WO2006045017A1 - Coating compositions for cans and methods of coating - Google Patents
Coating compositions for cans and methods of coating Download PDFInfo
- Publication number
- WO2006045017A1 WO2006045017A1 PCT/US2005/037750 US2005037750W WO2006045017A1 WO 2006045017 A1 WO2006045017 A1 WO 2006045017A1 US 2005037750 W US2005037750 W US 2005037750W WO 2006045017 A1 WO2006045017 A1 WO 2006045017A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acid
- functional
- amine
- parts
- anhydride
- Prior art date
Links
- 239000008199 coating composition Substances 0.000 title claims abstract description 47
- 238000000576 coating method Methods 0.000 title claims description 127
- 239000011248 coating agent Substances 0.000 title claims description 82
- 238000000034 method Methods 0.000 title claims description 70
- 239000000178 monomer Substances 0.000 claims abstract description 170
- 229920000642 polymer Polymers 0.000 claims abstract description 97
- 235000013361 beverage Nutrition 0.000 claims abstract description 81
- 239000000839 emulsion Substances 0.000 claims abstract description 69
- 150000003839 salts Chemical class 0.000 claims abstract description 50
- 235000013305 food Nutrition 0.000 claims abstract description 49
- 229920001002 functional polymer Polymers 0.000 claims abstract description 45
- 229920000126 latex Polymers 0.000 claims abstract description 45
- 239000004816 latex Substances 0.000 claims abstract description 40
- 239000006185 dispersion Substances 0.000 claims abstract description 31
- 150000001412 amines Chemical class 0.000 claims abstract description 22
- 150000003512 tertiary amines Chemical class 0.000 claims abstract description 15
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 126
- 239000000203 mixture Substances 0.000 claims description 116
- 229910052751 metal Inorganic materials 0.000 claims description 59
- 239000002184 metal Substances 0.000 claims description 59
- 239000000758 substrate Substances 0.000 claims description 57
- -1 aromatic glycidyl ether compounds Chemical class 0.000 claims description 41
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 claims description 38
- ISAOCJYIOMOJEB-UHFFFAOYSA-N benzoin Chemical compound C=1C=CC=CC=1C(O)C(=O)C1=CC=CC=C1 ISAOCJYIOMOJEB-UHFFFAOYSA-N 0.000 claims description 36
- 125000000466 oxiranyl group Chemical group 0.000 claims description 25
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 claims description 22
- 229910052782 aluminium Inorganic materials 0.000 claims description 21
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 claims description 20
- 229960002887 deanol Drugs 0.000 claims description 20
- 244000028419 Styrax benzoin Species 0.000 claims description 18
- 235000000126 Styrax benzoin Nutrition 0.000 claims description 18
- 235000008411 Sumatra benzointree Nutrition 0.000 claims description 18
- 229960002130 benzoin Drugs 0.000 claims description 18
- 239000012972 dimethylethanolamine Substances 0.000 claims description 18
- 235000019382 gum benzoic Nutrition 0.000 claims description 18
- 239000003999 initiator Substances 0.000 claims description 18
- 239000003054 catalyst Substances 0.000 claims description 16
- 150000003254 radicals Chemical class 0.000 claims description 16
- 239000003795 chemical substances by application Substances 0.000 claims description 14
- 239000004971 Cross linker Substances 0.000 claims description 13
- 125000000962 organic group Chemical group 0.000 claims description 13
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 12
- 229920000058 polyacrylate Polymers 0.000 claims description 12
- 239000004094 surface-active agent Substances 0.000 claims description 12
- 239000000314 lubricant Substances 0.000 claims description 11
- 239000003960 organic solvent Substances 0.000 claims description 11
- 229920000728 polyester Polymers 0.000 claims description 10
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 claims description 9
- 229910000831 Steel Inorganic materials 0.000 claims description 9
- 239000010959 steel Substances 0.000 claims description 9
- 125000003118 aryl group Chemical group 0.000 claims description 8
- 239000000945 filler Substances 0.000 claims description 8
- 230000000379 polymerizing effect Effects 0.000 claims description 7
- 239000000049 pigment Substances 0.000 claims description 6
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 claims description 6
- 229920000180 alkyd Polymers 0.000 claims description 5
- 239000004606 Fillers/Extenders Substances 0.000 claims description 4
- 239000002318 adhesion promoter Substances 0.000 claims description 4
- 239000003963 antioxidant agent Substances 0.000 claims description 4
- 239000002270 dispersing agent Substances 0.000 claims description 4
- 239000000975 dye Substances 0.000 claims description 4
- 125000005549 heteroarylene group Chemical group 0.000 claims description 4
- 239000004611 light stabiliser Substances 0.000 claims description 4
- WGYKZJWCGVVSQN-UHFFFAOYSA-N mono-n-propyl amine Natural products CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 claims description 4
- 239000004814 polyurethane Substances 0.000 claims description 4
- 229920002635 polyurethane Polymers 0.000 claims description 4
- 239000013008 thixotropic agent Substances 0.000 claims description 4
- 229940086542 triethylamine Drugs 0.000 claims description 4
- MSXVEPNJUHWQHW-UHFFFAOYSA-N 2-methylbutan-2-ol Chemical compound CCC(C)(C)O MSXVEPNJUHWQHW-UHFFFAOYSA-N 0.000 claims description 3
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 claims description 3
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 claims description 3
- XXBDWLFCJWSEKW-UHFFFAOYSA-N dimethylbenzylamine Chemical compound CN(C)CC1=CC=CC=C1 XXBDWLFCJWSEKW-UHFFFAOYSA-N 0.000 claims description 3
- 229940031098 ethanolamine Drugs 0.000 claims description 3
- CRVGTESFCCXCTH-UHFFFAOYSA-N methyl diethanolamine Chemical compound OCCN(C)CCO CRVGTESFCCXCTH-UHFFFAOYSA-N 0.000 claims description 3
- HZAXFHJVJLSVMW-UHFFFAOYSA-N monoethanolamine hydrochloride Natural products NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 claims description 3
- DAZXVJBJRMWXJP-UHFFFAOYSA-N n,n-dimethylethylamine Chemical compound CCN(C)C DAZXVJBJRMWXJP-UHFFFAOYSA-N 0.000 claims description 3
- ZUHZZVMEUAUWHY-UHFFFAOYSA-N n,n-dimethylpropan-1-amine Chemical compound CCCN(C)C ZUHZZVMEUAUWHY-UHFFFAOYSA-N 0.000 claims description 3
- GNVRJGIVDSQCOP-UHFFFAOYSA-N n-ethyl-n-methylethanamine Chemical compound CCN(C)CC GNVRJGIVDSQCOP-UHFFFAOYSA-N 0.000 claims description 3
- 150000002978 peroxides Chemical class 0.000 claims description 3
- 229920001225 polyester resin Polymers 0.000 claims description 3
- 239000004645 polyester resin Substances 0.000 claims description 3
- 239000012966 redox initiator Substances 0.000 claims description 3
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 claims description 3
- 229960004418 trolamine Drugs 0.000 claims description 3
- 239000008367 deionised water Substances 0.000 description 76
- 229910021641 deionized water Inorganic materials 0.000 description 76
- 239000002253 acid Substances 0.000 description 67
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 54
- 239000000463 material Substances 0.000 description 43
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 41
- 150000001875 compounds Chemical class 0.000 description 40
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 34
- 239000007787 solid Substances 0.000 description 30
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 28
- 238000010438 heat treatment Methods 0.000 description 28
- 229910052757 nitrogen Inorganic materials 0.000 description 28
- 238000012360 testing method Methods 0.000 description 25
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 24
- 238000010992 reflux Methods 0.000 description 24
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical compound C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 description 23
- 125000004432 carbon atom Chemical group C* 0.000 description 21
- VOZRXNHHFUQHIL-UHFFFAOYSA-N glycidyl methacrylate Chemical compound CC(=C)C(=O)OCC1CO1 VOZRXNHHFUQHIL-UHFFFAOYSA-N 0.000 description 19
- SOGAXMICEFXMKE-UHFFFAOYSA-N Butylmethacrylate Chemical compound CCCCOC(=O)C(C)=C SOGAXMICEFXMKE-UHFFFAOYSA-N 0.000 description 18
- 239000000243 solution Substances 0.000 description 17
- 238000001723 curing Methods 0.000 description 16
- 238000009928 pasteurization Methods 0.000 description 16
- 125000000217 alkyl group Chemical group 0.000 description 15
- 230000008569 process Effects 0.000 description 15
- 238000009472 formulation Methods 0.000 description 14
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 13
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 13
- 150000008064 anhydrides Chemical group 0.000 description 13
- 238000006243 chemical reaction Methods 0.000 description 13
- 150000002148 esters Chemical class 0.000 description 13
- 239000000047 product Substances 0.000 description 13
- 239000007921 spray Substances 0.000 description 13
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 12
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 12
- 238000006116 polymerization reaction Methods 0.000 description 12
- 229920005989 resin Polymers 0.000 description 12
- 239000011347 resin Substances 0.000 description 12
- 239000004593 Epoxy Substances 0.000 description 11
- 229910052739 hydrogen Inorganic materials 0.000 description 11
- 239000001257 hydrogen Substances 0.000 description 11
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 11
- 238000002360 preparation method Methods 0.000 description 11
- 238000000605 extraction Methods 0.000 description 10
- 239000004615 ingredient Substances 0.000 description 10
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 10
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 10
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 9
- 150000007513 acids Chemical class 0.000 description 9
- CQEYYJKEWSMYFG-UHFFFAOYSA-N butyl acrylate Chemical compound CCCCOC(=O)C=C CQEYYJKEWSMYFG-UHFFFAOYSA-N 0.000 description 9
- 239000000126 substance Substances 0.000 description 9
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 8
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 8
- 125000001931 aliphatic group Chemical group 0.000 description 8
- 238000004806 packaging method and process Methods 0.000 description 8
- 239000003381 stabilizer Substances 0.000 description 8
- IAXXETNIOYFMLW-UHFFFAOYSA-N (4,7,7-trimethyl-3-bicyclo[2.2.1]heptanyl) 2-methylprop-2-enoate Chemical compound C1CC2(C)C(OC(=O)C(=C)C)CC1C2(C)C IAXXETNIOYFMLW-UHFFFAOYSA-N 0.000 description 7
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 7
- 125000002947 alkylene group Chemical group 0.000 description 7
- 238000004132 cross linking Methods 0.000 description 7
- 239000007789 gas Substances 0.000 description 7
- 150000002430 hydrocarbons Chemical group 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- 238000004519 manufacturing process Methods 0.000 description 7
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 6
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 6
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 6
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 6
- 125000002723 alicyclic group Chemical group 0.000 description 6
- 238000013461 design Methods 0.000 description 6
- 239000003599 detergent Substances 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 6
- 239000011976 maleic acid Substances 0.000 description 6
- 230000004048 modification Effects 0.000 description 6
- 238000012986 modification Methods 0.000 description 6
- 230000002441 reversible effect Effects 0.000 description 6
- CIHOLLKRGTVIJN-UHFFFAOYSA-N tert‐butyl hydroperoxide Chemical compound CC(C)(C)OO CIHOLLKRGTVIJN-UHFFFAOYSA-N 0.000 description 6
- 229920002554 vinyl polymer Polymers 0.000 description 6
- 229920002818 (Hydroxyethyl)methacrylate Polymers 0.000 description 5
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical class OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 5
- CIWBSHSKHKDKBQ-DUZGATOHSA-N D-araboascorbic acid Chemical class OC[C@@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-DUZGATOHSA-N 0.000 description 5
- JIGUQPWFLRLWPJ-UHFFFAOYSA-N Ethyl acrylate Chemical compound CCOC(=O)C=C JIGUQPWFLRLWPJ-UHFFFAOYSA-N 0.000 description 5
- WOBHKFSMXKNTIM-UHFFFAOYSA-N Hydroxyethyl methacrylate Chemical compound CC(=C)C(=O)OCCO WOBHKFSMXKNTIM-UHFFFAOYSA-N 0.000 description 5
- 229920000877 Melamine resin Polymers 0.000 description 5
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 5
- 239000012736 aqueous medium Substances 0.000 description 5
- 125000000732 arylene group Chemical group 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- 239000008151 electrolyte solution Substances 0.000 description 5
- 235000010350 erythorbic acid Nutrition 0.000 description 5
- 239000004318 erythorbic acid Chemical class 0.000 description 5
- 238000011156 evaluation Methods 0.000 description 5
- 229940026239 isoascorbic acid Drugs 0.000 description 5
- PNJWIWWMYCMZRO-UHFFFAOYSA-N pent‐4‐en‐2‐one Natural products CC(=O)CC=C PNJWIWWMYCMZRO-UHFFFAOYSA-N 0.000 description 5
- 229920001568 phenolic resin Polymers 0.000 description 5
- 150000002989 phenols Chemical class 0.000 description 5
- 229920000570 polyether Polymers 0.000 description 5
- 238000005507 spraying Methods 0.000 description 5
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 5
- JAHNSTQSQJOJLO-UHFFFAOYSA-N 2-(3-fluorophenyl)-1h-imidazole Chemical compound FC1=CC=CC(C=2NC=CN=2)=C1 JAHNSTQSQJOJLO-UHFFFAOYSA-N 0.000 description 4
- BRLQWZUYTZBJKN-UHFFFAOYSA-N Epichlorohydrin Chemical compound ClCC1CO1 BRLQWZUYTZBJKN-UHFFFAOYSA-N 0.000 description 4
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 4
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 4
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 229920006397 acrylic thermoplastic Polymers 0.000 description 4
- 230000002411 adverse Effects 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- 235000013405 beer Nutrition 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 229910052799 carbon Inorganic materials 0.000 description 4
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 4
- HNEGQIOMVPPMNR-IHWYPQMZSA-N citraconic acid Chemical compound OC(=O)C(/C)=C\C(O)=O HNEGQIOMVPPMNR-IHWYPQMZSA-N 0.000 description 4
- LDHQCZJRKDOVOX-NSCUHMNNSA-N crotonic acid Chemical compound C\C=C\C(O)=O LDHQCZJRKDOVOX-NSCUHMNNSA-N 0.000 description 4
- 125000004122 cyclic group Chemical group 0.000 description 4
- 238000004821 distillation Methods 0.000 description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 4
- 239000001530 fumaric acid Substances 0.000 description 4
- 125000003055 glycidyl group Chemical group C(C1CO1)* 0.000 description 4
- 230000003301 hydrolyzing effect Effects 0.000 description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 4
- 229910052742 iron Inorganic materials 0.000 description 4
- LVHBHZANLOWSRM-UHFFFAOYSA-N methylenebutanedioic acid Natural products OC(=O)CC(=C)C(O)=O LVHBHZANLOWSRM-UHFFFAOYSA-N 0.000 description 4
- HNEGQIOMVPPMNR-UHFFFAOYSA-N methylfumaric acid Natural products OC(=O)C(C)=CC(O)=O HNEGQIOMVPPMNR-UHFFFAOYSA-N 0.000 description 4
- 229910052760 oxygen Inorganic materials 0.000 description 4
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 description 4
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- ISXSCDLOGDJUNJ-UHFFFAOYSA-N tert-butyl prop-2-enoate Chemical compound CC(C)(C)OC(=O)C=C ISXSCDLOGDJUNJ-UHFFFAOYSA-N 0.000 description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 4
- LDHQCZJRKDOVOX-UHFFFAOYSA-N trans-crotonic acid Natural products CC=CC(O)=O LDHQCZJRKDOVOX-UHFFFAOYSA-N 0.000 description 4
- 239000004342 Benzoyl peroxide Substances 0.000 description 3
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 3
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 3
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 3
- YIMQCDZDWXUDCA-UHFFFAOYSA-N [4-(hydroxymethyl)cyclohexyl]methanol Chemical compound OCC1CCC(CO)CC1 YIMQCDZDWXUDCA-UHFFFAOYSA-N 0.000 description 3
- 150000001299 aldehydes Chemical class 0.000 description 3
- 125000003545 alkoxy group Chemical group 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 235000019400 benzoyl peroxide Nutrition 0.000 description 3
- PXKLMJQFEQBVLD-UHFFFAOYSA-N bisphenol F Chemical compound C1=CC(O)=CC=C1CC1=CC=C(O)C=C1 PXKLMJQFEQBVLD-UHFFFAOYSA-N 0.000 description 3
- XUCHXOAWJMEFLF-UHFFFAOYSA-N bisphenol F diglycidyl ether Chemical compound C1OC1COC(C=C1)=CC=C1CC(C=C1)=CC=C1OCC1CO1 XUCHXOAWJMEFLF-UHFFFAOYSA-N 0.000 description 3
- 238000005886 esterification reaction Methods 0.000 description 3
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 3
- 238000009863 impact test Methods 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- KCTAWXVAICEBSD-UHFFFAOYSA-N prop-2-enoyloxy prop-2-eneperoxoate Chemical compound C=CC(=O)OOOC(=O)C=C KCTAWXVAICEBSD-UHFFFAOYSA-N 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 230000001954 sterilising effect Effects 0.000 description 3
- 238000004659 sterilization and disinfection Methods 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 238000003466 welding Methods 0.000 description 3
- 239000008096 xylene Substances 0.000 description 3
- ZINGPVGWKVTAAC-IAROGAJJSA-N (2z,4e)-2-chlorohexa-2,4-dienoic acid Chemical compound C\C=C\C=C(/Cl)C(O)=O ZINGPVGWKVTAAC-IAROGAJJSA-N 0.000 description 2
- XVOUMQNXTGKGMA-OWOJBTEDSA-N (E)-glutaconic acid Chemical compound OC(=O)C\C=C\C(O)=O XVOUMQNXTGKGMA-OWOJBTEDSA-N 0.000 description 2
- 239000001124 (E)-prop-1-ene-1,2,3-tricarboxylic acid Substances 0.000 description 2
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 2
- MYRTYDVEIRVNKP-UHFFFAOYSA-N 1,2-Divinylbenzene Chemical compound C=CC1=CC=CC=C1C=C MYRTYDVEIRVNKP-UHFFFAOYSA-N 0.000 description 2
- QBDAFARLDLCWAT-UHFFFAOYSA-N 2,3-dihydropyran-6-one Chemical compound O=C1OCCC=C1 QBDAFARLDLCWAT-UHFFFAOYSA-N 0.000 description 2
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 description 2
- KUBDPQJOLOUJRM-UHFFFAOYSA-N 2-(chloromethyl)oxirane;4-[2-(4-hydroxyphenyl)propan-2-yl]phenol Chemical compound ClCC1CO1.C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 KUBDPQJOLOUJRM-UHFFFAOYSA-N 0.000 description 2
- FALRKNHUBBKYCC-UHFFFAOYSA-N 2-(chloromethyl)pyridine-3-carbonitrile Chemical compound ClCC1=NC=CC=C1C#N FALRKNHUBBKYCC-UHFFFAOYSA-N 0.000 description 2
- HIGURUTWFKYJCH-UHFFFAOYSA-N 2-[[1-(oxiran-2-ylmethoxymethyl)cyclohexyl]methoxymethyl]oxirane Chemical compound C1OC1COCC1(COCC2OC2)CCCCC1 HIGURUTWFKYJCH-UHFFFAOYSA-N 0.000 description 2
- POAOYUHQDCAZBD-UHFFFAOYSA-N 2-butoxyethanol Chemical compound CCCCOCCO POAOYUHQDCAZBD-UHFFFAOYSA-N 0.000 description 2
- SZTBMYHIYNGYIA-UHFFFAOYSA-N 2-chloroacrylic acid Chemical compound OC(=O)C(Cl)=C SZTBMYHIYNGYIA-UHFFFAOYSA-N 0.000 description 2
- IJVRPNIWWODHHA-UHFFFAOYSA-N 2-cyanoprop-2-enoic acid Chemical compound OC(=O)C(=C)C#N IJVRPNIWWODHHA-UHFFFAOYSA-N 0.000 description 2
- OMIGHNLMNHATMP-UHFFFAOYSA-N 2-hydroxyethyl prop-2-enoate Chemical compound OCCOC(=O)C=C OMIGHNLMNHATMP-UHFFFAOYSA-N 0.000 description 2
- ONPJWQSDZCGSQM-UHFFFAOYSA-N 2-phenylprop-2-enoic acid Chemical compound OC(=O)C(=C)C1=CC=CC=C1 ONPJWQSDZCGSQM-UHFFFAOYSA-N 0.000 description 2
- CYUZOYPRAQASLN-UHFFFAOYSA-N 3-prop-2-enoyloxypropanoic acid Chemical compound OC(=O)CCOC(=O)C=C CYUZOYPRAQASLN-UHFFFAOYSA-N 0.000 description 2
- GXLIFJYFGMHYDY-ZZXKWVIFSA-N 4-chlorocinnamic acid Chemical compound OC(=O)\C=C\C1=CC=C(Cl)C=C1 GXLIFJYFGMHYDY-ZZXKWVIFSA-N 0.000 description 2
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 2
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 2
- UIERETOOQGIECD-UHFFFAOYSA-N Angelic acid Natural products CC=C(C)C(O)=O UIERETOOQGIECD-UHFFFAOYSA-N 0.000 description 2
- KAKZBPTYRLMSJV-UHFFFAOYSA-N Butadiene Chemical compound C=CC=C KAKZBPTYRLMSJV-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 2
- RRHGJUQNOFWUDK-UHFFFAOYSA-N Isoprene Chemical compound CC(=C)C=C RRHGJUQNOFWUDK-UHFFFAOYSA-N 0.000 description 2
- 239000004952 Polyamide Substances 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 229940091181 aconitic acid Drugs 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 125000003342 alkenyl group Chemical group 0.000 description 2
- 125000000304 alkynyl group Chemical group 0.000 description 2
- 229920003180 amino resin Polymers 0.000 description 2
- UIERETOOQGIECD-ARJAWSKDSA-N angelic acid Chemical compound C\C=C(\C)C(O)=O UIERETOOQGIECD-ARJAWSKDSA-N 0.000 description 2
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- 229930016911 cinnamic acid Natural products 0.000 description 2
- 235000013985 cinnamic acid Nutrition 0.000 description 2
- GTZCVFVGUGFEME-IWQZZHSRSA-N cis-aconitic acid Chemical compound OC(=O)C\C(C(O)=O)=C\C(O)=O GTZCVFVGUGFEME-IWQZZHSRSA-N 0.000 description 2
- 229940018557 citraconic acid Drugs 0.000 description 2
- 239000007859 condensation product Substances 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- QYMFNZIUDRQRSA-UHFFFAOYSA-N dimethyl butanedioate;dimethyl hexanedioate;dimethyl pentanedioate Chemical compound COC(=O)CCC(=O)OC.COC(=O)CCCC(=O)OC.COC(=O)CCCCC(=O)OC QYMFNZIUDRQRSA-UHFFFAOYSA-N 0.000 description 2
- 150000002009 diols Chemical class 0.000 description 2
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 238000007720 emulsion polymerization reaction Methods 0.000 description 2
- 230000032050 esterification Effects 0.000 description 2
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- IVJISJACKSSFGE-UHFFFAOYSA-N formaldehyde;1,3,5-triazine-2,4,6-triamine Chemical compound O=C.NC1=NC(N)=NC(N)=N1 IVJISJACKSSFGE-UHFFFAOYSA-N 0.000 description 2
- 229910052736 halogen Inorganic materials 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 150000002367 halogens Chemical class 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- CWVFNAOMKFUALX-UHFFFAOYSA-N henicos-2-enoic acid Chemical compound CCCCCCCCCCCCCCCCCCC=CC(O)=O CWVFNAOMKFUALX-UHFFFAOYSA-N 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 2
- QQVIHTHCMHWDBS-UHFFFAOYSA-N isophthalic acid Chemical compound OC(=O)C1=CC=CC(C(O)=O)=C1 QQVIHTHCMHWDBS-UHFFFAOYSA-N 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- JDSHMPZPIAZGSV-UHFFFAOYSA-N melamine Chemical compound NC1=NC(N)=NC(N)=N1 JDSHMPZPIAZGSV-UHFFFAOYSA-N 0.000 description 2
- HNEGQIOMVPPMNR-NSCUHMNNSA-N mesaconic acid Chemical compound OC(=O)C(/C)=C/C(O)=O HNEGQIOMVPPMNR-NSCUHMNNSA-N 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 2
- SNVLJLYUUXKWOJ-UHFFFAOYSA-N methylidenecarbene Chemical compound C=[C] SNVLJLYUUXKWOJ-UHFFFAOYSA-N 0.000 description 2
- KCTMTGOHHMRJHZ-UHFFFAOYSA-N n-(2-methylpropoxymethyl)prop-2-enamide Chemical compound CC(C)COCNC(=O)C=C KCTMTGOHHMRJHZ-UHFFFAOYSA-N 0.000 description 2
- SLCVBVWXLSEKPL-UHFFFAOYSA-N neopentyl glycol Chemical compound OCC(C)(C)CO SLCVBVWXLSEKPL-UHFFFAOYSA-N 0.000 description 2
- 230000003472 neutralizing effect Effects 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- 229920002647 polyamide Polymers 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 229920013730 reactive polymer Polymers 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 229930195734 saturated hydrocarbon Natural products 0.000 description 2
- CXMXRPHRNRROMY-UHFFFAOYSA-N sebacic acid Chemical compound OC(=O)CCCCCCCCC(O)=O CXMXRPHRNRROMY-UHFFFAOYSA-N 0.000 description 2
- 229910052710 silicon Inorganic materials 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 2
- 235000010199 sorbic acid Nutrition 0.000 description 2
- 239000004334 sorbic acid Substances 0.000 description 2
- 229940075582 sorbic acid Drugs 0.000 description 2
- 229940014800 succinic anhydride Drugs 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 238000010998 test method Methods 0.000 description 2
- GTZCVFVGUGFEME-UHFFFAOYSA-N trans-aconitic acid Natural products OC(=O)CC(C(O)=O)=CC(O)=O GTZCVFVGUGFEME-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- 238000004383 yellowing Methods 0.000 description 2
- MARCAKLHFUYDJE-UHFFFAOYSA-N 1,2-xylene;hydrate Chemical group O.CC1=CC=CC=C1C MARCAKLHFUYDJE-UHFFFAOYSA-N 0.000 description 1
- YQMXOIAIYXXXEE-UHFFFAOYSA-N 1-benzylpyrrolidin-3-ol Chemical compound C1C(O)CCN1CC1=CC=CC=C1 YQMXOIAIYXXXEE-UHFFFAOYSA-N 0.000 description 1
- LHENQXAPVKABON-UHFFFAOYSA-N 1-methoxypropan-1-ol Chemical compound CCC(O)OC LHENQXAPVKABON-UHFFFAOYSA-N 0.000 description 1
- IGGDKDTUCAWDAN-UHFFFAOYSA-N 1-vinylnaphthalene Chemical compound C1=CC=C2C(C=C)=CC=CC2=C1 IGGDKDTUCAWDAN-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- STMDPCBYJCIZOD-UHFFFAOYSA-N 2-(2,4-dinitroanilino)-4-methylpentanoic acid Chemical compound CC(C)CC(C(O)=O)NC1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O STMDPCBYJCIZOD-UHFFFAOYSA-N 0.000 description 1
- JJRUAPNVLBABCN-UHFFFAOYSA-N 2-(ethenoxymethyl)oxirane Chemical compound C=COCC1CO1 JJRUAPNVLBABCN-UHFFFAOYSA-N 0.000 description 1
- KUAUJXBLDYVELT-UHFFFAOYSA-N 2-[[2,2-dimethyl-3-(oxiran-2-ylmethoxy)propoxy]methyl]oxirane Chemical compound C1OC1COCC(C)(C)COCC1CO1 KUAUJXBLDYVELT-UHFFFAOYSA-N 0.000 description 1
- JHEKSKQMOBLXQS-UHFFFAOYSA-N 2-cyclopentylphenol Chemical compound OC1=CC=CC=C1C1CCCC1 JHEKSKQMOBLXQS-UHFFFAOYSA-N 0.000 description 1
- WBIQQQGBSDOWNP-UHFFFAOYSA-N 2-dodecylbenzenesulfonic acid Chemical compound CCCCCCCCCCCCC1=CC=CC=C1S(O)(=O)=O WBIQQQGBSDOWNP-UHFFFAOYSA-N 0.000 description 1
- BOZRCGLDOHDZBP-UHFFFAOYSA-N 2-ethylhexanoic acid;tin Chemical compound [Sn].CCCCC(CC)C(O)=O BOZRCGLDOHDZBP-UHFFFAOYSA-N 0.000 description 1
- UUODQIKUTGWMPT-UHFFFAOYSA-N 2-fluoro-5-(trifluoromethyl)pyridine Chemical compound FC1=CC=C(C(F)(F)F)C=N1 UUODQIKUTGWMPT-UHFFFAOYSA-N 0.000 description 1
- VHSHLMUCYSAUQU-UHFFFAOYSA-N 2-hydroxypropyl methacrylate Chemical compound CC(O)COC(=O)C(C)=C VHSHLMUCYSAUQU-UHFFFAOYSA-N 0.000 description 1
- CWNNYYIZGGDCHS-UHFFFAOYSA-N 2-methylideneglutaric acid Chemical compound OC(=O)CCC(=C)C(O)=O CWNNYYIZGGDCHS-UHFFFAOYSA-N 0.000 description 1
- QTWJRLJHJPIABL-UHFFFAOYSA-N 2-methylphenol;3-methylphenol;4-methylphenol Chemical compound CC1=CC=C(O)C=C1.CC1=CC=CC(O)=C1.CC1=CC=CC=C1O QTWJRLJHJPIABL-UHFFFAOYSA-N 0.000 description 1
- JLBJTVDPSNHSKJ-UHFFFAOYSA-N 4-Methylstyrene Chemical compound CC1=CC=C(C=C)C=C1 JLBJTVDPSNHSKJ-UHFFFAOYSA-N 0.000 description 1
- WWYFPDXEIFBNKE-UHFFFAOYSA-M 4-carboxybenzyl alcohol Chemical compound OCC1=CC=C(C([O-])=O)C=C1 WWYFPDXEIFBNKE-UHFFFAOYSA-M 0.000 description 1
- 229940090248 4-hydroxybenzoic acid Drugs 0.000 description 1
- QDBFCZCIBRFQBR-UHFFFAOYSA-N 4-hydroxybutyl benzoate Chemical compound OCCCCOC(=O)C1=CC=CC=C1 QDBFCZCIBRFQBR-UHFFFAOYSA-N 0.000 description 1
- QHPQWRBYOIRBIT-UHFFFAOYSA-N 4-tert-butylphenol Chemical compound CC(C)(C)C1=CC=C(O)C=C1 QHPQWRBYOIRBIT-UHFFFAOYSA-N 0.000 description 1
- GZVHEAJQGPRDLQ-UHFFFAOYSA-N 6-phenyl-1,3,5-triazine-2,4-diamine Chemical compound NC1=NC(N)=NC(C=2C=CC=CC=2)=N1 GZVHEAJQGPRDLQ-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 1
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 1
- 229920004688 Altek® Polymers 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 229920003270 Cymel® Polymers 0.000 description 1
- 239000004641 Diallyl-phthalate Substances 0.000 description 1
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Natural products CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 1
- CWYNVVGOOAEACU-UHFFFAOYSA-N Fe2+ Chemical class [Fe+2] CWYNVVGOOAEACU-UHFFFAOYSA-N 0.000 description 1
- 239000005057 Hexamethylene diisocyanate Substances 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- GYCMBHHDWRMZGG-UHFFFAOYSA-N Methylacrylonitrile Chemical compound CC(=C)C#N GYCMBHHDWRMZGG-UHFFFAOYSA-N 0.000 description 1
- IGFHQQFPSIBGKE-UHFFFAOYSA-N Nonylphenol Natural products CCCCCCCCCC1=CC=C(O)C=C1 IGFHQQFPSIBGKE-UHFFFAOYSA-N 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000004721 Polyphenylene oxide Substances 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- 229920001807 Urea-formaldehyde Polymers 0.000 description 1
- XTXRWKRVRITETP-UHFFFAOYSA-N Vinyl acetate Chemical compound CC(=O)OC=C XTXRWKRVRITETP-UHFFFAOYSA-N 0.000 description 1
- IKHGUXGNUITLKF-XPULMUKRSA-N acetaldehyde Chemical compound [14CH]([14CH3])=O IKHGUXGNUITLKF-XPULMUKRSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 125000005250 alkyl acrylate group Chemical group 0.000 description 1
- 125000002877 alkyl aryl group Chemical group 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- XYLMUPLGERFSHI-UHFFFAOYSA-N alpha-Methylstyrene Chemical compound CC(=C)C1=CC=CC=C1 XYLMUPLGERFSHI-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- ROOXNKNUYICQNP-UHFFFAOYSA-N ammonium peroxydisulfate Substances [NH4+].[NH4+].[O-]S(=O)(=O)OOS([O-])(=O)=O ROOXNKNUYICQNP-UHFFFAOYSA-N 0.000 description 1
- VAZSKTXWXKYQJF-UHFFFAOYSA-N ammonium persulfate Chemical compound [NH4+].[NH4+].[O-]S(=O)OOS([O-])=O VAZSKTXWXKYQJF-UHFFFAOYSA-N 0.000 description 1
- 229910001870 ammonium persulfate Inorganic materials 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- LPTWEDZIPSKWDG-UHFFFAOYSA-N benzenesulfonic acid;dodecane Chemical compound OS(=O)(=O)C1=CC=CC=C1.CCCCCCCCCCCC LPTWEDZIPSKWDG-UHFFFAOYSA-N 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- MTZQAGJQAFMTAQ-UHFFFAOYSA-N benzoic acid ethyl ester Natural products CCOC(=O)C1=CC=CC=C1 MTZQAGJQAFMTAQ-UHFFFAOYSA-N 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- YXVFYQXJAXKLAK-UHFFFAOYSA-N biphenyl-4-ol Chemical compound C1=CC(O)=CC=C1C1=CC=CC=C1 YXVFYQXJAXKLAK-UHFFFAOYSA-N 0.000 description 1
- 125000002529 biphenylenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3C12)* 0.000 description 1
- MLVSWIXRZNPEKF-UPHRSURJSA-N bis(oxiran-2-ylmethyl) (z)-but-2-enedioate Chemical compound C1OC1COC(=O)\C=C/C(=O)OCC1CO1 MLVSWIXRZNPEKF-UPHRSURJSA-N 0.000 description 1
- NRTSLUOVGBFANI-UHFFFAOYSA-N bis(oxiran-2-ylmethyl) 2-methylidenebutanedioate Chemical compound C1OC1COC(=O)C(=C)CC(=O)OCC1CO1 NRTSLUOVGBFANI-UHFFFAOYSA-N 0.000 description 1
- QUDWYFHPNIMBFC-UHFFFAOYSA-N bis(prop-2-enyl) benzene-1,2-dicarboxylate Chemical compound C=CCOC(=O)C1=CC=CC=C1C(=O)OCC=C QUDWYFHPNIMBFC-UHFFFAOYSA-N 0.000 description 1
- RLYNGYDVXRKEOO-SQQVDAMQSA-N but-2-enoic acid;(e)-but-2-enoic acid Chemical compound CC=CC(O)=O.C\C=C\C(O)=O RLYNGYDVXRKEOO-SQQVDAMQSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000004181 carboxyalkyl group Chemical group 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000004203 carnauba wax Substances 0.000 description 1
- 235000013869 carnauba wax Nutrition 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000013626 chemical specie Substances 0.000 description 1
- 230000015271 coagulation Effects 0.000 description 1
- 238000005345 coagulation Methods 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 230000007797 corrosion Effects 0.000 description 1
- 238000005260 corrosion Methods 0.000 description 1
- 238000005336 cracking Methods 0.000 description 1
- 229930003836 cresol Natural products 0.000 description 1
- MLUCVPSAIODCQM-NSCUHMNNSA-N crotonaldehyde Chemical compound C\C=C\C=O MLUCVPSAIODCQM-NSCUHMNNSA-N 0.000 description 1
- MLUCVPSAIODCQM-UHFFFAOYSA-N crotonaldehyde Natural products CC=CC=O MLUCVPSAIODCQM-UHFFFAOYSA-N 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000004956 cyclohexylene group Chemical group 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 210000003298 dental enamel Anatomy 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 150000001991 dicarboxylic acids Chemical class 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- GYZLOYUZLJXAJU-UHFFFAOYSA-N diglycidyl ether Chemical compound C1OC1COCC1CO1 GYZLOYUZLJXAJU-UHFFFAOYSA-N 0.000 description 1
- 230000003292 diminished effect Effects 0.000 description 1
- 238000007598 dipping method Methods 0.000 description 1
- SZXQTJUDPRGNJN-UHFFFAOYSA-N dipropylene glycol Chemical compound OCCCOCCCO SZXQTJUDPRGNJN-UHFFFAOYSA-N 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 238000004070 electrodeposition Methods 0.000 description 1
- MEGHWIAOTJPCHQ-UHFFFAOYSA-N ethenyl butanoate Chemical compound CCCC(=O)OC=C MEGHWIAOTJPCHQ-UHFFFAOYSA-N 0.000 description 1
- AFSIMBWBBOJPJG-UHFFFAOYSA-N ethenyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC=C AFSIMBWBBOJPJG-UHFFFAOYSA-N 0.000 description 1
- UIWXSTHGICQLQT-UHFFFAOYSA-N ethenyl propanoate Chemical compound CCC(=O)OC=C UIWXSTHGICQLQT-UHFFFAOYSA-N 0.000 description 1
- 125000001033 ether group Chemical group 0.000 description 1
- SLAFUPJSGFVWPP-UHFFFAOYSA-M ethyl(triphenyl)phosphanium;iodide Chemical compound [I-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(CC)C1=CC=CC=C1 SLAFUPJSGFVWPP-UHFFFAOYSA-M 0.000 description 1
- 238000007765 extrusion coating Methods 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 125000005567 fluorenylene group Chemical group 0.000 description 1
- MSYLJRIXVZCQHW-UHFFFAOYSA-N formaldehyde;6-phenyl-1,3,5-triazine-2,4-diamine Chemical compound O=C.NC1=NC(N)=NC(C=2C=CC=CC=2)=N1 MSYLJRIXVZCQHW-UHFFFAOYSA-N 0.000 description 1
- 238000010528 free radical solution polymerization reaction Methods 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 238000012812 general test Methods 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- RRAMGCGOFNQTLD-UHFFFAOYSA-N hexamethylene diisocyanate Chemical compound O=C=NCCCCCCN=C=O RRAMGCGOFNQTLD-UHFFFAOYSA-N 0.000 description 1
- XXMIOPMDWAUFGU-UHFFFAOYSA-N hexane-1,6-diol Chemical compound OCCCCCCO XXMIOPMDWAUFGU-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229920001519 homopolymer Polymers 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical class [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 238000005304 joining Methods 0.000 description 1
- 238000010030 laminating Methods 0.000 description 1
- 239000004850 liquid epoxy resins (LERs) Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 150000002734 metacrylic acid derivatives Chemical class 0.000 description 1
- FQPSGWSUVKBHSU-UHFFFAOYSA-N methacrylamide Chemical compound CC(=C)C(N)=O FQPSGWSUVKBHSU-UHFFFAOYSA-N 0.000 description 1
- DCUFMVPCXCSVNP-UHFFFAOYSA-N methacrylic anhydride Chemical compound CC(=C)C(=O)OC(=O)C(C)=C DCUFMVPCXCSVNP-UHFFFAOYSA-N 0.000 description 1
- WSFSSNUMVMOOMR-NJFSPNSNSA-N methanone Chemical compound O=[14CH2] WSFSSNUMVMOOMR-NJFSPNSNSA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 150000002763 monocarboxylic acids Chemical class 0.000 description 1
- UTSYWKJYFPPRAP-UHFFFAOYSA-N n-(butoxymethyl)prop-2-enamide Chemical compound CCCCOCNC(=O)C=C UTSYWKJYFPPRAP-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004957 naphthylene group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 125000004971 nitroalkyl group Chemical group 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- SNQQPOLDUKLAAF-UHFFFAOYSA-N nonylphenol Chemical compound CCCCCCCCCC1=CC=CC=C1O SNQQPOLDUKLAAF-UHFFFAOYSA-N 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000001151 other effect Effects 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- RPQRDASANLAFCM-UHFFFAOYSA-N oxiran-2-ylmethyl prop-2-enoate Chemical compound C=CC(=O)OCC1CO1 RPQRDASANLAFCM-UHFFFAOYSA-N 0.000 description 1
- NRZWYNLTFLDQQX-UHFFFAOYSA-N p-tert-Amylphenol Chemical compound CCC(C)(C)C1=CC=C(O)C=C1 NRZWYNLTFLDQQX-UHFFFAOYSA-N 0.000 description 1
- 239000005022 packaging material Substances 0.000 description 1
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- LSMAIBOZUPTNBR-UHFFFAOYSA-N phosphanium;iodide Chemical compound [PH4+].[I-] LSMAIBOZUPTNBR-UHFFFAOYSA-N 0.000 description 1
- XYFCBTPGUUZFHI-UHFFFAOYSA-O phosphonium Chemical compound [PH4+] XYFCBTPGUUZFHI-UHFFFAOYSA-O 0.000 description 1
- 125000005496 phosphonium group Chemical group 0.000 description 1
- 150000004714 phosphonium salts Chemical class 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920005749 polyurethane resin Polymers 0.000 description 1
- USHAGKDGDHPEEY-UHFFFAOYSA-L potassium persulfate Chemical compound [K+].[K+].[O-]S(=O)(=O)OOS([O-])(=O)=O USHAGKDGDHPEEY-UHFFFAOYSA-L 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- ARJOQCYCJMAIFR-UHFFFAOYSA-N prop-2-enoyl prop-2-enoate Chemical compound C=CC(=O)OC(=O)C=C ARJOQCYCJMAIFR-UHFFFAOYSA-N 0.000 description 1
- FBCQUCJYYPMKRO-UHFFFAOYSA-N prop-2-enyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCC=C FBCQUCJYYPMKRO-UHFFFAOYSA-N 0.000 description 1
- HJWLCRVIBGQPNF-UHFFFAOYSA-N prop-2-enylbenzene Chemical compound C=CCC1=CC=CC=C1 HJWLCRVIBGQPNF-UHFFFAOYSA-N 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000005551 pyridylene group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 238000005956 quaternization reaction Methods 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 238000010526 radical polymerization reaction Methods 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 239000013557 residual solvent Substances 0.000 description 1
- 238000005096 rolling process Methods 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 125000004079 stearyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004964 sulfoalkyl group Chemical group 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 1
- 125000005247 tetrazinyl group Chemical group N1=NN=NC(=C1)* 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- 238000005809 transesterification reaction Methods 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 1
- SRPWOOOHEPICQU-UHFFFAOYSA-N trimellitic anhydride Chemical compound OC(=O)C1=CC=C2C(=O)OC(=O)C2=C1 SRPWOOOHEPICQU-UHFFFAOYSA-N 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 230000002087 whitening effect Effects 0.000 description 1
- 150000003752 zinc compounds Chemical class 0.000 description 1
- CHJMFFKHPHCQIJ-UHFFFAOYSA-L zinc;octanoate Chemical compound [Zn+2].CCCCCCCC([O-])=O.CCCCCCCC([O-])=O CHJMFFKHPHCQIJ-UHFFFAOYSA-L 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D123/00—Coating compositions based on homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Coating compositions based on derivatives of such polymers
- C09D123/02—Coating compositions based on homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Coating compositions based on derivatives of such polymers not modified by chemical after-treatment
- C09D123/04—Homopolymers or copolymers of ethene
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B65—CONVEYING; PACKING; STORING; HANDLING THIN OR FILAMENTARY MATERIAL
- B65D—CONTAINERS FOR STORAGE OR TRANSPORT OF ARTICLES OR MATERIALS, e.g. BAGS, BARRELS, BOTTLES, BOXES, CANS, CARTONS, CRATES, DRUMS, JARS, TANKS, HOPPERS, FORWARDING CONTAINERS; ACCESSORIES, CLOSURES, OR FITTINGS THEREFOR; PACKAGING ELEMENTS; PACKAGES
- B65D17/00—Rigid or semi-rigid containers specially constructed to be opened by cutting or piercing, or by tearing of frangible members or portions
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B05—SPRAYING OR ATOMISING IN GENERAL; APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D—PROCESSES FOR APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D1/00—Processes for applying liquids or other fluent materials
- B05D1/02—Processes for applying liquids or other fluent materials performed by spraying
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B05—SPRAYING OR ATOMISING IN GENERAL; APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D—PROCESSES FOR APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D3/00—Pretreatment of surfaces to which liquids or other fluent materials are to be applied; After-treatment of applied coatings, e.g. intermediate treating of an applied coating preparatory to subsequent applications of liquids or other fluent materials
- B05D3/007—After-treatment
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B05—SPRAYING OR ATOMISING IN GENERAL; APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D—PROCESSES FOR APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D7/00—Processes, other than flocking, specially adapted for applying liquids or other fluent materials to particular surfaces or for applying particular liquids or other fluent materials
- B05D7/22—Processes, other than flocking, specially adapted for applying liquids or other fluent materials to particular surfaces or for applying particular liquids or other fluent materials to internal surfaces, e.g. of tubes
- B05D7/227—Processes, other than flocking, specially adapted for applying liquids or other fluent materials to particular surfaces or for applying particular liquids or other fluent materials to internal surfaces, e.g. of tubes of containers, cans or the like
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B65—CONVEYING; PACKING; STORING; HANDLING THIN OR FILAMENTARY MATERIAL
- B65D—CONTAINERS FOR STORAGE OR TRANSPORT OF ARTICLES OR MATERIALS, e.g. BAGS, BARRELS, BOTTLES, BOXES, CANS, CARTONS, CRATES, DRUMS, JARS, TANKS, HOPPERS, FORWARDING CONTAINERS; ACCESSORIES, CLOSURES, OR FITTINGS THEREFOR; PACKAGING ELEMENTS; PACKAGES
- B65D25/00—Details of other kinds or types of rigid or semi-rigid containers
- B65D25/14—Linings or internal coatings
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/12—Polymerisation in non-solvents
- C08F2/16—Aqueous medium
- C08F2/22—Emulsion polymerisation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/10—Esters
- C08F220/12—Esters of monohydric alcohols or phenols
- C08F220/16—Esters of monohydric alcohols or phenols of phenols or of alcohols containing two or more carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F265/00—Macromolecular compounds obtained by polymerising monomers on to polymers of unsaturated monocarboxylic acids or derivatives thereof as defined in group C08F20/00
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F265/00—Macromolecular compounds obtained by polymerising monomers on to polymers of unsaturated monocarboxylic acids or derivatives thereof as defined in group C08F20/00
- C08F265/02—Macromolecular compounds obtained by polymerising monomers on to polymers of unsaturated monocarboxylic acids or derivatives thereof as defined in group C08F20/00 on to polymers of acids, salts or anhydrides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F265/00—Macromolecular compounds obtained by polymerising monomers on to polymers of unsaturated monocarboxylic acids or derivatives thereof as defined in group C08F20/00
- C08F265/04—Macromolecular compounds obtained by polymerising monomers on to polymers of unsaturated monocarboxylic acids or derivatives thereof as defined in group C08F20/00 on to polymers of esters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F265/00—Macromolecular compounds obtained by polymerising monomers on to polymers of unsaturated monocarboxylic acids or derivatives thereof as defined in group C08F20/00
- C08F265/04—Macromolecular compounds obtained by polymerising monomers on to polymers of unsaturated monocarboxylic acids or derivatives thereof as defined in group C08F20/00 on to polymers of esters
- C08F265/06—Polymerisation of acrylate or methacrylate esters on to polymers thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F265/00—Macromolecular compounds obtained by polymerising monomers on to polymers of unsaturated monocarboxylic acids or derivatives thereof as defined in group C08F20/00
- C08F265/10—Macromolecular compounds obtained by polymerising monomers on to polymers of unsaturated monocarboxylic acids or derivatives thereof as defined in group C08F20/00 on to polymers of amides or imides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F283/00—Macromolecular compounds obtained by polymerising monomers on to polymers provided for in subclass C08G
- C08F283/006—Macromolecular compounds obtained by polymerising monomers on to polymers provided for in subclass C08G on to polymers provided for in C08G18/00
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F283/00—Macromolecular compounds obtained by polymerising monomers on to polymers provided for in subclass C08G
- C08F283/02—Macromolecular compounds obtained by polymerising monomers on to polymers provided for in subclass C08G on to polycarbonates or saturated polyesters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F291/00—Macromolecular compounds obtained by polymerising monomers on to macromolecular compounds according to more than one of the groups C08F251/00 - C08F289/00
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D123/00—Coating compositions based on homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Coating compositions based on derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D133/00—Coating compositions based on homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Coating compositions based on derivatives of such polymers
- C09D133/04—Homopolymers or copolymers of esters
- C09D133/06—Homopolymers or copolymers of esters of esters containing only carbon, hydrogen and oxygen, the oxygen atom being present only as part of the carboxyl radical
- C09D133/062—Copolymers with monomers not covered by C09D133/06
- C09D133/068—Copolymers with monomers not covered by C09D133/06 containing glycidyl groups
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D133/00—Coating compositions based on homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Coating compositions based on derivatives of such polymers
- C09D133/04—Homopolymers or copolymers of esters
- C09D133/14—Homopolymers or copolymers of esters of esters containing halogen, nitrogen, sulfur or oxygen atoms in addition to the carboxy oxygen
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D151/00—Coating compositions based on graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Coating compositions based on derivatives of such polymers
- C09D151/003—Coating compositions based on graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Coating compositions based on derivatives of such polymers grafted on to macromolecular compounds obtained by reactions only involving unsaturated carbon-to-carbon bonds
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D151/00—Coating compositions based on graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Coating compositions based on derivatives of such polymers
- C09D151/08—Coating compositions based on graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Coating compositions based on derivatives of such polymers grafted on to macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D4/00—Coating compositions, e.g. paints, varnishes or lacquers, based on organic non-macromolecular compounds having at least one polymerisable carbon-to-carbon unsaturated bond ; Coating compositions, based on monomers of macromolecular compounds of groups C09D183/00 - C09D183/16
- C09D4/06—Organic non-macromolecular compounds having at least one polymerisable carbon-to-carbon unsaturated bond in combination with a macromolecular compound other than an unsaturated polymer of groups C09D159/00 - C09D187/00
-
- C—CHEMISTRY; METALLURGY
- C23—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; CHEMICAL SURFACE TREATMENT; DIFFUSION TREATMENT OF METALLIC MATERIAL; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL; INHIBITING CORROSION OF METALLIC MATERIAL OR INCRUSTATION IN GENERAL
- C23C—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; SURFACE TREATMENT OF METALLIC MATERIAL BY DIFFUSION INTO THE SURFACE, BY CHEMICAL CONVERSION OR SUBSTITUTION; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL
- C23C4/00—Coating by spraying the coating material in the molten state, e.g. by flame, plasma or electric discharge
- C23C4/12—Coating by spraying the coating material in the molten state, e.g. by flame, plasma or electric discharge characterised by the method of spraying
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B05—SPRAYING OR ATOMISING IN GENERAL; APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D—PROCESSES FOR APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D2202/00—Metallic substrate
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B05—SPRAYING OR ATOMISING IN GENERAL; APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D—PROCESSES FOR APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D2202/00—Metallic substrate
- B05D2202/20—Metallic substrate based on light metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B05—SPRAYING OR ATOMISING IN GENERAL; APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D—PROCESSES FOR APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D2202/00—Metallic substrate
- B05D2202/20—Metallic substrate based on light metals
- B05D2202/25—Metallic substrate based on light metals based on Al
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B05—SPRAYING OR ATOMISING IN GENERAL; APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D—PROCESSES FOR APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D2254/00—Tubes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B05—SPRAYING OR ATOMISING IN GENERAL; APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D—PROCESSES FOR APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D2254/00—Tubes
- B05D2254/04—Applying the material on the interior of the tube
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B05—SPRAYING OR ATOMISING IN GENERAL; APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D—PROCESSES FOR APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D2259/00—Applying the material to the internal surface of hollow articles other than tubes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B05—SPRAYING OR ATOMISING IN GENERAL; APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D—PROCESSES FOR APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D2401/00—Form of the coating product, e.g. solution, water dispersion, powders or the like
- B05D2401/20—Aqueous dispersion or solution
- B05D2401/21—Mixture of organic solvent and water
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B05—SPRAYING OR ATOMISING IN GENERAL; APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D—PROCESSES FOR APPLYING FLUENT MATERIALS TO SURFACES, IN GENERAL
- B05D2520/00—Water-based dispersions
- B05D2520/05—Latex
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B15/00—Layered products comprising a layer of metal
- B32B15/04—Layered products comprising a layer of metal comprising metal as the main or only constituent of a layer, which is next to another layer of the same or of a different material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B15/00—Layered products comprising a layer of metal
- B32B15/04—Layered products comprising a layer of metal comprising metal as the main or only constituent of a layer, which is next to another layer of the same or of a different material
- B32B15/06—Layered products comprising a layer of metal comprising metal as the main or only constituent of a layer, which is next to another layer of the same or of a different material of natural rubber or synthetic rubber
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B15/00—Layered products comprising a layer of metal
- B32B15/20—Layered products comprising a layer of metal comprising aluminium or copper
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S525/00—Synthetic resins or natural rubbers -- part of the class 520 series
- Y10S525/922—Polyepoxide polymer having been reacted to yield terminal ethylenic unsaturation
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S525/00—Synthetic resins or natural rubbers -- part of the class 520 series
- Y10S525/93—Reaction product of a polyhydric phenol and epichlorohydrin or diepoxide, having a molecular weight of over 5,000, e.g. phenoxy resins
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/13—Hollow or container type article [e.g., tube, vase, etc.]
- Y10T428/1352—Polymer or resin containing [i.e., natural or synthetic]
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/13—Hollow or container type article [e.g., tube, vase, etc.]
- Y10T428/1352—Polymer or resin containing [i.e., natural or synthetic]
- Y10T428/1355—Elemental metal containing [e.g., substrate, foil, film, coating, etc.]
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/13—Hollow or container type article [e.g., tube, vase, etc.]
- Y10T428/1352—Polymer or resin containing [i.e., natural or synthetic]
- Y10T428/1386—Natural or synthetic rubber or rubber-like compound containing
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/13—Hollow or container type article [e.g., tube, vase, etc.]
- Y10T428/1352—Polymer or resin containing [i.e., natural or synthetic]
- Y10T428/139—Open-ended, self-supporting conduit, cylinder, or tube-type article
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/13—Hollow or container type article [e.g., tube, vase, etc.]
- Y10T428/1352—Polymer or resin containing [i.e., natural or synthetic]
- Y10T428/139—Open-ended, self-supporting conduit, cylinder, or tube-type article
- Y10T428/1393—Multilayer [continuous layer]
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/31504—Composite [nonstructural laminate]
- Y10T428/31551—Of polyamidoester [polyurethane, polyisocyanate, polycarbamate, etc.]
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/31504—Composite [nonstructural laminate]
- Y10T428/31678—Of metal
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/31504—Composite [nonstructural laminate]
- Y10T428/31678—Of metal
- Y10T428/31681—Next to polyester, polyamide or polyimide [e.g., alkyd, glue, or nylon, etc.]
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/31504—Composite [nonstructural laminate]
- Y10T428/31678—Of metal
- Y10T428/31688—Next to aldehyde or ketone condensation product
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/31504—Composite [nonstructural laminate]
- Y10T428/31678—Of metal
- Y10T428/31692—Next to addition polymer from unsaturated monomers
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/31504—Composite [nonstructural laminate]
- Y10T428/31678—Of metal
- Y10T428/31692—Next to addition polymer from unsaturated monomers
- Y10T428/31696—Including polyene monomers [e.g., butadiene, etc.]
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/31504—Composite [nonstructural laminate]
- Y10T428/31678—Of metal
- Y10T428/31692—Next to addition polymer from unsaturated monomers
- Y10T428/31699—Ester, halide or nitrile of addition polymer
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/31504—Composite [nonstructural laminate]
- Y10T428/31855—Of addition polymer from unsaturated monomers
Definitions
- a wide variety of coatings have been used to coat the surfaces of packaging articles (e.g., food and beverage cans).
- metal cans are sometimes coated using "coil coating” or "sheet coating” operations, i.e., a planar coil or sheet of a suitable substrate (e.g., steel or aluminum metal) is coated with a suitable composition and hardened (e.g., cured). The coated substrate then is formed into the can end or body.
- liquid coating compositions may be applied (e.g., by spraying, dipping, rolling, etc.) to the formed article and then hardened (e.g., cured).
- Packaging coatings should preferably be capable of high-speed application to the substrate and provide the necessary properties when hardened to perform in this demanding end use.
- the coating should be safe for food contact, have excellent adhesion to the substrate, and resist degradation over long periods of time, even when exposed to harsh environments.
- BPA bisphenol A
- PVC aromatic glycidyl ether compounds
- This invention provides a coating composition for a food or beverage can that includes an emulsion polymerized latex polymer.
- This polymer is formed by combining an ethylenically unsaturated monomer component with an aqueous dispersion of a salt of an acid- or anhydride-functional polymer (i.e., an acid group- or anhydride group-containing polymer) and an amine, preferably, a tertiary amine, and then polymerizing the monomer component.
- the ethylenically unsaturated monomer component is preferably a mixture of monomers. At least one of the monomers in the mixture is preferably an alpha, beta- unsaturated monomer, and at least one monomer is preferably an oxirane functional monomer. More preferably, at least one of the monomers in the mixture is an oxirane group-containing alpha, beta-ethylenically unsaturated monomer.
- a method of preparing a food or beverage can includes: forming a composition that includes an emulsion polymerized latex polymer, including: forming a salt of an acid- or anhydride- functional polymer and an amine in a carrier comprising water (and an optional organic solvent) to form an aqueous dispersion; combining an ethylenically unsaturated monomer component with the aqueous dispersion; and polymerizing the ethylenically unsaturated monomer component in the presence of the aqueous dispersion to form an emulsion polymerized latex polymer; and applying the composition including the emulsion polymerized latex polymer to a metal substrate prior to or after forming the metal substrate into a food or beverage can or portion thereof.
- the method includes: forming a composition including an emulsion polymerized latex polymer, including: forming a salt of an acid- or anhydride-functional polymer and a tertiary amine in a carrier comprising water (and an optional organic solvent) to form an aqueous dispersion; combining an ethylenically unsaturated monomer component comprising 0.1 wt-% to 30 wt-% of an oxirane-functional alpha, beta-ethylenically unsaturated monomer with the aqueous dispersion, based on the weight of the monomer component; and polymerizing the ethylenically unsaturated monomer component in the presence of the aqueous dispersion to form an emulsion polymerized latex polymer; and applying the composition comprising the emulsion polymerized latex polymer to a metal substrate prior to or after forming the metal substrate into a food or beverage can or portion thereof.
- the composition can include an organic solvent in the aqueous dispersion.
- the method can include removing at least a portion of the organic solvent, if present, from the aqueous dispersion.
- applying the composition to a metal substrate includes applying the composition to the metal substrate in the form of a planar coil or sheet, hardening the emulsion polymerized latex polymer, and forming the substrate into a food or beverage can or portions thereof.
- applying the composition to a metal substrate comprises applying the composition to the metal substrate after the metal substrate is formed into a can or portion thereof.
- forming the substrate into a can or portion thereof includes forming the substrate into a can end or a can body.
- the can is a 2-piece drawn food can, 3-piece food can, food can end, drawn and ironed food or beverage can, beverage can end, and the like.
- the metal substrate can be steel or aluminum.
- combining an ethylenically unsaturated monomer component with the aqueous dispersion includes adding the ethylenically unsaturated monomer component to the aqueous dispersion.
- the ethylenically unsaturated monomer component is added incrementally to the aqueous dispersion.
- the ethylenically unsaturated monomer component includes a mixture of monomers.
- the mixture of monomers includes at least one oxirane functional group-containing monomer, and more preferably, at least one oxirane functional group-containing alpha, beta-ethylenically unsaturated monomer.
- the oxirane functional group-containing monomer is present in the ethylenically unsaturated monomer component in an amount of at least 0.1 wt-%, based on the weight of the monomer mixture.
- the oxirane functional group-containing monomer is present in the ethylenically unsaturated monomer component in an amount of no greater than 30 wt-%, based on the weight of the monomer mixture.
- the methods of the present invention further include combining the emulsion polymerized latex polymer with one or more crosslinkers, fillers, catalysts, dyes, pigments, toners, extenders, lubricants, anticorrosion agents, flow control agents, thixotropic agents, dispersing agents, antioxidants, adhesion promoters, light stabilizers, organic solvents, surfactants or combinations thereof in the coating composition.
- the acid-functional polymer has a number average molecular weight of 1500 to 50,000.
- the composition is substantially free of mobile BPA and aromatic glycidyl ether compounds.
- the composition is substantially free of bound BPA and aromatic glycidyl ether compounds.
- the acid- or anhydride-functional polymer includes an acid- or anhydride-functional acrylic polymer, acid- or anhydride-functional alkyd resin, acid- or anhydride-functional polyester resin, acid- or anhydride- functional polyurethane, or combinations thereof.
- the acid- or anhydride-functional polymer includes an acid-functional acrylic polymer.
- the amine is a tertiary amine.
- the tertiary amine is selected from the group consisting of trimethyl amine, dimethylethanol amine (also known as dimethylamino ethanol), methyldiethanol amine, triethanol amine, ethyl methyl ethanol amine, dimethyl ethyl amine, dimethyl propyl amine, dimethyl 3 -hydroxy- 1 -propyl amine, dimethylbenzyl amine, dimethyl 2 -hydroxy- 1 -propyl amine, diethyl methyl amine, dimethyl l-hydroxy-2 -propyl amine, triethyl amine, tributyl amine, N-methyl morpholine, and mixtures thereof.
- the acid- or anhydride-functional polymer is at least 25% neutralized with the amine in water.
- the ethylenically unsaturated monomer component is polymerized in the presence of the aqueous dispersion with a water-soluble free radical initiator at a temperature of 0 0 C to 100 0 C.
- the free radical initiator includes a peroxide initiator.
- the free radical initiator includes hydrogen peroxide and benzoin.
- the free radical initiator includes a redox initiator system.
- the present invention also provides food cans and beverage cans prepared by a method described herein.
- the present invention provides a food or beverage can that includes: a body portion or an end portion including a metal substrate; and a coating composition disposed thereon, wherein the coating composition includes an emulsion polymerized latex polymer, wherein the emulsion polymerized latex polymer is prepared from a salt of an acid- or anhydride-functional polymer and an amine, an ethylenically unsaturated monomer component, and water.
- the coating composition includes an emulsion polymerized latex polymer, wherein the emulsion polymerized latex polymer is prepared from a salt of an acid- or anhydride-functional polymer and an amine, an ethylenically unsaturated monomer component, and water.
- the present invention provides a composition for use in coating a food or beverage can, wherein the composition includes an emulsion polymerized latex polymer, wherein the emulsion polymerized latex polymer is prepared from a salt of an acid- or anhydride-functional polymer and an amine, an ethylenically unsaturated monomer component, and water.
- compositions of the present invention contain less than 1000 parts per million (ppm) of the recited mobile compound.
- essentially free of a particular mobile compound means that the compositions of the present invention contain less than 100 parts per million (ppm) of the recited mobile compound.
- essentially completely free of a particular mobile compound means that the compositions of the present invention contain less than 5 parts per million (ppm) of the recited mobile compound.
- completely free of a particular mobile compound means that the compositions of the present invention contain less than 20 parts per billion (ppb) of the recited mobile compound.
- the term “mobile” means that the compound can be extracted from the cured coating when a coating (typically, approximate film weight of 1 mg/cm ) is exposed to a test medium for some defined set of conditions, depending on the end use.
- a coating typically, approximate film weight of 1 mg/cm
- An example of these testing conditions is exposure of the cured coating to 10 weight percent ethanol solution for two hours at 121 0 C followed by exposure for 10 days in the solution at 49°C.
- compositions of the present invention contain less than the aforementioned amount of the compound whether the compound is mobile in the coating or bound to a constituent of the coating.
- organic group means a hydrocarbon group (with optional elements other than carbon and hydrogen, such as oxygen, nitrogen, sulfur, and silicon) that is classified as an aliphatic group, cyclic group, or combination of aliphatic and cyclic groups (e.g., alkaryl and aralkyl groups).
- aliphatic group means a saturated or unsaturated linear or branched hydrocarbon group. This term is used to encompass alkyl, alkenyl, and alkynyl groups, for example.
- alkyl group means a saturated linear or branched hydrocarbon group including, for example, methyl, ethyl, isopropyl, t-butyl, heptyl, dodecyl, octadecyl, amyl, 2-ethylhexyl, and the like.
- alkenyl group means an unsaturated, linear or branched hydrocarbon group with one or more carbon-carbon double bonds, such as a vinyl group.
- alkynyl group means an unsaturated, linear or branched hydrocarbon group with one or more carbon-carbon triple bonds.
- cyclic group means a closed ring hydrocarbon group that is classified as an alicyclic group or an aromatic group, both of which can include heteroatoms.
- alicyclic group means a cyclic hydrocarbon group having properties resembling those of aliphatic groups.
- Ar refers to a divalent aryl group (i.e., an arylene group), which refers to a closed aromatic ring or ring system such as phenylene, naphthylene, biphenylene, fluorenylene, and indenyl, as well as heteroarylene groups (i.e., a closed ring hydrocarbon in which one or more of the atoms in the ring is an element other than carbon (e.g., nitrogen, oxygen, sulfur, etc.)).
- Suitable heteroaryl groups include furyl, thienyl, pyridyl, quinolinyl, isoquinolinyl, indolyl, isoindolyl, triazolyl, pyrrolyl, tetrazolyl, imidazolyl, pyrazolyl, oxazolyl, thiazolyl, benzofuranyl, benzothiophenyl, carbazolyl, benzoxazolyl, pyrimidinyl, benzimidazolyl, quinoxalinyl, benzothiazolyl, naphthyridinyl, isoxazolyl, isothiazolyl, purinyl, quinazolinyl, pyrazinyl, 1-oxidopyridyl, pyridazinyl, triazinyl, tetrazinyl, oxadiazolyl, thiadiazolyl, and so on. When such groups are divalent, they are typically
- a group that may be the same or different is referred to as being "independently" something. Substitution is anticipated on the organic groups of the compounds of the present invention.
- group and “moiety” are used to differentiate between chemical species that allow for substitution or that may be substituted and those that do not allow or may not be so substituted.
- group when the term “group” is used to describe a chemical substituent, the described chemical material includes the unsubstituted group and that group with O, N, Si, or S atoms, for example, in the chain (as in an alkoxy group) as well as carbonyl groups or other conventional substitution.
- alkyl group is intended to include not only pure open chain saturated hydrocarbon alkyl substituents, such as methyl, ethyl, propyl, t- butyl, and the like, but also alkyl substituents bearing further substituents known in the art, such as hydroxy, alkoxy, alkylsulfonyl, halogen atoms, cyano, nitro, amino, carboxyl, etc.
- alkyl group includes ether groups, haloalkyls, nitroalkyls, carboxyalkyls, hydroxyalkyls, sulfoalkyls, etc.
- alkyl moiety is limited to the inclusion of only pure open chain saturated hydrocarbon alkyl substituents, such as methyl, ethyl, propyl, t-butyl, and the like.
- a coating composition that comprises “a” polymer can be interpreted to mean that the coating composition includes “one or more” polymers.
- This invention provides a coating composition for use on food and beverage cans that includes a latex polymer.
- the polymer is prepared in an emulsion polymerization process, preferably a free radical initiated polymerization process.
- the latex polymer can be applied to a metal substrate either before or after the substrate is formed into a food or beverage can (e.g., two-piece cans, three-piece cans) or portions thereof, whether it be a can end or can body.
- the latex polymers of the present invention are suitable for use in food contact situations and may be used on the inside of such cans. They are particularly useful on the interior of two- piece drawn and ironed beverage cans and on beverage can ends.
- the latex polymer is prepared by polymerizing an ethylenically unsaturated monomer component in an aqueous medium in the presence of the salt of an acid group- or anhydride group-containing polymer and an amine, preferably, a tertiary amine.
- the ethylenically unsaturated monomer component is preferably a mixture of monomers.
- at least one of the monomers in the mixture is an alpha, beta-ethylenically unsaturated monomer, and preferably at least one of the monomers contains an oxirane groups. More preferably, at least one of the monomers is an oxirane group-containing alpha, beta-ethylenically unsaturated monomer.
- the composition may optionally include crosslinkers, fillers, catalysts, dyes, pigments, toners, extenders, lubricants, anticorrosion agents, flow control agents, thixotropic agents, dispersing agents, antioxidants, adhesion promoters, light stabilizers, surfactants, organic solvents, and mixtures thereof as required to provide the desired film properties.
- the coating composition is prepared by: forming a salt of an acid-functional or anhydride-functional polymer and an amine; dispersing the salt in a carrier that includes water and an optional organic solvent to form an aqueous dispersion; optionally removing the organic solvent, if present, from the aqueous dispersion; combining an ethylenically unsaturated monomer component with the aqueous dispersion (preferably, the ethylenically unsaturated monomer component is added to the aqueous dispersion); and polymerizing the ethylenically unsaturated monomer component in the presence of the aqueous dispersion to form an emulsion polymerized latex polymer.
- compositions are substantially free of mobile bisphenol A (BPA) and aromatic glycidyl ether compounds (e.g., BADGE, BFDGE, and epoxy novalacs), more preferably essentially free of these compounds, even more preferably essentially completely free of these compounds, and most preferably completely free of these compounds.
- BPA mobile bisphenol A
- aromatic glycidyl ether compounds e.g., BADGE, BFDGE, and epoxy novalacs
- the coating composition is also preferably substantially free of bound BPA and aromatic glycidyl ether compounds, more preferably essentially free of these compounds, most preferably essentially completely free of these compounds, and optimally completely free of these compounds.
- the ethylenically unsaturated monomer component is preferably a mixture of monomers that is capable of free radical initiated polymerization in aqueous medium.
- the monomer mixture preferably contains at least one oxirane functional monomer, and more preferably, at least one oxirane group-containing alpha, beta- ethylenically unsaturated monomer.
- the monomer mixture preferably contains at least 0.1 percent by weight (wt- %), more preferably at least 1 wt-%, of an oxirane group-containing monomer, based on the weight of the monomer mixture. Typically, at least 0.1 wt-% of the oxirane group-containing monomer contributes to the stability of the latex.
- the monomer mixture preferably contains no greater than 30 wt-%, more preferably no greater than 20 wt-%, even more preferably no greater than 10 wt-%, and optimally no greater than 9 wt-%, of the oxirane group-containing monomer, based on the weight of the monomer mixture.
- greater than 30 wt-% of the oxirane group-containing monomer in the monomer mixture can contribute to diminished film properties. Although not intended to be limited by theory, it is believed that this is due to embrittlement caused by an overabundance of crosslinking.
- Suitable oxirane-functional monomers include monomers having a reactive carbon-carbon double bond and an oxirane (i.e., a glycidyl) group.
- the monomer is a glycidyl ester of an alpha, beta-unsaturated acid, or anhydride thereof (i.e., an oxirane group-containing alpha, beta-ethylenically unsaturated monomer).
- Suitable alpha, beta-unsaturated acids include monocarboxylic acids or dicarboxylic acids.
- carboxylic acids include, but are not limited to, acrylic acid, methacrylic acid, alpha-chloroacrylic acid, alpha-cyanoacrylic acid, beta- methylacrylic acid (crotonic acid), alpha-phenylacrylic acid, beta-acryloxypropionic acid, sorbic acid, alpha-chlorosorbic acid, angelic acid, cinnamic acid, p- chlorocinnamic acid, beta-stearylacrylic acid, itaconic acid, citraconic acid, mesaconic acid, glutaconic acid, aconitic acid, maleic acid, fumaric acid, tricarboxyethylene, maleic anhydride, and mixtures thereof.
- Suitable monomers containing a glycidyl group are glycidyl (meth)acrylate (i.e., glycidyl methacrylate and glycidyl acrylate), mono- and di-glycidyl itaconate, mono- and di-glycidyl maleate, and mono- and di-glycidyl formate. It also is envisioned that allyl glycidyl ether and vinyl glycidyl ether can be used as the oxirane-functional monomer.
- a preferred monomer is glycidyl methacrylate ("GMA").
- the oxirane-functional monomer is preferably reacted with suitable other monomers within the monomer mixture. These can be ethylenically unsaturated monomer and hydroxy-functional monomers. Suitable ethylenically unsaturated monomers include alkyl (meth)acrylates, vinyl monomers, alkyl esters of maleic or fumaric acid, and the like.
- the R group can be substituted with one or more, and typically one to three, moieties such as hydroxy, halo, phenyl, and alkoxy, for example.
- Suitable alkyl (meth)acrylates therefore encompass hydroxy alkyl (meth)acrylates.
- the alkyl (meth)acrylate typically is an ester of acrylic or methacrylic acid.
- R 1 is hydrogen or methyl and R 2 is an alkyl group having two to eight carbon atoms.
- R 1 is hydrogen or methyl and R 2 is an alkyl group having two to four carbon atoms.
- alkyl (meth)acrylates include, but are not limited to, methyl (meth)acrylate, ethyl (meth)acrylate, propyl (meth)acrylate, isopropyl (mefh)acrylate, butyl (meth)acrylate, isobutyl (meth)acrylate, pentyl (meth)acrylate, isoamyl (meth)acrylate, hexyl (meth)acrylate, 2-ethylhexyl (meth)acrylate, cyclohexyl (meth)acrylate, decyl (meth)acrylate, isodecyl (meth)acrylate, benzyl (meth)acrylate, lauryl (meth)acrylate, isoborayl (meth)acrylate, octyl (meth)acrylate, nonyl (meth)acrylate, hydroxyethyl acrylate (HEA), hydroxyethyl methacrylate
- Difunctional (meth)acrylate monomers may be used in the monomer mixture as well. Examples include ethylene glycol di(meth)acrylate, 1,6-hexanediol di(meth)acrylate, allyl methacrylate, and the like.
- Suitable vinyl monomers include styrene, methyl styrene, halostyrene, isoprene, diallylphthalate, divinylbenzene, conjugated butadiene, alpha- methylstyrene, vinyl toluene, vinyl naphthalene, and mixtures thereof.
- the vinyl aromatic monomers described below in connection with the acid- or anhydride- functional polymer are also suitable for use in the ethylenically unsaturated monomer component used to make the latex polymer.
- Styrene is a presently preferred vinyl monomer, in part due to its relatively low cost.
- Suitable polymerizable vinyl monomers for use in the ethylenically unsaturated monomer component include acrylonitrile, acrylamide, methacrylamide, methacrylonitrile, vinyl acetate, vinyl propionate, vinyl butyrate, vinyl stearate, N- isobutoxymethyl acrylamide, N-butoxymethyl acrylamide, and the like.
- the oxirane group-containing monomer preferably constitutes 0.1 wt-% to
- the ethylenically unsaturated monomer component constitutes the remainder of the monomer component, that is, 70 wt-% to 99.9 wt-%, preferably 80 wt-% to 99 wt-%, based on total weight of the monomer mixture.
- at least 40 wt-% of the ethylenically unsaturated monomer component more preferably at least 50 wt-%, will be selected from alkyl acrylates and methacrylates.
- at least 20 wt-%, more preferably at least 30 wt-% will be selected from vinyl aromatic compounds.
- At least 5 wt-%, more preferably at least 25 wt-%, even more preferably at least 50 wt-%, and even more preferably at least 60 wt-%, of the ethylenically unsaturated monomer component is used in making the latex polymer.
- no greater than 95 wt-%, more preferably no greater than 90 wt-%, and even more preferably no greater than 85 wt-%, of the ethylenically unsaturated monomer component is used in making the latex polymer.
- Such percentages are based on total weight of ethylenically unsaturated monomer component and salt of the acid group-containing or anhydride group-containing polymer (i.e., acid- functional or anhydride-functional polymer).
- the choice of the acid-containing or anhydride-containing monomer(s) is dictated by the intended end use of the coating composition and is practically unlimited.
- the acid-containing polymer i.e., acid-functional polymer
- the acid-containing polymer preferably has an acid number of at least 40, and more preferably at least 100, milligrams (mg) KOH per gram resin.
- the acid-containing polymer preferably has an acid number of no greater than 400, and more preferably no greater than 300, mg KOH per gram resin.
- the anhydride-containing polymer when in water, preferably has similar acid number ranges. Low molecular weight polymers are preferred for certain applications of the present invention.
- the molecular weight of the acid- or anhydride- functional polymer is no greater than 50,000 on a number average molecular weight basis, and preferably no greater than 20,000.
- the molecular weight of the acid- or anhydride-functional polymer is at least 1500 on a number average molecular weight basis, and more preferably at least 2000.
- Preferred acid- or anhydride-functional polymers that may be employed include acid-functional or anhydride-functional acrylic polymers, alkyd resins, polyester polymers, and polyurethanes. Combinations of such polymers can be used if desired.
- the term polymer includes both homopolymers and copolymers (i.e., polymers of two or more different monomers).
- Preferred acid- or anhydride-functional polymers utilized in this invention include those prepared by conventional free radical polymerization techniques. Suitable examples include those prepared from unsaturated acid- or anhydride- functional monomers, or salts thereof, and other unsaturated monomers. Of these, preferred examples include those prepared from at least 15 wt-%, more preferably at least 20 wt-%, unsaturated acid- or anhydride-functional monomer, or salts thereof, and the balance other polymerizable unsaturated monomer. Examples of co- monomers described previously apply here as well.
- a variety of acid- or anhydride-functional monomers, or salts thereof, can be used; their selection is dependent on the desired final polymer properties.
- such monomers are ethylenically unsaturated, more preferably, alpha, beta-ethylenically unsaturated.
- Suitable ethylenically unsaturated acid- or anhydride-functional monomers for the present invention include monomers having a reactive carbon-carbon double bond and an acidic or anhydride group, or salts thereof. Preferred such monomers have from 3 to 20 carbons, at least 1 site of unsaturation, and at least 1 acid or anhydride group, or salt thereof.
- Suitable acid-functional monomers include ethylenically unsaturated acids
- mono-protic or diprotic anhydrides or monoesters of a dibasic acid, which are copolymerizable with the optional other monomer(s) used to prepare the polymer.
- Half-esters of these acids with alkanols of 1 to 8 carbon atoms are also suitable.
- Non-limiting examples of useful ethylenically unsaturated acid-functional monomers include acids such as, for example, acrylic acid, methacrylic acid, alpha- chloroacrylic acid, alpha-cyanoacrylic acid, crotonic acid, alpha-phenylacrylic acid, beta-acryloxypropionic acid, funiaric acid, maleic acid, sorbic acid, alpha- chlorosorbic acid, angelic acid, cinnamic acid, p-chlorocinnamic acid, beta- stearylacrylic acid, citraconic acid, mesaconic acid, glutaconic acid, aconitic acid, tricarboxyethylene, 2-methyl maleic acid, itaconic acid, 2-methyl itaconic acid, methyleneglutaric acid, and the like, or mixtures thereof.
- acids such as, for example, acrylic acid, methacrylic acid, alpha- chloroacrylic acid, alpha-cyanoacrylic acid, crotonic acid, alpha-
- Preferred unsaturated acid-functional monomers include acrylic acid, methacrylic acid, crotonic acid, fumaric acid, maleic acid, 2-methyl maleic acid, itaconic acid, 2-methyl itaconic acid and mixtures thereof. More preferred unsaturated acid-functional monomers include acrylic acid, methacrylic acid, crotonic acid, fumaric acid, maleic acid, itaconic acid, and mixtures thereof. Most preferred unsaturated acid-functional monomers include acrylic acid, methacrylic acid, maleic acid, crotonic acid, and mixtures thereof.
- Nonlimiting examples of suitable ethylenically unsaturated anhydride monomers include compounds derived from the above acids (e.g., as pure anhydride or mixtures of such).
- Preferred anhydrides include acrylic anhydride, methacrylic anhydride, and maleic anhydride. If desired, aqueous salts of the above acids may also be employed.
- Polymerization of the monomers to form an acid- or anhydride-functional polymer is usually conducted by organic solution polymerization techniques in the presence of a free radical initiator as is well known in the art. Although the preparation of the acid-functional or anhydride-functional polymer is conveniently carried out in solution, neat processes may be used if desired.
- acid- or anhydride-functional acrylic polymers can also be used in the practice of the invention.
- acid- or anhydride-functional alkyd polyester, polyurethane resins, or combinations thereof.
- polyester, polyurethane resins, or combinations thereof can also be used in the practice of the invention.
- Such polymers are described in U.S. Pat. Nos. 4,692,491; 3,479,310; and 4,147,679.
- the acid- or anhydride-functional polymers are acid-functional acrylic polymers.
- the acid- or anhydride-functional polymers are polyester polymers. Examples of such polyester polymers are disclosed in U.S. Provisional Patent Application Serial No. (Attorney Docket No.
- each Ar is independently a divalent aryl group (i.e., an arylene group) or heteroarylene group;
- R 1 is a divalent organic group;
- each R is independently a divalent organic group; and
- n is 0 or 1. Any one polymer can have a variety of such segments, which may be the same or different.
- R 1 provides hydrolytic stability to at least one of the adjacent ester linkages (-C(O)-O- and -O-C(O)-), and preferably to both of them.
- hydrolytic stability means that R 1 decreases the reactivity (preferably, by at least half) of the adjacent ester linkage with water compared to a -CH 2 -CH 2 - moiety under the same conditions. This can be accomplished by selection of R 1 that includes a sterically bulky group in proximity (preferably within two atoms distance) to the oxygen of the ester.
- the polymer preferably includes more than 70%, more preferably more than 80%, and even more preferably more than 90%, hydrolytically stable ester linkages (based on the total number of ester linkages).
- R ! is a divalent organic group, preferably, having at least 3 carbon atoms, more preferably, at least 4 carbon atoms, even more preferably, at least 5 carbon atoms, and even more preferably, at least 8 carbon atoms. It is envisioned that R 1 can be as large as desired for the particular application, which one of skill in the art can readily determine.
- R 1 is of the formula
- each R 2 is independently hydrogen or an organic group (e.g., an alicyclic group or a branched or unbranched alkyl group), Y is a divalent organic group, and t is 0 or 1 (preferably 1). In certain embodiments, each R 2 is independently hydrogen.
- Y can optionally include one or more ether or ester linkages.
- Y is a divalent saturated aliphatic group (i.e., a branched or unbranched alkylene group), a divalent alicyclic group, or a divalent aromatic group (i.e., an arylene group), or combinations thereof.
- Y is a divalent alkyl group (i.e., an alkylene group), which can be branched or unbranched, preferably having at least 1 carbon atom, more preferably having at least 2 carbon atoms, even more preferably having at least 3 carbon atoms, and even more preferably having at least 6 carbon atoms.
- Y is a divalent alicylic group, preferably cyclohexylene. It is envisioned that Y can be as large as desired for the particular application, which one of skill in the art can readily determine.
- Y provides hydrolytic stability to at least one of the ester linkages adjacent R 1 in Formula I. This can be accomplished by selection of Y that includes a sterically bulky group that is in proximity (preferably within two atoms) of at least one of the ester oxygen atoms in Formula I.
- R 1 has the formula -(C(R 2 )2) S - wherein s is at least 2, and preferably, s is at least 3, wherein each R 2 is as defined above.
- R 1 groups include, for example, neopentylene, butylethylpropylene, and -CH 2 -CH(CHa)-CH 2 -.
- Y has the formula -[Z W -C(R 2 ) 2 -O-C(O)-R 3 -C(O)-O-C(RV] V Z W -, wherein w is 0 or 1, v is 1 to 10, each R 2 is as defined above, each R 3 is independently a divalent organic group, and each Z is independently a divalent organic group.
- R 3 is a divalent saturated aliphatic group (i.e., branched or unbranched alkylene group), a divalent alicyclic group, an arylene group, or combinations thereof. In certain embodiments, R 3 is a (C3-C20)alkylene (branched or unbranched) group or a phenylene group.
- Z is a divalent saturated aliphatic group (i.e., branched or unbranched alkylene group), a divalent alicyclic group, a divalent aromatic group (i.e., an arylene group), or combinations thereof.
- Z provides hydrolytic stability to at least one of the ester linkages adjacent R 1 in Formula I and/or to an adjacent ester linkage contained within Y. This can be accomplished by selection of Z that includes a sterically bulky group that is in proximity (preferably within two atoms distance) of at least one of the ester oxygen atoms.
- n is preferably 0 (i.e., R is not present). If n is 1 and R is present, however, it is preferably a (Cl-C4)alkylene group, and more preferably a (Cl-C4)alkylene moiety.
- each Ar has less than 20 carbon atoms, more preferably less than 11 carbon atoms, and even more preferably less than 8 carbon atoms.
- Ar has at least 4 carbon atoms, more preferably at least 5 carbon atoms, and even more preferably, at least 6 carbon atoms.
- each Ar is a phenylene group.
- each Ar is a phenylene group of the formula -C 6 (R 4 ) 4 -, wherein each R 4 is independently hydrogen, a halogen, or an organic group, and wherein two R 4 groups can join to form a ring optionally containing one or more heteroatoms.
- R 4 is hydrogen or an organic group, wherein two R groups can join to form a 6-membered ring.
- R 4 is hydrogen.
- Polyester polymers such as these can be made by a variety of methods from compounds of Formula II:
- HO-Ar-R n -C(O)-O-R'-O-C(O)-R n -Ar-OH wherein Ar, R, R 1 , and n are as defined above.
- Such compounds can be made, for example, by the esterification reaction of one mole of a diol (e.g., HO-R'-OH such as, for example, 1 ,4-cyclohexane dimethanol, neopentyl glycol, 2-butyl-2-ethyl-l,3- propane diol, or 2-methyl-l ,3-propane diol) with two moles of an acid (e.g., A- hydroxy benzoic acid).
- a diol e.g., HO-R'-OH such as, for example, 1 ,4-cyclohexane dimethanol, neopentyl glycol, 2-butyl-2-ethyl-l,3- propane diol, or
- such compounds can be made, for example, by the transesterification reaction of one mole of a diol (e.g., 1 ,4-cyclohexane dimethanol, neopentyl glycol, 2-butyl-2-ethyl-l,3-propane diol, or 2-methyl-l, 3- propane diol) with two moles of an ester .(e.g., 4-hydroxy methyl benzoate, A- hydroxy ethyl benzoate, or 4-hydroxy butyl benzoate).
- a diol e.g., 1 ,4-cyclohexane dimethanol, neopentyl glycol, 2-butyl-2-ethyl-l,3-propane diol, or 2-methyl-l, 3- propane diol
- an ester e.g., 4-hydroxy methyl benzoate, A- hydroxy ethyl benzoate, or 4-hydroxy butyl benzoate.
- Polymers of Formula I can be prepared by methods that involve advancing the molecular weight of compounds of Formula II.
- compounds of Formula II e.g., dihydric phenols
- a diepoxide to advance the molecular weight.
- compounds of Formula II e.g., dihydric phenols
- non-BPA and non-BPF based diepoxides much in the same manner that Bisphenol A or Bisphenol F do, to create polymers that can be formulated with crosslinkers and additives for coatings for rigid packaging.
- compounds of Formula II can be reacted with a diepoxide to form a polymer that includes -CH 2 -CH(OH)-CH 2 - segments.
- compounds of Formula II can be reacted with epichlorohydrin to form a diepoxide analog of compounds of Formula II, which can then be reacted with other compounds of Formula II to form a polymer that includes -CH 2 -CH(OH)-CH 2 - segments.
- the diepoxide analogs of compounds of Formula II can be prepared by reacting the required proportions of a compound of Formula II (e.g., dihydric phenol) and epichlorohydrin in an alkaline medium.
- the desired alkalinity is obtained by adding basic substances, such as sodium or potassium hydroxide, preferably in stoichiometric excess to the epichlorohydrin.
- the reaction is preferably accomplished at temperatures of 50 0 C to 150 0 C. The heating is continued for several hours to effect the reaction and the product is then washed free of salt and base. Procedures for such reactions are generally well known and disclosed, for example, in U.S. Pat. No.
- suitable diepoxides are BPA- or BPF-free diepoxides, preferably with one or more ether linkages.
- Suitable diepoxides may be prepared by a variety of processes, for example, by the condensation of a dihydroxy compound and epichlorohydrin.
- Suitable diepoxides include, for example, 1,4- cyclohexanedimethanol diglycidyl ether (CHDMDGE), resorcinol diglycidyl ether, neopentyl glycol diglycidyl ether, and 2-methyl-l,3-propandiol diglycidyl ether.
- CHDMDGE 1,4- cyclohexanedimethanol diglycidyl ether
- resorcinol diglycidyl ether resorcinol diglycidyl ether
- neopentyl glycol diglycidyl ether 2-methyl-l,3-propandiol diglycidyl ether.
- the resultant polymers of Formula I may be epoxy terminated or phenoxy terminated, for example. They may be made in a variety of molecular weights, such as the molecular weights of commercially available BPA-based epoxy materials
- Preferred polymers of the present invention have a number average molecular weight (M n ) of at least 2,000, more preferably at least 3,000, and even more preferably at least 4,000.
- M n number average molecular weight
- the molecular weight of the polymer may be as high as is needed for the desired application. Advancement of the molecular weight of the polymer may be enhanced by the use of a catalyst in the reaction of a diepoxide (whether it be a diepoxide analog of Formula II or another diepoxide) with a compound of Formula (II).
- Typical catalysts usable in the advancement of the molecular weight of the epoxy material of the present invention include amines, hydroxides (e.g., potassium hydroxide), phosphonium salts, and the like.
- a presently preferred catalyst is a phosphonium catalyst.
- the phosphonium catalyst useful in the present invention is preferably present in an amount sufficient to facilitate the desired condensation reaction.
- the epoxy terminated polymers of Formula I may be reacted with fatty acids to form polymers having unsaturated (e.g., air oxidizable) reactive groups, or with acrylic acid or methacrylic acid to form free radically curable polymers.
- unsaturated e.g., air oxidizable
- Advancement of the molecular weight of the polymer may also be enhanced by the reaction of an epoxy terminated polymer of Formula I with a suitable diacid (such as adipic acid).
- a suitable diacid such as adipic acid
- a salt (which can be a full salt or partial salt) of the acid- or anhydride- functional polymer is formed by neutralizing or partially neutralizing the acid groups (whether present initially in the acid-functional polymer or formed upon addition of the anhydride-functional polymer to water) of the polymer with a suitable amine, preferably a tertiary amine.
- the degree of neutralization required to form the desired polymer salt may vary considerably depending upon the amount of acid included in the polymer, and the degree of solubility or dispersibility of the salt which is desired.
- the acidity of the polymer is at least 25% neutralized, preferably at least 30% neutralized, and more preferably at least 35% neutralized, with the amine in water.
- tertiary amines are trimethyl amine, dimethylethanol amine (also known as dimethylamino ethanol), methyldiethanol amine, triethanol amine, ethyl methyl ethanol amine, dimethyl ethyl amine, dimethyl propyl amine, dimethyl 3-hydroxy-l -propyl amine, dimethylbenzyl amine, dimethyl 2 -hydroxy- 1 -propyl amine, diethyl methyl amine, dimethyl l-hydroxy-2-propyl amine, triethyl amine, tributyl amine, N-methyl morpholine, and mixtures thereof. Most preferably triethyl amine or dimethyl ethanol amine is used as the tertiary amine.
- the amount of the salt of the acid-functional or anhydride-functional polymer that is used in the polymerization is preferably at least 5 wt-%, more preferably at least 10 wt-%, and eve'n more preferably at least 15 wt-%.
- the amount of the salt of the acid-functional or anhydride-functional polymer that is used in the polymerization is preferably no greater than 95 wt-%, preferably no greater than 50 wt-%, and even more preferably no greater than 40 wt-%. These percentages are based on total weight of polymerizable ethylenically unsaturated monomer component and the salt of the acid group-containing polymer.
- the reaction of tertiary amines with materials containing oxirane groups when carried out in the presence of water, can afford a product that contains both a hydroxyl group and a quaternary ammonium hydroxide.
- an acid group, an oxirane group, and an amine form a quaternary salt.
- This linkage is favored, as it not only links the polymers but promotes water dispersibility of the joined polymer. It should be noted that an acid group and an oxirane group may also form an ester. Some of this reaction is possible, though this linkage is less desirable when water dispersibility is sought.
- one reaction involves the tertiary amine neutralized acid-functional polymer reacting with an oxirane-functional monomer or polymer to form a quaternary ammonium salt.
- a second reaction involves esterification of the oxirane-functional monomer or polymer with a carboxylic acid or salt.
- the presence of water and level of amine favor formation of quaternary ammonium salts over ester linkages.
- a high level of quaternization improves water dispersability while a high level of esterification gives higher viscosity and possibly gel-like material.
- the ethylenically unsaturated monomer component is preferably polymerized in aqueous medium with a water-soluble free radical initiator in the presence of the salt of the acid- or anhydride-functional polymer.
- the temperature of polymerization is typically from O 0 C to 100 0 C, preferably from 5O 0 C to 9O 0 C, more preferably from 7O 0 C to 9O 0 C, and even more preferably from 80 0 C to 85°C.
- the pH of the aqueous medium is usually maintained at a pH of 5 to 12.
- the free radical initiator can be selected from one or more water-soluble peroxides which are known to act as free radical initiators.
- Examples include hydrogen peroxide and t-butyl hydroperoxide.
- Redox initiator systems well known in the art (e.g., t-butyl hydroperoxide, erythorbic acid, and ferrous complexes) can also be employed. It is especially preferred to use a mixture of benzoin and hydrogen peroxide.
- Persulfate initiators such as ammonium persulfate or potassium persulfate are not preferred, as they lead to poor water resistance properties of the cured coating.
- the polymerization reaction of the ethylenically unsaturated monomer component in the presence of the aqueous dispersion of the polymer salt may be conducted as a batch, intermittent, or continuous operation. While all of the polymerization ingredients may be charged initially to the polymerization vessel, better results normally are obtained with proportioning techniques.
- the reactor is charged with an appropriate amount of water, polymer salt, and free radical initiator.
- the reactor is then heated to the free radical initiation temperature and then charged with the ethylenically unsaturated monomer component.
- Preferably only water, initiator, polymer salt, and some portion of the ethylenically unsaturated monomer component are initially charged to the vessel. There may also be some water miscible solvent present.
- the remaining ethylenically unsaturated monomer component is added incrementally with the rate of addition being varied depending on the polymerization temperature, the particular initiator being employed, and the type and amount of monomers being polymerized.
- coating compositions using the aforementioned latices may be formulated using one or more optional curing agents (i.e., crosslinking resins, sometimes referred to as "crosslinkers")-
- crosslinkers i.e., crosslinking resins, sometimes referred to as "crosslinkers"
- crosslinkers typically depends on the particular product being formulated. For example, some coating compositions are highly colored (e.g., gold-colored coatings). These coatings may typically be formulated using crosslinkers that themselves tend to have a yellowish color. In contrast, white coatings are generally formulated using non-yellowing crosslinkers, or only a small amount of a yellowing crosslinker.
- Preferred curing agents are substantially free of mobile BPA and aromatic glycidyl ether compounds (e.g., BADGE, BFDGE and epoxy novalacs).
- hydroxy] -reactive curing resins can be used.
- phenoplast, and aminoplast curing agents may be used.
- Phenoplast resins include the condensation products of aldehydes with phenols. Formaldehyde and acetaldehyde are preferred aldehydes.
- Various phenols can be employed such as phenol, cresol, p-phenylphenol, p-tert-butylphenol, p-tert- amylphenol, and cyclopentylphenol.
- Aminoplast resins are the condensation products of aldehydes such as formaldehyde, acetaldehyde, crotonaldehyde, and benzaldehyde with amino or amido group-containing substances such as urea, melamine, and benzoguanamine.
- aldehydes such as formaldehyde, acetaldehyde, crotonaldehyde, and benzaldehyde with amino or amido group-containing substances such as urea, melamine, and benzoguanamine.
- crosslinking resins include, without limitation, benzoguanamine-formaldehyde resins, melamine-formaldehyde resins, esterified melamine-formaldehyde, and urea-formaldehyde resins.
- the crosslinker employed when practicing this invention includes a melamine-formaldehyde resin.
- a particularly useful crosslinker is the fully alkylated melamine-formaldehyde resin commercially available from Cytec Industries, Inc. under the trade name of CYMEL 303.
- curing agents are the blocked or non- blocked aliphatic, cycloaliphatic or aromatic di-, tri-, or poly-valent isocyanates, such as hexamethylene diisocyanate, cyclohexyl-l,4-diisocyanate, and the like.
- the level of curing agent i.e., crosslinker
- the crosslinker is typically present in an amount of up to 50 wt- %, preferably up to 30 wt-%, and more preferably up to 15 wt-%. These weight percentages are based upon the total weight of the resin solids in the coating composition.
- a coating composition of the present invention may also include other optional polymers that do not adversely affect the coating composition or a cured coating composition resulting therefrom. Such optional polymers are typically included in a coating composition as a filler material, although they can be included as a crosslinking material, or to provide desirable properties.
- One or more optional polymers e.g., filler polymers
- Such additional polymeric materials can be nonreactive, and hence, simply function as fillers.
- Such optional nonreactive filler polymers include, for example, polyesters, acrylics, polyamides, polyethers, and novalacs.
- additional polymeric materials or monomers can be reactive with other components of the composition (e.g., the acid-functional polymer).
- reactive polymers can be incorporated into the compositions of the present invention, to provide additional functionality for various purposes, including crosslinking. Examples of such reactive polymers include, for example, functionalized polyesters, acrylics, polyamides, and polyethers.
- Preferred optional polymers are substantially free of mobile BPA and aromatic glycidyl ether compounds (e.g., BADGE, BFDGE and epoxy novalacs)
- a coating composition of the present invention may also include other optional ingredients that do not adversely affect the coating composition or a cured coating composition resulting therefrom. Such optional ingredients are typically included in a coating composition to enhance composition esthetics, to facilitate manufacturing, processing, handling, and application of the composition, and to further improve a particular functional property of a coating composition or a cured coating composition resulting therefrom.
- Such optional ingredients include, for example, catalysts, dyes, pigments, toners, extenders, fillers, lubricants, anticorrosion agents, flow control agents, thixotropic agents, dispersing agents, antioxidants, adhesion promoters, light stabilizers, surfactants, and mixtures thereof.
- Each optional ingredient is included in a sufficient amount to serve its intended purpose, but not in such an amount to adversely affect a coating composition or a cured coating composition resulting therefrom.
- catalysts include, but are not limited to, strong acids (e.g., dodecylbenzene sulphonic acid (DDBSA, available as CYCAT 600 from Cytec), methane sulfonic acid (MSA), p-toluene sulfonic acid (pTSA), dinonylnaphthalene disulfonic acid (DNNDSA), and triflic acid), quaternary ammonium compounds, phosphorous compounds, and tin and zinc compounds.
- strong acids e.g., dodecylbenzene sulphonic acid (DDBSA, available as CYCAT 600 from Cytec
- MSA methane sulfonic acid
- pTSA p-toluene sulfonic acid
- DNNDSA dinonylnaphthalene disulfonic acid
- triflic acid triflic acid
- a catalyst is preferably present in an amount of at least 0.01 wt-%, and more preferably at least 0.1 wt-%, based on the weight of nonvolatile material. If used, a catalyst is preferably present in an amount of no greater than 3 wt-%, and more preferably no greater than 1 wt-%, based on the weight of nonvolatile material.
- a lubricant e.g., a wax
- Preferred lubricants include, for example, Carnauba wax and polyethylene type lubricants.
- a lubricant is preferably present in the coating composition in an amount of at least 0.1 wt-%, and preferably no greater than 2 wt- %, and more preferably no greater than 1 wt-%, based on the weight of nonvolatile material.
- a pigment such as titanium dioxide. If used, a pigment is present in the coating composition in an amount of no greater than 70 wt-%, more preferably no greater than 50 wt-%, and even more preferably no greater than 40 wt-%, based on the total weight of solids in the coating composition.
- Surfactants can be optionally added to the coating composition to aid in flow and wetting of the substrate.
- examples of surfactants include, but are not limited to, nonylphenol polyethers and salts and similar surfactants known to persons skilled in the art. If used, a surfactant is preferably present in an amount of at least 0.01 wt-%, and more preferably at least 0.1 wt-%, based on the weight of resin solids. If used, a surfactant is preferably present in an amount no greater than 10 wt-%, and more preferably no greater than 5 wt-%, based on the weight of resin solids.
- the coating compositions of the present invention are particularly well adapted for use on food and beverage cans (e.g., two-piece cans, three-piece cans, etc.).
- Two-piece cans are manufactured by joining a can body (typically a drawn metal body) with a can end (typically a drawn metal end).
- the coatings of the present invention are suitable for use in food or beverage contact situations and may be used on the inside of such cans. They are particularly suitable for spray applied, liquid coatings for the interior of two-piece drawn and ironed beverage cans and coil coatings for beverage can ends.
- the present invention also offers utility in other applications. These additional applications include, but are not limited to, wash coating, sheet coating, and side seam coatings (e.g., food can side seam coatings).
- Spray coating includes the introduction of the coated composition into the inside of a preformed packaging container.
- Typical preformed packaging containers suitable for spray coating include food cans, beer and beverage containers, and the like.
- the spray preferably utilizes a spray nozzle capable of uniformly coating the inside of the preformed packaging container.
- the sprayed preformed container is then subjected to heat to remove the residual solvents and harden the coating.
- a coil coating is described as the coating of a continuous coil composed of a metal (e.g., steel or aluminum). Once coated, the coating coil is subjected to a short thermal, ultraviolet, and/or electromagnetic curing cycle, for hardening (e.g., drying and curing) of the coating.
- Coil coatings provide coated metal (e.g., steel and/or aluminum) substrates that can be fabricated into formed articles, such as 2-piece drawn food cans, 3-piece food cans, food can ends, drawn and ironed cans, beverage can ends, and the like.
- coated metal e.g., steel and/or aluminum
- a wash coating is commercially described as the coating of the exterior of two-piece drawn and ironed ("D&I") cans with a thin layer of protectant coating.
- the exterior of these D&I cans are "wash-coated” by passing pre-formed two-piece D&I cans under a curtain of a coating composition.
- the cans are inverted, that is, the open end of the can is in the "down” position when passing through the curtain.
- This curtain of coating composition takes on a "waterfall-like" appearance. Once these cans pass under this curtain of coating composition, the liquid coating material effectively coats the exterior of each can.
- each can is passed through a thermal, ultraviolet, and/or electromagnetic curing oven to harden (e.g., dry and cure) the coating.
- the residence time of the coated can within the confines of the curing oven is typically from 1 minute to 5 minutes.
- the curing temperature within this oven will typically range from 15O 0 C to 22O 0 C.
- a sheet coating is described as the coating of separate pieces of a variety of materials (e.g., steel or aluminum) that have been pre-cut into square or rectangular "sheets." Typical dimensions of these sheets are approximately one square meter. Once coated, each sheet is cured. Once hardened (e.g., dried and cured), the sheets of the coated substrate are collected and prepared for subsequent fabrication. Sheet coatings provide coated metal (e.g., steel or aluminum) substrate that can be successfully fabricated into formed articles, such as 2-piece drawn food cans, 3- piece food cans, food can ends, drawn and ironed cans, beverage can ends, and the like.
- coated metal e.g., steel or aluminum
- a side seam coating is described as the spray application of a liquid coating over the welded area of formed three-piece food cans.
- a rectangular piece of coated substrate is formed into a cylinder.
- the formation of the cylinder is rendered permanent due to the welding of each side of the rectangle via thermal welding.
- each can typically requires a layer of liquid coating, which protects the exposed "weld” from subsequent corrosion or other effects to the contained foodstuff.
- the liquid coatings that function in this role are termed "side seam stripes.” Typical side seam stripes are spray applied and cured quickly via residual heat from the welding operation in addition to a small thermal, ultraviolet, and/or electromagnetic oven.
- Preferred coatings of the present invention display one or more of the properties described in the Examples Section. More preferred coatings of the present invention display one or more of the following properties: metal exposure value of less than 3 mA; metal exposure value after drop damage of less than 3.5 mA; global extraction results of less than 50 ppm; adhesion rating of 10; blush rating of at least 7; slight or no crazing in a reverse impact test; no craze (rating of 10) in a dome impact test; feathering below 0.2 inch; COF range of 0.055 to 0.095; and after paseurization or retort, a continuity of less than 20 mA.
- the curing conditions involve maintaining the temperature measured at the can dome at 188 0 C to 199 0 C for 30 seconds.
- the curing conditions involve the use of a temperature sufficient to provide a peak metal temperature within the specified time (e.g., 10 seconds at 204 0 C means 10 seconds, in the oven, for example, and a peak metal temperature achieved of 204 0 C).
- a temperature sufficient to provide a peak metal temperature within the specified time e.g., 10 seconds at 204 0 C means 10 seconds, in the oven, for example, and a peak metal temperature achieved of 204 0 C.
- This test method determines the amount the inside surface of the can that has not been effectively coated by the sprayed coating. This determination is made thorough the use of an electrically conductive solution (1% NaCl in deionized water). The coated can is filled with this conductive solution, and an electrical probe is attached in contact to the outside of the can (uncoated, electrically conducting). A second probe is immersed in the salt solution in the middle of the inside of the can. If any uncoated metal is present on the inside of the can, a current is passed between these two probes and registers as a value on an LED display. The LED displays the conveyed currents in milliamps (mA). The current that is passed is directly proportional to the amount of metal that has not been effectively covered with coating.
- an electrically conductive solution 1% NaCl in deionized water
- Preferred coatings give metal exposure values of less than 3 mA, more preferred values of less than 2 mA, and even more preferred values of less than 1 mA. Commercially acceptable metal exposure values are typically less than 2.0 mA on average.
- Drop damage resistance measures the ability of the coated container to resist cracks after being conditions simulating dropping of a filled can.
- the presence of cracks is measured by passing electrical current via an electrolyte solution, as previously described in the Metal Exposure section.
- a coated container is filled with the electrolyte solution and the initial metal exposure is recorded.
- the can is then filled with water and dropped through a tube from a specified height onto an inclined plane, causing a dent in the chime area.
- the can is then turned 180 degrees, and the process is repeated. Water is then removed from the can and metal exposure is again measured as described above. If there is no damage, no change in current (mA) will be observed. Typically, an average of 6 or 12 container runs is recorded. Both metal exposures results before and after the drop are reported. The lower the milliamp value, the better the resistance of the coating to drop damage.
- Preferred coatings give metal exposure values after drop damage of less than 3.5 mA, more preferred valued of less than 2.5 mA, and even more preferred values of
- Solvent Resistance The extent of "cure” or crosslinking of a coating is measured as a resistance to solvents, such as methyl ethyl ketone (MEK, available from Exxon, Newark, NJ) or isopropyl alcohol (IPA). This test is performed as described in ASTM D 5402 - 93. The number of double-rubs (i.e., one back-and forth motion) is reported.
- solvents such as methyl ethyl ketone (MEK, available from Exxon, Newark, NJ) or isopropyl alcohol (IPA).
- the global extraction test is designed to estimate the total amount of mobile material that can potentially migrate out of a coating and into food packed in a coated can.
- coated substrate is subjected to water or solvent blends under a variety of conditions to simulate a given end-use.
- Acceptable extraction conditions and media can be found in 21CFR 175.300 paragraphs (d) and (e).
- the allowable global extraction limit as defined by the FDA regulation is 50 parts per million (ppm).
- Adhesion testing is performed to assess whether the coating adheres to the coated substrate.
- the adhesion test was performed according to ASTM D 3359 - Test Method B, using SCOTCH 610 tape, available from 3M Company of Saint Paul, Minnesota.
- Adhesion is generally rated on a scale of 0-10 where a rating of "10" indicates no adhesion failure, a rating of "9” indicates 90% of the coating remains adhered, a rating of "8” indicates 80% of the coating remains adhered, and so on. Adhesion ratings of 10 are typically desired for commercially viable coatings. Blush Resistance
- Blush resistance measures the ability of a coating to resist attack by various solutions. Typically, blush is measured by the amount of water absorbed into a coated film. When the film absorbs water, it generally becomes cloudy or looks white. Blush is generally measured visually using a scale of 0-10 where a rating of " 10" indicates no blush and a rating of "0" indicates complete whitening of the film. Blush ratings of at least 7 are typically desired for commercially viable coatings and optimally 9 or above.
- the reverse impact measures the coated substrate's ability to withstand the deformation encountered when impacted by a steel punch with a hemispherical head.
- coated substrate was subjected to 12 in-lbs (1.36 N m) of force using BYK-Gardner "overall" Bend and Impact Tester and rated visually for micro-cracking or micro-fracturing - commonly referred to as crazing. Test pieces were impacted on the uncoated or reverse side. A rating of 10 indicates no craze and suggests sufficient flexibility and cure. A rating of 0 indicates complete failure. Commercially viable coatings preferably show slight or no crazing on a reverse impact test.
- Dome Dome impact was evaluated by subjecting the dome apex of a 12 oz. beverage can to a reverse impact as described in the previous section. Craze was evaluated after impact. A rating of 10 indicates no craze and suggests sufficient flexibility and cure. A rating of 0 indicates complete failure. Coatings for beverage can interiors preferably show no craze (rating of 10) on a dome impact.
- a 1 % solution of JOY Detergent (available from Procter & Gamble) in deionized water is prepared and heated to 82 0 C (18O 0 F). Coated panels are immersed in the heated solution for 10 minutes and are then removed, rinsed, and dried. Samples are then evaluated for adhesion and blush, as previously described. Commercially viable beverage interior coatings preferably give adhesion ratings of 10 and blush ratings of at least 7, optimally at least 9, in the detergent test.
- Feathering Feathering is a term used to describe the adhesion loss of a coating on the tab of a beverage can end. When a beverage can is opened, a portion of free film may be present across the opening of the can if the coating loses adhesion on the tab. This is feathering.
- a "tab" is scored on the backside of a coated panel, with the coated side of the panel facing downward.
- the test piece is then pasteurized as described under the Pasteurization section below.
- pliers are used to bend the cut “tab” to a 90 degree angle away from the coated side of the substrate.
- the test piece is then placed on a flat surface, coated side down.
- the cut "tab” is gripped using pliers and the “tab” is pulled from the test panel at an angle of 180 degrees until it is completely removed.
- any coating that extends into the opening on the test panel is measured. The distance of the greatest penetration (feathering) is reported in inches.
- Coatings for beverage ends preferably show feathering below 0.2 inch (0.508 cm), more preferably below 0.1 inch (0.254 cm), most preferably below 0.05 inch (0.127 cm), and optimally below 0.02 inch (0.051 cm).
- the "Dowfax” test is designed to measure the resistance of a coating to a boiling detergent solution. This is a general test run for beverage end coatings and is mainly used to evaluate adhesion. Historically, this test was used to indicate problems with the interaction of coating to substrate pretreatment.
- the solution is prepared by mixing 5 ml of Dowfax 2Al (product of Dow Chemical) into 3000 ml of deionized water. Typically, coated substrate strips are immersed into the boiling Dowfax solution for 15 minutes. The strips are then rinsed and cooled in deionized water, dried, and then tested and rated for blush and adhesion as described previously.
- Preferred beverage end coatings provide adhesion ratings of 10 and blush ratings of at least 4, more preferably 6 or above in the Dowfax detergent test.
- the sterilization or pasteurization test determines how a coating withstands the processing conditions for different types of food products packaged in a container.
- a coated substrate is immersed in a water bath and heated for 5-60 minutes at temperatures ranging from 65 0 C to 100 0 C.
- the coated substrate was immersed in a deionized water bath for 45 minutes at 85°C.
- the coated substrate was then removed from the water bath and tested for coating adhesion and blush as described above.
- Commercially viable coatings preferably provide adequate pasteurization resistance with perfect adhesion (rating of 10) and blush ratings of at least 5, optimally at least 9.
- Coefficient of friction is a measurement of lubricity of a coating and is used to give an indication of how a cured coating will perform on commercial fabrication equipment and presses. Typically, lubricants are added to coatings requiring aggressive post application fabrication to give the appropriate lubricity.
- an Altek Mobility / Lubricity Tester Model 9505 AE with a chart recorder was used to measure the COF of cure beverage end coatings on aluminum substrates. The instrument works by pulling a sled with steel balls attached to a loadbar across the surface of the coated substrate, and the COF is charted out as resistance on 0-10 scale chart paper. Each unit equals 0.25 COF units.
- Coatings of the present invention are formulated to give a preferred COF range of 0.055 to 0.095.
- WACO Enamel Rater II available from the Wilkens-Anderson Company, Chicago, IL, with an output voltage of 6.3 volts. The measured electrical current, in milliamps, is reported. End continuities are typically tested initially and then after the ends are subjected to pasteurization or retort. Preferred coatings of the present invention initially pass less than 10 milliamps (mA) when tested as described above, more preferably less than 5 rnA, most preferably less than 2 mA, and optimally less than 1 mA.
- preferred coatings give continuities of less than 20 mA, more preferably less than 10 mA, even more preferably less than 5 mA, and even more preferably less than 2 mA.
- Example 1 Run 1. Preparation of Acid-Functional Acrylic A premix of 512.6 parts glacial methacrylic acid (MAA), 512.6 parts butyl acrylate (BA), 114.0 parts styrene, and 73.2 parts benzoyl peroxide (70% water wet) was prepared in a separate vessel. A 3-liter flask was equipped with a stirrer, reflux condenser, thermocouple, heating mantle, and nitrogen blanket. Ten percent of the premix was added to the flask along with 405.9 parts butanol and 30.6 parts deionized water. To the remaining premix were added 496.1 parts butanol and 38.3 parts deionized water.
- MAA glacial methacrylic acid
- BA butyl acrylate
- benzoyl peroxide 70% water wet
- the contents were heated to 93°C.
- external heating was stopped and the material was allowed to increase in temperature for fifteen minutes.
- the batch was at 97 0 C, and the remaining premix was added uniformly over two hours maintaining 97°C to 100 0 C.
- the premix vessel was rinse with 5 parts butanol.
- the batch was held at temperature for two and a half hours.
- the heating was discontinued and 317.7 parts butyl cellosolve was added.
- the resulting acrylic prepolymer was 44.3% solids (NV), with an acid number of 313 and a Brookfield viscosity (as determined by ASTM D-2196) of 4,990 centipoise (cps).
- Example 1 Run 2. Preparation of Acid-Functional Acrylic A premix of 677.7 parts glacial methacrylic acid, 677.7 parts butyl methacrylate (BMA), 150.8 parts styrene, and 96.9 parts benzoyl peroxide (70% water wet) was prepared in a separate vessel. A 5-liter flask was equipped with a stirrer, reflux condenser, thermocouple, heating mantle, and nitrogen blanket. Ten percent of the premix was added to the flask along with 536.9 parts butanol and 40.7 parts deionized water. To the remaining premix were added 758.1 parts butanol and 50.6 parts deionized water.
- BMA butyl methacrylate
- benzoyl peroxide 70% water wet
- the contents were heated to 93°C.
- external heating was stopped, and the material was allowed to increase in temperature for ten minutes.
- the batch was at 98°C, and the remaining premix was added uniformly over two hours maintaining 97°C to 100 0 C.
- the batch was held at temperature for three hours.
- the heating was discontinued and the batch cooled.
- the resulting acrylic prepolymer was 49.9% NV, with an acid number of 304 and a Brookfield viscosity of 101,000 centipoise.
- a premix of 802.6 parts glacial methacrylic acid, 807 parts butyl methacrylate, 178.5 parts styrene, 80.3 parts t-butyl peroctoate, 838.5 parts butanol, and 59.9 parts deionized water was prepared in a separate vessel.
- a 5-liter flask was equipped with a stirrer, reflux condenser, thermocouple, heating mantle, and nitrogen blanket. Added to the 5 liter flask were 635.8 parts butanol and 48.1 parts deionized water. The flask was heated to 94°C. At 94°C 12.5 parts t-butyl peroctoate were added.
- the batch was held for five minutes after which the premix was added over two and a half hours.
- a second premix containing 59.2 parts butanol and 16.1 parts t-butyl peroctoate was prepared. When the addition of the first premix was complete the second premix was added over 30 minutes. Once complete, the batch was held for 30 minutes. A chase of 3.4 parts t-butyl peroctoate was added and the batch held for two hours. After the two-hour hold time, the heat was discontinued and the batch cooled.
- the resulting acrylic prepolymer was 50.1% NV, with an acid number of 292 and a Brookfield viscosity of 150,000 centipoise.
- Example 1 Run 4. Preparation of Acid-Functional Acrylic A premix of 802.6 parts glacial methacrylic acid, 445.9 parts ethyl acrylate, 535.1 parts styrene, 108.6 parts t-butyl peroctoate, 838.5 parts butanol, and 59.9 parts deionized water was prepared in a separate vessel. A 5-liter flask was equipped with a stirrer, reflux condenser, thermocouple, heating mantle, and nitrogen blanket. Added to the 5-liter flask was 635.8 parts butanol and 48.1 parts deionized water. The flask was heated to 94 0 C.
- Example 1 Run 12. Preparation of Acid-Functional Acrylic A premix of 803.4 parts glacial methacrylic acid, 446.3 parts ethyl acrylate (EA), 535.5 parts styrene, 153 parts benzoyl peroxide (70% water wet), 839.2 parts butanol, and 60 parts deionized water was prepared in a separate vessel. A 5-liter flask was equipped with a stirrer, reflux condenser, thermocouple, heating mantle and nitrogen blanket. To the flask, 636.3 parts butanol and 48.2 parts deionized water were added and heated to 97°C to 100 0 C with a nitrogen blanket flowing in the flask.
- the premix was added uniformly over two and a half hours maintaining 97°C to 100 0 C.
- the premix vessel was rinsed with 59.2 parts butanol and added to the flask.
- the batch was held at temperature for two hours. The heating was discontinued and the batch cooled.
- the resulting acrylic prepolymer was 50.2% NV, with an acid number of 301 and a Brookfield viscosity of 25,400 centipoise.
- Example 2 Run 1. Preparation of Salt of Acid-Functional Acrylic A 3-liter flask was equipped with a stirrer, reflux condenser, Dean Stark Tube, thermocouple, heating mantle, and nitrogen blanket. Into the flask was added 711.5 parts of Example 1: Run 1 acrylic, 762.9 parts deionized water, and 56.9 parts dimethyl ethanol amine (DMEA). The contents were heated to reflux and 553 parts were distilled from the flask. After distillation was complete, 598 parts of deionized water were added. The batch was cooled giving an acrylic solution at 20.3% solids and 307 acid number.
- Example 2 Run 2.
- Example 2 Run 3. Preparation of Salt of Acid-Functional Acrylic
- a 5-liter flask was equipped with a stirrer, reflux condenser, Dean Stark Tube, thermocouple, heating mantle, and nitrogen blanket.
- Into the flask was added 1852.3 parts of Example 1: Run 3 acrylic, 2219 parts deionized water, and 163 parts dimethyl ethanol amine. The contents were heated to reflux and 1463 parts were distilled from the flask. After distillation was complete, 1581 parts of deionized water were added. The batch was cooled giving an acrylic solution at 21.6% solids, 284 acid number, pH of 6.23 and a viscosity of 13 seconds (Number 4 Ford cup).
- Example 1 A 5-liter flask was equipped with a stirrer, reflux condenser, Dean Stark Tube, thermocouple, heating mantle, and nitrogen blanket. Into the flask was added 1799.2 parts of Example 1: Run 4 acrylic, 2155.9 parts deionized water, and 158.6 parts dimethyl ethanol amine. The contents were heated to reflux and 1541 parts were distilled from the flask. After distillation was complete, 1615 parts of deionized water were added. The batch was cooled giving an acrylic solution at 22.1% solids, 302 acid number, pH of 6.55 and a Brookfield viscosity of 2060 centipoise.
- Example 2 Using techniques from Example 2: Run 4 the systems shown in Table 3 were prepared. Each run of Example 2 used the correspondingly numbered run from Example 1. That is, Example 2: Run 5 used the acrylic prepolymer from Example 1: Run 5, etc.
- a 1 -liter flask was equipped with a stirrer, reflux condenser, thermocouple, heating mantle, and nitrogen blanket.
- Example 2 Run 3 salt and 267.3 parts deionized water.
- the contents of the flask were heated to 75°C at 280 revolutions per minute (RPM).
- RPM revolutions per minute
- a premix of 71.4 parts styrene, 116.3 parts butyl methacrylate, and 16.3 parts glycidyl methacrylate (GMA) was prepared. Once the flask was at 75°C, 10% of the premix was added followed by 2.04 parts benzoin and 20 parts deionized water. The flask was heated further to 79°C.
- a 0.5-liter flask was equipped with a stirrer, reflux condenser, thermocouple, heating mantle, and nitrogen blanket.
- Example 2 Run 4 salt and 120.6 parts deionized water.
- the contents of the flask were heated to 75°C at 240 RPM.
- a premix of 66.3 parts styrene, 19.6 parts ethyl acrylate, and 7.5 parts glycidyl methacrylate was prepared.
- 10% of the premix was added followed by 0.91 part benzoin and 9.4 parts deionized water.
- the flask was heated further to 79°C. At 79°C, 0.91 part of 35% hydrogen peroxide was added and held for five minutes.
- Example 3 Run 3. Emulsion A 1 -liter flask was equipped with a stirrer, reflux condenser, thermocouple, heating mantle, and nitrogen blanket. Into the flask was added 311.2 parts of Example 2: Run 4 salt and 241.2 parts deionized water. The contents of the flask were heated to 75°C at 270 RPM. In a separate vessel, a premix of 112.1 parts styrene, 59.8 parts ethyl acrylate, and 14.9 parts glycidyl methacrylate was prepared. Once the flask was at 75°C, 10% of the premix was added followed by 1.87 parts benzoin and 18.8 parts deionized water. The flask was heated further to 79°C.
- Example 3 Run 4. Emulsion A 5-liter flask was equipped with a stirrer, reflux condenser, thermocouple, heating mantle, and nitrogen blanket. Into the flask was added 1525.0 parts of Example 2: Run 4 salt and 1219.1 parts deionized water. The contents of the flask were heated to 70 0 C at 250 RPM. In a separate vessel a premix of 380.4 parts styrene, 278.3 parts butyl acrylate (BA), 194.9 parts butyl methacrylate, and 74.2 parts glycidyl methacrylate was prepared. Once the flask was at 70 0 C, 10% of the premix was added followed by 9.29 parts benzoin and 92.9 parts deionized water.
- BA butyl acrylate
- the flask was heated further to 79°C. At 79°C, 9.29 parts of 35% hydrogen peroxide were added and held for five minutes. After five minutes the temperature control was set at 81°C and the remaining premix was added over one hour. When the addition was complete, 92.9 parts deionized water were used to rinse the residual premix into the flask. The batch was held for ten minutes and then 1.59 parts benzoin, 92.9 parts deionized water, and 1.59 parts 35% hydrogen peroxide were added. The batch was held for 45 minutes and then 0.52 part benzoin and 0.52 part 35% hydrogen peroxide were added. After two hours the heat was removed and the batch cooled. This gave an emulsion at 31.4% solids, 64.1 acid number, pH of 6.95, and a viscosity of 22 seconds (Number 4 Ford cup).
- Example 3 Run 5. Emulsion A 12-liter flask was equipped with a stirrer, reflux condenser, thermocouple, heating mantle, and nitrogen blanket. Into the flask was added 3886.5 parts of
- Example 2 Run 4 salt and 3022.5 parts deionized water.
- the contents of the flask were heated to 70 0 C at 235 RPM.
- a premix of 771.25 parts styrene, 933.75 parts butyl acrylate, 537.5 parts butyl methacrylate, and 93.75 parts glycidyl methacrylate was prepared.
- the flask was heated further to 79°C. At 79 0 C, 23.38 parts of 35% hydrogen peroxide and 116.25 parts deionized water were added and held for five minutes.
- Example 2 A design experiment using Example 2: Run 4 as the acid functional acrylic salt and the process outlined above was prepared and is depicted in Table 7.
- Example 3 Run 5b was included as one of the variables and was a repeat of Run 5.
- a series of emulsions, shown in Table 11 were prepared using a monomer to acid functional acrylic ratio of 73/27 solids/solids. These systems were prepared using the process outlined in Example 3: Run 5 using Example 2: Run 4 as the acid functional acrylic salt.
- Results from Table 13 indicate the optimum level of IBMA in the emulsion monomer composition is around 5%, when used in conjunction with hydroxyl functionality in the acrylic polymer stabilizer.
- Run 4 was successfully formulated into a spray applied coating for the interior of beer/beverage aluminum cans.
- the product was formulated with or without additional surfactant, as described in Table 14.
- Example 3 The water-based emulsion of Example 3: Run 4 and Example 3: Run 7 were successfully formulated into spray applied coatings for the interior of beer/beverage aluminum cans. Coating compositions are shown in Table 16.
- the coatings of the present invention compare favorably to the commercial epoxy-acrylate coating, and there is a substantial benefit for retort resistance.
- Example 3 Run 5 emulsion was stirred with 16.75 parts SLIPAYD 404 wax. The mixture was stirred for 10 minutes to make it uniform. The mixture was then filtered. The mixture was approximately 31% solids. The mixture was applied at 7-8 milligrams per square inch (msi) (1.1- 1.25 mg/cm 2 ) on ALX Aleoa aluminum and baked for 10 seconds (sec) to achieve a 400 0 F (204 0 C) peak metal temperature in a coil oven.
- msi milligrams per square inch
- Valspar coded 13Q80AG Performed after a 45 minutes at 85 0 C (185°F) pasteurization. Measured in centimeters.
- Example 5 Using the process of Example 5: Run 1, the formulations shown in Table 19 were prepared to investigate the effect of GMA level on end continuities. Each formula was applied at 7-8 milligrams per square inch (msi) (1.1 - 1.25 mg/cm 2 ) on
- a phenol-formaldehyde phenolic at 50% in water prepared by reacting 2.3 moles of formaldehyde with 1 mole of phenol.
- Example 5 Using the process of Example 5: Run 1, the formulation shown in Table 21 was prepared. The formula was applied at 7-8 milligrams per square inch (msi) (1.1-1.25 mg/cm 2 ) on ALX Alcoa aluminum and baked for 10 seconds to achieve 400 0 F (204 0 C) and 42O 0 F (215 0 C) peak metal temperatures in a coil oven. Film and end performance properties are shown in Table 22. This material contains 4% GMA and 5% IBMA in the emulsion monomer mix and an acrylic composition with hydroxyl functionality.
- Results from Table 22 show that a beverage end formulation of the present invention can give similar performance to a commercial epoxy-based waterborne beverage end coating even with lower solvent resistance as measured by MEK double nibs. There is also an added benefit of improved feathering resistance.
- Example 6 is designed to illustrate the use of a different acid-functional polymer salt as the stabilizer for an emulsion of the present invention.
- a 2-liter flask was equipped with a stirrer, packed colomn, Dean Stark trap, reflux condenser, thermocouple, heating mantle and nitrogen blanket.
- To the flask 700.1 parts dipropylene glycol and 700.1 parts isophthalic acid were added. Under a nitrogen blanket, the contents were heated to 125 0 C. At 125 0 C, 1.05 parts FASCAT
- the material from Stage A (599.8 parts) was placed in a 2-liter flask. The temperature was set at 112 0 C and 82 parts trimellitic anhydride was added. The material was heated to 232 0 C, and water was removed. After an acid number of 48.4 was obtained, a portion of the material was used in Stage C.
- the material from Stage B (198.8 parts) was added to a 2-liter flask, and 40 parts of DOWANOL PNP were added. The material was adjusted to 74 0 C, and slow addition of deionized water (200 parts) was initiated. After about 30 parts of water were added, 7.6 parts dimethyl ethanolamine were introduced. When about 150 parts of the deionized water were in, heating was halted (the temperature was at 8O 0 C) and 2.4 parts dimethyl ethanolamine were added. After the entire charge of deionized water was complete, the viscosity was visually high and 200 additional parts deionized water was added. The material was allowed to slowly cool while additional dimethyl ethanolamine was added incrementally to increase the pH to 6.6. The resulting product was 29.7% solids with an acid number of 53.9.
- Stage D A 500-milliliter flask was equipped with a stirrer, reflux condenser, thermocouple, heating mantle, and nitrogen blanket. Into the flask was added 93.2 parts of the Stage C material and 179 parts deionized water. While the contents of the flask were being heated to 5O 0 C at 240 RPM, 2 drops of HAMP-OL 4.5% Iron and 1.11 parts erythorbic acid were added. In a separate vessel a premix of 28.8 parts styrene, 50.9 BA, 21.0 parts BMA, 5.6 parts IBMA, 4.5 parts GMA and 1.1 1 parts TRIGONOX A-W70 were premixed.
- Valspar coded 13Q80AG 2 30 minutes at 85 0 C (185 0 F). 3 90 minutes at 121 0 C (250 0 F).
- Example 7 Emulsion for Inside Spray A 3-liter flask was equipped with a stirrer, reflux condenser, thermocouple, heating mantle, and nitrogen blanket. Into the flask was added 392.2 parts of Example 1: Run 4 Acid Functional Acrylic, 86.4 parts deionized water, 34.6 parts DMEA, and 1120.8 parts deionized water. The contents of the flask were heated to 70 0 C. In a separate vessel a premix of 215.8 parts styrene, 302.7 parts butyl acrylate, and 42.0 parts glycidyl methacrylate was prepared.
- the batch was held for ten minutes and then 0.96 parts benzoin, 55.9 parts deionized water, and 0.95 parts 35% hydrogen peroxide were added. The batch was held for 45 minutes and then 0.31 parts benzoin and 0.31 part 35% hydrogen peroxide were added. After two hours the batch was cooled to 45°C. Once at 45°C, 0.46 part of HAMP-OL 4.5% Iron 2.98 parts TRIGONOX A- W70, and a premix of 2.1 parts erythorbic acid, 0.91 parts DMEA, and 18.0 parts deionized water, were added. The batch was held at 45°C for one hour. The material was then cooled to give an emulsion at 31.6% solids, 67.7 acid number, pH of 7O4, and a viscosity of 84 seconds (Number 4 Ford cup).
- Example 7 The water-based emulsion of Example 7 was successfully formulated into a spray applied coating for the interior of beer/beverage aluminum cans.
- the product was formulated as described in Table 24.
- This formulation was sprayed at typical laboratory conditions at 120 milligram per can (mg/can) to 130 mg/can coating weight for the application of interior beverage coatings, and cured at 188 0 C to 199 0 C (measured at the can dome) for 30 seconds through a gas oven conveyor at typical heat schedules for this application.
- the film properties shown in Table 25 were achieved.
- Valspar coded 10Q45AF 2 90 minutes at 25O 0 F (121 0 C)
- Example 9 Latex with Polyester-Polyether Stabilizer
- Example 9 illustrates the use of a different acid-functional polymer salt as the stabilizer for an emulsion of the present invention.
- a flask was equipped with a stirrer, packed column, Dean Stark trap, reflux condenser, thermocouple, heating mantle and nitrogen blanket.
- 809.8 parts sebacic acid and 1283.0 parts CHDM-90 (90% 1,4-cyclohexane dimethanol in water) were added. Under a nitrogen blanket, the contents were heated to distill the water from the CHDM-90. While the contents were heated at 165°C, 1.96 parts FASCAT 4100 was added. The temperature was increased to 220 0 C to remove water.
- a sample of the batch was tested and found to have an acid number of 0.5. The remainder of the batch was weighed, and to 1711.7 parts of this material were added 1040.2 parts of para-hydroxy benzoic acid. The batch was heated to 230 0 C to remove water. To aid in the removal of water xylene was added incrementally.
- the material from Stage A (1915.2 parts) was placed in a flask along with 823.8 parts ERISYS GE-22 (cyclohexanedimethanol diglycidyl ether, , 84.8 parts methyl isobutyl ketone (, and 2.63 parts Catalyst 1201 (ethyltriphenyl phosphonium iodide,).
- the temperature was set at 17O 0 C and the contents heated. After three hours at temperature, the epoxy value of the material was 0.003.
- the batch was adjusted to have 2684.2 parts of this material in the flask. Added to the flask were 145.0 parts methyl isobutyl ketone and 294.7 parts succinic anhydride.
- a 5-liter flask was equipped with a stirrer, reflux condenser, thermocouple, heating mantle, and nitrogen blanket.
- Into the flask was added 1183.4 parts of the Stage B material and 779.6 parts deionized water.
- a premix of 7.25 parts erythorbic acid, 6.5 parts DMEA, and 76.7 parts deionized water was prepared. This initial premix and 0.18 parts HAMP-OL 4.5% Iron were added to the flask.
- the contents of the flask were heated to 3O 0 C.
- a monomer premix of 249.0 parts styrene, 113.8 BA, 106.7 parts BMA, 177.8 parts Hydroxy Ethyl Methacrylate (HEMA), 35.6 parts IBMA, and 28.5 parts GMA was prepared.
- a third premix of 7.25 parts TRIGONOX A-W70 and 82.2 parts deionized water were made. Once all the premixes were prepared and the flask at 3O 0 C, the stirrer was set at 240 rpm and all of the monomer premix was added. The monomer premix vessel was rinsed with 81.6 parts deionized water, which was also added to the flask.
- the contents of the flask were stirred for 10 minutes, after which 10% of the third premix was added within one minute. Once the 10% of the third premix was in, the temperature was increased to 37 0 C and the batch was held for five minutes. After five minutes, the remaining amount of the third premix was added over 45 minutes. The temperature was allowed to increase with no external heat applied. During the addition the maximum temperature was 57 0 C. After the addition was complete the temperature was 51 0 C. Temperature control was set for 52 0 C. The third premix was rinsed with 108.4 parts deionized water and added to the batch. The batch was held for 1.5 hours and then cooled. This yielded an emulsion at 33.1% solids, 27.1 acid number, pH of 7.9, and a viscosity 12 sec (Number 4 Ford Cup).
- Example 10 illustrates the use of a different acid-functional polymer salt as the stabilizer for an emulsion of the present invention.
- BPA Approximately 1055 parts BPA is placed in a flask along with approximately 1684 parts of liquid epoxy resin (EPON 828), 85 parts methyl isobutyl ketone, and 2 to 3 parts Catalyst 1201. The temperature is set at 16O 0 C and the contents are then heated for approximately three hours to achieve an epoxy value of the material of approximately 0.003. The batch is then adjusted to have 2684.2 parts of this material in the flask. Added to the flask is 145.0 parts methyl isobutyl ketone and 294.7 parts succinic anhydride. The temperature is maintained at 120-135 0 C for two hours.
- liquid epoxy resin EPON 828
- methyl isobutyl ketone 85 parts methyl isobutyl ketone
- the temperature is set at 16O 0 C and the contents are then heated for approximately three hours to achieve an epoxy value of the material of approximately 0.003.
- the batch is then adjusted to have 2684.2 parts of this material in the flas
- a 5-liter flask is equipped with a stirrer, reflux condenser, thermocouple, heating mantle, and nitrogen blanket.
- Into the flask is added approximately 1183 parts of the Stage 1 material and 780 parts deionized water.
- a premix of 7.25 parts erythorbic acid, 6.5 parts DMEA, and 77 parts deionized water is prepared. This initial premix and 0.18 parts HAMP-OL 4.5% Iron are added to the flask.
- the contents of the flask are heated to 3O 0 C.
- a monomer premix of 249 parts styrene, 114 BA, 107 parts BMA, 178 parts HEMA, 36 parts IBMA, and 28 parts GMA is prepared.
- a third premix of 7.25 parts TRIGONOX A-W70 and 82.2 parts deionized water is made. Once all the premixes are prepared and the flask is at 3O 0 C, the stirrer is set at 240 rpm and all of the monomer premix is added. The monomer premix vessel is rinsed with 82 parts deionized water, which is also added to the flask. The contents of the flask are stirred for 10 minutes, after which 10% of the third premix is added within one minute. Once the 10% is in the temperature is increased to 37 0 C. The batch is held for five minutes. After five minutes, the remaining amount of the third premix is added over 45 minutes. The temperature is allowed to increase with no external heat applied.
- the Stage 3 composition may be applied to non-chrome aluminum panels and baked for 10 seconds to achieve a 217 0 C peak metal temperature.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Life Sciences & Earth Sciences (AREA)
- Wood Science & Technology (AREA)
- Mechanical Engineering (AREA)
- Plasma & Fusion (AREA)
- Metallurgy (AREA)
- Physics & Mathematics (AREA)
- Paints Or Removers (AREA)
- Application Of Or Painting With Fluid Materials (AREA)
- Details Of Rigid Or Semi-Rigid Containers (AREA)
- Rigid Containers With Two Or More Constituent Elements (AREA)
Abstract
Description









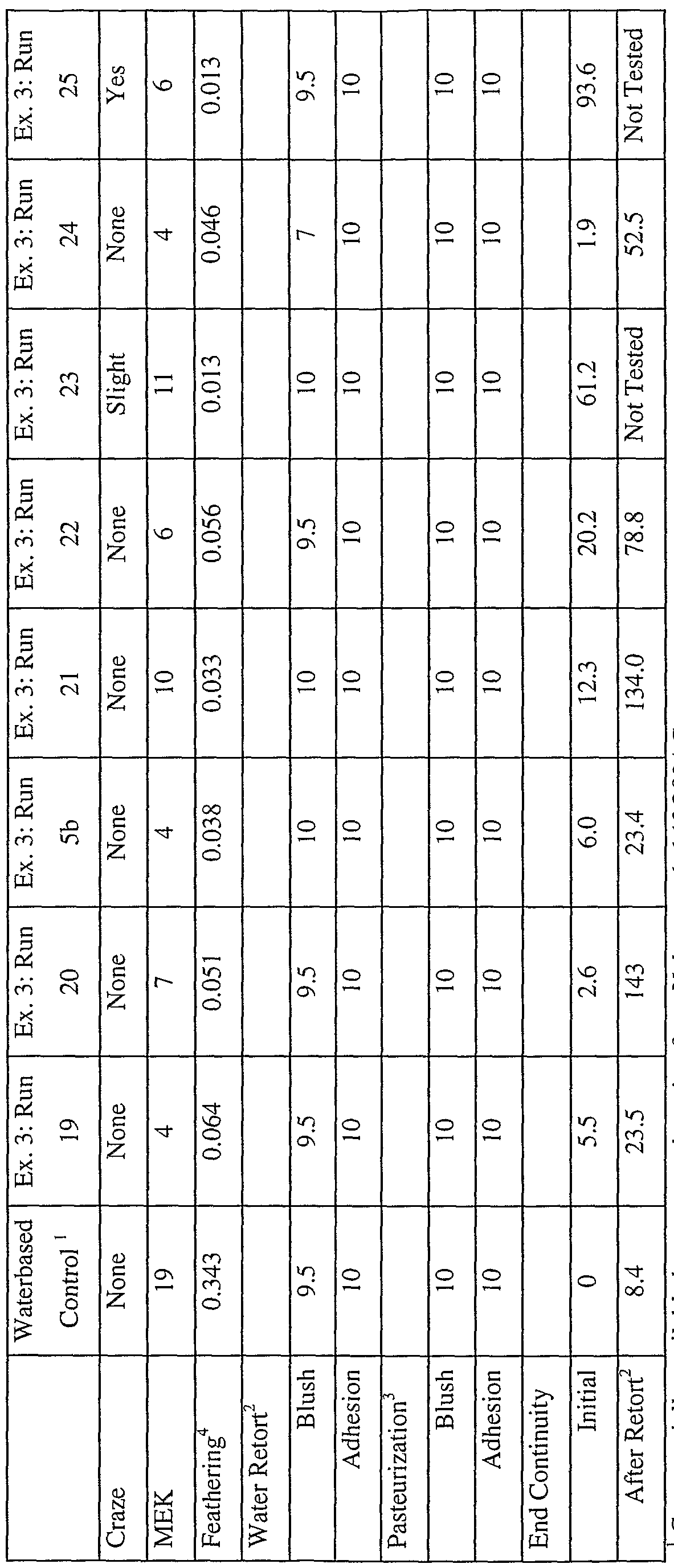



















Claims
Priority Applications (17)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP15199749.1A EP3037488B1 (en) | 2004-10-20 | 2005-10-18 | Coating compositions for cans and methods of coating |
| CA2579232A CA2579232C (en) | 2004-10-20 | 2005-10-18 | Coating compositions for cans and methods of coating |
| BRPI0517414A BRPI0517414B1 (en) | 2004-10-20 | 2005-10-18 | method for coating food or beverage can, food or beverage can, and spray-on composition for use in coating a food or beverage can |
| EP20174384.6A EP3733798B1 (en) | 2004-10-20 | 2005-10-18 | Coating compositions for cans and methods of coating |
| EP20174398.6A EP3778809B1 (en) | 2004-10-20 | 2005-10-18 | Coating compositions for cans and methods of coating |
| AU2005295228A AU2005295228B2 (en) | 2004-10-20 | 2005-10-18 | Coating compositions for cans and methods of coating |
| MX2007004463A MX2007004463A (en) | 2004-10-20 | 2005-10-18 | Coating compositions for cans and methods of coating. |
| BR122016007793-4A BR122016007793B1 (en) | 2004-10-20 | 2005-10-18 | Method for preparing an aluminum beverage can |
| CN2005800349149A CN101040016B (en) | 2004-10-20 | 2005-10-18 | Coating compositions for cans and methods of coating |
| EP11169891.6A EP2420542B2 (en) | 2004-10-20 | 2005-10-18 | Article and method of coating |
| JP2007538035A JP5027666B2 (en) | 2004-10-20 | 2005-10-18 | Coating composition for cans and jump coating method |
| EP05825629A EP1819789B1 (en) | 2004-10-20 | 2005-10-18 | Coating compositions for cans and methods of coating |
| EP22183883.2A EP4119626A1 (en) | 2004-10-20 | 2005-10-18 | Coating compositions for cans and methods of coating |
| PL05825629T PL1819789T3 (en) | 2004-10-20 | 2005-10-18 | Coating compositions for cans and methods of coating |
| KR1020127030357A KR20130003024A (en) | 2004-10-20 | 2005-10-18 | Coating compositions for cans and methods of coating |
| AT05825629T ATE513025T1 (en) | 2004-10-20 | 2005-10-18 | COATING COMPOSITIONS FOR CANS AND COATING PROCESSES |
| NO20071422A NO20071422L (en) | 2004-10-20 | 2007-03-16 | Coating mixtures for cans and coating methods |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US62063904P | 2004-10-20 | 2004-10-20 | |
| US60/620,639 | 2004-10-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006045017A1 true WO2006045017A1 (en) | 2006-04-27 |
Family
ID=35945336
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/037750 WO2006045017A1 (en) | 2004-10-20 | 2005-10-18 | Coating compositions for cans and methods of coating |
Country Status (14)
| Country | Link |
|---|---|
| US (11) | US7592047B2 (en) |
| EP (7) | EP3733798B1 (en) |
| JP (1) | JP5027666B2 (en) |
| KR (2) | KR20130003024A (en) |
| CN (2) | CN101040016B (en) |
| AT (1) | ATE513025T1 (en) |
| AU (1) | AU2005295228B2 (en) |
| BR (2) | BR122016007793B1 (en) |
| CA (1) | CA2579232C (en) |
| ES (3) | ES2565502T3 (en) |
| MX (1) | MX2007004463A (en) |
| NO (1) | NO20071422L (en) |
| PL (2) | PL1819789T3 (en) |
| WO (1) | WO2006045017A1 (en) |
Cited By (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007048094A2 (en) | 2005-10-18 | 2007-04-26 | Valspar Sourcing, Inc. | Coating compositions for containers and methods of coating |
| WO2011009040A1 (en) * | 2009-07-17 | 2011-01-20 | Valspar Sourcing, Inc. | Coating composition and articles coated therewith |
| US8227561B1 (en) | 2011-05-02 | 2012-07-24 | Empire Technology Development Llc | Bisphenol-A replacement materials |
| WO2012054691A3 (en) * | 2010-10-20 | 2012-08-09 | Valspar Sourcing, Inc. | Water-based coating system with improved adhesion to a wide range of coated and uncoated substrates including muffler grade stainless steel |
| EP2505625A1 (en) | 2011-03-31 | 2012-10-03 | Henkel AG & Co. KGaA | Varnishing formula for the interior surface of cans |
| WO2012166488A1 (en) * | 2011-05-27 | 2012-12-06 | Ppg Industries Ohio, Inc. | Grafted acrylic comprising water soluble and water insoluble portions and lattices and coatings comprising the same |
| DE102012223355A1 (en) | 2012-12-17 | 2014-06-18 | Henkel Ag & Co. Kgaa | Highly crosslinking paint formulation for inside can surfaces |
| US8802792B2 (en) | 2010-09-17 | 2014-08-12 | Empire Technology Development Llc | Partially hydrogenated bisphenol-A-based polymers as substitutes for bisphenol-A-based polymers |
| US8840966B2 (en) | 2009-09-18 | 2014-09-23 | Valspar Sourcing, Inc. | Polyurethane coating composition |
| WO2014186285A1 (en) * | 2013-05-16 | 2014-11-20 | The Coca-Cola Company | Polymer compositions and coatings for food and beverage packaging |
| WO2015002961A1 (en) * | 2013-07-02 | 2015-01-08 | Valspar Sourcing, Inc. | Coating compositions for packaging articles such as food and beverage containers |
| WO2015006522A1 (en) * | 2013-07-11 | 2015-01-15 | Ppg Industries Ohio, Inc. | Coating compositions with improved adhesion to containers |
| US9029470B2 (en) | 2009-02-24 | 2015-05-12 | Akzo Nobel Coatings International B.V. | Latex emulsions and coating compositions formed from latex emulsions |
| US9133292B2 (en) | 2009-03-05 | 2015-09-15 | Akzo Nobel Coatings International B.V. | Hydroxyl functional oil polyol acrylic graft copolymers |
| EP2420541B1 (en) | 2004-10-20 | 2015-12-30 | Valspar Sourcing, Inc. | Methods of coating a food or beverage can |
| US9260625B2 (en) | 2011-12-21 | 2016-02-16 | Akzo Nobel Coatings International B.V. | Water-based coating compositions |
| US9273226B2 (en) | 2011-12-21 | 2016-03-01 | Akzo Nobel Coatings International B.V. | Solvent-based coating compositions |
| US9394456B2 (en) | 2009-02-24 | 2016-07-19 | Akzo Nobel Coatings International B.V. | Latex emulsions and coating compositions formed from latex emulsions |
| US9458345B2 (en) | 2010-12-28 | 2016-10-04 | Akzo Nobel Coatings International B.V. | Coating compositions comprising latex emulsions and hydroxyl functional oil polyol graft copolymers |
| WO2016196174A1 (en) * | 2015-05-29 | 2016-12-08 | Ppg Industries Ohio, Inc. | Packaging coated with an emulsion polymerized latex polymer |
| EP2785604B1 (en) | 2011-12-02 | 2017-03-01 | PPG Industries Ohio Inc. | Coating composition for a food or beverage can |
| US9655912B2 (en) | 2012-02-16 | 2017-05-23 | Akeso Biomedical, Inc. | Reduction of gastrointestinal tract colonisation by campylobacter |
| WO2017106626A1 (en) * | 2015-12-16 | 2017-06-22 | Commonwealth Scientific And Industrial Research Organisation | Terpene derived coating composition |
| WO2017198936A1 (en) * | 2016-05-20 | 2017-11-23 | Constellium Neuf-Brisach | Metalloplastic strip for rigid food packaging, and manufacturing process |
| WO2017205110A1 (en) * | 2016-05-26 | 2017-11-30 | Ppg Industries Ohio, Inc. | Packaging coated with an emulsion polymerized latex polymer |
| WO2018013766A1 (en) * | 2016-07-15 | 2018-01-18 | Valspar Sourcing, Inc. | Latex coating composition having reduced flavor scalping properties |
| US9889468B2 (en) | 2012-12-17 | 2018-02-13 | Henkel Ag & Co. Kgaa | Method for manufacturing coated can lids |
| CN108794703A (en) * | 2018-06-27 | 2018-11-13 | 常州光辉化工有限公司 | A kind of preparation method of the polyacrylate anti-corrosion lotion with excellent salt spray resistance function |
| EP3498738A1 (en) * | 2017-12-15 | 2019-06-19 | PPG Industries Ohio, Inc. | A coating composition |
| JP2019518091A (en) * | 2016-04-15 | 2019-06-27 | エスダブリューアイエムシー・エルエルシー | Styrene-free copolymers, and coating compositions containing such copolymers |
| US10351714B2 (en) | 2013-07-02 | 2019-07-16 | Swimc Llc | Coating compositions for packaging articles such as food and beverage containers |
| US10526277B2 (en) | 2013-10-17 | 2020-01-07 | Swimc Llc | Food or beverage containers coated with polymers of di(amido(alkyl)phenol) compounds |
| US10563010B2 (en) | 2009-04-09 | 2020-02-18 | The Sherwin-Williams Company | Polymer having unsaturated cycloaliphatic functionality and coating compositions therefrom |
| US10793742B2 (en) | 2014-05-19 | 2020-10-06 | Valspar Sourcing, Inc. | Polyethers containing non-bisphenolic cyclic groups |
| US10800941B2 (en) | 2014-12-24 | 2020-10-13 | Valspar Sourcing, Inc. | Coating compositions for packaging articles such as food and beverage containers |
| US10968288B2 (en) | 2014-12-24 | 2021-04-06 | Swimc Llc | Styrene-free coating compositions for packaging articles such as food and beverage containers |
| US11059989B2 (en) | 2017-06-30 | 2021-07-13 | Valspar Sourcing, Inc. | Crosslinked coating compositions for packaging articles such as food and beverage containers |
| EP2658894B1 (en) | 2010-12-29 | 2022-10-19 | Akzo Nobel Coatings International B.V. | Adhesion promoter resin compositions and coating compositions having the adhesion promoter resin compositions |
| US11602768B2 (en) | 2016-10-19 | 2023-03-14 | Swimc, Llc | Acrylic polymers and compositions containing such polymers |
| US11981822B2 (en) | 2014-12-24 | 2024-05-14 | Swimc Llc | Crosslinked coating compositions for packaging articles such as food and beverage containers |
Families Citing this family (65)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2596077T5 (en) | 2003-04-02 | 2020-06-03 | Swimc Llc | Aqueous dispersions and coatings |
| US7644356B2 (en) * | 2005-06-10 | 2010-01-05 | Hewlett-Packard Development Company, L.P. | Constraint-based albuming of graphic elements |
| ATE453508T1 (en) | 2005-08-11 | 2010-01-15 | Valspar Sourcing Inc | COATINGS FREE OF BISPHENOL-A AND AROMATIC GLYCIDYL ETHER |
| US20090061219A1 (en) * | 2007-08-28 | 2009-03-05 | Valspar Sourcing, Inc. | Composition for Coating Glass |
| US8227027B2 (en) * | 2007-12-07 | 2012-07-24 | Presspart Gmbh & Co. Kg | Method for applying a polymer coating to an internal surface of a container |
| EP2227227B1 (en) * | 2007-12-07 | 2015-07-29 | Valspar Sourcing, Inc. | Coating suitable for medicament contact |
| US9801865B2 (en) | 2008-09-24 | 2017-10-31 | The United States Of America As Represented By The Department Of Veteran Affairs | Materials and methods for diagnosis, prevention and/or treatment of stress disorders and conditions associated with abeta peptide aggregation |
| WO2012051540A2 (en) * | 2010-10-15 | 2012-04-19 | Valspar Sourcing, Inc. | Polyester-based coating composition for metal substrates |
| EP3176201A1 (en) | 2009-04-09 | 2017-06-07 | Valspar Sourcing, Inc. | Polyester coating composition |
| WO2011009024A1 (en) * | 2009-07-17 | 2011-01-20 | Valspar Sourcing, Inc. | Coating compositions for cans and methods of coating |
| FR2953155B1 (en) * | 2009-12-02 | 2011-12-30 | Peugeot Citroen Automobiles Sa | METHOD FOR PREPARING THE BODY OF A MOTOR VEHICLE BEFORE PAINTING |
| JP5752784B2 (en) | 2010-04-16 | 2015-07-22 | ヴァルスパー・ソーシング・インコーポレーテッド | COATING COMPOSITION FOR PACKAGE ARTICLE AND COATING METHOD |
| EP2611951B1 (en) * | 2010-08-30 | 2019-08-21 | Ak Steel Properties, Inc. | Galvanized carbon steel with stainless steel-like finish |
| KR101830465B1 (en) | 2010-12-02 | 2018-02-20 | 디에스엠 아이피 어셋츠 비.브이. | Acrylic polymer |
| KR101864807B1 (en) * | 2010-12-29 | 2018-06-05 | 아크조노벨코팅스인터내셔널비.브이. | Latex emulsions and coating compositions formed from latex emulsions |
| RU2561969C2 (en) * | 2011-02-07 | 2015-09-10 | Вэлспар Сорсинг, Инк. | Compositions of coatings for containers and other products and methods of coating application |
| DK2705068T3 (en) | 2011-05-02 | 2015-06-22 | Ppg Ind Ohio Inc | Coating compositions containing 2,2'-biphenol |
| US9670378B2 (en) | 2011-05-23 | 2017-06-06 | Ppg Industries Ohio, Inc. | Coating compositions with improved adhesion to containers |
| US10442953B2 (en) | 2011-08-29 | 2019-10-15 | Ppg Industries Ohio, Inc. | Aqueous-based coating composition containing hydroxy-terminated polybutadiene |
| US9187221B2 (en) | 2011-09-27 | 2015-11-17 | Crown Packaging Technology, Inc. | Can ends having machine readable information |
| US9278776B2 (en) | 2011-09-27 | 2016-03-08 | Crown Packaging Technology, Inc. | Can ends having machine readable information |
| KR20140111679A (en) | 2011-12-28 | 2014-09-19 | 아크조노벨코팅스인터내셔널비.브이. | Acetoacetate functional latex emulsions cured with phenolic resins and coating compositions formed therefrom |
| CA2864122C (en) * | 2012-02-17 | 2020-07-21 | Valspar Sourcing, Inc. | Methods and materials for the functionalization of polymers and coatings including functionalized polymer |
| EP2838816B1 (en) | 2012-04-18 | 2021-09-08 | Swimc Llc | Low voc, water-based coating compositions suitable for protecting metal containing substrates including food and beverage packages |
| EP2861664B1 (en) * | 2012-06-19 | 2016-11-16 | Dow Global Technologies LLC | Aqueous based blend composition and method of producing the same |
| CN108528954B (en) | 2012-08-09 | 2020-05-12 | Swimc有限公司 | Compositions for containers and other articles and methods of using the same |
| WO2014025411A1 (en) | 2012-08-09 | 2014-02-13 | Valspar Sourcing, Inc. | Container coating system |
| US10358571B2 (en) | 2013-03-01 | 2019-07-23 | The Sherwin-Williams Company | Aqueous coating compositions including phenolic resin(s) |
| CN105189586B (en) * | 2013-03-15 | 2018-01-19 | 阿克佐诺贝尔国际涂料股份有限公司 | Mix moisture granular media, (poly-) ethene (methyl) acrylic copolymer compounded latex emulsion, mixing (poly-) ethene (methyl) acrylic acid organosilan compounded latex emulsion and the coating composition formed by it |
| CN103275656B (en) * | 2013-05-29 | 2015-07-08 | 北京化工大学 | Reactive pressure-sensitive adhesive having performance of structural adhesive after being cured, and preparation method thereof |
| US10308905B2 (en) | 2013-06-19 | 2019-06-04 | Isp Investments Llc | Method of removing phenols from a liquid |
| BR112016007095A2 (en) | 2013-10-02 | 2017-08-01 | Valspar Sourcing Inc | method for fabricating a removable closure system and removable closure and liner systems |
| EP4223659A1 (en) | 2013-10-02 | 2023-08-09 | Swimc Llc | Removable closure and coating system |
| US9464209B2 (en) | 2013-10-21 | 2016-10-11 | Ppg Industries Ohio, Inc. | Container coating compositions |
| CN106133235B (en) | 2014-04-07 | 2020-11-24 | 陶氏环球技术有限责任公司 | Sizing composition for carbon fibers |
| BR112016021583B1 (en) | 2014-04-14 | 2021-10-26 | Swimc Llc | METHOD FOR PRODUCING A HIGH MOLECULAR WEIGHT POLYETHER POLYMER AND FOOD AND BEVERAGE PACKAGING CONTAINER |
| US20160075855A1 (en) * | 2014-09-16 | 2016-03-17 | Eastman Chemical Company | Polymeric compositions with improved noise suppression |
| KR102696070B1 (en) * | 2014-10-23 | 2024-08-16 | 인프리아 코포레이션 | Organometallic solution based high resolution patterning compositions and corresponding methods |
| JP5968491B1 (en) * | 2015-04-20 | 2016-08-10 | 東洋製罐株式会社 | Structure having liquid film and method for producing the same |
| TWI614275B (en) | 2015-11-03 | 2018-02-11 | Valspar Sourcing Inc | Liquid epoxy resin composition for preparing a polymer |
| WO2017112837A1 (en) | 2015-12-23 | 2017-06-29 | Valspar Sourcing, Inc. | Latex polymers made using metallic-base-neutralized surfactant and blush-resistant coating compositions containing such polymers |
| MX2019001647A (en) * | 2016-08-10 | 2019-07-01 | Swimc Llc | Improvement of edge build and edge blister performance of coil coatings. |
| WO2018119175A1 (en) * | 2016-12-21 | 2018-06-28 | Valspar Sourcing, Inc. | Polymers containing reactive carbonyl groups and coating compositions containing such polymers |
| US10800866B2 (en) | 2017-01-17 | 2020-10-13 | Behr Process Corporation | Self-healing resin |
| EP3607014B1 (en) * | 2017-04-07 | 2021-06-02 | Akzo Nobel Coatings International B.V. | Coating compositions containing a hydroxyphenyl functional polymer and a latex polymer |
| CN109423129B (en) * | 2017-06-21 | 2020-08-28 | 广东华润涂料有限公司 | Aqueous coating composition suitable for forming side seam strip or coating on three-piece can |
| US11667809B2 (en) | 2017-08-25 | 2023-06-06 | Swimc Llc | Adhesion promoters and compositions for containers and other articles |
| CN117165157A (en) | 2017-09-01 | 2023-12-05 | 宣伟投资管理有限公司 | Multistage polymer latex, coating containing the latex and coated articles |
| KR102697657B1 (en) | 2017-09-01 | 2024-08-23 | 에스더블유아이엠씨 엘엘씨 | Multi-stage polymer latex, coating composition containing such latex, and article coated therewith |
| CN111448269B (en) * | 2017-10-16 | 2022-08-19 | 录象射流技术公司 | Continuous ink jet ink composition |
| WO2019118482A1 (en) | 2017-12-11 | 2019-06-20 | The Sherwin-Williams Company | Method for making water-dispersible and water-dispersed polymers |
| CN110922573A (en) * | 2017-12-28 | 2020-03-27 | 湖南辰砾新材料有限公司 | Organic fluorine unsaturated polyester resin emulsion and preparation method thereof |
| US10752799B2 (en) | 2018-02-07 | 2020-08-25 | Ppg Industries Ohio, Inc. | Self-curing coating compositions |
| JP7232022B2 (en) * | 2018-04-13 | 2023-03-02 | 東洋インキScホールディングス株式会社 | Aqueous paint composition, can member, and can |
| WO2019217641A1 (en) | 2018-05-11 | 2019-11-14 | Carbon, Inc. | Sustainable chemistry systems for recyclable dental models and other additively manufactured products |
| CN112384581A (en) * | 2018-06-11 | 2021-02-19 | 宣伟投资管理有限公司 | Packaging coatings comprising water-dispersible acrylic block copolymers |
| KR20210100701A (en) * | 2018-12-13 | 2021-08-17 | 피피지 인더스트리즈 오하이오 인코포레이티드 | Polyhydroxyalkylamide materials for use as crosslinking agents |
| EP3931227A1 (en) * | 2019-03-01 | 2022-01-05 | Dow Global Technologies LLC | Tackifier composition |
| BR112022009184A2 (en) | 2019-11-14 | 2022-07-26 | Swimc Llc | METAL PACKAGING POWDER COATING COMPOSITION, METHOD, METAL PACKAGING CONTAINER, COATED METALLIC SUBSTRATE, AND METHOD FOR MANUFACTURING METAL PACKAGING |
| JP2023519127A (en) * | 2020-03-26 | 2023-05-10 | マイケルマン,インコーポレーテッド | Low whitening coating formulations and related uses |
| US11597791B2 (en) * | 2020-03-27 | 2023-03-07 | Ppg Industries Ohio, Inc. | Crosslinking material and uses thereof |
| CN116964158A (en) * | 2021-01-29 | 2023-10-27 | Ppg工业俄亥俄公司 | Coating composition |
| JP2022148614A (en) * | 2021-03-24 | 2022-10-06 | 富士フイルムビジネスイノベーション株式会社 | Method for producing composite resin particle dispersion, method for producing pressure-sensitive adhesive, method for producing pressure-responsive resin, method for manufacturing toner for electrostatic charge image development, and composite resin particle dispersion |
| JP2024527454A (en) | 2021-05-19 | 2024-07-25 | エスダブリューアイエムシー・エルエルシー | Methods for coating metal substrates and making metal packaging, coated metal substrates, metal packaging, and powder coating composition systems |
| CN118284643A (en) | 2021-12-16 | 2024-07-02 | 湛新奥地利有限公司 | Coating composition |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3479310A (en) | 1963-09-19 | 1969-11-18 | Bayer Ag | Polyurethane plastics |
| US3991216A (en) * | 1974-05-16 | 1976-11-09 | Ppg Industries, Inc. | Beverage containers coated with a water based liner |
| GB1513866A (en) * | 1974-05-16 | 1978-06-14 | Ppg Industries Inc | Beverage containers |
| US4147679A (en) | 1976-06-02 | 1979-04-03 | Ppg Industries, Inc. | Water-reduced urethane coating compositions |
| GB2152065A (en) * | 1983-12-21 | 1985-07-31 | Inmont Corp | Water dilutable acrylated epoxy-phenolic coating compositions |
| US4600737A (en) * | 1983-12-21 | 1986-07-15 | Inmont Corporation | Water dilutable acrylated epoxy-phenolic coating compositions |
| US4647612A (en) * | 1985-12-30 | 1987-03-03 | Ppg Industries, Inc. | Polymer emulsion products |
| US4692491A (en) | 1985-12-30 | 1987-09-08 | Ppg Industries, Inc. | Polymer emulsion products |
| US5939482A (en) * | 1994-11-29 | 1999-08-17 | Vianova Resins Aktiengesellschaft | Method for the two-stage preparation of aqueous autocrosslinking copolymer dispersions and their use for coating materials |
Family Cites Families (344)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US1234060A (en) | 1913-04-30 | 1917-07-17 | Gen Electric | Incandescent electric lamp. |
| NL173809B (en) | 1951-11-17 | Rca Corp | IMAGE RECORDING DEVICE WITH COLOR CODING STRIP FILTER SYSTEM. | |
| US3242123A (en) | 1962-11-21 | 1966-03-22 | Union Carbide Corp | Glycidyl acrylate-styrene-ethyl acrylate-itaconic acid interpolymers |
| GB1096912A (en) | 1963-08-06 | 1967-12-29 | Ici Ltd | Synthetic polymers |
| US3335119A (en) * | 1963-08-08 | 1967-08-08 | Dal Mon Research Co | Fusible, convertible alkenyl aryl polymers |
| US3377406A (en) | 1963-12-16 | 1968-04-09 | Shell Oil Co | Process of esterification of polyepoxides with ethylenically unsaturated monocarboxylic acids in the presence of onium salts of inorganic acids |
| US3297621A (en) | 1965-01-04 | 1967-01-10 | Archer Daniels Midland Co | Two step polymerization of tetrapolymer |
| NL137295C (en) | 1965-11-03 | |||
| JPS4833015B1 (en) | 1967-06-02 | 1973-10-11 | Nippon Paint Co Ltd | |
| US3477990A (en) | 1967-12-07 | 1969-11-11 | Shell Oil Co | Process for reacting a phenol with an epoxy compound and resulting products |
| US3547885A (en) | 1968-03-08 | 1970-12-15 | Sun Oil Co | Process for curing polyepoxides with anhydrides and phosphonium halide catalysts therefor |
| CH515981A (en) | 1968-07-10 | 1971-11-30 | Bayer Ag | paint |
| US3568486A (en) | 1969-01-31 | 1971-03-09 | Montgomery H A Co | Preparation of metal for deforming operations |
| BE757936A (en) † | 1969-10-23 | 1971-04-01 | Bayer Ag | PROCESS FOR PREPARING ANIONIC MODIFIED POLYMERS IN EMULSION |
| CA955347A (en) | 1970-06-29 | 1974-09-24 | Nippon Oils And Fats Company Limited | Paint compositions capable of being cured by irradiation of electron ray |
| US3810859A (en) | 1970-07-22 | 1974-05-14 | Goodrich Co B F | Thickenable alkyl acrylate latices |
| US3694407A (en) | 1970-08-11 | 1972-09-26 | Shell Oil Co | Epoxy-containing condensates,their preparation and use |
| US3948855A (en) | 1971-09-16 | 1976-04-06 | The Dow Chemical Company | Process for reacting a phenol with a vicinal epoxy compound in the presence of phosphorus or carbon containing acid, ester or acid ester |
| US3738862A (en) | 1971-11-08 | 1973-06-12 | Shell Oil Co | Process for preparing reinforced laminates in situ with epoxy-polyhydric phenol condensates |
| US3819567A (en) | 1971-12-03 | 1974-06-25 | Du Pont | Sealer composition of an acrylic-epoxy ester graft copolymer and an epoxy resin |
| US3880793A (en) | 1972-03-30 | 1975-04-29 | Kansai Paint Co Ltd | Emulsifiers for emulsion polymerization of vinyl monomers |
| GB1421114A (en) | 1972-07-14 | 1976-01-14 | Canadian Ind | Thermosettable synthetic addition polymers |
| US3862914A (en) * | 1972-11-01 | 1975-01-28 | Mobil Oil Corp | Water-based coatings from polyepoxides and polycarboxylic acid monoanhydrides |
| JPS5339387Y2 (en) | 1973-07-26 | 1978-09-25 | ||
| GB1482969A (en) | 1973-10-12 | 1977-08-17 | Kansai Paint Co Ltd | Non-flammable binder compositions |
| US4247439A (en) | 1973-11-06 | 1981-01-27 | E. I. Du Pont De Nemours And Company | Water-borne coating composition made from epoxy resin, polymeric acid and tertiary amine |
| US4303488A (en) | 1973-11-06 | 1981-12-01 | E. I. Du Pont De Nemours And Company | Electrocoating with water-borne coating composition made from epoxy resin, polymeric acid and tertiary amine |
| US3945961A (en) | 1973-11-21 | 1976-03-23 | American Cyanamid Company | Novel cross-linking agents and their use in electrophoretic coating composition |
| US3943187A (en) | 1973-12-12 | 1976-03-09 | E. I. Du Pont De Nemours And Company | Ductile coating composition of an acrylic polymer having reactive sites and an epoxy resin |
| US4021396A (en) | 1973-12-12 | 1977-05-03 | E. I. Du Pont De Nemours And Company | Aqueous coating composition of an acrylic polymer having reactive sites and an epoxy resin |
| US3997694A (en) | 1973-12-12 | 1976-12-14 | E. I. Du Pont De Nemours And Company | Container coated with a ductile coating of an acrylic polymer having reactive sites and an epoxy resin |
| US4198330A (en) † | 1974-04-19 | 1980-04-15 | American Cyanamid Company | Polyurethane latices modified by a vinyl polymer |
| US4033920A (en) | 1974-06-14 | 1977-07-05 | Kansai Paint Company, Ltd. | Process for producing unsaturated resin emulsion |
| US4076676A (en) | 1975-02-04 | 1978-02-28 | E. I. Du Pont De Nemours And Company | Aqueous coating composition from aminoplast and reaction product of tertiary amine and polymer containing terminal epoxy groups |
| US4105811A (en) | 1975-02-07 | 1978-08-08 | Polygulf Associates | Method of protectively coating metallic aluminum containing substrate |
| JPS51146534A (en) * | 1975-06-12 | 1976-12-16 | Asahi Chem Ind Co Ltd | Powder coating composition |
| US4048141A (en) | 1975-11-06 | 1977-09-13 | The Dow Chemical Company | Latent catalysts for promoting reaction of epoxides with phenols and/or carboxylic acids |
| US4028294A (en) | 1975-11-10 | 1977-06-07 | Mobil Oil Corporation | Epoxy modified acrylic latices and method of producing same |
| US4064087A (en) | 1976-01-05 | 1977-12-20 | Ppg Industries, Inc. | Method for preparing polymers in aqueous medium |
| GB1574721A (en) | 1976-03-03 | 1980-09-10 | Canadian Ind | Process for preparing aqueous copolymer emulsions |
| JPS52108488A (en) | 1976-03-08 | 1977-09-10 | Kao Corp | Preparation of liquid resin dispersions |
| US4122052A (en) | 1976-03-09 | 1978-10-24 | Kansai Paint Company, Limited | Emulsion composition |
| US4148670A (en) | 1976-04-05 | 1979-04-10 | Amchem Products, Inc. | Coating solution for metal surface |
| US4308185A (en) | 1976-05-11 | 1981-12-29 | Scm Corporation | Graft polymer compositions of terminated epoxy resin, processes for making and using same, and substrates coated therewith |
| US4212781A (en) * | 1977-04-18 | 1980-07-15 | Scm Corporation | Modified epoxy resins, processes for making and using same and substrates coated therewith |
| US4129712A (en) | 1976-08-17 | 1978-12-12 | M&T Chemicals Inc. | Interpolymers, method of preparing the same and emulsions thereof, and metal cans coated with the interpolymers |
| CA1102465A (en) | 1976-08-26 | 1981-06-02 | Andreas Lindert | Protective coating for metal articles |
| JPS567367Y2 (en) | 1976-09-09 | 1981-02-18 | ||
| JPS5339387A (en) * | 1976-09-24 | 1978-04-11 | Nippon Jiyunyaku Kk | Method of making aquaous emulsion polymer |
| JPS5397083U (en) | 1977-01-11 | 1978-08-07 | ||
| JPS5397083A (en) | 1977-02-03 | 1978-08-24 | Nippon Paint Co Ltd | Production of aqueous resin dispersion for coating |
| US4136075A (en) | 1977-04-27 | 1979-01-23 | A. E. Staley Manufacturing Company | Acrylic copolymer coatings |
| US4144155A (en) | 1977-08-15 | 1979-03-13 | Japan Atomic Energy Research Institute | Radiation process for producing a reactive aqueous emulsion |
| US4151143A (en) | 1977-08-19 | 1979-04-24 | American Cyanamid Company | Surfactant-free polymer emulsion coating composition and method for preparing same |
| US4442246A (en) | 1978-06-12 | 1984-04-10 | Scm Corporation | Aqueous coating composition comprising self-emulsifiable ester of epoxy and acid containing addition polymer |
| US4482673A (en) | 1978-06-12 | 1984-11-13 | Scm Corporation | Aqueous coating composition comprising self-emulsifiable ester of epoxy and acid containing addition polymer and method of its preparation |
| US4191596A (en) | 1978-09-06 | 1980-03-04 | Union Carbide Corporation | Method and compositions for coating aluminum |
| US4212776A (en) | 1978-10-16 | 1980-07-15 | Mobil Oil Corporation | Self crosslinking water dispersible epoxy ester-acrylate polymers |
| NZ191816A (en) | 1978-10-24 | 1981-03-16 | Canadian Ind | Preparation of aqueous copolymer emulsion |
| US4174333A (en) | 1978-11-02 | 1979-11-13 | Ppg Industries, Inc. | Carboxylated amide polymers and coating compositions containing same |
| DE2850872A1 (en) | 1978-11-24 | 1980-06-04 | Bayer Ag | WATER-DUMBABLE VARNISH BINDERS FOR CHEMICAL-RESISTANT COATINGS |
| US4289674A (en) | 1978-11-29 | 1981-09-15 | Ppg Industries, Inc. | Base-solubilized acrylic polymers and aqueous resinous dispersions of acrylic polymers and epoxy resins |
| US4247659A (en) | 1979-03-09 | 1981-01-27 | Desoto, Inc. | Epoxy ester copolymer soluble in water with the aid of an amine |
| US4296011A (en) | 1979-03-09 | 1981-10-20 | Desoto, Inc. | Epoxy ester copolymer aqueous dispersions |
| US4285847A (en) | 1979-04-11 | 1981-08-25 | Scm Corporation | Polymerization process and product |
| US4304701A (en) | 1979-06-01 | 1981-12-08 | Ppg Industries, Inc. | Aqueous acrylic polymer dispersions |
| US4413015A (en) | 1979-06-21 | 1983-11-01 | Mobil Oil Corporation | Storage stable water-dilutable acid adducted epoxy based coating for metal food contact surfaces |
| US4329266A (en) | 1979-09-29 | 1982-05-11 | Kansai Paint Co., Ltd. | Water-dispersed coating composition comprising an acrylic graft polymer containing carboxyl groups, hydroxyl and/or amide groups |
| US4294737A (en) | 1979-12-28 | 1981-10-13 | Desoto, Inc. | Water soluble epoxy ester copolymers for interior can use |
| US4289811A (en) | 1980-02-21 | 1981-09-15 | Celanese Corporation | Stable aqueous dispersions of mixed resins and process for use as coating compositions |
| US4404336A (en) | 1980-05-27 | 1983-09-13 | Desoto, Inc. | Water soluble epoxy ester copolymers for interior can use |
| JPS6039283B2 (en) | 1980-06-12 | 1985-09-05 | 日立化成工業株式会社 | Method for producing polybutadiene-modified unsaturated polyester |
| US4347333A (en) | 1980-06-16 | 1982-08-31 | S. C. Johnson & Son, Inc. | Emulsion coating composition containing silicone and acrylic polymer |
| US4337185A (en) | 1980-06-23 | 1982-06-29 | The Dow Chemical Company | Process for making cationic structured particle latexes using reactive polymeric surfactants |
| US4480058A (en) | 1980-12-04 | 1984-10-30 | Scm Corporation | Aqueous epoxy ester emulsions |
| US4443568A (en) | 1981-10-26 | 1984-04-17 | Scm Corporation | Polymerization process and product |
| US4423165A (en) | 1982-04-16 | 1983-12-27 | E. I. Du Pont De Nemours And Company | Water-borne coating composition made from epoxy resin, first polymeric acid, tertiary amine and second polymeric acid |
| US4440897A (en) | 1982-06-01 | 1984-04-03 | Ppg Industries, Inc. | Process of making substantially external surfactant-free vinyl polymer emulsion products |
| US4477609A (en) | 1982-07-15 | 1984-10-16 | Inmont Corporation | Hydrosol coating compositions having improved stability |
| FI832883A (en) | 1982-08-12 | 1984-02-13 | Scm Corp | SUSTAINABLE POLYMER BEARING |
| US4499212A (en) | 1982-09-13 | 1985-02-12 | Scm Corporation | Thermosetting acrylic latexes |
| US4425451A (en) | 1982-09-29 | 1984-01-10 | Desoto, Inc. | Epoxy-phosphate aqueous dispersions |
| US4461857A (en) | 1982-09-29 | 1984-07-24 | Desoto, Inc. | Thermosetting aqueous coating compositions containing epoxy-phosphate dispersions |
| US4567246A (en) | 1982-12-16 | 1986-01-28 | Celanese Corporation | Water-swellable crosslinked polymeric microgel particles and aqueous dispersions of organic film-forming resins containing the same |
| US4560714A (en) | 1982-12-16 | 1985-12-24 | Celanese Corporation | Water-swellable crosslinked polymeric microgel particles and aqueous dispersions of organic film-forming resins containing the same |
| US4539348A (en) | 1982-12-16 | 1985-09-03 | Celanese Corporation | Water-swellable crosslinked polymeric microgel particles and aqueous dispersions of organic film-forming resins containing the same |
| US4546014A (en) | 1982-12-16 | 1985-10-08 | Celanese Corporation | Water-swellable crosslinked polymeric microgel particles and aqueous dispersions of organic film-forming resins containing the same |
| US4444923A (en) | 1982-12-30 | 1984-04-24 | Mobil Oil Corporation | Process for preparing aqueous coatings comprising dispersible epoxy resin-acid polymer ester |
| US4446258A (en) | 1982-12-30 | 1984-05-01 | Mobil Oil Corporation | Aqueous coating comprising dispersible epoxy resin-acid polymer ester and diluent polymer, and method of preparation |
| US4476262A (en) | 1982-12-30 | 1984-10-09 | Mobil Oil Corporation | Aqueous coatings comprising ionic polymer ester and diluent polymer with reduced monomer residue and method of preparation |
| US4497946A (en) | 1983-04-13 | 1985-02-05 | Desoto, Inc. | Epoxy phosphate-carboxyl copolymers and aqueous coatings containing the same |
| US4487859A (en) | 1983-11-21 | 1984-12-11 | Scm Corporation | Self-curing water dispersed polymers |
| US4487861A (en) | 1983-11-28 | 1984-12-11 | Scm Corporation | Aqueous polymeric blends |
| US4503173A (en) | 1983-12-19 | 1985-03-05 | Scm Corporation | Self-curing water dispersed polymer blends |
| US4629491A (en) | 1983-12-20 | 1986-12-16 | Allied Corporation | Oxidized sulfur derivatives of diaminophosphinyl compounds as urease inhibitors and urease inhibited urea based fertilizer compositions |
| US4501831A (en) | 1983-12-27 | 1985-02-26 | Chu Shaw C | Aqueous coating compositions and process for their preparation from epoxy acidic acrylic polymer and polyfunctional amine |
| US4507425A (en) | 1984-01-12 | 1985-03-26 | The B. F. Goodrich Company | Process for preparing stable poly(vinylidene halide) latices |
| US4579888A (en) | 1984-04-10 | 1986-04-01 | Toyo Ink Manufacturing Co., Ltd. | Aqueous resin dispersion |
| US4585814A (en) | 1984-05-14 | 1986-04-29 | Desoto, Inc. | Epoxy-maleate-phosphate copolymers |
| US4572610A (en) * | 1984-05-21 | 1986-02-25 | Desoto, Inc. | Optical fiber buffer coated with halogenated dihydroxy-terminated polybutadienes |
| JPS6145059A (en) | 1984-08-06 | 1986-03-04 | 株式会社アイジー技術研究所 | Siding board |
| US5344858A (en) | 1984-09-10 | 1994-09-06 | Ppg Industries, Inc. | Coating from phosphated epoxy and COOH-vinyl addition resins |
| US4638020A (en) | 1984-11-23 | 1987-01-20 | Ppg Industries, Inc. | Aqueous composition comprising a phosphated epoxy and non-self dispersible resin |
| US4644030A (en) * | 1985-02-01 | 1987-02-17 | Witco Corporation | Aqueous polyurethane - polyolefin compositions |
| FR2577935B1 (en) | 1985-02-26 | 1988-06-17 | Saint Gobain Vitrage | POLYURETHANE-BASED ADHESIVE LAYER AND ITS USE IN LAMINATED GLAZING |
| US4623680A (en) | 1985-06-03 | 1986-11-18 | Celanese Corporation | Aqueous epoxy resin dispersions for can coating use |
| DE3544337A1 (en) | 1985-12-14 | 1987-06-19 | Hoechst Ag | POLYMERISAT DISPERSIONS, METHOD FOR THE PRODUCTION THEREOF AND THEIR USE AS BINDERS |
| US4670500A (en) | 1985-12-20 | 1987-06-02 | Pennzoil Company | Surface coating composition |
| DE3601560A1 (en) | 1986-01-21 | 1987-07-23 | Herberts Gmbh | AQUEOUS, HEAT-CURABLE COATING AGENT, THE USE THEREOF AND ITEMS COATED ITEMS |
| US4703071A (en) | 1986-01-27 | 1987-10-27 | The Glidden Company | Stabilized aqueous coatings containing zinc oxide |
| US5051470A (en) | 1986-05-05 | 1991-09-24 | The Glidden Company | Epoxy-acrylic graft polymers |
| US5093392A (en) | 1986-05-05 | 1992-03-03 | The Glidden Company | Epoxy-acrylic graft polymers with aminoplast |
| US4683273A (en) | 1986-05-27 | 1987-07-28 | The Glidden Company | Polyester and epoxy resin coating |
| DE3627860A1 (en) | 1986-08-16 | 1988-02-18 | Basf Lacke & Farben | AQUEOUS COATING AGENT, METHOD FOR THE PRODUCTION THEREOF AND ITS USE FOR COATING CAN |
| AU590104B2 (en) | 1986-08-20 | 1989-10-26 | Glidden Company, The | Thermosetting emulsion polymers |
| US4898911A (en) | 1986-11-20 | 1990-02-06 | Kanegafuchi Kagaku Kogyo Kabushiki | Resin composition |
| US5087645A (en) | 1987-01-27 | 1992-02-11 | Toyo Seikan Kaisha Ltd. | Emulsion type water paint, process for its production, and process for applying same |
| EP0294599A3 (en) * | 1987-06-12 | 1990-05-09 | F. Hoffmann-La Roche Ag | Tricyclic pyridone derivatives |
| MX169357B (en) | 1987-08-13 | 1993-09-30 | Valspar Corp | COATING COMPOSITION IN AQUEOUS COMPOSITION |
| GB8721538D0 (en) | 1987-09-14 | 1987-10-21 | Polyvinyl Chemie Holland Bv | Aqueous dispersions |
| JPH0543830Y2 (en) | 1987-10-28 | 1993-11-05 | ||
| AU2376188A (en) | 1987-10-30 | 1989-05-04 | Ppg Architectural Finishes, Inc. | Alkyd based waterborne deck stain composition |
| US4871810A (en) | 1987-11-13 | 1989-10-03 | E. I. Du Pont De Nemours And Company | Composition comprising melt blended product of thermoplastic resin and two ethylene copolymers containing coreactive groups |
| US5087603A (en) | 1987-12-14 | 1992-02-11 | Nippon Shokubai Co., Ltd. | Heat-sensitive recording paper having an overcoat layer formed from an aqueous crosslinkable resin dispersion |
| US5087842A (en) | 1988-01-06 | 1992-02-11 | Digital Equipment Corporation | Delay circuit having one of a plurality of delay lines which may be selected to provide an operation of a ring oscillator |
| US4894397A (en) | 1988-04-21 | 1990-01-16 | S. C. Johnson & Son, Inc. | Stable emulsion polymers and methods of preparing same |
| FR2638466B1 (en) | 1988-11-03 | 1993-05-07 | Atochem | PROCESS FOR COATING METAL SUBSTRATES USING A POWDER PRIMER AND A DIP APPLIED COATING, POWDER PRIMER COMPOSITIONS USED AND COMPOSITE MATERIALS OBTAINED |
| US4906684A (en) | 1988-12-09 | 1990-03-06 | Rtz Chemicals, Ltd. | Ambient temperature curing polymer compositions containing acetoacetoxyethyl methacrylate, glycidyl methacrylate and a polymerizable acid |
| AU613550B2 (en) | 1989-01-17 | 1991-08-01 | Ppg Industries, Inc. | Use of mixed polymeric surfactants for improved properties |
| US5096992A (en) * | 1989-03-20 | 1992-03-17 | Reeves Brothers, Inc. | Use of modified diisocyanates for preparing linear thermoplastic polyurethane elastomers having improved properties |
| US4948834A (en) | 1989-03-27 | 1990-08-14 | Ppg Industries, Inc. | Vinyl chloride-olefin copolymers having good color stability and flexibility for container coatings |
| EP0394589B1 (en) | 1989-04-25 | 1994-03-09 | Toyo Ink Manufacturing Co., Ltd. | Aqueous coating composition for cans |
| US4946911A (en) | 1989-05-22 | 1990-08-07 | The Dow Chemical Company | Epoxy resin and water borne coatings therefrom |
| AU5383290A (en) * | 1989-06-12 | 1990-12-13 | Ppg Industries, Inc. | Vinyl chloride latex composition having improved stability and durability properties |
| JP2760102B2 (en) * | 1989-08-04 | 1998-05-28 | ジェイエスアール株式会社 | Aqueous copolymer dispersion for metal coating and composition thereof |
| US5116888A (en) | 1989-09-22 | 1992-05-26 | The Glidden Company | Epoxy-acrylic graft copolymers with phosphonium cocatalysts carbon-graft |
| US4963602A (en) | 1989-11-13 | 1990-10-16 | Hi-Tek Polymers, Inc. | Aqueous epoxy resin-acrylic resin coating compositions containing also phenoxy, novolac and resole resin combination |
| US5201436A (en) | 1989-11-13 | 1993-04-13 | The Glidden Company | Can coating of epoxy resin, acrylic copolymer and acrylic surfactant |
| US5196481A (en) | 1989-11-13 | 1993-03-23 | Glidden Company | Acrylic terpolymer surfactant for aqueous dispersed epoxy/acrylic binders |
| US5244960A (en) | 1990-04-02 | 1993-09-14 | Ppg Industries, Inc. | Thermosetting waterborne coating compositions prepared from aqueous emulsion polymers |
| US5166272A (en) | 1990-04-02 | 1992-11-24 | Ppg Industries, Inc. | Emulsion polymers and coating compositions prepared therefrom |
| US5157078A (en) | 1990-05-25 | 1992-10-20 | The Glidden Company | Glycidyl-epoxy-acrylic copolymers |
| US5212241A (en) | 1990-05-25 | 1993-05-18 | The Glidden Company | Glycidyl-epoxy-acrylic copolymers |
| US5082742A (en) | 1990-09-24 | 1992-01-21 | Monsanto Company | Composite of a styrenic polymer adhered to a polyolefin |
| US5043380A (en) | 1990-10-29 | 1991-08-27 | The Dexter Corporation | Metal container coating compositions comprising an acrylic polymer latex, melamine formaldehyde resin and an phenol formaldehyde resin |
| US5166289A (en) | 1990-12-19 | 1992-11-24 | Exxon Chemical Patents Inc. | Thermoset coating composition having improved hardness |
| US5196055A (en) | 1991-01-23 | 1993-03-23 | The Sherwin-Williams Company | VOC compliant pretreatment primers |
| US5500463A (en) | 1991-03-11 | 1996-03-19 | Nippon Paint Co., Ltd. | Aqueous resin composition and method for forming coating film on can body |
| US5177129A (en) | 1991-04-22 | 1993-01-05 | The Valspar Corporation | Interior can coating compositions containing cyclodextrins |
| US5173526A (en) | 1991-04-26 | 1992-12-22 | Air Products And Chemicals, Inc. | Aqueous polyurethane-vinyl polymer dispersions for coating applications |
| DE4113839A1 (en) | 1991-04-27 | 1992-10-29 | Basf Ag | METHOD FOR PRODUCING AN EMULSIFIER-FREE POLYMER DISPERSION |
| JP2808999B2 (en) | 1991-07-11 | 1998-10-08 | 東洋インキ製造株式会社 | Aqueous paint composition |
| DE4131706C2 (en) * | 1991-09-24 | 1994-10-20 | Basf Lacke & Farben | Process for the preparation of an aqueous coating composition, aqueous coating compositions and their use for coating packaging |
| US5264469A (en) | 1991-10-01 | 1993-11-23 | The Valspar Corporation | Aqueous epoxy resin-based coating compositions useful for coating metal containers |
| WO1993008154A1 (en) | 1991-10-18 | 1993-04-29 | Akzo Nobel N.V. | SYNTHESIS OF α,φ-BIS(p-HYDROXYBENZOYLOXY) ALKANE |
| GB9122594D0 (en) * | 1991-10-24 | 1991-12-04 | Ici Plc | Plastisol compositions |
| US5252669A (en) | 1991-12-19 | 1993-10-12 | Ppg Industries, Inc. | Grafted, thermoplastic, waterborne polymer and coating composition |
| AU658736B2 (en) | 1992-05-26 | 1995-04-27 | Nippon Paint Co., Ltd. | Coating composition process for forming a coating composite and a multi-layer coating composite |
| US5252637A (en) | 1992-06-12 | 1993-10-12 | The Glidden Company | Low VOC, high molecular weight epoxy emulsion coatings |
| JP2752862B2 (en) | 1992-08-28 | 1998-05-18 | 日本エヌエスシー株式会社 | One-component aqueous primer composition |
| JP2886007B2 (en) | 1992-10-23 | 1999-04-26 | 鐘淵化学工業株式会社 | Curable composition for paint |
| EP0672084B2 (en) | 1992-11-30 | 2007-03-07 | Bulk Chemicals, Inc. | A method and composition for treating metal surfaces |
| JP3503646B2 (en) | 1993-01-21 | 2004-03-08 | 日本ゼオン株式会社 | Aqueous dispersion of alkali-soluble oligomer and method for producing aqueous polymer dispersion using the same |
| US5308890A (en) | 1993-02-26 | 1994-05-03 | Rohm And Haas Company | Emulsion polymer blend of a multi-stage latex and a non-film forming latex |
| US5387625A (en) | 1993-05-18 | 1995-02-07 | The Dexter Corporation | Waterborne coating composition for metal containers |
| US5506328A (en) | 1993-05-24 | 1996-04-09 | Olin Corporation | Low VOC, moisture curable, two-component coating compositions based on organic polyisocyanates |
| US5290828A (en) | 1993-06-11 | 1994-03-01 | The Glidden Company | Aqueous dispersed acrylic grafted epoxy polyester protective coatings |
| US5726244A (en) * | 1995-08-10 | 1998-03-10 | Basf Corporation | Aqueous coating compositions for environmental etch resistant coatings |
| US5504145A (en) | 1993-08-31 | 1996-04-02 | The Thompson Minwax Company | Water-dispersible poly(urethane-urea) compositions |
| JP3804993B2 (en) * | 1993-09-20 | 2006-08-02 | キヤノン株式会社 | Inkjet recording method, recording liquid and apparatus used for the method |
| US5714539A (en) | 1993-10-04 | 1998-02-03 | Ppg Industries, Inc. | Polymeric surfactant and latex made therefrom |
| JP3503647B2 (en) | 1993-10-19 | 2004-03-08 | 日本ゼオン株式会社 | Aqueous coating composition |
| CA2177594C (en) * | 1993-12-23 | 2005-02-01 | Werner Wilfinger | Process for the preparation of water-dilutable coating binders based on acrylate copolymers, and their use |
| JPH07188353A (en) | 1993-12-27 | 1995-07-25 | Taisei Kako Kk | Normal temperature-curable resin composition based on aqueous acryl-urethane complex and coating agent or ink using the same |
| EP0669382A1 (en) * | 1994-02-28 | 1995-08-30 | Dsm N.V. | Use of a polyester in the preparation of coatings for the interior of can ends |
| US5428084A (en) | 1994-03-29 | 1995-06-27 | Ppg Industries, Inc. | Defunctionalized epoxy resins useful in coatings |
| US5723555A (en) * | 1994-03-29 | 1998-03-03 | Ppg Industries, Inc. | Vinyl resins containing alkoxyacrylamide-polyol reaction products and use in epoxy resin blends for coatings |
| US5464885A (en) | 1994-04-04 | 1995-11-07 | The Glidden Company | Low VOC, aqueous dispersed, epoxy-ester acrylic graft coatings |
| US6140386A (en) | 1994-04-19 | 2000-10-31 | Vanderhoff; John W. | Aqueous coating compositions, methods for making same and uses thereof |
| GB9408748D0 (en) | 1994-05-03 | 1994-06-22 | Zeneca Resins Bv | Production of aqueous polymer compositions |
| GB9408725D0 (en) | 1994-05-03 | 1994-06-22 | Zeneca Resins Bv | Production of aqueous polymer compositions |
| US5508325A (en) | 1995-01-19 | 1996-04-16 | The Glidden Company | Aqueous dispersed, acrylic grafted epoxy microgel protective coatings |
| US5554671A (en) * | 1994-05-25 | 1996-09-10 | The Glidden Company | Low VOC, aqueous dispersed acrylic epoxy microgels |
| US5576361A (en) | 1995-04-20 | 1996-11-19 | The Glidden Company | Zero VOC, aqueous dispersed, polyester modified acrylic-epoxy microgel polymers |
| JP3358873B2 (en) | 1994-06-28 | 2002-12-24 | 日本エヌエスシー株式会社 | Aqueous solution for primer |
| JP3412919B2 (en) | 1994-07-13 | 2003-06-03 | 関西ペイント株式会社 | Water-based paint for outer surface of cans |
| US5521246A (en) | 1994-07-25 | 1996-05-28 | Air Products And Chemicals, Inc. | Low temperature self-crosslinking aqueous urethane-vinyl polymers for coating applications |
| US5612415A (en) | 1994-07-28 | 1997-03-18 | Basf Corporation | Process for coating a substrate with coatings including high Tg acrylic polymers and coated article obtained thereby |
| US5623085A (en) | 1994-09-23 | 1997-04-22 | Rohm And Haas Company | Method for reducing microfoam in a spray-applied waterborne composition |
| US5527840B1 (en) * | 1994-10-04 | 1999-08-10 | Valspar Corp | Aqueous coating composition |
| AUPN023294A0 (en) | 1994-12-22 | 1995-01-27 | Commonwealth Scientific And Industrial Research Organisation | Polymerisable monomers and polymers |
| JP3392247B2 (en) * | 1995-02-02 | 2003-03-31 | 関西ペイント株式会社 | Aqueous coating composition |
| US5869590A (en) | 1995-04-12 | 1999-02-09 | Eastman Chemical Company | Waterborne polymers having pendant allyl groups |
| US5576063A (en) * | 1995-04-21 | 1996-11-19 | Basf Corporation | Multiple layer coating method |
| US5532297A (en) * | 1995-05-26 | 1996-07-02 | The Glidden Company | Divinyl benzene modified, aqueous dispersed, acrylic graft coatings |
| EP0747442B1 (en) † | 1995-06-07 | 2003-09-03 | National Starch and Chemical Investment Holding Corporation | Modified aqueous polyurethane dispersions and methods for making same |
| JP3817761B2 (en) * | 1995-09-18 | 2006-09-06 | 関西ペイント株式会社 | Aqueous coating composition |
| ES2180708T3 (en) | 1995-10-05 | 2003-02-16 | Rohm & Haas | COATING COMPOSITIONS. |
| JP3060091B2 (en) * | 1995-11-20 | 2000-07-04 | 三洋化成工業株式会社 | Overcoat composition for optical disc |
| US5830952A (en) * | 1996-02-22 | 1998-11-03 | The Dexter Corporation | Water-dispersible polymer and coating composition containing the same |
| JPH09227824A (en) | 1996-02-23 | 1997-09-02 | Kansai Paint Co Ltd | Water-based coating composition |
| US5907012A (en) | 1996-04-08 | 1999-05-25 | H.B. Fuller Licensing & Financing, Inc. | Water-based polyurethane-urea laminating adhesives and primers |
| TW377370B (en) | 1996-04-12 | 1999-12-21 | Du Pont | Waterborne fluoropolymer solutions for treating hard surfaces |
| US5672653A (en) * | 1996-05-13 | 1997-09-30 | Elf Atochem North America, Inc. | Anionic waterborne polyurethane dispersions |
| US5686511A (en) | 1996-06-28 | 1997-11-11 | The Valspar Corporation | Esterifying epoxy resin with carboxyl polymer and quenching |
| US5693723A (en) | 1996-07-01 | 1997-12-02 | Basf Corporation | Low voc curable coating composition utilizing carbamate-functional compound |
| DE69636600T2 (en) * | 1996-08-12 | 2007-08-16 | Toyo Ink Mfg. Co., Ltd. | AQUEOUS DISPERSION COMPOSITION |
| DE19633769A1 (en) * | 1996-08-22 | 1998-02-26 | Basf Lacke & Farben | Dispersion for the production of an electrophoretically depositable dip lacquer |
| US5993972A (en) | 1996-08-26 | 1999-11-30 | Tyndale Plains-Hunter, Ltd. | Hydrophilic and hydrophobic polyether polyurethanes and uses therefor |
| WO1998008884A1 (en) | 1996-08-26 | 1998-03-05 | Tyndale Plains-Hunter, Ltd. | Hydrophilic and hydrophobic polyether polyurethanes and uses therefor |
| US6048924A (en) * | 1996-09-10 | 2000-04-11 | Dainippon Ink And Chemicals, Inc. | Aqueous resin composition and aqueous paint |
| US6083585A (en) | 1996-09-23 | 2000-07-04 | Bp Amoco Corporation | Oxygen scavenging condensation copolymers for bottles and packaging articles |
| US5763012A (en) | 1996-10-16 | 1998-06-09 | Basf Aktiengesellschaft | Coating of substrates |
| DE19645663A1 (en) | 1996-11-06 | 1998-05-07 | Bayer Ag | Biodegradable and compostable moldings |
| JP2837142B2 (en) | 1996-11-14 | 1998-12-14 | 大成化工株式会社 | Room temperature curable aqueous resin composition which is an aqueous acrylic-urethane composite and coating agent or ink using the same |
| JP3155480B2 (en) | 1996-12-05 | 2001-04-09 | 日本エヌエスシー株式会社 | One-part cold crosslinking emulsion composition and production method thereof |
| JPH10273621A (en) * | 1997-01-28 | 1998-10-13 | Nkk Corp | Coating material composition for precoated steel sheet, precoated steel sheet and its production |
| US5733970A (en) * | 1997-01-28 | 1998-03-31 | The Glidden Company | Aqueous dispersed, epoxy crosslinked maleated oil microgel polymers for protective coatings |
| GB9708823D0 (en) * | 1997-05-01 | 1997-06-25 | Glidden Co | Aqueous dispersed,latex polymer modified acrylic-epoxy copolymer,microgel polymers for protective coatings |
| US6008273A (en) | 1997-05-09 | 1999-12-28 | The Dexter Corporation | Waterborne coating compositions for metal containers |
| DE69827547T2 (en) | 1997-05-19 | 2005-11-24 | Nisshinbo Industries, Inc. | Powder coating curing agent, powder coating composition containing this curing agent and powder coating film |
| US5976615A (en) | 1997-07-09 | 1999-11-02 | Basf Corporation | Carbamate curable coating composition and method for improved adhesion |
| US5942563A (en) * | 1997-07-18 | 1999-08-24 | The Glidden Company | Aqueous dispersed acrylic-epoxy, branched epoxy protective coatings |
| US6136927A (en) * | 1997-09-24 | 2000-10-24 | Ppg Industries Ohio, Inc. | Phosphatized amine chain-extended epoxy polymeric compounds |
| JP3726452B2 (en) * | 1997-10-28 | 2005-12-14 | 松下電工株式会社 | Thermal barrier for laminate molding |
| US6150429A (en) | 1997-10-30 | 2000-11-21 | Foster-Miller, Inc. | Polyester/vinyl dioxolane based coating compositions |
| BR9814212A (en) | 1997-11-17 | 2000-10-03 | Delta Airlines Inc | Coating process with low content of volatile organics, cyclic prepolymer, and process to manufacture a cyclic polyester prepolymer |
| US6262217B1 (en) | 1997-12-15 | 2001-07-17 | Lord Corporation | Polyurethane compositions |
| US6054208A (en) * | 1998-01-16 | 2000-04-25 | Avery Dennison Corporation | Film forming mixtures, image bearing films and image bearing retroreflective sheeting |
| US6087417A (en) | 1998-01-16 | 2000-07-11 | The Valspar Corporation | Epoxy resin/acid/tertiary amine reaction product with reactive diluent |
| DE19805421C1 (en) | 1998-02-11 | 1999-06-02 | Basf Coatings Ag | Scratch-resistant coating material with high reflow |
| AU2681399A (en) | 1998-02-18 | 1999-09-06 | Ppg Industries Ohio, Inc. | Low temperature cure waterborne coating compositions having improved appearance and humidity resistance and methods for coating substrates |
| FR2776662B1 (en) | 1998-03-25 | 2000-05-05 | Atochem Elf Sa | AQUEOUS POLYURETHANE DISPERSIONS AND THEIR PREPARATION PROCESS |
| GB9807213D0 (en) | 1998-04-04 | 1998-06-03 | Ici Ltd | Aqueous coating composition |
| US6126999A (en) * | 1998-04-13 | 2000-10-03 | Tomasino; Randolf R. | Urethane-acrylic rubber coating and method of forming a vehicle bed liner |
| IT1301690B1 (en) | 1998-06-11 | 2000-07-07 | Sinco Ricerche Spa | MIXTURES OF POLYESTER RESINS WITH HIGH PROPERTIES OF AIGAS BARRIER. |
| US6365673B1 (en) | 1998-06-22 | 2002-04-02 | E. I. Du Pont De Nemours And Company | Low viscosity imine reactive diluents and coating compositions made therefrom |
| JP4381510B2 (en) | 1998-07-01 | 2009-12-09 | 三井化学株式会社 | Aqueous dispersion composition and method for producing the same |
| KR100616797B1 (en) | 1998-08-20 | 2006-08-28 | 아사히 가라스 가부시키가이샤 | Aqueous dispersion of fluorocopolymer and composition for water-based coating material |
| US6730740B1 (en) | 1998-09-25 | 2004-05-04 | Akzo Nobel N.V. | Aqueous cross-linkable polymer composition for use in coatings and process for producing the same |
| US6251973B1 (en) | 1998-11-23 | 2001-06-26 | Akzo Nobel N.V. | Coatings and coating compositions of a reactive group-containing polymer, a hydrazide and a silane |
| US6458468B1 (en) | 1999-01-28 | 2002-10-01 | Eastman Chemical Company | Photocurable coatings for polyester articles |
| US7303797B1 (en) | 1999-02-16 | 2007-12-04 | E.I. Du Pont De Nemours And Company | Gas barrier coating system for polymeric films and rigid containers |
| US6359062B1 (en) * | 1999-03-02 | 2002-03-19 | The Valspar Corporation | Coating compositions |
| DE19912794A1 (en) | 1999-03-16 | 2000-09-21 | Grace W R & Co | BADGE-free can coating |
| US6420040B1 (en) | 1999-04-30 | 2002-07-16 | The Valspar Corporation | Coating composition for metal substrates |
| KR20020019089A (en) | 1999-06-16 | 2002-03-09 | 니혼 야마무라 글라스 가부시키가이샤 | Coating composition |
| JP2003506519A (en) | 1999-07-30 | 2003-02-18 | ピーピージー インダストリーズ オハイオ,インコーポレイティド | Coating compositions with improved scratch resistance, coated substrates and related methods |
| US6300422B1 (en) | 1999-08-25 | 2001-10-09 | Lilly Industries, Inc. | Tris(alkoxycarbonylamino)triazine crosslinked waterborne coating system |
| US6114430A (en) | 1999-08-25 | 2000-09-05 | H.B. Fuller Licensing & Financing, Inc. | Aqueous paint compositions comprising polyether amides |
| EP1081199A1 (en) | 1999-09-06 | 2001-03-07 | Dsm N.V. | Composition |
| US6194513B1 (en) | 1999-09-22 | 2001-02-27 | Olin Corporation | Tintable water-based coating composition |
| US6255366B1 (en) * | 1999-10-01 | 2001-07-03 | Eastman Chemical Company | Sulfopolymers as emulsion stabilizers with improved coagulum level |
| US6706794B1 (en) | 1999-10-04 | 2004-03-16 | Daikin Industries, Ltd. | Powder coating composition having high weather resistance |
| JP2001146572A (en) | 1999-11-19 | 2001-05-29 | Jsr Corp | Coating composition and cured film obtained therefrom |
| GB9927432D0 (en) | 1999-11-20 | 2000-01-19 | Avecia Bv | Aqueous polymer emulsions |
| DE10010405A1 (en) | 2000-03-03 | 2001-09-13 | Basf Coatings Ag | Aqueous (meth)acrylate copolymer dispersion, useful for coating agents, is prepared by (co)/graft polymerization in the presence of polymer dispersion using oil soluble free radical initiator |
| KR20010100849A (en) * | 2000-03-27 | 2001-11-14 | 사사키 요시오 | Aqueous Painting Resin Compositions and Aqueous Paint Compositions |
| CA2411001A1 (en) | 2000-05-25 | 2001-12-06 | Battelle Memorial Institute | Reversible crosslinked polymers, benzyl crosslinkers and method |
| DE10033851A1 (en) | 2000-07-12 | 2002-01-24 | Solutia Austria Gmbh Werndorf | Aqueous polyurethane dispersions containing polybutadiene building blocks |
| US7459167B1 (en) | 2000-07-27 | 2008-12-02 | 3M Innovative Properties Company | Biocidal polyurethane compositions and methods of use |
| US6514619B2 (en) * | 2000-08-30 | 2003-02-04 | Dainippon Ink And Chemicals, Inc. | Aqueous resin composition and coated metal material having cured coating of the same |
| GB0023514D0 (en) | 2000-09-26 | 2000-11-08 | Ici Plc | Aqueous dispersion of addition polymer particles |
| JP2002129095A (en) | 2000-10-20 | 2002-05-09 | Kansai Paint Co Ltd | Water-based film composition |
| US6339125B1 (en) | 2000-10-30 | 2002-01-15 | Crompton Corporation | Cationic polyurethane dispersion and composition containing same |
| US6465559B1 (en) | 2000-10-30 | 2002-10-15 | Crompton Corporation | Sizing composition |
| JP2002138245A (en) * | 2000-11-01 | 2002-05-14 | Kansai Paint Co Ltd | Aqueous coating composition |
| JP2002155234A (en) | 2000-11-24 | 2002-05-28 | Dainippon Ink & Chem Inc | Aqueous resin composition for coating for can manufacturing and metallic can using the same |
| US20020155235A1 (en) | 2001-02-13 | 2002-10-24 | Taylor James W. | Crosslinked coatings |
| US6800717B2 (en) | 2001-05-23 | 2004-10-05 | Air Products And Chemicals, Inc. | Acid catalyzed copolymerization of water and epoxy resin and uses thereof |
| JP3990667B2 (en) | 2001-07-25 | 2007-10-17 | ザ シャーウィン−ウィリアムズ カンパニー | Water repellent coating composition mainly composed of film-forming water |
| US7063895B2 (en) | 2001-08-01 | 2006-06-20 | National Starch And Chemical Investment Holding Corporation | Hydrophobically modified solution polymers and their use in surface protecting formulations |
| US6646041B2 (en) | 2001-09-25 | 2003-11-11 | Imperial Chemical Industries Plc | Aqueous dispersion of addition polymer particles |
| US6683145B2 (en) | 2001-10-26 | 2004-01-27 | Basf Corporation | Hydrophobic lattices and coating compositions containing them |
| US6930143B2 (en) | 2001-11-01 | 2005-08-16 | Arco Chemical Technology, L.P. | Acrylic latex composition |
| US6872789B2 (en) | 2001-11-07 | 2005-03-29 | Akzo Nobel N.V. | Cross-linkable polymer composition |
| WO2003068415A1 (en) | 2002-02-12 | 2003-08-21 | Valspar Sourcing, Inc. | Method of coating a packaging container using crosslinkable polyester-polyurethane |
| US6784248B2 (en) | 2002-02-15 | 2004-08-31 | Ppg Industries Ohio, Inc. | Thermosetting compositions containing alternating copolymers of isobutylene type monomers |
| DE60314734T2 (en) | 2002-03-08 | 2008-03-06 | Valspar Sourcing, Inc., Minneapolis | COATINGS WITH LOW CONTENT OF VOLATILE ORGANIC COMPOUNDS |
| US7001952B2 (en) | 2002-04-19 | 2006-02-21 | Ppg Industries Ohio, Inc. | Coating compositions containing polyurethane dispersions and highly crosslinked polymer particles |
| US6762240B2 (en) | 2002-04-19 | 2004-07-13 | Ppg Industries Ohio, Inc. | Highly crosslinked polymer particles and coating compositions containing the same |
| JP4228585B2 (en) * | 2002-05-01 | 2009-02-25 | 東洋製罐株式会社 | Water-based paint for metal packaging and metal packaging using the paint |
| US6790904B2 (en) | 2002-06-03 | 2004-09-14 | Ppg Industries Ohio, Inc. | Liquid coating of film-forming resin and particles chemically modified to lower surface tension |
| US20030232386A1 (en) | 2002-06-17 | 2003-12-18 | Shah Dinesh O. | Assay conjugate and uses thereof |
| JP4165130B2 (en) | 2002-06-24 | 2008-10-15 | 東洋インキ製造株式会社 | Paint composition |
| CA2493986C (en) | 2002-08-01 | 2014-06-03 | Valspar Sourcing, Inc. | Coating composition for metal substrates |
| GB0218125D0 (en) | 2002-08-05 | 2002-09-11 | Mckeown Colm | A robot harvesting system |
| US7745508B2 (en) | 2002-08-30 | 2010-06-29 | Ppg Industries Ohio, Inc. | Compositions and methods for coating food cans |
| DE10252627A1 (en) | 2002-11-11 | 2004-05-27 | Surface Specialties Germany Gmbh & Co. Kg | Binder and its application as a coating material for the coating of metal containers |
| US7268176B2 (en) | 2002-12-12 | 2007-09-11 | Ppg Industries Ohio, Inc. | Additives for imparting mar and scratch resistance and compositions comprising the same |
| GB2397578B (en) † | 2002-12-17 | 2004-12-08 | Ici Plc | Aqueous dispersions of polyurethane-addition polymer hybrid particles especially for use in coating compositions |
| JP3865693B2 (en) | 2002-12-26 | 2007-01-10 | 日本ペイント株式会社 | Aqueous resin composition for treatment of aluminum-zinc alloy plated steel sheet, coating method, and aluminum-zinc alloy plated steel sheet |
| US6831136B2 (en) | 2003-01-14 | 2004-12-14 | Sartomer Technology Company, Inc. | Amino-terminated polybutadienes |
| US6806314B2 (en) | 2003-02-03 | 2004-10-19 | Ppg Industries Ohio, Inc. | Coating of Hydroxy-functional polymer(s), crosslinker, and 1,3- and 1,4-cyclohexane dimethanols |
| DE10308103A1 (en) | 2003-02-26 | 2004-09-09 | Bayer Ag | Aqueous coating agents based on PUR-PAC hybrid dispersions |
| ES2596077T5 (en) | 2003-04-02 | 2020-06-03 | Swimc Llc | Aqueous dispersions and coatings |
| US20070043156A1 (en) | 2003-05-06 | 2007-02-22 | Nuplex Resines B.V. | Emulsion polymerization process, polymer dispersion and film-forming composition |
| US7728068B2 (en) | 2003-06-12 | 2010-06-01 | Valspar Sourcing, Inc. | Coating compositions containing reactive diluents and methods |
| CN1234060C (en) | 2003-06-12 | 2005-12-28 | 凌阳科技股份有限公司 | A music file production and playback method |
| DE10332723A1 (en) | 2003-07-18 | 2005-02-03 | Degussa Ag | Solvent-containing coating compositions |
| JP4591654B2 (en) | 2003-08-08 | 2010-12-01 | 東洋インキ製造株式会社 | Water-based coating composition and method for producing the coating composition |
| WO2005016999A1 (en) | 2003-08-13 | 2005-02-24 | The Valspar Corporation | Water-based polyurethane - polyethylene compositions |
| JP4344673B2 (en) | 2003-10-15 | 2009-10-14 | フタムラ化学株式会社 | Gas barrier film |
| US9006338B2 (en) | 2003-10-15 | 2015-04-14 | Mitsui Takeda Chemicals, Inc. | Aqueous resin composition having gas barrier properties and laminated film using the same |
| JP4661046B2 (en) | 2003-12-19 | 2011-03-30 | 東洋インキ製造株式会社 | Water-based paint composition containing polymer emulsion |
| US7867555B2 (en) | 2004-02-13 | 2011-01-11 | Valspar Sourcing Inc. | Dispersion-coated powder coloring system |
| US7375174B2 (en) | 2004-03-04 | 2008-05-20 | Basf Corporation | Acrylic composition and a curable coating composition including the same |
| WO2005087875A1 (en) | 2004-03-05 | 2005-09-22 | The Sherwin-Williams Company | High energy curable coatings comprising thermoplastic polymers |
| EP1584638A1 (en) | 2004-03-29 | 2005-10-12 | Imperial Chemical Industries Plc. | Coating composition based on modified epoxy resins |
| US7241830B2 (en) | 2004-05-07 | 2007-07-10 | Ppg Industries Ohio, Inc. | Organic solvent-free film-forming compositions, multi-layer composite coatings, and related methods |
| JP4621460B2 (en) | 2004-09-07 | 2011-01-26 | 出光サートマー株式会社 | Liquid polymer composition |
| EP3733798B1 (en) | 2004-10-20 | 2022-01-26 | Swimc Llc | Coating compositions for cans and methods of coating |
| US7479519B2 (en) | 2004-10-27 | 2009-01-20 | Mallard Creek Polymers, Inc. | Curable urethane polymer adhesives and coatings |
| US7666951B2 (en) | 2005-02-24 | 2010-02-23 | Ppg Industries Ohio, Inc. | Coating compositions that include a polyester polyol, related coated substrates, multi-layer coating and methods |
| EP1858947B1 (en) * | 2005-03-17 | 2009-12-02 | DSM IP Assets B.V. | Aqueous polyurethane compositions |
| AU2006247135A1 (en) | 2005-05-13 | 2006-11-23 | The Regents Of The University Of California | Vertical axis wind turbines |
| US7475786B2 (en) | 2005-08-03 | 2009-01-13 | Ppg Industries Ohio, Inc. | Can coatings, methods for coating can and cans coated thereby |
| WO2007048094A2 (en) | 2005-10-18 | 2007-04-26 | Valspar Sourcing, Inc. | Coating compositions for containers and methods of coating |
| JP2007238698A (en) | 2006-03-07 | 2007-09-20 | Toyo Ink Mfg Co Ltd | Polymer emulsion-containing aqueous coating composition |
| US7858162B2 (en) | 2006-04-06 | 2010-12-28 | Ppg Industries Ohio, Inc. | Food cans coated with a composition comprising an acrylic polymer |
| US7737243B2 (en) | 2006-05-16 | 2010-06-15 | E. I. Du Pont De Nemours And Company | Highly productive coating composition for automotive refinishing |
| US7981515B2 (en) | 2006-05-30 | 2011-07-19 | Ppg Industries Ohio, Inc. | Bis epoxy polyesters and food cans coated with a composition comprising same |
| EP2029681B1 (en) | 2006-06-01 | 2010-11-17 | Cytec Surface Specialties, S.A. | Pressure sensitive adhesives |
| CN101484490B (en) | 2006-07-05 | 2012-10-03 | 威士伯采购公司 | Water-dispersible polyurethane polymer |
| US20090247658A1 (en) | 2006-07-12 | 2009-10-01 | Mitsubishi Chemcial | Process for producing polyurethane and use of polyurethane obtained by the same |
| US8138262B2 (en) | 2006-11-21 | 2012-03-20 | Ppg Industries Ohio, Inc. | Waterborne, radiation-curable coating compositions and related methods |
| JP2008297379A (en) | 2007-05-30 | 2008-12-11 | Toyo Ink Mfg Co Ltd | Aqueous coating material and method for producing the same |
| US20090047531A1 (en) | 2007-08-17 | 2009-02-19 | Ppg Industries Ohio, Inc. | Packages having radiation-curable coatings |
| JP5065190B2 (en) | 2007-09-04 | 2012-10-31 | ローム アンド ハース カンパニー | Low corrosive curable composition |
| US8227027B2 (en) | 2007-12-07 | 2012-07-24 | Presspart Gmbh & Co. Kg | Method for applying a polymer coating to an internal surface of a container |
| US20090220795A1 (en) | 2008-02-29 | 2009-09-03 | Ppg Industries Ohio, Inc. | Composites comprising a multi-layer coating system |
| GB0822674D0 (en) | 2008-12-12 | 2009-01-21 | Nuplex Resins Bv | A crosslinkable polymer binder |
| US20100160561A1 (en) | 2008-12-24 | 2010-06-24 | Ppg Industries Ohio, Inc. | Copolymers of alpha-olefin type monomers and curable film-forming compositions containing them |
| BRPI1005142A2 (en) | 2009-01-20 | 2016-03-22 | Ppg Ind Ohio Inc | transparent and colorless composition absorbent infrared radiation |
| AU2010217673B2 (en) | 2009-02-24 | 2015-11-19 | Akzo Nobel Coatings International B.V. | Latex emulsions and coating compositions formed from latex emulsions |
| CN102482528B (en) | 2009-07-07 | 2014-06-25 | Ak钢铁产权公司 | Polymer coated metallic substrate and method for making |
| US8747979B2 (en) | 2009-07-17 | 2014-06-10 | Valspar Sourcing, Inc. | Coating compositions and articles coated therewith |
| EP2466294B1 (en) | 2009-08-10 | 2014-07-23 | Japan Science And Technology Agency | Method for measuring autophagy |
| US9708504B2 (en) | 2010-04-01 | 2017-07-18 | Ppg Industries Ohio, Inc. | Branched polyester polymers and coatings comprising the same |
| AU2011246885B2 (en) | 2010-04-29 | 2016-02-04 | Agricultural Research Organisation | Novel ruminant feed |
| US9346959B2 (en) | 2010-12-08 | 2016-05-24 | Ppg Industries Ohio, Inc. | Non-aqueous dispersions comprising a nonlinear acrylic stabilizer |
| KR101864807B1 (en) | 2010-12-29 | 2018-06-05 | 아크조노벨코팅스인터내셔널비.브이. | Latex emulsions and coating compositions formed from latex emulsions |
| US20120302690A1 (en) | 2011-05-27 | 2012-11-29 | Ppg Industries Ohio, Inc. | Grafted acrylic comprising water soluble and water insoluble portions and lattices and coatings comprising the same |
| US20130052380A1 (en) | 2011-08-29 | 2013-02-28 | Ppg Industries Ohio, Inc. | Coating compositions for food and beverage containers |
-
2005
- 2005-10-18 EP EP20174384.6A patent/EP3733798B1/en active Active
- 2005-10-18 KR KR1020127030357A patent/KR20130003024A/en not_active Application Discontinuation
- 2005-10-18 AT AT05825629T patent/ATE513025T1/en not_active IP Right Cessation
- 2005-10-18 JP JP2007538035A patent/JP5027666B2/en active Active
- 2005-10-18 PL PL05825629T patent/PL1819789T3/en unknown
- 2005-10-18 EP EP22183883.2A patent/EP4119626A1/en not_active Withdrawn
- 2005-10-18 ES ES11169890.8T patent/ES2565502T3/en active Active
- 2005-10-18 EP EP11169891.6A patent/EP2420542B2/en active Active
- 2005-10-18 EP EP05825629A patent/EP1819789B1/en not_active Revoked
- 2005-10-18 KR KR1020077008267A patent/KR101260526B1/en active IP Right Grant
- 2005-10-18 MX MX2007004463A patent/MX2007004463A/en active IP Right Grant
- 2005-10-18 PL PL15199749.1T patent/PL3037488T3/en unknown
- 2005-10-18 BR BR122016007793-4A patent/BR122016007793B1/en active IP Right Grant
- 2005-10-18 AU AU2005295228A patent/AU2005295228B2/en active Active
- 2005-10-18 BR BRPI0517414A patent/BRPI0517414B1/en active IP Right Grant
- 2005-10-18 EP EP15199749.1A patent/EP3037488B1/en not_active Revoked
- 2005-10-18 ES ES05825629T patent/ES2367516T3/en active Active
- 2005-10-18 EP EP20174398.6A patent/EP3778809B1/en not_active Revoked
- 2005-10-18 EP EP11169890.8A patent/EP2420541B1/en not_active Revoked
- 2005-10-18 WO PCT/US2005/037750 patent/WO2006045017A1/en active Application Filing
- 2005-10-18 ES ES11169891.6T patent/ES2488391T5/en active Active
- 2005-10-18 CA CA2579232A patent/CA2579232C/en active Active
- 2005-10-18 CN CN2005800349149A patent/CN101040016B/en active Active
- 2005-10-18 US US11/253,161 patent/US7592047B2/en active Active
- 2005-10-18 CN CN201110166266.XA patent/CN102390590B/en active Active
-
2007
- 2007-03-16 NO NO20071422A patent/NO20071422L/en not_active Application Discontinuation
-
2009
- 2009-07-17 US US12/505,250 patent/US8173265B2/en active Active
- 2009-07-17 US US12/505,236 patent/US8092876B2/en active Active
- 2009-07-17 US US12/505,255 patent/US8142868B2/en active Active
-
2012
- 2012-03-05 US US13/412,236 patent/US8617663B2/en active Active
-
2013
- 2013-03-13 US US13/801,133 patent/US8835012B2/en active Active
-
2014
- 2014-08-06 US US14/453,280 patent/US9242763B2/en active Active
-
2015
- 2015-10-20 US US14/918,258 patent/US9415900B2/en active Active
-
2016
- 2016-07-08 US US15/205,490 patent/US9862854B2/en active Active
-
2018
- 2018-01-05 US US15/862,967 patent/US10336909B2/en active Active
-
2019
- 2019-05-10 US US16/408,562 patent/US20200032096A1/en active Pending
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3479310A (en) | 1963-09-19 | 1969-11-18 | Bayer Ag | Polyurethane plastics |
| US3991216A (en) * | 1974-05-16 | 1976-11-09 | Ppg Industries, Inc. | Beverage containers coated with a water based liner |
| GB1513866A (en) * | 1974-05-16 | 1978-06-14 | Ppg Industries Inc | Beverage containers |
| US4147679A (en) | 1976-06-02 | 1979-04-03 | Ppg Industries, Inc. | Water-reduced urethane coating compositions |
| GB2152065A (en) * | 1983-12-21 | 1985-07-31 | Inmont Corp | Water dilutable acrylated epoxy-phenolic coating compositions |
| US4600737A (en) * | 1983-12-21 | 1986-07-15 | Inmont Corporation | Water dilutable acrylated epoxy-phenolic coating compositions |
| US4647612A (en) * | 1985-12-30 | 1987-03-03 | Ppg Industries, Inc. | Polymer emulsion products |
| US4692491A (en) | 1985-12-30 | 1987-09-08 | Ppg Industries, Inc. | Polymer emulsion products |
| US4647612B1 (en) * | 1985-12-30 | 1990-10-23 | Ppg Industries Inc | |
| US4692491B1 (en) | 1985-12-30 | 1990-10-30 | Ppg Industries Inc | |
| US5939482A (en) * | 1994-11-29 | 1999-08-17 | Vianova Resins Aktiengesellschaft | Method for the two-stage preparation of aqueous autocrosslinking copolymer dispersions and their use for coating materials |
Cited By (84)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3733798B1 (en) | 2004-10-20 | 2022-01-26 | Swimc Llc | Coating compositions for cans and methods of coating |
| EP3778809B1 (en) | 2004-10-20 | 2023-04-26 | Swimc Llc | Coating compositions for cans and methods of coating |
| EP2420542B2 (en) † | 2004-10-20 | 2018-09-19 | Valspar Sourcing, Inc. | Article and method of coating |
| EP2420541B1 (en) | 2004-10-20 | 2015-12-30 | Valspar Sourcing, Inc. | Methods of coating a food or beverage can |
| EP1937434A2 (en) * | 2005-10-18 | 2008-07-02 | Valspar Sourcing, Inc. | Coating compositions for containers and methods of coating |
| WO2007048094A2 (en) | 2005-10-18 | 2007-04-26 | Valspar Sourcing, Inc. | Coating compositions for containers and methods of coating |
| EP1937434A4 (en) * | 2005-10-18 | 2015-01-14 | Valspar Sourcing Inc | Coating compositions for containers and methods of coating |
| US9394456B2 (en) | 2009-02-24 | 2016-07-19 | Akzo Nobel Coatings International B.V. | Latex emulsions and coating compositions formed from latex emulsions |
| US9029470B2 (en) | 2009-02-24 | 2015-05-12 | Akzo Nobel Coatings International B.V. | Latex emulsions and coating compositions formed from latex emulsions |
| US9133292B2 (en) | 2009-03-05 | 2015-09-15 | Akzo Nobel Coatings International B.V. | Hydroxyl functional oil polyol acrylic graft copolymers |
| US10961344B2 (en) | 2009-04-09 | 2021-03-30 | The Sherwin-Williams Company | Polymer having unsaturated cycloaliphatic functionality and coating compositions therefrom |
| US10563010B2 (en) | 2009-04-09 | 2020-02-18 | The Sherwin-Williams Company | Polymer having unsaturated cycloaliphatic functionality and coating compositions therefrom |
| US8747979B2 (en) | 2009-07-17 | 2014-06-10 | Valspar Sourcing, Inc. | Coating compositions and articles coated therewith |
| WO2011009040A1 (en) * | 2009-07-17 | 2011-01-20 | Valspar Sourcing, Inc. | Coating composition and articles coated therewith |
| CN102712727B (en) * | 2009-07-17 | 2014-08-27 | 威士伯采购公司 | Coating composition and articles coated therewith |
| CN102712727A (en) * | 2009-07-17 | 2012-10-03 | 威士伯采购公司 | Coating composition and articles coated therewith |
| US9061798B2 (en) | 2009-07-17 | 2015-06-23 | Valspar Sourcing, Inc. | Coating composition and articles coated therewith |
| US9011999B2 (en) | 2009-09-18 | 2015-04-21 | Valspar Sourcing, Inc. | Coating composition including an unsaturated polymer |
| US8840966B2 (en) | 2009-09-18 | 2014-09-23 | Valspar Sourcing, Inc. | Polyurethane coating composition |
| US9206332B2 (en) | 2009-09-18 | 2015-12-08 | Valspar Sourcing, Inc. | Coating composition including an unsaturated polymer |
| US9487672B2 (en) | 2009-09-18 | 2016-11-08 | Valspar Sourcing, Inc. | Polyurethane coating composition |
| US8802792B2 (en) | 2010-09-17 | 2014-08-12 | Empire Technology Development Llc | Partially hydrogenated bisphenol-A-based polymers as substitutes for bisphenol-A-based polymers |
| WO2012054691A3 (en) * | 2010-10-20 | 2012-08-09 | Valspar Sourcing, Inc. | Water-based coating system with improved adhesion to a wide range of coated and uncoated substrates including muffler grade stainless steel |
| US9458345B2 (en) | 2010-12-28 | 2016-10-04 | Akzo Nobel Coatings International B.V. | Coating compositions comprising latex emulsions and hydroxyl functional oil polyol graft copolymers |
| EP2658894B1 (en) | 2010-12-29 | 2022-10-19 | Akzo Nobel Coatings International B.V. | Adhesion promoter resin compositions and coating compositions having the adhesion promoter resin compositions |
| WO2012130563A1 (en) | 2011-03-31 | 2012-10-04 | Henkel Ag & Co. Kgaa | Coating formulation for the interior surfaces of cans |
| EP2505625A1 (en) | 2011-03-31 | 2012-10-03 | Henkel AG & Co. KGaA | Varnishing formula for the interior surface of cans |
| US8329846B2 (en) | 2011-05-02 | 2012-12-11 | Empire Technology Development Llc | Bisphenol-A replacement materials |
| US8227561B1 (en) | 2011-05-02 | 2012-07-24 | Empire Technology Development Llc | Bisphenol-A replacement materials |
| WO2012166488A1 (en) * | 2011-05-27 | 2012-12-06 | Ppg Industries Ohio, Inc. | Grafted acrylic comprising water soluble and water insoluble portions and lattices and coatings comprising the same |
| EP2785604B1 (en) | 2011-12-02 | 2017-03-01 | PPG Industries Ohio Inc. | Coating composition for a food or beverage can |
| US10723906B2 (en) | 2011-12-02 | 2020-07-28 | Ppg Industries Ohio, Inc. | Coating composition for a food or beverage can |
| US9260625B2 (en) | 2011-12-21 | 2016-02-16 | Akzo Nobel Coatings International B.V. | Water-based coating compositions |
| US9273226B2 (en) | 2011-12-21 | 2016-03-01 | Akzo Nobel Coatings International B.V. | Solvent-based coating compositions |
| US9655912B2 (en) | 2012-02-16 | 2017-05-23 | Akeso Biomedical, Inc. | Reduction of gastrointestinal tract colonisation by campylobacter |
| US10195172B2 (en) | 2012-02-16 | 2019-02-05 | Akeso Biomedical, Inc. | Reduction of gastrointestinal tract colonisation by Campylobacter |
| DE102012223355A1 (en) | 2012-12-17 | 2014-06-18 | Henkel Ag & Co. Kgaa | Highly crosslinking paint formulation for inside can surfaces |
| US10315216B2 (en) | 2012-12-17 | 2019-06-11 | Henkel Ag & Co. Kgaa | Highly curable coating formulation for the inner surfaces of cans |
| US9889468B2 (en) | 2012-12-17 | 2018-02-13 | Henkel Ag & Co. Kgaa | Method for manufacturing coated can lids |
| US10266719B2 (en) | 2013-05-16 | 2019-04-23 | The Coca-Cola Company | Polymer compositions and coatings for food and beverage packaging |
| WO2014186285A1 (en) * | 2013-05-16 | 2014-11-20 | The Coca-Cola Company | Polymer compositions and coatings for food and beverage packaging |
| US10519337B2 (en) | 2013-07-02 | 2019-12-31 | The Sherwin-Williams Company | Coating compositions for packaging articles such as food and beverage containers |
| EP3998294A1 (en) * | 2013-07-02 | 2022-05-18 | Swimc, LLC | Coating compositions for packaging articles such as food and beverage containers |
| WO2015002961A1 (en) * | 2013-07-02 | 2015-01-08 | Valspar Sourcing, Inc. | Coating compositions for packaging articles such as food and beverage containers |
| EP3016988B1 (en) | 2013-07-02 | 2022-03-23 | Swimc Llc | Coating compositions for packaging articles such as food and beverage containers |
| US10829646B2 (en) | 2013-07-02 | 2020-11-10 | Valspar Sourcing, Inc. | Coating compositions for packaging articles such as food and beverage containers |
| EP3016868B1 (en) | 2013-07-02 | 2023-09-27 | Swimc Llc | Coating compositions for packaging articles such as food and beverage containers |
| US10351714B2 (en) | 2013-07-02 | 2019-07-16 | Swimc Llc | Coating compositions for packaging articles such as food and beverage containers |
| US11352520B2 (en) | 2013-07-02 | 2022-06-07 | The Sherwin-Williams Company | Coating compositions for packaging articles such as food and beverage containers |
| EP3019567B1 (en) | 2013-07-11 | 2017-05-24 | PPG Industries Ohio, Inc. | Coating compositions with improved adhesion to containers |
| WO2015006522A1 (en) * | 2013-07-11 | 2015-01-15 | Ppg Industries Ohio, Inc. | Coating compositions with improved adhesion to containers |
| RU2631304C2 (en) * | 2013-07-11 | 2017-09-20 | Ппг Индастриз Огайо, Инк. | Compositions of coatings with improved adhesion with containers |
| US10526277B2 (en) | 2013-10-17 | 2020-01-07 | Swimc Llc | Food or beverage containers coated with polymers of di(amido(alkyl)phenol) compounds |
| US10793742B2 (en) | 2014-05-19 | 2020-10-06 | Valspar Sourcing, Inc. | Polyethers containing non-bisphenolic cyclic groups |
| US11725067B2 (en) | 2014-12-24 | 2023-08-15 | Swimc Llc | Styrene-free coating compositions for packaging articles such as food and beverage containers |
| US10800941B2 (en) | 2014-12-24 | 2020-10-13 | Valspar Sourcing, Inc. | Coating compositions for packaging articles such as food and beverage containers |
| US11332636B2 (en) | 2014-12-24 | 2022-05-17 | Swimc Llc | Coating compositions for packaging articles such as food and beverage containers |
| US10968288B2 (en) | 2014-12-24 | 2021-04-06 | Swimc Llc | Styrene-free coating compositions for packaging articles such as food and beverage containers |
| US11981822B2 (en) | 2014-12-24 | 2024-05-14 | Swimc Llc | Crosslinked coating compositions for packaging articles such as food and beverage containers |
| EP3303490B1 (en) | 2015-05-29 | 2019-09-11 | PPG Industries Ohio, Inc. | Packaging coated with an emulsion polymerized latex polymer |
| US11091656B2 (en) | 2015-05-29 | 2021-08-17 | Ppg Industries Ohio, Inc. | Packaging coated with an emulsion polymerized latex polymer |
| WO2016196174A1 (en) * | 2015-05-29 | 2016-12-08 | Ppg Industries Ohio, Inc. | Packaging coated with an emulsion polymerized latex polymer |
| WO2017106626A1 (en) * | 2015-12-16 | 2017-06-22 | Commonwealth Scientific And Industrial Research Organisation | Terpene derived coating composition |
| JP2019518091A (en) * | 2016-04-15 | 2019-06-27 | エスダブリューアイエムシー・エルエルシー | Styrene-free copolymers, and coating compositions containing such copolymers |
| JP7110111B2 (en) | 2016-04-15 | 2022-08-01 | エスダブリューアイエムシー・エルエルシー | Styrene-free copolymers and coating compositions containing such copolymers |
| JP2022101555A (en) * | 2016-04-15 | 2022-07-06 | エスダブリューアイエムシー・エルエルシー | Copolymer containing no styrene, and coating composition containing the copolymer |
| JP7410997B2 (en) | 2016-04-15 | 2024-01-10 | エスダブリューアイエムシー・エルエルシー | Styrene-free copolymers and coating compositions containing such copolymers |
| US11623792B2 (en) | 2016-05-20 | 2023-04-11 | Constellium Neuf-Brisach | Metalloplastic strip for rigid food packaging and manufacturing method |
| WO2017198936A1 (en) * | 2016-05-20 | 2017-11-23 | Constellium Neuf-Brisach | Metalloplastic strip for rigid food packaging, and manufacturing process |
| FR3051395A1 (en) * | 2016-05-20 | 2017-11-24 | Constellium Neuf-Brisach | METALLOPLASTIC TAPE FOR RIGID FOOD PACKAGING AND METHOD OF MANUFACTURE |
| CN109219641A (en) * | 2016-05-26 | 2019-01-15 | Ppg工业俄亥俄公司 | The packaging of latex polymer coated with emulsion polymerization |
| CN109219641B (en) * | 2016-05-26 | 2021-06-29 | Ppg工业俄亥俄公司 | Package coated with emulsion polymerized latex polymer |
| WO2017205110A1 (en) * | 2016-05-26 | 2017-11-30 | Ppg Industries Ohio, Inc. | Packaging coated with an emulsion polymerized latex polymer |
| WO2018013766A1 (en) * | 2016-07-15 | 2018-01-18 | Valspar Sourcing, Inc. | Latex coating composition having reduced flavor scalping properties |
| EP4086314A1 (en) * | 2016-07-15 | 2022-11-09 | Swimc Llc | Latex coating composition having reduced flavor scalping properties |
| EP3484964B1 (en) | 2016-07-15 | 2022-04-13 | Swimc Llc | Latex coating composition having reduced flavor scalping properties |
| US11602768B2 (en) | 2016-10-19 | 2023-03-14 | Swimc, Llc | Acrylic polymers and compositions containing such polymers |
| US11717852B2 (en) | 2016-10-19 | 2023-08-08 | Swimc Llc | Alkali-soluble resin additives and coating compositions including such additives |
| US11059989B2 (en) | 2017-06-30 | 2021-07-13 | Valspar Sourcing, Inc. | Crosslinked coating compositions for packaging articles such as food and beverage containers |
| WO2019118697A1 (en) * | 2017-12-15 | 2019-06-20 | Ppg Industries Ohio, Inc. | A coating composition |
| US11787968B2 (en) | 2017-12-15 | 2023-10-17 | Ppg Industries Ohio, Inc. | Coating composition |
| EP3498738A1 (en) * | 2017-12-15 | 2019-06-19 | PPG Industries Ohio, Inc. | A coating composition |
| CN108794703A (en) * | 2018-06-27 | 2018-11-13 | 常州光辉化工有限公司 | A kind of preparation method of the polyacrylate anti-corrosion lotion with excellent salt spray resistance function |
| CN108794703B (en) * | 2018-06-27 | 2020-12-18 | 常州光辉化工有限公司 | Preparation method of polyacrylate anticorrosive emulsion with excellent salt mist resistance function |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7592047B2 (en) | Coating compositions for cans and methods of coating | |
| WO2011009024A1 (en) | Coating compositions for cans and methods of coating | |
| US7189787B2 (en) | Aqueous dispersions and coatings | |
| AU2012202815B2 (en) | Coating compositions for cans and methods of coating |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV LY MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2005295228 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2579232 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2005295228 Country of ref document: AU Date of ref document: 20051018 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005295228 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200580034914.9 Country of ref document: CN Ref document number: 1020077008267 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2007/004463 Country of ref document: MX Ref document number: 2007538035 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005825629 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005825629 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0517414 Country of ref document: BR |