WO2007018192A1 - 口腔内崩壊錠剤 - Google Patents
口腔内崩壊錠剤 Download PDFInfo
- Publication number
- WO2007018192A1 WO2007018192A1 PCT/JP2006/315620 JP2006315620W WO2007018192A1 WO 2007018192 A1 WO2007018192 A1 WO 2007018192A1 JP 2006315620 W JP2006315620 W JP 2006315620W WO 2007018192 A1 WO2007018192 A1 WO 2007018192A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- tablet
- tablet according
- lubricant
- weight
- carmellose
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
- A61K9/0056—Mouth soluble or dispersible forms; Suckable, eatable, chewable coherent forms; Forms rapidly disintegrating in the mouth; Lozenges; Lollipops; Bite capsules; Baked products; Baits or other oral forms for animals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
- A61K31/166—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide having the carbon of a carboxamide group directly attached to the aromatic ring, e.g. procainamide, procarbazine, metoclopramide, labetalol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/4152—1,2-Diazoles having oxo groups directly attached to the heterocyclic ring, e.g. antipyrine, phenylbutazone, sulfinpyrazone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4196—1,2,4-Triazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
- A61K31/542—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame ortho- or peri-condensed with heterocyclic ring systems
- A61K31/545—Compounds containing 5-thia-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. cephalosporins, cefaclor, or cephalexine
- A61K31/546—Compounds containing 5-thia-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. cephalosporins, cefaclor, or cephalexine containing further heterocyclic rings, e.g. cephalothin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/12—Carboxylic acids; Salts or anhydrides thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/38—Cellulose; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
Definitions
- a saccharide and Z or a disintegrant are generally used to ensure disintegration.
- Examples of documents disclosing oral disintegrating tablets containing saccharides and disintegrants include the following documents.
- Patent Document 1 describes an orally disintegrating tablet containing a pharmaceutical ingredient, erythritol, crystalline cellulose, and a disintegrant.
- Patent Document 2 describes an orally disintegrating tablet containing a pharmaceutical ingredient, D-mannitol, celluloses and a disintegrant. This document discloses that D-mannitol having an average particle diameter of 30 to 300 / ⁇ ⁇ is preferred.
- Patent Document 3 describes a method for producing an orally disintegrating tablet containing a granulated product obtained by spraying a saccharide having a low moldability to a saccharide having a low moldability as a binder.
- Patent Document 4 describes that an orally disintegrating tablet can be obtained by tableting a powder obtained by spray-drying a suspension in which an inorganic substance and a saccharide are uniformly dispersed together with crystalline cellulose and a disintegrant. Has been. On the other hand, it is described that tablets obtained by directly compressing a simple mixture having the same composition have poor hardness.
- Patent Document 5 a surface-modifying base such as light caustic anhydride is mixed with a medicinal component, surface-modified using a high-speed agitation granulator, etc., and the surface-modified powder thus obtained is obtained. It is described that an orally disintegrating tablet can be obtained by adding a disintegrant to the body and directly compressing it. The disintegrants are described as being partially alpha-ized starch and crospovidone are most suitable.
- Patent Document 6 a disintegrant is blended in granules containing a readily water-soluble drug, and after adding cellulose powder and Z or an inorganic additive, tablets are compressed to obtain tablets with good disintegration. It is stated that you can! RU
- Non-Patent Document 1 Korean Chemical Industry Co., Ltd. brochure (excipient anhydrous calcium hydrogen phosphate GS)) contains crystalline cellulose, anhydrous calcium hydrogen phosphate, carmellose, magnesium stearate 1 wt. 0/0 describes a data and formulation blended, Ru
- Patent Document 1 Japanese Patent Laid-Open No. 10-182436
- Patent Document 2 JP 2001-58944 A
- Patent Document 3 WO95Z20380
- Patent Document 4 JP 2000-86537
- Patent Document 5 WO00Z54752
- Patent Document 6 JP 2002-12540
- Non-Patent Document 1 Brochure of Kyowa Chemical Industry Co., Ltd. (Excipient for direct injection, anhydrous calcium phosphate phosphate GS)
- Patent Documents 1 and 2 are formulation examples in which a saccharide and a disintegrant, which are the most commonly used additives in the preparation of orally disintegrating tablets, are blended to increase the disintegration time.
- Non-Patent Document 1 As described above, the preparation of Non-Patent Document 1 is described for tablets containing anhydrous calcium hydrogen phosphate, crystalline cellulose, carmellose, and magnesium stearate, which is a lubricant, but magnesium stearate.
- the blending amount of 1% is 1%, and the disintegration property of the tablet may be lowered during heating and humidification.
- the present invention provides an orally disintegrating tablet that can be produced in existing equipment that has a broken feeling and good dosage, and does not require a special preparation machine.
- the blending amount of anhydrous calcium hydrogen phosphate as the inorganic excipient and metal stearate as the lubricant is not more than 0.8% by weight, preferably not more than 0.5% by weight, more preferably not more than 0.5% by weight per tablet.
- the present invention provides:
- a tablet characterized by containing an active ingredient, crystalline cellulose, an inorganic excipient, carmellose and a lubricant of 0.8% by weight or less per tablet,
- the tablet according to the above (14), comprising an active ingredient, crystalline cellulose, anhydrous calcium hydrogen phosphate, carmellose and 0.1 wt% or less magnesium stearate per tablet,
- the tablet of the present invention can be easily taken without water, rapidly disintegrates in the oral cavity, has an appropriate hardness, and has a good feeling. For this reason, it can be used as an orally disintegrating tablet. And the manufacturing method of the said tablet is simple.
- An "orally disintegrating tablet characterized by containing an active ingredient, crystalline cellulose, an inorganic excipient, carmellose and a lubricant of 0.8% by weight or less per tablet” means an active ingredient , Crystalline cellulose, inorganic excipient, carmellose and the lubricant of 0.8% by weight or less per tablet (the securing of good disintegration of the tablet and appropriate tablet hardness) Means a preparation that exhibits The active ingredient, crystalline cellulose, inorganic excipient, carmellose and 0.8% by weight or less of lubricant per tablet are essential constituents, and other additives are added so long as they do not affect the effect of the present invention. An agent may be included.
- crystalline cellulose used in the tablet of the present invention there are the following: THERAS PH101, CERAS PH102, CERAS PH301, CERAS PH302, Avicel PH-F20JP, CERAS K G802 (Asahi Kosei Kogyo Co., Ltd.), VIVAPUR (Grade 105, 101, 103, 301, 102, 112 ), ARBOCEL (grade M80, P290, A300), Prosolv SMCC50, Prosolv SM CC90 manufactured by QRS PHARMA) and the like. These crystalline celluloses may be used alone or in combination of two or more.
- the average particle diameter of crystalline cellulose before tablet production is preferably 10 to 150 m, more preferably 30 to 130 m, and particularly preferably 40 to 120 m. If it is larger or smaller than this average particle size, the hardness of the tablet may decrease or the disintegration time may be delayed.
- Ceraus PH102 Alignment PH102 (Asahi Kasei Kogyo Co., Ltd., average particle size of about 100 ⁇ m) is preferred.
- the inorganic excipient used in the tablet of the present invention is anhydrous calcium hydrogen phosphate, magnesium aluminate metasilicate, synthetic hydrotalcite, precipitated calcium carbonate, magnesium carbonate, and particularly preferably anhydrous phosphorus phosphate.
- Calcium hydrogen phosphate, anhydrous calcium hydrogen phosphate GS (manufactured by Kyowa Chemical Industry Co., Ltd.), Fujicalin (Fuji Chemical Industry Co., Ltd.), light anhydrous calcium hydrogen phosphate (manufactured by Kyowa Chemical Industry Co., Ltd.), hydrogen phosphate anhydrous Examples include calcium heavy (Kyowa Chemical Industry Co., Ltd.). These inorganic excipients may be used alone or in combination of two or more.
- the bulk density of the inorganic excipient before tablet production is 0.30 to: L Og / mL, more preferably 0.5 to 1. Og / mL, particularly preferably 0.6 to 1. Og / mL. If it is lower or higher than this bulk density, the hardness of the tablet may be reduced, and the disintegration time may be delayed.
- anhydrous calcium hydrogen phosphate GS manufactured by Kyowa Chemical Industry Co., Ltd., bulk density 0.71 ⁇ : L OgZmL is preferred.
- the content of crystalline cellulose and anhydrous calcium phosphate phosphate as an inorganic excipient can be easily determined. For example, it is easy to determine the suitability by mixing the desired amount of crystalline cellulose, anhydrous calcium hydrogen phosphate, carmellose with the active ingredient, and then compressing and confirming hardness and disintegration. I can do it.
- the contents of the crystalline cellulose and the inorganic excipient also depend on the physical properties of the active ingredient, it is preferable to determine appropriately as described above.
- the total tablet weight relative preferable to use from 30 to 99.9 by weight 0/0 of crystalline cellulose and inorganic excipients.
- the preparation of the present invention which is hardly affected by the physical properties of the active ingredient, exhibits a particularly good disintegration rate and tablet hardness.
- the blending ratio of crystalline cellulose and anhydrous calcium hydrogen phosphate is also preferably determined as described above.
- the weight specific force between crystalline cellulose and anhydrous calcium hydrogen phosphate may be in the range of 8: 2 to 2: 8. Within the above range, an intraoral rapidly disintegrating tablet having a good disintegration rate and tablet hardness can be obtained. If the blending ratio force S of crystalline cellulose is higher than this, the texture may be reduced due to the roughness of the crystalline cellulose, and if it is lower than this, the tablet hardness may be lowered.
- the amount of the active ingredient may be any amount, but it is 0.1 to 50% by weight, preferably 0.1 to 40% by weight, more preferably 0.1 to 30% by weight, based on the total weight of the tablet.
- the preparation of the present invention which is hardly affected by the physical properties of the active ingredient exhibits particularly good disintegration rate and tablet hardness.
- carmellose is preferable as the disintegrant used.
- Carmellose is also known as carboxymethylcellulose. Any carmellose may be used as long as it conforms to the 14th revision of the Japanese Pharmacopoeia. Specifically, NS-300 (Gotoku Pharmaceutical Co., Ltd.).
- the lubricant used in the tablet of the present invention includes sucrose fatty acid ester, talc, hydrous silicon dioxide, metal stearate, etc., preferably metal stearate.
- metal stearate examples include magnesium stearate and calcium stearate. Preferably, it is magnesium stearate.
- the content of the metal stearate is 0.8% by weight or less per tablet, preferably 0.5 wt% or less, more preferably 0.1 wt% or less. Specifically, 0. 001-0. 8 weight 0/0, preferably ⁇ or 0.001 to 0.5 wt 0/0, more preferably ⁇ or 0.001 to 0.1 wt 0/0. If it is more than this content, the disintegration time of the tablet may be prolonged.
- any active ingredient can be used.
- Any active ingredient that can be administered orally is not particularly limited.
- the tablet of the present invention may further contain various additive agents generally used in tablet production, if necessary.
- various additive agents generally used in tablet production, if necessary.
- 0.1 to 30% by weight (preferably 0.1 to: L0% by weight, particularly preferably 0.1 to 5.0% by weight) of the additive may be included with respect to the total weight of the tablet. .
- These substances may be used alone or in admixture at any ratio.
- the additive include sweeteners, flavoring agents, fragrances, lubricants, binders, fluidizing agents, coloring agents, and coating agents.
- the sweetener means sugars including sugars and sugar alcohols and other non-sugars. Since the preparation of the present invention does not contain saccharide and sugar alcohol as excipients, it is difficult to obtain sufficient sweetness using saccharide and sugar alcohol. For this reason, non-saccharide natural sweeteners and synthetic sweeteners are preferred for the tablets of the present invention, particularly those that disintegrate in the oral cavity, in which products that feel strong sweetness in a small amount are preferred compared to sugars and sugar alcohols. Specifically, when the sweetness of sucrose is 1, the sweetener has a sweetness of 50 times or more.
- Examples thereof include acesulfame potassium, aspartame, saccharin or a salt thereof, glycyrrhizic acid or a salt thereof, stevia or a salt thereof, sucralose, and thaumatin.
- the content of the sweetening agent is 10% by weight or less per tablet, preferably 0.1 to 10% by weight, more preferably 0.5 to 7.5% by weight.
- corrigent examples include ascorbic acid and its salt, glycine, sodium chloride, magnesium chloride, hydrochloric acid, dilute hydrochloric acid, succinic acid and its salt, succinic anhydride, L-glutamic acid and its salt, succinic acid and its salt , Acetic acid, tartaric acid and salts thereof, sodium hydrogencarbonate, fumaric acid and salts thereof, malic acid and salts thereof, glacial acetic acid, nitric acid inosinate, and honey.
- the fragrance includes what is called a flavoring agent, for example, orange essence, orange oil, carame nore, camphor, keihi oil, spearmint oil, strawberry essence, chocolate essence, cherry flavor, spruce oil, pine oil, vinegar oil. , Vanilla flavor, bitter essence, funole frano ichi, peno mint essence, mix flavono mint, mint flavor, menthol, lemon powder, lemon oil, rose oil and the like.
- a flavoring agent for example, orange essence, orange oil, carame nore, camphor, keihi oil, spearmint oil, strawberry essence, chocolate essence, cherry flavor, spruce oil, pine oil, vinegar oil.
- Vanilla flavor bitter essence, funole frano ichi, peno mint essence, mix flavono mint, mint flavor, menthol, lemon powder, lemon oil, rose oil and the like.
- binders include gum arabic, gum arabic powder, partially alpha-ized starch, gelatin, agar, dextrin, pullulan, povidone, polyvinyl alcohol, ethenoresenorelose, canoleboxymethylenoreethinorerose, canolemellose, strength Examples include noremerose sodium, hydroxyethinoresenorelose, hydroxyethylenomethenorescenellose, hydroxypropylcellulose, and hydroxypropylmethylcellulose.
- Examples of the fluidizing agent include hydrated sodium carbonate, light anhydrous caustic acid, heavy anhydrous caustic acid, and acidic titanium.
- the colorant examples include edible pigments such as edible red No. 3, edible yellow No. 5, edible blue No. 1, etc., yellow ferric oxide, ferric trioxide, brown iron oxide, black iron oxide, copper chlorophyll, copper chlorophyll Ilillin sodium, riboflavin, powdered green tea, etc.
- edible pigments such as edible red No. 3, edible yellow No. 5, edible blue No. 1, etc., yellow ferric oxide, ferric trioxide, brown iron oxide, black iron oxide, copper chlorophyll, copper chlorophyll Ilillin sodium, riboflavin, powdered green tea, etc.
- the coating agent examples include polybutyl alcohol, ethyl cellulose, carboxymethylenoresenorerose, canolemellose, canolemellose sodium, hydroxyetinoresenoreose, hydroxyetinoremethinoresenorelose, hydroxypropinolol Senorelose, hydroxypropyl methylcellulose, PVA copolymer, ethyl acrylate 'methyl methacrylate copolymer dispersion, aminoalkyl methacrylate copolymer, opdry, carnaparou, carboxyvinyl polymer, dry methacrylic acid copolymer, dimethylaminoethyl methacrylate T-methinoremethacrylate copolymer, stearyl alcohol, shellac, cetanol, hydroxypropyl methylcellulose acetate succinate, hydroxy Ropirumechiru Cellulose phthalate, fumaric acid 'stearic acid' polybulacetal jetyla
- crystalline cellulose z anhydrous calcium hydrogen phosphate Z carmellose Z magnesium stearate Z acesulfame potassium crystalline cellulose Z anhydrous calcium hydrogen phosphate Z carmellose Z magnesium stearate / acesulfame potassium / hatsu power oil / hydrous nitric acid ZC
- crystalline cellulose z anhydrous calcium hydrogen phosphate Z carmellose Z magnesium stearate z sucralose crystalline cellulose Z anhydrous calcium hydrogen phosphate Z carmellose Z magnesium stearate Z sucralose / hatsu power oil / hydrous nitric acid
- the orally disintegrating tablet of the present invention is an orally disintegrating tablet consisting essentially of an active ingredient, a crystal cell mouth and an inorganic excipient. That is, the orally disintegrating tablet is characterized in that it does not contain sugar as a commonly used excipient.
- saccharides such as sucrose, glucose, fructose, starch syrup and lactose are not used as excipients.
- the orally disintegrating tablet of the present invention is characterized by not containing a sugar alcohol as an excipient generally used in the orally disintegrating tablet.
- sugar alcohols such as erythritol, D-sorbitol, xylitol, D-mannitol and maltitol should not be used as excipients.
- the active ingredient and the raw material for the preparation are weighed, and a V-type mixer is used.
- a V-type mixer examples thereof include a method in which the mixed powder for tablets mixed with a proper mixer is directly compressed and compressed using a tableting machine described later.
- a binder When producing a mixed powder for tablets, a binder, a flavoring agent, a fluidizing agent, a lubricant, a fragrance, a sweetener, a coloring agent, and the like may be mixed as necessary.
- a small amount of lubricant (0.8% by weight or less per tablet, preferably 0.5% by weight or less, more preferably 0.1% by weight) is used in the formulation of the present invention. %) Or less), the usual tableting method (internal mixing method) or the external lubrication method in which a lubricant is attached to the mortar of a tableting machine can be used.
- EL SP1—Type III manufactured by Kikusui Seisakusho Co., Ltd. as an apparatus that performs the external lubrication method.
- the amount of lubricant added is decreased, the disintegration rate can be further increased, the tablet hardness can be improved, and the stability of the drug can be enhanced.
- the usual method and formulation for mixing lubricants in tablet powders require 1 to 3 mg of lubricant per lOOmg tablet, but in the present formulation, 0.8% by weight per tablet.
- tableting with a small amount of lubricant preferably 0.5% by weight or less, more preferably 0.1% by weight or less, is possible.
- the active ingredient, crystalline cellulose, anhydrous calcium hydrogen phosphate, carmellose and 0.1% by weight or less of magnesium stearate per tablet are blended, and the method of adding the lubricant is the external lubricant method. It is still preferable.
- the active ingredient crystalline cellulose, anhydrous calcium phosphate, carmellose (when adding a sweetener, acesulfame potassium or sucralose.
- the particle diameters of the active ingredient and the additive are not particularly limited.
- the mixed powder for tablets thus obtained is compression-molded at a tableting pressure of 200 kg to 1500 kg using, for example, an external sliding tableting device, a single tableting machine, a rotary tableting machine or the like. . If the pressure is lower than this, the tablet hardness is insufficient and sufficient hardness for handling cannot be secured, and the pressure is high V, which is preferable because disintegration is delayed.
- any shape can be adopted for molding the intraoral rapidly disintegrating tablet of the present invention, for example, round shape, elliptical shape, spherical shape, rod shape, donut shape, laminated tablet, dry-coated tablet, etc. Moreover, it can be coated by coating. In addition, marks for improving discrimination, characters, etc., and dividing lines for division may be attached.
- the tablet of the present invention is useful as an orally disintegrating tablet, which can be rapidly disintegrated in the oral cavity by saliva and can be taken smoothly without leaving any roughness.
- the dissolution of the orally disintegrating tablet of the present invention is usually 1 to 60 seconds, preferably 1 to 40 seconds, and more preferably about 1 to 30 seconds.
- the strength (measured with a tablet hardness meter) is generally known to be a value with no problem if it is about 30 to 70N.
- the orally disintegrating tablet of the present invention is 10 to 200N, preferably 30 to 150N. Degree.
- This preparation should be taken without being disintegrated in the oral cavity or taken with water.
- Tablets obtained in Examples and Comparative Examples were subjected to sensory tests by the following test methods, tablet hardness and disintegration time.
- Ethenzamid, disintegrant, crystalline cellulose, anhydrous calcium hydrogen phosphate, and sweetener were weighed according to the blending amounts shown in Table 1 and mixed in a plastic bag to prepare a tablet powder.
- a small experimental tablet machine VELA5 manufactured by Kikusui Seisakusho
- an external lubrication device ELSP1-Type III manufactured by Kikusui Seisakusho
- lmg of magnesium stearate per tablet In 1 tablet, lOOmg was prepared. At this time, the shape of the ridge was round and the diameter was 6.5 mm.
- Example 1 and Comparative Examples 1 to 5 tablet powders with different types of disintegrants were tableted, and the hardness, disintegration time, oral disintegration time and oral disintegration time of the tablets were changed. The feeling of quickness was evaluated.
- Ceraus PH102 manufactured by Asahi Kasei Corporation
- anhydrous calcium hydrogen phosphate GS manufactured by Kyowa Chemical Co., Ltd.
- acesulfame potassium manufactured by Niyu Trinova Japan
- the tableting pressure was 5-7 kN.
- Table 2 shows the experimental results. From these results, it was found that when carmellose was used, there was no crispness in the oral cavity where the tablet disintegration time was high and the oral disintegration time was fast.
- Example 1 the powder component of a tablet having a modified average particle diameter of crystalline cellulose was tableted, and the hardness, disintegration time, and oral disintegration time of the tablet were measured.
- NS-300 (made by Gotoku Pharmaceutical Co., Ltd.) as carmellose
- anhydrous calcium hydrogen phosphate GS made by Kyowa Chemical Co., Ltd.
- anhydrous hydrogen phosphate as sweetener Acesulfame potassium (Niyu Trinova Japan) was used.
- the tablet production method is the same as in Example 1.
- Example 1 CeraS ⁇ ⁇ — 1 0 2 (Asahi Kasei Corporation)
- Example 1 the tablet powder components with the average particle diameter of anhydrous calcium hydrogen phosphate changed were compressed, and the hardness, disintegration time, and oral disintegration time of the tablets were measured. It was measured.
- NS-300 (manufactured by Gotoku Pharmaceutical Co., Ltd.) was used as carmellose
- Cerath 102-102 manufactured by Asahi Kasei Co., Ltd.
- acesulfame potassium manufactured by Niyu Trinova Japan
- the tablet production method is the same as in Example 1.
- Table 4 shows the experimental results. From these results, the target values of hardness, disintegration time, and oral disintegration time were satisfied no matter which bulk density of anhydrous calcium hydrogen phosphate was used.
- anhydrous calcium hydrogen phosphate GS having a bulk density of 0.85 gZmL has high tablet hardness and fast disintegration time and oral disintegration time.
- Example 1 Anhydrous acid calcium GS (manufactured by Kyowa Chemical Co., Ltd.)
- Example 4 Anhydrous calcium hydrogen phosphate light (Kyowa Chemical Co., Ltd.)
- Example 6 to 9 and Example 30 as shown in Table 5, the powder components of tablets with different types of sweeteners were tableted, and the hardness, disintegration time, and oral disintegration time of the tablets were measured.
- NS-300 (manufactured by Gotoku Pharmaceutical Co., Ltd.) was used as carmellose, Ceraus PH102 (manufactured by Asahi Kasei Co., Ltd.) as crystalline cellulose, and anhydrous calcium hydrogen phosphate GS (manufactured by Kyowa Chemical Co., Ltd.) as anhydrous calcium hydrogen phosphate.
- the tablet production method is the same as in Example 1.
- Table 5 shows the experimental results. From these results, among these sweeteners, the disintegration time of the tablet containing dipotassium glycyrrhizinate of Example 30 and the disintegration time in the oral cavity were long and the disintegration property was poor. Tablets containing other sweeteners, acesulfame potassium, aspartame, stevia, sucralose, and thaumatin, had a high disintegration time and rapid oral disintegration time.
- Example 1 Example 6
- Example 7 Example «8 * Example 9
- Example 3 0 Sweetener Acesulfas palpartoso-machinsteia Sclaro-Glycyl-lym lym strength Li-mus stannic acid bismuth Hardness (N) 5 2.0 4 8. 2 5 1. 4 4 8. 8 4 7 .9 4 9 .5 Disintegration time (seconds) 0 9 9 1 0 1 0 3 0 i: l Disintegration time (seconds) 6 1 0 1 0 1 0 1 0 1 0 3 5
- Example 30 Dipotassium glycyrrhizinate (manufactured by Maruzumi Pharmaceutical Co., Ltd.) (Examination of tablet production method)
- the formulations for Examples 1 and 10 are shown in Table 6.
- An external lubrication method in which magnesium stearate is attached to the die the external lubricant method in which magnesium stearate is present only on the tablet surface, and the magnesium stearate and powder are mixed, and the internal mixing method in which magnesium stearate is present throughout the tablet Two ways were done. The hardness, disintegration time and oral disintegration time of the tablets prepared by these two methods were measured.
- Example 10 As a manufacturing method of the tablet of Example 10 in which magnesium stearate was blended inside the tablet (mixed internally), ethenzamide, carmellose, crystalline cellulose, anhydrous hydrogen phosphate lucum, acesulfame potassium, and magnesium stearate are listed in Table 6. Weighed according to the blending amount, mixed in a plastic bag, added magnesium stearate and mixed to prepare a powder for tablets. Next, a tablet of lOOmg per tablet was prepared with a small experimental tablet machine VELA5 (manufactured by Kikusui Seisakusho). At this time, the shape of the punch was round, the diameter was 6.5 mm, and tableting was performed at a tableting pressure of 6 kN.
- a small experimental tablet machine VELA5 manufactured by Kikusui Seisakusho
- Example 1 Example 1 0
- Table 7 shows the experimental results. From these results, both methods of adding magnesium stearate satisfied the target values for hardness, disintegration time, and oral disintegration time. In particular, the disintegration time and the disintegration time in the oral cavity of the tablets manufactured by the external lubrication method were fast.
- Table 8 shows the formulations of Examples 11 to 14 and Comparative Example 6.
- the tablet production methods of Examples 11 to 14 and Comparative Example 7 were the same as in Example 10, and tableting was performed so that the hardness was about 60N.
- Example 1 Male Example 1 2
- Example 1 3 3 ⁇ 4
- Example 1 4 Comparative Example 6
- Anhydrous 1 Calcium hydrogen phosphate 47. 6 47. 6 4 7.
- Acesulfame Carryum 1.
- 4 1. 4
- Table 9 shows the experimental results. From these results, the disintegration time and the oral disintegration time tended to be delayed as the amount of magnesium stearate added increased to 0.1 mg, 0.3 mg, 0.5 mg, 0.8 mg and 1 mg.
- Table 8 The components shown in Table 8 were tableted at a tableting pressure of 7 kN, and the tablets were left in an oven at 40 ° C. and a relative humidity of 75% for 9 days, and then tablet hardness and tablet disintegration time were measured.
- Table 10 shows the experimental results. From this result, when the addition power of magnesium stearate, a lubricant, is 0.1 mg, 0.3 mg, 0.5 mg, and 0.8 mg, the tablet hardness is 30 N or more and the disintegration time is 30 seconds or less. The target value has been achieved. On the other hand, in the case of 1. Omg, the tablet hardness was less than 30N, and the disintegration time was 30 seconds or more.
- Example 1 1 3 ⁇ 4 Example: 1 2
- Example 1 3 3 ⁇ 4 Example 14 Comparative Example 6
- Magnesium stearate (/ ⁇ ⁇ ga 0. 1 0, 3 0. 5 0. 8 1. 0)
- Examples 15- The formulation of 18 is shown in Table 11. The hardness, disintegration time, and oral disintegration time of tablets with different lubricant types were measured. In addition, the manufacturing method of the tablet of Examples 15-18 was the external lubrication method similarly to Example 1, and was manufactured with the tableting pressure of 6 kN.
- Table 12 shows the experimental results. From these results, even when the type of lubricant was changed, the disintegration time when the hardness was high and the disintegration time in the oral cavity were fast.
- Table 14 shows the experimental results. From these results, the tablets to which 1 mg, 5 mg, 10 mg and 3 mg of carmellose were added showed no problems in hardness, disintegration time and disintegration time in the oral cavity, and there was no crisp feeling.
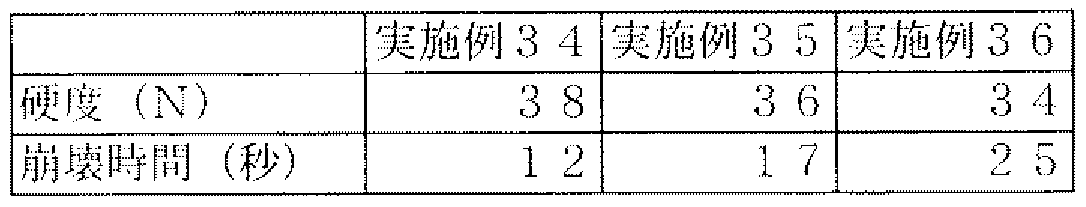
- Example 15 shows the formulations of Examples 1 and 22-25.
- the production methods of Examples 1 and 22 to 25 were the external lubrication method as in Example 1 and were produced at a tableting pressure of 6 kN.
- Example 15 Ingredients Example 1 Example 2 2 3 ⁇ 4 Example 2 3 Example 24 Example 2 5 Ethensamide 1 0 2 0 3 0 40 50 Carmellose] 0 1 0 1 0.1 .1 0 1 0 Crystalline cellulose 3 1 2 7 2 4 20 1 6 Anhydrous hydrogen phosphate 47. 6 1. 6 34. 6 28. 6 22. 6
- Table 16 shows the experimental results. From these results, even when the blending amount of ethenzamide, an active ingredient, was changed, the disintegration time in which the hardness of any tablet was high was rapid.
- Table 17 shows the formulation of this powder granule.
- Anhydrous calcium hydrogen phosphate and carmellose calcium were added to a type 2 high-speed mixer (Fukae Pututech), and a predetermined amount of isopropyl antipyrine, a drug, was suspended in an aqueous solution of 20 wZ w% hydroxypropylmethylcellulose 2910. The solution was stirred and granulated with stirring. The granulated product was dried with FL-MINI fluidized bed granulator (Freund Industries). The mixing ratio of the drug and the bitterness inhibiting base was set to 1: 3 by weight ratio (solid content).
- the drug is isopropylantipyrine (manufactured by Kongo Chemical Co., Ltd.), the bitterness-inhibiting base hydroxypropylmethylcellulose 2910 is TC 5EW (manufactured by Shin-Etsu Chemical Co., Ltd.), and the excipient is anhydrous calcium hydrogen phosphate ( Kyowa Chemical Co., Ltd.) was used.
- As the disintegrant carmellose calcium (manufactured by Gotoku Pharmaceutical Co., Ltd.) was used.
- the viscosity of a 20 wZw% hydroxypropyl methylcellulose 2910 aqueous solution at 20 ° C. is 50 to 14000 mPa's.
- tablet disintegration should be re-examined as tablet hardness may decrease, disintegration time and oral disintegration time may be delayed. It was.
- Example 26 Comparative Examples 8 to 12; tableting powders with the formulation shown in Table 17 as the basic formulation and the disintegrant type changed are compressed, and the tablet hardness, disintegration time, oral disintegration time, And the freshness in the oral cavity was evaluated.
- Table 18 shows the experimental results. From these results, when carmellose was used as in Table 2, the tablet had a high disintegration time and a rapid disintegration time in the oral cavity, and there was no quick feeling in the oral cavity. Other disintegrants had a longer disintegration time in the oral cavity and a quick feeling of power.
- Table 19 shows the formulation of the tablet of Example 27. Except for the addition of isopropylantipyrine, the same force radius as Example 26 was 7. Omm.
- Table 20 shows the experimental results. From this result, even when the amount of the active ingredient was increased, like the lmg tablet of Example 26, the hardness of the tablet increased, and the disintegration time and oral disintegration time increased rapidly.
- Table 21 shows the formulation of the tablets of Examples 28 and 29. The same as in Examples 26 and 27, except that a perfume herb oil and water-containing diacid oxide were blended.
- Table 22 shows the experimental results. From these results, even when a fragrance was added, the tablet hardness increased rapidly and the oral disintegration time increased rapidly.
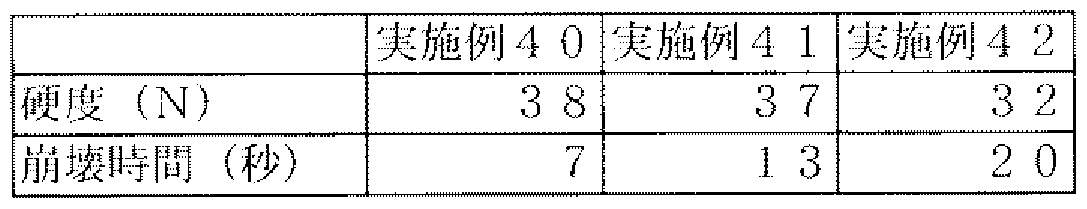
- Example 31 The tablet formulations of Examples 31, 32 and 33 are shown in Table 23. The same as in Examples 28 and 29, except that the amount of added calories of the drug and additive was changed.
- Example 3 1 3 ⁇ 4
- Example 32 Example 3 3 Milliform Isobropyr Antipyrine 0. ⁇ 3 3 m Hydroxy Lip Methyl Pill Methyl Cell]
- Table 25 shows the experimental results. From these results, as in Examples 28 and 29, all tablets had a hardness of 30 mm or more and the disintegration time was within 30 seconds.
- Tablet formulations are shown in Tables 26, 27 and 28.
- drugs acetaminophen, cefcapene pivoxil hydrochloride and rilmazafone hydrochloride were used.
- drugs, carmellose, crystalline cellulose, anhydrous calcium hydrogen phosphate, acesulfame potassium, and magnesium stearate are weighed according to the blending amounts shown in Tables 26, 27, and 28. And mixed with a plastic bag to prepare a powder for tablets.
- Tables 29, 30, and 31 show the experimental results for each drug. From this result, as the drug amount increased, the hardness of the tablet decreased and the disintegration time tended to be longer, but all tablets had a tablet hardness of 30 N or more and the disintegration time was within 30 seconds. there were.
- the tablet of the present invention has an easy production process and has strengths during production and storage, and has excellent strength and storage for a long period of time. In addition, since it disintegrates rapidly in the oral cavity, it is easy to take for the elderly and children, and as a safe formulation for the general public as well as various conventional oral preparations containing the same drug. It can be used for the treatment and prevention of other diseases.
Abstract
Description




















Claims
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11/990,109 US20080274178A1 (en) | 2005-08-10 | 2006-08-08 | Orally Disintegrating Tablet |
| CA2618977A CA2618977C (en) | 2005-08-10 | 2006-08-08 | Orally disintegratable tablet |
| CN2006800365470A CN101277721B (zh) | 2005-08-10 | 2006-08-08 | 口腔崩解片 |
| KR1020087003203A KR101465803B1 (ko) | 2005-08-10 | 2006-08-08 | 구강내 붕괴 정제 |
| JP2007529583A JP5366233B2 (ja) | 2005-08-10 | 2006-08-08 | 口腔内崩壊錠剤 |
| EP06782462A EP1923074A4 (en) | 2005-08-10 | 2006-08-08 | TABLET WHICH CAN BE ORALLY DISINTEGRATED |
| KR1020127033700A KR20130016386A (ko) | 2005-08-10 | 2006-08-08 | 구강내 붕괴 정제 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005-232083 | 2005-08-10 | ||
| JP2005232083 | 2005-08-10 | ||
| JP2005-367963 | 2005-12-21 | ||
| JP2005367963 | 2005-12-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007018192A1 true WO2007018192A1 (ja) | 2007-02-15 |
Family
ID=37727375
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2006/315620 WO2007018192A1 (ja) | 2005-08-10 | 2006-08-08 | 口腔内崩壊錠剤 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20080274178A1 (ja) |
| EP (1) | EP1923074A4 (ja) |
| JP (2) | JP5366233B2 (ja) |
| KR (2) | KR20130016386A (ja) |
| CA (1) | CA2618977C (ja) |
| TW (1) | TWI376243B (ja) |
| WO (1) | WO2007018192A1 (ja) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008285434A (ja) * | 2007-05-16 | 2008-11-27 | Taisho Pharm Ind Ltd | 口腔内速崩壊錠 |
| WO2009066773A1 (ja) | 2007-11-21 | 2009-05-28 | Dainippon Sumitomo Pharma Co., Ltd. | 口腔内崩壊錠 |
| JP2009256349A (ja) * | 2008-03-28 | 2009-11-05 | Kowa Co | 錠剤 |
| WO2010134540A1 (ja) | 2009-05-20 | 2010-11-25 | 大日本住友製薬株式会社 | 有核型の口腔内崩壊錠 |
| WO2010137716A1 (ja) * | 2009-05-29 | 2010-12-02 | マイラン製薬株式会社 | 沈降炭酸カルシウムを有効成分とする口腔内崩壊錠 |
| WO2013115171A1 (ja) * | 2012-02-03 | 2013-08-08 | 旭化成ケミカルズ株式会社 | 苦味マスク顆粒含有口腔内崩壊錠 |
| WO2015005241A1 (ja) * | 2013-07-06 | 2015-01-15 | 株式会社ダイセル | 超速崩壊錠剤及びその製造方法 |
| WO2016103904A1 (ja) * | 2014-12-25 | 2016-06-30 | 株式会社ダイセル | 超速崩壊錠剤及びその製造方法 |
| US9974738B2 (en) | 2013-09-27 | 2018-05-22 | Daicel Corporation | Disintegrating particle composition produced by two-stage wet granulation process, and intraorally disintegrating tablet containing same composition |
| US10398694B2 (en) | 2014-11-11 | 2019-09-03 | Shionogi & Co., Ltd. | Multi-layered tablet containing drug unstable to light |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI455733B (zh) * | 2009-03-30 | 2014-10-11 | Toray Industries | 口腔內崩壞性被覆錠劑 |
| KR101612073B1 (ko) * | 2009-10-30 | 2016-04-14 | 아이엑스 바이오파마 리미티드 | 신속 용해 고체 투약 제형 |
| US9433620B2 (en) | 2011-05-13 | 2016-09-06 | Cadila Healthcare Limited | Pharmaceutical compositions of lurasidone |
| US20150174075A2 (en) * | 2012-03-29 | 2015-06-25 | Daicel Corporation | Method for producing disintegrating particulate composition comprising acid-type carboxymethylcellulose, disintegrating particulate composition comprising acid-type carboxymethylcellulose, and orally disintegrating tablet including disintegrating particulate composition comprising acid-type carboxymethylcellulose |
| CN104010663B (zh) | 2012-09-20 | 2021-02-19 | 株式会社大赛璐 | 含酸式羧甲基纤维素和结晶纤维素的崩解性颗粒组合物及含该组合物的口腔内崩解片剂 |
| CN105555262A (zh) * | 2013-09-27 | 2016-05-04 | 株式会社大赛璐 | 含有崩解性颗粒组合物的口腔内崩解片剂 |
| GB201402070D0 (en) * | 2014-02-07 | 2014-03-26 | Galapagos Nv | Pharmaceutical compositions for the treatment of inflammatory disorders |
| JP6822619B1 (ja) * | 2019-04-03 | 2021-01-27 | アステラス製薬株式会社 | 医薬組成物 |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1203580A1 (en) | 1999-06-18 | 2002-05-08 | Takeda Chemical Industries, Ltd. | Quickly disintegrating solid preparations |
| JP2002179558A (ja) * | 2000-10-06 | 2002-06-26 | Takeda Chem Ind Ltd | 固形製剤 |
| JP2002188095A (ja) * | 2000-12-21 | 2002-07-05 | Fancl Corp | 植物性油脂粉末及び該粉末を含む食品組成物 |
| JP2003034655A (ja) * | 2001-05-15 | 2003-02-07 | Takeda Chem Ind Ltd | 速崩壊性固形製剤 |
| EP1329217A1 (en) | 2000-10-06 | 2003-07-23 | Takeda Chemical Industries, Ltd. | Solid preparations |
| EP1369109A1 (en) | 2001-03-06 | 2003-12-10 | Kyowa Hakko Kogyo Co., Ltd. | Preparations quickly disintegrating in oral cavity |
| US20040028741A1 (en) | 2000-09-22 | 2004-02-12 | Kazuyuki Fujihara | Oral preparations with favorable disintegration characteristics |
| JP2004123648A (ja) * | 2002-10-04 | 2004-04-22 | Taiyo Kagaku Co Ltd | 錠剤、打錠用改質剤 |
| JP2004155682A (ja) * | 2002-11-05 | 2004-06-03 | Dai Ichi Kogyo Seiyaku Co Ltd | 錠剤用滑沢剤組成物 |
| EP1523974A1 (en) | 2003-10-15 | 2005-04-20 | Fuji Chemical Industry Co., Ltd. | Composition for rapid disintegrating tablet in oral cavity |
| WO2005055955A2 (en) | 2003-12-09 | 2005-06-23 | Pharmasset, Inc. | DOSING METHODS FOR ß-D-2’,3’-DIDEOXY-2’,3’-DIDEHYDRO-5-FLUOROCYTIDINE ANTIVIRAL THERAPY |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4675188A (en) * | 1985-08-02 | 1987-06-23 | Stauffer Chemical Company | Granular anhydrous dicalcium phosphate compositions suitable for direct compression tableting |
| US5605889A (en) * | 1994-04-29 | 1997-02-25 | Pfizer Inc. | Method of administering azithromycin |
| ES2079327B1 (es) | 1994-12-13 | 1996-08-01 | Lilly Sa | Formulaciones farmaceuticas de cefaclor. |
| EP0839526A3 (en) * | 1996-10-31 | 1999-01-07 | Takeda Chemical Industries, Ltd. | Solid pharmaceutical preparation with fast buccal disintegration or dissolution |
| CA2327655C (en) * | 1998-04-08 | 2010-03-09 | Kyowa Hakko Kogyo Co., Ltd. | Tablet production method and tablet |
| JP2002029964A (ja) * | 2000-07-11 | 2002-01-29 | Lion Corp | 固形医薬組成物 |
| JP2002087965A (ja) * | 2000-09-14 | 2002-03-27 | Lion Corp | 口中崩壊性アスピリン含有錠剤 |
| US20020122823A1 (en) * | 2000-12-29 | 2002-09-05 | Bunick Frank J. | Soft tablet containing dextrose monohydrate |
| US20040265375A1 (en) * | 2003-04-16 | 2004-12-30 | Platteeuw Johannes J. | Orally disintegrating tablets |
-
2006
- 2006-08-08 JP JP2007529583A patent/JP5366233B2/ja active Active
- 2006-08-08 EP EP06782462A patent/EP1923074A4/en active Pending
- 2006-08-08 CA CA2618977A patent/CA2618977C/en not_active Expired - Fee Related
- 2006-08-08 KR KR1020127033700A patent/KR20130016386A/ko not_active Application Discontinuation
- 2006-08-08 KR KR1020087003203A patent/KR101465803B1/ko not_active IP Right Cessation
- 2006-08-08 WO PCT/JP2006/315620 patent/WO2007018192A1/ja active Application Filing
- 2006-08-08 US US11/990,109 patent/US20080274178A1/en not_active Abandoned
- 2006-08-09 TW TW095129136A patent/TWI376243B/zh not_active IP Right Cessation
-
2013
- 2013-07-02 JP JP2013138823A patent/JP2013224321A/ja active Pending
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1203580A1 (en) | 1999-06-18 | 2002-05-08 | Takeda Chemical Industries, Ltd. | Quickly disintegrating solid preparations |
| US20040028741A1 (en) | 2000-09-22 | 2004-02-12 | Kazuyuki Fujihara | Oral preparations with favorable disintegration characteristics |
| JP2002179558A (ja) * | 2000-10-06 | 2002-06-26 | Takeda Chem Ind Ltd | 固形製剤 |
| EP1329217A1 (en) | 2000-10-06 | 2003-07-23 | Takeda Chemical Industries, Ltd. | Solid preparations |
| JP2002188095A (ja) * | 2000-12-21 | 2002-07-05 | Fancl Corp | 植物性油脂粉末及び該粉末を含む食品組成物 |
| EP1369109A1 (en) | 2001-03-06 | 2003-12-10 | Kyowa Hakko Kogyo Co., Ltd. | Preparations quickly disintegrating in oral cavity |
| JP2003034655A (ja) * | 2001-05-15 | 2003-02-07 | Takeda Chem Ind Ltd | 速崩壊性固形製剤 |
| JP2004123648A (ja) * | 2002-10-04 | 2004-04-22 | Taiyo Kagaku Co Ltd | 錠剤、打錠用改質剤 |
| JP2004155682A (ja) * | 2002-11-05 | 2004-06-03 | Dai Ichi Kogyo Seiyaku Co Ltd | 錠剤用滑沢剤組成物 |
| EP1523974A1 (en) | 2003-10-15 | 2005-04-20 | Fuji Chemical Industry Co., Ltd. | Composition for rapid disintegrating tablet in oral cavity |
| WO2005055955A2 (en) | 2003-12-09 | 2005-06-23 | Pharmasset, Inc. | DOSING METHODS FOR ß-D-2’,3’-DIDEOXY-2’,3’-DIDEHYDRO-5-FLUOROCYTIDINE ANTIVIRAL THERAPY |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1923074A4 * |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008285434A (ja) * | 2007-05-16 | 2008-11-27 | Taisho Pharm Ind Ltd | 口腔内速崩壊錠 |
| WO2009066773A1 (ja) | 2007-11-21 | 2009-05-28 | Dainippon Sumitomo Pharma Co., Ltd. | 口腔内崩壊錠 |
| US8377995B2 (en) | 2007-11-21 | 2013-02-19 | Dainippon Sumitomo Pharma Co., Ltd. | Orally disintegrating tablet |
| JP2009256349A (ja) * | 2008-03-28 | 2009-11-05 | Kowa Co | 錠剤 |
| US8920839B2 (en) | 2009-05-20 | 2014-12-30 | Sumitomo Dainippon Pharma Co., Ltd. | Dry-coated orally-disintegrating tablet |
| WO2010134540A1 (ja) | 2009-05-20 | 2010-11-25 | 大日本住友製薬株式会社 | 有核型の口腔内崩壊錠 |
| JP5458096B2 (ja) * | 2009-05-20 | 2014-04-02 | 大日本住友製薬株式会社 | 有核型の口腔内崩壊錠 |
| WO2010137716A1 (ja) * | 2009-05-29 | 2010-12-02 | マイラン製薬株式会社 | 沈降炭酸カルシウムを有効成分とする口腔内崩壊錠 |
| JP2011006377A (ja) * | 2009-05-29 | 2011-01-13 | Mylan Seiyaku Ltd | 沈降炭酸カルシウムを有効成分とする口腔内崩壊錠 |
| US9744134B2 (en) | 2012-02-03 | 2017-08-29 | Asahi Kasei Chemicals Corporation | Orally disintegrating tablet containing bitterness-masking granules |
| JPWO2013115171A1 (ja) * | 2012-02-03 | 2015-05-11 | 旭化成ケミカルズ株式会社 | 苦味マスク顆粒含有口腔内崩壊錠 |
| WO2013115171A1 (ja) * | 2012-02-03 | 2013-08-08 | 旭化成ケミカルズ株式会社 | 苦味マスク顆粒含有口腔内崩壊錠 |
| WO2015005241A1 (ja) * | 2013-07-06 | 2015-01-15 | 株式会社ダイセル | 超速崩壊錠剤及びその製造方法 |
| EP3020416A4 (en) * | 2013-07-06 | 2016-12-07 | Daicel Corp | ULTRA-RAPID DISAGRÉGATION TABLE AND METHOD OF MANUFACTURE |
| JPWO2015005241A1 (ja) * | 2013-07-06 | 2017-03-02 | 株式会社ダイセル | 超速崩壊錠剤及びその製造方法 |
| US9974738B2 (en) | 2013-09-27 | 2018-05-22 | Daicel Corporation | Disintegrating particle composition produced by two-stage wet granulation process, and intraorally disintegrating tablet containing same composition |
| US10398694B2 (en) | 2014-11-11 | 2019-09-03 | Shionogi & Co., Ltd. | Multi-layered tablet containing drug unstable to light |
| WO2016103904A1 (ja) * | 2014-12-25 | 2016-06-30 | 株式会社ダイセル | 超速崩壊錠剤及びその製造方法 |
| JPWO2016103904A1 (ja) * | 2014-12-25 | 2017-10-05 | 株式会社ダイセル | 超速崩壊錠剤及びその製造方法 |
| JP2019131597A (ja) * | 2014-12-25 | 2019-08-08 | 株式会社ダイセル | 超速崩壊錠剤及びその製造方法 |
| US10426732B2 (en) | 2014-12-25 | 2019-10-01 | Daicel Corporation | Rapidly disintegrating tablet, and method for producing same |
| EP3238712B1 (en) * | 2014-12-25 | 2023-11-01 | Daicel Corporation | Very rapidly disintegrating tablet, and method for producing same |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2618977A1 (en) | 2007-02-15 |
| US20080274178A1 (en) | 2008-11-06 |
| EP1923074A4 (en) | 2011-09-14 |
| JPWO2007018192A1 (ja) | 2009-02-19 |
| TWI376243B (en) | 2012-11-11 |
| TW200726481A (en) | 2007-07-16 |
| KR20080039410A (ko) | 2008-05-07 |
| KR101465803B1 (ko) | 2014-11-26 |
| KR20130016386A (ko) | 2013-02-14 |
| CA2618977C (en) | 2014-10-21 |
| JP2013224321A (ja) | 2013-10-31 |
| JP5366233B2 (ja) | 2013-12-11 |
| EP1923074A1 (en) | 2008-05-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5366233B2 (ja) | 口腔内崩壊錠剤 | |
| US5958453A (en) | Solid pharmaceutical preparation with improved buccal disintegrability and/or dissolubility | |
| JP3996626B2 (ja) | 口腔内速崩壊錠 | |
| JP5752227B2 (ja) | 口腔内崩壊錠 | |
| JP5074190B2 (ja) | 口腔内速崩壊性錠剤 | |
| JP6521564B2 (ja) | 速崩性の固体被覆剤形 | |
| WO2000078292A1 (fr) | Preparations solides a desintegration rapide | |
| JP5656258B2 (ja) | ガランタミンを含有する口腔内崩壊錠剤 | |
| JP2003034655A (ja) | 速崩壊性固形製剤 | |
| JP2011513194A (ja) | 口腔内崩壊錠 | |
| JP2010270110A (ja) | ネオテームを含有する経口製剤 | |
| JP5204976B2 (ja) | イグラチモドを含有する速崩壊性錠剤 | |
| WO2007086846A1 (en) | Pharmaceutical formulations useful for inhibiting acid secretion and methods for making and using them | |
| JP2010241760A (ja) | 不快な味の軽減された口腔内速崩壊錠及びその製造方法 | |
| JP5275815B2 (ja) | リスペリドンを含有する口腔内崩壊錠剤および苦味抑制製剤 | |
| TWI651085B (zh) | N-[5-[2-(3,5-二甲氧基苯基)乙基]-2h-吡唑-3-基]-4-[(3r,5s)-3,5-二甲基哌-1-基]苯甲醯胺之醫藥調配物 | |
| KR20110007065A (ko) | 구강내 붕괴정 및 그 제조 방법 | |
| JP2016117652A (ja) | 小児への投与に適した速崩壊錠とその簡便な製造方法 | |
| JP2005029557A (ja) | 口腔内速崩壊性錠剤およびその製造方法 | |
| WO2016098459A1 (ja) | 遅延崩壊性粒子組成物 | |
| JP2002338500A (ja) | 苦味を低減した口腔内速崩壊錠および苦み低減化方法 | |
| WO2018147353A1 (ja) | 錠剤 | |
| JP2023163350A (ja) | チュアブル錠又は口腔内崩壊錠 | |
| JP2021113237A (ja) | 溶出が良好なイルベサルタン含有医薬組成物および口腔内崩壊錠 | |
| JP2005194225A (ja) | 胃内崩壊性錠剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680036547.0 Country of ref document: CN |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2007529583 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087003203 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11990109 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 693/CHENP/2008 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2006782462 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006782462 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2618977 Country of ref document: CA |