WO2007009578A1 - Heterocyclylamid-substituierte thiazole, pyrrole und thiophene - Google Patents
Heterocyclylamid-substituierte thiazole, pyrrole und thiophene Download PDFInfo
- Publication number
- WO2007009578A1 WO2007009578A1 PCT/EP2006/006434 EP2006006434W WO2007009578A1 WO 2007009578 A1 WO2007009578 A1 WO 2007009578A1 EP 2006006434 W EP2006006434 W EP 2006006434W WO 2007009578 A1 WO2007009578 A1 WO 2007009578A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- substituents
- phenyl
- alkyl
- substituted
- Prior art date
Links
- 150000003233 pyrroles Chemical class 0.000 title abstract description 4
- 150000003557 thiazoles Chemical class 0.000 title abstract description 4
- 229930192474 thiophene Chemical class 0.000 title abstract description 4
- 150000003577 thiophenes Chemical class 0.000 title abstract description 4
- 238000000034 method Methods 0.000 claims abstract description 52
- 238000011282 treatment Methods 0.000 claims abstract description 27
- 238000011321 prophylaxis Methods 0.000 claims abstract description 17
- 201000010099 disease Diseases 0.000 claims abstract description 14
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 14
- 239000003814 drug Substances 0.000 claims abstract description 10
- 238000004519 manufacturing process Methods 0.000 claims abstract description 6
- 150000001875 compounds Chemical class 0.000 claims description 84
- -1 trifluoromethoxy, difluoromethoxy, monofluoromethoxy Chemical group 0.000 claims description 69
- 125000001424 substituent group Chemical group 0.000 claims description 64
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 41
- 150000003839 salts Chemical class 0.000 claims description 29
- 238000002360 preparation method Methods 0.000 claims description 26
- 229910052736 halogen Inorganic materials 0.000 claims description 24
- 150000002367 halogens Chemical class 0.000 claims description 24
- 239000012453 solvate Substances 0.000 claims description 22
- 229910052757 nitrogen Inorganic materials 0.000 claims description 21
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 claims description 19
- 125000005034 trifluormethylthio group Chemical group FC(S*)(F)F 0.000 claims description 19
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 19
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 18
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 17
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 17
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 16
- 241000701024 Human betaherpesvirus 5 Species 0.000 claims description 15
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 15
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 15
- 125000003118 aryl group Chemical group 0.000 claims description 14
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 14
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 13
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 claims description 13
- 239000002585 base Substances 0.000 claims description 13
- 125000001072 heteroaryl group Chemical group 0.000 claims description 12
- 239000001257 hydrogen Substances 0.000 claims description 12
- 229910052739 hydrogen Inorganic materials 0.000 claims description 12
- ORTFAQDWJHRMNX-UHFFFAOYSA-N hydroxidooxidocarbon(.) Chemical group O[C]=O ORTFAQDWJHRMNX-UHFFFAOYSA-N 0.000 claims description 12
- 125000004043 oxo group Chemical group O=* 0.000 claims description 12
- 208000015181 infectious disease Diseases 0.000 claims description 11
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 9
- 230000009385 viral infection Effects 0.000 claims description 9
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 8
- 208000036142 Viral infection Diseases 0.000 claims description 8
- 125000000217 alkyl group Chemical group 0.000 claims description 8
- 239000004202 carbamide Substances 0.000 claims description 8
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 8
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 7
- 239000000460 chlorine Substances 0.000 claims description 7
- 229910052801 chlorine Inorganic materials 0.000 claims description 7
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 7
- 125000004076 pyridyl group Chemical group 0.000 claims description 7
- 125000004454 (C1-C6) alkoxycarbonyl group Chemical group 0.000 claims description 6
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 claims description 6
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 6
- 239000003638 chemical reducing agent Substances 0.000 claims description 6
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 6
- 241001465754 Metazoa Species 0.000 claims description 5
- 125000003545 alkoxy group Chemical group 0.000 claims description 5
- 150000004649 carbonic acid derivatives Chemical class 0.000 claims description 5
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 4
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 4
- 241000700586 Herpesviridae Species 0.000 claims description 4
- 125000004457 alkyl amino carbonyl group Chemical group 0.000 claims description 4
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 4
- 229910052794 bromium Inorganic materials 0.000 claims description 4
- 239000003153 chemical reaction reagent Substances 0.000 claims description 4
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 4
- 239000011737 fluorine Substances 0.000 claims description 4
- 229910052731 fluorine Inorganic materials 0.000 claims description 4
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 4
- 231100000252 nontoxic Toxicity 0.000 claims description 3
- 230000003000 nontoxic effect Effects 0.000 claims description 3
- 239000008194 pharmaceutical composition Substances 0.000 claims description 3
- 241000282412 Homo Species 0.000 claims description 2
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims 4
- 241000701022 Cytomegalovirus Species 0.000 abstract description 6
- 239000003443 antiviral agent Substances 0.000 abstract description 5
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 30
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 29
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 27
- 239000000203 mixture Substances 0.000 description 25
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 24
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 22
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 22
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 20
- 239000002904 solvent Substances 0.000 description 20
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 20
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 18
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 18
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 18
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 18
- 239000000243 solution Substances 0.000 description 18
- 238000005481 NMR spectroscopy Methods 0.000 description 17
- 210000004027 cell Anatomy 0.000 description 17
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 16
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 15
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 15
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 15
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 12
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 11
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 11
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical class Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 11
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 10
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 10
- 238000006243 chemical reaction Methods 0.000 description 10
- 239000012442 inert solvent Substances 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- 241000700605 Viruses Species 0.000 description 9
- QPFMBZIOSGYJDE-UHFFFAOYSA-N 1,1,2,2-tetrachloroethane Chemical compound ClC(Cl)C(Cl)Cl QPFMBZIOSGYJDE-UHFFFAOYSA-N 0.000 description 8
- 238000005160 1H NMR spectroscopy Methods 0.000 description 8
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 8
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 8
- 206010011831 Cytomegalovirus infection Diseases 0.000 description 8
- 125000004432 carbon atom Chemical group C* 0.000 description 8
- 235000019253 formic acid Nutrition 0.000 description 8
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 8
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 7
- 238000004128 high performance liquid chromatography Methods 0.000 description 7
- 239000002609 medium Substances 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- 239000003826 tablet Substances 0.000 description 7
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical class CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 6
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 6
- 239000003480 eluent Substances 0.000 description 6
- 150000002431 hydrogen Chemical class 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 5
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 5
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 5
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 5
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 5
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 5
- 230000037396 body weight Effects 0.000 description 5
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 5
- 150000002170 ethers Chemical class 0.000 description 5
- 239000012894 fetal calf serum Substances 0.000 description 5
- 229930195733 hydrocarbon Natural products 0.000 description 5
- 150000002430 hydrocarbons Chemical class 0.000 description 5
- WMFOQBRAJBCJND-UHFFFAOYSA-M lithium hydroxide Inorganic materials [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 5
- 229910052763 palladium Inorganic materials 0.000 description 5
- 239000003208 petroleum Substances 0.000 description 5
- 239000008096 xylene Substances 0.000 description 5
- UOCLXMDMGBRAIB-UHFFFAOYSA-N 1,1,1-trichloroethane Chemical compound CC(Cl)(Cl)Cl UOCLXMDMGBRAIB-UHFFFAOYSA-N 0.000 description 4
- PAMIQIKDUOTOBW-UHFFFAOYSA-N 1-methylpiperidine Chemical compound CN1CCCCC1 PAMIQIKDUOTOBW-UHFFFAOYSA-N 0.000 description 4
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 4
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical class OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- XSTXAVWGXDQKEL-UHFFFAOYSA-N Trichloroethylene Chemical group ClC=C(Cl)Cl XSTXAVWGXDQKEL-UHFFFAOYSA-N 0.000 description 4
- 229950005499 carbon tetrachloride Drugs 0.000 description 4
- 229960001701 chloroform Drugs 0.000 description 4
- 230000000120 cytopathologic effect Effects 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 229940052308 general anesthetics halogenated hydrocarbons Drugs 0.000 description 4
- 150000008282 halocarbons Chemical class 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 229940073584 methylene chloride Drugs 0.000 description 4
- ZJAOAACCNHFJAH-UHFFFAOYSA-N phosphonoformic acid Chemical compound OC(=O)P(O)(O)=O ZJAOAACCNHFJAH-UHFFFAOYSA-N 0.000 description 4
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 4
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 4
- 239000011541 reaction mixture Substances 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 238000004007 reversed phase HPLC Methods 0.000 description 4
- 238000010186 staining Methods 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 4
- 238000002054 transplantation Methods 0.000 description 4
- QDRKPHJFMGUXPN-UHFFFAOYSA-N 1-(5-methylpyridin-2-yl)piperazine Chemical compound N1=CC(C)=CC=C1N1CCNCC1 QDRKPHJFMGUXPN-UHFFFAOYSA-N 0.000 description 3
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- 239000007868 Raney catalyst Substances 0.000 description 3
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 3
- 229910000564 Raney nickel Inorganic materials 0.000 description 3
- 239000012317 TBTU Substances 0.000 description 3
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 3
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical class OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 3
- 238000011156 evaluation Methods 0.000 description 3
- IRSCQMHQWWYFCW-UHFFFAOYSA-N ganciclovir Chemical compound O=C1NC(N)=NC2=C1N=CN2COC(CO)CO IRSCQMHQWWYFCW-UHFFFAOYSA-N 0.000 description 3
- 229960002963 ganciclovir Drugs 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 238000001802 infusion Methods 0.000 description 3
- 229960004592 isopropanol Drugs 0.000 description 3
- 229910017604 nitric acid Inorganic materials 0.000 description 3
- 239000002953 phosphate buffered saline Substances 0.000 description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 description 3
- 235000011181 potassium carbonates Nutrition 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- CDBYLPFSWZWCQE-UHFFFAOYSA-L sodium carbonate Substances [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 238000000825 ultraviolet detection Methods 0.000 description 3
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- VWFCHDSQECPREK-LURJTMIESA-N Cidofovir Chemical compound NC=1C=CN(C[C@@H](CO)OCP(O)(O)=O)C(=O)N=1 VWFCHDSQECPREK-LURJTMIESA-N 0.000 description 2
- 208000035473 Communicable disease Diseases 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 102000003974 Fibroblast growth factor 2 Human genes 0.000 description 2
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 2
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical class OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 239000007821 HATU Substances 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical class CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical class OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 206010035664 Pneumonia Diseases 0.000 description 2
- 206010035742 Pneumonitis Diseases 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical class OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- WPVFJKSGQUFQAP-GKAPJAKFSA-N Valcyte Chemical compound N1C(N)=NC(=O)C2=C1N(COC(CO)COC(=O)[C@@H](N)C(C)C)C=N2 WPVFJKSGQUFQAP-GKAPJAKFSA-N 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- LOSFVIMHTOMZKT-UHFFFAOYSA-N [4-(trifluoromethoxy)phenyl]urea Chemical compound NC(=O)NC1=CC=C(OC(F)(F)F)C=C1 LOSFVIMHTOMZKT-UHFFFAOYSA-N 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 2
- 150000008041 alkali metal carbonates Chemical class 0.000 description 2
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 2
- 125000003282 alkyl amino group Chemical group 0.000 description 2
- 230000000840 anti-viral effect Effects 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical class OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- RROBIDXNTUAHFW-UHFFFAOYSA-N benzotriazol-1-yloxy-tris(dimethylamino)phosphanium Chemical compound C1=CC=C2N(O[P+](N(C)C)(N(C)C)N(C)C)N=NC2=C1 RROBIDXNTUAHFW-UHFFFAOYSA-N 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 229910000024 caesium carbonate Inorganic materials 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 229960000724 cidofovir Drugs 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 239000000975 dye Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- FOJSMLUSGLPFPR-UHFFFAOYSA-N ethyl 1-methyl-4-[[4-(trifluoromethoxy)phenyl]carbamoylamino]pyrrole-2-carboxylate Chemical compound CN1C(C(=O)OCC)=CC(NC(=O)NC=2C=CC(OC(F)(F)F)=CC=2)=C1 FOJSMLUSGLPFPR-UHFFFAOYSA-N 0.000 description 2
- 210000002950 fibroblast Anatomy 0.000 description 2
- 210000003953 foreskin Anatomy 0.000 description 2
- 229960005102 foscarnet Drugs 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 239000007943 implant Substances 0.000 description 2
- 229910052742 iron Inorganic materials 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical class CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 238000004949 mass spectrometry Methods 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- PGSADBUBUOPOJS-UHFFFAOYSA-N neutral red Chemical compound Cl.C1=C(C)C(N)=CC2=NC3=CC(N(C)C)=CC=C3N=C21 PGSADBUBUOPOJS-UHFFFAOYSA-N 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 229940068918 polyethylene glycol 400 Drugs 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 238000013207 serial dilution Methods 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 2
- CFVAYMJNEFGOFR-UHFFFAOYSA-N tert-butyl 4-(5-methylpyridin-2-yl)piperazine-1-carboxylate Chemical compound N1=CC(C)=CC=C1N1CCN(C(=O)OC(C)(C)C)CC1 CFVAYMJNEFGOFR-UHFFFAOYSA-N 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Chemical class OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 229960002149 valganciclovir Drugs 0.000 description 2
- 235000012431 wafers Nutrition 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 1
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical class OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- 0 *NC(N*C(O*)=O)=O Chemical compound *NC(N*C(O*)=O)=O 0.000 description 1
- WXDPLUNBGIWOHE-UHFFFAOYSA-N 1-(cyclopropylmethyl)-4-[[4-(trifluoromethoxy)phenyl]carbamoylamino]pyrrole-2-carboxylic acid Chemical compound C=1N(CC2CC2)C(C(=O)O)=CC=1NC(=O)NC1=CC=C(OC(F)(F)F)C=C1 WXDPLUNBGIWOHE-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- UFWHPKVVDRBABT-UHFFFAOYSA-N 1-[1-(cyclopropylmethyl)-5-(4-pyridin-2-ylpiperazine-1-carbonyl)pyrrol-3-yl]-3-[4-(trifluoromethoxy)phenyl]urea Chemical compound C1=CC(OC(F)(F)F)=CC=C1NC(=O)NC1=CN(CC2CC2)C(C(=O)N2CCN(CC2)C=2N=CC=CC=2)=C1 UFWHPKVVDRBABT-UHFFFAOYSA-N 0.000 description 1
- WCPHQKOASNSPIP-UHFFFAOYSA-N 1-[1-(cyclopropylmethyl)-5-[4-(5-methylpyridin-2-yl)piperazine-1-carbonyl]pyrrol-3-yl]-3-[4-(trifluoromethoxy)phenyl]urea Chemical compound N1=CC(C)=CC=C1N1CCN(C(=O)C=2N(C=C(NC(=O)NC=3C=CC(OC(F)(F)F)=CC=3)C=2)CC2CC2)CC1 WCPHQKOASNSPIP-UHFFFAOYSA-N 0.000 description 1
- ZNAPYPSWVPWJJH-UHFFFAOYSA-N 1-[1-butyl-5-(4-pyridin-2-ylpiperazine-1-carbonyl)pyrrol-3-yl]-3-[4-(trifluoromethoxy)phenyl]urea Chemical compound C1=C(C(=O)N2CCN(CC2)C=2N=CC=CC=2)N(CCCC)C=C1NC(=O)NC1=CC=C(OC(F)(F)F)C=C1 ZNAPYPSWVPWJJH-UHFFFAOYSA-N 0.000 description 1
- ZRFOVSYGVPFCFC-UHFFFAOYSA-N 1-[1-methyl-5-[4-(5-methylpyridin-2-yl)piperazine-1-carbonyl]pyrrol-3-yl]-3-[4-(trifluoromethoxy)phenyl]urea Chemical compound N1=CC(C)=CC=C1N1CCN(C(=O)C=2N(C=C(NC(=O)NC=3C=CC(OC(F)(F)F)=CC=3)C=2)C)CC1 ZRFOVSYGVPFCFC-UHFFFAOYSA-N 0.000 description 1
- QKAPPWMPTIOAMN-UHFFFAOYSA-N 1-butyl-4-[[4-(trifluoromethoxy)phenyl]carbamoylamino]pyrrole-2-carboxylic acid Chemical compound C1=C(C(O)=O)N(CCCC)C=C1NC(=O)NC1=CC=C(OC(F)(F)F)C=C1 QKAPPWMPTIOAMN-UHFFFAOYSA-N 0.000 description 1
- GJBKVSKPXVGBIF-UHFFFAOYSA-N 1-ethyl-4-[[4-(trifluoromethoxy)phenyl]carbamoylamino]pyrrole-2-carboxylic acid Chemical compound C1=C(C(O)=O)N(CC)C=C1NC(=O)NC1=CC=C(OC(F)(F)F)C=C1 GJBKVSKPXVGBIF-UHFFFAOYSA-N 0.000 description 1
- LGPKFIGMLPDYEA-UHFFFAOYSA-N 1-isocyanato-4-(trifluoromethoxy)benzene Chemical compound FC(F)(F)OC1=CC=C(N=C=O)C=C1 LGPKFIGMLPDYEA-UHFFFAOYSA-N 0.000 description 1
- MCTWTZJPVLRJOU-UHFFFAOYSA-N 1-methyl-1H-imidazole Chemical compound CN1C=CN=C1 MCTWTZJPVLRJOU-UHFFFAOYSA-N 0.000 description 1
- MQOJPWXJUMEQMS-UHFFFAOYSA-N 1h-pyrrol-2-ylurea Chemical class NC(=O)NC1=CC=CN1 MQOJPWXJUMEQMS-UHFFFAOYSA-N 0.000 description 1
- MUXGMFNQYPUQJO-UHFFFAOYSA-N 2,2,2-trichloro-1-(1-methyl-3-nitropyrrol-2-yl)ethanone Chemical compound CN1C=CC([N+]([O-])=O)=C1C(=O)C(Cl)(Cl)Cl MUXGMFNQYPUQJO-UHFFFAOYSA-N 0.000 description 1
- WASYNLWEPOHNBM-UHFFFAOYSA-N 2,2,2-trichloro-1-(1-methyl-4-nitropyrrol-2-yl)ethanone Chemical compound CN1C=C([N+]([O-])=O)C=C1C(=O)C(Cl)(Cl)Cl WASYNLWEPOHNBM-UHFFFAOYSA-N 0.000 description 1
- LWGNISUGCOYWRL-UHFFFAOYSA-N 2,2,2-trichloro-1-(1-methylpyrrol-2-yl)ethanone Chemical compound CN1C=CC=C1C(=O)C(Cl)(Cl)Cl LWGNISUGCOYWRL-UHFFFAOYSA-N 0.000 description 1
- YOYAIZYFCNQIRF-UHFFFAOYSA-N 2,6-dichlorobenzonitrile Chemical compound ClC1=CC=CC(Cl)=C1C#N YOYAIZYFCNQIRF-UHFFFAOYSA-N 0.000 description 1
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical class CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- ZUFZEPOUXIPZOO-UHFFFAOYSA-N 2-[[4-(trifluoromethoxy)phenyl]carbamoylamino]-1,3-thiazole-5-carboxylic acid Chemical compound S1C(C(=O)O)=CN=C1NC(=O)NC1=CC=C(OC(F)(F)F)C=C1 ZUFZEPOUXIPZOO-UHFFFAOYSA-N 0.000 description 1
- WEVYAHXRMPXWCK-MICDWDOJSA-N 2-deuterioacetonitrile Chemical compound [2H]CC#N WEVYAHXRMPXWCK-MICDWDOJSA-N 0.000 description 1
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 1
- MPSXGPCFLAGJOM-UHFFFAOYSA-M 2-tert-butyl-5-methyl-1,2-oxazol-2-ium;perchlorate Chemical compound [O-]Cl(=O)(=O)=O.CC1=CC=[N+](C(C)(C)C)O1 MPSXGPCFLAGJOM-UHFFFAOYSA-M 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- KLDLRDSRCMJKGM-UHFFFAOYSA-N 3-[chloro-(2-oxo-1,3-oxazolidin-3-yl)phosphoryl]-1,3-oxazolidin-2-one Chemical compound C1COC(=O)N1P(=O)(Cl)N1CCOC1=O KLDLRDSRCMJKGM-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical class NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- WRKZMXUBAKALML-UHFFFAOYSA-N 4-[[4-(trifluoromethoxy)phenyl]carbamoylamino]thiophene-2-carboxylic acid Chemical compound S1C(C(=O)O)=CC(NC(=O)NC=2C=CC(OC(F)(F)F)=CC=2)=C1 WRKZMXUBAKALML-UHFFFAOYSA-N 0.000 description 1
- OCKGFTQIICXDQW-ZEQRLZLVSA-N 5-[(1r)-1-hydroxy-2-[4-[(2r)-2-hydroxy-2-(4-methyl-1-oxo-3h-2-benzofuran-5-yl)ethyl]piperazin-1-yl]ethyl]-4-methyl-3h-2-benzofuran-1-one Chemical compound C1=C2C(=O)OCC2=C(C)C([C@@H](O)CN2CCN(CC2)C[C@H](O)C2=CC=C3C(=O)OCC3=C2C)=C1 OCKGFTQIICXDQW-ZEQRLZLVSA-N 0.000 description 1
- VINRASGOTTVGQC-UHFFFAOYSA-N 5-bromo-2-[[4-(trifluoromethoxy)phenyl]carbamoylamino]-1,3-thiazole-4-carboxylic acid Chemical compound S1C(Br)=C(C(=O)O)N=C1NC(=O)NC1=CC=C(OC(F)(F)F)C=C1 VINRASGOTTVGQC-UHFFFAOYSA-N 0.000 description 1
- DEMKNLXJQNYAFY-UHFFFAOYSA-N 5-chloro-2-methylpyridine Chemical compound CC1=CC=C(Cl)C=N1 DEMKNLXJQNYAFY-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- WSVLPVUVIUVCRA-KPKNDVKVSA-N Alpha-lactose monohydrate Chemical compound O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O WSVLPVUVIUVCRA-KPKNDVKVSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 239000005711 Benzoic acid Chemical class 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- INLBXDDZNIERLB-UHFFFAOYSA-N C[n]1c(C(O)=O)cc(NC(Nc(cc2)ccc2OC(F)(F)F)=O)c1 Chemical compound C[n]1c(C(O)=O)cc(NC(Nc(cc2)ccc2OC(F)(F)F)=O)c1 INLBXDDZNIERLB-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- BAMPVSWRQZNDQC-UHFFFAOYSA-N Cc1c(C)[s]c(C)n1 Chemical compound Cc1c(C)[s]c(C)n1 BAMPVSWRQZNDQC-UHFFFAOYSA-N 0.000 description 1
- ADDQOSJKHNLNJV-UHFFFAOYSA-N Cc1c(S)[s]c(C)n1 Chemical compound Cc1c(S)[s]c(C)n1 ADDQOSJKHNLNJV-UHFFFAOYSA-N 0.000 description 1
- 102000029816 Collagenase Human genes 0.000 description 1
- 108060005980 Collagenase Proteins 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical class OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- BWLUMTFWVZZZND-UHFFFAOYSA-N Dibenzylamine Chemical compound C=1C=CC=CC=1CNCC1=CC=CC=C1 BWLUMTFWVZZZND-UHFFFAOYSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- GKQLYSROISKDLL-UHFFFAOYSA-N EEDQ Chemical compound C1=CC=C2N(C(=O)OCC)C(OCC)C=CC2=C1 GKQLYSROISKDLL-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 206010017964 Gastrointestinal infection Diseases 0.000 description 1
- YQEZLKZALYSWHR-UHFFFAOYSA-N Ketamine Chemical compound C=1C=CC=C(Cl)C=1C1(NC)CCCCC1=O YQEZLKZALYSWHR-UHFFFAOYSA-N 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- 206010061309 Neoplasm progression Diseases 0.000 description 1
- 206010029155 Nephropathy toxic Diseases 0.000 description 1
- 229910021586 Nickel(II) chloride Inorganic materials 0.000 description 1
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Chemical class OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 206010038910 Retinitis Diseases 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 208000002847 Surgical Wound Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Chemical class [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- NOSIYYJFMPDDSA-UHFFFAOYSA-N acepromazine Chemical compound C1=C(C(C)=O)C=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 NOSIYYJFMPDDSA-UHFFFAOYSA-N 0.000 description 1
- 229960005054 acepromazine Drugs 0.000 description 1
- 229960004150 aciclovir Drugs 0.000 description 1
- MKUXAQIIEYXACX-UHFFFAOYSA-N aciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCO)C=N2 MKUXAQIIEYXACX-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000004442 acylamino group Chemical group 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Chemical class OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical class OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 239000006189 buccal tablet Substances 0.000 description 1
- HTRXGEPDTFSKLI-UHFFFAOYSA-N butanoic acid;ethyl acetate Chemical compound CCCC(O)=O.CCOC(C)=O HTRXGEPDTFSKLI-UHFFFAOYSA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 150000001728 carbonyl compounds Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 238000000451 chemical ionisation Methods 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- 239000012320 chlorinating reagent Substances 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000000515 collagen sponge Substances 0.000 description 1
- 229960002424 collagenase Drugs 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 230000001085 cytostatic effect Effects 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 229960002887 deanol Drugs 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 238000007872 degassing Methods 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- JMRYOSQOYJBDOI-UHFFFAOYSA-N dilithium;di(propan-2-yl)azanide Chemical compound [Li+].CC(C)[N-]C(C)C.CC(C)N([Li])C(C)C JMRYOSQOYJBDOI-UHFFFAOYSA-N 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- HCUYBXPSSCRKRF-UHFFFAOYSA-N diphosgene Chemical compound ClC(=O)OC(Cl)(Cl)Cl HCUYBXPSSCRKRF-UHFFFAOYSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 239000012154 double-distilled water Substances 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 206010014599 encephalitis Diseases 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical class CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- XOLGKCDCFJPOEY-UHFFFAOYSA-N ethyl 1-methyl-4-nitropyrrole-2-carboxylate Chemical compound CCOC(=O)C1=CC([N+]([O-])=O)=CN1C XOLGKCDCFJPOEY-UHFFFAOYSA-N 0.000 description 1
- JILQJAVCOAVWCC-UHFFFAOYSA-N ethyl 2-amino-5-chloro-1,3-thiazole-4-carboxylate Chemical compound CCOC(=O)C=1N=C(N)SC=1Cl JILQJAVCOAVWCC-UHFFFAOYSA-N 0.000 description 1
- 125000006260 ethylaminocarbonyl group Chemical group [H]N(C(*)=O)C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000010408 film Substances 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000013100 final test Methods 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000001530 fumaric acid Chemical class 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 1
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 239000001023 inorganic pigment Substances 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- UQSXHKLRYXJYBZ-UHFFFAOYSA-N iron oxide Inorganic materials [Fe]=O UQSXHKLRYXJYBZ-UHFFFAOYSA-N 0.000 description 1
- 235000013980 iron oxide Nutrition 0.000 description 1
- BAUYGSIQEAFULO-UHFFFAOYSA-L iron(2+) sulfate (anhydrous) Chemical compound [Fe+2].[O-]S([O-])(=O)=O BAUYGSIQEAFULO-UHFFFAOYSA-L 0.000 description 1
- VBMVTYDPPZVILR-UHFFFAOYSA-N iron(2+);oxygen(2-) Chemical class [O-2].[Fe+2] VBMVTYDPPZVILR-UHFFFAOYSA-N 0.000 description 1
- 229910000359 iron(II) sulfate Inorganic materials 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000005928 isopropyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(OC(*)=O)C([H])([H])[H] 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical class C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- 229960003299 ketamine Drugs 0.000 description 1
- 239000004310 lactic acid Chemical class 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical class OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Chemical class 0.000 description 1
- 239000001630 malic acid Chemical class 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- HCRQNZWIOUMCOW-UHFFFAOYSA-N methyl 4-aminothiophene-2-carboxylate Chemical compound COC(=O)C1=CC(N)=CS1 HCRQNZWIOUMCOW-UHFFFAOYSA-N 0.000 description 1
- YLGXILFCIXHCMC-JHGZEJCSSA-N methyl cellulose Chemical compound COC1C(OC)C(OC)C(COC)O[C@H]1O[C@H]1C(OC)C(OC)C(OC)OC1COC YLGXILFCIXHCMC-JHGZEJCSSA-N 0.000 description 1
- 125000004458 methylaminocarbonyl group Chemical group [H]N(C(*)=O)C([H])([H])[H] 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- BXGTVNLGPMZLAZ-UHFFFAOYSA-N n'-ethylmethanediimine;hydrochloride Chemical compound Cl.CCN=C=N BXGTVNLGPMZLAZ-UHFFFAOYSA-N 0.000 description 1
- WOOWBQQQJXZGIE-UHFFFAOYSA-N n-ethyl-n-propan-2-ylpropan-2-amine Chemical compound CCN(C(C)C)C(C)C.CCN(C(C)C)C(C)C WOOWBQQQJXZGIE-UHFFFAOYSA-N 0.000 description 1
- 125000001298 n-hexoxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- YZMHQCWXYHARLS-UHFFFAOYSA-N naphthalene-1,2-disulfonic acid Chemical class C1=CC=CC2=C(S(O)(=O)=O)C(S(=O)(=O)O)=CC=C21 YZMHQCWXYHARLS-UHFFFAOYSA-N 0.000 description 1
- 239000007923 nasal drop Substances 0.000 description 1
- 229940100662 nasal drops Drugs 0.000 description 1
- 229920005615 natural polymer Polymers 0.000 description 1
- 230000007694 nephrotoxicity Effects 0.000 description 1
- 231100000417 nephrotoxicity Toxicity 0.000 description 1
- 208000004235 neutropenia Diseases 0.000 description 1
- QMMRZOWCJAIUJA-UHFFFAOYSA-L nickel dichloride Chemical compound Cl[Ni]Cl QMMRZOWCJAIUJA-UHFFFAOYSA-L 0.000 description 1
- 230000000802 nitrating effect Effects 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 229940100692 oral suspension Drugs 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- AHWALFGBDFAJAI-UHFFFAOYSA-N phenyl carbonochloridate Chemical compound ClC(=O)OC1=CC=CC=C1 AHWALFGBDFAJAI-UHFFFAOYSA-N 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 125000004194 piperazin-1-yl group Chemical group [H]N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 1
- 239000000902 placebo Substances 0.000 description 1
- 229940068196 placebo Drugs 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229920001021 polysulfide Polymers 0.000 description 1
- 239000005077 polysulfide Substances 0.000 description 1
- 150000008117 polysulfides Polymers 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 229940086066 potassium hydrogencarbonate Drugs 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- GZRKXKUVVPSREJ-UHFFFAOYSA-N pyridinylpiperazine Chemical compound C1CNCCN1C1=CC=CC=N1 GZRKXKUVVPSREJ-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical class O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000015424 sodium Nutrition 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- SRRKNRDXURUMPP-UHFFFAOYSA-N sodium disulfide Chemical compound [Na+].[Na+].[S-][S-] SRRKNRDXURUMPP-UHFFFAOYSA-N 0.000 description 1
- JVBXVOWTABLYPX-UHFFFAOYSA-L sodium dithionite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])=O JVBXVOWTABLYPX-UHFFFAOYSA-L 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 229910052979 sodium sulfide Inorganic materials 0.000 description 1
- GRVFOGOEDUUMBP-UHFFFAOYSA-N sodium sulfide (anhydrous) Chemical compound [Na+].[Na+].[S-2] GRVFOGOEDUUMBP-UHFFFAOYSA-N 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 235000011150 stannous chloride Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 239000006190 sub-lingual tablet Substances 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000011975 tartaric acid Chemical class 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- CWXPZXBSDSIRCS-UHFFFAOYSA-N tert-butyl piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNCC1 CWXPZXBSDSIRCS-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 206010043554 thrombocytopenia Diseases 0.000 description 1
- AXZWODMDQAVCJE-UHFFFAOYSA-L tin(II) chloride (anhydrous) Chemical compound [Cl-].[Cl-].[Sn+2] AXZWODMDQAVCJE-UHFFFAOYSA-L 0.000 description 1
- 239000003106 tissue adhesive Substances 0.000 description 1
- YONPGGFAJWQGJC-UHFFFAOYSA-K titanium(iii) chloride Chemical compound Cl[Ti](Cl)Cl YONPGGFAJWQGJC-UHFFFAOYSA-K 0.000 description 1
- 125000005270 trialkylamine group Chemical group 0.000 description 1
- YFDSDPIBEUFTMI-UHFFFAOYSA-N tribromoethanol Chemical compound OCC(Br)(Br)Br YFDSDPIBEUFTMI-UHFFFAOYSA-N 0.000 description 1
- 229950004616 tribromoethanol Drugs 0.000 description 1
- PVFOMCVHYWHZJE-UHFFFAOYSA-N trichloroacetyl chloride Chemical compound ClC(=O)C(Cl)(Cl)Cl PVFOMCVHYWHZJE-UHFFFAOYSA-N 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 1
- 230000005751 tumor progression Effects 0.000 description 1
- 241001529453 unidentified herpesvirus Species 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
- BPICBUSOMSTKRF-UHFFFAOYSA-N xylazine Chemical compound CC1=CC=CC(C)=C1NC1=NCCCS1 BPICBUSOMSTKRF-UHFFFAOYSA-N 0.000 description 1
- 229960001600 xylazine Drugs 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/38—Heterocyclic compounds having sulfur as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the invention relates to heterocyclylamide-substituted thiazoles, pyrroles and thiophenes and processes for their preparation, their use for the treatment and / or prophylaxis of diseases and their use for the preparation of medicaments for the treatment and / or prophylaxis of diseases, in particular for use as antiviral agents, especially against cytomegaloviruses.
- WO 99/23091 describes aromatic heterocyclic compounds as antiinflammatory agents, which may, inter alia, also be suitable for the treatment of viral infections, and WO 04/052852 describes 3-pyrrolyl urea derivatives as antiviral agents which carry a carbocycle as a substituent on the urea.
- An object of the present invention is therefore to provide new compounds having the same or improved antiviral activity for the treatment of viral infectious diseases in humans and animals.
- the present invention relates to compounds of the formula
- R 1 is a group of the formula
- R 3 is phenyl or 5- or 6-membered heteroaryl
- phenyl and heteroaryl may be substituted by 1 to 3 substituents, where the substituents are independently selected from the group consisting of halogen, hydroxy, oxo, nitro, cyano, trifluoromethyl, difluoromethyl, trifluoromethoxy, difluoromethoxy, monofluoromethoxy, trifluoromethylthio, C 1 -C 6 -alkyl, C 1 -C 6 -alkoxy, hydroxycarbonyl, C 1 -C 6 -alkoxycarbonyl,
- R 4 is phenyl or 5- or 6-membered heteroaryl
- phenyl and heteroaryl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, hydroxy, oxo, nitro, cyano, trifluoromethyl,
- R 5 and R 6 independently of one another represent hydrogen, methyl or ethyl
- phenyl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, hydroxy, trifluoromethyl, difluoromethyl, trifluoromethoxy, difluoromethoxy, monofluoromethoxy, trifluoromethylthio, Ci-C 6 alkyl and QC 6- alkoxy, for a group of the formula
- # represents the point of attachment to the nitrogen atom of the urea
- R 7 is C 1 -C 6 -alkyl
- alkyl may be substituted with a substituent, whereby the substituent is selected from the group consisting of C 3 -C 6 cycloalkyl, C 6 -C 0 aryl and 5- or 6-membered heteroaryl,
- cycloalkyl, aryl and heteroaryl may be substituted by 1 to 3 substituents, where the substituents are independently selected from the group consisting of halogen, hydroxy, oxo, nitro, cyano, trifluoromethyl, difluoromethyl, trifluoromethoxy, difluoromethoxy, monofluoromethoxy, Trifluoromethylthio, C 1 -C 6 -alkyl, C 1 -C 6 -
- R 8 and R 9 independently of one another represent hydrogen, halogen or C 1 -C 6 -alkyl
- alkyl may be substituted with a substituent, whereby the substituent is selected from the group consisting of C 3 -C 6 cycloalkyl, C 6 -C 0 aryl and 5- or 6-membered heteroaryl, - -
- cycloalkyl, aryl and heteroaryl may be substituted by 1 to 3 substituents, where the substituents are independently selected from the group consisting of halogen, hydroxy, oxo, nitro, cyano, trifluoromethyl, difluoromethyl, trifluoromethoxy, difluoromethoxy, monofluoromethoxy, Trifluoromethylthio, C 1 -C 6 -alkyl, QC 6 -
- Compounds of the invention are the compounds of formula (I) and their salts, solvates and solvates of the salts; the compounds of the formula (I) mentioned below of the formulas and their salts, solvates and solvates of the salts and the compounds of formula (I), hereinafter referred to as exemplary compounds and their salts, solvates and solvates of the salts, as far as the of formula (I), compounds mentioned below are not already salts, solvates and solvates of the salts.
- the compounds of the invention may exist in stereoisomeric forms (enantiomers, diastereomers).
- the invention therefore relates to the enantiomers or diastereomers and their respective mixtures. From such mixtures of enantiomers and / or diastereomers, the stereoisomerically uniform components can be isolated in a known manner.
- the present invention encompasses all tautomeric forms.
- Salts used in the context of the present invention are physiologically acceptable salts of the compounds according to the invention. However, also included are salts which are not suitable for pharmaceutical applications themselves but can be used, for example, for the isolation or purification of the compounds according to the invention.
- Physiologically acceptable salts of the compounds according to the invention include acid addition salts of mineral acids, carboxylic acids and sulfonic acids, for example salts of hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, benzenesulfonic acid, naphthalenedisulfonic acid, acetic acid, trifluoroacetic acid, propionic acid, lactic acid, tartaric acid, Malic acid, citric acid, fumaric acid, maleic acid and benzoic acid.
- Physiologically acceptable salts of the compounds according to the invention also include salts of customary bases, such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts) and ammonium salts derived from ammonia or organic amines having 1 to 16 carbon atoms, such as, by way of example and by way of illustration, ethylamine, diethylamine, triethylamine, ethyldiisopropylamine, monoethanolamine, diethanolamine, triethanolamine, dicyclohexylamine, dimethylaminoethanol, procaine, dibenzylamine, N-methylmorpholine, arginine, lysine, ethylenediamine and N-methylpiperidine.
- customary bases such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts
- solvates are those forms of the compounds according to the invention which form a complex in the solid or liquid state by coordination with solvent molecules. Hydrates are a special form of solvates that coordinate with water.
- the present invention also includes prodrugs of the compounds of the invention.
- prodrugs includes compounds which may themselves be biologically active or inactive, but which are converted during their residence time in the body into compounds of the invention (for example metabolically or hydrolytically).
- Alkylamino, alkoxycarbonyl and alkyl amino carbonyl stand for a linear or branched alkyl radical having generally 1 to 6 (,, Ci-C 6 - alkyl "), preferably 1 to 4, more preferably 1 to 3 carbon atoms, for example and preferably methyl , Ethyl, n-propyl, isopropyl, tert -butyl, n -pentyl and n -hexyl.
- Alkoxy is, by way of example and by way of preference, methoxy, ethoxy, n-propoxy, isopropoxy, tert-butoxy, n-pentoxy and n-hexoxy.
- Alkylamino represents an alkylamino radical having one or two (independently selected) alkyl substituents, by way of example and by preference methylamino, ethylamino, n-propylamino, isopropylamino, tert-butylamino, n-pentylamino, n-hexylamino, N, N Dimethylamino, N, N-
- C r C 3 -alkylamino is, for example, a monoalkylamino radical having 1 to 3 carbon atoms or a dialkylamino radical having in each case 1 to 3 carbon atoms per alkyl substituent.
- Alkoxycarbonyl is, by way of example and by way of preference, methoxycarbonyl, ethoxycarbonyl, n-propoxycarbonyl, isopropoxycarbonyl, tert-butoxycarbonyl, n-pentoxycarbonyl and n-hexoxycarbonyl.
- Alkylaminocarbonyl is an alkylaminocarbonyl radical having one or two (independently selected) alkyl substituents, by way of example and preferably methylaminocarbonyl, ethylaminocarbonyl, n-propylaminocarbonyl, isopropylaminocarbonyl, tert-butylaminocarbonyl, n-pentylaminocarbonyl, n-hexylaminocarbonyl, N, N Dimethylaminocarbonyl, N, N-diethylaminocarbonyl, N-ethyl-N-methylaminocarbonyl, N-methyl-Nn-propylaminocarbonyl, N-isopropyl
- Ci-Ca-alkylaminocarbonyl is for example a
- Monoalkylaminocarbonyl radical having 1 to 3 carbon atoms or for a dialkylaminocarbonyl radical having in each case 1 to 3 carbon atoms per alkyl substituent.
- Aryl is a mono- or bicyclic aromatic, carbocyclic radical of usually 6 to 10 carbon atoms; by way of example and preferably phenyl and ⁇ aphthyl.
- 5- or 6-membered heteroaryl is in the context of the invention in general an aromatic, monocyclic radical having 5 or 6 ring atoms and up to 4 heteroatoms from the series S, O and / or ⁇ .
- the heteroaryl radical may be bonded via a carbon or heteroatom. Examples which may be mentioned are: thienyl, furyl, pyrrolyl, thiazolyl, oxazolyl, pyrazolyl, imidazolyl, pyridyl, pyrimidyl, pyridazinyl.
- Cycloalkyl represents a cycloalkyl group having usually 3 to 6 carbon atoms, by way of example and preferably cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl.
- Halogen is fluorine, chlorine, bromine and iodine.
- R 1 is a group of the formula
- R 3 is phenyl or 5- or 6-membered heteroaryl
- phenyl and heteroaryl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from
- R 2 is phenyl
- phenyl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, hydroxy, trifluoromethyl, difluoromethyl, trifluoromethoxy, difluoromethoxy, monofluoromethoxy, trifluoromethylthio, C r C 6 alkyl and Ci-C 6- alkoxy,
- A is a group of the formula
- # represents the point of attachment to the nitrogen atom of the urea
- R 7 is C r C 6 -alkyl, where alkyl may be substituted with a substituent, whereby the substituent is selected from the group consisting of C 3 -C 6 cycloalkyl, C 6 -C 0 aryl and 5- or 6-membered heteroaryl,
- cycloalkyl, aryl and heteroaryl may be substituted by 1 to 3 substituents, where the substituents are independently selected from the group consisting of halogen, hydroxy, oxo, nitro, cyano, trifluoromethyl, difluoromethyl, trifluoromethoxy, difluoromethoxy, monofluoromethoxy, trifluoromethylthio, C 1 -C 6 -alkyl, QC 6 - alkoxy, hydroxycarbonyl, Ci-C 6 alkoxycarbonyl, amino, Ci-C 6 alkyl amino, aminocarbonyl, and Ci-C 6 alkylaminocarbonyl,
- R 8 and R 9 independently of one another represent hydrogen, halogen or C 1 -C 6 -alkyl
- R 1 is a group of the formula
- R 3 is phenyl or pyridyl
- phenyl and pyridyl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, nitro, cyano, trifluoromethyl, difluoromethyl, trifluoromethoxy, difluoromethoxy, monofluoromethoxy, Ci-C 4 alkyl and C 1 -C 4 -alkoxy,
- R 2 is phenyl, wherein phenyl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of fluorine, chlorine, trifluoromethoxy, difluoromethoxy, trifluoromethylthio and methyl,
- A is a group of the formula
- # represents the point of attachment to the nitrogen atom of the urea
- R 7 is methyl, ethyl or n-butyl
- methyl, ethyl and n-butyl may be substituted with a substituent, wherein the substituent is selected from the group consisting of cyclopropyl and phenyl,
- R 8 and R 9 independently of one another represent hydrogen, bromine, chlorine, methyl or ethyl
- R 3 is phenyl or pyridyl
- phenyl and pyridyl may be substituted with 1 to 3 substituents, wherein the substituents are independently selected from the group consisting of halogen, nitro, cyano, trifluoromethyl, difluoromethyl, trifluoromethoxy, difluoromethoxy, monofluoromethoxy, Ci-C 4 alkyl and QC 4 -alkoxy.
- R 2 is phenyl
- phenyl may be substituted by 1 to 3 substituents, where the substituents are independently selected from the group consisting of fluorine, chlorine , Trifluoromethoxy, difluoromethoxy, trifluoromethylthio and methyl.
- R 7 is methyl, ethyl or n-butyl
- methyl, ethyl and n-butyl may be substituted with a substituent, wherein the substituent is selected from the group consisting of cyclopropyl and phenyl,
- R 8 is hydrogen, bromine, chlorine or methyl
- R 9 is hydrogen
- the invention further provides a process for the preparation of the compounds of the formula (I), wherein
- R 1 has the meaning given above
- R 2 has the meaning given above
- R 2 has the meaning given above
- R 2 has the meaning given above, and
- R 10 is methyl or ethyl
- R 1 has the meaning given above
- the reaction is generally carried out in inert solvents, preferably in a temperature range from 0 ° C. to the reflux of the solvents under atmospheric pressure to 3 bar.
- Reducing agents are, for example, palladium on activated carbon and hydrogen, formic acid / triethylamine / palladium on activated carbon, zinc, zinc / hydrochloric acid, iron, iron / hydrochloric acid, iron (II) sulfate / hydrochloric acid, sodium sulfide, sodium disulfide, sodium dithionite, ammonium polysulfide, Sodium borohydride / nickel chloride, tin dichloride, titanium trichloride or Raney nickel and aqueous hydrazine solution, preferably Raney nickel and aqueous hydrazine solution, palladium on activated carbon and hydrogen or formic acid / triethylamine / palladium on activated carbon.
- Inert solvents are, for example, ethers, such as diethyl ether, methyl tert-butyl ether, 1,2-dimethoxyethane, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, alcohols, such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert .-Butanol, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide, dimethylacetamide, acetonitrile or pyridine, in the case of water-miscible solvents, mixtures thereof with water, as the solvent is preferably methanol, ethanol, iso -Propanol or in the case of Raney nickel and aqueous hydrazine solution tetrahydro
- the reaction is generally carried out in inert solvents, preferably in a temperature range from room temperature to 40 ° C at atmospheric pressure.
- Carbonic acid derivatives are, for example, N, N-carbonyldiimidazole, phosgene, diphosgene, triphosgene, phenyl chloroformate or 4-nitrophenyl chloroformate, preferably N, N-carbonyldiimidazole.
- Inert solvents are, for example, halogenated hydrocarbons, such as methylene chloride, trichloromethane, tetrachloromethane, trichloroethane, tetrachloroethane, 1,2-dichloroethane or trichlorethylene, ethers, such as diethyl ether, methyl tert-butyl ether, 1,2-dimethoxyethane, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether , Hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as ethyl acetate, acetone, dimethylformamide, dimethylacetamide, 2-butanone, dimethyl sulfoxide, acetonitrile or pyridine, in the case of water-miscible solvents, mixture
- the reaction is generally carried out in inert solvents, if appropriate in the presence of a base, preferably in a temperature range from room temperature to reflux of the solvents under atmospheric pressure.
- Inert solvents are, for example, halogenated hydrocarbons such as methylene chloride, trichloromethane, tetrachloromethane, trichloroethane, tetrachloroethane, 1, 2-dichloroethane or Trichlorethylene, ethers such as diethyl ether, methyl tert-butyl ether, 1, 2-dimethoxyethane, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as ethyl acetate, acetone, dimethylformamide , Dimethylacetamide, 2-butanone, dimethyl sulfoxide, acetonitrile or pyridine, preferably tetrahydrofuran or methylene chloride.
- bases examples include alkali metal carbonates such as cesium carbonate, sodium or potassium carbonate, or potassium tert-butoxide, or other bases such as sodium hydride, DBU, triethylamine or diisopropylethylamine, preferably triethylamine.
- the reaction is generally carried out in inert solvents, preferably in a temperature range from 0 ° C. to the reflux of the solvents under normal pressure.
- bases are alkali metal hydroxides such as sodium, lithium or potassium hydroxide, or alkali metal carbonates such as cesium carbonate, sodium or potassium carbonate, preferably sodium hydroxide.
- Inert solvents are, for example, halogenated hydrocarbons such as methylene chloride, trichloromethane, tetrachloromethane, trichloroethane, tetrachloroethane, 1, 2-dichloroethane or trichlorethylene, ethers such as diethyl ether, methyl tert-butyl ether, 1,2-dimethoxyethane, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert-butanol, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide, dimethylacetamide, dimethylsulfoxide, aceton
- the reaction is generally carried out in inert solvents, if appropriate in the presence of a base, preferably in a temperature range from -70 0 C to 40 0 C at atmospheric pressure.
- dehydrating reagents include carbodiimides such as N, N'-diethyl, N, N, '- dipropyl, N, N'-diisopropyl, N, N'-dicyclohexylcarbodiimide, N- (3-dimethylaminoisopropyl) N'-ethylcarbodiimide hydrochloride (EDC), N-cyclohexylcarbodiimide-N'-propyl-oxymethyl-polystyrene (PS-carbodiimide) or carbonyl compounds such as carbonyldiimidazole, or 1,2-oxazolium compounds such as 2-ethyl-5-phenyl-1, 2-oxazolium-3-sulfate or 2-tert-butyl-5-methylisoxazolium perchlorate, or acylamino compounds such as 2-ethoxy-1-ethoxycarbonyl-1, 2 dihydroquinoline, or propanephosphonic an
- Bases are, for example, alkali carbonates, e.g. Sodium or potassium carbonate, or hydrogen carbonate, or organic bases such as trialkylamines e.g. Triethylamine, N-methylmorpholine, N-methylpiperidine, 4-dimethylaminopyridine (DMAP) or diisopropylethylamine, or DBU, DBN, pyridine, preferred is triethylamine.
- alkali carbonates e.g. Sodium or potassium carbonate
- hydrogen carbonate e.g. Sodium or potassium carbonate
- organic bases such as trialkylamines e.g. Triethylamine, N-methylmorpholine, N-methylpiperidine, 4-dimethylaminopyridine (DMAP) or diisopropylethylamine, or DBU, DBN, pyridine, preferred is triethylamine.
- DMAP 4-dimethylaminopyridine
- DBU diisopropylethylamine
- the condensation is carried out with TBTU and DMAP.
- Inert solvents are, for example, halogenated hydrocarbons such as methylene chloride, trichloromethane, tetrachloromethane, trichloroethane, tetrachloroethane, 1,2-dichloroethane or trichlorethylene, ethers such as diethyl ether, methyl tert-butyl ether, 1,2-dimethoxyethane, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as ethyl acetate, acetone, dimethylformamide, dimethylacetamide, 2-butanone, dimethylsulfoxide, acetonitrile or pyridine, in the case of water-miscible solvents, mixtures thereof with water
- the carboxylic acids obtained from the first stage of the process [C] in the second stage may first be converted with a chlorinating reagent such as thionyl chloride to the carboxylic acid chloride and then with compounds of the formula (VI) in the presence of a base to give compounds of the formula (I) be implemented.
- a chlorinating reagent such as thionyl chloride
- the compounds of formula (E) are known or can be prepared by reacting compounds of the formula
- R 10 has the meaning given above, - 1 -
- R 10 has the abovementioned meaning
- the compounds of the formula (VIII) are known or can be synthesized by known processes from the corresponding starting materials.
- the compounds of the formula (V) are known or can be prepared by reacting compounds of the formula (VII) in the first stage with a reducing agent and in the second stage in the presence of a carbonic acid derivative with compounds of the formula (IH) or in the second stage Compounds of formula (IV) are reacted.
- reaction is carried out as described in methods [A] and [B].
- the compounds of the general formula (I) according to the invention show an unforeseeable, surprising activity spectrum. They show an antiviral activity against representatives of the group of herpes viridae (herpesviruses), especially against cytomegaloviruses (CMV), in particular against the human cytomegalovirus (HCMV). They are thus suitable for the treatment and prophylaxis of diseases, especially infections with viruses, in particular the viruses mentioned above, and the infectious diseases caused thereby.
- a virus infection is understood below to mean both an infection with a virus and an infection caused by a virus.
- the compounds of the general formula (I) can be used because of their special properties for the preparation of medicaments which are suitable for the prophylaxis and / or treatment of diseases, in particular viral infections.
- HCMV infections Treatment and prophylaxis of HCMV infections in AIDS patients (retinitis, pneumonitis, gastrointestinal infections).
- the compounds according to the invention are preferably used for the preparation of medicaments which are suitable for the prophylaxis and / or treatment of infections with a member of the group of herpes viridae, in particular a cytomegalovirus, in particular the human cytomegalovirus.
- the compounds according to the invention can be used alone and, if required, also in combination with other active substances, in particular antiviral agents - -
- Agents such as ganciclovir, valganciclovir or acyclovir, for the treatment and / or prevention of viral infections, in particular of HCMV infections used.
- the present invention further provides for the use of the compounds according to the invention for the treatment and / or prophylaxis of diseases, preferably viral infections, in particular infections with the human cytomegalovirus (HCMV) or another member of the group of herpes viridae.
- diseases preferably viral infections, in particular infections with the human cytomegalovirus (HCMV) or another member of the group of herpes viridae.
- HCMV human cytomegalovirus
- Another object of the present invention is the use of the compounds of the invention for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases.
- Another object of the present invention is the use of the compounds of the invention for the manufacture of a medicament for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases.
- Another object of the present invention is a method for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases, using an antivirally effective amount of the compounds of the invention.
- the compounds according to the invention can act systemically and / or locally.
- they may be applied in a suitable manner, e.g. oral, parenteral, pulmonary, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctival, otic or as an implant or stent.
- the compounds according to the invention can be administered in suitable administration forms.
- parenteral administration can be done bypassing a resorption step (eg, intravenous, intraarterial, intracardiac, intraspinal, or intralumbar) or with involvement of resorption (eg, intramuscular, subcutaneous, intracutaneous, percutaneous, or intraperitoneal).
- a resorption step eg, intravenous, intraarterial, intracardiac, intraspinal, or intralumbar
- suitable application forms include injection and infusion treatment in the form of solutions, suspensions, emulsions, lyophilisates or sterile powders.
- Inhalation medicines including powder inhalers, nebulizers
- nasal drops solutions, sprays
- lingual, sublingual or buccal tablets films / wafers or capsules
- suppositories ear or ophthalmic preparations
- vaginal capsules aqueous suspensions (lotions, shake mixtures)
- lipophilic suspensions ointments, creams, transdermal therapeutic systems, milk, pastes , Foams, scattering powders, implants or stents.
- the compounds according to the invention can be converted into the stated administration forms. This can be done in a conventional manner by mixing with inert, non-toxic, pharmaceutically suitable excipients.
- excipients for example microcrystalline cellulose, lactose, mannitol
- solvents for example liquid polyethylene glycols
- emulsifiers and dispersants or wetting agents for example sodium dodecyl sulfate, polyoxysorbitanoleate
- binders for example polyvinylpyrrolidone
- synthetic and natural polymers for example albumin
- Stabilizers eg antioxidants such as ascorbic acid
- dyes eg inorganic pigments such as iron oxides
- flavor and / or odoriferous include, among others.
- Excipients for example microcrystalline cellulose, lactose, mannitol
- solvents for example liquid polyethylene glycols
- emulsifiers and dispersants or wetting agents for example sodium dodecyl
- compositions containing at least one compound of the invention usually together with one or more inert, non-toxic, pharmaceutically suitable excipients, and their use for the purposes mentioned above.
- Method 1 Device Type MS: Micromass ZQ; Device type HPLC: Waters Alliance 2795; Column: Phenomenex Synergi 2 ⁇ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 l water + 0.5 mL 50% formic acid, eluent B: 1 l acetonitrile + 0.5 mL 50% formic acid; Gradient: 0.0 min 90% A -> 2.5 min 30% A - »3.0 min 5% A -> 4.5 min 5% A; Flow: 0.0 min 1 mL / min, 2.5 min / 3.0 min / 4.5 min 2 mL / min; Oven: 50 ° C .; UV detection: 210 nm.
- Method 2 Instrument: Micromass Quattro LCZ with HPLC Agilent Series 1100; Column: Phenomenex Synergi 2 ⁇ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 l water + 0.5 mL 50% formic acid, eluent B: 1 l acetonitrile + 0.5 mL 50% formic acid; Gradient: 0.0 min 90% A -> 2.5 min 30% A -> 3.0 min 5% A - »4.5 min 5% A; Flow: 0.0 min 1 mL / min, 2.5 min / 3.0 min / 4.5 min 2 mL / min; Oven: 5O 0 C; UV detection: 208-400 nm.
- the preparation is analogous to Example 5A.
- the preparation is analogous to Example 5A.
- Example 4A and 5 A The preparation is analogous to Example 4A and 5 A starting from 4-aminothiophene-2-carboxylic acid methyl ester (synthesized according to A. A. Kiryano et al., Tetrahedron Lett. 2001, (42), 8797-8800).
- Example 4A and 5 A The preparation is analogous to Example 4A and 5 A starting from 2-amino-l, 3-thiazol-4-carboxylic acid ethyl ester (commercially available from ACROS).
- Example 4A and 5 A The preparation is carried out analogously to Example 4A and 5 A starting from 2-amino-5-methyl-l, 3-thiazole-4-carboxylic acid methyl ester (commercially available from Tyger Scientific).
- Example 4 A and 5 A The preparation is carried out analogously to Example 4 A and 5 A starting from ethyl 2-amino-5-chloro-1,3-thiazole-4-carboxylate (synthesis described in KJ Hodgetts et al., Org. Lett. 2002, (4) , 1363-1366).
- Example 4 A and 5 A The preparation is carried out analogously to Example 4 A and 5 A starting from 2-amino-5-bromo-l, 3-thiazol-4-carboxylic acid ethyl ester (synthesis described in JF Okonya et al., Tetrahedron Lett., 2002, (43) , 7051-7054).
- Example 5A 50 mg (0.15 mmol) of 1-methyl-4 - [( ⁇ [4- (trifluoromethoxy) phenyl] amino ⁇ carbonyl) amino] -H-pyrrole-2-carboxylic acid (Example 5A) are initially charged in 1 ml of DMF and washed with 56 mg (0.18 mmol) of O- (benzotriazol-1-yl) -N, N, N ', N'-tetramethyluronium tetrafluoroborate (TBTU) and 8.9 mg (0.07 mmol) of 4- (dimethylamino) -pyridine (DMAP).
- DMAP 4- (dimethylamino) -pyridine
- the preparation is carried out analogously to Example 1 from Example 5A and 6A.
- the preparation is analogous to Example 1 from Example 7A and 6A.
- the preparation is analogous to Example 1 from Example 7A and 6A.
- the preparation is carried out analogously to Example 1 from Example 8A.
- the preparation is analogous to Example 1 from Example 8A and 6A.
- the preparation is carried out analogously to Example 1 from Example 9A.
- the preparation is carried out analogously to Example 1 from Example 9A and 6A.
- the preparation is analogous to Example 1 from Example 10A and 6A.
- test compounds are used as 50 millimolar (mM) solutions in dimethylsulfoxide (DMSO).
- DMSO dimethylsulfoxide
- Ganciclovir, foscarnet and cidofovir serve as reference compounds.
- 2 .mu.l each of the 50, 5, 0.5 and 0.05 mM DMSO stock solutions of 98 .mu.l cell culture medium in the series 2 AH in duplicate, 1: 2 dilutions with 50 ul medium to 11 row of 96-well Plate.
- the wells in rows 1 and 12 each contain 50 ⁇ l medium.
- ⁇ l of a suspension of 1 ⁇ 10 4 cells human foreskin fibroblasts [NHDF]
- row 1 cell control
- MOI 0.001 - 0.002
- the series 12 (without substance) serves as a virus control.
- the final test concentrations are 250 - 0.0005 ⁇ M.
- the plates are incubated for 6 days at 37 ° C./5% CO 2 , ie until all cells are infected in the virus controls (100% cytopathogenic effect [CPE]).
- the wells are then fixed and stained by adding a mixture of formalin and Giemsa's dye (30 minutes), with double-distilled water. washed and dried in a drying oven at 5O 0 C. Thereafter, the plates are visually evaluated with an overhead microscope (plaque multiplier the company Technomara).
- CC 50 (NHDF) maximum substance concentration in ⁇ M, in which no visible cytostatic effects on the cells are recognizable in comparison to the untreated cell control;
- EC 50 substance concentration in ⁇ M, which inhibits the CPE (cytopathic effect) by 50% compared to the untreated virus control;
- SI (selectivity index) CC 50 (NHDF) / EC 50 (HCMV).
- mice 3-4 week old female immunodeficient mice (16-18 g), Fox Chase SCDD or Fox Chase SCID-NOD or SCID-beige are purchased from commercial breeders (Bomholtgaard, Jackson). The animals are kept in isolators under sterile conditions (including litter and food).
- HCMV Human cytomegalovirus
- MOI multiplicity of infection
- FCS fetal calf serum
- DMSO fetal calf serum
- 5 ng / ⁇ l basic fibroblast growth factor (bFGF) in 25 ⁇ l PBS / 0.1% BSA / 1 mM DTT are applied to the infected sponges after 12-13 hours and incubated for 1 hour.
- bFGF basic fibroblast growth factor
- the immuno-deficient mice are anesthetized with avertin or a mixture of azepromazine xylazine and ketamine, the back coat is removed with the help of a dry shaver, the epidermis is opened 1-2 cm, relieved and the wet sponges are transplanted under the dorsal skin. The surgical wound is closed with tissue glue. Twenty-four hours after transplantation, the mice are treated orally for a period of 8 days three times a day (7:00 am and 2:00 pm and 7:00 pm), twice a day (8:00 am and 5:00 pm), or once daily (2:00 pm). The dose is 3 or 10 or 30 or 100 mg / kg body weight, the application volume 10 mL / kg body weight.
- the formulation of the substances takes place in the form of a 0.5% Tylose suspension optionally with 2% DMSO. 9 days after transplantation and 16 hours after the last administration of the substance, the animals are killed without pain and the sponge removed.
- the virus-infected cells are released by collagenase digestion (330 U / 1.5 mL) from the sponge and stored in the presence of MEM, 10% fetal calf serum, 10% DMSO at -140 0 C.
- the evaluation is carried out after serial dilution of the virus-infected cells in steps of ten by titer determination on 24-well plates of confluent NHDF cells after vital staining with neutral red or after fixation and staining with a formalin-Giemsa mixture (as described above). The number of infectious virus particles after substance treatment is compared to the placebo-treated control group.
- the compounds according to the invention can be converted into pharmaceutical preparations as follows:
- Example 1 100 mg of the compound of Example 1, 50 mg of lactose (monohydrate), 50 mg of corn starch (native), 10 mg of polyvinylpyrrolidone (PVP 25) (BASF, Ludwigshafen, Germany) and 2 mg of magnesium stearate.
- the mixture of active ingredient, lactose and starch is granulated with a 5% solution (m / m) of the PVP in water.
- This mixture is compressed with a conventional tablet press (for the tablet format see above).
- a pressing force of 15 kN is used as a guideline for the compression.
- a single dose of 100 mg of the compound according to the invention corresponds to 10 ml of oral suspension.
- the rhodigel is suspended in ethanol, the active ingredient is added to the suspension. While stirring, the addition of water takes place. Until the swelling of the Rhodigels swirling is about 6 h stirred.
- Intravenously administrable solution
- the compound according to the invention is dissolved in the water together with polyethylene glycol 400 while stirring.
- the solution is sterile filtered (pore diameter 0.22 microns) and filled under aseptic conditions in heat sterilized infusion bottles. These are closed with infusion stoppers and crimp caps.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Virology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Pyrrole Compounds (AREA)
Abstract
Description




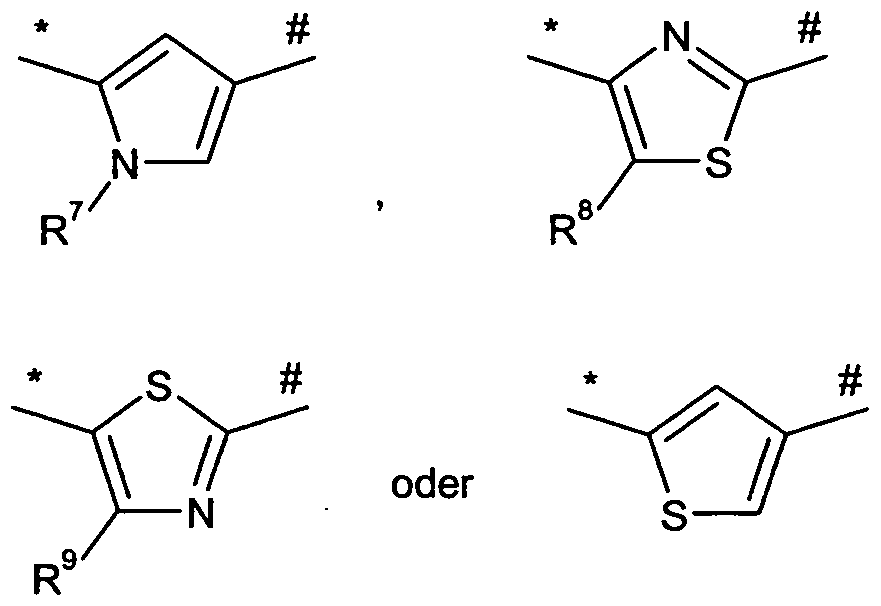













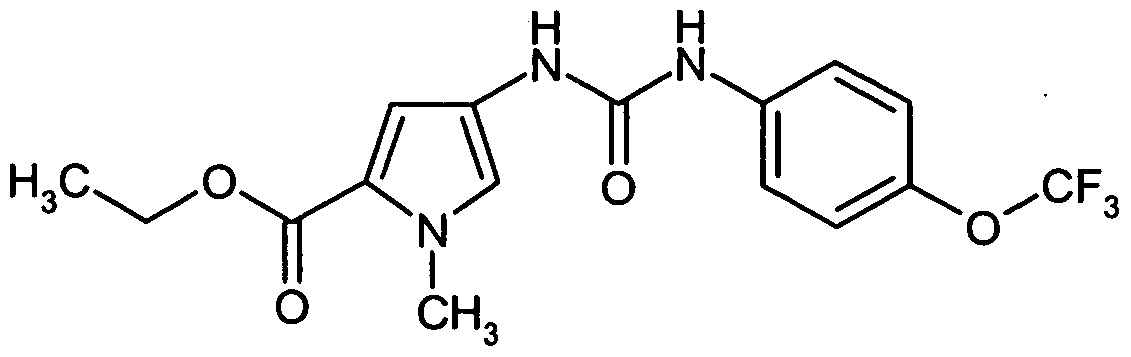


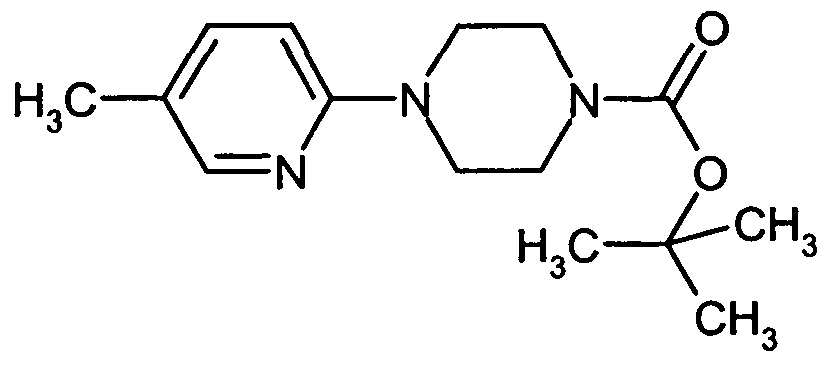



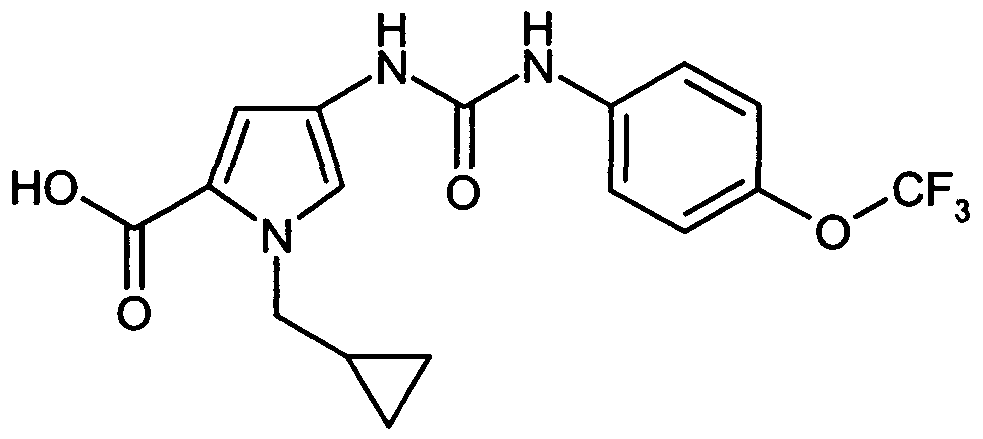

















Claims









Priority Applications (14)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2006800220467A CN101277946B (zh) | 2005-07-15 | 2006-07-01 | 杂环基酰胺-取代的噻唑,吡咯和噻吩 |
| MX2008000685A MX2008000685A (es) | 2005-07-15 | 2006-07-01 | Tiazoles, pirroles y tiofenos sustituidos con heterociclilamidas. |
| ES06754657.2T ES2518966T3 (es) | 2005-07-15 | 2006-07-01 | Tiazoles, pirroles y tiofenos sustituidos con heterociclilamida |
| AU2006272059A AU2006272059B2 (en) | 2005-07-15 | 2006-07-01 | Heterocyclylamide-substituted thiazoles, pyrroles and thiophenes |
| BRPI0615550-2A BRPI0615550A2 (pt) | 2005-07-15 | 2006-07-01 | tiazóis, pirróis e tiofenos substituìdos com heterociclilamida |
| KR1020087003556A KR101352646B1 (ko) | 2005-07-15 | 2006-07-01 | 헤테로시클릴아미드-치환된 티아졸, 피롤 및 티오펜 |
| EP06754657.2A EP1910330B1 (de) | 2005-07-15 | 2006-07-01 | Heterocyclylamid-substituierte thiazole, pyrrole und thiophene |
| JP2008520748A JP5140582B2 (ja) | 2005-07-15 | 2006-07-01 | ヘテロシクリルアミド置換チアゾール、ピロールおよびチオフェン |
| CA2615049A CA2615049C (en) | 2005-07-15 | 2006-07-01 | Heterocyclylamide-substituted thiazoles, pyrroles and thiophenes |
| US11/988,911 US8410090B2 (en) | 2005-07-15 | 2006-07-01 | Heterocyclylamide-substituted thiazoles, pyrroles and thiophenes |
| NZ564562A NZ564562A (en) | 2005-07-15 | 2006-07-01 | Heterocyclylamide-substituted thiazoles, pyrroles and thiophenes |
| IL188785A IL188785A (en) | 2005-07-15 | 2008-01-15 | Heterocyclylamide-substituted thiazoles, pyrroles and thiophenes, medicaments comprising them and use thereof for treating viral infection |
| NO20080740A NO340507B1 (no) | 2005-07-15 | 2008-02-11 | Heterocyklylamidsubstituerte tiazoler, pyrroler og tiofener |
| HK08111248.9A HK1115385A1 (en) | 2005-07-15 | 2008-10-10 | Heterocyclylamide substituted thiazoles, pyrroles and thiophenes |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102005033103.3 | 2005-07-15 | ||
| DE102005033103A DE102005033103A1 (de) | 2005-07-15 | 2005-07-15 | Heterocyclylamid-substituierte Thiazole, Pyrrole und Thiophene |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007009578A1 true WO2007009578A1 (de) | 2007-01-25 |
Family
ID=37056618
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/006434 WO2007009578A1 (de) | 2005-07-15 | 2006-07-01 | Heterocyclylamid-substituierte thiazole, pyrrole und thiophene |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US8410090B2 (de) |
| EP (1) | EP1910330B1 (de) |
| JP (1) | JP5140582B2 (de) |
| KR (1) | KR101352646B1 (de) |
| CN (1) | CN101277946B (de) |
| AU (1) | AU2006272059B2 (de) |
| BR (1) | BRPI0615550A2 (de) |
| CA (1) | CA2615049C (de) |
| DE (1) | DE102005033103A1 (de) |
| ES (1) | ES2518966T3 (de) |
| HK (1) | HK1115385A1 (de) |
| IL (1) | IL188785A (de) |
| MX (1) | MX2008000685A (de) |
| NO (1) | NO340507B1 (de) |
| NZ (1) | NZ564562A (de) |
| RU (1) | RU2425829C2 (de) |
| WO (1) | WO2007009578A1 (de) |
| ZA (1) | ZA200801149B (de) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016077232A2 (en) | 2014-11-10 | 2016-05-19 | Forge Life Science, Llc | Anti-hcmv compositions and methods |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102011113749A1 (de) * | 2011-09-14 | 2013-03-14 | Aicuris Gmbh & Co. Kg | Sulfonsäuresalze Heterocyclylamid-substituiertr Imidazole |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004002481A1 (en) * | 2002-06-27 | 2004-01-08 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| WO2004052852A1 (de) * | 2002-12-09 | 2004-06-24 | Bayer Healthcare Ag | 3-pyrrolyl-harnstoff-derivate und ihre verwendung als antivirale mittel |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE69217224T2 (de) * | 1991-07-03 | 1997-06-05 | Upjohn Co | Substituierte indole als anti-aids pharmaceutische zubereitungen |
| AU1367599A (en) | 1997-11-03 | 1999-05-24 | Boehringer Ingelheim Pharmaceuticals, Inc. | Aromatic heterocyclic compounds as anti-inflammatory agents |
| CA2366607A1 (en) * | 1999-03-08 | 2000-09-14 | Bayer Aktiengesellschaft | Thiazolyl urea derivatives and their utilization as antiviral agents |
| DE10131133A1 (de) | 2001-06-28 | 2003-01-16 | Bayer Ag | Pyridazinone |
| KR100574547B1 (ko) * | 2003-03-21 | 2006-04-27 | 율촌화학 주식회사 | 내수성이 개선된 생분해성 조성물 및 그 제조방법 |
| US8106048B2 (en) * | 2004-09-20 | 2012-01-31 | 4Sc Ag | Heterocyclic NF-κB inhibitors |
| EP1637529A1 (de) * | 2004-09-20 | 2006-03-22 | 4Sc Ag | Neue Piperidin-4-yl-Thiazol-Carboxamid-Analog als Inhibitoren für die Vermehrung von T-Zellen und ihre Verwendung |
-
2005
- 2005-07-15 DE DE102005033103A patent/DE102005033103A1/de not_active Ceased
-
2006
- 2006-07-01 CN CN2006800220467A patent/CN101277946B/zh not_active Expired - Fee Related
- 2006-07-01 EP EP06754657.2A patent/EP1910330B1/de not_active Not-in-force
- 2006-07-01 JP JP2008520748A patent/JP5140582B2/ja not_active Expired - Fee Related
- 2006-07-01 WO PCT/EP2006/006434 patent/WO2007009578A1/de active Application Filing
- 2006-07-01 MX MX2008000685A patent/MX2008000685A/es active IP Right Grant
- 2006-07-01 KR KR1020087003556A patent/KR101352646B1/ko active IP Right Grant
- 2006-07-01 ZA ZA200801149A patent/ZA200801149B/xx unknown
- 2006-07-01 AU AU2006272059A patent/AU2006272059B2/en not_active Ceased
- 2006-07-01 ES ES06754657.2T patent/ES2518966T3/es active Active
- 2006-07-01 US US11/988,911 patent/US8410090B2/en active Active
- 2006-07-01 BR BRPI0615550-2A patent/BRPI0615550A2/pt not_active Application Discontinuation
- 2006-07-01 CA CA2615049A patent/CA2615049C/en not_active Expired - Fee Related
- 2006-07-01 NZ NZ564562A patent/NZ564562A/en unknown
- 2006-07-01 RU RU2008105353/04A patent/RU2425829C2/ru active
-
2008
- 2008-01-15 IL IL188785A patent/IL188785A/en active IP Right Grant
- 2008-02-11 NO NO20080740A patent/NO340507B1/no not_active IP Right Cessation
- 2008-10-10 HK HK08111248.9A patent/HK1115385A1/xx not_active IP Right Cessation
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004002481A1 (en) * | 2002-06-27 | 2004-01-08 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| WO2004052852A1 (de) * | 2002-12-09 | 2004-06-24 | Bayer Healthcare Ag | 3-pyrrolyl-harnstoff-derivate und ihre verwendung als antivirale mittel |
Non-Patent Citations (1)
| Title |
|---|
| BURAK K ET AL: "SYNTHESIS OF ISOTHIAZOLE DERIVATIVES WITH POTENTIAL BIOLOGICAL ACTIVITY", PHARMAZIE, DIE, GOVI VERLAG, ESCHBORN, DE, vol. 47, no. 7, 1992, pages 492 - 495, XP000651576, ISSN: 0031-7144 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016077232A2 (en) | 2014-11-10 | 2016-05-19 | Forge Life Science, Llc | Anti-hcmv compositions and methods |
| EP3218373A4 (de) * | 2014-11-10 | 2018-06-13 | Forge Life Science, LLC | Anti-hcmv-zusammensetzungen und -verfahren |
| US10556894B2 (en) | 2014-11-10 | 2020-02-11 | Evrys Bio, Llc | Anti-HCMV compositions and methods |
Also Published As
| Publication number | Publication date |
|---|---|
| ZA200801149B (en) | 2009-07-29 |
| JP2009501170A (ja) | 2009-01-15 |
| KR101352646B1 (ko) | 2014-01-16 |
| CN101277946A (zh) | 2008-10-01 |
| EP1910330B1 (de) | 2014-07-23 |
| AU2006272059A1 (en) | 2007-01-25 |
| BRPI0615550A2 (pt) | 2011-05-24 |
| CA2615049A1 (en) | 2007-01-25 |
| NZ564562A (en) | 2010-12-24 |
| US8410090B2 (en) | 2013-04-02 |
| IL188785A0 (en) | 2008-11-03 |
| EP1910330A1 (de) | 2008-04-16 |
| CA2615049C (en) | 2013-12-03 |
| HK1115385A1 (en) | 2008-11-28 |
| MX2008000685A (es) | 2008-03-18 |
| DE102005033103A1 (de) | 2007-01-25 |
| KR20080091074A (ko) | 2008-10-09 |
| AU2006272059B2 (en) | 2012-05-17 |
| CN101277946B (zh) | 2013-01-02 |
| RU2425829C2 (ru) | 2011-08-10 |
| US20100144752A1 (en) | 2010-06-10 |
| IL188785A (en) | 2012-08-30 |
| NO20080740L (no) | 2008-02-11 |
| JP5140582B2 (ja) | 2013-02-06 |
| NO340507B1 (no) | 2017-05-02 |
| RU2008105353A (ru) | 2009-08-20 |
| ES2518966T3 (es) | 2014-11-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1853582B1 (de) | Heterocyclylamid-substituierte imidazole | |
| WO2004052852A1 (de) | 3-pyrrolyl-harnstoff-derivate und ihre verwendung als antivirale mittel | |
| WO2005087740A1 (de) | Fluorenone 1, 4,-dihydrpyridinderivate | |
| EP1910330B1 (de) | Heterocyclylamid-substituierte thiazole, pyrrole und thiophene | |
| EP1732901B1 (de) | 4-aminocarbonylamino-substitutierte imidazolverbindungen mit antiviraler wirkung | |
| DE102011113749A1 (de) | Sulfonsäuresalze Heterocyclylamid-substituiertr Imidazole | |
| EP1685123B1 (de) | Substituierte dihydrochinazoline ii | |
| DE10203086A1 (de) | 5-Ring Heterozyklen |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680022046.7 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006272059 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 564562 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 113/KOLNP/2008 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2006272059 Country of ref document: AU Date of ref document: 20060701 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006754657 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006272059 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12008500093 Country of ref document: PH Ref document number: 2615049 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/000685 Country of ref document: MX Ref document number: 188785 Country of ref document: IL Ref document number: 2008520748 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087003556 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008105353 Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006754657 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11988911 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0615550 Country of ref document: BR Kind code of ref document: A2 Effective date: 20080115 |