ル等が挙げられ
1-6
る。「ァリールォキシカルボ-ル」としては、例えばフエノキシカルボ-ル、 1 ナフトキ シカルボ-ル、 2—ナフトキシカルボ-ル等の C ァリールォキシカルボ-ル等が挙
7-12
げられる。「ァラルキルォキシカルボ-ル」としては、例えばべンジルォキシカルボ- ル、フエネチルォキシカルボ-ル等の C ァラルキルォキシカルボ-ル等(好ましく
7-15
は、 C ァリール—C アルコキシ カルボ-ル等)が挙げられる。
6-10 1-6
[0014] R2で示される COR5において、 R5で示される C ァリール基としては、フエ-ル基
6-14
、ナフチル基、アントラセニル基等が挙げられ、フエ-ル基が好まし 、。該 C ァリー
6-14 ル基が有していてもよい置換基としては、例えば C アルキル基、 C アルコキシ基
1-6 1-6
又はハロゲン原子等が挙げられる。「C アルキル基」としては直鎖又は分岐 C アル
1-6 1-6 キル基が挙げられ、例えばメチル、ェチル、 n—プロピル、イソプロピル、 n—ブチル、 イソブチル、 sec ブチル、 tert—ブチル、 n ペンチル、イソペンチル、 2—メチルブ チル、ネオペンチル、 1 ェチルプロピル、 n—へキシル、イソへキシル、 4ーメチルぺ ンチル、 3—メチルペンチル、 2—メチルペンチル、 1ーメチルペンチル、 3, 3 ジメ チルブチル、 2, 2—ジメチルブチル、 1, 1ージメチルブチル、 1, 2—ジメチルブチル 、 1, 3 ジメチルブチル、 2, 3 ジメチルブチル、 1ーェチルブチル、 2 ェチルブ チル等が挙げられる。「C アルコキシ基」としては直鎖又は分岐 C アルコキシ基が
1-6 1-6
挙げられ、例えばメトキシ、エトキシ、プロポキシ、イソプロポキシ、ブトキシ、 sec ブト キシ、 tert ブトキシ、 n—ペンチルォキシ等が挙げられる。ハロゲン原子としては、 例えば塩素原子、フッ素原子、臭素原子等が挙げられる。
[0015] R2で示される COR5において、 R5で示される複素環基としは、例えば酸素原子、 窒素原子及び Z又は硫黄原子から選択される 1〜5個のへテロ原子を含む複素環基 が挙げられる。複素環基としては具体的には、例えば、 1 ピロリル、 2 ピロリル、 3 —ピロリル、ピロリジル、 1 イミダゾリル、 2 イミダゾリル、 4 イミダゾリル、 1—ピラゾ リル、 3 ピラゾリル、 4 ピラゾリル、ビラゾリニル、 2 チアゾリル、 4 チアゾリル、 5 チアゾリル、 3 イソチアゾリル、 4 イソチアゾリル、 5 イソチアゾリル、 2 ォキサ ゾリル、 4ーォキサゾリル、 5—ォキサゾリル、 3—イソォキサゾリル、 4 イソォキサゾリ ル、 5 イソォキサゾリル、ピペリジノ、 2 ピペリジル、 3 ピペリジル、 4ーピペリジル 、 2 ピリジル、 3 ピリジル、 4 ピリジル、ピラジュル、 2 ピリミジ -ル、 4 ピリミジ -ル、 5 ピリミジ -ル、 3 ピリダジ -ル、 4 ピリダジ -ル、 2 モルホリニル、 3 モ ルホリニル、 2 フリル、 3 フリル、ビラ-ル、 2 チェ-ル又は 3 チェ-ル等が好 ましく挙げられる。中でも、 R5で示される複素環基としは、硫黄原子を 1個含む 5員の 複素環基が好ましぐ 2 チェニル又は 3 チェ-ル基がより好ましぐ 2 チェ-ル 基がさらに好ましい。該複素環基が有していてもよい置換基としては、上記 R5で示さ れる C ァリール基が有して 、てもよ 、置換基と同様の基が挙げられる。
6-14
[0016] R3で示されるハロゲン原子としては、例えば塩素原子、フッ素原子、臭素原子等が 挙げられる。
R3で示されるエステル化して 、てもよ 、カルボキシル基としては、上記 R1で示される カルボキシアルキル基における、「エステル化されて!/、てもよ!/、カルボキシル基」と同 様の基が挙げられる。
[0017] R4で示される C アルキル基としては、上記 R5で示される C ァリール基が有して
1-6 6-14
いてもよい置換基としての C アルキル基と同様の基が挙げられ、好ましくは直鎖 C
1-6 2-4 アルキル基であり、 n プロピルが最も好まし!/、。
[0018] 一般式 (I)で表される化合物の塩としては、薬学的に許容される塩が好ましぐ例え ば金属塩、アンモニゥム塩、有機塩基との塩、無機酸との塩、有機酸との塩、塩基性 又は酸性アミノ酸との塩等が挙げられる。金属塩の好適な例としては、例えばナトリウ ム塩、カリウム塩等のアルカリ金属塩;カルシウム塩、マグネシウム塩、バリウム塩等の アルカリ土類金属塩;アルミニウム塩等が挙げられる。有機塩基との塩の好適な例と
しては、例えばトリメチルァミン、トリェチルァミン、ピリジン、ピコリン、 2, 6—ルチジン 、エタノールァミン、ジエタノールァミン、トリエタノールァミン、シクロへキシルァミン、 ジシクロへキシルァミン、 N, Ν'—ジベンジルエチレンジァミン等との塩が挙げられる 。無機酸との塩の好適な例としては、例えば塩酸、臭化水素酸、硝酸、硫酸、リン酸 等との塩が挙げられる。有機酸との塩の好適な例としては、例えばギ酸、酢酸、トリフ ルォロ酢酸、フタル酸、フマル酸、シユウ酸、酒石酸、マレイン酸、クェン酸、コハク酸 、リンゴ酸、メタンスルホン酸、ベンゼンスルホン酸、 ρ—トルエンスルホン酸等との塩 が挙げられる。塩基性アミノ酸との塩の好適な例としては、例えばアルギニン、リジン 、オル-チン等との塩が挙げられ、酸性アミノ酸との塩の好適な例としては、例えばァ スパラギン酸、グルタミン酸等との塩が挙げられる。
[0019] 一般式 (I)で表される化合物のプロドラッグとしては、生体内において酵素や胃酸 等による反応により一般式 (I)で表される化合物に変換される化合物が挙げられる。 一般式 (I)で表される化合物のプロドラッグとしては、例えば、一般式 (I)で表される 化合物がカルボキシル基を有する場合、該カルボキシル基がエステル化、アミド化さ れた化合物、例えば、一般式 (I)で表される化合物のカルボキシル基が例えばェチ ルエステルイ匕、フエ-ルエステル化、カルボキシメチルエステル化、メチルアミド化さ れたィ匕合物等が挙げられるが、これらに限定されない。これらの化合物は公知の方 法によって製造することができる。一般式 (I)で表される化合物のプロドラッグは、廣 川書店 1990年刊「医薬品の開発」第 7卷「分子設計」 163〜198頁に記載されてい るような、生理的条件で一般式 (I)で表される化合物に変化するものであってもよ 、。 一般式 (I)で表される化合物の塩又はプロドラッグは水和物及び非水和物を包含 する。
[0020] また、一般式 (I)で表される化合物が、光学異性体、立体異性体、位置異性体、回 転異性体を含有する場合には、これらも一般式 (I)で表される化合物として包含され る。
[0021] 一般式 (I)で表される化合物としては、一般式 (II)
[化 6]

(式中、 R1'はエステル化していてもよいカルボキシアルキル基を示し、 R3'は水素原 子又はハロゲン原子を示し、 R5は上記と同意義である。)、又は一般式 (III)
[化 7]

(式中、 R3"はエステルイ匕していてもよいカルボキシル基を示し、 R4は上記と同意義で ある。 )が挙げられる。
一般式 (Π)における、 R1'で示されるエステル化していてもよいカルボキシアルキル 基は、上記 R1で示されるカルボキシアルキル基と同意義である。 R1'で示されるエステ ル化していてもよいカルボキシアルキル基としては、カルボキシェチルが好ましい。 R 3'で示されるハロゲン原子は、 R3で示されるハロゲン原子と同意義である。
一般式 (Π)で表される化合物としては、例えば以下の(1)〜(12)の化合物(以下、 それぞれ化合物 1,化合物 2,化合物 3,化合物 4,化合物 5,化合物 6,化合物 7,化 合物 8,化合物 9,化合物 10,化合物 11又は化合物 12という。):
(1)化合物 1
[化 8]

(2)化合物 2

[化 13]

(10)化合物 10 [化 17]


(12)化合物 12
[化 19]

等が好ましく挙げられる。
[0024] 一般式(III)における、 R3"で示されるエステル化して 、てもよレ、カルボキシル基は、
R3で示されるエステル化して 、てもよ L、カルボキシノレ基と同意義である。 R3"で示され るエステル化して 、てもよ 、カルボキシル基としては、カルボキシが好まし!/、。
[0025] 一般式 (III)で表される化合物としては、例えば
[化 20]

[0026] 上記化合物 1乃至 13は、公知の化合物であり、一般式 (I)で表される化合物は、公 知の方法又は自体公知の方法に基づき容易に製造することができる。
また、上記化合物の光学異性体は自体公知の方法により製造することができ、例え ば光学活性な合成中間体を用いるか、最終物のラセミ体を常法に従って光学分割す ることにより光学異性体を得ることができる。光学分割法としては、自体公知の方法、 例えば、分別再結晶法、キラルカラム法、ジァステレオマー法等が挙げられる。
[0027] 本発明に係る PPAR y作動剤に使用される一般式 (I)で表される化合物もしくはそ の塩又はプロドラッグ (以下、単に、一般式 (I)の化合物と略記することもある。)は核 内受容体に存在するシスティンと共有結合し得る。前記システィンとの共有結合の確 認は、例えば特開 2006— 30037号公報に記載の方法等により実施できる。
[0028] 本発明に係る PPAR γ作動剤は、細胞内のペルォキシゾームの増殖を活性ィ匕する 受容体に共有結合して該受容体を活性化 (作動)させ得る。 PPAR γがリガンドによ り活性化される (作動する)と、例えば骨格筋、脂肪又は肝臓等においてグルコース の取り込みが活性ィ匕され得る。このため、グルコースの取り込みが抑制されている状 態、例えばインスリン抵抗性の病態等を予防又は治療し得る。インスリン抵抗性は、 例えば骨格筋、脂肪又は肝臓等において血液からの糖の取り込みを促進させるイン スリンの作用が、これらの組織で低下又は抑制されている状態を含む。インスリン抵 抗性の病態としては、例えば II型糖尿病 (インスリン非依存性糖尿病)、肥満、高脂血 症又は動脈硬化症等が挙げられる。また、インスリン抵抗性の病態には、 II型糖尿病
、肥満又は高脂血症等の病気が複数同時に発症する状態、すなわちメタボリックシン ドロームや生活習慣病も包含される。
このため、本発明に係る PPAR γ作動剤は、 II型糖尿病 (インスリン非依存性糖尿 病)、肥満、高脂血症又は動脈硬化症の予防又は治療に使用し得る。該予防又は治 療には、メタボリックシンドロームや生活習慣病の予防、治療又は改善が含まれる。
[0029] 本発明にお ヽて II型糖尿病には、 II型糖尿病発病前症状の耐糖能異常が包含さ れる。 Π型糖尿病発病の予防又は治療には、血中グルコースレベルを低下させること が含まれる。また、 Π型糖尿病発病の予防又は治療には、耐糖能異常力も II型糖尿
病への進行の遅延又は防止が含まれ、さらにインスリン非依存性の π型糖尿病からィ ンスリン依存性の I型糖尿病への進行の遅延又は防止が含まれ、さらに、糖尿病に随 伴する糖尿病性合併症、例えば糖尿病性網膜症、糖尿病性腎症又は糖尿病性神 経障害の進行の遅延又は防止が含まれる。
[0030] 本発明において肥満には、内臓脂肪蓄積型肥満が包含される。肥満は身体に脂 肪が過剰に蓄積した状態をいい、インスリン抵抗性を呈し、 II型糖尿病や高血圧症を 誘発し得る。このため、肥満の予防又は治療には、 II型糖尿病や高血圧症の誘発抑 制又は防止が含まれる。
[0031] 本発明において高脂血症の予防又は治療には、血中の総コレステロール、 LDL ( 低比重リポ蛋白)コレステロール、中性脂肪 (TG)、遊離脂肪酸値もしくはレムナント様 リポ蛋白ーコレステロール (RLP—コレステロール)等が高値を示す状態、又は血中 HDLコレステロールが低値を示す状態の予防、治療又は改善等が含まれる。さらに 前記高脂血症の予防等は、高脂血症に起因する心臓や脳における動脈硬化を抑制 し又は防止することを含む。
[0032] 動脈硬化症は、動脈壁の内腔が狭窄や閉塞をきたし、血流の低下を伴って、例え ば一過性脳虚血発作、脳梗塞、心筋梗塞、狭心症等を伴い得る。動脈硬化には、ァ テローム硬化症を包含する。動脈硬化症の予防又は治療には、例えば脳梗塞のよう な脳動脈疾患や狭心症、心筋梗塞等の冠動脈疾患等を抑制し又は防止することを 含む。
[0033] 本発明に係る PPAR y作動剤は、ヒトのほか、ヒト以外の哺乳動物(例えば、サル、 ゥシ、ゥマ、ブタ、ヒッジ、ィヌ、ネコ、ラット、マウス、チンパンジー等)にも適用できる。 本発明に係る PPAR y作動剤には、ヒト PPAR y作動剤が包含され、 PPAR y作動 剤をヒトに適用する場合には、ヒト PPAR y作動剤が好ましい。
[0034] 本発明に係る PPAR γ作動剤は、一般式 (I)の化合物をそのままあるいは一般式( I)の化合物に薬理学的に許容される担体を配合して経口投与又は非経口投与に用 いられる剤形の製剤とすることができる。経口投与のための剤形としては、例えば錠 剤 (糖衣錠、フィルムコーティング錠を含む)、丸剤、顆粒剤、散剤、カプセル剤 (ソフ トカプセル剤、マイクロカプセル剤を含む)、シロップ剤、乳剤又は懸濁剤等が挙げら
れる。非経口投与のための剤形としては、例えば注射剤、注入剤、点滴剤又は坐剤 等が挙げられる。これら製剤は、例えば、酪酸の重合体、グリコール酸の重合体、酪 酸ーグリコール酸の共重合体、酪酸の重合体とグリコール酸の重合体との混合物又 はポリグリセロール脂肪酸エステル等の基剤と組み合わせて徐放性製剤とすることも できる。
[0035] 本発明に係る PPAR y作動剤における製剤中の一般式 (I)の化合物の含有量は、 製剤の形態に応じて適宜選択されることが好ましいが、通常、製剤全体に対して約 1 な!、し 99質量%が好まし 、。
[0036] 本発明に係る PPAR y作動剤の製造は、一般式 (I)の化合物を原料として、例え ば第 14改正日本薬局方、製剤総則に記載の方法又は当該分野で一般的に用いら れている公知の製造方法に従い、又は公知の製造方法に準じて行うことができる。ま た、 PPAR y作動剤を上記の剤形に製造する場合には、通常医薬品製剤に用いら れる添加物を適宜、適量を含有させて製造することが好ましい。前記添加物としては 、例えば、賦形剤(例えば乳糖、白糖、ブドウ糖、でんぷん、蔗糖、微結晶セルロース 、カンゾゥ末、マン-トール、炭酸水素ナトリウム、リン酸カルシウム、硫酸カルシウム 等)、結合剤(例えば、アラビアゴム、カルメロール又はその塩、ゼラチン、カルボキシ メチルセルロース、ヒドロキシプロピルセルロース、ポピドン等)、崩壊剤(例えば、カル メロース、でんぷん、炭酸カルシウム、結晶セルロース、低置換度ヒドロキシプロピル セルロース等)、滑沢剤(例えば、ケィ酸マグネシウム、ステアリン酸、ステアリン酸マ グネシゥム、ステアリン酸カルシウム、タルク、形質流動パラフィン、マクロゴール等)、 界面活性剤(例えば、ラウリル硫酸ナトリウム、ポリソルベート 80、ソルビタンモノ脂肪 酸エステル、ステアリン酸ポリオキシル 40、ポリオキシエチレン硬貨ヒマシ油等)、懸 濁化剤又は増粘剤(例えば、アラビアゴム、アルギン酸ナトリウム、カルボキシメチル セルロースナトリウム、メチノレセノレロース、ポピドン、ヒドロキシプロピルメチルセルロー ス、ベントナイト等)、乳化剤(例えば、アラビアゴム、コレステロール、ステアリン酸、ポ ビドン、ポリソルベート 80等)、溶剤 (例えば、生理食塩液、常水、精製水、滅菌精製 水、注射用水等の水溶性溶剤;ォリーブ油、ゴマ油等の植物油;イソプロパノール、 グリセリン、エタノール等の有機溶剤)、着色剤 (例えば、酸化チタン等)、保存剤 (例
えば、安息香酸、塩化ベンザルコ-ゥム、塩化べンゼトニゥム、クロロブタノール、パラ ベン類、ベンジルアルコール等)、芳香剤(例えば、サリチル酸メチル、ウイキヨゥ油、 チモール、メントール等)、矯味剤(例えば、果糖、単シロップ、ブドウ糖、 D—ソルビト ール、白糖等の甘味剤;塩酸、酒石酸等)、安定剤(例えば、ァスコルビン酸、チォ硫 酸ナトリウム、トコフエロール、亜硫酸水素ナトリウム等の抗酸化剤;ェデト酸ナトリウム 等のキレート剤等)、緩衝剤 (例えば、リン酸緩衝液、酢酸ナトリウム緩衝液等)、無痛 ィ匕剤(例えば、リドカイン、塩酸プロ力イン、ベンジルアルコール等)又は坐剤基剤(例 えば、カカオ脂、マクロゴール等)等が挙げられる。
[0037] 本発明に係る PPAR γ作動剤を例えば錠剤に製する場合には、例えば、一般式 (I )の化合物に上記賦形剤、結合剤、崩壊剤及び滑沢剤等を含有させて製造すること ができ、丸剤又は顆粒剤を製する場合には、例えば、一般式 (I)の化合物に上記賦 形剤、結合剤及び崩壊剤等を含有させて製造することができる。また、散剤及びカブ セル剤を製する場合には、例えば、一般式 (I)の化合物に上記賦形剤等を、シロップ 剤を製する場合には、例えば、一般式 (I)の化合物に矯味剤、芳香剤及び溶剤等を 、乳剤又は懸濁剤を製する場合には、例えば、一般式 (I)の化合物に懸濁化剤、界 面活性剤又は乳化剤及び溶剤等を、注射剤を製する場合には、例えば、一般式 (I) の化合物に溶剤及び無痛化剤等を、坐剤を製する場合には、例えば、一般式 (I)の 化合物に坐剤基剤等を含有させて製造することができる。また、錠剤又はカプセル剤 は、例えば、メタクリル酸コポリマー、ヒドロキシプロピルメチルセルロースアセテートサ クシネート又はヒドロキシプロピルメチルセルロースフタレート等の腸溶性コーティング 剤でコーティングしてもよい。また、カプセル剤は、腸溶性カプセルを用いてもよい。
[0038] 本発明に係る PPAR y作動剤は、安定かつ低毒性で安全に使用することができる 。その 1日の投与量は患者の状態や体重、化合物の種類、投与経路等によって異な る力 例えば、 II型糖尿病患者に経口投与する場合には、成人 (体重約 60kg) 1日当 りの投与量は有効成分 (一般式 (I)で表される化合物もしくはその塩又はそのプロドラ ッグ)として約 1 μ g〜500mg、好ましくは約 10 μ g〜50mgであり、これらを 1回又は 2な 、し 3回に分けて投与することが好ま 、。
[0039] 本発明に係る PPAR y作動剤は、本発明の作用を阻害しない範囲で、他の薬剤、
例えば、抗肥満剤、抗糖尿病薬、抗高脂血症剤、血圧降下剤等と組合せて用いるこ とちでさる。
[0040] 前記抗肥満剤としては、例えばレプチン、デキストロアンフェタミン、アンフェタミン、 フェンフルラミン、デキシフェンフルラミン、シブトラミン、マジンドール又はフエンテルミ ン等が挙げられる。
[0041] 抗糖尿病薬としては、インスリン、スルホニル尿素薬 (例えば、トルプタミド、ダリベン クラミド、ダリピジド等)、ビグアニド剤(例えば、メトホルミン等)、メグリチ-ド系薬 (例え ば、レパグリニド等)、 αダルコシダーゼ阻害剤(例えば、ミグリトーノレ、ァカノレボース 等)、ナテグリニド、トログリタゾン、ロシグリタゾン又はピオグリタゾン等が挙げられる。
[0042] 抗高脂血症剤としては、例えばコレスチラミン、コレスチポール、クロフイブラート、ジ ェムフイブ口ジル、フエノフイブレート、テサグリタザル、アトルバスタチン、フルパスタ チン、口パスタチン、プラバスタチン、シンパスタチン、セリバスタチン、ァシピモッタス 、プロブコール又はデキストロチロキシン等が挙げられる。
[0043] 血圧降下剤としては、例えばアルプレノロール、ァテノロール、チモロール、ピンドロ ール又はプロプラノロール等の 13ブロッカー;べナゼプリル、カプトプリル、ェナラブリ ル、フオシノプリル、リシノプリル、キナプリル又はラミプリル等のアンジォテンシン変換 酵素阻害剤; -フエジピン、フエロジピン、 -カルジピン、イスラジピン、 -モジピン、ジ ルチアゼム又はべラノミル等のカルシウムチャンネルブロッカー;及びドキサゾシン、 ゥラピジル、プラゾシン又はテラゾシン等の 1ブロッカー等が挙げられる。
[0044] 本発明はまた、インスリン抵抗性の病態の予防又は治療剤を製造するための一般 式 (I)で表される化合物もしくはその塩又はそのプロドラッグの使用を提供する。好ま しくは、 Π型糖尿病、肥満、高脂血症又は動脈硬化症の治療剤を製造するための一 般式 (I)で表される化合物もしくはその塩又はそのプロドラッグの使用である。一般式 (I)で表される化合物や、その好ましい形態としては、上述したのと同様である。一般 式 (I)で表される化合物もしくはその塩又はそのプロドラッグやその製造方法も、上述 したのと同様である。
[0045] 本発明はさらに、ヒトを含む哺乳動物に一般式 (I)で表される化合物もしくはその塩 又はそのプロドラッグを投与するインスリン抵抗性の病態を予防又は治療する方法で
もある。一般式 (I)で表される化合物や、その好ましい形態としては、上述したのと同 様である。一般式 (I)で表される化合物もしくはその塩又はそのプロドラッグやその製 造方法も、上述したのと同様である。
[0046] 本発明の予防または治療方法は、ヒトを含む哺乳動物、すなわち、ヒトのほか、ヒト 以外の哺乳動物、例えば、サル、ゥシ、ゥマ、ブタ、ヒッジ、ィヌ、ネコ、ラット、マウス、 チンパンジー等にも適用することができる。すなわちこれらの哺乳動物に一般式 (I) で表される化合物もしくはその塩又はそのプロドラッグを投与することにより、インスリ ン抵抗性の病態を予防又は治療する。本発明の予防または治療方法は、ヒトに適用 することが好ましぐインスリン抵抗性の病態を呈する患者に適用することが好ましい 。また、インスリン抵抗性の病態の中でも、 II型糖尿病、肥満、高脂血症又は動脈硬 化症の患者に適用することが好ましい。本発明の予防または治療方法をヒトに適用 する場合には、ヒト PPAR y作動剤を投与することが好ましい。
[0047] 一般式 (I)で表される化合物もしくはその塩又はそのプロドラッグをヒト等に投与す る方法としては特に限定されず、その製剤形態に応じた適当な投与経路により投与 することができる。また、本発明の作用を阻害しない範囲で、一般式 (I)で表される化 合物もしくはその塩又はそのプロドラッグと、他の薬剤、例えば、上述した抗肥満剤、 抗糖尿病薬、抗高脂血症剤、血圧降下剤等とを組合せて用いることもできる。
[0048] ¾細1
以下に実施例を用いて本発明を説明するが、本発明はこれらに限定されるもので はない。
実施例 1
[0049] 組み換え体核内受容体リガンド結合ドメイン (PPAR y LBD)におけるローダミンマ レイミドを用いたリガンドの転写活性の測定
(1)組み換え ppAR y LBDの大腸菌における発現と精製
Novagen社製の pET28bの NdelZBamHI部位にヒト PPAR y (配列番号 1)のし BD (aa. 195— 477)の N端及び C端部に制限酵素部位 (Ndel及び BamHI)の配列 を付加した DNAをクローユングし、シーケンスにより塩基配列を確認した。大腸菌 BL 21 (DE3)に开質転換し、 1Lの LB (Luria- Bertani)培地(1質量% Bacto Tripton 0.5
質量% Yeast Extract, 1質量% NaCl)で吸光度(OD600nm)が 0. 8に達するまで 37 °Cで培養を行った後に、 18°Cに冷却し、 ImM IPTG (イソプロピル— 1—チォ— β —D—ガラタトピラノシド)を加え 18時間培養を行った。大腸菌は遠心分離により回収 し、 20mM Hepes (pH7. 4)、 200mM NaCl、 25mMイミダゾール溶液に懸濁し、 — 20°Cに保存した。
[0050] 細胞を超音波破砕し、遠心分離により不溶性タンパク質を取り除いた後ニッケル力 ラムを用いたァフィユティークロマトグラフィー(溶出液: 20mM Tris (pH7. 4) , 250 mM NaCl, 250mMイミダゾール)により PPAR y LBDの精製を行った。さらに Sup erdex200ゲル濾過クロマトグラフィーにより精製と同時にァフィユティークロマトダラ フィ一の溶出液から 20mM Tris (pH7. 4)、 150mM NaClにバッファー交換を行つ た。得られた PPAR y LBDは限外濾過により PPAR y LBDの濃度が約 5mgZmLと なるまで濃縮し、 PPAR y LBD溶液とした。
[0051] (2)ローダミンマレイミドを用いたリガンドの転写活性測定
0. 1 M PPAR y LBD溶液と 10 μ Μのリガンド(ィ匕合物 12)のジメチルスルホキ シド (DMSO)溶液を混合し、室温で 30分インキュベートした後に、ドデシル硫酸ナト リウム(SDS)を 0. 5WZV%、ローダミンマレイミド(Molecular Probe社)を 200 M、 TCEP (トリス [2—カルボキシェチル]ホスフィン)を ImMになるように加え、室温 で 30分インキュベートした。 SDS-PAGE (ポリアクリルアミドゲル電気泳動法)を行 つた後に蛍光イメージヤー (日立製作所製 FMBIO II)によりローダミンマレイミドによ りラベルされた PPAR y LBDの量を測定した。
なお、対照として、リガンドの DMSO溶液のかわりに DMSOを用いた。また、陽性 対照として化合物 12のかわりに PPAR yと結合しな!ヽ BRL49653 (口シグリタゾン; J. Biol. Chem., 2005, Vol. 280 (14), 14145- 14153)及び MCC555 (J. Biol. Chem., 19 98, Vol. 273 (49), 32679-32684)、並びに PPAR γと共有結合する 15d— PGj及び
2
GW9662 (2—クロ口一 5—二トロ一 N—フエ二ノレ -ベンズアミド; PPAR yアンタゴ- スト; Biochemistry; 2002; 41(21); 6640- 6650)を用いた。
[0052] 結果を図 1に示す。図 1において、ローダミンマレイミドによりラベルされた PPAR γ LBDのシグナルが弱!、場合リガンドが共有結合して 、ることを示す。 BRL49653又
は MCC555と PPAR y LBD溶液をインキュベートした場合では、対照と同程度に P PAR γ LBDのシグナルが強く認められ、 PPAR y LBDが BRL49653又は MCC5 55と共有結合していないことを示した。一方、 15d— PGi又は GW9662と PPAR y
2
LBD溶液をインキュベートした場合では、対照と比較して PPAR y LBDのシグナル が非常に弱ぐ 15d-PGj又は GW9662が PPAR y LBDとリガンドが共有結合して
2
いることを示した。化合物 12と PPAR y LBD溶液をインキュベートした場合、 PPAR y LBDのシグナルは 15d—PGjのシグナルとほぼ同じ位であった。これは、化合物
2
12が 15d— PGiと同程度の PPAR γ LBDと共有結合することを示す。
2
実施例 2
[0053] 培養 COS— 7細胞におけるルシフェラーゼ活性
化合物 1〜13の転写活性を培養 COS— 7細胞における、ルシフェラーゼ活性を評 価した。すなわち、 COS— 7細胞は培養液 [10%FCS (子ゥシ胎児血清)を含むダ ルべッコ改変イーグル培地(DMEM) ]で培養した。トランスフエクシヨンの前日に培 養した COS— 7細胞を 24穴プレートに 5 X 104個 Zゥェルになるように播き、翌日 UA S -tk-Luc, pEYFP— Cl、 pCMX— gal— PPAR γのプラスミド(shiraki, T. et al. , J. Biol. Chem., 280: 14145-14153, 2005)をトランスフエクシヨンし、 6時間後培養液 を交換し、 DMSOに溶解したリガンド (化合物 1〜13)を最終濃度が 10 Mとなるよ う添加した。トランスフ クシヨンの 24時間後に細胞を回収し、発現したルシフェラー ゼの酵素活性を、ルシフェリンを基質とするキット(ピツカジーン;東洋インキ社製)を 用い、励起波長 485nm蛍光波長 530nmにおける YFP (黄色蛍光)の蛍光を測定す ることにより評価した。ルシフェラーゼ活性はフオトンカウンティングにより測定し、 YF Pの蛍光強度で割った値として表した。対照として、リガンドの DMSO溶液のかわりに DMSOを用い、陽性対照として 15d— PGjを用いた。
2
リガンド (化合物 1〜 13)のルシフェラーゼ活性を図 2に示す。いずれの化合物もル シフェラーゼ活性は対照よりも高ぐ転写活性を有することを示す。
産業上の利用可能性
[0054] 本発明の PPAR γ作動剤は、インスリン抵抗性の病態の予防又は治療剤として有 用である。
Claims
請求の範囲
一般式 (I)
[化 1]

(式中、 Α環は複素環を示し、 R1は水素原子又はエステルイ匕していてもよいカルボキ シアルキル基を示し、 R2は— COR5 (式中、 R5は置換基を有していてもよい C ァリー
6-14 ル基又は置換基を有していてもよい複素環基を示す。)又は— NOを示し、 R3は水
2
素原子、ハロゲン原子又はエステルイ匕していてもよいカルボキシル基を示し、 R4は水 素原子又は C アルキル基を示す。 )で表される化合物もしくはその塩又はそのプロ
1-6
ドラッグを含有することを特徴とするペルォキシゾーム増殖剤活性化受容体ガンマ (P PAR γ )作動剤。
[2] Α環がフラン環又はピラゾール環である請求の範囲第 1項に記載の剤。
[3] 化合物が、一般式 (Π)
[化 2]

(式中、 R1'はエステル化していてもよいカルボキシアルキル基を示し、 R3は水素原 子又はハロゲン原子を示し、 R5は置換基を有して 、てもよ 、C ァリール基又は置
6-14
換基を有して 、てもよ 、複素環基を示す。 )で表される化合物である請求の範囲第 1 項に記載の剤。
[4] R5で示されるァリール基がフエ-ル基である請求の範囲第 3項に記載の剤。
[5] R5で示される複素環基が 2—チェニル基である請求の範囲第 3項に記載の剤。
[6] 化合物が、
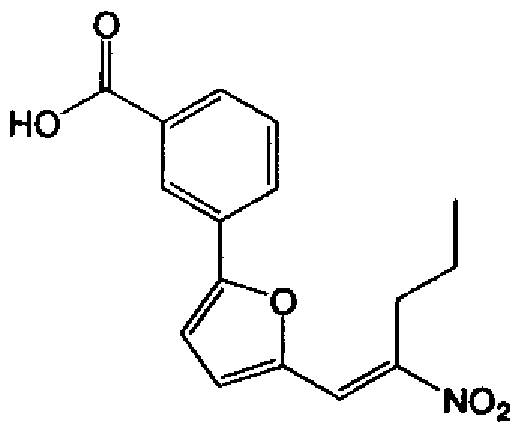
[化 3]

である請求の範囲第 3項に記載の剤。 化合物が、一般式 (III)
[化 4]

(式中、 R はエステルイ匕していてもよいカルボキシル基を示し、 Rは水素原子又は C アルキル基を示す。 )で表される化合物である請求の範囲第 1項に記載の剤。
1-6
[8] 化合物が、
[化 5]
[9] インスリン抵抗性の病態の予防又は治療剤である請求の範囲第 1〜8項のいずれ かに記載の剤。
[10] インスリン抵抗性の病態力 1型糖尿病、肥満、高脂血症又は動脈硬化症である請 求の範囲第 9項に記載の剤。
[11] インスリン抵抗性の病態の予防又は治療剤を製造するための一般式 (I)
[化 6]

[12] ヒトを含む哺乳動物に一般式 (I)
[化 7]

で表される化合物もしくはその塩を投与することを特徴とするインスリン抵抗性の病態 を予防又は治療する方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006117239 | 2006-04-20 | ||
| JP2006-117239 | 2006-04-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007122970A1 true WO2007122970A1 (ja) | 2007-11-01 |
Family
ID=38624878
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2007/056780 WO2007122970A1 (ja) | 2006-04-20 | 2007-03-29 | 核内受容体に結合するリガンド |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2007122970A1 (ja) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| DE102010015123A1 (de) | 2010-04-16 | 2011-10-20 | Sanofi-Aventis Deutschland Gmbh | Benzylamidische Diphenylazetidinone, diese Verbindungen enthaltende Arzneimittel und deren Verwendung |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013068486A1 (en) | 2011-11-08 | 2013-05-16 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the diagnosis and treatment of male infertility |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002050048A1 (en) * | 2000-12-20 | 2002-06-27 | Glaxo Group Limited | Thia- and oxazoles and their use as ppars activators |
| WO2003016291A1 (fr) * | 2001-08-10 | 2003-02-27 | Nippon Chemiphar Co., Ltd. | Activateur du recepteur $g(d) sensible au proliferateur de peroxysome |
| WO2004043951A1 (en) * | 2002-10-24 | 2004-05-27 | Carex S.A. | Pyrazole derivatives as modulators of peroxisome proliferator activated receptors |
-
2007
- 2007-03-29 WO PCT/JP2007/056780 patent/WO2007122970A1/ja active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002050048A1 (en) * | 2000-12-20 | 2002-06-27 | Glaxo Group Limited | Thia- and oxazoles and their use as ppars activators |
| WO2003016291A1 (fr) * | 2001-08-10 | 2003-02-27 | Nippon Chemiphar Co., Ltd. | Activateur du recepteur $g(d) sensible au proliferateur de peroxysome |
| WO2004043951A1 (en) * | 2002-10-24 | 2004-05-27 | Carex S.A. | Pyrazole derivatives as modulators of peroxisome proliferator activated receptors |
Non-Patent Citations (1)
| Title |
|---|
| HULIN B. ET AL.: "Novel thiazolidine-2,4-diones as potent euglycemic agents", J. MED. CHEM., vol. 35, no. 10, 1992, pages 1853 - 1864, XP002049116 * |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| DE102010015123A1 (de) | 2010-04-16 | 2011-10-20 | Sanofi-Aventis Deutschland Gmbh | Benzylamidische Diphenylazetidinone, diese Verbindungen enthaltende Arzneimittel und deren Verwendung |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013068486A1 (en) | 2011-11-08 | 2013-05-16 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the diagnosis and treatment of male infertility |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007122970A1 (ja) | 核内受容体に結合するリガンド | |
| US20070293538A1 (en) | Pharmaceutical Composition And Methods For Treating Neurodegenerative Disorders | |
| JP2010507672A (ja) | アルツハイマー病を治療するための方法および組み合わせ治療 | |
| US20070105959A1 (en) | Cynnamyl alcohol derivative compounds and drugs containing the compounds as active ingredient | |
| KR20010080144A (ko) | 카르복실산 유도체 및 이 유도체를 활성 성분으로서포함하는 약제 | |
| ITMI20130647A1 (it) | Composti con attività inibente sulle sirtuine | |
| EP1865778A2 (en) | Methods for avoiding edema in the treatment of metabolic, inflammatory, and cardiovascular disorders | |
| KR100635315B1 (ko) | 디히드로나프탈렌 유도체 화합물 및 이 화합물을 유효성분으로 하는 약제 | |
| WO2006132196A1 (ja) | β3作動薬を含有する新規な医薬 | |
| JPWO2002102780A1 (ja) | テトラヒドロキノリン誘導体化合物およびその化合物を有効成分として含有する薬剤 | |
| JP4813800B2 (ja) | 糖尿病を治療するためのキヌレニン3−ヒドロキシラーゼ阻害剤 | |
| JPH03500641A (ja) | 精神分裂症用薬剤 | |
| JP4661595B2 (ja) | フェニル酢酸誘導体、その製造方法および用途 | |
| CA2865845C (en) | An iloperidone metabolite for use in the treatment of psychiatric disorders | |
| US20100056460A1 (en) | Combination of organic compounds | |
| JP2004531454A (ja) | PPARδ(β)のレギュレーターならびに肥満およびインスリン抵抗性の治療におけるその使用 | |
| JP2005336173A (ja) | 骨粗鬆症治療剤 | |
| JPWO2008099907A1 (ja) | 尿排出障害治療剤 | |
| JP5559696B2 (ja) | 糖尿病性腎症の治療剤 | |
| JP2019506414A (ja) | 心血管疾患の治療のための新規クラスの化合物 | |
| ZA200403594B (en) | Carboxylic acid derivative compounds and drugs containing the same as the active ingredient. | |
| JP2021534141A (ja) | アルコール使用障害の治療剤 | |
| JP2009234946A (ja) | 併用医薬 | |
| JPH04145023A (ja) | ヒダントイン誘導体を有効成分とする循環器系疾患の予防及び治療剤 | |
| JP2005511654A (ja) | 炎症治療における4−オキソブタン酸誘導体の使用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07740218 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07740218 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |