抗癌性を示す化合物およびその中間体ならびにそれらの製造方法 技術分野
[0001] 本発明は、 2 ァセチルー 2,3 ジヒドロ一 5 ヒドロキシナフト [2, 3— b]フラン一 4 , 9ージオンおよびその製造方法、ならびに該化合物力 抗癌活性を有する 2— (1 ーヒドロキシェチル)ー5 ヒドロキシナフト [2, 3—b]フラン 4, 9ージオンを製造す る方法に関する。本発明はさらに、 2 - (1—ヒドロキシェチル) 5 ヒドロキシナフト[ 2, 3— b]フラン 4, 9ージオンのラセミ体または α体を有効成分とする、抗癌剤に 関する。
背景技術
[0002] 下記式:
[化 1]
で示される 2—(1ーヒドロキシェチル)ー5 ヒドロキシナフト [2, 3—b]フラン 4, 9 —ジオンは、ノウゼンカズラ科の植物、タヒボ(Tabebuia avellanedae Lorentz ex Grise b)に含まれる光学活性ィ匕合物であり、この化合物は β体であって、優れた抗癌作用 を有することが知られている(たとえば、特許文献 1参照)。し力しながら、 2- (1—ヒド ロキシェチル) 5 ヒドロキシナフト [2, 3—b]フラン 4, 9ージオンの入手方法は 当該植物からの抽出以外に知られておらず、当該植物の希少価値が高いこと、およ び収率が 0. 05%と極めて低いものあること (たとえば、非特許文献 1参照)により、 2 — (1 ヒドロキシェチル) 5 ヒドロキシナフト [2, 3—b]フラン一 4, 9 ジオンの医 薬としての使用は充分ではな 、。
特許文献 1:特許第 2669762号明細書
非特許文献 1 :ゥエダ'シンイチら(Shinichi Ueda et al)、「ファイトケミストリー(Phytoch
emistry)」、 1994年、第 36卷、第 2号、 p323— 325
発明の開示
発明が解決しょうとする課題
[0003] したがって、医薬として有用な 2—(1ーヒドロキシェチル)ー5 ヒドロキシナフト [2, 3— b]フラン 4, 9ージオンを安価かつ簡便に製造することが望まれる。
課題を解決するための手段
[0004] 本発明者らは、 5 ヒドロキシナフタレン一 1,4 ジオン (別名:ュグロン)が化学品と して比較的安価に入手可能であることに着目し、これを出発物質として 2—(1ーヒドロ キシェチル) 5 ヒドロキシナフト [2, 3— b]フラン 4, 9ージオンを安価かつ簡便 に、高収率で合成できることを見いだし、この知見に基づいて本発明を完成するに至 つた。すなわち、本発明は、ュグロン力 簡便に誘導できる 2 ァセチルー 5 ヒドロ キシナフト [2, 3—b]フラン 4, 9ージオンを中間体として経由する 2—(1ーヒドロキ シェチル)ー5 ヒドロキシナフト [2, 3—b]フラン 4, 9ージオンの製造方法に関す る。本発明者らはまた、このように製造された 2— (1—ヒドロキシェチル) 5 ヒドロ キシナフト [2, 3—b]フラン 4, 9ージオンがラセミ体の形で得られること、それらは 常法により光学分割することにより OC体と β体に分離できること、さらに該ラセミ体およ び a体が j8体に比し、安全性に優れることを知り、本発明を完成するに至った。 発明の効果
[0005] 本発明により、安価かつ簡便な 2—(1ーヒドロキシェチル) 5 ヒドロキシナフト [2 , 3—b]フラン一 4, 9 ジオンの製造方法が提供される。また、植物力 抽出される 2 — (1 ヒドロキシェチル) 5 ヒドロキシナフト [2, 3—b]フラン一 4, 9 ジオンは j8 体であるのに対して、本発明の製造方法はラセミ混合物を提供することが可能である 。さらに、当該ラセミ混合物のキラル分離、たとえばキラルカラムクロマトグラフィーで の分取または光学分割などにより、植物からは得ることのできない j8体のェナンチォ マー(以後、本明細書にぉ 、て「 OC体」と 、う)を得ることも可能である。
発明を実施するための最良の形態
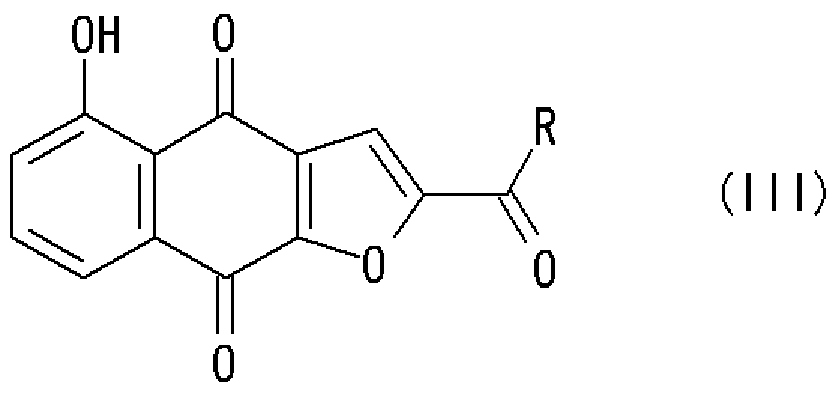
[0006] 第一の態様において、本発明は、式 (I):
[化 2]
で示される化合物を、式 (n) :
〔式中、 Rは C— Cアルキルであり、 X1および X2はそれぞれ独立してハロゲンである
1 6
]
で示される化合物と塩基の存在下で反応させることを特徴とする、式 (III):
[化 4]
〔式中、 Rは前記と同意義である〕
で示される化合物の製造方法を提供する。
さらなる態様において、本発明は、式 (I):
[化 5]
0
で示される化合物を、式 (π) :
〔式中、 Rは C— Cアルキルであり、 X1および X2はそれぞれ独立してハロゲンである
1 6
]
で示される化合物と塩基の存在下で反応させ、式 (VII):
[化 7]
〔式中、 Rは前記と同意義である〕
で示される化合物を製造し、次いで、得られた化合物 (VII)を酸化剤により酸ィ匕する ことを特徴とする、式 (III) :
[化 8]
〔式中、 Rは前記と同意義である〕
で示される化合物の製造方法を提供する。
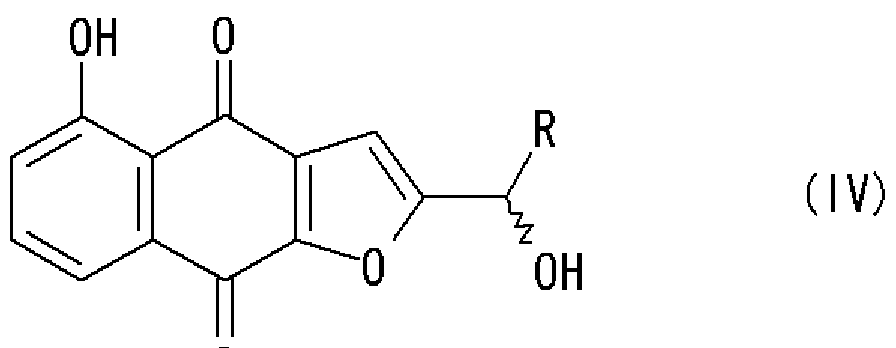
好ましい態様において、本発明は、上記のいずれかの方法で得られた式 (in)の化 合物を還元剤で還元し、式 (IV):
[化 9]
o o
〔式中、 Rは前記と同意義であり、波線はラセミ体であることを示す〕
で示される化合物を得ることをさらに含む、製造方法を提供する。
しかして、本発明は、式 (I)の化合物から直接または式 (VII)の化合物を経由して 製造される式 (III) :
〔式中、 Rは前記と同意義である〕
で示される化合物を還元剤で還元し、式 (IV):
[化 11]
〔式中、 Rは前記と同意義であり、波線はラセミ体であることを示す〕
で示されるラセミ化合物を製造する方法を提供する。
別の態様において、本発明は、式 (IV):
[化 12]
o o
〔式中、 Rは前記と同意義であり、波線はラセミ体であることを示す〕
で示される化合物を分割して、 2— ( 1—ヒドロキシェチル) 5 ヒドロキシナフト [2, 3— b]フラン 4, 9ージオンの a体および j8体を製造する方法を提供する。
さらに好ましい態様において、本発明は、溶媒に溶かしたジメチルァミンを、式 (V)
[化 13]
で示される化合物と反応させて式 (VI):
[化 14]
で示される化合物を得、次いで、得られた式 (VI)の化合物を、 5〜15% (wZw)の 酸水溶液と反応させて式 (I)の化合物を得、次 、で、得られた式 (I)の化合物を用い て上記の!/、ずれかの製造方法を行うことを含む、製造方法を提供する。
また、別の態様において、本発明は、式 (VII):
〔式中、 Rは前記と同意義である〕
で示される化合物を提供する。当該化合物は、 2— (1—ヒドロキシェチル)—5 ヒド ロキシナフト [2, 3—b]フラン 4, 9ージオンを製造するための中間体として使用さ れ得る。
[0013] さらに別の態様において、本発明は、メタノールお 0.25, 25°C)中で(+ )の旋光度 を示す 2—(1ーヒドロキシェチル) 5 ヒドロキシナフト [2, 3—b]フラン 4, 9ージ オン、すなわち (X体を提供する。
別の態様において、本発明は、ラセミ体の 2— (1—ヒドロキシェチル) 5 ヒドロキ シナフト [2, 3— b]フラン 4, 9ージオンを有効成分とする、抗癌剤を提供する。 別の態様において、本発明は、 α体の 2— (1—ヒドロキシェチル)—5 ヒドロキシ ナフト [2, 3— b]フラン 4, 9ージオンを有効成分とする、抗癌剤を提供する。
[0014] 本明細書において使用される「酸化剤」には、特に限定されるわけではないが、マ ンガンィ匕合物(たとえば二酸ィ匕マンガン、過マンガン酸カリウム)、クロム化合物(たと えば CrO 、 Na Cr O )、鉛化合物(たとえば PbO、 PbO 、 Pb (OCOCH ) )、その
3 2 2 7 2 3 4 他の金属化合物(たとえば HgO、 AgO、 Ag 0、 AgNO 、 CuCl 、 FeCl )、ハロゲン
2 3 2 3 およびハロゲン化物(たとえば CI、: Br 、 I 、 NaC10、 KBrO 、 KIO )、酸素、オゾン
2 2 2 3 4
、過酸ィ匕物(たとえば H O 、 Na O 、(C H CO) O )、過酸およびその塩 (たとえば
2 2 2 2 6 5 2 2
CH CO H、 C H CO H、 K S O )が含まれる。
3 3 6 5 3 2 2 8
[0015] 本明細書において使用される「ハロゲン」には、フッ素、塩素、臭素およびヨウ素が 含まれる。好ましくは、「ハロゲン」は臭素である。
[0016] 本明細書において使用される「C -C アルキル」は、直鎖または分枝鎖のいず
1 6
れであってもよぐたとえばメチル、ェチル、 n—プロピル、 iso プロピル、 n—ブチル 、 tert—ブチル、 n—ペンチル、 n—へキシルである。本発明において、好適な C C アルキルはメチルである。
[0017] 本明細書にぉ 、て使用される「塩基」なる語は、有機塩基または無機塩基の 、ずれ でもよい。有機塩基とは、たとえばピリジン、 DMAP (4—ジメチルァミノピリジン)、キノ リン、イソキノリン、トリェチルァミン、ジイソプロピルェチルァミン、 DBU (1, 8 ジァザ ビシクロ [5. 4. 0]ゥンデセンー7)または0 ?^ (1, 5 ジァザビシクロ [4. 3. 0]ノネ ン 5)を含むが、これらに限定されるわけではない。また、無機塩基とは、アルカリ金 属またはアルカリ土類金属の水酸ィ匕物塩、炭酸塩、重炭酸塩など、たとえば水酸ィ匕 ナトリウム、水酸ィ匕カリウム、水酸ィ匕カルシウム、炭酸ナトリウム、または炭酸水素ナトリ ゥムなどを意味する力 これらに限定されるわけではない。本発明において、好適な 塩基は DBUである。
[0018] 本明細書において使用される「還元剤」なる語は、たとえば水素化ホウ素ナトリウム( NaBH )、水素化ホウ素カリウム、水素化ホウ素リチウム、水素化アルミニウムナトリウ
4
ム、水素化アルミニウムカリウム、水素化アルミニウムリチウム、水素化ホウ素亜鉛、トリ ァセトキシ水素化ホウ素ナトリウム、ピリジン Zボラン、シァノ水素化ホウ素ナトリウム、 ナトリウムアマルガム、 H ZPd、 H ZPd— C、 H P H /PtO、 H R およ
2 2 2 2 2 2
ひ zラネ一一ニッケルを含む力 これらに限定されるわけではない。好適な還元
2
剤は水素化ホウ素ナトリウムである。
[0019] 所望により、上で得られた式 (VI)の化合物を、当分野において既知の方法、たとえ ば分別晶出またはキラルカラムクロマトグラフィーにより分離してもよい。また、上記の 還元剤に代えて当分野において既知のキラル還元剤、たとえば不斉ボラン誘導体( たとえば(一)もしくは( + ) B クロロジイソピノカンフェイルボラン)または BINAP ( ( R)もしくは(S)— 2, 2'—ビス(ジフエ-ルホスフイノ)一 1, 1 '—ビナフチル)などを用 いて光学活性の 2— (1—ヒドロキシェチル) 5 ヒドロキシナフト [2, 3— b]フラン一 4, 9—ジオンを得ることも自体公知の方法を適用することにより得ることができる。
[0020] 本発明の製造方法は、以下のように図示され得る。
〔式中、 R、 X1および X2は前記と同意義である〕
[0021] 上記スキームの工程 1は、好ましくは、チヤカーらの文献(Chaker, L.; Pautet, F.; Fi llion, H., Chem. Pharm. Bull, 1994, 42, 2238-2240)に記載された方法にしたがって 行われる。また、上記スキームの工程 1の出発物質である 5 ヒドロキシナフタレン 1,4 ジオン (別名:ュグロン)は、たとえば東京化成工業株式会社(日本、東京)から 市販されている。さらに、ュグロンについては、メルク'インデックス(Merck Index)第 1 3版第 5288頁およびその中の引用文献において詳細に記載されている。
本発明者らは、驚くべきことに、当該文献におけるジメチルァミン (沸点: 6°C)の 添加を溶媒に溶カゝしたジメチルァミンの添カ卩に代えても、同程度の結果 (置換基選択 性および収率)が得られることを見いだした。したがって、本発明はまた、溶媒に溶か したジメチルァミンを、ュグロンのトルエン溶液に添加することを特徴とする、工程 1に 記載の製造方法を提供する。
[0022] 上記スキームの工程 1において、ュグロンの溶媒は、当分野において通常使用され
る溶媒であれば、特に限定されるものではない。好ましい溶媒としては、トルエンが挙 げられる。また、ジメチルァミンの溶媒も、当分野において通常使用される溶媒であ れば、特に限定されるものではない。好ましい溶媒としては、水 (H 0)、へキサン、テ
2
トラヒドロフラン(THF)、ジェチルエーテル、トルエン、メタノールおよびエタノールが 挙げられる。
工程 1の反応は、—78°C力 溶媒の還流温度で行なうことが可能である力 好まし くは— 40°C〜室温の範囲で行われる。特に、— 40〜0°Cの反応温度は、選択性お よび操作の簡便性の点で有利である。
[0023] 上記スキームの工程 2は、好ましくは、ド 'オリベイラらの文献(De Oliveira, A.; Ferr eira, D. T.; Raslan, D. S., Tetrahedron Lett., 1988, 29, 155-158)に記載された方法 にしたがって行われる。本発明者らは、驚くべきことに、当該文献において使用され ている濃塩酸を、 5〜 15%の酸性水溶液に代えても同程度の収率が達成されること を見いだした。酸性水溶液は、一般に加水分解反応に用いることができる酸であれ ばいずれでもよいが、好ましくは塩酸または硫酸の水溶液が好ましい。さら〖こ、上記 水溶液に、適宜ジォキサンを添加してもよい。また、酸の濃度は、加水分解が可能な 濃度であれば特に限定されないが、安全性および扱いやすさの点から 5〜15%が好 ましい。したがって、本発明は、また、 5〜 15%の酸性水溶液、好ましくは 5〜 15%の 塩酸を用いることを特徴とする、工程 2に記載の製造方法を提供する。また、工程 2の 反応は、特に制限されるわけではないが、好ましくは加熱還流下で行われる。
[0024] 上記スキームの工程 3は、ハギワラらの文献(Hagiwara, H.; Sato, K.; Nishino, D.;
Hoshi, T.; Suzuki, T.; Ando, M., J. Chem. Soc. Perkin Trans. 1, 2001, 2946—2957) に記載された方法と同様に行われ得る。しかしながら、当該文献で報告されている収 率は 60%であるのに対して、当該文献に記載された出発物質を 2—ヒドロキシュグロ ンに代えた場合の反応の収率は極めて低 、ものであった。これに鑑み鋭意検討した 結果、蒸留精製後 24時間以内の、好ましくは 3時間以内のメチルビ-ルケトンを用い ることにより、工程 3の収率が飛躍的に上昇することを見いだした。したがって、本発 明はまた、蒸留精製直後の、好ましくは蒸留精製後 1時間以内のメチルビ-ルケトン を用いることを特徴とする、工程 3に記載の製造方法を提供する。この工程 3では、化
合物 (ΠΙ)および化合物 (VII)の混合物として得られる。化合物 (VII)は酸化剤を用 いて反応させることにより、化合物 (ΠΙ)に導くことができる(工程 4)。
[0025] また、工程 3におけるメチルビ二ルケトンおよび臭素の溶媒として使用できる溶媒は 、特に制限されるわけではないが、好ましくはペンタンまたはへキサンである。他方、 2—ヒドロキシュダロンの溶媒として使用できる溶媒は、特に制限されるわけではない 力 好ましくは THFまたはジェチルエーテルである。さらに、工程 3の反応温度は、 特に制限されるわけではないが、好ましくは室温である。
[0026] 上記スキームの工程 4および 5は、当分野において自体既知の方法、好ましくは、 ハギワラらの文献(Hagiwara, H.; Sato, K.; Nishino, D.; Hoshi, T.; Suzuki, T.; Ando, M., J. Chem. Soc. Perkin Trans. 1, 2001, 2946-2957)に記載された方法にしたがつ て行われる。
なお、工程 4における反応溶媒は、特に制限されるわけではないが、好ましくはハロ ゲン化炭化水素、たとえばクロ口ホルムまたは塩化メチレンである。また、工程 4の反 応は、特に制限されるわけではないが、好ましくは加熱還流下で行われる。
[0027] 工程 5における反応溶媒は、特に制限されるわけではないが、好ましくはクロ口ホル ムおよびエタノールの混合溶媒であり、特に好ましくはクロ口ホルム:エタノール =4: 1 (νΖν)の混合溶媒である。また、工程 5の反応は、特に制限されるわけではないが 、好ましくは 0°Cで行われる。
[0028] 以下の実施例により本発明を説明するが、本発明はこれらの実施例により制限され るわけではない。
以下の実施例において、以下の装置等を使用した。
1H核磁気共鳴スペクトル (^H— NMR): UNITY INOVA 500 (Varian社製)、 NM R測定溶媒: CDC1 (内部標準物質:テトラメチルシラン (TMS) );
3
融点測定器: Mp-J3 (ャナコ機器開発研究所)
実施例 1
[0029] 2—ジメチルアミノュグロンの製造
[0030] 5 ヒドロキシナフタレン 1,4ージオン(ュグロンとも呼ばれる)(171mg、 lmmol) のトルエン(5mL)溶液に、ジメチルァミン(0. 75mL、THF中 2. 0M溶液、 1. 5mm ol)を— 20°Cにて添加する。 20°Cにて 1時間撹拌後、ジメチルァミン(0. 75mL、 THF中 2. 0M溶液、 1. 5mmol)を添カ卩し、—20°Cにてさらに 30分間撹拌し、次い で溶媒を減圧留去する。残渣をシリカゲルカラムクロマトグラフィー(クロ口ホルム Z酢 酸ェチル = 20Zl (vZv) )により単離精製すると、 2—ジメチルアミノュグロン (87. 2 mg、 40%)および 3 ジメチルアミノュグロン(28. 8mg、 13%)を得る。
[0031] 2 ジメチルアミノュグロン
融点: 147〜148°C
JH-NMR (CDCl ): δ 3.25 (s, 6Η), 5.72 (s, IH), 7.20 (dd, IH, J = 1.2, 8.3 Hz), 7.45
3
-7.51 (m, 2H), 13.0 (s, IH).
3 -ジメチルアミノュグロン
JH-NMR (CDCl ): δ 3.23 (s, 6H), 5.84 (s, IH), 7.15 (dd, IH, J = 3.7, 6.1 Hz), 7.56
3
-7.59 (m, 2H), 11.9 (s, IH).
実施例 2
[0032] — 20°Cを— 40°Cに変更すること以外は実施例 1と同様に反応を行うと、 2 ジメチ ルアミノュグロン(104mg、 48%)および 3 ジメチルアミノュグロン(20mg、 10%)を 得る。
実施例 3
[0033] ジメチルァミンの溶媒を THFから水に変更して 0. 15mLのジメチルァミン水溶液( 50%水溶液、 1. 5mmol)を用い、そして反応温度を 0°Cとすること以外は、実施例 1 と同様に反応を行うと、 2 ジメチルアミノュグロン(97mg、 45%)および 3 ジメチル
アミノュグロン (67mg、 31%)を得る。この方法は、有機溶媒の代わりに水を用いると V、う点で、実施例 1の方法よりも環境面および安全面で有利である。
実施例 4
2 -ヒドロキシュグロンの製造
[化 18]
[0035] 2—ジメチルアミノュグロン(1. 95g、 9mmol)のジォキサン(45mL)の溶液に、 10 %塩酸(10mL)を添加し、 30分間加熱還流する。室温まで冷却後、反応液をクロ口 ホルムで抽出する。合わせた有機層をブラインで洗い、硫酸ナトリウムで乾燥し、濾過 し、そして溶媒を減圧留去すると、褐色固体として 2—ヒドロキシュダロン(1. 67g、 97 %)を得る。
融点: 220〜221°C
JH-NMR (CDC1 ): δ 6.31 (1Η, s), 7.35 (1H, dd, J = 8.5, 1.2 Hz), 7.44 (1H, s), 7.59
3
(1H, t, J = 8.5 Hz), 7.69 (1H, dd, J = 8.5, 1.2 Hz), 12.33 (1H, s).
実施例 5
[0036] 2—ァセチルー 5—ヒドロキシナフト [2, 3— b]フラン一 4, 9—ジオンおよび 2—ァセ チル一 2,3—ジヒドロ一 5—ヒドロキシナフト [2, 3— b]フラン一 4, 9—ジオンの製造 [化 19]
副生成物 主生成物
[0037] メチルビ-ルケトン(10. 5g、 150mmol)のペンタン(150mL)溶液に、—15°C
て臭素(25g、 156mmol)のペンタン(30mL)溶液を加える。 15°Cにて 10分間撹 拌した後に、溶媒を減圧留去すると、無色のオイルを得、次いでこれを 2—ヒドロキシ ュグロン(4. 75g、 25mmol)の THF (250mL)溶液に添カ卩する。これに DBUを 0°C にて添加し、そして室温にて一晩撹拌する。これに 10%塩酸を添加し、そして反応 混合液をクロ口ホルムにて抽出する。合わせた有機層をブラインで洗い、硫酸ナトリウ ムで乾燥し、濾過し、そして溶媒を減圧留去する。残渣をシリカゲルカラムクロマトダラ フィー(溶離液:クロ口ホルム Z酢酸ェチル = 9Z1 (v/v) )により精製すると、 1: 5の 比率で 2 ァセチルー 5 ヒドロキシナフト [2, 3—b]フラン 4, 9ージオンおよび 2 —ァセチル一 2,3 ジヒドロ一 5 ヒドロキシナフト [2, 3—b]フラン一 4, 9 ジオンを 含有する橙色の固体混合物を得る(6. 14g、 95%) oこの固体混合物を、シリカゲル カラムクロマトグラフィー (溶離液:クロ口ホルム)により分離して、それぞれ、 2—ァセチ ルー 5 ヒドロキシナフト [2, 3—b]フラン 4, 9ージオンおよび 2 ァセチルー 2,3 —ジヒドロ一 5 ヒドロキシナフト [2, 3—b]フラン一 4, 9 ジオンを得た。
[0038] 2 ァセチルー 2,3 ジヒドロ一 5 ヒドロキシナフト [2, 3—b]フラン一 4, 9 ジォ ン
融点: 175〜182°C (分解)
JH-NMR (CDC1 ): δ 2.39 (3Η, s), 3.39 (2H, d, J = 9.5 Hz), 5.30 (1H, t, J = 9.5 Hz)
3
, 7.26 (1H, dd, J = 8.0, 1.0 Hz), 7.56 (1H, t, J = 8.0 Hz), 7.65 (1H, dd, J = 8.0, 1.0 Hz), 12.18 (1H, s).
2 ァセチル一 5 ヒドロキシナフト [2, 3—b]フラン一 4, 9 ジオン
融点: 208〜220°C (分解)
1H-NMR (CDC1 ): δ 2.67 (3Η, s), 7.33 (1H, dd, J = 8.5, 1.0 Hz), 7.60 (1H, s), 7.67
3
(1H, t, J = 8.3 Hz), 7.81 (1H, dd, J = 7.4, 1.0 Hz), 12.13 (1H, s).
実施例 6
[0039] 2 ァセチル一 5 ヒドロキシナフト [2, 3—b]フラン一 4, 9 ジオンの製造
[0040] 2 ァセチルー 2,3 ジヒドロ一 5 ヒドロキシナフト [2, 3— b]フラン一 4, 9 ジォ ン(2. 4g、 9. 4mmol)のクロ口ホルム(50mL)溶液に、 20gの二酸化マンガン(アル ドリツチ社製、 85%活性二酸ィ匕マンガン、く 5ミクロン)を添加し、得られた懸濁液を 1 日間加熱還流する。室温まで冷却後、混合物を濾過する。濾液を減圧留去し、そし て残渣を、シリカゲルカラムクロマトグラフィー (溶離液:クロ口ホルム)により精製すると 、 2 ァセチル一 5 ヒドロキシナフト [2, 3— b]フラン一 4, 9 ジオンを得る(0. 718 g、 33%) o
融点: 208〜220°C (分解)
JH-NMR (CDC1 ): δ 2.67 (3Η, s), 7.33 (1H, dd, J = 8.5, 1.0 Hz), 7.60 (1H, s), 7.67
3
(1H, t, J = 8.3 Hz), 7.81 (1H, dd, J = 7.4, 1.0 Hz), 12.13 (1H, s).
[0041] なお、別法として、実施例 6における二酸化マンガン (アルドリッチ社製、 85%活性 二酸化マンガン、く 5ミクロン)を二酸化マンガン(アルドリッチ社製、 90%二酸化マン ガン、電池用、く 10ミクロン)に代えることも可能である(下記の実施例 7参照)。
実施例 7
[0042] 2 ァセチルー 2,3 ジヒドロ一 5 ヒドロキシナフト [2, 3— b]フラン一 4, 9 ジォ ン(0. 5g、 1. 95mmol)のクロ口ホルム(50mL)溶液に、 10gの二酸化マンガン(ァ ルドリッチ社製、 90%二酸ィ匕マンガン、電池用、く 10ミクロン)を添加し、得られた懸 濁液を 3日間加熱還流する。室温まで冷却後、混合物を濾過する。濾液を減圧留去 し、そして残渣を、シリカゲルカラムクロマトグラフィー (溶離液:クロ口ホルム)により精 製すると、 2 ァセチルー 5 ヒドロキシナフト [2, 3— b]フラン一 4, 9 ジオン(0. 2 16g、 44%)および 2 ァセチルー 2,3 ジヒドロ一 5 ヒドロキシナフト [2, 3— b]フ ラン— 4, 9 ジオン(0. 255g、 51%)を得る。
本方法により、実施例 6よりも低コストの 2 ァセチルー 5 ヒドロキシナフト [2, 3— b]フラン 4, 9ージオンの製造方法が提供される。
実施例 8
2— (1—ヒドロキシェチル) 5 ヒドロキシナフト [2, 3— b]フラン一 4, 9 ジオン の製造
[化 21]
[0044] 実施例 5で得られた 2 ァセチルー 5 ヒドロキシナフト [2, 3—b]フラン—4, 9 ジオン(694mg、 2. 73mmol)のクロ口ホルム(lOOmL)およびエタノール(25mL) 溶液に水素化ホウ素ナトリウム(515mg、 13. 7mmol)を 0°Cにて添加する。 30分間 撹拌後、反応を 10%塩酸の添カ卩によりタエンチする。水層をクロ口ホルムで 2回抽出 し、そして抽出液を水およびブラインで連続的に洗う。混合液を減圧留去し、シリカゲ ルカラムクロマトグラフィー (溶離液:クロ口ホルム)により精製すると、黄色の結晶のラ セミ混合物として 2— (1—ヒドロキシェチル) 5 ヒドロキシナフト [2, 3—b]フラン一 4, 9 ジオンを得る(516mg、 74%) 0
融点: 148〜149°C
1H-NMR (CDC1 ): δ 1.66 (3Η, d, J = 6.8 Hz), 2.31 (IH, brs), 5.05 (IH, m), 6.84 (1
3
H, s), 7.27 (IH, dd, J = 8.3, 1.0 Hz), 7.62 (IH, t, J = 8.0 Hz), 7.75 (IH, dd, J = 8.0, 0.9 Hz), 12.18 (IH, s).
[0045] 上で得られたラセミ混合物を、下記の条件のキラルカラムクロマトグラフィーにより、 ェナンチォマーを分離することができる。
カラム: SUMICHIRAL OA— 4500 (4. 6mm X 250mm)
移動相:へキサン Z2—プロパノール Zメタノール = 95 :4: 1
検出: UV 254nm
流速: 1. OmLZ分
温度:室温(25°C付近の一定温度)
注入量: 5 L (0. lmg/mL メタノーノレ)
保持時間: 30. 8および 34. 4分。
[0046] なお、上記条件における移動相を「へキサン Z2—プロパノール Zメタノール = 95 :
4: 1」から「へキサン/エタノール = 95 : 5」に代えても 2—( 1 ヒドロキシェチル) 5 ーヒドロキシナフト [2, 3— b]フラン 4, 9ージオンのェナンチォマーを互いに分離 することができ、その場合の保持時間は、 24. 9および 27. 4分である。同じ条件でキ ラルカラムクロマトグラフィーに付すと、天然由来の 2—(1ーヒドロキシェチル) - 5- ヒドロキシナフト [2, 3—b]フラン 4, 9ージオン、すなわち j8体の保持時間は 27. 4 分である。
光学活性カラムを用いた上記条件による合成 2—(1ーヒドロキシェチル) 5 ヒド ロキシナフト [2, 3— b]フラン 4, 9ージオンの HPLC分離により得られたそれぞれ のェナンチォマーの保持時間から、最初に溶出するものが非天然型(ひ体)、後から 溶出するものが天然型( ι8体)であることが判明した。それぞれの物性を以下の表 1 に示す。
[0047] 実施例 8において得られた 2—(1ーヒドロキシェチル) 5 ヒドロキシナフト [2, 3 b]フラン 4, 9ージオンのラセミ体、 α体および j8体を用いて、以下に示すように 抗腫瘍活性をおよび正常細胞に対する毒性を調べる。
[0048] [試験例 1]抗腫瘍活性
( 1) PC— 3細胞 (前立腺癌細胞)に対する抗腫瘍活性 1
PC— 3細胞 (大日本製薬 (株)、ラボラトリ'プロダクツ部)を、予めゥシ胎児血清 (FB S) 20%含むダルベッコ変法イーグル培地(DMEM)を入れた 35mmシャーレに 1 X 105/mlの濃度にて播種する。これを 5%CO中で 36°Cにて 1日間インキュベートし
2
、シャーレ底に細胞密着していることを確認する。得られた PC— 3細胞を 3群に分け 、第 1の群には(士) - 2- (1—ヒドロキシェチル) 5 ヒドロキシナフト [2, 3—b]フ ラン一 4, 9 ジオン(ラセミ体)を 0. 5mM、0. 05mMおよび 0. 005mMの濃度にて
添加し、第 2の群には対照としてアドリアマイシン (和研薬 (株))を 0. 5mM、 0. 05m Mおよび 0. 005mMの濃度にて添カ卩する。これらを 36°Cにて 3日間インキュベーショ ンし、生存細胞を計数し、生存率を算出する。結果を表 2に示す。
[表 2]
(2) PC- 3細胞に対する抗腫瘍活性 2
上記( 1 )と同様の処置をして得られた PC— 3細胞を 3群に分け、第 1の群には(士) — 2— (1—ヒドロキシェチル) 5 ヒドロキシナフト [2, 3— b]フラン一 4, 9 ジオン (ラセ ^体)を 0. 5mM、 0. 05mM、 0. 005mMおよび 0. 0005mMの濃度にて添加 し、第 2の群には非天然型 2— (1—ヒドロキシェチル) 5 ヒドロキシナフト [2, 3— b ]フラン— 4, 9 ジオン 体)を 0. 5mM、 0. 05mM、 0. 005mMおよび 0. 0005 mMの濃度にて添加し、第 3の群には天然型 2— (1—ヒドロキシェチル) 5 ヒドロ キシナフ卜 [2, 3— b]フラン— 4, 9 ジオン(j8体)を 0. 5mM、 0. 05mM、 0. 005 mMおよび 0. 0005mMの濃度にて添カ卩し、第 4の群には対照としてマイトマイシン を 0. 5mM、 0. 05mM、 0. 005mMおよび 0. 0005mMの濃度にて添加する。これ らを 36°Cにて 3日間インキュベーションし、生存細胞を計数し、生存率を算出する。 結果を表 3に示す。
[表 3]
(3) Α— 549細胞 (肺癌細胞)に対する抗腫瘍活性
A— 549細胞について上記(2)と同様の処置を行い、抗腫瘍活性を検討した。結 果を表 4に示す。
[表 4]
(4) MCF— 7細胞 (乳癌細胞)に対する抗腫瘍活性
MCF— 7細胞について上記(2)と同様の処置を行い、抗腫瘍活性を検討した。結 果を表 5に示す。
[表 5]
[試験例 2]ヒト皮膚正常細胞(Cell System Fb cells)、ヒト肝正常細胞(Cell Systems He cells)、ヒト小腸正常細胞 (Cell Systems IE cells)、ヒト肺正常細胞(MRC- 5)に対 する細胞毒性試験法
各ヒト正常細胞を、それぞれ予めゥシ胎児血清 (FBS) 20%含むダルベッコ変法ィ ーグル培地(DMEM)を入れた 35mmシャーレに 1 X 105Zmlの濃度にて播種する 。これらを 5%CO中で 36°Cにて 1日間インキュベートし、シャーレ底に細胞密着して
2
いることを確認する。ラセミ体、 α体および j8体を、それぞれ、 DMSOに溶かし、 0. 5 mM、 0. 05mM、 0. 005mMおよび 0. 0005mMの濃度の溶液を得、これを各細 胞に 2 μ Lずつ添加する。これらを 36°Cにて 3日間インキュベーションし、 0. 25%トリ パンブルーを用いて生存細胞を計数し、生存率を算出する。得られた結果を表 6〜9
に示す。なお、ヒト皮膚正常細胞に対する試験では、対照として市販の抗癌剤マイト マイシンについても試験し、その結果を合わせて表 6に示す。
[表 7]
[表 8]
[表 9]
表 6に示すように、既存の代表的抗腫瘍剤であるマイトマイシン Cは正常細胞に対
してもかなり強 、毒性を示す。