WO2006087321A1 - Selektive spaltung von substituierten bisbenzylamiden und -aminen - Google Patents
Selektive spaltung von substituierten bisbenzylamiden und -aminen Download PDFInfo
- Publication number
- WO2006087321A1 WO2006087321A1 PCT/EP2006/050915 EP2006050915W WO2006087321A1 WO 2006087321 A1 WO2006087321 A1 WO 2006087321A1 EP 2006050915 W EP2006050915 W EP 2006050915W WO 2006087321 A1 WO2006087321 A1 WO 2006087321A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- oxidation
- solvent
- electrolysis
- amide
- nucleophile
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/62—Preparation of compounds containing amino groups bound to a carbon skeleton by cleaving carbon-to-nitrogen, sulfur-to-nitrogen, or phosphorus-to-nitrogen bonds, e.g. hydrolysis of amides, N-dealkylation of amines or quaternary ammonium compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C211/00—Compounds containing amino groups bound to a carbon skeleton
- C07C211/01—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms
- C07C211/02—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C211/03—Monoamines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/02—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having nitrogen atoms of carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals
- C07C233/04—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having nitrogen atoms of carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals with carbon atoms of carboxamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C233/05—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having nitrogen atoms of carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals with carbon atoms of carboxamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton having the nitrogen atoms of the carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25B—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES FOR THE PRODUCTION OF COMPOUNDS OR NON-METALS; APPARATUS THEREFOR
- C25B3/00—Electrolytic production of organic compounds
- C25B3/20—Processes
- C25B3/23—Oxidation
Definitions
- the present invention relates to a process for the regioselective cleavage of secondary amines or amides to give primary amines.
- Primary amines are important starting materials or intermediates for industrial chemistry. For the recovery of amines, a number of reactions are available. Examples are the Hofmann alkylation, the reductive amination of carbonyl compounds, the reduction of nitro compounds or the Gabriel synthesis.
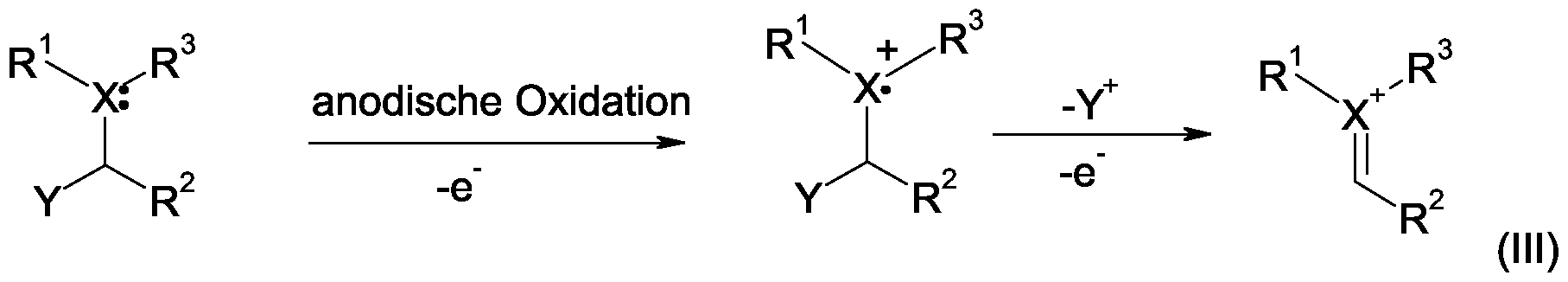
- a reactive cation radical (II) is formed as an intermediate, which is converted into a second cationic intermediate (IM) by further elimination.
- the cationic intermediate (III) is trapped with a nucleophile, for example, methanol.
- a nucleophile for example, methanol.
- N-acetylated amides of the general formula (IV) which are converted under anodic oxidation and subsequent trapping with the nucleophile methanol into N-acetylated aminals of the general formula (V):
- the present invention to provide a process for the production of primary amines from secondary amines or amides by electrochemical anodic oxidation.
- the process according to the invention should preferably be designed in such a way that it proceeds with the use of optically active starting materials while retaining the optical activity, so that the diastereomeric purity of the educt can be transferred to the product.
- the solution to this problem is based on a process for the regioselective cleavage of secondary amines or amides.
- the process according to the invention makes it possible to produce primary amines in an efficient manner.
- At least one bisbenzylamine or bisbenzylamide with at least one benzylic hydrogen atom is provided in a solvent.
- the amine or amide used in the process according to the invention is, for example, a bisbenzylamine of the general formula (VI) or a bisbenzylamide of the general formula (VII): PF0000056345 / Wa
- a bisbenzylamine is preferably used, and the unsubstituted nitrogen function of bisbenzylamine is provided with a protective group.
- the protective group is preferably selected from the group consisting of an acyl group, sulfone group, phosphoryl group or silyl group.
- an acyl group is used as the protective group, the bisbenzylamides of general formula (VII) already mentioned are used as starting materials.
- At least one of the benzyl rings of bisbenzylamines or bisbenzylamides is preferably substituted, and it is further preferred that the at least one substituted benzyl ring is substituted with an electron donating substituent.
- an electron donating substituent is meant when the substituent exerts a + I effect and / or + M effect on the benzyl ring.
- the electron-donating substituent is preferably selected from the group consisting of alkoxy groups, alkyl groups, thiolalkyl groups or halogen, wherein the alkyl groups are preferably selected from the group of Ci-Cs-alkyls.
- an alkoxy group is used as the electron donating substituent, it is preferably selected from the group consisting of methoxy, ethoxy, propoxy, isopropoxy, butoxy or tert-butoxy groups.
- the second benzyl radical of the bisbenzylamines or bisbenzylamides is preferably either unsubstituted or substituted with an electron-withdrawing group.
- an electron-withdrawing substituent is meant when the substituent exerts an -I effect and / or -M effect on the phenyl ring.
- suitable electron-withdrawing substituents are selected from the group consisting of cyano groups, nitro groups, ester groups and the halides fluorine, chlorine, bromine and iodine. PF0000056345 / Wa
- the secondary amine or amide is provided in a solvent.
- a solvent In a particularly preferred embodiment, it is an organic solvent, preferably an organic nucleophilic solvent. It is further preferred that the solvent is selected from the group consisting of protic polar solvents such as alcohols, aliphatic carboxylic acids such as acetic acid and water.
- an alcohol used as the solvent, these are, for example, methanol, ethanol, n- or / -propanol or butanols. Preference is given to methanol.
- mixtures of the abovementioned solvents can also be used.
- the electrolysis solution is added to customary cosolvents.
- these are the inert solvents generally used in organic chemistry with a high oxidation potential. Examples which may be mentioned are dimethyl carbonate, propylene carbonate, ethylene carbonate, tetrahydrofuran, dimethoxyethane, di-, tri-, tetrachloromethane or acetone itril.
- process step (b 1 ) an electrochemical oxidation of the secondary amine or amide and a trapping of the electrolysis intermediate with a nucleophile take place, whereby an electrolysis effluent is obtained.
- a conductive salt is required as a depolarizer to which the solution prepared in process step (a 1 ) is added.
- conducting salts which are contained in the electrolysis solution, it is generally at alkali metal, alkaline earth metal, tetra (C r to C 6 alkyl) ammonium, tri preferably (d- to C 6 alkyl) - methyl ammonium salts.
- Suitable counterions are sulfate, hydrogen sulfate, alkyl sulfates, aryl sulfates, halides, phosphates, carbonates, alkyl phosphates, alkyl carbonates, nitrate, alcoholates, tetrafluoroborate, hexafluorophosphate or perchlorate.
- acids derived from the abovementioned anions are suitable as conductive salts. PF0000056345 / Wa
- suitable electrolyte salts are ionic liquids. Suitable ionic liquids are described in "Lonic Liquids in Synthesis”, ed. Peter Wasserscheid, Tom Welton, Verlag Wiley VCH, 2003, Chap. 1 to 3 and DE-A-10 2004 011427.
- strong mineral acids and sulfonic acids are suitable as conductive salts in the present invention.
- Examples are H 2 SO 4 , H 3 PO 4 .
- the use of H 2 SO 4 is particularly preferred.
- the concentration of the conducting salt is generally 0.0001-5 mol / l, preferably 0.001-1 mol / l, more preferably 0.001-0.1 mol / l, most preferably 0.005-0.05 mol / l.
- the process conditions of the electrochemical oxidation with respect to temperature, duration of electrolysis, current strength and concentration of the secondary amines or amides depend on the educt used, in particular bisbenzylamine or bisbenzylamide, and on the solvent used.
- the electrolysis is carried out in the usual, known in the art electrolysis cells. Suitable electrolysis cells are known to the person skilled in the art. Preferably, one works continuously with undivided flow cells or discontinuously in beaker cells at reaction volumes ⁇ 100 mL.
- bipolar switched capillary gap cells or plate stacked cells in which the electrodes are designed as plates and are arranged plane-parallel (see Ullmann's Encyclopedia of Industrial Chemistry, 1999 electronic release, Sixth Edition, VCH-Verlag Weinheim, Volume Electrochemistry, Chapter 3.5 cell designs and Chapter 5, Organic Electrochemistry, Subchapter 5.4.3.2 Cell
- the current densities at which the process is carried out are generally 1 to 1.
- Suitable anode materials are, for example, graphite, carbon, noble metals such as platinum, metal oxides such as ruthenium or chromium oxide, mixed oxides of the type RuO x TiO x and diamond electrodes. Preference is given to graphite or carbon electrodes. PF0000056345 / Wa
- cathode materials are iron, steel, stainless steel, nickel, noble metals such as platinum, graphite, carbon materials and diamond electrodes.
- the systems are preferably graphite as the anode and cathode, graphite as the anode and nickel, stainless steel or steel as the cathode and platinum as the anode and cathode.
- the electrochemical oxidation is carried out until the benzylamide used as starting material has reacted completely or for the most part.
- the term "for the most part” means a degree of conversion of preferably greater than 90% .
- the progress of the reaction is monitored by laboratory methods (eg gas or thin-layer chromatography) Amide required.
- the concentration of the starting material to be oxidized in the solution to be electrolyzed is preferably 0.00001 to 5 mol / l, more preferably 0.0001 to 3 mol / l, especially 0.001 to 2 mol / l.
- step (b 1 ) provided for electrochemical oxidation the nitrogen function of the secondary amine or amide is oxidized, wherein a radical
- the electrochemical oxidation is preferably carried out on the side of
- Amines or amides on which the more stable radical is formed This is the side of the secondary amine or amide at which the benzyl ring with the electron donating
- Substituents is located.
- the regioselectivity is achieved in particular with substrates having alkoxy, thioalkyl or alkyl groups.
- step (b 1 ) the reaction of the electrolysis intermediate with a nucleophile is carried out immediately after the electrochemical oxidation.
- the electrolysis effluent is preferably reacted with a nucleophile selected from the group consisting of methanol, acetic acid and water.
- the nucleophile used is preferably the solvent used in process step (a 1 ), so that the addition of a further nucleophile can be dispensed with.
- the process according to the invention according to the first embodiment is preferably suitable for the electrochemical oxidation of optically active, ie diastereomeric bisbenzylamines or bisbenzylamides, since the stereochemical purity of the resulting products is essentially not changed by the electrochemical oxidation.
- optically active ie diastereomeric bisbenzylamines or bisbenzylamides
- optical purity of the product of the optical purity of the starting material by at most 10%, more preferably at most 5%, in particular at most 3%, deviates.
- process step (c 1 ) there is a work-up of the electrolysis discharge.
- (c'1) removing the solvent and adding water, an organic solvent selected from the group consisting of dichloromethane; Chloroform; Ethers, such as diethyl ether, tert-butyl methyl ether; Esters, such as ethyl acetate; Hydrocarbons, such as toluene, xylene or cyclohexane and an acid;
- an organic solvent selected from the group consisting of dichloromethane; Chloroform; Ethers, such as diethyl ether, tert-butyl methyl ether; Esters, such as ethyl acetate; Hydrocarbons, such as toluene, xylene or cyclohexane and an acid;
- any suitable acid can be used.
- Suitable acids are known to the person skilled in the art. Examples are hydrochloric acid, sulfuric acid,
- Phosphoric acid or nitric acid Preference is given to the use of 10% hydrochloric acid.
- process step (c'3) takes place, for example, over sodium carbonate or sodium sulfate. Alternatively, however, all other conventional drying agents can be used.
- the organic solvent is preferably removed by distillation.
- the hydrolysis of the worked-up electrolysis effluent is preferably carried out with a mixture of sodium hydroxide solution or potassium hydroxide solution and triethanolamine. Preference is given to the use of 50% sodium or potassium hydroxide solution. The content of 50% sodium hydroxide solution in the PF0000056345 / Wa
- Mixture preferably 10 to 50 wt .-%, particularly preferably 20 to 40 wt .-%, in particular 25 to 35 wt .-%.
- the content of triethanolamine in the mixture is preferably 10 to 50 wt .-%, particularly preferably 20 to 40 wt .-%, in particular 25 to 35 wt .-%.
- the primary amine forms.
- the present invention relates to a "wet-chemical" process for the regioselective cleavage of secondary amines or amides.
- At least one secondary amine or amide is provided in a solvent, wherein the solvent optionally comprises a nucleophile.
- Alkoxysubstituent carries. It is particularly preferred if the alkoxy substituent is selected from the group consisting of methoxy, ethoxy, propoxy, isopropoxy, butoxy and tert-butoxy.
- the solvent used in process step (a ") is preferably selected from the group consisting of dichloromethane, chloroform, 1,2-dichloroethane, tert-butyl methyl ether, acetonitrile, toluene and xylene.
- the solvent be used in admixture with a nucleophile such as water, for example, distilled water, an alcohol, for example, methanol, ethanol, n-propanol, i-propanol, or butanol.
- a nucleophile such as water, for example, distilled water, an alcohol, for example, methanol, ethanol, n-propanol, i-propanol, or butanol.
- Suitable examples are mixtures of 1,2-dichloroethane with water in a ratio of 100: 1 to 1: 1, particularly preferably 20: 1 to 5: 1, in particular 12: 1 to 8: 1. PF0000056345 / Wa
- Process step (b ") involves the oxidation of the secondary amine or amide by 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to give an oxidation effluent
- DDQ 2,3-dichloro-5,6-dicyano-1,4-benzoquinone
- the oxidation provided in process step (b") of the secondary amine or amide is preferably carried out by addition of DDQ to the educt to be oxidized, which is present in the solvent described above, optionally in the presence of a nucleophile.
- the oxidation is preferably carried out with stirring at a temperature of -10 0 C to 150 0 C, more preferably 20 to 100 ° C, in particular 60 to 90 0 C performed.
- the reaction time under these preferred conditions is preferably 0.2 to 24 hours, more preferably 1 to 12 hours, especially 5 to 10 hours.
- process step (c ") the reaction of the oxidation product with a nucleophile takes place, which is carried out by adding the optionally dissolved nucleophile to the oxidation product.
- Suitable nucleophiles are, for example, water, alcohols, such as methanol, ethanol or propanol.
- process step (a ) the amine or amide to be oxidized can be provided in a solvent in admixture with a nucleophile, whereby the oxidized amine or amide immediately in process step (b") after its oxidation from the nucleophile present intercepted, so that can be dispensed with the further addition of the nucleophile.
- the workup of the product resulting from process step (c ") can be carried out, for example, by washing with saturated sodium carbonate solution and / or saturated sodium chloride solution, extraction of the resulting aqueous phases with an organic solvent, drying of the combined organic phases, for example over sodium sulfate, and Concentration, for example in a vacuum, take place.
- the process according to the invention according to the second embodiment also leads to a regioselective cleavage of the secondary amines or amides used as starting materials.
- the regioselective oxidation is carried out according to the method of the invention second embodiment of the electron-rich benzyl ring.
- inventive method according to the second embodiment is also preferably suitable for the electrochemical oxidation of optically active, ie PF0000056345 / Wa
- substantially unchanged means that the optical purity of the product deviates from the optical purity of the starting material by at most 10%, particularly preferably at most 5%, in particular at most 2%.
- Another object of the present invention is the use of 2,3-dichloro-5,6-benzoquinone for the regioselective oxidation of secondary Bisbenzylaminen or bisbenzylamides having at least one benzylic hydrogen atom.
- the secondary bisbenzylamides used as starting material in the process according to the invention in the first and second embodiments can be prepared, for example, by reacting secondary bisbenzylamines with acetic anhydride.
- reaction mixture I (1) adding the acetic anhydride to the secondary amine or adding the secondary amine to the acetic anhydride to give a reaction mixture I;
- reaction mixture II (2) stirring the resulting reaction mixture I for a period of preferably from 0.5 to 24 hours, more preferably from 1 to 15 hours, especially from 1 to 2 hours, to give a reaction mixture II;
- the addition of the acetic anhydride to the secondary amine can take place at a temperature of preferably 0 to 100 ° C., particularly preferably 10 to 50 ° C., in particular 20 to 30 ° C.
- the resulting reaction mixture I is stirred at a temperature of preferably 0 to 150 0 C, particularly preferably 50 to 120 0 C, in particular 80 to 100 ° C. PF0000056345 / Wa
- the removal of acetic anhydride in the organic phase can be carried out, for example, by adding a base, the base preferably being present in an aqueous solution.
- the base may be, for example, sodium carbonate.
- process steps (5) and (6) can be followed by process step (3) or optionally process step (4):
- Another object of the present invention is the use of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone for the regioselective oxidation of secondary Bisbenzylaminen or bisbenzylamides having at least one benzylic hydrogen atom.
- the diastereomeric purity is determined by gas chromatography.
- the methanol is removed under reduced pressure and treated with 25 ml of water, 50 ml of dichloromethane and 10 ml of 10% hydrochloric acid.
- the aqueous phase is extracted several times with dichloromethane (3 ⁇ 20 ml).
- the combined extracts are dried over Na 2 SO 4 and the solvent is removed.
- the crude yield of amide is 80%.
- the residue consists of a mixture of N-1-phenylethylacetamide (37 GC-FI.%) And 4-methoxyacetophenone (63 GC-FI.%).
- Phenylethylacetamide is:
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- Materials Engineering (AREA)
- Metallurgy (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Electrolytic Production Of Non-Metals, Compounds, Apparatuses Therefor (AREA)
- Pyridine Compounds (AREA)
Abstract
Description




Claims
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06708252A EP1853549A1 (de) | 2005-02-15 | 2006-02-14 | Selektive spaltung von substituierten bisbenzylamiden und -aminen |
| CA002596998A CA2596998A1 (en) | 2005-02-15 | 2006-02-14 | Selective splitting of substituted bisbenzylamides and bisbenzylamines |
| US11/815,930 US20080073221A1 (en) | 2005-02-15 | 2006-02-14 | Selective Splitting of Substituted Bisbenzylamides and Bisbenzylamines |
| JP2007555593A JP2008530368A (ja) | 2005-02-15 | 2006-02-14 | 置換ベンジルアミドおよび置換ベンジルアミンの選択的分解 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102005007025A DE102005007025A1 (de) | 2005-02-15 | 2005-02-15 | Selektive Spaltung von substituierten Bisbenzylamiden und aminen |
| DE102005007025.6 | 2005-02-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006087321A1 true WO2006087321A1 (de) | 2006-08-24 |
Family
ID=36587065
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/050915 WO2006087321A1 (de) | 2005-02-15 | 2006-02-14 | Selektive spaltung von substituierten bisbenzylamiden und -aminen |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US20080073221A1 (de) |
| EP (1) | EP1853549A1 (de) |
| JP (1) | JP2008530368A (de) |
| KR (1) | KR20070103031A (de) |
| CN (1) | CN101119962A (de) |
| CA (1) | CA2596998A1 (de) |
| DE (1) | DE102005007025A1 (de) |
| WO (1) | WO2006087321A1 (de) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108658784B (zh) * | 2018-04-26 | 2020-12-18 | 联化科技股份有限公司 | (r)-1-(4-甲基苯基)乙胺的合成方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3907652A (en) * | 1974-10-30 | 1975-09-23 | Monsanto Co | Electrooxidation of phosphonomethyl amines |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4606041B2 (ja) * | 2004-03-05 | 2011-01-05 | セントラル硝子株式会社 | 光学活性1−アリール−2−フルオロ置換エチルアミン類およびその製造方法 |
-
2005
- 2005-02-15 DE DE102005007025A patent/DE102005007025A1/de not_active Withdrawn
-
2006
- 2006-02-14 CN CNA2006800049242A patent/CN101119962A/zh active Pending
- 2006-02-14 JP JP2007555593A patent/JP2008530368A/ja not_active Withdrawn
- 2006-02-14 KR KR1020077018649A patent/KR20070103031A/ko not_active Application Discontinuation
- 2006-02-14 US US11/815,930 patent/US20080073221A1/en not_active Abandoned
- 2006-02-14 EP EP06708252A patent/EP1853549A1/de not_active Withdrawn
- 2006-02-14 WO PCT/EP2006/050915 patent/WO2006087321A1/de active Application Filing
- 2006-02-14 CA CA002596998A patent/CA2596998A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3907652A (en) * | 1974-10-30 | 1975-09-23 | Monsanto Co | Electrooxidation of phosphonomethyl amines |
Non-Patent Citations (4)
| Title |
|---|
| DEINHAMMER ET AL: "Electrochemical Oxidation of Amine-Containing Compounds", LANGMUIR, vol. 10, no. 4, 1994, pages 1306 - 1313, XP009068224 * |
| E.LEE-RUFF ET AL: "Oxidation of allyl and benzyl ethers by 2,3-dichloro-5,6-dicyanobenzoquinone", CAN.J.CHEM., vol. 67, 1989, pages 699 - 702, XP009068244 * |
| M.FLEISCHMANN ET AL: "The Oxidation of Organic Compounds at a Nickel Anode in Alkaline Solution", J.ELECTROANAL.CHEM., vol. 31, 1971, pages 39 - 49, XP009068220 * |
| M.MASUI ET AL: "Anodic Oxidation of Amines", PHYS.ORG. J.CHEM.SOC. (B), 1971, pages 1593 - 1596, XP009068245 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2008530368A (ja) | 2008-08-07 |
| CA2596998A1 (en) | 2006-08-24 |
| KR20070103031A (ko) | 2007-10-22 |
| CN101119962A (zh) | 2008-02-06 |
| DE102005007025A1 (de) | 2006-08-24 |
| US20080073221A1 (en) | 2008-03-27 |
| EP1853549A1 (de) | 2007-11-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2010139687A1 (de) | Verfahren zur herstellung von unsymmetrischen biarylalkoholen | |
| JP2012501383A (ja) | 置換アリールアルコールのアノード脱水素二量化のための方法 | |
| EP2616424B1 (de) | Verfahren zur herstellung von 2-methyl-3-(4-tert-butylphenyl)-propanal mit hoher para-isomerenreinheit | |
| DE10058304A1 (de) | Verfahren zur Herstellung von alkoxylierten Carbonylverbindungen durch ein anodisches Oxidationsverfahren unter Nutzung der kathodischen Koppelreaktion zur organischen Synthese | |
| WO2006087321A1 (de) | Selektive spaltung von substituierten bisbenzylamiden und -aminen | |
| WO2009071478A1 (de) | Verfahren zur reduktiven hydrodimerisierung von ungesättigten organischen verbindungen mittels einer diamantelektrode | |
| WO2010108874A1 (de) | Elektrochemisches verfahern zur herstellung von 3-tert.-butylbenzaldehyd-dimethylacetal | |
| EP1769103B1 (de) | Elektrochemisches verfahren zur herstellung cyclopropylbenzylaminen | |
| EP0009697B1 (de) | Verfahren zur Herstellung von N-(Alpha-Methoxy-alkyl)-urethanen und neue N-(Alpha-methoxy-alkyl)-urethane | |
| US20080228009A1 (en) | Process for Preparing 1,1,4,4-Tetraalkoxybut-2-Ene Derivatives | |
| EP2534281A2 (de) | Verfahren zur herstellung von 4-isopropylcyclohexylmethanol | |
| EP0711851B1 (de) | Verfahren zur Herstellung von p-Hydroxybenzaldehyden | |
| EP0554564A1 (de) | Verfahren zur Herstellung von Benzaldehydacetalen | |
| DE10317878A1 (de) | Verfahren zur Herstellung von alpha-methylierten Cystein und Serin-Derivaten | |
| EP3774719A1 (de) | Verfahren zur herstellung von 2,6-dialkylphenyl-essigsäuren | |
| DE2331711A1 (de) | Verfahren zur selektiven elektrolytischen entbromierung | |
| DE2434845C3 (de) | Elektrochemische Herstellung aromatischer oder aromatisch-heterocyclischer Alkansäureester | |
| DE10146566A1 (de) | Verfahren zur Herstellung von Orthocarbonsäuretrialkylestern | |
| EP0085158A2 (de) | Verfahren zur Herstellung von Cycloalkenonderivaten | |
| EP0167097A1 (de) | Verfahren zur Herstellung von Chlorolefinen | |
| DE102004045029A1 (de) | Verfahren zur Herstellung von Glyoxalsäurealkylesterdialkylacetal | |
| EP0179377A1 (de) | Verfahren zur Herstellung von 1-Alkoxyisochromanen und neue 1-Alkoxy-alkylisochromane | |
| JPS6253506B2 (de) | ||
| DE4116147A1 (de) | Verfahren zur herstellung von 1-alkoxyisochromanen |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2596998 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11815930 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680004924.2 Country of ref document: CN Ref document number: 3554/CHENP/2007 Country of ref document: IN Ref document number: 1020077018649 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007555593 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006708252 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006708252 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 11815930 Country of ref document: US |